Химические соединения
Настоящее изобретение относится к некоторым новым соединениям. В частности, настоящее изобретение относится к соединениям, которые активируют рецепторы человека, активируемые пероксисомными пролифераторами (hPPARs). Настоящее изобретение также относится к способу получения соединений, их применению в медицине, фармацевтическим композициям, содержащим их, и к способам предупреждения или лечения PPAR-опосредованных заболеваний или состояний.
С сердечно-сосудистым заболеванием связывают несколько независимых факторов риска. Они включают гипертензию, повышенные уровни фибриногена, высокие уровни триглицеридов, повышенный LDL-холестерин (холестерин липопротеинов низкой плотности), повышенный общий холестерин и низкие уровни HDL-холестерина (холестерина липопротеинов высокой плотности). Ингибиторы HMG-CoA-редуктазы (3-гидрокси-3-метилглютарил-коэнзим А-редуктазы) ("статины") полезны для лечения состояний, характеризующихся высокими уровнями LDL-c (холестерина липопротеинов низкой плотности). Было показано, что уменьшение LDL-c не является достаточным для снижения риска сердечно-сосудистого заболевания у некоторых пациентов, в частности у пациентов с нормальными уровнями LDL-c. Эту популяцию определяют по независимому фактору риска - низкому HDLc (холестерину липопротеинов высокой плотности). Лекарственная терапия до сих пор была недостаточно успешно направлена на повышенный риск сердечно-сосудистого заболевания, связанного с низкими уровнями HDL-c (Bisgaier, C.L; Pape, M.E. Curr. Pharm. Des. 1998, 4, 53-70).
Синдром X (включая метаболический синдром) широко определяется как совокупность отклонений от нормы, включая гиперинсулинемию, ожирение, повышенные уровни следующих соединений: триглицеридов, мочевой кислоты, фибриногена, LDL-частиц небольшой плотности и ингибитора активатора плазминогена-1 (PAI-1), и пониженные уровни HDL-c.
NIDDM (инсулиннезависимый сахарный диабет) описывают как резистентность к инсулину, которая в свою очередь является причиной аномального продуцирования глюкозы и снижения потребления глюкозы скелетной мышцей. Эти факторы в итоге приводят к ухудшенной толерантности к глюкозе (IGT) и гиперинсулинемии.
Рецепторы, активируемые пероксисомными пролифераторами (PPARs), являются рецепторами-сиротами, принадлежащими к суперсемейству стероидных/ретиноидных рецепторов лигандактивируемых факторов транскрипции. Смотри, например, Willson T.M. and Wahli, W., Curr. Opin. Chem. Biol., 1, pp.235-241 (1997), и Willson T.M. et al., J. Med. Chem., 43, p.527-549 (2000). Связывание агонистов-лигандов с рецептором приводит к изменениям в уровне экспрессии мРНК, кодируемых генами-мишенями PPAR.
У млекопитающих были выделены три типа рецепторов, активируемых пероксисомными пролифераторами, и обозначены как PPAR-альфа, PPAR-гамма и PPAR-дельта (также известен как NUC1 или PPAR-бета). Эти PPARs регулируют экспрессию генов-мишеней посредством связывания с элементами последовательности ДНК, называемыми PPAR-отвечающими элементами (PPRE). До настоящего времени PPREs были идентифицированы в энхансерах ряда генов, кодирующих белки, которые регулируют метаболизм липидов, что позволяет предположить, что PPAR играют центральную роль в адипогенном сигнальном каскаде и липидном гомеостазе (Н.Keller and W.Wahli, Trends Endocrinol. Metab., 291-296, 4 (1993)).
В настоящее время сообщается о том, что лекарственные средства из класса тиазолидиндионов являются сильнодействующими и селективными активаторами PPAR-гамма и связываются непосредственно с PPAR-гамма рецептором (J.M.Lehmann et al., J.Biol. Chem., 12953-12956, 270 (1995)), что свидетельствует о том, что PPAR-гамма является возможной мишенью для терапевтических воздействий тиазолидиндионов.
В условиях клиники показано, что активаторы ядерного рецептора PPARγ, например розиглитазон, усиливают действие инсулина, понижают содержание глюкозы в сыворотке и оказывают небольшие, но значимые воздействия на снижение уровней триглицеридов в сыворотке у пациентов с диабетом 2 типа. Смотри, например, D.Е.Kelly et. al., Curr. Opin. Endocrinol. Diabetes., 90-96, 5 (2), (1998); M.D.Johnson et. al., Ann. Pharmacother., 337-348, 32 (3), (1997), и M.Leutenegger et al., Curr. Ther. Res., 403-416, 58 (7), (1997).
По-видимому, механизм этого понижающего уровень триглицеридов действия преимущественно заключается в повышенном клиренсе липопротеинов очень низкой плотности (VLDL) через индукцию экспрессии гена липопротеинлипазы (LPL). Смотри, например, В.Staels et al., Arterioscler. Thromb., Vasc., Biol., 1756-1764, 17 (9), (1997).
Фибраты представляют собой класс лекарственных средств, которые могут снижать триглицериды в сыворотке на 20-50%, снижать LDL-c на 10-15%, сдвигать размер LDL-частиц от более атерогенной малой плотности к LDL нормальной плотности и увеличивать HDLc на 10-15%. Экспериментальные данные указывают на то, что действия фибратов на липиды сыворотки опосредованы активацией PPARα. Смотри, например, В.Staels et al., Curr. Pharm. Des., 1-14, 3 (1), (1997). Активация PPARα вызывает транскрипцию ферментов, усиливающих катаболизм жирных кислот и уменьшающих синтез жирных кислот в печени de-novo, что ведет к уменьшению синтеза триглицеридов и к уменьшению продуцирования/секреции VLDL. Кроме того, активация PPARα уменьшает продуцирование ароС-III. Снижение уровня ароС-III, ингибитора активности LPL, повышает кпиренс VLDL. Смотри, например, J.Auwerx et al., Atherosclerosis (Shannon, Irel), S29-S37,124 (Suppl), (1996).
Некоторые соединения, которые активируют или иным образом взаимодействуют с одним или более PPARs, были вовлечены в регуляцию уровней триглицеридов или холестерина в моделях на животных. Смотри, например, патенты США 5847008 (Doebber et al.) и 5859051 (Adams et al.) и публикации PCT WO 97/28149 (Leibowitz et al.), WO 99/04815 (Shimokawa et al.) и WO 01/00603 (Glaxo Group Ltd.). В Oliver et al., Proc. Natl. Acad. Sci., 98, 5306-5311 (2001), сообщается об увеличении HDLc и понижении триглицеридов сыворотки у страдающей ожирением макаки-резус после введения агониста PPAR-дельта.
Согласно данному изобретению предложено соединение формулы (I) и его фармацевтически приемлемые соли и сольваты и гидролизуемые сложные эфиры

где R1 и R2 независимо представляют собой водород или С1-3алкил;
X представляет собой связь, СН2 или О;
R3 и R4 независимо представляют собой водород, C1-6алкил, ОСН3, CF3, аллил или галоген;
X1 представляет собой СН2, SO2 или СО;
R5 представляет собой -С1-6алкил (возможно замещенный С1-6алкокси или С1-6алкилтио), -С2-6алкенил, -С0-6алкилфенил (где фенил возможно замещен одним или более чем одним CF3, галогеном, C1-3алкилом, С1-3алкокси), -СОС1-6алкил, -SO2С1-6алкил;
R6 представляет собой фенил или 6-членную гетероарильную группу, содержащую 1, 2 или 3 атома N, при этом фенил или гетероарильная группа возможно замещена 1, 2 или 3 группировками, выбранными из группы, состоящей из С1-6алкила, галогена, -ОС1-6алкила, -SO2С1-3алкила, фенила (возможно замещенного одной или более группами, выбранными из галогена, CF3, C1-3алкила, ОС1-3алкила, ацетила, CN).
В другом аспекте настоящего изобретения описан способ предупреждения или лечения заболевания или состояния, опосредованного одним или более PPAR-альфа, -гамма или -дельта человека (DPPARs), включающий введение терапевтически эффективного количества соединения по данному изобретению. hPPAR-опосредованные заболевания или состояния включают дислипидемию, в том числе дислипидемию, связанную с диабетом, и смешанную дислипидемию, синдром Х (как определено в данной заявке, он включает метаболический синдром), сердечную недостаточность, гиперхолестеринемию, сердечно-сосудистое заболевание, включая атеросклероз, артериосклероз и гипертриглицеридемию, сахарный диабет II типа, диабет I типа, резистентность к инсулину, гиперлипидемию, воспаление, эпителиальные гиперпролиферативные заболевания, включая экзему и псориаз, и состояния, связанные с легким и кишечником и регуляцией аппетита и потребления пищи у субъектов, страдающих такими расстройствами, как ожирение, булимическая анорексия и нервная анорексия, рак, болезнь Альцгеймера и другие когнитивные нарушения. В частности, соединения по данному изобретению полезны в лечении и предупреждении диабета, ожирения и сердечно-сосудистых заболеваний и состояний, включая атеросклероз, артериосклероз, гипертриглицеридемию и смешанную дислипидемию.
В другом аспекте настоящего изобретения предложены фармацевтические композиции, содержащие соединение по изобретению, предпочтительно объединенное с фармацевтически приемлемым разбавителем или носителем.
В другом аспекте настоящего изобретения предложено соединение по изобретению для применения в терапии и, в частности, в терапии людей.
В другом аспекте настоящего изобретения предложено применение соединения по изобретению для изготовления лекарства для лечения hPPAR-опосредованного заболевания или состояния.
При использованиии в данном описании, "соединение по изобретению" означает соединение формулы (I) или его фармацевтически приемлемую соль, или сольват, или гидролизуемый сложный эфир.
Хотя гидролизуемые сложные эфиры включены в объем данного изобретения, предпочтительными являются кислоты, так как определенные данные позволяют предположить, что хотя сложные эфиры являются полезными соединениями, в действительности активными соединениями могут быть кислоты, до которых они гидролизуются. Из сложных эфиров, которые легко гидролизуются, в условиях анализа или in vivo может получаться карбоновая кислота. Как правило, карбоновая кислота активна как в анализах связывания, так и в анализах с временной трансфекцией, тогда как сложный эфир обычно не очень хорошо связывается, но активен в анализе с временной трансфекцией, предположительно вследствие гидролиза. Предпочтительными гидролизуемыми сложными эфирами являются С1-6алкильные сложные эфиры, в которых алкильная группа может быть прямой или разветвленной. Более предпочтительными являются метиловые или этиловые сложные эфиры.
Предпочтительно каждый R1 и R2 независимо представляет собой Н или метил. Более предпочтительно R1 и R2 оба представляют собой Н или оба представляют собой Me. Еще более предпочтительно R1 и R2 оба представляют собой Н.
Предпочтительно Х представляет собой О.
Предпочтительно R3 и R4 независимо представляют собой Н или C1-3алкил. Более предпочтительно по меньшей мере один из R3 и R4 представляет собой водород и, когда один из R4 и R3 представляет собой водород, а другой не является водородом, тогда тот радикал, который не является водородом, предпочтительно находится в орто-положении к изображенной группировке X. Более предпочтительно радикал, который не является водородом, представляет собой метил.
Предпочтительно X1 представляет собой СН2.
Предпочтительно R5 представляет собой бутил или метоксиэтил.
Предпочтительно R6 представляет собой фенил или 6-членный гетероцикл, выбранный из пиримидина, пиридина, пиридазина, пиразина, каждый из которых замещен фенилом (возможно замещенным одним или более чем одним CF3, C1-3алкилом, галогеном, CN) и возможно дополнительным C1-3алкильным заместителем. Предпочтительно этот фенильный заместитель находится в мета-положении к изображенной группе N. Более предпочтительно заместитель на фениле или 6-членном гетероцикле представляет собой пара-С6H4C3, C6H4Me или С6Н4Cl.
Предпочтительными соединениями по изобретению являются следующие:
2-метил-2-{2-метил-4-[([4-(трифторметил)бензил]{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)метил]фенокси}пропановая кислота;
2-{4-[(бутил{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)метил]-2-метилфенокси}-2-метилпропановая кислота;
{4-[(бутил{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)метил]-2-метилфенокси}уксусная кислота;
[4-({бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]-уксусная кислота;
[4-({(2-метоксиэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусная кислота;
[2-метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-фенокси]уксусная кислота;
[4-({(2-циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусная кислота;
[2-метил-4-({пропил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-фенокси]уксусная кислота;
[2-метил-4-({[2-(метилтио)этил][4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-метил)фенокси]уксусная кислота;
[4-({бутил[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусная кислота;
[4-({(2-метоксиэтил)[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-метил)-2-метилфенокси]уксусная кислота;
[4-({бутирил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусная кислота;
[2-метил-4-({(пропилсульфонил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-метил)фенокси]уксусная кислота;
[4-({бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-2-метилфенокси]уксусная кислота;
[2-метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-фенокси]уксусная кислота;
[4-({(2-циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-сульфонил)-2-метилфенокси]уксусная кислота;
{4-[(бутил{4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]-2-метилфенокси}уксусная кислота;
[4-({бутил[4-(4-хлорфенил)пиримидин-2-ил]амино}метил)-2-метилфенокси]-уксусная кислота;
{4-[((2-метоксиэтил){4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]-2-метилфенокси}уксусная кислота;
(4-{[[4-(4-хлорфенил)пиримидин-2-ил](2-метоксиэтил)амино]метил}-2-метилфенокси)уксусная кислота;
{2-метил-4-[(пропил{4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]-фенокси}уксусная кислота;
{4-[(бутил{6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]-2-метилфенокси}уксусная кислота;
[4-({бутил[6-(4-метилфенил)пиразин-2-ил]амино}метил)-2-метилфенокси]-уксусная кислота;
{4-[{(2-метоксиэтил){6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]-2-метилфенокси}уксусная кислота;
(4-{[бутил(2,4′-диметил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)-уксусная кислота;
(4-{[бутил(4′-фтор-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)-уксусная кислота;
(4-{[бутил(4′-циано-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)-уксусная кислота;
(4-{[бутил(4′-метокси-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)уксусная кислота;
(4-{[бутил(4′-хлор-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)-уксусная кислота;
(4-{[(4′-хлор-2-метил-1,1′-бифенил-3-ил)(2-метоксиэтил)амино]метил}-2-метилфенокси)уксусная кислота;
(4-{[(2,4′-диметил-1,1′-бифенил-3-ил)(2-метоксиэтил)амино]метил}-2-метилфенокси)уксусная кислота;
(4-{[(2-метоксиэтил)(4′-метокси-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)уксусная кислота;
(2-метил-4-{[[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил](пропил)амино]-метил}фенокси)уксусная кислота;
(4-{[(4′-хлор-2-метил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)-уксусная кислота;
(4-{[(2,4′-диметил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)-уксусная кислота;
(4-{[(4′-фтор-2-метил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)уксусная кислота;
(4-{[(4′-циано-2-метил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)уксусная кислота;
(4-{[(4′-метокси-2-метил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)уксусная кислота;
{4-[(бутил{5-метило-6-[4-(трифторметил)фенил]пиримидин-4-ил}амино)метил]-2-метилфенокси}уксусная кислота;
[4-({бутил[6-(4-метоксифенил)-5-метилпиримидин-4-ил]амино}метил)-2-метилфенокси]уксусная кислота;
[4-({бутил[5-метил-6-(4-метилфенил)пиримидин-4-ил]амино}метил)-2-метилфенокси]уксусная кислота;
[4-({бутил[6-(4-хлорфенил)-5-метилпиримидин-4-ил]амино}метил)-2-метилфенокси]уксусная кислота;
[4-({бутил[6-(4-хлорфенил)-2-пиразин-2-ил]амино}метил)-2-метилфенокси]-уксусная кислота;
[4-({[6-(4-хлорфенил)пиразин-2-ил][2-(метилокси)этил]амино}метил)-2-метилфенокси]уксусная кислота;
{2-метил-4-[(пропил{6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]-фенокси}уксусная кислота;
(2-метил-4-{[{5-метил-6-[4-(трифторметил)фенил]пиримидин-4-ил}(пропил)-амино]метил}фенокси)уксусная кислота;
(4-{[[6-(4-хлорфенил)-5-метилпиримидин-4-ил](пропил)амино]метил}-2-метилфенокси)уксусная кислота;
(2-метил-4-{[[5-метил-6-(4-метилфенил)пиримидин-4-ил](пропил)амино]метил}-фенокси)уксусная кислота;
(2-метил-4-{[{5-метил-6-[4-(метилокси)фенил]пиримидин-4-ил}(пропил)амино]-метил}фенокси)уксусная кислота;
{4-[(бутил{6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]-2-этилфенокси}уксусная кислота;
{2-этил-4-[(2-метилоксиэтил){6-[4-(трифторметил)фенил]пиразин-2-ил}амино)-метил]фенокси}уксусная кислота;
[4-({бутил[5-метил-6-(4-метилфенил)пиримидин-4-ил]амино}метил)-2-этилфенокси]уксусная кислота;
[4-({бутил[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-2-метилфенокси]уксусная кислота;
[2-метил-4-({(пропилсульфонил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-метил)фенокси]уксусная кислота;
[4-({бутил[6-(4-хлорфенил)-5-метилпиримидин-4-ил]амино}метил)-2-этилфенокси]уксусная кислота;
{4-[(бутил{5-метил-6-[4-(трифторметил)фенил]пиримидин-4-ил}амино)метил]-2-этилфенокси}уксусная кислота;
{2-этил-4-[([2-(метилокси)этил]{4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]фенокси}уксусная кислота;
{2-метил-4-[(2-пропен-1-ил{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)-метил]фенокси}уксусная кислота.
Хотя предпочтительные группы для каждой переменной в общем виде перечислены выше отдельно для каждой переменной, предпочтительные соединения по данному изобретению включают такие соединения, в которых несколько или каждая переменная в Формуле (I) выбрана из групп предпочтительных, более предпочтительных или наиболее предпочтительных для каждой переменной. Следовательно, подразумевается, что данное изобретение включает все комбинации предпочтительных и наиболее предпочтительных групп.
Специалистам в данной области техники очевидно, что в соединениях формулы (I) имеются стереоцентры. Соответственно, настоящее изобретение включает все возможные стереоизомеры и геометрические изомеры формулы (I) и включает не только рацемические соединения, но также подразумевается, что данное изобретение охватывает каждый из этих изомеров в их рацемической, обогащенной или очищенной формах. Если желательно иметь соединение формулы (I) в виде единственного энантиомера, его можно получить либо путем разделения конечного продукта, либо путем стереоспецифического синтеза, используя оптически активный катализатор или каталитическую систему с оптически активными лигандами, или изомерно чистое исходное вещество, или любое подходящее промежуточное соединение. Разделение конечного продукта, промежуточного соединения или исходного вещества может быть осуществлено любым подходящим способом, известным в данной области техники. Смотри, например, "Stereochemistry of Carbon Compounds" (E.L.Eliel; Mcgraw Hill, 1962) и "Tables of Resolving Agents" (S.H.Wilen). Кроме того, в ситуациях, когда возможны таутомеры соединений формулы (I), подразумевается, что настоящее изобретение охватывает все таутомерные формы соединений. У некоторых из этих хиральных соединений активности S- или R-изомеров в отношении различных PPAR-рецепторов различаются. Какой из этих изомеров является предпочтительным, зависит от конкретной целевой применимости соединения. Другими словами, даже для одного и того же соединения возможно, что для одних применений будет предпочтителен S-изомер, в то время как для других будет предпочтителен R-изомер.
hPPAR-агонисты формулы (I) могут быть агонистами только одного типа ("селективные агонисты"), агонистами двух подтипов PPAR ("двойные агонисты") или агонистами всех трех подтипов ("пан-агонисты"). При использовании здесь, под "агонистом" или "активирующим соединением" или "активатором" или подобным понимают такие соединения, которые имеют pKi по меньшей мере 6,0, предпочтительно по меньшей мере 7,0 в отношении релевантного PPAR, например hPPARδ, в анализе связывания, описанном ниже, и с помощью которых добиваются по меньшей мере 50%-ной активации релевантного PPAR относительно соответствующего указанного положительного контроля в описанном ниже трансфекционном анализе при концентрациях 10-5 М или ниже. Более предпочтительно, когда с помощью агонистов до данному изобретению добиваются 50%-ной активации по меньшей мере одного PPAR человека в релевантном трансфекционном анализе при концентрациях 10-6 М или ниже. Предпочтительно соединения формулы (I) являются агонистами hPPAR. Более предпочтительно данные соединения являются агонистами hPPARδ. Более предпочтительно они являются селективными агонистами hPPARδ.
Кроме того, специалистам в данной области техники очевидно, что соединения по настоящему изобретению также можно использовать в форме их фармацевтически приемлемой соли или сольвата. Физиологически приемлемые соли соединений формулы (I) включают традиционные соли, образованные фармацевтически приемлемыми неорганическими или органическими кислотами или основаниями, а также соли присоединения кислот и четвертичного аммония. Более конкретные примеры подходящих солей кислот включают соли соляной, бромистоводородной, серной, фосфорной, азотной, перхлорной, фумаровой, уксусной, пропионовой, янтарной, гликолевой, муравьиной, молочной, малеиновой, винной, лимонной, пальмовой, малоновой, оксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, толуолсульфоновой, метансульфоновой, нафталин-2-сульфоновой, бензолсульфоновой, оксинафтойной, иодистоводородной, яблочной, стеариновой, дубильной кислоты и подобных. Другие кислоты, как, например, щавелевая кислота, несмотря на то, что сами по себе не являются фармацевтически приемлемыми, могут быть использованы в получении солей, полезных в качестве промежуточных соединений при получении соединений по изобретению и их фармацевтически приемлемых солей. Более конкретные примеры подходящих солей оснований включают соли натрия, лития, калия, магния, алюминия, кальция, цинка, N,N′-дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, N-метилглюкамина и прокаина. Специалистам в области органической химии очевидно, что многие органические соединения могут образовывать комплексы с растворителями, в которых они взаимодействуют или из которых они осаждаются или кристаллизуются. Эти комплексы известны как "сольваты". Например, комплекс с водой известен как "гидрат". Сольваты соединения формулы (I) входят в объем данного изобретения. Ниже приведенные ссылки на соединение по изобретению включают как соединения формулы (I), так и их фармацевтически приемлемые соли и сольваты.
Соединения по изобретению и их фармацевтически приемлемые производные удобно вводить в форме фармацевтических композиций. Такие композиции удобно могут быть представлены для использования традиционным образом в смеси с одним или более физиологически приемлемыми носителемями или эксципиентами.
Хотя возможно, что соединения по настоящему изобретению могут быть терапевтическим образом введены в виде неочищенного химического средства, предпочтительно представить активный ингредиент в виде фармацевтического препарата. Носитель(и) должен быть "приемлемым(и)" в смысле совместимости с другими ингредиентами препарата и не оказывать вредного воздействия на их реципиента.
Таким образом, в настоящем изобретении также предложен фармацевтический препарат, содержащий соединение формулы (I) или его фармацевтически приемлемую соль или сольват вместе с одним или более фармацевтически приемлемыми носителями и, возможно, другими терапевтическими и/или профилактическими ингредиентами.
Данные препараты включают препараты, подходящие для перорального, парентерального (в том числе подкожного, например путем инъекции или посредством депо-таблетки, интрадермального, интратекального, внутримышечного, например путем депо, или внутривенного), ректального или местного (в том числе дермального, трансбуккального и сублингвального) введения, хотя наиболее подходящий путь может зависеть, например, от состояния и нарушения у реципиента. Данные препараты удобным образом могут быть представлены в стандартной лекарственной форме и могут быть изготовлены любым способом, хорошо известным специалистам в области фармацевтики. Все способы включают стадию объединения соединений ("активного ингредиента") с носителем, который образуется одним или более вспомогательными ингредиентами. Обычно препараты изготовляют путем равномерного и близкого объединения активного ингредиента с жидкими носителями или тонко измельченными твердыми носителями, или с обоими, и затем, если необходимо, придания продукту формы желаемого препарата.
Препараты, подходящие для перорального введения, могут быть представлены в виде отдельных единиц, таких как капсулы, облатки или таблетки (например жевательные таблетки, в частности для педиатрического введения), каждая из которых содержит предварительно определенное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной жидкости или неводной жидкости; или в виде жидкой эмульсии масло-в-воде, или жидкой эмульсии вода-в-масле. Активный ингредиент также может быть представлен в виде болюса, электуария или пасты.
Таблетка может быть изготовлена путем прессования или формования, возможно с одним или более вспомогательными ингредиентами. Прессованные таблетки могут быть изготовлены путем прессования в подходящей машине активного ингредиента в свободно текучей форме, такой как порошок или гранулы, возможно смешанного с другими традиционными эксципиентами, такими как связывающие агенты (например сироп, аравийская камедь, желатин, сорбит, трагакант, крахмальный клейстер или поливинилпирролидон), наполнители (например, лактоза, сахар, микрокристаллическая целлюлоза, кукурузный крахмал, фосфат кальция или сорбит), смазывающие вещества (например, стеарат магния, стеариновая кислота, тальк, полиэтиленгликоль или диоксид кремния), разрыхлители (например картофельный крахмал или натрия крахмала гликолят) или увлажняющие агенты, такие как лаурилсульфат натрия. Формованные таблетки могут быть изготовлены путем формования в подходящей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки возможно могут иметь покрытие или насечки и могут быть изготовлены в виде препарата, обеспечивающего медленное или регулируемое высвобождение активного ингредиента. Таблетки могут быть покрыты оболочкой способами, хорошо известными в данной области техники.
Альтернативно, соединения по настоящему изобретению могут входить в состав пероральных жидких препаратов, таких как, например, водные или масляные суспензии, растворы, эмульсии, сиропы или эликсиры. Кроме того, препараты, содержащие эти соединения, могут быть представлены в виде сухого продукта для разбавления водой или другим подходящим растворителем перед использованием. Такие жидкие препараты могут содержать традиционные добавки, такие как суспендирующие агенты, например сироп сорбита, метилцеллюлозу, глюкозный/сахарный сироп, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрированные пищевые жиры; эмульгирующие агенты, такие как лецитин, сорбитанмоноолеат или аравийская камедь; неводные наполнители (которые могут включать пищевые масла), такие как миндальное масло, ректифицированное кокосовое масло, сложные эфиры масел, пропиленгликоль или этиловый спирт; и консерванты, такие как метил- или пропил-пара-гидроксибензоаты или сорбиновая кислота. Такие препараты также могут быть изготовлены в виде суппозиториев, например, содержащих традиционные основы для суппозиториев, такие как какао-масло или другие глицериды.
Препараты для парентерального введения включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатические средства и растворенные вещества, делающие препарат изотоничным крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загущающие агенты.
Препараты могут быть представлены в однодозовых и многодозовых контейнерах, например герметично закрытых ампулах и флаконах, и могут храниться в высушенном вымораживанием (лиофилизованном) состоянии, нуждаясь только в добавлении стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием. Инъекционные растворы и суспензии для применения в нужный момент могут быть приготовлены из ранее описанных стерильных порошков, гранул и таблеток.
Препараты для ректального введения могут быть представлены в виде суппозитория с обычными носителями, такими как какао-масло, твердый жир или полиэтиленгликоль.
Препараты для местного введения в полость рта, например трансбуккально или сублингвально, включают таблетки, содержащие активный ингредиент в основе с добавлением корригентов, такой как сахароза и аравийская камедь или трагакант, и пастилки, содержащие активный ингредиент в такой основе, как желатин и глицерин или сахароза и аравийская камедь.
Кроме того, соединения могут быть изготовлены в виде депо-препаратов. Такие препараты с продолжительным действием могут быть введены путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Так, например, соединения могут быть изготовлены в виде препаратов с подходящими полимерными или гидрофобными веществами (например, в виде эмульсии в подходящем масле) или ионообменными смолами, или в виде умеренно растворимых производных, например в виде умеренно растворимой соли.
В дополнение к ингредиентам, отдельно упомянутым выше, данные препараты могут включать другие традиционно используемые в данной области техники агенты, имеющие отношение к рассматриваемому типу препарата, например препарат, подходящий для перорального введения, может включать корригирующие агенты.
Специалистам в данной области техники очевидно, что ссылка в данном описании на лечение распространяется на профилактику, а также на лечение установленных заболеваний или симптомов. Более того, очевидно, что количество соединения по изобретению, необходимое для применения в лечении, будет меняться в зависимости от природы состояния, которое лечат, и возраста и состояния пациента, и в конечном счете будет находиться на усмотрении лечащего врача или ветеринара. В общем случае, однако, дозы, используемые для лечения взрослого человека, в типичном случае будут находиться в интервале 0,02-5000 мг в сутки, предпочтительно 1-1500 мг в сутки. Требуемая доза удобным образом может быть представлена в виде разовой дозы или разделенных доз, вводимых через подходящие интервалы, например в виде двух, трех, четырех или более субдоз в сутки. Препараты согласно изобретению могут содержать 0,1-99% активного ингредиента; для таблеток и капсул подходит 30-95% и для жидких препаратов подходит 3-50%.
Соединение формулы (I) для применения в настоящем изобретении могут быть использованы в комбинации с другими терапевтическими агентами, например статинами и/или другими снижающими содержание липидов лекарственными средствами, например ингибиторами МТР (микросомального переносящего белка) и позитивными регуляторами LDLR. Соединения по изобретению также могут быть использованы в комбинации с антидиабетическими агентами, например метформином, сульфонилмочевинами и/или агонистами PPAR-гамма, PPAR-альфа или PPAR-альфа/гамма (например, с тиазолидиндионами, такими как, например, пиоглитазон и розиглитазон). Эти соединения также можно использовать в комбинации с антигипертензивными агентами, такими как антагонисты ангиотензина, например телмисартаном, с антагонистами кальциевых каналов, например лацидипином, и с ингибиторами АСЕ, например эналаприлом. Таким образом, в следующем аспекте данного изобретения предложено применение комбинации, содержащей соединение формулы (I) с еще одним терапевтическим агентом, в лечении hPPAR-опосредованного заболевания.
Когда соединения формулы (I) используют в комбинации с другими терапевтическими агентами, эти соединения могут быть введены либо последовательно, либо одновременно любым удобным путем.
Указанные выше комбинации удобным образом могут быть представлены для применения в форме фармацевтического препарата, и таким образом фармацевтические препараты, содержащие комбинацию, как она определена выше, оптимально вместе с фармацевтически приемлемым носителем или эксципиентом, составляют еще один аспект данного изобретения. Индивидуальные компоненты таких комбинаций могут быть введены либо последовательно, либо одновременно в виде отдельных или объединенных фармацевтических препаратов.
Очевидно, что при объединении в одном и том же препарате два соединения должны быть стабильны и совместимы друг с другом и другими компонентами этого препарата и могут быть приготовлены в виде препарата для введения. В случае раздельных препаратов они могут быть представлены в виде любого подходящего препарата, подходящего так, как подходят для таких соединений известные в данной области техники препараты.
Если соединение формулы (I) используют в комбинации со вторым терапевтическим агентом, активным в отношении того же hPPAR-опосредованного заболевания, доза каждого соединения может отличаться от дозы, когда соединение используют само по себе. Соответствующие дозы очевидны специалистам в данной области техники.
Соединения по данному изобретению могут быть получены удобным образом согласно общим способам, представленным ниже на схемах.
Схема 1

Промежуточные соединения (А) могут быть получены алкилированием подходящего 4-гидроксибензальдегида, с получаем бензальдегидов (промежуточное соединение С), с последующим восстановительным аминированием альдегидной группировки.
Схема 2

Промежуточные соединения (В) могут быть получены путем восстановления подходящего 4-гидроксибензальдегида до спирта. В результате алкилирования фенольной группировки подходящим бромацетатом с последующим галогенированием бензилового спирта получают промежуточные соединения (В).
Схема 3
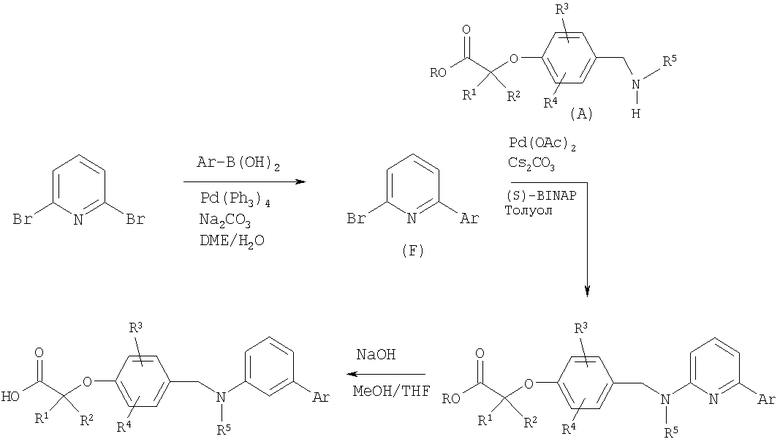
Имеющийся в продаже 2,6-дибромпиридин можно подвергнуть реакции сочетания с бороновыми кислотами в условиях сочетания Сузуки. Полученные монобромиды затем можно обработать амином (промежуточное соединение А) в реакции Бухвальда (Buchwald) с получением продуктов сочетания. Эфирную группу затем можно гидролизовать до соответствующих карбоновых кислот в стандартных условиях.
Схема 4
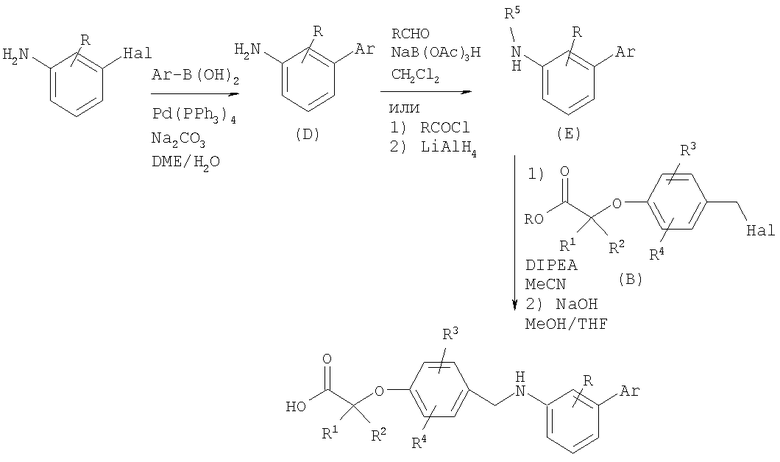
Подходящие галогенированные анилины можно подвергнуть реакции сочетания с бороновыми кислотами в условиях сочетания Сузуки с получением продуктов сочетания (промежуточное соединение D). В результате восстановительного аминирования или ацилирования и восстановления полученных соединений получают вторичные анилины (промежуточное соединение Е), которые алкилируют галогенидами (промежуточное соединение В). В результате гидролиза сложных эфиров карбоновых кислот в стандартных условиях получают карбоновые кислоты.
Схема 5
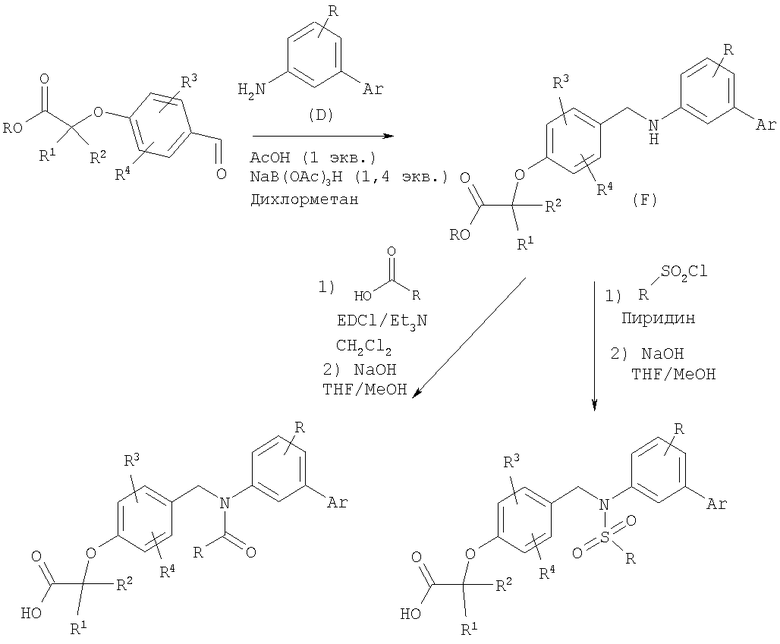
В результате восстановительного аминирования анилина (промежуточное соединение D) альдегидом (промежуточное соединение С) получают вторичный анилин (промежуточное соединение F), который может быть превращен в амид путем сочетания с подходящей карбоновой кислотой, а в сульфонамид путем взаимодействия с соответствующим алкилсульфонилхлоридом. Полученные сложные эфиры гидролизуют в стандартных условиях с получением карбоновых кислот.
Схема 6

В результате взаимодействия вторичного анилина (промежуточное соединение Е) с сульфонилхлоридом (промежуточное соединение G) получают связанные через сульфонамид соединения. В результате гидролиза сложных эфиров в стандартных условиях получают карбоновые кислоты.
Схема 7
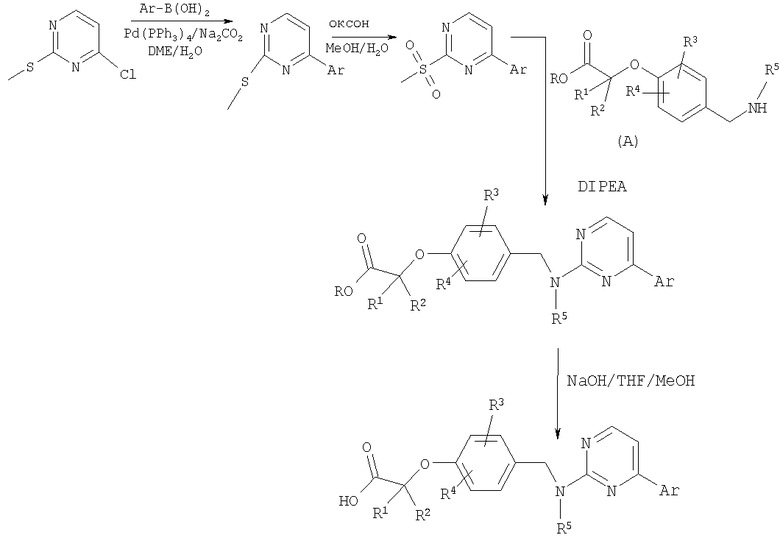
Имеющиеся в продаже 4-хлор-2-(метилтио)пиримидин и бороновые кислоты можно подвергнуть реакции сочетания в условиях Сузуки. В результате окисления тиолов получают соответствующие сульфоны. Сульфоны затем могут быть приведены во взаимодействие с аминами (промежуточные соединения А). Полученные в результате сочетания соединения гидролизуют в стандартных условиях с получением карбоновых кислот.
Схема 8
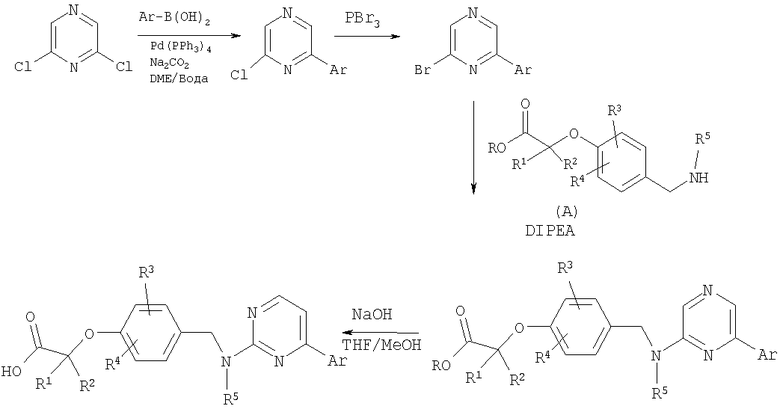
Имеющийся в продаже дихлорпиразин можно подвергнуть реакции сочетания с бороновыми кислотами в условиях сочетания Сузуки. Полученный монохлорпиразин может быть превращен в бромид путем обработки трибромидом фосфора. В результате замены бромида амином (промежуточное соединение А) в присутствии N,N-диизопропилэтиламина получают сложные эфиры, которые гидролизуют до кислот в стандартных условиях.
Схема 9
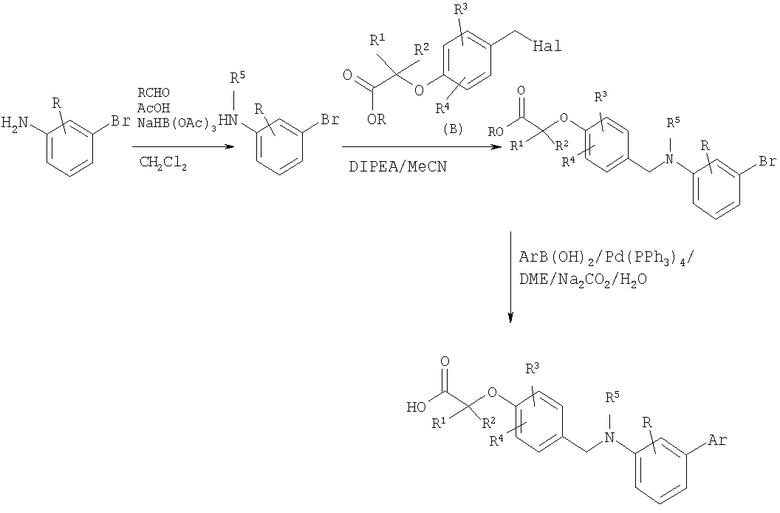
В результате восстановительного аминирования подходящего анилина альдегидом получают вторичный анилин, который затем может быть алкилирован подходящим галогенидом (промежуточное соединение В). Этот продукт можно подвергнуть реакции сочетания с соответствующей бороновой кислотой в стандартных условиях сочетания Сузуки с непосредственным получением карбоновых кислот.
Схема 10
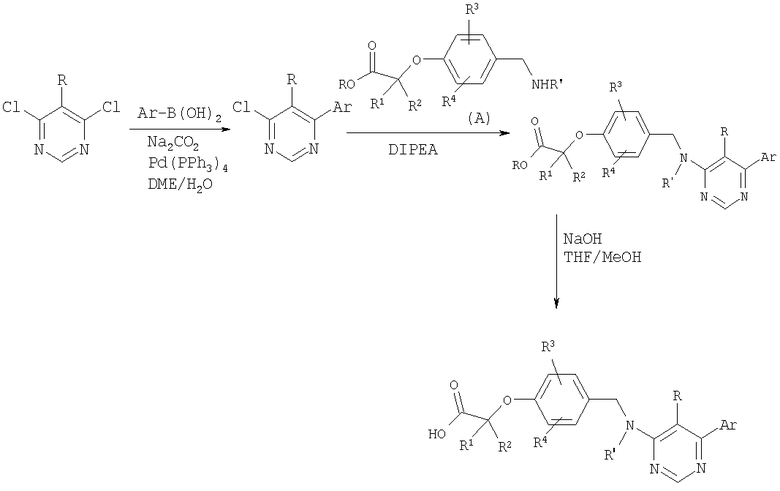
Подходящий дихлорпиридин можно подвергнуть реакции сочетания с бороновой кислотой в условиях сочетания Сузуки. Полученные монохлорпиримидины могут быть обработаны амином (промежуточное соединение А) в присутствии N,N-диизопропилэтиламина с получением сложных эфиров, которые гидролизуют в стандартных условиях до карбоновых кислот.
Ниже рассмотрены Промежуточные соединения и Примеры соединений Формулы 1, которые не следует истолковывать как представляющие собой ограничение.
Общие методы очистки и анализа
LC/MS (жидкостная хроматография/масс-спектрометрия) относится к анализу посредством аналитической HPLC (высокоэффективной жидкостной хроматографии), которую проводили на колонке Supelcosil LCABZ+PLUS (3 мкм; 3,3 см ×4,6 мм внутр. диам.), элюируя 0,1%-ной HCO2H и 0,01 М ацетатом аммония в воде (растворитель А) и 95%-ным ацетонитрилом и 0,05%-ной НСО2Н в воде (растворитель Б), используя следующий градиент элюирования: 0-0,7 минуты 0%-ный Б; 0,7-4,2 минуты 0→100%-ный Б; 4,2-5,3 минуты 100%-ный Б; 5,3-5,5 минуты 100→0%-ный Б, при скорости потока 3 мл/минуту. Масс-спектры (MS) регистрировали на Fisons VG Platform масс-спектрометре с использованием ионизации электрораспылением в режиме положительных ионов [(ES+ve с получением молекулярных ионов [М+Н]+ и [M+NH4]+] или ионизации электрораспылением в режиме отрицательных ионов [(ES-ve с получением молекулярного иона [М-Н]-].
1H ЯМР-спектры регистрировали, используя спектрометр Bruker DPX400 МГц.
Biotage™-хроматография относится к очистке, которую проводили, используя прибор, который продается у Dyax Corporation (либо Flash 40i, либо Flash 150i), и картриджи, предварительно упакованные KP-Sil™ диоксидом кремния.
Используемые реакционные сосуды поставлялись Perbio Science UK Ltd.
Аутопрепаративная HPLC с управлением по массе относится к способу, при котором вещество очищали высокоэффективной жидкостной хроматографией на колонке HPLCABZ+ (5 мкм; 5 см × 10 мм внутр. диам.), используя 0,1% HCO2Н в воде и 95% MeCN, 5% воды (0,5% HCO2Н), используя следующие условия градиентного элюирования: 0-1,0 минута 5%-ный Б; 1,0-8,0 минут 5→30%-ный Б; 8,0-8,9 минут 30%-ный Б; 8,9-9,0 минут 30→95%-ный Б; 9,0-9,9 минут 95%-ный Б; 9,9-10,0 минут 95→0%-ный Б, при скорости потока 8 мл/мин. Коллектор фракций Gilson 202 запускался VG Platform масс-спектрометром при детектировании интересующей массы.
Гидрофобные фритты относятся к фильтрационным трубкам, продающимся у Wathman.
SPE (твердофазная экстракция) относится к использованию картриджей, продаваемых International Sorbent Technology Ltd. SCX представляет собой стационарную фазу с бензолсульфоновой кислотой.
TLC (тонкослойная хроматография) относится к использованию TLC-пластинок, продаваемых Merck, покрытых силикагелем 60 F254.
Сокращения:
Промежуточное соединение 1: 2-бром-6-[4-(трифторметил)фенил]-пиридин
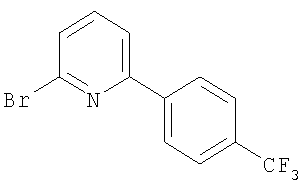
К раствору 2,6-дибромпиридина (1,25 г; 5,28 ммоль) в диметоксиэтане (100 мл) и воде (50 мл) добавляли 4-трифторметилфенилбороновую кислоту (1 г; 5,27 ммоль) и карбонат натрия (1,45 г; 13,7 ммоль). Полученную смесь продували азотом перед добавлением тетракис(трифенилфосфин)палладия(0) (60 мг; 0,05 ммоль). Реакционную смесь нагревали до кипения с обратным холодильником в атмосфере азота в течение 5 ч и затем оставляли охлаждаться до комнатной температуры. Растворители удаляли под вакуумом и остаток разбавляли водой (50 мл) и затем экстрагировали EtOAc (2×100 мл). Органический раствор сушили (MgSO4), фильтровали и растворители удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью циклогексан:хлороформ (4:1), получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,35 г).
1Н ЯМР (400 МГц; CDCl3) δ: 7.49 (1Н, d, J 7,5 Гц), 7.65 (1Н, t, J 8 Гц), 7.73 (3H, m), 8.11 (2H, d, J 8,5 Гц).
Промежуточное соединение 2: этил-2-метил-2-{2-метил-4-[([4-(трифторметил)бензил]{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)-метил]фенокси}пропаноат

Смесь этил-2-метил-2-[2-метил-4-({[4-(трифторметил)бензил]амино}-метил)фенокси]пропаноата (0,13 г; 0,318 ммоль), 2-бром-6-[4-(трифторметил)-фенил]пиридина (78,5 мг; 0,26 ммоль), ацетата палладия(II) (4,8 мг; 0,021 ммоль), (R)-2,2'-бис(дифенилфосфино)-1,1′-бинафтила (19,8 мг; 0,032 ммоль) и карбоната цезия (0,13 г; 0,397 ммоль) в толуоле (1,8 мл) нагревали при 70°С в реакционном сосуде в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали и растворитель удаляли под вакуумом. В результате очистки посредством SPE (диоксид кремния, 10 г), элюируя смесью циклогексан:EtOAc (99:1-19:1), получали указанное в заголовке соединение в виде бесцветного масла (75 мг).
LC/MS: m/z 631,3 [M+H]+, Rt 4,7 мин.
Промежуточное соединение 3: этил-2-{4-[(бутил{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)метил]-2-метилфенокси}-2-метилпропаноат
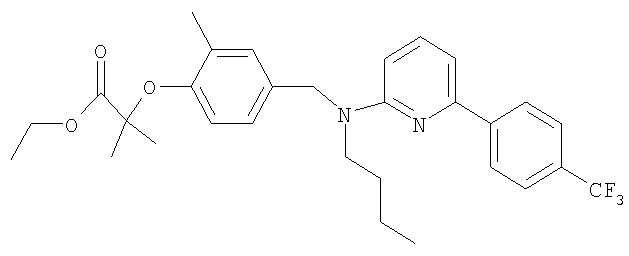
Ацетат палладия(II) (1,4 мг; 0,006 ммоль) и (S)-2,2'-бис(дифенилфосфино)-1,1′-бинафтил (5,9 мг; 0,0095 ммоль) в безводном толуоле (0,32 мл) вместе перемешивали при 70°С в герметично закрытом реакционном сосуде в течение 30 минут. Сосуд затем оставляли достигать комнатной температуры перед добавлением этил-2-{4-[(бутиламино)метил]-2-метилфенокси}-2-метилпропаноата (29 мг; 0,095 ммоль), 2-бром-6-[4-(трифторметил)фенил]пиридина (24 мг; 0,08 ммоль) и карбоната цезия (36 мг; 0,11 ммоль). Сосуд затем герметично закрывали и нагревали при 80°С в течение 18 ч, затем оставляли охлаждаться до комнатной температуры, реакционную смесь фильтровали и растворитель удаляли под вакуумом. В результате очистки посредством SPE (диоксид кремния, 10 г), элюируя смесью циклогексан:EtOAc (99:1), получали указанное в заголовке соединение в виде бесцветного масла (18 мг).
LC/MS: m/z 529,2 [M+H]+, Rt 4,7 мин.
Промежуточное соединение 4: метил-{4-[(бутил{6-[4-(трифторметил)-фенил]пиридин-2-ил}амино)-метил]-2-метилфенокси}ацетат

Ацетат палладия(II) (5,4 мг; 0,024 ммоль) и (S)-2,2'-бис(дифенилфосфино)-1,1′-бинафтил (22 мг; 0,036 ммоль) в безводном толуоле (1,2 мл) вместе перемешивали при 70°С в герметично закрытом реакционном сосуде в течение 30 минут. Сосуд затем оставляли достигать комнатной температуры перед добавлением метил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата (96 мг; 0,36 ммоль), 2-бром-6-[4-(трифторметил)фенил]-пиридина (90,6 мг; 0,3 ммоль) и карбоната цезия (137 мг; 0,42 ммоль). Сосуд затем герметично закрывали и нагревали при 80°С в течение 18 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры, реакционную смесь фильтровали и растворитель удаляли под вакуумом. В результате очистки посредством SPE (диоксид кремния, 10 г), элюируя смесью циклогексан:EtOAc (99:1-50:1), получали указанное в заголовке соединение в виде басцветного масла (50 мг).
LC/MS: m/z 487,3 [М+Н]+, Rt 4,5 мин.
Промежуточное соединение 5: этил-2-(4-формил-2-метилфенокси)-2-метилпропаноат

К раствору 4-гидрокси-3-метилбензальдегида (3 г; 22 ммоль) и этил-2-бром-2-метилпропионата (6,5 мл; 44 ммоль) в DMF (100 мл) добавляли карбонат цезия (15,8 г; 48 ммоль). Полученную смесь нагревали при 40°С в течение 42 ч, затем оставляли охлаждаться до комнатной температуры. Реакционную смесь разбавляли водой (200 мл) и экстрагировали EtOAc (2×150 мл). Органический раствор сушили (MgSO4), фильтровали и упаривали. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бледно-желтого масла (3,5 г).
1H ЯМР (400 МГц; CDCl3) δ: 1.21 (3Н, t, J 7 Гц), 1.68 (6Н, s) 2.29 (3H, s), 4.23 (2H, q, J 7 Гц), 6.68 (1Н, d, J 8 Гц), 7.59 (1Н, dd, J 8, 2 Гц), 7.70 (1Н, d, J 2 Гц), 9.85 (1Н, s).
Промежуточное соединение 6: этил-(4-формил-2-метилфенокси)-ацетат
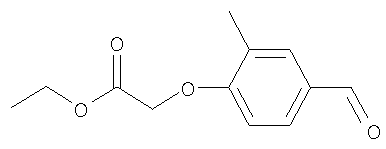
К раствору 4-гидрокси-3-метилбензальдегида (8 г; 58,8 ммоль) в безводном ацетонитриле (300 мл) в атмосфере азота при 0°С добавляли карбонат цезия (21 г; 64,6 ммоль) и этилбромацетат (6,52 мл; 58,8 ммоль). Полученную смесь оставляли достигать комнатной температуры и перемешивали в течение 16 ч. Реакционную смесь разбавляли водой (400 мл) и экстрагировали EtOAc (2×500 мл). Объединенные органические экстракты сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде коричневого твердого вещества (11,5 г).
LC/MS: m/z 223,2 [M+H]+, Rt 2,8 мин.
Промежуточное соединение 7: этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетат
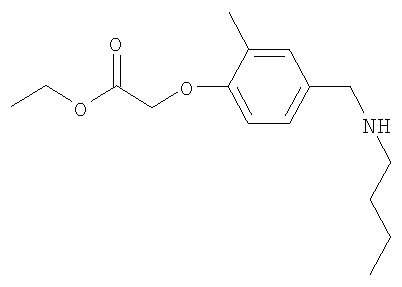
К раствору этил-(4-формил-2-метилфенокси)ацетата (3 г; 13,5 ммоль). в безводном этаноле (50 мл) при комнатной температуре добавляли н-бутиламин (1,6 мл; 16,2 ммоль) и 4Å молекулярные сита. Полученную смесь перемешивали при комнатной температуре в течение 18 ч в атмосфере азота перед добавлением порциями триацетоксиборогидрида натрия (3,43 г; 16,2 ммоль). После дополнительного перемешивания в течение 3 ч при комнатной температуре реакционную смесь гасили путем осторожного добавления насыщенного раствора бикарбоната натрия (40 мл) и затем разбавляли водой (50 мл), потом экстрагировали EtOAc (2 х 100 мл). Объединенные органические экстракты сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде коричневого масла (2,6 г).
LC/MS: m/z 280,2 [M+H]+, Rt 2,2 мин.
Промежуточное соединение 8: этил-{2-метил-4-[(пропиламино)-метил]фенокси}ацетат
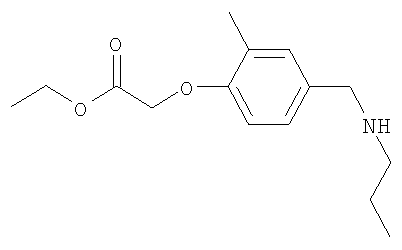
Получали, используя н-пропиламин (1,67 мл; 20,3 ммоль) и методику синтеза, описанную для Промежуточного соединения 7. В результате очистки неочищенного продукта посредством SCX SPE (5×10 г) с загрузкой и промыванием EtOH и последующим элюированием продукта 5%-ным NH3 в EtOH, и удалением растворителя под вакуумом, получали указанное в заголовке соединение в виде коричневого масла (2,4 г).
LC/MS: m/z 266,2 [М+Н]+, Rt 2,08 мин.
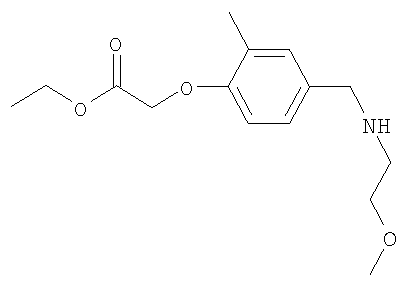
Промежуточное соединение 9: этил-(4-{[(2-метоксиэтил)амино]-метил}-2-метилфенокси)ацетат
Получали, используя 2-метоксиэтиламин (1,4 мл; 16,2 ммоль) и методику синтеза, описанную для Промежуточного соединения 7. В результате очистки неочищенного продукта посредством SCX SPE (5×10 г) с загрузкой и промыванием EtOH и затем элюированием продукта 5%-ным NH3 в EtOH и удалением растворителя под вакуумом получали указанное в заголовке соединение в виде коричневого масла (1,8 г).
LC/MS: m/z 282,1 [М+Н]+, Rt 2,0 мин.
Промежуточное соединение 10: этил-2-метил-2-[2-метил-4-({[4-(трифторметил)бензил]амино}метил)-фенокси]пропаноат
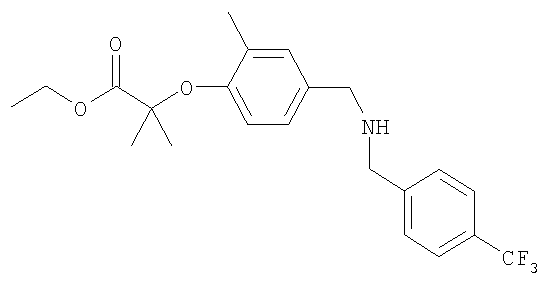
К раствору этил-2-(4-формил-2-метилфенокси)-2-метилпропаноата (0,563 г; 2,25 ммоль) в CH2Cl2 (10 мл) в атмосфере азота при комнатной температуре добавляли 4-трифторметилбензиламин (0,34 мл; 2,37 ммоль). Полученный раствор перемешивали в течение 30 минут перед добавлением триацетоксиборогидрида натрия (0,668 г; 3,15 ммоль). После перемешивания в течение 18 ч реакционную смесь гасили осторожным добавлением насыщенного водного бикарбоната натрия (30 мл) и экстрагировали CH2Cl2 (2×50 мл). Органический раствор сушили (MgSO4) и растворитель удаляли под вакуумом. В результате очистки посредством SPE (диоксид кремния, 20 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бесцветного масла (0,485 г).
LC/MS: m/z 410,2 [M+H]+, Rt 2,9 мин.
Промежуточное соединение 11: этил-2-{4-[(бутиламино)метил]-2-метилфенокси}-2-метилпропаноат

К раствору этил-2-(4-формил-2-метилфенокси)-2-метилпропаноата (0,56 г; 2,25 ммоль) в безводном CH2Cl2 (10 мл) добавляли н-бутиламин (0,33 мл; 3,38 ммоль). После перемешивания в течение 90 мин в атмосфере азота при комнатной температуре добавляли триацетоксиборогидрид натрия (0,668 г; 3,15 ммоль) и перемешивание продолжали в течение 18 ч. Реакционную смесь гасили путем осторожного добавления насыщенного водного карбоната натрия (20 мл) и затем экстрагировали CH2Cl2 (2×40 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде бледно-желтого масла (0,48 г).
LC/MS: m/z 308,2 [М+Н]+, Rt 2,5 мин.
Промежуточное соединение 12: метил-{4-[(бутиламино)метил]-2-метилфенокси}ацетат
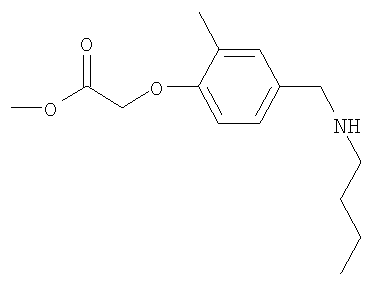
К раствору этил-(4-формил-2-метилфенокси)ацетата (0,65 г; 2,93 ммоль) в безводном метаноле (12 мл) в атмосфере азота добавляли н-бутиламин (0,3 мл; 3,07 ммоль) и полученную смесь перемешивали в течение 18 ч при комнатной температуре перед добавлением порциями борогидрида натрия (0,144 г; 3,8 ммоль). После 30 минут перемешивания при комнатной температуре реакционную смесь гасили осторожным добавлением насыщенного водного бикарбоната натрия и затем экстрагировали в этилацетат (2×40 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде бледно-желтого масла (0,65 г).
LC/MS: m/z 266,1 [M+H]+, Rt 2,1 мин.
Промежуточное соединение 13: 4-(гидроксиметил)-2-метилфенол

К раствору 4-гидрокси-3-метилбензальдегида (10 г; 73,5 ммоль) в безводном CH2Cl2 (550 мл) в атмосфере азота при 5°С добавляли тетра-N-бутиламмония борогидрид (20,58 г; 80 ммоль) и полученную смесь перемешивали в течение 1,25 ч при 5°С. К реакционной смеси добавляли насыщенный раствор хлорида аммония (30 мл) и полученную смесь перемешивали в течение 1 ч при 5°С. Добавляли насыщенный раствор хлорида аммония (60 мл), и реакционную смесь экстрагировали CH2Cl2, сушили (Na2SO4) и растворители удаляли под вакуумом. В результате очистки пропусканием через диоксид кремния (2×150 г), элюируя EtOAc, получали указанное в заголовке соединение в виде маслянистого твердого вещества желтого цвета (8,01 г).
1H ЯМР (400 МГц; MeOH-d4) δ: 2.19 (3Н, s), 4.46 (2H, s), 6.71 (1H, d, J 8 Гц), 6,99 (1H, dd, J 8 Гц, 2 Гц), 7.06 (1H, d, J 2 Гц), ОН не наблюдался.
Промежуточное соединение 14: этил-[4-(гидроксиметил)-2-метилфенокси]ацетат (Способ А)
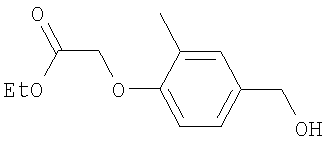
К раствору 4-(гидроксиметил)-2-метилфенола (7,95 г; 57 ммоль) в безводном ацетонитриле (300 мл) при 5°С добавляли карбонат цезия (20,42 г; 62 ммоль) и этилбромацетат (6,38 мл; 57 ммоль). Полученную смесь перемешивали в течение 3 ч при 5°С в атмосфере азота. Реакционную смесь разбавляли водой (500 мл), экстрагировали EtOAc (2 х 750 мл), сушили (Na2SO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде желтого масла (12,3 г).
1H ЯМР (400 МГц; CDCl3) δ: 1.3 (3Н, t, J 7 Гц), 2.3 (3Н, s), 4.26 (2H, q, J 7 Гц), 4.59 (2H, s), 4.64 (2H, s), 6.68 (1H, d, J 8,5 Гц), 7.12 (1H, dd, J 8,5 Гц, 1,5 Гц), 7.18 (1H, d, J 1,5 Гц), ОН не наблюдался.
Промежуточное соединение 14: этил-[4-(гидроксиметил)-2-метилфенокси]ацетат (Способ Б)
Этил-(4-формил-2-метилфенокси)ацетат (29,2 г; 0,131 моль) растворяли в этаноле (450 мл) и охлаждали до 10°С в атмосфере азота. В течение 10 мин к перемешиваемой реакционной смеси добавляли борогидрид натрия (5,45 г; 0,144 моль) и перемешивали при 10°С в течение еще 20 мин. Реакционную смесь осторожно гасили смесью вода/насыщенный NaHCO3 (3:1, 25 мл), затем разбавляли смесью вода/соляной раствор (3:1, 730 мл) и экстрагировали EtOAc (2×730 мл). Объединенный органический экстракт промывали соляным раствором (1 л), сушили (Na2SO4) и концентрировали под вакуумом. Остаток разбавляли дихлорметаном (400 мл), промывали водой (250 мл) и соляным раствором (250 мл), сушили (Na2SO4) и концентрировали под вакуумом с получением указанного в заголовке соединения в виде коричневого масла (28,2 г).
1H ЯМР (400 МГц; CDCl3) δ: 1.30 (3Н, t, J 7 Гц), 2.31 (3Н, s), 4.27 (2Н, q, J 7 Гц), 4.60 (2Н, s), 4.64 (2Н, s), 6.69 (1H, d, J 8,5 Гц), 7.13 (1Н, dd, J 8,5 Гц, 1,5 Гц), 7.18 (1H, d, J 1,5 Гц), ОН не наблюдался.
Промежуточное соединение 15: этил-[4-(бромметил)-2-метилфенокси]ацетат

К раствору этил-[4-(гидроксиметил)-2-метилфенокси]ацетата (4,27 г; 19 ммоль) в безводном СН2Cl2 (150 мл) в атмосфере азота при 5°С добавляли тетрабромметан (6,91 г; 20,9 ммоль), затем трифенилфосфин (5,71 г; 21,8 ммоль) порциями в течение 0,5 ч. Полученную смесь затем перемешивали в течение 16 ч при комнатной температуре. Растворители удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 2×90 г), элюируя смесью циклогексан:EtOAc (30:1), получали указанное в заголовке соединение в виде твердого вещества желтого цвета (2,95 г).
1H ЯМР (400 МГц; CDCl3) δ: 1.3 (3Н, t, J 7 Гц), 2.28 (3Н, s), 4.26 (2Н, q, J 7 Гц), 4.47 (2Н, s), 4.63 (2Н, s), 6.64 (1H, d, J 8,5 Гц), 7.16 (1H, dd, J 8,5 Гц, 2 Гц), 7.2 (1H, d, J 2 Гц).
Промежуточное соединение 16: 4′-(трифторметил)-1,1′-бифенил-3-иламин

К раствору 3-иоданилина в DME (40 мл) и воде (20 мл) добавляли пара-трифторметилфенилбороновую кислоту (2,6 г; 13,7 ммоль) и карбонат натрия (2,5 г; 23,6 ммоль). Аппарат заполняли азотом перед добавлением тетракис(трифенилфосфин)палладия(0) (0,312 г; 0,27 ммоль). Реакционную смесь перемешивали при 95°С в течение 17 ч в атмосфере азота, затем оставляли охлаждаться до комнатной температуры и растворители удаляли под вакуумом. Остаток распределяли между EtOAc (2 х 75 мл) и водой (75 мл). Органический раствор сушили (MgSO4) и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (колонка с 90 г диоксида кремния), элюируя смесью EtOAc:циклогексан (1:4), получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,33 г).
1H ЯМР (400 МГц; CDCl3) δ: 6.73 (1Н, add, J 8 Гц, 2,5 Гц, 1 Гц), 6.90 (1Н, m), 6.99 (1Н, dm, J 7,5 Гц), 7.26 (1Н, m), 7.66 (4H, s), NH2 не наблюдали.
Промежуточное соединение 17: N-бутил-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амин
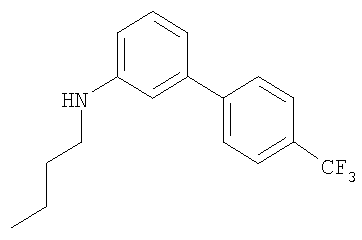
К раствору масляного альдегида (0,114 мл; 1,26 ммоль) в безводном CH2Cl2 в атмосфере азота добавляли 4′-(трифторметил)-1,1′-бифенил-3-иламин (0,3 г; 1,26 ммоль). Через 15 мин порциями добавляли триацетоксиборогидрид натрия (0,375 г; 1,77 ммоль), затем уксусную кислоту (0,072 мл; 1,26 ммоль). После перемешивания смеси в течение 18 ч реакционную смесь гасили осторожным добавлением насыщенного водного бикарбоната натрия (20 мл) и экстрагировали СН2Cl2 (2×30 мл). Органический экстракт разделяли с помощью гидрофобной фритты и растворители удаляли под вакуумом. В результате очистки посредством SPE (диоксид кремния, 10 г), элюируя смесью циклогексан:EtOAc (99:1), получали указанное в заголовке соединение в виде бесцветного масла (0,25 г).
LC/MS: m/z 293,8 [M+H]+, Rt 4,1 мин.
Промежуточное соединение 18: N-(2-метоксиэтил)-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амин
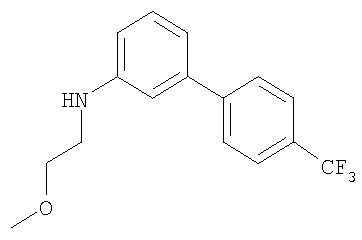
Раствор метоксиацетальдегида диметилацеталя (0,162 мл; 1,26 ммоль) в 0,5 М водной HCl (2,52 мл; 1,26 ммоль) нагревали до 50°С и перемешивали в атмосфере азота в течение 1 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и экстрагировали CH2Cl2 (10 мл). Органический экстракт разделяли с помощью гидрофобной фритты и сушили, выдерживая над молекулярными ситами в течение 2 ч. К этому раствору добавляли 4′-(трифторметил)-1,1′-бифенил-3-иламин (0,3 г; 1,26 ммоль), затем через 20 минут добавляли триацетоксиборогидрид натрия (0,375 г; 1,77 ммоль) и уксусную кислоту (0,072 мл; 1,26 ммоль). Полученную смесь перемешивали при комнатной температуре в атмосфере азота в течение 18 ч. Реакционную смесь гасили путем осторожного добавления насыщенного водного NaHCO3 (30 мл) и экстрагировали в CH2Cl2 (2×30 мл). Органический экстракт пропускали через гидрофобную фритту и растворители удаляли под вакуумом. В результате очистки Biotage™-хроматографией, элюируя смесью циклогексан:EtOAc (19:1), получали указанное в заголовке соединение в виде бледно-желтого масла (0,1 г).
1H ЯМР (400 МГц; CDCl3) δ: 3.35 (2Н, t, J 5 Гц), 3.41 (3Н, s), 3.64 (2H, t, J 5 Гц), 4.2 (1Н, br. s), 6.68 (1Н, dm, J 8 Гц), 6.83 (1Н, t, J 2 Гц), 6.94 (1Н, dm, J 8 Гц), 7.27 (1Н, t, 8 Гц), 7.97 (4Н, s).
Промежуточное соединение 19: N-пентил-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амин

К раствору 4′-(трифторметил)-1,1′-бифенил-3-иламина (0,3 г; 1,26 ммоль) в безводном CH2Cl2 (10 мл) в атмосфере азота при комнатной температуре добавляли валериановый альдегид (0,134 мл; 1,26 ммоль), триацетоксиборогидрид натрия (0,374 г; 1,76 ммоль) и уксусную кислоту (72 мкл; 1,26 ммоль). Полученную смесь перемешивали в течение 17 ч при комнатной температуре. Реакционную смесь гасили насыщенным водным NaHCO3 (15 мл), затем экстрагировали CH2Cl2 (2×20 мл). Органический раствор сушили (Na3SO4) и растворитель удаляли под вакуумом. В результате очистки посредством SPE (колонка с 20 г диоксида кремния), элюируя смесью EtOAc: циклогексан (1:200), получали указанное в заголовке соединение в виде бесцветного масла (0,17 г).
LC/MS: m/z 308,1 [M+H]+, Rt 4,3 мин.
Промежуточное соединение 20: N-пропил-N-[′-(трифторметил)-1,1′-бифенил-3-ил]амин

Получали, используя пропионовый альдегид (91 мкл; 1,26 ммоль) и методику синтеза, описанную для Промежуточного соединения 19. В результате очистки Biotage™-хроматографией (колонка с 40 г диоксида кремния), элюируя смесью EtOAc:циклогексан (1:50), получали указанное в заголовке соединение в виде белого кристаллического твердого вещества (0,22 г).
LC/MS: m/z 279,9 [M+H]+, Rt 4,0 мин.
Промежуточное соединение 21: N-[2-(метилтио)этил]-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амин

Раствор 2-(метилтио)-ацетальдегида диэтилацеталя (0,465 г; 2,83 ммоль) в 0,5 М HCl (5,66 мл; 2,83 ммоль) нагревали до 50°С и перемешивали в атмосфере азота в течение 1 ч. Реакционную смесь оставляли охлаждаться и экстрагировали СН2Cl2 (20 мл). Органический раствор сушили (гидрофобная фритта) и выдерживали над молекулярными ситами в течение 2 ч, затем декантировали в чистую колбу. К этому раствору добавляли 4′-(трифторметил)-1,1′-бифенил-3-иламин (0,67 г; 2,83 ммоль) с последующим добавлением триацетоксиборогидрида натрия (1,68 г; 7,93 ммоль) и уксусной кислоты (0,162 мл; 2,83 ммоль). Полученную смесь перемешивали при комнатной температуре в атмосфере азота в течение 16 ч. Реакционную смесь гасили насыщенным водным NaHCO3 (30 мл) и экстрагировали CH2Cl2 (2×40 мл), органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (колонка с 90 г диоксида кремния), элюируя смесью EtOAc:циклогексан (1:19), получали указанное в заголовке соединение в виде бледно-желтого масла (397 мг).
1H ЯМР (400 МГц; CDCl3) δ: 2.13 (3Н, s), 2.81 (2H, t, J 6,5 Гц), 3.40 (2Н, t, J 6,5 Гц), 4.23 (1Н, br. s), 6.68 (1H, d, J 8 Гц), 6.83 (1Н, t, J 2 Гц), 6.95 (1Н, d, J 7,5 Гц), 7.28 (1Н, t, J 8 Гц), 7.67 (4Н, s).
Промежуточное соединение 22: N-(2-циклопропилэтил)-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амин

К раствору оксалилхлорида (0,17 мл; 1,95 ммоль) в безводном CH2Cl2 (5 мл) в атмосфере азота при -78°С по каплям добавляли раствор безводного DMSO (0,18 мл; 2,6 ммоль) в безводном CH2Cl2 (2,5 мл), обеспечивая, чтобы температура оставалась ниже -60°С. Туда же добавляли раствор 2-циклопропилэтанола (0,132 г; 1,3 ммоль) в безводном CH2Cl2 (2,5 мл), снова обеспечивая, чтобы температура оставалась ниже -60°С. Реакционную смесь перемешивали при -78°С в течение 20 мин, затем обрабатывали триэтиламином (0,543 мл; 3,9 ммоль) в безводном CH2Cl2 (3 мл). Полученную смесь оставляли постепенно нагреваться до комнатной температуры и перемешивали в течение 16 ч. Реакционную смесь разбавляли CH2Cl2 (10 мл) и водой (5 мл), затем добавляли 2 М HCl (0,5 мл). Смесь энергично перемешивали в течение 20 мин, затем пропускали через гидрофобную фритту. Органический раствор выдерживали над молекулярными ситами в течение 2 ч, затем декантировали в чистую колбу. К этому раствору добавляли 4′-(трифторметил)-1,1′-бифенил-3-иламин (0,31 г; 1,3 ммоль) с последующим добавлением триацетоксиборогидрида натрия (0,386 г; 1,82 ммоль) и уксусной кислоты (74,3 мкл; 1,3 ммоль). Полученную смесь перемешивали при комнатной температуре в атмосфере азота в течение 20 ч. Реакционную смесь гасили насыщенным водным NaHCO3 (15 мл) и экстрагировали CH2Cl2 (2×20 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки посредством SPE (колонка с 20 г диоксида кремния), элюируя смесью EtOAc:циклогексан (1:200), получали указанное в заголовке соединение в виде бесцветного масла (255 мг).
LC/MS: m/z 306,2 [M+H]+, Rt 4,1 мин.
Промежуточное соединение 23: этил-[4-({бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетат

К раствору N-бутил-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амина (73 мг; 0,25 ммоль) в безводном ацетонитриле (5 мл) добавляли этил-[4-(бромметил)-2-метилфенокси]ацетат (75 мг; 0,25 ммоль) и N,N-диизопропилэтиламин (43 мкл; 0,25 ммоль). Полученный раствор нагревали с обратным холодильником в атмосфере азота в течение 2 ч и затем охлаждали до комнатной температуры. Растворитель удаляли под вакуумом, остаток разбавляли водой (10 мл) и экстрагировали СН2Cl2 (30 мл). Органический экстракт разделяли с помощью гидрофобной фритты и растворитель удаляли под вакуумом. В результате очистки посредством SPE (диоксид кремния, 5 г), элюируя смесью циклогексан:EtOAc (99:1), получали указанное в заголовке соединение в виде бледно-желтого масла (105 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.96 (3Н, t, J 7,5 Гц), 1.28 (3Н, t, J 7 Гц), 1.38 (2Н, m), 1.67 (2H, m), 2.27 (3Н, s), 3.43 (2H, t, 7,5 Гц), 4.25 (2Н, q, J 7 Гц), 4.50 (2Н, s), 4.61 (2H, s), 6.64 (1H, d, J 8 Гц), 6.71 (1Н, dd, J 8 Гц, 2 Гц), 6.80-6.90 (2H, m), 6.98 (1H, d, J 8 Гц), 7.04 (1H, m), 7.26 (1H, m), 7.59 (2H, d, J 8,5 Гц), 7.64 (2H, d, J 8,5 Гц).
LC/MS: m/z 500,2 [M+H]+, Rt 4,5 мин.
Промежуточное соединение 24: этил-[4-({(2-метоксиэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетат
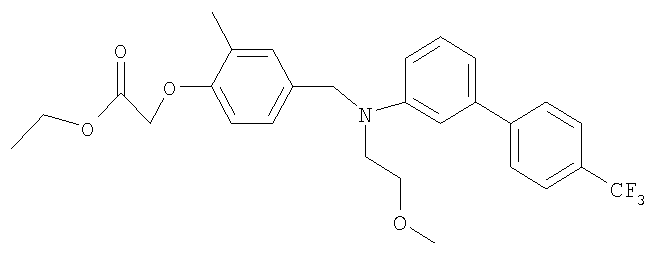
К раствору N-(2-метоксиэтил)-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амина (0,1 г; 0,34 ммоль) в безводном ацетонитриле (10 мл) добавляли этил-[4-(бромметил)-2-метилфенокси]ацетат (0,097 г; 0,34 ммоль) и N,N-диизопропилэтиламин (0,059 мл; 0,34 ммоль). Полученную смесь нагревали до кипения с обратным холодильником и перемешивали в атмосфере азота в течение 3 ч. Реакционную смесь концентрировали под вакуумом, остаток распределяли между водой (20 мл) и CH2Cl2 (40 мл), органический экстракт разделяли с помощью гидрофобной фритты и растворители удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 12 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бесцветного масла (90 мг).
1H ЯМР (400 МГц; CDCl3) δ: 1.28 (3Н, t J 7 Гц), 2.27 (3Н, s), 3.36 (3H, s), 3.60-3.70 (4H, m), 4.24 (2H, q, J 7 Гц), 4.57 (2Н, s), 4.61 (2H, s), 6.64 (1H, d, J 8,5 Гц), 6.76 (1H, d, J 8,5 Гц), 6.87-6.93 (2H, m), 6.99 (1H, d, J 8 Гц), 7.05 (1H, s), 7.26 (1H, m), 7.59 (2H, d, J 8 Гц), 7.64 (2H, d, J 8 Гц).
Промежуточное соединение 25: этил-[2-метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетат

К раствору N-пентил-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амина (70 мг; 0,23 ммоль) в безводном MeCN (6 мл) в атмосфере азота при комнатной температуре добавляли этил-[4-(бромметил)-2-метилфенокси]ацетат (65,4 мг; 0,23 ммоль) и N,N-диизопропилэтиламин (39,6 мкл; 0,23 ммоль). Полученный раствор нагревали до кипения с обратным холодильником и перемешивали в течение 4 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и растворитель удаляли под вакуумом. Остаток распределяли между CHCl3 (20 мл) и водой (10 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки посредством SPE (картридж с диоксидом кремния, 5 г), элюируя смесью EtOAc:циклогексан (1:200-1:100), получали указанное в заголовке соединение в виде бесцветного масла (66 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.91 (3Н, t, J 7 Гц), 1.28 (3Н, t, J 7 Гц), 1.31-1.38 (4Н, m), 1.68 (2H, m), 2.27 (3Н, s), 3.41 (2H, t, J 7,5 Гц), 4.25 (2Н, q, J 7 Гц), 4.49 (2H, s), 4.61 (2H, s), 6.64 (1H, d, J 8 Гц), 6.71 (1Н, dd, J 8,5 Гц, 2,5 Гц), 6.82-6.88 (2H, m), 6.98 (1H, m), 7.05 (1H, s), 7.26 (1H, m), 7.59 (2H, d, J 8,5 Гц), 7.64 (2H, d, J 8,5 Гц).
Промежуточное соединение 26: этил-[4-({(2-циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетат

К раствору N-(2-циклопропилэтил)-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амина (100 мг; 0,33 ммоль) в безводном MeCN (7 мл) в атмосфере азота при комнатной температуре добавляли этил-[4-(бромметил)-2-метилфенокси]ацетат (94 мг; 0,33 ммоль) и N,N-диизопропилэтиламин (57,5 мкл; 0,33 ммоль). Полученный раствор нагревали до кипения с обратным холодильником и перемешивали в течение 2,5 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и растворитель удаляли под вакуумом. Остаток распределяли между CHCl3 (2×10 мл) и водой (10 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки посредством SPE (картридж с диоксидом кремния, 5 г), элюируя смесью EtOAc:циклогексан (1:200-1:100), получали указанное в заголовке соединение в виде бесцветного масла (141 мг).
1H ЯМР (400 МГц; CDCl3) 6: 0.08 (2Н, m), 0.47 (2H, m), 0.69 (1H, m), 1.28 (3H, t, J 7,5 Гц), 1.56 (2H, m), 2.28 (3H, s), 3.55 (2H, t, J 7,5 Гц), 4.25 (2H, q, J 7 Гц), 4.51 (2H, s), 4.61 (2H, s), 6.64 (1H, d, J 8,5 Гц), 6.71 (1H, m), 6.83-6.88 (2H, m), 6.99 (1H, m), 7.05 (1H, s), 7.25 (1H, m), 7.59 (2H, d, J 8,5 Гц), 7.65 (2H, d, J 8,5 Гц).
Промежуточное соединение 27: этил-[2-метил-4-({пропил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетат

К раствору N-пропил-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амина (69,8 мг; 0,25 ммоль) в безводном MeCN (6 мл) в атмосфере азота при комнатной температуре добавляли этил-[4-(бромметил)-2-метилфенокси]ацетат (71,8 мг; 0,25 ммоль) и N,N-диизопропилэтиламин (43,5 мкл; 0,25 ммоль). Полученную смесь нагревали до кипения с обратным холодильником и перемешивали в течение 3 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и растворитель удаляли под вакуумом. Остаток распределяли между CHCl3 (2×10 мл) и водой (10 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки посредством SPE (картридж с диоксидом кремния, 5 г), элюируя смесью EtOAc:циклогексан (1:200), получали указанное в заголовке соединение в виде бесцветного масла (88 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.95 (3H, t, J 7,5 Гц), 1.28 (3H, t, J 7 Гц), 1.71 (2H, m), 2.27 (3H, s), 3.39 (2H, t, J 8 Гц), 4.25 (2H, q, J 7 Гц), 4.51 (2H, s), 4.61 (2H, s), 6.64 (1H, d, J 8,5 Гц), 6.71 (1H, dd, J 8 Гц, 2,5 Гц), 6.84 (1H, m), 6.86 (1H, d, J 7,5 Гц), 6.99 (1H, dd, J 8,5 Гц, 2 Гц), 7.05 (1H, d, J 1,5 Гц), 7.26 (1H, m), 7.59 (2H, d, J 8 Гц), 7.64 (2H, d, J 8 Гц).
Промежуточное соединение 28: метил-[2-метил-4-({[2-(метилтио)этил][4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-фенокси]ацетат
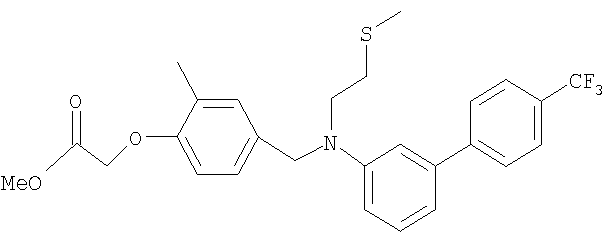
К раствору N-[2-(метилтио)этил]-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амина (0,27 г; 0,864 ммоль) в безводном MeCN (22 мл) в атмосфере азота при комнатной температуре добавляли метил-[4-(бромметил)-2-метилфенокси]ацетат (0,248 г; 0,864 ммоль) и N,N-диизопропилэтиламин (0,15 мл; 0,864 ммоль). Полученную смесь нагревали до кипения с обратным холодильником и перемешивали в течение 3 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и растворитель удаляли под вакуумом. Остаток распределяли между CHCl3 (2×30 мл) и водой (30 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки посредством SPE (картридж с диоксидом кремния, 20 г), элюируя смесью EtOAc:циклогексан (1:99-1:33), получали указанное в заголовке соединение в виде желтого масла (249 мг).
LC/MS: m/z 504,4 [М+Н]+, Rt 4,2 мин.
Промежуточное соединение 29: 2-метил-4′-(трифторметил)-1,1′-бифенил-3-иламин
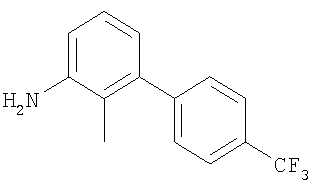
К раствору 3-бром-2-метиланилина (1,19 г; 6,4 ммоль) в DME (30 мл) и воде (15 мл) добавляли пара-трифторметилфенилбороновую кислоту (1,82 г; 9,6 ммоль) и карбонат натрия (1,76 г; 16,6 ммоль). Аппарат заполняли азотом перед добавлением тетракис(трифенилфосфин)палладия(0) (0,22 г; 0,19 ммоль). Полученную смесь нагревали до кипения с обратным холодильником и перемешивали в атмосфере азота в течение 17 ч, затем оставляли охлаждаться до комнатной температуры и растворители удаляли под вакуумом. Остаток распределяли между EtOAc (2 х 60 мл) и водой (60 мл). Органический раствор сушили (MgSO4) и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (колонка с 90 г диоксида кремния), элюируя смесью EtOAc:циклогексан (1:4), получали указанное в заголовке соединение в виде твердого вещества кремовой окраски (1,27 г).
LC/MS: m/z 252,2 [M+H]+; m/z 293,3 [M+CH3CN]+, Rt 3,6 мин.
Промежуточное соединение 30: N-бутил-N-[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амин
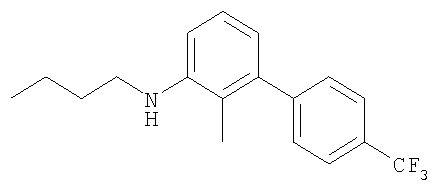
К раствору 2-метил-4′-(трифторметил)-1,1′-бифенил-3-иламина (0,32 г; 1,26 ммоль) в безводном CH2Cl2 (10 мл) в атмосфере азота при комнатной температуре добавляли масляный альдегид (0,114 мл; 1,26 ммоль), затем через 10 мин добавляли триацетоксиборогидрид натрия (0,374 г; 1,76 ммоль) и уксусную кислоту (72 мкл; 1,26 ммоль). Полученную смесь перемешивали в течение 22 ч при комнатной температуре. Реакционную смесь гасили насыщенным водным NaHCO3 (15 мл), затем экстрагировали CH2Cl2 (2×20 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки посредством SPE (картридж с диоксидом кремния, 20 г), элюируя смесью EtOAc:циклогексан (1:200), получали указанное в заголовке соединение в виде бесцветного масла (221 мг).
LC/MS: m/z 308 [М+Н]+, Rt 4,3 мин.
Промежуточное соединение 31: N-(2-метоксиэтил)-2-метил-4′-(трифторметил)-1,1′-бифенил-3-амин

1,1,2-Триметоксиэтан (0,162 мл; 1,26 ммоль) добавляли к 0,5 н. HCl (2,52 мл; 1,26 ммоль) и полученный раствор нагревали в течение 1 ч при 50°С. Охлажденную реакционную смесь экстрагировали CH2Cl2. Органический раствор пропускали через гидрофобную фритту, сушили в течение 2 часов (3Å молекулярные сита). К органическому раствору добавляли 2-метил-4′-(трифторметил)-1,1′-бифенил-3-иламин (0,32 г; 1,26 ммоль) и раствор перемешивали в течение 20 мин при комнатной температуре. Затем добавляли триацетоксиборогидрид натрия (0,325 г; 1,77 ммоль) и уксусную кислоту (0,072 мл; 1,26 ммоль) и полученную смесь перемешивали в течение 16 ч при комнатной температуре. К реакционной смеси добавляли насыщенный бикарбонат натрия и по завершении выделения газа смесь экстрагировали CH2Cl2. Органический раствор пропускали через гидрофобную фритту и растворители удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью 10% EtOAc:циклогексан, получали указанное в заголовке соединение (42 мг).
1H ЯМР (400 МГц; CDCl3) δ: 2.02 (3Н, s), 3.38 (2H, t, J 5,5 Гц), 3.42 (3Н, s), 3.69 (2H, t, J 5,5 Гц), 4.0 (1Н, s, широкий), 6.63 (1Н, d, J 8 Гц), 6.68 (1Н, d, J 8 Гц), 7.18 (1H, t, J 8 Гц), 7.41 (2H, d, J 8 Гц), 7.65 (2H, d, J 8 Гц).
Промежуточное соединение 32: этил-[4-({бутил[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетат
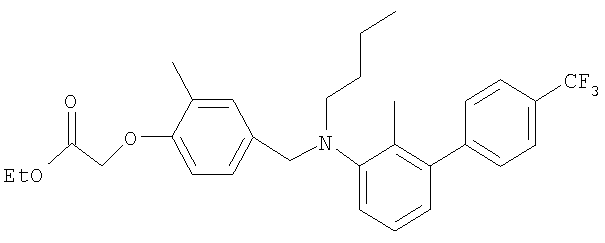
К раствору N-бутил-N-[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амина (102 мг; 0,33 ммоль) в безводном MeCN (8 мл) в атмосфере азота при комнатной температуре добавляли этил-[4-(бромметил)-2-метилфенокси]ацетат (95,3 мг; 0,33 ммоль) и N,N-диизопропилэтиламин (57,8 мкл; 0,33 ммоль). Полученный раствор нагревали до кипения с обратным холодильником и перемешивали в течение 3 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и растворитель удаляли под вакуумом. Остаток распределяли между CHCl3 (2×10 мл) и водой (10 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (колонка с 40 г диоксида кремния), элюируя смесью EtOAc:циклогексан (1:99-1:49), получали указанное в заголовке соединение в виде бесцветного масла (110 мг).
Термораспыление (положительный ион) m/z 514 [M+H]+.
Промежуточное соединение 33: этил-[4-({(2-метоксиэтил)[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетат
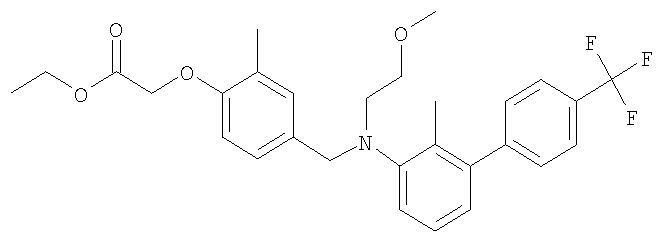
К раствору N-(2-метоксиэтил)-2-метил-4′-(трифторметил)-1,1′-бифенил-3-амина (500 мг; 1,62 ммоль) в ацетонитриле (30 мл) добавляли этил-[4-(бромметил)-2-метилфенокси]ацетат (488 мг; 1,7 ммоль) и N,N-диизопропилэтиламин (295 мкл; 1,7 ммоль). Полученную смесь нагревали в течение 4,5 ч при 80°С. После охлаждения растворители удаляли под вакуумом, остаток разбавляли водой и экстрагировали CH2Cl2. Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью 5% EtOAc:циклогексан, получали указанное в заголовке соединение в виде бесцветного масла (503 мг).
1H ЯМР (400 МГц; CDCl3) δ: 1.29 (3Н, t, J 7 Гц), 2.22 (3Н, s), 2.26 (3H, s), 3.16 (2H, t, J 6,5 Гц), 3.25 (3Н, s), 3.39 (2H, t, J 6,5 Гц), 4.10 (2Н, s), 4.26 (2H, q, J 7 Гц), 4.61 (2H, s), 6.62 (1H, d, J 8 Гц), 6.95 (1Н, dd, J 7 Гц, 1,5 Гц), 7.05-7.09 (2H, m), 7.15-7.22 (2H, m), 7.42 (2H, d, J 8 Гц), 7.66 (2H, d, J 8 Гц).
Промежуточное соединение 34: этил-[2-метил-4-({[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетат
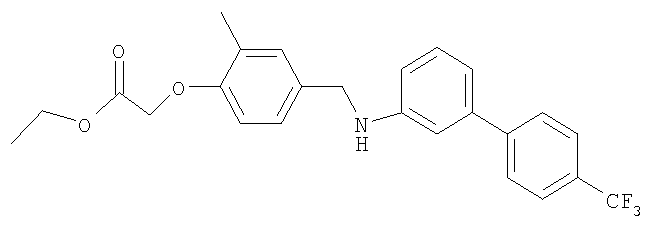
К раствору этил-(4-формил-2-метилфенокси)ацетата (0,2 г; 0,9 ммоль) в безводном СН2Cl2 (8 мл) добавляли 4′-(трифторметил)-1,1′-бифенил-3-иламин (0,214 г; 0,9 ммоль). Полученную смесь перемешивали в атмосфере азота при комнатной температуре в течение 15 мин, затем добавляли триацетоксиборогидрид натрия (0,267 г; 1,26 ммоль) и уксусную кислоту (0,052 мл; 0,9 ммоль). Полученную смесь перемешивали в течение 18 ч, затем гасили осторожным добавлением насыщенного раствора бикарбоната натрия (20 мл), затем реакционную смесь экстрагировали CH2Cl2 (20 мл). Органический экстракт разделяли с помощью гидрофобной фритты и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бесцветного масла (0,342 г).
LC/MS: m/z 444,3 [M+H]+, Rt 4,1 мин.
Промежуточное соединение 35: этил-[4-({бутирил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетат

К раствору этил-[2-метил-4-({[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетата (0,07 г; 0,16 ммоль) в безводном CH2Cl2 (2 мл) при 0°С в атмосфере азота добавляли масляную кислоту (0,016 мл; 0,17 ммоль), затем порциями добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (33 мг; 0,17 ммоль) с последующим добавлением триэтиламина (0,022 мл; 0,16 ммоль). После перемешивания в течение 15 мин при 0°С реакционную смесь оставляли достигать комнатной температуры и перемешивали в течение 18 ч. Реакционную смесь разбавляли CH2Cl2 (20 мл) и встряхивали с 1 М HCl (5 мл). Органический экстракт разделяли с помощью гидрофобной фритты и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 12 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бесцветного масла (40 мг).
LC/MS: m/z 513,9 [M+H]+, Rt 4,0 мин.
Промежуточное соединение 36: этил-[2-метил-4-({(пропилсульфонил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-фенокси]ацетат
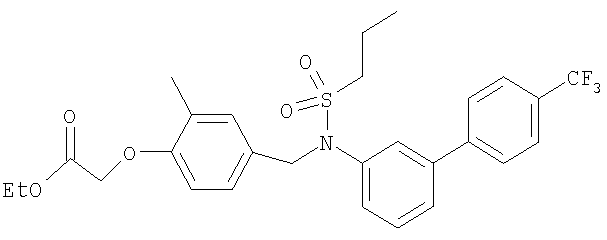
В реакционный сосуд, содержащий этил-[2-метил-4-({[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетат (100 мг; 0,225 ммоль), добавляли пиридин (0,321 мл; 3,97 ммоль) и 1-пропансульфонилхлорид (25,3 мкл; 0,225 ммоль). Полученную смесь перемешивали в герметично закрытом реакционном сосуде при 80°С в течение 16 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и распределяли между СН2Cl2 (2×10 мл) и водой (10 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (колонка с 12 г диоксида кремния), элюируя смесью EtOAc:циклогексан (1:9), получали указанное в заголовке соединение в виде желтого масла (20 мг).
LC/MS: m/z 567,1 [М+NH4]+, Rt 3,9 мин.
Промежуточное соединение 37: этил-[4-({бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-2-метилфенокси]ацетат
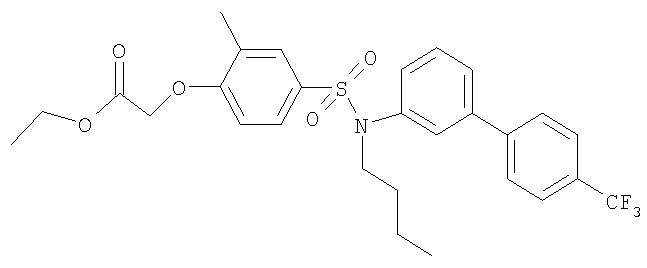
К раствору N-бутил-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амина (0,128 г; 0,44 ммоль) в безводном CH2Cl2 (10 мл) добавляли этил-[4-(хлорсульфонил)-2-метилфенокси]ацетат (0,121 г; 0,41 ммоль) и триэтиламин (0,122 мл; 0,87 ммоль). Полученную смесь перемешивали в течение 24 ч при комнатной температуре, затем добавляли сульфонилхлорид (0,121 мг; 0,41 ммоль) и триэтиламин (0,122 мл; 0,87 ммоль). После еще 18 ч перемешивания при комнатной температуре реакционную смесь разбавляли водой (10 мл) и экстрагировали CH2Cl2 (2×10 мл). Органический раствор сушили (Na2SO4) и растворители удаляли под вакуумом. В результате очистки посредством SPE (диоксид кремния, 10 г), элюируя смесью циклогексан:EtOAc (49:1-19:1), получали указанное в заголовке соединение в виде бледно-желтого масла (0,182 г).
LC/MS: m/z 550,3 [M+H]+, Rt 4,2 мин.
Промежуточное соединение 38: этил-[2-метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)фенокси]ацетат
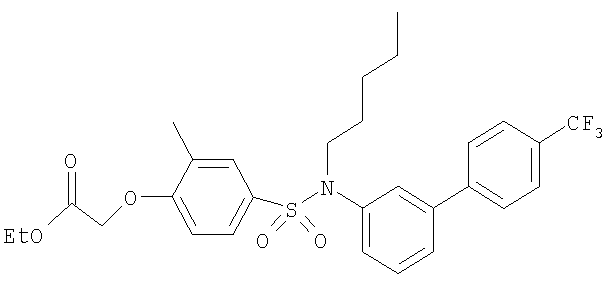
К раствору N-пентил-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амина (70,6 мг; 0,23 ммоль) в CH2Cl2 (5 мл) в атмосфере азота при комнатной температуре добавляли этил-[4-(хлорсульфонил)-2-метилфенокси]ацетат (101 мг; 0,35 ммоль) и триэтиламин (96 мкл; 0,69 ммоль). Полученную смесь перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь экстрагировали CH2Cl2 (15 мл) и водой (10 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки посредством SPE (картридж с диоксидом кремния, 5 г), элюируя смесью EtOAc:циклогексан (1:49-1:19), получали указанное в заголовке соединение в виде белого кристаллического твердого вещества (76 мг).
LC/MS: m/z 564,4 [М+H]+, Rt 4,3 мин.
Промежуточное соединение 39: этил-[4-({(2-циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-2-метилфенокси]-ацетат

К раствору N-(2-циклопропилэтил)-N-[4′-(трифторметил)-1,1′-бифенил-3-ил]амина (155 мг; 0,51 ммоль) в безводном CH2Cl2 (10 мл) в атмосфере азота при комнатной температуре добавляли этил-[4-(хлорсульфонил)-2-метилфенокси]ацетат (223 мг; 0,76 ммоль) и триэтиламин (0,213 мл; 1,53 ммоль). Полученную смесь перемешивали в течение 40 ч при комнатной температуре. Реакционную смесь разбавляли водой (15 мл) и экстрагировали CH2Cl2 (30 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (колонка с 40 г диоксида кремния), элюируя смесью EtOAc:циклогексан (1:19-1:9), получали указанное в заголовке соединение в виде белого кристаллического твердого вещества (156 мг).
LC/MS: m/z 561,8 [М+Н]+, Rt 4,3 мин.
Промежуточное соединение 40: 2-(метилтио)-4-[4-(трифторметил)-фенил]пиримидин

К раствору 4-хлор-2-метилтиопиримидина (2,17 мл; 18,7 ммоль) в диметоксиэтане (140 мл) и воде (70 мл) добавляли карбонат натрия (5,15 г; 48,5 ммоль), 4-трифторметилфенилбороновую кислоту и тетракис(трифенилфосфин)палладий(0) (0,43 г). Полученную смесь нагревали с обратным холодильником в атмосфере азота в течение 18 ч, затем оставляли охлаждаться до комнатной температуры и диметоксиэтан удаляли под вакуумом. Остаток разбавляли водой (30 мл) и экстрагировали CH2Cl2 (2×100 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью циклогексан:EtOAc (19:1), получали указанное в заголовке соединение в виде бледно-желтого твердого вещества (4,47 г).
LC/MS: m/z 471,0 [M+H]+, Rt 3,7 мин.
Промежуточное соединение 41: 4-(4-хлорфенил)-2-(метилтио)-пиримидин
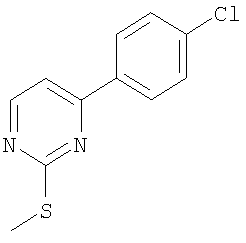
Смесь 4-хлор-2-(метилтио)пиримидина (3 г; 18,7 ммоль), 4-хлорфенилбороновой кислоты (3,5 г; 22,3 ммоль), тетракис(трифенилфосфин)палладия(0) (0,43 г; 0,37 ммоль), карбоната натрия (5,15 г; 48,5 ммоль) в 1,2-диметоксиэтане (140 мл) и воде (70 мл) нагревали при 90°С в течение 20 ч в атмосфере азота. Охлажденную реакционную смесь концентрировали под вакуумом, разбавляли водой (50 мл), экстрагировали CH2Cl2 (2×100 мл), сушили (Na2SO4) и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью циклогексан:EtOAc (от 10:1 до 2:1), получали указанное в заголовке соединение в виде твердого вещества белого цвета (3,98 г).
LC/MS: m/z 237 [M+H]+, Rt 3,56 мин.
Промежуточное соединение 42: 2-(метилсульфонил)-4-[4-(трифторметил)фенил]пиримидин
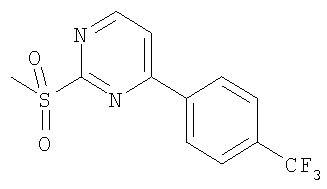
К суспензии 2-(метилтио)-4-[4-(трифторметил)фенил]пиримидина (2,24 г; 8,4 ммоль) в метаноле (85 мл) и воде (85 мл) при 0°С добавляли оксон™ (15,25 г; 24,8 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 ч и затем при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли водой (200 мл) и экстрагировали хлороформом (2×250 мл). Органический раствор сушили (Na2SO4), фильтровали и упаривали с получением указанного в заголовке соединения в виде белого твердого вещества (2,235 г).
LC/MS: m/z 302,8 [М+Н]+, Rt 3,1 мин.
Промежуточное соединение 43: 4-(4-хлорфенил)-2-(метилсульфонил)пиримидин
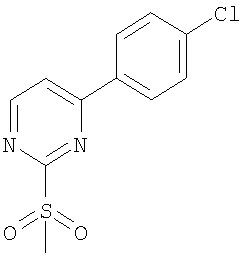
Раствор 4-(4-хлорфенил)-2-(метилтио)пиримидина (3 г; 12,71 ммоль) в метаноле (125 мл) и воде (125 мл) в атмосфере азота при 4°С обрабатывали оксоном™ (23,44 г; 38,13 ммоль), перемешивали и оставляли нагреваться до комнатной температуры в течение 18 ч. Реакционную смесь разбавляли водой (220 мл), экстрагировали CHCl3 (250 мл), сушили (Na2SO4) и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде желтого твердого вещества (3,36 г).
LC/MS: m/z 269 [М+Н]+, Rt 2,82 мин.
Промежуточное соединение 44: этил-{4-[(бутил{4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]-2-метилфенокси}-ацетат

Смесь 2-(метилсульфонил)-4-[4-(трифторметил)фенил]пиримидина (0,3, г; 1 ммоль), N,N-диизопропилэтиламина (0,18 мл; 1 ммоль) и этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата (0,424 г; 1,53 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 18 ч. Остаток растворяли в CH2Cl2 и очищали Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью EtOAc:циклогексан (3:97), с получением указанного в заголовке соединения в виде бледно-желтого масла (110 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.94 (3Н, t, J 7 Гц), 1.28 (3Н, t, J 7 Гц), 1.37 (2Н, m), 1.64 (2H, m), 2.26 (3Н, s), 3.63 (2H, t, J 7,5 Гц), 4.25 (2Н, q, J 7 Гц), 4.61 (2Н, s), 4.88 (2H, s), 6.63 (1H, d, J 8,5 Гц), 6.94 (1Н, d, J 5,5 Гц), 7.04 (1Н, d, J 8,5 Гц), 7.09 (1Н, s), 7.70 (2H, d, J 8 Гц), 8.13 (2H, br d, J 8 Гц), 8.42 (1Н, d, J 5,5 Гц).
Промежуточное соединение 45: этил-[4-({бутил[4-(4-хлорфенил)-пиримидин-2-ил]амино}метил)-2-метилфенокси]ацетат
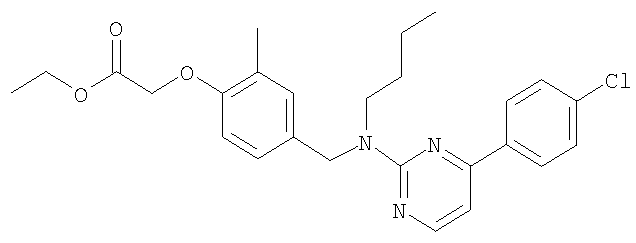
К смеси этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата (686 мг; 2,46 ммоль) и N,N-диизопропилэтиламина (285 мкл; 1,64 ммоль) добавляли 4-(4-хлорфенил)-2-(метилсульфонил)пиримидин (439 мг; 1,64 ммоль) и полученную смесь нагревали при 100°С в течение 16 ч в реакционном сосуде. Охлажденную реакционную смесь затем обрабатывали N,N-диизопропилэтиламином (285 мкл; 1,64 ммоль) и нагревали в течение еще 16 ч при 100°С в реакционном сосуде. Охлажденные реакционные смеси разбавляли CH2Cl2 (30 мл), промывали водой (20 мл), водную фазу экстрагировали СН2Cl2 (30 мл), СН2Cl2 экстракты объединяли и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью циклогексан:EtOAc (10:1), получали указанное в заголовке соединение в виде бесцветной смолы (178 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.95 (3Н, t, J 7,5 Гц), 1.3 (3Н, t, J 7 Гц), 1.37 (2H, m), 1.64 (2H, m), 2.27 (3Н, s), 3.63 (2H, t, J 7,5 Гц), 4.26 (2H, q, J 7 Гц), 4.62 (2H, s), 4.89 (2H, s), 6.64 (1Н, d, J 8,5 Гц), 6.90 (1Н, d, J 5 Гц), 7.03-7.13 (2H, m), 7.43 (2H, d, J 8,0 Гц), 8.00 (2H, d, J 8,0 Гц), 8.39 (1Н, d, J 5 Гц).
LC/MS: m/z 468 [М+Н]+, Rt 4,34 мин.
Промежуточное соединение 46: этил-[4-[((2-метоксиэтил){4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]-2-метилфенокси}-ацетат
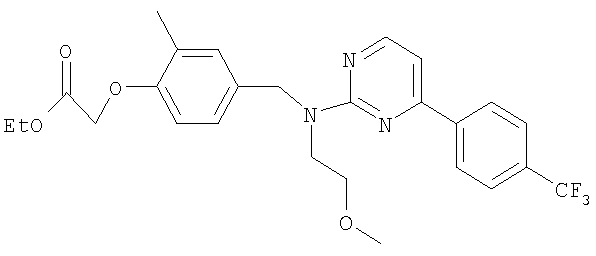
Смесь 2-(метилсульфонил)-4-[4-(трифторметил)фенил]пиримидина (0,2 г; 0,66 ммоль), N,N-диизопропилэтиламина (0,11 мл; 0,66 ммоль) и этил-(4-{[(2-метоксиэтил)амино]метил}-2-метилфенокси)ацетата (0,28 г; 0,99 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 20 часов. Реакционную смесь затем оставляли достигать комнатной температуры и остаток растворяли в СН2Cl2 (30 мл), органический экстракт промывали водой (20 мл) и пропускали через гидрофобную фритту, затем концентрировали под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью EtOAc:циклогексан (1:9), получали указанное в заголовке соединение в виде бледно-желтого масла (0,08 г).
LC/MS: m/z 503,8 [M+H]+, Rt 4,1 мин.
Промежуточное соединение 47: этил-(4-{[[4-(4-хлорфенил)-пиримидин-2-ил](2-метоксиэтил)амино]метил}-2-метилфенокси)ацетат

Получали из этил-(4-{[(2-метоксиэтил)амино]метил}-2-метилфенокси)-ацетата, используя методику синтеза, описанную для Промежуточного соединения 45. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью циклогексан:EtOAc (от 100:15 до 100:25), получали указанное в заголовке соединение в виде бесцветной смолы (165 мг).
1H ЯМР (400 МГц; CDCl3) δ:1.28 (3Н, t, J 7 Гц), 2.26 (3Н, s), 3.34 (3H, s), 3.63 (2H, t, J 5,5 Гц), 3.83 (2Н, t, J 5,5 Гц), 4.25 (2Н, q, J 7 Гц), 4.6 (2Н, s), 4.96 (2H, s), 6.63 (1H, d, J 8 Гц), 6.92 (1Н, d, J 5 Гц), 7.06 (1Н, d, J 8 Гц), 7.11 (1Н, s), 7.42 (2H, d, J 8,5 Гц), 7.98 (2H, d, J 8,5 Гц), 8.39 (1Н, d, 5 Гц).
LC/MS: m/z 470 [M+H]+, Rt 3,98 мин.
Промежуточное соединение 48: этил-{2-метил-4-[(пропил{4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]фенокси}ацетат
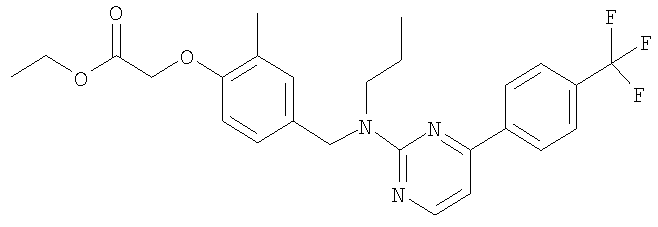
К смеси этил-{2-метил-4-[(пропиламино)метил]фенокси}ацетата (652 мг; 2,46 ммоль) и N,N-диизопропилэтиламина (570 мкл; 3,28 ммоль) добавляли 2-(метилсульфонил)-4-[4-(трифторметил)фенил]пиримидин (495 мг; 1,64 ммоль) и полученную смесь нагревали при 100°С в течение 16 ч в реакционном сосуде. Охлажденную реакционную смесь разбавляли СН2Cl2 (30 мл), промывали водой (20 мл), водную фазу экстрагировали СН2Cl2 (30 мл), CH2Cl2 экстракты объединяли и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью циклогексан:EtOAc (10:1), получали указанное в заголовке соединение в виде бесцветной смолы (139 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.95 (3Н, t, J 7,5 Гц), 1.30 (3Н, t, J 7 Гц), 1.69 (2Н, m), 2.28 (3Н, s), 3.61 (2H, t, J 7,5 Гц), 4.26 (2Н. q, J 7 Гц), 4.62 (2Н, s), 4.9 (2H, s), 6.65 (1H, d, J 8 Гц), 6.95 (1Н, d, J 5 Гц), 7.06 (1Н, d, J 8 Гц), 7.11 (1Н, s), 7.72 (2H, d, J 8 Гц), 8.145 (2H, d, J 8 Гц), 8.44 (1Н, d, J 5 Гц).
LC/MS: m/z 488 [M+H]+, Rt 4,24 мин.
Промежуточное соединение 49: 2-хлор-6-[4-(трифторметил)фенил]-пиразин
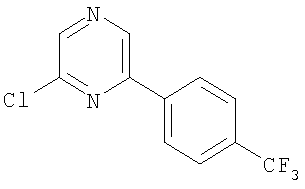
К смеси 2,6-дихлорпиразина (0,5 г; 3,36 ммоль), карбоната натрия (0,925 г; 8,73 ммоль) и 4-трифторметилфенилбороновой кислоты (0,638 г; 3,36 ммоль) в диметоксиэтане (15 мл) и воде (7,5 мл) в атмосфере азота добавляли тетракис(трифенилфосфин)палладий(0) (0,078 г). Полученную смесь нагревали с обратным холодильником в течение 5 ч в атмосфере азота, затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (50 мл) и экстрагировали хлороформом (2×75 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), цикпогексан:EtOAc (10:1-6:1), получали указанное в заголовке соединение в виде белого твердого вещества (0,317 г).
1Н ЯМР (400 МГц; CDCl3) δ: 7.78 (2Н, d, J 8 Гц), 8.16 (2Н, d, J 8 Гц), 8.61 (1H, s), 8.98 (1H, s).
Промежуточное соединение 50: 2-хлор-6-(4-метилфенил)пиразин
К смеси 2,6-дихлорпиразина (3 г; 20 ммоль), карбоната натрия (5,55 г; 52 ммоль) и 4-метилфенилбороновой кислоты (2,74 г; 20 ммоль) в диметоксиэтане (90 мл) и воде (45 мл) в атмосфере азота добавляли тетракис(трифенилфосфин)палладий(0) (0,46 г). Полученную смесь нагревали с обратным холодильником в течение 18 ч в атмосфере азота. После охлаждения до комнатной температуры реакционную смесь разбавляли водой (100 мл) и экстрагировали хлороформом (2×150 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде белого твердого вещества (2,3 г).
LC/MS: m/z 205,1 [М+Н]+, Rt 3,4 мин.
Промежуточное соединение 51: 2-бром-6-[4-(трифторметил)фенил]-пиразин
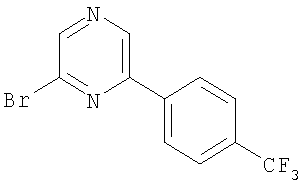
В колбу, содержащую 2-хлор-6-[4-(трифторметил)фенил]пиразин (0,8 г; 3,1 ммоль), добавляли трибромид фосфора (20 мл) и смесь нагревали с обратным холодильником в течение 6 ч в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, затем аккуратно вливали в ледяную воду (400 мл), полученную смесь доводили до рН 6-7, используя водный аммиак, и экстрагировали диэтиловым эфиром (3×250 мл). Органический раствор сушили (MgSO4) и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде бледно-желтого твердого кристаллическиго вещества (0,97 г).
1H ЯМР (400 МГц; CDCl3) δ: 7.78 (2Н, d, J 8 Гц), 8.15 (2Н, J 8 Гц), 8.69 (1Н, s), 8.99 (1H, s).
Промежуточное соединение 52: 2-бром-6-(4-метилфенил)пиразин

В колбу, содержащую 2-хлор-6-(4-метилфенил)пиразин (2,5 г; 12,3 ммоль), добавляли трибромид фосфора (20 мл) и смесь нагревали с обратным холодильником в течение 6 ч в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, затем аккуратно вливали в ледяную воду (600 мл). Полученный раствор доводили до рН 6-7, используя водный аммиак, и экстрагировали Et2O (2×250 мл). Органический раствор сушили (MgSO4) и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде твердого вещества белого цвета (2,32 г).
LC/MS: m/z 249,0; 251,0 [M+H]+, Rt 3,4 мин.
Промежуточное соединение 53: этил-{4-[(бутил{6-[4-(трифторметил)фенил]-пиразин-2-ил}амино)метил]-2-метилфенокси}-ацетат
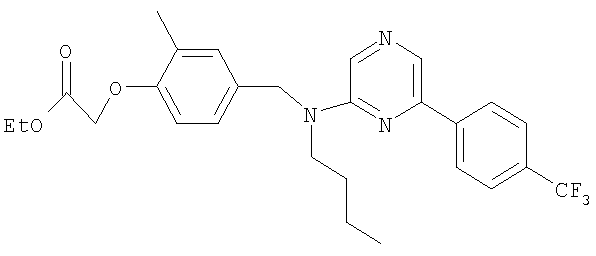
Смесь 2-бром-6-[4-(трифторметил)фенил]пиразина (0,5 г; 1,6 ммоль), N,N-диизопропилэтиламина (0,29 мл; 1,6 ммоль) и этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата (0,5 г; 1,8 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 18 ч. Реакционную смесь затем оставляли достигать комнатной температуры, остаток растворяли в СН2Cl2 (130 мл), органический экстракт промывали водой (30 мл), пропускали через гидрофобную фритту и концентрировали под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью EtOAc:циклогексан (1:19-1:4), получали указанное в заголовке соединение в виде бледно-желтого масла (0,15 г).
LC/MS: m/z 501,8 [М+Н]+, Rt 4,3 мин.
Промежуточное соединение 54: этил-[4-({бутил[6-(4-метилфенил)-пиразин-2-ил]амино}метил)-2-метилфенокси]ацетат
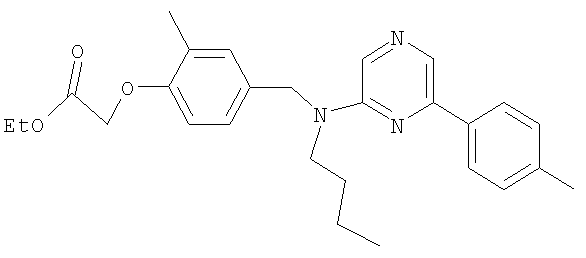
Смесь 2-бром-6-(4-метилфенил)пиразина (0,25 г; 1 ммоль), N,N-диизопропилэтиламина (0,35 мл; 2 ммоль) и этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата (0,42 г; 1,5 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 18 ч. Реакционную смесь затем оставляли достигать комнатной температуры, остаток растворяли в CH2Cl2 (30 мл), органический экстракт промывали водой (15 мл) и пропускали через гидрофобную фритту, затем концентрировали под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью EtOAc:циклогексан (1:19-1:4), получали указанное в заголовке соединение в виде бледно-желтого масла (37 мг).
LC/MS: m/z 448,2 [M+H]+, Rt 4,1 мин.
Промежуточное соединение 55: этил-{4-[((2-метоксиэтил){6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]-2-метилфенокси}ацетат
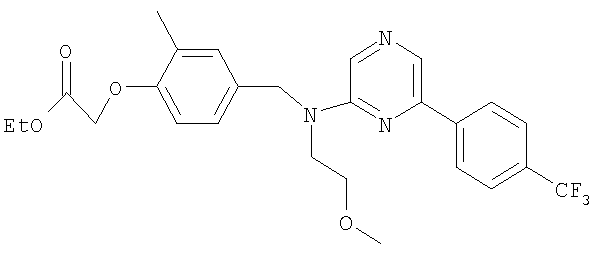
Смесь 2-бром-6-[4-(трифторметил)фенил]пиразина (0,5 г; 1,65 ммоль), N,N-диизопропилэтиламина (0,29 мл; 1,65 ммоль) и этил-(4-{[(2-метоксиэтил)-амино]метил}-2-метилфенокси)ацетата (0,7 г; 2,47 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 20 ч. Реакционную смесь затем оставляли достигать комнатной температуры, остаток разбавляли водой (40 мл) и экстрагировали СН2Cl2 (2×40 мл). Органический экстракт пропускали через гидрофобную фритту и концентрировали под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью EtOAc:циклогексан (1:19-1:4), получали указанное в заголовке соединение в виде бледно-желтого масла (0,16 г).
LC/MS: m/z 503,8 [М+Н]+, Rt 4,0 мин.
Промежуточное соединение 56: 3-бром-2-метил-N-пропиланилин
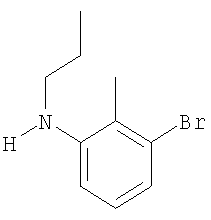
К раствору 3-бром-2-метилфениламина (1,5 г; 8,06 ммоль) в безводном CH2Cl2 с 3Å молекулярными ситами добавляли пропионовый альдегид (580 мкл; 8,06 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 20 мин в атмосфере азота. Добавляли уксусную кислоту (561 мкл; 8,06 ммоль) и триацетоксиборогидрид натрия (4,78 г; 22,56 ммоль) и полученную смесь перемешивали в атмосфере азота при комнатной температуре в течение 16 ч. Порциями добавляли насыщенный бикарбонат натрия (60 мл), полученную смесь перемешивали в течение 0,5 ч и экстрагировали CH2Cl2 (2×100 мл). Органический раствор сушили (Na2SO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде желтого масла (1,8 г).
LC/MS: m/z 230 [М+Н]+, Rt 3,64 мин.
Промежуточное соединение 57: 3-бром-N-бутил-2-метиланилин
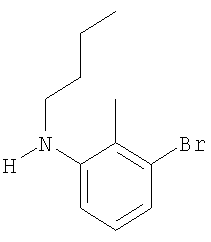
Получали из масляного альдегида и 3-бром-2-метилфениламина, используя методику, описанную для Промежуточного соединения 56.
LC/MS: m/z 244 [M+H]+, Rt 3,82 мин.
Промежуточное соединение 58: 3-бром-N-(2-метоксиэтил)-2-метиланилин
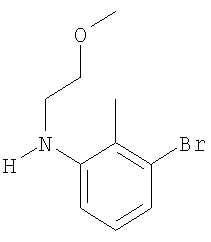
Раствор 1,1,2-триметоксиэтана (2,07 мл; 16,07 ммоль) в 0,5 М HCl (32 мл) нагревали при 100°С в течение 2 ч. Охлажденную смесь экстрагировали CH2Cl2 (30 мл) и органический раствор сушили (3Å молекулярные сита). Добавляли 3-бром-2-метилфениламин и полученный раствор перемешивали в атмосфере азота в течение 20 мин. Добавляли уксусную кислоту (461 мкл; 8,05 ммоль) и триацетоксиборогидрид натрия (4,78 г; 22,56 ммоль) и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 16 ч. Порциями добавляли насыщенный бикарбонат натрия (60 мл), полученную смесь перемешивали в течение 0,5 ч и экстрагировали CH2Cl2 (2×100 мл). Органический раствор сушили (Na2SO4) и растворители удаляли под вакуумом. В результате очистки посредством SPE (диоксид кремния, 2×20 г), элюируя смесью циклогексан:EtOAc (от 100:2 до 100:5), получали указанное в заголовке соединение в виде желтого масла (687 мг).
LC/MS: m/z 246 [М+Н]+, Rt 3,27 мин.
Промежуточное соединение 59: этил-(4-{[(3-бром-2-метилфенил)-(бутил)амино]метил}-2-метилфенокси)ацетат
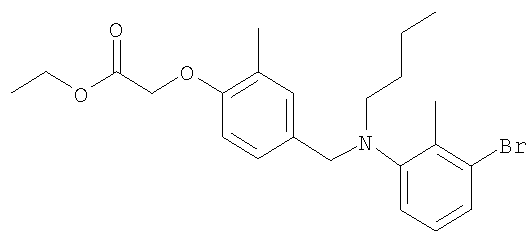
К раствору 3-бром-N-бутил-2-метиланилина (700 мг; 2,89 ммоль) в безводном ацетонитриле (75 мл) добавляли этил-[4-(бромметил)-2-метилфенокси]ацетат (830 мг; 2,89 ммоль) и N,N-диизопропилэтиламин (331 мкл; 1,9 ммоль). Полученную смесь нагревали при 80°С в течение 3 ч. Реакционную смесь охлаждали, растворители удаляли под вакуумом, разбавляли водой (50 мл) и экстрагировали CH2Cl2 (2×100 мл). Органический раствор сушили (Na2SO4) и растворители удаляли под вакуумом. В результате очистки посредством SPE (диоксид кремния, 20 г), элюируя смесью циклогексан:EtOAc (от 100:2 до 100:5), получали указанное в заголовке соединение в виде бесцветного масла (749 мг).
LC/MS: m/z 450 [M+H]+, Rt 4,32 мин.
Промежуточное соединение 60: этил-(4-{[(3-бром-2-метилфенил)(пропил)амино]метил}-2-метилфенокси)ацетат
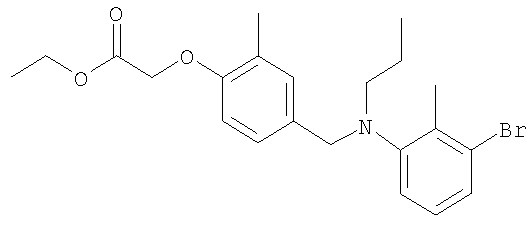
Получали из 3-бром-2-метил-N-пропиланилина, используя методику, описанную для Промежуточного соединения 59.
LC/MS: m/z 436 [М+Н]+, Rt 4,24 мин.
Промежуточное соединение 61: этил-(4-{[{3-бром-2-метилфенил)(2-метоксиэтил)амино]метил}-2-метилфенокси)ацетат

Получали из 3-бром-N-(2-метоксиэтил)-2-метиланилина, используя методику, описанную для Промежуточного соединения 59.
LC/MS: m/z 452 [M+H]+, Rt 3,92 мин.
Промежуточное соединение 62: 4-хлор-5-метил-6-[4-(трифторметил)фенил]пиримидин
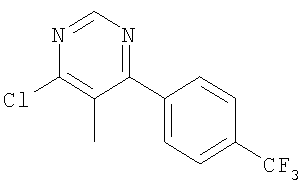
К раствору 4,6-дихлор-5-метилпиримидина (1 г; 6,13 ммоль) в диметоксиэтане (28 мл) и воде (14 мл) добавляли карбонат натрия (1,69 г; 16,0 ммоль), 4-трифторметилфенилбороновую кислоту (1,165 г; 6,13 ммоль) и тетракис(трифенилфосфин)палладий(0) (0,14 г). Полученную смесь нагревали с обратным холодильником в атмосфере азота в течение 18 ч, реакционную смесь оставляли охлаждаться до комнатной температуры и разбавляли водой (100 мл), затем экстрагировали в CHCl3 (2×100 мл). Органический раствор разделяли с помощью гидрофобной фритты и растворители удаляли под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью циклогексан:EtOAc (19:1), получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,1 г).
LC/MS: m/z 273,1 [M+H]+, Rt 3,3 мин.
Промежуточное соединение 63: 4-хлор-6-(4-хлорфенил)-5-метилпиримидин
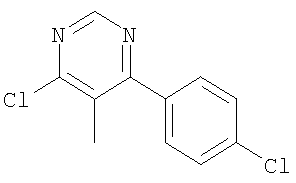
Получали из 4-хлорфенилбороновой кислоты и 4,6-дихлор-5-метилпиримидина, используя методику, описанную для Промежуточного соединения 62.
LC/MS: m/z 239,1 [M+H]+, Rt 3,2 мин.
Промежуточное соединение 64: 4-хлор-5-метил-6-(4-метилфенил)-пиримидин

Получали из 4-метилфенилбороновой кислоты и 4,6-дихлор-5-метилпиримидина, используя методику, описанную для Промежуточного соединения 62.
LC/MS: m/z 219,1 [М+Н]+, Rt 3,2 мин.
Промежуточное соединение 65: 4-хлор-6-(4-метоксифенил)-5-метилпиримидин
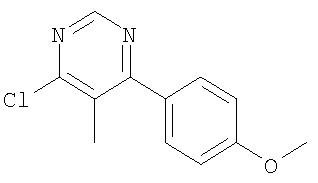
Получали из 4-метоксифенилбороновой кислоты (0,93 г; 6,13 ммоль) и 4,6-дихлор-5-метилпиримидина, используя методику, описанную для Промежуточного соединения 62.
LC/MS: m/z 235,1 [М+Н]+, Rt 3,0 мин.
Промежуточное соединение 66: этил-{4-[(бутил{5-метил-6-[4-(трифторметил)фенил]пиримидин-4-ил}амино)метил]-2-метилфенокси}-ацетат
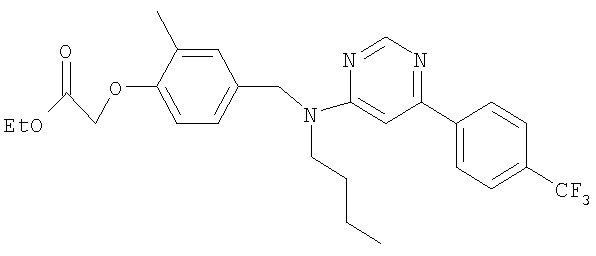
Смесь 4-хлор-5-метил-6-[4-(трифторметил)фенил]пиримидина (0,229 г; 0,84 ммоль), N,N-диизопропилэтиламина (0,293 мл; 1,68 ммоль) и этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата (0,35 г; 1,26 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 18 ч. Реакционную смесь затем оставляли достигать комнатной температуры, остаток растворяли в CH2Cl2 (50 мл), органический экстракт промывали водой (20 мл) и пропускали через гидрофобную фритгу, затем концентрировали под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью EtOAc:циклогексан (1:19-1:9), получали указанное в заголовке соединение в виде бесцветного масла (0,19 г).
LC/MS: m/z 516,2 [М+Н]+, Rt 3,9 мин.
Промежуточное соединение 67: этил-[4-({бутил[6-(4-метоксифенил)-5-метилпиримидин-4-ил]амино}метил)-2-метилфенокси]ацетат
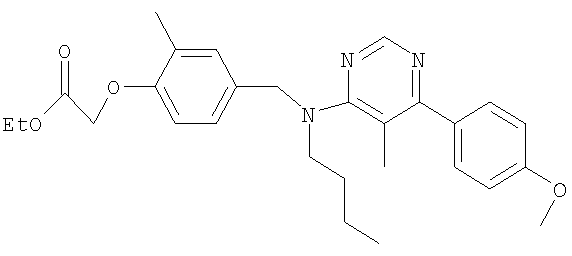
Смесь 4-хлор-6-(4-метоксифенил)-5-метилпиримидина (0,164 г; 0,7 ммоль), диизопропилэтиламина (0,24 мл; 1,4 ммоль) и этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата (0,29 г; 1,05 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 18 ч. Реакционную смесь затем оставляли достигать комнатной температуры. Остаток растворяли в CH2Cl2 (50 мл), органический раствор промывали водой (20 мл) и пропускали через гидрофобную фритту, затем концентрировали под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью EtOAc:циклогексан (3:17-1:4), получали указанное в заголовке соединение в виде бледно-желтого масла (62 мг).
LC/MS: m/z 478,3 [М+Н]+, Rt 3,2 мин.
Промежуточное соединение 68: этил-[4-({бутил[6-(4-хлорфенил)-5-метилпиримидин-4-ил]амино}метил)-2-метилфенокси]ацетат
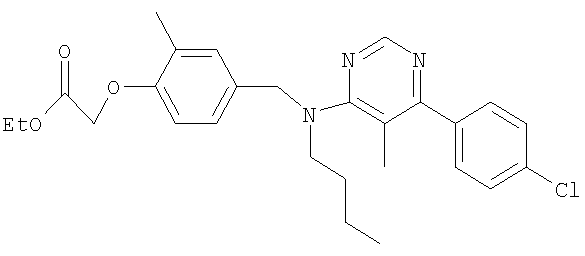
Получали из 4-хлор-6-(4-хлорфенил)-5-метилпиримидина и этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата, используя методику получения Промежуточного соединения 67.
LC/MS: m/z 482,2 [M+H]+, Rt 3,7 мин.
Промежуточное соединение 69: этил-[4-({бутил[5-метил-6-(4-метилфенил)пиримидин-4-ил]амино}метил)-2-метилфенокси]ацетат
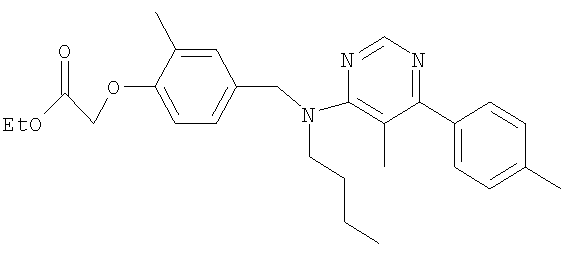
Получали из 4-хлор-5-метил-6-(4-метилфенил)пиримидина и этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата, используя методику получения Промежуточного соединения 67.
LC/MS: m/z 462,3 [M+H]+, Rt 3,4 мин.
Промежуточное соединение 70: 2,4′-диметил-1,1′-бифенил-3-иламин
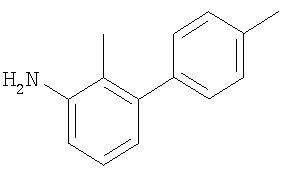
К раствору 3-бром-2-метиланилина (15 г; 0,0806 моль) в 1,2-диметоксиэтане (330 мл) и воде (165 мл) добавляли 4-метилфенилбороновую кислоту (12,05 г; 0,0886 моль) и карбонат натрия (22,2 г; 0,2095 моль). Аппарат заполняли азотом перед добавлением тетракис(трифенилфосфин)палладия(0) (1,86 г; 1,61 ммоль). Полученную смесь нагревали при 90°С и перемешивали в атмосфере азота в течение 22 ч, затем оставляли охлаждаться до комнатной температуры и растворители удаляли под вакуумом. Остаток распределяли между EtOAc (2×400 мл) и водой (400 мл). Органический раствор сушили (Na2SO4) и растворитель удаляли под вакуумом. В результате очистки Biotage™-хроматографией (Si-колонка 4×90 г), элюируя смесью EtOAcщиклогексан (от 9:1 до 8:1), получали указанное в заголовке соединение в виде желтого масла (14,78 г).
LC/MS: m/z 198 [М+Н]+, Rt 3,22 мин.
Промежуточное соединение 71: N-(2,4′-диметил-1,1′-бифенил-3-ил)-бутанамид
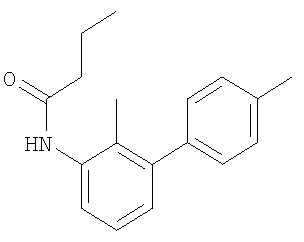
К раствору 2,4′-диметил-1,1′-бифенил-3-иламина (20 г; 0,1015 моль) в безводном СН2Cl2 (250 мл) при 0°С в атмосфере азота добавляли триэтиламин (28,29 мл; 0,203 моль). По каплям в течение 0,5 ч добавляли бутирилхлорид (11,07 мл; 0,1066 моль), скорость добавления обеспечивала поддержание температуры реакционной смеси ниже 10°С. Реакционную смесь затем перемешивали при комнатной температуре в течение 2 ч в атмосфере азота, разбавляли CH2Cl2 (200 мл), промывали водой (400 мл), насыщенным бикарбонатом натрия (400 мл) и насыщенным хлоридом натрия (400 мл). Органический раствор сушили (Na2SO4) и растворитель удаляли под вакуумом с получением твердого вещества кремовой окраски. К твердому веществу добавляли циклогексан (400 мл), смесь перемешивали в течение 2 ч и фильтровали. Твердые остатки растворяли в CH2Cl2 и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде твердого вещества кремовой окраски (25 г).
LC/MS: m/z 268 [М+Н]+, Rt 3,37 мин.
Промежуточное соединение 72: N-бутил-N-(2,4′-диметил-1,1′-бифенил-3-ил)амин
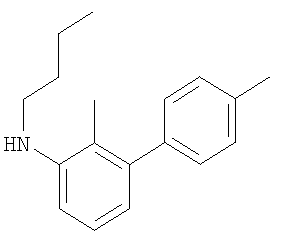
К раствору N-(2,4′-диметил-1,1′-бифенил-3-ил)бутанамида (15 г; 0,056 моль) в безводном тетрагидрофуране (500 мл) добавляли по каплям в атмосфере азота литийалюминийгидрид (1 М раствор в эфире; 112 мл). Полученную смесь нагревали при 75°С в течение 5,5 ч, затем оставляли охлаждаться до комнатной температуры в течение 16 ч. К реакционной смеси добавляли по каплям насыщенный водой эфир (50 мл), затем 2 М NaOH (50 мл) и воду (50 мл). Смесь фильтровали, твердые остатки промывали эфиром, фильтрат сушили (Na2SO4) и растворители удаляли под вакуумом. Остаток распределяли между CH2Cl2 (400 мл) и водой (20 мл). СН2Cl2-слой сушили (Na2SO4) и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде желтого масла (13,12 г).
LC/MS: m/z 254 [М+Н]+, Rt 4,07 мин.
Промежуточное соединение 73: этил-[4-(хлорметил)-2-метилфенокси]ацетат
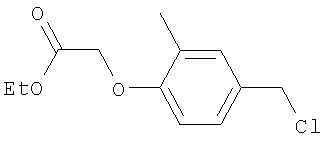
Этил-[4-(гидроксиметил)-2-метилфенол (54,1 г; 0,241 моль) растворяли в безводном DCM (540 мл) и охлаждали до -6°С в атмосфере азота. Триэтиламин (67 мл; 0,48 моль) затем добавляли в течение 15 мин. Затем в течение 40 мин добавляли мезилхлорид (28 мл; 0,362 моль), поддерживая температуру реакционной смеси ниже 5°С. По завершении добавления смесь перемешивали при 0°С в течение 30 мин, затем нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь гасили добавлением воды (22 мл). Реакционную смесь промывали водой (370 мл) и водные промывки экстрагировали DCM (370 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали под вакуумом с получением указанного в заголовке соединения в виде масла, из которого при хранении образовывалось низкоплавкое твердое вещество оранжевого цвета при +5°С (52,05 г).
1H ЯМР (400 МГц; CDCl3) δ: 1.3 (3Н, t, J 7 Гц), 2.29 (3Н, s), 4.27 (2H, q, J 7 Гц), 4.54 (2H, s), 4.64 (2H, s), 6.66 (1H, d, J 8,5 Гц), 7.15 (1Н, dd, J 8,5 Гц, 2,5 Гц) 7.20 (1H, d, J 2,5 Гц).
Промежуточное соединение 74: этил-(4-{[бутил(2,4′-диметил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)ацетат

Этил-[4-(хлорметил)-2-метилфенокси]ацетат (35,9 г; 0,148 моль) растворяли в безводном ацетонитриле (285 мл). Добавляли в виде одной порции иодид натрия (20,1 г; 0,135 моль; 1 экв.) и смесь перемешивали в течение 5 мин. В течение 10 мин добавляли раствор N-бутил-N-(2,4′-диметил-1,1′-бифенил-3-ил)амина (34 г; 0,135 моль) и диизопропилэтиламина (23,5 мл; 0,135 моль) в безводном ацетонитриле (285 мл) и использовали охлаждение, чтобы в процессе добавления поддерживать температуру реакции приблизительно 16°С. Реакционную смесь концентрировали под вакуумом и полученное оранжевое масло распределяли между DCM (700 мл) и водой (700 мл). Водный слой экстрагировали DCM (500 мл) и объединенные органические экстракты осторожно промывали соляным раствором (300 мл). После высушивания (Na2SO4) смесь концентрировали под вакуумом с получением темно-оранжевого масла. В результате очистки с использованием флэш-хроматографии (колонка с 2 кг диоксида кремния), элюируя смесью циклогексан: DCM (1:1-0:1), получали указанное в заголовке соединение в виде желтого масла (48,8 г).
LC/MS: m/z 460,2 [М+Н]+, Rt 4,42 мин.
Промежуточное соединение 75: 2-хлор-6-(4-хлорфенил)пиразин
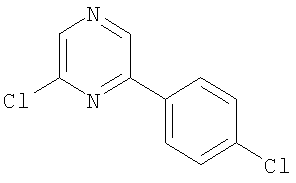
К смеси 2,6-дихлорпиразина (3 г; 20 ммоль), карбоната натрия (5,5 г; 52 ммоль) и 4-хлорфенилбороновой кислоты (3,79 г; 3,36 ммоль) в диметоксиэтане (140 мл) и воде (70 мл) в атмосфере азота добавляли тетракис(трифенилфосфин)палладий(0) (0,46 г). Полученную смесь нагревали с обратным холодильником в течение 18 ч в атмосфере азота. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли водой (150 мл) и экстрагировали дихлорметаном (2×200 мл). Органический раствор пропускали через гидрофобную фритту и растворители удаляли под вакуумом. В результате очистки посредством Biotage™ (Si, 90 г), циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде белого твердого вещества (2,33 г).
LC/MS: m/z 225,0 [M+H]+, Rt 3,45 мин.
Промежуточное соединение 76: 2-бром-6-(4-хлорфенил)пиразин
В колбу, содержащую 2-хлор-6-(4-хлорфенил)пиразин (2,35 г; 10,4 ммоль), добавляли трибромид фосфора (17 мл) и смесь нагревали с обратным холодильником в течение 6 ч в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, затем осторожно вливали в ледяную воду (600 мл), рН полученной смеси доводили до 6-7, используя водный аммиак, и экстрагировали диэтиловым эфиром (2×250 мл). Органический раствор сушили (MgSO4) и растворитель удаляли под вакуумом. Реакция не завершилась, поэтому остаток обрабатывали добавленным трибромидом фосфора (17 мл) и нагревали с обратным холодильником в течение еще 5 ч в атмосфере азота. В результате вышеописанной обработки получали указанное в заголовке соединение в виде бледно-желтого твердого вещества (0,97 г).
LC/MS: m/z 270 [M+H]+, Rt 3,53 мин.
Промежуточное соединение 77: этил-[4-({бутил[6-(4-хлорфенил)-пиразин-2-ил]амино}метил)-2-метилфенокси]ацетат

Смесь 2-бром-6-(4-хлорфенил)пиразина (0,27 г; 1 ммоль), N,N-диизопропилэтиламина (0,18 мл; 1 ммоль) и этил-{4-[(бутиламино)метил]-2-метилфенокси}ацетата (0,42 г; 1,5 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 18 ч. Реакционную смесь затем оставляли достигать комнатной температуры и остаток растворяли в CH2Cl2 (50 мл), органический экстракт промывали водой (15 мл) и пропускали через гидрофобную фритту, затем концентрировали под вакуумом. В результате очистки Biotage™-хроматографией (Si, 40 г), элюируя смесью EtOAcщиклогексан (1:9-1:1,5), получали указанное в заголовке соединение в виде бледно-желтого масла (78 мг).
LC/MS: m/z 468 [M+H]+, Rt 4,2 мин.
Промежуточное соединение 78: этил-[4-({[6-(4-хлорфенил)пиразин-2-ил][2-(метилокси)этил]амино}метил)-2-метилфенокси]ацетат

Использовали этил-(4-{[(2-метоксиэтил)амино]метил}-2-метилфенокси)-ацетат (0,42 г; 1,5 ммоль) и методику синтеза, описанную для Промежуточного соединения 77. В результате очистки Biotage™-хроматографией (Si, 40 г), элюируя смесью EtOAc:циклогексан (1:4-1:1), получали указанное в заголовке соединение в виде бледно-желтого масла (81 мг).
LC/MS: m/z 470 [M+H]+, Rt 3,8 мин.
Промежуточное соединение 79: этил-{2-метил-4-[(пропил{6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]фенокси}ацетат

Использовали 2-хлор-6-[4-(трифторметил)фенил]пиразина (0,303 г; 1 ммоль) и этил-{2-метил-4-[(пропиламино)метил]фенокси}ацетат (0,4 г; 1,5 ммоль) и методику синтеза, описанную для Промежуточного соединения 77. В результате очистки Biotage™-хроматографией (Si, 40 г), элюируя смесью циклогексан:EtOAc (9:1-7:3), получали указанное в заголовке соединение в виде бледно-желтого масла (56 мг).
LC/MS: m/z 488 [M+H]+, Rt 4,1 мин.
Промежуточное соединение 80: этил-(2-метил-4-{[{5-метил-6-[4-(трифторметил)фенил]пиримидин-4-ил}(пропил)амино]метил}фенокси)-ацетат

Смесь этил-{2-метил-4-[(пропиламино)метил]фенокси}ацетата (0,279 г; 1,05 ммоль), N,N-диизопропилэтиламина (0,24 мл; 1,4 ммоль) и 4-хлор-5-метил-6-[4-(трифторметил)фенил]пиримидина (0,191 г; 0,7 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 18 ч. После этого реакционный сосуд оставляли охлаждаться до комнатной температуры, остаток растворяли в СН2Cl2 (50 мл) и промывали водой (20 мл). Органический экстракт разделяли с помощью гидрофобной фритты и упаривали. В результате очистки Biotage™-хроматографией (Si, 40 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бледно-желтого масла (159 мг).
LC/MS: m/z 502,2 [M+H]+, Rt 3,8 мин.
Промежуточное соединение 81: этил-(4-{[[6-(4-хлорфенил)-5-метилпиримидин-4-ил](пропил)амино]метил}-2-метилфенокси)ацетат

Использовали 4-хлор-6-(4-хлорфенил)-5-метилпиримидин (167 мг; 0,7 ммоль) и методику синтеза, описанную для Промежуточного соединения 80. В результате очистки Biotage™-хроматографией (Si, 40 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бледно-желтого масла (116 мг).
LC/MS: m/z 468,1 [М+Н]+, Rt 3,6 мин.
Промежуточное соединение 82: этил-(2-метил-4-{[[5-метил-6-(4-метилфенил)пиримидин-4-ил](пропил)амино]метил}фенокси)ацетат
Использовали 4-хлор-5-метил-6-(4-метилфенил)пиримидин (153 мг; 0,7 ммоль) и методику синтеза, описанную для Промежуточного соединения 80. В результате очистки Biotage™-хроматографией (Si, 40 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бледно-желтого масла (80 мг).
LC/MS: m/z 448,2 [М+Н]+, Rt 3,2 мин.
Промежуточное соединение 83: этил-(2-метил-4-{[{5-метил-6-[4-(метилокси)фенил]пиримидин-4-ил}(пропил)амино]метил}фенокси)ацетат
Использовали 4-хлор-6-(4-метоксифенил)-5-метилпиримидин (164 мг; 0,7 ммоль) и методику синтеза, описанную для Промежуточного соединения 80. В результате очистки Biotage™-хроматографией (Si, 40 г), элюируя смесью циклогексан:EtOAc (4:1), получали указанное в заголовке соединение в виде бледно-желтого масла (60 мг).
LC/MS: m/z 464,2 [М+Н]+, Rt 3,1 мин.
Промежуточное соединение 84: 2-этил-4-(гидроксиметил)фенол
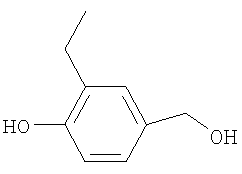
К раствору 3-этил-4-гидроксибензальдегида (4,7 г; 26 ммоль) в безводном THF (150 мл) при 0°С в атмосфере N2 добавляли по каплям 1 М LiAlH4 в Et2O (52 мл; 52,2 ммоль). Полученную смесь перемешивали при 0°С в течение 2 ч, затем гасили осторожным добавлением воды и перемешивали в течение 15 минут. Реакционную смесь затем абсорбировали на диоксиде кремния и наносили на набивку диоксида кремния, которую промывали EtOAc. EtOAc промывки собирали и концентрировали под вакуумом с получением указанного в заголовке соединения в виде бесцветного масла (3,67 г).
LC/MS: m/z 151,1 [М-Н]-, Rt 2,1 мин.
Промежуточное соединение 85: этил-[2-этил-4-(гидроксиметил)-фенокси]ацетат

К раствору 2-этил-4-(гидроксиметил)фенола (3,67 г; 24,1 ммоль) в безводном ацетонитриле (125 мл) добавляли карбонат цезия (8,65 г; 26,6 ммоль), реакционную смесь затем продували азотом и охлаждали до 0°С, затем добавляли этилбромацетат (2,68 мл; 24,1 ммоль). Реакционную смесь оставляли постепенно достигать комнатной температуры и перемешивали в течение 18 ч. Добавляли дополнительно этилбромацетат (0,26 мл; 2,4 ммоль) и перемешивание продолжали в течение 4 ч. Реакционную смесь разбавляли водой (100 мл) и затем экстрагировали EtOAc (2×100 мл). Объединенные органические экстракты сушили (MgSO4), фильтровали и упаривали. В результате очистки Biotage™-хроматографией (диоксид кремния, 90 г), элюируя смесью циклогексан:EtOAc (4:1), затем EtOAc, получали указанное в заголовке соединение в виде бледно-желтой смолы (3,54 г).
1H ЯМР (400 МГц; CDCl3) δ:1.23 (3Н, t, J 7,5 Гц), 1.29 (3Н, t, J 7,5 Гц), 2.72 (2Н, q, J 7,5 Гц), 4.26 (2Н, q, J 7,5 Гц), 4.58-4.68 (4Н, m), 6.69 (1H, d, J 8,5 Гц), 7.13 (1H, dd, J 8,5, 2 Гц), 7.19 (1H, d, J 2 Гц).
Промежуточное соединение 86: этил-(2-этил-4-формилфенокси)-ацетат
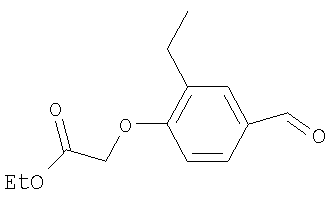
К раствору этил-[2-этил-4-(гидроксиметил)фенокси]ацетата (2,74 г; 11,5 ммоль) в хлороформе (250 мл) добавляли диоксид марганца (10 г; 11,5 ммоль). Полученную смесь перемешивали в течение 18 ч при комнатной температуре. Смесь фильтровали через набивку целита и затем концентрировали под вакуумом с получением указанного в заголовке соединения в виде бледно-желтого масла (2,7 г).
LC/MS: m/z 237,2 [M+H]+, Rt 3,1 мин.
Промежуточное соединение 87: этил-[2-этил-4-({[4'-(трифторметил)-1,1'-бифенил-3-ил]амино}метил)фенокси]ацетат
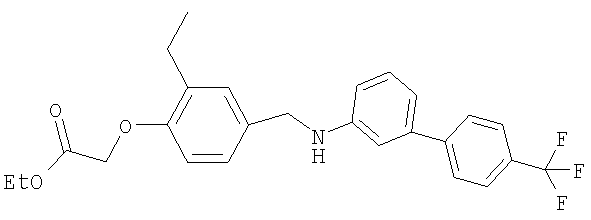
К раствору этил-(2-этил-4-формилфенокси)ацетата (0,3 г; 1,27 ммоль) в безводном СН2Cl2 (12 мл) добавляли 4′-(трифторметил)-1,1′-бифен-3-иламин (0,3 г; 1,27 ммоль). Полученный раствор перемешивали при комнатной температуре в атмосфере N2 в течение 15 мин и затем добавляли триацетоксиборогидрид натрия (0,376 г; 1,77 ммоль) и ледяную уксусную кислоту (0,0725 мл; 1,27 ммоль). Через 20 ч перемешивания при комнатной температуре в атмосфере азота добавляли по каплям насыщенный водный NaHCO3 (40 мл) и смесь перемешивали в течение 20 мин. Смесь затем экстрагировали CH2Cl2 (2×50 мл), органические экстракты разделяли с помощью гидрофобной фритты и концентрировали под вакуумом. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бледно-желтого кристаллического твердого вещества (0,47 г).
LC/MS: m/z 458,2 [M+H]+, Rt 4,1 мин.
Промежуточное соединение 88: этил-{4-[(бутиламино)метил]-2-этилфенокси}ацетат
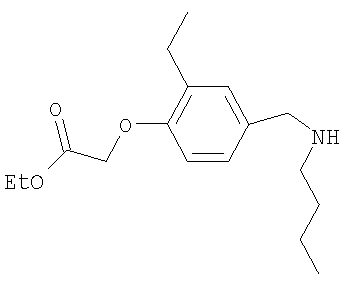
К раствору этил-(2-этил-4-формилфенокси)ацетата (0,52 г; 2,2 ммоль) в безводном этаноле (10 мл) добавляли бутиламин (0,26 мл; 2,6 ммоль). Полученный раствор перемешивали при комнатной температуре в атмосфере N2 в течение 18 ч над 4Å молекулярными ситами. Реакционную смесь гасили осторожным добавлением насыщенного водного NaHCO3 (30 мл) и смесь перемешивали в течение 30 мин. Смесь затем экстрагировали EtOAc (2×100 мл). Объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали под вакуумом. Проводили очистку посредством SPE (SCX, 20 г), обрабатывая картридж этанолом, затем наносили соединение и промывали CH2Cl2, затем этанолом, потом элюировали указанное в заголовке соединение, используя 10%-ный NH3 в этаноле, выпаривали 10%-ный NH3 в этанольном экстракте с получением указанного в заголовке соединения в виде бледно-желтого масла (0,364 г).
LC/MS: m/z 294,3 [М+Н]+, Rt 2,43 мин.
Промежуточное соединение 89: этил-[2-этил-4-{[2-(метоксиэтил)-амино]метил}фенокси]ацетат

Использовали 2-метоксиэтиламин (0,23 мл; 0,26 ммоль) и методику синтеза, описанную для синтеза Промежуточного соединения 88. Проводили очистку посредством SPE (SCX, 20 г), обрабатывая картридж этанолом, затем наносили соединение и промывали CH2Cl2, затем этанолом, потом элюировали указанное в заголовке соединение, используя 10%-ный NH3 в этаноле, выпаривали 10%-ный NH3 в этанольном экстракте с получением указанного в заголовке соединения в виде бледно-желтого масла (0,341 г).
LC/MS: m/z 296,2 [M+H]+, Rt 2,2 мин.
Промежуточное соединение 90: этил-{4-[(бутил{6-[4-(трифторметил)-фенил]пиразин-2-ил}амино)метил]-2-этилфенокси}ацетат

Смесь 2-хлор-6-[4-(трифторметил)фенил]пиразина (0,172 г; 0,56 ммоль), этил-{4-[(бутиламино)метил]-2-этилфенокси}ацетата (0,25 г; 0,85 ммоль) и N,N-диизопропилэтиламина (0,099 мл; 0,56 ммоль) нагревали вместе в реакционном сосуде при 100°С в течение 18 ч. Сосуд затем оставляли охлаждаться до комнатной температуры, остаток растворяли в CH2Cl2 (50 мл) и органический раствор промывали водой (15 мл) и соляным раствором (10 мл). Раствор затем пропускали через гидрофобную фритту и упаривали. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бледно-желтого масла (50 мг).
LC/MS: m/z 516,3 [M+H]+, Rt 4,3 мин.
Промежуточное соединение 91: этил-{2-этил-4-[(2-метилоксиэтил){6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]фенокси}ацетат

Смесь 2-хлор-6-[4-(трифторметил)фенил]пиразина (0,109 г; 0,36 ммоль), этил-[2-этил-4-{[2-(метоксиэтил)амино]метил}фенокси]ацетата (0,159 г; 0,54 ммоль) и N,N-диизопропилэтиламина (0,063 мл; 0,36 ммоль) нагревали вместе в реакционном сосуде при 100°С в течение 18 ч. Сосуд затем оставляли охлаждаться до комнатной температуры, остаток растворяли в СН2Cl2 (50 мл) и органический раствор промывали водой (15 мл) и соляным раствором (10 мл). Раствор пропускали через гидрофобную фритту и упаривали. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью циклогексан:EtOAc (9:1-4:1), получали указанное в заголовке соединение в виде бледно-желтого масла (48 мг).
LC/MS: m/z 518,3 [M+H]+, Rt 3,9 мин.
Промежуточное соединение 92: этил-[4-({бутил[5-метил-6-(4-метилфенил)пиримидин-4-ил]амино}метил)-2-этилфенокси]ацетат
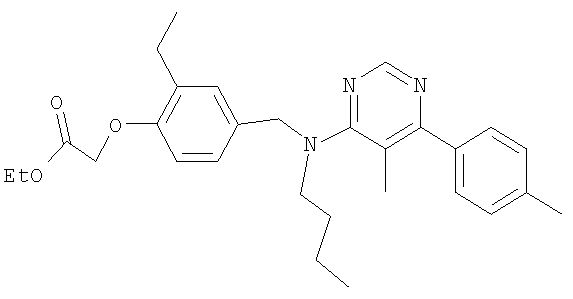
Смесь этил-{4-[(бутиламино)метил]-2-этилфенокси}ацетата (0,23 г; 0,78 ммоль), N,N-диизопропилэтиламина (0,18 мл; 1,05 ммоль) и 4-хлор-5-метил-6-(4-метилфенил)пиримидина (0,114 г; 0,52 ммоль) нагревали при 100°С в герметично закрытом реакционном сосуде в течение 18 ч. После этого реакционный сосуд оставляли охлаждаться до комнатной температуры, остаток растворяли в CH2Cl2 (50 мл) и промывали водой (10 мл) и соляным раствором (10 мл). Органический экстракт разделяли с помощью гидрофобной фритты и упаривали. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью циклогексан:EtOAc (9:1), получали указанное в заголовке соединение в виде бледно-желтого масла (46 мг).
LC/MS: m/z 476,3 [M+H]+, Rt 3,6 мин.
Промежуточное соединение 93: этил-[4-({бутил[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-2-метилфенокси]-ацетат

К раствору N-бутил-N-[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амина (58 мг; 0,19 ммоль) в безводном СН2Cl2 (5 мл) добавляли этил-[4-(хлорсульфонил)-2-метилфенокси]ацетат (83 мг; 0,28 ммоль) и триэтиламин (0,052 мл; 0,38 ммоль). Полученный раствор перемешивали 20 ч при комнатной температуре в атмосфере азота. Реакционную смесь разбавляли СН2Cl2 (50 мл) и органический раствор промывали насыщенным водным NaHCO3 (10 мл) и соляным раствором (10 мл). Органический экстракт разделяли с помощью гидрофобной фритты и упаривали. В результате очистки аутопрепаративной HPLC с управлением по массе получали указанное в заголовке соединение в виде бесцветного масла (15 мг).
LC/MS: m/z 564,2 [M+H]+, Rt 4,2 мин.
Промежуточное соединение 94: этил-[2-этил-4-({(пропилсульфонил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-фенокси]ацетат

К раствору этил-[2-этил-4-({[4′-(трифторметил}-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетата (0,1 г; 0,22 ммоль) в безводном пиридине (0,3 мл) добавляли 1-пропансульфонилхлорид (0,037 мл; 0,3 ммоль). Реакционную смесь затем нагревали при 80°С в герметично закрытом реакционном сосуде в течение 18 ч. Как только реакционная смесь охладилась до комнатной температуры, сосуд открывали и остаток растворяли в CH2Cl2 (30 мл) и промывали водой (20 мл) и соляным раствором (20 мл). Органический экстракт разделяли с помощью гидрофобной фритты и упаривали. В результате очистки Biotage™-хроматографией (диоксид кремния, 12 г), элюируя смесью циклогексан:EtOAc (9:1-17:3), получали указанное в заголовке соединение в виде твердого вещества желтого цвета (65 мг).
LC/MS: m/z 581,3 [M+NH4]+, Rt 4,0 мин.
Промежуточное соединение 95: этил-[4-({бутил[6-(4-хлорфенил)-5-метилпиримидин-4-ил]амино}метил)-2-этилфенокси]ацетат

Использовали 4-хлор-6-(4-хлорфенил)-5-метилпиримидин (0,125 г; 0,52 ммоль) и методику синтеза, описанную для Примера 92. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью циклогексан:EtOAc (9:1) с последующей очисткой с использованием OPTIX (диоксид кремния, 4 г) и элюируя смесью циклогексан:EtOAc (19:1), получали указанное в заголовке соединение в виде бледно-желтого масла (64 мг).
LC/MS: m/z 496,2 [М+Н]+, Rt 4,0 мин.
Промежуточное соединение 96: этил-{4-[(бутил{5-метил-6-[4-(трифторметил)фенил]пиримидин-4-ил}амино)метил]-2-этилфенокси}-ацетат
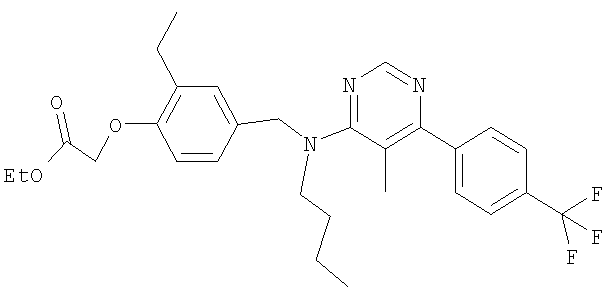
Использовали 4-хлор-5-метил-6-[4-(трифторметил)фенил]пиримидин (0,143 г; 0,52 ммоль) и методику синтеза, описанную для соединения Примера 92. В результате очистки Biotage™-хроматографией (диоксид кремния, 40 г), элюируя смесью циклогексан:EtOAc (9:1) с последующей очисткой с использованием OPTIX (диоксид кремния, 4 г), элюируя смесью циклогексан:EtOAc (19:1), получали указанное в заголовке соединение в виде бледно-желтого масла (64 мг).
LC/MS: m/z 430,2 [M+H]+, Rt 4,2 мин.
Промежуточное соединение 97: этил-{2-этил-4-[([2-(метилокси)этил]{4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)-метил]фенокси}ацетат
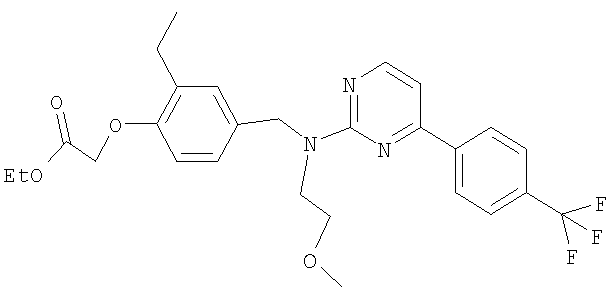
Смесь пиримидин-2-(метилсульфонил)-4-[4-(трифторметил)фенил]-пиримидина (0,109 г; 0,36 ммоль), этил-[2-этил-4-{[2-(метоксиэтил)амино]-метил}фенокси]ацетата (0,159 г; 0,54 ммоль) и N,N-диизопропилэтиламина (0,063 мл; 0,36 ммоль) нагревали вместе в реакционном сосуде при 100°С в течение 18 ч. Сосуд затем оставляли охлаждаться до комнатной температуры и остаток растворяли в CH2Cl2 (50 мл). Органический раствор промывали водой (15 мл) и соляным раствором (10 мл). Раствор затем пропускали через гидрофобную фритту и упаривали. В результате очистки посредством SPE (диоксид кремния, 10 г), элюируя смесью циклогексан:EtOAc (9:1-4:1), получали указанное в заголовке соединение в виде бледно-желтого масла (52 мг).
LC/MS: m/z 518,3 [M+H]+, Rt 4,1 мин.
Промежуточное соединение 98: N-2-пропен-1-ил-6-[4-(трифторметил)фенил]-2-пиридинамин
Смесь аллиламина (0,447 мл; 5,96 ммоль) и 2-бром-6-[4-(трифторметил)-фенил]пиридина (0,1 г; 0,298 ммоль) в этаноле (5 мл) нагревали при 100°С в 25 мл сосуде высокого давления. После нагревания в течение ночи реакционную смесь охлаждали и добавляли сульфат меди, нагревание продолжали при 100°С в течение ночи. После этого реакционную смесь оставляли охлаждаться до комнатной температуры и концентрировали под вакуумом. В результате очистки посредством SPE (диоксид кремния, 10 г), элюируя смесью циклогексан:хлороформ (3:1), получали указанное в заголовке соединение в виде темно-желтого твердого вещества (0,062 г).
LC/MS: m/z 279,1 [М+Н]+, Rt 3,7 мин.
Пример 1. 2-Метил-2-{2-метил-4-[([4-(трифторметил)бензил]{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)метил]фенокси}пропановая кислота
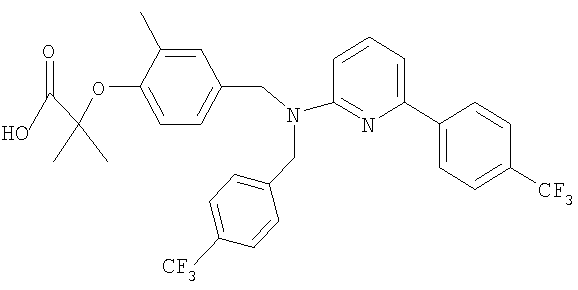
К раствору этил-2-метил-2-{2-метил-4-[([4-(трифторметил)бензил]{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)метил]фенокси}пропаноата (0,07 г; 0,11 ммоль) в тетрагидрофуране (2 мл) и метаноле (2 мл) добавляли 2 М водный NaOH (1 мл). Полученный раствор перемешивали при комнатной температуре в течение 20 ч, затем концентрировали под вакуумом. Остаток подкисляли 2 М водным HCl и экстрагировали CH2Cl2 (2×30 мл). Объединенные органические экстракты сушили (Na2SO4), фильтровали и упаривали с получением указанного в заголовке соединения в виде бесцветного масла (60 мг).
1H ЯМР (400 МГц; CDCl3) δ: 1.60 (6Н, s), 2.20 (3H, s), 4.77 (2H, s), 4.93 (2H, s), 6.47 (1H, d, J 8,5 Гц), 6.77 (1Н, d, J 8 Гц), 7.00 (1Н, dd, J 8,5, 2 Гц), 7.07 (1Н, d, J 2 Гц), 7.14 (1Н, d, J 7,5 Гц), 7.37 (2H, d, J 8,5 Гц), 7.47-7.58 (3H, m), 7.64 (2H, d, J 8,5 Гц), 8.05 (2H, d, J 8,5 Гц), СО2Н не наблюдался.
LC/MS: m/z 603,2 [M+H]+, Rt 4,6 мин.
Пример 2. 2-{4-[(Бутил{6-[4-(трифторметил)фенил]пиридин-2-ил}-амино)метил]-2-метилфенокси}-2-метилпропановая кислота
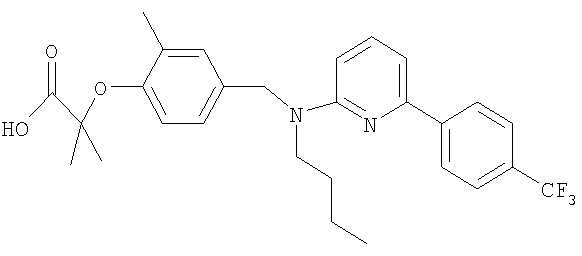
К раствору этил-2-{4-[(бутил{-6-[4-(трифторметил)фенил]пиридин-2 ил}амино)метил]-2-метилфенокси}-2-метилпропаноата (45 мг; 0,085 ммоль) в метаноле (1,5 мл) и тетрагидрофуране (1,5 мл) добавляли 2 М водный NaOH (1 мл). Полученную смесь нагревали до 70°С в течение 1 ч и затем перемешивали при комнатной температуре в течение 18 ч, после чего нагревали до 70°С в течение следующих 4 ч. Реакционную смесь оставляли достигать комнатной температуры и растворители удаляли под вакуумом. Остаток подкисляли 2 М HCl и экстрагировали EtOAc (2×10 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом. Очищали посредством аминопропил-SPE (5 г) с обработкой картриджа нанесением соединения и промыванием метанолом перед элюированием с использованием 10%-ного NH3 в метаноле. В результате выпаривания метанольного раствора аммиака получали белое твердое вещество, которое обрабатывали 1 М HCl (5 мл) и затем экстрагировали в СН2Cl2 (20 мл). Органический экстракт сушили (Na2SO4) и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде бесцветного масла (19 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.95 (3Н, t, J 7 Гц), 1.37 (2Н, m), 1.56 (6H, s), 1.64 (2Н, m), 2.19 (3Н, s), 3.55 (2Н, t, J 7,5 Гц), 4.74 (2Н, s), 6.44 (1H, d, J 8,5 Гц), 6.75 (1H, d, J 8,5 Гц), 6.98 (1H, dd, J 8,5, 1,5 Гц), 7.04 (1H, d, J 7,5 Гц), 7.08 (1H, d, J 1,5 Гц), 7.48 (1H, dd, J 8,5, 7,5 Гц), 7.64 (2Н, d, J 8 Гц), 8.08 (2Н, d, J 8Гц), CO2Н не наблюдался.
LC/MS: m/z 501,2 [M+H]+, Rt 4,7 мин.
Пример 3. {4-[(Бутил{6-[4-(трифторметил)фенил]пиридин-2-ил}-амино)метил]-2-метилфенокси}уксусная кислота

К раствору метил-{4-[(бутил{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)метил]-2-метилфенокси}ацетата (40 мг; 0,082 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) добавляли 2 М водный NaOH (2 мл). После перемешивания в течение 1 ч при комнатной температуре растворитель удаляли под вакуумом и остаток подкисляли 2 М HCl перед экстрагированием в этилацетат (2×15 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде твердого вещества белого цвета (35 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.96 (3Н, t, J 7,5 Гц), 1.39 (2Н, m), 1.66 (2H, m), 2.26 (3Н, s), 3.57 (2H, t, J 8 Гц), 4.66 (2H, s), 4.76 (2H, s), 6.47 (1H, d, J 8,5 Гц), 6.67 (1H, d, J 8,5 Гц), 7.02-7.14 (3Н, m), 7.50 (1H, m), 7.66 (2H, d, J 8 Гц), 8.08 (2H, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 473,2 [М+Н]+, Rt 5,0 мин.
Пример 4. [4-({Бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-метил)-2-метилфенокси]уксусная кислота
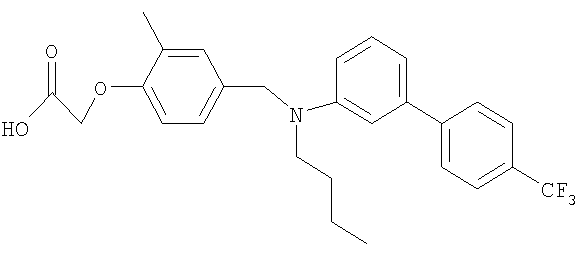
К раствору этил-[4-({бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-метил)-2-метилфенокси]ацетата (105 мг; 0,21 ммоль) в метаноле (4 мл) и тетрагидрофуране (4 мл) добавляли 2 М водный NaOH (2 мл). После перемешивания в течение 1 ч при комнатной температуре растворитель удаляли под вакуумом и остаток подкисляли 2 М HCl, затем экстрагировали в этилацетат (2×20 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде бледно-желтой пены (90 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 0.97 (3Н, t, J 7,5 Гц), 1.42 (2H, m), 1.60 (2H, m), 2.20 (3Н, s), 3.69 (2H, m), 4.66-4.70 (4H, m), 6.77 (1H, d, J 8 Гц), 7.00-7.07 (2H, m), 7.10-7.57 (4H, m), 7.71 (2H, d, J 8 Гц), 7.76 (2H, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 472,1 [М+Н]+, Rt 4,6 мин.
Пример 5. [4-({(2-Метоксиэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусная кислота
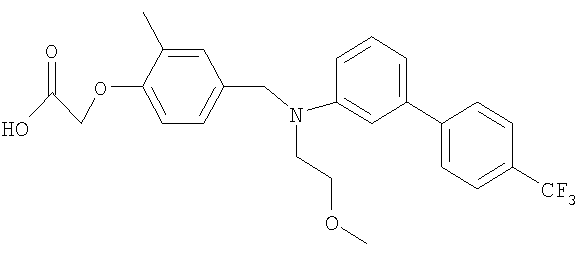
К раствору этил-[4-({(2-метоксиэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетата (90 мг; 0,18 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) добавляли 2 М водный NaOH (1 мл). После перемешивания в течение 1 ч при комнатной температуре растворитель удаляли под вакуумом, остаток разбавляли водой (10 мл), подкисляли 2 М HCl, затем экстрагировали в этилацетат (2×20 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (28 мг).
1H ЯМР (400 МГц; CDCl3) δ: 2.22 (3Н, s), 3.33 (3Н, s), 3.68 (4H, s), 4.58 (2H, s), 4.65 (2H, s), 6.63 (1H, d, J 9 Гц), 6.88-7.15 (5Н, m), 7.31 (1H, t, J 8 Гц), 7.58 (2H, d, J 8 Гц), 7.65 (2H, d, J 8 Гц), СО2Н не наблюдался.
LC/MS: m/z 471,9 [M-H]-, Rt 4,1 мин.
Пример 6. [2-Метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]-амино}метил)фенокси]уксусная кислота
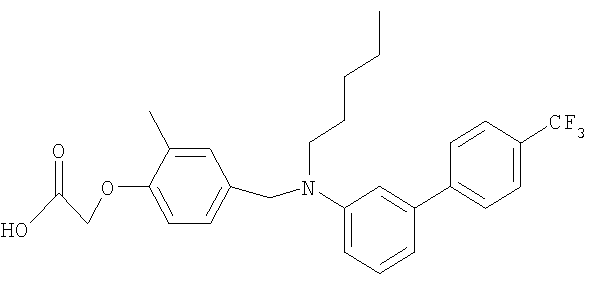
К раствору этил-[2-метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетата (54 мг; 0,11 ммоль) в МеОН (2 мл) и THF (2 мл) при комнатной температуре добавляли 2 М NaOH (1 мл; 2 ммоль). Полученную смесь перемешивали в течение 1 ч и затем растворители удаляли под вакуумом. Остаток распределяли между СН2Cl2 (2×10 мл) и 2 М HCl (10 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли с получением указанного в заголовке соединения в виде белого твердого вещества (59 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 0.91 (3Н, t, J 7 Гц), 1.35 (4Н, m), 1.61 (2H, br. s), 2.19 (3Н, s), 3.73 (2H, m), 4.70 (4Н, s), 6,76 (1H, d, J 8,5 Гц), 7.04 (2H, m), 7.25-7.43 (2H, m), 7.51-7.63 (2H, m), 7.72 (2H, d, J 8 Гц), 7.78 (2H, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 486,3 [M+H]+, Rt 4,9 мин.
Пример 7. [4-({(2-Циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусная кислота
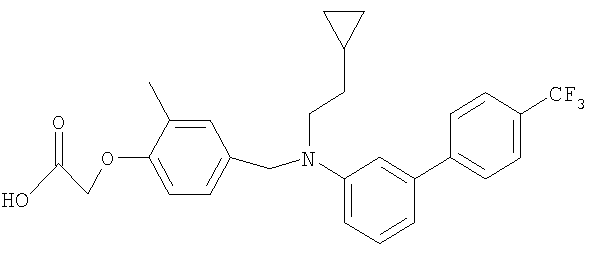
К раствору этил-[4-({(2-циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетата (141 мг; 0,28 ммоль) в МеОН (2 мл) и THF (2 мл) при комнатной температуре добавляли 2 M NaOH (1 мл, 2 ммоль). Полученную смесь перемешивали в течение 1,5 ч, затем растворители удаляли под вакуумом. Остаток распределяли между СН2Cl2 (2×20 мл) и 2 M HCl (20 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли с получением указанного в заголовке соединения в виде белого твердого вещества (115 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 0.07 (2H, m), 0.50 (2H, dm, J 8 Гц), 0.70 (1H, m), 1.46 (2H, m), 2.15 (3Н, s), 3.89 (2H, t, J 7,5 Гц), 4.68 (2H, s), 4.74 (2H, br s), 6.75 (1H, d, J 8,5 Гц), 6.97 (1H, d, J 1,5 Гц), 7.04 (1H, dd, J 8,5 Гц, 2,5 Гц), 7.42 (1H, br s), 7.50 (1H, br s), 7.63 (1H, t, J 8 Гц), 7.68-7.81 (5H, m), CO2Н не наблюдался.
LC/MS: m/z 484,3 [M+H]+, Rt 4,6 мин.
Пример 8. [2-Метил-4-({пропил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]уксусная кислота
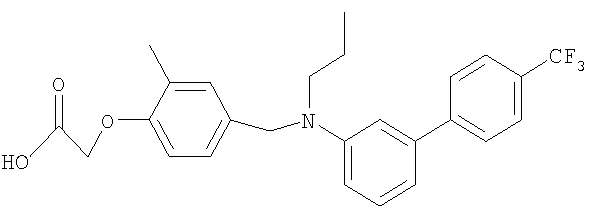
К раствору этил-[2-метил-4-({пропил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетата (88 мг; 0,18 ммоль) в МеОН (2 мл) и THF (2 мл) при комнатной температуре добавляли 2 М NaOH (1 мл, 2 ммоль). Полученный раствор перемешивали в течение 1 ч, затем растворители удаляли под вакуумом. Остаток разбавляли HCl (10 мл) и экстрагировали CH2Cl2 (2×10 мл), затем EtOAc (2×10 мл). Органические растворы сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде белого кристаллического твердого вещества (64,1 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 0.95 (3Н, t, J 7,5 Гц), 1.69 (2Н, m), 2.21 (3Н, s), 3.42 (2Н, t, J 7,5 Гц), 4.50 (2Н, s), 4.56 (2Н, s), 6.68-6.75 (2Н, m), 6.82-6.88 (2Н, m), 6.98 (1H, dd, J 8,5 Гц, 2 Гц), 7.03 (1Н, s), 7.21 (1H, t, J 8,5 Гц), 7.64 (4Н, m), СО2Н не наблюдался.
LC/MS: m/z 455,9 [M-Н]-, Rt 4,4 мин.
Пример 9. [2-Метил-4-({[2-(метилтио)этил][4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]уксусная кислота

К раствору метил-[2-метил-4-({[2-(метилтио)этил][4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетата (96,4 мг; 0,19 ммоль) в МеОН (2 мл) и THF (2 мл) при комнатной температуре добавляли 2 М NaOH (1 мл, 2 ммоль). Полученную смесь перемешивали в течение 16 ч и затем растворители удаляли под вакуумом. Остаток распределяли между CH2Cl2 (2×20 мл) и 2 М HCl (20 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде желтого твердого вещества (94 мг).
1H ЯМР (400 МГц; CDCl3) δ: 2.15 (3Н, s), 2.27 (3Н, s), 2.76 (2Н, t, J 7,5 Гц), 3.66 (2Н, t, J 7,5 Гц), 4.55 (2Н, s), 4.67 (2Н, s), 6.68 (1H, d, J 8,5 Гц), 6.74 (1H, d, J 7,5 Гц), 6.87 (1H, s), 6.91 (1H, d, J 7 Гц), 7.02 (1H, d, J 8 Гц), 7.07 (1H, s), 7.29 (1H, m), 7.59 (2Н, d, J 8 Гц), 7.65 (2Н, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 487,8 [М-Н]-, Rt 4,3 мин.
Пример 10. [4-({Бутил[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]-амино}метил)-2-метилфенокси]уксусная кислота
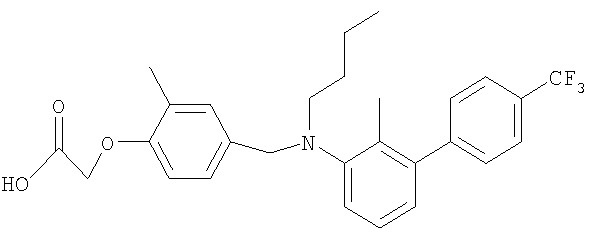
К раствору этил-[4-({бутил[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетата (110 мг; 0,21 ммоль) в МеОН (2 мл) и THF (2 мл) при комнатной температуре добавляли 2 М NaOH (1 мл, 2 ммоль). Полученную смесь перемешивали в течение 2 ч. Растворители удаляли под вакуумом и остаток распределяли между CH2Cl2 (2×20 мл) и 2 М HCl (20 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (103 мг).
1H ЯМР (400 МГц; DMSO-d6+DCl, 85°С) δ: 0.82 (3Н, t, J 7,5 Гц), 1.28 (2Н, m), 1.42 (2Н, m), 1.89 (3Н, s), 2.03 (3Н, s), 3.69 (2Н, t, J 7,5 Гц), 4.59 (2Н, br s), 4.61 (2Н, s), 6.69 (1H, d, J 8,5 Гц), 6.98 (1Н, d, J 2 Гц), 7.08 (1Н, dd, J 8,5 Гц, 2 Гц), 7.21 (1Н, d, J 7,5 Гц), 7.27 (2Н, d, J 8 Гц), 7.47 (1Н, t, J 8 Гц), 7.72 (2Н, d, J 8 Гц), 7.82 (1Н, d, J 8,5 Гц), CO2Н не наблюдался.
LC/MS: m/z 486,4 [M+H]+, Rt 4,6 мин.
Пример 11. [4-({(2-Метоксиэтил)[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусная кислота

К раствору этил-[4-({(2-метоксиэтил)[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетата (500 мг; 0,97 ммоль) в THF (6 мл) и МеОН (6 мл) добавляли NaOH (2 М, 3 мл) и полученный раствор перемешивали в течение 2 ч при комнатной температуре. Смесь концентрировали под вакуумом, подкисляли 2 М HCl и экстрагировали CH2Cl2. Органический раствор сушили (Na2SO4) и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде бесцветного масла (453 мг).
1H ЯМР (400 МГц; DMSO-d6) δ: 2.09 (3Н, s), 2.14 (3H, s), 3.05 (2H, m), 3.12 (3H, m), 3.32 (2H, m), 4.05 (2H, s), 4.6 (2H, m), 6.67 (1H, d, J 8,5 Гц), 6.89 (1Н, m), 6.99-7.02 (2H, m), 7.17 (2H, m), 7.52 (2H, d, J 8 Гц), 7.73 (2H, d, J 8 Гц), 12.9 (1H, s, широкий).
LC/MS: m/z 488 [M+H]+, Rt 4,06 мин.
Пример 12. [4-({Бутирил[4′-(трифторметил)-1,1′-бифенил-3-ил]-амино}метил)-2-метилфенокси]уксусная кислота
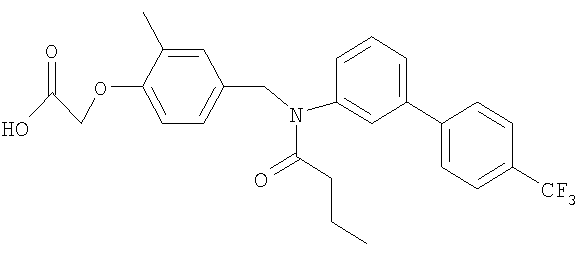
К раствору этил-[4-({бутирил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]ацетата (40 мг; 0,078 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) добавляли 2 М гидроксид натрия (1 мл). После перемешивания в течение 1 ч при комнатной температуре растворитель удаляли под вакуумом и остаток подкисляли 2 M HCl перед экстрагированием в этилацетат (2×15 мл). Органический раствор сушили (MgSO4), и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде твердого вещества белого цвета (28 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.84 (3Н, t, J 7,5 Гц), 1.64 (2H, m), 2.08 (2H, m), 2.24 (3H, s), 4.67 (2H, s), 4.84 (2H, s), 6.64 (1H, d, J 8 Гц), 6.98-7.05 (3Н, m), 7.12 (1H, br s), 7.45 (1H, t, J 8 Гц), 7.55 (3H, d, J 8 Гц), 7.69 (2H, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 485,9 [М+Н]+, Rt 3,9 мин.
Пример 13. [2-Метил-4-({(пропилсульфонил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]уксусная кислота
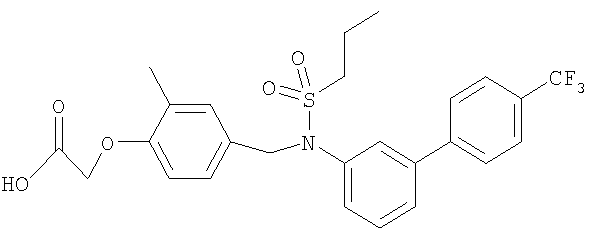
К раствору этил-[2-метил-4-({(пропилсульфонил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетата (20 мг; 0,036 ммоль) в МеОН (1 мл) и THF (1 мл) при комнатной температуре добавляли 2 М NaOH (0,5 мл; 1 ммоль). Полученную смесь перемешивали в течение 2 ч. Растворители удаляли под вакуумом, остаток распределяли между CH2Cl2 (2×10 мл) и 2 М HCl (10 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом. Очищали аутопрепаративной HPLC, проводимой на колонке Supelco ABZ+ (5 мкм, 10 см ×21,2 мм внутр. диам.), с элюированием при скорости потока 8 мл/мин с градиентом 50-95% Б в течение 20 мин и выдерживанием в условиях 95% Б в течение 10 мин. При этом растворитель А=0,1%-ная HCO2Н в Н2О и растворитель Б=0,05%-ная HCO2Н в MeCN. Указанное в заголовке соединение получали в виде желтого масла (10,7 мг).
1H ЯМР (400 МГц; CDCl3) δ: 1.06 (3Н, t, J 7,5 Гц), 1.93 (2Н, m), 2.23 (3H, s), 3.04 (2Н, m), 4.64 (2Н, s), 4.82 (2Н, s), 6.61 (1H, d, J 8,5 Гц), 7.03 (1Н, dd, J 8,5 Гц, 2 Гц), 7.09 (1H, d, J 2 Гц), 7.28 (1H, m), 7.39-7.50 (3H, m), 7.56 (2Н, d, J 8 Гц), 7.68 (2Н, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 520,1 [М-Н]-, Rt 3,6 мин.
Пример 14. [4-({Бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-сульфонил)-2-метилфенокси]уксусная кислота
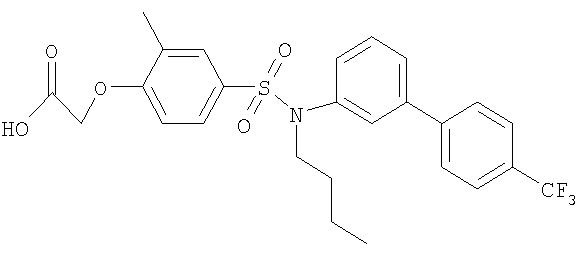
К раствору этил-[4-({бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-сульфонил)-2-метилфенокси]ацетата (100 мг; 0,18 ммоль) в метаноле (4 мл) и тетрагидрофуране (4 мл) добавляли 2 М водный NaOH (2 мл). После перемешивания в течение 1 ч при комнатной температуре растворитель удаляли под вакуумом и остаток подкисляли 2 М HCl, затем экстрагировали в этилацетат (2×20 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (85 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.87 (3H, t, J 7 Гц), 1.30-1.45 (4Н, m), 2.28 (3Н, s), 3.55 (2H, t, J 7 Гц), 4.78 (2Н, s), 6.74 (1H, d, J 8,5 Гц), 7.10 (1Н, dm, J 8 Гц), 7.25 (1H, t, J 2 Гц), 7.39-7.47 (3H, m), 7.51-7.60 (3H, m), 7.69 (2H, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 522,3 [M+H]+, Rt 4,5 мин.
Пример 15. [2-Метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)фенокси] уксусная кислота
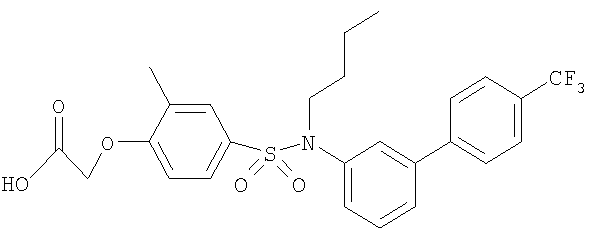
К раствору этил-[2-метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)фенокси]ацетата (76 мг; 0,13 ммоль) в МеОН (2 мл) и THF (2 мл) при комнатной температуре добавляли 2 М водный NaOH (1 мл, 2 ммоль). Полученную смесь перемешивали в течение 1 ч. Растворители удаляли под вакуумом и остаток распределяли между CH2Cl2 (2×15 мл) и 2 М HCl (10 мл). Органический раствор пропускали через гидрофобную фритту и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (75 мг).
1H ЯМР (400 МГц; MeOH-d4) δ:0.85 (3Н, t, J 7,5 Гц), 1.22-1.45 (6Н, m), 2.23 (3H, s), 3.60 (2H, t, J 7 Гц), 4.81 (2H, s), 6.96 (1H, d, J 8,5 Гц), 7.14 (1H, dm, J 7 Гц), 7.23 (1H, t, J 2 Гц), 7.32 (1H, d, J 1,5 Гц), 7.40-7.50 (2H, m), 7.62-7.68 (3Н, m), 7.75 (2H, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 536,3 [M+H]+, Rt 4,5 мин.
Пример 16. [4-({(2-Циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-2-метилфенокси]уксусная кислота
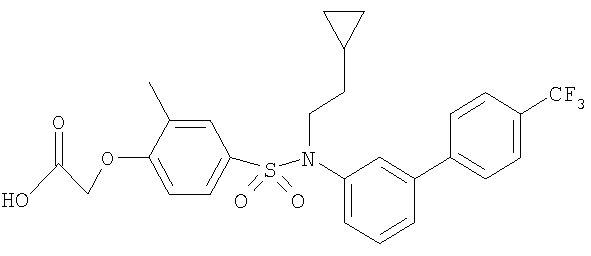
К раствору этил-[4-({(2-циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-2-метилфенокси]ацетата (156 мг; 0,28 ммоль) в МеОН (2 мл) и THF (2 мл) при комнатной температуре добавляли 2 М водный NaOH (1 мл, 2 ммоль). Полученную смесь перемешивали в течение 2 ч. Растворители удаляли под вакуумом и остаток распределяли между EtOAc (2×20 мл) и 2 М HCl (20 мл). Органический раствор сушили (MgSO4) и растворитель удаляли под вакуумом. Очищали посредством SPE (5 г аминопропильный картридж) с нанесением неочищенного вещества в CH2Cl2 и последующим промыванием МеОН перед элюированием 5%-ным NH3 в МеОН. Экстракт в 5%-ном NH3 в МеОН концентрировали под вакуумом и остаток распределяли между CH2Cl2 (20 мл) и 2 М HCl (2 мл). Органический раствор сушили (MgSO4) и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (64,4 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: -0.03 (2Н, m), 0.40 (2H, m), 0.70 (1H, m), 1.32 (2H, q, J 7 Гц), 2.23 (3Н, s), 3.68 (2H, t, J 7 Гц), 4.82 (2H, s), 6.96 (1H, d, J 9 Гц), 7.14 (1H, dm, J 8 Гц), 7.24 (1H, m), 7.32 (1H, m) 7.42-7.50 (2H, m), 7.62-7.68 (3Н, m), 7.74 (2H, d, J 8,5 Гц), CO2Н не наблюдался.
LC/MS: m/z 531,9 [М-Н]-, Rt 4,3 мин.
Пример 17. {4-[(Бутил{4-[4-(трифторметил)фенил]пиримидин-2-ил}-амино)метил]-2-метилфенокси}уксусная кислота
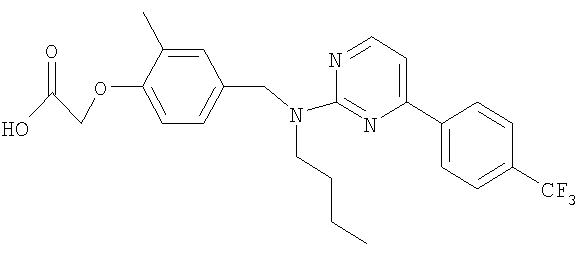
К раствору этил-{4-[(бутил{4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]-2-метилфенокси}ацетата (100 мг; 0,2 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) добавляли 2 М гидроксид натрия (2 мл). После перемешивания в течение 1 ч при комнатной температуре растворитель удаляли под вакуумом, остаток разбавляли водой (10 мл) и подкисляли 2 М HCl, затем экстрагировали в CH2Cl2 (2×40 мл). Органический раствор разделяли с помощью гидрофобной фритты и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде твердого вещества кремовой окраски (85 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.95 (3Н, t, J 7 Гц), 1.42 (2Н, m), 1.68 (2H, m), 2.26 (3Н, s), 3.74 (2H, m), 4.65 (2H, s), 4.96 (2H, s), 6.68 (1H, d, J 8,5 Гц), 7.05-7.15 (3Н, m), 7.75 (2H, d, J 8 Гц), 8.15 (2H, d, J 8 Гц), 8.50 (1H, br s), CO2H не наблюдался.
LC/MS, m/z 474,0 [M+H]+, Rt 4,3 мин.
Пример 18. [4-({Бутил[4-(4-хлорфенил)пиримидин-2-ил]амино}-метил)-2-метилфенокси]уксусная кислота

К раствору этил-[4-({бутил[4-(4-хлорфенил)пиримидин-2-ил]амино}-метил)-2-метилфенокси]ацетата (178 мг; 0,38 ммоль) в THF (3 мл) и МеОН (3 мл) добавляли NaOH (2 M, 2 мл) и полученную смесь перемешивали в течение 1,5 ч. Смесь упаривали и подкисляли 2 M HCl, экстрагировали CH2Cl2 (10 мл), EtOAc (10 мл). Водный слой фильтровали, отфильтрованные твердые вещества и экстракты в растворителе объединяли и растворитель удаляли под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (161 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 0.99 (3Н, t, J 7 Гц), 1.38-1.48 (2H, m), 1.63-1.75 (2H, m), 2.26 (3Н, s), 3.6-3.8 (2H, широкий s), 4.7 (2H, s), 4.91-5.09 (2H, широкий s), 6.81 (1H, d, J 8,5 Гц), 7.09-7.2 (2H, m), 7.50 (1H, d, J 6 Гц), 7.61 (2H, d, J 8,5 Гц), 8.24 (2H, d, J 8,5 Гц), 8.35 (1H, d, J 6 Гц), CO2Н не наблюдался.
LC/MS: m/z 440 [M+H]+, Rt 4,35 мин.
Пример 19. {4-[((2-Метоксиэтил){4-[4-(трифторметил)фенил]-пиримидин-2-ил}амино)метил]-2-метилфенокси}уксусная кислота
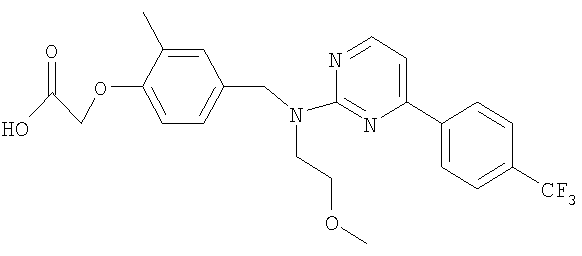
К раствору этил-{4-[((2-метоксиэтил){4-[4-(трифторметил)фенил]-пиримидин-2-ил}амино)метил]-2-метилфенокси}ацетата (80 мг; 0,16 ммоль) в метаноле (1,5 мл) и тетрагидрофуране (1,5 мл) добавляли 2 М гидроксид натрия (1,5 мл). Полученную смесь перемешивали при комнатной температуре в течение 1 ч. Растворители удаляли под вакуумом и остаток разбавляли водой (10 мл), подкисляли 2 М HCl и экстрагировали этилацетатом (2×30 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде твердого вещества кремовой окраски (72 мг).
1H ЯМР (400 МГц; CDCl3) δ: 2.26 (3Н, s), 3.36 (3H, s), 3.70 (2H, m), 3.93 (2H, m), 4.65 (2H, s), 5.04 (2H, br s), 6.68 (1H, d, J 8 Гц), 7.04-7.14 (3Н, m), 7.74 (2H, d, J 8 Гц), 8.14 (2H, d, J 8 Гц), 8.48 (1H, J 5 Гц), (CO2Н не наблюдался).
LC/MS: m/z 476,3 [М+H]+, Rt 3,9 мин.
Пример 20. (4-{[[4-(4-Хлорфенил)пиримидин-2-ил](2-метоксиэтил)-амино]метил}-2-метилфенокси)уксусная кислота

Получали из этил-(4-{[[4-(4-хлорфенил)пиримидин-2-ил](2-метоксиэтил)-амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 18.
1H ЯМР (400 МГц; MeOH-d4) δ: 2.21 (3Н, s), 3.32 (3Н, s), 3.62 (2H, t, J 5,5 Гц), 3.83 (2H, t, J 5,5 Гц), 4.64 (2H, s), 4.95 (2H, s), 6.70 (1H, d, J 8 Гц), 7.00-7.10 (2H, m), 7.18 (1H, d, J 5,5 Гц), 7.48 (2H, d, J 8,5 Гц), 8.10 (2H, d, J 8,5 Гц), 8.35 (1H, d, J 5,5 Гц), СО2Н не наблюдался.
LC/MS: m/z 442 [M+H]+, Rt 3,89 мин.
Пример 21. {2-Метил-4-[(пропил{4-[4-(трифторметил)фенил]-пиримидин-2-ил}амино)метил]фенокси}уксусная кислота
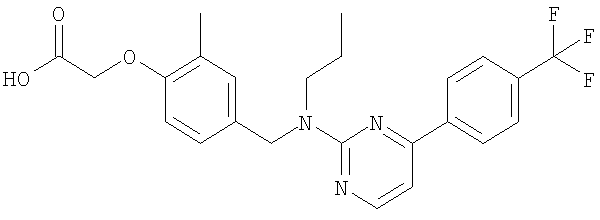
Получали из этил-{2-метил-4-[(пропил{4-[4-(трифторметил)фенил]-пиримидин-2-ил}амино)метил]фенокси}ацетата, используя методику, описанную для Примера 18.
1H ЯМР (400 МГц; MeOH-d4) δ: 0.96 (3Н, m), 1.7 (2H, m), 2.25 (3H, s), 3.63 (2H, m), 4.68 (2H, s), 4.9 (2H, s), 6.75 (1H, dm, J 8 Гц), 7.02-7.24 (3Н, m), 7.73-7.84 (2H, m), 8.21-8.34 (2H, m), 8.38-8.47 (1H, m), CO2H не наблюдался.
LC/MS: m/z 460 [М+Н]+, Rt 4,25 мин.
Пример 22. {4-[(Бутил{6-[4-(трифторметил)фенил]пиразин-2-ил}-амино)метил]-2-метилфенокси}уксусная кислота

К раствору этил-{4-[(бутил{6-[4-(трифторметил)фенил]пиразин-2-ил}-амино)метил]-2-метилфенокси}ацетата (150 мг; 0,3 ммоль) в метаноле (4 мл) и тетрагидрофуране (4 мл) добавляли 2 M NaOH (2 мл). После перемешивания в течение 1 ч при комнатной температуре растворитель удаляли под вакуумом и остаток подкисляли 2 M HCl, затем экстрагировали в этилацетат (2×50 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде твердого вещества ярко-желтой окраски (115 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.96 (3Н, t, J 7,5 Гц), 1.40 (2H, m), 1.66 (2H, m), 2.26 (3Н, s), 3.61 (2H, t, J 7,5 Гц), 4.66 (2H, s), 4.73 (2H, s), 6.68 (1H, d, J 8,5 Гц), 6.99 (1H, dm, J 8,5 Гц), 7.05 (1H, s), 7.70 (2H, d, J 8 Гц), 7.91 (1H, s), 8.09 (2H, d, J 8 Гц), 8.29 (1H, s), CO2H не наблюдался.
LC/MS: m/z 473,9 [M+H]+, Rt 4,4 мин.
Пример 23. 4-({Бутил[6-(4-метилфенил)пиразин-2-ил]амино}метил)-2-метилфенокси]уксусная кислота

К раствору этил-[4-({бутил[6-(4-метилфенил)пиразин-2-ил]амино}метил)-2-метилфенокси]ацетата (39 мг; 0,087 ммоль) в метаноле (1 мл) и тетрагидрофуране (1 мл) добавляли 2 М гидроксид натрия (1 мл). После встряхивания в течение 1 ч при комнатной температуре растворитель выпаривали и остаток затем подкисляли 2 М HCl и экстрагировали CH2Cl2 (2×10 мл). Органический раствор разделяли с помощью гидрофобной фритты и растворители удалили под вакуумом с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (34 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 0.98 (3Н, t, J 7 Гц), 1.43 (2Н, m), 1.69 (2H, m), 2.24 (3Н, s), 2.41 (3Н, s), 3.72 (2H, t, J 7,5 Гц), 4.66 (2H, s), 4.84 (2H, s), 6.76 (1H, d, J 8,5 Гц), 7.08 (1Н, dm, J 8,5 Гц), 7.13 (1Н, s), 7.34 (2H, d, J 8 Гц), 7.92 (1Н, s), 7.98 (2H, d, J 8 Гц), 8.29 (1Н, s), CO2H не наблюдался.
LC/MS: m/z 420,2 [M+H]+, Rt 4,2 мин.
Пример 24. {4-[{(2-Метоксиэтил){6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]-2-метилфенокси}уксусная кислота

К раствору этил-{4-[((2-метоксиэтил){6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]-2-метилфенокси}ацетата (160 мг; 0,32 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) добавляли 2 М гидроксид натрия (2 мл). Полученную смесь перемешивали при комнатной температуре в течение 1 ч. Растворители удаляли под вакуумом и остаток разбавляли водой (10 мл), подкисляли 2 М HCl и экстрагировали этилацетатом (2×30 мл). Органический раствор сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (140 мг).
1H ЯМР (400 МГц; CDCl3) δ: 2.26 (3Н, s), 3.34 (3Н, s), 3.66 (2H, t, J 5,5 Гц), 3.86 (2H, t, J 5,5 Гц), 4.67 (2H, s), 4.82 (2H, s), 6.68 (1Н, d, J 8,5 Гц), 7.00 (1Н, dd, J 8,5, 2 Гц), 7.06 (1Н, m), 7.72 (2H, d, J 8 Гц), 7.99 (1Н, s), 8.07 (2H, d, J 8 Гц), 8.30 (1Н, s), CO2H не наблюдался.
LC/MS: m/z 476,3 [M+H]+, Rt 3,8 мин.
Пример 25. (4-{[Бутил(2,4′-диметил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)уксусная кислота (Способ А)
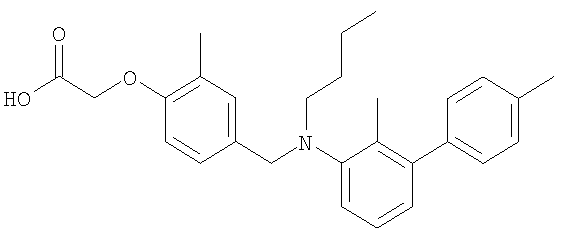
Смесь этил-(4-{[(3-бром-2-метилфенил)(бутил)амино]метил}-2-метилфенокси)ацетата (90 мг; 0,2 ммоль), 4-метилфенилбороновой кислоты (32 мг; 0,23 ммоль), тетракис(трифенилфосфин)палладия(0) (5 мг; 4,32 ммоль), карбоната натрия (55 мг; 0,52 ммоль) в 1,2-диметоксиэтане (10 мл) и воде (5 мл) нагревали при 90°С в течение 18 ч в атмосфере азота. Реакционную смесь охлаждали и растворители удаляли под вакуумом. Очищали посредством SPE (C18, 10 г), элюируя 20 миллилитрами каждого из следующих растворителей: 5%-ный, 10%-ный, 30%-ный, 50%-ный ацетонитрил/вода, ацетонитрил, МеОН. В результате дополнительной очистки посредством SPE (аминопропил, 10 г), элюируя CH2Cl2 (40 мл), CHCl3 (20 мл), эфиром (20 мл), EtOAc (40 мл), МеОН (20 мл), 10%-ным NH3/МеОН (40 мл), получали указанное в заголовке соединение в виде бесцветного масла (50 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.67 (3Н, t, J 7 Гц), 1.07 (2Н, m), 1.25 (2H, m), 1.91 (3Н, s), 2.18 (3Н, s), 2.33 (3Н, s), 2.72 (2H, m), 3.76 (2H, s), 4.09 (2H, s), 6.48 (1H, d, J 8 Гц), 6.79-6.90 (4Н, m), 7.02 (1H, t, J 7,5 Гц), 7.12 (4Н, s), CO2H не наблюдался.
LC/MS: m/z 432 [M+H]+, Rt 4,33 мин.
Пример 25. (4-{[Бутил(2,4′-диметил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)уксусная кислота (Способ Б)
Этил-(4-{[бутил(2,4′-диметил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)ацетат (12,7 г; 27,6 ммоль) растворяли в THF (210 мл) и добавляли воду (70 мл), затем 2 М водный NaOH (61 мл; 122 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч и затем упаривали с получением белой суспензии. Суспензию разбавляли водой (100 мл) и доводили до рН 7 2 М водной HCl. EtOAc (200 мл) водный слои разделяли и экстрагировали ETOAc (2×100 мл). Объединенные органические экстракты промывали соляным раствором (200 мл), сушили (Na2SO4) и концентрировали под вакуумом с получением указанного в заголовке соединения в виде белой пены (11,1 г).
1H ЯМР (400 МГц; DMSO-d6) δ: 0.79 (3Н, t, J 7 Гц), 1.20 (2Н, m), 1.36 (2H, m), 2.14 (3Н, s), 2.21 (3Н, s), 2.34 (3Н, s), 2.87 (2H, m), 3.92 (2H, s), 4.64 (2H, s), 6.72 (1H, d, J 8 Гц), 6.87 (1Н, d, J 7,5 Гц), 7.03-7.17 (4Н, m), 7.22 (4H, s), 12.95 (1H, s, широкий).
Пример 26. {4-{[Бутил(4′-фтор-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)уксусная кислота

Получали из этил-(4-{[(3-бром-2-метилфенил)(бутил)амино]метил}-2-метилфенокси)ацетата и 4-фторфенилбороновой кислоты, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 0.73 (3Н, t, J 7,5 Гц), 1.13 (2H, m), 1.3 (2H, m), 2.01 (3Н, s), 2.16 (3Н, s), 2.78 (2H, t, J 7,5 Гц), 3.84 (2H, s), 4.17 (2H, s), 6.50 (1H, d, J 8 Гц), 6.85-7.10 (7Н, m), 7.18 (2H, m), СО2Н не наблюдался.
LC/MS: m/z 436 [М+Н]+, Rt 4,25 мин.
Пример 27. (4-{[Бутил(4′-циано-2-метил-1,1′-бифенил-3-ил)амино]-метил}-2-метилфенокси)уксусная кислота

Получали из этил-(4-{[(3-бром-2-метилфенил)(бутил)амино]метил}-2-метилфенокси)ацетата и 4-цианофенилбороновой кислоты, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 0.75 (3Н, t, J 7,5 Гц), 1.14 (2Н, m), 1.31 (2H, m), 2.03 (3Н, s), 2.15 (3Н, s), 2.8 (2H, t, J 7 Гц), 3.86 (2H, s), 4.17 (2H, s), 6.51 (1H, d, J 8,5 Гц), 6.84-6.89 (2H, m), 6.93 (1H, s), 7.02 (1H, d, J 8 Гц), 7.11 (1H, t, J 8 Гц), 7.33 (2H, d, J 8,5 Гц), 7.61 (2H, d, J 8,5 Гц), СО2Н не наблюдался.
LC/MS: m/z 443 [М+Н]+, Rt 4,13 мин.
Пример 28. (4-{[Бутил(4′-метокси-2-метил-1,1′-бифенил-3-ил)амино]-метил}-2-метилфенокси)уксусная кислота

Получали из этил-(4-{[(3-бром-2-метилфенил)(бутил)амино]метил}-2-метилфенокси)ацетата и 4-метоксифенилбороновой кислоты, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 0.71 (3Н, t, J 7,5 Гц), 1.11 (2H, m), 1.29 (2H, m), 1.99 (3Н, s), 2.2 (3Н, s), 2.76 (2H, t, J 7,5 Гц), 3.78 (3Н, s), 3.82 (2H, s), 4.14 (2H, s), 6.49 (1H, d, J 8,5 Гц), 6.82-6.95 (6Н, m), 7.05 (1H, t, J 7,5 Гц), 7.17 (2H, d, J 8,5 Гц), CO2Н не наблюдался.
LC/MS: m/z 448 [М+H]+, Rt 4,01 мин.
Пример 29. (4-{[Бутил(4′-хлор-2-метил-1,1′-бифенил-3-ил)амино]-метил}-2-метилфенокси)уксусная кислота

Получали из этил-(4-{[(3-бром-2-метилфенил)(бутил)амино]метил}-2-метилфенокси)ацетата и 4-хлорфенилбороновой кислоты, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 0.71 (ЗН, t, J 7,5 Гц), 1.10 (2Н, m), 1.28 (2H, m), 1.96 (3Н, s), 2.15 (3Н, s), 2.76 (2H, t, J 7 Гц), 3.8 (2H, s), 4.1 (2H, s), 6.47 (1H, d, J 8,5 Гц), 6.8-6.96 (4Н, m), 7.05 (1H, t, J 7,5 Гц), 7.14 (2H, d, J 8,5 Гц), 7.28 (2H, d, J 8,5 Гц), СО2Н не наблюдался.
LC/MS: m/z 452 [M+H]+, Rt 4,48 мин.
Пример 30. (4-{[(4′-Хлор-2-метил-1,1′-бифенил-3-ил)(2-метоксиэтил)-амино]метил}-2-метилфенокси)уксусная кислота
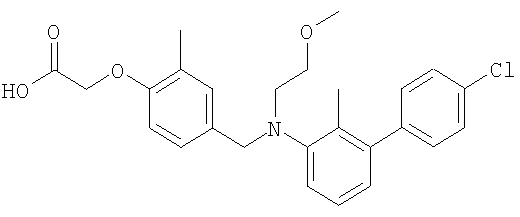
Получали из 4-хлорфенилбороновой кислоты и этил-(4-{[(3-бром-2-метилфенил)(2-метоксиэтил)амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 2.03 (3Н, s), 2.16 (3Н, s), 3.01 (2H, t, J 6 Гц), 3.11 (3Н, s), 3.25 (2H, t, J 6 Гц), 3.92 (2H, s), 4.18 (2H, s) 6.53 (1H, d, J 8,5 Гц), 6.84-6.96 (3Н, m), 7.0-7.1 (2H, m), 7.15 (2H, d, J 8,5 Гц), 7.29 (2H, d, J 8,5 Гц), CO2Н не наблюдался.
LC/MS: m/z 454 [М+Н]+, Rt 4,06 мин.
Пример 31. (4-{[(2,4′-Диметил-1,1′-бифенил-3-ил)(2-метоксиэтил)-амино]метил}-2-метилфенокси)уксусная кислота
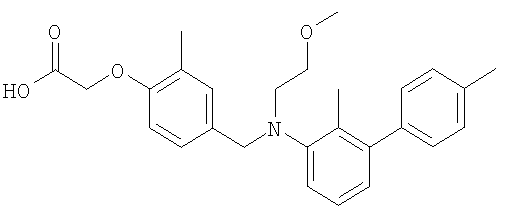
Получали из 4-метилфенилбороновой кислоты и этил-(4-{[(3-бром-2-метилфенил)(2-метоксиэтил)амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 2.10 (3Н, s), 2.22 (3Н, s), 2.36 (3Н, s), 3.04 (2H, t, J 6 Гц), 3.14 (3Н, s), 3.29 (2H, t, J 6 Гц), 3.96 (2H, s), 4.27 (2H, s), 6.60 (1H, m), 6.9-7.14 (5H, m), 7.16 (4Н, s), CO2H не наблюдался.
LC/MS: m/z 434 [M+H]+, Rt 3,94 мин.
Пример 32. (4-{[(2-Метоксиэтил)(4′-метокси-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)уксусная кислота

Получали из 4-метоксифенилбороновой кислоты и этил-(4-{[(3-бром-2-метилфенил)(2-метоксиэтил)амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 2.04 (3Н, s), 2.2 (3H, s), 3.01 (2H, t, J 6 Гц), 3.1 (3Н, s), 3.25 (2H, t, J 6 Гц), 3.79 (3Н, s), 3.92 (2H, s), 4.19 (2H, s), 6.56 (1H, d, J 8,5 Гц), 6.86-7.09 (7Н, m), 7.165 (2H, J 8,5 Гц), CO2H не наблюдался.
LC/MS: m/z 450 [M+H]+, Rt 3,68 мин.
Пример 33. 2-Метил-4-{[[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил](пропил)амино]метил}фенокси)уксусная кислота

Получали из 4-(трифторметил)фенилбороновой кислоты и этил-(4-{[(3-бром-2-метилфенил)(пропил)амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 0.7 (3Н, t, J 7 Гц), 1.34 (2H, m), 2.04 (3Н, s), 2.16 (3Н, s), 2.76 (2H, t, J 7 Гц), 3.86 (2H, s), 4.2 (2H, s), 6.52 (1H, d, J 8,5 Гц), 6.83-7.1 (5Н, m), 7.33 (2H, d, J 8 Гц), 7.57 (2H, d, J 8 Гц), СО2Н не наблюдался.
LC/MS: m/z 472 [М+Н]+, Rt 4,35 мин.
Пример 34. (4-{[(4′-Хлор-2-метил-1,1′-бифенил-3-ил)(пропил)амино]-метил}-2-метилфенокси)уксусная кислота

Получали из 4-хлорфенилбороновой кислоты и этил-(4-{[(3-бром-2-метилфенил)(пропил)амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 25.
1Н ЯМР (400 МГц; CDCl3) δ: 0.71 (3Н, t, J 7 Гц), 1.34 (2Н, m), 2.04 (3H, s), 2.17 (3H, s), 2.75 (2H, t, J 7 Гц), 3.85 (2Н, s), 4.21 (2H, s), 6.5 (1H, d, J 8,5 Гц), 6.83-7.0 (4Н, m), 7.07 (1H, t, J 8 Гц), 7.16 (2H, d, J 8,5 Гц), 7.29 (2H, d, J 8,5 Гц), CO2H не наблюдался.
LC/MS: m/z 438 [M+H]+, Rt 4,39 мин.
Пример 35. (4-{[(2,4′-Диметил-1,1′-бифенил-3-ил)(пропил)амино]-метил}-2-метилфенокси)уксусная кислота
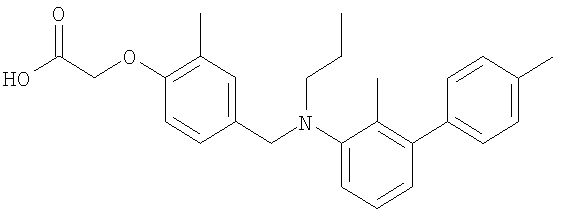
Получали из 4-метилфенилбороновой кислоты и этил-(4-{[(3-бром-2-метилфенил)(пропил)амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 0.7 (3Н, t, J 7 Гц), 1.33 (2H, m), 2.03 (3Н, s), 2.21 (3Н, s), 2.35 (3Н, s), 2.74 (2H, t, J 7 Гц), 3.85 (2H, s), 4.19 (2H, s), 6.51 (1H, d, J 8,5 Гц), 6.85-6.97 (4Н, m), 7.06 (1H, t, J 8 Гц), 7.14 (4Н, s), CO2H не наблюдался.
LC/MS: m/z 418 [M+H]+, Rt 4,15 мин.
Пример 36. (4-{[(4′-Фтор-2-метил-1,1′-бифенил-3-ил)(пропил)амино]-метил}-2-метилфенокси)уксусная кислота
Получали из 4-фторфенилбороновой кислоты и этил-(4-{[(3-бром-2-метилфенил)(пропил)амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 0.69 (3H, t, J 7 Гц), 1.31 (2Н, m), 2.01 (3H, s), 2.16 (3H, s), 2.74 (2Н, J 7 Гц), 3.83 (2Н, s), 4.16 (2Н, s), 6.49 (1H, d, J 8 Гц), 6.83-7.09 (7Н, m), 7.17 (2Н, m), CO2H не наблюдался.
LC/MS: m/z 422 [M+H]+, Rt 4,08 мин.
Пример 37. (4-{[(4′-Циано-2-метил-1,1′-бифенил-3-ил)(пропил)амино]-метил}-2-метилфенокси)уксусная кислота
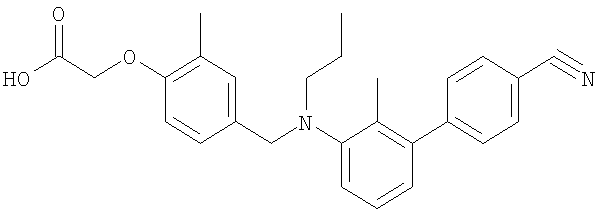
Получали из 4-цианофенилбороновой кислоты и этил-(4-{[(3-бром-2-метилфенил)(пропил)амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 0.8 (3H, t, J 7 Гц), 1.43 (2Н, m), 2.22 (6H, s), 2.86 (2Н, t, J 7 Гц), 3.99 (2Н, s), 4.57 (2Н, s), 6.63 (1H, d, J 8 Гц), 6.905 (1H, d, J 7,5 Гц), 7.02-7.22 (4Н, m), 7.41 (2Н, d, J 8 Гц), 7.68 (2Н, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 429 [М+Н]+, Rt 4,01 мин.
Пример 38. (4-{[(4′-Метокси-2-метил-1,1′-бифенил-3-ил)(пропил)-амино]метил}-2-метилфенокси)уксусная кислота
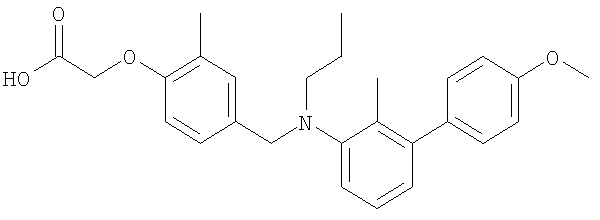
Получали из 4-метоксифенилбороновой кислоты и этил-(4-{[(3-бром-2-метилфенил)(пропил)амино]метил}-2-метилфенокси)ацетата, используя методику, описанную для Примера 25 (Способ А).
1H ЯМР (400 МГц; CDCl3) δ: 0.7 (3H, t, J 7 Гц), 1.33 (2Н, m), 2.04 (3H, s), 2.22 (3H, s), 2.74 (2Н, t, J 7 Гц), 3.79 (3H, s), 3.85 (2Н, s), 4.21 (2Н, s), 6.52 (1H, d, J 8 Гц), 6.84-6.98 (6H, m), 7.06 (1H, t, J 8 Гц), 7.18 (2Н, J 8,5 Гц), CO2H не наблюдался.
LC/MS: m/z 434 [M+H]+, Rt 3,86 мин.
Пример 39. {4-[(Бутил{5-метил-6-[4-(трифторметил)фенил]-пиримидин-4-ил}амино)метил]-2-метилфенокси}уксусная кислота
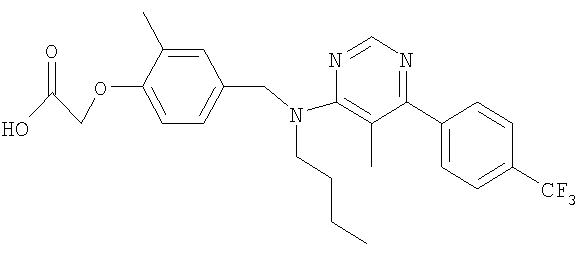
К раствору этил-{4-[(бутил{5-метил-6-[4-(трифторметил)фенил]-пиримидин-4-ил}амино)метил]-2-метилфенокси}ацетата (190 мг; 0,37 ммоль) в метаноле (4 мл) и тетрагидрофуране (4 мл) добавляли 2 М гидроксид натрия (4 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Растворители удаляли под вакуумом и остаток разбавляли водой (20 мл), подкисляли 2 М HCl и экстрагировали этилацетатом (50 мл). Органический раствор промывали соляным раствором (20 мл), сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде белой пены (180 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.94 (3Н, t, J 7,5 Гц), 1.34 (2Н, m), 1.70 (2H, m), 2.17 (3Н, s), 2.26 (3Н, s), 3.60 (2H, t, J 7,5 Гц), 4.63 (2H, s), 4.83 (2H, s), 6.68 (1H, d, J 8 Гц), 6.96 (1Н, d, J 8 Гц), 7.02 (1Н, s), 7.69 (2H, d, J 8 Гц), 7.75 (2H, d, J 8 Гц), 8.71 (1 H, s), CO2H не наблюдался.
LC/MS: m/z 488,1 [M+H]+, Rt 3,6 мин.
Пример 40. [4-({Бутил[6-(4-метоксифенил)-5-метилпиримидин-4-ил]-амино}метил)-2-метилфенокси]уксусная кислота

К раствору этил-[4-({бутил[6-(4-метоксифенил)-5-метилпиримидин-4-ил]амино}метил)-2-метилфенокси]ацетата (62 мг; 0,13 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) добавляли 2 М гидроксид натрия (1 мл). После встряхивания в течение 1 ч при комнатной температуре растворитель удаляли. Остаток затем подкисляли 0,5 М водной HCl (20 мл), после чего экстрагировали в EtOAc (2×25 мл). Органический раствор промывали соляным раствором, сушили (MgSO4) и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (58 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.95 (3Н, t, J 7,5 Гц), 1.33 (2Н, m), 1.69 (2H, m), 2.23 (3Н, s), 2.20 (3H, s), 3.03 (2H, t, J 7 Гц), 3.84 (3H, s), 4.66 (2H, s), 4.88 (2H, s), 6.70 (1H, d, J 8 Гц), 6.90-7.05 (4H, m), 7.58 (2H, d, J 8,5 Гц), 8.69 (1Н, s), CO2H не наблюдался.
LC/MS: m/z 450,2 [M+H]+, Rt 2,9 мин.
Пример 41. [4-({Бутил[5-метил-6-(4-метилфенил)пиримидин-4-ил]-амино}метил)-2-метилфенокси]уксусная кислота
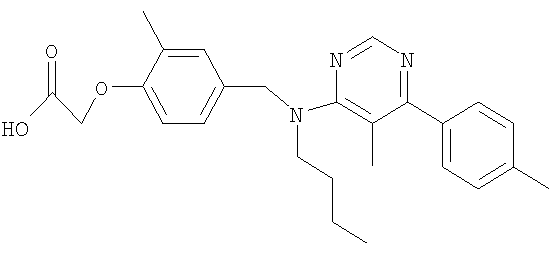
Получали из этил-[4-({бутил[5-метил-6-(4-метилфенил)пиримидин-4-ил]амино}метил)-2-метилфенокси]ацетата (58 мг; 0,126 ммоль), используя методику, описанную для Примера 40.
1H ЯМР (400 МГц; CDCl3) δ: 0.95 (3Н, t, J 7,5 Гц), 1.34 (2H, m), 1.70 (2H, m), 2.22 (3Н, s), 2.26 (3Н, s), 2.40 (3Н, s), 3.64 (2H, m), 4.65 (2H, s), 4.89 (2H, s), 6.71 (1H, d, J 8 Гц), 6.95 (1H, d, J 8 Гц), 7.00 (1H, s), 7.32 (2H, d, J 8 Гц), 7.48 (2H, d, J 8 Гц), 8.70 (1H, s), CO2H не наблюдался.
LC/MS: m/z 434,3 [М+Н]+, Rt 3,1 мин.
Пример 42. [4-({Бутил[6-(4-хлорфенил)-5-метилпиримидин-4-ил]-амино}метил)-2-метилфенокси]уксусная кислота

Получали из этил-[4-({бутил[6-(4-хлорфенил)-5-метилпиримидин-4-ил]амино}метил)-2-метилфенокси]ацетата (98 мг; 0,203 ммоль), используя методику, описанную для Примера 40.
1H ЯМР (400 МГц; CDCl3) δ: 0.95 (3Н, t, J 7 Гц), 1.34 (2Н, m), 1.70 (2H, m), 2.18 (3Н, s), 2.26 (3Н, s), 3.62 (2H, t, J 7,5 Гц), 4.64 (2H, s), 4.85 (2H, s), 6.69 (1H, d, J 8 Гц), 6.95 (1H, d), 7.01 (1H, s), 7.45-7.54 (4H, m), 8.69 (1H, s), CO2H не наолюдался.
LC/MS: m/z 454,2 [M+H]+, Rt 3,5 мин.
Пример 43. [4-({Бутил[6-(4-хлорфенил)-2-пиразин-2-ил]амино}метил)-2-метилфенокси]уксусная кислота
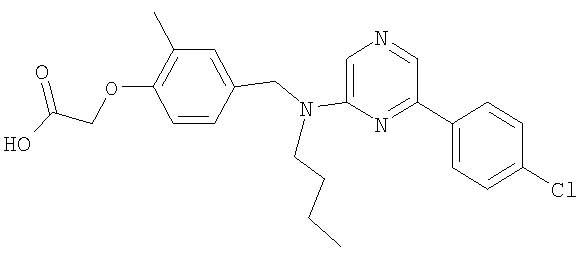
К раствору этил-[4-({бутил[6-(4-хлорфенил)пиразин-2-ил]амино}метил)-2-метилфенокси]ацетата (78 мг; 0,17 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) добавляли 2 M NaOH (1 мл). После перемешивания в течение 1 ч при комнатной температуре растворитель удаляли под вакуумом и остаток подкисляли 1 M HCl (5 мл), затем экстрагировали в этилацетат (2×25 мл). Органический раствор сушили и растворители удаляли под вакуумом с получением указанного в заголовке соединения в виде ярко-желтого твердого вещества (70 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 0.98 (3Н, t, J 7,5 Гц), 1.43 (2H, m), 1.70 (2H, m), 2.23 (3Н, s), 3.74 (2H, t, J 6 Гц), 4.66 (2H, s), 4.88 (2H, s), 6.77 (1H, d, J 8,5 Гц), 7.09 (1H, dd, J 8,5, 1,5 Гц), 7.14 (1H, s), 7.55 (2H, d, J 8,5 Гц), 8.06-8.11 (3Н, m), 8.39 (1H, s), CO2H не наблюдался.
LC/MS: m/z 440 [M+H]+, Rt 4,3 мин.
Пример 44. [4-({[6-(4-Хлорфенил)пиразин-2-ил][2-(метилокси)этил]-амино}метил)-2-метилфенокси]уксусная кислота
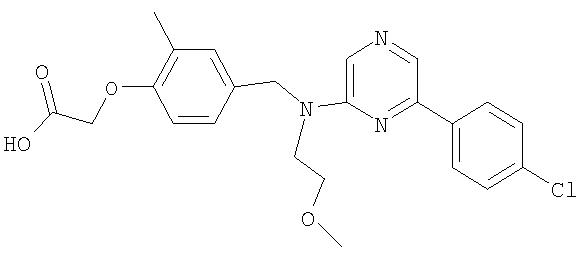
Использовали этил-[4-({[6-(4-хлорфенил)пиразин-2-ил][2-(метилокси)-этил]амино}метил)-2-метилфенокси]ацетат (81 мг; 0,17 ммоль) и методику синтеза, описанную для Примера 43, с получением указанного в заголовке соединения в виде ярко-желтого твердого вещества (77 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 2.19 (3Н, s), 3.30 (3H, s), 3.65 (2H, t, J 5 Гц), 3.91 (2H, t, J 5 Гц), 4.63 (2H, s), 4.92 (2H, s), 6.73 (1H, d, J 8,5 Гц), 7.07 (1Н, d, J 8,5 Гц), 7.11 (1H, s), 7.51 (2Н, d, J 8,5 Гц), 8.03 (2H, d, J 8 Гц), 8.14 (1H, s), 8.36 (1H, s), CO2H не наблюдался.
LC/MS: m/z 442,1 [M+H]+, Rt 3,7 мин.
Пример 45. {2-Метил-4-[(пропил{6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]фенокси}уксусная кислота

Использовали этил-{2-метил-4-[(пропил{6-[4-(трифторметил)фенил]-пиразин-2-ил}амино)метил]фенокси}ацетат (56 мг; 0,12 ммоль) и методику синтеза, описанную для Примера 43, с получением указанного в заголовке соединения в виде ярко-желтого твердого вещества (50 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 1.01 (3Н, t, J 7,5 Гц), 1.76 (2H, m), 2.23 (3Н, s), 3.72 (2H, t, J 8 Гц), 4.66 (2H, s), 4.90 (2H, s), 6.78 (1H, d, J 8,5 Гц), 7.10 (1H, d, J 8,5 Гц), 7.15 (1H, s), 7.85 (2H, d, J 8 Гц), 8.15 (1H, s), 8.28 (2H, d, J 8 Гц), 8.46 (1H, s), CO2H не наблюдался.
LC/MS: m/z 460,1 [M+H]+, Rt 4,1 мин.
Пример 46. (2-Метил-4-{[{5-метил-6-[4-(трифторметил)фенил]-пиримидин-4-ил}(пропил)амино]метил}фенокси)уксусная кислота

Использовали этил-(2-метил-4-{[{5-метил-6-[4-(трифторметил)фенил]-пиримидин-4-ил}(пропил)амино]метил}фенокси)ацетат (159 мг; 0,32 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде белого твердого вещества (145 мг).
1H ЯМР (400 МГц; DMSO-d6) δ: 0.87 (3Н, t, J 7 Гц), 1.71 (2Н, m), 2.15 (3Н, s), 2.18 (3Н, s), 3.51 (2Н, m), 4.69 (2Н, s), 4.87 (2Н, s), 6.80 (1H, d, J 8,5 Гц), 7.08 (1H, d, J 8,5 Гц), 7.13 (1H, s), 7.88 (2Н, d, J 8 Гц), 7.97 (2H, d, J 8 Гц), 8.77 (1H, s), CO2H не наблюдался.
LC/MS: m/z 474,2 [М+Н]+, Rt 3,5 мин.
Пример 47. (4-{[[6-(4-Хлорфенил)-5-метилпиримидин-4-ил](пропил)-амино]метил}-2-метилфенокси)уксусная кислота
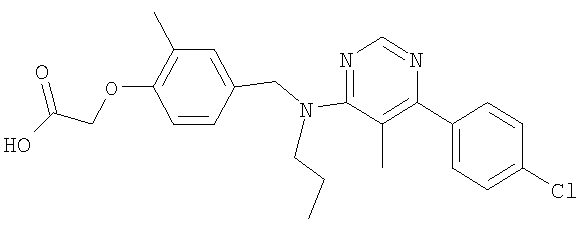
Использовали этил-(4-{[[6-(4-хлорфенил)-5-метилпиримидин-4-ил]-(пропил)амино]метил}-2-метилфенокси)ацетат (116 мг; 0,25 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде белого твердого вещества (56 мг).
1H ЯМР (400 МГц; DMSO-d6) δ: 0.86 (3Н, t, J 7,5 Гц), 1.69 (2Н, m), 2.16 (3Н, s), 2.18 (3Н, s), 3.52 (2Н, t, J 7,5 Гц), 4.69 (2Н, s), 4.87 (2Н, s), 6.79 (1H, d, J 8,5 Гц), 7.08 (1H, d, J 8,5 Гц), 7.12 (1H, s), 7.68 (4H, m), 8.75 (1H, s), CO2H не наблюдался.
LC/MS: m/z 440,1 [М+Н]+, Rt 3,3 мин.
Пример 48. (2-Метил-4-{[[5-метил-6-(4-метилфенил)пиримидин-4-ил](пропил)амино]метил}фенокси)уксусная кислота

Использовали этил-(2-метил-4-{[[5-метил-6-(4-метилфенил)пиримидин-4-ил](пропил)амино]метил}фенокси)ацетат (50 мг; 0,11 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде белого твердого вещества (20 мг).
1H ЯМР (400 МГц; DMSO-d6) δ: 0.87 (3Н, t, J 7,5 Гц), 1.71 (2Н, m), 2.17 (3Н, s), 2.18 (3Н, s), 2.41 (3Н, s), 3.55 (2Н, t, J 7,5 Гц), 4.69 (2Н, s), 4.90 (2Н, s), 6.80 (1H, d, J 8,5 Гц), 7.08 (1Н, dd, J 8,5, 1,5 Гц), 7.13 (1Н, s), 7.41 (2Н, d, J 8 Гц), 7.55 (2Н, d, J 8 Гц), 8.76 (1Н, s), CO2H не наблюдался.
LC/MS: m/z 420,2 [M+H]+, Rt 2,9 мин.
Пример 49. (2-Метил-4-{[{5-метил-6-[4-(метилокси)фенил]пиримидин-4-ил}(пропил)амино]метил}фенокси)уксусная кислота
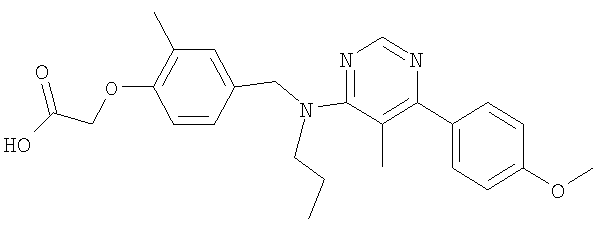
Использовали этил-(2-метил-4-{[{5-метил-6-[4-(метилокси)фенил]-пиримидин-4-ил}(пропил)амино]метил}фенокси)ацетат (60 мг; 0,13 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде белого твердого вещества (42 мг).
1H ЯМР (400 МГц; DMSO-d6) δ: 0.87 (3Н, t, J 7,5 Гц), 1.72 (2Н, m), 2.18 (3Н, s), 2.19 (3Н, s), 3.55 (2Н, t, J 7,5 Гц), 3.86 (3Н, s), 4.69 (2Н, s), 4.91 (2Н, s), 6.80 (1Н, d, J 8 Гц), 7.09 (1Н, dd, J 8, 1,5 Гц), 7.12-7.19 (3Н, m), 7.62 (2Н, d, J 8,5 Гц), 8.75 (1Н, s), CO2H не наблюдался.
LC/MS: m/z 436,2 [M+H]+, Rt 2,8 мин.
Пример 50. {4-[(Бутил{6-[4-(трифторметил)фенил]пиразин-2-ил}-амино)метил]-2-этилфенокси}уксусная кислота
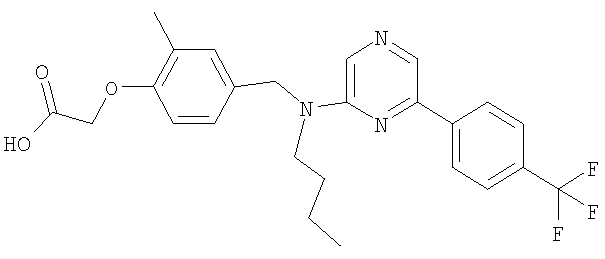
Использовали этил-{4-[(бутил{6-[4-(трифторметил)фенил]пиразин-2-ил}-амино)метил]-2-этилфенокси}ацетат (50 мг; 0,10 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде ярко-желтого твердого вещества (45 мг).
1H ЯМР (400 МГц; DMSO-d6) δ: 0.91 (3Н, t, J 7.5 Гц), 1.09 (3Н, t, J 7,5 Гц), 1.36 (2Н, m), 1.60 (2H, m), 2.56 (2H, q, J 7,5 Гц), 3.62 (2Н, t, J 7,5 Гц), 4.65 (2Н, s), 4.77 (2H, s), 6.76 (1H, d, J 8 Гц), 7.03 (1Н, dd, J 8, 2 Гц), 7.13 (1Н, s), 7.85 (2H, d, J 8 Гц), 8.13 (1Н, s), 8.26 (2H, d, J 8 Гц), 8.46 (1Н, s), CO2H не наблюдался.
LC/MS: m/z 488,3 [M+H]+, Rt 4,4 мин.
Пример 51. {2-Этил-4-[(2-метилоксиэтил){6-[4-(трифторметил)фенил]-пиразин-2-ил}амино)метил]фенокси}уксусная кислота
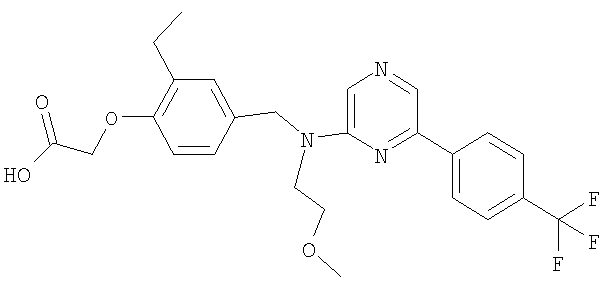
Использовали этил-{2-этил-4-[(2-метилоксиэтил){6-[4-(трифторметил)-фенил]пиразин-2-ил}амино)метил]фенокси}ацетат (48 мг; 0,09 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде ярко-желтого твердого вещества (40 мг).
1H ЯМР (400 МГц; DMSO-d6) δ: 1.09 (3Н, t, J 7,5 Гц), 2.56 (2H, q, J 7,5 Гц), 3.26 (3Н, s), 3.60 (2H, t, J 5,5 Гц), 3.84 (2H, t, J 5,5 Гц), 4.65 (2H, s), 4.80 (2H, s), 6.76 (1Н, d, J 8,5 Гц), 7.03 (1Н, dd, J 8,5, 2 Гц), 7.13 (1Н, d, J 2 Гц), 7.84 (2H, d, J 8,5 Гц), 8.16 (1Н, s), 8.25 (2H, d, J 8,5 Гц), 8.47 (1Н, s), CO2H не наблюдался.
LC/MS: m/z 490,3 [М+Н]+, Rt 3,9 мин.
Пример 52. [4-({Бутил[5-метил-6-(4-метилфенил)пиримидин-4-ил]-амино}метил)-2-этилфенокси]уксусная кислота

Использовали этил-[4-({бутил[5-метил-6-(4-метилфенил)пиримидин-4-ил]-амино}метил)-2-этилфенокси]ацетат (46 мг; 0,10 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде бесцветного масла (39 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.95 (3Н, t, J 7,5 Гц), 1.19 (3Н, t, J 7,5 Гц), 1.34 (2Н, m), 1.70 (2H, m), 2.21 (3Н, s), 2.40 (3Н, s), 2.69 (2H, q, J 7,5 Гц), 3.65 (2Н, t, J 7,5 Гц), 4.66 (2H, s), 4.89 (2H, s), 6.71 (1H, d, J 8 Гц), 6.95 (1Н, d, J 8 Гц), 7.03 (1Н, s), 7.32 (2H, d, J 8 Гц), 7.46 (2H, d, J 8 Гц), 8.73 (1Н, s), CO2H не наблюдался.
LC/MS: m/z 448,3 [M+Н]+, Rt 3,2 мин.
Пример 53. [4-({Бутил[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]-амино}сульфонил)-2-метилфенокси]уксусная кислота

Использовали этил-[4-({бутил[2-метил-4′-(трифторметил}-1,1′-бифенил-3-ил]амино}сульфонил)-2-метилфенокси]ацетат (15 мг; 0,03 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде бесцветного масла (13 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.87 (3Н, t, J 7 Гц), 1.22-1.55 (4Н. m), 2.26 (3Н, s), 2.33 (3Н, s), 3.20 (1Н, m), 3.74 (1Н, m), 4.80 (2H, s), 6.68 (1Н, dd, J 7,5, 1,5 Гц), 6.79 (1Н, m), 7.10-7.21 (2H, m), 7.46 (2H, d, J 8 Гц), 7.54 (2H, m), 7.69 (2H, d, J 8 Гц), CO2Н не наблюдался.
LC/MS: m/z 536,3 [M+H]+, Rt 4,3 мин.
Пример 54. [2-Метил-4-({(пропилсульфонил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]уксусная кислота
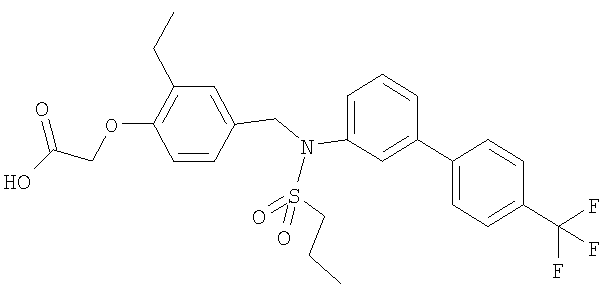
Использовали этил-[2-этил-4-({(пропилсульфонил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)фенокси]ацетат (65 мг; 0,12 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде желтого твердого вещества (56 мг).
1H ЯМР (400 МГц; CDCl3) δ: 1.06 (3Н, t, J 7,5 Гц), 1.12 (3Н, t, J 7,5 Гц), 1.93 (2Н, m), 2.63 (2H, q, J 7,5 Гц), 3.05 (2Н, m), 4.65 (2H, s), 4.84 (2H, s), 6.63 (1H, d, J 8,5 Гц), 7.05 (1H, d, J 8,5 Гц), 7.07 (1H, s), 7.29 (1H, m), 7.40-7.50 (3Н, m), 7.56 (2H, d, J 8 Гц), 7.68 (2H, d, J 8 Гц), СО2Н не наблюдался.
LC/MS: m/z 534,2 [M-H]-, Rt 3,9 мин.
Пример 55. [4-({Бутил[6-(4-хлорфенил)-5-метилпиримидин-4-ил]-амино}метил)-2-этилфенокси]уксусная кислота

Использовали этил-[4-({бутил[6-(4-хлорфенил)-5-метилпиримидин-4-ил]-амино}метил)-2-этилфенокси]ацетат (64 мг; 0,13 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде бесцветного масла (61 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.96 (3Н, t, J 5,5 Гц), 1.19 (3Н, t, J 7,5 Гц), 1.36 (2H, m), 1.72 (2H, m), 2.17 (3Н, s), 2.69 (2H, q, J 7,5 Гц), 3.67 (2H, t, J 8 Гц), 4.67 (2H, s), 4.90 (2H, s), 6.71 (1H, d, J 8,5 Гц), 6.93 (1H, d, J 8,5 Гц), 7.02 (1H, s), 7.45-7.55 (4H, m), 8.69 (1H, s), CO2H не наблюдался.
LC/MS: m/z 468,3 [M+H]+, Rt 3,7 мин.
Пример 56. {4-[(Бутил{5-метил-6-[4-(трифторметил)фенил]-пиримидин-4-ил}амино)метил]-2-этилфенокси}уксусная кислота

Использовали этил-{4-[(бутил{5-метил-6-[4-(трифторметил)фенил]-пиримидин-4-ил}амино)метил]-2-этилфенокси}ацетат (82 мг; 0,15 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде бесцветного масла (78 мг).
1H ЯМР (400 МГц; CDCl3) δ: 0.96 (3Н, t, J 7,5 Гц), 1.20 (3Н, t, J 7,5 Гц), 1.36 (2Н, m), 1.73 (2H, m), 2.18 (3Н, s), 2.69 (2H, квартет, J 7,5 Гц), 3.67 (2Н, t, J 7 Гц), 4.66 (2H, s), 4.90 (2H, s), 6.70 (1H, d, J 8 Гц), 6.94 (1Н, d, J 8 Гц), 7.04 (1Н, s), 7.70 (2H, d, J 8 Гц), 7.77 (2H, d, J 8 Гц), 8.72 (1Н, s), CO2H не наблюдался.
LC/MS: m/z 502,3 [M+H]+, Rt 3,9 мин.
Пример 57. {2-Этил-4-[([2-(метилокси)этил]{4-[4-(трифторметил)-фенил]пиримидин-2-ил}амино)метил]фенокси}уксусная кислота
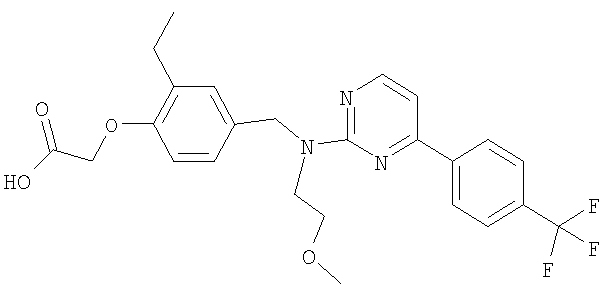
Использовали этил-{2-этил-4-[([2-(метилокси)этил]{4-[4-(трифторметил)-фенил]пиримидин-2-ил}амино)метил]фенокси}ацетат (52 мг; 0,10 ммоль) и методику, описанную для Примера 40, с получением указанного в заголовке соединения в виде бесцветной смолы (47 мг).
1H ЯМР (400 МГц; DMSO-d6) δ: 1.09 (3Н, br s), 2.58 (2H, m), 3.25 (3Н, s), 3.55 (2H, brs), 3.79 (2H, brs), 4.65 (2H, s), 4.88 (2H, s), 6.75 (1Н, d, J 8,5 Гц), 6.95-7.2 (2H, m), 7.30 (1Н, d, J 5 Гц), 7.88 (2H, J 6,5 Гц), 8.31 (2H, d, J 6,5 Гц), 8.51 (1Н, d, J 5 Гц), CO2Н не наблюдался.
LC/MS: m/z 490,3 [M+H]+, Rt 4,1 мин.
Пример 58. {2-Метил-4-[(2-пропен-1-ил{6-[4-(трифторметил)фенил]-пиридин-2-ил}амино)метил]фенокси}уксусная кислота
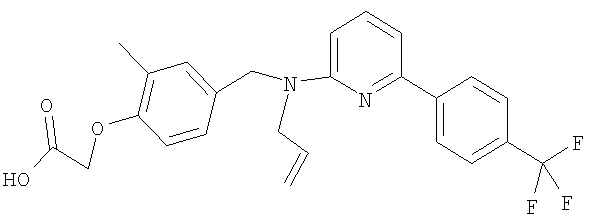
Раствор N-2-пропен-1-ил-6-[4-(трифторметил)фенил]-2-пиридинамина (0,062 г; 0,223 ммоль) в безводном DMF (3 мл) охлаждали до 0°С в атмосфере азота и обрабатывали NaH (0,012 г; 0,29 ммоль). К этой смеси добавляли этил-[4-(бромметил)-2-метилфенокси)ацетат (0,07 г; 0,245 ммоль), реакционную смесь оставляли достигать комнатной температуры и перемешивали в течение ночи. Реакционную смесь гасили метанолом и концентрировали под вакуумом. Остаток распределяли между соляным раствором/хлороформом, органический экстракт отделяли и концентрировали. Остаток затем обрабатывали дополнительным количеством гидрида натрия и этил-[4-(бромметил)-2-метилфенокси)ацетата и перемешивали в течение 70 ч. В результате обработки, как описано ранее, и очистки посредством SPE (диоксид кремния, 10 г), элюируя смесью циклогексан/этилацетат (9:1), получали бесцветное масло 75%-ной чистоты (0,098 г). Этот вещество обрабатывали 2 М NaOH (5 мл) в THF (5 мл). Через 3 ч перемешивания при комнатной температуре реакционную смесь подкисляли 2 М HCl и экстрагировали в этилацетат. Органический раствор промывали соляным раствором, сушили (MgSO4) и упаривали. В результате очистки аутопрепаративной HPLC получали указанное в заголовке соединение в виде белого твердого вещества (14 мг).
1H ЯМР (400 МГц; MeOH-d4) δ: 2.25 (3Н, s), 4.30 (2H, d, J 5,5 Гц), 4.68 (2Н, s), 4.82 (2H, s), 5.22 (1H, s), 5.26 (1H, d, J 4,5 Гц), 5.90-5.99 (1Н, m), 6.78 (2H, d, J 8,5 Гц), 7.10 (1H, m), 7.14 (1H, brs), 7.20 (1H, d, J 7 Гц), 7.69 (1H, t, J 8,5 Гц), 7.75 (2H, d, J 8,5 Гц), 8.09 (2H, d, J 8,5 Гц), CO2Н не наблюдался.
LC/MS: m/z 457,2 [M+H]+, Rt 4,3 мин.
Следующие промежуточные соединения и лиганды получали для описанных ниже анализов связывания и трансфекции:
(1) 2-{2-метил-4-[({4-метил-2-[4-(трифторметил)фенил]-1,3-тиазол-5-ил}-метил)сульфанил]фенокси}уксусная кислота.
Это соединение использовали в качестве PPAR-дельта эталона в описанных ниже трансфекционных анализах и получали способом, описанным в WO 200100603-A1;
(2) 2-метил-2-[4-{[(4-метил-2-[4-трифторметилфенил]-тиазол-5-илкарбонил)амино]метил}-фенокси]пропионовая кислота.
Это соединение использовали в качестве PPAR-альфа эталона в описанном ниже трансфекционном анализе и получали способом, описанным в WO 200140207-A1;
(3) 5-{4-[2-(метилпиридин-2-иламино)-этокси]-бензил}-тиазолидин-2,4-дион.
Это соединение использовали в качестве PPAR-гамма эталона в описанном ниже трансфекционном анализе и получали способом, описанным в J. Med. Chem. 1994, 37(23), 3977.
Анализ связывания
Соединения тестировали на их способность связываться с hPPAR-гамма, hPPAR-альфа или hPPAR-дельта, используя анализ сцинтилляционной близости (Scintillation Proximity Assay (SPA)). PPAR лигандсвязывающий домен (LBD) экспрессировали в Е. coli в виде поли-His-меченых слитых белков и был очищен. LBD затем метили биотином и иммобилизовали на модифицированных стрептавидином гранулах для анализа сцинтилляционной близости. Гранулы затем инкубировали с постоянным количеством соответствующего радиолиганда (3H-BRL 49653 для PPAR-гамма и меченый GW 2433 (смотри в Brown, Р. J. et al., Chem. Biol., 4, 909-918 (1997), структуру и синтез этого лиганда) для PPAR-альфа и для PPAR-дельта) и переменными концентрациями текстируемого соединения, и после установления равновесия связанную на гранулах радиоактивность измеряли с помощью сцинтилляционного счетчика. Для каждого тестируемого соединения строили графики зависимости СРМ (количества импульсов в минуту) связанного радиолиганда от концентрации лиганда, оценивали наблюдаемые Ki величины путем экстраполяции данных нелинейным методом наименьших квадратов, предполагая простое конкурентное связывание. Подробности данного анализа описаны в других источниках (смотри Blanchard, S. G. et al., Development of a Scintillation Proximity Assay for Peroxisome Proliferator-Activated Receptor gamma Ligand Binding Domain. Anal. Biochem., 257, 112-119 (1998)).
Трансфекционный анализ
Проводили скрининг соединений на функциональную эффективность в анализах со временной трансфекции в CV-1-клетках по их способности активировать подтипы PPAR (анализ трансактивации). Использовали созданную заранее химерную рецепторную систему, чтобы обеспечить сравнение относительной транскрипционной активности рецепторных подтипов на одном и том же гене-мишени и чтобы предотвратить усложнение интерпретации результатов из-за эндогенной рецепторной активации. Смотри, например, Lehmanh, J.M.; Moore, L.В.; Smith-Oliver, Т.A.; Wilkison, W.Q.; Willson, Т.M.; Kliewer, S.A., An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPARgamma), J. Biol. Chem., 270, 12953-6 (1995). Каждый из лигандсвязывающих доменов мышиных и человеческих PPAR-альфа, PPAR-гамма и PPAR-дельта каждый был слит с ДНК-связывающим доменом транскрипционного фактора дрожжей GAL4. CV-1-клетки временно трансфицировали векторами экспрессии соответствующей PPAR-химеры вместе с репортерной конструкцией, содержащей пять копий GAL4 ДНК-связывающего сайта, управляющего экспрессией секреторной плацентарной щелочной фосфатазы (SPAP) и бета-галактозидазы. Через 16 ч среду заменяли на DME-среду с добавлением 10%-ной делипидированной фетальной телячьей сывороткой и тестируемого соединения в соответствующей концентрации. Через еще 24 ч клеточные экстракты готовили и анализировали на активность щелочной фосфатазы и бета-галактозидазы. Активность щелочной фосфатазы корректировали с учетом эффективности трансфекции, используя активность бета-галактозидазы в качестве внутреннего стандарта (смотри, например, Kliewer, S.A., et. al. Cell 83, 813-819 (1995)). В качестве положительного контроля в hPPAR-гамма анализе использовали розиглитазон (BRL 49653). Положительным контролем в hPPAR-альфа анализах была 2-метил-2-[4-{[(4-метил-2-[4-трифторметилфенил]-тиазол-5-илкарбонил)амино]метил}-фенокси]пропионовая кислота. Положительным контролем в PPAR-дельта анализах была 2-{2-метил-4-[({4-метил-2-(трифторметил)фенил]-1,3-тиазол-5-ил}метил)сульфанил]фенокси}уксусная кислота.
Все приведенные выше примеры кислот демонстрировали по меньшей мере 50%-ную активацию PPARδ по сравнению с положительным контролем в концентрациях 10-7 М или менее.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ФЕНОКСИПИРИДИНИЛАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ PDE4-ОПОСРЕДОВАННЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2009 |
|
RU2509077C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2827641C1 |
| ЗАМЕЩЕННЫЕ ФЕНОКСИУКСУСНЫЕ КИСЛОТЫ, ОБЛАДАЮЩИЕ МОДУЛИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРОВ CRTh2 | 2004 |
|
RU2372330C2 |
| 2, 5-ДИОКСОИМИДАЗОЛИДИН-4-ИЛ-МЕТИЛКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2003 |
|
RU2326117C2 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ ПРЕПАРАТАХ | 2014 |
|
RU2694254C1 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2006 |
|
RU2408586C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| ПРОИЗВОДНЫЕ 3-ЗАМЕЩЕННОГО 4-АРИЛХИНОЛИН-2-ОНА В КАЧЕСТВЕ МОДУЛЯТОРОВ КАЛИЕВЫХ КАНАЛОВ | 1999 |
|
RU2240998C2 |
Соединение формулы (I) или его фармацевтически приемлемая соль, сольват или гидролизуемый сложный эфир, где R1 и R2 независимо представляют собой водород или С1-3алкил; Х представляет собой О; один из R3 и R4 независимо представляет собой водород, а другой представляет собой C1-3алкил; X1 представляет собой CH2 или SO2; R5 представляет собой -C1-6алкил (возможно замещенный С1-6алкокси или C1-6алкилтио), -С2-6алкенил, -С0-6алкилфенил (где фенил возможно замещен одним или более чем одним CF3, галогеном, C1-3алкилом, С1-3алкокси), -COC1-6алкил, SO2С1-6алкил; R6 представляет собой фенил или 6-членную гетероарильную группу, содержащую 1 или 2 атома N, причем фенил или гетероарильная группа возможно замещена 1, 2 или 3 группировками, выбранными из группы, состоящей из C1-6алкила, фенила (возможно замещенного одной или более чем одной группой, выбранной из галогена, CF3, С1-3алкила, OC1-3алкила, CN). Соединение формулы (I) предназначено для применения в качестве активатора рецепторов человека, активируемых пероксисомными пролифераторами (hPPAR). Технический результат - производные пропионовых кислот, активирующие рецепторы человека, активируемые пероксисомными пролифераторами (hPPAR). 2 н. и 12 з.п. ф-лы.


где R1 и R2 независимо представляют собой водород или C1-3алкил;
X представляет собой О;
один из R3 и R4 независимо представляет собой водород, а другой представляет собой С1-3алкил;
X1 представляет собой СН2 или SO2;
R5 представляет собой -С1-6алкил (возможно замещенный С1-6алкокси или C1-6алкилтио), -С2-6алкенил, -С0-6алкилфенил (где фенил возможно замещен одним или более чем одним CF3, галогеном, C1-3алкилом, C1-3алкокси), -COC1-6алкил, SO2С1-6алкил;
R6 представляет собой фенил или 6-членную гетероарильную группу, содержащую 1 или 2 атома N, причем фенил или гетероарильная группа возможно замещены 1, 2 или 3 группировками, выбранными из группы, состоящей из C1-6алкила, фенила (возможно замещенного одной или более чем одной группой, выбранной из галогена, CF3, C1-3алкила, OC1-3алкила, CN).
2-метил-2-{2-метил-4-[([4-(трифторметил)бензил]{6-[4-(трифторметил)фенил]-пиридин-2-ил}амино)метил]фенокси}пропановой кислоты;
2-{4-[(бутил{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)метил]-2-метилфенокси}-2-метилпропановой кислоты;
{4-[(бутил{6-[4-(трифторметил)фенил]пиридин-2-ил}амино)метил]-2-метилфенокси} уксусной кислоты;
[4-({бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]-уксусной кислоты;
[4-({(2-метоксиэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусной кислоты;
[2-метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-фенокси]уксусной кислоты;
[4-({(2-циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусной кислоты;
[2-метил-4-({пропил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-фенокси]уксусной кислоты;
[2-метил-4-({[2-(метилтио)этил][4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-метил)фенокси]уксусной кислоты;
[4-({бутил[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусной кислоты;
[4-({(2-метоксиэтил)[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-метил)-2-метилфенокси]уксусной кислоты;
[4-({бутирил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}метил)-2-метилфенокси]уксусной кислоты;
[2-метил-4-({(пропилсульфонил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-метил)фенокси]уксусной кислоты;
[4-({бутил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-2-метилфенокси]уксусной кислоты;
[2-метил-4-({пентил[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}сульфонил)-фенокси]уксусной кислоты;
[4-({(2-циклопропилэтил)[4′-(трифторметил)-1,1′-бифенил-3-ил]амино}-сульфонил)-2-метилфенокси]уксусной кислоты;
{4-[(бутил{4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]-2-метилфенокси}уксусной кислоты;
[4-({бутил[4-(4-хлорфенил)пиримидин-2-ил]амино}метил)-2-метилфенокси]-уксусной кислоты;
{4-[((2-метоксиэтил){4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]-2-метилфенокси}уксусной кислоты;
(4-{[[4-(4-хлорфенил)пиримидин-2-ил](2-метоксиэтил)амино]метил}-2-метилфенокси)уксусной кислоты;
{2-метил-4-[(пропил{4-[4-(трифторметил)фенил]пиримидин-2-ил}амино)метил]-фенокси}уксусной кислоты;
{4-[(бутил{6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]-2-метилфенокси}уксусной кислоты;
[4-({бутил[6-(4-метилфенил)пиразин-2-ил]амино}метил)-2-метилфенокси]-уксусной кислоты;
{4-[((2-метоксиэтил){6-[4-(трифторметил)фенил]пиразин-2-ил}амино)метил]-2-метилфенокси}уксусной кислоты;
(4-{[бутил(2,4′-диметил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)-уксусной кислоты;
(4-{[бутил(4′-фтор-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)-уксусной кислоты;
(4-{[бутил(4′-циано-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)-уксусной кислоты;
(4-{[бутил(4-метокси-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)уксусной кислоты;
(4-{[бутил(4′-хлор-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)-уксусной кислоты;
(4-{[(4′-хлор-2-метил-1,1′-бифенил-3-ил)(2-метоксиэтил)амино]метил}-2-метилфенокси)уксусной кислоты;
(4-{[(2,4′-диметил-1,1′-бифенил-3-ил)(2-метоксиэтил)амино]метил}-2-метилфенокси)уксусной кислоты;
(4-{[(2-метоксиэтил)(4′-метокси-2-метил-1,1′-бифенил-3-ил)амино]метил}-2-метилфенокси)уксусной кислоты;
(2-метил-4-{[[2-метил-4′-(трифторметил)-1,1′-бифенил-3-ил](пропил)амино]-метил}фенокси)уксусной кислоты;
(4-{[(4′-хлор-2-метил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)уксусной кислоты;
(4-{[(2,4′-диметил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)-уксусной кислоты;
(4-{[(4′-фтор-2-метил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)уксусной кислоты;
(4-{[(4′-циано-2-метил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)уксусной кислоты;
(4-[[(4′-метокси-2-метил-1,1′-бифенил-3-ил)(пропил)амино]метил}-2-метилфенокси)уксусной кислоты;
{4-[(бутил{5-метил-6-[4-(трифторметил)фенил]пиримидин-4-ил} амино)метил]-2-метилфенокси}уксусной кислоты;
[4-({бутил[6-(4-метоксифенил)-5-метилпиримидин-4-ил]амино}метил)-2-метилфенокси]уксусной кислоты;
[4-({бутил[5-метил-6-(4-метилфенил)пиримидин-4-ил]амино}метил)-2-метилфенокси]уксусной кислоты;
[4-({бутил[6-(4-хлорфенил)-5-метилпиримидин-4-ил]амино}метил)-2-метилфенокси]уксусной кислоты.
| ПИПЕТКА ДЛЯ ВЗЯТИЯ ПРОБ ЖИДКОСТИ С РЕГУЛИРУЕМЫМ СБРАСЫВАТЕЛЕМ | 2001 |
|
RU2273518C2 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Смачиватель для мерсеризации | 1982 |
|
SU1067109A1 |
| WO 00/23407 A, 27.04.2000 | |||
| US 3912756 A, 14.10.1975 | |||
| ПРОИЗВОДНЫЕ ПИРИДИНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ ВЕЩЕСТВА | 1994 |
|
RU2142944C1 |

