Область техники
Настоящее изобретение относится к области медицинской химии. В частности, настоящее изобретение относится к новому типу производных, содержащих трициклический гетероарил, способу его синтеза и применению в качестве ингибитора различных киназ, включая CDK и/или TRK, при получении лекарственных средств для лечения опухолей и других подобных заболеваний.
Уровень техники
Злокачественные новообразования, или злокачественные опухоли, представляют собой патологию с одним из самых высоких в мире показателей заболеваемости и летальности. Они характеризуются неконтролируемой пролиферацией клеток и метастазами, которые распространяются по организму в течение короткого или относительно короткого времени после начала заболевания. Традиционные варианты лечения включают хирургическое вмешательство (при возможности резекции), лучевую терапию и химиотерапию. Варианты таргетной терапии, разработанные в последние годы, имеют преимущества в виде снижения токсичности и выраженности побочных эффектов, а также улучшения выживаемости. Однако после применения таргетных препаратов в течение определенного периода времени развивается устойчивость к лекарственным средствам, и тогда рост и распространение опухолевых клеток происходит чрезвычайно быстро. Распространенными видами онкологических заболеваний являются: гемобластозы, рак легкого, рак молочной железы, рак печени, рак мочевого пузыря, рак прямой кишки, рак желудка и т.д.
Регуляцию клеточного цикла осуществляет главным образом ряд серин/треониновых киназ. Этот класс серин/треониновых киназ также называется циклин-зависимой киназой (CDK). Они участвуют в регуляции клеточного цикла, транскрипции генетической информации и нормального деления, а также пролиферации клеток, функционируя вместе со своей соответствующей регуляторной субъединицей циклином. Семейство CDK человека насчитывает более 20 подтипов, которые делятся на две основные категории в соответствии с различными функциями: один тип CDK регулирует клеточный цикл, например, CDK2/4/6 и т. д; другой тип CDK участвует в регуляции транскрипции/процессинге РНК, например, CDK7/9 и т.д. Киназа CDK связывается с белком циклином, образуя специфический комплекс, и таким образом активируется. При многих видах злокачественных новообразований человека наблюдается избыточная активация CDK или отсутствие функции белка, ингибирующего CDK, что приводит к аномальной пролиферации и размножению опухолевых клеток. Поэтому CDK является важной мишенью противоопухолевыхсредств.
CDK2 и CDK4/6 являются ключевыми регуляторами клеточного цикла. В состоянии покоя клетки (фаза G0) транскрипционная активность фактора транскрипции E2F ингибируется белком ретинобластомы (Rb). При стимуляции клетки сигналом к делению она переходит в фазу G1. В фазе G1 циклин D (CyclinD) связывается с CDK4/6 и активирует его, после чего активированный CDK4/6 фосфорилирует Rb, что приводит к активации E2F. В это время E2F остается связанным с белком Rb, но может запускать транскрипцию таких белков, как CCNE1, CCNA2, CCNB1 и CDK2. В поздней фазе G1 (после точки рестрикции) CyclinE связывается с CDK2 и активирует его, после чего CDK2 дополнительно фосфорилирует Rb, что приводит к полному высвобождению Rb и активации E2F. Затем E2F индуцирует транскрипцию белков S-фазы, таких как CyclinA и CyclinE. CDK2/CyclinA и CDK1 поддерживают фосфорилирование белка Rb для обеспечения процесса деления клетки. CDK2/CyclinA способствует процессу преобразования фазы S/G2. Поэтому ингибирование киназной активности CDK2 и CDK4/6 может блокировать клеточный цикл и реализовать желаемый эффект подавления пролиферации опухоли. Препараты-ингибиторы CDK4/6 палбоциклиб, рибоциклиб и абемациклиб присутствуют на рынке, а эффективного ингибитора CDK2 в настоящее время не существует. CDK9 в основном регулирует процесс транскрипции РНК. CDK9 и соответствующий циклин образуют комплекс позитивного фактора элонгации транскрипции-b (P-TEFb). Большая часть CDK9 связывается с CyclinT1, и лишь небольшое количество - с CyclinT2a, CyclinT2b и CyclinK. CDK9 является каталитической субъединицей комплекса P-TEF, который может фосфорилировать С-концевой домен РНК-полимеразы II и способствовать процессу элонгации транскрипции РНК различных онкогенов, таких как MYC и MCL-1. Наблюдается патологическая гиперэкспрессия MYC и MCL-1 при различных типах злокачественных опухолей, при этом эффективных целенаправленных ингибиторов данных онкогенов пока не существует. Кроме того, в условиях репликации клеток комплекс CDK9/CyclinK также играет важную роль в поддержании стабильности генома.
CDK16 экспрессируется в различных клетках и типах тканей человека, в наибольшей степени — в головном мозге и яичках. Активация CDK16 зависит от CyclinY. Выключение гена CDK16 не влияет на нормальный рост мышей, но приводит к бесплодию у самцов мышей, что указывает на важную роль CDK16 в выработке спермы. Гиперэкспрессия CDK16 может способствовать росту и инвазии различных опухолевых клеток, например, при раке легких или гепатокарциноме. Этот эффект может быть связан со снижением экспрессии опухолевого супрессора p27 под действием CDK16.
CDK5 представляет собой особый белок семейства CDK. Хотя последовательность белка во многом сходна с другими представителями семейства CDK, основная функция CDK5 зависит от нециклиновых белков, таких как p35 и p39. CDK5 экспрессируется во многих тканях человека, но его активирующие факторы p35 и p39 в основном экспрессируются в нейронах, поэтому его активность сосредоточена преимущественно в нервной системе. Фосфорилируя различные нейронные белки, такие как Tau, Axin, CRMP2 и нейрофиламент, CDK5 может регулировать физиологические функции нейронов, например, миграцию нейронов, рост аксона, образование синапсов, формирование памяти, восприятие боли и т.д. В нейронах пациентов с нейродегенеративными заболеваниями CDK5 более устойчиво связывается с p25 - продуктом сплайсинга p35, и происходит патологическая активация, приводящая к дегенерации и гибели нервных клеток. Кроме того, CDK5 также играет роль в иммунном ответе, ангиогенезе, регуляции клеточного цикла, реакции на повреждение ДНК, старении клеток и апоптозе. Поэтому CDK5 постепенно становится важной мишенью в борьбе с нейродегенеративными и онкологическими заболеваниями.
Семейство киназ рецептора нейротрофина (NTRK) включает TRKA, TRKB и TRKC, которые кодируются генами NTRK1, NTRK2 и NTRK3 соответственно, и обычно экспрессируются в тканях нервной системы. Рецепторы TRK могут активироваться под действием различных нейротрофических факторов. Из них NGF преимущественно активирует TRKA; BDNF и NT-4/5 преимущественно активируют TRKB, а BT-3 активирует TRKC. После связывания с соответствующим лигандом происходит димеризация и фосфорилирование TRK, далее активируются последующие сигнальные пути, такие как PI3K/AKT, RAS/RAF/MEK и PLC-гамма, способствующие пролиферации и сохранению жизнеспособности клеток.
Показано, что мутации слияния генов NTRK связаны с различными видами злокачественных опухолей. Гены NTRK (в основном NTRK1 и NTRK3) могут сливаться с другими генами, а затем транскрибироваться и транслироваться в постоянно активированный белок TRK, что приводит к росту и пролиферации опухолевых клеток с мутациями слияния TRK. Мутации слияния NTRK наблюдаются примерно в 0,31 % от общего числа злокачественных опухолей у взрослых и в 0,34 % от общего числа злокачественных опухолей у детей. Мутации слияния NTRK3 чаще встречаются в некоторых редких опухолях, таких как секреторный рак молочной железы, фибросаркома и рак слюнных желез. Мутации NTRK1 преимущественно встречаются в некоторых распространенных типах рака, таких как аденокарцинома легких и рак толстой кишки, хотя частота мутаций относительно низкая. Кроме того, чрезмерная активация сигнального пути NGF-TRKA также играет важную роль в патогенезе воспалительной боли и боли при злокачественных новообразованиях.
Таким образом, разработка новых типов ингибиторов киназ, направленных на подтипы CDK и TRK, имеет большое значение.
Сущность изобретения
Настоящее изобретение представляет новый ингибитор киназы, который способен ингибировать несколько киназ, таких как CDK и TRK.
В первом аспекте настоящего изобретения описано соединение по следующей формуле (I), или его оптические изомеры, фармацевтически приемлемые соли, пролекарства, дейтерированные формы, гидраты или сольваты:
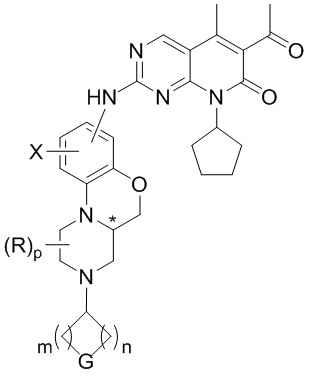
(I)
Знаком «*» обозначен хиральный центр;
X представляет собой водород, дейтерий, галоген, C1–4 алкил, OR1, NR1R2 или N(R1)C(O)R3;
все R независимо друг от друга представляют собой водород или C1–4 алкил; или когда два R одновременно присоединены к одному атому углерода, эти два R и атом углерода, к которому они присоединены, могут опционально образовывать карбонильную группу (C=O);
G представляет собой NRf, O, S, S(O), S(O)2 или CRgRg;
p равно 0, 1, 2 или 3;
m и n независимо друг от друга равны 0, 1, 2 или 3; при этом m и n не могут быть одновременно равны 0;
R1 и R2 независимо друг от друга представляют собой водород или C1–4 алкил;
R3 представляет собой C1–4 алкил, C2–4 алкенил или C2–4 алкинил;
Rf представляет собой водород, C1–4 алкил, C1–4 галоалкил, цианозамещенный C1–4 алкил, C2–4 алкенил, C2–4 алкинил, C3–8 циклоалкил, 4–8-членный гетероциклил, арил, гетероарил, C(O)R4, C(O)OR1, C(O)NR1R2, S(O)2R4 или S(O)2NR1R2;
все Rg независимо друг от друга выбирают из группы, состоящей из водорода, галогена или C1–4 алкила; или два Rg вместе с атомом углерода, к которому они присоединены, образуют карбонильную группу (C=O); или два Rg вместе с атомом углерода, к которому они присоединены, образуют 3–8-членную циклическую структуру, которая опционально содержит 0, 1 или 2 гетероатома, выбранных из N, O или S;
R4 представляет собой C1–4 алкил, C2–4 алкенил, C2–4 алкинил, C3–8 циклоалкил, 4–8-членную гетероциклическую группу, арил или гетероарил;
где каждый из указанных выше алкилов, алкенилов, алкинилов, циклоалкилов, гетероциклилов, арилов и гетероарилов необязательно и независимо замещен 1–3 заместителями, которые независимо друг от друга выбирают из группы, включающей галоген, C1–4 алкил, C1–4 галогеналкил, C2–4 алкенил, C2–4 алкинил, C3–8 циклоалкил, 3–8-членную гетероциклическую группу, арил, гетероарил, CN, NO2, OR1, SR1, NR1R2, C(O)R4, C(O)OR1, C(O)NR1R2, NR1C(O)R4 или S(O)2R4, при условии, что образовавшаяся химическая структура стабильна и логически обоснована; R1, R2 и R4 определены выше;
Если не указано иное, арил представляет собой ароматическую группу, содержащую от 6 до 12 атомов углерода; гетероарил представляет собой 5–15-членную гетероароматическую группу; а циклическая структура представляет собой насыщенную или ненасыщенную циклическую группу с гетероатомами или без них.
В другом предпочтительном варианте осуществления знаком «*» обозначен хиральный центр;
X представляет собой водород, дейтерий, галоген, C1–4 алкил, OR1, NR1R2 или NR1C(O)R3;
все R независимо друг от друга представляют собой водород или C1–4 алкил; или когда два R одновременно присоединены к одному атому углерода, эти два R и атом углерода, к которому они присоединены, могут опционально образовывать карбонильную группу (C=O);
G представляет собой NRf, O, S, S(O), S(O)2 или CRgRg;
p равно 0, 1, 2 или 3;
m и n каждый независимо равен 1, 2 или 3;
R1 и R2 независимо друг от друга представляют собой водород или C1–4 алкил;
R3 представляет собой C1–4 алкил, C2–4 алкенил или C2–4 алкинил;
Rf представляет собой водород, C1–4 алкил, C1–4 галоалкил, C2–4 алкенил, C2–4 алкинил, C3–8 циклоалкил, 4–8-членный гетероциклил, арил, гетероарил, C(O)R4, C(O)OR1, C(O)NR1R2 или S(O)2R4;
все Rg независимо друг от друга выбирают из группы, состоящей из водорода, галогена или C1–4 алкила; или два Rg вместе с атомом углерода, к которому они присоединены, образуют карбонильную группу (C=O); или два Rg вместе с атомом углерода, к которому они присоединены, образуют 3–8-членную циклическую структуру, которая опционально содержит 0, 1 или 2 гетероатома, выбранных из N, O или S;
R4 представляет собой C1–4 алкил, C2–4 алкенил, C2–4 алкинил, C3–8 циклоалкил, 4–8-членную гетероциклическую группу, арил или гетероарил;
где каждый из указанных выше алкилов, алкенилов, алкинилов, циклоалкилов, гетероциклилов, арилов и гетероарилов необязательно и независимо замещен 1–3 заместителями, которые независимо друг от друга выбирают из группы, включающей галоген, C1–4 алкил, C1–4 галогеналкил, C2–4 алкенил, C2–4 алкинил, C3–8 циклоалкил, 3–8-членную гетероциклическую группу, арил, гетероарил, CN, NO2, OR1, SR1, NR1R2, C(O)R4, C(O)OR1, C(O)NR1R2, NR1C(O)R4 или S(O)2R4, при условии, что образовавшаяся химическая структура стабильна и логически обоснована; R1, R2 и R4 определены выше;
Если не указано иное, арил представляет собой ароматические группы, содержащие от 6 до 12 атомов углерода; гетероарил представляет собой 5–15-членные гетероароматические группы; а циклическая структура представляет собой насыщенные или ненасыщенные циклические группы с гетероатомами или без них.
В другом предпочтительном варианте осуществления 4–8-членная гетероциклическая группа представляет собой 4–6-членную гетероциклическую группу.
В другом предпочтительном варианте осуществления соединение по формуле (I) представляет собой:
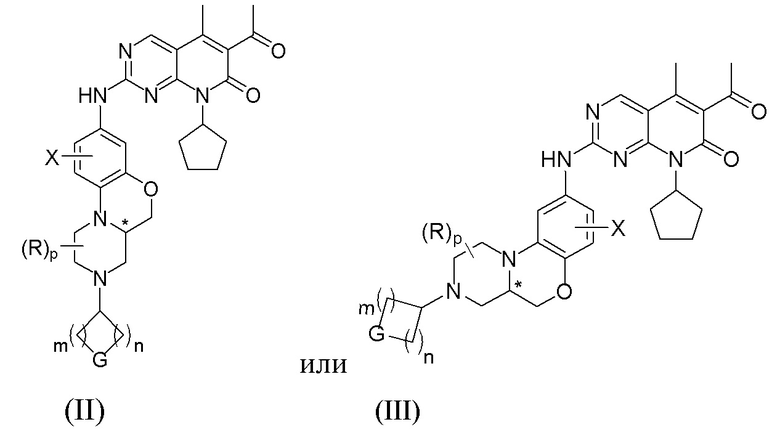
Знаком «*» обозначен хиральный центр;
X, R, G, p, m и n определены в первом аспекте настоящего изобретения.
В другом предпочтительном варианте осуществления, X представляет собой водород, галоген или C1–4 алкил; R представляет собой водород, или два R и атом углерода, к которому они присоединены, образуют карбонильную группу (C=O).
В другом варианте осуществления G представляет собой NRf, O или CRgRg; m и n независимо друг от друга равны 1 или 2; где Rf представляет собой водород, C1–4 алкил, C1–4 галогеналкил, C3–8 циклоалкил, 4-8-членную гетероциклическую группу, арил, гетероарил, C(O)R4 или S(O)2R4; где R4 представляет собой C1–4 алкил, C2–4 алкенил, C2–4 алкинил, C3–8 циклоалкил, 4–8-членную гетероциклическую группу.
В другом предпочтительном варианте осуществления  представляет собой 4–6-членное кольцо.
представляет собой 4–6-членное кольцо.
В другом реальном варианте осуществления соединение по формуле (I) представляет собой:
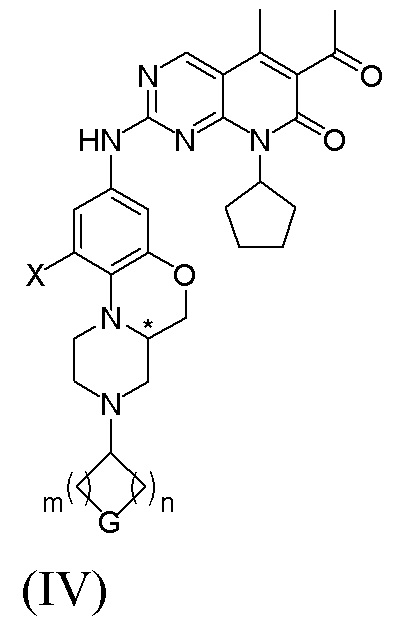
Знаком «*» обозначен хиральный центр;
X представляет собой водород, галоген или C1–4 алкил;
G представляет собой NRf, O или CRgRg; m и n независимо друг от друга равны 1 или 2; где Rf представляет собой водород, C1–4 алкил, C1–4 галогеналкил, C3–8 циклоалкил, 4-8-членную гетероциклическую группу, арил, гетероарил, C(O)R4 или S(O)2R4; где R4 представляет собой C1–4 алкил, C2–4 алкенил, C2–4 алкинил, C3–8 циклоалкил, 4–8-членную гетероциклическую группу.
В другом реальном варианте осуществления соединение по формуле (I) представляет собой:
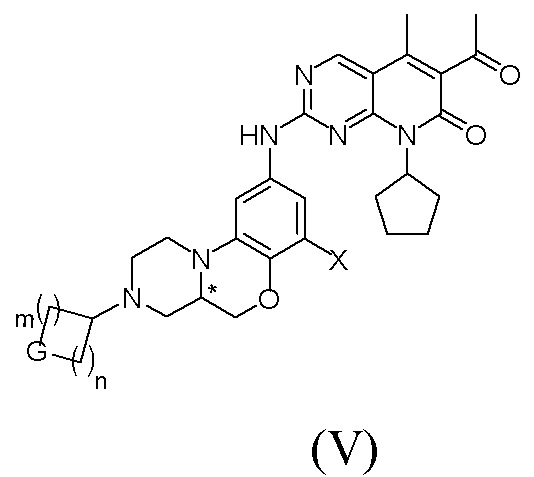
Знаком «*» обозначен хиральный центр;
X представляет собой водород, галоген или C1–4 алкил;
G представляет собой NRf, O или CRgRg; m и n независимо друг от друга равны 1 или 2; где Rf представляет собой водород, C1–4 алкил, C1–4 галогеналкил, C3–8 циклоалкил, 4-8-членную гетероциклическую группу, арил, гетероарил, C(O)R4 или S(O)2R4; где R4 представляет собой C1–4 алкил, C2–4 алкенил, C2–4 алкинил, C3–8 циклоалкил, 4–8-членную гетероциклическую группу.
В другом предпочтительном варианте осуществления, Rf представляет собой водород, C1–4 алкил, C1–4 галогеналкил, цианозамещенный C1–4 алкил, C3–8 циклоалкил, C(O)R4 или S(O)2R4; где R4 представляет собой C1–4 алкил.
В другом предпочтительном варианте осуществления, все Rg независимо друг от друга представляют собой водород или галоген.
В другом реализуемом варианте осуществления в формуле (IV) или формуле (V):
Знаком «*» обозначен хиральный центр;
X представляет собой водород, фтор или метил;
G представляет собой NRf, O или CRgRg; m и n независимо друг от друга равны 1 или 2; где Rf представляет собой водород, метил, этил, CH2CF3, CH2CN, циклопропил, C(O)CH3 или S(O)2CH3; все Rg независимо друг от друга представляют собой водород или фтор.
В другом предпочтительном варианте осуществления соединение по формуле (I) представляет собой соединение, выбираемое из следующих соединений, или смесь с соответствующим его энантиомером:
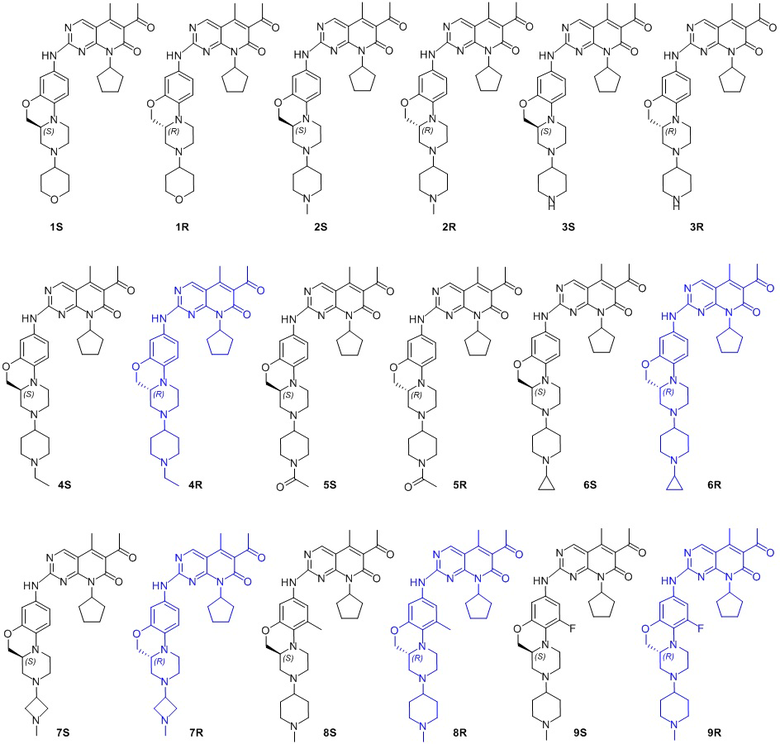
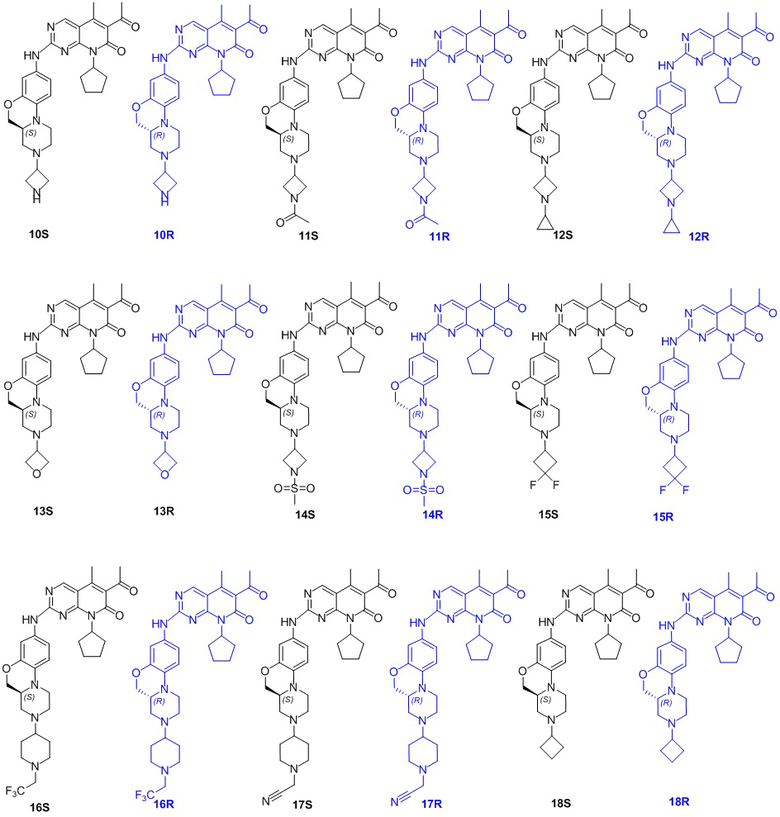
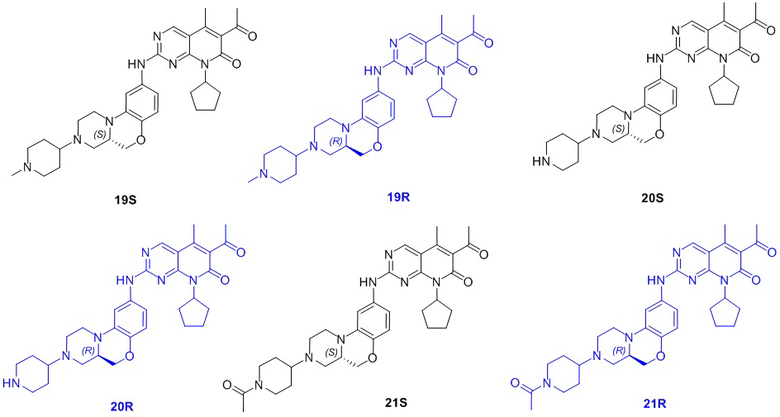
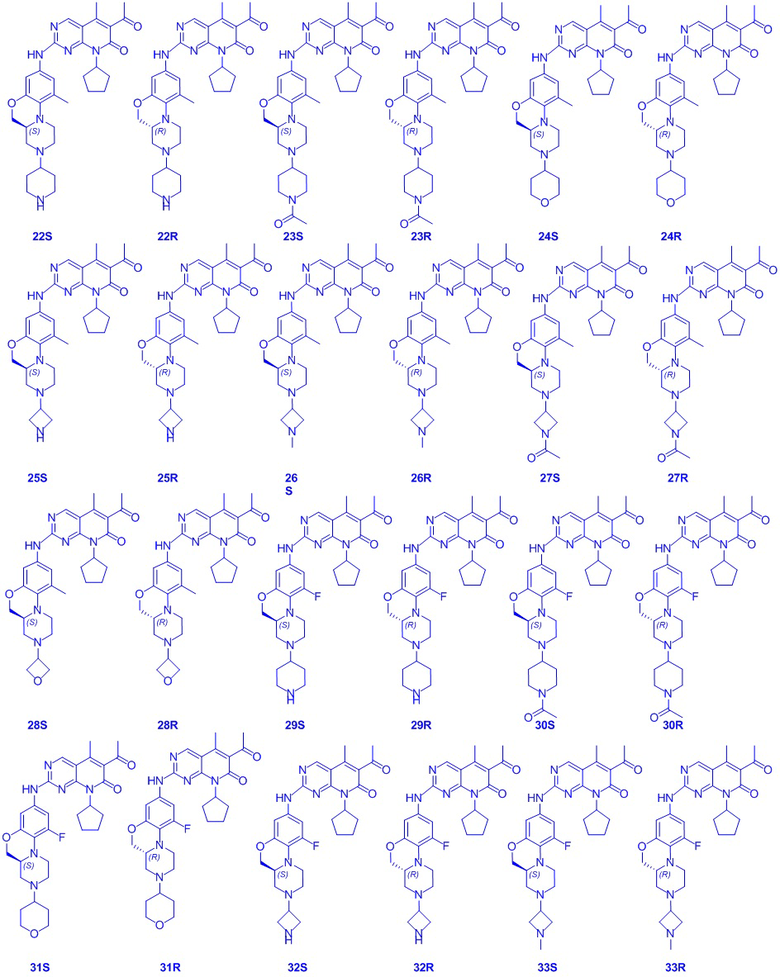
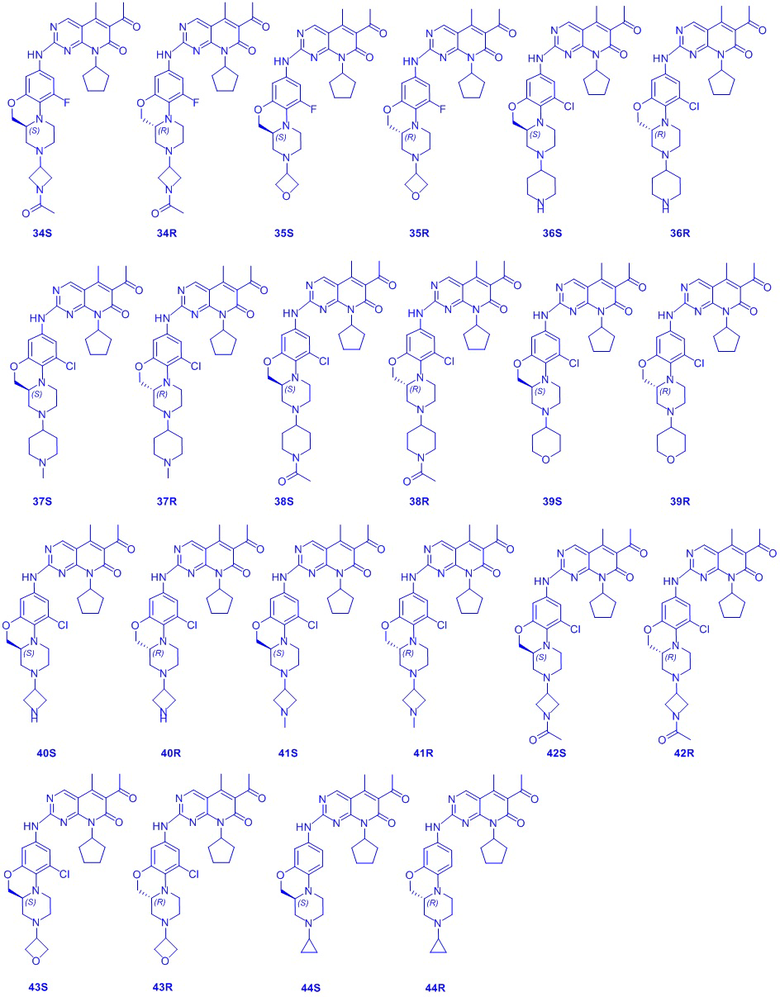 .
.
В другом предподчительном варианте осуществления соль представляет собой гидрохлорид.
Во втором аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая эффективное количество соединения, описанного в первом аспекте настоящего изобретения, или его оптических изомеров, фармацевтически приемлемых солей, пролекарств, дейтерированных форм, гидратов или сольватов.
В другом предподчительном варианте осуществления фармацевтическая композиция содержит: (i) терапевтически эффективное количество соединения формулы (I) по первому аспекту изобретения или его оптических изомеров, фармацевтически приемлемых солей, пролекарств, дейтерированных производных, гидратов или сольватов, и (ii) фармацевтически приемлемый носитель.
В третьем аспекте настоящего изобретения представлены следующие варианты применения соединения формулы (I) по первому аспекту изобретения или его оптических изомеров, фармацевтически приемлемых солей, пролекарств, дейтерированных производных, гидратов или сольватов или фармацевтической композиции по второму аспекту изобретения:
(а) получение лекарственного средства для лечения заболеваний, связанных с активностью киназы или уровнем экспрессии;
(b) получение таргетного ингибитора киназы; и/или
(c) нетерапевтическое ингибирование активности киназы in vitro;
при этом киназу выбирают из CDK и/или TRK.
В четвертом аспекте изобретения предложено применение соединений по первому аспекту изобретения или их оптических изомеров, фармацевтически приемлемых солей, пролекарств, дейтерированных форм, гидратов или сольватов или фармацевтической композиции по второму аспекту изобретения в качестве ингибиторов киназы или для лечения заболеваний, связанных с высокой экспрессией киназы; при этом киназу выбирают из группы, включающей CDK и/или TRK.
В пятом аспекте настоящего изобретения предложен способ ингибирования активности киназы, который включает следующую стадию: введение эффективного ингибирующего количества соединения формулы I по первому аспекту изобретения или оптических изомеров, фармацевтически приемлемых солей, пролекарств, дейтерированных форм, их гидратов или сольватов или фармацевтической композиции по второму аспекту изобретения субъекту, нуждающемуся в этом; при этом киназу выбирают из группы, включающей CDK и/или TRK.
В другом предпочтительном варианте осуществления заболевание выбирают из группы, включающей ДНК- и РНК-вирусные инфекций, В-клеточную лимфому, моноцитарный лейкоз, мегаполиспленическую полицитемию, синдром эозинофильного лейкоцитоза, идиопатическую тромбоцитопеническую пурпуру, системную гигантоклеточную болезнь, гемобластозы, солидные опухоли, нейродегенеративные заболевания.
В другом предпочтительном варианте осуществления, заболевание выбирают из группы, включающей аллергическую астму, миелофиброз, ревматоидный артрит, воспалительную боль, боль при злокачественном новообразовании, СПИД, вирус герпеса и вирус гриппа, секреторный рак молочной железы, фибросаркому, рак слюнных желез, рак печени, рак прямой кишки, рак мочевого пузыря, рак глотки и гортани, немелкоклеточный рак легкого, мелкоклеточный рак легкого, аденокарцинома легкого, плоскоклеточный рак легкого, рак молочной железы, рак предстательной железы, нейроглиоцитому, рак яичников, плоскоклеточный рак головы и шеи, рак шейки матки, рак пищевода, рак почки, рак поджелудочной железы, рак толстой кишки, рак кожи, лимфому, рак желудка, множественную миелому, опухоль головного мозга, рак легкого, болезнь Альцгеймера, болезнь Паркинсона.
В шестом аспекте настоящего изобретения предложен способ получения соединения по первому аспекту настоящего изобретения, который включает следующие стадии:
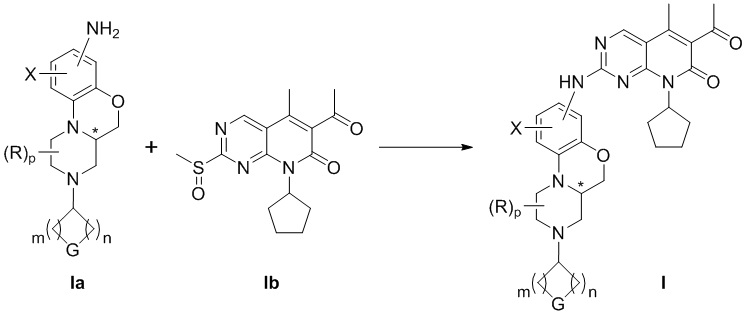
реакция соединения формулы Ia и соединения формулы Ib в инертном растворителе приводит к образованию соединения формулы I.
В другом предпочтительном варианте осуществления способ получения дополнительно включает следующие стадии:
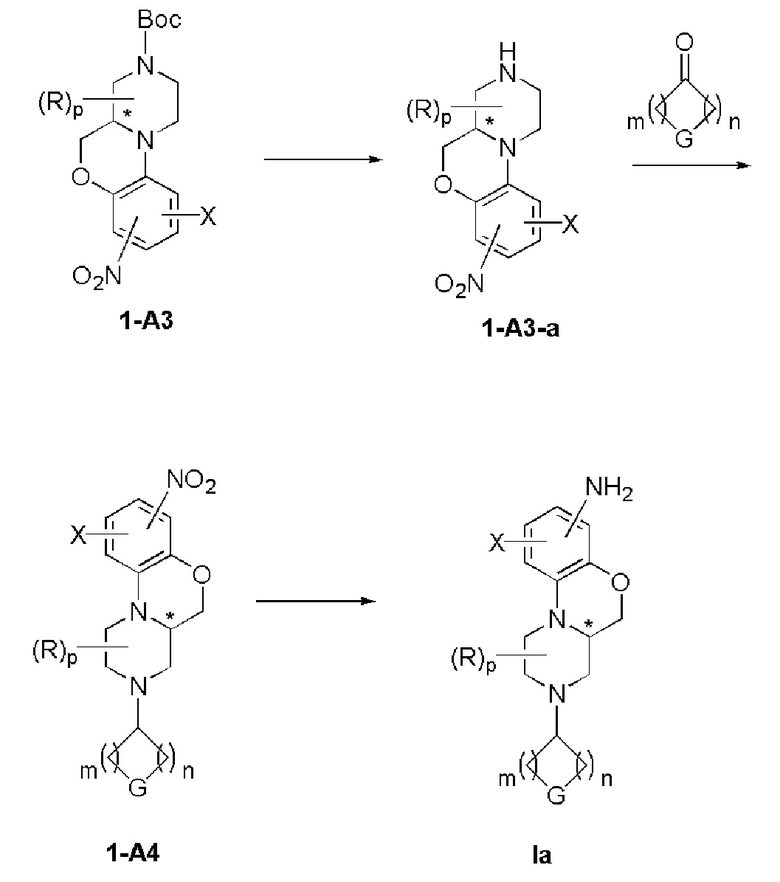
(1) снятие защиты с соединения формулы 1-А3 в инертном растворителе с получением соединения формулы 1-А3-а;
(2) реакция восстановительного аминирования соединения формулы 1-A3-a с соединением формулы 1-A3-b в инертном растворителе с получением соединения формулы 1-A4;
(i) восстановление соединения формулы 1-A4 в инертном растворителе с получением соединения формулы Ia.
Следует понимать, что в рамках настоящего изобретения различные технические признаки настоящего изобретения и различные технические признаки, описанные далее (например, в вариантах осуществления), могут быть объединены друг с другом с целью создания нового или предпочтительного технического решения. Ввиду ограничений по объему мы не будем повторно приводить их здесь.
Подробности осуществления изобретения
После длительных и интенсивных исследований авторы настоящего изобретения неожиданно открыли новый класс полициклических соединений со свойствами ингибиторов протеинкиназ, а также способы их получения и применения. Соединения изобретения могут применяться для лечения различных заболеваний, связанных с активностью киназ, включая CDK и TRK. На основании вышеизложенного изобретатели совершили настоящее изобретение.
Терминология
Если не указано иное, термин «или», используемый здесь, имеет то же значение, что и «и/или» (относится к «или» и «и»).
Если не указано иное, у всех соединений по настоящему изобретению каждый хиральный атом углерода (хиральный центр) может опционально находиться в R-конфигурации или S-конфигурации или в смеси R-конфигурации и S-конфигурации.
Используемый в настоящем документе термин «алкил», отдельно или в составе другого заместителя, относится к прямой (т. е. неразветвленной) или разветвленной насыщенной углеводородной группе, содержащей только атомы углерода, или к комбинации прямой и разветвленной цепей. Когда алкильная группа имеет ограничение по числу атомов углерода (например, C1–10), это означает, что алкильная группа имеет от 1 до 10 атомов углерода. Например, C1–8 алкил относится к алкильной группе, содержащей от 1 до 8 атомов углерода, включая метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил или тому подобное.
Используемый в настоящем документе термин «алкенил», отдельно или в составе другого заместителя, относится к группе с прямой или разветвленной углеродной цепью, имеющей как минимум одну двойную связь между атомами углерода. Алкенильные группы могут быть замещенными или незамещенными. Когда алкенильная группа имеет ограничение по количеству атомов углерода (например, C2–8), это означает, что алкенильная группа имеет от 2 до 8 атомов углерода. Например, C2–8 алкенил относится к алкенильным группам, имеющим 2–8 атомов углерода, включая этенил, пропенил, 1,2-бутенил, 2,3-бутенил, бутадиенил и тому подобное.
Используемый в настоящем документе термин «алкинил», отдельно или в составе другого заместителя, относится к алифатической углеводородной группе, имеющей как минимум одну тройную связь между атомами углерода. Алкинильная группа может быть прямой или разветвленной, или их комбинацией. Когда алкинильная группа имеет ограничение по количеству атомов углерода (например, C2–8 алкинильная группа), это означает, что алкинильная группа имеет от 2 до 8 атомов углерода. Например, термин «C2–8 алкинил» относится к прямой или разветвленной алкинильной группе, имеющей 2–8 атомов углерода, включая этинил, пропинил, изопропинил, бутинил, изобутинил, втор-бутинил, трет-бутинил или тому подобное.
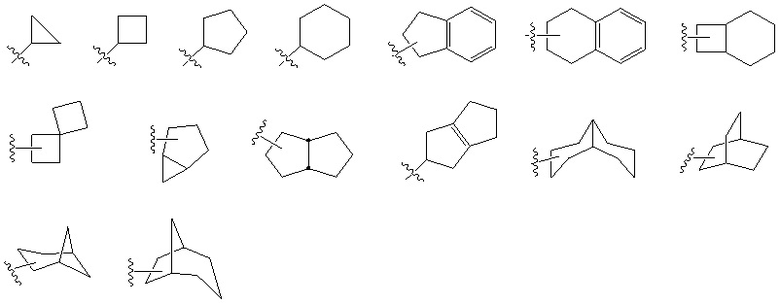
Используемый в настоящем документе термин «циклоалкил», отдельно или в составе другого заместителя, относится к группе, имеющей насыщенное или частично насыщенное кольцо, бициклическую или полициклическую (конденсированное кольцо, мостиковое или спиро) кольцевую систему. Когда определенная циклоалкильная группа имеет ограничение по числу атомов углерода (например, C3–10), это означает, что циклоалкильная группа имеет от 3 до 10 атомов углерода. В некоторых предпочтительных вариантах осуществления термин «C3–8 циклоалкил» относится к насыщенной или частично насыщенной моноциклической или бициклической алкильной группе, имеющей от 3 до 8 атомов углерода, включая циклопропил, циклобутил, циклопентил, циклогептил или тому подобное. «Спироциклоалкил» относится к бициклической или полициклической группе, которая имеет общий атом углерода (называемый спиро-атомом) между моноциклическими кольцами. Эти группы могут содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. «Конденсированнный циклоалкил» относится к бициклической или полициклической группе, состоящей только из атомов углерода, в которой в каждом кольце есть два соседних атома углерода, общих с другим кольцом (кольцами), которые могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. «Мостиковый циклоалкил» относится к полициклической группе, состоящей только из атомов углерода, в которой два кольца имеют два общих атома углерода, не связанных напрямую, которые могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Все атомы, входящие в циклоалкильную группу, представляют собой атомы углерода. Ниже приведены некоторые примеры циклоалкильных групп, но настоящее изобретение не ограничивается следующими циклоалкильными группами.
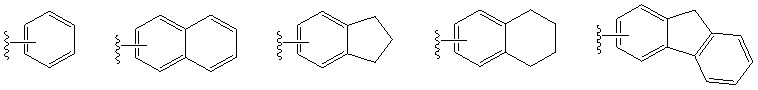
Если не указано иное, следующие термины, используемые в спецификации и формуле изобретения, имеют следующие значения. «Арил» означает моноциклические или конденсированные полициклические (т.е. кольца, имеющие пару соседних атомов углерода) группы, состоящие только из атомов углерода, имеющие сопряженную π-электронную систему, такие как фенил и нафтил. Арильное кольцо может соединяться с другими циклическими группами (включая насыщенные и ненасыщенные кольца), но не может содержать гетероатомы, такие как азот, кислород или сера, а точка присоединения к исходному кольцу должна находиться на атомах углерода кольца в сопряженной π-электронной системы. Арильная группа может быть замещенной или незамещенной. Ниже приведены некоторые примеры арильных групп, но настоящее изобретение не ограничивается описанными ниже арильными группами.
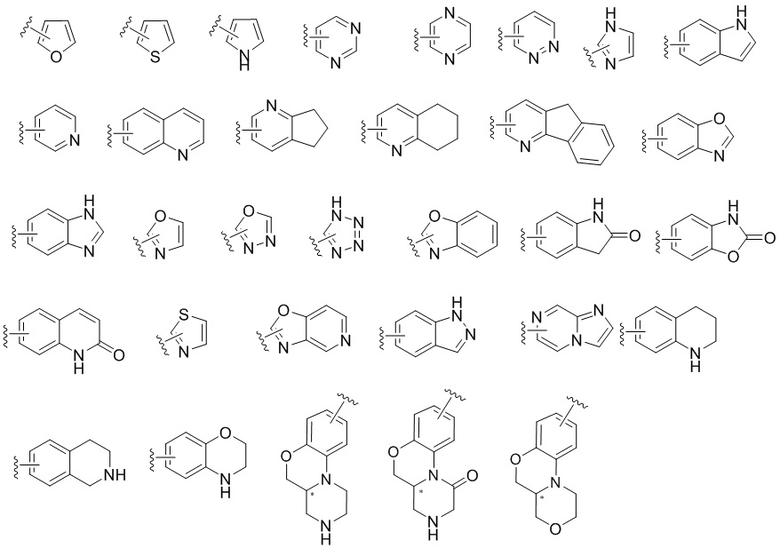
«Гетероарил» означает ароматическую моноциклическую или полициклическую группу, содержащую от одного до нескольких гетероатомов (опционально из азота, кислорода и серы), или полициклическую группу, образованную путем конденсации гетероциклической группы (содержащей от одного до нескольких гетероатомов, опционально из азота, кислорода и серы) с арильной группой, а место присоединения расположено на арильной группе. Гетероарильная группа может быть опционально замещенной или незамещенной. Ниже приведены некоторые примеры гетероарильных групп, но настоящее изобретение не ограничивается следующими гетероарильными группами.
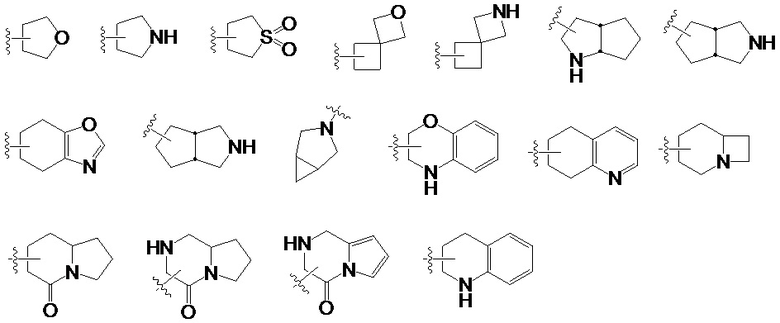
«Гетероциклил» означает насыщенный или частично ненасыщенный моноциклический или полициклический циклический углеводородный заместитель, в котором один или несколько атомов кольца выбирают из азота, кислорода или серы, а остальные атомы кольца представляют собой углерод. Неограничивающие примеры моноциклических гетероциклических групп включают пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил. Полициклическая гетероциклическая группа относится к гетероциклической группе, включающей спирокольцо, конденсированное кольцо и мостиковое кольцо. «Спироциклический гетероциклил» относится к полициклической гетероциклической группе, в которой каждое кольцо системы имеет общий атом (называемый спиро-атомом) с другими кольцами системы, где один или более атомов кольца выбирают из группы, состоящей из азота, кислорода или серы, остальные атомы кольца представляют собой углерод. «Гетероциклил с конденсированным кольцом» относится к полициклической гетероциклической группе, в которой каждое кольцо системы имеет общую соседнюю пару атомов с другими кольцами в системе, и одно или несколько колец могут содержать одну или несколько двойных связей, но ни одно кольцо не имеет полностью сопряженной π-электронной системы, и в которой один или более атомов в кольце выбирают из азота, кислорода или серы, а остальные атомы в кольце представляют собой углерод. «Мостиковый гетероциклил» относится к полициклической гетероциклической группе, в которой любые два кольца имеют два общих атома, которые не связаны напрямую, они могут содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы, и где один или большее количество кольцевых атомов выбирают из азота, кислорода или серы, а остальные атомы кольца представляют собой углерод. Если гетероциклическая группа имеет как насыщенное кольцо, так и ароматическое кольцо (например, насыщенное кольцо и ароматическое кольцо конденсированы), точка присоединения к исходному кольцу, должна находиться на насыщенном кольце. Примечание. Когда точка присоединения к исходному кольцу находится на ароматическом кольце, такая группа называется гетероарильной, а не гетероциклической. Ниже приведены некоторые примеры гетероциклической группы, но настоящее изобретение не ограничивается следующей гетероциклической группой.
Используемый здесь термин «галоген», отдельно или как часть другого заместителя, обозначает F, Cl, Br или I.
Используемый здесь термин «замещенный» (с «опционально» или без него) означает, что один или несколько атомов водорода в определенной группе замещены определенным заместителем. Определенные заместители представляют собой заместители, описанные выше в соответствующих абзацах, или заместители, которые указаны в примерах. Если не указано иное, опционально замещенная группа может иметь заместитель, выбранный из определенной группы, в любом замещаемом положении группы, и заместители могут быть одинаковыми или разными в каждом положении. Циклический заместитель, такой как гетероциклическая группа, может быть присоединен к другому кольцу, такому как циклоалкильная группа, с образованием спиробициклической кольцевой системы, т.е. два кольца имеют общий атом углерода. Специалистам в данной области техники будет понятно, что комбинации заместителей, предусмотренные настоящим изобретением, должны быть стабильными или химически достижимыми. Заместители представляют собой, помимо прочих, C1–8 алкил, C2–8 алкенил, C2–8 алкинил, C3–8 циклоалкил, 3–12-членный гетероциклил, арил, гетероарил, галоген, гидрокси, карбокси (-COOH), C1–8 альдегид, C2–10 ацил, C2–10 сложный эфир, аминогруппа.
Для удобства и в соответствии с общепринятым пониманием термин «опционально замещенный» или «необязательно замещенный» применяется только к участкам, которые могут быть замещены заместителем, и не включает химически недостижимые.
Если не указано иное, используемый здесь термин «фармацевтически приемлемая соль» относится к соли, которая пригодна для контакта с тканью субъекта (например, человека), не вызывая неприятных побочных эффектов. В некоторых вариантах осуществления фармацевтически приемлемая соль соединения изобретения включает соль (например, калиевую соль, натриевую соль, магниевую соль, кальциевую соль) соединения изобретения, имеющего кислотную группу, или основную соль A соединения изобретения (например, сульфат, гидрохлорид, фосфат, нитрат, карбонат).
Применение:
Настоящее изобретение относится к классу соединений формулы (I) или их дейтерированных производных, фармацевтически приемлемых солей, оптических изомеров (энантиомеров или диастереомеров, если таковые имеются), гидратов, сольватов или фармацевтических комбинаций, содержащих соединение, представленное формулой (I), его оптические изомеры, фармацевтически приемлемые соли, пролекарства, дейтерированные формы, гидраты и сольваты для ингибирования активности киназ, где киназа включает, помимо прочих, CDK и/или TRK.
Соединение по настоящему изобретению можно использовать в качестве ингибитора киназы. Предпочтительно киназа представляет собой CDK и/или TRK.
У пациентов с онкологическими заболеваниями экспрессия или активность различных упомянутых выше протеинкиназ в значительной степени повышены. Эта гиперэкспрессия и/или патологически высокая активность протеинкиназы напрямую связаны с возникновением и прогрессированием опухолей. Соединения по настоящему изобретению являются одиночными и/или двойными ингибиторами этих протеинкиназ. Заболевания можно предотвратить, облегчить или вылечить, модулируя активность этих протеинкиназ. Упомянутые заболевания включают аллергическую астму, миелофиброз, ревматоидный артрит, боль, связанную с воспалением и злокачественным новообразованием, СПИД, ДНК- и РНК-вирусные инфекции, такие как вирус герпеса и вирус гриппа, В-клеточную лимфому, моноцитарный лейкоз, полицитемию, спленомегалию, синдром эозинофильного лейкоцитоза, эссенциальную тромбоцитопению, системную гигантоклеточную болезнь, секреторный рак молочной железы, фибросаркому, рак слюнных желез, рак печени, рак прямой кишки, рак мочевого пузыря, рак гортани, немелкоклеточный рак, мелкоклеточный рак легкого, аденокарцинома легкого, плоскоклеточный рак легкого, рак молочной железы, рак предстательной железы, глиобластому, рак яичников, плоскоклеточный рак головы и шеи, рак шейки матки, рак пищевода, рак почки, рак поджелудочной железы, рак толстой кишки, рак кожи, лимфому, рак желудка, опухоль головного мозга, рак легких, множественную миелому и другие гемобластозы и солидные опухоли, а также нейродегенеративные заболевания, такие как болезнь Альцгеймера и болезнь Паркинсона.
С определенной точки зрения, многоцелевые ингибиторы киназ взаимодействуют с несколькими различными киназами одновременно, и производимые противоопухолевые эффекты часто являются аддитивными, поэтому они могут более эффективно лечить различные виды онкологических заболеваний.
Соединения по настоящему изобретению можно использовать в качестве комбинированных лекарственных средств с другими низкомолекулярными лекарственными средствами или биологическими агентами, такими как ингибиторы PD-1 (например, Опдиво® и Китруда®), для лечения различных видов онкологических и других заболеваний.
Соединения по настоящему изобретению и их дейтерированные формы, а также фармацевтически приемлемые соли или изомеры (если они присутствуют) или их гидраты и/или сочетания можно смешивать с фармацевтически приемлемыми вспомогательными веществами или носителями, полученную композицию можно вводить людям или животным для лечения нарушений, симптомов и заболеваний. Состав может представлять собой таблетки, пилюли, суспензии, растворы, эмульсии, капсулы, аэрозоли, стерильные растворы для инъекций, стерильные порошки и тому подобное. В предпочтительном варианте осуществления фармацевтическая композиция представляет собой лекарственную форму, пригодную для приема внутрь, включая, помимо прочих, таблетки, растворы, суспензии, капсулы, гранулы и порошки. Количество соединения или фармацевтической композиции, вводимой пациенту, не является фиксированным и обычно вводится в фармацевтически эффективном количестве. В то же время количество фактически вводимого соединения может быть определено врачом в соответствии с реальной ситуацией, включая заболевание, которое необходимо лечить, выбранный путь введения, фактическое вводимое соединение, индивидуальное состояние пациента и так далее. Дозировка соединения по настоящему изобретению зависит от конкретного применения для лечения, способа введения, состояния пациента и решения врача. Соотношение или концентрация соединения по настоящему изобретению в фармацевтической композиции зависит от множества факторов, включая дозировку, физические и химические свойства, способ введения и тому подобное.
Следует понимать, что в рамках настоящего изобретения различные технические признаки настоящего изобретения и различные технические признаки, описанные далее (например, в вариантах осуществления), могут быть объединены друг с другом с целью создания нового или предпочтительного технического решения.
Общие схемы синтеза
Соединение формулы I по настоящему изобретению можно получить следующим способом:
Схема 1:
В инертном растворителе соединение (Ia) взаимодействует с соединением (Ib) с получением соединения (I);
В приведенных выше формулах определение каждой группы соответствует приведенным выше. Реактивы и условия каждой стадии могут быть выбраны из обычных реактивов или условий, характерных для этого типа способов получения в данной области техники. После раскрытия структуры соединения по настоящему изобретению специалисты в данной области техники могут сделать этот выбор в соответствии со знаниями в данной области.
Другими словами, соединение, представленное общей формулой I по настоящему изобретению, может быть получено следующим способом, но условия способа, такие как реактивы, растворитель, основание, количество используемого соединения, температура реакции, требуемое время реакции, и т.д. не ограничиваются указанными в следующем объяснении. Соединения по настоящему изобретению также могут быть легко получены путем комбинирования различных способов синтеза, приведенных в данном описании или известных в данной области техники, и такие комбинации могут быть легко осуществлены специалистами в области, к которой относится настоящее изобретение.
В способе получения по настоящему изобретению каждую реакцию обычно проводят в инертном растворителе, а температура реакции обычно составляет от -20 до 150° C (предпочтительно от 0 до 120° C). Время реакции на каждой стадии обычно составляет от 0,5 до 48 часов, предпочтительно от 2 до 12 часов.
Соединения IIa и IIIa являются частью соединения I. Схема 2 иллюстрирует общий синтез соединений IIa и IIIa:
Схема 2:
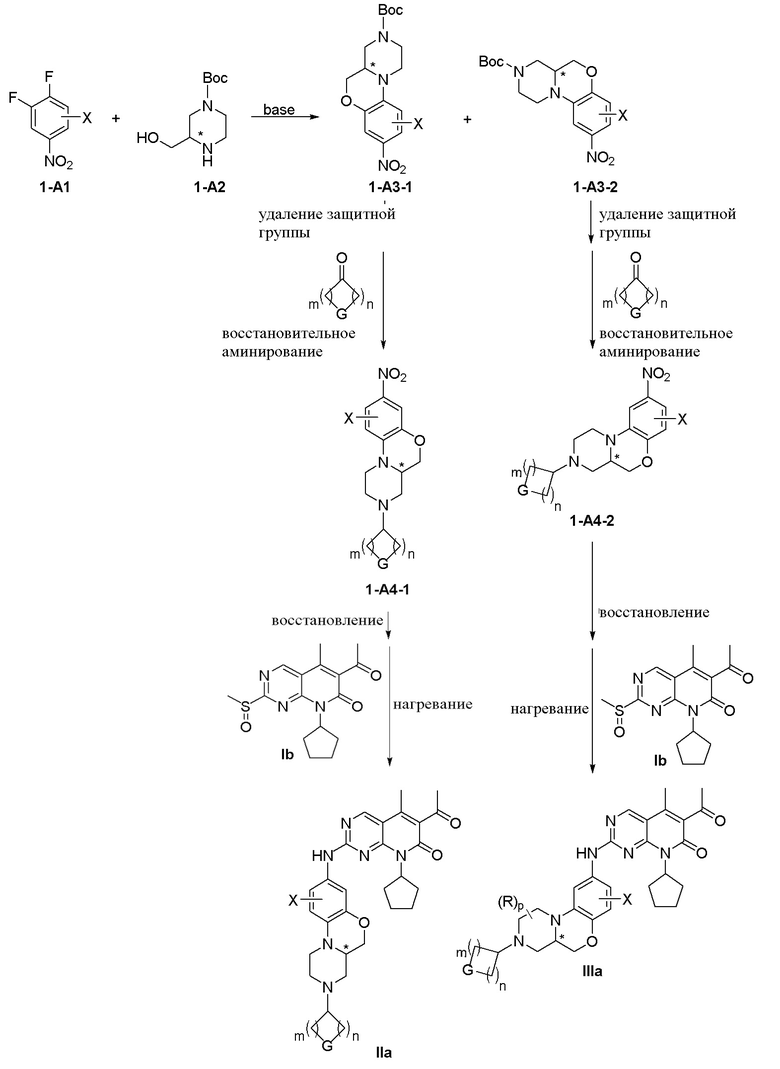
Определения X, R, G, m, n и p в приведенных выше схемах 1–2 те же, что и в п.1.
Промежуточное соединение Ib получают в соответствии с Journal of Medicinal Chemistry, 2005, 2371-2387 и указанными ссылками.
Фармацевтическая композиция и способ применения
Поскольку соединение по настоящему изобретению обладает превосходной ингибирующей активностью в отношении ряда протеинкиназ, соединение по настоящему изобретению и его различные кристаллические формы, оптические изомеры, фармацевтически приемлемые неорганические или органические соли, пролекарства, дейтерированные формы, гидраты или сольваты и фармацевтические композиции, содержащие соединения по настоящему изобретению в качестве основных действующих веществ, могут применяться для лечения, профилактики и облегчения заболеваний, связанных с активностью или экспрессией CDK, TRK и других киназ.
Фармацевтические композиции по настоящему изобретению содержат безопасное и эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли и фармацевтически приемлемое вспомогательное вещество или носитель. «Безопасное и эффективное количество» относится к такому количеству соединения, которого достаточно для существенного улучшения состояния без появления серьезных побочных эффектов. Обычно фармацевтические композиции содержат от 1 до 2000 мг соединения по настоящему изобретению на каждое действующее вещество, более предпочтительно от 5 до 200 мг соединения по изобретению на каждое действующее вещество. Предпочтительно «одна доза» представляет собой капсулу или таблетку.
«Фармацевтически приемлемый носитель» означает один или несколько совместимых твердых или жидких наполнителей или гелевых материалов, которые подходят для использования человеком и которые должны иметь достаточную чистоту и достаточно низкую токсичность. Под «совместимостью» здесь подразумевается, что компоненты композиции способны смешиваться с соединениями по настоящему изобретению и друг с другом без значительного снижения эффективности соединения. Примеры фармацевтически приемлемых носителей включают, помимо прочих, наполнитель (или разбавитель), разрыхлитель, смазывающее вещество, связывающее вещество, матрицу, эмульгаторы, смачиватели, красители, ароматизаторы, стабилизаторы, антиоксиданты, консерванты, апирогенную воду и тому подобное.
Способ введения соединения или фармацевтической композиции по настоящему изобретению не имеет конкретных ограничений, и репрезентативные способы введения включают, помимо прочих, пероральный, внутриопухолевый, ректальный, парентеральный (внутривенный, внутримышечный или подкожный) и местный.
Твердые лекарственные формы для введения внутрь включают капсулы, таблетки, пилюли, порошки и гранулы. В этих твердых лекарственных формах активное соединение смешивают как минимум с одним обычным инертным вспомогательным веществом (или носителем). В капсулах, таблетках и пилюлях лекарственные формы также могут содержать буферные агенты.
Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, могут быть приготовлены с покрытиями и оболочками, такими как растворимые в кишечнике покрытия и другие материалы, известные в этой области. Они могут содержать замутнители, и высвобождение активного соединения или соединений в таких композициях может происходить с задержкой в определенной части пищеварительного тракта. Примерами фиксирующих компонентов, которые можно использовать, являются полимерные и восковые материалы. При необходимости активное соединение может быть также в микрокапсулированной форме с одним или несколькими из вышеупомянутых вспомогательных веществ.
Кроме этих инертных разбавителей, композиции могут содержать вспомогательные вещества, такие как смачиватели, эмульгаторы и суспендирующие вещества, подсластители, ароматизаторы и вкусовые добавки.
В дополнение к активному соединению суспензия может содержать суспендирующие агенты.
Композиции для парентерального введения могут включать физиологически приемлемый стерильный водный или неводный раствор, дисперсию, суспензию или эмульсию и стерильный порошок для восстановления с получением стерильного раствора или дисперсию для инъекций. Подходящие водные и неводные носители, разбавители, растворители или носители включают воду, этанол, полиолы и их подходящие смеси.
Лекарственные формы соединений по настоящему изобретению для местного применения включают мази, порошки, пластыри, пропелленты и препараты для ингаляций. Действующее вещество смешивают в стерильных условиях с физиологически приемлемым носителем и любыми консервантами, буферными средствами или, при необходимости, пропеллентами.
Соединения по настоящему изобретению можно принимать самостоятельно или в сочетании с другими фармацевтически приемлемыми соединениями.
При использовании фармацевтической композиции безопасное и эффективное количество соединения по настоящему изобретению вводят млекопитающему (например, человеку), нуждающемуся в лечении, при этом доза представляет собой фармацевтически эффективную дозу для массы тела 60 кг. Вводимая доза обычно составляет от 1 до 2000 мг, предпочтительно от 5 до 500 мг. Конечно, конкретные дозы должны также учитывать такие факторы, как путь введения, состояние здоровья пациента и т.д., которые находятся в компетенции квалифицированного врача.
Основные преимущества данного изобретения включают:
1. Предложено соединение формулы I.
2. Предложена новая структура ингибиторов CDK, TRK (включая CDK2, CDK4, CDK5, CDK6, CDK9, CDK16, TRKA, TRKB и TRKC и т.д.), а также их получение и применение. Эти вещества ингибируют активность вышеуказанных протеинкиназ в очень низких концентрациях.
3. Предложен класс фармацевтических композиций для лечения заболеваний, связанных с активностью CDK, TRK и других киназ.
4. Предложен тип ингибиторов киназы, таких как CDK и TRK, с хорошей всасываемостью при приеме внутрь.
Ниже описание изобретения приводится вместе со специфическими вариантами осуществления. Следует понимать, что данные примеры не предназначены для ограничения объема изобретения. Экспериментальные методы в следующих примерах, в которых не указаны конкретные условия, обычно соответствуют традиционным условиям или условиям, рекомендованным производителем. Проценты и части приведены по массе, если не указано иное.
Пример 1: получение соединения 1S
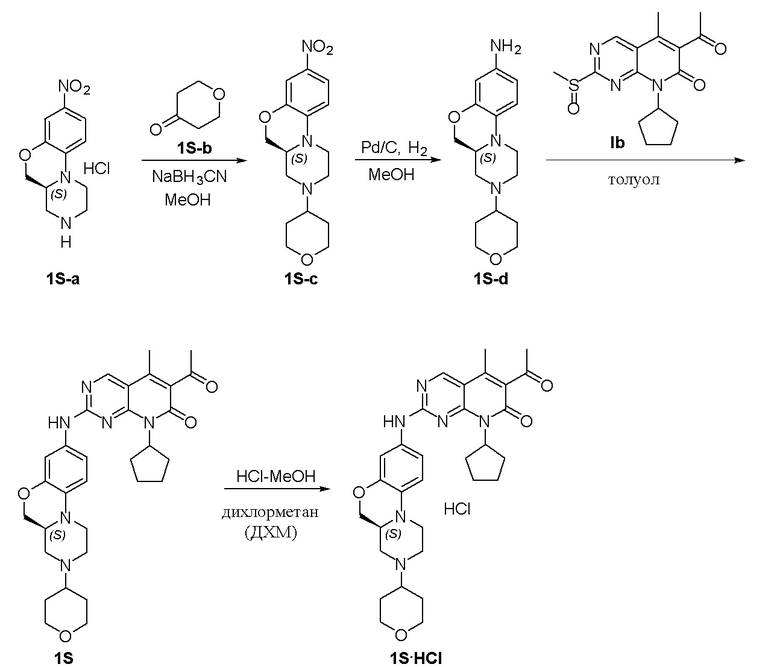
Соединение 1S-a (600 мг, 2,21 ммоль) растворяли в метаноле (20 мл). Добавляли тетрагидропиранон (1S-b, 265 мг, 2,65 ммоль) и триэтиламин (224 мг, 2,21 ммоль). Реакционную смесь перемешивали при 50 °C в течение 2 часов. Добавляли натрия цианоборгидрид (208 мг, 3,31 ммоль) при комнатной температуре и перемешивали реакционную смесь в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении для удаления метанола. Добавляли воду (10 мл) и полученную смесь экстрагировали дихлорметаном (3 × 20 мл). Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:EtOAc = 1:1) с получением соединения 1S-c (580 мг, выход 82 %) в виде твердого вещества желтого цвета. МС m/z 320,4 [M+H]+.
К раствору соединения 1S-c (580 мг, 1,82 ммоль) в метаноле (15 мл) добавляли Pd на угле (10 %, 70 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 3 часов. Завершение реакции контролировали методом ТСХ. Фильтровали через целит. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 50:1) с получением соединения 1S-d (460 мг, выход 88 %) в виде твердого вещества коричневого цвета. МС m/z 290,4 [M+H]+.
Соединение 1S-d (200 мг, 0,69 ммоль) и соединение Ib (230 мг, 0,69 ммоль) растворяли в толуоле (6 мл). Реакционную смесь перемешивали при 90 °C в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 80:1) с получением соединения 1S (290 мг, выход 75 %) в виде твердого вещества желтого цвета. МС m/z 559,8 [M+H]+.
Соединение 1S (290 мг, 0,52 ммоль) растворяли в дихлорметане (10 мл). Добавляли раствор HCl в метаноле (4,0 М, 0,13 мл, 0,52 ммоль) на ледяной бане. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. Добавляли воду (5 мл) и полученную смесь высушивали путем лиофилизации с получением гидрохлоридной соли соединения 1S (290 мг) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CD3OD) δ 8,77 (синглет, 1H), 7,16 (синглет, 1H), 6,99 (синглет, 2H), 5,90–5,75 (мультиплет, 1H), 4,37 (двойной дублет, J = 10,9, 2,4 Гц, 1H), 4,17 (дублет, J = 13,2 Гц, 1H), 4,11–4,04 (мультиплет, 3H), 3,82–3,70 (мультиплет, 2H), 3,63–3,49 (мультиплет, 2H), 3,45 (триплет, J = 11,5 Гц, 2H), 3,29–3,22 (мультиплет, 1H), 3,20–3,06 (мультиплет, 1H), 2,96 (триплет, J = 11,8 Гц, 1H), 2,47 (синглет, 3H), 2,33 (синглет, 3H), 2,30–2,19 (мультиплет, 2H), 2,18–2,10 (мультиплет, 2H), 2,01–1,76 (мультиплет, 6H), 1,67–1,53 (мультиплет, 2H) м.д. МС m/z 559,8 [M+H]+.
Пример 2: получение соединения 2S
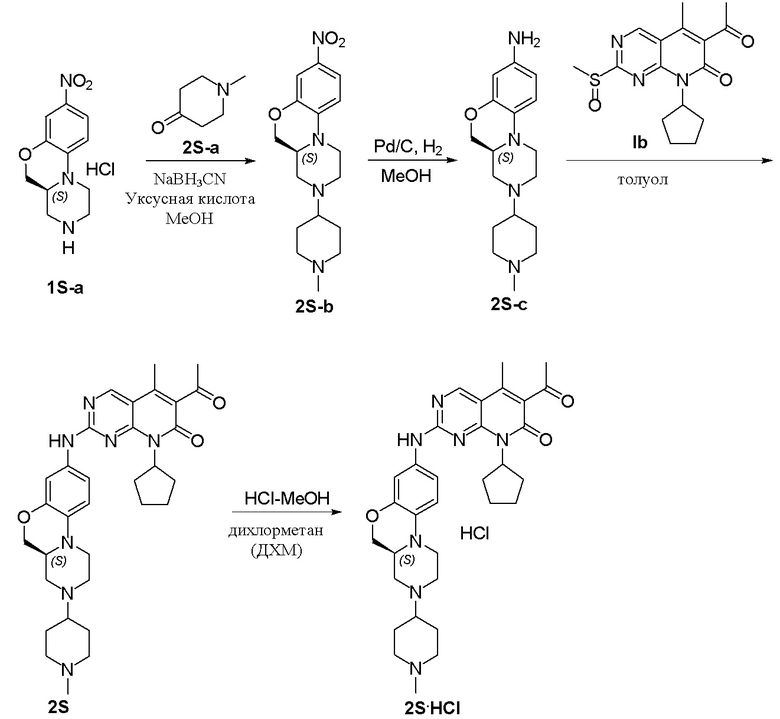
К метанолу (15 мл) добавляли соединение 1S-а (500 мг, 1,84 ммоль), N-метилпиперидон (2S-а, 416 мг, 3,68 ммоль) и уксусную кислоту (2 капли). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли натрия цианоборгидрид (231 мг, 3,68 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении для удаления метанола. Добавляли воду (10 мл) и полученную смесь экстрагировали дихлорметаном (3 × 20 мл). Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 30:1) с получением соединения 2S-b (500 мг) в виде твердого вещества желтого цвета, используемого на следующей стадии.
К метанолу (15 мл) добавляли соединение 2S-b (500 мг, 1,50 ммоль) и Pd на угле (10 %, 70 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1) с получением соединения 2S-c (280 мг, выход 62 %) в виде твердого вещества коричневого цвета.
Соединение 2S-c (280 мг, 0,93 ммоль) и соединение Ib (310 мг, 0,93 ммоль) растворяли в толуоле (8 мл). Реакционную смесь перемешивали при 90 °C в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1) с получением соединения 2S (180 мг, выход 34 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,86 (широкий синглет, 1H), 8,90 (синглет, 1H), 7,16 (дублет, J = 2,1 Гц, 1H), 7,06 (дублет, J = 8,9 Гц, 1H), 6,81 (дублет, J = 8,9 Гц, 1H), 5,91–5,70 (мультиплет, 1H), 4,23 (двойной дублет, J = 10,5, 2,4 Гц, 1H), 3,89 (двойной дублет, J = 10,5, 9,0 Гц, 1H), 3,69 (дублет, J = 11,3 Гц, 1H), 3,00–2,88 (мультиплет, 3H), 2,79 (дублет, J = 11,2 Гц, 2H), 2,59–2,53 (мультиплет, 1H), 2,41 (синглет, 3H), 2,34–2,16 (мультиплет, 7H), 2,14 (синглет, 3H), 1,94–1,73 (мультиплет, 9H), 1,62–1,51(мультиплет, 2H), 1,48–1,36 (мультиплет, 2H) м.д. МС m/z 572,8 [M+H]+.
Соединение 2S (180 мг, 0,31 ммоль) растворяли в дихлорметане (10 мл). Добавляли раствор HCl в метаноле (4,0 М, 0,08 мл, 0,32 ммоль) на ледяной бане. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. Добавляли воду (5 мл) и полученную смесь высушивали путем лиофилизации с получением гидрохлоридной соли соединения 2S (185 мг) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CD3OD) δ 8,79 (синглет, 1H), 7,21 (синглет, 1H), 7,05–6,97 (мультиплет, 2H), 5,92–5,80 (мультиплет, 1H), 4,42–4,35 (мультиплет, 1H), 4,19 (дублет, J = 13,0 Гц, 1H), 4,11–4,07 (мультиплет, 1H), 3,81–3,71 (мультиплет, 4H), 3,68–3,54 (мультиплет, 2H), 3,21–3,11 (мультиплет, 4H), 3,06–3,00 (мультиплет, 1H), 2,92 (синглет, 3H), 2,58–2,50 (мультиплет, 2H), 2,47 (синглет, 3H), 2,34 (синглет, 3H), 2,32–2,23 (мультиплет, 2H), 2,22–2,11 (мультиплет, 2H), 1,98–1,77 (мультиплет, 4H), 1,67–1,56 (мультиплет, 2H) м.д. МС m/z 572,8 [M+H]+.
Пример 3: получение соединения 2R
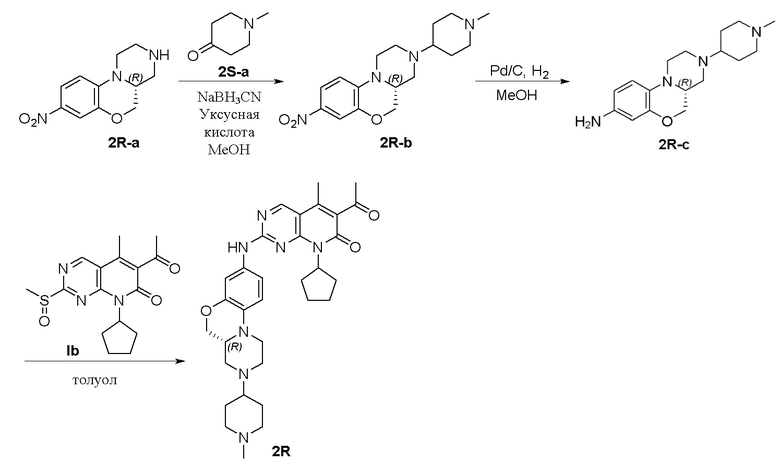
Смесь соединения 2R-а (100 мг, 0,43 ммоль), N-метил-4-пиперидона (2S-а, 144 мг, 1,28 ммоль) и уксусной кислоты (2 капли) в метаноле (6 мл) перемешивали при комнатной температуре в течение 1 часа. Добавляли натрия цианоборгидрид (80 мг, 1,28 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении и остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 2R-b (200 мг) в виде твердого вещества желтого цвета, используемого на следующей стадии. МС m/z 333,5 [M+H]+.
К метанолу (6 мл) при комнатной температуре добавляли соединение 2R-b (200 мг, 0,60 ммоль) и Pd на угле (10 %, 80 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 2R-c (93 мг, выход двух стадий 72 %) в виде маслянистой жидкости желтого цвета. МС m/z 303,5 [M+H]+.
Смесь соединения 2R-c (50 мг, 0,17 ммоль) и соединения Ib (55 мг, 0,17 ммоль) в толуоле (1 мл) в закрытой пробирке нагревали до 100 °C и перемешивали в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1) с получением соединения 2R (43 мг, выход 45 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,87 (широкий синглет, 1H), 8,90 (синглет, 1H), 7,16 (дублет, J = 2,0 Гц, 1H), 7,06 (дублет, J = 8,0 Гц, 1H), 6,81 (дублет, J = 8,9 Гц, 1H), 5,91–5,70 (мультиплет, 1H), 4,23 (двойной дублет, J = 10,5, 2,4 Гц, 1H), 3,89 (двойной дублет, J = 10,4, 9,1 Гц, 1H), 3,69 (дублет, J = 11,3 Гц, 1H), 3,02–2,89 (мультиплет, 3H), 2,80 (дублет, J = 11,0 Гц, 2H), 2,58–2,51 (мультиплет, 1H), 2,41 (синглет, 3H), 2,34–2,15 (мультиплет, 7H), 2,14 (синглет, 3H), 1,95–1,68 (мультиплет, 9H), 1,63–1,52 (мультиплет, 2H), 1,48–1,36 (мультиплет, 2H) м.д. МС m/z 572,8 [M+H]+.
Пример 4: получение соединения 3S
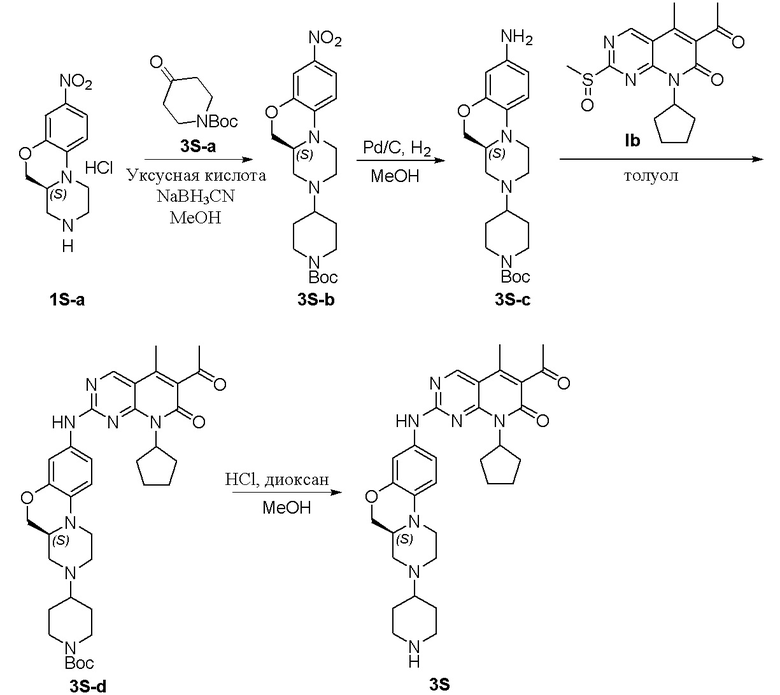
Соединение 1S-a (1 г, 4,25 ммоль) растворяли в метаноле (20 мл). Добавляли N-бутилоксикарбонил-4-пиперидон (3S-a, 2,54 г, 12,75 ммоль) и уксусную кислоту (4 капли). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли натрия цианоборгидрид (801 мг, 12,75 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении для удаления метанола. Добавляли воду (10 мл) и полученную смесь экстрагировали дихлорметаном (3 × 20 мл). Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1) с получением соединения 3S-b (1,39 г, выход 78 %) в виде твердого вещества желтого цвета. МС m/z 419,5 [M+H]+.
К метанолу (5 мл) при комнатной температуре добавляли соединение 3S-b (190 мг, 0,45 ммоль) и Pd на угле (10 %, 60 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 40:1) с получением соединения 3S-c (80 мг, выход 45 %) в виде твердого вещества желтого цвета. МС m/z 389,5 [M+H]+.
Соединение 3S-c (80 мг, 0,21 ммоль) и соединение Ib (69 мг, 0,21 ммоль) растворяли в толуоле (1 мл). Реакционную смесь перемешивали в течение ночи при температуре 90 °C. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 30:1) с получением соединения 3S-d (30 мг, выход 22 %) в виде твердого вещества желтого цвета. МС m/z 658,8 [M+H]+.
Соединение 3S-d (30 мг, 0,45 ммоль) растворяли в метаноле (2 мл). Добавляли раствор HCl в 1,4-диоксане (4,0 М, 1,1 мл, 4,4 ммоль) на ледяной бане. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении. Остаток нейтрализовали насыщенным водным раствором NaHCO3 до pH 7–8 и экстрагировали дихлорметаном (3 × 10 мл). Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали методом ТСХ (CH2Cl2:CH3OH = 20:1) с получением соединения 3S (13 мг, выход 51 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CDCl3) δ 8,70 (синглет, 1H), 7,11 (широкий синглет, 1H), 7,11 (дублет, J = 2,5 Гц, 1H), 6,97 (двойной дублет, J = 9,0, 2,5 Гц, 1H), 6,76 (дублет, J = 9,0 Гц, 1H), 5,88–5,80 (мультиплет, 1H), 4,20 (двойной дублет, J = 10,0, 2,0 Гц, 1H), 4,03 (триплет, J = 10,0 Гц, 1H), 3,67 (дублет, J = 12,0 Гц, 1H), 3,22–3,10 (мультиплет, 3H), 3,03 (дублет, J = 11,0 Гц, 1H), 2,89 (дублет, J = 10,5 Гц, 1H), 2,82–2,75 (мультиплет, 1H), 2,61 (триплет, J = 11,5 Гц, 2H), 2,54 (синглет, 3H), 2,54–2,47 (мультиплет, 1H), 2,44–2,39 (мультиплет, 1H), 2,34 (синглет, 3H), 2,34–2,25 (мультиплет, 2H), 2,07 (триплет, J = 10,8 Гц, 1H), 1,99–1,89 (мультиплет, 2H), 1,86–1,79 (мультиплет, 4H), 1,67–1,56 (мультиплет, 2H), 1,49–1,38 (мультиплет, 2H) м.д. МС m/z 558,8 [M+H]+.
Пример 5: получение соединения 4S
толуол
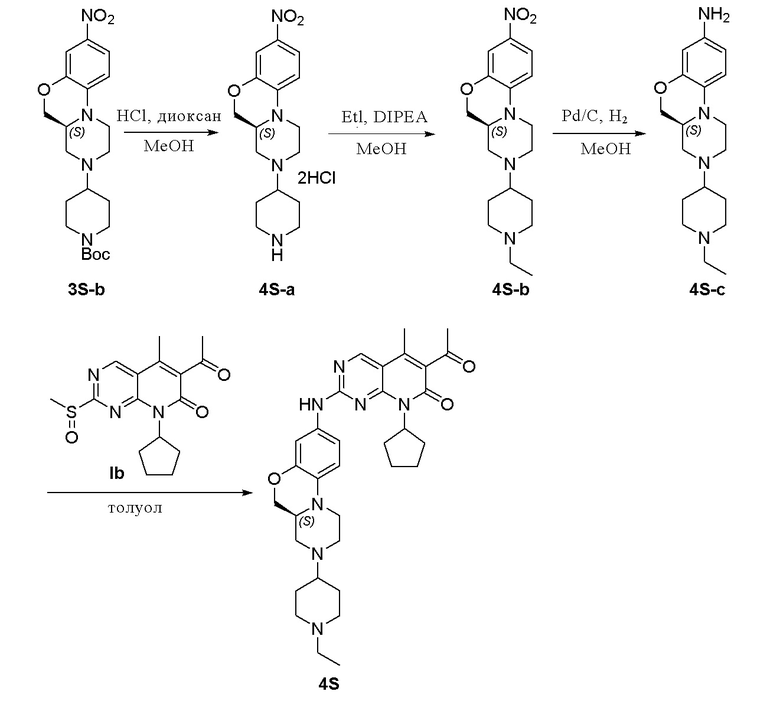
Соединение 3S-b (500 мг, 1,19 ммоль) растворяли в метаноле (10 мл). Добавляли раствор HCl в 1,4-диоксане (4,0 М, 3 мл, 12 ммоль) на ледяной бане. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении с получением соединения 4S-a (500 мг) в виде твердого вещества желтого цвета, используемого на следующей стадии. МС m/z 319,5 [M+H]+.
Соединение 4S-a (140 мг, 0,36 ммоль) растворяли в метаноле (5 мл) и добавляли DIPEA (0,2 мл, 1,32 ммоль) и йодэтан (137 мг, 0,88 ммоль). Реакционную смесь перемешивали при 50 °C в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1) с получением соединения 4S-b (56 мг, выход 45 %) в виде маслянистой жидкости желтого цвета. МС m/z 347,5 [M+H]+.
К метанолу (3 мл) при комнатной температуре добавляли соединение 4S-b (56 мг, 0,16 ммоль) и Pd на угле (10 %, 30 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1) с получением соединения 4S-c (36 мг, выход 70 %) в виде твердого вещества желтого цвета. МС m/z 317,5 [M+H]+.
Соединение 4S-c (36 мг, 0,11 ммоль) и соединение Ib (45 мг, 0,14 ммоль) растворяли в толуоле (1 мл). Реакционную смесь перемешивали в течение ночи при температуре 90 °C. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали методом препаративной ТСХ (CH2Cl2:CH3OH = 30:1) с получением соединения 4S (25 мг, выход 38 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CDCl3) δ 8,70 (синглет, 1H), 7,23 (широкий синглет, 1H), 7,11 (дублет, J = 2,5 Гц, 1H), 6,95 (двойной дублет, J = 9,0, 2,5 Гц, 1H), 6,76 (дублет, J = 9,0 Гц, 1H), 5,88–5,80 (мультиплет, 1H), 4,19 (двойной дублет, J = 10,5, 2,5 Гц, 1H), 4,02 (двойной дублет, J = 10,5, 9,5 Гц, 1H), 3,66 (дублет, J = 11,5 Гц, 1H), 3,17–3,10 (мультиплет, 1H), 3,07 (дублет, J = 10,5 Гц, 2H), 3,01 (дублет, J = 9,5 Гц, 1H), 2,86 (дублет, J = 10,0 Гц, 1H), 2,81–2,75 (мультиплет, 1H), 2,54 (синглет, 3H), 2,54–2,42 (мультиплет, 3H), 2,39–2,27 (мультиплет, 3H), 2,34 (синглет, 3H), 2,08 (триплет, J = 10,8 Гц, 1H), 2,05–1,75 (мультиплет, 8H), 1,70–1,56 (мультиплет, 4H), 1,13 (триплет, J = 7,0 Гц, 3H) м.д. МС m/z 586,8 [M+H]+.
Пример 6: получение соединения 5S
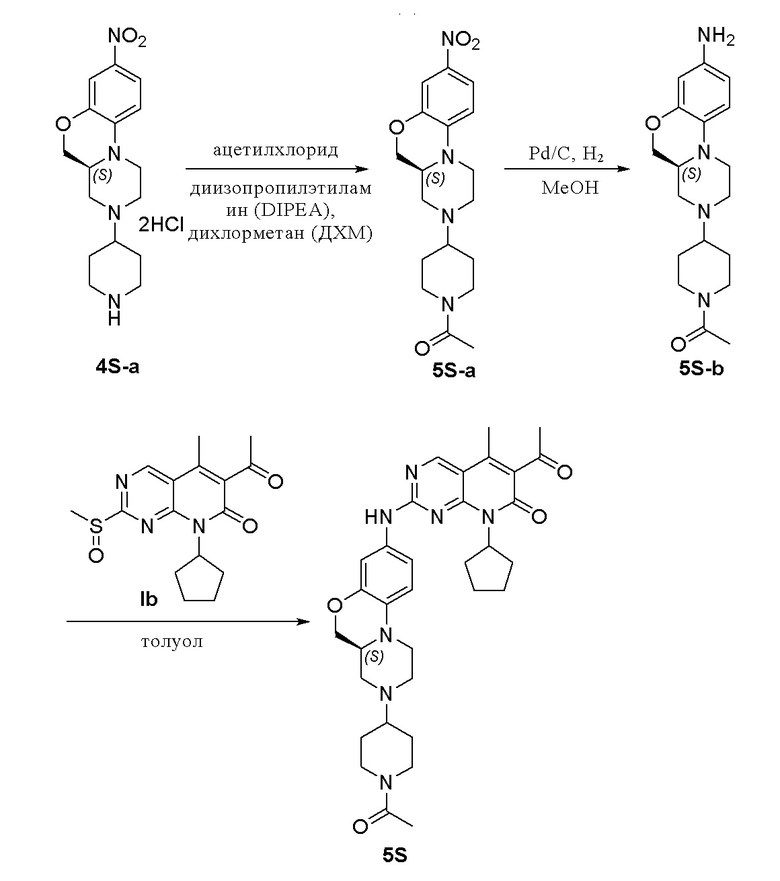
К раствору соединения 4S-a (200 мг, 0,63 ммоль) в дихлорметане (5 мл) добавляли DIPEA (0,3 мл, 1,88 ммоль) при 0 °C и добавляли раствор ацетилхлорида (98 мг, 1,26 ммоль) в дихлорметане (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Завершение реакции контролировали методом ТСХ. Добавляли воду (10 мл) и полученную смесь экстрагировали дихлорметаном (3 × 10 мл). Объединенные органические слои промывали рассолом (20 мл), сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 30:1) с получением соединения 5S-a (170 мг, выход 92 %) в виде твердого вещества желтого цвета. МС m/z 361,5 [M+H]+.
К метанолу (3 мл) при комнатной температуре добавляли соединение 5S-a (170 мг, 0,47 ммоль) и Pd на угле (10 %, 60 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1) с получением соединения 5S-b (130 мг, выход 83 %) в виде твердого вещества желтого цвета. МС m/z 331,5 [M+H]+.
Соединение 5S-b (65 мг, 0,20 ммоль) и соединение Ib (79 мг, 0,24 ммоль) растворяли в толуоле (1 мл). 中. Реакционную смесь перемешивали в течение ночи при температуре 90 °C. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали методом препаративной ТСХ (CH2Cl2:CH3OH = 20:1) с получением соединения 5S (15 мг, выход 13 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CD3OD) δ 8,81 (синглет, 1H), 7,18 (дублет, J = 2,0 Гц, 1H), 6,97 (дублет, J = 8,5 Гц, 1H), 6,83 (дублет, J = 8,5 Гц, 1H), 6,02–5,83 (мультиплет, 1H), 4,57 (дублет, J = 12,5 Гц, 1H), 4,25 (дублет, J = 10,0 Гц, 1H), 4,04–3,92 (мультиплет, 2H), 3,78 (дублет, J = 11,0 Гц, 1H), 3,19–2,99 (мультиплет, 4H), 2,78–2,59 (мультиплет, 3H), 2,58–2,50 (мультиплет, 1H), 2,47 (синглет, 3H), 2,38–2,25 (мультиплет, 2H), 2,35 (синглет, 3H), 2,17–2,08 (мультиплет, 1H), 2,11 (синглет, 3H), 2,05–1,89 (мультиплет, 4H), 1,87–1,78 (мультиплет, 2H), 1,68–1,59 (мультиплет, 2H), 1,56–1,49 (мультиплет, 1H), 1,45–1,37 (мультиплет, 1H) м.д. МС m/z 600,8 [M+H]+.
Пример 7: получение соединения 6S
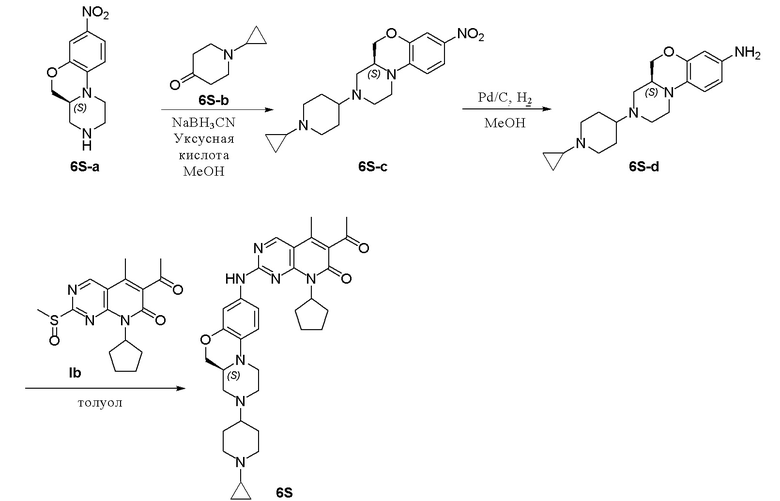
Смесь соединения 6S-a (250 мг, 1,06 ммоль), 1-циклопропил-4-пиперидона (6S-b, 444 мг, 3,19 ммоль) и уксусной кислоты (2 капли) в метаноле (15 мл) перемешивали при комнатной температуре в течение 1 часа. Добавляли натрия цианоборгидрид (200 мг, 3,19 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 6S-c (200 мг, выход 53 %) в виде твердого вещества желтого цвета. МС m/z 359,4 [M+H]+.
К метанолу (8 мл) при комнатной температуре добавляли соединение 6S-c (200 мг, 0,56 ммоль) и Pd на угле (10 %, 80 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (дихлорметан:метанол = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 6S-d (128 мг, выход 70 %) в виде маслянистой жидкости желтого цвета. МС m/z 329,5 [M+H]+.
Смесь соединения 6S-d (88 мг, 0,27 ммоль) и соединения Ib (89 мг, 0,27 ммоль) в толуоле (1,5 мл) в закрытой пробирке нагревали до 100 °C и перемешивали в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1) с получением соединения 6S (96 мг, выход 60 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,87 (широкий синглет, 1H), 8,90 (синглет, 1H), 7,16 (дублет, J = 1,9 Гц, 1H), 7,06 (дублет, J = 8,3 Гц, 1H), 6,81 (дублет, J = 8,9 Гц, 1H), 5,91–5,76 (мультиплет, 1H), 4,22 (двойной дублет, J = 10,5, 2,2 Гц, 1H), 3,93–3,84 (мультиплет, 1H), 3,69 (дублет, J = 11,0 Гц, 1H), 3,04–2,78 (мультиплет, 5H), 2,58–2,51 (мультиплет, 1H), 2,41 (синглет, 3H), 2,37–2,06 (мультиплет, 6H), 2,29 (синглет, 3H), 1,95–1,83 (мультиплет, 3H), 1,81–1,68 (мультиплет, 4H), 1,61–1,52 (мультиплет, 3H), 1,41–1,27 (мультиплет, 2H), 0,43–0,34 (мультиплет, 2H), 0,32–0,21 (мультиплет, 2H) м.д. МС m/z 598,8 [M+H]+.
Пример 8: получение соединения 7S
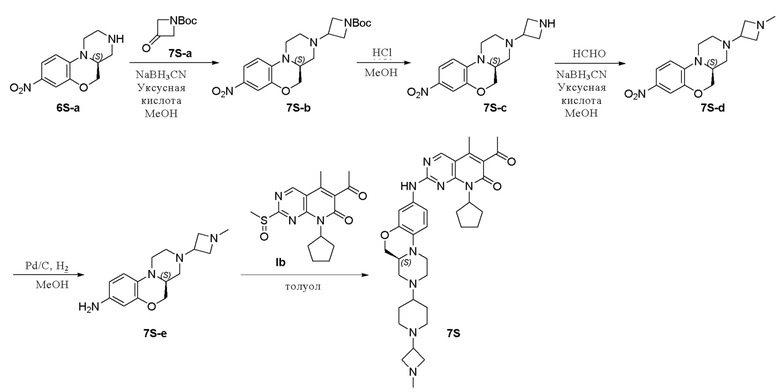
Смесь соединения 6S-a (500 мг, 2,13 ммоль), 1-бутилоксикарбонил-3-азциклобутанона (7S-a, 1,09 г, 6,38 ммоль) и уксусной кислоты (4 капли) в метаноле (10 мл) перемешивали при комнатной температуре в течение 1 часа. Добавляли натрия цианоборгидрид (400 мг, 6,38 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 7S-b (1,24 г) в виде твердого вещества желтого цвета, используемого на следующей стадии. МС m/z 391,4 [M+H]+.
Соединение 7S-b (1,24 г, 3,18 ммоль) растворяли в метаноле (15 мл) при комнатной температуре и добавляли раствор HCl в метаноле (4,0 М, 4 мл). Реакционную смесь перемешивали при 60 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, нейтрализовали насыщенным водным раствором NaHCO3 до рН = 7–8 и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 10:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 7S-c (410 мг, выход двух стадий 66 %) в виде твердого вещества желтого цвета. МС m/z 291,3 [M+H]+.
Смесь соединения 7S-c (410 мг, 1,41 ммоль), параформальдегида (127 мг, 4,24 ммоль) и уксусной кислоты (3 капли) в метаноле (8 мл) перемешивали при комнатной температуре в течение 1 часа. Добавляли натрия цианоборгидрид (266 мг, 4,24 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (дихлорметан:метанол = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 7S-d (254 мг, чистота 80 %, выход 47 %) в виде твердого вещества желтого цвета, используемого на следующей стадии. МС m/z 305,3 [M+H]+.
К метанолу (3 мл) при комнатной температуре добавляли соединение 7S-d (100 мг, 0,28 ммоль) и Pd на угле (10 %, 80 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (дихлорметан:метанол = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 7S-e (47 мг, выход 51 %) в виде твердого вещества желтого цвета. МС m/z 275,4 [M+H]+.
Смесь соединения 7S-e (47 мг, 0,17 ммоль) и соединения Ib (57 мг, 0,17 ммоль) в толуоле (1,5 мл) в закрытой пробирке нагревали до 100 °C в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали методом препаративной ТСХ (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидроксида) с получением соединения 7S (5 мг, выход 5 %) в виде твердого вещества желтого цвета. МС m/z 544,7 [M+H]+.
Пример 9: получение соединения 8S
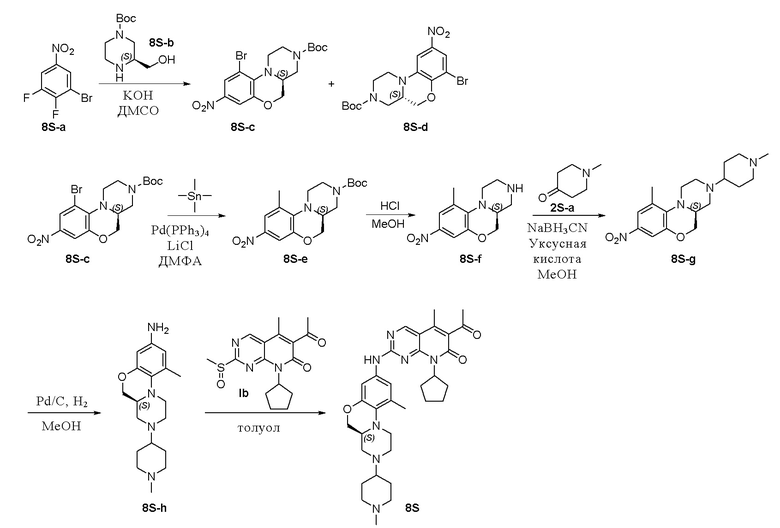
Соединение 8S-a (100 мг, 0,42 ммоль), соединение 8S-b (91 мг, 0,42 ммоль) и КОН (71 мг, 1,26 ммоль) растворяли в диметилсульфоксиде (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем при температуре 60 °C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и выливали в ледяную воду. Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Проводили экстракцию дихлорметаном (3 × 25 мл). Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (петролейный эфир:EtOAc = 10:1) с получением соединения 8S-c (64 мг, выход 37 %) в виде твердого вещества желтого цвета и соединения 8S-d (64 мг, выход 37 %) в виде твердого вещества желтого цвета. 8S-c: 1H-ЯМР (500 МГц, CDCl3) δ 8,03 (дублет, J = 2,5 Гц, 1H), 7,71 (дублет, J = 2,6 Гц, 1H), 4,26–4,15 (мультиплет, 2H), 3,86–3,78 (мультиплет, 2H), 3,60 (двойной дублет, J = 13,8, 4,1 Гц, 1H), 3,52–3,36 (мультиплет, 2H), 3,29–3,15 (мультиплет, 2H), 1,48 (синглет, 9H); 8S-d: 1H-ЯМР (500 МГц, CDCl3) δ 7,94 (дублет, J = 2,5 Гц, 1H), 7,63 (дублет, J = 2,5 Гц, 1H), 4,49–4,44 (мультиплет, 1H), 4,28–4,04 (мультиплет, 3H), 3,71 (дублет, J = 11,3 Гц, 1H), 3,22–3,15 (мультиплет, 1H), 3,10–3,01 (мультиплет, 1H), 2,88–2,79 (мультиплет, 1H), 2,66–2,57 (мультиплет, 1H), 1,49 (синглет, 9H) м.д.
Смесь соединения 8S-c (64 мг, 0,16 ммоль), тетраметилолова (56 мг, 0,31 ммоль), тетракис(трифенилфосфин)палладия (9 мг, 0,01 ммоль) и LiCl (13 мг, 0,31 ммоль) в ДМФА (2 мл) нагревали до 90 °C и перемешивали в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (петролейный эфир:EtOAc = 10:1) с получением соединения 8S-e (33 мг, выход 60 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CDCl3) δ 7,65 (дублет, J = 2,5 Гц, 1H), 7,59 (дублет, J = 2,6 Гц, 1H), 4,24–4,14 (мультиплет, 2H), 3,89–3,78 (мультиплет, 2H), 3,60–3,49 (мультиплет, 1H), 3,39–3,30 (мультиплет, 1H), 3,28–3,18 (мультиплет, 1H), 3,10–2,98 (мультиплет, 2H), 2,37 (синглет, 3H), 1,48 (синглет, 9H).
Соединение 8S-e (200 мг, 0,57 ммоль) растворяли в метаноле (2 мл). Добавляли раствор HCl в метаноле (1 мл, 4 М) на ледяной бане. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, нейтрализовали насыщенным водным раствором NaHCO3 (10 мл) и экстрагировали этилацетатом (3 × 10 мл). Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении с получением соединения 8S-f (100 мг, выход 70 %) в виде твердого вещества желтого цвета. МС m/z 250,3 [M+H]+.
Смесь соединения 8S-f (100 мг, 0,40 ммоль), N-метилпиперидона (2S-а, 227 мг, 2,00 ммоль) и уксусной кислоты (2 капли) в метаноле (5 мл) перемешивали при комнатной температуре в течение 1 часа. Добавляли натрия цианоборгидрид (75 мг, 1,21 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Ее концентрировали при пониженном давлении, экстрагировали дихлорметаном (3 × 15 мл). Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (дихлорметан:метанол = 30:1) с получением соединения 8S-g (140 мг) в виде твердого вещества желтого цвета, используемого на следующей стадии. МС m/z 347,4 [M+H]+.
К метанолу (5 мл) при комнатной температуре добавляли соединение 8S-g (101 мг, 0,29 ммоль) и Pd на угле (10 %, 20 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (дихлорметан:метанол = 50:1) с получением соединения 8S-h (63 мг, выход 68 %) в виде твердого вещества коричневого цвета. МС m/z 317,4 [M+H]+.
Соединение 8S-h (53 мг, 0,17 ммоль) и соединение Ib (56 мг, 0,17 ммоль) растворяли в толуоле (1 мл). Реакционную смесь перемешивали в течение ночи при температуре 100 °C. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали методом препаративной ТСХ (CH2Cl2:CH3OH = 20:1) с получением соединения 8S (12 мг, выход 12 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CDCl3) δ 8,72 (синглет, 1H), 7,15 (широкий синглет, 1H), 7,03 (синглет, 1H), 6,96 (синглет, 1H), 5,93–5,84 (мультиплет, 1H), 4,60 (триплет, J = 10,7 Гц, 1H), 4,00 (двойной дублет, J = 10,6, 2,8 Гц, 1H), 3,19 (дублет, J = 10,5 Гц, 1H), 3,13–3,01 (мультиплет, 3H), 2,92–2,85 (мультиплет, 2H), 2,82 (дублет, J = 11,5 Гц, 2H), 2,62–2,55 (мультиплет, 1H), 2,54 (синглет, 3H), 2,46–2,39 (мультиплет, 3H), 2,35 (синглет, 3H), 2,34–2,30 (мультиплет, 3H), 2,29 (синглет, 3H), 2,04–1,73 (мультиплет, 9H), 1,69–1,55 (мультиплет, 3H) м.д. МС m/z 586,8 [M+H]+.
Пример 10: получение соединения 9S
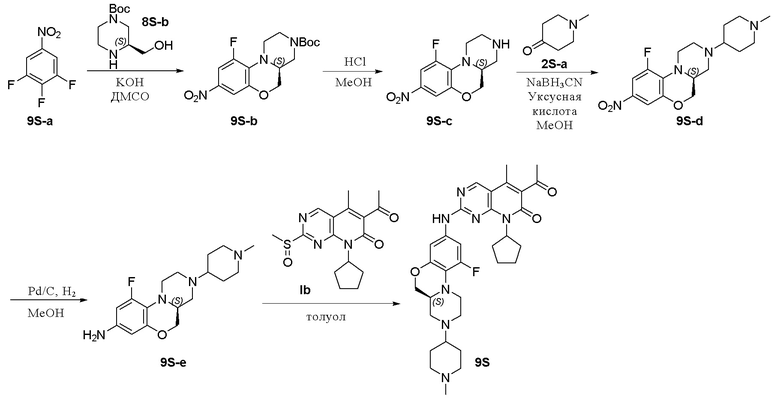
Соединение 9S-a (1,0 г, 5,65 ммоль), соединение 8S-b (1,22 г, 5,65 ммоль) и KОН (950 мг, 16,94 ммоль) растворяли в ДМСО (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем при температуре 60 °C в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и выливали в ледяную воду. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и экстрагировали дихлорметаном (3 × 30 мл). Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (петролейный эфир:EtOAc:CH2Cl2 = 5:1:1) с получением соединения 9S-b (1,45 г, выход 73 %) в виде твердого вещества желтого цвета. МС m/z 354,4 [M+H]+.
Соединение 9S-b (1,45 г, 4,10 ммоль) растворяли в метаноле (15 мл). Добавляли раствор HCl в метаноле (4,0 М, 4 мл). Полученную смесь перемешивали при 60 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (дихлорметан:метанол = 10:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 9S-c (1,25 г, выход 100 %) в виде твердого вещества желтого цвета. МС m/z 254,3 [M+H]+.
Смесь соединения 9S-c (500 мг, 1,73 ммоль), N-метил-4-пиперидона (2S-а, 587 мг, 5,18 ммоль) и уксусной кислоты (4 капли) в метаноле (8 мл) перемешивали при комнатной температуре в течение 1 часа. Добавляли натрия цианоборгидрид (326 мг, 5,18 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 9S-d (704 мг) в виде твердого вещества желтого цвета, используемого на следующей стадии. МС m/z 351,4 [M+H]+.
К метанолу (3 мл) при комнатной температуре добавляли соединение 9S-d (80 мг, 0,23 ммоль) и Pd на угле (10 %, 80 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 9S-e (50 мг, выход 68 %) в виде маслянистой жидкости желтого цвета. МС m/z 321,5 [M+H]+.
Смесь соединения 9S-e (50 мг, 0,16 ммоль) и соединения Ib (52 мг, 0,16 ммоль) в толуоле (1,5 мл) в закрытой пробирке нагревали до 100 °C и перемешивали в течение ночи. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 9S (13 мг, выход 14 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CDCl3) δ 8,72 (синглет, 1H), 7,13 (синглет, 1H), 7,07 (двойной дублет, J = 14,6, 2,5 Гц, 1H), 6,83 (синглет, 1H), 5,91–5,79 (мультиплет, 1H), 4,17–4,08 (мультиплет, 2H), 3,79–3,68 (мультиплет, 1H), 3,27–3,19 (мультиплет, 1H), 3,13–3,03 (мультиплет, 3H), 2,89–2,77 (мультиплет, 2H), 2,66–2,58 (мультиплет, 1H), 2,54 (синглет, 3H), 2,49–2,27 (мультиплет, 7H), 2,36 (синглет, 3H), 2,04–1,93 (мультиплет, 3H), 1,92–1,80 (мультиплет, 6H), 1,73–1,59 (мультиплет, 3H) м.д. МС m/z 590,8 [M+H]+.
Пример 11: получение соединения 10S
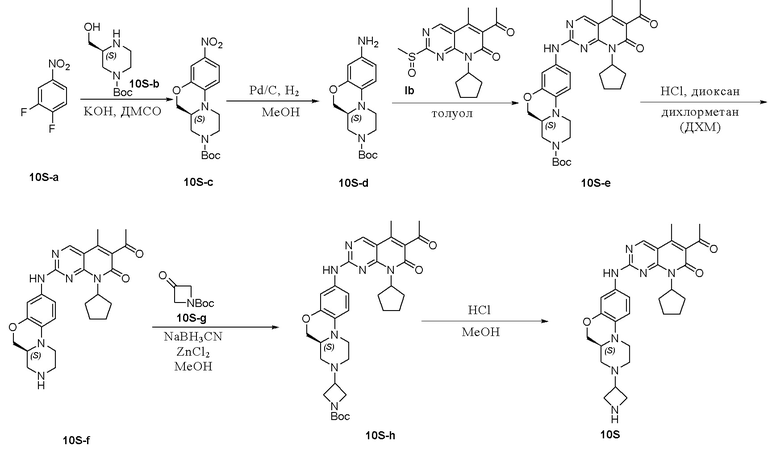
Соединение 10S-a (2,95 г, 13,6 ммоль) и соединение 10S-b (2,17 г, 13,6 ммоль) растворяли в ДМСО (30 мл). Добавляли KОН (2,30 г, 40,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи до исчезновения соединения 10S-a. Перемешивали при 60 ℃ в течение ночи. Завершение реакции контролировали методом ТСХ. Реакцию гасили водой. Полученную смесь экстрагировали этилацетатом. Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (петролейный эфир:EtOAc = 1:1) с получением соединения 10S-c (2,30 г, 50 %). МС m/z 336,4 [M+H]+, 280,4[M-55]+. 1H-ЯМР (500 МГц, CDCl3) δ 7,83–7,75 (мультиплет, 1H), 7,65 (дублет, J = 2,6 Гц, 1H), 6,75 (дублет, J = 9,1 Гц, 1H), 4,29 (двойной дублет, J = 11,0, 3,0 Гц, 1H), 4,27–4,01 (мультиплет, 2H), 3,98 (двойной дублет, J = 11,0, 8,0 Гц, 1H), 3,78 (дублет, J = 11,4 Гц, 1H), 3,36–3,27 (мультиплет, 1H), 3,12–2,99 (мультиплет, 1H), 2,98–2,89 (мультиплет, 1H), 2,76–2,53 (мультиплет, 1H), 1,48 (синглет, 9H) м.д.
Соединение 10S-c (6,10 г, 18,19 ммоль) растворяли в метаноле (100 мл). Добавляли Pd на угле (7 %, 500 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 1 часа. Завершение реакции контролировали методом ТСХ, реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH:водный раствор аммония гидроксида = 30:1:0,3) с получением соединения 10S-d (4,70 г, 85 %). МС m/z 306,4 [M+H]+. 1H-ЯМР (500 МГц, CDCl3) δ 6,64 (дублет, J = 8,5 Гц, 1H), 6,32 (двойной дублет, J = 8,5, 2,6 Гц, 1H), 6,29 (дублет, J = 2,5 Гц, 1H), 4,23–4,01 (мультиплет, 3H), 3,96 (двойной дублет, J = 10,6, 9,0 Гц, 1H), 3,56 (дублет, J = 11,3 Гц, 1H), 3,15–2,90 (мультиплет, 2H), 2,71–2,49 (мультиплет, 2H), 1,48 (синглет, 9H) м.д.
Соединение 10S-d (4,7 г, 15,39 ммоль) и соединение Ib (5,64 г, 16,93 ммоль) растворяли в толуоле (30 мл). Реакционную смесь перемешивали в течение ночи при температуре 90–100 °C. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрационный осадок промывали EtOAc и высушивали с получением твердого вещества желтого цвета. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH:водный раствор аммония гидроксида = 50:1:0,5) с получением соединения 10S-e (5,30 г, 60 %) в виде твердого вещества желтого цвета. МС m/z 575,7 [M+H]+.
Соединение 10S-e (5,30 г, 9,22 ммоль) растворяли в метаноле (60 мл). Добавляли раствор HCl в метаноле (4 М, 10 мл). Реакционную смесь перемешивали при 40 °C в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали дихлорметаном. Объединенные органические слои промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении с получением соединения 10S-f (4,30 г, 98 %). МС m/z 475,6 [M+H]+.
Соединение 10S-f (200 мг, 0,42 ммоль), 1-бутилоксикарбонил-3-азетидинон 10S-g (108 мг, 0,63 ммоль) и цинка хлорид (172 мг, 1,26 ммоль) растворяли в метаноле (7 мл). Добавляли натрия цианоборгидрид (80 мг, 1,26 ммоль). Реакционную смесь перемешивали при 75 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 10S-h (250 мг, выход 94 %) в виде твердого вещества желтого цвета. МС m/z 630,6 [M+H]+.
Соединение 10S-h (250 мг, 0,40 ммоль) растворяли в метаноле (8 мл). Добавляли раствор HCl в 1,4-диоксана (4,0 М, 1,5 мл). Полученную смесь перемешивали при 40 °C в течение 2 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Осадок растворяли в небольшом количестве метанола, нейтрализовали водным раствором аммония гидроксида и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 6:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 10S (173 мг, выход 82 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CD3OD) δ 8,82 (синглет, 1H), 7,19 (дублет, J = 2,4 Гц, 1H), 6,98 (двойной дублет, J = 8,7, 2,0 Гц, 1H), 6,84 (дублет, J = 8,8 Гц, 1H), 5,98–5,89 (мультиплет, 1H), 4,25 (двойной дублет, J = 10,6, 2,6 Гц, 1H), 4,06–3,93 (мультиплет, 4H), 3,79 (дублет, J = 11,8 Гц, 1H), 3,44–3,37 (мультиплет, 1H), 3,14–3,08 (мультиплет, 1H), 2,98–2,94 (мультиплет, 1H), 2,90–2,85 (мультиплет, 1H), 2,78–2,72 (мультиплет, 1H), 2,48 (синглет, 3H), 2,47–2,44 (мультиплет, 1H), 2,35 (синглет, 3H), 2,34–2,27 (мультиплет, 2H), 2,24–2,16 (мультиплет, 1H), 1,99–1,90 (мультиплет, 2H), 1,88–1,77 (мультиплет, 3H), 1,68–1,59 (мультиплет, 2H) м.д. МС m/z 530,4 [M+H]+.
Пример 12: получение соединения 11S
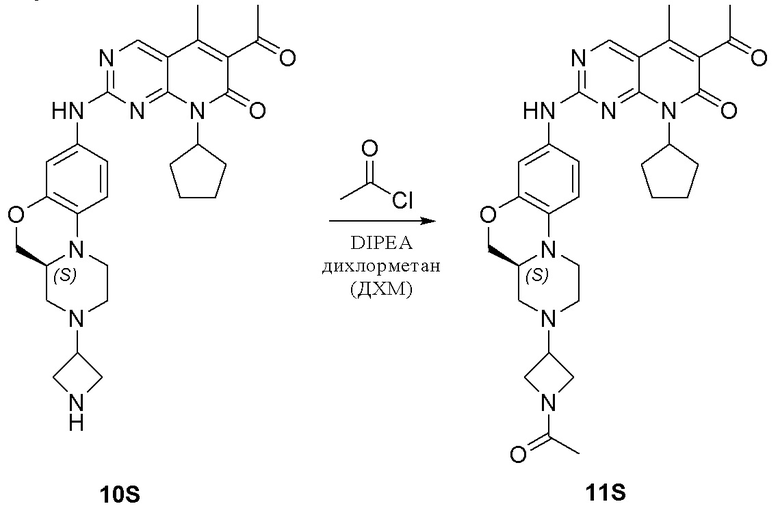
Соединение 10S (20 мг, 0,04 ммоль) и N,N-диизопропилэтиламин (7 мг, 0,06 ммоль) растворяли в дихлорметане (4 мл), затем добавляли раствор ацетилхлорида (3 мг, 0,04 ммоль) в дихлорметане (1 мл) при 0 °C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 11S (20 мг, выход 93 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CD3OD) δ 8,82 (синглет, 1H), 7,19–7,17 (мультиплет, 1H), 6,97 (дублет, J = 8,6 Гц, 1H), 6,84 (дублет, J = 8,0 Гц, 1H), 5,99–5,88 (мультиплет, 1H), 4,31–4,22 (мультиплет, 2H), 4,11 (двойной дублет, J = 9,1, 5,0 Гц, 1H), 4,08–4,02 (мультиплет, 1H), 4,01–3,96 (мультиплет, 1H), 3,87 (двойной дублет, J = 10,3, 5,1 Гц, 1H), 3,78 (дублет, J = 12,1 Гц, 1H), 3,27–3,21 (мультиплет, 1H), 3,14–3,08 (мультиплет, 1H), 3,03–2,97 (мультиплет, 1H), 2,95–2,88 (мультиплет, 1H), 2,78–2,71 (мультиплет, 1H), 2,48 (синглет, 3H), 2,35 (синглет, 3H), 2,34–2,28 (мультиплет, 2H), 2,25–2,18 (мультиплет, 1H), 1,96–1,92 (мультиплет, 2H), 1,90 (синглет, 3H), 1,88–1,78 (мультиплет, 3H), 1,68–1,60 (мультиплет, 2H) м.д. МС m/z 572,7 [M+H]+.
Пример 13: получение соединения 12S
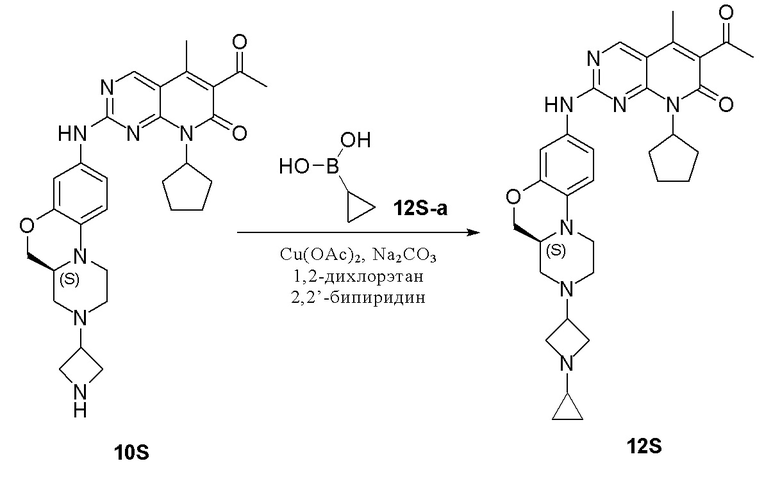
Соединение 10S (10 мг, 0,02 ммоль), циклопропилбороновую кислоту 12S-a (3 мг, 0,04 ммоль), меди ацетат (3 мг, 0,02 ммоль), 2,2'-бипиридин (3 мг, 0,081 ммоль) и натрия карбонат (3 мг, 0,037 ммоль) растворяли в 1,2-дихлорэтане (3 мл). Реакционную смесь нагревали до 70 ℃ в течение 2 часов на воздухе. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 12S (1,7 мг, выход 16 %) в виде твердого вещества желтого цвета. МС m/z 570,6 [M+H]+.
Пример 14: получение соединения 13S
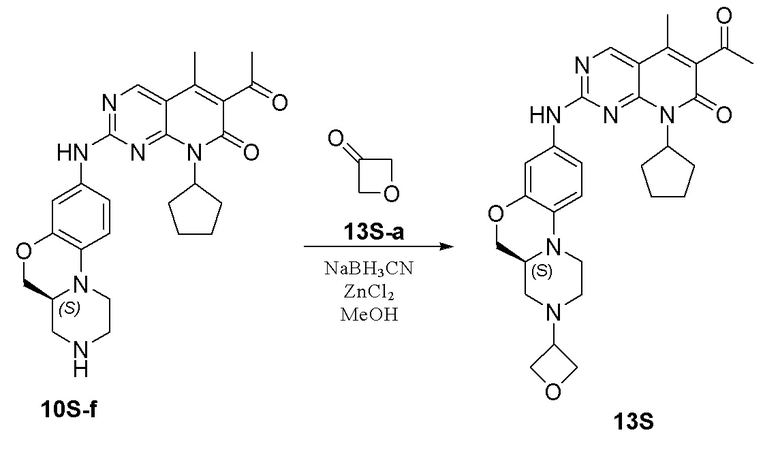
Соединение 10S-f (30 мг, 0,06 ммоль), 3-оксэтанон 13S-a (5 мг, 0,06 ммоль) и цинка хлорид (17 мг, 0,13 ммоль) растворяли в метаноле (5 мл) и добавляли натрия цианоборогидрид (12 мг, 0,19 ммоль). Реакционную смесь перемешивали при 75 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 13S (18 мг, выход 54 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,88 (синглет, 1H), 8,90 (синглет, 1H), 7,17 (синглет, 1H), 7,08 (дублет, J = 7,5 Гц, 1H), 6,84 (дублет, J = 8,9 Гц, 1H), 5,88–5,80 (мультиплет, 1H), 4,62–4,52 (мультиплет, 2H), 4,50–4,40 (мультиплет, 2H), 4,24 (двойной дублет, J = 10,5, 2,6 Гц, 1H), 3,94–3,85 (мультиплет, 1H), 3,73 (дублет, J = 11,8 Гц, 1H), 3,50–3,40 (мультиплет, 1H), 3,07–2,96 (мультиплет, 1H), 2,81 (двойной дублет, J = 19,5, 10,4 Гц, 2H), 2,67–2,56 (мультиплет, 1H), 2,39 (синглет, 3H), 2,28 (синглет, 3H), 2,26–2,17 (мультиплет, 2H), 2,09–2,00 (мультиплет, 1H), 1,88 (синглет, 2H), 1,76 (синглет, 2H), 1,64 (триплет, J = 10,6 Гц, 1H), 1,60–1,51 (мультиплет, 2H) м.д. МС m/z 531,6 [M+H]+.
Пример 15: получение соединения 14S
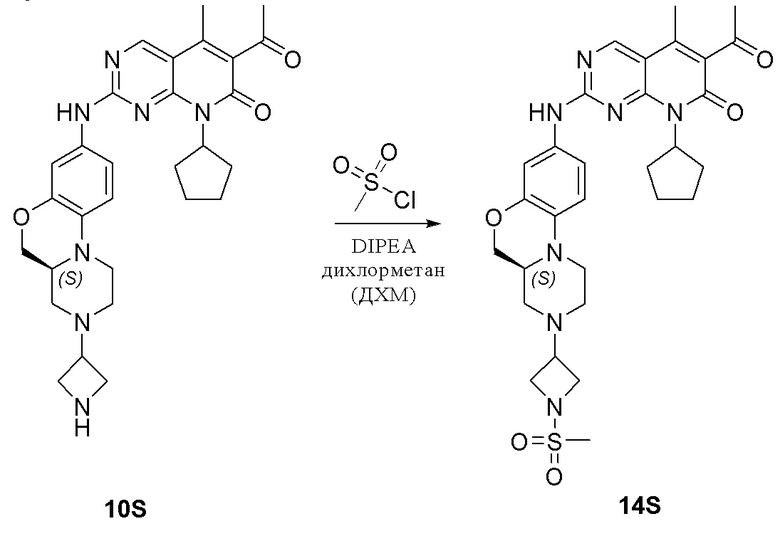
Соединение 10S (20 мг, 0,04 ммоль) и N,N-диизопропилэтиламин (7 мг, 0,06 ммоль) растворяли в дихлорметане (4 мл). Добавляли раствор метилсульфонилхлорида (4 мг, 0,04 ммоль) в дихлорметане (1 мл) при 0 °C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 14S (21 мг, выход 92 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,88 (синглет, 1H), 8,90 (синглет, 1H), 7,18 (дублет, J = 1,9 Гц, 1H), 7,08 (дублет, J = 8,2 Гц, 1H), 6,85 (дублет, J = 8,9 Гц, 1H), 5,89–5,79 (мультиплет, 1H), 4,24 (двойной дублет, J = 10,6, 2,6 Гц, 1H), 3,94–3,71 (мультиплет, 6H), 3,25–3,18 (мультиплет, 1H), 3,01 (синглет, 3H), 2,99–2,97 (мультиплет, 1H), 2,87 (двойной дублет, J = 20,6, 10,5 Гц, 2H), 2,64–2,57 (мультиплет, 1H), 2,42 (синглет, 3H), 2,29 (синглет, 3H), 2,27–2,19 (мультиплет, 2H), 2,14–2,06 (мультиплет, 1H), 1,93–1,84 (мультиплет, 2H), 1,81–1,68 (мультиплет, 3H), 1,62–1,55 (мультиплет, 2H) м.д. МС m/z 608,8 [M+H]+.
Пример 16: получение соединения 15S
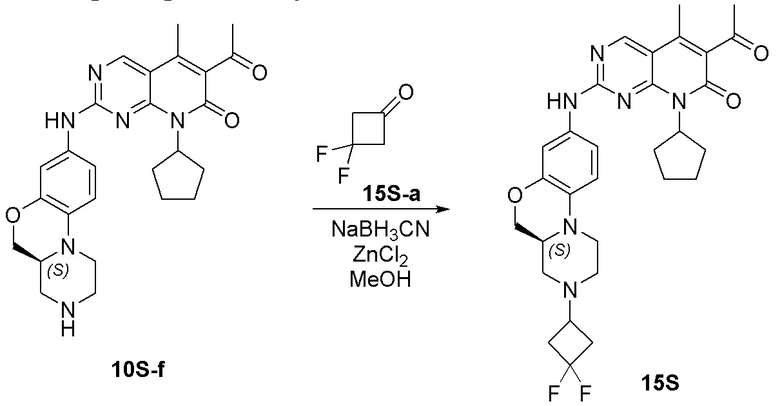
Соединение 10S-f (15 мг, 0,03 ммоль), 3,3-дифторциклобутанон 15S-a (5 мг, 0,05 ммоль) и цинка хлорид (13 мг, 0,09 ммоль) растворяли в метаноле (4 мл) и добавляли натрия цианоборогидрид (6 мг, 0,09 ммоль). Реакционную смесь перемешивали при 80 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 30:1) с получением соединения 15S (2 мг, выход 11 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CDCl3) δ 8,71 (синглет, 1H), 7,16 (синглет, 2H), 6,97 (дублет, J = 8,8 Гц, 1H), 6,77 (дублет, J = 8,6 Гц, 1H), 5,89–5,80 (мультиплет, 1H), 4,23 (дублет, J = 10,3 Гц, 1H), 4,08–3,97 (мультиплет, 1H), 3,78–3,65 (мультиплет, 1H), 3,24–3,11 (мультиплет, 1H), 3,04–2,92 (мультиплет, 1H), 2,87–2,66 (мультиплет, 4H), 2,54 (синглет, 3H), 2,35 (синглет, 3H), 2,33–2,26 (мультиплет, 2H), 2,25–2,13 (мультиплет, 1H), 2,00–1,90 (мультиплет, 2H), 1,88–1,79 (мультиплет, 2H), 1,68–1,51 (мультиплет, 6H) м.д. МС m/z 565,6 [M+H]+.
Пример 17: получение соединения 16S
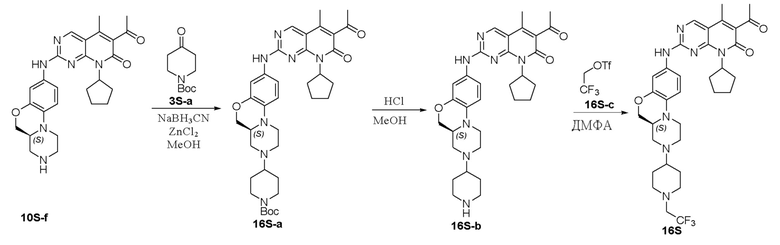
Соединение 10S-f (80 мг, 0,17 ммоль), N-бутилоксикарбонил-4-пиперидон 3S-a (50 мг, 0,25 ммоль) и цинка хлорид (69 мг, 0,51 ммоль) растворяли в метаноле (5 мл) и добавляли натрия цианоборогидрид (32 мг, 0,51 ммоль). Реакционную смесь перемешивали при 80 °C в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 15:1) с получением соединения 16S-a (91 мг, выход 82 %) в виде твердого вещества желтого цвета. МС m/z 658,7 [M+H]+.
Соединение 16S-a (91 г, 0,14 ммоль) растворяли в метаноле (4 мл) и добавляли раствор HCl в 1,4-диоксане (4,0 М, 1 мл). Реакционную смесь перемешивали при 40 °C в течение 2 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Осадок растворяли в небольшом количестве метанола, нейтрализовали водным раствором аммония гидроксида и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 15:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 16S-b (58 мг, выход 75 %) в виде твердого вещества желтого цвета. МС m/z 558,6 [M+H]+.
Соединение 16S-b (20 мг, 0,04 ммоль), соединение 16S-c (10 мг, 0,04 ммоль) и N,N-диизопропилэтиламин (9 мг, 0,07 ммоль) растворяли в N,N-диметилформамиде (2 мл). Реакционную смесь перемешивали при 80 °C в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 15:1) с получением соединения 16S (16 мг, выход 70 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CD3OD) δ 8,81 (синглет, 1H), 7,17 (дублет, J = 2,4 Гц, 1H), 6,96 (двойной дублет, J = 8,7, 2,0 Гц, 1H), 6,82 (дублет, J = 8,9 Гц, 1H), 5,97–5,89 (мультиплет, 1H), 4,24 (двойной дублет, J = 10,5, 2,6 Гц, 1H), 3,97 (двойной дублет, J = 10,5, 9,0 Гц, 1H), 3,75 (дублет, J = 11,7 Гц, 1H), 3,14–2,98 (мультиплет, 7H), 2,75–2,67 (мультиплет, 1H), 2,47 (синглет, 3H), 2,46–2,42 (мультиплет, 1H), 2,39 (триплет, J = 11,1 Гц, 2H), 2,35 (синглет, 3H), 2,33–2,27 (мультиплет, 3H), 2,03 (триплет, J = 10,7 Гц, 1H), 1,96–1,78 (мультиплет, 6H), 1,68–1,55 (мультиплет, 4H) м.д. МС m/z 640,8 [M+H]+.
Пример 18: получение соединения 17S
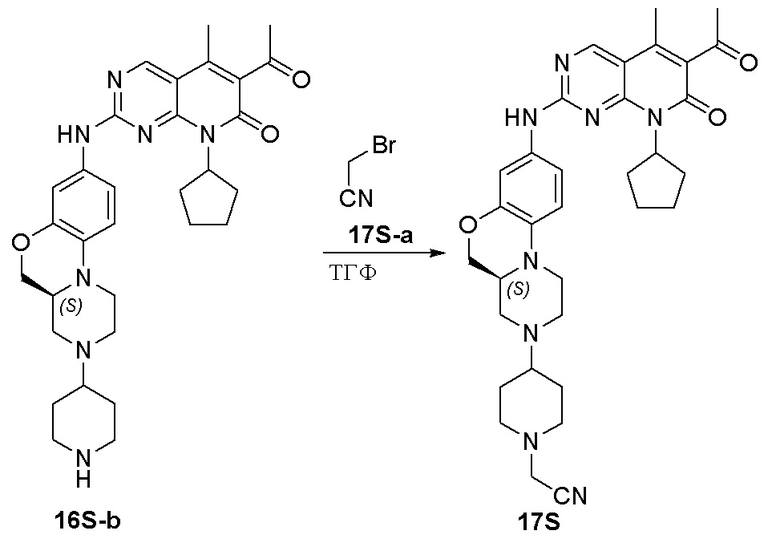
Соединение 16S-b (20 мг, 0,04 ммоль) и бромацетонитрил 17S-a (5 мг, 0,04 ммоль) растворяли в ТГФ (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1) с получением соединения 17S (12 мг, выход 56 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,87 (синглет, 1H), 8,90 (синглет, 1H), 7,16 (дублет, J = 2,0 Гц, 1H), 7,07 (дублет, J = 8,0 Гц, 1H), 6,82 (дублет, J = 8,9 Гц, 1H), 5,90–5,77 (мультиплет, 1H), 4,24 (двойной дублет, J = 10,6, 2,4 Гц, 1H), 3,90 (двойной дублет, J = 10,5, 9,0 Гц, 1H), 3,72–3,66 (мультиплет, 3H), 3,01–2,89 (мультиплет, 3H), 2,83 (дублет, J = 11,2 Гц, 2H), 2,58–2,52 (мультиплет, 1H), 2,41 (синглет, 3H), 2,35–2,31 (мультиплет, 1H), 2,29 (синглет, 3H), 2,26–2,20 (мультиплет, 3H), 2,16 (триплет, J = 10,8 Гц, 2H), 1,93–1,84 (мультиплет, 3H), 1,82–1,73 (мультиплет, 4H), 1,62–1,54 (мультиплет, 2H), 1,51–1,39 (мультиплет, 2H) м.д. МС m/z 597,8 [M+H]+.
Пример 19: получение соединения 18S
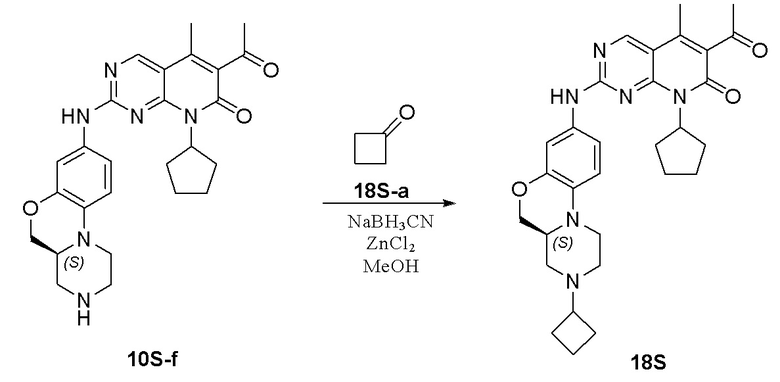
Соединение 10S-f (15 мг, 0,03 ммоль), циклобутанон 18S-a (3 мг, 0,05 ммоль) и цинка хлорид (13 мг, 0,09 ммоль) растворяли в метаноле (4 мл) и добавляли натрия цианоборогидрид (6 мг, 0,09 ммоль). Реакционную смесь перемешивали при 80 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 15:1) с получением соединения 18S (11 мг, выход 66 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,87 (синглет, 1H), 8,90 (синглет, 1H), 7,16 (дублет, J = 2,0 Гц, 1H), 7,07 (дублет, J = 7,8 Гц, 1H), 6,82 (дублет, J = 8,9 Гц, 1H), 5,87–5,78 (мультиплет, 1H), 4,25 (двойной дублет, J = 10,6, 2,6 Гц, 1H), 3,89 (двойной дублет, J = 10,5, 9,1 Гц, 1H), 3,70 (дублет, J = 11,6 Гц, 1H), 2,99–2,92 (мультиплет, 1H), 2,86 (дублет, J = 11,7 Гц, 1H), 2,81 (дублет, J = 10,2 Гц, 1H), 2,77–2,69 (мультиплет, 1H), 2,59–2,52 (мультиплет, 1H), 2,41 (синглет, 3H), 2,29 (синглет, 3H), 2,26–2,19 (мультиплет, 2H), 2,02–1,73 (мультиплет, 8H), 1,69–1,50 (мультиплет, 5H), 1,27–1,22 (мультиплет, 1H) м.д. МС m/z 529,7 [M+H]+.
Пример 20: получение соединения 19S
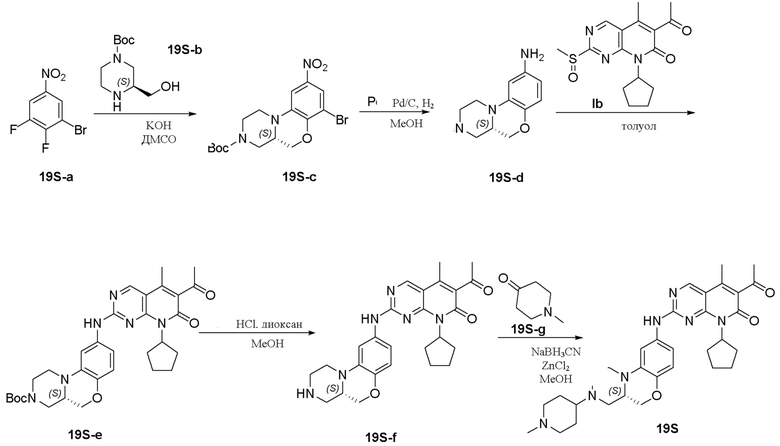
Соединение 19S-a (3,0 г, 12,61 ммоль), соединение 19S-b (2,73 г, 12,61 ммоль) и KОН (2,12 г, 37,82 ммоль) растворяли в диметилсульфоне (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем при температуре 60 °C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, выливали в ледяную воду и перемешивали в течение 1 часа. Смесь экстрагировали дихлорметаном (3 × 40 мл), промывали рассолом, сушили над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (петролейный эфир:EtOAc:CH2Cl2 = 15:1:1) с получением соединения 19S-b (1,07 г, выход 20 %) в виде твердого вещества желтого цвета.
К метанолу (6 мл) добавляли соединение 19S-c (500 мг, 1,21 ммоль) и Pd на угле (10 %, 150 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н2 при давлении 1 атм. в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:EtOAc = 4:1) с получением соединения 19S-d (235 мг, выход 64 %) в виде твердого вещества серого цвета. МС m/z 306,4 [M+H]+.
Смесь соединения 19S-d (235 мг, 0,77 ммоль) и соединения Ib (257 мг, 0,77 ммоль) в толуоле (6 мл) в закрытой пробирке нагревали до 100 °C и перемешивали в течение 5 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:EtOAc = 2:1) с получением соединения 19S-e (200 мг, выход 45 %) в виде твердого вещества желтого цвета. МС m/z 575,8 [M+H]+.
Соединение 19S-e (200 г, 0,35 ммоль) растворяли в метаноле (2 мл) и добавляли раствор HCl в 1,4-диоксане (4,0 М, 2 мл). Реакционную смесь перемешивали при 40 °C в течение 2 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Осадок растворяли в небольшом количестве метанола, нейтрализовали водным раствором аммония гидроксида и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 15:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 19S-f (140 мг, выход 85 %) в виде твердого вещества желтого цвета.
Соединение 19S-f (20 мг, 0,04 ммоль), соединение 19S-g (5 мг, 0,04 ммоль) и цинка хлорид (12 мг, 0,08 ммоль) растворяли в метаноле (5 мл) и добавляли натрия цианоборогидрида (8 мг, 0,13 ммоль). Реакционную смесь перемешивали при 75 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 19S (10,44 мг, выход 43 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CDCl3) δ 8,71 (синглет, 1H), 7,13 (синглет, 1H), 7,09 (синглет, 1H), 6,77 (синглет, 2H), 5,92–5,83 (мультиплет, 1H), 4,19 (двойной дублет, J = 10,5, 2,7 Гц, 1H), 4,03–3,95 (мультиплет, 1H), 3,65 (дублет, J = 11,2 Гц, 1H), 3,22–3,15 (мультиплет, 2H), 3,11–3,01 (мультиплет, 1H), 2,99 (дублет, J = 10,2 Гц, 1H), 2,90–2,78 (мультиплет, 2H), 2,54 (синглет, 3H), 2,50–2,45 (мультиплет, 1H), 2,44–2,37 (мультиплет, 3H), 2,36 (синглет, 3H), 2,32–2,22 (мультиплет, 4H), 2,11–2,02 (мультиплет, 2H), 2,01–1,71 (мультиплет, 10H) м.д. МС m/z 572,7 [M+H]+.
Пример 21: получение соединения 20S
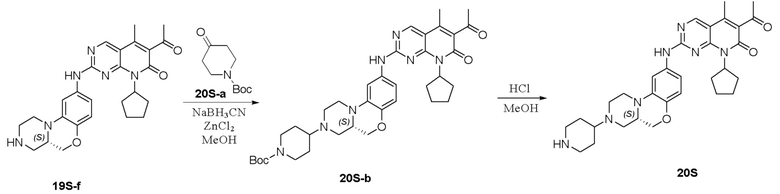
Соединение 19S-f (30 мг, 0,06 ммоль), N-бутилоксикарбонил-4-пиперидон (13 мг, 0,06 ммоль) и цинка хлорид (17 мг, 0,13 ммоль) растворяли в метаноле (5 мл) и добавляли натрия цианоборогидрид (12 мг, 0,19 ммоль). Реакционную смесь перемешивали при 75 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 20S-b (25 мг, выход 60 %) в виде твердого вещества желтого цвета. МС m/z 658,3 [M+H]+.
Соединение 20S-b (25 г, 0,04 ммоль) растворяли в метаноле (2 мл) и добавляли раствор HCl в 1,4-диоксане (4,0 М, 1,5 мл). Реакционную смесь перемешивали при 40 °C в течение 2 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Осадок растворяли в небольшом количестве метанола, нейтрализовали водным раствором аммония гидроксида и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 15:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 20S (12,8 мг, выход 60 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CD3OD) δ 8,82 (синглет, 1H), 7,17 (дублет, J = 2,0 Гц, 1H), 6,88–6,81 (мультиплет, 1H), 6,69 (дублет, J = 8,5 Гц, 1H), 6,01–5,90 (мультиплет, 1H), 4,23 (двойной дублет, J = 10,5, 2,6 Гц, 1H), 3,99–3,91 (мультиплет, 1H), 3,73 (дублет, J = 11,5 Гц, 1H), 3,41–3,35 (мультиплет, 3H), 3,13–3,06 (мультиплет, 2H), 3,01 (дублет, J = 10,5 Гц, 1H), 2,95–2,86 (мультиплет, 2H), 2,79–2,72 (мультиплет, 1H), 2,62–2,55 (мультиплет, 1H), 2,47 (синглет, 3H), 2,35 (синглет, 3H), 2,33–2,23 (мультиплет, 2H), 2,10–2,01 (мультиплет, 3H), 2,01–1,88 (мультиплет, 2H), 1,81 (дублет, J = 4,8 Гц, 2H), 1,75–1,65 (мультиплет, 2H), 1,63–1,55 (мультиплет, 2H) м.д. МС m/z 558,6 [M+H]+.
Пример 22: получение соединения 21S
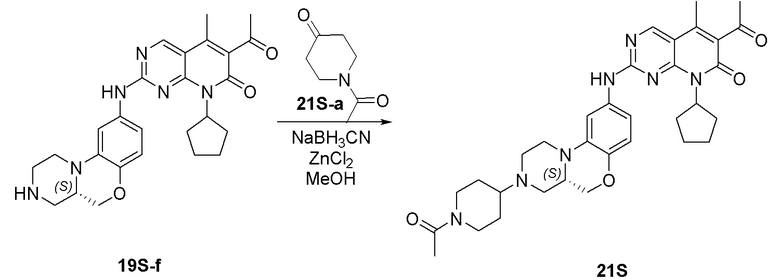
Соединение 19S-f (20 мг, 0,04 ммоль), соединение 21S-a (6 мг, 0,04 ммоль) и цинка хлорид (12 мг, 0,08 ммоль) растворяли в метаноле (5 мл) и добавляли натрия цианоборогидрида (8 мг, 0,13 ммоль). Реакционную смесь перемешивали при 75 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 21S (15,30 мг, выход 60 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CDCl3) δ 8,71 (синглет, 1H), 7,15 (синглет, 1H), 7,11 (синглет, 1H), 6,86–6,72 (мультиплет, 2H), 5,93–5,71 (мультиплет, 1H), 4,74–4,65 (мультиплет, 1H), 4,21 (дублет, J = 9,6 Гц, 1H), 4,06–3,96 (мультиплет, 1H), 3,90 (дублет, J = 12,8 Гц, 1H), 3,76–3,68 (мультиплет, 1H), 3,28–2,80 (мультиплет, 5H), 2,69–2,55 (мультиплет, 2H), 2,54 (синглет, 3H), 2,35 (синглет, 3H), 2,34–2,25 (мультиплет, 2H), 2,10 (синглет, 3H), 2,04–1,77 (мультиплет, 7H), 1,78–1,54 (мультиплет, 5H) м.д. МС m/z 600,8 [M+H]+.
Пример 23: получение соединения 1R
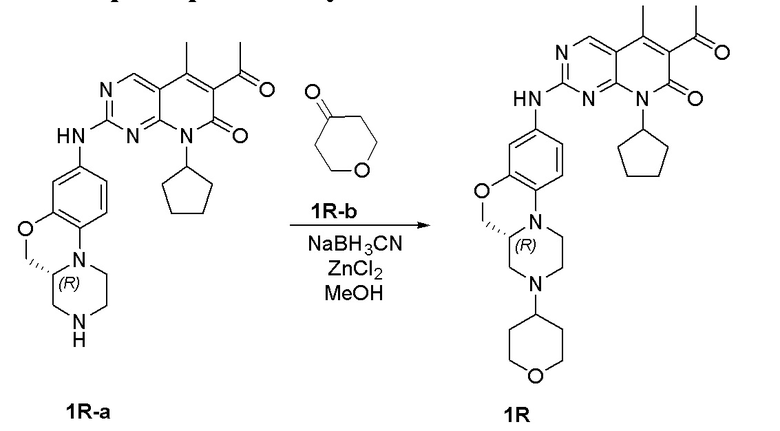
Соединение 1R-a было получено по способу, описанному в патенте WO2017101763.
Соединение 1R-a (30 мг, 0,06 ммоль), тетрагидро-4H-пиран-4-он 1R-b (9 мг, 0,06 ммоль) и цинка хлорид (17 мг, 0,13 ммоль) растворяли в метаноле (5 мл) и добавляли натрия цианоборогидрид (12 мг, 0,19 ммоль). Реакционную смесь перемешивали при 75 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 1R (15 мг, выход 42 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, ДМСО-d6) δ 9,88 (синглет, 1H), 8,90 (синглет, 1H), 7,16 (дублет, J = 2,0 Гц, 1H), 7,07 (дублет, J = 7,7 Гц, 1H), 6,82 (дублет, J = 8,9 Гц, 1H), 5,88–5,80 (мультиплет, 1H), 4,24 (двойной дублет, J = 10,5, 2,4 Гц, 1H), 4,02–3,85 (мультиплет, 3H), 3,71 (дублет, J = 11,7 Гц, 1H), 3,33–3,23 (мультиплет, 2H), 3,06–2,86 (мультиплет, 3H), 2,63–2,54 (мультиплет, 2H), 2,43 (синглет, 3H), 2,29 (синглет, 3H), 2,26–2,17 (мультиплет, 2H), 2,01–1,85 (мультиплет, 4H), 1,82–1,68 (мультиплет, 4H), 1,65–1,52 (мультиплет, 2H), 1,51–1,34 (мультиплет, 2H) м.д. МС m/z 559,8 [M+H]+.
Пример 24: получение соединения 3R
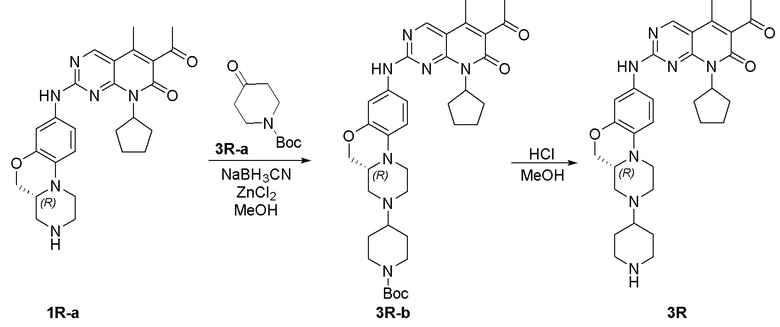
Соединение 1R-a (30 мг, 0,06 ммоль), 1-бутилоксикарбонил-4-пиперидон 3R-a (12 мг, 0,06 ммоль) и цинка хлорид (17 мг, 0,13 ммоль) растворяли в метаноле (5 мл) и добавляли натрия цианоборогидрид (12 мг, 0,19 ммоль). Реакционную смесь перемешивали при 75 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 3R-b (25 мг, выход 60 %) в виде твердого вещества желтого цвета. МС m/z 658,3 [M+H]+.
Соединение 3R-b (25 г, 0,04 ммоль) растворяли в метаноле (2 мл) и добавляли раствор HCl в 1,4-диоксане (4,0 М, 1,5 мл). Реакционную смесь перемешивали при 40 °C в течение 2 часов. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении. Полученную смесь разбавляли в небольшом количестве метанола, нейтрализовали водным раствором аммония гидроксида и концентрировали при пониженном давлении. Остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 15:1, 2 % водный раствор аммония гидрохлорида) с получением соединения 3R (9 мг, выход 42 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, MeOD–d4) δ 8,72 (синглет, 1H), 7,07 (дублет, J = 2,0 Гц, 1H), 6,86 (двойной дублет, J = 9,0, 2,0 Гц, 1H), 6,73 (дублет, J = 9,0 Гц, 1H), 5,92–5,75 (мультиплет, 1H), 4,14 (двойной дублет, J = 10,5, 2,6 Гц, 1H), 3,98–3,82 (мультиплет, 1H), 3,66 (дублет, J = 11,6 Гц, 1H), 3,16–3,06 (мультиплет, 2H), 3,04–2,87 (мультиплет, 3H), 2,66–2,55 (мультиплет, 3H), 2,40 (синглет, 3H), 2,36–2,31 (мультиплет, 1H), 2,25 (синглет, 3H), 2,24–2,15 (мультиплет, 2H), 1,95 (триплет, J = 21,3, 10,6 Гц, 2H), 1,92–1,78 (мультиплет, 4H), 1,77–1,67 (мультиплет, 2H), 1,61–1,49 (мультиплет, 2H), 1,49–1,37 (мультиплет, 2H) м.д. МС m/z 558,6 [M+H]+.
Пример 25: получение соединения 5R
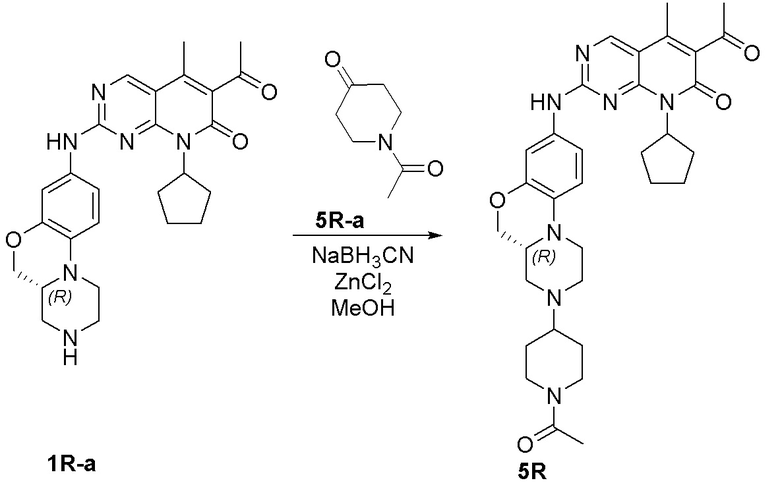
Соединение 1R-a (30 мг, 0,06 ммоль), N-ацетил-4-пиперидон 5R-a (9 мг, 0,06 ммоль) и цинка хлорид (17 мг, 0,13 ммоль) растворяли в метаноле (5 мл) и добавляли натрия цианоборогидрид (12 мг, 0,19 ммоль). Полученную реакционную смесь перемешивали при 75 °C в течение 1 часа. Завершение реакции контролировали методом ТСХ. Реакционную смесь концентрировали при пониженном давлении, а остаток очищали хроматографированием на SiO2 (CH2Cl2:CH3OH = 20:1, 2 % водный раствор NH4OH) с получением соединения 5R (14 мг, выход 37 %) в виде твердого вещества желтого цвета. 1H-ЯМР (500 МГц, CD3OD) δ 8,81 (синглет, 1H), 7,17 (дублет, J = 2,4 Гц, 1H), 6,96 (двойной дублет, J = 8,7, 1,9 Гц, 1H), 6,82 (дублет, J = 8,9 Гц,1H), 5,99–5,87 (мультиплет, 1H), 4,53 (дублет, J = 12,4 Гц, 1H), 4,24 (дублет, J = 9,8 Гц, 1H), 4,05–3,92 (мультиплет, 2H), 3,76 (дублет, J = 10,7 Гц, 1H), 3,15–2,95 (мультиплет,4H), 2,76–2,55 (мультиплет, 3H), 2,54–2,50 (мультиплет, 1H), 2,47 (синглет, 3H), 2,35–2,26 (мультиплет, 5H), 2,11–2,02(мультиплет, 4H), 2,02–1,88 (мультиплет, 4H), 1,88–1,77 (мультиплет, 2H), 1,70–1,58 (мультиплет, 2H), 1,55–1,48(мультиплет,1H), 1,43–1,36 (мультиплет, 1H) м.д. МС m/z 600,8 [M+H]+.
Пример 26:
1) анализ ингибирования активности киназ CDK2, CDK4 и CDK6
In vitro ферментативную активность изоформ CDK CDK2/CycA2, CDK4/CycD3 и CDK6/cycD3 измеряли с использованием анализа сдвига подвижности Caliper. Растворяют соединения в ДМСО и разбавляют полученный раствор соответствующим буферным раствором для киназ (CDK2/CycA2 и CDK6/CycD3 анализировали в условиях буферизации 50 мМ раствором HEPES (pH 7,5), 10 мМ MgCl2, 0,0015 % Brij-35 и 2 мМ дитиотреитола; CDK4/CycD3 анализировали в условиях буферизации 20 мМ раствором HEPES (pH 7,5), 10 мМ MgCl2, 0,01 % тритона X-100 и 2 мМ дитиотреитола). Добавляют 5 мкл соединения (10 % ДМСО) с 5-кратной конечной концентрацией в реакционной смеси в 384-луночный планшет. Добавляют 10 мкл 2,5-кратного раствора фермента и выдерживают при комнатной температуре в течение 10 минут, затем добавляют 10 мкл 2,5-кратного раствора субстрата (для каждой изоформы дозировка фермента и АТФ составляет CDK2/CycA2 12 нМ, АТФ Km 39 мкМ; CDK4/CycD3 10 нМ, АТФ Km 221 мкМ; CDK6/cycD3 15 нМ, АТФ Km 800 мкМ). После выдерживания в течение 60 мин, 180 мин и 60 мин CDK2, CDK4 и CDK6 соответственно при 28 °C, реакции были остановлены путем добавления 25 мкл стоп-раствора (100 мМ HEPES (pH 7,5), 0,015 % Brij-35, 0,2 % покровного реактива № 3, 50 мМ ЭДТА). Собирают данные о конверсии на считывателе Caliper EZ Reader II (Caliper Life Sciences). Преобразуют коэффициент конверсии в данные о степени ингибирования (степень ингибирования, % = (макс. коэффициент конверсии) / (макс. – мин.)*100). Макс. — коэффициент конверсии контроля (ДМСО); мин. — коэффициенту конверсии неактивного контроля. Строят кривую концентрации и степени ингибирования соединения по осям абсцисс и ординат соответственно. Для аппроксимации кривой и расчета IC50 используют программу-надстройку XLFit для Excel версии 4.3.1. Активность некоторых репрезентативных соединений показана в таблице 1.
2) Анализ ингибирования активности киназы TRKA
Активность протеинкиназы TRKA измеряли методом анализа сдвига подвижности Caliper. Соединения растворяли в ДМСО для получения исходных растворов с концентрацией 10 мМ. Готовят 1х киназный реакционный буферный раствор для серийного разведения соединений. По 250 мкл 100× конечной концентрации испытуемых соединений переносили в целевой планшет 3573 с помощью дозатора Echo 550. Готовят раствор киназы конечной концентрации 2,5× с 1× киназным буферным раствором. Добавляют по 10 мкл 2,5× конечного раствора киназы в лунки с соединениями и лунки положительного контроля; добавляют по 10 мкл 1× киназного буферного раствора в лунки отрицательного контроля. Центрифугируют при 1000 об/мин в течение 30 с, встряхивают и перемешивают реакционный планшет и выдерживают при комнатной температуре в течение 10 мин. Готовят смесь раствора АТФ и киназного субстрата в конечной концентрации 5/3× с 1× киназным буферным раствором. Реакцию запускали добавлением 15 мкл смешанного раствора. Центрифугируют 384-луночный планшет при 1000 об/мин в течение 30 с, энергично встряхивают и перемешивают, выдерживают при комнатной температуре в течение необходимого времени. Добавляют 30 мкл стоп-раствора для остановки киназной реакции, центрифугируют при 1000 об/мин в течение 30 с, перемешивают встряхиванием и считывают данные о коэффициенте конверсии с помощью Caliper EZ Reader. Преобразуют конверсию в данные по ингибированию (% ингибирования = (максимальная конверсия лунки с ДМСО – конверсия лунки с испытуемым образцом) / (максимальная конверсия лунки с ДМСО – минимальная конверсия отрицательного контроля)*100). Где: макс. — коэффициент конверсии контроля (ДМСО); мин. — коэффициенту конверсии неактивного контроля. Строят кривую концентрации и степени ингибирования соединения по осям абсцисс и ординат соответственно. Для аппроксимации кривой и расчета IC50 используют программу-надстройку XLFit для Excel версии 4.3.1. Активность некоторых репрезентативных соединений показана в таблице 1.
3) Анализ ингибирования активности киназ CDK5, CDK9 и CDK16
Активность протеинкиназ CDK5/p35NCK, CDK9/CycT1 и CDK16/CycY определяли с помощью анализа киназ ADP-Glo. Разбавляют положительное соединение и испытуемое соединение (исходный раствор 10 мМ) в 25 раз 100 % ДМСО, готовят 4-кратное серийное разведение в 96-луночном планшете для разведения и добавляют 1 мкл соединения к 49 мкл киназного реакционного буферного раствора (1 мМ Трис, 20 мМ MgCl2, 0,10 % бычьего сывороточного альбумина и 0,5 мМ дитиотреитола) на шейкере для микропланшетов в течение 20 мин. Переносят 2 мкл киназы 2× в 384-луночный реакционный планшет, добавляют 1 мкл испытуемого соединения в 384-луночный реакционный планшет, центрифугируют при 1000 об/мин в течение 1 мин и инкубируют при 25° C в течение 10 мин. Переносят 1 мкл 4-кратной смеси субстратов в 384-луночный реакционный планшет, центрифугируют при 1000 об/мин в течение 1 мин и инкубируют при 25 °C в течение 60 мин. Переносят 4 мкл ADP-Glo в 384-луночный реакционный планшет, центрифугируют при 1000 об/мин в течение 1 мин и инкубируют при 25 °C в течение 40 мин. Переносят 8 мкл раствора для обнаружения в 384-луночный реакционный планшет, центрифугируют при 1000 об/мин в течение 1 мин и инкубируют при 25 °C в течение 40 мин. Сигнал RLU (относительная единица люминесценции) считывают с помощью многофункционального считывателя планшета Biotek. Интенсивность сигнала используется для характеристики степени активности киназы. Данные по степени ингибирования соединения: степень ингибирования, % = [1–(среднее значение RLU для соединения – среднее значение RLU для положительного контроля) / (среднее значение RLU для отрицательного контроля – среднее значение RLU для положительного контроля)]*100. Приняв логарифмическое значение концентрации по оси X и степень ингибирования (%) по оси Y, log(ингибитор) в зависимости от тангенса угла наклона переменной отклика аналитического программного обеспечения GraphPad Prism 5 использовали для подбора кривой доза-ответа для получения значение IC50 каждого соединения на активность фермента. Активность некоторых репрезентативных соединений показана в таблице 1.
Таблица 1. Ингибирующая активность в отношении киназ CDK2, CDK4, CDK6, CDK5, CDK9, CDK16 и TRKA (IC50, нМ)
a Соединение представляет собой гидрохлоридную соль.
Результаты указывают на то, что соединение по настоящему изобретению обладает такой же ингибирующей активностью в отношении CDK4 и CDK6, как и селективный ингибитор CDK4/6 палбоциклиб (Станд.-A) в предшествующем уровне техники, а также обладает мощной ингибирующей активностью в отношении киназ CDK2, CDK5, CDK9, CDK16 и TRKA и является мультитаргетным ингибитором киназ.
Пример 27. Фармакокинетическое исследование на крысах
Оборудование: прибор XEVO TQ-S ЖХ/МС производства Waters. Все результаты измерений собирают и обрабатывают с помощью программного обеспечения Masslynx V4.1, а данные рассчитывают и обрабатывают с помощью программного обеспечения Microsoft Excel. Для расчета фармакокинетических параметров использовали метод статистических моментов, расчеты проводили с помощью программного обеспечения WinNonLin 8.0. Главным образом включали кинетические параметры Tmax, T1/2, Cmax, AUClast и т.д. Колонка: ACQUITY UPLC BEH C18 (2,1 мм × 50 мм, 1,7 мкм); температура колонки 40 °C; подвижная фаза А — вода (0,1 % раствор муравьиной кислоты), подвижная фаза В — ацетонитрил, скорость потока 0,350 мл/мин, включен градиент элюирования 0,50 мин: 10 % В; 1,50 мин: 10 % В; 2,30 мин: 95 % В; 2,31 мин: 10 % В; 3,00 мин: остановка. Объем введения: 5 мкл.
Животные: 3 самца крыс Спрег-Доули массой 200–220 г. После покупки они будут храниться в лаборатории Центра подопытных животных 2 дня, а затем использоваться. Они будут голодать в течение 12 часов до и 4 часа после введения испытуемого соединения. Во время испытания предоставляется неограниченный доступ к питьевой воде. После введения испытуемого соединения крысам через желудочный зонд брали образцы крови в соответствии с установленными временными точками.
Растворитель: 0,5 % метилцеллюлоза (водный раствор, содержащий 0,4 % Твин 80 и 1 % этанола). Приготовление раствора для внутрижелудочного введения: готовят точную навеску соединения, добавляют его в растворитель и обрабатывают ультразвуком при комнатной температуре в течение 5 минут до полного растворения и готовят раствор лекарственного препарата 0,3 мг/мл.
Фармацевтические образцы: репрезентативные соединения структуры, показанной в запатентованной формуле (I) настоящего изобретения, как правило, берут несколько образцов со сходной структурой (с разницей молекулярной массы более 2 единиц), готовят точные навески и вводят вместе (кассета ФК). Таким образом, можно одновременно исследовать несколько соединений и сравнивать скорость их всасывания при введении внутрь. Однократное введение также использовали для изучения фармакокинетики испытуемого образца лекарственного препарата у крыс.
После внутрижелудочного введения кровь забирали из орбиты через 0,25, 0,5, 1, 2, 4, 9, 12 и 24 часа и помещали в пластиковую центрифужную пробирку, предварительно обработанную гепарином натрия. После центрифугирования плазмы использовали надосадочный слой для анализа методом ЖХ-МС/МС.
Готовят точную навеску соединений для получения растворов различных концентраций, проводят количественный анализ методом масс-спектрометрии для построения стандартной кривой, а затем определяют концентрацию вышеупомянутого соединения в плазме крови в разные моменты времени. Все данные измерений собираются и обрабатываются соответствующим программным обеспечением, а метод статистических моментов используется для расчета фармакокинетических параметров (главным образом кинетические параметры Tmax, T1/2, Cmax, AUClast и т.д.). Кинетические параметры некоторых репрезентативных соединений показаны в таблице 2.
Таблица 2. Фармакокинетические параметры соединений у крыс Спрег-Доули
a Соединение представляет собой гидрохлоридную соль.
Результаты показывают, что всасываемость соединений по настоящему изобретению при пероральном введении у крыс значительно выше, чем у соединения 7S (Станд.-B) в предшествующем патенте (WO2017101763).
Станд.-A и Станд.-B имеют следующие структуры:
Станд.-A(Палбоциклиб) Станд.-B (WO2017101763, соединение 7S)
Соединения по настоящему изобретению не только обладают ингибирующей активностью в отношении CDK4/CDK6, но также обладают высокой ингибирующей активностью в отношении других подтипов CDK (включая CDK2, CDK5, CDK9 и CDK16), поэтому они относятся к типу пан-CDK-ингибиторов и также обладают высокой ингибирующей активностью в отношении киназы TRK. В предшествующем уровне техники нет других сообщений о том, что эти соединения обладают ингибирующей активностью в отношении вышеуказанных подтипов CDK и киназ TRK.
Кроме того, по сравнению с существующими в данной области соединениями, используемыми в качестве ингибиторов CDK, соединения по настоящему изобретению также имеют значительно улучшенные фармакокинетические свойства. После введения пиковая концентрация в плазме и экспозиция соединений по настоящему изобретению у крыс были значительно выше, что позволяет предположить, что соединения по настоящему изобретению можно вводить в более низкой дозе.
Все литературные источники, упомянутые в настоящей заявке, включены в настоящую заявку посредством ссылки, и каждый документ индивидуально включен посредством ссылки. Кроме того, следует понимать, что после прочтения вышеизложенного специалисты в данной области могут вносить различные изменения и модификации в настоящее изобретение. Эти эквиваленты также входят в объем, определенный прилагаемой формулой изобретения.
Настоящее изобретение относится к области биотехнологии и может быть использовано в медицине. Изобретение раскрывает новые непептидные ингибиторы протеинкиназ. Соединения согласно настоящему изобретению характеризуются общей формулой (I) и представляют класс органических соединений, содержащих трициклические гетероарильные группы. Изобретение может быть использовано при лечении заболеваний, при которых наблюдается повышенная активность киназ CDK и/или TRK. 4 н. и 14 з.п. ф-лы, 27 пр., 2 табл.
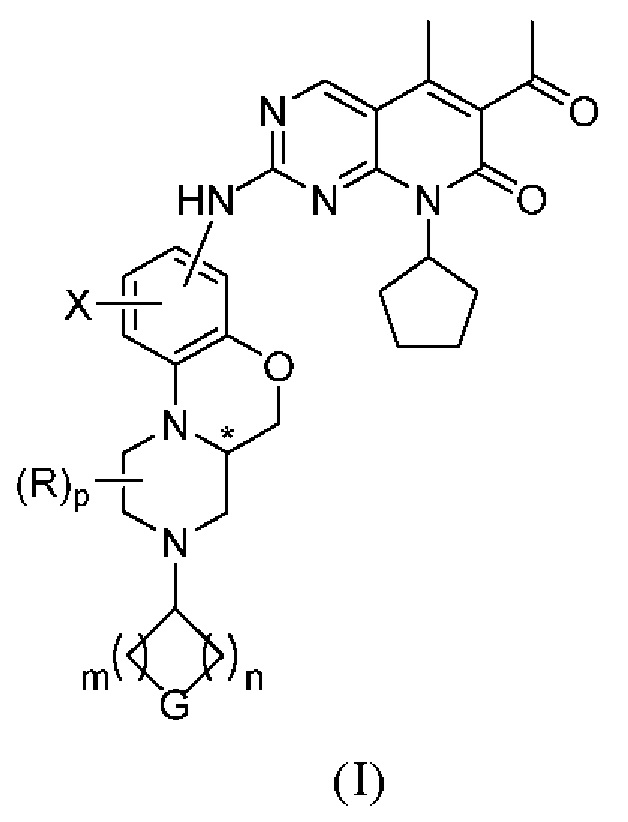
1. Соединение по следующей формуле (I), или его фармацевтически приемлемая соль:
(I)
знаком «*» обозначен хиральный центр;
X представляет собой водород, дейтерий, галоген, C1-4 алкил, OR1, NR1R2 или N(R1)C(O)R3;
все R независимо друг от друга представляют собой водород или C1–4 алкил; или когда два R одновременно присоединены к одному атому углерода, эти два R и атом углерода, к которому они присоединены, могут опционально образовывать карбонильную группу (C=O);
G представляет собой NRf, O, S, S(O), S(O)2 или CRgRg;
p равно 0, 1, 2 или 3;
m и n независимо друг от друга равны 0, 1, 2 или 3; при условии, что m и n не могут быть одновременно равны 0;
R1 и R2 независимо друг от друга представляют собой водород или C1-4 алкил;
R3 представляет собой C1-4 алкил, C2-4 алкенил или C2-4 алкинил;
Rf представляет собой водород, C1–4 алкил, C1–4 галоалкил, цианозамещенный C1–4 алкил, C2-4 алкенил, C2-4 алкинил, C3–8 циклоалкил, 4–8-членный гетероциклил, арил, гетероарил, C(O)R4, C(O)OR1, C(O)NR1R2, S(O)2R4 или S(O)2NR1R2;
все Rg независимо друг от друга выбирают из группы, состоящей из водорода, галогена или C1-4 алкила; или два Rg вместе с атомом углерода, к которому они присоединены, образуют карбонильную группу (C=O); или два Rg вместе с атомом углерода, к которому они присоединены, образуют 3–8-членную циклическую структуру, которая опционально содержит 0, 1 или 2 гетероатома, выбранных из N, O или S;
R4 представляет собой C1-4 алкил, C2-4 алкенил, C2-4 алкинил, C3-8 циклоалкил, 4–8-членную гетероциклическую группу, арил или гетероарил;
где каждый из указанных выше алкилов, алкенилов, алкинилов, циклоалкилов, гетероциклилов, арилов и гетероарилов необязательно и независимо замещен 1–3 заместителями, которые независимо друг от друга выбирают из группы, включающей галоген, C1-4 алкил, C1-4 галогеналкил, C2-4 алкенил, C2-4 алкинил, C3-8 циклоалкил, 3–8-членную гетероциклическую группу, арил, гетероарил, CN, NO2, OR1, SR1, NR1R2, C(O)R4, C(O)OR1, C(O)NR1R2, NR1C(O)R4 или S(O)2R4, при условии, что образовавшаяся химическая структура стабильна и логически обоснована; R1, R2 и R4 определены выше;
если не указано иное, арил представляет собой ароматические группы, содержащие от 6 до 12 атомов углерода; гетероарил представляет собой 5–15-членные гетероароматические группы; а циклическая структура представляет собой насыщенные или ненасыщенные циклические группы с гетероатомами или без них.
2. Соединение по п.1, или его фармацевтически приемлемая соль, где знаком «*» обозначен хиральный центр;
X представляет собой водород, дейтерий, галоген, C1-4 алкил, OR1, NR1R2 или NR1C(O)R3;
все R независимо друг от друга представляют собой водород или C1-4 алкил; или когда два R одновременно присоединены к одному атому углерода, эти два R и атом углерода, к которому они присоединены, могут опционально образовывать карбонильную группу (C=O);
G представляет собой NRf, O, S, S(O), S(O)2 или CRgRg;
p равно 0, 1, 2 или 3;
m и n каждый независимо равен 1, 2 или 3;
R1 и R2 независимо друг от друга представляют собой водород или C1-4 алкил;
R3 представляет собой C1-4 алкил, C2-4 алкенил или C2-4 алкинил;
Rf представляет собой водород, C1-4 алкил, C1-4 галоалкил, C2-4 алкенил, C2-4 алкинил, C3-8 циклоалкил, 4–8-членный гетероциклил, арил, гетероарил, C(O)R4, C(O)OR1, C(O)NR1R2 или S(O)2R4;
Rg независимо друг от друга выбирают из группы, состоящей из водорода, галогена или C1–4 алкила; или два Rg вместе с атомом углерода, к которому они присоединены, образуют карбонильную группу (C=O); или два Rg вместе с атомом углерода, к которому они присоединены, образуют 3–8-членную циклическую структуру, которая опционально содержит 0, 1 или 2 гетероатома, выбранных из N, O или S;
R4 представляет собой C1-4 алкил, C2-4 алкенил, C2-4 алкинил, C3-8 циклоалкил, 4–8-членную гетероциклическую группу, арил или гетероарил;
где каждый из указанных выше алкилов, алкенилов, алкинилов, циклоалкилов, гетероциклилов, арилов и гетероарилов необязательно и независимо замещен 1–3 заместителями, которые независимо друг от друга выбирают из группы, включающей галоген, C1-4 алкил, C1-4 галогеналкил, C2-4 алкенил, C2-4 алкинил, C3-8 циклоалкил, 3–8-членную гетероциклическую группу, арил, гетероарил, CN, NO2, OR1, SR1, NR1R2, C(O)R4, C(O)OR1, C(O)NR1R2, NR1C(O)R4 или S(O)2R4, при условии, что образовавшаяся химическая структура стабильна и логически обоснована; R1, R2 и R4 определены выше;
если не указано иное, арил представляет собой ароматические группы, содержащие от 6 до 12 атомов углерода; гетероарил представляет собой 5–15-членные гетероароматические группы; а циклическая структура представляет собой насыщенные или ненасыщенные циклические группы с гетероатомами или без них.
3. Соединение по п. 1, или его фармацевтически приемлемая соль, где формула (I) представляет собой:
знаком «*» обозначен хиральный центр;
где X, R, G, p, m и n определены в п.1.
4. Соединение по любому из пп. 1–3, или его фармацевтически приемлемая соль, где X представляет собой водород, галоген, C1-4 алкил; R представляет собой водород, или два R и атом углерода, к которому они присоединены, образуют карбонильную группу (C=O).
5. Соединение по любому из пп. 1–4 или его фармацевтически приемлемая соль, где G представляет собой NRf, O или CRgRg; m и n независимо друг от друга равны 1 или 2; где Rf представляет собой водород, C1-4 алкил, C1-4 галогеналкил, C3-8 циклоалкил, 4-8-членную гетероциклическую группу, арил, гетероарил, C(O)R4 или S(O)2R4; где R4 представляет собой C1-4 алкил, C2-4 алкенил, C2-4 алкинил, C3-8 циклоалкил, 4–8-членную гетероциклическую группу.
6. Соединение по любому из пп. 1–5 или его фармацевтически приемлемая соль, где формула (I) представляет собой:
(IV)
знаком «*» обозначен хиральный центр;
X представляет собой водород, галоген или C1-4 алкил;
G представляет собой NRf, O или CRgRg; m и n независимо друг от друга равны 1 или 2; где Rf представляет собой водород, C1-4 алкил, C1-4 галогеналкил, C3-8 циклоалкил, 4-8-членную гетероциклическую группу, арил, гетероарил, C(O)R4 или S(O)2R4; где R4 представляет собой C1-4 алкил, C2-4 алкенил, C2-4 алкинил, C3-8 циклоалкил, 4–8-членную гетероциклическую группу.
7. Соединение по любому из пп. 1–5, или его фармацевтически приемлемая соль, где формула (I) представляет собой:
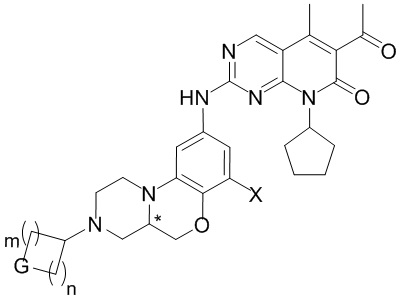
(V)
знаком «*» обозначен хиральный центр;
X представляет собой водород, галоген или C1–4 алкил;
G представляет собой NRf, O или CRgRg; m и n независимо друг от друга равны 1 или 2; где Rf представляет собой водород, C1-4 алкил, C1-4 галогеналкил, C3-8 циклоалкил, 4-8-членную гетероциклическую группу, арил, гетероарил, C(O)R4 или S(O)2R4; где R4 представляет собой C1-4 алкил, C2-4 алкенил, C2-4 алкинил, C3-8 циклоалкил или 4–8-членную гетероциклическую группу.
8. Соединение по любому из пп. 6, 7, или его фармацевтически приемлемая соль, где Rf представляет собой водород, C1-4 алкил, C1–4 галогеналкил, цианозамещенный C1-4 алкил, C3-8 циклоалкил, C(O)R4 или S(O)2R4; где R4 представляет собой C1-4 алкил.
9. Соединение по любому из пп. 5–7, или его фармацевтически приемлемая соль, где все Rg независимо друг от друга представляют собой водород или галоген.
10. Соединение по п. 6 или 7, или его фармацевтически приемлемая соль, где:
в формуле (IV) или формуле (V):
Знаком «*» обозначен хиральный центр;
X представляет собой водород, фтор или метил;
G представляет собой NRf, O или CRgRg; m и n независимо друг от друга равны 1 или 2; где Rf представляет собой водород, метил, этил, CH2CF3, CH2CN, циклопропил, C(O)CH3 или S(O)2CH3; все Rg независимо друг от друга представляют собой водород или фтор.
11. Соединение по п. 1 или его фармацевтически приемлемая соль, где формула (I) представляет собой соединение, выбираемое из следующей группы, или смесь с его соответствующим энантиомером:
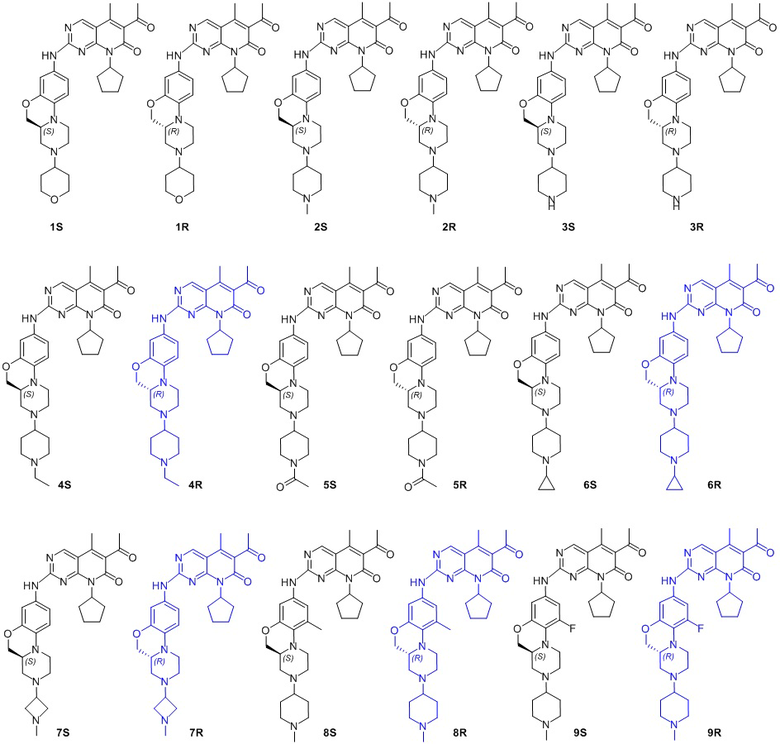
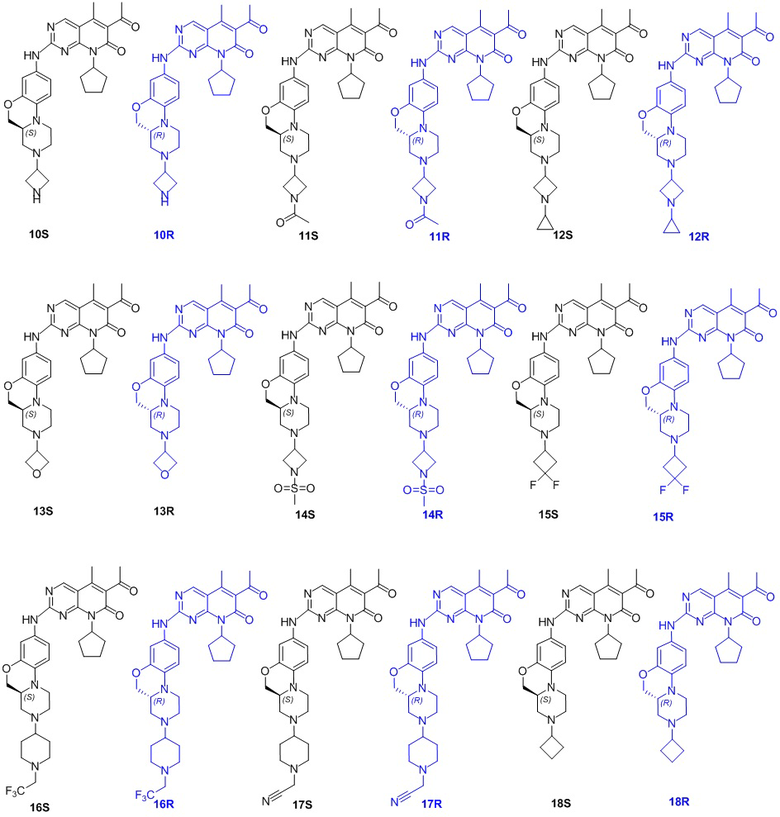
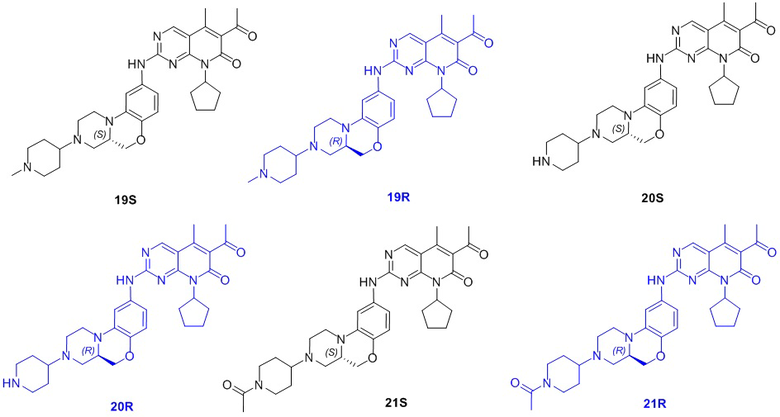
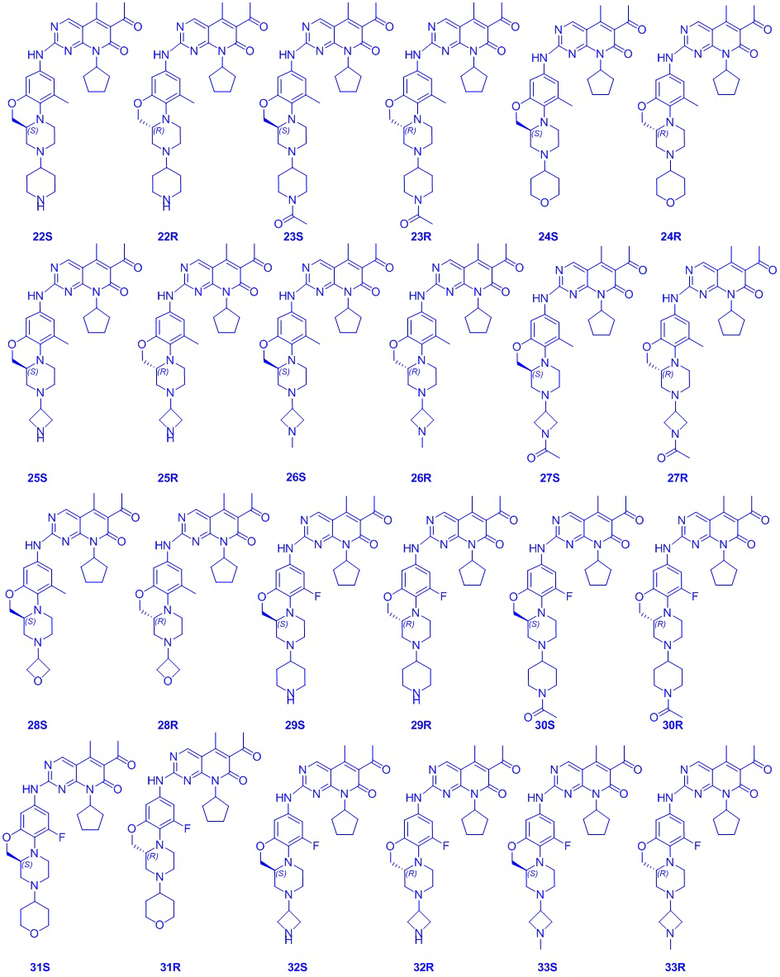
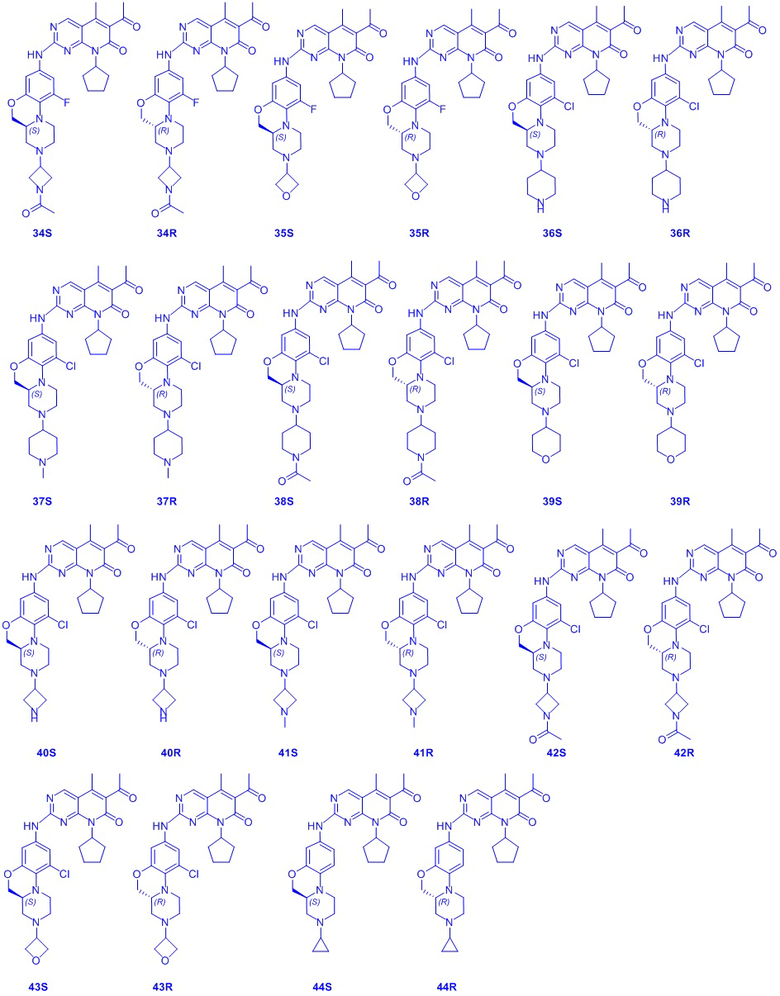 .
.
12. Соединение по любому из пп. 1–11 или его фармацевтически приемлемая соль, где соль представляет собой гидрохлорид.
13. Фармацевтическая композиция, содержащая: эффективное количество соединения по любому из пп. 1–12 или его фармацевтически приемлемой соли; фармацевтическая композиция предназначена для лечения заболеваний, связанных со сверхактивацией или гиперэкспрессией киназы; где киназа выбрана из CDK и/или TRK; где CDK означает циклинзависимые киназы, а TRK означает киназы тирозинового рецептора.
14. Применение соединения по любому из пп. 1–12 или его фармацевтически приемлемой соли, для получения лекарственного средства для лечения заболеваний, связанных со сверхактивацией или гиперэкспрессией киназы;
при этом киназу выбирают из CDK и/или TRK.
15. Применение по п.14, где заболевание выбирают из группы, включающей ДНК- и РНК-вирусные инфекций, В-клеточную лимфому, моноцитарный лейкоз, мегаполиспленическую полицитемию, синдром эозинофильного лейкоцитоза, идиопатическую тромбоцитопеническую пурпуру, системную гигантоклеточную болезнь, гемобластозы, солидные опухоли и нейродегенеративные заболевания.
16. Применение по п.14, где заболевание выбирают из группы, включающей аллергическую астму, миелофиброз, ревматоидный артрит, воспалительную боль, боль при злокачественном новообразовании, СПИД, вирус герпеса и вирус гриппа, секреторный рак молочной железы, фибросаркому, рак слюнных желез, рак печени, рак прямой кишки, рак мочевого пузыря, рак глотки и гортани, немелкоклеточный рак легкого, мелкоклеточный рак легкого, аденокарцинома легкого, плоскоклеточный рак легкого, рак молочной железы, рак предстательной железы, нейроглиоцитому, рак яичников, плоскоклеточный рак головы и шеи, рак шейки матки, рак пищевода, рак почки, рак поджелудочной железы, рак толстой кишки, рак кожи, лимфому, рак желудка, множественную миелому, опухоль головного мозга, рак легкого, болезнь Альцгеймера, болезнь Паркинсона.
17. Способ получения соединения по п.1 или его фармацевтически приемлемой соли, включающий следующие стадии:
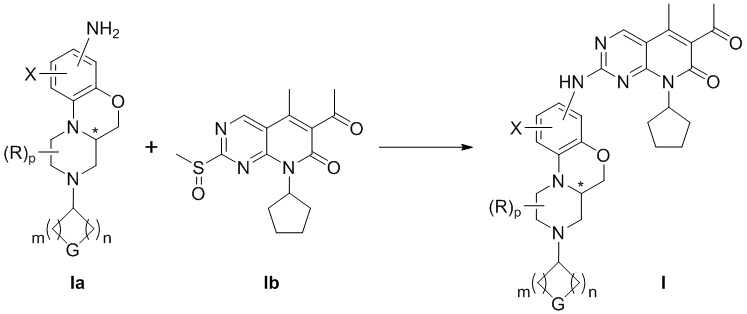
взаимодействие соединения формулы Ia и соединения формулы Ib в инертном растворителе с получением соединения формулы I.
18. Способ по п.17, где соединение Ia получают следующим способом:
1-A3-b
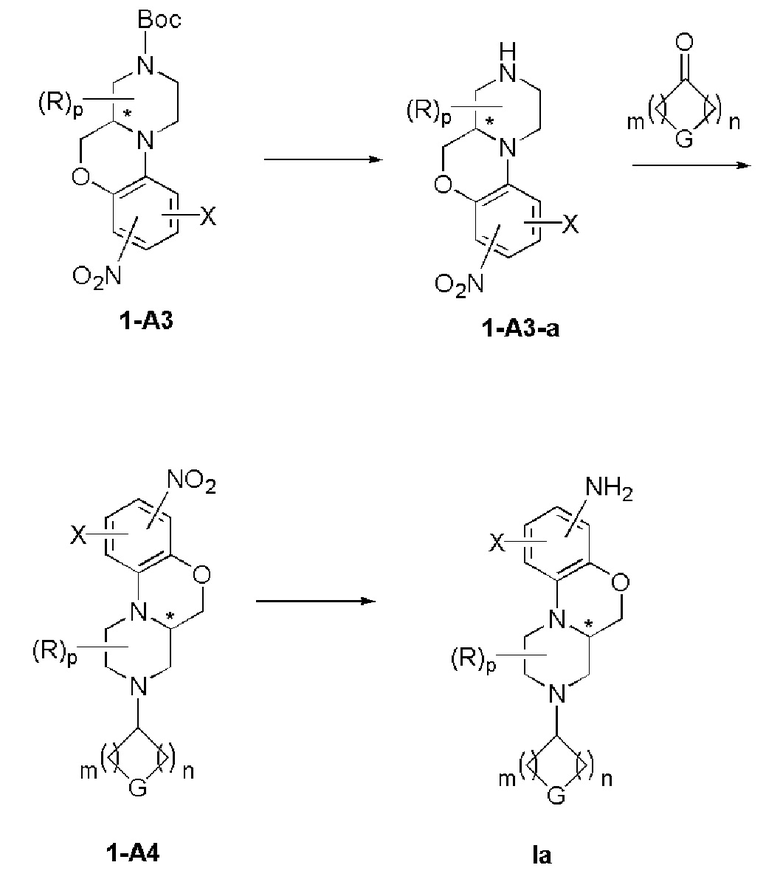
(1) снятие защиты с соединения формулы 1-А3 в инертном растворителе с получением соединения формулы 1-А3-а;
(2) реакция восстановительного аминирования соединения формулы 1-A3-a с соединением формулы 1-A3-b в инертном растворителе с получением соединения формулы 1-A4;
(i) восстановление соединения формулы 1-A4 в инертном растворителе с получением соединения формулы Ia.
| WO 2017101763 A1, 22.06.2017 | |||
| WO 2018108084 A1, 21.06.2018 | |||
| WO 2016030439 A1, 03.03.2016 | |||
| ИНГИБИРУЮЩИЕ CDK-КИНАЗЫ ПИРИМИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2002 |
|
RU2330024C2 |
| Приспособление к подъемникам для улавливания оборвавшихся концов грузовых канатов | 1933 |
|
SU32693A1 |

