ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка претендует на преимущества предварительной заявки на патент США No. 60/361759, поданной 4 марта 2002 года. Полное содержание предварительной заявки включено в описание в качестве ссылки.
ЗАЯВЛЕНИЕ ОБ УЧАСТИИ В РАЗРАБОТКАХ ПРАВИТЕЛЬСТВА
Настоящее изобретение выполнено полностью или частично при правительственной поддержке в рамках гранта номер 1R21 CA 096228-01, выданного National Cancer Institute. Правительство может иметь определенные права на настоящее изобретение.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способам селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток и/или ингибированию гистондезацетилаз (HDAC) путем введения фармацевтических композиций, содержащих ингибиторы HDAC. Пероральные формы фармацевтических композиций имеют благоприятные фармакокинетические профили по таким позициям, как высокая биодоступность, и неожиданно позволяют достигать высокого содержания активных соединений в крови в течение длительного периода времени.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Приведенные в заявке ссылки на различные публикации обозначены арабскими цифрами в скобках. Полное цитирование указанных публикаций приведено в конце описания непосредственно перед формулой изобретения. Полное содержание приведенных публикаций включено в настоящую заявку в качестве ссылки для более полной характеристики состояния в той области техники, к которой относится изобретение.
Злокачественная опухоль представляет собой заболевание, при котором популяция клеток становится в различной степени невосприимчивой к контрольным механизмам, которые в норме регулируют пролиферацию и дифференцировку клеток. В течение многих лет имелись две принципиальные стратегии химиотерапевтического лечения злокачественной опухоли: а) блокирование пролиферации гормон-зависимых опухолевых клеток путем создания препятствий для образования или периферического действия половых гормонов и b) лизис злокачественных клеток путем непосредственного воздействия на них цитотоксических веществ, которые повреждают популяции как опухолевых, так и нормальных клеток.
Терапия злокачественных опухолей также ставила целью индукцию конечной дифференцировки опухолевых клеток (1). Дифференцировка на моделях клеточных культур была показана в случае воздействия на клетки множества стимулов, включая циклический АМФ и ретиноевую кислоту (2,3), акларубицин и другие антрациклины (4).
Несмотря на многие достижения в области онкологии, большинство солидных опухолей все еще не поддается лечению на поздних стадиях заболевания. В большинстве случаев используют цитотоксическую терапию, однако она зачастую вызывает значительное болезненное состояние у пациента без значимого клинического успеха. Исходя из этого проводятся поиски менее токсичных и более специфичных средств для лечения и подавления развернутых злокачественных новообразований.
Накоплено множество доказательств того, что неопластическая трансформация необязательно разрушает потенциал злокачественных клеток к дифференцировке (1,5,6). Имеется немало примеров опухолевых клеток, которые не реагируют на нормальные регуляторы пролиферации, так что создается впечатление, что в них блокирована экспрессия программы дифференцировки, и тем не менее в таких клетках можно индуцировать дифференцировку и остановить репликацию. Множество средств, включая некоторые относительно простые полярные соединения (5,7-9), производные витамина D и ретиноевой кислоты (10-12), стероидные гормоны (13), ростовые факторы (6,14), протеазы (15,16), опухолевые промоторы (17,18) и ингибиторы синтеза ДНК или РНК (4, 19-24), могут индуцировать в различных трансформированных клеточных линиях и первичных эксплантатах человеческих опухолей экспрессию более дифференцированных характеристик.
В более ранних исследованиях была идентифицирована серия полярных соединений, которые являются эффективными индукторами дифференцировки для большого числа трансформированных клеточных линий (8,9). Среди них наиболее эффективным индуктором является гибридное полярное/неполярное соединение бисацетамид N,N'-гексаметилена (БАГМ (HMBA)) (9). Использование указанного полярного/неполярного соединения для индукции эритродной дифференцировки в эритролейкозных клетках мышей (MELC) с подавлением онкогенности привело к созданию модели, применимой для изучения опосредованной индуктором дифференцировки трансформированных клеток (5,7-9). БАГМ-индуцированная конечная дифференцировка эритроидов в MELC представляет собой многоступенчатый процесс. При добавлении БАГМ в культуре к MELC (745А-DS19) наблюдается латентный период в течение 10-12 часов перед началом конечной дифференцировки. Детерминация определяется как способность клеток экспрессировать конечную дифференцировку, несмотря на удаление индуктора (25). При продолжении воздействия БАГМ наблюдается прогрессирующий рекрутинг клеток к дифференцировке. Настоящие авторы показали, что клеточные линии MELC, приобретшие резистентность к относительно низким содержанием винкристина, становятся заметно более чувствительными к индуцирующему действию БАГМ и могут быть индуцированы к дифференцировке, при наличии небольшого латентного периода или вообще в его отсутствие (26).
БАГМ способен индуцировать соответствующие дифференцировке фенотипические изменения в самых различных клеточных линиях (5). Характеристики эффекта, индуцируемого лекарственным соединением, широко изучались с использованием системы эритролейкозных клеток мышей (MELC) (5,25,27,28). Индукция MELC к дифференцировке зависит от времени и концентрации. Минимальная концентрация, необходимая для демонстрации эффекта in vitro у большинства штаммов, составляет 2-3 мМ; минимальная длительность непрерывной экспозиции, необходимой в основном для индукции дифференцировки в значительной части (>20%) клеток без продолжения воздействия лекарственного соединения, составляет примерно 36 часов.
Первичная мишень действия БАГМ неизвестна. Имеются данные, свидетельствующие о том, что протеинкиназа С участвует в механизме дифференцировки, опосредованной индуктором (29). Исследования in vitro дали основания рассматривать БАГМ в качестве средства, способствующего клеточной дифференцировке, применимого для лечения злокачественных опухолей человека (30). К настоящему времени завершен ряд клинический испытаний, проведенных с применением БАГМ на фазе I (31-36). Результаты проведенных клинических испытаний показали, что данное соединение может индуцировать терапевтический ответ у больных со злокачественной опухолью (35,36). Однако указанные клинические испытания на фазе I также показали, что потенциальная эффективность БАГМ ограничена, частично в связи с его токсичностью, зависимой от дозы, которая мешает достичь оптимальных содержаний данного соединения в крови, и в связи с необходимостью внутривенного введения больших количеств данного средства в течение длительных периодов времени.
Было показано, что множество близких к БАГМ соединений с полярными группами, разделенными неполярными связями, характеризуются, на молярной основе, такой же активностью (37) или в 100 раз большей активностью, чем БАГМ (38). Однако, что касается класса, то было показано, что симметричные димеры, такие как БАГМ и родственные соединения, являются не лучшими клеточными дифференцирующими средствами.
Неожиданно было обнаружено, что наилучшие соединения содержат две полярные концевые группы, разделенные гибкой цепью метиленовых групп, где одна или обе полярные концевые группы представляют собой крупную гидрофобную группу. Предпочтительно обе полярные концевые группы являются разными и лишь одна из них представляет собой крупную гидрофобную группу. Данные соединения, как неожиданно оказалось, обладают в тысячи раз большей активностью, чем БАГМ, и в десять раз большей активностью, чем соединения, родственные БАГМ.
Ингибиторы гистондезацетилазы, такие как субероиланилид гидроксамовой кислоты (САГК (SAHA)), принадлежат к классу соединений, которые обладают способностью индуцировать остановку роста опухолевых клеток, дифференцировку и/или апоптоз (39). Указанные соединения направлены на механизмы, придающие опухолевым клеткам способность становиться злокачественными, и, по всей видимости, не обладают токсичностью в дозах, эффективных для ингибирования опухолевого роста у животных (40). Получено несколько доказательств того, что ацетилирование и дезацетилирование гистонов представляют собой механизмы, посредством которых в клетке достигается регуляция транскрипции (41). Считается, что указанные эффекты осуществляются через изменения структуры хроматина за счет изменения афинности гистоновых белков к скрученной ДНК в нуклеосоме. В нуклеосомах было идентифицировано пять типов гистонов (обозначенных как H1, H2A, H2B, H3 и H4). Каждая нуклеосома содержит в своем ядре два представителя каждого типа гистонов, за исключением Н1, который присутствует только во внешней части нуклеосомной структуры. Считается, что когда гистоновые белки гипоацетилированы, то такие гистоны обладают большей афинностью к фосфатному скелету ДНК. Афинность способствует более тесной связи ДНК с гистоном и делает такую ДНК недоступной для регуляторных элементов и механизмов транскрипции. Регуляция уровня ацетилирования происходит через баланс активности двух ферментных комплексов: гистонацетилтрансферазы (ГАТ (НАТ)) и гистондезацетилазы (HDAC). Считается, что гипоацетилированное состояние ингибирует транскрипцию соответствующей ДНК. Такое гипоацетилированное состояние достигается за счет катализа крупными мультибелковыми комплексами, которые содержат HDAC ферменты. В частности, было показано, что HDAC катализирует удаление ацетильных групп из ядерных гистонов хроматина.
Ингибирование HDAC под действием САГК происходит, как показано в исследованиях с использованием рентгеновской кристаллографии (42), через непосредственное взаимодействие с каталитическим сайтом фермента. Считается, что достигаемое ингибирование HDAC не оказывает генерализованного эффекта на весь геном, а воздействует, скорее всего, на небольшую часть генома (43). Результаты, полученные с микрочипами ДНК при использовании линий злокачественных клеток, культивируемых с ингибитором HDAC, указывают на то, что имеется определенное (1-2%) число генов, продукты которых содержат изменения. Например, клетки, которые подвергают обработке ингибиторами HDAC в культуре, демонстрируют индукцию ингибитора циклин-зависимой киназы р21 (44). Данный белок играет важную роль в остановке клеточного цикла. Считается, что ингибиторы HDAC повышают скорость транскрипции р21 за счет распространения гиперацетилированного состояния гистона на участке р21 гена, что делает данный ген доступным для механизмов транскрипции. Гены, экспрессия которых не подвергается воздействию ингибиторов HDAC, не демонстрируют изменений в уровне ацетилирования региональных ассоциированных гистонов (45).
В ряде случаев было показано, что потеря активности ГАТ или HDAC включается в развитие злокачественного фенотипа. Например, при остром промиелоцитарном лейкозе онкопротеин, образуемый при слиянии PML и RAR-альфа, по всей видимости, подавляет транскрипцию специфичного гена путем рекрутинга HDAC (46). В такой ситуации опухолевая клетка не способна завершить дифференцировку, что ведет к избыточной пролиферации клеток в лейкозной клеточной линии.
В патентах США NoNo. 5369108, 5932616, 5700811, 6087367 и 6511990, принадлежащих авторам настоящего изобретения, описываются соединения, применимые для целей селективной индукции конечной дифференцировки опухолевых клеток, где указанные соединения имеют две полярные концевые группы, разделенные гибкой цепью метиленовых групп или ригидной фенильной группой, причем одна или обе концевые полярные группы представляют крупную гидрофобную группу. Некоторые из таких соединений содержат дополнительную крупную гидрофобную группу на том же конце молекулы, что и первая гидрофобная группа, что также повышает дифференцирующую активность как в ферментном тесте, примерно в 100 раз, так и в тесте на клеточную дифференцировку, примерно в 50 раз. Способы синтеза соединений, используемых в способах и фармацевтических композициях настоящего изобретения, подробно раскрыты в указанных выше патентах, полное содержание которых включено в настоящее описание в качестве ссылки.
В указанных выше патентах не описываются конкретные пероральные композиции ингибиторов HDAC или конкретные дозировки и режим дозирования приведенных соединений. Важно, что указанные выше патенты не описывают пероральные композиции, которые обладали бы благоприятными фармакокинетическими профилями, такими как высокая биодоступность, которая позволяет достигать высокого содержания активных соединений в крови в течение длительного периода времени.
Класс, содержащий соединения по настоящему изобретению, может рассматриваться для использования с целью селективной индукции конечной дифференцировки опухолевых клеток и в этой связи для лечения опухолей у пациентов. Таким образом, имеется острая потребность в определении подходящих дозировок, режима дозирования указанных соединений и в разработке композиций, предпочтительно пероральных композиций, которые бы позволили поддерживать стойкое терапевтическое эффективное содержание активных соединений в крови в течение длительного периода времени.
КРАТКОЕ ОПИСАНИЯ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам достижения в плазме крови средних значений концентрации ингибитора гистондезацетилазы (HDAC), способных ингибировать дезацетилирование гистонов in vivo у субъекта в течение длительного периода времени, равного по меньшей мере двум часам после введения, которые предусматривают введение указанному субъекту эффективного количества фармацевтической композиции, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Настоящее изобретение также относится к способам селективной индукции конечной дифференцировки, остановки роста клеток/или апоптоза опухолевых клеток, за счет чего достигается ингибирование пролиферации таких клеток, и к способам индукции дифференцировки опухолевых клеток за счет достижения средних значений концентрации в плазме крови ингибитора гистондезацетилазы (HDAC), способных ингибировать гистондезацетилазу in vivo у субъекта в течение длительного периода времени, равного по меньшей мере двум часам после введения, посредством введения указанному субъекту эффективного количества фармацевтической композиции, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Настоящее изобретение также относится к способам достижения в плазме крови среднего значения концентрации, равного по меньшей мере 10 нМ, субероиланилида гидроксамовой кислоты (САГК (SAHA)) in vivo у субъекта в течение длительного периода времени, равного по меньшей мере двум часам после введения, которые предусматривают введение указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Настоящее изобретение также относится к способам селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток, за счет чего достигается ингибирование пролиферации таких клеток, к способам индукции дифференцировки опухолевых клеток за счет достижения в плазме крови средних значений концентрации, равных по меньшей мере 10 нМ САГК, in vivo у субъекта в течение длительного периода времени, равного по меньшей мере двум часам после введения, которые предусматривают введение указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Настоящее изобретение относится к фармацевтическим композициям, подходящим для перорального введения, которые содержат соединение, применимое для селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток, и/или которое относится к мощному ингибитору гистондезацетилазы (HDAC). Фармацевтические композиции также содержат микрокристаллическую целлюлозу, натрийкросскармелозу и стеарат магния. Настоящее изобретение также относится к фармацевтическим композициям для перорального введения, содержащим САГК, микрокристаллическую целлюлозу, натрийкросскармелозу и стеарат магния. Пероральная биодоступность активных соединений в композициях по настоящему изобретению удивительно высокая. Кроме того, композиции неожиданно позволяют достичь высоких терапевтически эффективных содержаний активных соединений в крови в течение длительного периода времени. Настоящее изобретение также относится к безопасному режиму ежедневного дозирования данных композиций, который легко применять и соблюдать.
Как показано в настоящем описании, авторы неожиданно обнаружили, что пероральные композиции, содержащие ингибиторы HDAC, в особенности субероиланилид гидроксамовой кислоты (САГК), характеризуются в случае перорального введения очень высокой общей биодоступностью активного соединения in vivo. Кроме того, композиции позволяют достичь высокого содержания активного соединения в крови, которые сохраняются неожиданно высокими в течение длительного периода времени, например до 10-12 часов. Пероральные композиции по настоящему изобретению имеют массу преимуществ, особенно в сравнении с парентеральными композициями, поскольку они, с одной стороны, обеспечивают достижение высоких, стабильных и длительных терапевтически эффективных уровней ингибиторов HDAC в крови, а с другой стороны, могут легко применяться пациентами в обычном режиме перорального введения.
Соответственно, настоящее изобретение относится к фармацевтической композиции для перорального введения, содержащей ингибитор гистондезацетилазы (HDAC) или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, так что указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения. В предпочтительном варианте осуществления изобретения концентрация ингибитора HDAC эффективна для ингибирования HDAC в течение периода времени, равного по меньшей мере 10 часам после введения.
В предпочтительном варианте настоящее изобретение относится к фармацевтической композиции для перорального введения, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови САГК, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения. В предпочтительном варианте настоящего изобретения концентрация САГК эффективна для ингибирования HDAC в течение периода времени, равного по меньшей мере 10 часам после введения.
Композиции по настоящему изобретению используются для селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток и в этой связи способствуют лечению опухолей у пациентов.
Соответственно, настоящее изобретение также относится к способу селективной индукции конечной дифференцировки опухолевых клеток у субъекта и в этой связи к ингибированию пролиферации таких клеток у субъекта, предусматривающему стадию перорального введения субъекту эффективного количества фармацевтической композиции, содержащей ингибитор гистондезацетилазы (HDAC) или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, так что указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения.
Кроме того, настоящее изобретение также относится к способу селективной индукции, остановки роста опухолевых клеток у субъекта и достижения при этом ингибирования пролиферации таких клеток у субъекта, предусматривающему стадию перорального введения субъекту эффективного количества фармацевтической композиции, содержащей ингибитор гистондезацетилазы (HDAC) или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения.
Кроме того, настоящее изобретение также относится к способу селективной индукции апоптоза опухолевых клеток у субъекта и в этой связи к ингибированию пролиферации таких клеток у субъекта, предусматривающему стадию перорального введения субъекту эффективного количества фармацевтической композиции, содержащей ингибитор гистондезацетилазы (HDAC) или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения.
Кроме того, настоящее изобретение также относится к способу индукции дифференцировки опухолевых клеток у субъекта с опухолью, предусматривающему стадию перорального введения субъекту эффективного количества фармацевтической композиции, содержащей ингибитор гистондезацетилазы (HDAC) или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения.
Кроме того, настоящее изобретение также относится к способу ингибирования активности гистондезацетилазы у субъекта, предусматривающему стадию перорального введения субъекту эффективного количества фармацевтической композиции, содержащей ингибитор гистондезацетилазы (HDAC) или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения.
Кроме того, настоящее изобретение также относится к способу селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток у субъекта и в этой связи к ингибированию пролиферации таких клеток у субъекта, предусматривающему стадию введения субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови САГК, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения.
Кроме того, настоящее изобретение также относится к способу индукции дифференцировки опухолевых клеток у субъекта с опухолью, предусматривающему стадию введения субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови САГК, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения.
Кроме того, настоящее изобретение относится к способу ингибирования активности гистондезацетилазы, предусматривающему стадию перорального введения субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови САГК, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo в течение периода времени, равного по меньшей мере 2 часам после введения.
В предпочтительном варианте осуществления изобретения САГК или любой другой ингибитор HDAC вводят пациенту в суммарной ежедневной дозе, равной 25-4000 мг/м2. В другом предпочтительном варианте осуществления изобретения САГК или любой другой ингибитор HDAC вводят пациенту в суммарной ежедневной дозе 200 мг. САГК или любой другой ингибитор HDAC вводят пациенту в суммарной ежедневной дозе 400 мг.
В одном предпочтительном варианте осуществления изобретения композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC (например, САГК), способной ингибировать гистондезацетилазу в течение периода времени, равного по меньшей мере 2 часам после введения, причем предпочтительно указанная концентрация составляет по меньшей мере примерно 10 нМ. В еще одном предпочтительном варианте осуществления изобретения данная композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, равной по меньшей мере примерно 10 нМ, в течение периода времени, равного по меньшей мере 10 часам после введения.
В одном предпочтительном варианте осуществления настоящего изобретения композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC (например, САГК), способной селективно индуцировать конечную дифференцировку, остановку роста клеток и/или апоптоз опухолевых клеток или способной индуцировать дифференцировку опухолевых клеток в опухоли, где указанная концентрация поддерживается в течение периода времени, равного по меньшей мере 2 часам после введения, причем предпочтительно указанная концентрация составляет по меньшей мере примерно 2,5 мкМ. В еще одном предпочтительном варианте осуществления настоящего изобретения композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, равной по меньшей мере 2,5 мкМ, в течение периода времени, равного по меньшей мере 10 часам после введения.
Композиции по настоящему изобретению могут изготавливаться в любой стандартной дозированной форме (жидкой или твердой), подходящей для перорального введения, например, в виде гранулы, таблетки, таблетки, покрытой оболочкой, капсулы, желатиновой капсулы, раствора, суспензии или дисперсии. В предпочтительном варианте осуществления настоящего изобретения композиция имеет форму желатиновой капсулы.
В композициях может использоваться любой инертный наполнитель, который обычно выполняет функцию носителя или разбавителя, такой как, например, камедь, крахмал, сахар, целлюлозный материал, акрилат или их смеси. Предпочтительным разбавителем является микрокристаллическая целлюлоза. Композиции могут также содержать разрыхлитель (например, натрийкросскармелозу) и смазывающее вещество (например, стеарат магния) и, кроме того, они могут содержать одну или более добавок, выбранных из связующего вещества, буфера, ингибитора протеазы, поверхностно-активного вещества, солюбилизирующего средства, пластификатора, эмульгатора, стабилизатора, средства повышения вязкости, подсластителя, пленкообразующего средства или любого их сочетания. Кроме того, композиции по настоящему изобретению могут быть изготовлены в форме композиции с контролируемым высвобождением или с немедленным высвобождением.
Широкое множество ингибиторов HDAC пригодно для использования в композициях по настоящему изобретению. В предпочтительном варианте осуществления настоящего изобретения ингибитор HDAC представляет собой субероиланилид гидроксамидной кислоты (САГК (SAHA))
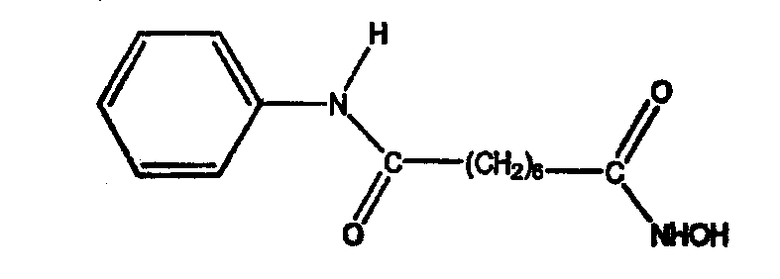
Другие неограничивающие примеры ингибиторов HDAC, которые подходят для использования в композициях по настоящему изобретению, содержат следующие соединения.
Пироксамид, описываемый структурой
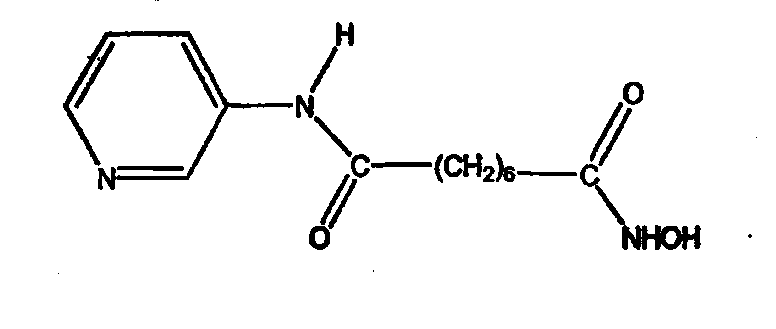
Соединение, описываемое структурой
где R3 и R4 обозначают независимо замещенную или незамещенную, разветвленную или неразветвленную алкильную, алкенильную, циклоалкильную, арильную, алкилокси, арилокси, арилалкилокси или пиридиновую группу или R3 и R4, связанные вместе, образуют пиперидиновую группу; R2 обозначает гидроксиламиногруппу и n равно целому числу от 5 до примерно 8.
Соединение, описываемое структурой
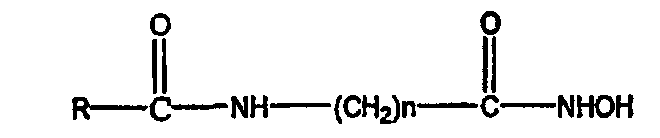
где R обозначает замещенный или незамещенный фенил, пиперидин, тиазол, 2-пиридин, 3-пиридин или 4-пиридин и n равно целому числу от 4 до примерно 8.
Соединение описываемое структурой
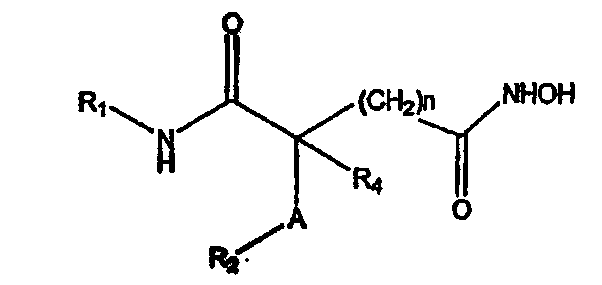
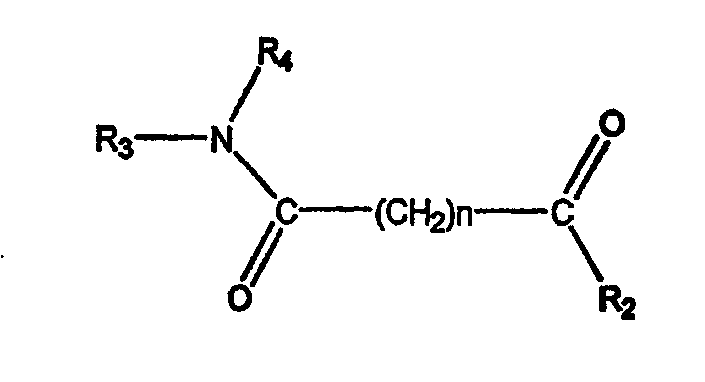
где А обозначает амидный фрагмент, R1 и R2, каждый, выбирают из замещенного или незамещенного арила (например, фенила), арилалкила (например, бензила), нафтила, пиридинамино, 9-пурин-6-амино, тиазоламино, арилокси, арилалкилокси, пиридила, хинолинила или изохинолинила; R4 обозначает водород, галоген, фенильный или циклоалкильный фрагмент и n равно целому числу от 3 до 10.
Кроме того, в соответствии с конкретными вариантами осуществления настоящего изобретения в нем предлагается фармацевтическая композиция для перорального введения, содержащая ингибитор гистондезацетилазы (HDAC) или его фармацевтически приемлемую соль или гидрат, микрокристаллическую целлюлозу в качестве носителя или разбавителя, натрийкросскармелозу в качестве разрыхлителя и стеарат магния в качестве смазывающего вещества, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, эффективной для ингибирования гистондезацетилазы in vivo в течение периода времени, равного по меньшей мере 2 часам после введения. В предпочтительном варианте осуществления настоящего изобретения ингибитор HDAC представляет собой субероиланилид гидроксамовой кислоты (САГК).
Кроме того, в соответствии с конкретными вариантами осуществления настоящего изобретения в нем предлагается фармацевтическая композиция для перорального введения, содержащая субероиланилид гидроксамовой кислоты (САГК) или его фармацевтически приемлемую соль или гидрат, микрокристаллическую целлюлозу в качестве носителя или разбавителя, натрийкросскармелозу в качестве разрыхлителя и стеарат магния в качестве смазывающего вещества, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме крови ингибитора HDAC, эффективной для ингибирования гистондезацетилазы in vivo в течение периода времени, равного по меньшей мере 2 часам после введения. В предпочтительном варианте осуществления настоящего изобретения композиция содержит 50-70 мас.% САГК или его фармацевтически приемлемой соли или гидрата, 20-40 мас.% микрокристаллической целлюлозы в качестве носителя или разбавителя, 5-15 мас.% натрийкросскармелозы в качестве разрыхлителя и 0,1-5 мас.% стеарата магния в качестве смазывающего вещества. В другом предпочтительном варианте осуществления настоящего изобретения композиция содержит примерно 50-200 мг САГК. В особенно предпочтительном варианте осуществления настоящего изобретения композиция имеет форму желатиновой капсулы.
Настоящее изобретение также относится к безопасному режиму ежедневного введения доз указанных композиций, который легко применять и соблюдать. Композиции по настоящему изобретению применимы для селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток и в этой связи способствуют лечению опухолей у пациентов.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Приведенные выше и другие цели, особенности и преимущества настоящего изобретения станут понятными из прилагаемого ниже более подробного описания предпочтительных вариантов осуществления настоящего изобретения, проиллюстрированных в прилагаемых чертежах, в которых указанные цифры относятся к тем же частям в разных планах. Приведенные чертежи необязательно относятся к полному описанию изобретения, они не акцентируют внимание на каких-то его деталях, а даны лишь для иллюстрации принципов настоящего изобретения.
Фиг. 1 представляет собой иллюстрацию вестерн-блоттинга (верхний план), показывающую количества ацетилированного гистона-4 (α-AcH4) в плазме крови у пациента после перорального или внутривенного (в/в) введения дозы САГК. В/в САГК вводят в дозе 200 мг инфузией в течение двух часов. Перорально САГК вводят однократно в виде капсул по 200 мг. Количество α-AcH4 приведено в указанные временные точки. Нижний план: окрашивание красителем кумасси-блю.
Фиг. 2 представляет собой иллюстрацию вестерн-блоттинга (верхние планы), показывающую количества ацетилированного гистона-4 (α-AcH4) в плазме крови у пациентов, имеющих солидные опухоли, после перорального или внутривенного (в/в) введения дозы САГК. В/в и пероральное введение САГК проводят по методике, проиллюстрированной на фиг. 1. Количество (α-AcH4) приведено в указанные временные точки. Эксперимент проведен с двойным повтором (фиг. 2А и фиг. 2B). Нижние планы: окрашивание красителем кумасси-блю.
Фиг. 3 представляет собой иллюстрацию вестерн-блоттинга (верхние планы), показывающую количества ацетилированного гистона-4 (α-AcH4) (фиг. 3А) и ацетилированного гистона-3 (α-AcH3) (фиг. 3B-E) в плазме крови у пациентов после перорального или внутривенного (в/в) введения дозы САГК на день 1 и день 21. В/в и пероральное введение САГК проводят по методике, проиллюстрированной на фиг. 1. Количество α-AcH4 или α-AcH3 приведено в указанные временные точки. Нижние планы: окрашивание красителем кумасси-блю.
Фиг. 4представляет собой иллюстрацию вестерн-блоттинга (верхние планы), показывающую количества ацетилированного гистона-3 (α-AcH3) в плазме крови у пациентов, имеющих солидные опухоли, после перорального или внутривенного (в/в) введения дозы САГК. В/в и пероральное введение САГК проводят по методике, проиллюстрированной на фиг. 1. Количество (α-AcH3) приведено в указанные временные точки. Нижний план: окрашивание красителем кумасси-блю.
Фиг. 5 представляет собой иллюстрацию вестерн-блоттинга (верхние планы), показывающую количества ацетилированного гистона-3 (α-AcH3) в плазме крови у пациентов после перорального или внутривенного (в/в) введения дозы САГК. В/в САГК вводят в дозе 400 мг в течение двух часов. Пероральный САГК вводят однократно в виде капсулы с дозой 400 мг. Количество α-AcH4 приведено в указанные временные точки. Эксперимент проиллюстрирован в двух вариантах (фиг. 5А и B). Нижние планы: окрашивание красителем кумасси-блю.
Фиг. 6 представляет собой иллюстрацию вестерн-блоттинга (верхний план), показывающую количества ацетилированного гистона-3 (α-AcH3) в плазме крови у пациентов, имеющих солидные опухоли, после перорального или внутривенного (в/в) введения дозы САГК. В/в и пероральное введение САГК проводят по методике, проиллюстрированной на фиг. 5. Количество α-AcH3 приведено в указанные временные точки. Нижний план: окрашивание красителем кумасси-блю.
Фиг. 7представляет собой иллюстрацию вестерн-блоттинга (верхние планы), показывающую количества ацетилированного гистона-3 (α-AcH3) в плазме крови у пациентов, имеющих солидные опухоли, после перорального или внутривенного (в/в) введения дозы САГК на день 1 и день 21. В/в и пероральное введение САГК проводят по методике, проиллюстрированной на фиг. 4. Количество α-AcH4 или α-AcH3 приведено в указанные временные точки. Эксперимент проиллюстрирован в трех вариантах (фиг. 7А-С). Нижние планы: окрашивание красителем кумасси-блю.
Фиг. 8 представляет собой иллюстрацию вестерн-блоттинга (верхние планы), показывающую количества ацетилированного гистона-3 (α-AcH3) в плазме крови пациента после перорального или внутривенного (в/в) введения дозы САГК. В/в и пероральное введение САГК проводят по методике, проиллюстрированной на фиг. 5. Количество α-AcH3 приведено в указанные временные точки. Нижние планы: окрашивание красителем кумасси-блю.
Фиг. 9А-С представляет собой графики, показывающие среднее значение концентрации в плазме крови САГК (нг/мл) в указанные временные точки после введения: Фиг. 9А - пероральная доза (200 мг и 400 мг), принимаемая натощак, на 8 день; фиг. 9B - пероральная доза, принимаемая с пищей, на 9 день; фиг. 9С - в/в доза на день 1.
Фиг. 10 иллюстрирует кажущийся период полураспада перорального САГК в дозе 200 мг и 400 мг в дни 8, 9 и 22.
Фиг. 11 иллюстрирует ППК (нг/мл/час) для пероральной дозы САГК в 200 мг и 400 мг в дни 8, 9 и 22.
Фиг. 12 иллюстрирует биодоступность САГК после перорального приема в дозе 200 мг и 400 мг в дни 8, 9 и 22.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам достижения среднего значения концентрации в плазме крови ингибитора гистондезацетилазы (HDAC), способной ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часом после введения, которые предусматривают введение указанному субъекту эффективного количества фармацевтической композиции, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Настоящее изобретение также относится к способам селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток с достижением при этом ингибирования пролиферации таких клеток и к способам индукции дифференцировки опухолевых клеток путем создания в плазме крови средней концентрации ингибитора гистондезацетилазы (HDAC), способной ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения указанному субъекту эффективного количество фармацевтической композиции, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Настоящее изобретение также относится к способам достижения среднего значения в плазме крови концентрации, равной 10 нМ, субероиланилида гидроксамовой кислоты (САГК) in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, которые предусматривают введение указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Настоящее изобретение также относится к способам селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток с достижением при этом ингибирования пролиферации таких клеток и к способам индукции дифференцировки опухолевых клеток за счет создания в плазме крови средней концентрации, равной по меньшей мере 10 нМ САГК, in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Настоящее изобретение также относится к фармацевтическим композициям, подходящим для перорального введения, которые содержат соединение, используемое для селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток и/или которое является мощным ингибитором гистондезацетилазы (HDAC). Фармацевтические композиции также содержат микрокристаллическую целлюлозу, натрийкросскармелозу и стеарат магния. Настоящее изобретение также относится к фармацевтическим композициям для перорального введения, содержащим САГК, микрокристаллическую целлюлозу, натрийкросскармелозу и стеарат магния. Пероральная биодоступность активных соединений в композициях по настоящему изобретению удивительно высокая. Кроме того, композиции позволяют поддерживать неожиданно высокие терапевтически эффективные уровни активных соединений в крови в течение длительного периода времени. Настоящее изобретение также относится к безопасному режиму ежедневного введения доз данных композиций, который легко применять и соблюдать.
Пероральная биодоступность активных соединений в композициях по настоящему изобретению удивительно высокая. Кроме того, композиции позволяют поддерживать неожиданно высокие терапевтически эффективные содержания активных соединений в крови в течение длительного периода времени. Настоящее изобретение также относится к безопасному режиму ежедневного введения доз данных композиций, который легко применять и который дает возможность поддерживать терапевтически эффективное количество указанных соединений in vivo. Композиции по настоящему изобретению применимы для селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток и в этой связи, способствуют лечению опухолей у пациентов.
Как показано в настоящем описании, фармацевтические композиции, предлагаемые в настоящем изобретении, позволяют создавать начальную среднюю концентрацию в плазме (то есть концентрацию, которая достигается сразу после введения композиции), которая остается неожиданно высокой в течение длительного периода времени. В сравнении с родительскими композициями (такими как в/в композиции) в той же дозе, из которых активное соединение выводится из организма почти сразу же, данные пероральные композиции поддерживают высокое значение средней концентрации в плазме крови активного соединения в течение длительного периода времени, равного по меньшей мере 2 часам, но в типичном случае по меньшей мере в течение 10 или 12 часов после введения. В типичном случае средняя концентрация в плазме крови пероральных композиций не падает ниже 50% от исходной средней концентрации в плазме в течение периода времени до 12 часов или еще дольше.
До открытия, сделанного в настоящем изобретении, считалось, что внутривенное введение ингибиторов HDAC, описанных в настоящем изобретении, является наиболее эффективным. Внутривенное введение соединения может быть осуществлено непрерывно, но ежедневно в течение длительного периода времени, например, по меньшей мере в течение примерно 3 дней и предпочтительно более 5 дней. Очевидно, что такой режим ложится тяжелым бременем для пациента, проходящего такое лечение. Неожиданное и удивительное открытие, сделанное авторами настоящего изобретения, дает возможность создавать пероральные дозированные формы, поддерживающие высокое и стойкое содержание активных соединений in vivo без необходимости непрерывного введения лекарственных средств путем в/в инфузии, что является огромным преимуществом для пациента, проходящего такое лечение.
Соответственно, настоящее изобретение относится к фармацевтической композиции для перорального введения, содержащей ингибитор гистондезацетилазы (HDAC) или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo у субъекта в течение периода времени, равного по меньшей мере 2 часам после введения. В предпочтительном варианте концентрация ингибитора HDAC эффективна для ингибирования HDAC в течение периода времени, равного по меньшей мере 8 часам после введения. В другом предпочтительном варианте концентрация ингибитора HDAC эффективна для ингибирования HDAC в течение периода времени, равного по меньшей мере 10 часам после введения. В другом предпочтительном варианте концентрация ингибитора HDAC эффективна для ингибирования HDAC в течение периода времени, равного по меньшей мере 12 часам после введения.
В предпочтительном варианте настоящее изобретение относится к фармацевтической композиции для перорального введения, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель, где указанная композиция обеспечивает достижение среднего значения концентрации в плазме САГК, эффективной для ингибирования гистондезацетилазы (HDAC) in vivo у субъекта в течение периода времени, равного по меньшей мере 2 часам после введения. В предпочтительном варианте концентрация САГК эффективна для ингибирования HDAC в течение периода времени, равного по меньшей мере 8 часам после введения. В другом предпочтительном варианте концентрация САГК эффективна для ингибирования HDAC в течение периода времени, равного по меньшей мере 10 часам после введения. В другом предпочтительном варианте концентрация САГК эффективна для ингибирования HDAC в течение периода времени, равного по меньшей мере 12 часам после введения.
Композиции по настоящему изобретению применимы для селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток и в этой связи, способствуют лечению опухолей у пациентов.
Соответственно, настоящее изобретение также относится к способу селективной индукции конечной дифференцировки опухолевых клеток у субъекта и в этой связи ингибирования пролиферации таких клеток у субъекта, предусматривающему достижение среднего значения концентрации в плазме ингибитора гистондезацетилазы (HDAC), способной ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения указанному субъекту эффективного количества фармацевтической композиции, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение также относится к способу селективной индукции остановки роста опухолевых клеток у субъекта и в этой связи ингибирования пролиферации таких клеток у субъекта, предусматривающему достижение среднего значения концентрации в плазме ингибитора гистондезацетилазы (HDAC), способной ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения такому субъекту эффективного количества фармацевтической композиции, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение также относится к способу селективной индукции апоптоза опухолевых клеток у субъекта и в этой связи, ингибирования пролиферации таких клеток у субъекта, предусматривающему достижение среднего значения концентрации в плазме ингибитора HDAC, способной ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения такому субъекту эффективного количества фармацевтической композиции, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение также относится к способу индукции дифференцировки опухолевых клеток у субъекта с опухолью, достижения среднего значения концентрации в плазме ингибитора HDAC, способной ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения такому субъекту эффективного количества фармацевтической композиции, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение относится к способу ингибирования активности гистондезацетилазы у субъекта, предусматривающему достижение среднего значения концентрации в плазме ингибитора HDAC, способной ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения указанному субъекту эффективного количества фармацевтической композиции, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение также относится к способу селективной индукции конечной дифференцировки, остановки роста клетки и/или апоптоза опухолевых клеток у субъекта с достижением при этом ингибирования пролиферации таких клеток у субъекта, предусматривающему создание средних концентраций в плазме САГК, способных ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения путем введения указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение также относится к способу селективной индукции конечной дифференцировки, остановки роста клетки и/или апоптоза опухолевых клеток у субъекта с достижением при этом ингибирования пролиферации таких клеток у субъекта, предусматривающему достижение средней концентрации в плазме САГК, равной по меньшей мере 10 нМ, in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение также относится к способу индукции дифференцировки опухолевых клеток у субъекта, имеющего опухоль, предусматривающему достижение среднего значения концентрации в плазме САГК, способной ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение также относится к способу индукции дифференцировки опухолевых клеток у субъекта, имеющего опухоль, предусматривающему достижение среднего значения концентрации в плазме САГК, равной по меньшей мере 10 нМ, in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение относится к способу ингибирования активности гистондезацетилазы у субъекта, предусматривающему достижение среднего значения концентрации в плазме САГК, способной ингибировать гистондезацетилазу in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
Кроме того, настоящее изобретение относится к способу ингибирования активности гистондезацетилазы у субъекта, предусматривающему достижение среднего значения концентрации в плазме САГК, равной по меньшей мере 10 нМ, in vivo у субъекта в течение периода времени, равного по меньшей мере двум часам после введения, путем введения указанному субъекту эффективного количества фармацевтической композиции, содержащей САГК или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или разбавитель.
В другом предпочтительном варианте настоящее изобретение относится к средней концентрации в плазме ингибитора HDAC (например, САГК), способной ингибировать гистондезацетилазу в течение периода времени, равного по меньшей мере двум часам после введения, где предпочтительно указанная концентрация составляет по меньшей мере 10 нМ. В другом варианте указанная композиция обеспечивает достижение среднего значения концентрации в плазме HDAC, равной по меньшей мере примерно 10 нМ, в течение периода времени, равного по меньшей мере 8 часам после введения. В еще одном предпочтительном варианте осуществления настоящего изобретения указанная композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC, равной по меньшей мере примерно 10 нМ, в течение периода времени, равного по меньшей мере 10 часам после введения. В еще одном предпочтительном варианте осуществления настоящего изобретения композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC, равной по меньшей мере примерно 10 нМ, в течение периода времени, равного по меньшей мере 12 часам после введения. Неограничивающие примеры создаваемых в плазме средних концентраций составляют примерно 10 нМ, 25 нМ, 40 нМ, 45 нМ, 50 нМ, 100 нМ, 1 мкМ, 2 мкМ, 2,5 мкМ, 5 мкМ, 10 мкМ, 25 мкМ, 50 мкМ, 100 мкМ и т.п. Специалисту в данной области понятно, что указанные дозы никоим образом не ограничивают область настоящего изобретения и что любая средняя концентрация в плазме, которая способна ингибировать гистондезацетилазу, будет приемлема.
В одном предпочтительном варианте осуществления настоящего изобретения композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC (например, САГК), способной селективно индуцировать конечную дифференцировку, остановку роста и/или апоптоз опухолевых клеток или индуцировать дифференцировку опухолевых клеток в опухоли, где указанная концентрация поддерживается в течение периода времени, равного по меньшей мере двум часам после введения, и где указанная концентрация предпочтительно равна по меньшей мере примерно 2,5 мкМ. В другом варианте осуществления настоящего изобретения композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC, равной по меньшей мере примерно 2,5 мкМ, в течение периода времени, равного по меньшей мере 8 часам после введения. В еще одном предпочтительном варианте осуществления настоящего изобретения данная композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC, равной по меньшей мере примерно 2,5 мкМ, в течение периода времени, равного по меньшей мере 10 часам после введения. В еще одном предпочтительном варианте осуществления настоящего изобретения композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC, равной по меньшей мере примерно 2,5 мкМ, в течение периода времени, равного по меньшей мере 12 часам после введения. Неограничивающие примеры достигаемых в плазме средних концентраций содержат примерно 10 нМ, 25 нМ, 40 нМ, 45 нМ, 50 нМ, 100 нМ, 1 мкМ, 2 мкМ, 2,5 мкМ, 5 мкМ, 10 мкМ, 25 мкМ, 50 мкМ, 100 мкМ и т.п. Специалисту в данной области понятно, что указанные дозы никоим образом не ограничивают область настоящего изобретения и что любая средняя концентрация в плазме, которая способна индуцировать конечную дифференцировку, остановку роста клеток и/или апоптоз опухолевых клеток, будет приемлема.
В другом предпочтительном варианте осуществления настоящего изобретения композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC (например, САГК), эффективной для индукции дифференцировки опухолевых клеток у субъекта, имеющего опухоль, где указанное количество поддерживается в течение периода времени, равного по меньшей мере двум часам после введения субъекту. В другом предпочтительном варианте композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC, эффективной для индукции дифференцировки опухолевых клеток у субъекта, имеющего опухоль, где указанное количество поддерживается в течение периода времени, равного по меньшей мере примерно 8 часам после введения субъекту. В другом предпочтительном варианте данная композиция обеспечивает достижение среднего значения концентрации в плазме ингибитора HDAC, эффективной для индукции дифференцировки опухолевых клеток у субъекта, имеющего опухоль, где указанное количество поддерживается в течение периода, равного по меньшей мере примерно 10 часам после введения субъекту. Неограничивающие примеры достигаемых в плазме средних концентраций составляют примерно 10 нМ, 25 нМ, 40 нМ, 45 нМ, 50 нМ, 100 нМ, 1 мкМ, 2 мкМ, 2,5 мкМ, 5 мкМ, 10 мкМ, 25 мкМ, 50 мкМ, 100 мкМ и т.п. Специалисту в данной области понятно, что указанные дозы никоим образом не ограничивают область настоящего изобретения и что любая средняя концентрация в плазме, которая способна индуцировать дифференцировку опухолевых клеток, будет приемлема.
Способы по настоящему изобретению пригодны для осуществления настоящего изобретения как in vitro, так и in vivo. Если указанные способы осуществляют in vitro, указанный контакт может быть достигнут при инкубировании клеток с данным соединением. Концентрация соединения, вступающего в контакт с клетками, должна составлять от примерно 1 нМ до примерно 25 мМ, например от примерно 10 нМ до примерно 1 мМ, от примерно 40 нМ до примерно 0,5 мМ. Неограничивающие примеры конкретных дозировок содержат примерно 10 нМ, 25 нМ, 40 нМ, 45 нМ, 50 нМ, 100 нМ, 1 мкМ, 2 мкМ, 2,5 мкМ, 5 мкМ, 10 мкМ, 25 мкМ, 50 мкМ, 100 мкМ и т.п. Указанная концентрация зависит от конкретного соединения и от состояния опухолевых клеток.
Несмотря на то, что способы по настоящему изобретению могут быть осуществлены in vitro, считается, что предпочтительный вариант осуществления способов селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза опухолевых клеток предусматривают контакт клеток in vivo, например, посредством введения соединений нуждающемуся в лечении субъекту, имеющему опухолевые клетки или злокачественные клетки.
Способы по настоящему изобретению могут также предусматривать первоначальное введение субъекту противоопухолевого средства, так чтобы придать опухолевым клеткам такого субъекта устойчивость к противоопухолевому средству, с последующим введением эффективного количества любой из композиций по настоящему изобретению, эффективной для селективной индукции конечной дифференцировки, остановки роста клеток и/или апоптоза таких клеток.
Противоопухолевое средство может содержать один из многочисленных химиотерапевтических препаратов, таких как алкилирующее средство, антиметаболит, гормональное средство, антибиотик, колхицин, алкалоид винки, L-аспарагиназа, прокарбазин, гидроксимочевина, митотан, нитрозомочевина или карбоксамид имидазола. Подходящими средствами являются такие средства, которые ускоряют деполяризацию тубулина. Предпочтительно противоопухолевое средство представляет колхицин или алкалоид винки, особенно предпочтительными являются винбластин и винкристин. В тех вариантах, когда противоопухолевое средство представляет собой винкристин, клетки предпочтительно обрабатывают таким образом, чтобы они стали устойчивыми к винкристину в концентрации примерно 5 мг/мл. Обработка клеток, которая придает им устойчивость к противоопухолевому средству, может быть осуществлена путем контакта клеток с указанным средством в течение периода времени, равного по меньшей мере 3-5 дням. Контакт полученных клеток с любым соединением из указанного выше перечня проводится по описанной выше методике. Введение данных соединений может быть объединено не только с указанными выше химиотерапевтическими средствами, но и с проведением лучевой терапии.
Настоящее изобретение также относится к способу лечения пациента, имеющего опухоль, которая характеризуется пролиферацией опухолевых клеток, где указанный способ предусматривает введение пациенту эффективного количества любой композиции по настоящему изобретению из числа указанных выше, эффективной для селективной индукции конечной дифференцировки таких опухолевых клеток с достижением при этом ингибирования их пролиферации.
Способ по настоящему изобретению предназначен для лечения больных людей с опухолями. Однако, вполне вероятно, что данный способ может быть эффективным при лечении опухолей у других млекопитающих. Термин "опухоль" в контексте настоящего описания включает любой вид злокачественной опухоли, вызванный пролиферацией опухолевых клеток, такой как злокачественная опухоль легкого, острая лимфоидная миелома, белознь Ходжкина, не-ходжкинская лимфома, меланома мочевого пузыря, злокачественная опухоль почки, злокачественная опухоль молочной железы, злокачественная опухоль предстательной железы, злокачественная опухоль яичников и колоректальная злокачественная опухоль.
Введение фармацевтических композиций может осуществляться с использованием стандартных дозированных форм, которые могут вводиться перорально один раз в день, дважды в день, три раза в день и т.п. Предпочтительные в настоящее время варианты содержат однократное введение, двукратное введение каждый день и трехкратное введение каждый день.
Гистондезацетилазы и ингибиторы гистондезацетилазы
Термин "гистондезацетилаза" (HDAC) в контексте настоящего описания включает ферменты, которые катализируют удаление ацетильных групп из лизиновых остатков на фрагментах аминоконца нуклеосомных ядерных гистонов. Как таковые HDAC вместе с гистонацетилтрансферазами (ГАТ) регулируют содержание ацетилирования гистонов. Гистонацетилирование воздействует на экспрессию генов, и ингибиторы HDAC, такие как гибридное полярное соединение на основе гидроксамовой кислоты - субероиланилид гидроксамовой кислоты (САГК), индуцируют остановку роста, дифференцировку и/или апоптоз трансформированных клеток in vitro и ингибируют рост опухоли in vivo. HDAC могут быть подразделены на три класса на основе их структурной гомологии. HDAC класса I (HDAC 1,2,3 и 8), имеющие сходство с дрожжевым белком RPD3, расположены в ядре и обнаруживаются в виде комплексов, связанных с ко-репрессорами транскрипции. HDAC класса II (4, 5, 6, 7 и 9) близки к дрожжевому белку HDA1 и характеризуются как ядерной, так и цитоплазматической локализацией в клетке. HDAC класса I и II ингибируются ингибиторами HDAC на основе гидроксамовой кислоты, такими как САГК. HDAC класса III образуют структурно далекий класс НАД-зависимых ферментов, которые близки к дрожжевым белкам SIR2 и не ингибируются ингибиторами HDAC на основе гидроксамовой кислоты.
Термины "ингибиторы гистондезацетилазы" или "ингибиторы HDAC" в контексте настоящего описания обозначают соединения, которые способны ингибировать дезацетилирование гистонов in vivo, in vitro или в обоих случаях. Как таковые ингибиторы HDAC ингибируют активность по меньшей мере одной гистондезацетилазы. В результате ингибирования дезацетилирования по меньшей мере одного гистона происходит повышение уровня ацетилирования гистона, и накопление ацетилированного гистона представляет собой приемлемый биологический маркер для оценки активности ингибитора HDAC. В этой связи процедуры, которые могут использоваться для анализа накопления ацетилированных гистонов, могут также использоваться для определения ингибирующей активности интересующих соединений в отношении HDAC. Следует понимать, что соединения, которые могут ингибировать активность гистондезацетилазы, могут связываться также с другими субстратами и как таковые могут ингибировать другие биологически активные молекулы, такие как ферменты. Следует также понимать, что соединения по настоящему изобретению способны ингибировать любые гистондезацетилазы, указанные выше, или любые другие гистондезацетилазы.
Например, у пациентов принимавших для лечения ингибиторы HDAC, накопление ацетилированных гистонов в периферических мононуклеарных клетках, а также в тканях после лечения ингибиторами HDAC может быть определено с использованием соответствующего контроля.
Ингибирующая активность конкретного соединения в отношении HDAC может быть определена in vitro с использованием, например, ферментативных тестов, которые выявляют ингибирование по меньшей мере одной гистондезацетилазы. Кроме того, определение накопления ацетилированных гистонов в клетках, обработанных конкретной композицией, может служить указанием на HDAC-ингибирующую активность такого соединения.
Тесты для оценки накопления ацетилированных гистонов известны в литературе. См., например, Marks, P.A. et al., J. Natl. Cancer Inst., 92: 1210-1215, 2000, Butler, L.M. et al., Cancer Res. 60: 5165-5170 (2000), Richon, V.M. et al., Proc. Natl. Acad. Sci., USA, 95: 3003-3007, 1998, and Yoshida, M. et al., J. Biol. Chem., 265:17174-17179, 1990.
Например, ферментативный тест для оценки активности ингибитора гистондезацетилазы может быть проведен следующим образом. Вкратце, процедура состоит в том, что эффект ингибитора HDAC на афинность очищенного человеческого меченого эпитопа (Flag) к HDAC 1 может быть оценен при инкубировании ферментного препарата на льду в отсутствие субстрата в течение примерно 20 минут с указанным количеством ингибирующего соединения. Добавляют субстрат ([3H]ацетил-меченый гистон из эритролейкозных клеток мышей) и образец инкубируют в течение 20 минут при 37°С в общем объеме 30 мкл. Затем реакцию останавливают, экстрагируют высвобожденный ацетат и определяют количество высвобожденной радиоактивности путем измерения в сцинтилляционном счетчике. Альтернативный тест, используемый для определения активности ингибитора гистондезацетилазы, содержит набор для определения активности HDAC на основе флуоресценции ("HDAC Fluorescent Activity Assay; Drug Discovery Kit-AK-500"), доступный от BIOMOL® Research Laboratories, Inc., Plymouth Meeting, PA.
Анализ in vivo может быть проведен следующим образом. Животным, например мышам, может быть инъецирован внутрибрюшинно ингибитор HDAC. Выбранные ткани, например мозг, селезенка, печень и т.п., могут быть выделены в заданные временные точки после введения. Гистоны могут быть выделены из тканей по методике, описанной по существу Yoshida et al., J. Biol. Chem., 265:17174-17179, 1990. Равные количества гистонов (примерно 1 мкг) могут быть подвергнуты разделению посредством электрофореза в полиакриламидном геле, содержащем 15% ДСН, с последующим переносом на фильтры Hybond-P (доступны от Amersham). Фильтры могут быть блокированы 3% молоком, далее проводят зондирование с использованием очищенного поликлонального антитела кролика против ацетилированного гистона Н4 (αAc-H4) и антитела против ацетилированного гистона Н3 (αAc-H3) (Upstate Biotechnology, Inc.). Уровни ацетилирования гистона могут быть определены с помощью конъюгированного с пероксидазой хрена козьего антитела против кроличьего иммуноглобулина (1:5000) и хемилюминисцентного субстрата SuperSignal (Pierce). В качестве соответствующего контроля, имитирующего гистоновый белок, может быть проведено параллельное разделение с использованием гелей, окрашенных кумасси-блю (КБ).
Кроме того, было показано, что ингибиторы HDAC на основе гидроксамовой кислоты регулируют экспрессию гена p21WAF1. Синтез белка p21WAF1 может быть индуцирован ингибиторами HDAC по процедуре стандартных методик в течение 2 часов в культурах множества трансформированных клеток. Индукция гена p21WAF1 сопровождается накоплением ацетилированных гистонов в хроматиновой области данного гена. Таким образом, можно полагать, что индукция p21WAF1 участвует в механизме остановки клеточного цикла на стадии G1, вызванной ингибиторами HDAC в трансформированных клетках.
В типичном случае ингибиторы HDAC могут быть разделены на пять основных классов: 1) производные гидроксамовой кислоты; 2) короткоцепочечные жирные кислоты (КЦЖК); 3) циклические тетрапептиды; 4) бензамиды и 5) электрофильные кетоны.
Таким образом, настоящее изобретение характеризуется широкой областью, охватывая композиции, содержащие ингибиторы HDAC, которые представляют собой: 1) производные гидроксамовой кислоты; 2) короткоцепочечные жирные кислоты (КЦЖК); 3) циклические тетрапептиды; 4) бензамиды и 5) электрофильные кетоны и/или соединения любого другого класса, способные ингибировать гистондезацетилазы, с целью применения их для ингибирования гистондезацетилазы, включая достижение конечной дифференцировки опухолевых клеток и/или индукцию дифференцировки опухолевых клеток в опухоли.
Примеры таких ингибиторов HDAC содержат, без ограничения, следующие.
А. Производные гидроксамовой кислоты, такие как субероиланилид гидроксамовой кислоты (САГК) (Richon et al., Proc. Natl. Acad. Sci. USA, 95: 3003-3007, 1998); бисгидроксамид м-карбоксикоричной кислоты (БАКК (CBHA)) (Richon et al., supra); пироксамид; аналоги трихостатина, такие как трихостатин А (ТСА (TSA)) и трихостатин С (Koghe et al., 1998, Biochem. Pharmacol. 56: 1359-1364); салицилгидроксамовая кислота (СБГК (SBHA)) (Andrews et al., International J. Parasitology 30, 761-768 (2000)); субероил-бисгидроксамовая кислота (СБГК (SBHA)) (патент США No. 5 608 108); азелаин-бисгидроксамовая кислота (АБГК (ABHA)) (Andrews et al., supra); азелаин-1-гидроксамат-9-анилид (ААГА (AAHA)) (Qiu et al., Mol. Biol, Cell 11, 2069-2083 (2000)); 6-(3-хлорфенилуреидо) капроновой гидроксамовой кислоты (3Cl-UCHA); оксамфлатин [(2E)-5-[3-[(фенилсульфонил)аминолфенил]пент-2-ен-4-иногидроксамовая кислота] (Kim et al., Oncogene, 18: 2461-2470 (1999)); A-161906, Scriptaid (Su et al., 2000 Cancer Research, 60: 3137-3142); PXD-101 (Prolifix); LAQ-824; CHAP; MW2796 (Andrews et al., supra); MW2996 (Andrews et al., supra) или любая другая из числа гидроксамовых кислот, описанных в патентах США NoNo. 5369108, 5932616, 5700811, 6087367 и 6511990.
B. Циклические тетрапептиды, такие как трапоксин А (TPX)-циклический тетрапептид (цикло(L-фенилаланил-L-фенилаланил-D-пипеколинил-L-2-амино-8-оксо-9,10-эпокси-деканоил)) (Kijima et al., J. Biol. Chem. 268, 22429-22435 (1993)); ER901228 (FK 228, депсипептид) (Nakajima et al., Ex. Cell Res. 241, 126-133 (1998)); FR225497 циклический тетрапептид (H. Mori et al., Заявка PCT WO 00/08048 (17 февраля 2000 года)); апицидиновый циклический тетрапептид [цикло(N-O-метил-L-триптофанил-L-изолейцинил-D-пипеколинил-L-2-амино-8-оксодеканоил)] (Darkin-Rattray et al., Proc. Natl. Acad. Sci. USA 93, 13143-13147 (1996)); апицидин Ia, апицидин Ib, апицидин Ic, апицидин IIa и апицидин IIb (P. Dulski et al., Заявка PCT WO 97/11366); CHAP; циклический тетрапептид HC-токсина (Bosch et al., Plant Cell 7, 1941-1950 (1995)); WF27082 циклический тетрапептид (Заявка PCT WO 98/48825) и хламидоцин (Bosch et al., supra).
С. Производные короткоцепочечных жирных кислот (КЦЖК), такие как бутират натрия (Cousens et al., J. Biol. Chem. 254, 1716-1723 (1979)); изовалерат (McBain et al., Biochem. Pharm. 53: 1357-1368 (1997)); валерат (McBain et al., supra); 4-фенилбутират (4-PBA) (Lea and Tulsyan, Anticancer Research, 15, 879-873 (1995)); фенилбутират (ФБ) (Wang et al., Cancer Research, 59, 2766-2799 (1999)); пропионат (McBain et al., supra); бутирамид (Lea and Tulsyan, supra), изобутирамид (Lea and Tulsyan, supra), фенилацетат (Lea and Tulsyan, supra), 3-бромпропионат (Lea and Tulsyan, supra), трибутирин (Guan et al., Cancer Research, 60, 749-755 (2000)); валпроиновая кислота и валпроат.
D. Производные бензамида, такие как CI-994; MS-27-275 [N-(2-аминофенил)-4-[N-(пиридин-3-илметоксикарбонил)аминометил]бензамид] (Saito et al., Proc. Natl. Acad. Sci. USA 96, 4592-4597 (1999)) и 3'-аминопроизводное MS-27-275 (Saito et al., supra).
E. Электрофильные кетоновые производные, такие как трифторметилкетоны (Frey et al., Bioorganic & Med. Chem. Lett. (2002), 12, 3443-3447; US 6511990) и α-кетоамиды, такие как N-метил-α-кетоамиды.
F. Другие ингибиторы HDAC, такие как депудецин (Kwon et al., 1998. PNAS 95:3356-3361).
Предпочтительные ингибиторы HDAC на основе гидраксамовой кислоты содержат субероиланилид гидроксамовой кислоты (САГК), бисгидроксамид м-карбоксикоричной кислоты (БАКК) и пироксамид. Было показано, что САГК связывается непосредственно в каталитическом участке фермента гистондезацетилаза. САГК индуцирует остановку клеточного цикла, дифференцировку и/или апоптоз трансформированных клеток в культуре и ингибирует рост опухоли у грызунов. САГК эффективен с точки зрения индукции указанных эффектов в отношении солидных опухолей и лейкозов. Было показано, что САГК эффективен с точки зрения ингибирования опухолей у животных и не проявляет токсичности для животных. САГК-индуцированное ингибирование роста опухоли сопровождается накоплением ацетилированных гистонов в опухоли. САГК эффективен в плане ингибирования развития и безостановочного роста индуцированных канцерогеном (N-метилнитрозомочевиной) опухолей молочной железы у крыс. САГК вводят крысам в составе пищевого рациона в течение 130 дней исследования. Таким образом, САГК представляет собой нетоксичный активный при пероральном введении противоопухолевый агент, механизм действия которого содержит ингибирование активности гистондезацетилазы.
Предпочтительные ингибиторы HDAC представляют собой соединения, описанные в патентах США NoNo. 5369108, 5932616, 5700811, 6087367 и 6511990, принадлежащих некоторым авторам настоящего изобретения, полное содержание которых включено в настоящее описание в качестве ссылок, ниже приведены неограничивающие примеры данных соединений.
Таким образом, согласно одному варианту осуществления настоящего изобретения предлагается фармацевтическая композиция, содержащая соединение, описываемое структурной формулой I, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
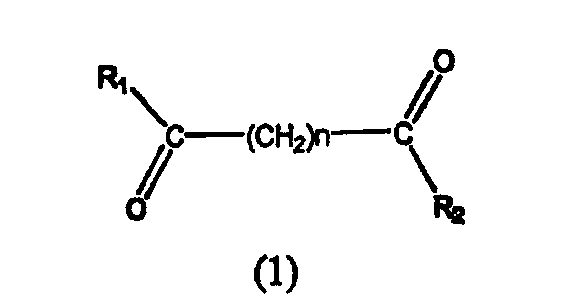
где R1 и R2 могут быть одинаковыми или различными, когда R1 и R2 одинаковые, каждый из них обозначает замещенный или незамещенный ариламино, циклоалкиламино, пиридинамино, пиперидино, 9-пурин-6-амин или тиазоламиногруппу; когда R1 и R2 различаются, R1= R3-N-R4, где R3 и R4 могут быть одинаковыми или могут отличаться друг от друга и обозначает атом водорода, гидроксильную группу, замещенную или незамещенную, разветвленную или неразветвленную алкильную, алкенильную, циклоалкильную, арильную, алкилокси, арилокси, арилалкилокси или пиридиновую группу, или R3 и R4 соединяются вместе с образованием пиперидиновой группы, R2 обозначает гидроксиламино, гидроксильную, амино, алкиламино, диалкиламино или алкилоксигруппу и n равно целому числу от примерно 4 до примерно 8.
В конкретном варианте соединения формулы 1 R1 и R2 имеют одинаковые значения и обозначают замещенную или незамещенную тиазоламиногруппу и n равно целому числу от примерно 4 до примерно 8.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 2, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
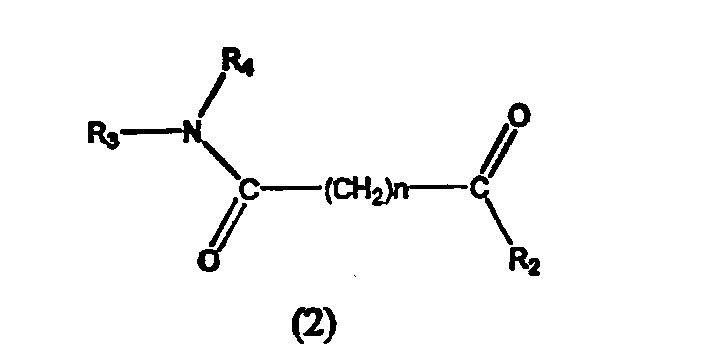
где R3 и R4 могут быть одинаковыми или разными и каждый независимо обозначает атом водорода, гидроксильную группу, замещенную или незамещенную, разветвленную или неразветвленную алкильную, алкенильную, циклоалкильную, арилалкилокси, арилокси, арилалкилокси или пиридиновую группу, или R3 и R4 соединяются вместе с образованием пиперидиновой группы, R2 обозначает гидроксиламино, гидроксильную, амино, алкиламино, диалкиламино или алкилоксигруппу и n равно целому числу от примерно 4 до примерно 8.
В предпочтительном варианте формулы 2 R3 и R4 независимо могут быть одинаковыми или могут отличаться друг от друга и каждый обозначает атом водорода, гидроксильную группу, замещенную или незамещенную, разветвленную или неразветвленную алкильную, алкенильную, циклоалкильную, арильную, алкилокси, арилокси, арилалкилокси или пиридиновую группу, или R3 и R4 соединяются вместе с образованием пиперидиновой группы, R2 обозначает гидроксиламино, гидроксильную, амино, алкиламино или алкилокси группу, n равно целому числу от 5 до 7 и R3-N-R4 и R2 отличаются друг от друга.
В другом конкретном варианте формулы 2 n равно 6. В еще одном варианте формулы II R4 обозначает атом водорода, R3 обозначает замещенный или незамещенный фенил и n равно 6.
В еще одном варианте формулы II R4 обозначает атом водорода, R3 обозначает замещенный фенил и n равно 6, где фенильный заместитель выбирают из группы, состоящей из метильной, циано, нитро, трифторметильной, амино, аминокарбонильной, метилцианогруппы, атомов хлора, фтора, брома, йода, 2,3-дифтор-, 2,4-дифтор-, 2,5-дифтор-, 3,4-дифтор-, 3,5-дифтор-, 2,6-дифтор-, 1,2,3-трифтор-, 2,3,6-трифтор-, 2,4,6-трифтор-, 3,4,5-трифтор-, 2,3,5,6-тетрафтор-, 2,3,4,5,6-пентафтор-, азидо, гексильной, трет-бутильной, фенильной, карбоксильной, гидроксильной, метокси, фенилокси, бензилокси, фениламиноокси, фениламинокарбонильной, метоксикарбонильной, метиламинокарбонильной, диметиламино, диметиламинокарбонильной или гидроксиламинокарбонильной группы.
В еще одном варианте формулы 2 n равно 6, R4 обозначает атом водорода и R3 обозначает циклогексильную группу. В другом варианте формулы 2 n равно 6, R4 обозначает атом водорода и R3 обозначает метоксигруппу. В другом варианте формулы 2 n равно 6 и R3 и R4 связываются вместе с образованием пиперидиновой группы. В другом варианте формулы 2 n равно 6, R4 обозначает атом водорода и R3 обозначает бензилоксигруппу. В другом варианте формулы 2 R4 обозначает атом водорода и R3 обозначает γ-пиридиновую группу. В другом варианте формулы 2 R4 обозначает атом водорода и R3 обозначает β-пиридиновую группу. В другом варианте формулы 2 R4 обозначает атом водорода и R3 обозначает α-пиридиновую группу. В другом варианте формулы 2 n равно 6, R3 и R4 обозначают, оба, метильные группы. В другом варианте формулы II n равно 6, R4 обозначает метильную группу и R3 обозначает фенильную группу.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 3, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
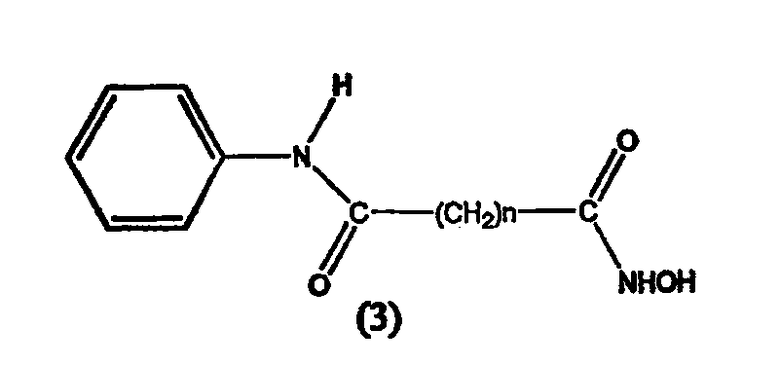
где n равно целому числу от 5 до примерно 8.
В предпочтительном варианте формулы 3 n равно 6. В соответствии с данным вариантом осуществления настоящее изобретение относится к фармацевтической композиции, содержащей САГК (4) или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель. САГК может иметь следующие структурные формулы
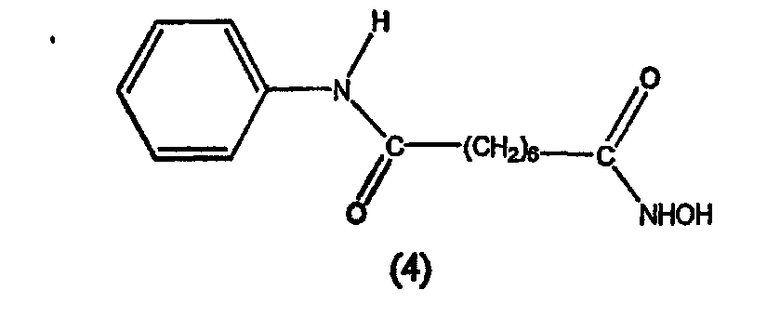
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 5, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
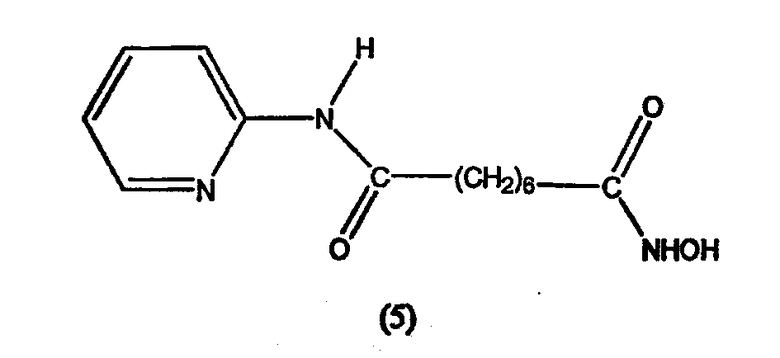
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 6 (пироксамид), или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
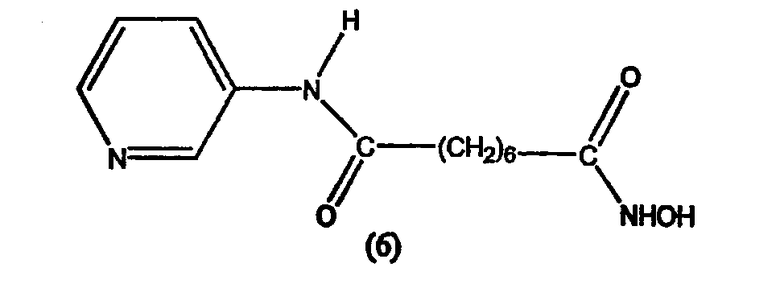
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 7, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
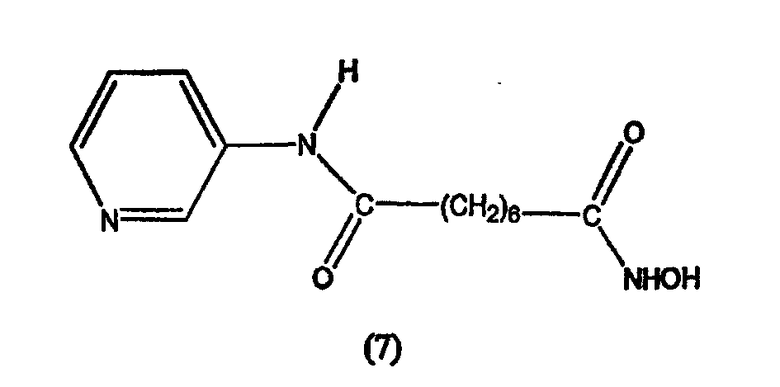
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 8, или его фармацевтически приемлемую соль или гидрат или фармацевтически приемлемый носитель или наполнитель

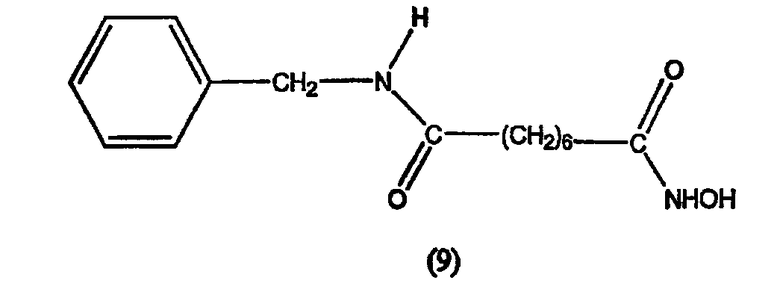
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 9, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 10, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
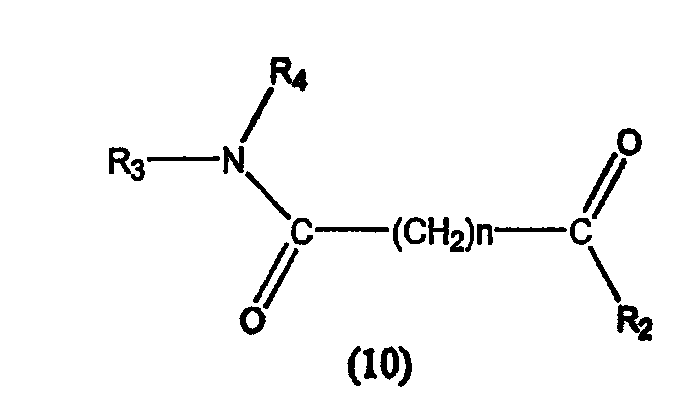
где R3 обозначает атом водорода и R4 обозначает циклоалкильную, арильную, арилокси, арилалкилокси или пиридиновую группу или R3 и R4 соединяются вместе с образованием пиперидиновой группы, R2 обозначает гидроксиламиногруппу и n равно целому числу от 5 до примерно 8.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 11, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель

где R3 и R4 обозначают независимо замещенную или незамещенную, разветвленную или неразветвленную алкильную, алкенильную, циклоалкильную, арильную, алкилокси, арилокси, арилалкилокси или пиридиновую группу, циклоалкильную, арильную, арилокси, арилалкилокси или пиридиновую группу или R3 и R4 соединяются вместе с образованием пиперидиновой группы; R2 обозначает гидроксиламиногруппу и n равно целому числу от 5 до примерно 8.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 12, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
где X и Y независимо могут быть одинаковыми или отличаться друг от друга и каждый обозначает гидроксильную, амино или гидроксиламиногруппу; замещенную или незамещенную алкилокси, алкиламино, диалкиламино, ариламино, алкилариламино, алкилоксиамино, арилоксиамино, алкилоксиалкиламино или арилоксиалкиламиногруппу; R обозначает атом водорода, гидроксильную группу; замещенную или незамещенную алкильную, арилалкилокси или арилоксигруппу; и m и n могут быть независимо одинаковыми или отличаться друг от друга и каждый равен целому числу от примерно 0 до примерно 8.
В конкретном варианте ингибитор HDAC представляет собой соединение формулы XI, где X, Y и R, каждый, обозначает гидроксил и m и n равны 5.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 13, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
где X и Y независимо могут быть одинаковыми или отличаться друг от друга и каждый обозначает гидроксильную, амино или гидроксиламиногруппу; замещенную или незамещенную алкилокси, алкиламино, диалкиламино, ариламино, алкилариламино, алкилоксиамино, арилоксиамино, алкилоксиалкиламино или арилоксиалкиламино группу; R1 и R2 могут быть независимо одинаковыми или отличаться друг от друга и каждый обозначает атом водорода, гидроксильную группу; замещенную или незамещенную алкильную, арильную, алкилокси или арилоксигруппу и m, n и о независимо могут быть одинаковыми или отличаться друг от друга и каждый равен целому числу от примерно 0 до примерно 8.
В одном конкретном варианте формулы 13 каждый из X и Y обозначают гидроксильную группу и каждый из R1 и R2 обозначает метильную группу. В другом конкретном варианте формулы 13 каждый из X и Y обозначает гидроксильную группу и каждый из R1 и R2 обозначает метильную группу, каждый из n и o равен 6 и m равно 2.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 14, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
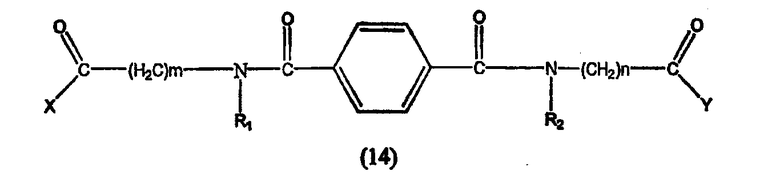
где X и Y независимо могут быть одинаковыми или могут отличаться друг от друга и каждый обозначает гидроксильную, амино или гидроксиламиногруппу; замещенную или незамещенную алкилокси, алкиламино, диалкиламино, ариламино, алкилариламино, алкилоксиамино, арилоксиамино, алкилоксиалкиламино или арилоксиалкиламиногруппу; R1 и R2 могут быть независимо одинаковыми или могут отличаться друг от друга и каждый обозначает атом водорода, гидроксильную группу, замещенную или незамещенную алкильную, арильную, алкилокси или арилоксигруппу; m и n независимо могут быть одинаковыми или могут отличаться друг от друга и каждый равен целому числу от примерно 0 до примерно 8.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 15, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
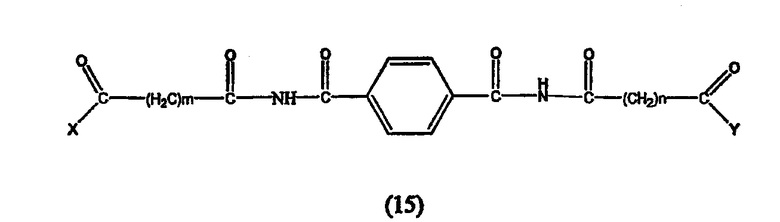
где X и Y независимо могут быть одинаковыми или могут отличаться друг от друга и каждый обозначает гидроксильную, амино или гидроксиламиногруппу; замещенную или незамещенную алкилокси, алкиламино, диалкиламино, ариламино, алкилариламино, алкилоксиамино, арилоксиамино, алкилоксиалкиламино или арилоксиалкиламиногруппу; m и n независимо могут быть одинаковыми или могут отличаться друг от друга и каждый равен целому числу от примерно 0 до примерно 8.
В одном конкретном варианте формулы 1 каждый из X и Y обозначает гидроксильную группу и каждый из m и n равен 5.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 16, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
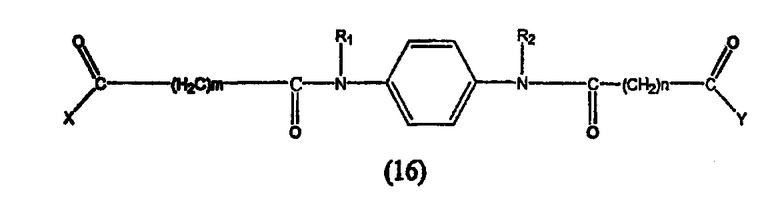
где X и Y независимо могут быть одинаковыми или могут отличаться друг от друга и каждый обозначает гидроксильную, амино или гидроксиламиногруппу, замещенную или незамещенную алкилокси, алкиламино, диалкиламино, ариламино, алкилариламино, алкилоксиамино, арилоксиамино, алкилоксиалкиламино или арилоксиалкиламиногруппу; R1 и R2 могут быть независимо одинаковыми или могут отличаться друг от друга и каждый обозначает атом водорода, гидроксильную группу, замещенную или незамещенную алкильную, арилалкилокси или арилоксигруппу; m и n независимо могут быть одинаковыми или могут отличаться друг от друга и каждый равен целому числу от примерно 0 до примерно 8.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 17, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
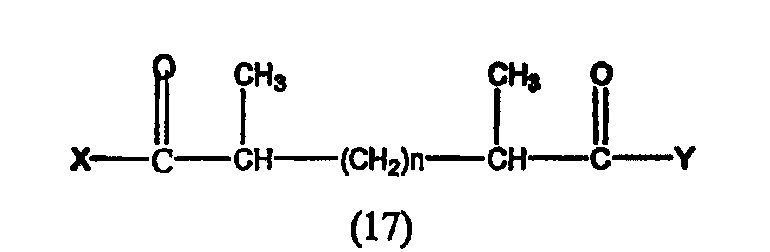
где X и Y независимо могут быть одинаковыми или могут отличаться друг от друга и каждый обозначает гидроксильную, амино или гидроксиламиногруппу, замещенную или незамещенную алкилокси, алкиламино, диалкиламино, ариламино, алкилариламино или арилоксиалкиламиногруппу и n равно целому числу от примерно 0 до примерно 8.
В одном конкретном варианте формулы 17 каждый из X и Y обозначает гидроксиламиногруппу, R1 обозначает метильную группу, R2 обозначает атом водорода и каждый из m и n равен 2. В другом конкретном варианте формулы 17 каждый из X и Y обозначает гидроксиламиногруппу, R1 обозначает карбонилгидроксиламиногруппу, R2 обозначает атом водорода и каждый из m и n равен 5. В другом конкретном варианте формулы 17 каждый из X и Y обозначает гидроксиламиногруппу, каждый из R1 и R2 обозначает фтор и каждый из m и n равен 2.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 18, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
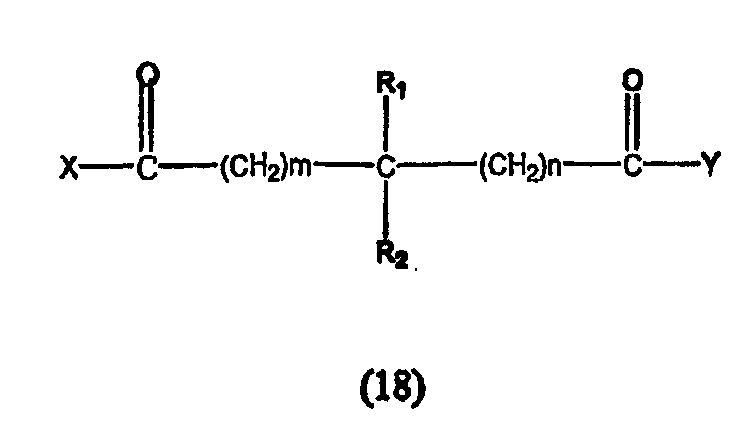
где X и Y независимо могут быть одинаковыми или могут отличаться друг от друга и каждый обозначает гидроксильную, амино или гидроксиламиногруппу, замещенную или незамещенную алкилокси, алкиламино, диалкиламино, ариламино, алкилариламино, алкилоксиамино, арилоксиамино, алкилоксиалкиламино или арилоксиалкиламино группу, R1 и R2 могут быть независимо одинаковыми или могут отличаться друг от друга и каждый обозначает атом водорода, гидроксильную группу; замещенную или незамещенную алкильную, арильную, алкилокси, арилокси, карбонилгидроксиламиногруппу или атом фтора; m и n независимо могут быть одинаковыми или могут отличаться друг от друга и каждый равен целому числу от примерно 0 до примерно 8.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 19, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
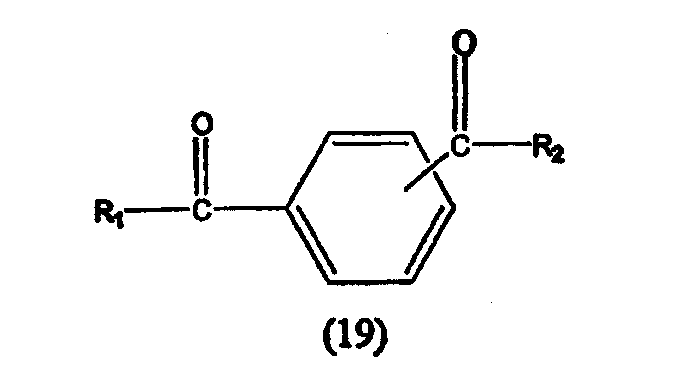
где R1 и R2 могут быть одинаковыми или могут отличаться друг от друга и каждый обозначает гидроксильную, алкилокси, амино, гидроксиламино, алкиламино, диалкиламино, ариламино, алкилариламино, алкилоксиамино, арилоксиамино, алкилоксиалкиламино или арилоксиалкиламиногруппу. В конкретном варианте настоящего изобретения ингибитор HDAC представляет собой соединение структурной формулы X, где R1 и R2 обозначают, оба, гидроксиламиногруппу.
В одном конкретном варианте формулы 19 R1 обозначает фениламиногруппу и R2 обозначает гидроксиламиногруппу.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 20, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
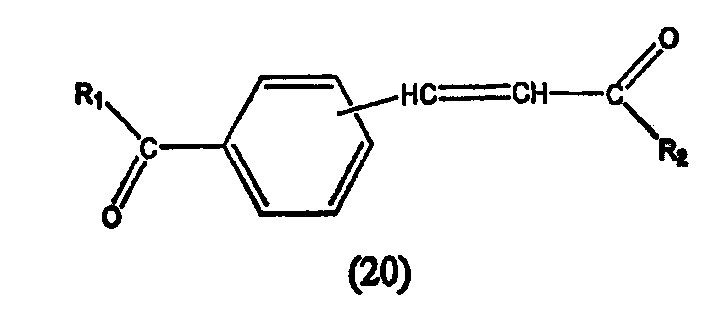
где R1 и R2 независимо могут быть одинаковыми или могут отличаться друг от друга и каждый обозначает гидроксильную, алкилокси, амино, гидроксиламино, алкиламино, диалкиламино, ариламино, алкилариламино, алкилоксиамино, арилоксиамино, алкилоксиалкиламино или арилоксиалкиламиногруппу. В конкретном варианте настоящего изобретения ингибитор HDAC представляет собой соединение структурной формулы 11, где R1 и R2 обозначают, оба, гидроксиламиногруппу.
В одном конкретном варианте формулы 18 R1 обозначает гидроксиламиногруппу. В другом варианте формулы 21 R2 обозначает гидроксиламиногруппу.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 22, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
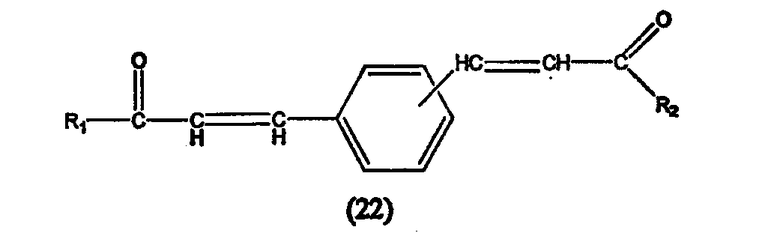
где R1 и R2 независимо могут быть одинаковыми или могут отличаться друг от друга и каждый обозначает гидроксильную, алкилокси, амино, гидроксиламино, алкиламино, диалкиламино, ариламино, алкилариламино, алкилоксиамино, арилоксиамино, алкилоксиалкиламино или арилоксиалкиламиногруппу. В конкретном варианте настоящего изобретения ингибитор HDAC представляет собой соединение структурной формулы 12, где R1 и R2 обозначают, оба, гидроксиламиногруппу.
В одном конкретном варианте формулы 23 R1 обозначает фениламиногруппу и R2 обозначает гидроксиламиногруппу.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 24, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
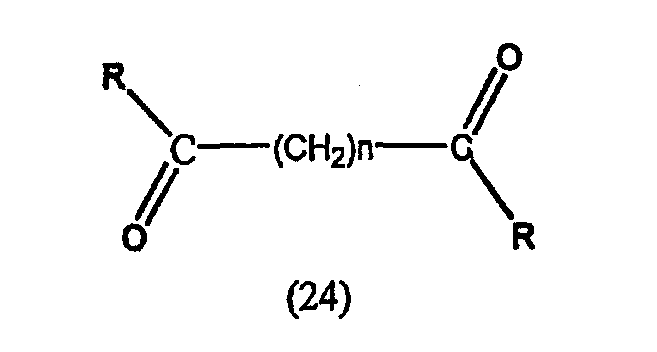
где R обозначает фениламиногруппу, замещенную циано, метилциано, нитро, карбоксильной, аминокарбонильной, метиламинокарбонильной, диметиламинокарбонильной, трифторметильной, гидроксиламинокарбонильной, N-гидроксиламинокарбонильной, метоксикарбонильной группами, атомами хлора, фтора, метильной, метокси, 2,3-дифтор-, 2,4-дифтор-, 2,5-дифтор-, 2,6-дифтор-, 3,5-дифтор-, 2,3,6-трифтор-, 2,4,6-трифтор-, 1,2,3-трифтор-, 3,4,5-трифтор-, 2,3,4,5-тетрафтор или 2,3,4,5,6-пентафторгруппой, и n равно целому числу от 4 до 8.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 25 (БАКК) (CBHA), или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
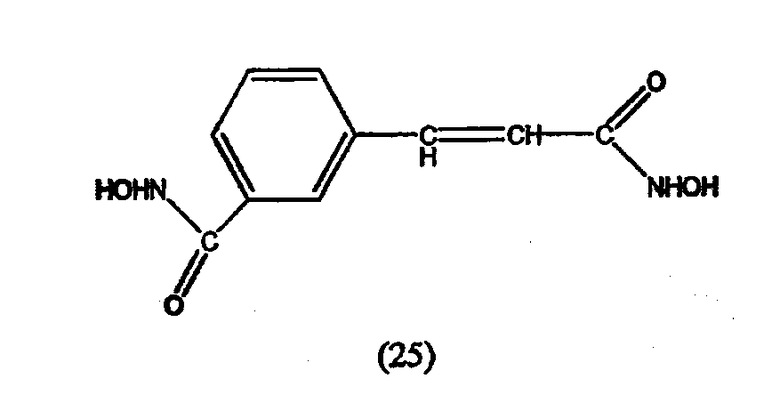
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 26, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
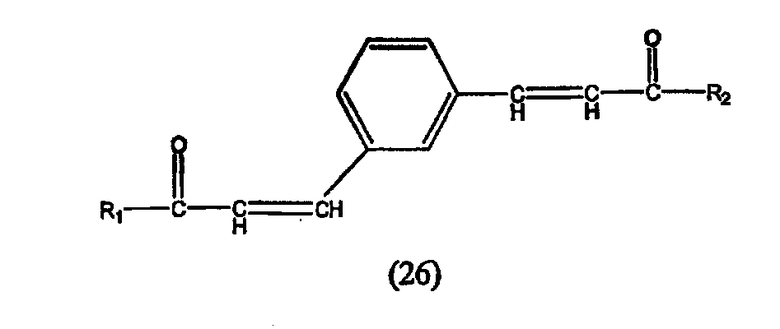
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 27, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель

где R обозначает замещенный или незамещенный фенил, пиперидин, тиазол, 2-пиридин, 3-пиридин или 4-пиридин и n равно целому числу от примерно 4 до примерно 8.
В одном конкретном варианте формулы 27 R обозначает замещенную фенильную группу. В другом конкретном варианте формулы 27 R обозначает замещенную фенильную группу, где заместитель выбирают из группы, состоящей из метильной, циано, нитро, тио, трифторметильной, амино, аминокарбонильной, метилцианогруппы, атома хлора, фтора, брома, йода, 2,3-дифтор-, 2,4-дифтор-, 2,5-дифтор-, 3,4-дифтор-, 3,5-дифтор-, 2,6-дифтор-, 1,2,3-трифтор-, 2,3,6-трифтор-, 2,4,6-трифтор-, 3,4,5-трифтор-, 2,3,5,6-тетрафтор-, 2,3,4,5,6-пентафтор-, азидо, гексильной, трет-бутильной, фенильной, карбоксильной, гидроксильной, метилокси, фенилокси, бензилокси, фениламиноокси, фениламинокарбонильной, метилоксикарбонильной, метиламинокарбонильной, диметиламино, диметиламинокарбонильной или гидроксиаминокарбонильной группы.
В другом конкретном варианте формулы 27 R обозначает замещенный или незамещенный 2-пиридин, 3-пиридин или 4-пиридин и n равно целому числу от примерно 4 до примерно 8.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 28, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
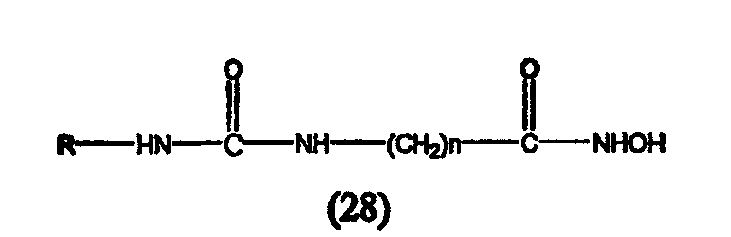
где R обозначает замещенную или незамещенную фенильную, пиридиновую, пиперидиновую или тиазоловую группу и n равно целому числу от примерно 4 до примерно 8.
В одном конкретном варианте формулы 28 R обозначает замещенную фенильную группу. В другом конкретном варианте формулы 28 R обозначает замещенную фенильную группу, где заместитель выбирают из группы, состоящей из метильной, циано, нитро, тио, трифторметильной, амино, аминокарбонильной, метилцианогруппы, атома хлора, фтора, брома, йода, 2,3-дифтор-, 2,4-дифтор-, 2,5-дифтор-, 3,4-дифтор-, 3,5-дифтор-, 2,6-дифтор-, 1,2,3-трифтор-, 2,3,6-трифтор-, 2,4,6-трифтор-, 3,4,5-трифтор-, 2,3,5,6-тетрафтор-, 2,3,4,5,6-пентафтор-, азидо, гексильной, трет-бутильной, фенильной, карбоксильной, гидроксильной, метилокси, фенилокси, бензилокси, фениламиноокси, фениламинокарбонильной, метилоксикарбонильной, метиламинокарбонильной, диметиламино, диметиламинокарбонильной или гидроксиламинокарбонильной группы.
В другом конкретном варианте формулы 28 R обозначает фенил и n равно 5. В другом варианте n равно 5, а R обозначает 3-хлорфенил.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 29, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
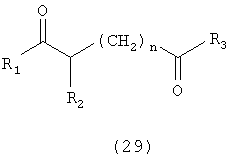
где каждый из R1 и R2 присоединен непосредственно или через линкер и обозначает замещенный или незамещенный арил (например, фенил), арилалкил (например, бензил), нафтил, циклоалкил, циклоалкиламино, пиридинамино, пиперидино, 9-пурин-6-амино, тиазоламино, гидроксил, разветвленный или неразветвленный алкил, алкенил, алкилокси, арилокси, арилалкилокси, пиридил, хинолинил или изохинолинил, n равно целому числу от примерно 3 до примерно 10 и R3 обозначает гидроксамовую кислоту, гидроксиламино, гидроксильную, амино, алкиламино или алкилоксигруппу. Линкер может представлять собой компонент амидного фрагмента, например O-, -S-, -NH-, NR5, -CH2-, -(CH2)m-, -(CH=CH)-, фенилен, циклоалкилен или любое их сочетание, где R5 обозначает замещенный или незамещенный С1-С5 алкил.
В некоторых вариантах формулы 29 R1 обозначает -NH-R4, где R4 обозначает замещенный или незамещенный арил (например, фенил), арилалкил (например, бензил), нафтил, циклоалкил, циклоалкиламино, пиридинамино, пиперидино, 9-пурин-6-амино, тиазоламино, гидроксил, разветвленный или неразветвленный алкил, алкенил, алкилокси, арилокси, арилалкилокси, пиридил, хинолинил или изохинолинил.
В другом варианте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, описываемое структурной формулой 30, или его фармацевтически приемлемую соль или гидрат и фармацевтически приемлемый носитель или наполнитель
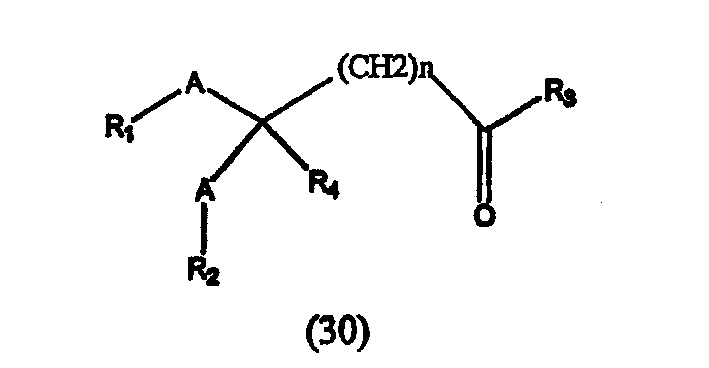
где каждый из R1 и R2 обозначает замещенный или незамещенный арил (например, фенил), арилалкил (например, бензил), нафтил, циклоалкил, циклоалкиламино, пиридинамино, пиперидино, 9-пурин-6-амино, тиазоламино, гидроксил, разветвленный или неразветвленный алкил, алкенил, алкилокси, арилокси, арилалкилокси, пиридил, хинолинил или изохинолинил; R3 обозначает гидроксамовую кислоту, гидроксиламино, гидроксильную, амино, алкиламино или алкилоксигруппу; R4 обозначает атом водорода, атом галогена, фенильный или циклоалкильный фрагмент и А может быть одинаковым или различным и представляет собой амидный фрагмент, O-, -S-, -NH-, NR5, -CH2-, -(CH2)m-, -(CH=CH)-, фенилен, циклоалкилен или любое их сочетание, где R5 обозначает замещенный или незамещенный С1-С5 алкил, аn и m, каждый, равен целому числу от 3 до 10.
В еще одном конкретном варианте соединения, имеющие более специфическую структуру и относящиеся к категории соединений формулы 29 и 30, представляют собой следующие.
Соединение, описываемое структурной формулой 31
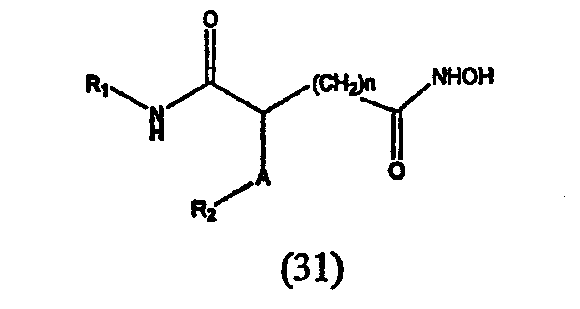
где А обозначает амидный фрагмент, R1 и R2 выбирают, каждый, из замещенного или незамещенного арила (например, фенила), арилалкила (например, бензила), нафтила, пиридинамино, 9-пурин-6-амино, тиазоламино, арилокси, арилалкилокси, пиридила, хинолинила или изохинолинила и n равно целому числу от 3 до 10.
Например, соединение формулы 30 может иметь структуру 31 или 32
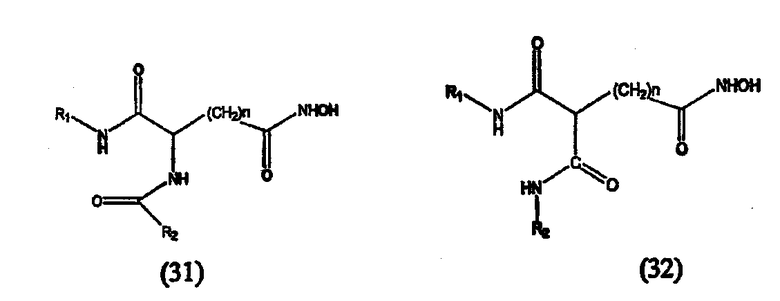
где R1, R2 и n имеют значения, описанные для формулы 30.
Соединение, описываемое структурной формулой 33
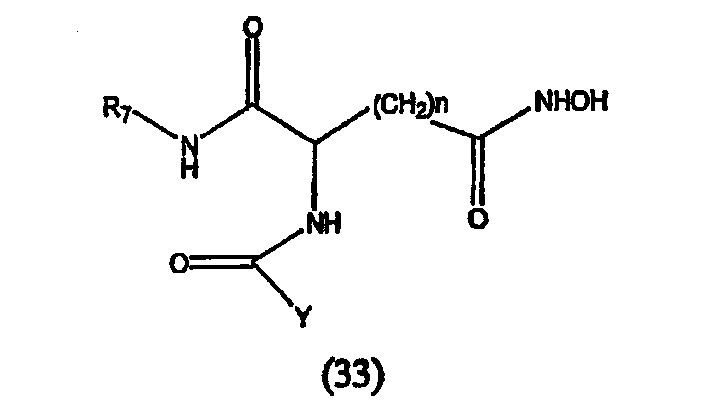
где R7 выбирают из замещенного или незамещенного арила (например, фенила), арилалкила (например, бензила), нафтила, пиридинамино, 9-пурин-6-амино, тиазоламино, арилокси, арилалкилокси, пиридила, хинолинила или изохинолинила; n равно целому числу от 3 до 10 и Y выбирают из
Соединение, описываемое структурной формулой 34
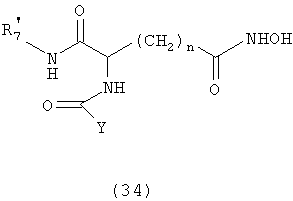
де n равно целому числу от 3 до 10, Y выбирают из соединений формулы
и R7' выбирают из соединений формулы
Соединение, описываемое структурной формулой 35
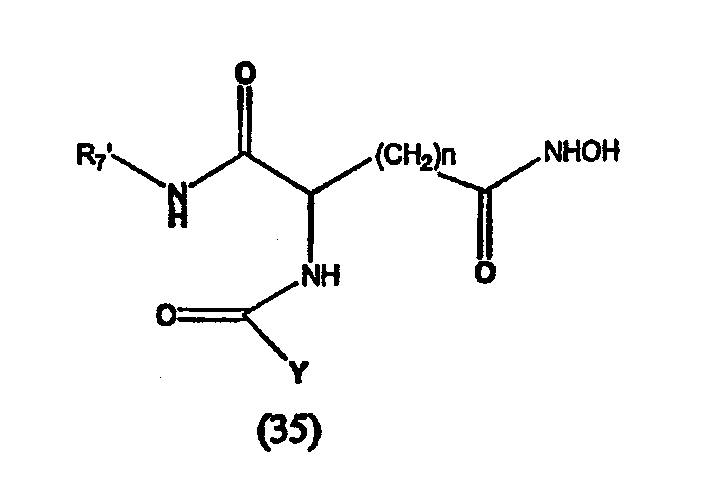
Y выбирают из арила (например, фенила), арилалкила (например, бензила), нафтила, пиридинамино, 9-пурин-6-амино, тиазоламино, арилокси, арилалкилокси, пиридила, хинолинила или изохинолинила; n равно целому числу от 3 до 10, и R7' выбирают из соединений формулы
Соединение, описываемое структурной формулой 36

где А обозначает амидный фрагмент, R1 и R2 выбирают, каждый, из замещенного или незамещенного арила (например, фенила), арилалкила (например, бензила), нафтила, пиридинамино, 9-пурин-6-амино, тиазоламино, арилокси, арилалкилокси, пиридила, хинолинила или изохинолинила, R4 обозначает атом водорода, атом галогена, фенильный или циклоалкильный фрагмент и n равно целому числу от 3 до 10.
Например, соединение формулы 36 может иметь структуру 37 или 38
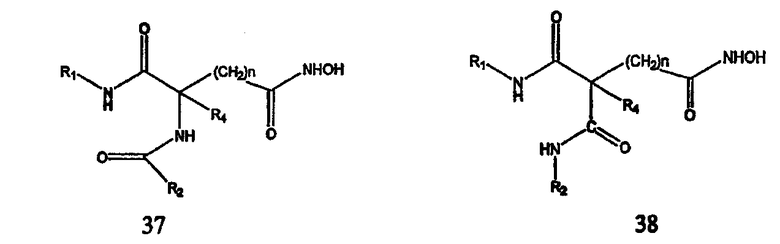
где R1, R2, R4и n имеют значения, описанные для формулы 36.
Соединение, описываемое структурной формулой 39
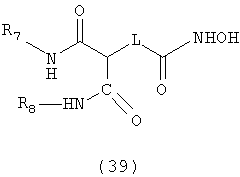
где L обозначает линкер, выбираемый из группы, состоящей из амидного фрагмента, -O-, -S-, -NH-, NR5, -CH2-, -(CH2)m-, -(CH=CH)-, фенилена, циклоалкилена или любого их сочетания, где R5 обозначает замещенный или незамещенный С1-С5 алкил и где каждый из R7 и R8 независимо обозначает замещенный или незамещенный арил (например, фенил), арилалкил (например, бензил), нафтил, пиридинамино, 9-пурин-6-амино, тиазоламино, арилокси, арилалкилокси, пиридил, хинолинил или изохинолинил; n равно целому числу от 3 до 10 и m равно целому числу от 0 до 10.
Например, соединение формулы 39 может иметь структуру
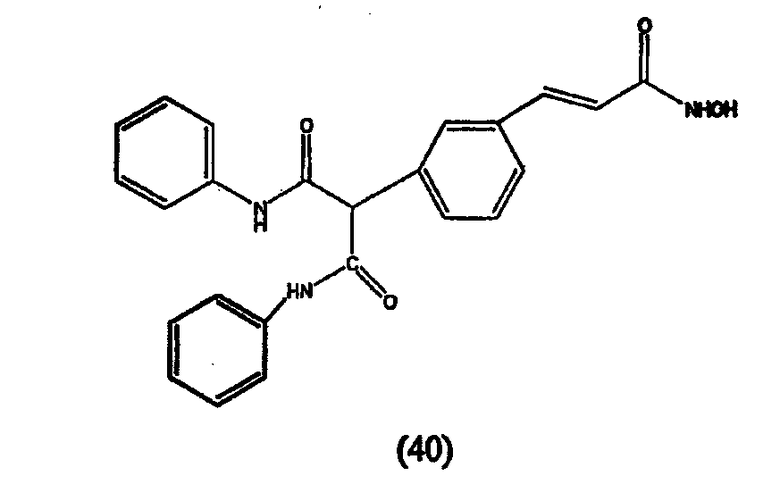
Другие ингибиторы HDAC, подходящие для использования в настоящем изобретении, содержат те соединения, которые описываются представленными ниже более специфичными формулами
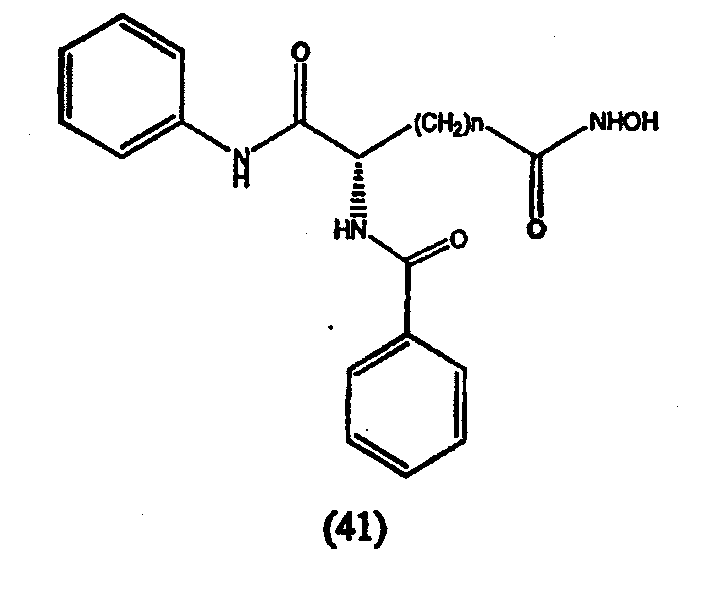
где n равно целому числу от 3 до 10, или их энантиомеры. В одном конкретном варианте формулы 41 n=5.
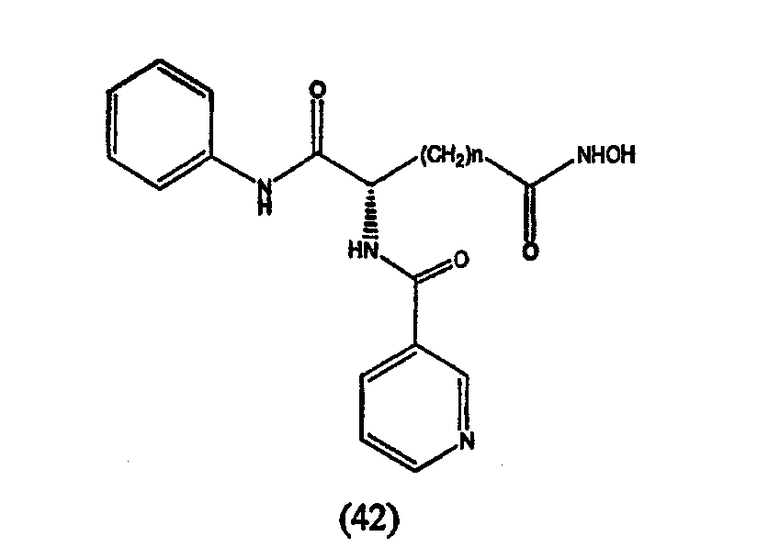
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 42 n=5.
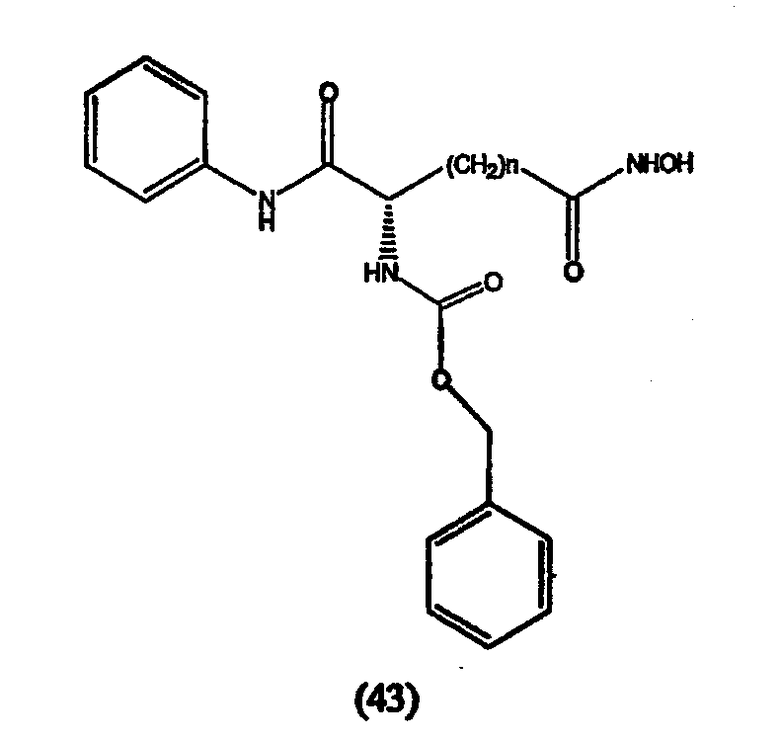
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 43 n=5.
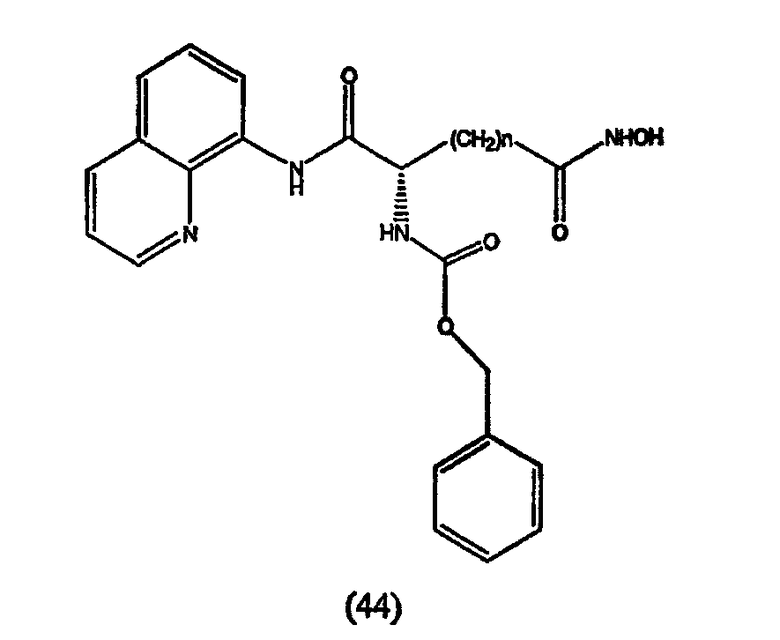
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 44 n=5.
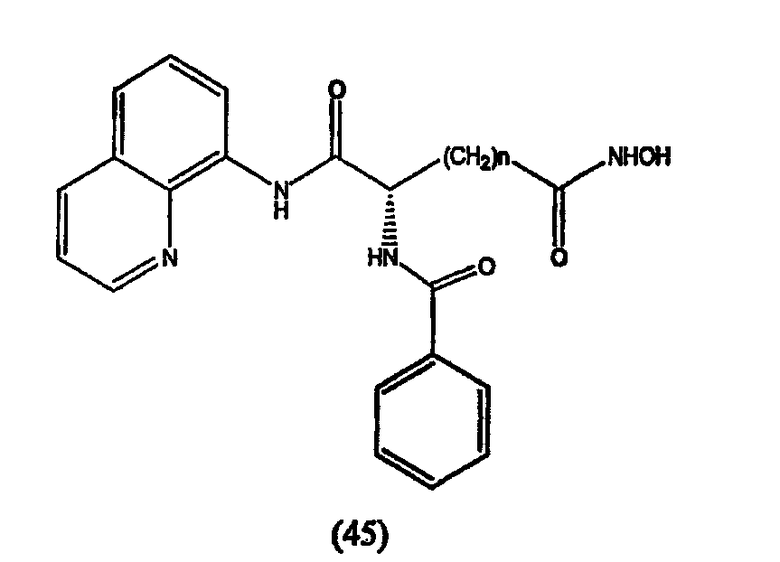
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 45 n=5.
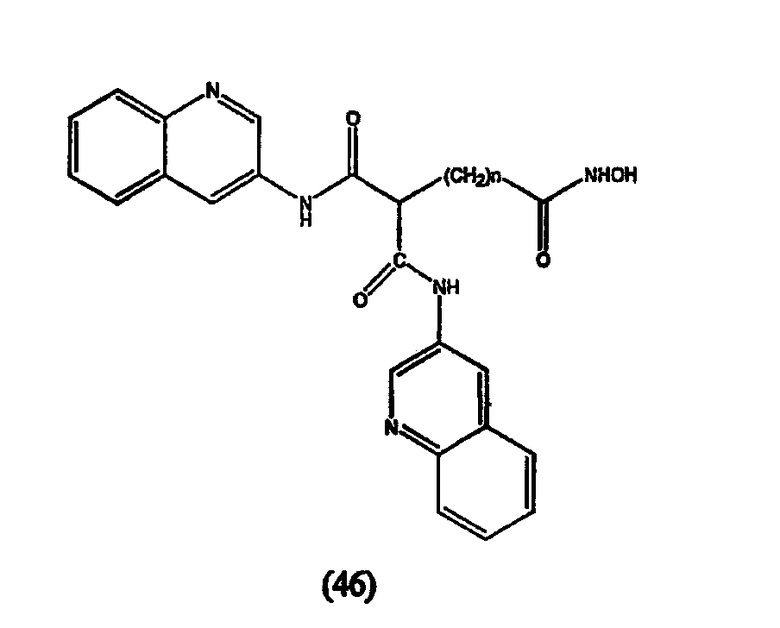
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 46 n=5.
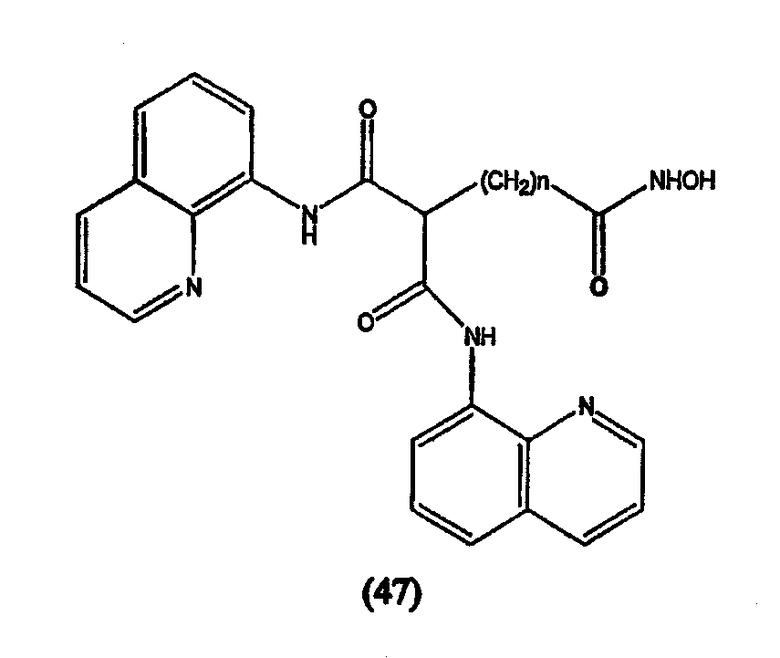
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 47 n=5.
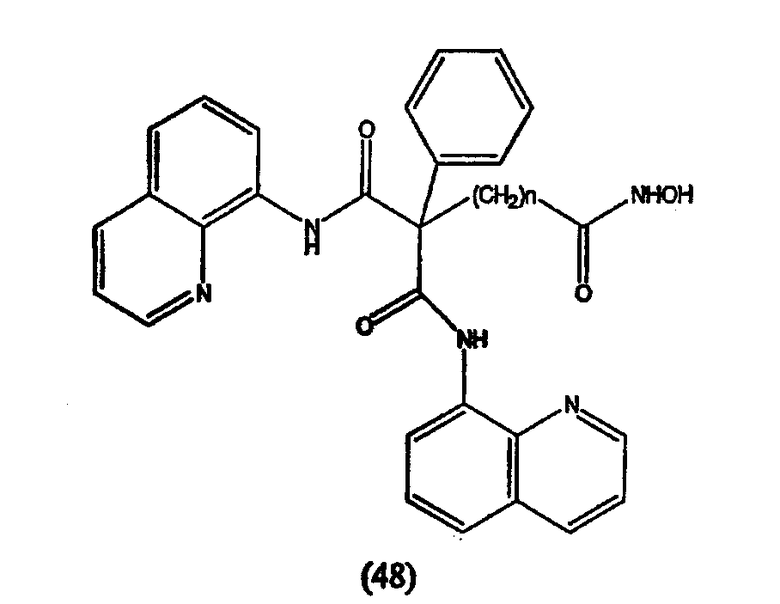
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 48 n=5.
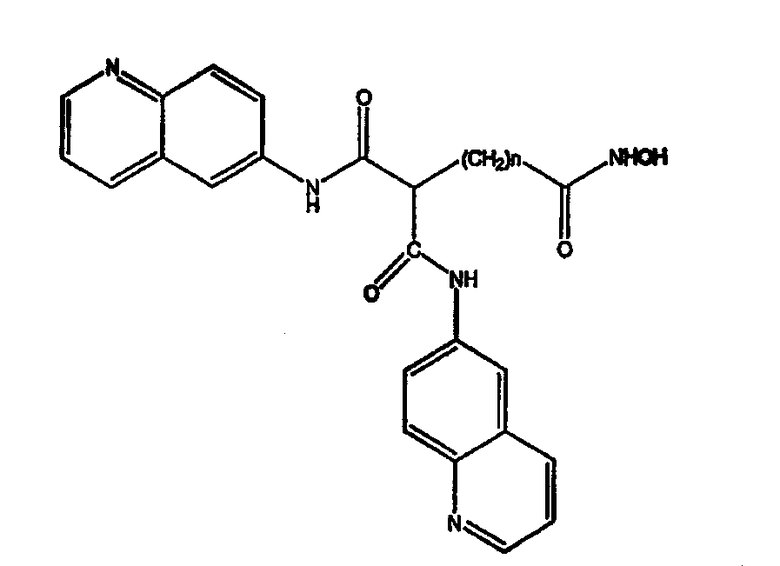
(49)
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 49 n=5.
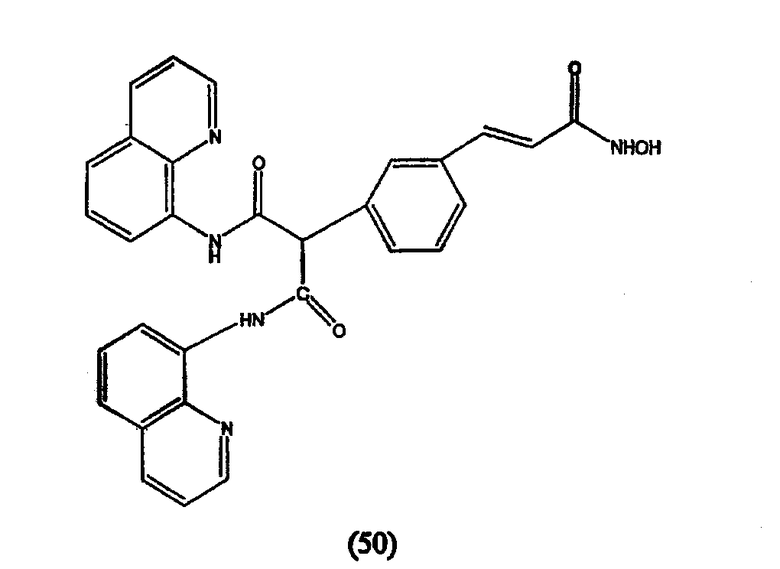
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 50 n=5.
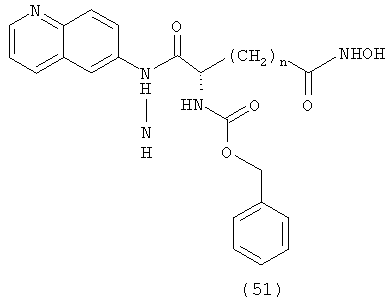
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 51 n=5.
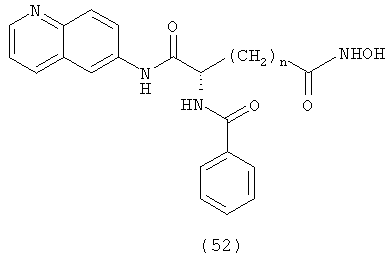
где n равно целому числу от 3 до 10, или его энантиомер. В одном конкретном варианте формулы 52 n=5.
Другие примеры таких соединений и других ингибиторов HDAC описаны в патентной и научной литературе: патент США No. 5369108, выданный 29 ноября 1994 г., патент США No. 5700811, выданный 23 декабря 1997 г., патент США No. 5773474, выданный 30 июня 1998 г., патент США No. 5932616, выданный 3 августа 1999 г., и патент США No. 6511990, выданный 28 января 2003 г., все принадлежащие Breslow и соавт.; патент США No. 5055608, выданный 8 октября 1991 г., патент США No. 5175191, выданный 29 декабря 1992 г., и патент США No. 5608108, выданный 4 марта 1997 г., все принадлежащие Marks с соавт., а также Yoshida, M. et al. Bioassays 17, 423-430 (1995); Saito, A. et al.,PNAS USA 96, 4592-4597, (1999); Furamai R. et al.,PNAS USA 98 (1), 87-92 (2001); Komatsu, Y. et al., Cancer Res. 61(11), 4459-4466 (2001); Su, G.H. et al, Cancer Res. 60, 3137-3142 (2000); Lee, B.I. et al., Cancer Res., 61(3), 931-934; Suzuki, T. et al., J. Med. Chem. 42(15), 3001-3003 (1999); публикация по заявке PCT WO 01/18171, опубликованная 15 марта 2001 г., правами на которую владеют Институт по исследованию злокачественной опухоли (Sloan-Kettering Institute for Cancer Research) и Трастовая компания Колумбийского Университета; публикация по заявке PCT WO 02/246144, принадлежащей Hoffinann-La Roche; публикация по заявке PCT WO 02/22577, принадлежащей Novartis; публикация по заявке PCT WO 02/30879, принадлежащей Prolifix; публикации по заявке PCT WO 01/38322 (опубликована 31 мая 2001 г.), WO 01/70675 (опубликована 27 сентября 2001 г.) и WO 00/71703 (опубликована 30 ноября 2000 г.), все принадлежащие компании Methylgene, Inc.; опубликованная 8 октября 1999 г. публикация по заявке PCT WO 00/21979, принадлежащей компании Fujisawa Pharmaceutical Co., Ltd.; опубликованная 11 марта 1998 г., публикация по заявке PCT WO 98/40080, принадлежащей Beacon Laboratories, L.L.C., а также Curtin M. (Current patent status of histone deacetylase inhibitors Expert Opin. Ther. Patents (2002) 12(9): 1375-1384 и содержащиеся в работе ссылки).
САГК или любое другое соединений из группы HDAC может быть синтезировано в соответствии с методиками, приведенными в экспериментальном разделе настоящей заявки, или по методике, описанной в патентах США NoNo. 5369108, 5700811, 5932616 и 6511990, содержание которых включено в настоящее описание полностью в качестве ссылки, или в соответствии с любой другой методикой, известной специалистам в данной области.
Настоящее изобретение, в дополнение к перечисленным выше соединениям, охватывает использование гомологов и аналогов таких соединений. В данном контексте гомологи представляют собой молекулы, обладающие существенным структурным сходством с описанными выше соединениями, а аналоги представляют собой молекулы, обладающие существенным биологическим сходством, независимо от их структурного сходства.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим фармацевтически приемлемые соли ингибиторов HDAC на основе органических и неорганических кислот, например аддитивные соли кислоты, которая может быть, к примеру, хлористоводородной кислотой, серной кислотой, метансульфоновой кислотой, фумаровой кислотой, малеиновой кислотой, янтарной кислотой, уксусной кислотой, бензойной кислотой, щавелевой кислотой, лимонной кислотой, винной кислотой, угольной кислотой, фосфорной кислотой и т.п. Фармацевтически приемлемые соли могут быть также приготовлены путем обработки неорганическими основаниями, например гидроксидами натрия, калия, аммония, кальция или железа, и такими органическими основаниями, как изопропиламин, триметиламин, 2-этиламиноэтанол, гистидин, прокаин и т.п.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим гидраты ингибиторов HDAC. Термин «гидрат» включает, без ограничения, полугидрат, моногидрат, дигидрат, тригидрат и т.п.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим любую твердую или жидкую физическую форму САГК или любого другого из ингибиторов HDAC. Например, ингибиторы HDAC могут иметь кристаллическую форму, аморфную форму и могут иметь любой размер частиц. Частицы ингибитора HDAC могут быть микронизированы или могут быть агломерированы, а также могут представлять собой гранулированные частицы, порошки, масла, масляные суспензии или любую другую твердую или жидкую физическую форму.
Фармацевтические композиции
Соединения по настоящему изобретению, их производные, фрагменты, аналоги, гомологи, их фармацевтически приемлемые соли или гидраты могут быть включены в состав фармацевтических композиций, приемлемых для перорального введения, вместе с фармацевтически приемлемым носителем или наполнителем. Такие композиции в типичном случае содержат фармацевтически эффективное количество любого из указанных выше соединений и фармацевтически приемлемый носитель. Предпочтительно эффективное количество представляет собой количество, эффективное для селективной индукции конечной дифференцировки соответствующих опухолевых клеток, которое меньше количества, оказывающего токсическое воздействие на пациента.
Любой инертный наполнитель, который обычно используют в качестве носителя или разбавителя, может быть использован в композициях по настоящему изобретению, такой как, например, камедь, крахмал, сахар, целлюлозный материал, акрилат или их смесь. Предпочтительным разбавителем является микрокристаллическая целлюлоза. Композиции могут также содержать разрыхлитель (например, натрийкросскармелозу) и смазывающее вещество (например, стеарат магния) и вдобавок могут содержать одно или более вспомогательных средств, выбираемых из связующего вещества, буфера, ингибитора протеазы, поверхностно-активного вещества, солюбилизирующего средства, пластификатора, эмульгатора, стабилизатора, средства повышения вязкости, подсластителя, пленкообразующего средства или любого их сочетания. Кроме того, композиции по настоящему изобретению могут быть приготовлены в форме композиций с контролируемым высвобождением или с немедленным высвобождением.
Один вариант настоящего изобретения относится к фармацевтической композиции для перорального введения, содержащей ингибитор HDAC или его фармацевтически приемлемую соль или гидрат, микрокристаллическую целлюлозу, натрийкросскармелозу и стеарат магния. Другой вариант относится к САГК в качестве ингибитора HDAC. Другой вариант содержит 50-70 мас.% ингибитора HDAC или его фармацевтически приемлемой соли или гидрата, 20-40 мас.% микрокристаллической целлюлозы, 5-15 мас.% натрийкросскармелозы и 0,1-5 мас.% стеарата магния. Другой вариант содержит примерно 50-200 мг ингибитора HDAC.
В одном варианте фармацевтические композиции вводят перорально, и в этой связи их изготавливают в форме, приемлемой для перорального введения, например в виде твердого или жидкого препарата. Подходящие твердые пероральные композиции содержат таблетки, капсулы, пилюли, гранулы, порошки и т.п. Подходящие жидкие пероральные композиции содержат растворы, суспензии, дисперсии, эмульсии, масла и т.п. В одном варианте осуществления настоящего изобретения композиция изготавливается в виде капсул. В соответствии с данным вариантом композиции по настоящему изобретению содержат, в дополнение к ингибитору HDAC, активному соединению и инертному носителю или разбавителю, твердую желатиновую капсулу.
В контексте настоящего описания термин «фармацевтически приемлемый носитель» содержит любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства, средства для поддержания изотоничности и средства, задерживающие поглощение, и т.п., совместимые с выбранным способом введения фармацевтического препарата, такие как стерильная апирогенная вода. Приемлемые носители описаны в последнем издании руководства Ремингтона (Remington's Pharmaceutical Sciences), представляющего собой стандартное руководство в данной области, которое включено в настоящее описание в качестве ссылки. Предпочтительные примеры таких носителей или разбавителей содержат, без ограничения, воду, солевой раствор, раствор финджера, раствор декстрозы и 5% раствор человеческого сывороточного альбумина. Могут также использоваться липосомы и неводные носители, такие как жирные масла. Использование таких сред и агентов в сочетании с фармацевтически активными веществами хорошо известно в данной области техники. За исключением тех случаев, когда какие-либо традиционные агенты или среды несовместимы с данным активным соединением, использование всех остальных сред и агентов приемлемо для получения настоящих композиций. В композиции могут быть также включены дополнительные активные соединения.
Твердые носители/разбавители содержат, без ограничения, камедь, крахмал (например, кукурузный крахмал, предварительно желированный крахмал), сахар (например, лактозу, манит, сахарозу, декстрозу), целлюлозный материал (например, микрокристаллическую целлюлозу), акрилат (например, полиметилакрилат), карбонат кальция, оксид магния, тальк или их смеси.
Фармацевтически приемлемые носители для жидких композиций могут содержать водные или неводные растворы, суспензии, эмульсии или масла. Примеры неводных растворителей содержат пропиленгликоль, полиэтиленгликоль и инъецируемые сложные органические эфиры, такие как этилолеат. Водные носители содержат воду, спиртовые/водные растворы, эмульсии или суспензии, включая солевой раствор и забуференные среды. Примеры масел содержат такие масла, как масла нефтяного, животного растительного или синтетического происхождения, например, арахисовое масло, соевое масло, минеральное масло, оливковое масло, подсолнечное масло и масло из печени рыб. Растворы или суспензии могут также содержать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, солевой раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетраэтилацетат (ЭДТА); буферные вещества, такие как ацетаты, цитраты или фосфаты, и средства для корректировки тонуса, такие как хлорид натрия или декстроза. Значение рН может быть скорректировано с использованием кислот и оснований, таких как хлористоводородная кислота или гидроксид натрия.
Дополнительно композиции могут также содержать связующие вещества (например, аравийскую камедь, кукурузный крахмал, желатин, карбомер, этилцеллюлозу, гуаровую камедь, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, повидон), разрыхлитель (например, кукурузный крахмал, картофельный крахмал, альгиновую кислоту, диоксид кремния, натрийкросскармелозу, кроссповидон, гуаровую камедь, натрийгликолят крахмала, Примогель (Primogel)), буферы (например, трис-HCl, ацетатный, фосфатный) с различными значениями рН и ионной силы, добавки, такие как альбумин или желатин, для предупреждения адсорбции на поверхности, детергенты (например, Твин-20, Твин-80, Плуроник F68 (Pluronic F68), соли желчных кислот), ингибиторы протеазы, поверхностно-активные вещества (например, лаурилсульфат натрия), усилители проницаемости, солюбилизирующие средства (например, глицерин, полиэтиленглицерин), средства, способствующие скольжению (например, коллоидный диоксид кремния), антиоксиданты (например, аскорбиновая кислота, метабисульфит натрия, бутилированный гидроксианизол), стабилизаторы (например, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза), средства повышения вязкости (например, карбомер, коллоидный диоксид кремния, этилцеллюлоза, гуаровая камедь), подсластители (например, сахароза, аспартам, лимонная кислота), вкусовые вещества (например, добавки со вкусом перечной мяты, метилсалицилат или добавки со вкусом апельсина), консерванты (например, Тимеросал (Thimerosal), бензиловый спирт, парабены), смазывающее вещество (например, стеариновая кислота, стеарат магния, полиэтиленгликоль, лаурилсульфат натрия), средства, способствующие текучести (например, коллоидный диоксид кремния), пластификаторы (например, диэтилфталат, триэтилцитрат), эмульгаторы (например, карбомер, гидроксипропилцеллюлоза, лаурилсульфат натрия), полимерные покрытия (например, полоксамеры или полоксамины), покрытия и средства, образующие пленки (например, этилцеллюлоза, акрилаты, полиметакрилаты) и/или адъюванты.
В одном варианте активные соединения получают с использованием носителей, которые защищают соединение от быстрого удаления из организма, с получением таких композиций, как композиции с контролируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. Могут использоваться биодеградируемые биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Способы получения таких композиций известны специалистам в данной области. Указанные материалы могут быть также получены в коммерческом варианте от компаний Alza Corporation и Nova Pharmaceuticals, Inc. Липосомальные суспензии (содержащие липосомы направленного воздействия на инфицированные клетки с моноклональными антителами к вирусным антигенам) также могут быть использованы в качестве фармацевтически приемлемых носителей. Указанные средства могут быть получены с использованием методик, известных специалистам в данной области, например, как описано в патенте США No. 4522811.
Особенно выгодно изготавливать пероральные композиции в стандартной дозированной форме в связи с легкостью их введения и достигаемой однородностью дозировки. Стандартные дозированные формы в контексте настоящего описания относятся к физически дискретным единицам, применяемым в качестве однократных дозировок для субъектов, подлежащих лечению; каждая такая единица содержит заданное количество активного соединения, которое по расчетам будет оказывать желательный терапевтический эффект, в сочетании с нужным фармацевтическим носителем. Выбор стандартных дозированных форм по настоящему изобретению диктуется и непосредственно зависит от уникальных характеристик активного соединения и от конкретного терапевтического эффекта, которого необходимо достичь, и ограничивается теми рамками, которые известны в области составления фармацевтических композиций, такими как выбор активного соединения для лечения конкретных индивидуумов.
Фармацевтические композиции могут быть включены в состав контейнера, пакета или диспенсера вместе с инструкцией по применению.
Ежедневное введение повторяют непрерывно в течение периода времени от нескольких дней до нескольких лет. Пероральное лечение может длиться непрерывно от одной недели до всей продолжительности жизни пациента. Предпочтительно введение проводят в течение пяти последовательный дней, после чего оценивают состояние пациента для определения необходимости дальнейшего введения. Введение может быть непрерывным или перемежающимся, например лечение может проводиться в течение нескольких последовательных дней, после чего делают период отдыха.
Соединения по настоящему изобретению могут вводиться внутривенно в первый день лечения с последующим пероральным введением на второй и во все последующие дни.
Соединения по настоящему изобретению могут вводиться с целью предотвращения прогрессирования заболевания и стабилизации роста опухоли.
Получение фармацевтических композиций, которые содержат активный компонент, хорошо известно в данной области и осуществляется, например, путем смешивания, гранулирования или путем таблетирования. Активный терапевтический ингредиент часто смешивают с дополнительными ингредиентами, которые являются фармацевтически приемлемыми и совместимыми с активным ингредиентом. Для перорального введения активные агенты смешивают с обычными для данной цели добавками, такими как носители, стабилизаторы или инертные разбавители, и превращают обычными способами в формы, приемлемые для введения, такие как таблетки, покрытые таблетки, твердые или мягкие желатиновые капсулы, водные, спиртовые или масляные растворы и т.п., как было описано выше.
Соединения по настоящему изобретению могут вводиться перорально в суммарной дневной дозе от 25 до 4000 мг/м2, например примерно от 25 до 1000 мг, в дозе 50-1000 мг, 100 мг, 200 мг, 300 мг, 400 мг, 600 мг, 800 мг, 1000 мг и т.п. В типичном случае соединение вводят в виде однократной дозы в случае введения пациенту до 400 мг. В случае более высоких дозировок (например, выше 400 мг), общую дозу разбивают на многократные дозировки, вводимые, например, дважды в день, три раза в день, или подобным образом, предпочтительно распределяя на равные периоды времени в течение дня. Так, например, две дозы, например, по 500 мг каждая, могут вводиться через 12 часов для достижения суммарной дневной дозы в 1000 мг.
В одном предпочтительном варианте САГК или любые другие ингибиторы HDAC вводят пациенту в суммарной дневной дозе 200 мг. В другом предпочтительном варианте САГК или любые другие ингибиторы HDAC вводят пациенту в суммарной дневной дозе 400 мг. В другом предпочтительном варианте САГК или любые другие ингибиторы HDAC вводят пациенту в суммарной дневной дозе 600 мг.
Количество соединения, вводимого пациенту, является меньшим, чем то количество, которое может оказать токсическое воздействие на пациента. В некоторых вариантах количество соединения, которое вводят пациенту, является меньшим, чем то количество, которое приводит к созданию в плазме пациента концентрации искомого соединения, равной или превышающей токсическое содержание данного соединения. Предпочтительно концентрация указанного соединения в плазме пациента поддерживается на уровне примерно 10 нМ. В другом варианте концентрацию соединения в плазме пациента поддерживают на уровне примерно 25 нМ. В другом варианте концентрацию соединения в плазме пациента поддерживают на уровне примерно 50 нМ. В другом варианте концентрацию соединения в плазме пациента поддерживают на уровне примерно 100 нМ. В другом варианте концентрацию соединения в плазме пациента поддерживают на уровне примерно 500 нМ. В другом варианте концентрацию соединения в плазме пациента поддерживают на уровне примерно 1000 нМ. В другом варианте концентрацию соединения в плазме пациента поддерживают на уровне примерно 2500 нМ. В другом варианте концентрацию соединения в плазме пациента поддерживают на уровне примерно 5000 нМ. С использованием БАГМ было показано, что введение указанного соединения в количестве от примерно 5 г/м2/день до примерно 30 г/м2/день, особенно примерно 20 г/м2/день, является эффективным и не приводит к развитию токсического эффекта у пациента. Оптимальное количество соединения, которое должно вводиться пациенту при осуществлении настоящего изобретения, зависит от конкретно используемого соединения и от типа злокачественной опухоли, подлежащей лечению.
В предпочтительном варианте осуществления настоящего изобретения фармацевтическая композиция содержит ингибитор гистондезацетилазы (HDAC), микрокристаллическую целлюлозу в качестве носителя или разбавителя, натрийкросскармелозу в качестве разрыхлителя и стеарат магния в качестве смазывающего вещества. В особенно предпочтительном варианте ингибитор HDAC представляет собой субероиланилид гидроксамовой кислоты (САГК).
Процентное соотношение активного ингредиента и различных наполнителей в указанных композициях можно варьировать. Например, композиция может содержать от 20 до 90 мас.%, предпочтительно 50-70 мас.%, гистондезацетилазы (HDAC). Кроме того, композиция может содержать от 10 до 70 мас.%, предпочтительно 20-40 мас.%, микрокристаллической целлюлозы в качестве носителя или разбавителя. Кроме того, композиция может содержать от 1 до 30 мас.%, предпочтительно 5-15 мас.%, натрийкросскармелозы в качестве разрыхлителя. Кроме того, композиция может содержать 0,1-5 мас.% стеарата магния в качестве смазывающего вещества. В другом предпочтительном варианте композиция содержит примерно 50-200 мг ингибитора HDAC (например, 50 мг, 100 мг и 200 мг ингибитора HDAC, например САГК). В особенно предпочтительном варианте указанная композиция имеет вид желатиновой капсулы.
Предпочтительный вариант осуществления настоящего изобретения относится к твердой композиции САГК с микрокристаллической целлюлозой, НФ (NF, Avicel Ph 101), натрийкросскармелозой, НФ (NF, AC-Di-Sol) и стеаратом магния, НФ (NF), помещенными в желатиновую капсулу. Другой предпочтительный вариант содержит 200 мг твердого САГК с 89,5 мг микрокристаллической целлюлозы, 9 мг натрийкросскармелозы и 1,5 мг стеарата магния, помещенных в желатиновую капсулу.
Специалисту в данной области должно быть понятно, что фармацевтические композиции по настоящему изобретению могут использоваться не только для ингибирования пролиферации опухолевых клеток и лечения злокачественной опухоли, но и что указанные композиции применимы также для лечения широкого перечня заболеваний, при которых могут быть применимы ингибиторы HDAC.
Так, например, было показано, что ингибиторы HDAC, в частности САГК, применимы при лечении множества острых и хронических воспалительных заболеваний, аутоиммунных заболеваний, аллергических заболеваний, заболеваний, связанных с окислительным стрессом, и заболеваний, характеризующихся гиперпролиферацией клеток. Не ограничивающие область применения примеры содержат воспалительные состояния суставов, в том числе ревматоидный артрит (РА) и псориатический артрит; воспалительные заболевания кишечника, такие как болезнь Крона и язвенный колит; спондилоартропатии; склеродерму, псориаз (включая псориаз, опосредованный Т-клетками) и воспалительные дерматозы, такие как дерматит, экзема, атопический дерматит, аллергический контактный дерматит, крапивница, васкулит (например, некротизирующий, кожный и гиперчувствительный васкулит); эозинофильный миозит, эозинофильный фасцит; формы злокачественной опухоли с инфильтрацией лейкоцитов в кожу или органы, ишемическое повреждение, включающее церебральную ишемию (например, повреждение мозга в результате травмы, эпилепсии, геморрагии или инсульта, каждое из которых может привести к нейродегенерации); ВИЧ; сердечная недостаточность, хронические, острые или злокачественные заболевания печени, аутоиммунный тиреоидит; системная красная волчанка, синдром Шегрена, легочные заболевания (например, РДСВ); острый панкреатит; амиотрофический боковой склероз (АБС); болезнь Альцгеймера; кахексия/анорексия; астма; атеросклероз; синдром хронической усталости, лихорадка; диабет (например, инсулиновый диабет или приступ ювенильного диабета); гломерулонефрит; отторжение трансплантатов (например, при трансплантации); геморрагический шок; гиперальгезия; воспалительное заболевание кишечника; рассеянный склероз; миопатии (например, нарушение метаболизма мышечного белка, особенно при сепсисе); остеопороз; болезнь Паркинсона; болевой синдром; преждевременные роды; псориаз; реперфузионное поражение; цитокин-индуцированная токсичность (например, септический шок, эндотоксический шок); побочные эффекты лучевой терапии; заболевания височно-нижнечелюстного сустава, опухолевые метастазы; или при воспалительных состояниях, вызванных перегрузками, растяжением, повреждением хрящей, травмами, такими как ожог, при ортопедических операциях, при инфекции и других болезненных процессах. Аллергические заболевания и состояния содержат, не ограничиваясь приведенным перечнем, респираторные аллергические заболевания, такие как астма, аллергический ринит, гиперчувствительное состояние легких, аллергическая пневмония, эозинофильная пневмония (например, синдром Лефлера, хроническая эозинофильная пневмония), гиперчувствительность задержанного типа, интерстициальная болезнь легкого (ИЛБ) (например, идиопатический легочный фиброз или ИЛБ, связанная с ревматоидным артритом, системной красной волчанкой, анкилозирующим спондилоартритом, системным склерозом, синдромом Шегрена, полимиозитом или дерматомиозитом); системная анафилаксия или реакция гиперчувствительности, лекарственная аллергия (например, на пенициллин, цефалоспорины), аллергия на укусы насекомых и т.п.
Например, было показано, что ингибиторы HDAC, в частности САГК, применимы при лечении множества нейродегенеративных заболеваний, не исчерпывающий область применения перечень которых содержит следующие.
I. Заболевания, характеризующиеся прогрессирующей деменцией при отсутствии других выраженных неврологических признаков, такие как болезнь Альцгеймера, сенильная деменция типа Альцгеймера и болезнь Пика (долевая атрофия).
II. Синдромы, объединяющие прогрессирующую деменцию с другими выраженными неврологическими аномалиями, такие как А) синдромы, возникающие в основном у взрослых (например, болезнь Хантингтона, множественная системная атрофия, объединяющая деменцию с атаксией и/или проявлениями болезни Паркинсона), прогрессирующий супрануклеарный паралич (синдром Стила-Ричардсона-Ольшевского), диффузное заболевание телец Леви и кортикодентатонигральная дегенерация); Б) синдромы, возникающие в основном у детей или у взрослых в молодом возрасте (например, болезнь Халлевордена-Шпатса и прогрессирующая семейная миоклоническая эпилепсия).
III. Синдромы постепенно развивающихся аномалий осанки и движений, такие как ажитированный паралич (болезнь Паркинсона), стриатонигральная дегенерация, прогрессирующий супрануклеарный паралич, торсионная дистония (торсионный спазм, деформирующая дистония мышц), спастическая кривошея и другие дискинезии, наследственный тремор и синдром Жиль де ля Туретта.
IV. Синдромы прогрессирующей атаксии, такие как мозжечковые дегенерации (например, мозжечково-корковая дегенерация и оливомостомозжечковая атрофия (ОММА (OPCA)), и спиномозжечковая дегенерация (атаксия Фридриха и родственные расстройства).
V. Синдром недостаточности центральной автономной нервной системы (синдром Шая-Дреджера).
VI. Синдромы мышечной слабости и истощения без сенсорных изменений (заболевания двигательных нейронов, такие как амиотрофический боковой склероз, спинальная мышечная атрофия (например, детская спинальная мышечная атрофия (атрофия Верднига-Хоффмана), юношеская спинальная мышечная атрофия (атрофия Вольфарта-Кугельберга-Веландера) и другие формы наследственной спинальной мышечной атрофии), первичный боковой склероз и наследственная спастическая параплегия.
VII. Синдромы, объединяющие мышечную слабость и истощение с сенсорными изменениями (прогрессирующая нервно-мышечная атрофия; хронические семейные полиневропатии), такие как мышечная атрофия перонеального типа (болезнь Шарко-Мари-Тута); гипертрофическая интерстициальная полиневропатия (болезнь Дежерина-Сотта); другие формы хронической прогрессирующей невропатии.
VIII. Синдромы прогрессирующей потери зрения, такие как пигментная дегенерация сетчатки (пигментный ретинит) и наследственная атрофия зрительных нервов (болезнь Лебера).
Далее, в рамках экспериментального раздела настоящее изобретение иллюстрируется примерами. Данный раздел приведен для пояснения настоящего изобретения, но он никоим образом не направлен на ограничение области настоящего изобретения, которое определяется приведенной в нем формулой изобретения.
ПОДРОБНЫЙ ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
ПРИМЕР 1
Синтез САГК
САГК может быть синтезирован по описанной ниже методике или в соответствии с методикой, описанной в патенте США No. 5369108, содержание которого включено в настоящее описание полностью в качестве ссылки, или по любой другой методике.
Синтез САГК
Стадия 1. Синтез суберанилиновой кислоты
В колбу на 22 л вносят 3500 г (20,09 моль) субериновой кислоты и расплавляют кислоту при нагревании. Температуру повышают до 175°С и затем добавляют 2040 г (21,92 моль) анилина. Температуру повышают до 190°С и выдерживают смесь при этой температуре в течение 20 минут. Затем расплав переливают в сосуд Нальгена, содержащий 4017 г гидроксида калия, растворенного в 50 л воды. Полученную смесь перемешивают в течение 20 минут после добавления расплава. Реакционную смесь воспроизводят в тех же масштабах и вторую порцию расплава выливают в тот же раствор гидроксида калия. После тщательного перемешивания смеси мешалку убирают и оставляют смесь для осаждения. Далее смесь фильтруют через подушку целлита (4200 г), полученный продукт фильтруют для удаления нейтрального побочного продукта (для предупреждения воздействия анилина на оба конца субериновой кислоты). Фильтрат содержит соль продукта, а также соль непрореагировавшей субериновой кислоты. Полученную смесь оставляют для осаждения, поскольку фильтрование идет очень медленно и занимает несколько дней. Полученный фильтрат подкисляют с использованием 5 л концентрированной хлористоводородной кислоты; указанную смесь перемешивают в течение одного часа и затем оставляют на ночь для осаждения. Полученный продукт собирают фильтрованием и промывают на воронке деионизированной водой (4х5 л). Влажную фильтр-прессную лепешку переносят с фильтра в колбу на 72 л, содержащую 44 л деионизированной воды, нагревают полученную смесь до 50°С и отделяют твердый материал горячим фильтрованием (желательный продукт загрязнен субериновой кислотой, которая обладает гораздо большей растворимостью в горячей воде. Делают несколько растираний в горячей воде для удаления субериновой кислоты. Продукт анализируют методом ЯМР [D6-ДМСО] для контроля удаления субериновой кислоты). Горячее растирание повторяют с использованием 44 л воды при температуре 50°С. Продукт снова отделяют фильтрованием и промывают 4 л горячей воды. Далее его высушивают в течение выходных дней в вакуумном шкафу при 65°С с использованием насоса Нэша в качестве источника вакуума. Насос Нэша представляет собой жидкостный кольцевой насос (водяной), который создает вакуум на уровне примерно 29 дюймов ртутного столба. Для удаления воды в качестве вспомогательного средства используют периодическое пропускание аргона, получая в итоге 4182,8 г суберанилиновой кислоты.
Указанный продукт все еще содержит небольшое количество субериновой кислоты; в этой связи, проводят горячее растирание порций полученного продукта при 65°С, используя каждый раз примерно 300 г продукта. Каждую порцию фильтруют и тщательно промывают дополнительным количеством горячей воды (всего используют примерно 6 л). Указанную процедуру повторяют до полной очистки данной партии. Такая процедура приводит к полному удалению субериновой кислоты из продукта. Полученный твердый продукт объединяют в колбе и перемешивают с 6 л смеси метанол/вода (1:2) и затем отделяют фильтрованием и высушивают на воздухе прямо на фильтре в течение выходных дней. Затем продукт помещают на подносы и сушат в вакуумном шкафу при 65°С в течение 45 часов с использованием насоса Нэша и продувки аргоном. Готовый продукт имеет вес 3278,4 г (выход продукта 32,7%).
Стадия 2. Синтез метилсуберанилата
В колбу на 50 л, присоединенную к механической мешалке и конденсатору, вносят 3229 г суберанилиновой кислоты, полученной на предыдущей стадии, 20 л метанола и 398,7 г смолы Dowex 50WX2-400. Полученную смесь нагревают до температуры кипения с обратным холодильником и выдерживают при температуре кипения с обратным холодильником в течение 18 часов. Смесь фильтруют для удаления гранул смолы и собирают фильтрат для переноса в роторный испаритель.
Остаток, полученный после выпаривания на роторном испарителе, переносят в колбу на 50 л, снабженную конденсатором и механической мешалкой. В указанную колбу добавляют 6 л метанола и нагревают полученную смесь до получения раствора. Затем добавляют 2 л деионизированной воды и прекращают нагревание. Смеси дают охладиться при перемешивании, а затем помещают колбу на ледяную баню и дают реакционной смеси охладиться. Полученный твердый продукт отделяют фильтрованием и промывают фильтрпресную лепешку на фильтре с использованием 4 л холодной смеси метанол/вода (1:1). Полученный продукт сушат при 45°С в вакуумном шкафу с использованием насоса Нэша в общей сложности в течение 64 часов, получая в итоге 2850,2 г (выход продукта 84%) метилсуберанилата, CSL лот #98-794-92-31.
Стадия 3. Синтез неочищенного САГК
В колбу на 50 л, снабженную механической мешалкой, термопарой и вводом для создания инертной атмосферы, помещают 1451,9 г гидрохлорида гидроксиламина, 19 г безводного метанола и 3,93 л 30% раствора метоксида натрия в метаноле. Затем в колбу вносят 2748,0 г метилсуберанилата и затем 1,9 л 30% раствора метоксида натрия в метаноле. Полученную смесь подвергают перемешиванию в течение 16 часов и 10 минут. Примерно половину реакционной смеси переносят из реакционной колбы (колба 1) в колбу на 50 л (колба 2), снабженную механической мешалкой. Затем в колбу 1 добавляют 27 л деионизированной воды и перемешивают полученную смесь в течение 10 минут. С помощью рН-метра измеряют значение рН, которое составляет 11,56. Значение рН смеси доводят до 12,02 путем добавления 100 мл 30% раствора метоксида натрия в метаноле; при этом образуется прозрачный раствор (реакционная смесь в этот момент содержит небольшое количество твердого материала. Значение рН корректируют до получения прозрачного раствора, из которого будет проводиться осаждение осаждаемого продукта). Реакционную смесь в колбе 2 разбавляют таким же образом; добавляют 27 л деионизированной воды и корректируют рН путем добавления к реакционной смеси 100 мл 30% раствора метоксида натрия в метаноле до достижения значения рН 12,01 (прозрачный раствор).
Реакционную смесь в каждой колбе подкисляют путем добавления ледяной уксусной кислоты, осаждая искомый продукт. В колбе 1 конечное значение рН составляет 8,98, а колба 2 имеет конечное значение рН 8,70. Полученный продукт из обеих колб отделяют фильтрованием с использованием воронки Бюхнера и тканевого фильтра. Фильтрпресную лепешку промывают 15 л деионизированной воды, после чего воронку закрывают и частично высушивают продукт прямо в воронке под вакуумом в течение 15,5 часов. Полученный продукт вынимают и размещают на пяти стеклянных подносах. Подносы помещают в вакуумный шкаф и сушат продукт до постоянного веса. Первый период сушки составляет 22 часа при 60°С с использованием насоса Нэша в качестве источника вакуума и с продувкой аргоном. После этого подносы вынимают из вакуумного шкафа и взвешивают. Затем подносы возвращают в шкаф и продукт сушат еще в течение 4 часов и 10 минут с использованием масляного насоса в качестве источника вакуума и без продувки аргоном. Полученный материал упаковывают в двойные (4-mill) полиэтиленовые пакеты и помещают во внешний пластиковый контейнер. Конечный вес после отбора образца составляет 2633,4 г (95,6%).
Стадия 4. Перекристаллизация неочищенного САГК
Неочищенный САГК перекристаллизовывают из смеси метанол/вода. В колбу на 50 л, снабженную механической мешалкой, термопарой, конденсатором и вводом для создания инертной атмосферы, помещают неочищенный САГК, подлежащий кристаллизации (2525,7 г), после чего добавляют 2625 мл деионизированной воды и 15755 мл метанола. Материал нагревают до температуры кипения с обратным холодильником с получением раствора. Затем в реакционную смесь добавляют 5250 мл деионизированной воды. Нагревание прекращают и оставляют смесь для остывания. Когда реакционная смесь остынет достаточно для того, чтобы с колбой можно было безопасно работать (28°С), колбу снимают с нагревательной сетки и помещают в ванну, используемую в качестве охлаждающей бани. В ванну помещают смесь лед/вода для охлаждения реакционной смеси до температуры -5°С. Смесь выдерживают ниже данной температуры в течение 2 часов. Искомый продукт отделяют фильтрованием и промывают фильтр-прессную лепешку на фильтре с помощью 1,5 л холодной смеси метанол/вода (2:1). Затем воронку накрывают и частично высушивают продукт под вакуумом в течение 1,75 часа. Затем продукт вынимают из воронки и размещают на 6 стеклянных подносах. Подносы помещают в вакуумный шкаф и сушат продукт в течение 64,75 часа при температуре 60°С с использованием насоса Нэша в качестве источника вакуума и с продувкой аргоном. Затем подносы вынимают для взвешивания, после чего снова возвращают в шкаф и сушат еще 4 часа при 60°С до достижения постоянного веса. Источником вакуума при повторном высушивании служит масляный насос, а продувка аргоном не используется. Полученный материал упаковывают в двойные (4-mill) полиэтиленовые пакеты и помещают во внешний пластиковый контейнер. Конечный вес после отбора образца составляет 2540,9 г (92,5%).
ПРИМЕР 2
Пероральное дозирование субероиланилида гидроксамовой кислоты (САГК)
Предпосылки. Лечение гибридными полярными средствами, воздействующими на клеточную дифференцировку, приводит к ингибированию роста клеточных линий, получаемых из солидных опухолей человека и ксенотрансплантатов. Указанный эффект частично опосредован ингибированием гистондезацетилазы. САГК является мощным ингибитором гистондезацетилазы, который, как было показано, обладает способностью индуцировать остановку роста опухолевых клеток, дифференцировку и апоптоз в лабораторных и доклинических испытаниях.
Цели. Определяется режим безопасной ежедневной пероральной дозировки САГК, которая может использоваться в испытаниях на фазе II. Кроме того, дается оценка фармакокинетического профиля пероральной композиции САГК. Отслеживается пероральная биологическая доступность САГК у людей натощак в сравнении с результатом приема не натощак, а также противоопухолевый эффект лечения. Дополнительно оценивается также биологическое воздействие САГК на нормальные ткани и опухолевые ткани и фиксируются ответные реакции в отношении уровня ацетилирования гистонов.
Пациенты. В испытания включены пациенты с гистологически доказанной развитой стадией первичных или метастазированных солидных опухолей взрослых, которые не поддаются стандартной терапии или для которых не существует стандартной излечивающей терапии. Пациенты должны иметь статус показателя Карновски (Karnofsky Performance Status) ≥70%, иметь адекватную гематологическую, печеночную и почечную функции. У пациентов должно пройти по меньшей мере четыре недели после предшествующего курса химиотерапии, лучевой терапии или употребления иных противораковых лекарственных средств.
Режим дозирования. На первый день пациенты получают вначале 200 мг внутривенно вводимого САГК. Начиная со второго дня, пациенты получают дневные дозы перорального САГК в соответствии с таблицей 1. Каждая когорта получает разную дозу САГК. "QD" указывает на однократное дозирование в день; "Q12 часов" указывает на дозирование два раза в день. Например, пациенты в когорте IV получают две дозы по 800 мг САГК в день. Дозы вводят пациентам ежедневно и непрерывно. Образцы крови для исследования отбирают на первый день и на 21 день после начала перорального лечения. В ряде случаев лечение пероральным САГК у некоторых пациентов было отменено в связи с прогрессированием заболевания, регрессией опухоли, развитием неприемлемых побочных эффектов или в связи с проведением другого курса лечения.
Режим дозирования перорального САГК
Результаты. Сравнение уровней в плазме крови указывает на высокую биологическую доступность САГК, вводимого перорально, как в случае приема натощак, так и в том случае, когда пациенты не воздерживались от еды, в сравнении с внутривенным введением САГК (в/в САГК). «ППК (AUC)» представляет собой оценку биологической доступности САГК в (нг/мл)мин, где 660 нг/мл соответствует 2,5 мкМ САГК. Величина ППК, взятая в сочетании с полупериодом выведения (t1/2), показывает, что суммарная биологическая доступность у перорального САГК лучше, чем у в/в САГК. Показатель Cmax представляет собой максимальную концентрацию САГК, наблюдаемую после введения. В/в САГК вводят в количестве 200 мг путем инфузии в течение двух часов. Пероральный САГК вводят в виде однократной капсулы в количестве 200 мг. В таблицах 2 и 3 суммированы результаты определений методом ВЭЖХ (LCMS с использованием дейтериевого стандарта), которые позволяют оценить количество САГК в плазме крови пациента в зависимости от времени, с использованием ацетилированного гистона-4 (α-AcH4) в качестве маркера.
Содержание перорально вводимого САГК в плазме крови
Пациент №1
(не натощак)
Содержание перорально вводимого САГК в плазме крови
Пациент №2
(не натощак)
Фиг. 1-8 представляют собой слайды, показывающие результаты ВЭЖХ в отношении количества α-AcH4 у пациентов в когортах I и II, измеряемого в течение 10 часов после получения пероральной дозы, в сравнении с уровнем α-AcH4 при внутривенном введении САГК. На фиг.9 показано среднее значение концентрации САГК в плазме (нг/мл) в указанные временные точки после введения. Фиг.9А: пероральная доза (200 мг и 400 мг), вводимая натощак, на день 8. Фиг.9В: пероральная доза, принимаемая с пищей, на день 9. Фиг.9С: в/в доза на день 1. Фиг.10 показывает кажущийся период полураспада перорального САГК в дозе 200 мг и 400 мг в дни 8, 9 и 22. Фиг.11 показывает величину ППК (нг/мл/час) перорально вводимого САГК в дозе 200 мг и 400 мг в дни 8, 9 и 22. Фиг.12 показывает биологическую доступность САГК после перорального приема дозы 200 мг и 400 мг в дни 8, 9 и 22.
ПРИМЕР 3
Введение пероральной дозы субероиланилида гидроксамовой кислоты (САГК) - повышение дозы
В другом эксперименте двадцать пять пациентов с солидными опухолями были включены в подгруппу А, тринадцать пациентов с болезнью Ходжкина и не-ходжкинской лимфомой были включены в подгруппу В, и один пациент с острым лейкозом и еще один пациент с миелодиспластическим синдромом были включены в подгруппу С, как показано в таблице 4.
Схема повышения дозы и количество пациентов, отобранных для каждого уровня дозирования
(подгруппа А/подгруппа В/подгруппа С)*
Результаты. Среди одиннадцати пациентов, проходивших лечение в когорте II, у одного пациента развились признаки DLT: диарея 3 стадии и обезвоживание 3 стадии в ходе первого цикла лечения. Девять пациентов были включены в когорту III. У двух пациентов не удалось оценить развитие токсичности на 28 день в связи с преждевременным прекращением исследования из-за быстрого прогрессирования заболевания. Из семи оставшихся пациентов у пяти появились признаки DLT в ходе первого цикла лечения: диарея/обезвоживание (n=1), слабость/обезвоживание (n=1), анорексия (n=1), обезвоживание (n=1) и анорексия/обезвоживание (n=1). Пятеро указанных пациентов восстановились примерно в течение одной недели после приостановки испытания. Далее они получали сниженные дозы по 400 мг один раз в день, которые они, по всей видимости, хорошо переносили. Средняя длительность приема дозы 400 мг дважды в день для всех пациентов в группе III составляла 21 день. На основании полученных результатов был сделан вывод, что режим дозирования, содержащий прием дозы 400 мг каждые 12 часов, превосходит максимально переносимую дозу. После изменения процедуры продолжают нарастающее введение дозы в группе IV в дозе 600 мг один раз в день. Из семи пациентов, включенных в группу IV, у двоих оценить состояние по развитию токсичности на 28 день оказалось невозможно из-за преждевременного прекращения исследования в связи с быстрым прогрессированием заболевания. У троих пациентов в ходе первого цикла лечения развились признаки DLT: анорексия/обезвоживание/утомляемость (n=1) и диарея/обезвоживание (n=2). В этой связи был сделан вывод о том, что доза в 600 мг превосходит максимально переносимую дозу, и в качестве максимально переносимой дозы для однократного ежедневного перорального приема была определена доза 400 мг. Процедура была изменена для дополнительной оценки уровней дозы, вводимой в непрерывном режиме каждый день в дозировке 200 мг два раза в день и 300 мг два раза в день.
Фармакокинетический анализ был проведен на промежуточной стадии у 18 пациентов, получавших лечение дозами по 200 мг один раз в день, 400 мг один раз в день и 400 мг два раза в день. В целом, средние оценки значений Cmax и ППКбескон (AUCinf) для вводимого перорально натощак или с приемом пищи САГК повышались пропорционально повышению дозы в диапазоне от 200 до 400 мг. В целом, фракция ППКбескон (AUCinf) по результатам экстраполяции составляет 1% и менее. Средние оценки кажущегося периода полураспада варьируют по группам дозирования как при приеме натощак, так и при приеме вместе с пищей в интервале от 61 до 114 минут. Средние оценки Cmax варьируют от 233 нг/мл (0,88 мкМ) до 570 нг/мл (2,3 мкМ). Биологически доступная фракция САГК, вычисленная на основании значений ППКбескон (AUCinf) после в/в инфузии и перорального введения, составляет, как было показано, примерно 0,48.
Мононуклеарные клетки периферической крови собирают до начала лечения, сразу после инфузии и в интервале от 2 до 10 часов после перорального введения капсул с САГК для оценки эффекта САГК на уровень ацетилирования гистонов в нормальных хозяйских клетках. Гистоны выделяют и зондируют антителами к ацетилированному гистону (Н3) с последующим введением конъюгированных с пероксидазой хрена вторичных антител. Предварительный анализ показывает увеличение в накоплении ацетилированных гистонов в периферических мононуклеарных клетках, что может быть обнаружено в период до 10 часов после перорального приема капсул с САГК с уровнем дозирования 400 мг в день.
Лечение в течение 3-12 месяцев продолжили тринадцать пациентов с развившимися или стабильными заболеваниями: щитовидной железы (n=3), потовых желез (n=1), почек (n=2), гортани (n=1), предстательной железы (n=1), болезнью Ходжкина (n=2), не-ходжкинской лимфомой (n=2) и лейкозом (n=1).
У шестерых пациентов было отмечено сморщивание опухоли по результатам КТ-сканирования. Трое из указанных шести пациентов удовлетворяли критерию частичной реакции (один пациент с метастазами злокачественной опухоли гортани и два пациента с не-ходжкинскими лимфомами). Указанные частичные реакции имели место при приеме дозы 400 мг два раза в день (n=2) и 600 мг один раз в день (n=1).
ПРИМЕР 4
Внутривенное введение доз САГК
В таблице 5 показан режим дозирования для пациентов, получающих внутривенный САГК. Пациенты, вначале объединенные в когорту I, начинают получать по 300 мг/м2 САГК в течение пяти последовательных дней недели в первую неделю до достижения общей дозы 1500 мг/м2. Затем пациенты наблюдаются в течение двух недель и затем продолжают лечение в когорте II, после чего переходят в последующие когорты, если лечение не прерывается в связи с прогрессированием заболевания, регрессией опухоли, неприемлемыми побочными эффектами или по причине получения пациентом другого курса лечения.
Стандартная процедура наращивания дозы при внутривенном введении САГК
(мг/м2)
Несмотря на то, что настоящее изобретение показано конкретно и описано со ссылками на предпочтительные варианты его осуществления, специалисту в данной области понятно, что могут быть внесены различные изменения по форме и в деталях без отхода от смысла настоящего изобретения. В значительной степени, область настоящего изобретения определяется прилагаемой ниже формулой изобретения.
Список цитированной литературы
1. Sporn, M. В., Roberts, А. В. and Driscoll, J. S. (1985) in Cancer Principles and Practice of Oncology, eds. Hellman, S., Roseaberg, S. A. and DeVita, V. Т., Jr., Bd. 2,(J. B. Lippincott, Philadelphia), P. 49.
2. Breitman, Т. R., Selonick, S. В. and Collins, S. J. (1980) Proc. Natl. Acad. Sci. USA 77:2936-2940.
3. Olsson, I. L. and Breitman, T. R. (1982) Cancer Res. 42:3924-3927.
4. Schwartz, E. L. and Sartorelli, А. С. (1982) Cancer Res. 42:2651-2655.
5. Marks, P. A., Sheffery, M. and Rifkind, R. A. (1987) Cancer Res. 47:659.
6. Sachs, L. (1978) Nature (Lond.) 274:535.
7. Friend, С., Scher, W., Holland, J. W. and Sato, T. (1971) Proc. Natl. Acad. Sci (USA) 68:378-382.
8. Tanaka, M., Levy, J., Terada, M., Breslow, R., Rifkind, R. A. and Marks, P. A. (1975) Proc. Natl. Acad. Sci. (USA) 72:1003-1006.
9. Reuben, R. С., Wife, R. L., Breslow, R., Rifkind, R. A. and Marks, P. A. (1976) Proc. Natl. Acad. Sci (USA) 73: 862-866.
10. Abe, E., Miyaura, C., Sakagami, H., Takeda, M., Konno, К., Yamazaki, Т., Yoshika, S. and Suda, T. (1981) Proc. Natl, Acad, Sci. (USA) 78:4990-4994.
11. Schwartz, E. L., Snoddy, J. R., Kreutter, D., Rasmussen, H. and Sartorelli, A. C. (1983) Proc. Am. Assoc. Cancer Res. 24:18.
12. Tanenaga, К., Hozumi, M. and Sakagami, Y. (1980) Cancer Res. 40:914-919.
13. Lotem, J. and Sachs, L. (1975) Int. J. Cancer 15:731-740.
14. Metcalf, D. (1985) Science, 229:16-22.
15. Scher, W, Scher, В. M. and Waxman, S. (1983) Exp. Hematol. 11:490-498.
16. Scher, W., Scher, В. M. and Waxman, S. (1982) Biochem. & Biophys. Res. Comm. 109:348-354.
17. Huberman, E. and Callaham, M. F. (1979) Proc. Natl. Acad. Sci. (USA) 76:1293-1297.
18. Lottem, J. and Sachs, L. (1979) Proc. Natl. Acad. Sci. (USA) 76:5158-5162.
19. Terada, M., Epner, E., Nudel, U., Salmon, J., Fibach, E., Rifkind, R. A. and Marks, P. A. (1978) Proc. Natl. Acad. Sci. (USA) 75:2795-2799.
20. Morin, M. J. and Sartorelli, A. C. (1984) Cancer Res. 44:2807-2812.
21. Schwartz, E. L, Brown, B. J., Nierenberg, M., Marsh, J. C. and Sartorelli, A. C. (1983) Cancer Res. 43:2725-2730.
22. Sugano,H., Furusawa, M., Kawaguchi, Т., and Ikawa, Y. (1973) Bibl. Hematol. 39:943-954.
23. Ebert, P. S., Wars, I. and Buell, D. N. (1976) Cancer Res. 36:1809-1813.
24. Hayashi, M., Okabe, J. and Hozumi, M. (1979) Gann 70:235-238.
25. Fibach, E., Reuben, R. C., Rifkind, R. A. and Marks, P. A. (1977) Cancer Res. 37: 440-444.
26. Melloni, E., Pontremoli, S., Damiani, G., Viotti, P., Weich, N., Rifkind, R. A. and Marks, P. A. (1988) Proc. Natl. Acad. Sci. (USA) 85:3835-3839.
27. Reuben, R., Khanna, P. L, Gazitt, Y., Breslow, R., Rifkind, R. A. and Marks, P. A. (1978) J. Biol. Chem. 253:4214-4218.
28. Marks, P. A. and Rifkind, R. A. (1988) International Journal of Cell Cloning 6: 230-240.
29. Melloni, F., Pontremoli, S., Michetti, M., Sacco, O., Cakiroglu, A. G., Jackson, J. F., Rifkind, R. A. and Marks, P. A. (1987) Proc. Natl. Acad. Sciences (USA) 84:5282-5286.
30. Marks, P. A. and Rifkind, R. A. (1984) Cancer 54:2766-2769.
31. Egorin, M. J., Sigman, L. M. VanEcho, D. A., Forrest, A., Whitacre, M. Y. and Aisner, J. (1987) Cancer. Res. 47:617-623.
32. Rowinsky, E. W., Ettinger, D. S., Grochow, L. В., Brundrett, R. В., Cates, А. E. and Donehower, R. C. (1986) J. Clin. Oncol. 4:1835-1844.
33. Rowinsky, E. L. Ettinger, D. S., McGuire, W. P., Noe, D. A., Grochow, L. В. and Donehower, R. C. (1987) Cancer Res. 47: 5788-5795.
34. Callery, P. S., Egorin, M. J., Geelhaar, L. A. and Nayer, M. S. B. (1986) Cancer Res. 46:4900-4903.
35. Young, C. W. Fanucchi, M. P., Walsh, Т. В., Blatzer, L., Yaldaie, S., Stevens, Y. W., Gordon, C., Tong, W., Rifkind, R. A. and Marks, P. A. (1988) Cancer Res. 48:7304-7309.
36. Andreeff, M., Young, C., Clarkson, В., Fetten, J., Rifkind, R. A. and Marks, P. A. (1988) Blood 72:186a.
37. Marks, P. A., Breslow, R., Rifkind, R. A., Ngo, L. and Singh, R. (1989) Proc. Natl. Acad. Sci. (USA) 86:6358-6362.
38. Breslow, R., Jursic, В., Yan, Z. F., Friedman, E., Leng, L., Ngo, L., Rifkind, R. A. and Marks, P. A. (1991) Proc. Natl. Acad. Sci. (USA) 88:5542-5546.
39. Richon, V.M., Webb, Y., Merger, R. et al. (1996) PNAS 93:5705-8.
40. Cohen, L.A., Amin, S., Marks, P.A., Rifkind, R.A., Desai, D. and Richon, V.M. (1999) Anticancer Research 19:4999-5006.
41. Grunstein, M. (1997) Nature 389:349-52.
42. Finnin, M.S., Donigian, J.R., Cohen, A. et al. (1999) Nature 401:188-193.
43. Van Lint,C., Emiliani, S., Verdin, E. (1996) Gene Expression 5:245-53.
44. Archer, S. Shufen, M. Shei, A., Hodin, R. (1998) PNAS 95:6791-96.
45. Dressel, U., Renkawitz, R., Baniahmad, A. (2000) Anticancer Research 20(2A):1017-22.
46. Lin, R.J., Nagy, L., Inoue, S. et al. (1998) Nature 391:811-14.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ИНДУКЦИИ КОНЕЧНОЙ ДИФФЕРЕНЦИРОВКИ | 2003 |
|
RU2394022C2 |
| СПОСОБЫ ИНДУКЦИИ КОНЕЧНОЙ ДИФФЕРЕНЦИРОВКИ | 2010 |
|
RU2530648C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКА С ИСПОЛЬЗОВАНИЕМ ИНГИБИТОРОВ HDAC | 2004 |
|
RU2356547C2 |
| ИНГИБИТОРЫ ГИСТОНДЕЗАЦЕТИЛАЗЫ ИЗ НОВЫХ ПРОИЗВОДНЫХ БЕНЗАМИДА С СИЛЬНОЙ ДИФФЕРЕНЦИРОВОЧНОЙ И АНТИПРОЛИФЕРАТИВНОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2364589C2 |
| КОМБИНАЦИЯ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ И ИНГИБИТОРА ПРОТЕИНКИНАЗЫ И ЕЕ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2770754C1 |
| КОМБИНАЦИИ СПЕЦИФИЧНЫХ ИНГИБИТОРОВ ГИСТОНОВЫХ ДЕАЦЕТИЛАЗ КЛАССА I С ИНГИБИТОРАМИ ПРОТЕАСОМ | 2007 |
|
RU2456990C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ ИНГИБИТОРА ГИСТОНДИАЦЕТИЛАЗЫ В КОМБИНАЦИИ С БЕНДАМУТИНОМ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2609833C2 |
| Фармацевтическая композиция, содержащая ингибитор HDAC и антитело к PD1 или антитело к PD-L1 | 2020 |
|
RU2824577C1 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ГИСТОНДЕАЦЕТИЛАЗ | 2011 |
|
RU2565076C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗ И ИНГИБИТОРА ПРОТЕАСОМ ИЛИ ИММУНОМОДУЛИРУЮЩЕГО ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ РАКА КРОВИ | 2017 |
|
RU2721409C1 |
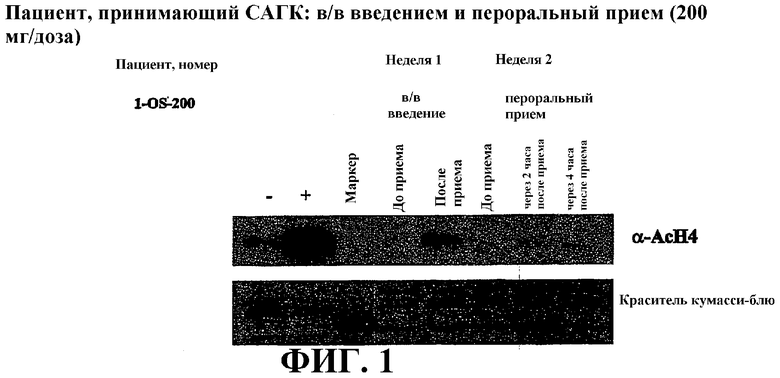
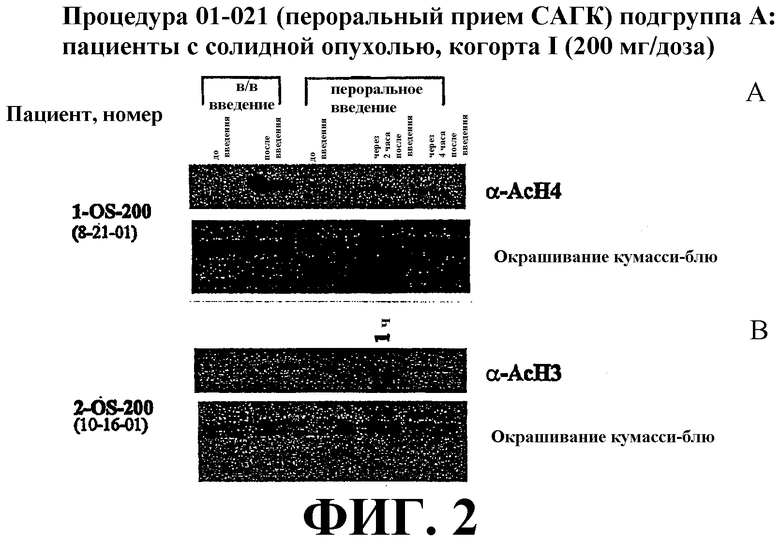
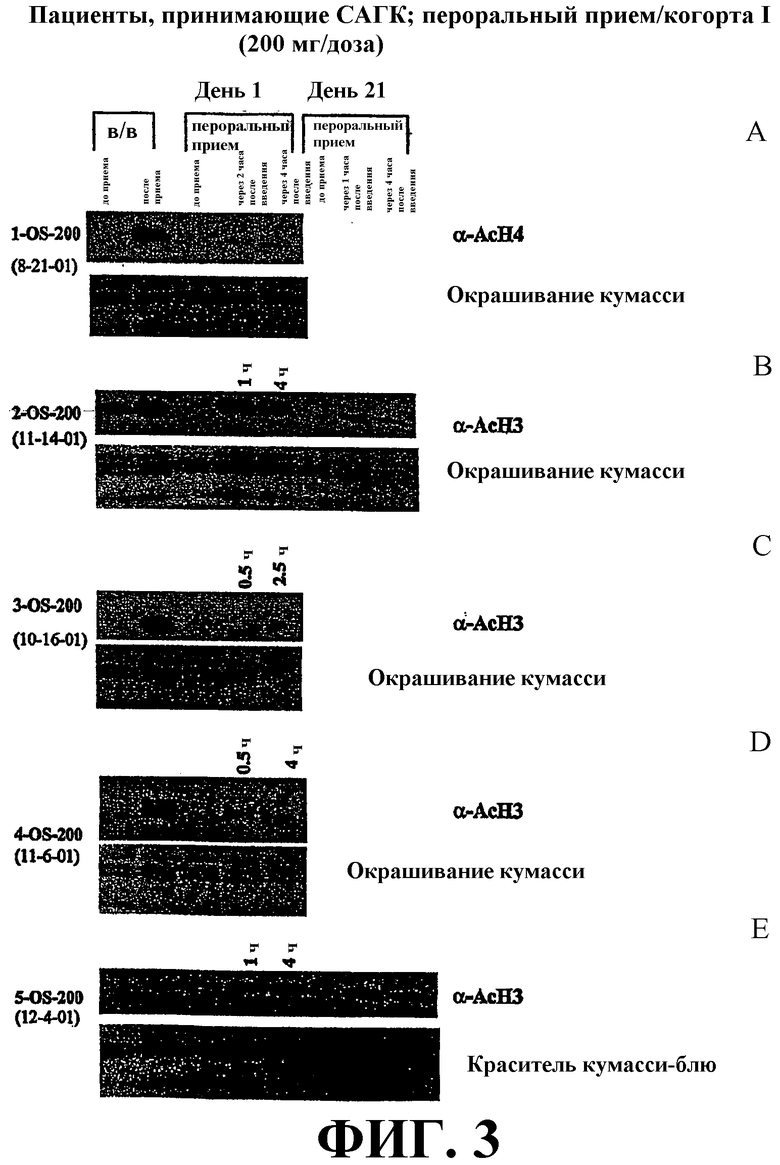

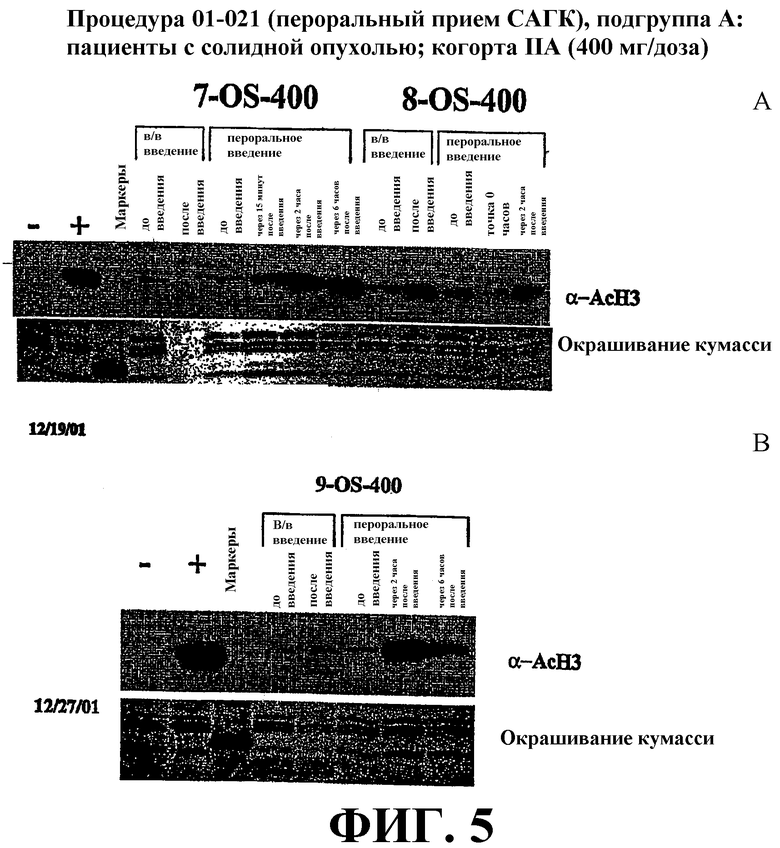
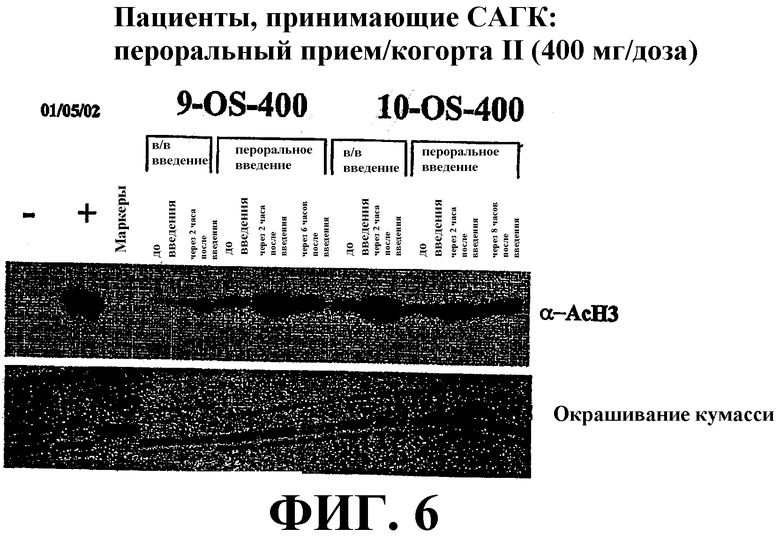
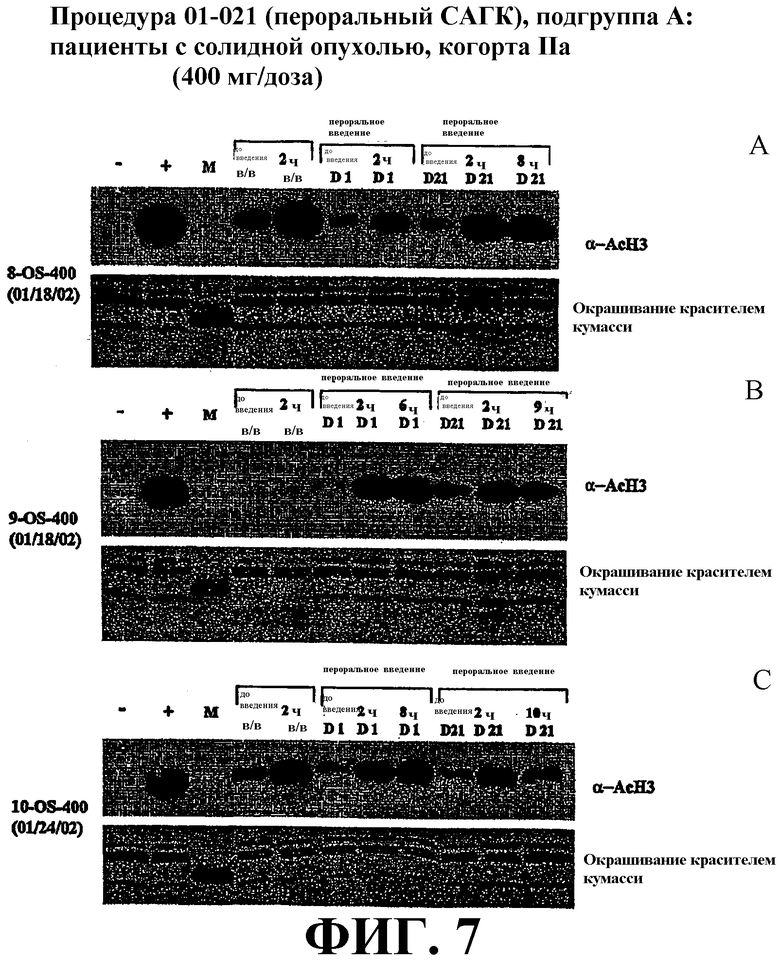
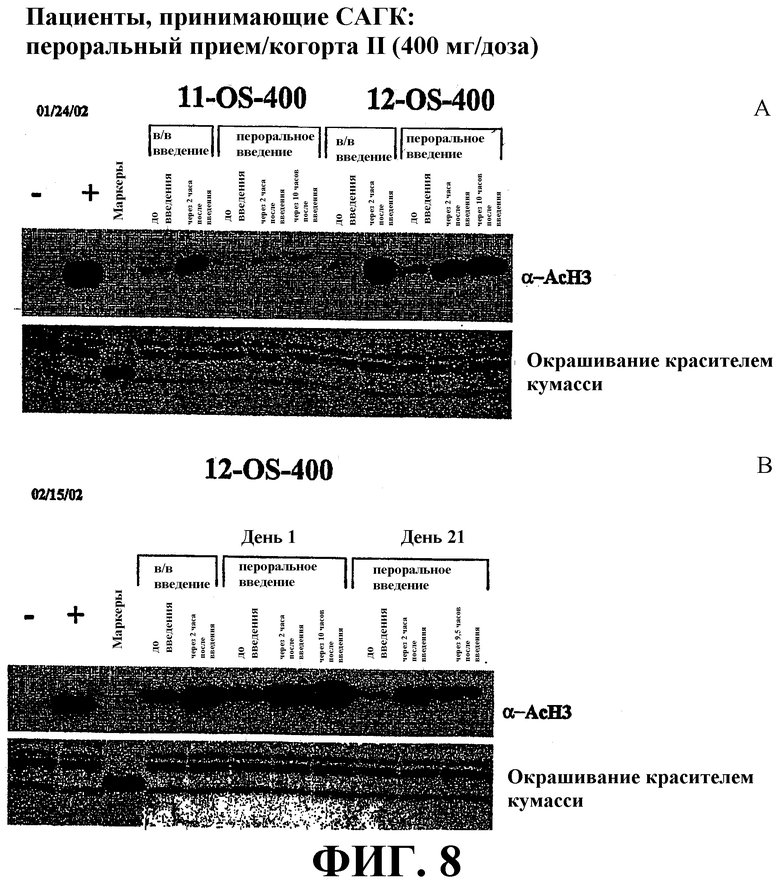
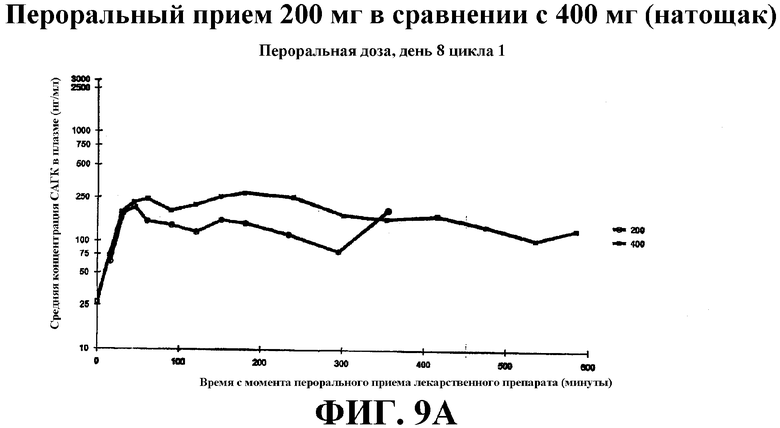
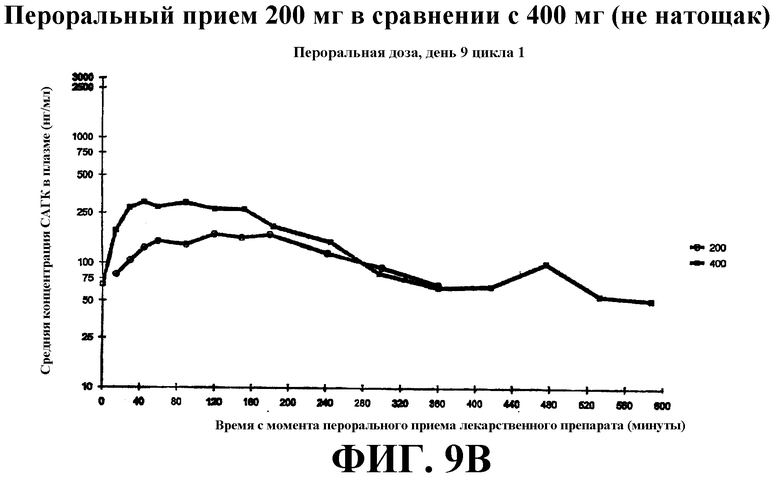
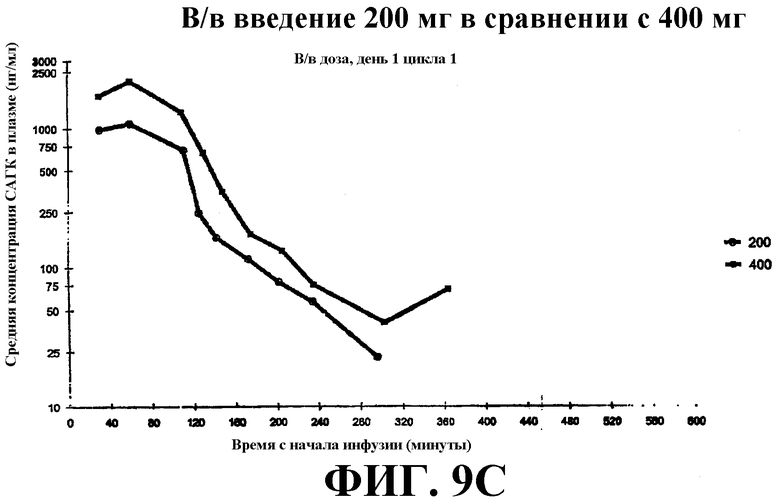
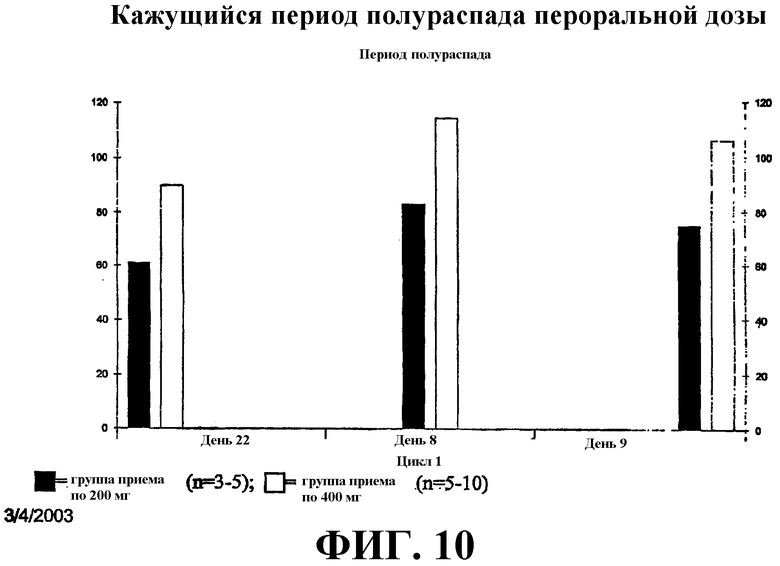
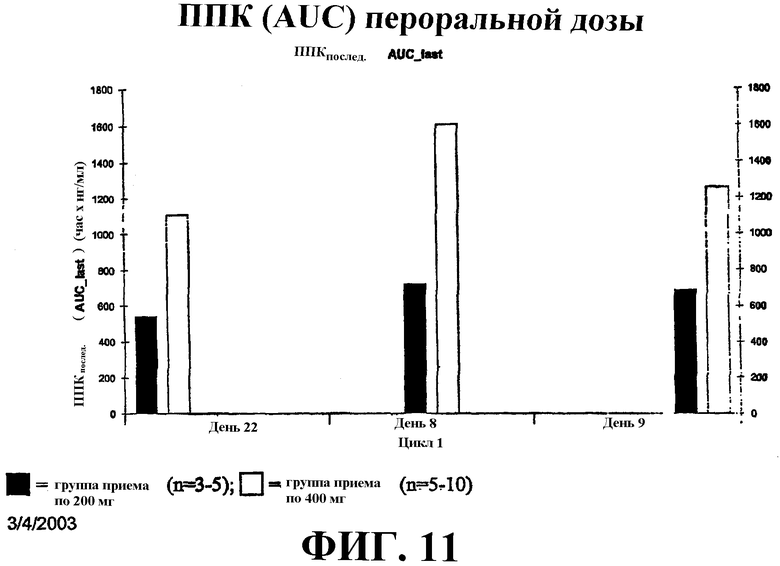
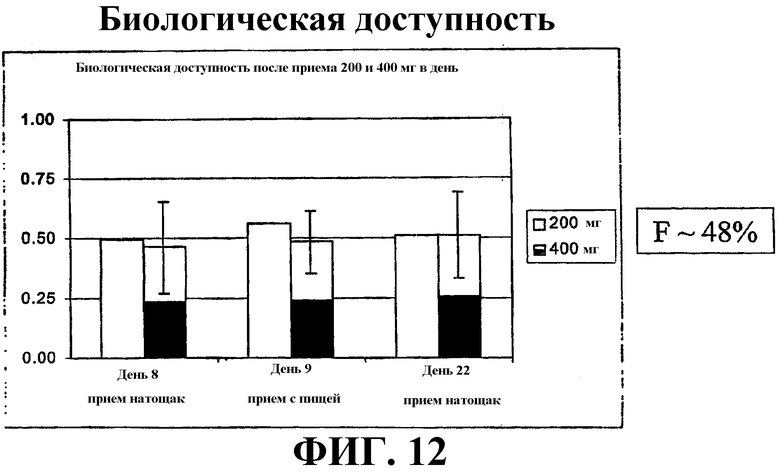
Изобретение относится к способам селективной индукции, остановки клеточного роста и/или апоптоза опухолевых клеток и/или ингибирования гистондезацетилазы (HDAC) путем введения фармацевтической композиции, включающей сильнодействующие ингибиторы HDAC. Изобретение обеспечивает удивительно высокую биологическая доступность активных соединений в фармацевтических композициях при пероральном приеме, где фармацевтические композиции неожиданно дают возможность поддерживать высокие терапевтически эффективные уровни активных соединений в крови в течение длительного периода времени. Изобретение также обеспечивает безопасный режим ежедневного дозирования указанных фармацевтических композиций, который легко соблюдать и который приводит к поддержанию терапевтически эффективного количества ингибиторов HDAC in vivo, 35 н. и 9 з.п. ф-лы, 5 табл., 12 ил.
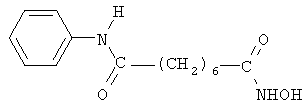
или его фармацевтически приемлемую соль или гидрат; и, необязательно, 20-40% по массе микрокристаллической целлюлозы, 5-15% по массе натрий-кросскармеллозы и 0,1-5% по массе стеарата магния.
или его фармацевтически приемлемую соль или гидрат; и 20-40% по массе микрокристаллической целлюлозы, 5-15% по массе натрий-кросскармеллозы и 0,1-5% по массе стеарата магния.
или его фармацевтически приемлемую соль или гидрат; и, необязательно, 20-40% по массе микрокристаллической целлюлозы, 5-15% по массе натрий-кросскармеллозы и 0,1-5% по массе стеарата магния.
полученного в виде соли с использованием основания, выбранного из группы, состоящей из гидроксидов железа, изопропиламина, триметиламина, гистидина и прокаина.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения 400 мг в день САГК или его фармацевтически приемлемой соли или гидрата для лечения не-ходжкинской лимфомы.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения 400 мг в день САГК или его фармацевтически приемлемой соли или гидрата для лечения миелодиспластического синдрома.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения 400 мг в день САГК или его фармацевтически приемлемой соли или гидрата для лечения лейкоза.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения два раза в день 200 мг САГК или его фармацевтически приемлемой соли или гидрата, где пероральное введение осуществляют в течение последовательных дней с последующим периодом отдыха для лечения миелодиспластического синдрома.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения три раза в день 200 мг САГК или его фармацевтически приемлемой соли или гидрата, где пероральное введение осуществляют в течение последовательных дней с последующим периодом отдыха для лечения миелодиспластического синдрома.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения два раза в день 200 мг САГК или его фармацевтически приемлемой соли или гидрата, где пероральное введение осуществляют в течение последовательных дней с последующим периодом отдыха для лечения лейкоза.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения три раза в день 200 мг САГК или его фармацевтически приемлемой соли или гидрата, где пероральное введение осуществляют в течение последовательных дней с последующим периодом отдыха для лечения лейкоза.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения 400 мг в день САГК или его фармацевтически приемлемой соли или гидрата, где пероральное введение является продолжительным, для лечения злокачественных опухолей с инфильтрацией лейкоцитов.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 400 мг, и инструкции для лечения не-ходжкикской лимфомы путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата в количестве, равном 400 мг в день.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 400 мг, и инструкции для лечения миелодиспластического синдрома путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата в количестве, равном 400 мг в день.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 400 мг, и инструкции для лечения лейкоза путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата в количестве, равном 400 мг в день.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 200 мг, и инструкции для лечения миелодиспластического синдрома путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата два раза в день в количестве 200 мг в течение последовательных дней с последующим периодом отдыха.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 200 мг, и инструкции для лечения миелодиспластического синдрома путей перорального введения САГК или его фармацевтически приемлемой соли или гидрата три раза в день в количестве 200 мг в течение последовательных дней с последующим периодом отдыха.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 200 мг, и инструкции для лечения лейкоза путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата два раза в день в количестве 200 мг в течение последовательных дней с последующим периодом отдыха.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 200 мг, и инструкции для лечения лейкоза путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата три раза в день в количестве 200 мг в течение последовательных дней с последующим периодом отдыха.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 400 мг, и инструкции для лечения злокачественных опухолей с инфильтрацией лейкоцитов путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата в количестве 400 мг в день, где пероральное введение является продолжительным.
а. взаимодействия субериновой кислоты с анилином с образованием суберанилиновой кислоты, имеющей структуру
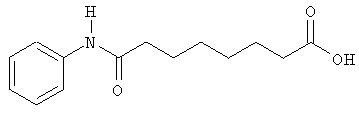
или ее соли;
b. взаимодействия суберанилиновой кислоты или ее соли с метанолом (образованием метилсуберанилата, имеющего структуру
с. взаимодействия метилсуберанилата с гидрохлоридом гидроксиламина с образованием неочищенного субероиланилида гидроксамовой кислоты в реакционной смеси;
d. добавления метоксида натрия к реакционной смеси с получением прозрачного раствора;
е. добавления ледяной уксусной кислоты к прозрачному раствору с образованием осадка, содержащего неочищенный субероиланилид гидроксамовой кислоты; и
f. перекристаллизации неочищенного препарата САГК из смеси метанола и воды в соотношении примерно 2:1.
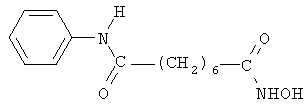
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для лечения злокачественной опухоли у субъекта, где лекарственное средство вызывает среднюю концентрацию САГК или его фармацевтически приемлемой соли или гидрата в плазме субъекта, равную, по меньшей мере, примерно 100 нМ через два часа после введения.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения один раз в день 300 мг САГК или его фармацевтически приемлемой соли или гидрата для лечения не-ходжкинской лимфомы.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения два раза в день 300 мг САГК или его фармацевтически приемлемой соли или гидрата, где пероральное введение является периодическим для лечения не-ходжкинской лимфомы.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения два раза в день 300 мг САГК или его фармацевтически приемлемой соли или гидрата, где пероральное введение осуществляется в течение последовательных дней с последующим периодом отдыха для лечения не-ходжкинской лимфомы.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения один раз в день 400 мг САГК или его фармацевтически приемлемой соли или гидрата для лечения не-ходжкинской лимфомы.
или его фармацевтически приемлемой соли или гидрата для получения лекарственного средства для перорального введения два раза в день 200 мг САГК или его фармацевтически приемлемой соли или гидрата для лечения не-ходжкинской лимфомы.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 300 мг, и инструкции для лечения не-ходжкинской лимфомы путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата один раз в день в количестве 300 мг.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 300 мг, и инструкции для лечения не-ходжкикской лимфомы путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата два раза в день периодически в количестве 300 мг.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 300 мг, и инструкции для лечения не-ходжкинской лимфомы путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата три раза в день в количестве 300 мг в течение последовательных дней с последующим периодом отдыха.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 400 мг, и инструкции для лечения не-ходжкикской лимфомы путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата один раз в день в количестве 400 мг.
или его фармацевтически приемлемой соли или гидрата, где фармацевтически эффективная единичная доза составляет примерно 200 мг, и инструкции для лечения не-ходжкинской лимфомы путем перорального введения САГК или его фармацевтически приемлемой соли или гидрата два раза в день в количестве 200 мг.
| RU 2001112763 А, 27.05.2003 | |||
| US 5932616 А, 03.08.1999 | |||
| US 6451334 В2, 17.09.2002 | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

