Изобретение относится к новым двойным антитромботическим ингибиторам, содержащим остаток биотина или производное биотина, способу получения, фармацевтическим композициям, содержащим данные соединения в качестве активных ингредиентов, а также к применению вышеуказанных соединений для изготовления лекарственных средств.
Достигнутый в последнее время прогресс в поиске синтетических активных фармацевтических веществ, имеющих такие же или превосходящие гепарин антитромботические свойства, привел к разработке новых двойных ингибиторов, таких например, как описаны в WO 99/65934 и WO 01/42262. Эти соединения обычно являются конъюгатами остатка олигосахарида, связанного с непосредственным ингибитором тромбина посредством по существу фармакологически неактивного спейсера. Остаток олигосахарида молекулы демонстрирует анти-тромбин III (АТ-III)-опосредованную анти-Ха активность. Таким образом, новые конъюгаты имеют двойную активность, антитромботическую активность и антикоагулянтную активность.
Прекрасным примером нового класса двойных ингибиторов является соединение, обозначаемое кодом Org 42675, в котором пентасахарид соединен с непосредственным ингибитором тромбина, имеющее следующую формулу:
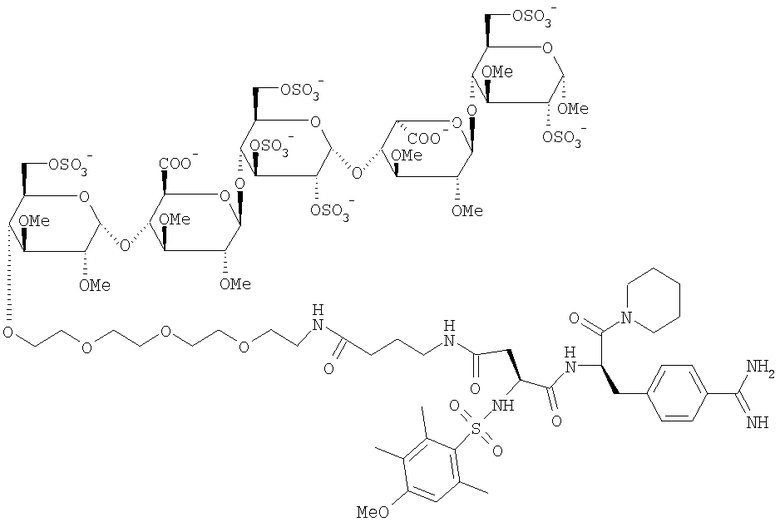
Исследования на экспериментальных моделях тромбоза продемонстрировали, что данное соединение, в дополнение к антитромботической активности и антикоагулянтной активности, также ингибирует активность тромбосвязанного тромбина. Кроме того, Org 42675 оказался высокоэффективным для предотвращения реокклюзий тромбозов, следующих за тромболизом окклюзивных артериальных тромбов. Соединение имеет в 10 раз увеличенное время полужизни по сравнению с неконъюгированным непосредственным ингибитором тромбина, полученным из NAPAP. По сравнению с аргатробаном, гепарином и фондапаринуксом, Org 42675 продемонстрировал более высокую эффективность (Journal of Thrombosis and Haemostasis, Volume 1, Issue 9, Page 1945, 2003).
Клинический потенциал нового двойного ингибитора признан существенным и, поэтому, клинические исследования уже были начаты.
В качестве предупредительной меры, в области антикоагулянтной и антитромботической терапии существует потребность в антидоте, способном эффективно нейтрализовать или минимизировать активность используемого антикоагулянтного или антитромботического лекарства. Это обусловлено тем, что хорошо известно, что кровоизлияние у проходящих лечение пациентов может быть вызвано любой случайной причиной. Далее, для пациентов, находящихся под воздействием антитромботической или антикоагулянтной терапии, может возникнуть потребность в хирургическом вмешательстве. К тому же, во время некоторых хирургических процедур антикоагулянты могут быть использованы в высоких дозах для предотвращения сворачивания крови, и необходимо нейтрализовать их после завершения операции. Таким образом, полезно иметь в доступности антитромботические/антикоагулянтные агенты, которые могут быть нейтрализованы для остановки антитромботической/антикоагулянтной активности в любое время.
В US 2004/0024197 раскрыто, что, в экстренных ситуациях, антитромботическая активность некоторых полисахаридов может быть снижена при использовании авидина, если эти полисахариды имеют как минимум одну ковалентную связь с биотином или производным биотина.
Настоящее изобретение касается новых нейтрализуемых двойных ингибиторов, происходящих из двойных ингибиторов, описанных в WO99/65934 и WO 01/42262. Было обнаружено, что определенный биотиновый маркер, являющийся группой 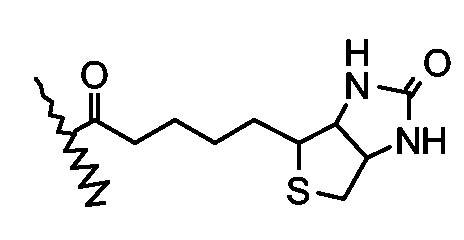 , также обозначаемый в этом документе как «BT» (полученный из гексагидро-2-оксо-1Н-тиено[3,4-d]имидазол-4-пентановой кислоты, предпочтительно D(+)-изомера) или его аналог, может быть присоединен или введен в структуру соединений, описанных в WO 99/65934 и WO 01/42262, что приводит к образованию нейтрализуемых двойных ингибиторов.
, также обозначаемый в этом документе как «BT» (полученный из гексагидро-2-оксо-1Н-тиено[3,4-d]имидазол-4-пентановой кислоты, предпочтительно D(+)-изомера) или его аналог, может быть присоединен или введен в структуру соединений, описанных в WO 99/65934 и WO 01/42262, что приводит к образованию нейтрализуемых двойных ингибиторов.
Таким образом, настоящее изобретение относится к соединениям формулы (I)
олигосахарид-спейсер-А (I),
где олигосахаридом является отрицательно заряженный остаток олигосахарида, содержащий от двух до двадцати пяти остатков моносахарида; заряд компенсируется положительно заряженным противоионом, и где остаток олигосахарида получен из олигосахарида, имеющего (АТ-III-опосредованную) анти-Ха активность;
спейсером является по существу фармакологически неактивный гибкий соединяющий остаток с длиной цепи от 10 до 70 атомов;
А является остатком CH[NH-SO2-R1][CO-NR2-CH(4-бензамидин)-CO-NR3R4],
где R1 является фенильной, нафтильной, 1,2,3,4-тетрагидронафтильной, (изо)хинолинильной, тетрагидро(изо)хинолинильной, 3,4-дигидро-1Н-изохинолинильной, хроманильной или камфорной группой, каковые группы могут быть необязательно замещены одним или более заместителями, выбираемыми из (1-8С)алкила или (1-8С)алкокси; и где R2 и R3 являются независимо Н или (1-8С)алкилом; R4 является (1-8С)алкилом или (3-8С)циклоалкилом; или R3 и R4 вместе с атомом азота, с которым они связаны, образуют неароматическое (4-8)членное кольцо, необязательно содержащее еще один гетероатом; кольцо необязательно замещается (1-8С)алкилом или SO2-(1-8C)алкилом;
или их фармакологически приемлемые соли, или пролекарства, или их сольваты;
где соединение формулы I далее содержит как минимум одну ковалентную связь с остатком биотина или его аналогом.
Соединения изобретения являются двойными ингибиторами, имеющими настраиваемый смешанный профиль непосредственной антитромбин (фактор IIA) активности и антитромбин (АТ-III)-опосредованной анти-Xa активности. Смешанный профиль соединения изобретения может быть настроен при помощи изменения природы остатка олигосахарида и силы непосредственного ингибитора тромбина (NAPAP аналоги). Соединения изобретения имеют долгий период полужизни в плазме крови и, как результат, они обладают пролонгированной антитромбиновой активностью по сравнению с ранее опубликованными в литературе данными о NAPAP или его производных. К тому же, соединения изобретения могут избегать нейтрализующего воздействия тромбоцитарного фактора 4 (PF4). Низкая токсичность также является выгодным свойством соединений данного изобретения.
Соединения данного изобретения являются полезными для лечения и предотвращения тромбин-опосредованных или тромбин-связанных заболеваний. Они включают некоторые тромбозные или протромбозные состояния, в которых активирован коагулянтный каскад, к которым относятся, но не только, глубокий тромбоз вен, эмболия сосудов легких, тромбофлебит, закупорка артерии вследствие тромбоза или эмболии, острый или хронический атеросклероз, инсульт, инфаркт миокарда, рак и метастазы и нейродегенеративные заболевания. Соединения данного изобретения могут быть также использованы как антикоагулянты при искусственном кровообращении, что необходимо при диализе или хирургическом вмешательстве.
Соединения изобретения могут быть использованы как in vitro коагулянты.
Биотиновый маркер (или его аналоги) в соединении данного изобретения может быть быстро узнан и связан специфическим антидотом, являющимся авидином (Merck Index, Twelfth edition, 1996, M.N. 920, 151-152) или стрептавидином, двумя тертрамерными белками с относительной массой примерно в 66000 и 60000 Да соответственно, имеющими очень высокое сродство к биотину. Таким образом, в экстренных ситуациях, активность двойного ингибитора может быть нейтрализована при использовании авидина или стрептавидина, например, при инъекции фармацевтического раствора, их содержащего. Аналоги авидина или стрептавидина, имеющие высокое сродство к биотину, могут быть использованы сходным образом. Получающийся в результате неактивный комплекс антидот-ингибитор вычищается из кровообращения.
Аналоги биотина, которые могут быть использованы в качестве маркера, в соответствии с данным изобретением, могут быть выбраны из, но не только, аналогов биотина, указанных в Pierce catalogue, 1999-2000, стр. 62-81, например, 6-биотинамидогексаноат, 6-(6-биотинамидогексанамидо)гексаноат и 2-биотинамидоэтанэтиол и т.д. В таких аналогах остаток биотина BT, как определено ранее, является характеристической частью молекулы. Другими аналогами являются, например, аналоги биотина, алкилированные по биотинамидной связи (где алкилом является (1-4С)алкил, предпочтительно метил) и которые устойчивы к расщеплению биотинидазами (Bioconjugate Chem., Vol. 11, 2000, 569-583; Bioconjugate Chem., Vol. 11, 2000, 584-598), или другие аналоги биотина, содержащие, например, гидроксиметилен, карбоксилат или ацетат в альфа-положении по отношению к биотинамидной связи, как описано в Bioconjugate Chem., Vol. 12, 2001, 616-623.
Предпочтительный остаток аналогов биотина имеет формулу -(NH-CO)n-(CH2)p-NR-BT, где n равно 0 или 1 (в особенности n равно 0), p равно 4 или 5 (в особенности p равно 4), R=H или (1-4С)алкилу и BT определен ранее. В предпочтительном варианте осуществления R является Н.
В сравнительных исследованиях с соответствующими небиотинилированными соединениями было показано, что введение биотинового маркера в двойные ингибиторы данного изобретения существенно не влияет на их непосредственную силу как ингибиторов тромбина и их антитромбин III (AT-III)-опосредованную анти-Ха активность. Кроме того, антитромботическая активность соединений формулы I является (по существу) полностью нейтрализуемой при введении авидина или стрептавидина.
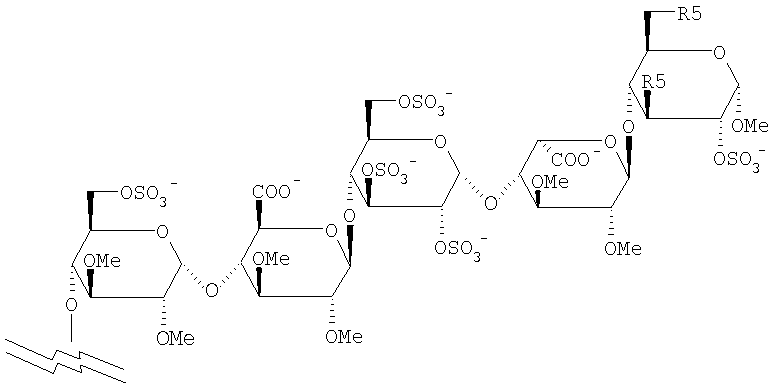
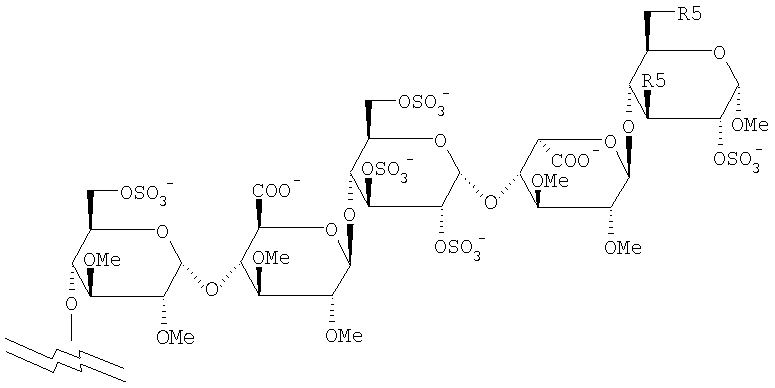
Любой отрицательно заряженный остаток олигосахарида от двух до двадцати пяти моносахаридных остатков длиной может быть использован в соединении данного изобретения. Подходящими соединениями изобретения являются соединения, где олигосахаридом является сульфатированный остаток олигосахарида. Предпочтительно, остаток олигосахарида получают из олигосахарида, который сам по себе имеет (AT-III опосредованную) анти-Ха активность, таких как олигосахариды, раскрытые в EP 0454220, EP 0529715, WO 97/47659, WO 99/36443 и WO 99/36428. Более предпочтительны остатки олигосахарида, имеющие от двух до шестнадцати, особенно от двух до шести, остатков моносахарида. Наиболее предпочтительным олигосахаридом является сульфатированный пентасахаридный остаток. Предпочтительный остаток пентасахарида имеет формулу (II)
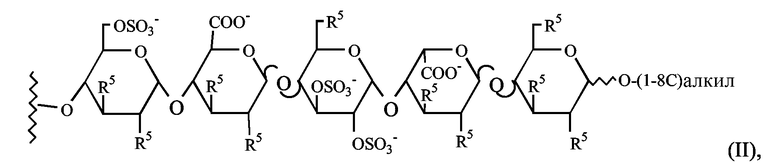
где R5 является независимо остатком биотина или его аналогом, OSO3- или (1-8С)алкокси. В предпочтительном варианте остатка пентасахарида общее количество сульфатных групп равняется 4, 5, 6 или 7.
Предпочтительными соединениями, соответствующими изобретению, являются соединения, в которых остаток пентасахарида имеет структуру:
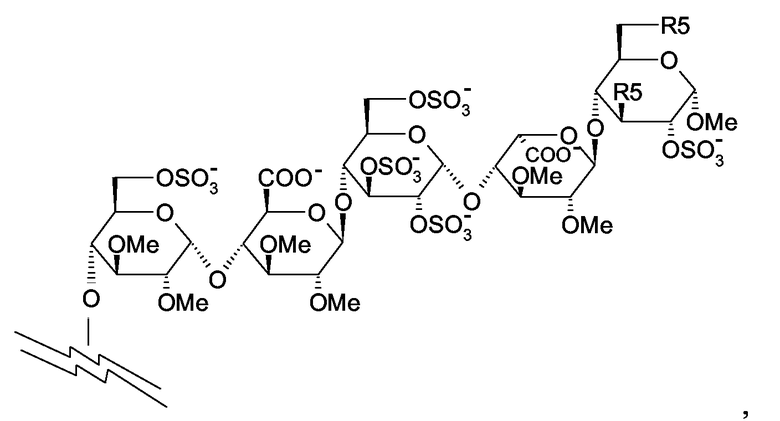
где R5 является OCH3 или OSO3-.
Особенно, предпочтительным остатком пентасахарида является
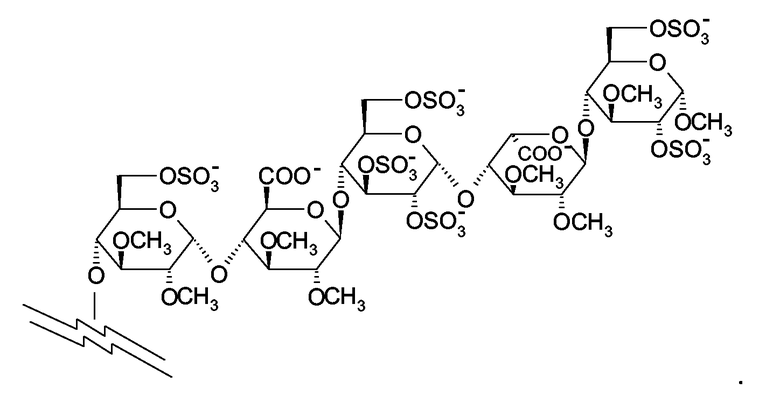
Далее, предпочтительными соединениями изобретения являются соединения формулы I, где R1 является фенилом или нафтилом, по желанию замещенными одним или более заместителями, выбираемыми из метил- или метоксигрупп. Более предпочтительно, R1 является 4-метокси-2,3,6-триметилфенил. В предпочтительных соединениях, NR3R4 представляет пиперидинилгруппу. Предпочтительно, R2 является Н.
Спейсер является существенно фармакологически неактивным гибким соединяющим остатком, предпочтительно имеющим 10-50 атомов, считая вдоль остова спейсера, атом кислорода остатка олигосахарида не считается. Далее, предпочтительна длина в 13-25 атомов, предпочтительно 16-22, и наиболее предпочтительно 19 атомов.
Химическая природа спейсера имеет небольшое значение для антитромботической активности соединения изобретения. Спейсер может содержать (относительно) жесткие элементы, такие как кольцевые структуры и ненасыщенные связи. Спейсеры, имеющие большую гибкость, более предпочтительны, чем другие. Подходящие спейсеры могут быть легко разработаны специалистом в данной области. Исходя из соображений синтеза, более длинные спейсеры рассматриваются как менее подходящие, однако, более длинные спейсеры могут быть успешно применены в соединениях данного изобретения. Предпочтительные спейсеры имеют как минимум один -(CH2CH2O)- элемент.
Предпочтительные соединения, соответствующие изобретению, содержат одну ковалентную связь с биотином или его аналогом.
Биотиновый (или ему аналогичный) маркер может присутствовать во всех частях соединения формулы I. Таким образом, осуществлениями данного изобретения являются соединения, в которых (a) остаток олигосахарида соединения формулы I содержит ковалентную связь с остатком биотина или его аналогом, (b) спейсер соединения формулы I содержит ковалентную связь с биотином или его аналогом и (c) остаток A соединения формулы I содержит ковалентную связь с остатком биотина или его аналогом (что означает, что атом водорода или заместитель в определении A заменяется на остаток биотина).
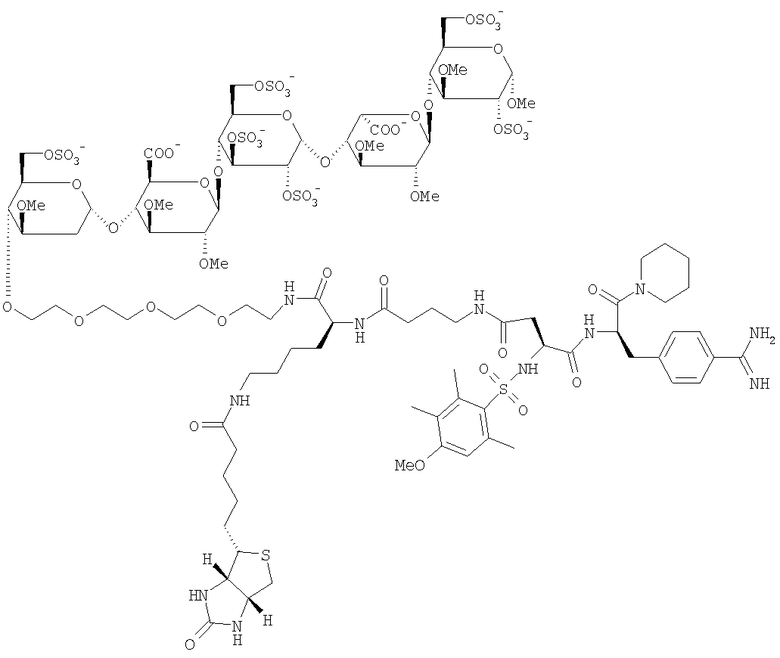
Примером биотинилированного двойного ингибитора настоящего изобретения является:
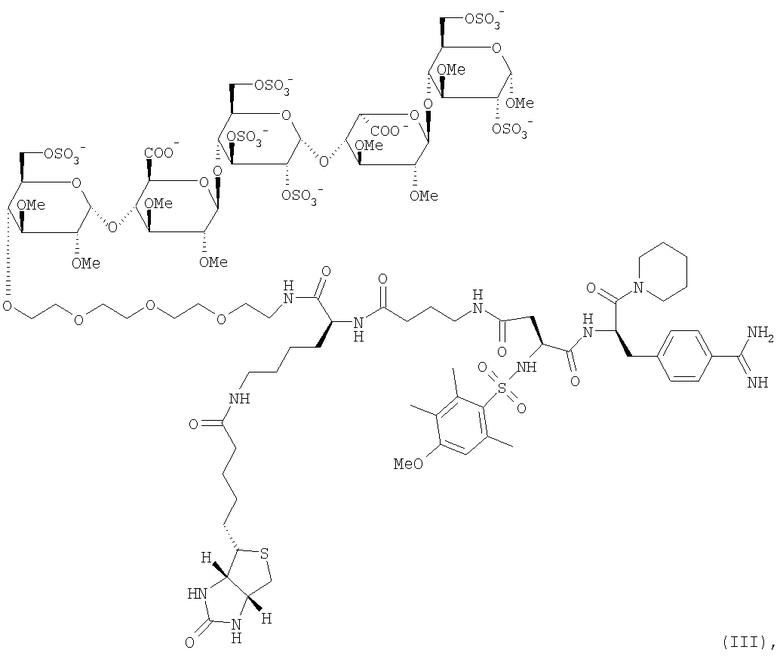
и
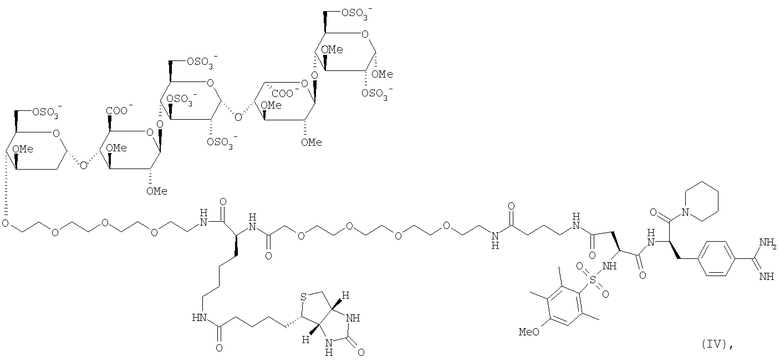
и их соли, но также соединения формулы I, где спейсер связан с олигосахаридом в другом положении и/или соединения, где остаток биотина (или его аналог) находится в другом положении молекулы. Предпочтительна натриевая соль.
Соединение формулы III является предпочтительным примером данного изобретения.
В описании соединений формулы (I) используются следующие определения.
Термины (1-4С)алкил и (1-8С)алкил означают разветвленную или неразветвленную алкильную группу, имеющую 1-4 или 1-8 атомов углерода соответственно, например, метил, этил, пропил, изопропил, бутил, вторичный бутил, трет-бутил, гексил и октил. Метил и этил являются предпочтительными алкильными группами.
Термин (1-8С)алкокси означает, что алкоксигруппа имеет 1-8 атомов углерода, алкильная группа имеет ранее определенное значение. Метокси является предпочтительной алкоксигруппой.
Термин (3-8С)циклоалкил означает циклоалкильную группу, имеющую 3-8 атомов углерода, являющуюся циклопропилом, циклобутилом, циклопентилом, циклогексилом, циклогептилом или циклооктилом. Циклопентил и циклогексил являются предпочтительными циклоалкильными группами.
Длиной спейсера является количество атомов в спейсере, считая по наиболее короткой цепи между остатком олигосахарида и пептидной частью молекулы, не считая атом кислорода остатка олигосахарида, связанный со спейсером.
Термин «пролекарство» означает соединение изобретения, в котором, например, аминогруппа амидино-компонента является защищенной, например, гидрокси или (1-6С)алкоксикарбонильной группой.
Сольваты, соответствующие данному изобретению, включают гидраты.
В отношении пути синтеза присоединения биотина к соединению формулы I химическая литература предлагает несколько вариантов, которые могут быть использованы и в которые могут быть вовлечены разные наборы защитных групп, хорошо известных специалисту в данной области. Остаток биотина, содержащий как реактивные группы типа, например, активированного эфира, малеимида, иодоацетила или первичного амина, будут предпочтительно реагировать с аминной функциональной группой, или тиоловой функциональной группой, или функциональной группой карбоновых кислот, или альдегидной функциональной группой в реакции, протекающей при условиях, описанных в литературе (cf. Savage et al., Avidin-Biotin Chemistry: A Handbook; Pierce Chemical Company, 1992).
Остаток биотина может быть, например, непосредственно связан с (негативно заряженным) остатком олигосахарида или через - необязательно - N-(1-4С)-алкилированную функциональную группу остатка олигосахарид-спейсера или через - необязательно - N-(1-4С)-алкилированный остаток аминокислоты к - необязательно - N-(1-4C)-алкилированной аминной функциональной группе остатка олигосахарида соединения формулы I.
В другом аспекте данного изобретения остаток биотина может быть, например, напрямую связан с остатком А или через - необязательно - N-(1-4C)-алкилированную аминную функциональную группу соединяющего остатка или через - необязательно - N-(1-4C)-алкилированный остаток аминокислоты к - необязательно - N-(1-4C)-алкилированной аминной функциональной группе остатка А соединения формулы I.
Еще в другом аспекте данного изобретения остаток биотина может быть, например, введен поэтапно: во-первых, связываясь непосредственно с остатком А или через - необязательно - N-(1-4C)-алкилированную аминную функциональную группу части спейсера формулы I или через - необязательно - N-(1-4C)-алкилированный остаток аминокислоты к - необязательно - N-(1-4C)-алкилированной аминной функциональной группе остатка А соединения формулы I и, во-вторых, связываясь непосредственно с олигосахаридом или через - необязательно - N-(1-4C)-алкилированную аминную функциональную группу части спейсера формулы I или через - необязательно - N-(1-4C)-алкилированный остаток аминокислоты к - необязательно - N-(1-4C)-алкилированной аминной функциональной группе (негативно заряженного) олигосахарида соединения формулы I, или наоборот.
В другом аспекте изобретения - необязательно - N-алкилированный остаток аминокислоты или α-N-замещенные (бета-) аналоги аминокислоты, такие как описано в [Bioconjugate Chem., Vol. 12, № 4, 2001, 616-623], могут быть введены через пептидное соединение с использованием известных в данной области методов. Азидная группа является подходящей скрытой аминной функциональной группой, которая может быть использована в предшественниках соединений формулы I для последующего введения остатка биотина.
Предпочтительный процесс подготовки соединений формулы I содержит стадию, где бензамидиновый компонент остатка А присутствует в форме предшественника, предпочтительно являясь 1,2,4-оксадиозалин-5-оновой группой, и впоследствии конвертируется в бензамидин при депротекции, особенно при гидрогенировании (Bolton, R.E. et al., Tetrahedron Letters, Vol 36, № 25, 1995, pp 4471-4474).
Соединения настоящего изобретения далее получают при модификации NAPAP (или аналога NAPAP) по глицину с цистеином или лизином или аспартатом, используя всем известные в данной области методы, каковые соединение далее (а) связывают с остатком олигосахарид-спейсера или (b) связывают со спейсером, который затем модифицируют с тиоловой функциональной группой или функциональной группой карбоновых кислот и затем связывают с остатком олигосахарида. Любой подходящий олигосахарид может быть использован для данных целей, например, известные в литературе олигосахариды (например, из EP 0454220 или EP 0529715, но не ограниченные данными источниками) или коммерчески доступные олигосахариды. Связывание спейсера с олигосахаридом может, например, быть осуществлено с помощью методов, описанных в EP 0649854.
Пептидное связывание, техническая стадия в вышеописанном методе получения соединений изобретения, может быть осуществлена методами, широко известными в области связывания, или конденсации, пептидных фрагментов, такими как азидный метод, смешанно-ангидридный метод, активированно-эфирный метод, или, предпочтительно, при воздействии аммонийно/уронийных солей, например TBTU, особенно при добавлении каталитических компонентов или компонентов, подавляющих рацемизацию, таких как N-гидроксисукцинимид, N-гидроксибензотриазол или 7-аза-N-гидроксибензотриазол. Обзор дан в The Peptides, Analysis, Synthesis, Biology, Vol 3, E. Gross and J. Meienhofer, eds. (Academic Press, New York, 1981) и Peptides: Chemistry and Biology, N. Sewald and H.-D. Jakubke (Wiley-VCH, Weinheim, 2002).
Аминные группы, присутствующие в соединении, могут быть защищены во время синтеза с помощью N-защитной группы, под которой понимается группа, широко используемая в пептидной химии для защиты α-амино группы, такой как трет-бутилоксикарбонил (Boc) группы, бензокарбонил (Z) группы, 9-фторометилоксикарбонил (Fmoc) группы или фталоил (Phth) группы, или могут быть введены при демаскировании азидного компонента. Обзор аминопротектирующих групп и методов их удаления дан в вышеуказанном The Peptides, Analysis, Synthesis, Biology, Vol. 3 и Peptides: Chemistry and Biology.
Соединения согласно изобретению, которые могут быть в виде свободного основания, могут быть изолированы из реакционной смеси в форме фармацевтически приемлемой соли. Фармацевтически приемлемая соль может быть получена при обработке свободного основания формулы I с органическими и неорганическими кислотами, такими как хлороводородная кислота, бромоводородная кислота, иодоводородная кислота, серная кислота, фосфорная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, метансульфоновая кислота, фумаровая кислота, янтарная кислота, винная кислота, лимонная кислота, бензойная кислота, аскорбиновая кислота и им подобные.
Соединения этого изобретения или их интерпретации имеют хиральный атом углерода и, таким образом, могут быть получены в виде чистых энантиомеров, или в виде смеси энантиомеров, или как смесь, содержащая диастереомеры. Методы получения чистых энантиомеров хорошо известны в данной области, например, кристаллизация солей, которые получены из оптически активных кислот и рацемических смесей, или хроматография с использованием хиральных колонок. Для диастереомеров также могут быть использованы прямо- и обратнофазные хроматографические колонки.
Соединения изобретения могут применяться энтерально или парентерально. Точная доза и режим этих компонентов и их композиций будут обязательно зависимы от потребностей индивидуального субъекта, принимающего лекарственное средство, степени болезни или потребностей и решения практикующего врача. В общем, парентеральное применение требует меньших доз, чем другие методы применения, более зависимые от абсорбции. Однако дневные дозы для человека предпочтительны в диапазоне 0,001-100 мг на кг массы тела, более предпочтительно 0,01-10 мг на кг массы тела.
Медикамент, произведенный с соединениями этого изобретения, может также быть использован в качестве адъюванта при острой антикоагулянтной терапии. В таком случае, медикамент применяется с другими компонентами, полезными при таких болезненных состояниях.
Смешанные с фармацевтически приемлемыми вспомогательными веществами, например, как описано в стандартной ссылке, Gennaro et al., Remington's Pharmaceutical Sciences, (18th ed., Mack Publishing Company, 1990, см. особенно Part 8 Pharmaceutical Preparations and Their Manufacture), соединения могут быть сформированы в твердые дозированные единицы, такие как пилюли, таблетки или преобразованы в капсулы или суппозитории. Посредством фармацевтически приемлемых жидкостей, соединения также могут быть применены в виде раствором, суспензий, эмульсий, например, для использования при инъекциях, или в виде спрея, например, для использования в виде спрея для носа.
Для производства дозированных единиц, например, таблеток, использование обычных добавок, таких как наполнители, красители, полимерные связующие вещества и им подобные рассматриваются. В общем, любая фармацевтически доступная добавка, не интерферирующая с функцией активных соединений, может быть использована.
Подходящие носители, с которыми композиции могут применяться, включают лактозу, крахмал, производные целлюлозы и им подобные, или их смеси, использованные в подходящих количествах.
Подписи к чертежам:
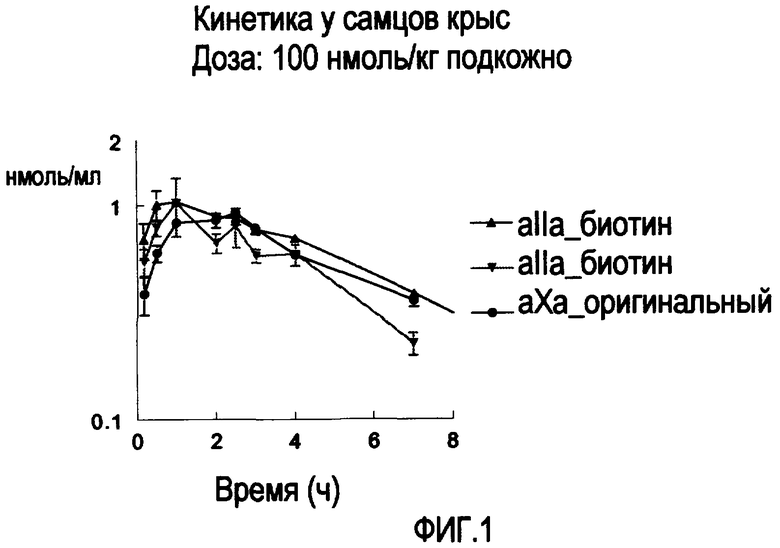
Фиг.1 Средний уровень в плазме, определенный при измерении анти-Ха или анти-IIa активности после подкожного применения 100 нмоль/кг биотинилированного соединения данного изобретения («биотин»). Кроме того, дан уровень в плазме для Org 42675 («оригинальный») после определения анти-Ха активности.
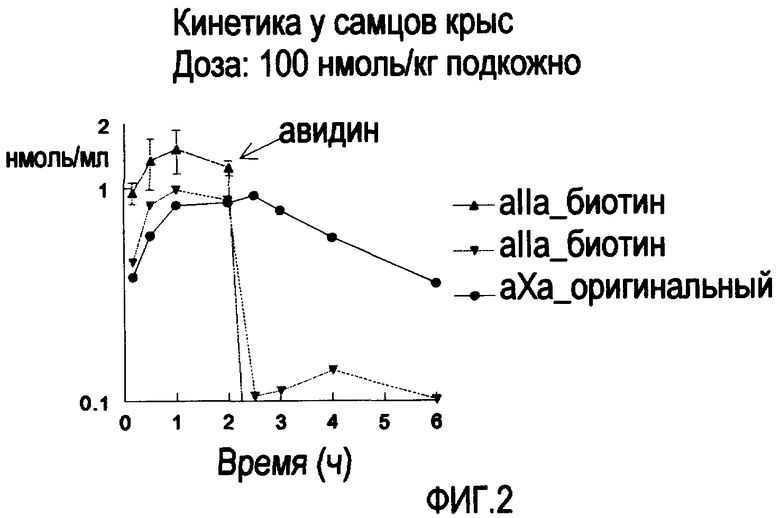
Фиг.2 Показан средний уровень в плазме ± стандартное отклонение после подкожного применения 100 нмоль/кг соединения. В точке t=2 часам авидин (10 мг/кг) был применен внутривенно к крысам, получавшим соединение III данного изобретения («биотин») или Org 42675 («оригинальный»). Фармакокинетика соединения III данного изобретения подвергается влиянию авидина, по сравнению с фармакокинетикой оригинального соединения.
Изобретение далее проиллюстрировано следующими примерами.
Примеры
Используемые сокращения
Aq.=водный
ATIII=антитромбин III
Boc=трет-бутилоксикарбонил
DCM=дихлорметан
DiPEA=N,N-диизопропилэтиламин
DMF=N,N-диметилформамид
Et=этил
fmoc=9-флуоренилметилоксикарбонил
NMM=N-метилморфолин
Me=метил
sat.=насыщенный
PRP=богатая тромбоцитами плазма
PPP=бедная тромбоцитами плазма
RT=комнатная температура
TBTU=2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафтороборат
TFA=трифторуксусная кислота
THF=тетрагидрофуран
Пример 1
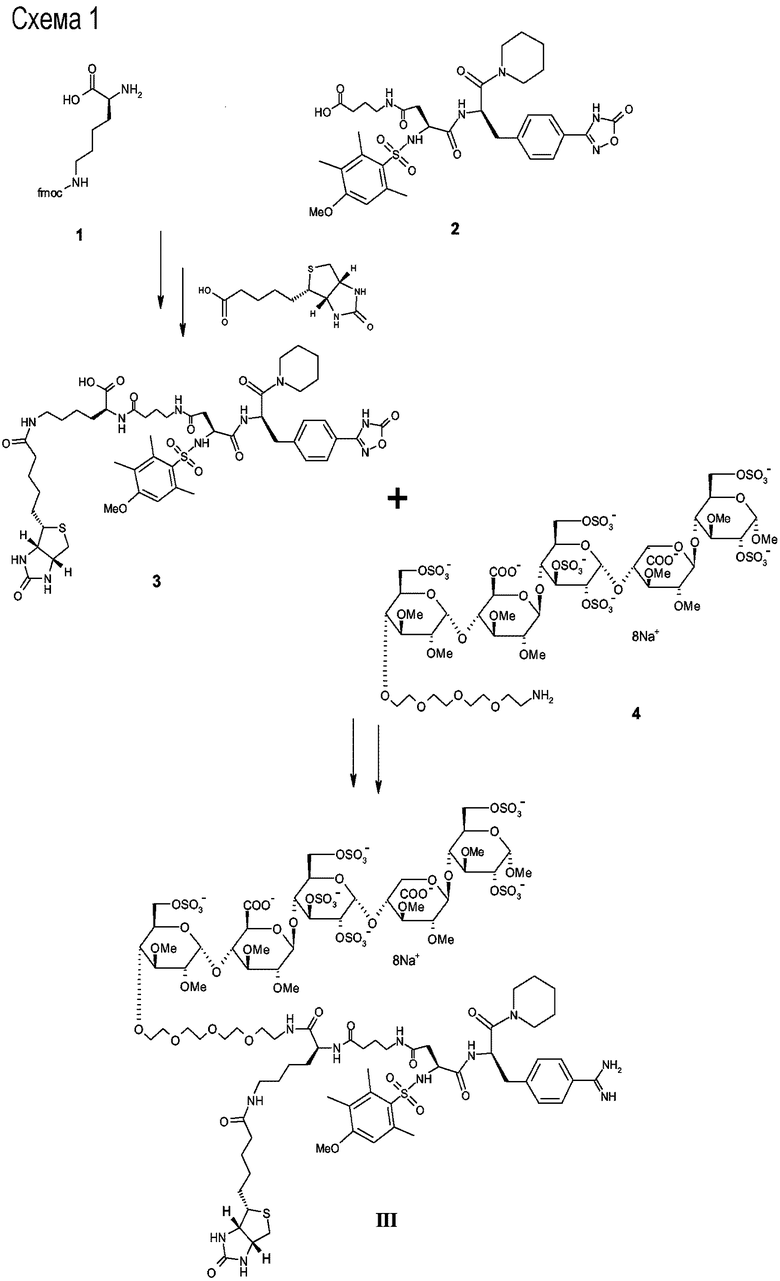
Схема 1
N ε -(D-(+)-биотинил)-N-{4-[[4-[[(1R)-1-[[4-(1,2,4-оксадиазол-5-онил)фенил]метил]-2-оксо-2-(1-пиперидинил)этил]амино]-3-[[(4-метокси-2,3,6-триметилфенил)сульфонил]амино]-1,4-(S)-диоксо-бутил]бутаноил}-L-лизин (3)
Соединение 2 (0,20 г, 0,27 ммоль), которое получено, как описано в WO 01/42262, растворено в DCM (10 мл). TBTU (86 мг, 0,27 ммоль) добавляли вслед за DiPEA (0,14 мл, 0,81 ммоль). После 1 ч перемешивания в атмосфере азота, соединение 1 (Nε-fmoc-Lys-OH, 0,27 ммоль, Fluka) было добавлено в твердом виде. DMF (5 мл) добавлено к раствору Nε-fmoc-Lys-OH. Смесь взбалтывали 16 ч и наливали в 0,5 н. раствор HCl. Раствор экстрагировали с DCM (3×). Объединенные органические слои промывали с солью, высушивали (MgSO4) и концентрировали при пониженном давлении (850 мбар, 45°С). Неочищенный продукт очищали с помощью хроматографии на силика-колонках (DCM/MeOH/AcOH, 99/0/1→89/10/1, об./об./об.), оставшийся AcOH удаляли повторным концентрированием в толуоле. Дальнейшая очистка выполнялась с помощью ВЭЖХ (AСN/H2O/0,1 Н TFA, 20:78:3→95:2:3) и давала 0,15 г (53%) чистого соединения. TLC: Rf 0,5 (DCM/MeOH/AcOH, 90/9/1, об./об./об.). LC-MS: m/z 1089 [M+H]1+.
Защищенное fmoc промежуточное соединение (0,15 г, 0,14 ммоль) растворяли в THF (5 мл) и добавляли Et2NH (2 мл). Смесь оставляли для перемешивания на 45 минут при 45°С. Раствор концентрировали при пониженном давлении и в толуоле. ESI-MS: m/z 857 [M+H]1+.
Nε-депротектированное лизиновое производное (0,14 ммоль) растворяли в DMF (3 мл) и добавляли в раствор D-(+)-биотина (34 мг, 1 эквив.), TBTU (45 мг, 1 эквив.) и DiPEA (61 мкл, 2,5 эквив.) в DМF (4 мл), который перемешивали в атмосфере азота в течение 1 ч. Полученную смесь перемешивали в течение 16 ч. Растворитель удаляли при пониженном давлении. Добавляли EtOAc (30 мл) и перемешивали. Твердый продукт 3 собирали фильтрацией и промывали MeOH и EtOAc и высушивали в вакууме. Выход вещества: 86 мг (57%). Rf 0,15 (DCM/MeOH, 9/1, об./об.). ESI-MS: 1083,6 [M+H]+.
Основная процедура приготовления соединений III и IV:
Производное карбоновой кислоты (33 мкмоль) (т.е. соединение 3 или 11) высушивали повторным концентрированием в безводном DMF (2×2 мл), растворяли в DMF (1 мл) и перемешивали в присутствии TBTU (11 мг, 33 мкмоль) и DiPEA (5,7 мкл, 33 мкмоль) в атмосфере азота. Через 1 ч добавляли пентасахарид 4 (31 мкмоль). Реакционную смесь перемешивали 16 ч при комнатной температуре и анализировали с помощью ионообменной (Mono-Q) хроматографии. Реакционную смесь концентрировали (<50°С, 15 мм рт.ст.). (Неочищенный) продукт (10 мг/мл в H2O/t-BuOH, 1/1, об./об.) депротектировали гидрогенизацией (Н2) свыше 10% Pd/C (добавлено количество, эквивалентное весу неочищенного вещества). Через 16 ч раствор дегазировали, фильтровали через 0,45 мкМ ВЭЖХ фильтр и концентрировали при пониженном давлении (<50°С, 15 мм рт.ст.). Конъюгаты очищали при помощи ионообменной хроматографии (Q-сефароза, буфер: H2O→2M NaCl), далее обессоливали с помощью колонок Sephadex G25 (H2O) и лиофилизировали.
Метил О-2,3-ди-О-метил-4-О-<<<12-N ε -<<N-(D-(+)-биотинил)-N-<)]-{4-[[4-[[(1R)-1-[[4-(аминоиминометил)фенил]метил]-2-оксо-2-(1-пиперидинил)этил]амино]-3-[[(4-метокси-2,3,6-триметилфенил)сульфонил]амино]-1,4-(S)-диоксо-бутил]амино]бутаноил}-L-лизил>>-12-аза-3,6,9-триокса-додецил>>>-6-О-сульфо-альфа-D-глюкопиранозил-(1->4)-O-2,3-ди-О-метил-бета-D-глюкопирануронозил-(1->4)-O-2,3,6-три-О-сульфо-альфа-D-глюкопиранозил-(1->4)-O-2,3-ди-О-метил-альфа-L-идопирануронозил-(1->4)-3-О-метил-2,6-ди-О-сульфо-альфа-D-глюкопиранозида четвертичная натриевая соль (III)
Соединение III приготавливается и очищается конъюгацией соединения 3 (86 мг, 81 мкмоль) с соединением 4 (0,14 г, 80 мкмоль), которые приготавливают, как описано в WO 01/42262, в соответствии с основной процедурой. Выход вещества 0,14 г (60%). ESI/TOF-MS: 880,91 [M-3H]3-, 888,57 [M-3H+Na]3-, 660,43 [M-4H]4-.
1H-ЯМР (D2O, 600 МГц); референсный водный сигнал на 4,71 ч/млн препятствующий достоверной интеграции сигналов между 4,80-4,50 ч/млн. δ 7,66 (м, 2H), 7,30 (м, 2H), 6,74 (м, 1H), 5,36 (м, 1H), 5,25 (м, 1H), 5,05 (м, 1H), 4,98 (м, 1H), 4,84 (м, 1H), 4,46 (м, 2H), 4,31 (м, 1H), 4,27-4,13 (5H), 4,13-3,99 (5H), 3,92 (м, 1H), 3,88-3,70 (8H), 3,70-3,07 (49H±5), 3,07-2,92 (4H), 2,85 (м, 1H), 2,76 (м, 1H), 2,65 (м, 1H), 2,51 (c, 3H), 2,40 (c, 3H), 2,36-2,22 (м, 2H), 2,21-2,11 (м, 2H), 2,08 (м, 1H), 2,06-1,91 (4H), 1,83-1,03 (20H±2).
Пример 2
Схема 2
трет -Бутил 15-N-(9-флуоренилметилоксикарбонил)-15-аза-3,6,9,12-тетраокса-пентадеканоат (6)
трет-Бутил 15-амино-3,6,9,12-тетраокса-пентадеканоат (5) (0,50 г, 1,45 ммоль) растворяли в THF (7,5 мл) и Н2О (5 мл). Добавляли 4 н. раствор NaOH до рН приблизительно 9. N-9-флуоренилметилкарбаматсукцинимид (FmocOSu, 0,54 г, 1,60 ммоль, 1,1 экв.) добавляли порциями. Через 10 мин дополнительно добавляли раствор 4 н. NaOH для доведения рН приблизительно до 9. Через 3 ч реакционную смесь подкисляли 1 н. раствором HCl до рН 6-7. Добавляли в реакционную смесь Н2О, которую затем экстрагировали 3 раза с EtOAc. Органическую фазу промывали с солью и высушивали над MgSO4. После фильтрации растворитель удаляли при пониженном давлении (50 мбар, 50°С). Неочищенное масло очищали с помощью хроматографии на силика-колонках (DCM/MeOH, 1/0→95/5, об./об.), что давало соединение 6 в виде желтоватого масла (0,61 г, 79%). Rf 0,64 (DCM/MeOH, 95/5, об./об.).
15-N-(9-флуоренилметилоксикарбонил)-15-аза-3,6,9,12-тетраокса-пентадеканоат (7)
Соединение 6 растворяли в DCM (3.5 мл) и добавляли TFA (3.5 мл) в атмосфере азота. Через 1.5 ч перемешивания реакционную смесь концентрировали при пониженном давлении. Затем избыток TFA удаляли повторным концентрированием в толуоле. Добавляли DCM/Et2O (100 мл, 1/2, об./об.) и промывали 1 н. HCl. Водный слой экстрагировали DCM/Et2O (100 мл, 1/2, об./об.). Объединенные органические слои промывали с солевым раствором, высушивали над MgSO4. После фильтрации растворитель удаляли при атмосферном давлении (50°С). Неочищенное масло очищали с помощью хроматографии на силика-колонках (DCM/MeOH/AcOH, 99/0/1→89/10/1, об./об./об.), что давало соединение 7. Оставшийся AcOH удаляли растворением неочищенного масляного продукта в DCM/Et2O (1/2, об./об.) и промывали Н2О (3 раза) и солевым раствором, затем высушивали над MgSO4. После фильтрации растворитель удаляли при атмосферном давлении (50°С), что давало соединение 7 в виде желтоватого масла (0,37 г, 67%). Rf 0,32 (DCM/MeOH, 89/10/1, об./об.).
трет-Бутил 15-N-(9-флуоренилметилоксикарбонил)-15-аза-3,6,9,12-тетраокса-пентадеканоил-ε-N-(Z)-L-лизин (8)
Соединение 7 (0,37 мг, 0,77 ммоль) растворяли DCM (18 мл). DiPEA (0,40 мкл, 2,31 ммоль, 3 экв.) и TBTU (0,25 г, 0,77 ммоль) последовательно добавляли в атмосфере N2, и оставляли раствор для перемешивания на 10 мин. Затем добавляли ε-(Z)-L-Lys-OtBu.HCl (0,29 г, 0,77 ммоль), и перемешивали реакционную смесь еще 1,5 ч. Реакционную смесь разбавляли DCM и промывали Н2О, 0,1 н. HCl, насыщ. NaHCO3-раств. и солевым раствором. Органическую фазу высушивали (MgSO4) и концентрировали при атмосферном давлении. Очистка производилась с помощью хроматографии на колонках с силикагелем (DCM/MeOH, 1/0→9/1, об./об.), что давало соединение 8 в виде желтоватого масла (0,51 г, 83%). Rf 0,85 (DCM/MeOH, 9/1, об./об.). ESI/MS: 792,6 [M+H]+, 814,6 [M+Na]+, 736,4 [M-tBu+H]+.
трет-Бутил 15-N-трет-бутилоксикарбонил-15-аза-3,6,9,12-тетраокса-пентадеканоил-ε-N-(Z)-L-лизин (9)
Соединение 8 (0,26 г, 0,32 ммоль) растворяли в TНF (5 мл). Добавляли Et2N (1 мл), и оставляли раствор для перемешивания на 24 ч. Избыток Et2N и растворитель удаляли при пониженном давлении (50°С). Добавляли толуол и удаляли при пониженном давлении (50°С, 65 мбар), что давало N-депротектированный промежуточный продукт (0,21 г, 0,32 ммоль), Rf 0,23 (DCM/MeOH, 9/1, об./об.). ESI/MS: 570,4 [M+H]+, 514,4 [M-tBu+H]+. Неочищенный продукт растворяли в DCM (3 мл). Et3N (0,11 мл) добавляли после ди-трет-бутилдикарбоната (73 мг, 0,34 ммоль, 1,1 экв.) в атмосфере N2. После перемешивания в течение 5 ч смесь добавляли в холодный (5°С) раствор 0,1 н. HCl и экстрагировали с EtOAc. Органический слой промывали с солевым раствором и высушивали (MgSO4). После фильтрации растворители удаляли при пониженном давлении (180 мбар, 50°С). Очистка производилась с помощью хроматографии на колонках с силикагелем (DCM/MeOH, 1/0→95/5, об./об.), что давало соединение 9 в виде бесцветного масла (0,17 г, 82%). Rf 0,5 (DCM/MeOH, 9/1, об./об.). ESI/MS: 670,6 [M+H]+, 692,4 [M+Na]+, 570,4 [M-Boc+H]+, 514,1 [M-Boc-tBu+H]+.
15-аза-3,6,9,12-тетраокса-пентадеканоил-ε-[D-(+)-биотинил]-L-лизин (10)
Соединение 9 (0,23 г, 0,34 ммоль) растворяли в EtOH (8 мл) и Н2О (1,2 мл). После промывания раствора азотом в течение 5 минут, добавляли Pd/C 10% (0,11 г). Водород пропускали через раствор в течение 4 ч. Азот пропускали через раствор в течение 10 мин, чтобы удалить весь водород. Смесь фильтровали через decalite и концентрировали при пониженном давлении (170 мбар, 50°С), что давало N-L-лизиновое депротектированное промежуточное соединение в виде бесцветного масла (0,15 г, 81%). Rf 0,02 (DCM/EtOAc, 9/1, об./об.).
D-(+)-Биотин (75 мг, 0,31 ммоль) суспендировали в DCM (7 мл). DiPEA (0,11 мл, 0,62 ммоль, 2 экв.) и TBTU (0,10 г, 0,31 ммоль) последовательно добавляли в атмосфере N2, и оставляли раствор для перемешивания на 1 ч. Раствор ранее описанного N-L-лизинового депротектированного промежуточного соединения в DCM (3 мл) добавляли в реакционную смесь. Смесь оставляли для перемешивания на 16 ч. Добавляли Н2О и экстрагировали DCM (3×). Органический слой высушивали (MgSO4), фильтровали и концентрировали при пониженном давлении (850 мбар, 50°С). Очистка производилась с помощью хроматографии на колонках с силикагелем (элюенты: DCM/MeOH, 1/0→9/1, об./об.), что давало масло (0,13 г, 60%). Rf 0,48 (DCM/MeOH, 9/1, об./об.). ESI/MS: 762,6 [M+H]+, 784,6 [M+Na]+, 662,4 [M-Boc+H], 606,4 [M-Boc-tBu+H]. Масло растворяли в обезвоженном 4 н. растворе HCl в диоксане (4 мл) и перемешивали. Через 1 ч появлялось нерастворимое масло, после чего растворитель удаляли при пониженном давлении (100 мбар, 50°С), что давало соединение 10 в количественном выходе. ESI/MS: 606,4 [M+H]+, 628,4 [M+Na]+.
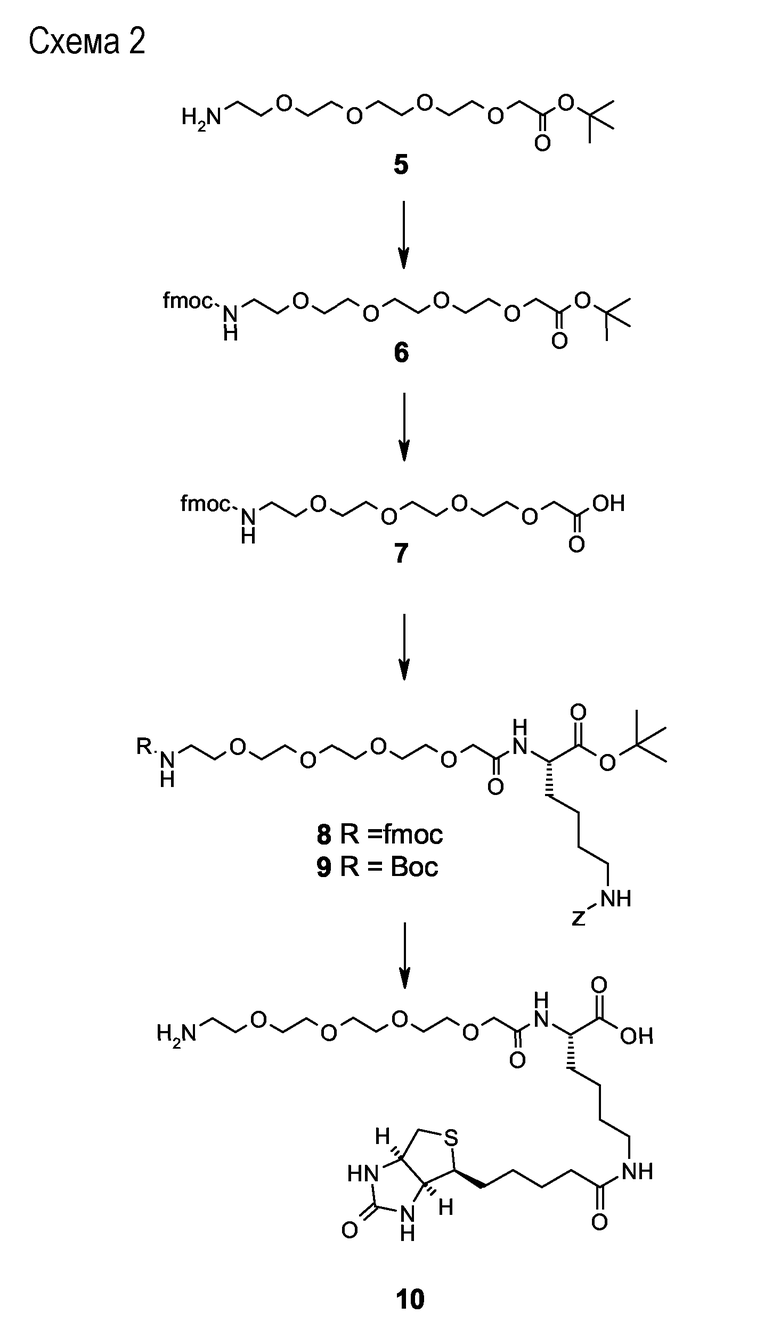
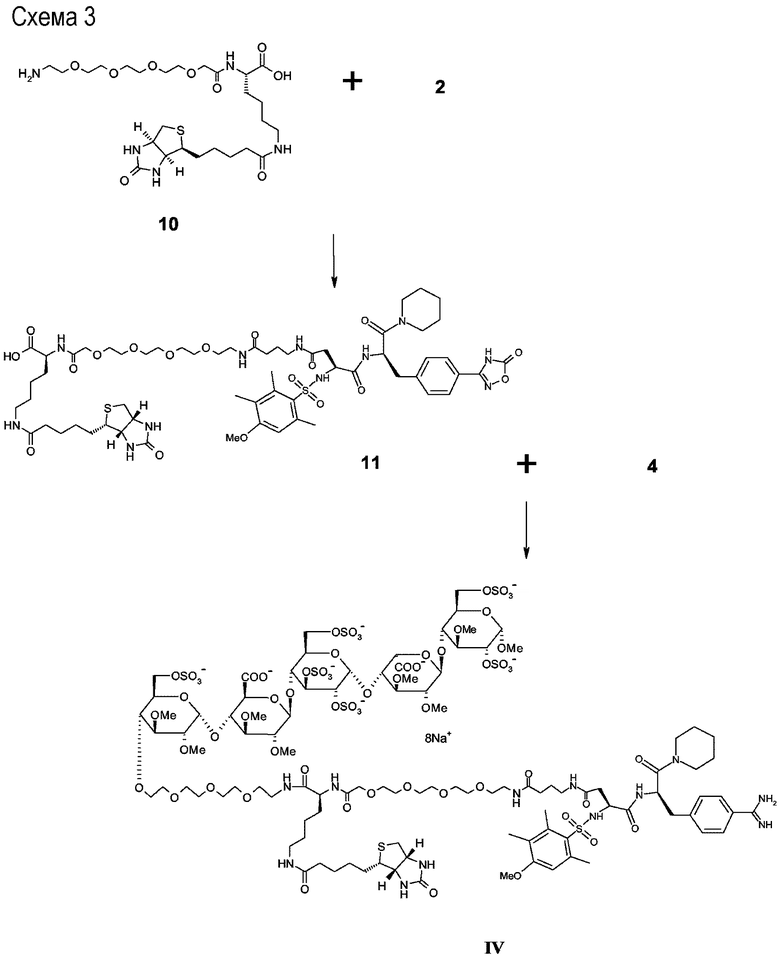
Схема 3
N ε -(D-(+)-биотинил)-N-{{4-[[4-[[(1R)-1-[[4-(1,2,4-оксадиазол-5-онил)фенил]метил]-2-оксо-2-(1-пиперидинил)этил]амино]-3-[[(4-метокси-2,3,6-триметилфенил)сульфонил]амино]-1,4-(S)-диоксо-бутил]амино]бутаноил}-15-N-(15-аза-1-оксо-3,6,9,12-тетраокса-пентадецил)]}-L-лизин (11)
Соединение 10 (0,12 г, 0,21 ммоль) связывалось с соединением 2 (0,15 г, 0,21 ммоль), как описано для приготовления соединения 3. Неочищенный продукт очищали предварительной ВЭЖХ (C18, ACN/H2O, 0,01% TFA), что давало соединение 11 в чистом виде. Выход вещества 60 мг (22%). ESI-MS: 1316,8 [M+H]+.
Метил О-2,3-ди-О-метил-4-О-<<<12-N ε -<<N-(D-(+)-биотинил)-N-<[15-N-(15-аза-1-оксо-3,6,9,12-тетраокса-пентадецил)]-{4-[[4-[[(1R)-1-[[4-(аминоиминометил)фенил]метил]-2-оксо-2-(1-пиперидинил)этил]амино]-3-[[(4-метокси-2,3,6-триметилфенил)сульфонил]амино]-1,4-(S)-диоксо-бутил]амино]бутаноил}>-L-лизил>>-12-аза-3,6,9-триокса-додецил>>>-6-O-сульфо-альфа-D-глюкопиранозил-(1->4)-O-2,3-ди-О-метил-бета-D-глюкопирануронозил-(1->4)-О-2,3,6-три-О-сульфо-альфа-D-глюкопиранозил-(1->4)-O-2,3-ди-О-метил-альфа-L-идопирануронозил-(1->4)-3-О-метил-2,6-ди-О-сульфо-альфа-D-глюкопиранозида четвертичная натриевая соль (IV)
Соединение 11 связывалось с соединением 4, и промежуточный конъюгат был депротектирован в соответствии с основной процедурой с получением соединения IV.
Выход вещества 9,2 мг (7,6%)
1H-ЯМР (D2O, 600 МГц); референсный водный сигнал на 4,71 ч/млн, препятствующий достоверной интеграции сигналов между 4,80-4,54 ч/млн. δ 7,59 (д, 2H), 7,23 (д, 2H), 6,68 (c, 1H), 5,32 (м, 1H), 5,19 (м, 1H), 4,91 (м, 1H), 4,85 (м, 1H), 4,76 (м), 4,53-4,31 (3H), 4,30-4,05 (9H), 4,04-3,90 (7H), 3,89-3,62 (7H), 3,61-3,42 (42H±4), 3,42-3,31 (18H±2), 3,31-3,14 (12H), 3,14-3,05 (5H), 3,03-2,94 (3H), 2,93-2,84 (2H), 2,81 (дд, 1H), 2,70 (дд, 1H), 2,59 (д, 1H), 2,44 (c, 3H), 2,34 (c, 3H), 2,22-2,08 (4H), 2,06 (т, 2H), 1,96 (c, 3H), 1,72-1,01 (18H±2).
ESI/TOF-MS: m/z 574,72 [M-5H]5-, при m/z 718,66 [M-4H]4-, при m/z 958,56 [M-3H]3-.
Пример 3
Фармакология
In vitro тест для определения анти-фактор Ха и фактор IIа активности в человеческой плазме
Анти-фактор Ха и фактор IIа активность тестируемых соединений в человеческой плазме измеряли амидолитически с использованием S2222 или S2238 (Chromogenix, Chromogenics Ltd., Molndal, Швеция) в качестве субстратов, соответственно. Протоколы основывались на методе, описанном Teien и Lie (Teien AN, Lie M. Evaluation of an amidolytic heparin assay method increased sensitivity by adding purified antithrombin III. Thromb. Res. 1977, 10: 399-410). Обе активности выражали в IC-50 (моль/л).
Краткая характеристика антитромботической активности in vitro
Человеческая плазма рН 7,4 IC-50(М)
Человеческая плазма рН 7,4 IC-50(М)
3.1 Фармакокинетика
Фармакокинетические свойства Org 42675 и соединения III этого изобретения изучали на самцах крыс линии Вистар весом 300-400 г. Крысы были анестезированы ингаляционно смесью O2/N2O/изофлуран, после чего была канюлирована правая яремная вена. На следующий день крысам подкожно вводили вещества в дозах 100 нмоль/кг. После подкожного введения отбирали кровь в нескольких временных интервалах. Кровь центрифугировали, после чего плазму отбирали и хранили при -20°С до использования. Концентрацию тестируемых соединений измеряли амидолитически с использованием S-2222 или S-2238 в качестве субстратов (Chromogenix, Chromogenics Ltd., Molndal, Швеция) для определения анти-Ха и анти-IIа активности, соответственно. Обе процедуры основывались на методе Teien и Lie (Teien AN, Lie M. Evaluation of an amidolytic heparin assay method increased sensitivity by adding purified antithrombin III. Thromb. Res. 1977, 10: 399-410). Концентрации в полученных образцах плазмы определяли по калибровочной кривой, которая была построена с использованием маточных растворов тестируемых соединений. Концентрации в образцах представляли в нмоль/мл и кинетические параметры вычисляли с помощью некомпартментной модели WinNonlin. (см. фиг. 1).
Фармакокинетические параметры после подкожного введения соединения III этого изобретения или Org 42675 (100 нмоль/кг) крысам. Эксперименты выполнены при n=3 в группе
анти-Ха
анти-IIа
анти-Ха
Можно заключить, что в пределах ошибки эксперимента соединение III этого изобретения и Org 42675 показывают одинаковое фармакокинетическое поведение у крыс.
3.2 Фармакокинетика - эксперимент по нейтрализации
Крысы получали соединение III данного изобретения или Org 42675 в дозе 100 нмоль/кг подкожно, образцы крови отбирали через 2 ч, и 10 мг/кг Авидина (из белка яиц, Sigma) вводили внутривенно крысам, получавшим соединение III этого изобретения или Org 42675. Кровь отбирали через 0,17, 0,5, 1, 2, 3 и 7 часов. Кровь подготавливали, как описано для фармакокинетического эксперимента, и концентрацию в образцах определяли измерением (остаточной) анти-Ха или анти IIа активности (см. фиг. 2).
Площадь под кривой (AUC), вычисленная после подкожного введения 100 нмоль/кг соединения III данного изобретения или Org 42675 и авидина (10 мг/кг) через 2 ч. Данные подсчитаны для Т=2 ч. Эксперименты выполнены при n=3 в группе
Анти-Ха
анти-IIа
анти-Ха
Можно заключить, что после подкожного введения соединения III данного изобретения (100 нмоль/кг) антитромботическая активность, определенная измерением (остаточной) анти-Ха или анти IIа активности, может быть нейтрализована внутривенным введением 10 мг/кг авидина. Нейтрализация соединения III данного изобретения авидином отражается в быстром снижении его концентрации в плазме и выраженным снижением AUCinf. соединения III данного изобретения после введения авидина по сравнению с соединением Org 42675. Кроме того, фармакокинетическое поведение небиотинилированного эквивалента соединения Org 42675 не изменяется добавлением авидина. Последнее подтверждает, что нейтрализация ассоциирована с присутствием биотинового остатка и это не изменяет фармакокинетическое поведение двойного ингибитора.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИТРОМБОТИЧЕСКИЕ ДВОЙНЫЕ ИНГИБИТОРЫ, ВКЛЮЧАЮЩИЕ БИОТИНОВУЮ МЕТКУ | 2006 |
|
RU2434876C2 |
| ПЕНТАСАХАРИДНЫЙ КОНЪЮГАТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2000 |
|
RU2266913C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОЗАЩИЩЕННЫХ α,ω-ДИАМИНОАЛКАНОВ | 2017 |
|
RU2747400C2 |
| КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ ПИРРОЛИДИНА ИЛИ ПИПЕРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ АГРЕГАЦИИ ТРОМБОЦИТОВ | 1997 |
|
RU2194038C2 |
| ВОДОРАСТВОРИМЫЕ АНАЛОГИ СС-1065 И ИХ КОНЪЮГАТЫ | 2007 |
|
RU2489423C2 |
| КОНТРАСТНЫЕ АГЕНТЫ | 2016 |
|
RU2743167C2 |
| УГЛЕВОДНЫЕ ЛИГАНДЫ, КОТОРЫЕ СВЯЗЫВАЮТСЯ С IgM АНТИТЕЛАМИ ПРОТИВ МИЕЛИН-АССОЦИИРОВАННОГО ГЛИКОПРОТЕИНА | 2015 |
|
RU2760005C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ИМИНОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2018 |
|
RU2801302C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ИНГИБИТОРЫ С-FMS КИНАЗЫ | 2007 |
|
RU2475483C2 |
Изобретение относится к антитромботическому соединению формулы (I) (олигосахарид-спейсер-А (I)), где олигосахарид является отрицательно заряженным пентасахаридным остатком формулы, приведенной ниже, где R5 - ОСН3 или OSO3 -, заряд компенсируется положительно заряженными противоионами, и где пентасахаридный остаток получен из пентасахарида, который имеет АТ-III опосредованную анти-Ха активность per se; спейсер - по существу фармакологически неактивный подвижно связанный остаток, имеющий цепь длиной от 10 до 50 атомов; А - остаток -CH[NH-SO2-R1][CO-NR2-CH(4-бензамидин)-CO-NR3R4], где R1 - 4- метокси-2,3,6-триметилфенил; R2 - Н; NR3R4 является пиперидинильной группой или его фармацевтически приемлемая соль или производное, где аминогруппа амидинового остатка защищена гидроксилом или (1-6С)алкоксикарбонильной группой; где спейсер соединения формулы I содержит одну ковалентную связь с остатком биотина формулы -(CH2)4-NR-ВТ, где R - Н или (1-4С)алкил и ВТ - остаток
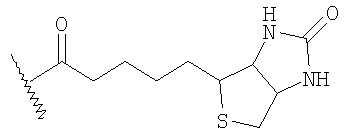
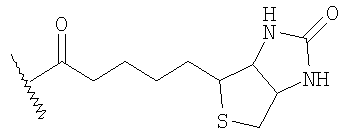
Изобретение относится также к фармацевтической композиции на основе соединений формулы I для лечения или предупреждения тромбоза или других, связанных с тромбином заболеваний. 2 н. и 4 з.п.ф-лы, 3 табл., 2 ил.
1. Антитромботическое соединение формулы (I)
олигосахарид-спейсер-А (I),
где олигосахарид является отрицательно заряженным пентасахаридным остатком формулы
где R5 - ОСН3 или OSO3-, заряд компенсируется положительно заряженными противоионами, и где пентасахаридный остаток получен из пентасахарида, который имеет АТ-III опосредованную анти-Ха активность per se;
спейсер - по существу, фармакологически неактивный подвижно связанный остаток, имеющий цепь длиной от 10 до 50 атомов;
А - остаток -CH[NH-SO2-R1][СО-NR2-CH(4-бензамидин)-СО-NR3R4], где R1 - 4-метокси-2,3,6-триметилфенил; R2 - Н; NR3R4 является пиперидинильной группой, или его фармацевтически приемлемая соль, или производное, где аминогруппа амидинового остатка защищена гидроксилом или (1-6С)алкоксикарбонильной группой;
где спейсер соединения формулы I содержит одну ковалентную связь с остатком биотина формулы -(CH2)4-NR-BT, где R - Н или (1-4С)алкил и ВТ - остаток
2. Соединение по п.1, где пентасахаридный остаток имеет структуру
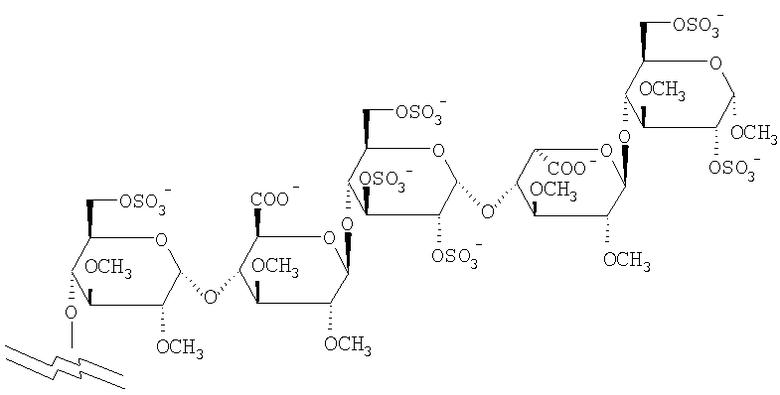
3. Соединение по п.2, которое представляет собой
или его фармацевтически приемлемая соль, или производное, где аминогруппа амидинового остатка защищена гидроксилом или (1-6С)алкоксикарбонильной группой.
4. Соединение по п.2, представляющее собой
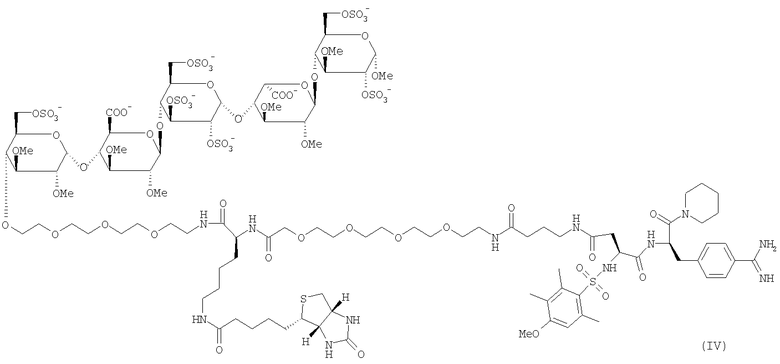
5. Соединение по пп.1-4 в форме его натриевой соли.
6. Фармацевтическая композиция для лечения или предупреждения тромбоза или других связанных с тромбином заболеваний, содержащая соединение по любому из пп.1-4 и фармацевтически приемлемые вспомогательные вещества.
| ПЕНТАСАХАРИДНЫЙ КОНЪЮГАТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2000 |
|
RU2266913C2 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Rogier С.Buijsman et al | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| АНТИТРОМБОТИЧЕСКИЕ СОЕДИНЕНИЯ | 1999 |
|
RU2225869C2 |

