Приоритет указан в данной заявке из предварительной заявки США Сер. №. 60/447915, опубликованной 14 февраля 2003.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к получению и применению новых производных бензамида в качестве ингибиторов гистондезацетилазы для лечения расстройств, связанных с дифференцировкой и/или пролиферацией, таких как рак и псориаз.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Нарушенная экспрессия генов играет значительную роль в патогенезе или патологических нарушениях при раке, эндокринных расстройствах, иммунных/воспалительных расстройствах, генетических расстройствах и неврологических расстройствах. Геном человека упакован в хроматин, который состоит из ДНК, гистонов и негистоновых белков. Структура хроматина является важным фактором, определяющим то, будет ли экспрессироваться тот или иной ген или нет. В общем, конденсированный хроматин опосредует подавление транскрипции, тогда как транскрипционно активные гены находятся в областях открытого хроматина. Нуклеосомы образуют основной повторяющийся элемент хроматина и состоят из ДНК, обернутой вокруг октамера гистонов, который образован четырьмя блоками гистонов, а именно тетрамером H3-H4 и двумя димерами H2A-H2B. Гистон H1 действует наподобие линкера пространственной стабилизации упаковки более высокого порядка путем электростатической нейтрализации линкерных сегментов ДНК посредством положительно заряженного карбоксиконцевого домена. Следовательно, динамическая структура нуклеосом более высокого порядка определяет различные уровни организации хроматина и, следовательно, активацию генов. Ricky W. Johnstone, "Histone deacetylase inhibitors: novel drugs for the treatment of cancer", Nature Reviews Drug Discovery 2002, 1: 287. На способность октомера гистонов компактизировать ДНК влияет ряд посттранскрипционных модификаций, которые проявляются в N-концевых частях гистонов. Одна модификация включает в себя обратимое ацетилирование и дезацетилирование эпсилон-аминогрупп лизиновых компонентов, обнаруженных в концевых участках гистонов. Суммарный уровень ацетилирования N-терминальных концевых фрагментов гистонов регулируется активностью двух семейств ферментов, гистонацетилтрансфераз (HAT) и гистондезацетилаз (HDAC). В добавление к HAT и HDAC другие факторы также участвуют в определении структуры хроматина, включая метил-CpG-связывающий белок и аденозинтрифосфатзависимые комплексы, ремоделирующие хроматин, которые могут непосредственно укреплять HDAC, что приводит к подавлению активации генов (см. обзор Current Opinion in Oncology 2001, 13:477-483).
Определение комплексов коактиваторов, которые обладают внутренней активностью HAT, сильно поддерживает связь между ацетилированием гистона и активацией транскрипции. Подобным образом, было показано, как комплексы, подавляющие транскрипцию, привлекают HDAC к промотору генов-мишеней (Bioassays 1998, 20:615). Специфические в отношении последовательности транскрипционные факторы, такие как суперсемейство ядерных рецепторов гормонов, белок, связывающий энхансер, связанный с циклическим аденозин-3',5'-монофосфатом (CREB), преобразователь сигнала и активатор транскрипции-1 (STAT-1) взаимодействуют с определенными коактиваторами и корепрессорами внутри комплекса аппарата транскрипции селективным образом в окружении ДНК и ткани, приводя к селективной регуляции сети экспрессии гена. Такая регуляторная сеть управляет гомеостазом физиологических функций нашего организма, и расстройство такой сети вызывает расстройства и/или глубоко влияет на прогрессирование заболевания. Следовательно, комплексные взаимодействия аппарата транскрипции обеспечивают новые стратегии вмешательства для лечения рака, эндокринных расстройств, иммунных/воспалительных расстройств, генетических заболеваний и нейродегенерации (Korzus, E., et al., Transcription Factor-specific Requirements for Coactivator and Their Acetyltransferase Functions. Science 1998, 279: 703-707; McKenna, N.J. and B.W. O'Malley, Combinatorial Control of Gene Expression by Nuclear Receptors and Coregulators. Cell 2002, 108(4):465-474; Pazin, M.J. and J.T. Kadonaga, What's Up and Down with Histone Deacetylation and Transcription? Cell 1997, 89(3):325-328; Zhong, H., R.E. Voll, and S. Ghosh, Phosphorylation of NF-B p65 by PKA Stimulates Transcriptional Activity by Promoting a Novel Bivalent Interaction with the Coactivator CBP/p300. Molecular Cell 1998, 1(5):661-671; Steffan JS. et al., Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila, Nature 2001, 413:691-694; HDAC inhibitor VX-563 from Vertex Pharmaceuticals proceeds for genetic disorders, 2002 EDGAR online News, US20020115716A1, WO0056153A1).
Например, развитие и дифференцировка клеток управляется иерархическим порядком последовательной активации генов, который регулируется на уровне структуры хроматина. Генетические нарушения или мутации, которые вызывают нерегулируемую активацию онкопротеинов, таких как RAS, или инактивацию супрессоров опухоли, таких как p53, влияют на большое число молекулярных программ, включая транскрипцию. Кроме того, генетические нарушения, которые приводят к неправильному нацеливанию HAT и HDAC на определенные локусы, функциональная инактивация HAT, повышенная экспрессия HDAC или эпигенетические изменения из-за гипер- или гипометилирования ДНК могут сместить баланс между программами клеточного развития и дифференцировки, что часто приводит к развитию и прогрессированию опухоли (см. обзор Current Opinion Genet. Development 1999, 9: 40-48 и 175-184). Некоторые виды человеческого рака ассоциированы с нарушением активности HAT и HDAC. Одним примером является транслокация хромосом 15 и 17, наблюдаемая у большинства пациентов с острым промиелоцитарным лейкозом (ОПЛ). При ОПЛ хромосомные транслокации вызывают образование слитых белков, которые содержат RARальфа, PML (белок промиелоцитарного лейкоза) и PLZF (промиелоцитарный цинковый палец). Такие аберрантные белки связываются с чувствительными к ретиноевой кислоте элементами, привлекают HDAC с высокой афинностью посредством усиленного связывания с корепрессором SMRT и не отвечают на ретиноиды, приводя к конститутивному подавлению генов, целевых для RAR (Oncogene 2001, 20:7204-7215). Рецептор ретиноевой кислоты (RAR) представляет собой лиганд-активируемый транскрипционный модулятор, который важен для миелоидной дифференцировки. RAR, гетеродимеризованный со своим партнером RXR, связывается с элементом, чувствительным к ретиноевой кислоте, расположенным в участке промотора целевых генов, и, в отсутствие ретиноидов, подавляет транскрипцию привлечением SIN3/HDAC посредством корепрессоров NCOR и SMRT. Добавление лиганда высвобождает комплексы HDAC из RAR/RXR и позволяет последующему связыванию HAT, таких как TIF2 и CBP, активировать транскрипцию. Следовательно, согласованная активация и подавление генов, которые содержат функциональные чувствительные к ретиноевой кислоте элементы, являются необходимыми для дифференцировки миелоидных клеток. Кроме того, добавление ингибиторов HDAC может восстановить чувствительность клеток ОПЛ к дифференцировке миелоидных клеток, индуцированной ретиноидами, показывая, что нарушенное дезацетилирование гистонов является ключевым процессом развития лейкозов.
Существуют сообщения, что гистондезацетилазы при повышенной экспрессии подавляют экспрессию генов, подавляющих опухоль, которые являются естественным препятствием для роста опухоли. Например, p53, критический регулятор пролиферации клеток, передает сигналы генам, которые регулируют клеточный цикл и апоптоз, когда клетки находятся в состоянии стресса. Функции являются преимущественно регулируемыми способностью p53 связываться с ДНК со специфичностью к последовательности и активировать транскрипцию. Инактивация этого свойства p53, по большей части мутациями, которые появляются в центральном ДНК-связывающем домене, часто приводит к злокачественному новообразованию. Было продемонстрировано, что CBP/p300 может потенцировать p53 посредством ацетилирования ядра гистонов и ацетилирования p53. (W. Gu и R.G. Roeder, Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 1997, 90(4): 595-606.) Напротив, показано, что HDAC-1, HDAC-2 и HDAC-3 млекопитающих способны ингибировать функцию p53 дезацетилированием и ядерного гистона, и p53 (Juan, L.-J., et al., Histone Deacetylases Specifically Down-regulate p53-dependent Gene Activation. The Journal of Biological Chemistry 2000, 275(27): 20436-20443).
Эти данные показывают, что несоответствующее подавление транскрипции, опосредованное HDAC, является одним из обычных молекулярных механизмов, которые используется онкобелками, и нарушения в структуре хроматина могут вторгаться в нормальную дифференцировку, что приводит к образованию опухоли и другим гиперпролиферативным расстройствам. Следовательно, ингибирование активности HDAC, видимо, является рациональной терапевтической возможностью для рака и других гиперпорлиферативных расстройств.
Было идентифицировано несколько классов ингибиторов HDAC, включая (1) короткоцепочечные жирные кислоты, например бутират и фенилбутират; (2) органические гидроксамовые кислоты, например субероиланилидгидроксамовая кислота (SAHA) и трихостатин A (TSA); (3) циклические тетрапептиды, содержащие 2-амино-8-оксо-9,10-экспоксидеканоиловый компонент (AOE), например трапоксин и HC-токсин; (4) циклические пептиды без AOE-компонента, например апицидин и FK228; и (5) бензамиды, например MS-275 (EP 0847992 A1, US 2002/0103192 A1, WO 02/26696 A1, WO 01/70675 A2, WO 01/18171 A2).
Сложные эфиры масляной кислоты действуют как ингибиторы пролиферации клеток и индукторы клеточной дифференцировки, главным образом, посредством их активности ингибирования гистондезацетилазы (A. Nudelman and A. Rephaeli, Novel Mutual Prodrug of Retinoic и Butyric Acids with Enhanced Anticancer Activity. J. Med. Chem. 2000, 43(15): 2962-2966.) Фенилбутират используется в качестве единственного агента в лечении β-талассемии, токсоплазмоза и малярии. Также сообщают, что он полезен в лечении резистентного ОПЛ в комбинации с РК (ретиноевой кислотой) (R.P. Warrell. et al., Therapeutic targeting of transcription in acute promyelocytic leukemia by use of an inhibitor of histone deacetylase. J. Natl. Cancer Inst. 1998, 90(21): 1621-1625). Другая жирная кислота, вальпроевая кислота, которая является сильным противосудорожным средством, стабилизатором настроения и тератогеном, также является прямым ингибитором гистондезацетилазы (C.J. Phiel et al., Histone Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, и Teratogen. The Journal of Biological Chemistry 2001,276(39): 36734-36741; EP1170008A1).
Было обнаружено, что группа бензамидов имеет ингибирующую HDAC активность в низком микромолярном диапазоне. Было исследовано ведущее вещество из этой группы бензамидов, MS-275, Mitsui Chemicals, Inc., и оно является первым ингибитором HDAC, который продемонстрировал пероральную противораковую активность в моделях на животных с отсутствием тяжелых побочных эффектов (A. Saito et al., A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proceedings of the National Academy of Sciences of the United States of America 1999, 96(8): 4592-4597; EP 0847992 Al). В настоящее время MS-275 проходит стадию клинических исследований в Центре Рака Greenebaum для больных с лейкозами Университета штата Мэриленд и Национальном институте рака США для запущенных опухолей (E.B. Levit, Clinical Trials in Leukemia focus on New Treatment Approaches. 2001 Release - University of Maryland Medical News 2001 Maryland http://www.umm.edu/news/releases/karp.html, A Phase I Study of an Oral Histone Deacetylase Inhibitor, MS-275, in Refractory Solid Tumors и Lymphomas. 2001, National Cancer Institute). Однако до настоящего времени все еще остается необходимость в открытии новых соединений с улучшенными профилями, такими как более сильная активность ингибирования HDAC.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает соединения, которые проявляют эффекты индуцирования дифференцировки и ингибирования пролиферации и применимы в качестве терапевтического лечения или улучшающих агентов для расстройств, связанных с дифференцировкой и/или пролиферацией, таких как рак и псориаз. В частности, они являются высокоэффективными против гематологических злокачественных новообразований и солидных карцином.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На чертеже графически показана активация транскрипции различных ядерных рецепторов гормонов на примере ингибиторов HDAC, т.е. трихостатина A, MS-275, а также на примере соединения согласно изобретению.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В описании настоящей заявки цитированы различные публикации. Содержание этих публикаций и содержание документов, цитированных в этих публикациях, включены в данное описание в виде ссылки.
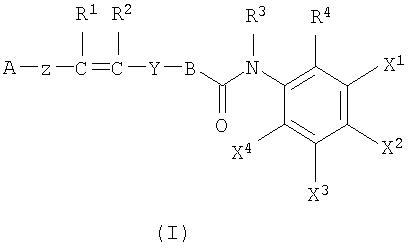
Настоящее изобретение обеспечивает соединения, представленные формулой (I), или их стереоизомеры, энантиомеры, диастереомеры, гидраты или фармацевтически приемлемые соли:
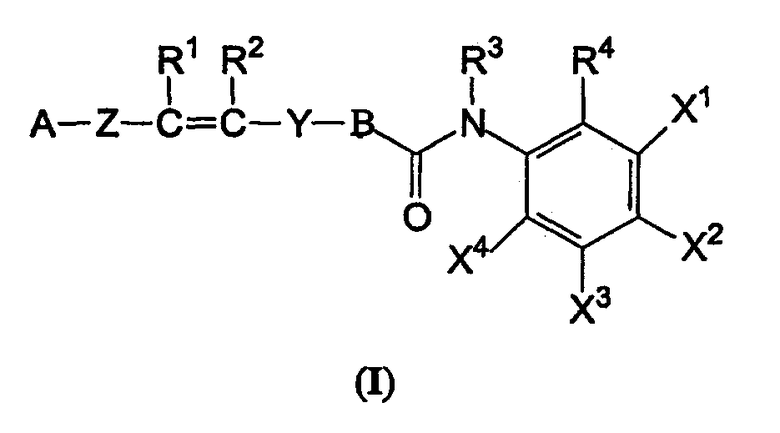
где A представляет собой фенильную или гетероциклическую группу, необязательно замещенную 1-4 заместителями, выбранными из группы, состоящей из атома галогена, гидроксильной группы, аминогруппы, нитрогруппы, цианогруппы, алкильной группы, имеющей от 1 до 4 атомов углерода, алкоксигруппы, имеющей от 1 до 4 атомов углерода, аминоалкильной группы, имеющей от 1 до 4 атомов углерода, алкиламиногруппы, имеющей от 1 до 4 атомов углерода, ацильной группы, имеющей от 2 до 4 атомов углерода, ациламиногруппы, имеющей от 2 до 4 атомов углерода, алкитиогруппы, имеющей от 1 до 4 атомов углерода, перфторалкильной группы, имеющей от 1 до 4 атомов углерода, перфторалкилоксигруппы, имеющей от 1 до 4 атомов углерода, карбоксильной группы, алкоксикарбонильной группы, имеющей от 1 до 4 атомов углерода, фенильной группы и гетероциклической группы;
В представляет собой фенильную или гетероциклическую группу, необязательно замещенную 1-3 заместителями, выбранными из группы, состоящей из атома галогена, гидроксильной группы, аминогруппы, нитрогруппы, цианогруппы, алкильной группы, имеющей от 1 до 4 атомов углерода, алкоксигруппы, имеющей от 1 до 4 атомов углерода, аминоалкильной группы, имеющей от 1 до 4 атомов углерода, алкиламиногруппы, имеющей от 1 до 4 атомов углерода, ацильной группы, имеющей от 2 до 4 атомов углерода, ациламиногруппы, имеющей от 2 до 4 атомов углерода, алкитиогруппы, имеющей от 1 до 4 атомов углерода, перфторалкильной группы, имеющей от 1 до 4 атомов углерода, перфторалкилоксигруппы, имеющей от 1 до 4 атомов углерода, карбоксильной группы, алкоксикарбонильной группы, имеющей от 1 до 4 атомов углерода, фенильной группы и гетероциклической группы;
Z представляет собой связь, необязательно замещенный алкилен, имеющий от 1 до 4 атомов углерода, или фрагмент, имеющий -O-, -S-, -NH-, -СО-, -CS-, -SO- или -SO2-, который является линейным, циклическим или их комбинацией;
Y представляет собой фрагмент, имеющий -СО-, -CS-, -SO- или -SO2-, который является линейным, циклическим или их комбинацией; и в котором расстояния между центром тяжести цикла В (W1), центром тяжести цикла A (W2) и атомом кислорода или серы, в качестве акцептора водородной связи во фрагменте Y (W3) может, например, быть следующим: W1-W2=6,0-12,0 Å, W1-W3=3,0-6.0 Å и W2-W3=4,0-8,0 Å; предпочтительно W1-W2=8,0-10,0 Å, W1-W3=3,0-5,0 Å, W2-W3=5,0-8,0 Å (соединения согласно изобретению, описанные в настоящем описании, однако, необязательно ограничены этими размерами);
R1 и R2 являются независимо водородом или необязательно замещенным алкилом, имеющим от 1 до 4 атомов углерода; или R1 и R2 могут образовывать связь;
R3 представляет собой водород или необязательно замещенный алкил, имеющий от 1 до 4 атомов углерода; R4 представляет собой атом водорода, атом галогена, гидроксильную группу, аминогруппу, нитрогруппу, цианогруппу, алкильную группу, имеющую от 1 до 4 атомов углерода, алкоксигруппу, имеющую от 1 до 4 атомов углерода, аминоалкильную группу, имеющую от 1 до 4 атомов углерода, алкиламиногруппу, имеющую от 1 до 4 атомов углерода, ацильную группу, имеющую от 1 до 4 атомов углерода, ациламиногруппу, имеющую от 1 до 4 атомов углерода, алкилтиогруппу, имеющую от 1 до 4 атомов углерода, перфторалкильную группу, имеющую от 1 до 4 атомов углерода, перфторалкилоксигруппу, имеющую от 1 до 4 атомов углерода, карбоксильную группу или алкоксикарбонильную группу, имеющую от 1 до 4 атомов углерода.
Один из X1, X2, X3 или X4 является атомом галогена, гидроксильной группой, аминогруппой, нитрогруппой, цианогруппой, алкильной группой, имеющей от 1 до 4 атомов углерода, алкоксигруппой, имеющей от 1 до 4 атомов углерода, аминоалкильной группой, имеющей от 1 до 4 атомов углерода, алкиламиногруппой, имеющей от 1 до 4 атомов углерода, ацильной группой, имеющей от 1 до 4 атомов углерода, ациламиногруппой, имеющей от 1 до 4 атомов углерода, алкилтиогруппой, имеющей от 1 до 4 атомов углерода, перфторалкильной группой, имеющей от 1 до 4 атомов углерода, перфторалкилоксигруппой, имеющей от 1 до 4 атомов углерода, карбоксильной группы или алкоксикарбонильной группы, имеющей от 1 до 4 атомов углерода, тогда как другие из X1, X2, X3 или X4 независимо являются атомом водорода, атомом галогена, гидроксильной группой, аминогруппой, нитрогруппой, цианогруппой, алкильной группой, имеющей от 1 до 4 атомов углерода, алкоксигруппой, имеющей от 1 до 4 атомов углерода, аминоалкильной группой, имеющей от 1 до 4 атомов углерода, алкиламиногруппой, имеющей от 1 до 4 атомов углерода, ацильной группой, имеющей от 1 до 4 атомов углерода, ациламиногруппой, имеющей от 1 до 4 атомов углерода, алкилтиогруппой, имеющей от 1 до 4 атомов углерода, перфторалкильной группой, имеющей от 1 до 4 атомов углерода, перфторалкилоксигруппой, имеющей от 1 до 4 атомов углерода, карбоксильной группой или алкоксикарбонильной группой, имеющей от 1 до 4 атомов углерода.
В вышеуказанной структурной формуле (I) и на протяжении настоящего описания следующие термины имеют указанные значения.
Термин "гетероциклил" в данном описании означает одновалентную насыщенную или ненасыщенную группу, являющуюся моноциклической и содержащую один или более гетероатомов, такую как пирролидин, пирролин, пиразолин, имидазолидин, имидазолин, пиперидин, морфолин и подобные.
Термин "галоген" в данном описании означает фтор, хлор, бром или йод.
Термин "алкил, имеющий от 1 до 4 атомов углерода" в данном описании включает в себя метил, этил, н-пропил, изо-пропил, бутил, изо-бутил, втор-бутил и трет-бутил.
Термин "алкокси, имеющий от 1 до 4 атомов углерода" в данном описании включает в себя метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси и подобные.
Термин "аминоалкил, имеющий от 1 до 4 атомов углерода" в данном описании включает в себя аминометил, 1-аминопропил, 2-аминопропил и подобные.
Термин "алкиламино, имеющий от 1 до 4 атомов углерода" в данном описании включает в себя N-метиламино, N-этиламино, N-изопропиламино и подобные.
Термин "ацил, имеющий от 2 до 4 атомов углерода" в данном описании включает в себя ацетил, пропионил, бутирил, изобутирил и подобные.
Термин "ациламино, имеющий от 2 до 4 атомов углерода" в данном описании включает в себя ацетиламино, пропиониламино, бутириламино, изобутириламино и подобные.
Термин "алкилтио, имеющий от 2 до 4 атомов углерода" в данном описании включает в себя метилтио, этилтио, пропилтио и подобные.
Термин "перфторалкил, имеющий от 2 до 4 атомов углерода" в данном описании включает в себя трифторметил, пентафторэтил и подобные.
Термин "перфторалкилокси, имеющий от 2 до 4 атомов углерода" в данном описании включает в себя трифторметокси, пентафторэтокси и подобные.
Термин "алкилен, имеющий от 1 до 4 атомов углерода" в данном описании включает в себя метилен, этилен и подобные.
Термин "центр тяжести цикла", используемый в определении пространственной конфигурации, может быть определен как среднее из осей X, Y и Z атомов, образующих цикл.
Соединения согласно изобретению получали, как описано ниже.
(а) Соединение, представленное формулой (II), конденсировали с соединением, представленным формулой (III), для получения соединения, представленного формулой (IV)
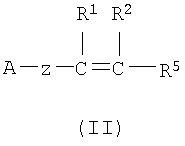
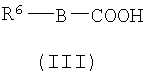
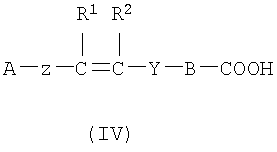
где А, Z, Y, В, R1 и R2 соответствуют тому, как определено выше; R5 представляет собой фрагмент, имеющий -C(=Q)OH (Q представляет собой атом кислорода или серы), или фрагмент, имеющий -NH2; R6 представляет собой фрагмент, имеющий -NH2, когда R5 представляет собой фрагмент, имеющий -C(=Q)OH (Q представляет собой атом кислорода или серы) и фрагмент, имеющий -С(=Q)ОН (Q представляет собой атом кислорода или серы), когда R5 представляет собой фрагмент, имеющий -NH2.
(b) Соединение, представленное формулой (IV), конденсировали с соединением, представленным формулой (V), для получения соединения согласно изобретению.
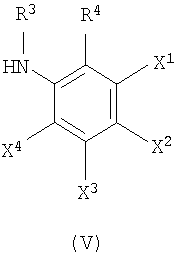
где R3, R4, X1, X2, X3 и X4 соответствуют тому, как определено выше.
Вышеуказанные реакции конденсации (а) и (b) проводили с использованием пептидного конденсирующего агента, такого как дициклогексилкарбодиимид, N,N'-карбонилдиимидазол, дифенилфосфорный азид, диэтилфосфорилцианид и др.
Реакцию можно было проводить при 0-80°С в течение 4-72 часов. Растворителями, которые могут быть использованы, являются обычные растворители, такие как бензол, толуол, тетрагидрофуран, диоксан, дихлорметан, хлороформ, N,N-диметилформамид и др. Если необходимо, основание, такое как гидроксид натрия, триэтиламин и пиридин, или кислота, такая как соляная кислота, уксусная кислота и трифторуксусная кислота, могут быть добавлены в реакционную систему.
Соединение согласно изобретению и промежуточные продукты, представленные формулой (I), могут быть очищены или выделены обычными методами разделения, такими как экстракция, рекристаллизация, колоночная хроматография.
Новые соединения согласно изобретению имеют эффекты, индуцирующие дифференцировку, и, следовательно, применимы в качестве терапевтического лечения или улучшающих агентов, относящихся к расстройствам, связанным с дифференцировкой и/или пролиферацией, таким как рак и псориаз. В частности, они являются высокоэффективными в качестве канцеростатических агентов для гематологических злокачественных новообразований и солидных карцином.
Активный ингредиент согласно изобретению, применимый в качестве лекарственного средства, может быть применимым в виде обычной фармацевтической композиции. Фармацевтическая композиция может быть в форме, обычно применяемой, такой как таблетки, капсулы, порошки, сиропы, растворы, аэрозоли и пр., может содержать ароматизаторы, подсластители и др. в подходящих твердых или жидких носителях или разбавителях или в подходящей стерильной среде для создания инъекционных растворов или суспензий. Такие композиции обычно содержат от 1 до 70%, предпочтительно от 5 до 50% по массе активного соединения, остальной частью композиции являются фармацевтически приемлемые носители, разбавители или растворители, или солевые растворы.
Соединения согласно изобретению являются клинически вводимыми млекопитающим, включая человека и животных, посредством перорального, назального, трансдермального, легочного или парентерального путей. Введение пероральным путем предпочтительно является более обычным и позволяет избежать возможной боли и раздражения от инъекции. При любом пути введения доза составляет в диапазоне примерно от 0,0001 до 200 мг/кг массы тела в день, вводимых однократно, или в виде разделенной дозы. Однако оптимальная доза для отдельного субъекта, получающего лечение, будет определяться человеком, ответственным за лечение, обычно начинают с меньших доз, а затем их увеличивают, подбирая наиболее подходящую дозу.
Следующие примеры даны в качестве специфических иллюстраций изобретения. Необходимо понимать, однако, что изобретение не ограничено специфическими деталями, указанными в примерах. Все части и процентные отношения в примерах, а также в остальной части описания, представлены по массе, если не указано иначе.
Кроме того, любой диапазон чисел, представленный в описании или в разделах настоящего описания ниже, связан с описанием различных аспектов изобретения, представляющих собой определенный ряд свойств, единиц измерения, условий, физических состояний или процентных отношений, и предназначен для включения в данное описание в виде ссылки или, иначе, любого числа, попадающего в такой диапазон, включая любую подгруппу чисел или диапазонов, включенных в любой диапазон, таким образом указанный. Термин "примерно", когда используется в качестве модификатора в связи с переменной, предназначен для передачи того, что числа и диапазоны, описанные в данном описании, являются гибкими и что применение настоящего изобретения специалистом в области техники с использованием температуры, концентраций, количеств, содержания, количества атомов углерода и свойств, которые находятся вне диапазона или отличаются от отдельного значения, позволит достичь желаемого результата.
Пример 1
Получение 4-[N-(Пиридин-3-илакрилоил)аминометил]бензойной кислоты
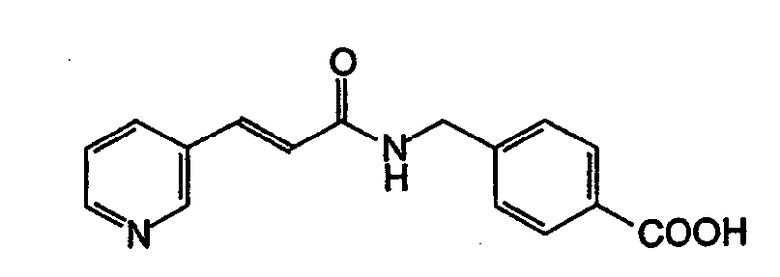
К суспензии 0,33 г (2,01 ммоль) N,N'-карбонилдиимидазола в тетрагидрофуране (10 мл) добавляли по каплям раствор 0,30 г (2,01 ммоль) 3-пиридинакриловой кислоты при 0°C. Затем смесь перемешивали при комнатной температуре в течение 3 часов и добавляли по каплям к отдельно приготовленным 2,0 мл (2,00 ммоль) 1Н водного раствора гидроксида натрия, включающего 0,30 г (2,00 ммоль) 4-аминометилбензойной кислоты, затем перемешивали при комнатной температуре в течение 8 часов. Реакционную смесь выпаривали в вакууме. К остатку добавляли насыщенный раствор хлорида натрия (2 мл), затем смесь нейтрализовали концентрированной соляной кислотой до pH 5. Осажденное белое твердое вещество собирали фильтрацией, промывали ледяной водой и затем сушили для получения указанного в заголовке соединения (0,46 г, 82%). МСВР рассчит. для C16H14N2O3: 282,2988. Обнаружено: 282,2990. MA рассчит. для: C16H14N2O3: C, 68,07%; H, 5,00%; N, 9,92%. Обнаружено: C, 68,21%; H, 5,03%; N, 9,90%.
Пример 2
Получение N-(2-амино-4-фторфенил)-4-[N-(пиридин-3-илакрилоил)аминометил]бензамида
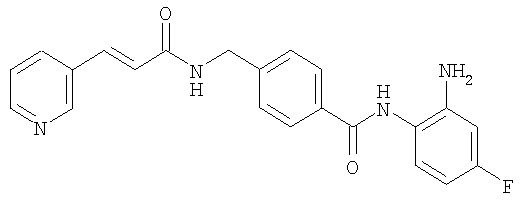
К суспензии 0,29 г (1,78 ммоль) N,N'-карбонилдиимидазола в тетрагидрофуране (15 мл) добавляли 0,50 г (1,78 ммоль) 4-[N-(пиридин-3-илакрилоил)аминометил]бензойной кислоты, затем перемешивали при 45°С в течение 1 часа. После охлаждения реакционную смесь добавляли к отдельно полученному раствору тетрагидрофурана (10 мл), включающему 0,28 г (2,22 ммоль) 4-фтор-1,2-фенилендиамина и 0,20 г (1,78 ммоль) трифторуксусной кислоты при комнатной температуре. После реакции при комнатной температуре в течение 24 часов осажденное белое твердое вещество собирали фильтрацией, промывали тетрагидрофураном и затем сушили для получения указанного в заголовке соединения (0,40 г, 57%). 1H ЯМР (300 МГц, ДМСО-d6): δ ч/млн: 4,49 (2Н, д), 4,84 (2Н, шир.с), 6,60 (1Н, т), 6,80 (2Н, м), 6,96 (1Н, т), 7,18 (1Н, д), 7,42 (2Н, д), 7,52 (1Н, д), 7,95 (2Н, д), 8,02 (1Н, д), 8,56 (1Н, д), 8,72 (1Н, шир.т), 8,78 (1Н, с), 9,60 (1Н, шир.с). ИК (КВr) см-1: 3310, 1655, 1631, 1524, 1305, 750. МСВР рассчит. для C22H19N4O2F: 390,4170. Обнаружено: 390,4172. МА рассчит. для C22H19N4O2F: С, 67,68%; Н, 4,40%; N, 14,35%. Обнаружено: С, 67,52%; Н, 4,38%; N, 14,42%.
Пример 3
Получение 4-[N-циннамоиламинометил]бензойной кислоты
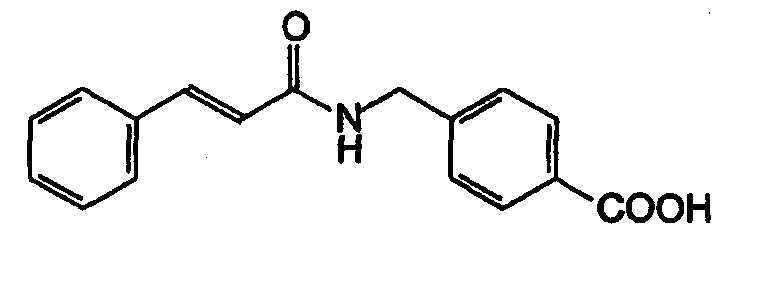
К суспензии 0,33 г (2,01 ммоль) N,N'-карбонилдиимидазола в тетрагидрофуране (10 мл) добавляли по каплям раствор 0,30 г (2,01 ммоль) коричной кислоты при 0°C. Затем смесь перемешивали при комнатной температуре в течение 3 часов и добавляли по каплям к отдельно полученным 2,0 мл (2,00 ммоль) 1Н водного раствора гидроксида натрия, включающего 0,30 г (2,00 ммоль) 4-аминометилбензойной кислоты, затем перемешивали при комнатной температуре в течение 8 часов. Реакционную смесь выпаривали в вакууме. К остатку добавляли насыщенный раствор хлорида натрия (2 мл), затем смесь нейтрализовали концентрированной соляной кислотой до pH 7. Осажденное белое твердое вещество собирали фильтрацией, промывали ледяной водой и затем сушили для получения указанного в заголовке соединения (0,51 г, 91%). МСВР рассчит. для C17H15NO3: 281,3242. Обнаружено: 281,3240. MA рассчит. для C17H15NO3: C, 72,58%; H, 5,38%; N, 4,98%. Обнаружено: C, 72,42%; H, 5,37%; N, 4,87%.
Пример 4
Получение N-(2-амино-4-фторфенил)-4-[N-циннамоиламинометил]бензамида
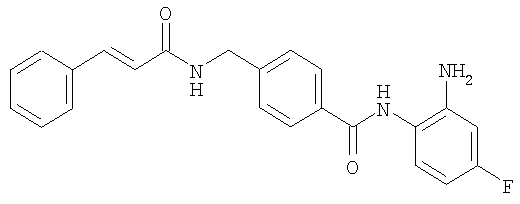
К суспензии 0,29 г (1,78 ммоль) N,N'-карбонилдиимидазола в тетрагидрофуране (15 мл) добавляли 0,50 г (1,78 ммоль) 4-[N-циннамоиламинометил]бензойной кислоты, затем перемешивали при 45°С в течение 1 часа. После охлаждения реакционную смесь добавляли к отдельно полученному раствору тетрагидрофурана (10 мл), включающему 0,28 г (2,22 ммоль) 4-фтор-1,2-фенилендиамина и 0,20 г (1,78 ммоль) трифторуксусной кислоты при комнатной температуре. После реакции при комнатной температуре в течение 16 часов осажденное белое твердое вещество собирали фильтрацией, промывали тетрагидрофураном и затем сушили для получения указанного в заголовке соединения (0,45 г, 64%). 1H ЯМР (300 МГц, ДМСО-d6): δ ч/млн: 4,42 (2Н, д), 4,92 (2Н, шир.с), 6,62 (1Н, т), 6,78 (2Н, м), 7,01 (1Н, т), 7,32 (5Н, м), 7,54 (5Н, м), 8,76 (1Н, шир.т), 9,58 (1Н, шир.с). ИК (КВr) см-1: 3306, 1618, 1517, 1308, 745. МСВР рассчит. для С23Н20N3O2F: 389,4292. Обнаружено: 389,4294. МА рассчит. для С23Н20N3O2F: С, 70,94%; Н, 5,18%; N, 10,79%. Обнаружено: С, 70,72%; Н, 5,18%; N, 10,88%.
Пример 5
Ингибирование in vitro ферментативной активности HDAC N-(2-амино-4-фторфенил)-4-[N-(пиридин-3-илакрилоил)аминометил]бензамидом (соединение CS02100055), N-(2-аминофенил)-4-[N-(4-фторфенил)аминометил]бензамидом (соединение CS02100019) и N-(2-аминофенил)-4-[N-(пиридин-3-илметоксикарбонил)аминометил]бензамидом (MS-275, EP 0847992).
Ингибирующие эффекты MS-275 и соединений CS02100055 и CS 02100019 на HDAC исследовали посредством набора для исследования колориметрической активности HDAC (BIOMOL Research Laboratories, PA, USA) в соответствии с инструкциями производителя. Коротко, тестируемые соединения в различных концентрациях добавляли в 96-луночные планшеты, затем смешивали с экстрактом клеток HeLa, содержащим ферментную активность HDAC, обеспеченную производителем. Реакции HDAC инициировали добавлением субстрата. Через 10 минут реакции останавливали добавлением Color De Lys Developer. Микропланшеты считывали в спектрофотометре для прочтения планшетов при 405 нм. Ингибирование активности HDAC рассчитывали, следуя инструкциям. Результаты тестирования перечислены в таблице 1.
Ингибирование активности HDAC in vitro MS-275, CS02100055 и CS02100019
Пример 6
Эффект ингибирования роста N-(2-амино-4-фторфенил)-4-[N-(пиридин-3-илакрилоил)аминометил]бензамида (соединение CS02100055), N-(2-аминофенил)-4-[N-(4-фторфенил)аминометил]бензамида (соединение CS02100019) и N-(2-аминофенил)-4-[N-(пиридин-3-илметоксикарбонил)аминометил]бензамида (MS-275, EP 0847992) на различные опухолевые клеточные линии in vitro.
Тесты ингибирования роста проводили методом MTS. Приблизительно за 72 часа до анализа выживаемости клетки высевали в 96-луночные планшеты в концентрации 5-10×103 клеток/ячейку (в соответствии со скоростью роста отдельных используемых клеточных линий). Через 24 часа добавляли тестируемые соединения в различных концентрациях, и клетки культивировали в течение 48 часов, затем добавляли 20 мкл/ячейку раствора реагента CellTiter 96 AQueous One, содержащего соединение тетразолия (Promega), в каждую ячейку. MTS последовательно добавляли к культуральной среде. После инкубации планшетов в течение 2 часов примерно при 37°С записывали поглощение при 490 нм при помощи спектрофотометра для считывания 96-луночных планшетов. Выживаемость клеток рассчитывали по Алечение/Аконтроль × 100% (А представляет собой поглощение при 490 нм). Концентрацию, которая ингибировала рост клеток на 50% больше контроля, определяли как GI50. Все соединения растворяли в ДМСО и добавляли в культуру в разведении 1:1000 для получения конечной концентрации ДМСО ≤0,1%. Все образцы исследовали в двойном экземпляре, и каждый эксперимент повторяли по меньшей мере три раза. Результаты исследования суммированы в таблице 2.
Ингибирование роста опухолевых клеточных линий MS-275, CS02100055 и CS02100019
Пример 7
Активация транскрипции ядерных рецепторов гормонов под действием N-(2-амино-4-фторфенил)-4-[N-(пиридин-3-илакрилоил)аминометил]бензамидом (соединение CS02100055), N-(2-аминофенил)-4-[N-(пиридин-3-илметоксикарбонил)аминометил]бензамидом (MS-275, EP 0847992) и трихостатина A (TSA).
Активацию транскрипции некоторых ядерных рецепторов гормонов тестируемыми соединениями, как показано на чертеже, проводили в экспериментах по исследованию гена-репортера. Коротко, клетки U2OS высевали в 96-луночные планшеты за день до трансфекции для получения слияния 50-80%. Клетки трансфицировали одной из плазмид экспрессии, содержащей кДНК, кодирующие рецептор глюкокортикоидов (GR), активированный рецептор пролифератора пероксисом γ (PPARγ), рецептор эстрогенов α (ER α) или рецептор эстрогенов β (ER β), в комбинации с рецептором ретиноида X α (RXR α), и их соответствующими плазмидами гена-репортера люциферазы с использованием реагента трансфекции FuGene6, в соответствии с инструкциями производителя (Roche). Клеткам давали возможность экспрессировать белок в течение 24 часов, затем добавляли отдельные соединения или растворитель (ДМСО). Через 24 часа клетки собирали и проводили люциферазный анализ с использованием набора для люциферазного анализа в соответствии с инструкциями производителя (Promega). Для нормализации данных люциферазных анализов измеряли β-галактозидазную активность трансфицированных клеток с использованием набора (Promega), как указано производителем. Чувствительными элементами для отдельного неясного рецептора были следующие:
GR (5'-GATCTTGTACAGGATGTTCTCTAGCGATGTACAGGATGTTCTCTAGCGATGTACAGGATGTTCTCTAG-3') (SEQ ID No. 1), PPAR (5'-CGCGTTCCTTTCCGAACGTGACCTTTGTCCTGGTCCCCTTTTGCT-3') (SEQ ID No. 2), и ER (5'-TCGAGTCAGGTCACAGTGACCTGATC-3') (SEQ ID No. 3). Результаты исследования суммированы на чертеже. На чертеже показана активация транскрипции ядерных рецепторов гормонов различными ингибиторами HDAC трихостатином A, MS-275 и CS02100055. Эксперименты проводили, как описано выше. LD относится к соответствующим лигандам для каждого рецептора, а CS55 - для CS02100055 в каждой панели фигуры. Концентрации тестируемых соединений во всех экспериментах составляли для TSA 0,2 мкM, MS-275 1 мкM и CS55 1 мкM. Дексаметазон (0,1 мкM), Росиглитазон (10 мкM) и E2 (0,01 мкM) использовали в качестве лигандов для GR, PPARγ и ER соответственно. Проводили три независимых эксперимента, и результаты репрезентативного эксперимента показаны на чертеже.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ НАФТАЛИНКАРБОКСАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ И ГИСТОНДЕАЦЕТИЛАЗЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2010 |
|
RU2497809C2 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ДЕАЦЕТИЛАЗЫ ГИСТОНОВ | 2006 |
|
RU2448965C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ГИСТОНДЕАЦЕТИЛАЗ | 2011 |
|
RU2565076C2 |
| ИНГИБИТОРЫ HDAC | 2014 |
|
RU2665554C2 |
| 4-(3-((2-Адамантил)амино)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамид - новое средство для лечения болезни Альцгеймера | 2022 |
|
RU2783995C1 |
| ИНГИБИТОРЫ ГИСТОНДЕЗАЦЕТИЛАЗЫ | 2008 |
|
RU2453536C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ АЛКИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ГИСТОНДЕАЦЕТИЛАЗЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2016 |
|
RU2683022C1 |
| СПОСОБЫ УМЕНЬШЕНИЯ КАХЕКСИИ, СВЯЗАННОЙ С РАКОМ | 2014 |
|
RU2692785C1 |
| Гидроксамовые кислоты, производные 4-аминохиназолин-7-карбоновой кислоты как ингибиторы гистондеацетилазы и способ их получения | 2021 |
|
RU2779981C1 |
| N1-(4-(5-(ЦИКЛОПРОПИЛМЕТИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-ИЛ)ПИРИДИН-2-ИЛ)ЦИКЛОГЕКСАН-1,4-ДИАМИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ СK1 И/ИЛИ IRAK1 ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2761457C2 |
Настоящее изобретение относится к соединениям формулы I и их фармацевтически приемлемым солям. Соединения настоящего изобретения могут найти применение в качестве лекарственного средства для лечения расстройств, связанных с пролиферацией клеток или опосредованных ядерными рецепторами. В общей формуле (I)
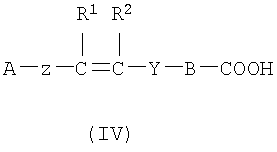
А представляет собой фенильную или ненасыщенную моноциклическую 6-членную группу, содержащую атом азота в качестве гетероатома; В представляет собой фенильную группу; Z представляет собой связь; Y представляет собой группу -CO-NH-CH2-; R1, R2, R3 независимо представляют собой водород; R4 представляет собой аминогруппу; и один из Х1, X2, X3 или X4 представляет собой атом галогена, тогда как другие из X1, X2, X3 или X4 независимо представляют собой атом водорода. Изобретение также относится к способу получения, фармацевтическим композициям, к применению соединений изобретения, а также к соединению формулы IV. 8 н. и 14 з.п. ф-лы, 2 табл., 1 ил.
1. Соединение формулы I:
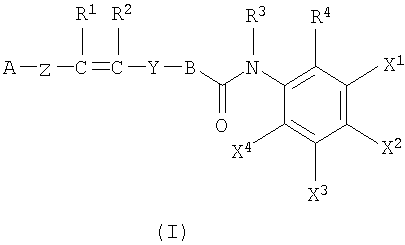
или его фармацевтически приемлемая соль;
где А представляет собой фенильную или ненасыщенную моноциклическую 6-членную группу, содержащую атом азота в качестве гетероатома;
В представляет собой фенильную группу;
Z представляет собой связь;
Y представляет собой группу -CO-NH-CH2-;
R1, R2, R3 независимо представляют собой водород;
R4 представляет собой аминогруппу; и
один из X1, X2, X3 или X4 представляет собой атом галогена, тогда как другие из X1, X2, X3 или X4 независимо представляют собой атом водорода.
2. Соединение формулы IV:
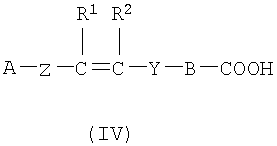
или его фармацевтически приемлемая соль;
где А представляет собой фенильную или ненасыщенную моноциклическую 6-членную группу, содержащую атом азота в качестве гетероатома;
В представляет собой фенильную группу;
Z представляет собой связь;
Y представляет собой группу -CO-NH-CH2-; и R1 и R2 независимо являются водородом.
3. Способ получения соединения по п.1 или его фармацевтически приемлемой соли, включающий в себя стадии
(a) конденсирования соединения, представленного формулой (II), с соединением, представленным формулой (III), с получением соединения, представленного формулой (IV):
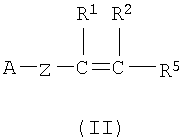
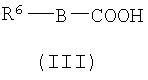
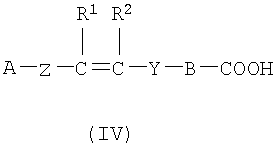
R5 представляет собой группу -C(=Q)OH, где Q представляет собой атом кислорода;
R6 представляет собой группу -CH2-NH2; и
(b) конденсирование соединения, представленного формулой (IV), с соединением, представленным формулой (V), с получением соединения формулы (I)
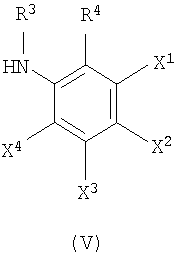
где один из X1, X2, X3 или X4 представляет собой атом галогена, тогда как другие из Х1, X2, X3 или X4 независимо представляют собой атом водорода;
R3 представляет собой атом водорода; и
R4 представляет собой аминогруппу.
4. Способ по п.3, где реакции конденсации на стадиях (а) и (b) проводят с использованием пептидного конденсирующего агента.
5. Способ по п.4, где указанным пептидным конденсирующим агентом является дициклогексилкарбодиимид, N,N'-карбонилдиимидазол, дифенилфосфорный азид, или диэтилфосфорилцианид.
6. Способ по п.3, где указанные реакции конденсации на стадиях (а) и (b) проводят при температуре примерно от 0 до 80°С.
7. Фармацевтическая композиция для лечения расстройств, связанных с пролиферацией клеток, содержащая эффективное количество соединения по п.1 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, носитель или разбавитель.
8. Фармацевтическая композиция по п.7, где указанное расстройство, связанное с пролиферацией, выбрано из группы, состоящей из псориаза, гематологического злокачественного заболевания и солидной карциномы.
9. Фармацевтическая композиция по п.7 в виде дозированной формы, содержащей примерно от 0,0001 до около 200 мг указанного соединения по п.1.
10. Фармацевтическая композиция по п.7 для введения пероральным, назальным, трансдермальным, легочным или парентеральным путем.
11. Применение соединения по п.1 для получения лекарственного средства для лечения клеточных пролиферативных заболеваний,
12. Применение по п.11, где указанное клеточное пролиферативное заболевание выбрано из группы, состоящей из злокачественной опухоли и псориаза.
13. Применение по п.11, где указанное лекарственное средство содержит примерно от 0,0001 до 200 мг указанного соединения по п.1.
14. Фармацевтическая композиция для активации ядерных рецепторов, включающая в себя эффективное количество соединения по п.1 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, носитель или разбавитель.
15. Фармацевтическая композиция по п.14 в виде дозированной формы, содержащей примерно от 0,0001 до 200 мг указанного соединения по п.1.
16. Фармацевтическая композиция по п.14 для введения пероральным, назальным, трансдермальным, легочным или парентеральным путем.
17. Применение соединения по п.1 для получения лекарственного средства для лечения или предотвращения состояния, опосредованного ядерными рецепторами.
18. Применение соединения по п.1 для получения лекарственного средства для лечения или предотвращения состояния, опосредованного аномально низкой активностью ядерного рецептора.
19. Применение по п.17, где состояние выбрано из группы, состоящей из эндокринных расстройств, расстройств иммунной системы или воспалительных расстройств, генетических расстройств и нейродегенерации.
20. Применение по п.18, где состояние выбрано из группы, состоящей из эндокринных расстройств, расстройств иммунной системы или воспалительных расстройств, генетических расстройств и нейродегенерации.
21. Применение по п.17, где указанное лекарственное средство содержит примерно от 0,0001 до 200 мг указанного соединения по п.1.
22. Применение по п.18, где указанное лекарственное средство содержит примерно от 0,0001 до 200 мг указанного соединения по п.1.
| Способ получения производных 1-фенокси-3-аминопропан2-ола или их солей | 1973 |
|
SU563120A3 |
| Способ получения пикрата 5-окситриптамина | 1958 |
|
SU116106A1 |
| Устройство для анализа времениОТКРыТОгО и зАКРыТОгО СОСТОяНияглАз | 1979 |
|
SU847992A1 |

