Область техники, к которой относится изобретение
Настоящее изобретение относится к новому циклическому пептидному соединению или его соли, обладающему ингибирующей активностью против репликации РНК репликона вируса гепатита С (в дальнейшем упоминается как HCV). В частности, настоящее изобретение относится к новому пептидному соединению или его соли, к способу его получения, к фармацевтической композиции, содержащей новое циклическое пептидное соединение или его соль, и к способу профилактического и/или терапевтического лечения гепатита С у человека или животного.
Предпосылки создания изобретения
Приблизительно подсчитанное количество носителей HCV составляет примерно 170 млн во всем мире (примерно 3%) и примерно 1,5 млн в Японии. Даже в случае комбинированной терапии с использованием интерферона (в дальнейшем упоминается как IFN) и рибавирина (виразол, Virazole), доступной как первый вариант лечения, ее эффективность составляет 40% для всех типов HCV. Кроме того, ее эффективность составляет лишь 15-20% для вируса, резистентного к IFN (генотип 1b), особенно распространенного в Японии. С другой стороны, комбинированная терапия часто имеет побочные эффекты. Таким образом, трудно избавиться от вируса полностью путем использования доступных в настоящее время способов лечения. В случае, когда хронический гепатит не может быть вылечен полностью, гепатит гарантированно развивается до циррозного гепатита (30%) или гепатоцеллюлярной карциномы (25%). В Европе и в Соединенных Штатах Америки гепатит С является главным критерием для трансплантации печени. Однако повторное развитие HCV часто встречается даже в трансплантированной печени. По этим причинам в обществе в очень сильной степени существует необходимость в новых средствах, улучшенных как в отношении эффективности, так и в отношении безопасности, обладающих высокими антивирусными эффектами и способных ингибировать гепатит С.
Новое циклическое пептидное соединение или его соль, обладающее ингибирующей активностью к РНК-репликации HCV, описано в Международной заявке WO-2007/049803, которая опубликована после даты приоритета настоящей заявки.
HCV представляет собой вирус, имеющий РНК "плюс-цепь" в качестве гена, и классифицирован в Flaviviridae в соответствии с анализом последовательности оснований гена. Согласно Fields Virology, четвертое изд., D. Knipe et al ed., Филадельфия, Lippincott Williams & Wilkins, 2001, 1127-1161, несмотря на то, что существование HCV было предвидено в 1970-х годах, открытие HCV было очень затруднительным. HCV в течение многих лет называли вирусом гепатита «не-А не-В». В 1989, в соответствии с Choo Q-L и др., Science, 244, 359-362 (1989), часть гена этого вируса клонировали из сыворотки инфицированного лабораторного животного и его последовательность ДНК идентифицировали и подтвердили, вследствие чего вирус назвали «HCV».
Раскрытие данного изобретения
Циклоспорин А используют в качестве иммунодепрессанта для трансплантации органа. M. Thali и др., Nature, 372, 363-365 (1994) сообщали, что циклоспорин А обладает анти-ВИЧ-активностью путем ингибирования взаимодействия между циклоспорином А и вирусной частицей, образующей белок вируса иммунодефицита человека типа 1 (HIV-1). Кроме того, K. Inoue et al., 6th International Symposium on Hepatitis C and Related Virus, 3-6 июня (2000), Bethesda, MD, USA, сообщали, что циклоспорин А обладает анти-HCV-активностью. Однако сообщения в отношении поддержки этого открытия не были представлены другими группами вплоть до настоящего времени.
M. Berenguer и др., J. Hepatol, 32, 673-684 (2000), сообщали, что клиническое применение циклоспорина А, служащего в качестве иммунодепрессанта, провоцировало HCV многократно у трансплантированных пациентов.
Следовательно, вследствие вышеуказанных причин требуется средство против гепатита С, улучшенное в отношении активности, перехода в кровь, селективности и побочных эффектов, например, по сравнению с циклоспорином А.
Целевое циклическое пептидное соединение согласно настоящему изобретению представляет собой новое соединение и может быть представлено следующей общей формулой (I):
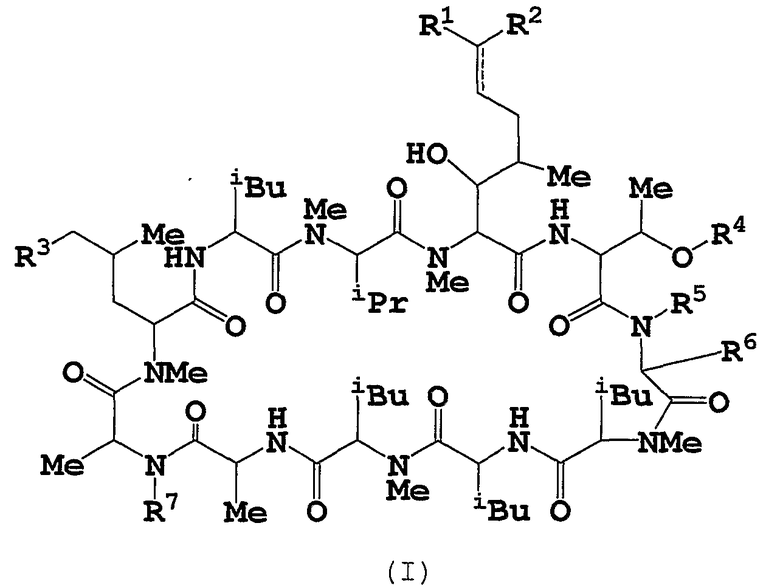
где
R1 и R2, независимо, означают водород, низший алкил, -О-(низший алкил), -NH-(низший алкил), -S-(низший алкил), арил или гетероарил;
R3 означает:
(1) -ОН или SO2Ph;
(2) гетероциклическую группу, которая может иметь один или более подходящий(их) заместитель(ей);
(3) -NR8R9, где R8 и R9, независимо, означают водород, низший алкил, гетероциклическую группу или ацил, каждый из которых может иметь один или более подходящий(их) заместитель(ей); или, альтернативно, R8 и R9 вместе с атомом азота, к которому они присоединены, представляют собой содержащую азот гетероциклическую группу, которая может иметь один или более подходящий(их) заместитель(ей);
(4) -ОС(О)-NR10R11, где R10 и R11, независимо, означают водород, низший алкил, цикло(низший)алкил, арил или гетероциклическую группу, каждый из которых может иметь один или более подходящий(их) заместитель(ей); или, альтернативно, R10 и R11 вместе с атомом азота, к которому они присоединены, представляют собой содержащую азот гетероциклическую группу, которая может иметь один или более подходящий(их) заместитель(ей);
(5) -О-R12, где R12 означает низший алкил или арил, каждый из которых может иметь один или более подходящий(их) заместитель(ей); или
(6) -S-R13, где R13 означает низший алкил, ацил или гетероциклическую группу, каждый из которых может иметь один или более подходящий(их) заместитель(ей);
R4 означает водород или низший алкил;
R5 означает низший алкил;
R6 означает водород, низший алкил или низший алкенил, каждый из которых может иметь один или более подходящий(их) заместитель(ей);
R7 означает водород или низший алкил; и
---- представляет собой одинарную связь или двойную связь;
или его соль.
Предпочтительные воплощения соединения, объекта изобретения, (I) представлены ниже:
1) соединение общей формулы (I),
где
R4 означает водород или метил;
R5 означает метил или этил; и
R7 означает водород, метил или этил;
или его соль;
2) соединение по п.1),
где
R4 означает водород; и
R7 означает водород;
или его соль;
3) соединение по пп.1)-2),
где
R1 означает метил; и
R2 означает водород;
или его соль;
4) соединение по пп.1)-3),
где
часть ---- означает двойную связь;
или его соль;
5) соединение по пп.1)-4),
где
R6 означает водород или низший алкил, который может иметь один или более подходящий(их) заместитель(ей);
или его соль.
Более предпочтительные воплощения соединения (I), объекта изобретения, представлены ниже:
а) соединение общей формулы (I),
где
R1 означает метил;
R2 означает водород;
R4 означает водород;
R5 означает метил или этил;
R7 означает водород; и
часть ---- означает двойную связь;
или его соль;
b) соединение по п.а),
где
R6 означает -CH2OH, -CH2OMe, -CH2OEt, -CH2OC(O)Ме или -CH2Ph;
или его соль;
с) соединение по п.b),
где
R3 означает:
(1) гетероциклическую группу, которая может иметь один или более подходящий(их) заместитель(ей);
(2) -NR8R9, где R8 и R9, независимо, означают водород, или низший алкил, или ацил, каждый из которых может иметь один или более подходящий(их) заместитель(ей);
или, альтернативно, R8 и R9 вместе с атомом азота, к которому они присоединены, представляют собой содержащую азот гетероциклическую группу, которая может иметь один или более подходящий(их) заместитель(ей);
(3) -ОС(О)-NR10R11, где R10 и R11, независимо, означают водород или низший алкил, цикло(низший)алкил, арил или гетероциклическую группу, каждый из которых может иметь один или более подходящий(их) заместитель(ей);
или, альтернативно, R10 и R11 вместе с атомом азота, к которому они присоединены, представляют собой содержащую азот гетероциклическую группу, которая может иметь один или более подходящий(их) заместитель(ей);
или его соль.
Соединение (I) или его соль согласно настоящему изобретению можно получать при использовании способов, как проиллюстрировано на следующих реакционных схемах, способы 1-6.
И исходные соединения или их соли согласно настоящему изобретению можно получать, например, при использовании способов, как проиллюстрировано на следующих реакционных схемах, способы А-Н.
Способ 1
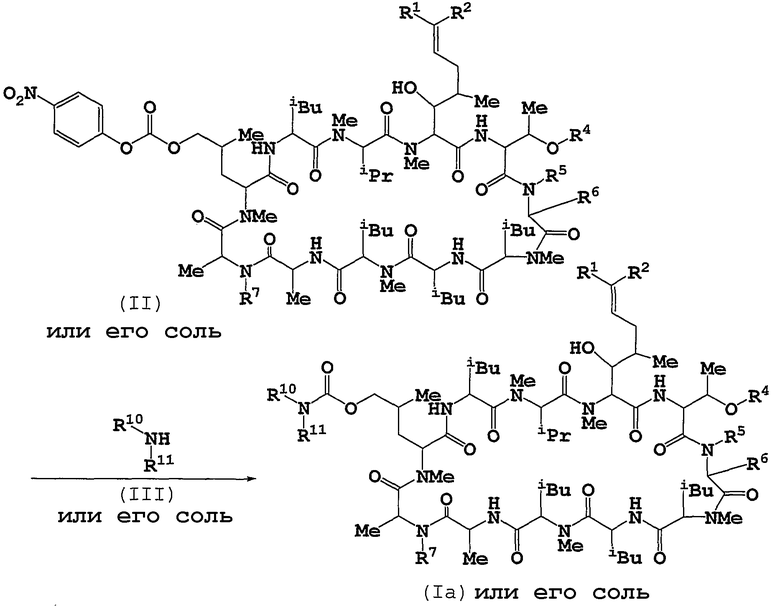
Способ 2
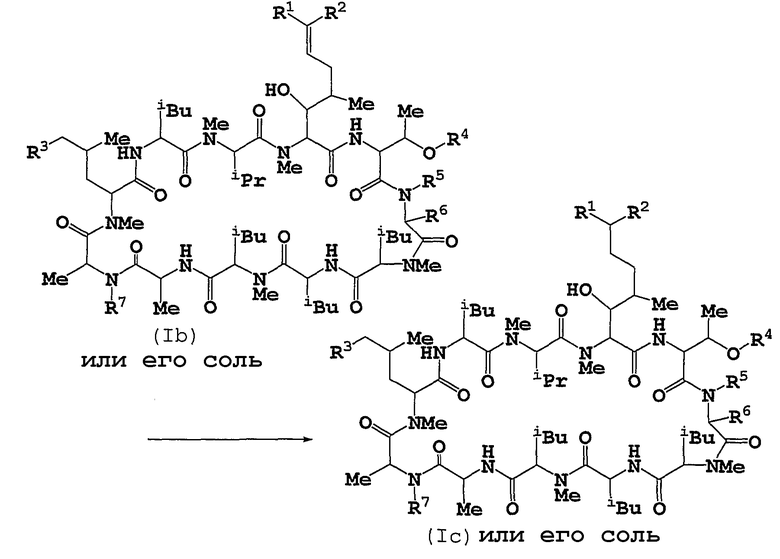
Способ 3
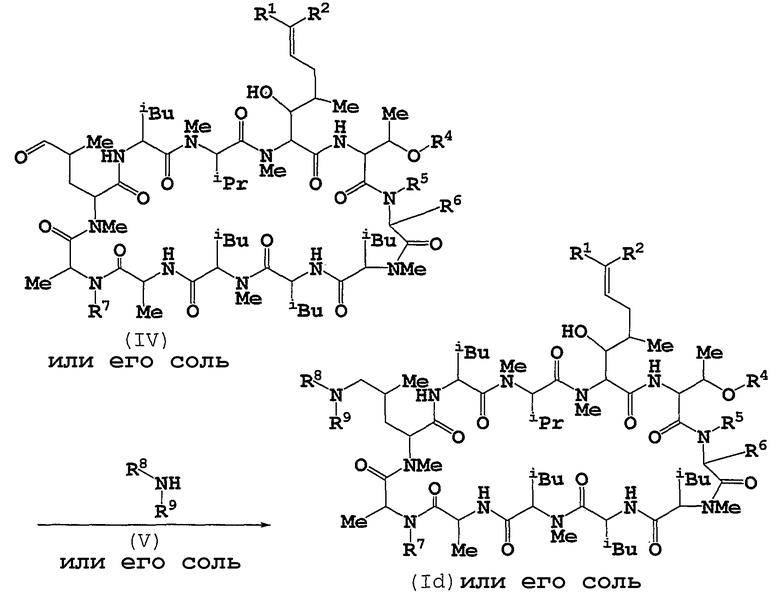
Способ 4
Способ 5
Способ 6
Способ 7
Способ А
Способ В
Способ С
Способ Е
Способ F
Способ G
Способ Н
где
R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11 и R13 имеют значения, как описано выше, и
L представляет собой удаляемую группу;
R6 a имеет значение, такое же, как R6, как описано выше, за исключением водорода;
R7 a имеет значение, такое же, как R7, как описано выше, за исключением водорода;
Р1 означает защитную для гидроксила группу; и
Р2 означает аминозащитную группу.
Способы получения соединений, объекта изобретения, и исходных соединений описаны ниже.
Способ 1
Соединение (Ia) или его соль можно получать путем введения во взаимодействие соединения (II) или его соли с соединением (III) или его солью.
Реакцию обычно осуществляют в стандартном растворителе, таком как тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Эту реакцию (особенно, когда соединение (II) и/или соединение (III) находится в форме соли) обычно осуществляют в присутствии неорганического или органического основания. Подходящим неорганическим основанием может быть щелочной металл [например, натрий или калий], гидроксид щелочного металла [например, гидроксид натрия или гидроксид калия], гидрокарбонат щелочного металла [например, гидрокарбонат натрия или гидрокарбонат калия], карбонат щелочного металла [например, карбонат натрия или карбонат калия], карбонат щелочноземельного металла [например, карбонат кальция или карбонат магния], гидрид щелочного металла [например, гидрид натрия или гидрид калия] или т.п. Подходящим органическим основанием может быть три(низший)алкиламин [например, триэтиламин или N,N-диизопропилэтиламин], алкилмагнийбромид [например, метилмагнийбромид или этилмагнийбромид], алкиллитий [например, метиллитий или бутиллитий], диизопропиламид лития, гексаметилдисилазид лития или т.п.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
Способ 2
Соединение (Ic) или его соль можно получать путем подвергания соединения (Ib) или его соли восстановлению.
Пригодным способом восстановления является каталитическое гидрирование.
Пригодными катализаторами, используемыми при каталитическом гидрировании, являются стандартные катализаторы, такие как катализаторы на основе платины (например, платиновая пластинка, губчатая платина, платиновая чернь, коллоидная платина, оксид платины, платиновая проволока и т.д.), катализаторы на основе палладия (например, губчатый палладий, палладиевая чернь, оксид палладия, палладий-на-угле, гидроксид палладия-на-угле, коллоидный палладий, палладий-на-сульфате бария, палладий-на-карбонате бария и т.д.) или т.п.
Гидрирование обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
Способ 3
Соединение (Id) или его соль можно получать путем подвергания соединения (IV) или его соли реакции восстановительного аминирования с соединением (V) или его солью.
Эту реакцию обычно осуществляют в присутствии восстановителя, такого как триацетоксиборгидрид натрия или т.п.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
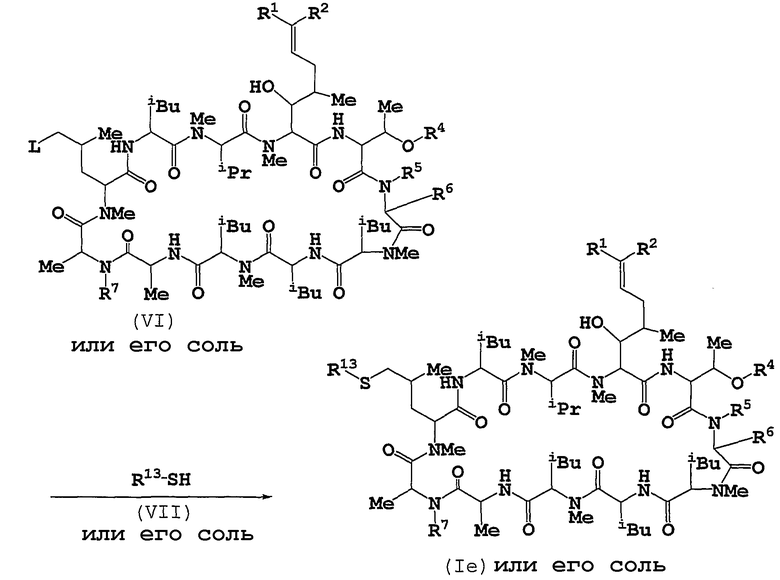
Способ 4
Соединение (Ie) или его соль можно получать путем введения во взаимодействие соединения (VI) или его соли с соединением (VII) или его солью.
L представляет собой удаляемую группу. Примеры удаляемых групп включают галоген, алкансульфонил, необязательно замещенный одним или более атомами галогена, арилсульфонил и т.п.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
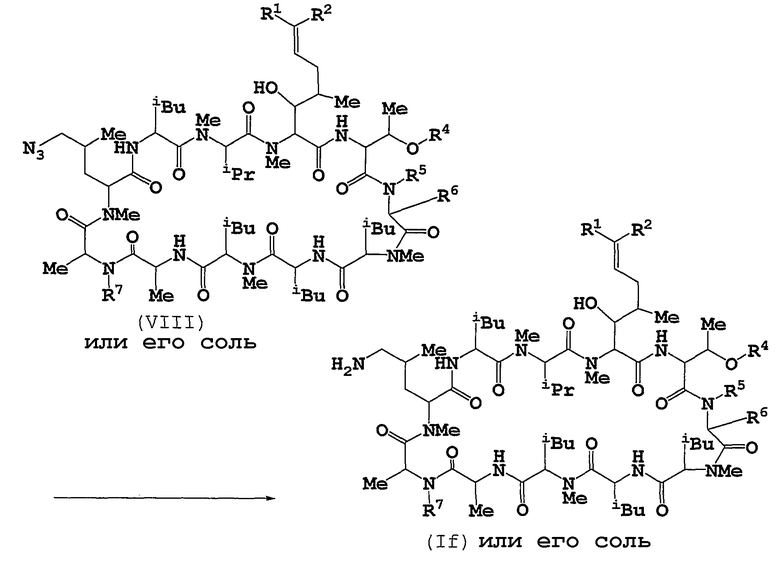
Способ 5
Соединение (If) или его соль можно получать путем подвергания соединения (VIII) или его соли восстановлению.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
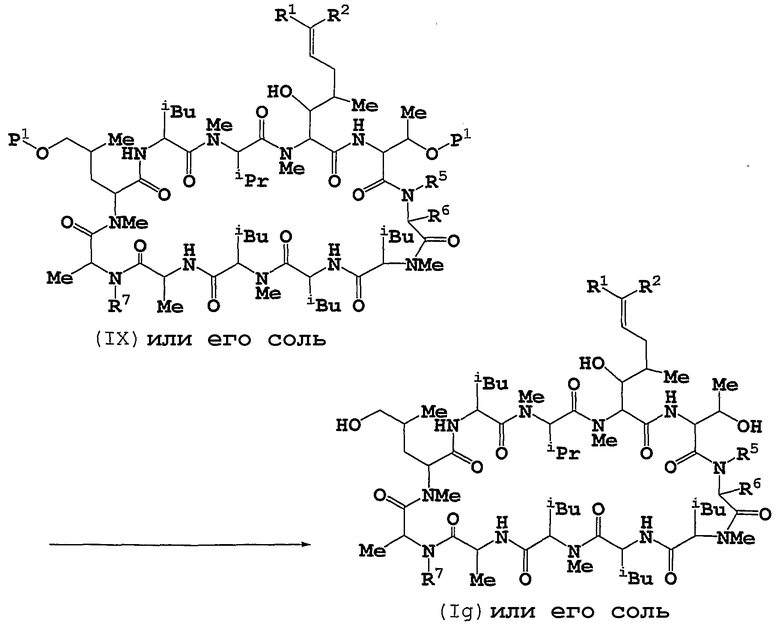
Способ 6
Соединение (Ig) можно получать путем подвергания соединения (IX) снятию защиты.
Эту реакцию осуществляют в соответствии со стандартным способом, таким как гидролиз, восстановление или т.п.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
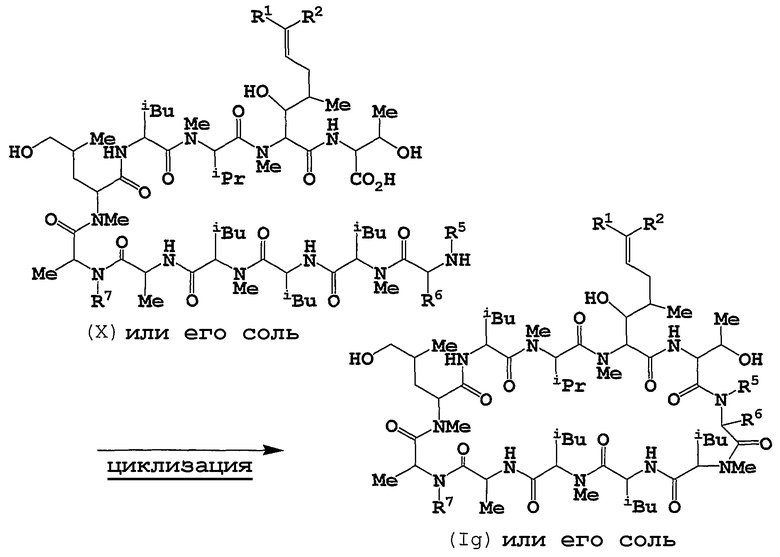
Способ 7
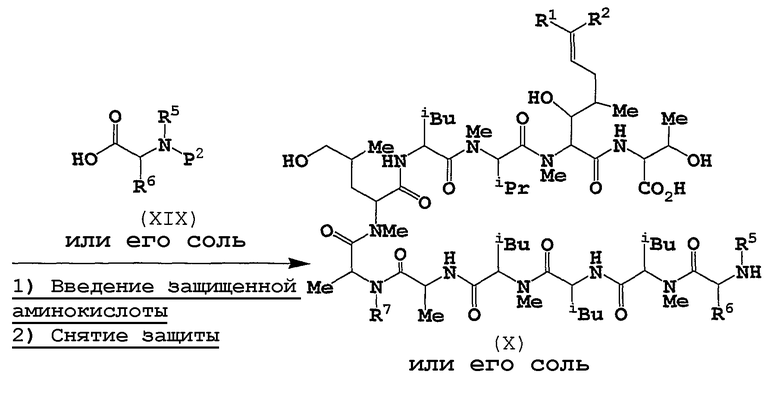
Данную циклизацию осуществляют путем амидирования соединения (Х) или его соли.
Эту реакцию предпочтительно осуществляют в присутствии агента конденсации (включая карбодиимид (например, N,N-диизопропилкарбодиимид, N,N'-дициклогексилкарбодиимид, 1-[3-(диметиламино)пропил]-3-этилкарбодиимид и т.п.), дифенилфосфиназид, дифенилфосфонхлорид или т.п.).
Эту реакцию обычно осуществляют в присутствии добавки, такой как N-гидроксибензотриазол (HOBt), 1-гидрокси-7-азабензотриазол (HOAt), бис(2-оксо-3-оксазолидинил)фосфохлорид и т.п.
Эту реакцию также можно осуществлять в присутствии органического или неорганического основания, такого как бикарбонат щелочного металла, три(низший)алкиламин, пиридин, N-(низший)алкилморфолин, N,N-ди(низший)алкилбензиламин или т.п.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как вода, ацетон, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, N,N-диметилформамид, или в любых других органических растворителях, которые не оказывают вредного влияния на реакцию, или в их смесях.
Температура реакции не является лимитированной и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
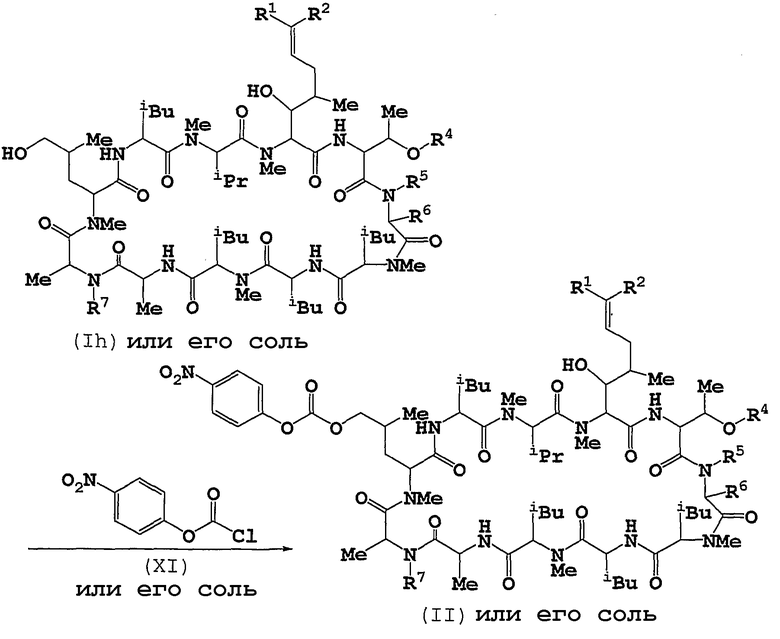
Способ А
Соединение (II) или его соль можно получать путем введения во взаимодействие соединения (Ih) или его соли с соединением (XI) или его солью.
Реакцию обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол и т.п.), ацетон, диоксан, ацетонитрил, хлороформ, метиленхлорид, этиленхлорид, тетрагидрофуран, этилацетат, N,N-диметилформамид, пиридин, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию. Эти стандартные растворители также можно использовать в смеси с водой.
Реакцию также можно осуществлять в присутствии неорганического или органического основания, такого как карбонат щелочного металла (например, карбонат калия и т.д.), бикарбонат щелочного металла, три(низший)алкиламин, пиридин, N-(низший)алкилморфолин, N,N-ди(низший)алкилэтиламин (например, N,N-диизопропилэтиламин и т.д.), N,N-ди(низший)алкилбензиламин или т.п.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
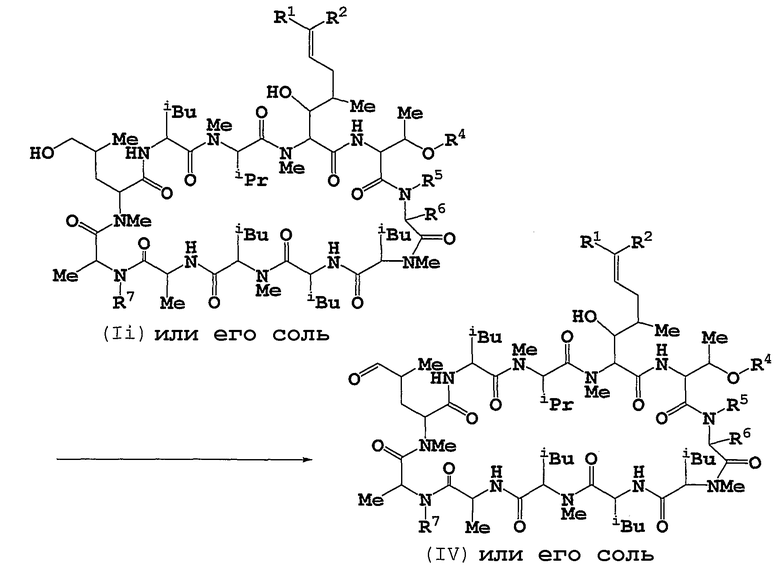
Способ В
Соединение (IV) или его соль можно получать путем подвергания соединения (Ii) окислению.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
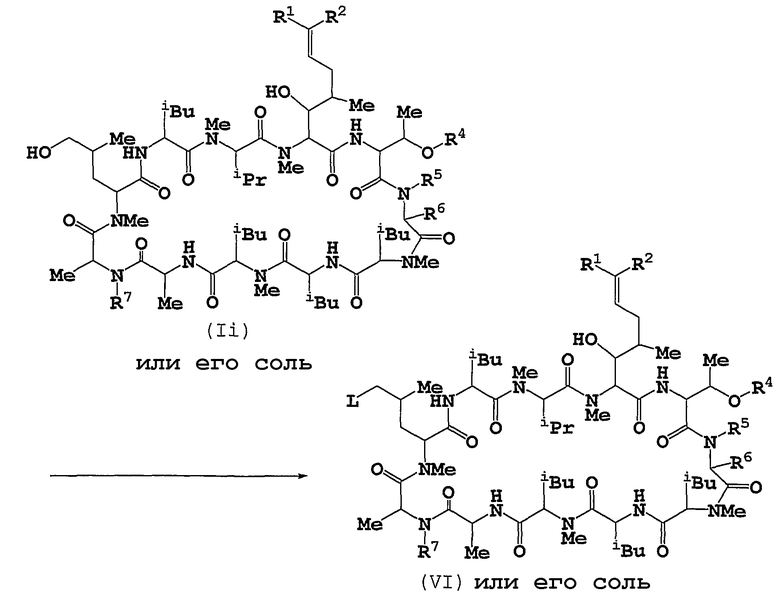
Способ С
Соединение (VI) или его соль можно получать путем подвергания соединения (Ii) или его соли реакции введения удаляемой группы.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
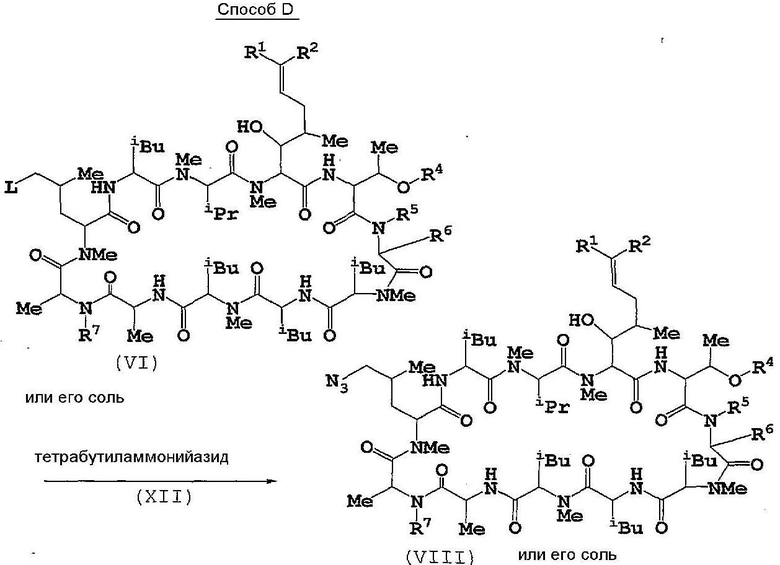
Способ D
Соединение (VIII) или его соль можно получать путем введения во взаимодействие соединения (VI) или его соли с соединением (XII) или его солью.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
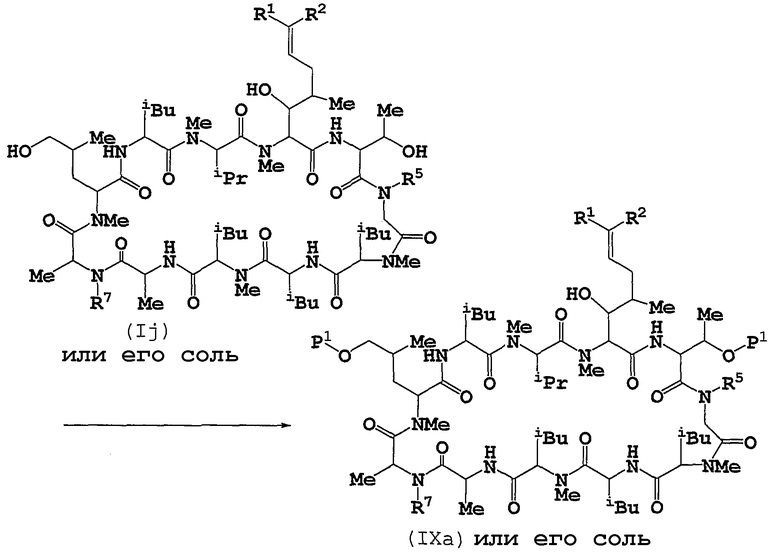
Способ Е
Соединение (IXa) или его соль можно получать путем подвергания соединения (Ij) или его соли реакции защиты гидроксильной группы.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
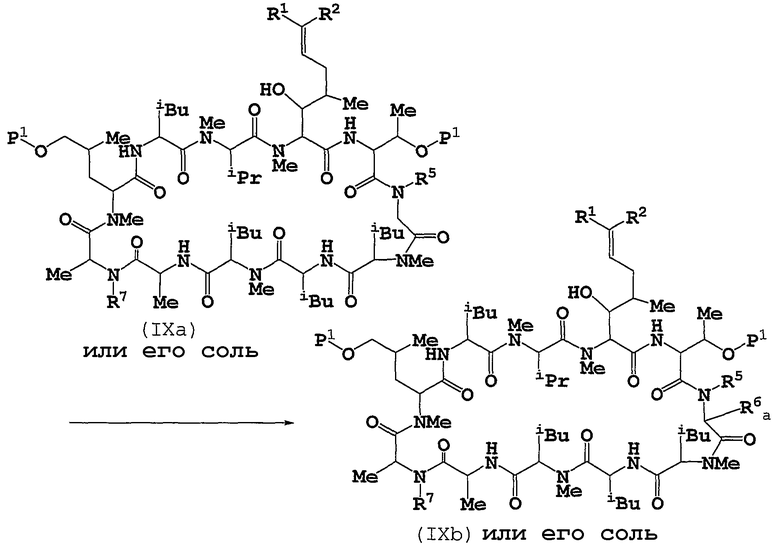
Способ F
Соединение (IXb) или его соль можно получать путем подвергания соединения (IXa) или его соли алкилированию.
Эту реакцию обычно осуществляют в стандартном растворителе, таком как тетрагидрофуран, диоксан, толуол, диэтиловый эфир, диизопропиловый эфир, циклопентилметиловый эфир, или в любом другом органическом растворителе, который не оказывает вредного влияния на взаимодействие, или в их смесях. Основания, используемые в этом способе, представляют собой такие, как диизопропиламид лития, гексаметилдисилазид лития, гексаметилдисилазид натрия, гексаметилдисилазид калия, амид натрия, амид лития, 2,2,4,4-тетраметилпиперидиновая соль лития, н-битуллитий, N-метиланилид лития.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
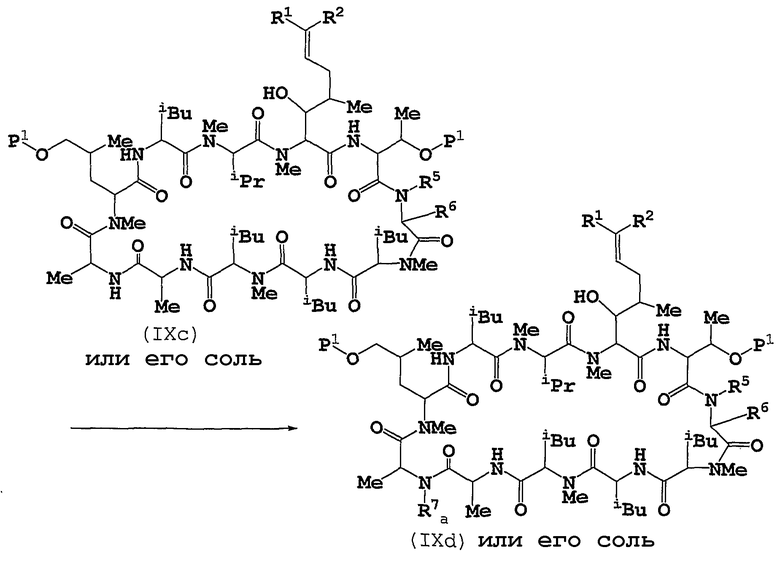
Способ G
Соединение (IXc) или его соль можно получать путем подвергания соединения (IXd) или его соли алкилированию.
Данную реакцию можно осуществлять в растворителе, таком как вода, фосфатный буфер, ацетон, хлороформ, ацетонитрил, нитробензол, метиленхлорид, этиленхлорид, формамид, N,N-диметилформамид, метанол, этанол, втор-бутанол, амиловый спирт, диэтиловый эфир, диоксан, тетрагидрофуран, диметилсульфоксид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, предпочтительно, в растворителях, имеющих сильные полярности. Из растворителей, гидрофильные растворители можно использовать в смеси с водой.
Реакцию предпочтительно проводят в присутствии основания, например неорганического основания, такого как гидроксид щелочного металла, карбонат щелочного металла, бикарбонат щелочного металла, гидрид щелочного металла (например, гидрид натрия и т.д.), органического основания, такого как триалкиламин, и т.п.
Температура реакции не является критической и введение во взаимодействие обычно осуществляют при температуре окружающей среды, при нагреве или при подогреве.
Данную реакцию согласно настоящему изобретению предпочтительно осуществляют в присутствии галогенида щелочного металла (например, иодид натрия, иодид калия и т.д.), тиоцианата щелочного металла (например, тиоцианат натрия, тиоцианат калия и т.д.), ди(низший)алкилазодикарбоксилата (например, диэтилазодикарбоксилат, диизопропилазодикарбоксилат и т.д.) или т.п.
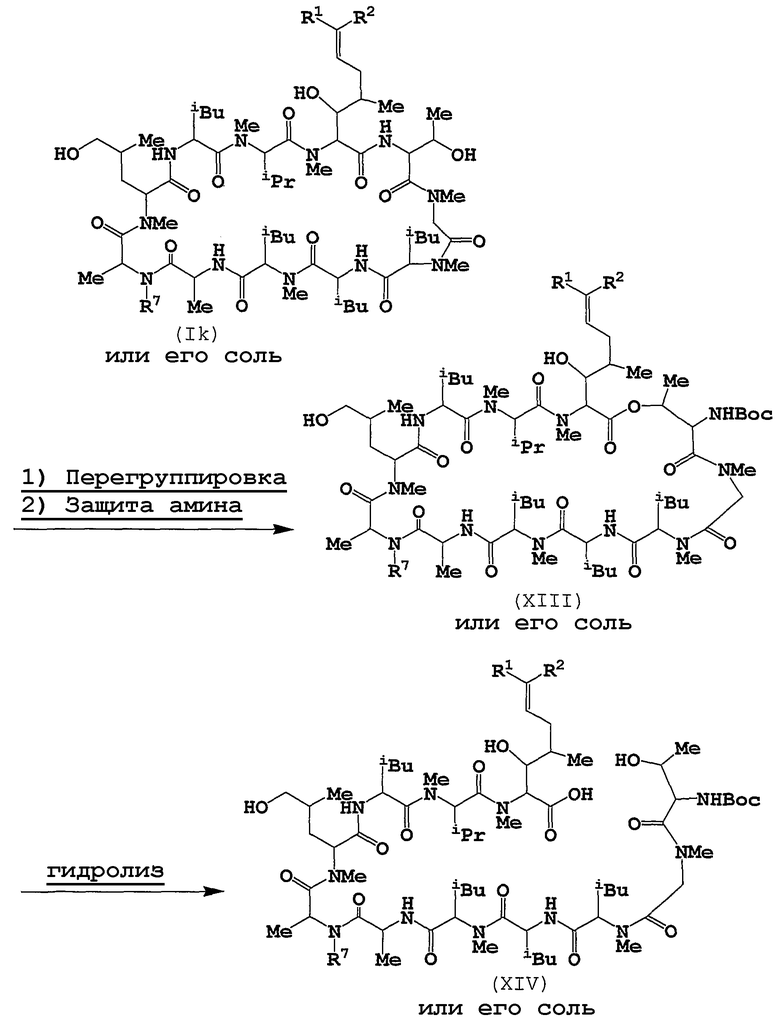
Способ Н
Соединение (Х) или его соль можно получать из соединения (Ik) или его соли при использовании следующих способов:
а) Перегруппировка
Эта реакция представляет собой перегруппировку соединения (Ik).
Данную реакцию обычно осуществляют в присутствии кислоты (такой как трифторуксусная кислота, серная кислота, метансульфоновая кислота или т.п.).
Данную реакцию обычно осуществляют в стандартном растворителе, таком как вода, ацетон, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, N,N-диметилформамид, или в любых других органических растворителях, которые не оказывают вредного влияния на реакцию, или в их смесях.
Температура реакции не является лимитированной и реакцию обычно осуществляют в условиях от охлаждения до нагревания.
Эту реакцию согласно настоящему изобретению, из-за субстрата и благодаря этому, можно осуществлять в мягких условиях, таких как слабая кислота (п-толуолсульфоновая кислота) и при невысокой температуре (от температуры окружающей среды до нагревания), получая соединение, селективно подвергнутое реакции перегруппировки.
b) Аминозащита
Эта реакция представляет собой защиту аминогруппы, которая появляется за счет реакции перегруппировки.
Данную реакцию обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, N,N-диметилформамид, или в любых других органических растворителях, которые не оказывают вредного влияния на реакцию, или в их смесях.
Температура реакции не является лимитированной и реакцию обычно осуществляют в условиях от охлаждения до нагревания.
с) Гидролиз
Гидролиз предпочтительно осуществляют в присутствии основания (включая неорганическое основание и органическое основание, такое как щелочной металл (например, натрий, калий и т.д.), щелочноземельный металл (например, магний, кальций и т.д.), гидроксид или карбонат или бикарбонат щелочного металла или щелочноземельного металла, триалкиламин (например, триметиламин и т.д.), гидразин, пиколин, 1,5-диазабицикло[4.3.0]нон-5-ен, 1,8- диазабицикло[5.4.0]ундец-7-ен, или т.п.), или в присутствии кислоты (включая органическую кислоту (как, например, муравьиная кислота, уксусная кислота, пропановая кислота, трифторуксусная кислота и т.д.), неорганическую кислоту (как, например, бромоводородная кислота, серная кислота, соляная кислота и т.д.) и кислоту Льюиса (например, трибромид бора, хлорид алюминия, трихлорид титана и т.д.)).
Реакцию обычно осуществляют в стандартном растворителе, таком как вода, спирт (например, метанол, этанол, изопропиловый спирт и т.д.), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, N,N-диметилформамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Жидкое основание или кислоту также можно использовать в качестве растворителя.
Температура реакции не является лимитированной и введение во взаимодействие обычно осуществляют в условиях от охлаждения до нагревания.
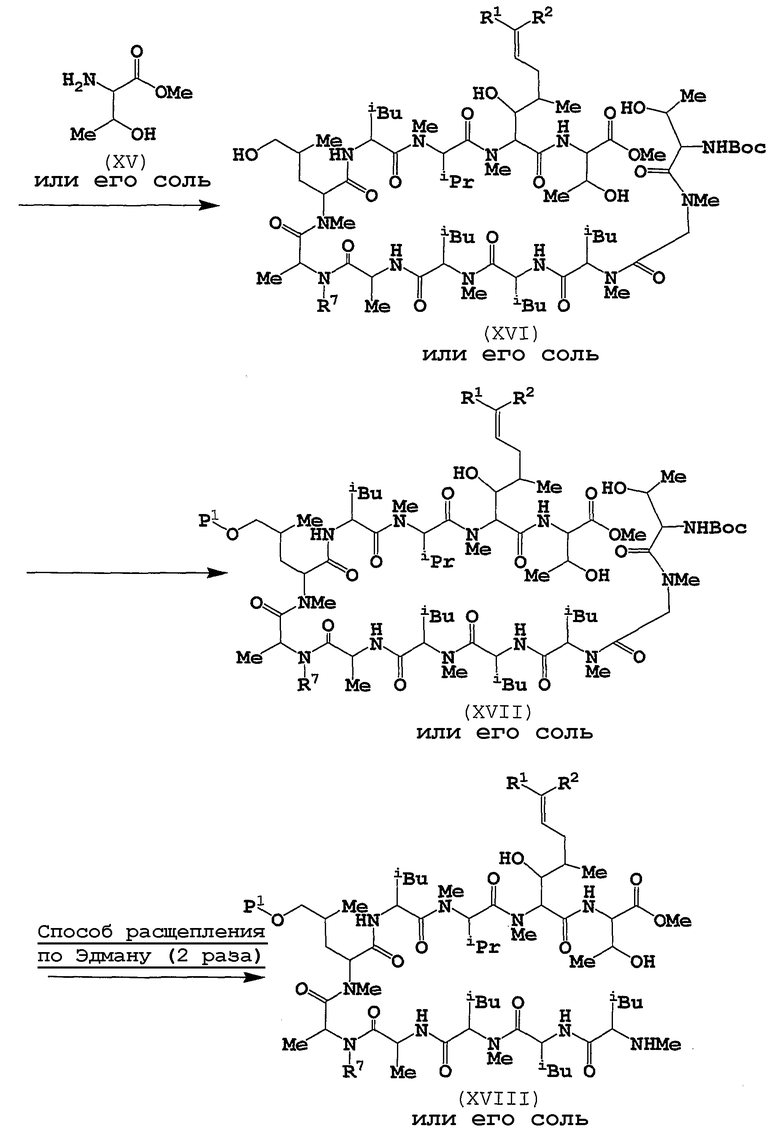
d) Ваимодействие с соединением (XV)
Эта реакция представляет собой амидирование соединения (XIV) или его соли с помощью соединения (XV) или его соли; так, эту реакцию можно осуществлять таким же образом, как в случае вышеуказанного способа 7, и, кроме того, используемые реагенты и реакционные условия (например, растворитель, температура реакции и т.д.), могут относиться к таковым способа 7.
е) Защита гидроксильной группы
Данную реакцию обычно осуществляют в стандартном растворителе, таком как вода, ацетонитрил, ацетон, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, этилацетат, N,N-диметилформамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является лимитированной и реакцию обычно осуществляют в условиях от охлаждения до нагревания.
f) Способ расщепления по Эдману
Эту реакцию обычно осуществляют в стандартном растворителе, таком как вода, ацетонитрил, ацетон, спирт (например, метанол, этанол, изопропиловый спирт и т.п.), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, этилацетат, N,N-диметилформамид, или в любом другом органическом растворителе, который не оказывает вредного влияния на реакцию, или в их смесях.
Температура реакции не является лимитированной и реакцию обычно осуществляют в условиях от охлаждения до нагревания.
И данную реакцию осуществляют дважды.
Эту реакцию можно осуществлять подобными способами, описанными в литературе, например, M.K. Eberle и др., J. Org. Chem., 59, 7249-7258 (1994).
g) Взаимодействие с соединением (XIX)
Эта реакция представляет собой амидирование соединения (XIX) или его соли с помощью соединения (XVII) или его соли; так, эту реакцию можно осуществлять таким же образом, как описано выше в п.d), и, кроме того, используемые реагенты и реакционные условия (например, растворитель, температура реакции и т.д.) могут относится к таковым п.d).
h) Снятие защиты
Эту реакцию осуществляют в соответствии со стандартным способом, таким как гидролиз, восстановление или т.п.
Более конкретно, соединение, объект данного изобретения, можно получать способами, описанными в примерах согласно настоящей заявке, или подобными способами.
Соединения, получаемые с помощью вышеуказанных способов 1-6 и способов А-Н можно выделять и очищать стандартным методом, таким как распыление, перекристаллизация, колоночная хроматография, высокоэффективная жидкостная хроматография, переосаждение и колоночная хроматография при использовании деминерализованной смолы.
Пригодные соли соединения (I), объекта данного изобретения, представляют собой обычные фармацевтически приемлемые и нетоксичные соли и могут представлять собой аддитивную соль основания или аддитивную соль кислоты, например соль с неорганическим основанием (такая как соль щелочного металла, например соль натрия, соль калия и т.д., соль щелочноземельного металла, например соль кальция, соль магния и т.д., соль аммония), соль с органическим основанием (такая как соль органического амина, например соль триэтиламина, соль диизопропилэтиламина, соль пиридина, соль пиколина, соль этаноламина, соль триэтаноламина, соль дициклогексиламина, соль N,N'-дибензилэтилендиамина и т.д.), аддитивная соль неорганической кислоты (такая как гидрохлорид, гидробромид, сульфат, фосфат и т.д.), аддитивная соль органической карбоновой кислоты или аддитивная соль сульфоновой кислоты (такая как формиат, ацетат, трифторацетат, малеат, тартрат, глюконат, фумарат, метансульфонат, бензолсульфонат, толуолсульфонат и т.д.), соль с основной или кислой аминокислотой (такой как аргинин, аспарагиновая кислота, глутаминовая кислота и т.д.), и т.п.
В вышеприведенных и последующих описаниях согласно настоящему описанию изобретения, подходящие примеры и пояснения различных определений, включенные в рамки данного изобретения, разъяснены детально следующим образом.
Термин «низший» означает группу, имеющую 1-6, предпочтительно 1-4 атома(ов), за исключением иначе указанного.
Подходящие примеры термина «низший алкил» и «низший алкильный остаток» могут включать линейный или разветвленный остаток, имеющий 1-6 атома(ов) углерода, как, например, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, трет-пентил, неопентил, гексил, изогексил и т.п.
Подходящие примеры термина «цикло(низший)алкил» могут включать циклический алкил, имеющий 3-6 атомов углерода, такой как циклопропил, циклобутил, циклопентил, циклогексил и т.п.
Подходящие примеры термина «низший алкенил» могут включать таковой с линейной или разветвленной цепью, имеющей 2-6 атомов углерода, такой как винил, 1- или 2-пропенил, изопропенил, 1-, или 2-, или 3-бутенил, изобутенил, втор-бутенил, трет-бутенил, пентенил, трет-пентенил, неопентенил, гексенил, изогексенил и т.п.
Подходящие примеры термина «цикло(низший)алкенил» могут включать циклоалкенил, имеющий 3-6 атомов углерода, такой как циклопропенил, циклобутенил, циклопентенил и циклогексенил и т.п.
Подходящие примеры «арила» и «арильного» остатка могут включать фенил, который может быть замещен с помощью низшего алкила (например, фенил, мезитил, толил и т.д.), нафтил, антрил, тетрагидронафтил, инденил, тетрагидроинденил и т.п.
Подходящие примеры термина «галоген» представляют собой фтор, хлор, бром и иод.
Термин «гетероциклическая группа», как используется в данном контексте, относится как к гетероарильной, так и к гетероциклоалкильной группам, и, другими словами, подходящие примеры «гетероциклической группы» могут представлять собой насыщенную или ненасыщенную, моноциклическую или полициклическую группу, содержащую, по меньшей мере, один гетероатом, такой как атом азота, атом кислорода или атом серы, например, которая может включать:
ненасыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую 1-4 атома азота, например пирролил, пирролинил, имидазолил, пиразолил, пиридил, дигидропиридил, пиримидинил, пиразинил, пиридазинил, триазолил (например, 4Н-1,2,4-триазолил, 1Н-1,2,3-триазолил, 2Н-1,2,3-триазолил и т.д.), тетразолил (например, 1Н-тетразолил, 2Н-тетразолил и т.д.), азепинил и т.д.;
насыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую 1-4 атома азота, например азиридинил, азетидинил, пирролидинил, имидазолидинил, пиперидил, пиперазинил, 2,5-метанопиперазинил, гексагидроазепинил и т.д.;
ненасыщенную конденсированную гетероциклическую группу, содержащую 1-4 атома азота, например индолил, изоиндолил, индолинил, индолизинил, бензимидазолил, хинолил, изохинолил, индазолил, бензотриазолил, тетрагидрохинолил, тетрагидроизохинолил, тетрагидроиндолил, дигидроиндазолил, дигидроимидазопиразинил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода и 1-3 атома азота, например оксазолил, изоксазолил, оксадиазолил (например, 1,2,4-оксадиазолил, 1,3,4-оксадиазолил, 1,2,5-оксадиазолил и т.д.) и т.д.;
насыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода и 1-3 атома азота, например, морфолинил, сиднонил и т.д.;
ненасыщенную конденсированную гетероциклическую группу, содержащую 1 или 2 атома кислорода и 1-3 атома азота, например бензоксазолил, бензоксадиазолил, дигидропиридооксазинил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома серы и 1-3 атома азота, например тиазолил, изотиазолил, тиадиазолил (например, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил и т.д.), дигидротиазинил и т.д.;
насыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома серы и 1-3 атома азота, например тиазолидинил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома серы, например тиенил, дигидродитиинил, дигидродитионил и т.д.;
ненасыщенную конденсированную гетероциклическую группу, содержащую 1 или 2 атома серы и 1-3 атома азота, например бензотиазолил, бензотиадиазолил, имидазотиадиазолил, дигидротиазолопиридинил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую атом кислорода, например фурил, и т.д.;
насыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода, например оксиранил, 1,3-диоксоланил, тетрагидрофуранил, тетрагидропиранил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно, 5- или 6-членную) гетеромоноциклическую группу, содержащую атом кислорода и 1 или 2 атома серы, например дигидрооксатиинил и т.д.;
ненасыщенную конденсированную гетероциклическую группу, содержащую 1 или 2 атома серы, например бензотиенил, бензодитиинил и т.д.;
ненасыщенную конденсированную гетероциклическую группу, содержащую атом кислорода и 1 или 2 атома серы, например бензооксатиинил и т.д.;
насыщенную конденсированную гетеромоноциклическую группу, содержащую 1-3 атома азота, например тетрагидропиридопирролидинил и т.д.;
и тому подобное.
Подходящий «гетероарил» может относиться к таковым, как указанные выше, где гетероциклическая группа имеет ароматическую циклическую систему.
Подходящая «содержащая азот гетероциклическая группа» может относиться к таковым, как указанные выше, где гетероциклическая группа содержит, по меньшей мере, один атом азота в качестве члена цикла, как, например, пирролидинил, пиперидил, пиперазинил, морфолинил, тиазолил, оксазолил и т.п.
Подходящие примеры «подходящего(их) заместителя(ей)» могут включать гидрокси, низший алкил, -(низший алкил)-О-(низший алкил), -S(=О)2-(низший алкил), -С(=О)NH2, цикло(низший)алкил, -О-(низший алкил), галоген, амино, арил, гетероциклическую группу, арил(низший)алкил, ацил и т.п., и каждый из которых может иметь один или более подходящий(их) заместитель(ей).
Подходящие примеры «ацила» могут включать низший алканоил, низший алкеноил, цикло(низший)алкенилкарбонил, ароил, гетероциклокарбонил, гетероцикло(низший)алканоил, гетероцикло(низший)алкеноил, низший алкилсульфинил, низший алкенилсульфинил, арилсульфинил, гетероциклосульфинил, низший алкилсульфонил, низший алкенилсульфонил, арилсульфонил, гетероциклосульфонил, карбокси, защищенный карбокси и т.п.
Подходящие примеры вышеуказанного «низшего алканоила» могут представлять собой формил, ацетил, пропионил, бутирил, изобутирил, пивалоил, гексаноил и т.п.
Подходящий вышеуказанный «низший алкеноил» может представлять собой акрилоил, метакрилоил, кротоноил, циннамоил и т.п.
Подходящий вышеуказанный «ароил» может представлять собой бензоил, толиоил, нафтоил и т.п.
Подходящие примеры вышеуказанного «защищенного карбокси» могут представлять собой:
i) этерефицированный карбокси, в случае которого пригодный этерифицированный карбокси может включать -О-(низший алкил)карбонил (например, метоксикарбонил, этоксикарбонил, пропоксикарбонил, бутоксикарбонил, трет-бутоксикарбонил, пентилоксикарбонил, гексилоксикарбонил и т.д.), арил-О-(низший алкил)карбонил (например, бензилоксикарбонил, фенетилоксикарбонил, 2-фенилпропоксикарбонил, 4-фенилбутоксикарбонил, 4-фенилпентилоксикарбонил, 1,3-дифенилгексилоксикарбонил и т.д.) и т.п.
ii) амидированный карбокси, в случае которого пригодный амидированный карбокси может включать карбамоил, N-(низший)алкилкарбамоил (например, N-метилкарбамоил, N-этилкарбамоил, N-изопропилкарбамоил, N-бутилкарбамоил, N-пентилкарбамоил, N-гексилкарбамоил и т.д.), N,N-ди(низший)алкилкарбамоил [например, N,N-диметилкарбамоил, N,N-диэтилкарбамоил, N-метил-N-этилкарбамоил, N,N-дипропилкарбамоил, N,N-ди(трет-бутил)карбамоил, N-пентил-N-гексилкарбамоил и т.д.], N-низший алкил-N-ар(низший)алкилкарбамоил (например, N-метил-N-бензилкарбамоил и т.д.) и т.п.
«Гетероциклический» остаток в случае вышеуказанных гетероциклокарбонила, гетероцикло(низшего)алканоила, гетероцикло(низшего)алкеноила, гетероциклосульфинила и гетероциклосульфонила, может включать карбонильную группу, замещенную гетероциклической группой, как указано выше, например морфолинилкарбонил, пиперидилкарбонил, пиперазинилкарбонил, имидазолилкарбонил, пиразолилкарбонил, пирролидинилкарбонил, пиразинилкарбонил, никотиноил, изоникотиноил, фуроил, теноил и т.п.
V. Lohmann и др., Science, 285, 110-113 (1999), сообщалось, что ими получены клеточные линии гепатоаденомы человека (Huh-7), в которые встроены молекулы субгеномной РНК HCV, и обнаружено, что субгеномная РНК HCV реплицируется в клетках с высокой скоростью. Полагают, что механизм репликации субгеномной РНК HCV в случае этих клеточных линий очень подобен репликации полноразмерной РНК генома HCV в гепатоцитах, инфицированных HCV. Поэтому метод оценки активности соединения формулы (I) в отношении ингибирования репликации РНК согласно настоящему изобретению базируется на методе клеточного анализа с использованием клеток Huh-7, в которые встроена субгеномная РНК HCV.
Для того чтобы продемонстрировать пригодность соединения формулы (I) или его соли согласно настоящему изобретению, ниже приводится фармакологический тест-пример типичного соединения согласно настоящей заявке.
Тест-пример
Анализ репортер-репликона HCV
Ингибирующую активность тестируемых соединений против репликации репликона HCV оценивали путем количественного определения активности люциферазы, вырабатываемого репортер-геном продукта, кодируемого в репликон-системе, как описано Yokota et al., EMBO J., 4, 602-608 (2003). Ферментный анализ осуществляли в соответствии с техническим руководством системы анализа люциферазы Steady-Glo (торговая марка) (Promega). Анализ репликона осуществляли с помощью модифицированного метода, описанного Lohmann и др., Science, 285: 110 (1999). Подробности приводятся ниже.
1) Добавление агента к клеткам
6×103 клеток репликона HCV в модифицированной по способу Дульбекко среде Игла (DMEM), содержащей 5% фетальной телячьей сыворотки, высевали в каждую лунку 96-луночного титрационного микропланшета (Corning Inc.). После инкубации клеток при температуре 37°С в течение 16 часов в присутствии 5% СО2 добавляли тестируемое соединение.
2) Методика анализа люциферазы
После культивирования в течение более двух суток культуральную среду удаляли и в каждую лунку добавляли 25 мкл буфера для лизиса Glo и инкубировали в течение 5 минут. При дальнейшем протекании лизиса в каждую лунку добавляли 25 мкл реагента для анализа Steady-Glo (торговая марка). После инкубации в течение 5 минут измеряли люминесценцию при использовании люминометра Mithras LB940 (BERTHOLD, TECHNOLOGIES Gmbh and Co.KG), следуя инструкциям изготовителя.
Результат теста
Люциферазные активности в репликоновых клетках, обработанных каждыми концентрациями соединения, использовали для расчета значения ЕС50 каждого соединения, которое представляет собой концентрацию соединения, показывающую 50%-ный уровень ферментной активности по отношению к контролю (группа без лекарственного средства, содержащая только ДМСО).
Из результата вышеприведенного тест-примера понятно, что соединение формулы (I) или его соль, согласно настоящему изобретению, обладает активностью против вируса гепатита С.
Некоторые из соединений согласно настоящему изобретению также проявили ингибирующую репликацию репликона HCV активность в случае человеческой сыворотки (вместо фетальной телячьей сыворотки).
Кроме того, некоторые из соединений согласно настоящему изобретению показали предпочтительный фармакокинетический профиль.
Средство против HCV согласно настоящему изобретению, содержащее соединение формулы (I) или его соль в качестве активного ингредиента, может быть использовано в виде фармацевтического препарата, например, в твердой, полутвердой или жидкой форме, в смеси с органическим или неорганическим носителем или эксципиентом, пригодным для перорального; сублингвального; буккального; назального; респираторного; парентерального (внутрикожного, во внутренний орган, подкожного, интрадермального, внутримышечного, внутрисуставного, центрально-венного, печеночно-венного, периферически-венного, лимфатического, сердечно-сосудистого, артериального, глазного, включая окологлазную инъекцию или окологлазное капельное внутривенное вливание); путем капельного внутривенного вливания в глазное яблоко, глазную структуру или глазной слой; ушного, включая слуховой канал, папиллярную полость, внешний и внутренний слуховые каналы, барабанную перепонку, барабанную полость среднего уха, внутренний слуховой канал, включая ганглий спиральной улитки, лабиринт и т.д.; интестинального; ректального; вагинального; мочеточникового; и везикального введения. Что касается внутриматочных и перинатальных адаптационных заболеваний, предпочтительным является парентеральное введение, так как введение осуществляют в материнские кровеносные сосуды или в пустоты, как например материнские органы, включая матку, шейку матки и влагалище; плодный эмбрион, плод, новорожденного и комбинированную ткань; и амнион, пуповину, пупочную артерию и вену; плаценту и т.п. Использование этих путей введения изменяется в зависимости от состояния каждого пациента.
Соединение формулы (I) или его соль может быть введено независимо в качестве терапевтического агента или может быть желательно использование в качестве части прописанных лекарственных средств. «Средство против HCV» в соответствии с настоящим изобретением может быть использовано в виде фармацевтического препарата, например, в твердой, полутвердой или жидкой форме, в смеси, по меньшей мере, с одним или несколькими подходящими органическими или неорганическими носителями или эксципиентами или другими фармакологическими терапевтическими агентами. Активный ингредиент может быть смешан, например, с обычными фармакологически приемлемыми и нетоксичными носителями в твердой форме, как например гранулы, таблетки, пилюли, пастилки, капсулы или суппозитории; кремы; мази; аэрозоли; порошки для инсуффляции; в жидкой форме, как например растворы, эмульсии или суспензии для инъекции; в форме для перорального приема внутрь; глазные капли; и любые другие формы, пригодные для использования. И, если необходимо, в вышеуказанные препараты могут быть включены вспомогательные вещества, такие как стабилизаторы, загустители, смачиватели, отвердители и красители; отдушки или буферы; или обычно используемые любые другие добавки. Соединение формулы (I) или его фармацевтически приемлемую соль включают в фармацевтическую композицию в количестве, достаточном для продуцирования желательного эффекта против гепатита С во время процесса или состояния заболеваний.
Комбинированное использование IFN и/или рибавирина с соединением формулы (I) или его солью является эффективным против гепатита С.
В случае применения композиции для людей, предпочтительным является ее применение путем внутривенного, внутримышечного, пульмонального, перорального введения, введения в виде глазных капель или инсуффляции. Хотя дозировка терапевтически эффективного количества соединения формулы (I) изменяется в зависимости от, и, следовательно, зависит от возраста и состояния каждого индивидуального пациента, подвергаемого лечению, обычно для лечения или предотвращения гепатита С в случае внутривенного введения суточная доза составляет 0,001-400 мг соединения формулы (I) на 1 кг массы человека; в случае внутримышечного введения суточная доза составляет 0,1-20 мг соединения формулы (I) на 1 кг массы человека; в случае перорального введения суточная доза составляет 0,5-50 мг соединения формулы (I) на 1 кг массы человека. Однако в случае этих доз может быть необходимо превышение их предела для достижения терапевтических результатов.
Количество липопептидного соединения формулы (I) или его фармацевтически приемлемой соли, содержащееся в композиции в виде разовой унифицированной дозы, согласно настоящему изобретению, составляет 0,1-400 мг, более предпочтительно, 1-200 мг, еще более предпочтительно, 5-100 мг, особенно 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 и 100 мг.
Настоящее изобретение может включать промышленное изделие, содержащее упаковочный материал и соединение формулы (I), идентифицированное выше, содержащееся внутри вышеуказанного упаковочного материала, где вышеуказанное соединение формулы (I) является эффективным для предотвращения или лечения гепатита С и где вышеуказанный упаковочный материал включает этикетку или надпись, на которой указывается, что вышеуказанное соединение формулы (I) может или должно быть использовано для предотвращения или лечения гепатита С.
И настоящее изобретение может включать коммерчески доступную упаковку, включающую фармацевтическую композицию, содержащую соединение формулы (I), идентифицированное выше, и связанную с этим надпись, где в надписи сообщается, что соединение форомулы (I) может или должно быть использовано для предотвращения или лечения гепатита С.
Нужно указать, что соединение формулы (I) или его соль может включать один или более стереоизомеров, таких как оптический изомер (оптические изомеры) и геометрический изомер (геометические изомеры), что обусловлено асимметрическим(ми) атомом(ами) углерода и двойной(ными) связью(ями), и что все такие изомеры и их смеси включены в рамки настоящего изобретения.
Соединение формулы (I) или его соль может включать сольватированное соединение (например, гидрат, этанолят и т.д.).
Соединение формулы (I) или его соль может включать как кристаллическую форму, так и некристаллическую форму.
Соединение формулы (I) или его соль может включать пролекарственную форму.
Описания изобретений к патентам и публикации, цитированные в данном контексте, включены в данное описание путем ссылки.
Нижеследующие Получения и Примеры приводятся в целях пояснения настоящего изобретения. Однако настоящее изобретение не ограничено этими Получениями и Примерами.
Используемые исходные соединения и целевые соединения, синтезируемые согласно нижеследующим Примерам 1-265, получают как указано ниже.
Аббревиатуры, символы и термины, используемые в Получениях, Примерах и формулах в вышеприведенном и последующем контекстах настоящего описания (включая таблицы), имеют следующие значения.
Получение 1
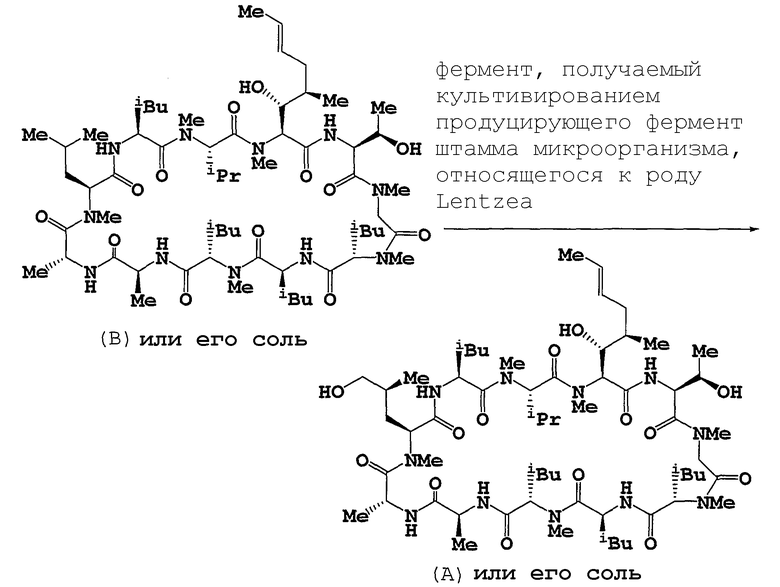
Соединение (А) или его соль может быть получено(на) из соединения (В) или его соли в соответствии со способом, описанным в Международной публикации WO 2006/054801 (соединение (А) или его соль, при использовании фермента, который может быть получен путем ферментации продуцирующего фермент штамма микроорганизма, относящегося к роду Lentzea sp., депозитный номер FERM BP-10079), и соединение (В) или его соль может быть получено(на) путем ферментации грибка (Stachybotrys chartarum, № 19392: депозитный номер FERM BP-3364), в соответствии со способом, описанным, например, в выложенной патентной заявке Японии Hei 5-271267. Оба микроорганизма хранятся в Международном депозитарии запатентованных микроорганизмов (IPOD) Государственного института современной промышленной науки и технологии, AIST Tsukuba Central 6, 1-1, Higashi 1-chome, Tsukuba-shi, IBARAKI, 305-8566, JAPAN.
Получение 2
К раствору соединения (А) (500 мг) в CH2Cl2 (25 мл) добавляют 4-нитрофенилхлорформиат (98 мг) и N-метилморфолин (89 мкл). Затем смесь перемешивают в течение ночи, добавляют три последующие порции 4-нитрофенилхлорформиата (98 мг) и N-метилморфолина (89 мкл) с интервалами в 1 час. После того как исходное соединение израсходуется, смесь разбавляют с помощью EtOAc, промывают с помощью 1 н. водного раствора HCl и водного раствора NaHCO3, сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (н-гексан:EtOAc = 1:4 и затем CH2Cl2:МеОН = 9:1), получая 30 мг неосновного целевого соединения (Получение 2-В) и 438 мг основного целевого соединения (Получение 2-А).
Неосновное соединение: MS: 1564,51.
Основное соединение: MS: 1399,25.
Получение 3
К раствору (3R,5R)-1-бензил-3,5-диметилпиперазина (810 мг) в CH2Cl2 (20 мл) и 1 н. растворе NaOH (8 мл) добавляют Вос2О (865 мг) и смесь перемешивают при комнатной температуре в течение 3 часов. Органическую фазу отделяют, сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (н-гексан:EtOAc = 9:1), получая трет-бутил-(2R,6R)-4-бензил-2,6-диметил-1-пиперазинкарбоксилат (930 мг).
1Н-ЯМР: 1,29 (6Н, д, J=6,4 Гц), 1,46 (9Н, с), 2,10-2,60 (4Н, м), 3,30-4,10 (4Н, м), 7,10-7,40 (5Н, м).
MS: 305,4.
Получение 4
Раствор целевого соединения из Получения 3 (930 мг) в МеОН гидрируют при использовании 10%-ного Pd/C (влажность 50%; 180 мг) в течение 2 часов. Смесь отфильтровывают и фильтрат концентрируют. К раствору остатка в CH2Cl2 (10 мл) и МеОН (2 мл) добавляют 37%-ный водный раствор формальдегида (0,2 мл) и затем триацетоксиборгидрид натрия (1,94 г), и смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь гасят с помощью водного раствора NaHCO3 и дважды экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (н-гексан:EtOAc = от 8:1 до 0:100), получая трет-бутил-(2R,6R)-2,4,6-триметил-1-пиперазинкарбоксилат (460 мг).
1Н-ЯМР: 1,32 (6Н, д, J=6,5 Гц), 1,49 (9Н, с), 2,10-2,60 (7Н, м), 3,89 (2Н, м).
MS: 229,3.
Получение 5
К раствору целевого соединения из Получения 4 (440 мг) в EtOAc (5 мл), по каплям, добавляют 4 н. раствор хлористого водорода в EtOAc (5 мл) и смесь перемешивают при комнатной температуре в течение ночи. Полученный порошок собирают, промывают с помощью CH2Cl2 и высушивают в вакууме, получая (3R,5R)-1,3,5-триметилпиперазиндигидрохлорид (375 мг).
1Н-ЯМР (ДМСО-d6): 1,00-1,70 (6Н, м), 2,77 (3Н, с), 2,80-4,20 (6Н, м), 9,83 (1Н, уш.с), 10,20 (1Н, уш.с).
MS (свободный): 129,4.
Получение 6
К раствору 2,6-цис-диметилпиперазина (3 г) в CH2Cl2 (15 мл) добавляют ТЕА (3,7 мл) и Вос2О (5,73 г), при охлаждении льдом, и смесь перемешивают при этой температуре в течение 2 часов. Затем смесь обрабатывают водой и три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2:ацетон = от 1:1 до 2:3), получая трет-бутил-цис-3,5-диметил-1-пиперазинкарбоксилат (5,47 г).
1Н-ЯМР: 1,06 (6Н, д, J=6,2 Гц), 1,46 (9Н, с), 2,20-4,30 (6Н, м).
Получение 7
К раствору целевого соединения из Получения 6 (5,43 г) в CH2Cl2 (55 мл) добавляют бензальдегид (3,09 мл) и затем триацетоксиборгидрид натрия (16,11 г), и смесь перемешивают при комнатной температуре в течение ночи. Затем смесь обрабатывают с помощью насыщенного водного раствора NaHCO3 и дважды экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (н-гексан:EtOAc = от 19:1 до 4:1), получая трет-бутил-цис-4-бензил-3,5-диметил-1-пиперазинкарбоксилат (7,30 г).
1Н-ЯМР: 1,08 (6Н, д, J=4,5 Гц), 1,44 (9Н, с), 2,30-3,00 (4Н, м), 3,70-4,10 (4Н, м), 7,10-7,60 (5Н, м).
MS: 305,4.
Получение 8
К раствору целевого соединения из Получения 7 (7,25 г) в EtOAc (25 мл) по каплям добавляют 4 н. раствор хлористого водорода в EtOAc (25 мл) и смесь перемешивают при комнатной температуре в течение ночи. Смесь концентрируют, совместно выпаривают с МеОН и растирают в порошок при использовании ацетона. Полученный порошок собирают, промывают ацетоном и высушивают в вакууме, получая цис-1-бензил-2,6-диметилпиперазиндигидрохлорид (4,48 г).
1Н-ЯМР (ДМСО-d6): 1,20-4,80 (14Н, м), 7,30-7,80 (5Н, м).
MS (свободный): 205,3.
Получение 9
К раствору целевого соединения из Получения 8 (1,89 г) в смешанном растворителе из CH2Cl2 (20 мл) и МеОН (5 мл) добавляют DIPEA (4,75 мл), 35%-ный водный раствор формальдегида (0,6 мл) и затем триацетоксиборгидрид натрия (4,34 г), и смесь перемешивают при комнатной температуре в течение 2 часов. Смесь подщелачивают с помощью насыщенного водного раствора NaHCO3 и четыре раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2:МеОН=97,5:2,5), получая цис-1-бензил-2,4,6-триметилпиперазин (0,98 г).
1Н-ЯМР: 1,04 (6Н, д, J=6,0 Гц), 1,80-2,90 (9Н, м), 3,83 (2Н, с), 7,10-7,50 (5Н, м).
MS: 219,4.
Получение 10
Раствор целевого соединения из Получения 9 (940 мг) в МеОН гидрируют при использовании 10%-ного Pd/C (влажность 50%) в течение 2 часов. Смесь отфильтровывают и концентрируют. Остаток растворяют в CH2Cl2, обрабатывают с помощью 4 н раствора хлористого водорода в EtOAc (4 мл) и концентрируют, получая цис-1,3,5-триметилпиперазиндигидрохлорид (870 мг).
1Н-ЯМР (ДМСО-d6, δ): 1,32 (6Н, д, J=6,5 Гц), 2,79 (3Н, с), 2,80-4,40 (6Н, м).
MS (свободный): 129,4.
Получение 11
К раствору 4-нитропиразола (2,0 г) в ДМФА добавляют NaH (60%-ная дисперсия в масле), при охлаждении льдом. После перемешивания при той же самой температуре в течение 10 минут добавляют 2-бромэтилметиловый эфир (2,00 мл) и NaI (2,92 г). Смесь затем перемешивают при комнатной температуре в течение 3 часов, гасят с помощью раствора фосфатного буфера (рН 7) и экстрагируют с помощью EtOAc. Экстракт промывают водой и насыщенным солевым раствором, сушат над MgSO4 и выпаривают в вакууме. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля, получая 1-(2-метоксиэтил)-4-нитро-1Н-пиразол (2,15 г).
1Н-ЯМР: 3,36 (3Н, с), 3,75 (3Н, т, J=2,5 Гц), 4,32 (3Н, т, J=2,5 Гц), 8,07 (1Н, с), 8,23 (1Н, с).
Получение 12
4-[2-(4-Нитро-1Н-пиразол-1-ил)этил]морфолин получают в соответствии с подобной методикой, как таковая в случае Получения 11.
1Н-ЯМР: 2,40-2,55 (4Н, м), 2,82 (2Н, т, J=6,0 Гц), 3,60-3,80 (4Н, м), 4,26 (2Н, т, J=6,0 Гц), 8,06 (1Н, с), 8,27 (1Н, с).
Получение 13
Раствор целевого соединения из Получения 11 (2,14 г) в МеОН и ТГФ гидрируют при использовании 20%-ного Pd/C (влажность 50%; 1,1 г), при давлении, равном 3 атм, в атмосфере водорода, при температуре 50°С, в течение 1,5 часов. Смесь отфильтровывают. К фильтрату добавляют Вос2О (2,87 г) и смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают и остаток хроматографируют на силикагеле, получая трет-бутил[1-(2-метоксиэтил)-1Н-пиразол-4-ил]карбамат (2,97 г).
1Н-ЯМР: 1,50 (9Н, с), 3,33 (3Н, с), 3,72 (2Н, т, J=5,3 Гц), 4,22 (2Н, т, J=5,3 Гц), 6,10-6,40 (1Н, уш.), 7,35 (1Н, с), 7,69 (1Н, уш.с).
Получение 14
трет-Бутил{1-[2-(4-морфолинил)этил]-1Н-пиразол-4-ил}карбамат получают в соответствии с подобной методикой, как таковая в Получении 13.
1Н-ЯМР: 1,50 (9Н, с), 2,40-2,55 (4Н, м), 2,81 (2Н, т, J=6,8 Гц), 3,65-3,75 (4Н, м), 4,19 (2Н, т, J=6,8 Гц), 6,25 (1Н, уш.с), 7,32 (1Н, с), 7,69 (1Н, уш.с).
Получение 15
К раствору целевого соединения из Получения 13 (1,33 г) в ДМФА добавляют NaH (60%-ная дисперсия в масле, 223 мг), при охлаждении льдом. После перемешивания при той же самой температуре в течение 10 минут по каплям добавляют MeI (0,45 мл). Смесь затем перемешивают при комнатной температуре в течение 3 часов, гасят с помощью раствора фосфатного буфера (рН 7) и экстрагируют с помощью EtOAc. Экстракт промывают водой и насыщенным солевым раствором, сушат над MgSO4 и выпаривают в вакууме. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля, получая трет-бутил[1-(2-метоксиэтил)-1Н-пиразол-4-ил]метилкарбамат (2,98 г).
1Н-ЯМР: 1,52 (9Н, с), 3,21 (3Н, с), 3,34 (3Н, с), 3,73 (2Н, т, J=5,4 Гц), 4,23 (2Н, т, J=5,4 Гц), 7,44 (1Н, уш.с), 7,50-8,00 (1Н, уш.).
Получение 16
трет-Бутилметил{1-[2-(4-морфолинил)этил]-1Н-пиразол-4-ил}карбамат получают в соответствии с подобной методикой, как таковая в Получении 15.
1Н-ЯМР: 1,52 (9Н, с), 2,45-2,55 (4Н, м), 2,82 (2Н, т, J=6,8 Гц), 3,21 (3Н, с), 3,65-3,75 (4Н, м), 4,20 (2Н, т, J=6,8 Гц), 7,41 (1Н, с), 7,45-7,95 (1Н, уш.).
Получение 17
К раствору целевого соединения из Получения 15 (1,18 г) в МеОН по каплям добавляют 4 н. раствор хлористого водорода в диоксане и смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают и остаток растирают в Et2O. Твердое вещество собирают и промывают с помощью Et2O и высушивают в вакууме, получая 1-(2-метоксиэтил)-N-метил-1Н-пиразол-4-аминдигидрохлорид.
1Н-ЯМР (ДМСО-d6): 2,83 (3Н, с), 3,22 (3Н, с), 3,67 (2Н, т, J=5,2 Гц), 4,28 (2Н, т, J=5,2 Гц), 5,80-6,30 (3Н, уш.), 7,69 (1Н, с), 8,08 (1Н, с).
Получение 18
N-Метил-1-[2-(4-морфолинил)этил]-1Н-пиразол-4-аминтригидрохлорид получают в соответствии с подобной методикой, как таковая в Получении 17.
1Н-ЯМР: 2,84 (3Н, с), 2,95-3,50 (8Н, м), 3,57 (2Н, т, J=6,8 Гц), 4,68 (2Н, т, J=6,8 Гц), 7,80 (1Н, с), 8,24 (1Н, с), 11,0-12,0 (4Н, уш.).
Получение 19
трет-Бутилэтил(1-метил-1Н-пиразол-4-ил)карбамат получают в соответствии с подобной методикой, как таковая в Получении 15.
1Н-ЯМР: 1,18 (3Н, т, J=7,1 Гц), 1,50 (9Н, с), 3,62 (2Н, кв., J=7,1 Гц), 3,86 (3Н, с), 7,37 (1Н, с), 7,37-7,90 (1Н, уш.).
Получение 20
N-Этил-1-метил-1Н-пиразол-4-аминдигидрохлорид получают в соответствии с подобной методикой, как таковая в Получении 17.
1Н-ЯМР (ДМСО-d6): 1,23 (3Н, т, J=7,2 Гц), 3,20 (2Н, кв., J=7,2 Гц), 3,86 (3Н, с), 7,65 (1Н, с), 8,06 (1Н, с), 8,30-9,00 (2Н, уш.).
Получение 21
К раствору соединения (А) (3,0 г) и DIPEA (3 мл) в CH2Cl2 добавляют трет-бутилдиметилсилилхлорид (1,6 г) и смесь перемешивают при комнатной температуре в течение 2 дней. Смесь разбавляют с помощью EtOAc, промывают три раза водой и насыщенным солевым раствором, сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2:ацетон = 1:1), получая 3,28 г целевого соединения.
MS: 1348,88.
Получение 22
К раствору целевого соединения из Получения 21 (3,27 г) в пиридине (31 мл) добавляют уксусный ангидрид (22,9 мл) и DMAP (148 мг) и смесь перемешивают при комнатной температуре в течение ночи. Смесь разбавляют водой и экстрагируют с помощью EtOAc. Органическую фазу дважды промывают водой и концентрируют. Остаток растворяют в МеОН (40 мл) и обрабатывают с помощью 1 н. раствора HCl (15 мл). После перемешивания в течение 4 часов, смесь разбавляют водой и дважды экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2:ацетон = 1:1), получая 2,91 г целевого соединения.
MS: 1318,62.
Получение 23
К раствору целевого соединения из Получения 22 (720 мг) и димера ацетата родия (241 мг) в CH2Cl2 (14 мл) по каплям добавляют этилдиазоацетат (287 мкл). Добавляют две последующие порции димера ацетата родия (120 мг) и этилдиазоацетата (145 мкл), с интервалами в 2 часа. После того как исходное соединение израсходуется, смесь отфильтровывают и фильтрат концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2:ацетон = 1:1), получая 601 мг целевого соединения.
MS: 1404,54.
Получение 24
К раствору целевого соединения из Получения 23 (76 мг) в МеОН (8 мл) добавляют 1 н. водный раствор NaOH (4 мл) и смесь перемешивают при температуре 50°С в течение 2 часов. Смесь подкисляют с помощью 1 н. раствора HCl и три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2:МеОН = 9:1), получая 39 мг целевого соединения.
MS: 1292,73.
Получение 25
К раствору целевого соединения из Получения 24 (230 мг) и ТЕА (50 мкл) в ТГФ (10 мл) добавляют изобутилхлорформиат (35 мкл) и смесь перемешивают при комнатной температуре в течение 0,5 часа. Затем смесь отфильтровывают, к фильтрату, в виде порций, добавляют боргидрид натрия (20 мг). После перемешивания в течение 1 часа, смесь подкисляют с помощью 1 н. раствора HCl и три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток подвергают очистке при использовании силикагеля, модифицированного октадецильными группами (ODS-очистка), получая 85 мг целевого соединения.
MS: 1278,53.
Получение 26
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 2.
MS: 1443,15.
Получение 27
К раствору целевого соединения из Получения 25 (82 мг) в пиридине (3 мл) добавляют 4-метилбензолсульфонилхлорид (60 мг), и смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь гасят водой и экстрагируют с помощью EtOAc. Органическую фазу дважды промывают с помощью 1 н. раствора HCl и дважды промывают с помощью водного раствора NaHCO3, сушат над MgSO4 и концентрируют, получая 83 мг сырого продукта, который используют на следующей стадии без дальнейшей очистки.
MS: 1432,07.
Получение 28
К двухфазному раствору соединения (А) (1,0 г), N,N,N,N-тетрабутиламмонийхлорида (45 мг) и 2,2,6,6-тетраметил-1-пиперидинилокси, свободный радикал, (ТЕМРО, 25 мг) в CH2Cl2 (10 мл) и смешанному водному раствору NaHCO3 (0,5 М) и карбоната калия (0,05 М) (10 мл) порциями добавляют N-хлорсукцинимид (162 мг). Затем смесь перемешивают при комнатной температуре в течение ночи, добавляют две последующие порции N,N,N,N-тетрабутиламмонийхлорида (18 мг), ТЕМРО (25 мг) и N-хлорсукцинимида (160 мг), с интервалами в 2 часа. После того как исходное соединение израсходуется, смесь дважды экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2:ацетон = от 4:1 до 2:3), получая 86 мг целевого соединения.
MS: 1232,31.
Получение 29
Целевое соединение получают в соответствии с подобной методикой, как таковая в Примере 43.
MS: 1247,25.
Получение 30
К раствору соединения (А) (1,23 г) в пиридине (8 мл) добавляют п-толуолсульфонилхлорид (760 мг). Затем реакционную смесь перемешивают при комнатной температуре в течение 4 часов, смесь разбавляют этилацетатом и смесью воды со льдом. Раствор обрабатывают с помощью 6 н. раствора HCl и органическую фазу отделяют. После экстракции с помощью EtOAc объединенную органическую фазу промывают водой и насыщенным солевым раствором. Раствор сушат над MgSO4 и концентрируют в вакууме. Остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 1:1), получая 1,09 г целевого соединения.
ESI (M+H2O)+: 1405,50.
Получение 31
Смесь целевого соединения из Получения 30 (2,93 г) с ДМФА (30 мл) обрабатывают имидазолом (718 мг) и 2,0 М толуольным раствором трет-бутилдиметилсилилхлорида (4,22 мл), при комнатной температуре, в течение 4 часов. Реакционную смесь разбавляют растворяющей смесью из EtOAc и гексана (в соотношении 1:1) и промывают водой и насыщенным солевым раствором. Экстракт сушат над MgSO4 и концентрируют в вакууме. Остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 3:7), получая 996 мг побочного целевого соединения (Получение 31-В) и 1,87 г основного целевого соединения (Получение 31-А).
Неосновное соединение: MS: 1366,55.
Основное соединение: ESI (M+H2O)+: 1519,60.
Получение 32
Смесь побочного целевого соединения из Получения 31 (100 мг) в ДМФА (1,4 мл) обрабатывают с помощью тиофенола (11 мкл) и карбоната калия (20 мг), при комнатной температуре, в течение 12 часов. Реакционную смесь разбавляют с помощью EtOAc и промывают водой и насыщенным солевым раствором. Экстракт сушат над MgSO4 и концентрируют в вакууме. Остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 3:7), получая 93,8 мг целевого соединения.
MS: 1440,63.
Получение 33
Смесь целевого соединения из Получения 32 (144 мг) с CH2Cl2 (2 мл) обрабатывают с помощью м-хлорнадбензойной кислоты (52 мг), при температуре 0°С, в течение 1 часа. Реакционную смесь разбавляют с помощью EtOAc и промывают с помощью насыщенного раствора сульфита натрия, NaHCO3, воды и насыщенного солевого раствора. Экстракт сушат над MgSO4 и концентрируют в вакууме. Остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 4:6), получая 64,1 мг целевого соединения.
MS: 1472,48.
Получение 34
Смесь целевого соединения из Примера 49 (468 мг) в EtOH (9 мл) обрабатывают с помощью 2,6 М раствора этоксида натрия в EtOH (29 мкл), при комнатной температуре, в течение 30 минут. После обработки с помощью уксусной кислоты (30 мкл), смесь концентрируют и остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 45:55), получая 354 мг целевого соединения.
MS: 1250,22.
Получение 35
Смесь основного целевого соединения из Получения 31 (225 мг) с ДМФА (4,5 мл) обрабатывают с помощью 2,4-дифторфенола (43 мкл) и NaH (12 мг), при комнатной температуре, в течение 12 часов. Реакционную смесь разбавляют с помощью EtOAc и промывают водой и насыщенным солевым раствором. Экстракт сушат над MgSO4 и концентрируют в вакууме. Остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 40:60), получая 129 мг целевого соединения.
MS: 1460,66.
Получение 36
Смесь целевого соединения из Получения 30 (2,778 г) в ДМФА (20 мл) обрабатывают с помощью тетрабутиламмонийазида (1,71 г), при температуре 60°С, в течение 1 часа. Реакционную смесь разбавляют с помощью EtOAc и промывают водой и насыщенным солевым раствором. Экстракт сушат над MgSO4 и концентрируют в вакууме. Остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 1:1), получая 2,249 г целевого соединения.
MS: 1259,47.
Получение 37
К смеси целевого соединения из Получения 36 (2,08 г) и 2,6-лутидина (893 мкл) в CH2Cl2 (30 мл) добавляют, при температуре 0°С, трет-бутилдиметилсилилтрифторметансульфонат (1,17 мл). После перемешивания при комнатной температуре в течение 30 минут, реакционную смесь разбавляют EtOAc и гексаном и промывают водой и насыщенным солевым раствором. Экстракт сушат над MgSO4 и концентрируют в вакууме. Остаток хроматографируют на силикагеле (ацетон:гексан = от 1:9 до 3:7), получая 1,39 г целевого соединения.
MS: 1373,64.
Получение 38
Смесь целевого соединения из Получения 37 (120 мг) с толуолом (1,2 мл) обрабатывают с помощью бензальдегида (18 мкл) и PPh3 (34 мг), при температуре 100°С, в течение 1,5 часов. Реакционную смесь выливают в раствор боргидрида натрия (18 мг) в EtOH (1,2 мл), при температуре 0°С. После перемешивания при комнатной температуре в течение 30 минут, смесь обрабатывают с помощью уксусной кислоты (100 мкл) и концентрируют. Остаток хроматографируют на силикагеле (МеОН:CH2Cl2 (0,2% ТЕА) = от 2:98 до 10:90), получая 57 мг целевого соединения.
MS: 1437,74.
Получение 39
Смесь целевого соединения из Получения 36 (1,26 г) с толуолом (5 мл) и EtOH (5 мл) обрабатывают с помощью PPh3 (390 мг), при температуре 90°С, в течение 2 часов. Смесь концентрируют. Остаток хроматографируют на силикагеле (МеОН:CH2Cl2 (0,2% ТЕА) = от 2:98 до 10:90), получая 1,03 г целевого соединения.
MS: 1233,85.
Получение 40
Раствор целевого соединения из Получения 36 (0,93 г) в МеОН (10 мл) гидрируют при использовании 10%-ного Pd/C (влажность 50%; 0,78 г) в атмосфере водорода в течение 2 часов. Смесь отфильтровывают. Растворитель выпаривают и остаток хроматографируют на силикагеле (МеОН:CH2Cl2 (0,2% ТЕА) = от 2:98 до 8:92), получая целевое соединение.
MS: 1235,86.
Получение 41
К раствору целевого соединения из Получения 30 (800 мг) в ацетоне (40 мл) добавляют NaI (432 мг) и смесь перемешивают при температуре 60°С в течение 6 часов. После охлаждения до комнатной температуры, реакционную смесь гасят водой и экстрагируют с помощью EtOAc. Органическую фазу дважды промывают с помощью 5%-ного водного раствора NaHCO3, сушат над MgSO4 и концентрируют, получая 750 мг сырого продукта, который используют на следующей стадии без дальнейшей очистки.
MS: 1344,46.
Получение 42
К раствору трет-бутил-4-{[(трифторметил)сульфонил]окси}-3,6-дигидропиридин-1(2Н)-карбоксилата (610 мг) в 1,2-диметоксиэтане (15 мл) добавляют 3-тиенилбороновую кислоту (353 мг), тетракис(трифенилфосфин)палладий (106 мг) и 2 М водный раствор карбоната цезия (2,7 мл) и смесь кипятят с обратным холодильником в течение 2 часов в атмосфере азота. После удаления растворителя в вакууме, остаток экстрагируют с помощью EtOAc. Экстракт промывают насыщенным солевым раствором, сушат над MgSO4 и концентрируют в вакууме. Полученный остаток очищают с помощью колоночной хроматографии при использовании силикагеля (н-гексан:EtOAc = 80:20), получая трет-бутил-4-(3-тиенил)-3,6-дигидропиридин-1(2Н)-карбоксилат (350 мг) в виде бесцветного масла.
1Н-ЯМР (ДМСО-d6, δ): 1,42 (9Н, с), 2,43 (2Н, уш.с), 3,51 (2Н, т, J=5,6 Гц), 3,97 (2Н, уш.с), 6,17 (1Н, уш.с), 7,34 (1Н, дд, J=5,2, 1,4 Гц), 7,41 (1Н, уш.с), 7,51 (1Н, дд, J=5,2, 3,0 Гц).
Получение 43
К раствору трет-бутил-4-(3-тиенил)-3,6-дигидропиридин-1(2Н)-карбоксилата (170 мг) в EtOAc (1,7 мл) и воды (0,085 мл) добавляют 4 н. раствор хлористого водорода в EtOAc (1,7 мл) и смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют в вакууме, получая 4-(3-тиенил)-1,2,3,6-тетрагидропиридингидрохлорид (120 мг) в виде порошка темно-серого цвета. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
1Н-ЯМР (ДМСО-d6, δ): 2,66 (2Н, уш.с), 3,27 (2Н, уш.с), 3,71 (2Н, уш.с), 6,20 (1Н, уш.с), 7,39 (1Н, дд, J=5,0, 1,4 Гц), 7,54 (1Н, м), 7,56 (1Н, дд, J=5,0, 2,8 Гц), 9,12 (1Н, уш.с).
Получение 44
К раствору 1-метил-1Н-имидазола (2 г) в ТГФ (15 мл) медленно добавляют 1,59 М раствор н-бутиллития в н-гексане (18,4 мл), при охлаждении на ледяной бане, и смесь перемешивают при комнатной температуре в течение 3 часов. К этой реакционной смеси добавляют раствор трет-бутил-4-оксопиперидин-1-карбоксилата (5,8 г) в ТГФ (15 мл) и смесь перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь экстрагируют с помощью EtOAc и промывают насыщенным солевым раствором и сушат над MgSO4. Растворитель удаляют в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля (CHCl3:МеОН = от 95:5 до 90:10), получая трет-бутил-4-гидрокси-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (4,02 г) в виде порошка бледно-желтого цвета.
1Н-ЯМР (ДМСО-d6, δ): 1,40 (9Н, с), 1,79 (2Н, д, J=13,6 Гц), 1,88-1,98 (2Н, м), 3,23 (2Н, уш.с), 3,63-3,66 (2Н, м), 3,77 (3Н, с), 5,40 (1Н, с), 6,71 (1Н, д, J=1,1 Гц), 7,02 (1Н, д, J=1,1 Гц).
Получение 45
К раствору трет-бутил-4-гидрокси-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (300 мг) в ДМФА (3 мл) добавляют NaH (60%-ная дисперсия в минеральном масле; 77 мг) и MeI (120 мкл), при охлаждении на ледяной бане, и смесь перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь экстрагируют с помощью EtOAc и промывают водой и насыщенным солевым раствором и сушат над MgSO4. Растворитель удаляют в вакууме и остаток очищают путем колоночной хроматографии при использовании силикагеля (н-гексан:EtOAc = от 50:50 до только EtOAc), получая трет-бутил-4-метокси-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (182 мг), в виде бесцветного масла.
1Н-ЯМР: 1,46 (9Н, с), 2,11 (4Н, уш.д), 3,02 (3Н, с), 3,22-3,29 (2Н, м), 3,82 (3Н, с), 3,79-3,89 (2Н, м), 6,86 (1Н, д, J=1,2 Гц), 6,95 (1Н, д, J=1,2 Гц).
Получение 46
К раствору трет-бутил-4-метокси-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (182 мг) в EtOAc (0,7 мл) и воде (0,09 мл) добавляют 4 н. раствор хлористого водорода в EtOAc (0,7 мл) и смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель удаляют в вакууме, получая 4-метокси-4-(1-метил-1Н-имидазол-2-ил)пиперидиндигидрохлорид (122 мг) в виде бесцветного порошка. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
1Н-ЯМР (ДМСО-d6, δ): 1,46 (9Н, с), 2,39-2,45 (4Н, м), 2,98-3,10 (2Н, м), 3,07 (3Н, с), 3,26 (2Н, д, J=13 Гц), 7,01 (1Н, д, J=2,0 Гц), 7,83 (1Н, д, J=2,0 Гц), 9,35 (1Н, уш.с), 9,53 (1Н, уш.с).
Получение 47
К раствору трет-бутил-4-гидрокси-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (800 мг) в CH2Cl2 (8 мл) добавляют метансульфонилхлорид (0,44 мл) и ТЕА (1,6 мл), при охлаждении на ледяной бане, и смесь перемешивают при комнатной температуре в течение 4 часов. Реакционную смесь гасят с помощью водного раствора NaHCO3 и экстрагируют с помощью EtOAc. Органическую фазу промывают насыщенным солевым раствором и сушат над MgSO4. Растворитель удаляют в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля (CHCl3:МеОН = от 95:5 до 90:10), получая трет-бутил-4-(1-метил-1Н-имидазол-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (398 мг) в виде бесцветного масла.
1Н-ЯМР (ДМСО-d6, δ): 1,36-1,45 (2Н, м), 1,42 (9Н, с), 3,50 (2Н, д, J=5,6 Гц), 3,67 (3Н, с), 3,98-4,04 (2Н, м), 6,05 (1Н, уш.с), 6,85 (1Н, д, J=1,0 Гц), 7,10 (1Н, д, J=1,0 Гц).
Получение 48
К раствору трет-бутил-4-(1-метил-1Н-имидазол-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилата (398 мг) в МеОН (4,0 мл) добавляют 5%-ный Pd/C (влажный; 40 мг) и смесь перемешивают при комнатной температуре в течение 7 часов в атмосфере водорода (1 атм). После удаления Pd/C с помощью церита, фильтрат концентрируют в вакууме. Полученный остаток очищают с помощью колоночной хроматографии при использовании силикагеля (н-гексан:EtOAc = от 80:20 до только EtOAc), получая трет-бутил-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (132 мг) в виде бесцветного масла.
1Н-ЯМР (ДМСО-d6, δ): 1,41 (9Н, с), 1,53 (2Н, м), 1,75 (2Н, дд, J=13,4, 2,8 Гц), 2,86-2,97 (3Н, м), 3,58 (3Н, с), 3,98 (2Н, д, J=13,3 Гц), 6,72 (1Н, д, J=1,2 Гц), 6,97 (1Н, д, J=1,2 Гц).
Получение 49
К раствору трет-бутил-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (132 мг) в EtOAc (1,3 мл) добавляют 4 н. раствор хлористого водорода в EtOAc (1,3 мл) и смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель удаляют в вакууме, получая 4-(1-метил-1Н-имидазол-2-ил)пиперидиндигидрохлорид (108 мг) в виде бесцветного порошка. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
1Н-ЯМР (ДМСО-d6, δ): 2,07-2,11 (4Н, м), 2,96-3,09 (2Н, м), 3,37 (2Н, д, J=12,7 Гц), 3,52-3,62 (1Н, м), 3,85 (3Н, с), 7,58 (1Н, д, J=2,0 Гц), 7,66 (1Н, д, J=2,0 Гц), 9,31 (1Н, уш.с), 9,58 (1Н, уш.с), 14,82 (1Н, уш.с).
Получение 50
Азетидин-3-карбоновую кислоту (2,0 г) растворяют в смешанном растворителе из ТГФ (20 мл) и воды (10 мл). Устанавливают значение рН смеси равным 9 с помощью 1 н. раствора NaOH, при охлаждении на ледяной бане. К смеси добавляют Вос2О и всё вместе перемешивают в течение 1,5 часа при поддерживании рН 9. Смесь подкисляют с помощью 0,5 н. раствора HCl и экстрагируют с помощью EtOAc и экстракт промывают водой и насыщенным солевым раствором и сушат над MgSO4. Концентрируют при пониженном давлении, получая 1-(трет-бутоксикарбонил)азетидин-3-карбоновую кислоту (2,7 г).
ESI (M+Na)+: 224,2.
Получение 51
К раствору 1-(трет-бутоксикарбонил)азетидин-3-карбоновой кислоты (2,0 г) в ТГФ (30 мл) добавляют комплекс боран-диметилсульфид (4,0 мл; 10,0 М в расчете на боран), при охлаждении на ледяной бане, и смесь перемешивают в течение 8 часов при комнатной температуре. К смеси по каплям добавляют 1 н. раствор HCl (10 мл) при охлаждении на ледяной бане и ТГФ удаляют при пониженном давлении. Весь остаток экстрагируют с помощью EtOAc и экстракт промывают насыщенным солевым раствором и сушат над MgSO4. Концентрируют при пониженном давлении, получая трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилат (1,6 г).
ESI (M+Na)+: 210,4.
Получение 52
К раствору трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилата (1,0 г) в ДМФА (20 мл) добавляют NaH (235 мг; 60%-ная суспензия в масле), в атмосфере азота, и смесь перемешивают в течение 30 минут при комнатной температуре. К смеси добавляют MeI (665 мкл) и всё вместе перемешивают в течение 2 часов. К смеси добавляют воду и всё экстрагируют с помощью EtOAc. Экстракт промывают водой и насыщенным солевым раствором и сушат над MgSO4. Концентрируют при пониженном давлении, получая остаток, который очищают с помощью колоночной хроматографии при использовании силикагеля, элюируя смесью н-гексан:EtOAc = 5:1, получая трет-бутил-3-(метоксиметил)азетидин-1-карбоксилат (1,04 г).
MS: 202,08.
Получение 53
трет-Бутил-3-(метоксиметил)азетидин-1-карбоксилат растворяют в 4 н растворе хлористого водорода в EtOAc (13 мл), при охлаждении на ледяной бане, и всё вместе перемешивают в течение 2 часов при комнатной температуре. Концентрируют при пониженном давлении, получая 3-(метоксиметил)азетидингидрохлорид (559 мг).
MS: 102,2.
Получение 54
К перемешиваемой смеси (2R)-1-аминопропан-2-ола (1,2 г) и ди-трет-бутилдикарбоната (3,7 г) в ТГФ (24 мл) добавляют ТЕА (3,3 мл) и всё вместе перемешивают в течение ночи. К смеси добавляют воду и всё подкисляют с помощью лимонной кислоты. Смесь экстрагируют с помощью EtOAc и экстракт промывают водой, раствором NaHCO3, водой и насыщенным солевым раствором и сушат над MgSO4. Концентрируют при пониженном давлении, получая трет-бутил[(2R)-2-гидроксипропил]карбамат (2,7 г).
ESI (M+Na)+: 198,2.
Получение 55
К раствору трет-бутил[(2R)-2-гидроксипропил]карбамата (2,7 г) в CH2Cl2 (41 мл) добавляют N,N,N',N'-тетраметилнафталин-1,8-диамин (3,63 г) и триметилоксонийтетрафторборат (2,51 г), и всю совокупность перемешивают в течение 3 часов при комнатной температуре. К смеси добавляют воду и всё экстрагируют с помощью EtOAc и экстракт промывают водой и насыщенным солевым раствором и сушат над MgSO4. Концентрируют при пониженном давлении, получая трет-бутил[(2R)-2-метоксипропил]карбамат (2,73 г).
ESI (M+Na)+: 212,4.
Получение 56
К раствору трет-бутил[(2R)-2-метоксипропил]карбамата (2,72 г) в ДМФА (41 мл) добавляют NaH (862 мг; 60%-ная суспензия в масле), в атмосфере азота, и смесь перемешивают при комнатной температуре в течение 30 минут. К смеси добавляют MeI (1,79 мл) и всё вместе перемешивают в течение 3 часов при той же температуре. Добавляют воду и всю смесь экстрагируют с помощью EtOAc и экстракт промывают водой и насыщенным солевым раствором и сушат над MgSO4. Концентрируют при пониженном давлении, получая остаточное масло, которое очищают с помощью колоночной хроматографии при использовании силикагеля, элюируя смесью EtOAc:н-гексан = 1:5, получая трет-бутил[(2R)-2-метоксипропил]метилкарбамат (1,94 г).
ESI (M+Na)+: 226,3.
Получение 57
Смесь трет-бутил[(2R)-2-метоксипропил]метилкарбамата (1,93 г) и 4 н раствора хлористого водорода в EtOAc (4,75 мл) перемешивают в течение 3 часов при комнатной температуре. Растворитель удаляют при пониженном давлении, получая (2R)-2-метокси-N-метилпропан-1-амингидрохлорид (1,35 г).
MS: 104,3.
Получение 58
трет-Бутил[(2S)-2-гидроксипропил]карбамат получают в соответствии с подобной методикой, как таковая в Получении 54.
ESI (M+Na)+: 198,2.
Получение 59
трет-Бутил[(2S)-2-метоксипропил]карбамат получают в соответствии с подобной методикой, как таковая в Получении 55.
ESI (M+Na)+: 212,4.
Получение 60
трет-Бутил[(2S)-2-метоксипропил]метилкарбамат получают в соответствии с подобной методикой, как таковая в Получении 56.
ESI (M+Na)+: 226,3.
Получение 61
(2S)-2-Метокси-N-метилпропан-1-амингидрохлорид получают в соответствии с подобной методикой, как таковая в Получении 57.
MS: 104,3.
Получение 62
К перемешиваемому раствору диизопропиламина (88 мл) в ТГФ (650 мл) добавляют н-бутиллитий (238 мл, 2,64 М раствор в гексане), при температуре -60°С, в атмосфере азота, и смесь перемешивают в течение 30 минут при нагревании до температуры -10°С. Смесь снова охлаждают до температуры -60°С и к смеси добавляют 4-бензилморфолин-3-он в ТГФ (100 мл). Всё перемешивают в течение 30 минут при той же самой температуре. К перемешиваемому раствору этилхлоркарбоната (60 мл) в ТГФ (50 мл), в атмосфере азота и при температуре -60°С, добавляют вышеуказанную смесь при той же самой температуре, и всё перемешивают в течение 30 минут при температуре -40°С. Смесь гасят с помощью буфера с рН 7 и экстрагируют с помощью EtOAc. Экстракт промывают водой и насыщенным солевым раствором и сушат над MgSO4. Концентрируют при пониженном давлении, получая остаточное масло, которое очищают с помощью колоночной хроматографии при использовании силикагеля, элюируя смесью EtOAc:н-гексан = 1:4-1:3, получая диэтил-4-бензил-3-оксоморфолин-2,2-дикарбоксилат (25,0 г).
ESI (M+Na)+: 358,1.
Получение 63
К перемешиваемой суспензии литийалюминийгидрида (8,49 г) в ТГФ (500 мл) добавляют диэтил-4-бензил-3-оксоморфолин-2,2-дикарбоксилат (25 г), при охлаждении на ледяной бане, и смесь перемешивают в течение 1 часа при комнатной температуре. Затем всё вместе нагревают до температуры 65°С (внутренняя температура) и смесь перемешивают в течение 2 часов. Всю реакционную смесь охлаждают на ледяной бане и к смеси, по каплям, добавляют воду (8,5 мл). Всю реакционную смесь перемешивают в течение 15 минут. К смеси добавляют 4 н раствор NaOH (8,5 мл) и всё перемешивают в течение 15 минут. Затем снова к смеси добавляют воду (25,5 мл) и всю реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Осадок белого цвета отфильтровывают и фильтрат концентрируют при пониженном давлении. Остаток растворяют в диизопропиловом эфире и растирают в порошок до получения твердого вещества белого цвета, которое собирают путем фильтрации, получая (4-бензилморфолин-2,2-диил)диметанол (10,3 г).
ESI (M+Na)+: 260,2.
Получение 64
К перемешиваемому раствору (4-бензилморфолин-2,2-диил)диметанола (10,3 г) в ДМФА (103 мл) добавляют NaH (3,82 г; 60%-ная суспензия в масле), при охлаждении на ледяной бане. Смеси дают нагреться до комнатной температуры и перемешивают в течение 20 минут. Всю реакционную смесь охлаждают до температуры 10°С (внутренняя температура) снова при использовании ледяной бани и к смеси, по каплям, добавляют MeI (6,09 мл). Смесь перемешивают в течение 45 минут и гасят с помощью буфера с рН 7. Всё экстрагируют с помощью EtOAc и экстракт промывают с помощью 1 н. раствора HCl (100 мл). Водный слой подщелачивают с помощью водного раствора NaHCO3 и всё экстрагируют с помощью EtOAc. Экстракт промывают водой и насыщенным солевым раствором и сушат над MgSO4. Концентрируют при пониженном давлении, получая остаточное масло (10,1 г), которое очищают с помощью хроматографии при использовании силикагеля, элюируя смесью н-гексан:EtOAc = 20:1 → 4:1, получая 4-бензил-2,2-бис(метоксиметил)морфолин (6,88 г).
MS: 266,4.
Получение 65
К раствору 4-бензил-2,2-бис(метоксиметил)морфолина (6 г) и концентрированной HCl (2,41 мл) в МеОН (60 мл) добавляют 20%-ный гидроксид палладия-на-угле (1,2 г) и всю смесь перемешивают в течение 2 часов при комнатной температуре в атмосфере водорода (2,0 атм). Катализатор отфильтровывают и фильтрат концентрируют при пониженном давлении, получая 2,2-бис(метоксиметил)морфолингидрохлорид (3,6 г). Полученный гидрохлорид разбавляют с помощью CH2Cl2 и промывают с помощью 1 н. раствора NaOH. Органическую фазу концентрируют при пониженном давлении, получая свободную форму, которую используют на следующей реакционной стадии.
MS: 176,2.
Получение 66
К раствору трет-бутил-4-(2-гидроксиэтил)-4-(гидроксиметил)пиперидин-1-карбоксилата (500 мг) в ДМФА (5 мл) добавляют NaH (60%-ная суспензия в масле, 185 мг) и MeI (600 мкл), при охлаждении на ледяной бане, и смесь перемешивают при температуре окружающей среды в течение 2 часов. Реакционную смесь экстрагируют с помощью EtOAc и промывают насыщенным солевым раствором и сушат над MgSO4. Растворитель удаляют в вакууме, получая трет-бутил-4-(2-метоксиэтил)-4-(метоксиметил)пиперидин-1-карбоксилат (460 мг) в виде порошка белого цвета. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
ESI (M+Na)+: 310,3.
Получение 67
К раствору трет-бутил-4-(2-метоксиэтил)-4-(метоксиметил)-пиперидин-1-карбоксилата (400 мг) в EtOAc (7 мл) добавляют 1 н. раствор HCl (7 мл), при охлаждении на ледяной бане, и смесь перемешивают при температуре окружающей среды в течение 3 часов. Реакционную смесь концентрируют в вакууме, получая 4-(2-метоксиэтил)-4-(метоксиметил)пиперидингидрохлорид (260 мг) в виде порошка белого цвета. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
1Н-ЯМР: 1,50-1,74 (4Н, м), 2,49 (4Н, 5 тг, J=1,8 Гц), 2,90-3,07 (3Н, м), 3,20 (2Н, с), 3,21 (3Н, с), 3,26 (3Н, с), 3,34 (2Н, т, J=6,8 Гц), 8,73 (1Н, уш.с).
MS: 188,4.
Получение 68
трет-Бутил-4-метокси-4-(3-метоксипропил)пиперидин-1-карбоксилат получают в соответствии с подобной методикой, как таковая в Получении 66.
1Н-ЯМР: 1,32-1,42 (2Н, м), 1,45 (9Н, с), 1,47-1,64 (2Н, м), 1,69-1,77 (2Н, м), 2,98-3,11 (2Н, м), 3,14 (3Н, с), 3,33 (3Н, с), 3,38 (2Н, т, J=6,0 Гц), 3,68-3,85 (2Н, м).
ESI (M+Na)+: 310,3.
Получение 69
4-Метокси-4-(3-метоксипропил)пиперидингидрохлорид получают в соответствии с подобной методикой, как таковая в Получении 67.
1Н-ЯМР (ДМСО-d6, δ): 1,37-1,51 (2Н, м), 1,53-1,68 (2Н, м), 1,78-1,89 (2Н, м), 2,78-2,93 (2Н, м), 2,98-3,11 (2Н, м), 3,05 (3Н, с), 3,22 (3Н, с), 3,31 (2Н, т, J=5,5 Гц), 8,90 (2Н, уш.с).
MS: 188,4.
Получение 70
К раствору 1-трет-бутил-4-этилпиперидин-1,4-дикарбоксилата (2,57 г) в ТГФ (26 мл) добавляют 0,5 М раствор бис(триметилсилил)амида калия в толуоле (2,40 мл), при температуре -70°С. Спустя 15 минут добавляют хлорметилметиловый эфир (1,2 мл) и смесь перемешивают при температуре 0°С в течение 2 часов. Добавляют воду и экстрагируют с помощью EtOAc. Органическую фазу промывают водой и насыщенным солевым раствором, сушат над MgSO4, выпаривают при пониженном давлении. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля, получая 1-трет-бутил-4-этил-4-(метоксиметил)пиперидин-1,4-дикарбоксилат (2,05 г) в виде масла.
1Н-ЯМР: 1,27 (3Н, т, J=7,2 Гц), 1,46 (9Н, с), 2,04-2,13 (2Н, м), 2,88-3,03 (2Н, м), 3,30 (3Н, с), 3,38 (3Н, с), 3,79-3,91 (2Н, м), 4,20 (2Н, кв., J=7,2 Гц).
ESI (M+Na)+: 324,2.
Получение 71
К раствору 1-трет-бутил-4-этил-4-(метоксиметил)пиперидин-1,4-дикарбоксилата (2,05 г) в толуоле (21 мл) добавляют 0,99 М раствор диизобутилалюминийгидрида в толуоле (16,5 мл), при температуре 0°С. Смесь перемешивают при комнатной температуре в течение 2 часов. Добавляют воду и экстрагируют с помощью EtOAc. Органическую фазу промывают водой и насыщенным солевым раствором, сушат над MgSO4, выпаривают. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля, получая трет-бутил-4-(гидроксиметил)-4-(метоксиметил)пиперидин-1-карбоксилат (0,95 г) в виде масла.
1Н-ЯМР: 1,26-1,57 (4Н, м), 1,45 (9Н, с), 2,20-2,90 (1Н, уш.с), 3,27-3,36 (2Н, м), 3,36 (3Н, с), 3,37 (3Н, с), 3,40-3,55 (2Н, м), 3,61 (2Н, с).
ESI (M+Na)+: 282,3.
Получение 72
трет-Бутил-4,4-бис(метоксиметил)пиперидин-1-карбоксилат получают в соответствии с подобной методикой, как таковая в Получении 66.
1Н-ЯМР: 1,42-1,49 (4Н, м), 1,45 (9Н, с), 3,26 (4Н, с), 3,33 (6Н, с), 3,35-3,41 (4Н, м).
ESI (M+Na)+: 296,4.
Получение 73
4,4-бис(Метоксиметил)пиперидингидрохлорид получают в соответствии с подобной методикой, как таковая в Получении 67.
1Н-ЯМР (ДМСО-d6, δ): 1,59 (4Н, т, J=6,0 Гц), 2,95-3,06 (4Н, м), 3,22 (4Н, с), 3,26 (6Н, с), 8,87 (2Н, уш.с).
MS: 174,4.
Получение 74
К раствору трет-бутил[(2R)-2,3-дигидроксипропил]карбамата (1,05 г) в CH2Cl2 (21 мл) добавляют N,N,N',N'-тетраметилнафталин-1,8-диамин (4,12 г) и молекулярные сита 3Å (2,89 г). К этому раствору, порциями и при охлаждении льдом, добавляют триметилоксонийтетрафторборат (2,84 г). И всю смесь перемешивают при комнатной температуре в течение ночи. К этой смеси добавляют насыщенный водный раствор NaHCO3, нерастворимые вещества отфильтровывают и фильтрат экстрагируют с помощью EtOAc. Органическую фазу промывают два раза с помощью 10%-ного раствора гидросульфата калия с помощью насыщенного водного раствора NaHCO3 и насыщенного солевого раствора, сушат над MgSO4, отфильтровывают и выпаривают. Сырой остаток, содержащий трет-бутил[(2R)-2,3-диметоксипропил]карбамат, используют на следующей стадии без дальнейшей очистки.
1Н-ЯМР: 1,45 (9Н, с), 3,15-3,48 (6Н, м), 3,37 (3Н, с), 3,43 (3Н, с), 4,70-5,00 (1Н, уш.).
Получение 75
трет-Бутил[(2R)-2,3-диметоксипропил]метилкарбамат получают в соответствии с подобной методикой, как таковая в Получении 66.
1Н-ЯМР: 1,46 (9Н, с), 2,92 (3Н, с), 3,22-3,61 (5Н, м), 3,37 (3Н, с), 3,48 (3Н, с).
Получение 76
(2R)-2,3-Диметокси-N-метилпропан-1-амингидрохлорид получают в соответствии с подобной методикой, как таковая в Получении 67.
1Н-ЯМР (ДМСО-d6, δ): 2,52 (3Н, с), 2,87-3,08 (2Н, м), 3,28 (3Н, с), 3,36 (3Н, с), 3,42 (2Н, дд, J=10,6, 4,4 Гц), 3,47 (2Н, дд, J=10,6, 4,6 Гц), 3,68-3,74 (1Н, м), 8,69 (1Н, уш.с), 9,19 (1Н, уш.с).
MS: 134,4.
Получение 77
трет-Бутил[(2S)-2,3-диметоксипропил]карбамат получают в соответствии с подобной методикой, как таковая в Получении 74.
1Н-ЯМР: 1,45 (9Н, с), 3,15-3,48 (6Н, м), 3,37 (3Н, с), 3,43 (3Н, с), 4,70-5,00 (1Н, уш.).
Получение 78
трет-Бутил[(2S)-2,3-диметоксипропил]метилкарбамат получают в соответствии с подобной методикой, как таковая в Получении 66.
1Н-ЯМР: 1,46 (9Н, с), 2,92 (3Н, с), 3,22-3,61 (5Н, м), 3,37 (3Н, с), 3,48 (3Н, с).
Получение 79
(2S)-2,3-Диметокси-N-метилпропан-1-амингидрохлорид получают в соответствии с подобной методикой, как таковая в Получении 67.
1Н-ЯМР (ДМСО-d6, δ): 2,52 (3Н, с), 2,87-3,08 (2Н, м), 3,28 (3Н, с), 3,36 (3Н, с), 3,42 (2Н, дд, J=10,6, 4,4 Гц), 3,47 (2Н, дд, J=10,6, 4,6 Гц), 3,68-3,74 (1Н, м), 8,70 (1Н, уш.с), 9,20 (1Н, уш.с).
MS: 134,4.
Получение 80
К раствору соединения (А) (12,4 г) в ТГФ (60 мл) добавляют моногидрат 4-метилбензолсульфоновой кислоты (10,5 г) и смесь перемешивают при температуре 55°С в течение 22 часов. К смеси добавляют 1 н. раствор NaOH и нейтрализуют, при охлаждении на ледяной бане, и добавляют Вос2О (10,8 г). Значение рН смеси доводят до 8 с помощью 1 н. раствора NaOH, при охлаждении на ледяной бане. Смесь перемешивают при температуре окружающей среды в течение 5 часов. Полученную смесь концентрируют в вакууме и экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью насыщенного солевого раствора и сушат над сульфатом магния. Растворитель удаляют в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля (ацетон:н-гексан = 50:50), получая целевое соединение (7,7 г) в виде аморфного порошка.
MS: 1334,59.
Получение 81
К раствору целевого соединения из Получения 80 (7,7 г) в МеОН (110 мл) добавляют 1 н. раствор NaOH (57 мл), при охлаждении на ледяной бане. После перемешивания в течение 4 часов, при температуре окружающей среды, раствор подкисляют с помощью 1 н. раствора HCl до рН 6,8, концентрируют в вакууме для удаления МеОН и экстрагируют с помощью EtOAc. Органический слой промывают насыщенным солевым раствором, сушат над MgSO4 и концентрируют в вакууме, получая целевое соединение (6,4 г) в виде аморфного порошка. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
MS: 1352,74.
Получение 82
К раствору целевого соединения из Получения 81 (6,4 г) в CH2Cl2 (120 мл) добавляют гидрохлорид метилового эфира L-треонина (1,5 г), HOAt (1,5 г) и WSC (1,7 мл), при охлаждении на ледяной бане, и смесь перемешивают в течение 7 часов при температуре окружающей среды. Полученную смесь промывают с помощью 5%-ного раствора лимонной кислоты и насыщенного солевого раствора и сушат над MgSO4. Растворитель удаляют в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля (ацетон:CH2Cl2 = 50:50), получая целевое соединение (6,5 г) в виде аморфного порошка.
MS: 1467,63.
Получение 83
К раствору целевого соединения из Получения 82 (6,5 г) в CH2Cl2 (120 мл) добавляют коллидин (6 мл) и ацетилхлорид (1,6 мл), при охлаждении на ледяной бане, и смесь перемешивают в течение 3,5 часов при температуре окружающей среды. Полученную смесь промывают с помощью 0,5 н. раствора HCl и насыщенного солевого раствора и сушат над MgSO4. Растворитель удаляют в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля (МеОН:CH2Cl2 = 4:96), получая целевое соединение (5,6 г) в виде аморфного порошка.
MS: 1509,71.
Получение 84
К раствору целевого соединения из Получения 83 (5,5 г) в CH2Cl2 (60 мл) добавляют трифторуксусную кислоту (14 мл), при охлаждении на ледяной бане, и смесь перемешивают в течение 2,5 часов при охлаждении на ледяной бане. Полученную смесь нейтрализуют с помощью водного раствора карбоната калия, при охлаждении на ледяной бане, и концентрируют в вакууме. К остаточному раствору добавляют насыщенный водный раствор NaHCO3 до достижения рН 8 и смесь экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью насыщенного водного раствора NaHCO3 и насыщенного солевого раствора и сушат над MgSO4. Растворитель удаляют в вакууме, получая целевое соединение (5,6 г) в виде твердого вещества. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
MS: 1409,64.
Получение 85
К раствору целевого соединения из Получения 84 (5,5 г) в EtOAc (70 мл) добавляют раствор изотиоцианатобензола (940 мкл) и DIPEA (2,0 мл), при температуре окружающей среды. После перемешивания в течение 3 часов, при той же самой температуре, к раствору добавляют N,N-диметиламинопропиламин (1,24 мл). Раствор перемешивают в течение 0,5 часа. Раствор промывают с помощью 0,5 н. раствора HCl, водного раствора NaHCO3 и насыщенного солевого раствора, сушат над MgSO4 и концентрируют в вакууме, получая целевое соединение (5,5 г) в виде твердого вещества. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
MS: 1544,95.
Получение 86
К целевому соединению из Получения 85 (2,8 г), растворенному в MeCN (30 мл), добавляют 1 н. раствор HCl (18 мл), при охлаждении на ледяной бане. После перемешивания при температуре 30°С в течение 2 часов, раствор нейтрализуют с помощью 1 н. раствора NaOH (19 мл), концентрируют в вакууме для удаления MeCN и экстрагируют с помощью EtOAc (150 мл). Органическую фазу промывают с помощью водного раствора NaHCO3 и насыщенного солевого раствора, сушат над MgSO4 и концентрируют в вакууме. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля (МеОН:CH2Cl2 = 10:90), получая целевое соединение (2,1 г) в виде аморфного порошка.
MS: 1308,42.
Получение 87
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 85.
MS: 1443,62.
Получение 88
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 86.
MS: 1237,58.
Получение 89
К раствору целевого соединения из Получения 88 (670 мг) в CH2Cl2 (15 мл) добавляют (2R)-2-{[(9H-флуорен-9-илметокси)карбонил](метил)амино}пропановую кислоту (265 мг), 1-гидрокси-7-азабензотриазол (221 мг) и WSC (194 мкл), при охлаждении на ледяной бане, и смесь перемешивают при температуре 5°С в течение 4 часов. Реакционную смесь концентрируют в вакууме и остаток экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью 1 н. раствора HCl, насыщенного водного раствора NaHCO3 и насыщенного солевого раствора и сушат над MgSO4. Растворитель удаляют в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля (ацетон:гексан = 45:55), получая целевое соединение (574 мг) в виде аморфного порошка.
MS: 1544,57.
Получение 90
К раствору целевого соединения из Получения 89 (574 мг) в диоксане (6 мл) добавляют 1 н. раствор гидроксида лития (1,5 мл), при температуре окружающей среды, и смесь перемешивают в течение 2 часов. К реакционной смеси добавляют 5%-ный водный раствор лимонной кислоты для достижения рН 5 и раствор экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью насыщенного солевого раствора и сушат над MgSO4. Растворитель удаляют в вакууме, получая целевое соединение (200 мг) в виде твердого вещества. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
MS: 1266,60.
Получение 91
К раствору целевого соединения из Получения 90 (178 мг) в CH2Cl2 (141 мл) добавляют 1-гидрокси-7-азабензотриазол (23 мг) и WSC (27 мкл), при охлаждении на ледяной бане, и смесь перемешивают при температуре окружающей среды в течение 15 часов. Реакционную смесь промывают с помощью 0,5 н. раствора HCl, насыщенного водного раствора NaHCO3 и насыщенного солевого раствора и сушат над MgSO4. Растворитель удаляют в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля (ацетон:CH2Cl2 = 50:50), получая целевое соединение (102 мг) в виде аморфного порошка.
MS: 1248,59.
Время удерживания: 5,4 мин.
(ВЭЖХ, колонка: Shiseido UG120 C18, 100 мм × 4,6 мм - внутренний диаметр, элюент: 60% MeCN/Н2О, объемная скорость потока: 1,0 мл/мин).
Получение 92
К раствору целевого соединения из Получения 91 (102 мг) в CH2Cl2 (5 мл) добавляют 4-нитрофенилхлорформиат (131 мг) и N-метилморфолин (72 мкл), при охлаждении на ледяной бане. Затем смесь перемешивают при комнатной температуре в течение ночи, смесь разбавляют с помощью EtOAc, промывают с помощью 1 н. раствора HCl и водного раствора NaHCO3, сушат над MgSO4 и концентрируют. Растворитель удаляют в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля (ацетон:CH2Cl2 = 50:50), получая целевое соединение (97 мг) в виде аморфного порошка.
MS: 1413,34.
Получение 93
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 28.
MS: 1246,99.
Получение 94
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 28.
MS: 1277,01.
Получение 95
Целевое соединение, которое используют на следующей стадии без дальнейшей очистки, получают в соответствии с подобной методикой, как таковая в Получении 119.
MS: 1490,48.
Получение 96
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 121.
Конфигурацию в положении 3 этого соединения определяют как (R) при сравнении со спектром ВЭЖХ аутентичного образца, чья конфигурация в положении 3 подтверждена как (R), синтезированного альтернативным способом синтеза, как описано ниже (Получение 154).
1Н-ЯМР: 0,70 (3H, д, J=6,8 Гц), 0,81 (3H, д, J=6,5 Гц), 0,83 (3H, д, J=6,5 Гц), 0,84-0,92 (9H, м), 0,92-1,08 (8H, м), 1,12 (6H, т-подобный, J=6,6 Гц), 1,20-1,48 (10H, м), 1,48-1,65 (3H, м), 1,66 (3H, д, J=5,2 Гц), 1,67-2,40 (23H, м), 2,43 (1H, д, J=4,9 Гц), 2,91 (3H, с), 3,00 (3H, с), 3,03 (3H, с), 3,07 (3H, с), 3,08 (3H, с), 3,22 (3H, с), 3,35-3,45 (2H, м), 3,45-3,55 (2H, м), 3,73 (1H, м), 4,05-4,40 (2H, м), 4,52 (1H, т-подобный, J=7,0 Гц), 4,70 (1H, т-подобный, J=7,0 Гц), 4,70-4,90 (2H, м), 4,90-5,02 (2H, м), 5,02-5,12 (3H, м), 5,12-5,58 (3H, м), 5,65 (1H, д, J=3,1 Гц), 6,69 (1H, д, J=8,0 Гц), 6,89 (1H, д, J=7,5 Гц), 7,00 (1H, д, J=9,0 Гц), 7,73 (1H, д, J=9,0 Гц), 9,03 (1H, д, J=9,2 Гц).
MS: 1262,30.
Время удерживания: 5,9 мин.
(чистота 94%; ВЭЖХ, колонка: Shiseido UG120 C18, 100 мм × 4,6 мм - внутренний диаметр, элюент: 60% MeCN/Н2О, объемная скорость потока: 1,0 мл/мин).
Получение 97
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 2.
MS: 1427,37.
Получение 98
К раствору N,N-диизопропиламина (0,96 мл) в ТГФ (15 мл) по каплям добавляют 1,5 М раствор н-бутиллития в гексане (4,6 мл), при температуре -20°С, и смесь перемешивают в течение 5 минут при той же самой температуре. После охлаждения до температуры -78°С, к смеси по каплям добавляют раствор исходного соединения (1,0 г) в ТГФ (10 мл), в течение 10 минут, и всю смесь перемешивают в течение 15 минут. К полученному раствору желтого цвета порциями добавляют аллилиодид (0,63 мл), при той же самой температуре, и смесь постепенно нагревают до температуры 5°С. После перемешивания при температуре 5°С в течение 5 минут, реакционную смесь гасят водой и экстрагируют с помощью EtOAc. Органическую фазу сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (гексан/ацетон = 7/3), получая 0,52 г целевого промежуточного продукта в виде смеси с исходным соединением. Этот сырой продукт растворяют в МеОН (15 мл) и обрабатывают с помощью 1 н. водного раствора HCl (7 мл). После перемешивания при комнатной температуре в течение 2 часов, смесь разбавляют водой и три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2/ацетон = 1/1), получая 116 мг целевого соединения.
MS: 1274,32;
Время удерживания: 6,2 мин;
(чистота: 95%, ВЭЖХ, колонка: Shiseido UG120 C18, 100 мм × 4,6 мм - внутренний диаметр, элюент: 60%-ная смесь MeCN/Н2О, объемная скорость потока: 1,0 мл/мин).
Получение 99
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 2.
MS: 1439,91.
Получение 100
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 89.
MS: 1498,9.
Получение 101
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 90.
MS: 1342,7.
Получение 102
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 91.
MS: 1324,7.
Получение 103
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 92.
MS: 1489,8.
Получение 104
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 89.
MS: 1466,9.
Получение 105
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 90.
MS: 1310,7.
Получение 106
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 91.
MS: 1292,7.
Получение 107
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 92.
MS: 1457,8.
Получение 108
К раствору целевого соединения из Получения 119 (500 мг) в пиридине (10 мл) по каплям добавляют уксусный ангидрид (158 мкл) и смесь перемешивают при комнатной температуре в течение ночи. Смесь разбавляют водой и экстрагируют с помощью EtOAc. Органическую фазу дважды промывают водой, сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (гексан/ацетон = 7/3), получая 116 мг целевого соединения.
MS: 1535,01.
Получение 109
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 121.
MS: 1306,79.
Получение 110
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 2.
MS: 1471,84.
Получение 111
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 119 (используют иодэтан вместо параформальдегида).
MS: 1519,70.
Получение 112
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 121.
MS: 1291,74.
Получение 113
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 2.
MS: 1456,64.
Получение 114
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 30.
MS: 1401,46.
Получение 115
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 36.
MS: 1272,45.
Получение 116
К раствору целевого соединения из Получения 115 в ТГФ (15 мл) добавляют воду (0,2 мл) и PPh3 (326 мг) и смесь нагревают при температуре 70°С в течение 2 часов. Смесь охлаждают, концентрируют и выпаривают совместно с толуолом. Остаток хроматографируют на силикагеле (CH2Cl2/МеОН = от 10/0 до 4/1), получая 461 мг целевого соединения.
MS: 1247,84.
Получение 117
К раствору целевого соединения из Получения 116 (46 мг) в EtOH (10 мл) добавляют ацетальдегид (0,1 мл) и смесь перемешивают при комнатной температуре в течение 0,5 часа. После концентрирования в вакууме до 1/10 объема, смесь разбавляют с помощью EtOH (5 мл). К смеси порциями добавляют боргидрид натрия (10 мг) и всё вместе перемешивают при комнатной температуре в течение 0,5 часа. Реакционную смесь гасят с помощью водного раствора NaHCO3 и шесть раз экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют, получая 33 мг целевого соединения, которое используют на следующей стадии без дальнейшей очистки.
MS: 1275,83.
Получение 118
К раствору соединения (А) (10,0 г) и 1Н-имидазола (5,5 г) в ДМФА (100 мл) добавляют, при комнатной температуре, трет-бутилхлордиметилсилан (9,8 г). После перемешивания в течение 21 часа при комнатной температуре, реакционную смесь выливают в смесь EtOAc и воды. Органический слой последовательно промывают насыщенным солевым раствором и сушат над MgSO4. Растворитель выпаривают в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля, элюируя смесью гексана и ацетона (от 100:0 → 50:50). Элюированные фракции, содержащие желательный продукт, собирают и выпаривают в вакууме, получая целевое соединение (11,5 г) в виде бесцветного аморфного вещества.
Получение 119
К раствору N,N-диизопропиламина (4,8 мл) в ТГФ (100 мл) по каплям добавляют 1,5 М раствор н-бутиллития в гексане (4,8 мл), при температуре -20°С, и смесь перемешивают в течение 5 минут при той же самой температуре. После охлаждения до температуры -78°С, к смеси добавляют по каплям раствор целевого соединения из Получения 118 (5,0 г) в ТГФ (20 мл), в течение 10 минут, и всю совокупность перемешивают в течение 15 минут. К полученному раствору желтого цвета добавляют, в виде порций, параформальдегид (616 мг), при той же самой температуре, и смесь постепенно нагревают до комнатной температуры. После перемешивания при комнатной температуре в течение 1,5 часов, реакционную смесь гасят водой и экстрагируют с помощью EtOAc. Органическую фазу сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (гексан/ацетон = 7/3), получая 2,20 г целевого соединения, в виде смеси с соответствующим бисгидроксиметилированным соединением, которое используют на следующей стадии без дальнейшей очистки.
MS: 1492,39;
Время удерживания: 6,8 мин.
(чистота: 60%; ВЭЖХ, колонка: YMC-C8 AS-202, 150 мм × 4,6 мм - внутренний диаметр, элюент: 100% MeCN, объемная скорость потока: 1,0 мл/мин).
Получение 120
К раствору целевого соединения из Получения 119 (101,7 г) в CH2Cl2 (2,03 л) добавляют N,N,N',N'-тетраметилнафталин-1,8-диамин (87,6 г) и молекулярные сита 3 Å (55 г). К этой смеси, порциями и при охлаждении льдом, добавляют триметилоксонийтетрафторборат (50,4 г). И всё перемешивают при комнатной температуре в течение 5 часов. Затем нерастворимые вещества отфильтровывают, к фильтрату добавляют насыщенный водный раствор NaHCO3 и CH2Cl2 выпаривают. Остаток экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью 5%-ного водного раствора лимонной кислоты (2 раза), насыщенного водного раствора NaHCO3 и насыщенного солевого раствора, сушат над MgSO4, отфильтровывают и выпаривают. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля (CH2Cl2/ацетон = CH2Cl2 только до 70/30), получая целевое соединение (51,8 г) в виде порошка белого цвета.
MS: 1507,00.
Получение 121
К раствору целевого соединения из Получения 120 (5,9 г) в МеОН (59 мл) добавляют 1 н. раствор HCl (29,5 мл) и смесь перемешивают при комнатной температуре в течение 5 часов. После удаления метанола в вакууме, остаток экстрагируют с помощью CH2Cl2. Экстракт промывают насыщенным солевым раствором, сушат над MgSO4 и концентрируют в вакууме. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля (CH2Cl2:ацетон = CH2Cl2 только до 50:50), получая целевое соединение (3,5 г) в виде порошка белого цвета.
Конфигурацию в положении 3 целевого соединения определяют как (R) при сравнении со спектром ВЭЖХ аутентичного образца, чья конфигурация в положении 3 подтверждена как (R), синтезированного путем альтернативного способа синтеза, как описано ниже (Получение 158).
1Н-ЯМР: 0,70 (3H, д, J=6,7 Гц), 0,75 (1H, т, J=5,6 Гц), 0,81 (6H, д, J=6,4 Гц), 0,87 (3H, д, J=6,8 Гц), 0,89 (3H, д, J=6,8 Гц), 0,90 (3H, д, J=7,0 Гц), 0,93 (3H, д, J=6,2 Гц), 0,94 (3H, д, J=6,4 Гц), 0,97-1,03 (9H, м), 1,10 (3H, д, J=6,8 Гц), 1,12 (3H, д, J=7,0 Гц), 1,26 (3H, с), 1,30-1,36 (5H, м), 1,52-1,65 (2H, м), 1,66 (3H, д, J=5,5 Гц), 1,70-2,15 (4H, м), 2,15-2,22 (6H, м), 2,44 (1H, д, J=4,8 Гц), 2,63 (2H, с), 2,91 (3H, с), 3,01 (3H, с), 3,06 (3H, с), 3,08 (3H, с), 3,09 (3H, с), 3,17 (3H, с), 3,37 (6H, с), 3,44-3,51 (1H, м), 3,55 (1H, дд, J=6,4, 9,8 Гц), 3,59-3,63 (1H, м), 3,75 (2H, с), 4,17-4,31 (2H, м), 4,52 (1H, дд, J=7,0, 9,0 Гц), 4,70 (тН, 6,9), 4,75-4,89 (2H, м), 4,91-5,03 (3H, м), 5,08 (1H, дд, J=5,0, 10,1 Гц), 5,15 (1H, дд, J=4,2, 11,8 Гц), 5,27-5,56 (4H, м), 5,65 (1H, д, J=3,2 Гц), 6,76 (1H, д, J=7,7 Гц), 6,86 (1H, д, J=7,7 Гц), 7,00 (1H, д, J=9,2 Гц), 7,67 (1H, д, J=9,2 Гц), 8,92 (1H, д, J=9,4 Гц)
MS: 1278,63.
Время удерживания: 5,0 мин.
(ВЭЖХ, колонка: Shiseido UG120 C18, 100 мм × 4,6 мм - внутренний диаметр, элюент: 60% MeCN/Н2О, объемная скорость потока: 1,0 мл/мин).
Получение 122
К перемешиваемому раствору целевого соединения из Получения 119 (13,5 г) в MeCN (270 мл) добавляют DMAP (9,94 г) и О-(4-фторфенил)хлортиокарбонат (7,76 г) и смесь перемешивают в течение 4 часов. Растворитель удаляют при пониженном давлении. Остаток разбавляют с помощью EtOAc и промывают с помощью 1 н. раствора HCl, воды, водного раствора NaHCO3 и насыщенного солевого раствора и сушат над MgSO4. Концентрируют при пониженном давлении, получая остаток, который очищают с помощью колоночной хроматографии при использовании силикагеля, элюируя смесью ацетон/CH2Cl2 = 0/100 → 50/50, получая целевое соединение (6,04 г).
MS: 1647,16.
Получение 123
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 92.
MS: 1413,34.
Получение 124
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 92.
MS: 1443,87.
Получение 125
К раствору соединения (А) (20 г) в 1,2-диметоксиэтане (200 мл) добавляют 4-метилбензолсульфоновую кислоту (7,7 г) и смесь перемешивают при температуре 50°С в течение 14 часов. К смеси добавляют 1 н. раствор NaOH и нейтрализуют, при охлаждении на ледяной бане, и добавляют ди-трет-бутилдикарбонат (8,8 г). Значение рН смеси доводят до 8 с помощью 1 н. раствора NaOH, при охлаждении на ледяной бане. Смесь перемешивают при температуре окружающей среды в течение 2,5 часов. Полученную смесь концентрируют в вакууме и экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью насыщенного водного раствора карбоната натрия, 0,1 н. раствора HCl и насыщенного солевого раствора и сушат над сульфатом натрия. Растворитель удаляют в вакууме, получая целевое соединение (12,2 г) в виде пены коричневого цвета. Полученный сырой продукт используют на следующей реакционной стадии без дальнейшей очистки.
MS: 1334,7.
Получение 126
К раствору целевого соединения из Получения 125 (5,1 г) в МеОН (50 мл) добавляют метоксид натрия (1,24 г) в МеОН (50 мл) и смесь перемешивают в течение 14 часов. К смеси добавляют 10%-ный водный раствор лимонной кислоты, экстрагируют с помощью EtOAc. Органическую фазу промывают насыщенным солевым раствором и сушат над сульфатом натрия. Растворитель удаляют в вакууме, получая пену коричневого цвета. Полученный сырой продукт очищают с помощью хроматографии при использовании силикагеля (CH2Cl2:ацетон = 60:40), получая целевое соединение (5,23 г).
MS: 1366,7.
Получение 127
К раствору целевого соединения из Получения 126 (6,3 г) в CH2Cl2 (64 мл) добавляют 2,4,6-триметилпиридин (1,24 г) и ацетилхлорид (0,5 мл) и смесь перемешивают в течение 14 часов. Смесь разбавляют с помощью CH2Cl2 и промывают с помощью 10%-ного водного раствора лимонной кислоты и насыщенного солевого раствора и сушат над сульфатом натрия. Растворитель удаляют в вакууме, получая целевое соединение (6,28 г).
MS: 1408,70.
Получение 128
Целевое соединение из Получения 127 (6,28 г) растворяют в 10%-ном растворе трифторуксусной кислоты в CH2Cl2 (63 мл), при охлаждении на ледяной бане. После перемешивания при той же самой температуре в течение 2 часов, к реакционному раствору добавляют 1 М водный раствор NaHCO3 до достижения рН 8. Реакционную смесь экстрагируют с помощью CHCl3 и органический слой промывают с помощью насыщенного водного раствора NaHCO3 и насыщенного солевого раствора, сушат над MgSO4 и концентрируют в вакууме, получая целевое соединение (5,65 г).
MS: 1308,69.
Получение 129
К раствору целевого соединения из Получения 128 (3 г) в MeCN (45 мл) добавляют изотиоцианатобензол (0,41 мл), при температуре окружающей среды, и для достижения значения рН смеси, равного 7,5, добавляют DIPEA (0,12 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 1,5 часов. К полученному раствору добавляют N,N-диметилпропандиамин (0,19 мл) и перемешивают в течение 5 минут, затем добавляют 1 н. раствор HCl (45 мл), и смесь перемешивают при температуре 30°С в течение 2 часов. Полученную смесь нейтрализуют с помощью раствора карбоната натрия (3,8 г в 100 мл воды) и концентрируют в вакууме. Значение рН остаточного раствора доводят до 8 с помощью насыщенного водного раствора NaHCO3 и раствор экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью насыщенного водного раствора NaHCO3 и насыщенного солевого раствора и сушат над сульфатом натрия. Растворитель удаляют в вакууме, получая целевое соединение (1,82 г).
MS: 1207,58.
Получение 130
К раствору целевого соединения из Получения 129 (1,58 г) в MeCN (23 мл) добавляют изотиоцианатобензол (0,23 мл), при температуре окружающей среды, и значение рН смеси доводят до 7,5 с помощью диизопропилэтиламина (0,068 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 1,5 часов. К полученному раствору добавляют N,N-диметилпропандиамин (0,33 мл) и перемешивают в течение 5 минут, затем добавляют 1 н. раствор HCl (23 мл) и смесь перемешивают при температуре 30°С в течение 2 часов. Полученную смесь нейтрализуют с помощью раствора карбоната натрия (3,8 г в 100 мл воды) и концентрируют в вакууме. Значение рН остаточного раствора доводят до 8 с помощью насыщенного водного раствора NaHCO3 и раствор экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью насыщенного водного раствора NaHCO3 и насыщенного солевого раствора и сушат над сульфатом натрия. Растворитель удаляют в вакууме, получая целевое соединение (1,1 г) в виде пены бледно-желтого цвета.
MS: 1136,5.
Получение 131
К раствору целевого соединения из Получения 130 (390 мг) добавляют N-(трет-бутоксикарбонил)-N-этил-D-аланин (149 мг), бис(2-оксо-3-оксазолидинил)фосфинийхлорид (262 мг) и диизопропилэтиламин (358 мкл), при охлаждении на ледяной бане. Смесь перемешивают в течение 13 часов при температуре окружающей среды и экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью 10%-ного водного раствора лимонной кислоты, насыщенного водного раствора NaHCO3 и насыщенного солевого раствора и сушат над сульфатом натрия. Растворитель удаляют в вакууме и остаток растворяют в 10%-ном растворе трифторуксусной кислоты в CH2Cl2 (5,7 мл), при охлаждении на ледяной бане. После перемешивания при той же самой температуре в течение 2 часов, к реакционному раствору добавляют 1 М водный раствор NaHCO3 до достижения значения рН 8. Реакционную смесь экстрагируют с помощью CHCl3 (50 мл) и органический слой промывают с помощью насыщенного водного раствора NaHCO3 и насыщенного солевого раствора, сушат над MgSO4 и концентрируют в вакууме, получая целевое соединение (0,38 г).
MS: 1235,6.
Получение 132
К раствору целевого соединения из Получения 131 (106 мг) добавляют 1-гидрокси-7-азабензотриазол (23 мг), N-(трет-бутоксикарбонил)-L-треонил-N-этил-D-аланин (37,6 мг) и О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (65 мг) и диизопропилэтиламин (276 мкл), при охлаждении на ледяной бане. Смесь перемешивают в течение 13 часов при температуре окружающей среды и экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью 10%-ного водного раствора лимонной кислоты, насыщенного водного раствора NaHCO3 и насыщенного солевого раствора и сушат над сульфатом натрия. Растворитель удаляют в вакууме и остаток очищают с помощью препаративной тонкослойной хроматографии (CHCl3:МеОН = 90:10), получая целевое соединение (0,13 г).
MS: 1436,86.
Получение 133
Целевое соединение из Получения 132 (130 мг) растворяют в 10%-ном растворе трифторуксусной кислоты в CH2Cl2 (2,6 мл), при охлаждении на ледяной бане. После перемешивания при той же самой температуре в течение 2 часов, к реакционному раствору добавляют 1 М водный раствор NaHCO3 до достижения значения рН 8. Реакционную смесь экстрагируют с помощью CHCl3 (50 мл) и органический слой промывают с помощью насыщенного водного раствора NaHCO3 и насыщенного солевого раствора, сушат над MgSO4 и концентрируют в вакууме. К полученному остатку в ТГФ (1,5 мл) добавляют 1 н. раствор NaOH (0,015 мл), при температуре окружающей среды, и смесь перемешивают в течение 2 часов. К реакционной смеси добавляют 10%-ный водный раствор лимонной кислоты до достижения рН=4 и раствор экстрагируют с помощью EtOAc. Органическую фазу промывают насыщенным солевым раствором и сушат над сульфатом натрия. Растворитель удаляют в вакууме и остаток растирают в Et2O, получая целевое соединении (0,1 г).
MS: 1280,7.
Получение 134
К раствору целевого соединения из Получения 133 (100 мг) в CH2Cl2 (80 мл) добавляют 1-гидрокси-7-азабензотриазол (21 мг) и WSC (30 мг), при охлаждении на ледяной бане, и смесь перемешивают при температуре 5°С в течение 13 часов. Реакционную смесь концентрируют в вакууме и остаток экстрагируют с помощью EtOAc. Органическую фазу промывают водой, 10%-ным водным раствором лимонной кислоты, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором и сушат над сульфатом натрия. Растворитель удаляют в вакууме и остаток очищают с помощью препаративной тонкослойной хроматографии (CHCl3:МеОН = 95:5), получая целевое соединение (46 мг).
MS: 1262,66.
Получение 135
К раствору целевого соединения из Получения 134 (46 мг) в CH2Cl2 (1 мл) добавляют 4-нитрофенилхлорформиат (14 мг) и N-метилморфолин (9 мкл). Затем смесь перемешивают в течение ночи. После того как исходное соединение израсходуется, смесь разбавляют с помощью EtOAc, промывают 1 н. водным раствором HCl и водным раствором NaHCO3, сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (гексан/EtOAc = 1/4 и затем CH2Cl2/МеОН = 9/1), получая целевое соединение (50 мг).
MS: 1427,2.
Получение 136
Целевое соединение, которое используют на следующей стадии без дальнейшей очистки, получают в соответствии с подобной методикой, как таковая в Получении 92, затем непрерывно, как в Примере 74.
Получение 137
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 81.
MS: 1478,51.
Получение 138
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 82.
MS: 1593,65.
Получение 139
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 84, затем непрерывно, как в Получении 85.
MS: 1629,09.
Получение 140
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 86, затем непрерывно, как в Получении 85.
MS: 1528,13.
Получение 141
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 86.
MS: 1322,04.
Получение 142
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 89.
MS: 1628,88.
Получение 143
Целевое соединение, которое используют на следующей стадии без дальнейшей очистки, получают в соответствии с подобной методикой, как таковая в Получении 90 (NaOH используют вместо гидроксида лития).
Получение 144
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 89.
ESI MS (М+2Н+)/2: 822,10.
Получение 145
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 90 (NaOH используют вместо гидроксида лития).
MS: 1407,02.
Получение 146
К раствору целевого соединения из Получения 118 (5,0 г) и MeI (426 мкл) в ДМФА (100 мл) добавляют NaH (273 мг), при охлаждении льдом. После перемешивания в течение 3 часов, при охлаждении льдом, реакционную смесь выливают в смесь EtOAc и воды. Органический слой последовательно промывают насыщенным солевым раствором и сушат над MgSO4. Растворитель выпаривают в вакууме и остаток очищают с помощью колоночной хроматографии при использовании силикагеля, элюируя смесью гексана и EtOAc (100:0 → 50:50). Элюированные фракции, содержащие желательный продукт, собирают и выпаривают в вакууме, получая целевое соединение (4,23 г) в виде бесцветного аморфного вещества.
MS: 1477,09.
Получение 147
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 119.
MS: 1507,10.
Получение 148
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 120.
MS: 1538,27.
Получение 149
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 121.
MS: 1292,99.
Получение 150
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 92.
MS: 1457,99.
Получение 151
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 89.
MS: 1437,07.
Получение 152
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 157.
MS: 1336,78.
Получение 153
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 90.
MS: 1280,82.
Получение 154
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 91.
MS: 1262,27.
Время удерживания: 5,9 мин.
(ВЭЖХ, колонка: Shiseido UG120 C18, 100 мм × 4,6 мм - внутренний диаметр, элюент: 60% MeCN/Н2О, объемная скорость потока: 1,0 мл/мин).
Получение 155
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 89.
MS: 1452,90.
Получение 156
К раствору целевого соединения из Получения 155 (46 мг) в CH2Cl2 (1,6 мл) добавляют трифторуксусную кислоту (0,4 мл), при температуре 0°С, и смесь перемешивают при той же самой температуре в течение 3 часов. Смесь разбавляют водой, устанавливают рН 9 с помощью водного раствора NaHCO3 и три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют, получая целевое соединение (31 мг).
MS: 1352,82.
Получение 157
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 90.
MS: 1296,29.
Получение 158
Целевое соединение получают в соответствии с подобной методикой, как таковая в Получении 91.
MS: 1278,29.
Время удерживания: 5,0 мин.
(ВЭЖХ, колонка: Shiseido UG120 C18, 100 мм × 4,6 мм - внутренний диаметр, элюент: 60% MeCN/Н2О, объемная скорость потока: 1,0 мл/мин).
Пример 1
К раствору основного целевого соединения из Получения 2 (50 мг) в ДМФА (1 мл) добавляют морфолин (16 мкл) и смесь перемешивают при комнатной температуре в течение ночи. Смесь подвергают ODS-очистке, получая 35 мг целевого соединения.
Пример 12
К раствору основного целевого соединения из Получения 2 (50 мг) в ДМФА (2 мл) добавляют N-этилметиламин (15 мкл) и смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь гасят с помощью водного раствора NaHCO3 и экстрагируют с помощью EtOAc. Органическую фазу дважды промывают с помощью водного раствора NaHCO3, сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2/МеОН = от 95/5 до 90/10), получая 46 мг целевого соединения.
Пример 18
К раствору основного целевого соединения из Получения 2 (50 мг) в ДМФА (0,4 мл) добавляют 5,6,7,8-тетрагидроимидазо[1,2-a]пиразиндигидрохлорид (21 мг) и DIPEA (31 мкл) и смесь перемешивают при комнатной температуре в течение ночи. Смесь подвергают ODS-очистке, получая 33 мг целевого соединения.
Пример 21
К раствору целевого соединения из Примера 40 (его структура представлена в Таблице 3) (379 мг) в EtOAc по каплям добавляют 4 н. раствор хлористого водорода в диоксане и смесь перемешивают при комнатной температуре в течение 1,5 часов. Смесь подщелачивают с помощью водного раствора NaHCO3 и три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток подвергают ODS-очистке, получая 92 мг целевого соединения.
Пример 22
К раствору целевого соединения из Примера 41 (73 мг) в ДМФА (3 мл) последовательно добавляют (диметиламино)уксусную кислоту (20 мг), О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний-гексафторфосфат (31 мг) и DIPEA (30 мкл) и смесь перемешивают при комнатной температуре в течение 1 часа. Смесь разбавляют с помощью EtOAc, три раза промывают водой, сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2/МеОН = от 100/0 до 95/5), получая 57 мг целевого соединения.
Пример 34
К раствору целевого соединения из Примера 13 (его структура представлена в Таблице 1) (184 мг) в смешанном растворителе из CH2Cl2 (8 мл) и МеОН (2 мл) последовательно добавляют 35%-ный водный раствор формальдегида (90 мкл) и триацетоксиборгидрид натрия (85 мл) и смесь перемешивают при комнатной температуре в течение 2,5 часов. Смесь подщелачивают с помощью насыщенного водного раствора NaHCO3 и три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2/МеОН = от 100:0 до 90:10), получая 180 мг целевого соединения.
Пример 43
К раствору целевого соединения из Получения 28 (38 мг) и морфолина (10 мкл) в CH2Cl2 добавляют триацетоксиборгидрид натрия (30 мг) и смесь перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь гасят с помощью водного раствора NaHCO3 и три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток подвергают ODS-очистке, получая 20 мг целевого соединения.
Пример 44
Смесь целевого соединения из Получения 32 (90 мг) в EtOH (1,8 мл) обрабатывают с помощью 1 н. раствора HCl (600 мкл) при комнатной температуре в течение 12 часов. Реакционную смесь концентрируют в вакууме. Остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 1:1), получая 55,8 мг целевого соединения.
Пример 45
Смесь целевого соединения из Получения 35 (125 мг) в EtOH (2,1 мл) обрабатывают с помощью 6 н. раствора HCl (700 мкл) при комнатной температуре в течение 12 часов. Реакционную смесь концентрируют в вакууме. Остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 1:1), получая 107 мг целевого соединения.
Пример 46
Смесь целевого соединения из Получения 38 (57 мг) в МеОН (2,1 мл) обрабатывают с помощью 6 н. раствора HCl (0,7 мл) при комнатной температуре в течение 30 минут. Реакционную смесь выливают в раствор боргидрида натрия (18 мг) в EtOH (1,2 мл), при температуре 0°С. К смеси добавляют ТЕА (200 мкл) и раствор концентрируют. Остаток хроматографируют на силикагеле (МеОН:CH2Cl2 (0,2% ТЕА) = от 0:100 до 1:9), получая 46 мг целевого соединения.
Пример 47
Смесь целевого соединения из Получения 33 (64 мг) в EtOH (1,8 мл) обрабатывают с помощью 1 н. раствора HCl (600 мкл) при комнатной температуре в течение 12 часов. После обработки с помощью ТЕА (100 мкл), смесь концентрируют. Остаток очищают с помощью препаративной ВЭЖХ (MeCN:вода = от 40:60 до 25:75), получая 44,5 мг целевого соединения.
Пример 48
Смесь целевого соединения из Получения 30 (69,4 мг) в ДМФА (1,0 мл) обрабатывают с помощью 1,2,3-тиадиазол-5-тиолята натрия (27 мг) при комнатной температуре в течение 12 часов. После обработки уксусной кислотой (10 мкл), смесь концентрируют. Остаток очищают с помощью препаративной ВЭЖХ (CapcellPak UG; MeCN/вода = от 2/8 до 7/3), получая 53,7 мг целевого соединения.
Пример 49
Смесь целевого соединения из Получения 30 (694 мг) в ДМФА (10 мл) обрабатывают с помощью тиобензойной кислоты (207 мг) и карбоната калия (207 мг) при комнатной температуре в течение 12 часов. После обработки уксусной кислотой (70 мкл), смесь концентрируют. Остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 45:55), получая 607 мг целевого соединения.
Пример 50
Смесь целевого соединения из Примера 49 (68 мг) в EtOH (1,3 мл) обрабатывают с помощью 2,6 М раствора этоксида натрия в EtOH (29 мкл) при комнатной температуре в течение 30 минут. К раствору добавляют бензилбромид (18 мкл). После перемешивания в течение 1 часа, смесь обрабатывают уксусной кислотой (10 мкл). Смесь концентрируют и остаток хроматографируют на силикагеле (ацетон:н-гексан = от 1:9 до 45:55), получая 59,8 мг целевого соединения.
Пример 51
К смеси целевого соединения из Получения 34 (64 мг) в CH2Cl2 (0,5 мл) при температуре 0°С добавляют 1,0 М раствор диметиламинопиридина в CH2Cl2 (100 мкл) и 1,0 М раствор 4-нитрофенилхлорформиата в CH2Cl2 (100 мкл). После перемешивания при температуре 0°С в течение 30 минут, смесь обрабатывают с помощью морфолина (44 мкл) при комнатной температуре в течение 30 мин. К смеси добавляют уксусную кислоту (30 мкл) и раствор концентрируют. Остаток очищают с помощью препаративной ВЭЖХ (CapcellPak UG; MeCN/вода = от 2/8 до 7/3), получая 36,3 мг целевого соединения.
Пример 54
К раствору целевого соединения из Получения 39 (37 мг), 4-метоксибензойной кислоты (23 мг) и 1-гидроксибензотриазола (24 мг) в N-метилпирролидиноне (0,6 мл) добавляют DIPEA (21 мкл) и этил-WSC-гидрохлорид (29 мг) и смесь перемешивают при комнатной температуре в течение 4 часов. Смесь разбавляют с помощью EtOAc, промывают водой и насыщенным солевым раствором, сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (МеОН:CH2Cl2 (0,2% ТЕА) = от 2:98 до 8:92), получая 36 мг целевого соединения.
Пример 64
К раствору целевого соединения из Получения 22 (200 мг) в MeCN (5 мл) последовательно, при температуре 0°С, добавляют (триметилсилил)диазометан (2,0 М раствор в гексане; 0,2 мл) и 42%-ный водный раствор тетрафторборной кислоты (22 мкл). Добавляют шесть последующих порций (триметилсилил)диазометана (2,0 М раствор в гексане; 0,2 мл) и 42%-ного водного раствора тетрафторборной кислоты (22 мкл), через интервалы по 20 минут, при той же самой температуре. После перемешивания в течение 6 часов, реакционную смесь гасят с помощью насыщенного водного раствора NaHCO3 и экстрагируют с помощью EtOAc. Экстракт три раза промывают водой, сушат над MgSO4 и концентрируют. К суспензии остатка в МеОН добавляют 1 н. водный раствор NaOH (5 мл). После перемешивания при комнатной температуре в течение ночи, смесь три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток подвергают ODS-очистке, получая 22 мг целевого соединения.
Пример 66
К раствору целевого соединения из Получения 27 (42 мг) в ДМФА (3 мл) добавляют морфолин (13 мкл) и смесь перемешивают при температуре 60°С в течение 6 часов. Реакционную смесь гасят водой и экстрагируют с помощью EtOAc. Экстракт дважды промывают водой, сушат над MgSO4 и концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2:ацетон = от 2:3 до 0:100), получая 23 мг целевого соединения.
Пример 67
К раствору целевого соединения из Получения 41 (50 мг) в ДМСО (2 мл) добавляют целевое соединение из Получения 65 (23 мг) и смесь перемешивают при температуре 40°С в течение 12 часов. Реакционную смесь гасят водой и экстрагируют с помощью EtOAc. Экстракт дважды промывают водой, сушат над MgSO4 и концентрируют. Остаток очищают с помощью препаративной ВЭЖХ с обращенными фазами (YMC Pack pro C8; MeCN:вода = 75:25), получая 40 мг целевого соединения.
Пример 74
К раствору целевого соединения из Получения 92 (20 мг) в ТГФ (0,5 мл) добавляют (2-метоксиэтил)метиламин (5 мкл) и смесь перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь гасят с помощью водного раствора карбоната калия и экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью водного раствора NaHCO3, сушат над MgSO4 и концентрируют. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля (CH2Cl2:ацетон = от 75:25 до 60:40), получая целевое соединение (18 мг) в виде порошка белого цвета.
Пример 80
К раствору целевого соединения из Примера 78 (32 мг) в МеОН (5 мл) добавляют метоксид натрия (2 мг) и смесь перемешивают при комнатной температуре в течение 1 часа. Смесь разбавляют водой и три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют, получая целевое соединение (27 мг).
Пример 82
К раствору целевого соединения из Получения 124 (30 мг) в ДМФА (1 мл) добавляют морфолин (20 мкл) и смесь перемешивают при комнатной температуре в течение 0,5 часов. Смесь подвергают очистке с помощью колоночной хроматографии при использовании силикагеля, модифицированного октадецильными группами (ODS), получая 18 мг целевого соединения.
Пример 98
К раствору целевого соединения из Получения 92 (42 мг) в ДМФА (1,0 мл) добавляют (3R,5S)-3,5-диметилморфолингидрохлорид (90 мг), DIPEA (160 мкл) и смесь перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь гасят с помощью водного раствора карбоната калия и экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью водного раствора NaHCO3, сушат над MgSO4 и концентрируют. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля (CH2Cl2:ацетон = от 75:25 до 60:40), получая целевое соединение (7 мг) в виде порошка белого цвета.
Пример 102
Раствор целевого соединения из Примера 76 (15 мг) в МеОН (1 мл) гидрируют при использовании 10%-ного Pd/C (влажность 50%; 5 мг), при комнатной температуре, в течение 5 часов. Смесь отфильтровывают и фильтрат выпаривают, получая целевое соединение (10 мг) в виде порошка белого цвета.
Пример 204
Раствор 1,1,1,3,3,3-гексаметил-2-(триметилсилил)трисилана (755 мг) в толуоле (20 мл) нагревают до температуры 120°С и по каплям добавляют, при перемешивании, смесь целевого соединения из Получения 122 (1,0 г) и α,α'-азобисизобутиронитрила (99,7 мг) в толуоле (10 мл). Всю реакционную смесь перемешивают в течение 1 часа при той же самой температуре и охлаждают до комнатной температуры. Растворитель удаляют при пониженном давлении. Затем остаток растворяют в МеОН (10 мл) и добавляют 1 н. раствор HCl (4,25 мл). Смесь перемешивают при комнатной температуре в течение ночи. Всю смесь подщелачивают с помощью водного раствора NaHCO3 и экстрагируют с помощью CH2Cl2 (три раза). Объединенные экстракты сушат над MgSO4 и концентрируют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля, элюируя смесью ацетон:CH2Cl2 = от 0:100 до 50:50, получая целевое соединение (360 мг).
Конфигурацию в положении 3 данного соединения определяют как (R) при сравнении со спектром ВЭЖХ аутентичного образца, чья конфигурация в положении подтверждена как (R), синтезированного путем альтернативного способа синтеза, как описано выше (Получение 91).
Пример 205
К раствору целевого соединения из Примера 204 (150 мг), молекулярных сит 3 Å (порошок, 0,3 г) и N,N,N',N'-тетраметилнафталин-1,8-диамина (103 мг) в CH2Cl2 (10 мл) добавляют, в виде порций, триметилоксонийтетрафторборат (35 мг), при температуре 0°С, и смесь перемешивают при той же самой температуре в течение 1,5 часов. Реакционную смесь гасят с помощью водного раствора NaHCO3 и экстрагируют с помощью EtOAc. Органический экстракт промывают с помощью 1 н. водного раствора HCl, сушат над MgSO4 и концентрируют. Остаток подвергают ODS-очистке, получая 32 мг основного целевого соединения (Пример 205-А) и 7 мг побочного целевого соединения (Пример 205-В).
Пример 207
К раствору целевого соединения из Получения 117 (46 мг) в смешанном растворителе из ТГФ (5 мл) и насыщенного водного раствора NaHCO3 (1 мл) добавляют изопропилхлоркарбонат (15 мкл) и смесь перемешивают при комнатной температуре в течение 30 минут. Смесь разбавляют водой, три раза экстрагируют с помощью CH2Cl2. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток подвергают ODS-очистке, получая 18 мг целевого соединения.
Пример 210
К раствору целевого соединения из Получения 117 (50 мг) в CH2Cl2 добавляют DIPEA (100 мкл) и хлорангидрид диметилкарбаминовой кислоты (50 мкл) и смесь перемешивают при комнатной температуре в течение ночи. Смесь разбавляют водой, экстрагируют с помощью EtOAc. Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток подвергают ODS-очистке, получая 18 мг целевого соединения.
Пример 233
К раствору целевого соединения из Получения 135 (50 мг) в ДМФА (1 мл) добавляют морфолин (30 мкл) и смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь гасят с помощью водного раствора карбоната калия и экстрагируют с помощью EtOAc. Органическую фазу промывают с помощью водного раствора NaHCO3, сушат над MgSO4 и концентрируют. Остаток очищают с помощью колоночной хроматографии при использовании силикагеля (CH2Cl2:ацетон = от 75:25 до 60:40), получая целевое соединение (13 мг) в виде порошка белого цвета.
Пример 238
Раствор целевого соединения из Примера 21 (его структура приведена в Таблице 2) (111 мг) в МеОН гидрируют при использовании 20%-ного Pd/C (влажность 50%; 20 мг) при комнатной температуре в течение 1,5 часов. Эту смесь отфильтровывают и фильтрат концентрируют. Остаток хроматографируют на силикагеле (CH2Cl2:МеОН = от 97:3 до 90:10), получая 108 мг целевого соединения.
Пример 259
Раствор целевого соединения из Примера 227 (38 мг) в МеОН гидрируют при использовании 10%-ного Pd/C (влажность 50%; 20 мг) при комнатной температуре в течение 1,5 часов. Смесь отфильтровывают и фильтрат концентрируют. Остаток растворяют в CH2Cl2, сушат над MgSO4, отфильтровывают и концентрируют, получая 35 мг целевого соединения.
Структура, данные [физические данные; MS: ESI (масс-спектрометрия: ионизация электронным распылением) (M+H)+, за исключением иначе указанного; ЯМР: пик δ (м.д.) в случае данных 1Н-ЯМР (дейтерированный хлороформ в качестве растворителя для измерения, за исключением иначе указанного, при использовании (СН3)4Si в качестве внутреннего стандарта] и синтез [способ получения (номер указывает номер примера, соответствующий получению)] для соединений Примеров представлены в нижеприводимых Таблицах. И структуры соединений из Получений 2, 21-41 и 80-158 также представлены в нижеприводимых Таблицах.
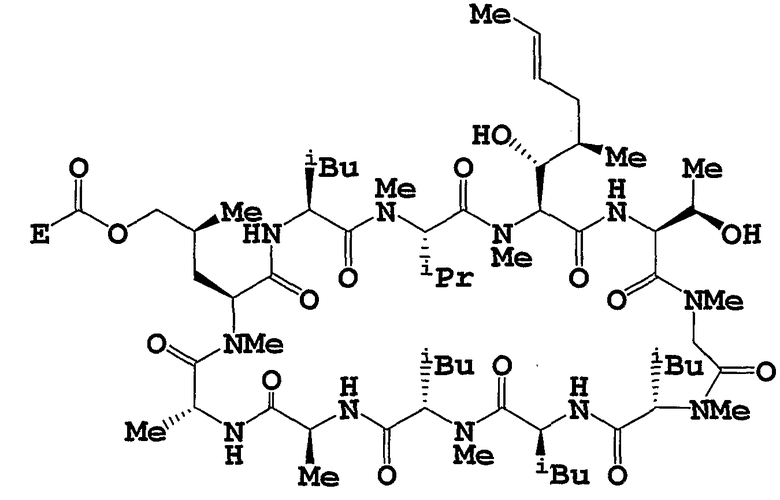
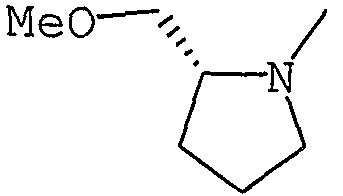
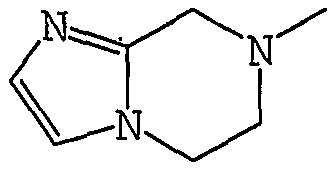
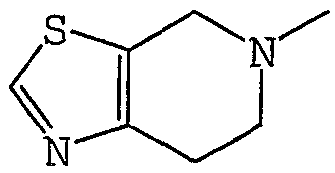
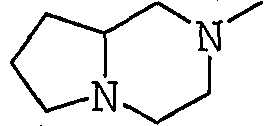
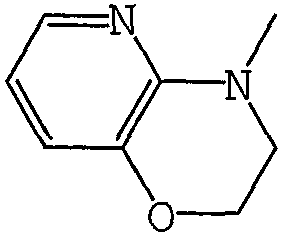
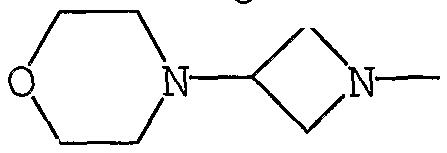
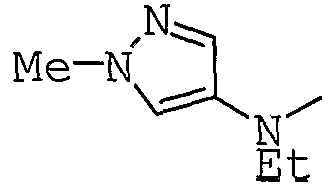
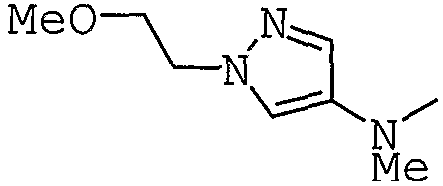
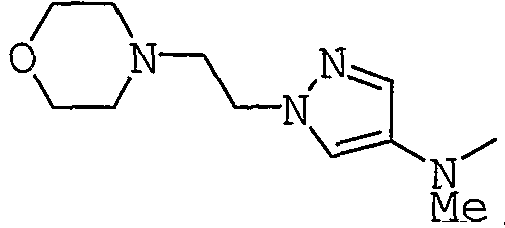
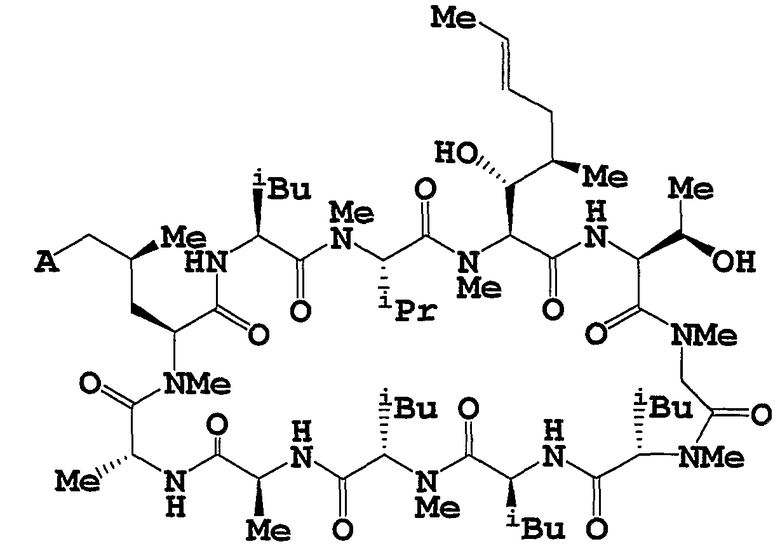
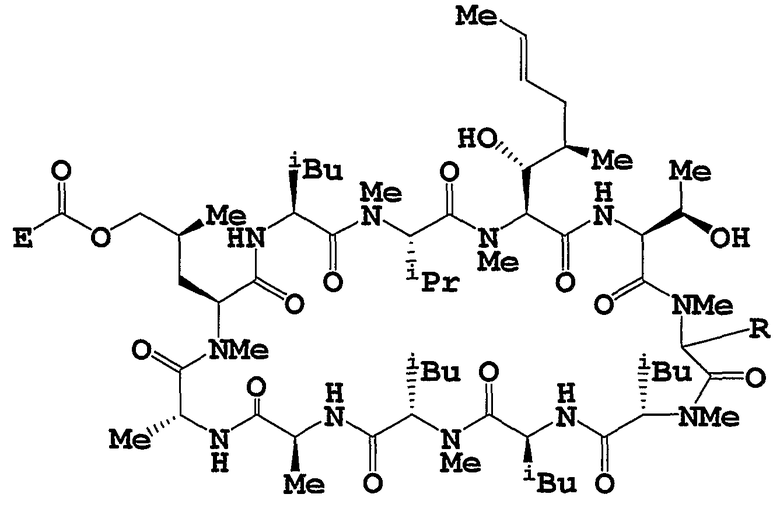
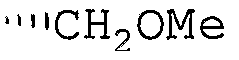
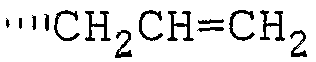
 смесь эпимеров
смесь эпимеров
 смесь эпимеров
смесь эпимеров
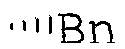
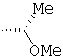
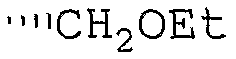
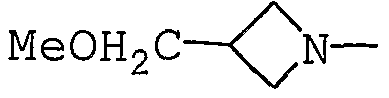
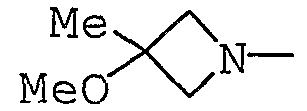
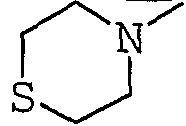
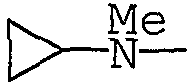
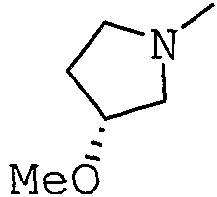
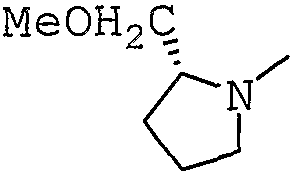
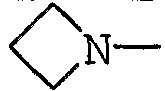
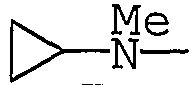
 смесь эпимеров
смесь эпимеров
 смесь эпимеров
смесь эпимеров
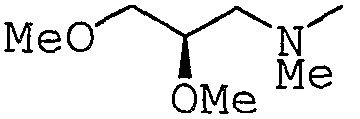
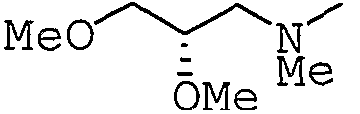
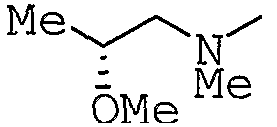
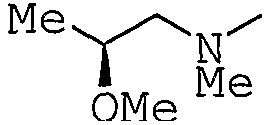
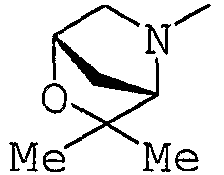
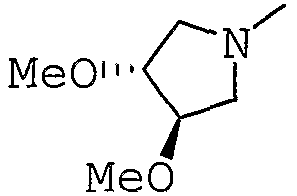
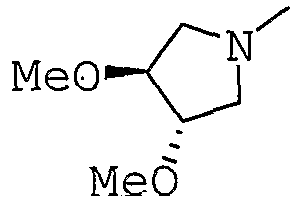
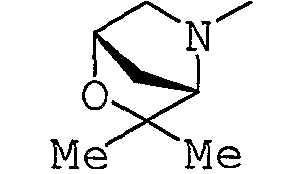
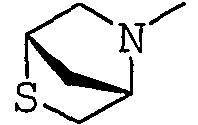
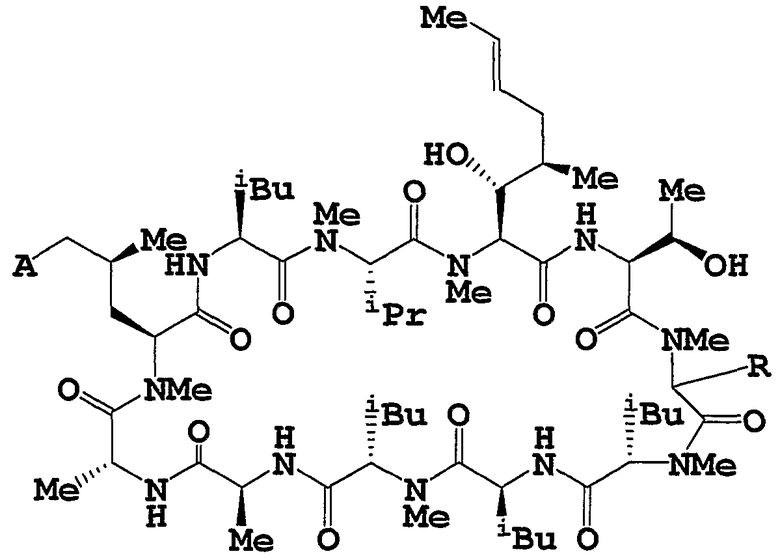
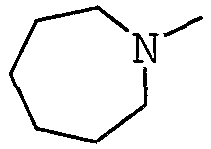
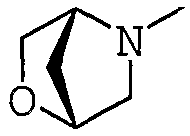
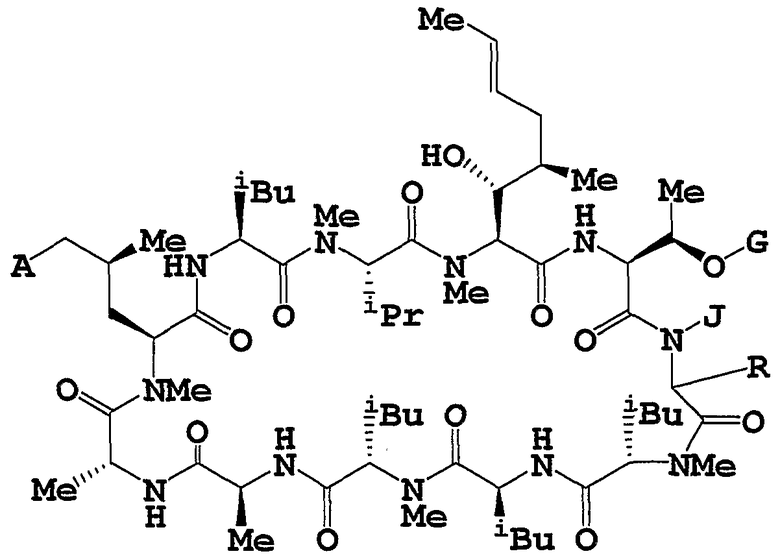
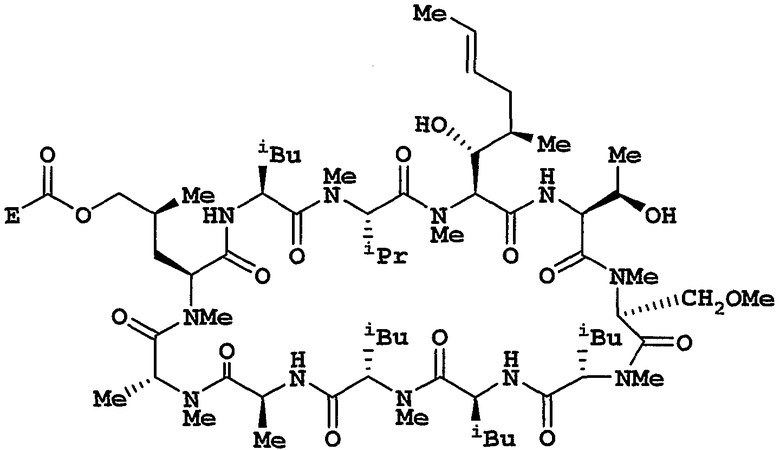
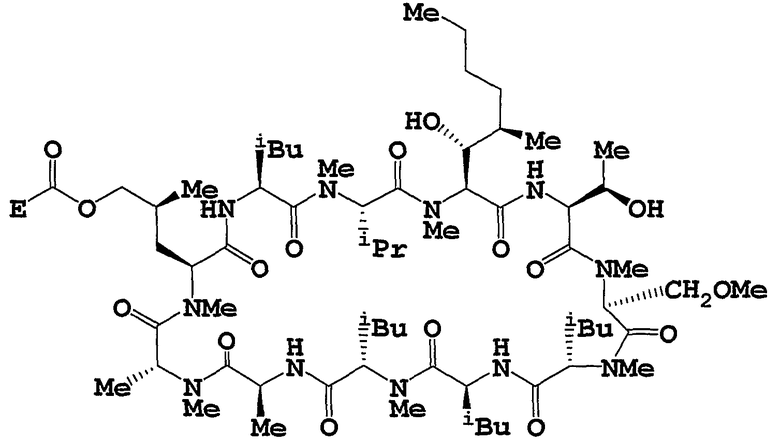
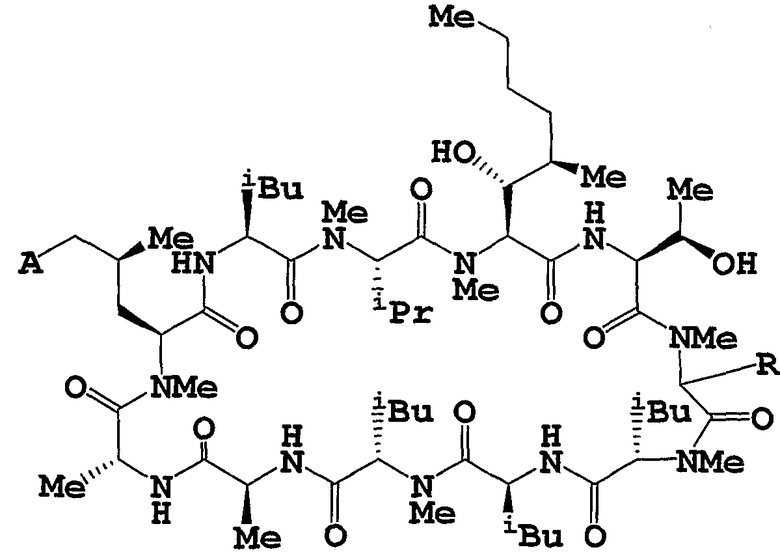
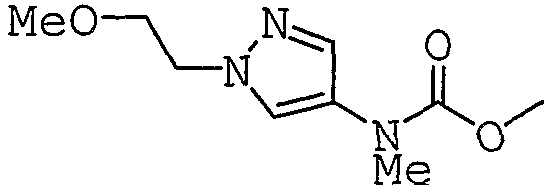
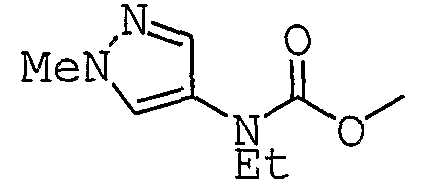
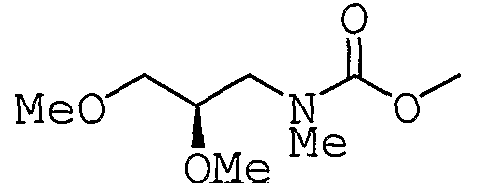
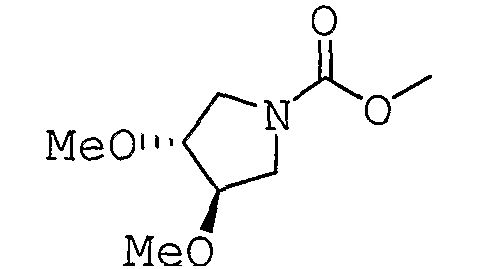
MS: 1346,74
MS: 1348,78
MS: 1361,00
MS: 1319,51
MS: 1346,11
MS: 1303,95
MS: 1326,59, ESI (M+H2O)+: 1343,60
MS: 1346,57
MS: 1323,59
MS: 1358,88
MS: 1334,88
MS: 1354,95
MS: 1340,79
MS: 1364,25
MS: 1367,09
MS: 1369,23
MS: 1248,45
MS: 1391,20
MS: 1347,16
MS: 1391,80
MS: 1346,74
ESI (M+H2O)+: 1408,35
MS: 1404,35
MS: 1393,35
MS: 1389,42
MS: 1402,43
MS: 1389,45
MS: 1389,43
MS: 1389,47
MS: 1363,34
MS: 1418,08
MS: 1435,92
MS: 1471,03
MS: 1392,07
MS: 1378,81
MS: 1396,01
MS: 1390,64
MS: 1388,48
MS: 1406,56
MS: 1406,50
MS: 1320,14
MS: 1393,05
MS: 1376,09
MS: 1378,86,
MS: 1399,92
MS: 1439,34
MS: 1466,98
MS: 1408,16
MS: 1408,16
MS: 1402,85
MS: 1378,46
MS: 1406,00
MS: 1437,92
MS: 1248,80
Время удерживания: 5,4 мин
(ВЭЖХ, колонка: Shiseido UG120 C18, 100 мм × 4,6 мм - внутренний диаметр, элюент: 60% MeCN/Н2О, объемная скорость потока: 1,0 мл/мин)
MS: 1317,82
MS: 1362,05
MS: 1407,05
MS: 1333,96
MS: 1436,11
MS: 1348,01
MS: 1408,16, ESI (M+Na)+: 1392,8
MS: 1450,00
MS: 1410,26
MS: 1350,09
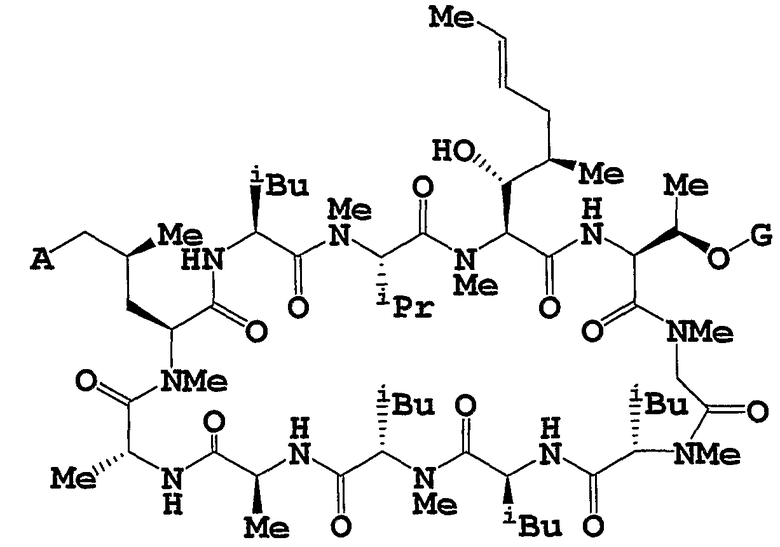
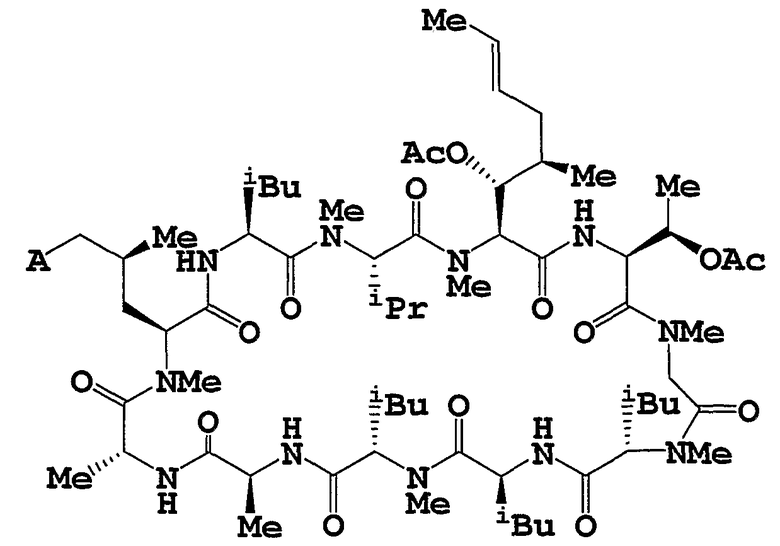
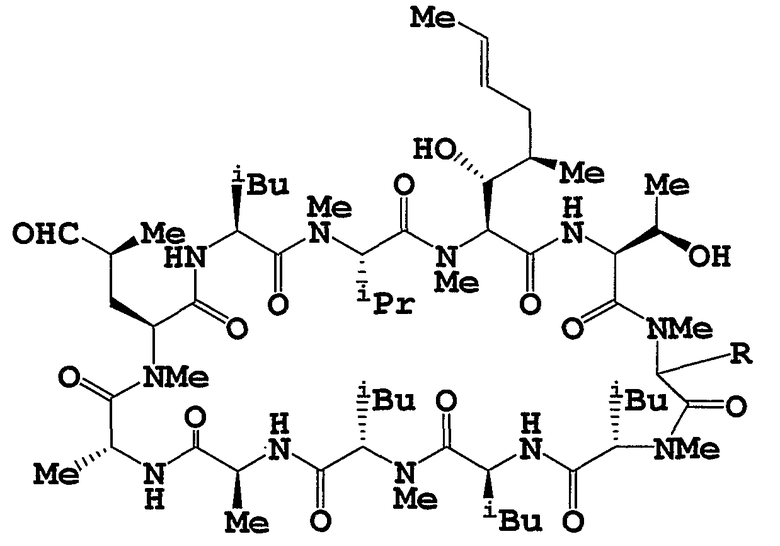
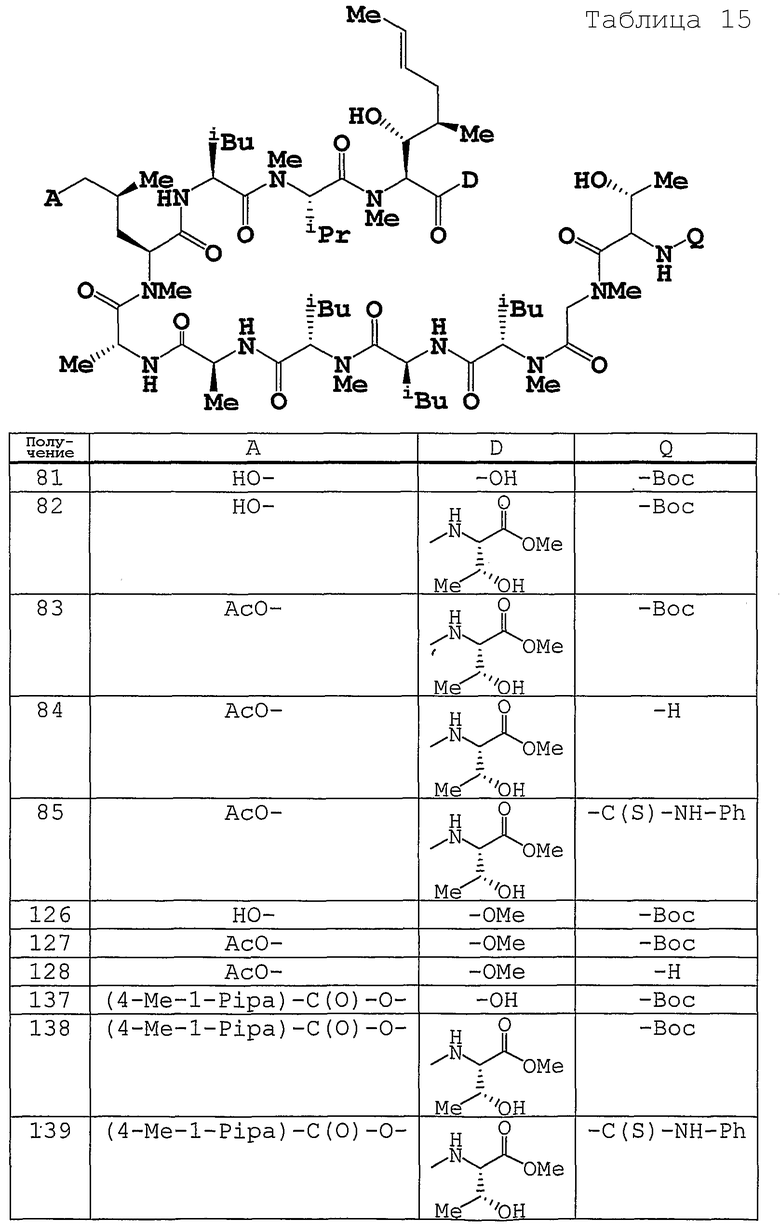
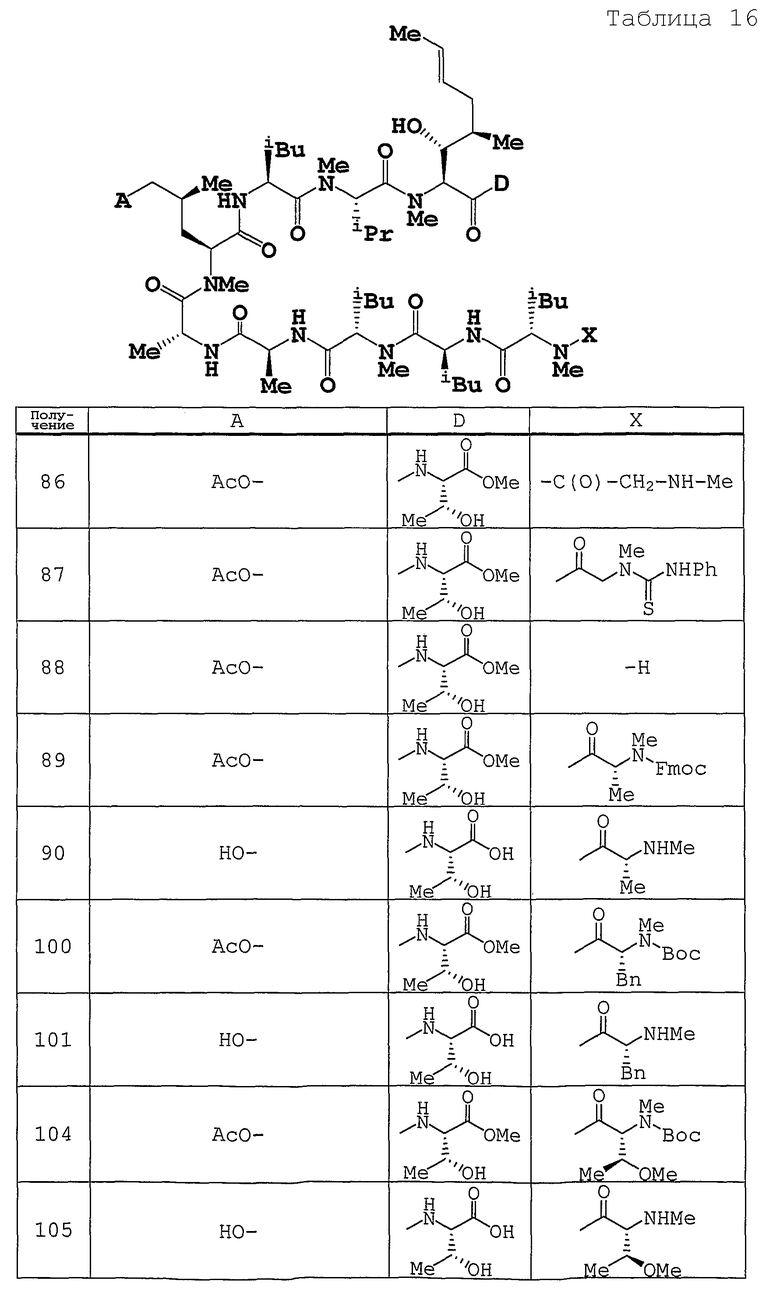
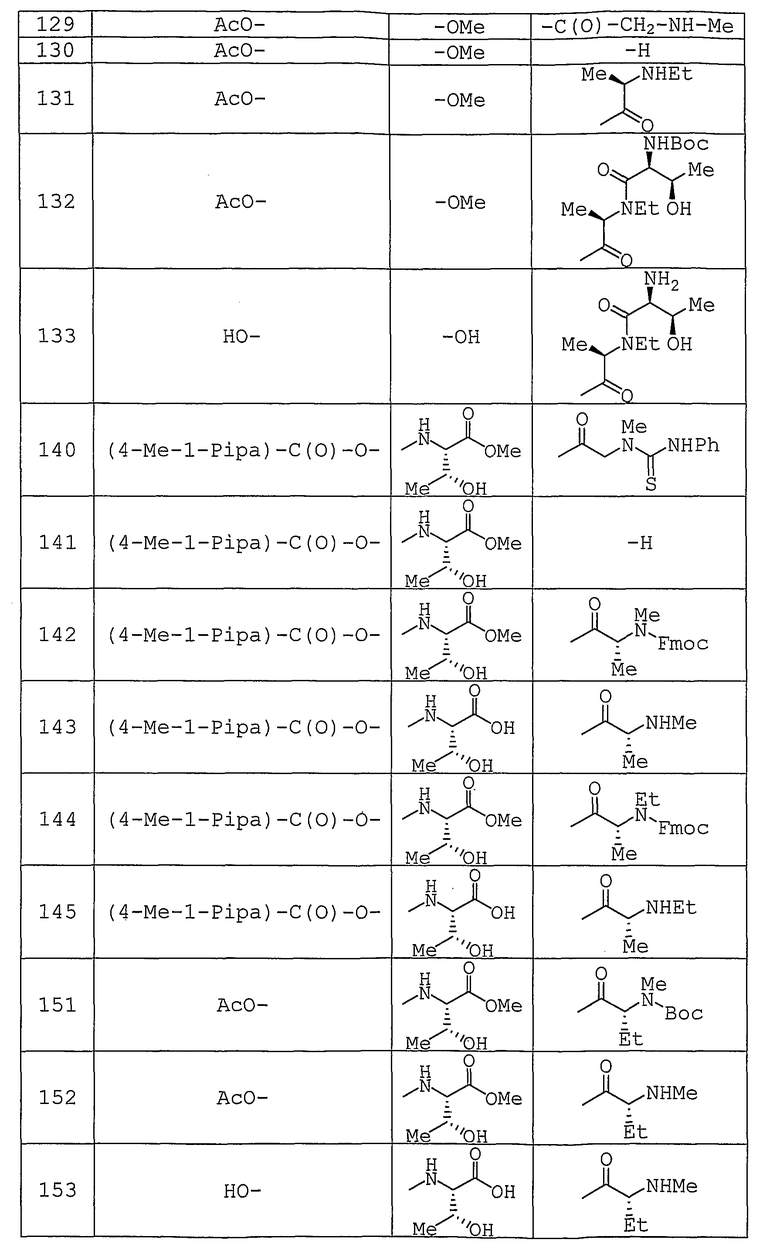
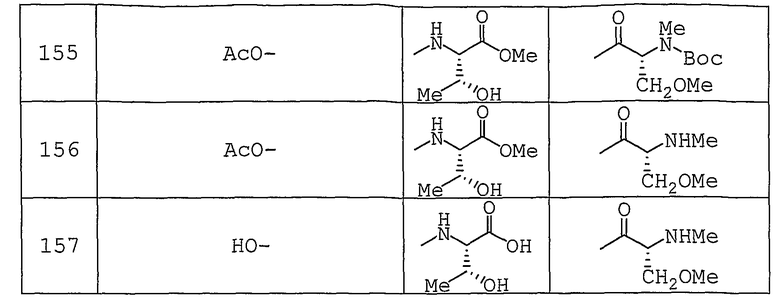
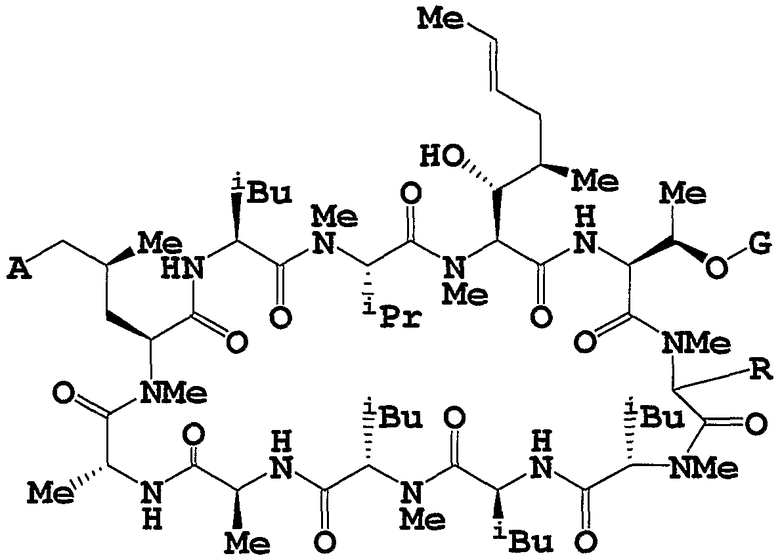
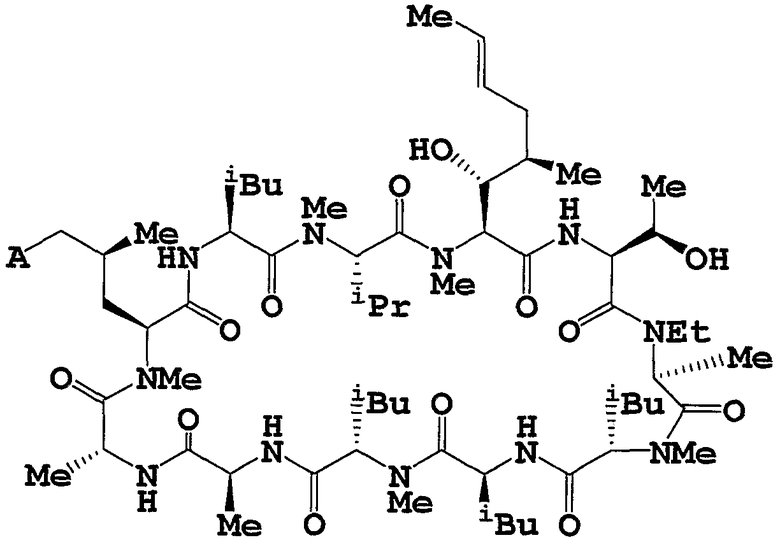
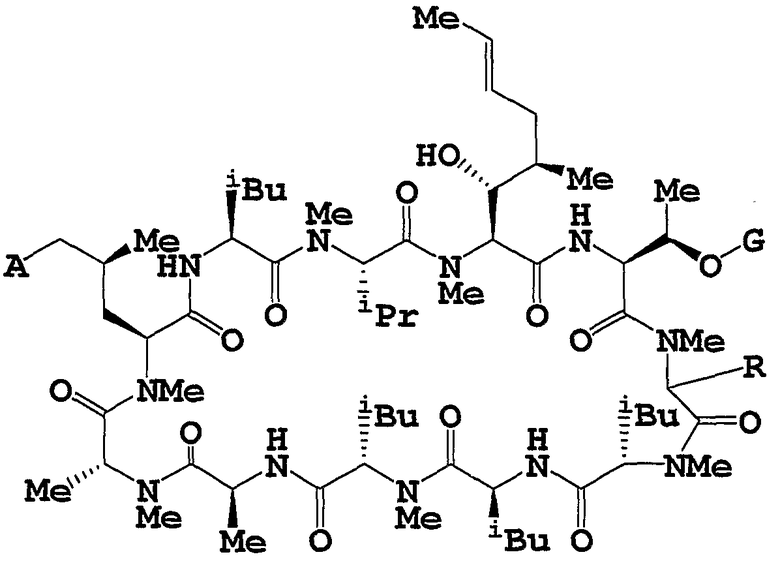
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2001 |
|
RU2275373C2 |
| ЗАМЕЩЕННЫЕ 3-АМИНОХИНУКЛИДИНЫ | 1992 |
|
RU2092486C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, МОДЕЛИРУЮЩИЕ АКТИВНОСТЬ ХЕМОКИНОВОГО РЕЦЕПТОРА, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2297413C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| СУЛЬФОНАМИДНЫЕ ПЕРИ-ЗАМЕЩЕННЫЕ БИЦИКЛЫ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ПОРАЖЕНИЯ АРТЕРИЙ | 2005 |
|
RU2403240C2 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| НИТРОИМИДАЗООКСАЗИНОВЫЕ И НИТРОИМИДАЗООКСАЗОЛЬНЫЕ АНАЛОГИ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2540860C2 |
| СПОСОБ МОДУЛЯЦИИ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2525115C2 |
Настоящее изобретение относится к новому циклическому пептидному соединению или его фармацевтически приемлемой соли, обладающему активностью против вируса гепатита С, основанной на ингибирующей активности против репликации РНК репликона вируса гепатита С, фармацевтической композиции, включающей указанное соединение или его фармацевтически приемлемую соль, и к применению соединения или его фармацевтически приемлемой соли для приготовления лекарственного средства, обладающего анти-HCV активностью. 3 н. и 6 з.п. ф-лы, 19 табл.
1. Циклическое пептидное соединение следующей общей формулы (I):
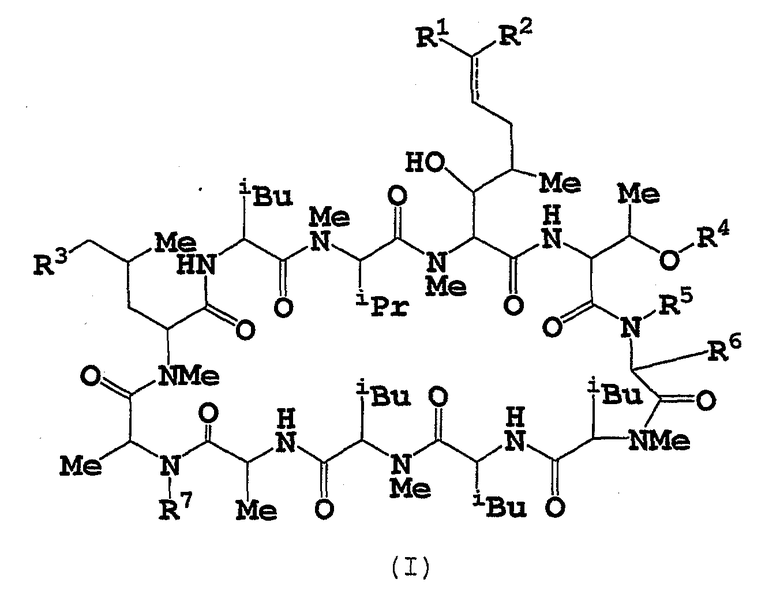
где R1 и R2, независимо, означают водород или метил;
R3 означает:
(1) -ОН или SO2Ph;
(2) гетероциклическую группу, выбранную из: насыщенной 3-8-членной гетеромоноциклической группы, содержащей 1 или 2 атома кислорода и 1-3 атома азота (которая может быть замещена одной или более группой -CH2-O-Me, Me); насыщенной 3-8-членной гетеромоноциклической группы, содержащей 1-4 атома азота/насыщенной 3-8-членной гетеромоноциклической группы, содержащей 1 или 2 атома серы и 1-3 атома азота; или
(3) -NR8R9, где R8 и R9, независимо, означают водород, низший алкил (возможно замещенный пиридилом, фенилом, ОМе) или группу -C(O)-(Ph-OMe), -C(O)-(Ph-N(Me)2), -С(О)-(Ph-4-морфолинил), -С(O)-ОРri, -С(O)-Рri, -С(О)-(4-морфолинил);
(4) -ОС (О) -NR10R11, где R10 и R11, независимо, означают водород, низший алкил (возможно замещенный -N(Me)2, -N(Et)2, морфолинилом, пиридилом, 1-метил-4-пиразолилом, -O-Ме, -Me), гетероциклическую группу, представляющую собой ненасыщенную 3-8-членную гетеромоноциклическую группу, содержащую 1-4 атома азота (возможно замещенную -Me, - (CH2)2-ОМе, - (СН2)2-морфолинилом), ненасыщенную 3-8-членную гетеромоноциклическую группу, содержащую 1 или 2 атома серы и 1-3 атома азота, насыщенную 3-8-членную конденсированную гетеромоноциклическую группу, содержащую 1-3 атома азота, цикло(низший)алкил, фенил;
или, альтернативно, R10 и R11 вместе с атомом азота, к которому они присоединены, представляют собой содержащую азотнасыщенную 3-8-членную гетеромоноциклическую группу, содержащую 1-4 атома азота [возможно замещенную группой, выбранной из -Me, -Et, iPr, -O-Ме, -OEt, -CH2-O-Me, - (СН2)2-O-Me, -(CH2)3-О-Ме, -(CH2)2-O-Et, -SO2-Et, -СН2-С(О)-(4-морфолинил), 4-Вос-, пиридила, пиримидинила, пиразолила, имидазолила, пиразинила, -С(O)-СН2-N(Me)2, -(CH2)2-N(Me)2, -(СН2)3-N (Me) 2, морфолинила,-nBu, галогена, СF3. -СН2-пиперидила, -(СН2)2-пиперидила, -СН2-СН=СН2, -пиперидила (возможно замещенного Me), -СН2-тетрагидрофуранила, насыщенной 3-8-членной гетеромоноциклической группы, содержащей 1 или 2 атома кислорода и 1-3 атома азота (возможно замещенной Me, -CH2-O-Me), пиридила (возможно замещенного Me)] или группу, выбранную из:
(5) -O-R12, где R12 означает низший алкил (возможно замещенный группой -О-С(О)-(4-морфолинил)) или фенил (возможно замещенный галогеном); или
(6) -S-R13, где R13 означает низший алкил (возможно замещенный фенилом, группой -С(О)-[Ph-4-N(Me)2], -Ph-4-OMe, -Ph-4-(NH-Ac)), -C(O)-Ph, -С(О)-морфолинил, -С(О)-пиперазинил [где пиперазинил возможно замещен группой -СН2-С(О)-(4-морфолинил)], или ненасыщенную 3-8-членную гетеромоноциклическую группу, содержащую 1 или 2 атома серы и 1-3 атома азота;
R4 означает водород или низший алкил;
R5 означает низший алкил;
R6 означает водород, низший алкил (возможно замещенный -ОАс, -ОН, -ОМе, -OEt) или низший алкенил;
R7 означает водород или низший алкил; и
 представляет собой ординарную связь или двойную связь;
представляет собой ординарную связь или двойную связь;
при условии, что, когда R3 означает -ОН, тогда R6 означает низший алкил или низший алкенил, каждый из которых может иметь один или более подходящий(их) заместитель(ей);
или его фармацевтически приемлемая соль.
2. Соединение по п.1,
где R4 означает водород или метил;
R5 означает метил или этил; и
R7 означает водород, метил или этил,
или его фармацевтически приемлемая соль.
3. Соединение по п.2,
где R4 означает водород; и
R7 означает водород,
или его фармацевтически приемлемая соль.
4. Соединение по п.3
где R1 означает метил;
R2 означает водород,
или его фармацевтически приемлемая соль.
5. Соединение по п.4, где
 означает двойную связь,
означает двойную связь,
или его фармацевтически приемлемая соль.
6. Соединение по п.5, где
R6 означает водород или низший алкил (необязательно замещенный -ОAс, -ОН, -ОМе, -OEt) или низший алкенил;
или его фармацевтически приемлемая соль.
7. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении репликации HCV, включающая, в качестве активного ингредиента, соединение по п.1 или его фармацевтически приемлемую соль в терапевтически эффективном количестве в смеси с фармацевтически приемлемыми носителями или эксципиентами.
8. Применение соединения по п.1 или его фармацевтически приемлемой соли для получения лекарственного средства, обладающего анти-НСV-активностью.
9. Соединение по п.1 или его фармацевтически приемлемая соль, обладающее(ая) анти-НСV-активностью.
| WO 2006054801 A1, 26.05.2006 | |||
| WO 2005032576 A1, 14.04.2005 | |||
| WO 2005021028 A1, 10.03.2005 | |||
| RU 2004110941 A, 10.08.2005 | |||
| WATASHI К | |||
| ЕТ AL | |||
| CYCLOSPORIN A SUPPRESSES REPLICATION OF HEPATITIS С VIRUS GENOME IN CULTURED HEPATOCYTES" HEPATOLOGY, 2003, vol | |||
| Способ сужения чугунных изделий | 1922 |
|
SU38A1 |
| Приспособление для равномерного распределения по цилиндрам горючей жидкости в многоцилиндровых двигателях внутреннего горения | 1924 |
|
SU1282A1 |
| NAKAGAWA M | |||
| ЕТ AL | |||
| Specific inhibition of hepatitis С virus replication by | |||

