Настоящее изобретение относится к способу введения 1,2-двойной связи в 3-оксо-4-азастероиды путем дегидрирования 3-оксо-4-азастероидов, насыщенных в 1,2-положении, в частности, путем дегидрирования 17β-замещенных 3-оксо-4-азастероидов, для получения соответствующих 17β-замещенных 3-оксо-4-азастероидов, которые в 1,2-положении имеют одну двойную связь.
Из патента EP 0 155 096 известно получение 17β-замещенных 4-аза-5-альфа-андростанов с 1,2-двойной связью путем окисления соответствующего 1,2-дигидросоединения смесью бензола и ангидрида селеновой кислоты. Другие способы введения 1,2-двойной связи в 17β-замещенные 4-аза-5-альфа-андростаны описаны, например, также в патентах EP 0298652, EP 0428366 и EP 0473225. 17β-замещенные 4-аза-5-альфа-андростаны с 1,2-двойной связью являются фармацевтически важными соединениями с широким спектром действия. Важное значение имеет, например, соединение 17β-(N-трет-бутилкарбамоил)-4-аза-андрост-1-ен-3-он (финастерид), которое применяется, например, как ингибитор 5-альфа-редуктазы для лечения доброкачественной гиперплазии предстательной железы или андрогенетической алопеции. Важное значение имеет, например, также 17β-{N-[2,5-бис(трифторметил)фенил]}-4-аза-андрост-1-ен-3-он (дутастерид). Известные способы получения этих соединений имеют свои особые недостатки, так что существует потребность в улучшенном альтернативном способе.
Настоящее изобретение относится к такому альтернативному способу получения.
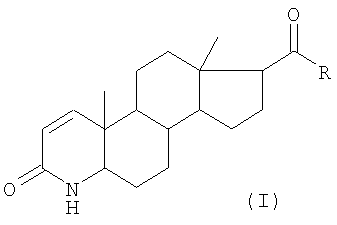
Настоящее изобретение раскрывается в формуле изобретения. Настоящее изобретение относится к способу получения 17β-замещенных 4-аза-андрост-1-ен-3-онов общей формулы (I):

в которой означают:
R гидроксил, возможно замещенный, линейный или разветвленный (C1-C12)-алкил или (C1-C12)-алкенил; фенил или бензил; остаток
-OR1, или остаток -NHR1, или остаток -NR1R2;
R1 водород, возможно, замещенный, линейный или разветвленный (C1-C12)-алкил или (C1-C12)-алкенил, или фенил, возможно замещенный;
R2 водород, метил, этил или пропил; или
-NR1R2 5- или 6-членный гетероцикл, и для R = гидроксил также его фармацевтически приемлемая соль,
отличающемуся тем, что
(A) в 3-кето-4-аза-группу (лактамную группу) соединения общей формулы (II):
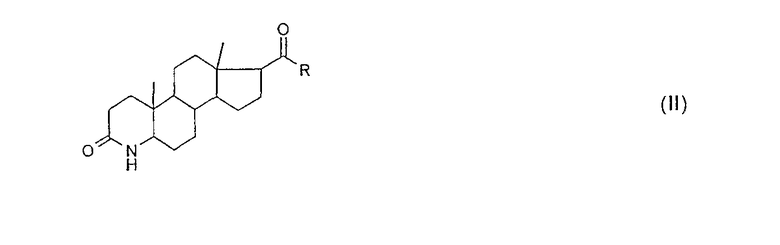
вводят защитные группы, так что образуется соединение общей формулы (III):
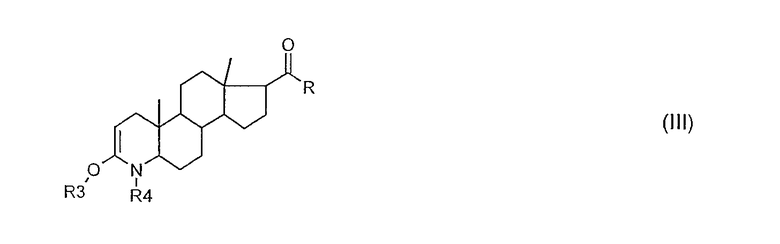
в котором означают:
R3 триалкилсилил, или вместе с R4 остаток -C(О)-C(О)- или
-C(О)-Y-C(О)-;
R4 алкилоксикарбонил или фенилоксикарбонил, предпочтительно Boc (= трет-бутилоксикарбонил); или триалкилсилил, или вместе с R3 остаток -C(O)-C(O)- или -C(O)-Y-C(O)-;
Y -[C(R5)(R6)]n-, или -CH(R5)=CH(R6)-, или ортофенилен;
R5 и R6 - независимо друг от друга водород, линейный или разветвленный (C1-С8)-алкил или -алкенил, фенил, возможно замещенный, или бензил; и
n целое число от 1 до 4;
и в случае, если R означает гидроксил, оно, возможно, прореагировало с защитной группой;
(B) полученное [согласно стадии (A)] соединение реагирует (i) в присутствии катализатора дегидрирования и (ii) в присутствии, возможно замещенного, бензохинона, аллилметилкарбоната, аллилэтилкарбоната и/или аллилпропилкарбоната, причем в 1/2-положение вводится Δ1-двойная связь, и
(C) защитные группы R3 и R4 удаляют, и при R = гидроксил полученное соединение возможно преобразуют в соль.
R предпочтительно означает линейный или разветвленный (C1-C6)-алкил, предпочтительно метил, этил, пропил или н-бутил, втор-бутил или трет-бутил, предпочтительно трет-бутил; или остаток
-OR1, или остаток -NHR1, или остаток -NR1R2. Предпочтительно остаток означает -NHR1.
Если R означает гидроксил (или остаток -C(О)R означает карбоксил), согласно изобретению может также быть получена фармацевтически приемлемая соль соединения формулы (I), предпочтительно соль щелочного металла, соль щелочно-земельного металла или аммонийная соль, предпочтительно соль натрия, калия или аммония, предпочтительно соль натрия или калия.
R1 означает предпочтительно линейный или разветвленный (C1-C6)-алкил, или фенил, возможно замещенный. R1 как (C1-C6)-алкил означает предпочтительно метил, этил, пропил, н-бутил, втор-бутил или трет-бутил, предпочтительно трет-бутил. R1 как фенил, возможно замещенный, означает предпочтительно моно(трифторметил)фенил или бис(трифторметил)фенил, предпочтительно 2,5-бис(трифторметил)фенил.
В остатке -NR1R2 R2 предпочтительно означает метил.
Заместитель -NR1R2 как 5- или 6-членный гетероцикл означает предпочтительно остаток пиперидина или пирролидина.
Предпочтительно заместитель является -NHR1, в котором R1 означает трет-бутил или 2,5-бис(трифторметил)фенил.
R3 предпочтительно означает триметилсилил или вместе с R4 остаток -C(О)-C(О)- или -C(O)-Y-C(O)-.
R4 предпочтительно означает BOC, триметилсилил или вместе с R3 остаток -C(О)-C(O)- или -C(О)-Y-C(О)-. Предпочтительно R4 означает Boc или вместе с R3 остаток -C(O)-C(О)- или -C(О)-Y-C(О)-.
R4 как алкилоксикарбонил означает предпочтительно изобутилоксикарбонил, трет-бутилоксикарбонил, трет-амилоксикарбонил, циклобутилоксикарбонил, 1-метилциклобутилоксикарбонил, циклопентилоксикарбонил, циклогексилоксикарбонил, 1-метилциклогексил, предпочтительно трет-бутилоксикарбонил.
R5 и R6 независимо друг от друга предпочтительно означают водород, линейный или разветвленный (C1-С4)-алкил, или фенил, предпочтительно водород, метил, этил или пропил или фенил.
n означает предпочтительно 1 или 2, предпочтительно 1.
Предпочтительно Y означает остаток -CH(R5)- или ортофенилен, предпочтительно метилен.
Для введения защитной триалкилсилильной группы, т.е. для силилирования NH-группы и/или атома кислорода или OH-группы [согласно стадии (A)] предпочтительно применяют (алкил)3Si(галоген), например, (CH3)3SiCl, или бистриметилсилилтригалогенацетамид, бистриметилсилилацетамид, гексаметилдисилазан и/или бистриметилмочевину, предпочтительно бистриметилсилилтрифторацетамид, или триалкилсилилтрифторметансульфонат, предпочтительно триметилсилилтрифторметансульфонат. Условия реакции силилирования известны из патента EP 0473226.
Для введения защитной группы, когда R3 вместе с R4 являются остатком -C(О)-C(О)- или -C(О)-Y-C(О)-, соединение общей формулы (II) или лактамную группу [согласно стадии (A)] подвергают взаимодействию с оксалилхлоридом (хлорангидрид щавелевой кислоты) или малонилхлоридом (хлорангидрид малоновой кислоты), причем оксалилхлорид предпочтителен. Условия реакции с оксалилхлоридом известны из патента EP 0428366, и с малонилхлоридом или аналогично реагирующими соединениями должны использоваться аналогичным образом.
Для введения защитной группы, когда R4 означает алкилоксикарбонил, например, трет-бутилоксикарбонил (Boc), используют известный способ, в котором соединение общей формулы (II) приводят во взаимодействие, например, с Boc-ангидридом (Boc-О-Boc) {[(CH3)3C-О-C(О)]2-О} или с Boc-карбаматом [(CH3)3C-O-C(О)-N(C1-4-алкил)2]. При этом Boc может здесь означать и другие аналогично реагирующие соединения, то есть соединения, в которых трет-бутиловый остаток замещен другим аналогично реагирующим остатком, как, например, названные остатки трет-амил, циклобутил, циклопентил или циклогексил. Такие аналогичные реакции в большом количестве описаны в литературе. Если R3 означает триалкилсилил, а R4 означает Boc, то сначала вводят защитную Boc-группу, а затем силилируют.
На стадии (B) соединение, полученное на стадии (A), превращают в присутствии (i) катализатора дегидрирования и в присутствии (ii) бензохинона, возможно замещенного, аллилметилкарбоната, аллилэтилкарбоната и/или аллилпропилкарбоната, при этом в 1-/2-положение вводится Δ1-двойная связь. Катализатор дегидрирования предпочтительно выбран из соединений (солей и комплексов) группы переходных металлов периодической системы элементов, в частности, выбран из соединений металлов VIII группы периодической системы, в частности, из железа (Fe), рутения (Ru) и осмия (Os); кобальта (Co), родия (Rh), и иридия (Ir); никеля (Ni), палладия (Pd) и платины (Pt), а также группы IB, т.е. меди (Cu), серебра (Ag) и золота (Au). Предпочтительны соединения металлов VIII группы периодической системы. Предпочтительны, в частности, соединения или катализаторы дегидрирования на основе родия (Rh), палладия (Pd) и платины (Pt). Предпочтительны соединения палладия. Примерами таких соединений палладия являются: Pd(0)-соединения, такие как комплекс трис(дибензилиденацетон)дипалладийхлороформ и Pd(II)-соединения, такие как PdCl2, Pd(dppe)2, [dppe = бис-(1,2-дифенилфосфин)этан], Pd(dppe)Cl2, Pd(OAc)2, Pd(dppe)(OAc)2, комплексы p-аллил-Pd, предпочтительно димер p-аллил-Pd-хлорида. Предпочтительны Pd(0)-соединения, в частности, комплекс трис(дибензилиденацетон)дипалладийхлороформ. Эти соединения, а также соли и комплексы известны и описаны в литературе.
Для термической стабилизации палладиевого комплекса можно использовать дополнительный комплексообразователь, как 2,2'-дипиридил или 1,10-фенантролин, предпочтительно 2,2'-дипиридил.
Для объяснения механизма катализа можно указать, что элемент Pd присоединяется к C-атому в 2-положение с отщеплением кислород-защищающей группы [например, группы -Si(CH3)3]. Последующее отщепление бета-водорода у C-атома в 1 положении приводит к образованию желаемой Δ1-двойной связи в 1-/2-положении, и высвобождается другой элемент палладия, который возвращается в каталитический цикл. Указания на этот механизм реакции приведены в Tetrahydron Letters, p. 4783 (1984). Однако настоящее изобретение не связано с этим объяснением.
В качестве хинона можно применять также замещенный хинон, например, хинон, замещенный C1-4-алкилом, галогеном, циано или нитро. Такие хиноны известны.
На стадии (C) полученное соединение превращают в соединение формулы (I) путем удаления введенных защитных групп. Это происходит предпочтительно обработкой подходящей кислотой, например, муравьиной кислотой, уксусной кислотой и/или трифторуксусной кислотой, предпочтительно муравьиной кислотой. Затем полученное соединение можно при необходимости известным путем перевести в фармацевтически приемлемую соль (для R = гидроксил).
Предпочтительно полученное соединение перекристаллизовывают. Эту перекристаллизацию можно проводить в неполярных растворителях, таких как бензин, гептан, гексан и толуол, предпочтительно в толуоле. Под соединением формулы (I) имеется в виду, в частности, упоминавшееся выше соединение 17β-(N-трет-бутилкарбамоил)-4-аза-андрост-1-ен-3-он (финастерид), которое встречается в двух полиморфных формах, а именно полиморфной форме I и полиморфной форме II, причем предпочтительна форма I. Форма I образуется, например, при перекристаллизации полученного согласно изобретению неочищенного финастерида из насыщенного раствора толуола (примерно одна часть неочищенного финастерида в шести частях толуола) при охлаждении до примерно 25°C. Полиморфная форма II образуется, например, при перекристиллизации неочищенного финастерида, полученного согласно изобретению, из раствора толуола (примерно одна часть неочищенного финастерида на примерно шесть частей толуола) при охлаждении до примерно 0°C.
Свойства 17β-{N-[2,5-бис(трифторметил)фенил]}-4-аза-андрост-1-ен-3-она (дутастерид) известны из литературы.
Для описанного способа со стадиями (A)-(C) в качестве растворителя могут применяться многочисленные органические безводные соединения, такие как, например, толуол, бензин, гексан, гептан, трет-бутиловый спирт, диэтиловый эфир, ацетон, бензол, диоксан, тетрагидрофуран, хлороформ, диметилформамид или пиридин. Следующие примеры поясняют изобретение.
Пример 1 (Замещение дигидрофинастерида группой Boc по атому азота 3-кето-4-аза-группы)
10 г (26,7 ммоль) дигидрофинастерида помещают в тетрагидрофуран (ТГФ) и охлаждают до -78°C. К полученной суспензии прибавляют 15 мл (30 ммоль) раствора диизопропиламида лития (Li-ДА-раствор), и прозрачный раствор перемешивают около 30 минут. Затем прибавляют раствор 6,7 г (30 ммоль) Boc-ангидрида в ТГФ. Раствор оставляют нагреваться до комнатной температуры. После обычной обработки получают влажный желтый порошок, который в течение ночи выдерживают в сушильном шкафу и непосредственно используют в примере 2.
Пример 2 (Силилирование соединения, полученного в примере 1)
1 г (2,1 ммоль) 4-Boc-дигидрофинастерида растворяют в ТГФ. К прозрачному желтому раствору добавляют при охлаждении смесью льда с метанолом 2,3 мл (4,6 ммоль) раствора Li-ДА. Суспензию перемешивают около 45 минут, после чего по каплям добавляют 0,46 г (4,2 ммоль) триметилхлорсилана (TMSCl) при 18-20°C. Прозрачный раствор концентрируют и остаток соединяют с гептаном. После фильтрации фильтрат концентрируют насколько возможно и полученное масло медово-коричневого цвета используют на следующих стадиях (пример 3 и пример 5).
Пример 3 (Введение Δ1-двойной связи в 4-бензилоксикарбонилфинастерид)
0,145 г (0,65 ммоль) ацетата палладия растворяют с 0,07 г (0,65 ммоль) бензохинона в ацетонитриле и оставляют. 0,8 г (1,5 ммоль) силильного соединения, полученного согласно примеру 3, соединяют с ацетонитрилом и добавляют по каплям при внутренней температуре (ВТ) 20-25°C. Реакционную смесь перемешивают 8 часов и очищают силикагелем. Слабоокрашенный прозрачный раствор концентрируют при температуре окружающей среды 55-60°C. Полученное твердое вещество используют в примере 4.
Пример 4 (Удаление защитных групп и кристаллизация)
a) 0,5 г твердого вещества из примера 3 смешивают с 20 г (0,175 моль) трифторуксусной кислоты и нагревают с обратным холодильником около 15 часов. При этом трифторуксусную кислоту используют и как реагент, и как растворитель. После охлаждения реакционную смесь выливают на смесь 300 г насыщенного раствора бикарбоната натрия и 50 г льда и экстрагируют 20 г эфира уксусной кислоты;
b) коричневый полупродукт, полученный на предыдущей стадии a), растворяют в толуоле при 90°C (массовое отношение толуол:сырье = 6:1), охлаждают до 20-25°C. Выпавшую в осадок серовато-белую массу отфильтровывают при 20-25°C и сушат. Получают финастерид полиморф I.
Пример 5 (Введение Δ1-двойной связи в 4-бензилоксикарбонилфинастерид)
2,0 г (3,7 ммоль) соединения из примера 2 смешивают с 1,29 г (11,1 ммоль) аллилметилкарбоната в ацетонитриле. Смесь добавляют по каплям к нагретому до 60-70°C раствору 166 мг (0,74 ммоль) ацетата палладия-II в ацетонитриле. Через 1-2 часа нагрева с обратным холодильником обрабатывают, как описано в примере 3. Получено 3 г твердого вещества.
Пример 6 (Введение Δ1-двойной связи)
A) 20 г (0,047 моль) оксалиленолового эфира дигидрофинастерида [соединение IIIa, с R = -NH-трет-бутил, R3 и R4 = -C(O)-C(О)-] нагревают вместе с 16,3 г (0,140 моль) аллилметилкарбоната и 76 г безводного ацетонитрила до температуры флегмообразования. Прибавляют поочередно 5 порций смеси, содержащей каждый раз по 18 г ксилола и по 0,049 г комплекса трис(дибензилиденацетон)дипалладий - хлороформ (полное мольное количество катализатора 0,284 ммоль). Каждый раз при добавлении наблюдается значительное газообразование. Через 12 ч нагрева с обратным холодильником реакцию возобновляют путем добавления второй порции горячей смеси, содержащей по 3 г ксилола и по 0,024 г катализатора дегидрирования (смесь медленно нагревают), при необходимости добавляют последующие порции. После фильтрации реакционную смесь по возможности концентрируют, после чего остается 24,5 г желтой, медообразной массы.
B) Медообразную массу соединяют с 105 г метанола и охлаждают до 0-5°C. Медленно прибавляют 11,3 г (0,0403 моль) 25%-ного раствора метоксида калия и перемешивают около 1 часа при внутренней температуре 0-5°C. Затем добавляют 20 г воды и удаляют охлаждающую ванну, внутреннюю температуру повышают до 15-20°C. Смесь концентрируют до сухого состояния, к твердому остатку добавляют 50 г воды, 90 г толуола и 12 г метанола и 1 час нагревают при температуре флегмообразования. После остановки мешалки без проблем разделяют органическую и водную фазы; органическую фазу выделяют горячей. Охлаждение в пределах 2-4 часов до 25°C приводит финастерид в полиморфной форме I к кристаллизации. После сушки получают 8,1 г белого порошка.
Пример 7
Осуществляют способ, аналогичный описанному в примерах с 1 по 6, когда вводят Δ1-двойную связь в дигидродутастерид, т.е. в соответствующее дигидросоединение формулы (I), в котором R означает остаток -NHR1, а R1 означает 2,5-бис(трифторметил)фенил, причем путем введения Δ1-двойной связи образуется дутастерид.
Пример 8 (Получение метилового эфира 3-оксо-4-аза-5α-андрост-1-ен-17β-карбоновой кислоты)
Стадия 1 (Получение соединения IIIb, т.е., соединения формулы (III), где R = -OMe, R3 и R4 = -C(O)-C(O)-)
2 г (0,005 моль, содержание >95%) метилового эфира 3-оксо-4-аза-5α-андрост-1-ен-17β-карбоновой кислоты смешивают с 30 г толуола и при охлаждении медленно добавляют 2,6 г (0,019 моль) оксалилхлорида. Постепенно начинается постоянное газообразование. Мутную смесь перемешивают в течение ночи. Из прозрачного реакционного раствора при комнатной температуре и пониженном давлении дистилляцией удаляют избыточный оксалилхлорид и толуол до половины первоначального объема. При этом осаждается белое твердое вещество, которое фильтруют и трижды интенсивно промывают гептаном по 15 г каждый раз. После сушки отсасыванием остается 1,6 г неочищенного метилового эфира. Его соединяют с приблизительно 20 г дихлорметана, мутный раствор интенсивно промывают 33 г 5%-ного раствора карбоната калия, смесь фильтруют и органическую фазу трижды повторно промывают, используя каждый раз по 10 г воды. Прозрачную бесцветную органическую фазу концентрируют насколько возможно и получают 0,9 г соединения III.
1H-ЯМР (200 МГц, CDCl3, δ): 4,95 (1H, т); 3,68 (3H, с); 3,62-3,5 (1H, м); 3,22-3,06 (1H, м); 2,41-0,80 (17H, м); 0,97 (3H, с); 0,68 (3H, с)
Стадия 2 (Введение Δ1-двойной связи)
0,2 г (0,5 ммоль) полученного на стадии 1 соединения IIIb нагревают вместе с 8 г абсолютного ацетонитрила, 1,5 г хлороформа, 0,18 г (1,5 ммоль) аллилметилкарбоната и 0,05 г (0,05 ммоль) палладиевого катализатора при температуре флегмообразования (70-80°C). Уже при нагревании наблюдается образование газа. Через примерно 30 минут нагрева с обратным холодильником реакционную смесь концентрируют насколько возможно, остаток соединяют со смесью 15 г метанола и 5 г толуола и нагревают до образования прозрачного раствора. После охлаждения до 0-5°C медленно добавляют раствор, содержащий 0,18 г (1 ммоль) 30%-ного раствора метилата натрия в 2 г метанола, прозрачный раствор перемешивают 1 час. После удаления охлаждающей ванны добавляют к этому 3 г воды и мутную смесь перемешивают при комнатной температуре еще 1 час. Затем концентрируют насколько возможно и к остатку добавляют 10 г толуола, а также 3 г воды. Как только при нагревании смесь разделится на две прозрачные фазы, органическую фазу отделяют и охлаждают. Добавление 2-4 г гептана приводит к кристаллизации продукта. После фильтрации, повторной промывки примерно 5 г гептана и сушки отсасыванием остается 34 мг метилового эфира 3-оксо-4-аза-5α-андрост-1-ен-17β-карбоновой кислоты.
1H-ЯМР (200 МГц, CDCl3, δ): 6,81 (1H, д); 5,82 (1H, д); 5,48 (1H, с, шир.); 3,69 (3H, с); 3,4-3,35 (1H, м); 2,45-1,0 (17H, м); 0,97 (3H, с); 0,66 (3H, с)
Пример 9 (Получение дутастерида)
Стадия 1 (получение 3-оксо-4-аза-5α-андростан-17β-карбоновой кислоты)
Суспензию из 100 г (0,26 моль) дигидрофинастерида, 480 г 20%-ного раствора HCl (2,63 моль) и 120 г метанола нагревают до температуры флегмообразования и 8-12 часов интенсивно кипятят. Эдукт при нагревании переходит в раствор, через 8 часов получают суспензию, которую можно легко отфильтровать. Осадок трижды интенсивно промывают на нутч-фильтре, используя каждый раз по 100 г воды, примерно 15 минут сушат отсасыванием и затем сушат в течение ночи. Выход: 60 г.
1Н-ЯМР (200 МГц, DMSO, δ): 11,95 (1H, с); 7,32 (1H, с); 2,95 (1H, м); 2,2 (2H, м); 2,0-0,85 (17H, м); 0,81 (3H, с); 0,62 (3H, с)
Стадия 2 (Получение соединения IIIc, т.е. соединения формулы (III), где R=Cl, R3 и R4 = -C(O)-C(O)-
К суспензии 40 г (0,12 моль) соединения со стадии 1 в 633 г бензола при охлаждении по каплям в пределах 20-30 минут добавляют 159 г (1,2 моль) оксалилхлорида, суспензию перемешивают 12 часов (газообразования незаметно). При пониженном давлении и комнатной температуре дистилляцией отделяют бензол и избыточный оксалилхлорид до тех пор, пока объем первоначального раствора не сократится наполовину. При этом в осадок выпадает серовато-белое твердое вещество, которое после фильтрации трижды промывают гептаном, используя каждый раз по 150 г, и примерно 15 минут сушат отсасыванием. Выход: 37,1 г соединения IIIc.
1H-ЯМР (200 МГц, CDCl3, δ): 4,93 (1H, т); 3,58 (1H, м); 3,12 (1H, м); 2,88 (1H, м); 2,31-0,72 (18H, м); 0,97 (3H, с); 0,80 (3H, с)
Стадия 3 (Получение соединения IIId (R= -NH-(2,5-(CF3)2-C6H3), R3 и R4 = -C(О)-C(O)-
Суспензию из 1,48 г (6 ммоль) бис-2,5-трифторметиланилина, 2,35 г (5,3 ммоль) соединения IIIc со стадии 2 и 50 г толуола примерно 8 часов нагревают при температуре флегмообразования (100-110°C) и затем охлаждают. При пониженном давлении и комнатной температуре толуол и анилин отделяют дистилляцией до тех пор, пока объем первоначального раствора не уменьшится наполовину. К суспензии добавляют 30 г гептана и нагревают до 60-70°C. Через один час интенсивного перемешивания фильтруют на нутче, остаток на нутч-фильтре четыре раза интенсивно промывают, каждый раз по 10 г гептана, и примерно 30-45 минут сушат отсасыванием. Выход: 1,7 г соединения III.
1H-ЯМР (200 МГц, CDCl3, δ): 8,79 (1H, с, шир.); 7,72 (1H, д); 7,49 (2H, м); 4,93 (1H, т); 3,59 (1H, м); 3,17 (1H, м); 2,38-1,0 (17H, м); 0,97 (3H, с); 0,81 (3H, с)
Стадия 4 (Получение дутастерида)
1 г (1,6 ммоль) соединения IIId со стадии 3 нагревают вместе с 8 г абсолютного ацетонитрила, 2 г хлороформа, 0,55 г (4,8 ммоль) аллилметилкарбоната и 0,17 г (0,16 ммоль) палладиевого катализатора до температуры флегмообразования (70-80°C). Уже при нагревании наблюдается образование газа. Через примерно 30 минут нагрева с обратным холодильником (образования газа больше не видно) реакционную смесь по возможности концентрируют и остаток соединяют с 5 г метанола. После охлаждения до 0-5°C медленно добавляют раствор, состоящий из 0,6 г (3,2 ммоль) 30%-ного раствора метилата натрия в 4 г метанола, и прозрачный раствор 1 час перемешивают также при внутренней температуре 0-5°C. После удаления охлаждающей ванны к этому прибавляют 3 г воды, снова перемешивают 1 час при комнатной температуре, мутную смесь концентрируют насколько возможно и к остатку добавляют 20 г толуола, а также 6 г воды. Смесь нагревают до температуры флегмообразования. Через 30 минут прозрачную органическую фазу отделяют горячей и охлаждают до комнатной температуры. Добавление 5-10 г гептана приводит к кристаллизации дутастерида. После фильтрования, трехкратного промывания гептаном, каждый раз по 4 г, и сушки отсасыванием выделяют 0,3 г дутастерида.
1H-ЯМР (200 МГц, CDCl3, δ): 8,80 (1H, с, шир.); 7,75 (1H, д); 7,49 (2H, м); 6,80 (1H, д); 5,82 (1H, д); 8,80 (1H, с, шир.); 5,46 (1H, с шир.); 3,35 (1H, м); 2,38-1,0 (17H, м); 0,97 (3H, с); 0,81 (3H, с).
Описывается улучшенный способ получения 17β-замещенных 4-аза-андрост-1-ен-3-онов общей формулы (I), где R-OH, С1-4алкил, С1-4алкенил, фенил, бензил и др. или их фармацевтически приемлемых солей, заключающийся в том, что в 3-кето-4-аза (лактамную группу) соединения общей формулы II, где R имеет указанные значения, вводят защитные группы с образованием соединения общей формулы III, где R3 триалкилсилил, или вместе с R4 остаток -С(O)-С(O)- или -C(O)-Y-C(O)-, где R4 - предпочтительно трет-бутилоксикарбонил, Y - (CH2)n n=1÷4, или ортофенилен, после чего полученное соединение преобразовывают в присутствии катализатора дегидрирования и в присутствии бензохинона, аллилметилкарбоната, аллилэтилкарбоната и/или аллилпропилкарбоната, при этом в 1,2-положение вводится Δ1-двойная связь, с последующим удалением защитных групп, и при необходимости (когда R-OH) переводом в соли. Способ дает высокий выход и степень чистоты продуктов. 20 з.п.ф-лы.

где R гидроксил, линейный или разветвленный (С1-С4)-алкил или (С1-C4)-алкенил; фенил или бензил; остаток -OR1, или остаток -NHR1, или остаток -NR1R2;
R1 водород, линейный или разветвленный (С1-С4)-алкил или (C1-C4)-алкенил, или фенил, возможно замещенный;
R2 водород, метил, этил или пропил; или -NR1R2 означает 5- или 6-членный гетероцикл, и в случае, когда R означает гидроксил, также их фармацевтически приемлемых солей, отличающийся тем, что
(А) в 3-кето-4-аза-группу (лактамную группу) соединения общей формулы (II)

вводят защитные группы, так что образуется соединение общей формулы (III)

в которой R3 триалкилсилил, или вместе с R4 остаток -С(O)-С(O)- или -C(O)-Y-C(O)-;
R4 алкилоксикарбонил или фенилоксикарбонил, предпочтительно Boc (=трет-бутилоксикарбонил); или триалкилсилил, или вместе с
R3 остаток -С(O)-С(O)- или -C(O)-Y-C(O)-;
Y-[С(R5)(R6)]n- или ортофенилен;
R5 и R6 независимо друг от друга водород, метил или этил; и n целое число от 1 до 4;
и в случае, если R означает гидроксил, возможно, реагирует с защитной группой;
(В) полученное [согласно стадии (А)] соединение преобразовывают в присутствии (i) катализатора дегидрирования, выбранного из соединений металлов группы VIII периодической системы элементов, и в присутствии (ii) бензохинона, возможно замещенного, аллилметилкарбоната, аллилэтилкарбоната и/или аллилпропилкарбоната, при этом в 1-/2-положение вводится Δ1-двойная связь, и
(С) защитные группы R3 и R4 удаляют, и при R=гидроксил полученное соединение при необходимости переводят в соль.
Приоритеты по пунктам:
| БИБЛИОТЕКА 1 | 0 |
|
SU298652A1 |
| УСТРОЙСТВО ДЛЯ РЕГУЛИРОВАНИЯ ПЕРЕМЕННОГО НАПРЯЖЕНИЯ | 1970 |
|
SU428366A1 |
| US 5710342 А, 20.01.1998. | |||

