Данное изобретение относится к новому фармацевтическому препарату с немедленным высвобождением, который предложен для доставки отдельных фармацевтических препаратов, к изготовлению такого препарата и к применению такого препарата в лечении или предупреждении тромбоза.
С целью обеспечения достаточной концентрации лекарственного средства в плазме в пределах временных рамок, необходимых для достижения требуемой реакции на терапию, часто требуется изготавливать фармацевтически активные соединения для немедленного высвобождения после перорального и/или парентерального введения в виде препаратов.
Немедленное высвобождение может быть особенно желательным в случаях, когда, например, необходима быстрая реакция на терапию (например, в лечении неотложных случаев), или в случае парентерального введения, когда пероральная доставка в желудочно-кишечный тракт не может обеспечить достаточного системного поглощения в пределах требуемых временных рамок.
В случае лечения или профилактики тромбоза препараты с немедленным высвобождением могут быть необходимы, чтобы гарантировать обеспечение достаточного количества лекарственного средства в плазме в пределах относительно короткого периода времени для того, чтобы сделать возможным быстрое начало действия. Препараты с немедленным высвобождением также обычно являются более простыми для разработки, чем препараты с модифицированным высвобождением, а также могут обеспечивать большую гибкость в отношении изменения доз, которые следует вводить пациентам. Препараты с немедленным высвобождением являются лучшими в случаях, когда не требуются множественные дозы и когда нет необходимости поддерживать уровень концентрации в плазме на постоянном уровне в течение продолжительного времени.
В международной заявке на патент № PCT/SE01/02657 (WO 02/44145, дата наиболее раннего приоритета 1 декабря 2000, дата подачи 30 ноября 2001, опубликованная 6 июня 2002) раскрыт ряд соединений, которые представляют собой соединения, или метаболизируются до соединений, являющихся конкурентными ингибиторами трипсиноподобных протеаз, таких как тромбин. Среди конкретно описанных соединений находятся следующие три соединения:
a) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe):
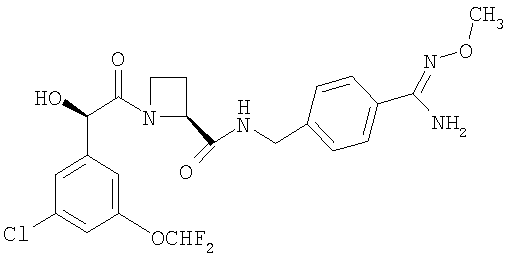
соединение, которое далее в настоящем описании изобретения упоминается как Соединение А;
б) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OMe):
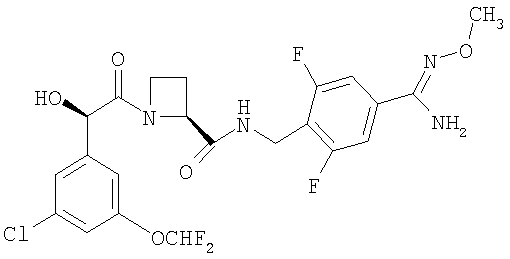
соединение, которое далее в настоящем описании изобретения упоминается как Соединение Б; и
в) Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe):

соединение, которое далее в настоящем описании изобретения упоминается как Соединение В.
Метоксиамидиновые соединения А, Б и В метаболизируются после перорального и/или парентерального введения до соответствующих свободных амидиновых соединений, причем последние соединения, как было установлено, представляют собой сильные ингибиторы тромбина. Таким образом:
- Соединение А метаболизируется до Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab (соединения, которое далее в настоящем описании изобретения упоминается как Соединение Г) через промежуточное пролекарство Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OH) (соединение, которое далее в настоящем описании изобретения упоминается как Соединение Ж);
- Соединение Б метаболизируется до Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF) (соединения, которое далее в настоящем описании изобретения упоминается как Соединение Д) через промежуточное пролекарство Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OH) (соединение, которое далее в настоящем описании изобретения упоминается как Соединение З); и
- Соединение В метаболизируется до Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab (соединения, которое далее в настоящем описании изобретения упоминается как Соединение Е) через промежуточное пролекарство Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OH) (соединение, которое далее в настоящем описании изобретения упоминается как Соединение К).
Способы синтеза Соединений А, Б, В, Г, Д, Е, Ж и К описаны в Примерах 12, 40, 22, 3, 39, 21, 2 и 31 (соответственно) международной заявки на патент № PCT/SE01/02657. Препараты с немедленным высвобождением этих соединений или их метаболитов, однако, должны быть описаны в литературе. Авторы изобретения обнаружили, что соединения формулы (I) и их соли могут быть изготовлены в виде фармацевтических препаратов с немедленным высвобождением, которые легко вводить, например, путем перорального или парентерального введения.
В первом аспекте изобретения предложен фармацевтический препарат с немедленным высвобождением, содержащий в качестве активного ингредиента соединение формулы (I):
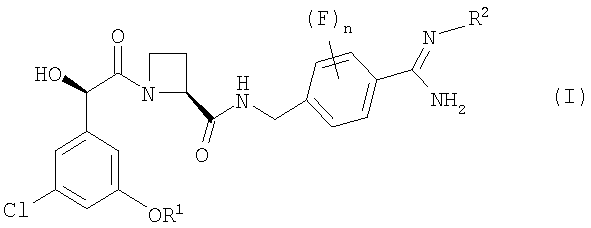
где
R1 представляет собой С1-2алкил, замещенный одним или более чем одним заместителем фторо;
R2 представляет собой водород, гидрокси, метокси или этокси; и
n равно 0, 1 или 2;
или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель;
при условии, что этот препарат не содержит исключительно:
- раствор одного активного ингредиента и воду;
- раствор одного активного ингредиента и диметилсульфоксид; или
- раствор одного активного ингредиента в смеси этанол:12-гидроксистеарат PEG 660:вода в соотношении 5:5:90;
такие препараты далее в настоящем описании изобретения упоминаются как "препараты по изобретению".
12-Гидроксистеарат PEG 660 представляет собой неионное поверхностно-активное вещество и более известен как Solutol K™.
Во втором аспекте настоящего изобретения предложено Соединение 3, Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OH), которое может быть получено способами, аналогичными описанным ниже для получения Соединений Ж и К.
Соединения формулы (I) или их фармацевтически приемлемые соли могут находиться в форме сольвата, гидрата, смешанного сольвата/гидрата или предпочтительно ансольвата, такого как ангидрат. Сольваты могут быть образованы одним или более органическими растворителями, такими как низшие (например, С1-4) алкиловые спирты (например, метанол, этанол или изопропанол), кетоны (такой как ацетон), сложные эфиры (такой как этилацетат) или их смеси.
В одном конкретном аспекте изобретения R1 представляет собой CHF2 или CH2CH2F.
Переменная n предпочтительно равна 0 или 2.
Более предпочтительные соединения формулы (I) включают соединения, в которых n равно 0, или соединения, в которых n равно 2, в случае когда два атома фтора расположены в положениях 2 и 6 (то есть в двух орто-положениях относительно места присоединения бензольного кольца к группе -NH-CH2-).
Соединение формулы (I) представляет собой главным образом Соединение А, Соединение Б или Соединение В.
Предпочтительные соли соединений формулы (I) представляют собой соли присоединения кислот. Соли присоединения кислот включают соли присоединения неорганических кислот, такие как соли серной кислоты, азотной кислоты, фосфорной кислоты и галогеноводородных кислот, таких как бромистоводородная кислота и соляная кислота. Более предпочтительные соли присоединения кислот включают соли органических кислот, такие как соли диметилфосфорной кислоты, сахариновой кислоты, циклогексилсульфаминовой кислоты; соли карбоновых кислот (таких как малеиновая кислота, фумаровая кислота, аспарагиновая кислота, янтарная кислота, малоновая кислота, уксусная кислота, бензойная кислота, терефталевая кислота, гиппуровая кислота, 1-гидрокси-2-нафтойная кислота, памовая кислота, гидроксибензойная кислота и подобные); соли оксикислот (таких как салициловая кислота, винная кислота, лимонная кислота, яблочная кислота (включая L-(-)-яблочную кислоту и D,L-яблочную кислоту), глюконовая кислота (включая D-глюконовую кислоту), гликолевая кислота, аскорбиновая кислота, молочная кислота и подобные); соли аминокислот (таких как глутаминовая кислота (включая D-глутаминовую, L-глутаминовую и D,L-глутаминовую кислоты), аргинин (включая L-аргинин), лизин (включая L-лизин и L-лизина гидрохлорид), глицин и подобные); и особенно соли сульфоновых кислот (таких как 1,2-этандисульфоновая кислота, камфорсульфоновые кислоты (включая 1S-(+)-10-камфорсульфоновую кислоту и (+/-)-камфорсульфоновые кислоты), этансульфоновая кислота, пропансульфоновая кислота (включая н-пропансульфоновую кислоту), бутансульфоновая кислота, пентансульфоновая кислота, толуолсульфоновая кислота, метансульфоновая кислота, пара-ксилолсульфоновая кислота, 2-мезитиленсульфоновая кислота, нафталинсульфоновые кислоты (включая 1,5-нафталинсульфоновую кислоту и нафталинсульфоновую кислоту), бензолсульфоновая кислота, гидроксибензолсульфоновые кислоты, 2-гидроксиэтансульфоновая кислота, 3-гидроксиэтансульфоновая кислота и подобные).
Особенно предпочтительные соли включают соли С1-0(например, С1-4)-алкансульфоновых кислот, таких как этансульфоновая кислота (эзилат) и пропансульфоновая кислота (например, н-пропансульфоновая кислота), и, возможно, замещенных (например, одной или более С1-2алкильными группами) арилсульфоновых кислот, таких как бензолсульфоновая кислота (безилат) и нафталиндисульфоновая кислота.
Подходящие стехиометрические соотношения кислоты и свободного основания находятся в пределах от 0,25:1,5 до 3,0:1, например от 0,45:1,25 до 1,25:1, включая от 0,50:1 до 1:1.
В еще одном аспекте изобретения предложен препарат, содержащий соединение формулы (I) в по существу кристаллической форме.
Хотя авторы обнаружили, что возможно получение соединений по изобретению в формах, которые являются кристаллическими более чем на 80%, термин «по существу кристаллическая» охватывает более чем на 20%, предпочтительно более чем на 30% и более предпочтительно более чем на 40% (например, любую из более чем на 50, 60, 70, 80 или 90%) кристаллическую форму.
В еще одном аспекте изобретения также предложено соединение по изобретению в частично кристаллической форме. Термин «частично кристаллическая» охватывает кристаллическую на 5% или между 5% и 20% форму.
Степень (%) кристалличности может быть определена специалистом в данной области техники с применением дифракции рентгеновских лучей на порошке (XRPD). Также могут быть использованы другие методики, такие как NMR в твердом состоянии, FT-IR, спектроскопия комбинационного рассеяния, дифференциальная сканирующая калориметрия (DSC) и микрокалориметрия.
Предпочтительные соединения формулы (I), которые могут быть получены в кристаллической форме, включают соли С1-6(например, С2-6, таких как С2-4)-алкансульфоновых кислот, таких как этансульфоновая кислота, пропансульфоновая кислота (например, н-пропансульфоновая кислота), и, возможно, замещенных арилсульфоновых кислот, таких как бензолсульфоновая кислота и нафталиндисульфоновая кислота.
Термин фармацевтический препарат "с немедленным высвобождением" включает в себя любой препарат, а котором скорость высвобождения лекарственного средства из препарата и/или абсорбции лекарственного средства ни значительно, ни преднамеренно не замедлена галеновыми манипуляциями. В данном случае немедленное высвобождение может быть обеспечено посредством подходящего фармацевтически приемлемого разбавителя или носителя, который не увеличивает в значительной степени скорость высвобождения и/или абсорбции лекарственного средства. Таким образом, термин исключает препараты, которые приспособлены обеспечивать "модифицированное", "контролируемое", "замедленное", "пролонгированное", "длительное" или "задержанное" высвобождения лекарственного средства.
В данном контексте термин "высвобождение" включает обеспечение (или подачу) лекарственного средства из препарата в желудочно-кишечный тракт, к тканям тела и/или в большой круг кровообращения. Для высвобождения в желудочно-кишечном тракте высвобождение происходит при определенных значениях рН, таких как рН 1-3, особенно при или приблизительно при рН 1. В одном аспекте изобретения из препарата, как он определен здесь, с соединением формулы (I) или с его солью присоединения кислоты в кристаллической форме лекарственное средство высвобождается в интервале значений рН. В другом аспекте изобретения из препарата, как он определен здесь, с соединением формулы (I) или с его солью присоединения кислоты лекарственное средство высвобождается при значениях рН, таких как рН 1-3, особенно при или приблизительно при рН 1. Таким образом, из препаратов по изобретению может высвобождаться по меньшей мере 70% (предпочтительно 80%) активного ингредиента в пределах 4 часов, например в пределах 3 часов, предпочтительно 2 часов, более предпочтительно в пределах 1,5 часов и особенно в пределах одного часа (например, в пределах 30 минут) после введения, независимо от того пероральное оно или парентеральное.
Препараты по изобретению могут быть изготовлены в соответствии с рядом известных методик, например, как описано M.E.Aulton в "Pharmaceutics: The Science of Dosage Form Design" (1988) (Churchill Livingstone), релевантные сообщения из этого документа включены в данное описание изобретения ссылкой.
Препараты по изобретению могут быть подходящими или могут быть приспособлены в соответствии со стандартными методиками, чтобы стать подходящими, для перорального введения, например в форме таблетки с немедленным высвобождением, капсулы с немедленным высвобождением или жидкой лекарственной формы, содержащей активный ингредиент. Такие виды препаратов хорошо известны специалисту в данной области техники и могут быть изготовлены в соответствии с методиками, известными в данной области техники.
Подходящие разбавители/носители (которые также могут именоваться "наполнители") для применения в пероральных препаратах по изобретению, например находящихся в форме таблеток с немедленным высвобождением, включают одноосновный фосфат кальция, двухосновный фосфат кальция (включая дигидрат двухосновного фосфата кальция и ангидрат двухосновного фосфата кальция), трехосновный фосфат кальция, лактозу, микрокристаллическую целлюлозу, силикатированную микрокристаллическую целлюлозу, маннит, сорбит, крахмал (такой как кукурузный, картофельный или рисовый), глюкозу, лактат кальция, карбонат кальция и подобные. Предпочтительные разбавители/носители включают двухосновный фосфат кальция и микрокристаллическую целлюлозу, которые можно использовать сами по себе или в сочетании с другим разбавителем/носителем, таким как маннит.
Препарат по изобретению в форме таблетки с немедленным высвобождением может включать в себя один или более эксципиентов для улучшения физических и/или химических свойств конечной композиции и/или для облегчения способа изготовления. Такие эксципиенты являются традиционными в изготовлении препаратов с немедленным высвобождением для пероральной доставки лекарственных средств и включают один или более эксципиент из следующих: одно или более смазывающее вещество (такое как стеарат магния, стеариновая кислота, стеарат кальция, стеариловый спирт или предпочтительно стеарилфумарат натрия); скользящий агент (такой как тальк или коллоидный диоксид кремния); одно или более связующее вещество (такое как поливинилпирролидон, микрокристаллическая целлюлоза, полиэтиленгликоль (PEG), полиэтиленоксид, низкомолекулярная гидроксипропилметилцеллюлоза (НРМС), низкомолекулярная метилцеллюлоза (МС), низкомолекулярная гидроксипропилцеллюлоза (НРС), низкомолекулярная гидроксиэтилцеллюлоза (НЕС), крахмал (такой как кукурузный, картофельный или рисовый) или низкомолекулярная карбоксиметилцеллюлоза натрия; (предпочтительными связывающими веществами являются поливинилпирролидон или низкомолекулярная НРМС); один или более рН-регулирующий агент (такой как органическая кислота (например, лимонная кислота) или ее соль щелочного металла (например, натрия), оксид магния, сульфат, метабисульфат, пропионат или сорбат щелочного или щелочноземельного металла (например, натрия, кальция или калия)); один или более разрыхлитель (например, натрия крахмала гликолят, сшитый поливинилпирролидон, сшитая карбоксиметилцеллюлоза натрия, крахмал (такой как кукурузный, картофельный или рисовый) или альгинат); краситель, ароматизатор, агент, изменяющий тоничность, покрывающий агент или консервант.
Очевидно, что некоторые из упомянутых выше эксципиентов, которые могут присутствовать в конечном пероральном (например, таблеточном) препарате по изобретению с немедленным высвобождением, могут иметь более чем одну из указанных выше функций.
В еще одном аспекте изобретения жидкий препарат по изобретению приспособлен для перорального введения.
Подходящие жидкие препараты, которые предназначены для перорального введения, включают препараты, в которых соединение формулы (I), особенно Соединение А, Соединение Б или Соединение В или их фармацевтически приемлемая соль, присутствует вместе с водным носителем, таким как вода. Однако следует упомянуть, что некоторые конкретные препараты не заявлены (смотри отдельные аспекты и формулу изобретения).
Препарат по настоящему изобретению, содержащий водный носитель, может, кроме того, содержать один или более эксципиент, такой как антибактериальный консервант; модификатор тоничности (например, хлорид натрия, маннит или глюкоза); регулирующее рН вещество (например, обычная неорганическая кислота или основание, включая соляную кислоту или гидроксид натрия); контролирующие рН вещества (то есть буфер, например винная кислота, уксусная кислота или лимонная кислота); поверхностно-активное вещество (например, додецилсульфат натрия (SDS) или Solutol™); солюбилизатор, который служит для облегчения растворения активного ингредиента (например, этанол, полиэтиленгликоль или гидроксипропил-β-циклодекстрин или поливинилхлорид (PVP)); или антиоксидант.
Жидкие пероральные препараты могут находиться в форме суспензий активного ингредиента в сочетании с водным растворителем или более предпочтительно в форме водных растворов (то есть растворов активного соединения, включая воду в качестве растворителя). В этом контексте термин "водный раствор" включает в себя препараты, в которых по меньшей мере 99% активного ингредиента находится в растворе приблизительно при 5°С и атмосферном давлении, а термин "суспензия" означает, что более чем 1% активного ингредиента находится не в растворе при таких условиях. Типичными диспергирующими веществами для суспензий являются гидроксипропилметилцеллюлоза, АОТ (диоктилсульфосукцинат), PVP и SDS. Могут быть возможны другие варианты.
В другом аспекте настоящего изобретения предложен жидкий пероральный препарат, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, воду и по меньшей мере один дополнительный агент. Дополнительные агенты включают:
1) полиэтиленгликоль (PEG) и, возможно, также этанол, и/или винную кислоту, и/или лимонную кислоту, и/или соляную кислоту; или
2) хлорид натрия (который будет растворен в препарате) и, возможно, также этанол; или
3) соляную кислоту и/или гидроксид натрия для доведения рН до подходящего значения (предпочтительно в интервале 3-8 для соединения формулы (I), где R2 представляет собой метокси или этокси, такого как Соединение А, Б или В); или
4) DMA (диметилацетамид) и, возможно, также триглицерид со средней длиной цепи (такой как миглиол); или
5) β-циклодекстрин (такой как гидроксипропил-β-циклодекстрин);
6) модификатор тоничности, такой как хлорид натрия и/или маннит.
В еще одном аспекте настоящего изобретения предложен пероральный раствор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, (предпочтительно Соединение А, Б или В), воду и по меньшей мере один дополнительный агент, как изложено выше в пунктах (1)-(6).
В другом аспекте изобретения предложен водный препарат соединения формулы (I) (такого как Соединение А, Б или В), содержащий солюбилизирующий агент, такой как полиэтиленгликоль, β-циклодекстрин (такой как гидроксипропил-β-циклодекстрин), сорбит или этанол.
В еще одном аспекте настоящего изобретения предложен препарат в виде перорального раствора, содержащий соединение формулы (I) и этанол. Этот препарат может, кроме того, содержать триглицерид со средней длиной цепи (такой как миглиол).
В еще одном аспекте настоящего изобретения предложен препарат в виде перорального раствора, содержащий соединение формулы (I) и DMA. Этот препарат может, кроме того, содержать триглицерид со средней длиной цепи (такой как миглиол).
В другом аспекте соединение формулы (I) является кристаллическим (особенно соль Соединения А, предпочтительно соль С1-6(например, С2-6, такой как С2-4)-алкансульфоновой кислоты, такой как этансульфоновая кислота, пропансульфоновая кислота (например, Н-пропансульфоновая кислота), или соль, возможно, замещенной арилсульфоновой кислоты, такой как соль бензолсульфоновой кислоты или нафталиндисульфоновой кислоты).
Предложен конкретный жидкий пероральный фармацевтический препарат с немедленным высвобождением по п.1, где активный ингредиент представляет собой:
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe),
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OMe),
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe),
или его фармацевтически приемлемая соль.
Кроме того, предложен конкретный жидкий пероральный фармацевтический препарат с немедленным высвобождением по п.1, где активный ингредиент представляет собой: Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe) или его соль С1-6алкансульфоновой кислоты или, возможно, замещенной арилсульфоновой кислоты.
Предложен еще один конкретный жидкий пероральный фармацевтический препарат с немедленным высвобождением по п.1, где активный ингредиент представляет собой: Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(ОМе) или его соль, возможно, замещенной арилсульфоновой кислоты (например, соль нафталин-1,5-дисульфоновой кислоты).
Однако следует отметить, что некоторые конкретные препараты не заявлены (смотри отдельные аспекты и формулу изобретения).
В еще одном аспекте изобретения препарат по изобретению приспособлен для парентерального введения. Термин "парентеральное" включает в себя любой способ введения, который не включает пероральное введение в желудочно-кишечный тракт и включает в себя введение подкожное, внутривенное, внутриартериальное, чрескожное, интраназальное, буккальное, внутрикожное, внутримышечное, внутрь жировой ткани, внутрибрюшинное, ректальное, сублингвальное, местное, ингаляционное или любым другим парентеральным путем.
Подходящие препараты по изобретению, которые следует вводить парентерально, включают препараты, в которых соединение формулы (I) или его фармацевтически приемлемая соль находится вместе с водным носителем, таким как вода.
Препарат по настоящему изобретению, содержащий водный носитель, может, кроме того, содержать один или более эксципиент, такой как противомикробный консервант; регулятор тоничности (например, хлорид натрия, маннит или глюкозу); рН-регулирующий агент (например, обычную неорганическую кислоту или основание, включая соляную кислоту или гидроксид натрия); рН-контролирующие агенты (то есть буфер; например, винную кислоту, уксусную кислоту или лимонную кислоту); поверхностно-активное вещество (например, додецилсульфат натрия (SDS) или Solutol™); солюбилизатор, который служит для облегчения растворения активного ингредиента (например, этанол, полиэтиленгликоль или гидроксипропил-β-циклодекстрин или поливинилхлорид (PVP)); или антиоксидант.
Парентеральные препараты могут находиться в форме суспензий активного ингредиента в сочетании с водным растворителем, или более предпочтительно водных растворов (то есть растворов активного соединения, включающих в качестве растворителя воду). В данном контексте термин "водный раствор" включает препараты, в которых по меньшей мере 99% активного ингредиента находится в растворе при температуре выше 5°С и атмосферном давлении, а термин "суспензия" означает, что более 1% активного ингредиента при таких условиях находится не в растворе. Типичными диспергирующими агентами для суспензий являются гидроксипропилметилцеллюлоза, АОТ (диоктилсульфосукцинат), PVP и SDS, но возможны и другие варианты.
Количество эксципиентов, используемых в пероральных и парентеральных препаратах по изобретению, зависит от многих факторов, таких как природа и количество присутствующего активного ингредиента и количество включенного разбавителя/носителя (водного растворителя или иного).
В еще одном аспекте настоящего изобретения предложен парентеральный препарат, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, воду и по меньшей мере один дополнительный агент. Дополнительные агенты включают:
1) полиэтиленгликоль (PEG) и, возможно, также этанол, и/или винную кислоту, и/или соляную кислоту; или
2) хлорид натрия (который будет растворен в препарате) и, возможно, также этанол; или
3) соляную кислоту и/или гидроксид натрия для доведения рН до подходящего значения (предпочтительно в интервале 3-8 для соединения формулы (I), где R2 представляет собой водород, такого как Соединение Г, Д или Е, или предпочтительно в интервале 3,5-8 для соединения формулы (I), где R2 представляет собой метокси или этокси, такого как Соединение А, Б или В); или
4) DMA (диметилацетамид) и, возможно, также триглицерид со средней длиной цепи (такой как миглиол); или
5) β-циклодекстрин (такой как гидроксипропил-β-циклодекстрин);
6) модификатор тоничности, такой как хлорид натрия и/или маннит.
В еще одном аспекте настоящего изобретения предложен инъекционный раствор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль (предпочтительно Соединение Г, Д или Е), воду и по меньшей мере один дополнительный агент, как изложено выше в пунктах (1)-(4).
В другом аспекте изобретения предложен водный препарат соединения формулы (I) (такого как Соединение Г, Д или Е), содержащий солюбилизирующий агент, такой как полиэтиленгликоль, β-циклодекстрин (такой как гидроксипропил-β-циклодекстрин), сорбит или этанол.
В еще одном аспекте настоящего изобретения предложен парентеральный препарат, содержащий соединение формулы (I) и этанол. Этот препарат может, кроме того, содержать триглицерид со средней длиной цепи (такой как миглиол).
В еще одном аспекте настоящего изобретения предложен парентеральный препарат, содержащий соединение формулы (I) и DMA. Этот препарат может, кроме того, содержать триглицерид со средней длиной цепи (такой как миглиол).
В другом аспекте соединение формулы (I) является кристаллическим (особенно соль Соединения А; предпочтительно соль С1-6(например, С2-6, такой как С2-4)-алкансульфоновой кислоты, такой как этансульфоновая кислота, пропансульфоновая кислота (например, н-пропансульфоновая кислота), или соль, возможно, замещенной арилсульфоновой кислоты, такой как соль бензолсульфоновой кислоты).
Еще в одном аспекте препарат по настоящему изобретению находится в твердой лекарственной форме, где R2 представляет собой гидрокси, метокси или этокси (предпочтительно метокси) (соединение формулы (I) представляет собой, в частности, Соединение А, Соединение Б или Соединение В).
В еще одном аспекте настоящего изобретения предложен парентеральный препарат (особенно инъекционный раствор на водной основе), содержащий соединение формулы (I) в форме свободного основания.
В еще одним аспекте настоящего изобретения предложен парентеральный препарат, содержащий соединение формулы (I) в форме свободного основания, где R2 представляет собой водород.
В еще одном аспекте настоящего изобретения предложен твердый препарат, содержащий микрокристаллическую целлюлозу и поливинилпирролидон (PVP); или содержащий микрокристаллическую целлюлозу и натрия крахмала гликолят.
Препараты по изобретению, такие как парентеральные препараты, содержащие соли, могут быть изготовлены посредством добавления разбавителя/носителя к подходящей предварительно приготовленной соли.
Композиции, включающие активный ингредиент, также могут быть получены в твердой форме, подходящей для применения в изготовлении препарата по изобретению (например, раствора, такого как водный раствор, например, для парентерального введения) в нужный момент. Такие композиции могут находиться в форме твердого вещества, содержащего активный ингредиент, возможно, в присутствии одного или более дополнительного эксципиента, как определено выше, и, возможно, до 10% включительно (мас./мас.) разбавителя и/или носителя, как определено здесь выше, эти композиции упоминаются далее как «твердые композиции по изобретению».
Твердые композиции по изобретению могут быть изготовлены путем удаления разбавителя/носителя (например, растворителя) из препарата по изобретению или концентрированного препарата по изобретению, который может находиться, например, в форме раствора, такого как водный раствор.
В другом аспекте настоящего изобретения предложен перорально вводимый препарат с немедленным высвобождением, содержащий соединение формулы (I) или его соль, носитель (такой как микрокристаллическая целлюлоза), разрыхлитель (такой как натрия крахмала гликолят), связующее вещество (такое как поливинилпирролидон) или смазывающее вещество (такое как стеарилфумарат натрия). Такой препарат может также содержать дополнительный носитель (или наполнитель), такой как маннит.
Препараты по изобретению, которые находятся в форме таблеток с немедленным высвобождением, могут быть приготовлены путем приведения в контакт активного ингредиента с разбавителем/носителем с использованием стандартных методик, и используя стандартное оборудование, известное специалисту в данной области техники, включая влажное и сухое гранулирование, прямое сжатие/прессование, сушку, измельчение, смешивание, таблетирование и нанесение покрытия, а также сочетания этих процессов, например, как описано далее. В одном аспекте изобретения соли присоединения кислот соединений формулы (I) в кристаллической форме изготавливают в виде таблеток.
Таким образом, предложен способ образования твердой композиции, подходящей для применения в изготовлении препарата по изобретению (например, раствора, такого как водный раствор) в нужный момент, который включает удаление разбавителя/носителя (например, растворителя) из препарата по изобретению или концентрированного препарата по изобретению.
Растворитель можно удалять рядом методов, известных специалистам в данной области техники, например, путем выпаривания (при пониженном давлении или иначе), сушки вымораживанием или любым способом удаления растворителя (сушки), посредством которого удаляют растворитель (такой как воду), и при этом сохраняют целостность активного ингредиента. Примером сушки служит сушка вымораживанием.
Таким образом, в соответствии с еще одним аспектом изобретения предложена высушенная вымораживанием (лиофилизированная) твердая композиция по изобретению.
Специалисту в данной области техники очевидно, что при изготовлении твердых композиций по изобретению подходящие дополнительные эксципиенты могут быть добавлены на подходящей стадии перед удалением разбавителя/носителя. Например, в случае водных растворов можно контролировать и/или регулировать рН, как описано выше. Кроме того, может быть добавлен подходящий дополнительный эксципиент, чтобы способствовать образованию твердой композиции по изобретению в процессе удаления разбавителя/носителя (например, маннит, сахароза, глюкоза, манноза или трегалоза).
Таким образом, твердая композиция соединения формулы (I) или его соли включает композицию, в которой содержание растворителя (например, воды), иного чем растворитель для кристаллизации, составляет не более 10%, например, менее 2% от несвязанного растворителя, такого как воде.
Препараты по изобретению можно стерилизовать, например, путем стерильной фильтрации или автоклавирования, и/или помещать в первичные упаковки, такие как ампулы, картриджы и предварительно наполняемые шприцы. Такие технологические стадии могут также иметь место перед сушкой с образованием твердой композиции по изобретению.
Перед введением высушенную твердую композицию можно перерастворить и/или развести, например, в воде, в физиологическом растворе, растворе глюкозы или любом другом подходящем растворе.
Количество разбавителя/носителя в пероральном (например, в таблетке с немедленным высвобождением) препарате по изобретению зависит от многих факторов, таких как природа и количество используемого активного ингредиента и природа и количества любых других составляющих (например, дополнительных эксципиентов), которые присутствуют в препарате, но обычно составляет до 40% (мас./мас.) включительно, предпочтительно до 30% включительно, более предпочтительно до 20% включительно и, в частности, до 10% (мас./мас.) включительно от массы конечной композиции. Количество дополнительных эксципиентов в таком пероральном препарате по изобретению также зависит от таких факторов, как природа и количество используемого активного ингредиента, а также природа и количество любых других составляющих (например, разбавителей/носителей и/или других дополнительных эксципиентов), которые присутствуют в препарате, но для смазывающих веществ и скользящих агентов обычно составляет до 5% (мас./мас.) включительно, а для связующих агентов и разрыхлителей обычно составляет до 10% (мас./мас.) включительно от массы конечной композиции.
Препараты по изобретению вводят пациентам-млекопитающим (включая людей), и соединения формулы (I), где R2 не является водородом, после этого метаболизируются в организме с образованием соединений формулы (I), где R2 является водородом, являющихся фармакологически активными.
Таким образом, в соответствии с еще одним аспектом изобретения предложен препарат по изобретению для применения в качестве фармацевтического средства.
В частности, соединения формулы (I) представляют собой сильные ингибиторы тромбина или метаболизируются после введения с образованием таких ингибиторов, например, как может быть продемонстрировано в тестах, описанных, в числе прочего, в международной заявке на патент № PCT/SE01/02657, а также в международных заявках на патент WO 02/14270, WO 01/87879 и WO 00/42059, релевантные описания изобретений которых включены в данное описание изобретения ссылкой.
Под термином «пролекарство ингибитора тромбина» авторы понимают соединения, которые метаболизируются после введения и образуют ингибитор тромбина в экспериментально обнаруживаемом количестве после введения.
Под термином "активный ингредиент" и "активное вещество" авторы понимают фармацевтический агент (включая ингибитор тромбина и его пролекарство), присутствующий в препарате.
Таким образом, ожидается, что препараты по изобретению будут полезны при таких состояниях, при которых требуется ингибирование тромбина, и/или при состояниях, при которых показана антикоагулянтная терапия, включая следующие.
Лечение и/или профилактика тромбоза и гиперкоагуляции в крови и/или тканях животных, включая человека. Известно, что гиперкоагуляция может приводить к тромбоэмболическим заболеваниям. Состояния, связанные с гиперкоагуляцией и тромбоэмболическими заболеваниями, которые можно упомянуть, включают наследственную или приобретенную резистентность к активированному протеину С, такую как мутация фактора V (фактора V Лейдена), и наследственные или приобретенные дефициты антитромбина III, протеина С, протеина S, кофактора гепарина II. Другие состояния, связь которых с гиперкоагуляцией и тромбоэмболическими заболеваниями известна, включают циркуляцию антифосфолипидных антител (волчаночный антикоагулянт), гомоцистеинемию, тромбоцитопению, индуцированную гепарином, и нарушения в фибринолизе, а также синдромы коагуляции (например, диссеминированную внутрисосудистую коагуляцию (DIC)) и сосудистые повреждения вообще (например, из-за хирургической операции).
Лечение состояний, при которых существует нежелательный избыток тромбина без признаков гиперкоагуляции, например, при нейродегенеративных заболеваниях, таких как болезнь Альцгеймера.
Конкретные болезненные состояния, которые могут быть упомянуты, включают терапевтическое и/или профилактическое лечение венозного тромбоза (например, DVT (тромбоза глубоких вен)) и эмболии легких, артериального тромбоза (например, при инфаркте миокарда, нестабильной стенокардии, инсульте в результате тромбоза и тромбоза периферических артерий) и системной эмболии, обычно от предсердия во время фибрилляции предсердий (например, неклапанного мерцания предсердий) или от левого желудочка после трансмурального инфаркта миокарда, или вызванной застойной сердечной недостаточностью; профилактику реокклюзии (то есть тромбоза) после тромболиза, чрескожной транслюминальной ангиопластики (РТА) и операции коронарного шунтирования; предупреждение повторного тромбоза после микрохирургии и сосудистой хирургии вообще.
Дополнительные показания включают терапевтическое и/или профилактическое лечение диссеминированной внутрисосудистой коагуляции, вызванной бактериями, множественной травмой, интоксикацией или любым другим механизмом; антикоагулянтное лечение, при котором кровь находится в контакте с инородными поверхностями в организме, такими как сосудистые трансплантаты, сосудистые стенты, сосудистые катетеры, механические и биологические искусственные клапаны или любое другое медицинское устройство; и антикоагулянтное лечение, при котором кровь находится в контакте с медицинскими устройствами вне тела, такими как использование аппарата искусственного кровообращения во время кардиососудистой хирургии или при гемодиализе; терапевтическое и/или профилактическое лечение идиопатического и респираторного дистресс-синдрома взрослых, фиброза легких, после радиологического лечения или химиотерапии, септического шока, сепсиса, воспалительных ответов, которые включают отек, острый или хронический атеросклероз, такой как заболевание коронарных артерий и образование атеросклеротических бляшек, заболевание артерий головного мозга, церебральный инфаркт, тромбоз сосудов головного мозга, эмболию сосудов головного мозга, заболевание периферических артерий, ишемию, стенокардию (включая нестабильную стенокардию), повреждение при реперфузии, рестеноз после чрескожной транслюминальной ангиопластики (РТА) и операции коронарного шунтирования, но не ограничиваются ими.
Препарат по настоящему изобретению может также содержать любой(ые) антитромботический(е) агент(ы) с иным механизмом действия, чем механизм соединений формулы (I), такой(ие) как один или более из следующих: антитромбоцитные агенты - ацетилсалициловая кислота, тиклопидин и клопидогрел; ингибиторы рецепторов тромбоксана и/или синтетазы; антагонисты рецепторов фибриногена; простациклин-миметики; ингибиторы фосфодиэстеразы; антагонисты ADP(аденозиндифосфат)-рецепторов (Р2Т); и ингибиторы карбоксипептидазы U (CPU).
Соединения формулы (I), которые ингибируют трипсин и/или тромбин, также могут быть полезны в лечении панкреатита.
Таким образом, соединения по изобретению показаны как в терапевтическом, так и/или в профилактическом лечении этих состояний.
Согласно еще одному аспекту настоящего изобретения предложен способ лечения состояния, при котором требуется ингибирование тромбина, включающий введение терапевтически эффективного количества соединения по изобретению субъекту, страдающему от такого состояния или подверженного ему.
Согласно еще одному аспекту настоящего изобретения предложен препарат по изобретению для изготовления лекарства для применения в лечении тромбоза,
Согласно еще одному аспекту изобретения предложен способ лечения тромбоза, включающий введение препарата по изобретению субъекту, страдающему от такого состояния или подверженного ему.
Во избежание неопределенности под термином "лечение" авторы понимают терапевтическое лечение, а также профилактику состояния.
Подходящие количества активного ингредиента в препаратах (пероральных или парентеральных), концентрированных препаратах и твердых композициях по изобретению зависят от многих факторов, таких как природа этого ингредиента (свободное основание/соль и так далее), доза, которая требуется в пероральном препарате или в конечном "готовом к употреблению" парентеральном препарате, который готовят или который должен быть изготовлен, и природа и количество других компонентов препарата. Однако типичная суточная доза соединения формулы (I) или его фармацевтически приемлемой соли находится в интервале 0,001-100 мг/кг массы тела при пероральном введении и 0,001-50 мг/кг массы тела при парентеральном введении, исключая массу любой кислоты-противоиона, независимо от числа отдельных доз, которые вводят в течение этих суток. В случае парентерального препарата с немедленным высвобождением введение может быть непрерывным (например, путем вливания). Предпочтительная суточная пероральная доза составляет 20-500 мг, и предпочтительная парентеральная доза находится в интервале 0,1-50 мг.
Общие методы
ТСХ проводили на силикагеле. Анализ методом хиральной HPLC проводили, используя колонку Chiralcel OD 46 мм × 250 мм с защитной колонкой 5 см. Температуру колонки поддерживали на уровне 35°С. Использовали скорость протока 1,0 мл/мин. Использовали Gilson 115 UV детектор при 228 нм. Подвижная фаза состояла из гексанов, этанола и трифторуксусной кислоты, и для каждого соединения приведены подходящие соотношения. Обычно продукт растворяли в минимальном количестве этанола и затем разбавляли подвижной фазой.
В Подготовительных примерах А-И LC-MS/MS (жидкостную хроматографию-тандемную масс-спектрометрию) проводили, используя прибор HP-1100, оснащенного инжектором CTC-PAL и 5 Tm, 4×100 мм ThermoQuest, Hypersil BDS-C18 колонкой. Использовали API-3000 (Sciex) MS детектор. Скорость протока составляла 1,2 мл/мин, а подвижная фаза (градиент) состояла из 10-90% ацетонитрила с 90-10% 4 мМ водного ацетата аммония, при этом оба растворителя содержали 0,2% муравьиной кислоты. Или записывали масс-спектры низкого разрешения (LRMS), используя Micromass ZQ спектрометр в режиме переключения положительных/отрицательных ионов с ESI (ионизацией в электроспрее) (диапазон масс m/z 100-800); а масс-спектры высокого разрешения (HRMS) записывали, используя спектрометр Micromass LCT в режиме отрицательной ионизации в ES (диапазон масс m/z 100-1000) с лейцин-энкефалином (C28H37N5O7) в качестве внутреннего стандарта массы.
Спектры 1H ЯМР записывали, используя в качестве внутреннего стандарта тетраметилсилан.
Способы синтеза соединений формулы (I) содержатся в международной заявке на патент № PCT/SE01/02657 (WO 02/44145, дата наиболее раннего приоритета 01 декабря 2000, дата подачи 30 ноября 2001, дата опубликования 06 июня 2002), релевантная информация из которой включена в данное описание изобретения.
Подготовительный пример А: Получение Соединения А
1) 3-Хлор-5-метоксибензальдегид
3,5-Дихлоранизол (74,0 г; 419 ммоль) в THF (200 мл) добавляли по каплям к металлическому магнию (14,2 г; 585 ммоль; предварительно промытому 0,5 н. HCl) в THF (100 мл) при 25°С. После добавления по каплям добавляли 1,2-дибромэтан (3,9 г; 20,8 ммоль). Полученную темно-коричневую смесь нагревали с обратным холодильником в течение 3 часов. Смесь охлаждали до 0°С и одной порцией добавляли N,N-диметилформамид (60 мл). Эту смесь распределяли между диэтиловым эфиром (3×400 мл) и 6 н. HCl (500 мл). Объединенные органические экстракты промывали рассолом (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением масла. После флэш-хроматографии (2×) на силикагеле с элюированием смесью Нех:EtOAc (4:1) получали указанное в подзаголовке соединение (38,9 г; 54%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3): δ 9.90 (s, 1H), 7.53 (s, 1H), 7.38 (s, 1H), 7.15 (s, 1H), 3.87 (s, 3H).
2) 3-Хлор-5-гидроксибензальдегид
Раствор 3-хлор-5-метоксибензальдегида (22,8 г; 134 ммоль; смотри стадию (1) выше) в CH2Cl2 (250 мл) охлаждали до 0°С. Трибромид бора (15,8 мл; 167 ммоль) добавляли по каплям в течение 15 минут. После перемешивания реакционной смеси в течение 2 часов медленно добавляли Н2O (50 мл). Раствор затем экстрагировали Et2O (2×100 мл). Органические слои объединяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. После флэш-хроматографии на силикагеле с элюированием смесью Нех:EtOAc (4:1) получали указанное в подзаголовке соединение (5,2 г; 25%).
1H ЯМР (300 МГц, CDCl2): δ 9.85 (s, 1H), 7.35 (s, 1Н), 7.20 (s, 1H), 7.10 (s, 1H), 3.68 (s, 1H).
3) 3-Хлор-5-дифторметоксибензальдегид
Раствор 3-хлор-5-гидроксибензальдегида (7,5 г; 48 ммоль; смотри стадию (2) выше) в 2-пропаноле (250 мл) и 30%-ном КОН (100 мл) нагревали до кипения с обратным холодильником. При перемешивании в реакционную смесь в течение 2 часов барботировали CHClF2. Реакционную смесь охлаждали, подкисляли 1 н. HCl и экстрагировали EtOAc (2×100 мл). Органические слои промывали рассолом (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. В результате флэш-хроматографии на силикагеле с элюированием смесью Hex: EtOAc (4:1) получали указанное в подзаголовке соединение (4,6 г; 46%).
1H ЯМР (300 МГц, CDCI3): δ 9.95 (s, 1H), 7.72 (s, 1H), 7.52 (s, 1H), 7.40 (s, 1H), 6.60 (t, JH-F=71,1 Гц, 1Н).
4) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OTMS)CN
Раствор 3-хлор-5-дифторметоксибензальдегида (4,6 г; 22,3 ммоль; смотри стадию (3) выше) в CH2Cl2 (200 мл) охлаждали до 0°С. Добавляли Znl2 (1,8 г; 5,6 ммоль) и триметилсилилцианид (2,8 г; 27,9 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 15 часов. Смесь частично концентрировали в вакууме с получением указанного в подзаголовке соединения в виде жидкости, которую непосредственно использовали на нижеприведенной стадии (5) без дополнительной очистки или характеристики.
5)Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt
Ph(3-Cl)(5-OCHF2)-(R,S)CH(OTMS)CN (6,82 г; предположительно 22,3 ммоль; смотри стадию (4) выше) добавляли по каплям к HCl/EtOH (500 мл). Реакционную смесь перемешивали в течение 15 часов, затем частично концентрировали в вакууме с получением указанного в подзаголовке соединения в виде жидкости, которую использовали на стадии (6) без дополнительной очистки или характеристики.
6)Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OEt
Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt (6,24 г; предположительно 22,3 ммоль; смотри стадию (5) выше) растворяли в THF (250 мл), добавляли 0,5 М Н2SO4 (400 мл) и реакционную смесь перемешивали при 40°С в течение 65 часов, охлаждали и затем частично концентрировали в вакууме для удаления большей части THF. Затем реакционную смесь экстрагировали Et2O (3×100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения в виде твердого вещества, которое использовали на стадии (7) без дополнительной очистки или характеристики.
7) Ph(3-Cl)(5-OCHF2)-(R,S)CH(ОН)С(О)OH
Раствор Ph(3-CI)(5-OCHF2)-(R,S)CH(OH)C(O)OEt (6,25 г; предположительно 22,3 ммоль; смотри стадию (6) выше) в 2-пропаноле (175 мл) и 20%-ном КОН (350 мл) перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь затем частично концентрировали в вакууме для удаления большей части 2-пропанола. Оставшуюся смесь подкисляли 1 М H2SO4, экстрагировали Et2O (3×100 мл), сушили (Na2SO4) и концентрировали в вакууме с получением твердого вещества. В результате флэш-хроматографии на силикагеле с элюированием смесью CHCl3:МеОН:концентрированный NH4OH (6:3:1) получали аммониевую соль соединения, указанного в подзаголовке. Аммониевую соль затем растворяли в смеси EtOAc (75 мл) и H2O (75 мл) и подкисляли 2 н. HCl. Органический слой отделяли и промывали рассолом (50 мл), сушили (Na2SO4) и концентрировали в вакууме с получением указанного в подзаголовке соединения (3,2 г; 57% со стадий (4)-(7)).
1H ЯМР (300 МГц, CD3OD): δ 7.38 (s, 1Н), 7.22 (s, 1H), 7.15 (s, 1H), 6.89 (t, JH-F=71,1 Гц, 1Н), 5.16 (з,1Н).
8) (a) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH и
(б)Ph(3-Cl)-(5-OCHF2)-(S)CH(ОАс)С(O)OH
Смесь Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OH (3,2 г; 12,7 ммоль; смотри стадию (7) выше) и липазу Lipase PS "Amano" (~2,0 г) в винилацетате (125 мл) и МТВЕ (125 мл) нагревали с обратным холодильником в течение 48 часов. Реакционную смесь охлаждали, фильтровали через Celite® и осадок на фильтре промывали EtOAc. Фильтрат концентрировали в вакууме и подвергали флэш-хроматографии на силикагеле, элюируя смесью CHCl3:МеОН:концентрированный NH4OH (6:3:1), с получением аммониевых солей соединений (а) и (б), указанных в подзаголовке. Соединение (а) в виде соли растворяли в H2O, подкисляли 2 н. HCl и экстрагировали EtOAc. Органический слой промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в "вакууме" с получением указанного в подзаголовке соединения (а) (1,2 г; 37%).
Для указанного в подзаголовке соединения (а):
1H ЯМР (300 МГц, CD3OD): δ 7.38 (s, 1Н), 7.22 (s, 1H), 7.15 (s, 1H), 6.89 (t, Jh-f=71,1 Гц, 1H), 5.17 (s, 1H).
9)Ph(3-CI)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (1,1 г; 4,4 ммоль; смотри стадию (8) выше) и H-Aze-Pab(Teoc) (смотри международную заявку на патент WO 00/42059; 2,6 г; 5,7 ммоль) в DMF (50 мл) при 0°С добавляли РуВОР (2,8 г; 5,3 ммоль) и коллидин (1,3 г; 10,6 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 часов и затем при комнатной температуре в течение дополнительных 15 часов. Реакционную смесь концентрировали в вакууме и подвергали флэш-хроматографии на силикагеле (3х), элюируя сначала смесью CHCl3:EtOH (9:1), затем смесью EtOAc:EtOH (20:1) и, наконец, элюируя смесью СН2Cl2:СН3ОН (95:5) с получением указанного в подзаголовке соединения (1,0 г; 37%) в виде белого твердого вещества.
1H ЯМР (300 МГц, CD3OD, смесь ротамеров): δ 7.79-7.85 (d, J=8,7 Гц, 2Н), 7.15-7.48 (m, 5Н), 6.89 и 6.91 (t, JH-F=71,1 Гц, 1H), 5.12 и 5.20 (s, 1H), 4.75-4.85 (m, 1H), 3.97-4.55 (m, 6H), 2.10-2.75 (m, 2H), 1.05-1.15 (m, 2H), 0.09 (s, 9H).
MS(m/z)611 (M+1)+
10) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe, Teoc)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,40 г; 0,65 ммоль; смотри стадию (9) выше) растворяли в 20 мл ацетонитрила и добавляли 0,50 г (6,0 ммоль) O-метилгидроксиламина гидрохлорида. Смесь нагревали при 70°С в течение 2 часов. Растворитель выпаривали и остаток распределяли между водой и этилацетатом. Водную фазу еще два раза экстрагировали этилацетатом и объединенную органическую фазу промывали водой, рассолом, сушили (Na2SO4), фильтровали и упаривали. Выход: 0,41 г (91%).
1H ЯМР (400 МГц, CDCl3): δ 7.83 (bt, 1H), 7.57 (bs, 1H), 7.47 (d, 2H), 7.30 (d, 2Н), 7.20 (m, 1H), 7.14 (m, 1H), 7.01 (m, 1H), 6.53 (t, 1H), 4.89 (s, 1H), 4.87 (m, 1H), 4.47 (m. 2H), 4.4-4.2 (b, 1H), 4.17-4.1 (m, 3Н), 3.95 (s, 3Н), 3.67 (m, 1H), 2.68 (m, 1H), 2.42 (m, 1H), 0.97 (m, 2H), 0.01 (s, 9H).
11) Соединение А
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe, Teoc) (0,40 г; 0,62 ммоль; смотри стадию (10) выше) растворяли в 5 мл TFA и оставляли для взаимодействия в течение 30 минут. TFA выпаривали, а остаток распределяли между этилацетатом и NaHCO3 (водн.). Водную фазу экстрагировали еще два раза этилацетатом и объединенную органическую фазу промывали водой, рассолом, сушили (Na2SO4), фильтровали и упаривали. Продукт лиофильно высушивали из смеси вода/ацетонитрил. Никакой очистки не требовалось. Выход: 0,28 г (85%).
1H ЯМР (600 МГц, CDCl3): δ 7.89 (bt, 1H), 7.57 (d, 2H), 7.28 (d, 2H), 7.18 (m, 1H), 7.13 (m, 1H), 6.99 (m, 1H), 6.51 (t. 1H), 4.88 (s, 1H), 4.87 (m, 1H), 4.80 (bs, 2H), 4.48 (dd, 1H), 4.43 (dd, 1H), 4.10 (m, 1H), 3.89 (s, 3Н), 3.68 (m, 1H), 2.68 (m, 1H), 2.40 (m,1H).
13C ЯМР (125 МГц, CD3Cl3): (карбонильные и/или амидиновые атомы углерода, ротамеры) δ 172.9, 170.8, 152.7, 152.6.
HRMS: рассчитано для С22Н23ClF2N4O5 (М-Н)- 495,1242, найдено 495,1247.
Подготовительный пример Б: Получение Соединения Б
1) 2,6-Дифтор-4[(метилсульфинил)(метилтио)метил]бензонитрил
(Метилсульфинил)(метилтио)метан (7,26 г; 0,0584 моль) растворяли в 100 мл обезвоженного THF в атмосфере аргона и охлаждали до -78°С. По каплям при перемешивании добавляли бутиллитий в гексане (16 мл; 1,6 M; 0,0256 моль). Смесь перемешивали в течение 15 минут. Между тем раствор 3,4,5-трифторбензонитрила (4,0 г; 0,025 ммоль) в 100 мл безводного THF охлаждали до -78°С в атмосфере аргона и в течение 35 минут первый раствор через канюлю добавляли к последнему раствору. Через 30 минут охлаждающую баню убирали, и когда реакционная смесь достигала комнатной температуры, ее вливали в 400 мл воды. THF выпаривали, а оставшийся водный слой трижды экстрагировали диэтиловым эфиром. Объединенную эфирную фазу промывали водой, сушили (Na2SO4) и упаривали. Выход: 2,0 г (30%).
1H ЯМР (500 МГц, CDCl3): δ 7.4-7.25 (m, 2H), 5.01 (s, 1H, диастереомер), 4.91 (s, 1H, диастереомер), 2.88 (s, 3Н, диастереомер), 2.52 (s, 3Н, диастереомер), 2.49 (s, 3Н, диастереомер), 2.34 (s, 3Н, диастереомер), 1.72 (широкий, 1H).
2} 2,6-Дифтор-4-формилбензонитрил
2,6-Дифтор-4[(метилсульфинил)(метилтио)метил]бензонитрил (2,17 г; 8,32 ммоль; смотри стадию (1) выше) растворяли в 90 мл THF и добавляли 3,5 мл концентрированной серной кислоты. Смесь оставляли при комнатной температуре в течение 3 суток и затем вливали в 450 мл воды. Трижды проводили экстракцию EtOAc и объединенную эфирную фазу дважды промывали водным бикарбонатом натрия и рассолом, сушили (Na2SO4) и упаривали. Выход: 1,36 г (98%). Положение формильной группы устанавливали посредством 13С ЯМР. Сигнал от фторированных углеродов при 162,7 м.д. явился доказательством ожидаемой картины взаимодействия с двумя константами взаимодействия порядка 260 Гц и 6,3 Гц соответственно, которые отвечают ипсо- и мета-связыванию атомов фтора.
1Н ЯМР (400 МГц, CDCl3): δ 10.35 (s, 1H), 7.33 (m, 2H).
3) 2,6-Дифтор-4-гидроксиметилбензонитрил
2,6-Дифтор-4-формилбензонитрил (1,36 г; 8,13 ммоль; смотри стадию (2) выше) растворяли в 25 мл метанола и охлаждали на ледяной бане. Порциями при перемешивании добавляли боргидрид натрия (0,307 г; 8,12 ммоль) и реакционную смесь оставляли на 65 минут. Растворитель выпаривали и остаток распределяли между диэтиловым эфиром и водным бикарбонатом натрия. Эфирный слой дополнительно промывали водным бикарбонатом натрия и рассолом, сушили (Na2SO4) и упаривали. Вскоре кристаллизовался неочищенный продукт, который можно было использовать без дополнительной очистки. Выход: 1,24 г (90%).
1H ЯМР (400 МГц, CDCl3): δ 7.24 (m, 2H), 4.81 (s, 2H), 2.10 (широкий, 1H).
4) 4-Циано-2,6-дифторбензила метансульфонат
К ледяному раствору 2,6-дифтор-4-гидроксиметилбензонитрила (1,24 г; 7,32 ммоль; смотри стадию (3) выше) и метансульфонилхлорида (0,93 г; 8,1 ммоль) в 60 мл метиленхлорида при перемешивании добавляли триэтиламин (0,81 г; 8,1 ммоль). После 3 часов выдерживания при 0°С смесь дважды промывали 1 М HCl и один раз водой, сушили (Na2SO4) и упаривали. Продукт можно было использовать без дополнительной очистки. Выход: 1,61 г (89%).
1H ЯМР (300 МГц, CDCl3): 8 7.29 (m, 2Н), 5.33 (s, 2H), 3.07 (s, 3Н).
5)4-Азидометил-2,6-дифторбензонитрил
Смесь 4-циано-2,6-дифторбензила метансульфоната (1,61 г; 6,51 ммоль; смотри стадию (4) выше) и азида натрия (0,72 г; 0,0111 моль) в 10 мл воды и 20 мл. DMF перемешивали в течение ночи при комнатной температуре. Полученную смесь затем вливали в 200 мл воды и трижды экстрагировали диэтиловым эфиром. Объединенную эфирную фазу пять раз промывали водой, сушили (Na2SO4) и упаривали. Небольшой образец упаривали для проведения ЯМР и кристаллизации продукта. Оставшуюся часть осторожно упаривали, но не до полного высушивания. Предполагалось на основании ЯМР и аналитической ВЭЖХ, что выход (теоретически 1,26 г) был почти количественным.
1H ЯМР (400 МГц, CDCl3): δ 7.29 (m, 2H), 4.46 (s, 2H).
6) 4-Аминометил-2,6-дифторбензонитрил
Это взаимодействие проводили в соответствии с методикой, описанной в J. Chem. Res. (M) (1992) 3128. К суспензии 520 мг 10%-го Pd/C (влажность 50%) в 20 мл воды добавляли раствор боргидрида натрия (0,834 г; 0,0221 моль) в 20 мл воды. Выделялось некоторое количество газа. 4-Азидометил-2,6-дифторбензонитрил (1,26 г; 6,49 ммоль; смотри стадию (5) выше) растворяли в 50 мл THF и добавляли к этой водной смеси на ледяной бане в течение 15 минут. Смесь перемешивали в течение 4 часов, после чего добавляли 20 мл 2 М HCl и смесь фильтровали через Celite. Celite дополнительно промывали водой и объединенную водную фазу промывали EtOAc и затем подщелачивали 2 М NaOH. Затем трижды экстрагировали метиленхлоридом и объединенную органическую фазу промывали водой, сушили (Na2SO4) и упаривали. Выход: 0,87 г (80%).
1H ЯМР (400 МГц, CDCl3): δ 7.20 (m, 2H), 3.96 (s, 2H), 1,51 (широкий, 2H).
7) 2,6-Дифтор-4-трет-бутоксикарбониламинометилбензонитрил
Раствор 4-аминометил-2,6-дифторбензонитрила (0,876 г; 5,21 ммоль; смотри стадию (6) выше) растворяли в 50 мл THF и добавляли ди-трет-бутилдикарбонат (1,14 г; 5,22 ммоль) в 10 мл THF. Смесь перемешивали в течение 3,5 часов. THF выпаривали и остаток распределяли между водой и EtOAc. Органический слой три раза промывали 0,5 М HCl и водой, сушили (Na2SO4) и упаривали. Продукт мог быть использован без дополнительной очистки. Выход: 1,38 г (99%).
1H ЯМР (300 МГц, CDCl3): δ 7.21 (m, 2H), 4.95 (широкий, 1Н), 4.43 (широкий, 2Н), 1.52 (s, 9H).
8) Вос-Pab(2.6-диF)(OH)
Смесь 2,6-дифтор-4-трет-бутоксикарбониламинометилбензонитрила (1,38 г; 5,16 ммоль; смотри стадию (7) выше), гидрохлорида гидроксиламина (1,08 г; 0,0155 моль) и триметиламина (1,57 г; 0,0155 моль) в 20 мл этанола перемешивали при комнатной температуре в течение 36 часов. Растворитель выпаривали, а остаток распределяли между водой и метиленхлоридом. Органический слой промывали водой, сушили (Na2SO4) и упаривали. Продукт можно было использовать без дополнительной очистки. Выход: 1,43 г (92%).
1H ЯМР (500 МГц, CD3OD): δ 7.14 (m, 2H), 4.97 (широкий, 1Н), 4.84 (широкий, 2H), 4.40 (широкий, 2H), 1.43 (s, 9H).
9) Вос-Pab(2.6-диF) × НОАс
Это взаимодействие проводили в соответствии с методикой, описанной Judkins et al, Synth. Comm. (1998) 4351. Вос-Pab(2,6-диF)(ОН) (1,32 г; 4,37 ммоль; смотри стадию (8) выше), уксусный ангидрид (0,477 г; 4,68 ммоль) и 442 мг 10%-го Pd/C (влажность 50%) в 100 мл уксусной кислоты гидрировали при давлении 5 атм (0,5 МПа) в течение 3,5 часов. Смесь фильтровали через Celite, промывали этанолом и упаривали. Остаток лиофильно высушивали из ацетонитрила и воды и нескольких капель этанола. Продукт, указанный в подзаголовке, можно было использовать без дополнительной очистки. Выход: 1,49 г (99%).
1H ЯМР (400 МГц, CD3OD): δ 7.45 (m, 2H), 4.34 (s, 2H), 1.90 (s, 3Н), 1.40 (s, 9H).
10) Вос-Pab(2.6-диF)(Теос)
К раствору Вос-Pab(2,6-диF) × НОАс (1,56 г; 5,49 ммоль; смотри стадию (9) выше) в 100 мл THF (250 мл) и 1 мл воды добавляли 2-(триметилсилил)этил-пара-нитрофенилкарбонат (1,67 г; 5,89 ммоль). Раствор карбоната калия (1,57 г; 0,0114 моль) в 20 мл воды добавляли по каплям за 5 минут. Смесь перемешивали в течение ночи. THF выпаривали и остаток распределяли между водой и метиленхлоридом. Водный слой экстрагировали метиленхлоридом и объединенную органическую фазу дважды промывали водным бикарбонатом натрия, сушили (Na2SO4) и упаривали. В результате флэш-хроматографии на силикагеле со смесью гептан/EtOAc=2/1 получали 1,71 г (73%) чистого соединения.
1H ЯМР (400 МГц, CDCl3): δ 7.43 (m, 2H), 4.97 (широкий, 1Н), 4.41 (широкий, 2H), 4.24 (m, 2H), 1.41 (s, 9Н), 1.11 (m, 2H), 0.06 (s, 9H).
11) Вос-Aze-Pab(2,6-диF)(Теос)
Вос-Pab(2,6-диF)(Теос) (1,009 г; 2,35 ммоль; смотри стадию (10) выше) растворяли в 50 мл EtOAc, насыщенного HCl (газ). Смесь оставляли на 10 минут, упаривали и растворяли в 18 мл DMF и затем охлаждали на ледяной бане. Добавляли Boc-Aze-OH (0,450 г; 2,24 ммоль), РуВОР (1,24 г; 2,35 ммоль) и в конце диизопропилэтиламин (1,158 г; 8,96 ммоль). Реакционную смесь перемешивали в течение 2 часов и затем вливали в 350 мл воды и три раза экстрагировали EtOAc. Объединенную органическую фазу промывали рассолом, сушили (Na2SO4) и упаривали. В результате флэш-хроматографии на силикагеле со смесью гептан:EtOAc (1:3) получали 1,097 г (96%) требуемого соединения.
1H ЯМР (500 МГц, CDCl3): δ 7.46 (m, 2H), 4.64-4.5 (m, 3Н), 4.23 (m, 2H), 3.87 (m, 1H), 3.74 (m, 1H), 2.45-2.3 (m, 2H), 1.40 (s, 9H), 1.10 (m, 2H), 0.05 (s, 9H).
12) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(Teoc)
Вос-Aze-Pab(2,6-диF)(Теос) (0,256 г; 0,500 ммоль; смотри стадию (11) выше) растворяли в 20 мл EtOAc, насыщенного HCl (газ). Смесь оставляли на 10 минут, упаривали и растворяли в 5 мл DMF. Добавляли Ph(3-CI)(5-OCHF2)-(R)CH(OH)C(O)OH (0,120 г; 0,475 ммоль; смотри Подготовительный пример А (8) выше), РуВОР (0,263 г; 0,498 ммоль) и, наконец, диизопропилэтиламин (0,245 г; 1,89 ммоль). Реакционную смесь перемешивали в течение 2 часов и затем вливали в 350 мл воды и три раза экстрагировали EtOAc. Объединенную органическую фазу промывали рассолом, сушили (Na2SO4) и упаривали. В результате флэш-хроматографии на силикагеле с EtOAc получали 0,184 г (60%) требуемого соединения, указанного в подзаголовке.
1H ЯМР (400 МГц, CD3OD, смесь ротамеров): δ 7.55-7.45 (m, 2H), 7.32 (m, 1H, основной ротамер), 7.27 (m, 1H, минорный ротамер), 7.2-7.1 (m, 2H), 6.90 (t, 1H, основной ротамер), 6.86 (t, 1Н, минорный ротамер), 5.15 (s, 1Н, основной ротамер), 5.12 (m, 1H, минорный ротамер), 5.06 (s, 1H, минорный ротамер), 4.72 (m, 1H, основной ротамер), 4.6-4.45 (m, 2H), 4.30 (m, 1H, основной ротамер), 4.24 (m, 2H), 4.13 (m, 1H, основной ротамер), 4.04 (m, 1H, минорный ротамер), 3.95 (m, 1H, минорный ротамер), 2.62 (m, 1H, минорный ротамер), 2.48 (m, 1H, основной ротамер), 2.22 (m, 1H, основной ротамер), 2.10 (m, 1H, минорный ротамер), 1.07 (m, 2H), 0.07 (m, 9H).
13) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-Pab(2.6-диF(ОМе, Теос)
Смесь Ph(3-CI)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(Teoc) (64 мг; 0,099 ммоль; смотри стадию (12) выше) и O-метилгидроксиламина гидрохлорида (50 мг; 0,60 ммоль) в 4 мл ацетонитрила нагревали при 70°С в течение 3 часов. Растворитель выпаривали и остаток распределяли между водой и EtOAc. Водный слой дважды экстрагировали EtOAc и объединенную органическую фазу промывали водой, сушили (Na2SO4) и упаривали. Продукт можно было использовать без дополнительной очистки. Выход: 58 мг (87%).
1H ЯМР (400 МГц, CD3Cl3): δ 7.90 (bt, 1H), 7.46 (m, 1H), 7.25-6.95 (m, 5H), 6.51 (t, 1H), 4.88 (s, 1H), 4.83 (m, 1H), 4.6-4.5 (m, 2H), 4.4-3.9 (m, 4Н), 3.95 (s, 3H), 3.63 (m, 1H), 2.67 (m, 1H), 2.38 (m, 1H), 1.87 (широкий, 1H), 0.98 (m, 2H), 0.01 (s, 9H).
14) Соединение Б
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(OMe, Teoc) (58 мг; 0,086 ммоль; смотри стадию (13) выше) растворяли в 3 мл TFA, охлаждали на ледяной бане и оставляли для взаимодействия на 2 часа. TFA выпаривали и остаток растворяли в EtOAc. Органический слой дважды промывали водным карбонатом натрия и водой, сушили (Na2SO4) и упаривали. Остаток лиофильно высушивали из воды и ацетонитрила с получением 42 мг (92%) соединения, указанного в заголовке.
1H ЯМР (300 МГц, CD3Cl3): δ 7.95 (bt, 1H), 7.2-7.1 (m, 4Н), 6.99 (m, 1H), 6.52 (t, 1H), 4.88 (s, 1H), 4.85-4.75 (m, 3Н), 4.6-4.45 (m, 2H), 4.29 (широкий, 1H), 4.09 (m, 1H), 3.89 (s, 3Н), 3.69 (m, 1H), 2.64 (m, 1H), 2.38 (m, 1H), 1.85 (широкий, 1H).
13С ЯМР (100 МГц, CDCl3): (карбонильный и/или амидиновый углероды) δ 172.1, 169.8, 151.9.
APCI-MS: (M+1)=533/535 m/z.
Подготовительный пример В: Получение Соединения В
1) (2-Монофторэтил)метансульфонат
К перемешиваемому магнитной мешалкой раствору 2-фторэтанола (5,0 г; 78,0 ммоль) в CH2Cl2 (90 мл) в атмосфере азота при 0°С добавляли триэтиламин (23,7 г; 234 ммоль) и метансульфонилхлорид (10,7 г; 93,7 ммоль). Смесь перемешивали при 0°С в течение 1,5 часов, разбавляли CH2Cl2 (100 мл) и промывали 2 н. HCl (100 мл). Водный слой экстрагировали CH2Cl2 (50 мл) и объединенные органические экстракты промывали рассолом (75 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения (9,7 г; 88%) в виде желтого масла, которое использовали без дополнительной очистки.
1H ЯМР (300 МГц, CDCl3): δ 4.76 (t, J=4 Гц, 1H), 4.64 (t, J=4 Гц, 1H), 4.52 (t, J=4 Гц, 1H), 4.43 (t, J=4 Гц, 1H), 3.09 (s, 3Н).
2) 3-Хлор-5-монофторэтоксибензальдегид
К раствору 3-хлор-5-гидроксибензальдегида (8,2 г; 52,5 ммоль; смотри Подготовительный пример А (2) выше) и карбоната калия (9,4 г; 68,2 ммоль) в DMF (10 мл) в атмосфере азота добавляли по каплям раствор (2-монофторэтил)метансульфоната (9,7 г; 68,2 ммоль; смотри стадию (1) выше) в DMF (120 мл) при комнатной температуре. Смесь нагревали до 100°С в течение 5 часов и затем перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°С, вливали в ледяную 2 н. HCl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Коричневое масло хроматографировали на силикагеле, элюируя смесью Нех:EtOAc (4:1) с получением указанного в подзаголовке соединения (7,6 г; 71%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3): δ 9.92 (s, 1H), 7.48 (s, 1Н), 7.32 (s, 1H), 7.21 (s, 1Н), 4.87 (t, J=4 Гц, 1H), 4.71 (t, J=3 Гц, 1H), 4.33 (t, J=3 Гц, 1H), 4.24 (t, J=3 Гц, 1H).
3)Ph(3-CI)(5-OCH2CH2F)-(R,S)CH(OTMS)CN
К раствору 3-хлор-5-монофторэтоксибензальдегида (7,6 г; 37,5 ммоль; смотри стадию (2) выше) и иодида цинка (3,0 г; 9,38 ммоль) в CH2Cl2 (310 мл) добавляли по каплям триметилсилилцианид (7,4 г; 75,0 ммоль) при 0°С в атмосфере азота. Смесь перемешивали при 0°С в течение 3 часов и при комнатной температуре в течение ночи. Реакционную смесь разбавляли H2O (300 мл), органический слой отделяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения (10,6 г; 94%) в виде коричневого масла, которое использовали без дополнительной очистки или характеристики.
4)Ph(3-Cl)-(5-ОСН2СН2F)-(R,S)CH(ОН)С(O)OH
Концентрированную соляную кислоту (100 мл) добавляли к Ph(3-CI)(5-OCH2CH2F)-(R,S)CH(OTMS)CN (10,6 г; 5,8 ммоль; смотри стадию (3) выше), и раствор перемешивали при 100°С в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь дополнительно охлаждали до 0°С, медленно подщелачивали 3 н. NaOH (~300 мл) и промывали Et2O (3×200 мл). Водный слой подкисляли 2 н. HCl (80 мл) и экстрагировали EtOAc (3×100 мл). Объединенные этилацетатные экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения (8,6 г; 98%) в виде бледно-желтого твердого вещества, которое использовали без дополнительной очистки.
Rf=0,28 (90:8:2 CHCl3:МеОН:концентрированный NH4OH).
1H ЯМР (300 МГц, CD3OD): δ 7.09 (s, 1H), 7.02 (s, 1H), 6.93 (s, 1H), 5.11 (s, 1H), 4.77-4.81 (m, 1H), 4.62-4.65 (m, 1H), 4.25-4.28 (m, 1H), 4.15-4.18 (m, 1H).
5) (a) Ph(3-Cl)(5-OCH2CH2F)-(S)CH(OAc)C(O)OH и
(б) Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH
Раствор Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OH)C(O)OH (8,6 г; 34,5 ммоль; смотри стадию (4) выше) и липазы Lipase PS "Amano" (4,0 г) в винилацетате (250 мл) и МТВЕ (250 мл) нагревали при 70°С в атмосфере азота в течение 3 суток. Реакционную смесь охлаждали до комнатной температуры и фермент удаляли фильтрацией через Celite®. Остаток на фильтре промывали EtOAc и фильтрат концентрировали в вакууме. В результате хроматографии на силикагеле с элюированием смесью CHCl3:МеОН:Et3N (90:8:2) получали триэтиламиновую соль указанного в подзаголовке соединения (а) в виде желтого масла. Кроме того, получали триэтиламиновую соль указанного в подзаголовке соединения (б) (4,0 г). Соль указанного в подзаголовке соединения (б) растворяли в Н2О (250 мл), подкисляли 2 н. HCl и экстрагировали EtOAc (3×200 мл). Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения (б) (2,8 г; 32%) в виде желтого масла.
Данные для указанного в подзаголовке соединения (б):
Rf=0,28 (90:8:2 CHCl3:МеОН:концентрированный NH4OH).
1H ЯМР (300 МГц, CD3OD): δ 7.09 (s, 1H), 7.02 (s, 1H), 6.93 (s, 1H), 5.11 (s, 1H), 4.77-4.81 (m, 1H), 4.62-4.65 (m, 1H), 4.25-4.28 (m, 1H), 4.15-4.18 (m, 1H).
6) Соединение В
К раствору Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (818 мг; 3,29 ммоль; смотри стадию (5) выше) в DMF (30 мл) в атмосфере азота при 0°С добавляли HAze-Pab(OMe)·2HCl (1,43 г; 4,27 ммоль; смотри международную заявку на патент WO 00/42059), РуВОР (1,89 г; 3,68 ммоль) и DIPEA (1,06 г; 8,23 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 часов и затем при комнатной температуре в течение ночи. Смесь концентрировали в вакууме и остаток дважды хроматографировали на силикагеле, элюируя первый раз смесью CHCl3:EtOH (15:1) и второй раз смесью EtOAc:EtOH (20:1), с получением указанного в заголовке соединения (880 мг; 54%).
Rf=0,60 (10:1 CHCl3:EtOH).
1H ЯМР (300 МГц, CD3OD, сложная смесь ротамеров): δ 7.58-7.60 (d, J=8 Гц, 2Н), 7.34 (d, J=7 Гц, 2Н), 7.05-7.08 (m, 2H), 6.95-6.99 (m, 1H), 5.08-5.13 (m, 1H), 4.77-4.82 (m, 1H), 4.60-4.68 (m, 1H), 3.99-4.51 (m, 7H), 3.82 (s, 3H), 2.10-2.75 (m, 2H).
13C ЯМР (150 МГц, CD3OD): (карбонильные и/или амидиновые углероды) δ 173.3, 170.8, 152.5.
APCI-MS:(M+1)=493 m/z.
Получение Соединения Г (Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab)
Соединение Г
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,045 г; 0,074 ммоль; смотри Подготовительный пример А (9) выше) растворяли в 3 мл TFA и оставляли для взаимодействия в течение 1 часа. TFA выпаривали и остаток лиофильно высушивали из смеси вода/ацетонитрил с получением 0,043 г (100%) указанного в подзаголовке соединения в виде его TFA соли.
1H ЯМР (400 МГц, CD3OD, ротамеры): δ 7.8-7.75 (m, 2H), 7.55-7.5 (m, 2H), 7.35 (m, 1H, главный ротамер), 7.31 (m, 1H, минорный ротамер), 7.19 (m, 1H, главный ротамер), 7.15 (m, 1H), 7.12 (m, 1H, минорный ротамер), 6.89 (t, 1H, главный ротамер), 6.87 (t, 1H, минорный ротамер), 5.22 (m, 1H, минорный ротамер), 5.20 (s, 1H, главный ротамер); 5.13 (s, 1H, минорный ротамер), 4.80 (m, 1H, главный ротамер), 4.6-4.4 (m, 2H), 4.37 (m, 1H, главный ротамер), 4.19 (m, 1H, главный ротамер), 4.07 (m, 1H, минорный ротамер), 3.98 (m, 1H, минорный ротамер), 2.70 (m, 1H, минорный ротамер), 2.55 (m, 1H, главный ротамер), 2.29 (m, 1H, главный ротамер), 2.15 (m, 1H, минорный ротамер).
13С ЯМР (100 МГц, CD3OD): (карбонильные и/или амидиновые углероды, ротамеры) δ 172.6, 172.5, 172.0, 171.7, 167.0.
MS (m/z) 465 (M-1)-; 467 (M+1)+.
Получение Соединения Д (Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2.6-диF))
Соединение Д
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(2,6-диF)(Теос) (81 мг; 0,127 ммоль; смотри Подготовительный пример Б (12) выше) растворяли в 0,5 мл метиленхлорида и охлаждали на ледяной бане. Добавляли TFA (3 мл) и реакционную смесь оставляли на 75 минут. TFA выпаривали и остаток лиофильно высушивали из воды и ацетонитрила. Неочищенный продукт очищали препаративной RPLC (жидкостной хроматографией на обращенной фазе) со смесью СН3CN:0,1 М NH4OAc (35:65) с получением 39 мг (55%) указанного в заголовке соединения в виде НОАс соли, чистота: 99%.
1H ЯМР (400 МГц, CD3OD, смесь ротамеров): δ 7.5-7.4 (m, 2H), 7.32 (m, 1H, главный ротамер), 7.28 (m, 1H, минорный ротамер), 7.2-7.1 (m, 3Н), 6.90 (t, 1H, главный ротамер), 6.86 (t, минорный ротамер), 5.15 (s, 1H, главный ротамер), 5.14 (m, 1H, минорный ротамер), 5.07 (s, 1H, минорный ротамер), 4.72 (m, 1H, главный ротамер), 4.65-4.45 (m, 2H), 4.30 (m, 1H, главный ротамер), 4.16 (m, 1H, главный ротамер), 4.03 (m, 1H, минорный ротамер), 3.95 (m, 1H, минорный ротамер), 2.63 (m, 1H, минорный ротамер), 2.48 (m, 1H, главный ротамер), 2.21 (m, 1H, главный ротамер), 2.07 (m, 1H, минорный ротамер), 1.89 (S, 3Н).
13С ЯМР (75 МГц, CD3OD): (карбонильные и/или амидиновые углероды, смесь ротамеров) δ 171.9, 171.2, 165.0, 162.8, 160.4.
APCI-MS: (M+1)=503/505 m/z.
Получение Соединения Е (Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab·TFA)
1)Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (940 мг; 3,78 ммоль; смотри Подготовительный пример В (5) выше) в DMF (30 мл) в атмосфере азота при 0°С добавляли HAze-Pab(Teoc)·HCl (2,21 г; 4,91 ммоль), РуВОР (2,16 г; 4,15 ммоль) и DlPEA (1,22 г; 9,45 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 часов и затем при комнатной температуре в течение 4 часов. Смесь концентрировали под вакуумом и остаток дважды хроматографировали на силикагеле, элюируя сначала смесью CHCl3:EtOH (15:1) и дополнительно смесью EtOAc:EtOH (20:1) с получением указанного в подзаголовке соединения (450 мг; 20%) в виде сминаемой белой пены.
Точка плавления: 80-88°С.
Rf=0,60 (10:1 CHCl3:EtOH).
1H ЯМР (300 МГц, CD3OD, сложная смесь ротамеров): δ 7.79 (d, J=8 Гц, 2H), 7.42 (d, J=8 Гц, 2H), 7.05-7.08 (m, 1H), 6.93-6.99 (m, 2H), 5.08-5.13 (m, 1H), 4.75-4.80 (m, 2H), 4.60-4.68 (m, 1H), 3.95-4.55 (m, 8H), 2.10-2.75 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9H).
APCl-MS: (M+1)=607 m/z.
2) Соединение Е
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,357 г; 0,589 ммоль; смотри выше стадию (1)) растворяли в 10 мл TFA и оставляли для взаимодействия в течение 40 минут. TFA выпаривали и остаток лиофильно высушивали из смеси вода/ацетонитрил с получением 0,33 г (93%) указанного в заголовке соединения в виде TFA соли.
1H ЯМР (600 МГц, CD3OD, ротамеры): δ 7.8-7.7 (m, 2H), 7.54 (d, 2H), 7.08 (s, 1H, главный ротамер), 7.04 (s, 1H, минорный ротамер), 6.99 (s, 1H, главный ротамер), 6.95 (s, 1H), 6.92 (s, 1H, минорный ротамер), 5.18 (m, 1H, минорный ротамер), 5.14 (s, 1H, главный ротамер), 5.08 (s, 1H, минорный ротамер), 4.80 (m, 1H, главный ротамер), 4.73 (m, 1H), 4.65 (m, 1H), 4.6-4.4 (m, 2H), 4.35 (m, 1H, главный ротамер), 4.21 (дублет мультиплетов, 2H), 4.12 (m, 1H, главный ротамер), 4.06 (m, 1H, минорный ротамер), 3.99 (m, 1H, минорный ротамер), 2.69 (m, 1H, минорный ротамер), 2.53 (m, 1H, главный ротамер), 2.29 (m, 1H, главный ротамер), 2.14 (m, 1H, минорный ротамер).
13С ЯМР (150 МГц, CD3OD): (карбонильные и/или амидиновые углероды) δ 172.8,172.1,167.4.
ESI-MS+: (M+1)=463 (m/z).
Получение Соединения Ж (Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH))
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH, Teoc)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,148 г; 0,24 ммоль; смотри выше стадию (9) Подготовительного примера А) растворяли в 9 мл ацетонитрила и добавляли 0,101 г (1,45 ммоль) гидрохлорида гидроксиламина. Смесь нагревали при 70°С в течение 2,5 часов, фильтровали через Celite® и упаривали. Неочищенный продукт (0,145 г; 75%-ная чистота) использовали непосредственно на следующей стадии без дополнительной очистки.
2)(Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(OH, Teoc) (0,145 г; 0,23 ммоль; смотри выше стадию (1)) растворяли в 0,5 мл CH2Cl2 и 9 мл TFA. Оставляли для взаимодействия в течение 60 минут. TFA выпаривали и остаток очищали, используя препаративную HPLC. Интересующие фракции объединяли и лиофильно высушивали (2×), получая 72 мг (выход с двух стадий 62%) указанного в заголовке соединения.
MS (m/z) 482 (M -1)-; 484 (M+1)+.
1H ЯМР (400 МГц, CD3OD): δ 7.58 (d, 2H), 7.33 (m, 3H), 7.15 (m, 2H), 6.89 (t, 1H главный ротамер), 6.86 (t, 1H минорный ротамер), 5.18 (s, 1H главный ротамер; и m, 1H минорный ротамер), 5.12 (s, 1H минорный ротамер), 4.77 (m, 1Н главный ротамер), 4.42 (m, 2H), 4.34 (m, 1H главный ротамер), 4.14 (m, 1H главный ротамер), 4.06 (m, 1H минорный ротамер), 3.95 (m, 1H минорный ротамер), 2.66 (m, 1H минорный ротамер), 2.50 (m, 1H главный ротамер), 2.27 (m, 1H главный ротамер), 2.14 (m, 1H минорный ротамер).
13С ЯМР (100 МГц, CD3OD). (карбонильные и/или амидиновые углероды, ротамеры) δ 172.4, 172.3, 172.0, 171.4, 152.3, 152.1.
Получение Соединения 3: Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF(OH)
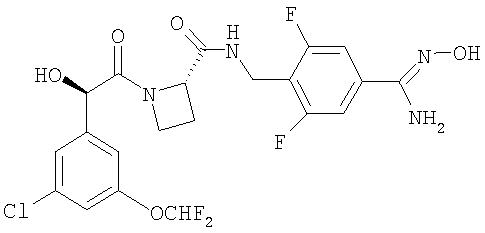
1) Вос-(S)Aze-NHCH2-Ph(2,6-диF, 4-CN)
Boc-(S)Aze-OH (1,14 г; 5,6 ммоль) растворяли в 45 мл DMF. Добавляли 4-аминометил-2,6-дифторбензонитрил (1,00 г; 5,95 моль; смотри выше Пример 1 (14)), РуВОР (3,10 г; 5,95 ммоль) и DIPEA (3,95 мл; 22,7 ммоль) и раствор перемешивали при комнатной температуре в течение 2 часов. Растворитель выпаривали и остаток распределяли между Н2О и EtOAc (75 мл каждого). Водную фазу экстрагировали 2×50 мл EtOAc и объединенную органическую фазу промывали рассолом и сушили над Na2SO4. После флэш-хроматографии (SiO2, EtOAc/гептан (3/1)) получали указанное в подзаголовке соединение (1,52 г; 77%) в виде масла, которое кристаллизовали в холодильнике.
1H ЯМР (400 МГц, CD3OD): δ 7.19 (m, 2H), 4.65-4.5 (m, 3Н), 3.86 (m, 1H), 3.73 (m, 1H), 2.45-2.3 (m, 2H), 1.39 (s, 9Н).
2) H-(S)Aze-NHCH2-Ph(2,6-диF, 4-CN)×HCl
Boc-(S)Aze-NHCH2-Ph(2,6-диF, 4-CN) (0,707 г; 2,01 ммоль; смотри выше стадию (1)) растворяли в 60 мл EtOAc, насыщенного HCl (газ). После перемешивания при комнатной температуре в течение 15 минут растворитель выпаривали. Остаток растворяли в смеси СН3CN/Н2О (1/1) и лиофильно высушивали с получением указанного в подзаголовке соединения (0,567 г; 98%) в виде не совсем белого аморфного порошка.
1H ЯМР (400 МГц, CD3OD): δ 7.49 (m, 2Н), 4.99 (m, 1H), 4.58 (m, 2H), 4.12 (m, 1H), 3.94 (m, 1H), 2.80 (m, 1H), 2.47 (m, 1H).
MS (m/z) 252,0 (M+1)+.
3)Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-(S)Aze-NHCH2-Ph(2.6-диF, 4-CN)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (0,40 г; 1,42 ммоль; смотри выше Пример 1 (8)) растворяли в 10 мл DMF и добавляли H-(S)Aze-NHCH2-Ph(2,6-диР, 4-CN) ×HCl (0,43 г; 1,50 ммоль; смотри выше стадию (2)) и РуВОР (0,779 г; 1,50 ммоль) с последующим добавлением DIPEA (1,0 мл; 5,7 ммоль). После перемешивания при комнатной температуре в течение 2 часов растворитель выпаривали. Остаток распределяли между H2O (200 мл) и EtOAc (75 мл). Водную фазу экстрагировали 2×75 мл EtOAc и объединенную органическую фазу промывали рассолом и сушили над Na2SO4. После флэш-хроматографии (SiO2, EtOAc/гептан (4/1)) получали указанное в подзаголовке соединение (0,56 г; 81%) в виде масла.
1H ЯМР (400 МГц, CD3OD) ротамеры: δ 7.43 (m, 2H), 7.31 (m, 1H, главный ротамер), 7.26 (m, 1H, минорный ротамер), 7.2-7.1 (m, 2H), 6.90 (t, 1H, главный ротамер), 6.86 (t, 1H, минорный ротамер), 5.14 (s, 1H, главный ротамер), 5.11 (s, 1H, минорный ротамер), 5.04 (s, 1H, минорный ротамер), 4.71 (m, 1H, главный ротамер), 4.6-4.45 (m, 2H), 4.30 (m, 1H, главный ротамер), 4.2-3.9 (m, 1H; и 1H, минорный ротамер), 2.62 (m, 1H, минорный ротамер), 2.48 (m, 1H, главный ротамер), 2.21 (m, 1H, главный ротамер), 2.09 (m, 1H, минорный ротамер).
13C ЯМР (100 МГц, CD3OD): (карбонильные углероды) δ 171.9, 171.8.
MS (m/z) 484,0, 485,9 (M-1)-; 486,0, 487,9 (M+1)+.
4)Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OH)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-NHCH2-Ph(2,6-диF,4-CN) (0,555 г; 1,14 ммоль; со стадии (3) выше) растворяли в 10 мл EtOH (95%). К этому раствору добавляли гидрохлорид гидроксиламина (0,238 г; 3,42 ммоль) и Et3N (0,48 мл; 3,4 ммоль). После перемешивания при комнатной температуре в течение 14 часов растворитель удаляли и остаток растворяли в EtOAc. Органическую фазу промывали рассолом и H2O и сушили над Na2SO4. Неочищенный продукт очищали препаративной RPLC со смесью СН3CN:0,1 М NH4OAc в качестве элюента с получением указанного в заголовке соединения в виде аморфного порошка (0,429 г; 72%) после лиофильной сушки.
1H ЯМР (400 МГц, CD3OD) ротамеры: δ 7.35-7.1 (m, 5Н), 6.90 (t, 1H, главный ротамер), 6.85 (t, 1H, минорный ротамер), 5.15 (s, 1H, главный ротамер), 5.12 (т, 1H, минорный ротамер), 5.08 (s, 1H, минорный ротамер), 4.72 (m, 1H, главный ротамер), 4.6-4.4 (m, 2H), 4.30 (m, 1H, главный ротамер), 4.12 (m, 1H, главный ротамер), 4.04 (m, 1H, минорный ротамер), 3.94 (m, 1H, минорный ротамер), 2.62 (m, 1H, минорный ротамер), 2.48 (m, 1H, главный ротамер), 2.22 (m, 1H, главный ротамер), 2.10 (m, 1H, минорный ротамер).
13С ЯМР (100 МГц, CD3OD): (карбонильные и амидиновые углероды, ротамеры) δ 172.4, 171.9, 171.0, 152.3, 151.5.
MS (m/z) 517.1, 519.0 (M-1)-; 519.1, 521.0 (M+1)+.
Получение Соединения К(Ph(3-Cl)(5-ОСН2CHF2)-(R)CH(OH)C(O)-Aze-Pab(OH))
1) Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Z)
Boc-Aze-Pab(Z) (смотри международную заявку на патент WO 97/02284, 92 мг, 0,197 ммоль) растворяли в 10 мл EtOAc, насыщенного HCl (газ) и оставляли для взаимодействия на 10 минут. Растворитель выпаривали и остаток смешивали с Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)OH (50 мг; 0,188 ммоль; смотри выше Подготовительный пример В (5)), РуВОР (109 мг; 0,209 ммоль) и в конце диизопропилэтиламин (96 мг; 0,75 ммоль) в 2 мл DMF. Смесь перемешивали в течение 2 часов и затем вливали в 50 мл воды и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водой, сушили (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле со смесью EtOAc:МеОН (9:1). Выход: 100 мг (87%).
1H ЯМР (300 МГц, CD3OD, смесь ротамеров):δ 7.85-7.75 (m, 2H), 7.45-7.25 (m, 7H), 7.11 (m, 1H, главный ротамер), 7.08 (m, 1H, минорный ротамер), 7.05-6.9 (m, 2H), 6.13 (bt, 1H), 5.25-5.05 (m, 3Н), 4.77 (m, 1H, частично скрыт сигналом от CD3ОН), 4.5-3.9 (m, 7H), 2.64 (m, 1H, минорный ротамер), 2.47 (m, 1H, главный ротамер), 2.25 (m, 1H, главный ротамер), 2.13 (m, 1H, минорный ротамер).
2)Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
Гидрохлорид гидроксиламина (65 мг; 0,94 ммоль) и триэтиламин (0,319 г; 3,16 ммоль) смешивали в 8 мл THF и обрабатывали ультразвуком в течении 1 часа при 40°С. Добавляли Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Z) (96 мг; 0,156 ммоль; смотри выше стадию (1)) с еще 8 мл THF. Смесь перемешивали при 40°С в течение 4,5 суток. Растворитель выпаривали и неочищенный продукт очищали препаративной RPLC со смесью СН3CN:0,1 М NH4OAc (40:60). Выход: 30 мг (38%). Чистота: 99%.
1H ЯМР (300 МГц, CD3OD, смесь ротамеров):δ 7.6-7.55 (m, 2H), 7.35-7.3 (m, 2H), 7.12 (m, 1Н, главный ротамер), 7.09 (m, 1H, минорный ротамер), 7.05-6.9 (m, 2H), 6.15 (триплет мультиплетов, 1H), 5.15 (m, 1H, минорный ротамер), 5.13 (s, 1H, главный ротамер), 5.08 (s, 1H, минорный ротамер), 4.77 (m, 1H, главный ротамер), 4.5-4.2 (m. 5H), 4.08 (m, 1H, главный ротамер), 3.97 (m, 1H, минорный ротамер), 2.66 (m, 1H, минорный ротамер), 2.50 (m, 1H, главный ротамер), 2.27 (m, 1H, главный ротамер), 2.14 (m, 1H, минорный ротамер).
13С ЯМР (100 МГц, CD3OD): (карбонильные и/или амидиновые углероды, смесь ротамеров) δ 172.8, 172.2, 171.4, 159.1, 158.9, 154.2.
APCl-MS: (M+1)=497/499 m/z.
Методы 1 и 2: Получение солей Соединения А
Метод 1: Общий способ получения солей
Следующий общий способ использовали для получения солей Соединения А: 200 мг Соединения А (смотри Подготовительный пример А выше) растворяли в 5 мл МеОН. К этому раствору добавляли раствор соответствующей кислоты (1,0 молярный эквивалент), растворенной в 5 мл МеОН. После перемешивания в течение 10 минут при комнатной температуре растворитель удаляли при помощи роторного испарителя. Оставшееся твердое вещество повторно растворяли в 8 мл смеси ацетонитрил:Н2О (1:1). В результате лиофильного высушивания в каждом случае получали бесцветное аморфное вещество.
Использованные кислоты:
(1S)-(+)-10-камфорсульфоновая
яблочная
циклогексилсульфаминовая
фосфорная
диметилфосфорная
пара-толуолсульфоновая
L-лизин
L-лизина гидрохлорид
сахариновая
метансульфоновая
соляная
Соответствующие характеристики показаны в таблице 1.
495.1
497.0
727.3
495.1
497.0
495.1
496.9
674.3
676.1
497,0
593.1
495.1
497.0
621.2
623.0
495.1
497.0
495.1
497.0
497.0
531.1
(HCl)
495.1
497.0
497.0
591.2
593.1
496.9
531.1
532.5
535.2
Все соли, образовавшиеся в этом Методе, были аморфными.
Метод 2
Следующие аморфные соли Соединения А получали, используя методики, аналогичные описанным в Методе 1 выше, из следующих кислот:
бромистоводородная кислота (соль 1:1)
соляная кислота (соль 1:1)
серная кислота (соль 1:0,5)
1,2-этандисульфоновая кислота (соль 1:0,5)
1S-камфорсульфоновая кислота (соль 1:1)
(+/-)-камфорсульфоновая кислота (соль 1:1)
этансульфоновая кислота (соль 1:1)
азотная кислота (соль 1:1)
толуолсульфоновая кислота (соль 1:1)
метансульфоновая кислота (соль 1:1)
пара-ксилолсульфоновая кислота (соль 1:1)
2-мезитиленсульфоновая кислота (соль 1:1)
1,5-нафталинсульфоновая кислота (соль 1:0,5)
нафталинсульфоновая кислота (соль 1:1)
бензолсульфоновая кислота (соль 1:1)
сахариновая кислота (соль 1:1)
малеиновая кислота (соль 1:1)
фосфорная кислота (соль 1:1)
D-глутаминовая кислота (соль 1:1)
L-глутаминовая кислота (соль 1:1)
D,L-глутаминовая кислота (соль 1:1)
L-аргинин (соль 1:1)
L-лизин (соль 1:1)
L-лизина гидрохлорид (соль 1:1)
глицин (соль 1:1)
салициловая кислота (соль 1:1)
винная кислота (соль 1:1)
фумаровая кислота (соль 1:1)
лимонная кислота (соль 1:1)
L-(-)-яблочная кислота (соль 1:1)
D,L-яблочная кислота (соль 1:1)
D-глюконовая кислота (соль 1:1)
Метод 3: получение аморфного Соединения А,
соли этансульфоновой кислоты
Соединение А (203 мг; смотри выше Подготовительный пример А) растворяли в этаноле (3 мл) и к этому раствору добавляли этансульфоновую кислоту (1 экв.; 95%; 35 мкл). Смесь перемешивали в течение нескольких минут и затем растворитель выпаривали. Полученное масло суспендировали в изооктане и упаривали досуха до получения твердого вещества. Наконец, вещество ресуспендировали в изооктане и растворитель снова выпаривали с получением белого, сухого, аморфного твердого вещества. Это вещество сушили в вакууме при 40°С в течение ночи.
Примеры 4-9: получение кристаллического Соединения А.
соли этансульфоновой кислоты
Пример 4: кристаллизация аморфного вещества
Аморфное Соединение А в виде соли этансульфоновой кислоты (17,8 мг; смотри Метод 3 выше) суспендировали в метилизобутилкетоне (600 мкл). Через 1 неделю обнаруживались игольчатые кристаллы, которые отфильтровывали и сушили на воздухе.
Методы 5-7: реактивные кристаллизации (без антирастворителя)
Метод 5
Соединение А (277 мг; смотри выше Подготовительный пример А) растворяли в метилизобутилкетоне (3,1 мл). Добавляли этансульфоновую кислоту (1 экв.; 95%; 48 мкл). Сразу же происходило осаждение аморфного этансульфоната. Добавляли дополнительно метилизобутилкетон (6 мл) и суспензию обрабатывали ультразвуком. Наконец, добавляли третью порцию метилизобутилкетона (3,6 мл) и затем суспензию оставляли на ночь при перемешивании (магнитная мешалка). На следующий день вещество превращалось в игольчатые кристаллы. Суспензию отфильтровывали, промывали метилизобутилкетоном (0,5 мл) и сушили на воздухе.
Метод 6
Соединение А (236 мг; смотри выше Подготовительный пример А) растворяли при комнатной температуре в метилизобутилкетоне (7 мл). Этансульфоновую кислоту (1 экв.; 41 мкл) в пробирке смешивали с 2 мл метилизобутилкетона. В раствор Соединения А вводили затравку кристаллического Соединения А в виде соли этансульфоновой кислоты (смотри Методы 4 и 5 выше). Затем порциями за 45 минут добавляли 250 мкл метилизобутилкетонового раствора этансульфоновой кислоты. В раствор снова вводили затравку и температуру повышали до 30°С. Затем добавляли 500 мкл метилизобутилкетонового раствора за приблизительно 1 час. Полученную суспензию оставляли на ночь, затем за 20 минут добавляли последнее количество метилизобутилкетонового раствора кислоты. Пробирку ополаскивали 1,5 мл метилизобутилкетона, который добавляли к суспензии. Через еще 6 часов кристаллы отфильтровывали, промывали метилизобутилкетоном (2 мл) и сушили при пониженном давлении при 40°С. Получали в общей сложности 258 мг кристаллической соли, что соответствует выходу приблизительно 87%.
Метод 7
Соединение А (2,36 г; смотри выше Подготовительный пример А) растворяли в метилизобутилкетоне (90 мл). К раствору добавляли затравочные кристаллы Соединения А (10 мг) в виде соли этансульфоновой кислоты (смотри Методы 4-6 выше) и затем двумя порциями добавляли этансульфоновую кислоту (40 мкл). Затем дополнительно добавляли затравочные кристаллы (12 мг) и две порции этансульфоновой кислоты (2×20 мкл). Суспензию разбавляли метилизобутилкетоном (15 мл), затем добавление этансульфоновой кислоты продолжали. Общее количество добавленной порциями в течение 1 часа этансульфоновой кислоты составляло 330 мкл. Добавляли небольшое количество затравочных кристаллов и, наконец, оставляли суспензию на ночь при перемешивании. На следующий день кристаллы отфильтровывали, промывали метилизобутилкетоном (2×6 мл) и сушили при пониженном давлении при 40°С. После сушки получали в общей сложности 2,57 г белого кристаллического продукта, что соответствует выходу 89%.
Методы 8 и 9: реактивные кристаллизации (с антирастворителем)
Метод 8
Соединение А (163 мг; смотри выше Подготовительный пример А) растворяли в изопропаноле (1,2 мл). Раствор нагревали до 35°С. Добавляли этансульфоновую кислоту (28 мкл). Затем добавляли этилацетат (4,8 мл) и в этот раствор вводили затравку кристаллического Соединения А в виде соли этансульфоновой кислоты (смотри Методы 4-7 выше). Почти сразу же начиналась кристаллизация. Суспензию оставляли в течение приблизительно 80 минут при 35°С, затем оставляли остывать до температуры окружающей среды (21°С). Через два часа кристаллы отфильтровывали, трижды промывали этилацетатом (3×0,4 мл) и сушили при пониженном давлении при 40°С. Получали в общей сложности 170 мг кристаллического продукта, указанного в заголовке, что соответствует выходу приблизительно 82%.
Метод 9
Соединение А (20,0 г; смотри выше Подготовительный пример А) растворяли в изопропаноле (146,6 мл) при 40°С и к раствору добавляли этансульфоновую кислоту (3,46 мл; 95%; 1 экв.). К полученному в результате прозрачному раствору добавляли затравочные кристаллы Соединения А в виде соли этансульфоновой кислоты (50 мг; смотри Методы 4-8 выше). Затем в течение 10 минут добавляли этилацетат (234 мл). В полученный слегка мутный раствор еще раз вводили затравку (70 мг) и оставляли в течение 1 часа при 40°С при перемешивании, чтобы началась кристаллизация. После этого добавляли этилацетат общим количеством 352 мл при постоянной скорости в течение 1 часа. Когда весь этилацетат был добавлен, суспензию оставляли на 1 час, затем охлаждали до 21°С в течение 2 часов. Кристаллизации давали возможность продолжаться в течение 1 часа при 21°С, затем кристаллы отфильтровывали, дважды промывали этилацетатом (50 мл + 60 мл) и, наконец, сушили при пониженном давлении при 40°С в течение ночи. Получали в общей сложности 21,6 г белой кристаллической соли, что соответствует выходу приблизительно 90%.
Соединение А в виде соли этансульфоновой кислоты было охарактеризовано посредством ЯМР, как изложено ниже: 23 мг соли растворяли в дейтерированном метаноле (0,7 мл) для спектроскопии. Использовали сочетание 1D (одномерных) (1H, 13C и селективная NOE (спектроскопия ЯМР с ядерным эффектом Оверхаузера)) и 2D (двумерных) (gCOSY, gHSQC и gHMBC) ЯМР-экспериментов. Все данные имели хорошую степень соответствия с теоретической структурой соли, приведенной ниже. Молекула в метаноле существует в двух конформациях. Исходя из интеграла пика, соответствующего Н5 (основной конформер), и пика, соответствующего Н5′ (другой конформер), было установлено, что соотношение между двумя конформерами составляет 70:30. Н22 нельзя было наблюдать, так как происходил быстрый обмен этих протонов с растворителем CD3OD.
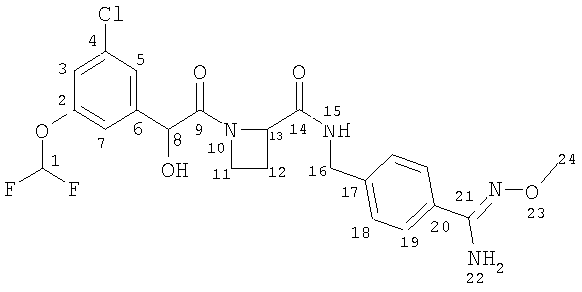
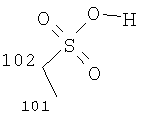
Как протонный, так и углеродный резонансы, соответствующие положению 1, расщепляются из-за спин-спинового взаимодействия с двумя ядрами фтора в этом положении. Константы взаимодействия составляют 2JHF=73 Гц и 1JCF=263 Гц.
Распределение химических сдвигов в спектре 1H и 13С ЯМР и протон-протонные корреляции представлены в таблице 2.
1′
117,5д
6.88 (t)
2′
153.5
3′
119.7
7.13 (s)
4′
135.9
5′
124.9
7.31 (s)
6′
145.3
7′
117.2
7.15 (s)
8′
74.0
5.12 (s)
9′
173.8
11′
49.0
б: 4.21 (m)
а: 4.06 (m)
б: 3.99 (m)
12′
23.2
б: 2.29 (m)
а: 2.70 (m)
б: 2.15 (m)
13′
66.2
5.22 (m)
14′
173.6
15′
8.79 (t, br)
5.2
16′
43.6
4.46 (AB-структура)
4.53 (AB-структура)
4.49 (AB-структура)
15.9
15.9
15.9
17′
147.0
18′
129.1
7.57 (d)
7.8
19′
129.4
7.70 (d)
7.8
20′
124.9
21′
162.3
б Относительно резонанса растворителя при 3.30 м.д.
HRMS: рассчитано для C24H29ClF2N4O8S (М-Н)- 605,1284, найдено 605,1296.
Кристаллы Соединения А в виде соли этансульфоновой кислоты (полученные одним или более из вышеприведенных Методов 4-9) были проанализированы посредством XRPD, и результаты приведены ниже в таблице (Таблица 3) и представлены на Фиг.1.
При DSC наблюдали эндотерму с экстраполированной температурой начала плавления приблизительно 131°С. TGA показал потерю массы приблизительно 0,2% (мас./мас.) вблизи точки плавлении. DSC анализ, повторенный с образцом, имеющим более низкое содержание растворителя, показал температуру начала плавления приблизительно 144°С.
Метод 10: получение аморфного Соединения А в виде соли бензолсульфоновой кислоты
Соединение А (199 мг; смотри выше Подготовительный пример А) растворяли в этаноле (2 мл). Бензолсульфоновую кислоту (1 экв. 90%; 70 мг) растворяли в пробирке в этаноле (1 мл). Этанольный раствор кислоты добавляли к раствору Соединения А, а пробирку ополаскивали 1 мл этанола, который затем добавляли к смеси. Смесь перемешивали в течение нескольких минут, а затем выпаривали этанол до образования масла. Добавляли этилацетат (3 мл) и растворитель снова выпаривали досуха. Образовывалось аморфное твердое вещество.
Методы 11-13: получение кристаллического Соединения А,
соли бензолсульфоновой кислоты
Метод 11: кристаллизация аморфного вещества
Аморфное Соединение А в виде соли бензолсульфоновой кислоты (20,7 мг; смотри Метод 10 выше) суспендировали в этилацетате (600 мкл). Через 5 суток в суспензии наблюдались игольчатые кристаллы.
Методы 12 и 13: реактивная кристаллизация
Метод 12
Соединение А (128 мг; смотри выше Подготовительный пример А) растворяли в этилацетате (3 мл). В раствор вводили затравку суспензии из вышеприведенного Метода 11. Затем добавляли бензолсульфоновую кислоту (1 экв., 90%; 45 мг). Сразу же происходило осаждение соли бензолсульфоновой кислоты. Изопропанол добавляли к суспензии (0,8 мл) и в смесь снова вводили затравку. Через 2 дня вещество превращалось в игольчатые кристаллы. Суспензию фильтровали, промывали этилацетатом (3×0,2 мл) и сушили в течение короткого периода в вакууме при 40°С. Получали в общей сложности приблизительно 140 мг белого твердого вещества.
Метод 13
Соединение А (246 мг; смотри Подготовительный пример А выше) растворяли в изопропаноле (1,52 мл). Добавляли бензолсульфоновую кислоту (88 мг; 90%). К прозрачному раствору добавляли этилацетат (3 мл), а затем в смесь вводили затравку, чтобы инициировать кристаллизацию. Через 1 час дополнительно добавляли этилацетат (2,77 мл). Наконец, суспензию оставляли кристаллизоваться в течение ночи, затем кристаллы отфильтровывали, промывали этилацетатом (3×0,3 мл) и сушили при 40°С в вакууме. Получали в общей сложности 279 мг соли, что соответствует выходу приблизительно 86%.
Соль бензолсульфоновой кислоты соединения А характеризовали посредством ЯМР, как изложено ниже: 20 мг соли растворяли в дейтерированном метаноле (0,7 мл). Использовали сочетание 1D (1Н,13С и селективная NOE) и 2D (gCOSY, gHSQC и gHMBC) ЯМР-экспериментов. Все данные хорошо согласовывались с теоретической структурой соли, показанной ниже. В метаноле молекула существует в двух конформациях. Было найдено исходя из интеграла пика, соответствующего Н12 (основной конформер), и пика, соответствующего Н12' (другой конформер), что соотношение между двумя конформерами составляло 70:30. Н22 нельзя было наблюдать, так как происходил быстрый обмен этих протонов с растворителем CD3OD.
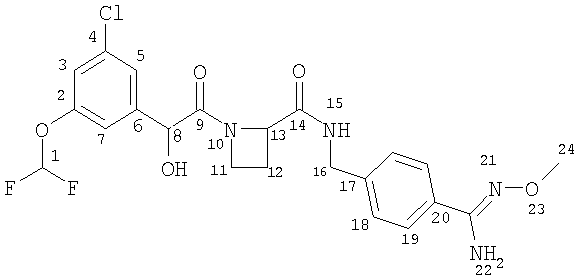
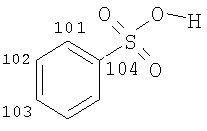
Как протонный, так и углеродный резонансы, соответствующие положению 1, расщеплены из-за спин-спинового взаимодействия с двумя ядрами фтора в этом положении. Константы взаимодействия составляют 2JHF=74 Гц и 1JCF=260 Гц.
Распределение химических сдвигов в спектре 1H и 13С ЯМР и протон-протонные корреляции представлены в таблице 4.
1′
117.5д
6.87 (t)
2′
153.5
3′
119.7
7.12 (s)
4′
135.9
5′
124.9
7.31 (s)
6′
145.3
7′
117.2
7.14 (s)
8′
74.0
5.12 (s)
9′
173.8
11′
49.0
б:4.20 (m)
a:4.05 (m)
б:3.98 (m)
12′
23.2
б:2.28 (m)
a:2.69 (m)
б:2.14 (m)
13′
66:2
5.22 (m)
14′
173.6
15′
8.78 (t, br)
5,3
16′
43.6
4.44 (AB-структура)
4.51 (AB-структура)
4.46 (AB-структура)
16,0 и 4,8
16,0
16,0
17′
147.0
18′
129.2
7.56 (d)
8,3
19′
129.4
7.69 (d)
8,3
20′
124.9
21′
162.4
HRMS: рассчитано для C28H29CIF2N4O8S (M-H)- 653,1284, найдено 653,1312.
Кристаллы Соединения А в виде соли бензолсульфоновой кислоты (полученные способом одним или более из вышеприведенных Методов 11-13) были проанализированы посредством XRPD, и результаты приведены ниже в таблице (Таблица 5) и представлены на Фиг.2.
При DSC наблюдали эндотерму с экстраполированной температурой начала плавления приблизительно 152°С. TGA показал потерю массы приблизительно 0,1% (мас./мас.) вблизи точки плавления.
Метод 14: получение аморфного Соединения А в виде соли н-пропансульфоновой кислоты
Соединение А (186 мг; смотри Подготовительный пример А выше) растворяли в изопропаноле (1,39 мл) и добавляли н-пропансульфоновую кислоту (1 экв.; 95%; 39 мкл). Добавляли этилацетат (5,6 мл) и растворитель выпаривали до образования аморфного твердого вещества.
Методы 15 и 16: получение кристаллического Соединения А.
соли н-пропансульфоновой кислоты
Метод 15: кристаллизация аморфного вещества
Аморфное Соединение А в виде соли н-пропансульфоновой кислоты (20 мг; смотри Метод 14 выше) растворяли в изопропаноле (60 мкл) и добавляли изопропилацетат (180 мкл). Через трое суток наблюдались игольчатые кристаллы.
Метод 16: реактивная кристаллизация
Соединение А (229 мг; смотри Подготовительный пример А выше) растворяли в изопропаноле (1,43 мл). Добавляли н-пропансульфоновую кислоту (1 экв., 95%; 48 мкл). Добавляли этилацетат (2 мл) и затем в раствор вводили затравку кристаллической соли из вышеприведенного Метода 15. Дополнительно добавляли этилацетат (5 мл) и суспензию оставляли в течение ночи для кристаллизации. Кристаллы отфильтровывали, промывали этилацетатом (3×0,3 мл) и сушили в вакууме при 40°С.
Соединение А в виде соли н-пропансульфоновой кислоты характеризовали посредством ЯМР, как изложено ниже: 13 мг соли растворяли в дейтерированном метаноле (0,7 мл) для спектроскопии. Использовали сочетание 1D (1Н, 13С) и 2D (gCOSY) ЯМР-экспериментов. Все данные хорошо согласовывались с теоретической структурой соли, показанной ниже. Молекула в метаноле существует в двух конформациях. Исходя из интеграла пика, соответствующего Н12 (основной конформер), и пика, соответствующего Н12′ (другой конформер), было установлено, что соотношение между двумя конформерами составляет 65:35. Н22 нельзя было наблюдать, так как происходил быстрый обмен этих протонов с растворителем CD3OD.
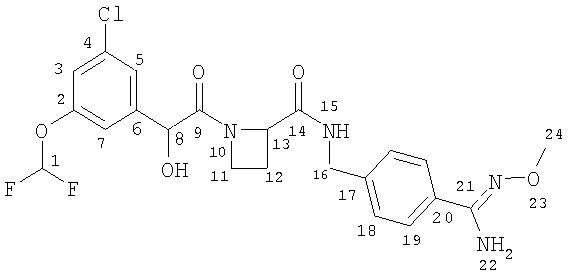
Как протонный, так и углеродный резонанс, соответствующие положению 1, расщеплены из-за спин-спинового взаимодействия с двумя ядрами фтора в этом положении. Константы взаимодействия составляют 2JHF=74 Гц и 1JCF=260 Гц.
Распределение химических сдвигов в спектре 1H и 13С ЯМР и протон-протонные корреляции представлены в Таблице 6.
б Относительно резонанса растворителя при 3.30 м.д.
HRMS: рассчитано для C25H31ClF2N4O8S (M-H)- 619,1441, найдено 619,1436.
Кристаллы Соединения А в виде соли н-пропансульфоновой кислоты (полученные одним или более вышеприведенным Методом 15 и 16) анализировали посредством XRPD, и результаты приведены ниже в таблице (Таблица 7) и представлены на Фиг.3.
При DSC наблюдали эндотерму с экстраполированной температурой начала плавления приблизительно 135°С. TGA показал отсутствие потери массы вблизи точки плавления.
Метод 17
Метод 17-А: получение аморфного Соединения А.
соли н-бутансульфоновой кислоты
Аморфное Соединение А (277 мг) растворяли в IPA (1,77 мл) и добавляли бутансульфоновую кислоту (приблизительно 1 экв.; 70 мкл). Добавляли этилацетат (6 мл) и растворитель выпаривали до образования сухого аморфного твердого вещества.
Метод 17-Б: получение кристаллического Соединения А.
соли бутансульфоновой кислоты
Аморфное Соединение А в виде соли бутансульфоновой кислоты (71,5 мг; смотри получение выше) суспендировали в этилацетате (500 мкл) в течение ночи. Кристаллы отфильтровывали и сушили на воздухе.
Соединение А в виде соли бутансульфоновой кислоты характеризовали посредством ЯМР, как изложено ниже: 21,6 мг соли растворяли в дейтерированном диметилсульфоксиде (0,7 мл) и исследовали при помощи 1H и 13С ЯМР спектроскопии.
Эти спектры очень похожи на спектры других солей этого соединения и хорошо согласуются со структурой, изображенной ниже. Большинство резонансов в спектрах представлены в виде совокупностей двух пиков из-за медленного вращения вокруг связи С9-N10, что приводит к одновременному существованию в растворе двух атропоизомеров. Это продемонстрировано и для других солей этого соединения.
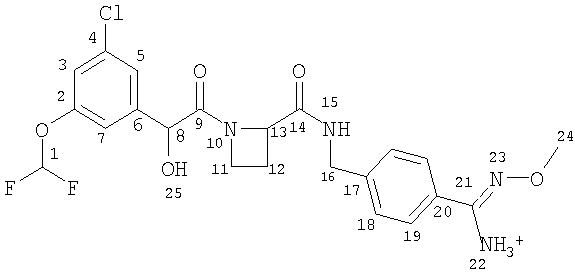

Два ядра фтора в положении 1 вызывают расщепление резонансов протона и углерода в этом положении. Константы взаимодействия составляют 2JHF=73 Гц и 1JCF=258 Гц.
Химические сдвиги протонов и атомов углерода представлены в таблице 8. Протоны в положении 22 и 24 не обнаруживаются из-за химического обмена. В протонном спектре присутствует очень широкий максимум между 8 и 9 м.д., соответствующий этим протонам.
1'
116.3г
7.28 (t)
73 (2JHF)
2′
151.3
na
na
3′
117.6
7.21 (t)д
nd
4′
133,4
na
na
5′
123.6
7.25 (t)д
nd
6′
145.2
na
na
7′
116.1
7.12 (t)д
nd
8′
71.2′
4.99 (s)
na
9′
171.1
na
na
11′
46.9
3.85 (m)
nd
12′
21.7
а:2.60 (m) б:2.02 (m)
nd
13′
63.9
5.12(m)
nd
14′
171.0
na
na
16′
42.0
4.38 (m)
nd
127.6
7.44
nd
24'
63.3
3.82 (s)
па
6 Относительно резонанса растворителя при 3.30 м.д.
HRMS: рассчитано для С26Н32ClF2N4O8S (М-Н)- 633,1597, найдено 633,1600.
Кристаллы соли н-бутансульфоновой кислоты Соединения А (полученные, как описано в Методе 17-Б выше) анализировали посредством XRPD, и результаты приведены ниже в таблице (таблица 9) и представлены на Фиг.4.
При DSC наблюдали эндотерму с экстраполированной температурой начала плавления приблизительно 118°С, a TGA показал приблизительно 0,04%-ную потерю массы.
Метод 18: получение солей Соединения Б
Метод 18-А: общий способ получения солей
Следующий общий способ использовали для получения солей Соединения Б: 200 мг Соединения Б (смотри выше Подготовительный пример Б) растворяли в 5 мл MIBK (метилизобутилкетона). К этому раствору добавляли раствор соответствующей кислоты (1,0 или 0,5 молярного эквивалента, как указано в таблице 10), растворенной в 1,0 мл MIBK. После перемешивания в течение 10 минут при комнатной температуре растворитель удаляли при помощи роторного испарителя. Оставшееся твердое вещество повторно растворяли приблизительно в 8 мл смеси ацетонитрил:Н2O (1:1). В результате лиофилизации в каждом случае получали бесцветное аморфное вещество.
Используемые кислоты:
эзилат (этансульфоновая кислота)
безилат (бензолсульфоновая кислота)
циклогексилсульфамат
сульфат
бромид
пара-толуолсульфонат
2-нафталинсульфонат
гемисульфат
метансульфонат
нитрат
гидрохлорид
Соответствующие характеристики показаны в таблице 10.
531,1
641,0
531,1
689,2
531,2
710,4
613,1
531,1
703,1
531,1
739,3
(1:2)
630,85
(1:1)
631,0
627,1
594,0
569,0
Все соли, образовавшиеся в этом Методе, являлись аморфными.
Метод 18-Б
Кроме того, аморфные соли Соединения Б получали, используя методики, аналогичные описанным в Методе 18-А выше, со следующими кислотами:
1,2-этандисульфоновая (0,5 соль)
1S-камфорсульфоновая
(+/-)-камфорсульфоновая
пара-ксилолсульфоновая
2-мезитиленсульфоновая
сахариновая
малеиновая
фосфорная
D-глутаминовая
L-аргинин
L-лизин
L-лизин ×HCl
Метод 18-В: получение аморфного Соединения Б.
соли геми-1,5-нафталиндисульсЬоновой кислоты
Аморфное Соединение Б (110,9 мг) растворяли в 2,5 мл 2-пропанола и добавляли 0,5 эквивалента тетрагидрата 1,5-нафталиндисульфоновой кислоты (растворенного в 1 мл 2-пропанола). Образец перемешивали в течение ночи. При помощи микроскопии наблюдали лишь небольшие частицы (аморфные) или капли масла. Образец упаривали досуха.
Метод 18-Г: получение кристаллического Соединения Б.
соли геми-1,5-нафталиндисульфоновой кислоты
Кристаллизацию проводили при температуре окружающей среды. Аморфное Соединение Б (0,4 г) растворяли в этаноле (1,5 мл) и добавляли 0,5 эквивалента тетрагидрата 1,5-нафталиндисульфоновой кислоты (1,35 г; 10% в этаноле). Затем добавляли гептан (0,7 мл) до тех пор, пока раствор не становился слегка замутненным. Приблизительно через 15 минут раствор становился мутным. Приблизительно через 30 минут получали жидкую суспензию и дополнительно добавляли гептан (1,3 мл). Затем суспензию оставляли в течение ночи для созревания. Для разбавления густой суспензии добавляли смесь этанола и гептана (1,5 мл и 1,0 мл соответственно). Приблизительно через 1 час суспензию фильтровали и кристаллы промывали смесью этанола и гептана (1,5:1) и окончательно чистым гептаном. Кристаллы сушили при температуре окружающей среды в течение 1 суток. Масса сухих кристаллов составляла 0,395 г.
Метод 18-Д: получение кристаллического Соединения Б.
соли геми-1,5-нафталиндисульфоновой кислоты
Аморфное Соединение Б (1,009 г) растворяли в 20 мл 2-пропанола и 20 мл этилацетата. По каплям добавляли 351,7 мг тетрагидрата 1,5-нафталиндисульфоновой кислоты, растворенного в 20 мл 2-пропанола. Приблизительно за 5 минут происходило осаждение. Суспензию перемешивали в течение ночи и затем фильтровали.
Метод 18-Е: получение кристаллического Соединения Б.
соли геми-1,5-нафталиндисульфоновой кислоты
430,7 мг соли 1,5-нафталиндисульфоновой кислоты растворяли в 30 мл 1-пропанола. Для растворения вещества раствор нагревали до кипения. Раствор оставляли в течение ночи при температуре окружающей среды для кристаллизации и затем кристаллы отфильтровывали.
Метод 18-Ж: получение кристаллического Соединения Б.
соли геми-1,5-нафталиндисульфоновой кислоты
Маточный раствор из Метода 18-Е упаривали и твердый остаток (61,2 мг) растворяли в 6 мл смеси ацетонитрил/1-пропанол в соотношении 2:1. Раствор оставляли в течение ночи при температуре окружающей среды для кристаллизации и затем кристаллы отфильтровывали.
Метод 18-3: получение кристаллического Соединения Б.
соли геми-1,5-нафталиндисульфоновой кислоты
Образец из Метода 18-В растворяли приблизительно в 2 мл метанола. Этанол (приблизительно 3 мл) добавляли в качестве антирастворителя при температуре окружающей среды и добавляли затравки. Кристаллизации не происходило, поэтому растворитель выпаривали (приблизительно половину количества) и добавляли новую порцию этанола (приблизительно 2 мл) и затравки. Кристаллические частицы образовывались при перемешивании при температуре окружающей среды в течение ночи.
Метод 18-И: получение кристаллического Соединения Б.
соли геми-1,5-нафталиндисульфоновой кислоты
Аморфное Соединение Б (104,1 мг) растворяли в 2-пропаноле (1,5 мл) и добавляли 1-эквивалент тетрагидрата 1,5-нафталиндисульфоновой кислоты, растворенного в 2-пропаноле. Общее количество 2-пропанола составляло приблизительно 2,5 мл. Раствор перемешивали при 44°С в течение приблизительно 80 минут, и образовывался осадок. Частицы были кристаллическими согласно микроскопии в поляризованном свете. Образец фильтровали.
Метод 18-К: получение кристаллического Соединения Б.
соли геми-1,5-нафталиндисульфоновой кислоты
Соединение Б в виде соли геми-1,5-нафталиндисульфоновой кислоты (56,4 мг), растворяли в 1,5 мл метанола. Добавляли метилэтилкетон (3 мл). К раствору добавляли затравки, и начиналась кристаллизация. Кристаллы отфильтровывали, промывали метилэтилкетоном и сушили на воздухе.
Метод 18-Л: получение кристаллического Соединения Б.
соли геми-1,5-нафталиндисульфоновой кислоты
Аморфное Соединение Б (161,0 мг) растворяли в 3,5 мл 1-бутанола и раствор нагревали до 40°С. В другом химическом стакане растворяли 57,4 мг тетрагидрата нафталиндисульфоновой кислоты в 3 мл 1-бутанола. Пару капель этого раствора кислоты добавляли к раствору Соединения Б. Затем к раствору добавляли затравки и через 2 часа медленно добавляли остаток раствора кислоты (при 40°С). Затем температуру медленно понижали до комнатной температуры и образец оставляли при перемешивании в течение ночи. Суспензию фильтровали, промывали 1-бутанолом и сушили в вакууме при 44°С в течение 2 часов. Выход составлял 83%.
Характеристика
Кристаллы соли геми-1,5-нафталиндисульфоновой кислоты Соединения Б, полученные приведенным выше Методом 18-Г, характеризовали посредством ЯМР, как изложено ниже.
21,3 мг соли растворяли в дейтерированном метаноле, 0,7 мл исследовали при помощи ЯМР-спектроскопии. Использовали сочетание 1D (1Н, 13С и селективных NOE) и 2D (gCOSY, gHSQC и gHMBC) ЯМР-экспериментов. Все данные хорошо согласуются с предполагаемой структурой, изображенной ниже. Определены все углероды и протоны, присоединенные к углеродам. Протоны, присоединенные к гетероатомам, обмениваются на дейтерий из растворителя и не обнаруживаются. Большинство резонансов в 1D 1H и 13С ЯМР спектрах представлены в виде совокупности двух пиков. Причиной этого служит медленное вращение вокруг C9-N10 связи, что приводит к одновременному существованию в растворе двух атропоизомеров, 1D NOE эксперимент является доказательством этого. Когда излучается резонансная частота одного атропоизомера, насыщение переносится к соответствующему пику другого атропоизомера. Резонансные частоты, соответствующие противоиону 1,5-нафталиндисульфоната, не демонстрируют атропоизомерии.
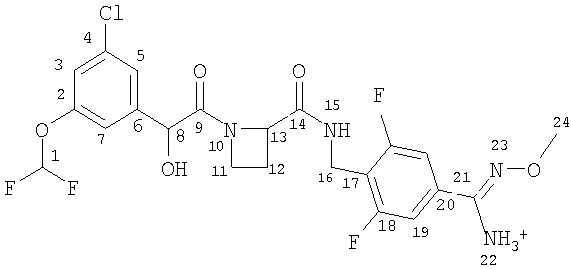
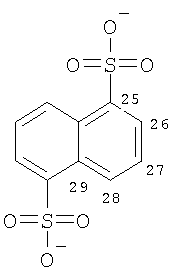
В молекуле присутствуют четыре атома фтора. Они вызывают расщепление резонансов для некоторых протонов и углеродов. Как протонный, так и углеродный резонанс, соответствующие положению 1, расщеплены из-за спин-спинового взаимодействия с двумя ядрами фтора в этом положении. Константы взаимодействия составляют 2JHF=73 Гц и 1JCF=263 Гц. Кроме того, протонный резонанс, соответствующий Н19, является искаженным дублетом с 3JHF=6,9 Гц из-за спин-спинового взаимодействия с ядрами фтора в положении 18. Углеродные резонансы, соответствующие С17, С18, С19 и С20, также демонстрируют взаимодействия с этими ядрами фтора. Резонансы С17 и С20 представляют собой триплет с 2JCF=19 Гц и 3JCF=11 Гц соответственно. Резонанс С18 представляет собой дублет дублетов с константами взаимодействия 1JCF=251 Гц и 3JCF=8 Гц. Резонанс С19 представляет собой мультиплет.
Сравнение значений резонансных интегралов, соответствующих противоиону 1,5-нафталиндисульфоната и исходному соединению, дает стехиометрическое соотношение: один 1,5-нафталиндисульфонатный противоион, кристаллизованный с двумя молекулами родительского соединения.
Распределение химических сдвигов в 1H и 13С ЯМР-спектрах и протон-протонные корреляции представлены в таблице 11.
1′
117.5д
6.87 (t)
73 (2JCF)
nd
2′
153.3
na
na
na
3′
119.6
7.11 (t)°
nd
5′, 7′
4′
135.8
na
na
na
5′
124.9
7.28 (t)°
nd
3′, 7′
6′
145.3
na
na
na
7′
117.1
7.12 (t)°
nd
3′, 5′
8′
73.6
5.07 (s)
na
nd
9′
173.5
na
na
na
11′
48.6
(m)
a:4.01 (m) б:3.93
(m)
nd
12′, 13′
12′
22.8
(m)
а:2.61 (m) б:2.03
(m)
nd
11′, 13′
13′
65.8
5.14 (dd)
9,4
5,6 и
9,1
12′
14′
173.2
na
na
na
16′
32.5
4.51 (m)
nd
nd
21′
159.9
na
na
na
24′
64.8
3.92 (s)
na
nd
7,2
б Относительно резонанса растворителя при 3.30 м.д.
Кристаллы соли геми-1,5-нафталиндисульфоновой кислоты Соединения Б (полученные приведенным выше Методом 18-И) анализировали посредством XRPD, и результаты приведены ниже в таблице (таблица 12) и представлены на Фиг.5.
При DSC наблюдали эндотерму с экстраполированной температурой начала плавления приблизительно 183°С, a TGA показал приблизительно 0,3%-ную потерю массы между 25 и 110°С.
Аббревиатуры
Ас = ацетил
APCI = химическая ионизация при атмосферном давлении (в отношении MS)
API = ионизация при атмосферном давлении (в отношении MS)
водн. = водный
АОТ = аэрозоль диизооктилсульфосукцината натрия
Aze(&(S)-Aze) = (S)-азетидин-2-карбоксилат (если не указано иного)
Boc = трет-бутилоксикарбонил
br = широкий (в отношении ЯМР)
Cl = химическая ионизация (в отношении MS)
d = сутки
а = дублет (в отношении ЯМР)
DCC = дициклогексилкарбодиимид
dd = дублет дублетов (в отношении ЯМР)
DIBAL-H = диизобутилалюминия гидрид
DIPEA = диизопропилэтиламин
DMAP = 4-(N,N-диметиламино)пиридин
DMF = N,N-диметилформамид
DMSO = диметилсульфоксид
DSC = дифференциальная сканирующая калориметрия
DVT = тромбоз глубоких вен
EDC = 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид
экв. = эквивалент
ES = электроспрей
ESI = электроспрей-интерфейс
Et = этил
эфир = диэтиловый эфир
EtOAc = этилацетат
EtOH = этанол
Et2O = диэтиловый эфир
FT-IR = инфракрасная спектроскопия с Фурье-преобразованием
gCOSY = градиент-селективная корреляционная спектроскопия
gHMBC = градиент-селективная гетероядерная корреляция кратных связей
gHSQC = градиент-селективная гетероядерная одноквантовая корреляция
HATU = O-(азабензотриазол-1-ил)-N,N,N′,N′-тетраметилурония гексафторфосфат
HBTU = [N,N,N′,N′-тетраметил-O-(бензотриазол-1-ил)урония гексафторфосфат]
HCl = соляная кислота, газообразный хлороводород или солянокислая соль (в зависимости от контекста)
Hex = гексаны
НОАс = уксусная кислота
HPLC = высокоэффективная жидкостная хроматография
IPA = изопропанол
LC = жидкостная хроматография
m = мультиплет (в отношении ЯМР)
Me = метил
МеОН = метанол
мин = минута(ы)
MS = масс-спектроскопия
МТВЕ = метил-трет-бутиловый эфир
МОЕ = ядерный эффект Оверхаузера
NMR (ЯМР) = ядерный магнитный резонанс
ОАс = ацетат
Pab = пара-амидинобензиламино
Н-Pab = пара-амидинобензиламин
Pd/C = палладий на угле
Ph = фенил
РуВОР = (бензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат
q = квартет (в отношении ЯМР)
QF = тетрабутиламмония фторид
rt/RT = комнатная температура
s = синглет (в отношении ЯМР)
solutol = 12-гидроксистеарат PEG 660 (неионное поверхностно-активное вещество)
t = триплет (в отношении ЯМР)
TBTU = [N,N,N′,N′-тетраметил-O-(бензотриазол-1-ил)урония тетрафторборат]
TEA = триэтиламин
Теос = 2-(триметилсилил)этоксикарбонил
TEMPO = 2,2,6,6-тетраметил-1-пиперидинилокси свободный радикал
TFA = трифторуксусная кислота
TGA = термогравиметрический анализ
THF = тетрагидрофуран
TLC = тонкослойная хроматография
UV = ультрафиолетовый
XRPD = дифракция рентгеновских лучей на порошке
Приставки n-, s-, i-, t- и трет- имеют свои обычные значения: нормальный, вторичный, изо и третичный.
Изобретение иллюстрируется посредством следующих Примеров. Пример 1
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 50/5/45 (мас./мас.)% с последующим осторожным перемешиванием. Эту композицию перорально вводили собакам посредством желудочного зонда один раз в сутки в течение 5 суток. Доза 150 мкмоль/кг давала максимальные концентрации в плазме в интервале 118-254 мкМ (118-254 мкмоль/л) после первой дозы и 186-286 мкМ (186-286 мкмоль/л) после пятой дозы.
Пример 2
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 50/5/45 (мас./мас.)% с последующим осторожным перемешиванием. Эту композицию перорально вводили крысам посредством желудочного зонда один раз в сутки в течение 5 суток. Доза 400 мкмоль/кг давала максимальные концентрации в плазме в интервале 3,17-6,91 мкМ (3,17-6,91 мкмоль/л) после первой дозы и 3,01-10,5 мкМ (3,01-10,5 мкмоль/л) после пятой дозы.
Пример 3
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 50/5/45 (мас./мас.)% с последующим осторожным перемешиванием. Эту композицию перорально вводили крысам посредством желудочного зонда один раз в сутки в течение 5 суток. Доза 800 мкмоль/кг давала максимальные концентрации в плазме в интервале 7,00-23,9 мкМ (7,00-23,9 мкмоль/л) после первой дозы и 10,3-32,8 мкМ (10,3-32,8 мкмоль/л) после пятой дозы.
Пример 4
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 50/5/45 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 1000 раз выше, чем в одной воде.
Пример 5
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 20/10/70 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 100 раз выше, чем в одной воде.
Пример 6
Вода содержала 50 мкмоль/мл винной кислоты
Препарат готовили путем растворения Соединения А в подкисленной смеси PEG 400/этанол/вода 20/10/70 (мас./мас.)% с последующим осторожным перемешиванием. Значение рН этого раствора составляло 3,6. Растворимость Соединения А в этом носителе по меньшей мере в 250 раз выше, чем в одной воде.
Пример 7
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 30/5/65 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшой мере в 200 раз выше, чем в одной воде.
Пример 8
Препарат готовили путем растворения Соединения А в подкисленной смеси PEG 400/этанол/вода 30/5/65 (мас./мас.)% с последующим осторожным перемешиванием. рН этого раствора доводили до 3,6 посредством добавления HCl. Растворимость Соединения А в этом носителе по меньшей мере в 400 раз выше, чем в одной воде.
Пример 9
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 40/5/55 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 600 раз выше, чем в одной воде.
Пример 10
Препарат готовили путем растворения Соединения А в подкисленной смеси PEG 400/этанол/вода 40/5/55 (мас./мас.)% с последующим осторожным перемешиванием. рН этого раствора доводили до 3,8 посредством добавления HCl. Растворимость Соединения А в этом носителе по меньшей мере в 1000 раз выше, чем в одной воде. Препараты Соединения А в этом носителе стабильны по меньшей мере в течение 3 месяцев при <-15°С.
Пример 11
до рН 3,7 сколько требуется Препарат готовили путем растворения Соединения А в смеси гидроксипропил-β-циклодекстрин/вода 40/60 (мас./мас.)% с последующим осторожным перемешиванием. рН этого раствора доводили до 4,7 добавлением HCl. Растворимость Соединения А в этом носителе по меньшей мере в 700 раз выше, чем в одной воде.
Пример 12
Препарат готовили путем растворения Соединения А в смеси гидроксипропил-β-циклодекстрин/вода 28/72 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 400 раз выше, чем в одной воде.
Пример 13
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/solutol™/вода 50/5/5/40 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 80 раз выше, чем в одной воде.
Пример 14
Препарат готовили путем растворения Соединения А в PEG 400 с последующим осторожным перемешиванием по меньшей мере в течение 1 часа, после этого добавляли воду до конечного объема. Растворимость Соединения А в этом носителе по меньшей мере в 200 раз выше, чем в одной воде.
Пример 15
Вода содержала 50 мкмоль/мл винной кислоты
Препарат готовили путем растворения Соединения А в PEG 400 с последующим осторожным перемешиванием по меньшей мере в течение 1 часа, после этого добавляли воду до конечного объема. Растворимость Соединения А в этом носителе по меньшей мере в 250 раз выше, чем в одной воде.
Пример 16
Препарат готовили путем растворения Соединения А в PEG 400 с последующим осторожным перемешиванием по меньшей мере в течение 1 часа, после этого добавляли воду до конечного объема. Растворимость Соединения А в этом носителе по меньшей мере в 300 раз выше, чем в одной воде.
Пример 17
Препарат готовили путем растворения Соединения А в PEG 400 с последующим осторожным перемешиванием по меньшей мере в течение 1 часа, после этого добавляли воду до конечного объема. Растворимость Соединения А в этом носителе по меньшей мере в 400 раз выше, чем в одной воде.
Пример 18
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 45/1/54 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 450 раз выше, чем в одной воде.
Пример 19
Препарат готовили путем растворения Соединения А в подкисленной смеси PEG 400/этанол/вода 45/1/54 (мас./мас.)% с последующим осторожным перемешиванием. рН этого раствора доводили до 4,2 при помощи HCl. Растворимость Соединения А в этом носителе по меньшей мере в 800 раз выше, чем в одной воде.
Пример 20
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 45/2/53 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 500 раз выше, чем в одной воде.
Пример 21
Препарат готовили путем растворения Соединения А в подкисленной смеси PEG 400/этанол/вода 45/2/53 (мас./мас.)% с последующим осторожным перемешиванием. рН этого раствора доводили до 4,3 добавлением HCl. Растворимость Соединения А в этом носителе по меньшей мере в 800 раз выше, чем в одной воде.
Пример 22
Препарат готовили путем растворения Соединения А в носителе с последующим осторожным перемешиванием по меньшей мере в течение 1 часа. Растворимость Соединения А в этом носителе по меньшей мере в 230 раз выше, чем в одной воде.
Пример 23
Препарат готовили путем растворения Соединения А в носителе с последующим осторожным перемешиванием по меньшей мере в течение 1 часа. Растворимость Соединения А в этом носителе по меньшей мере в 150 раз выше, чем в одной воде.
Пример 24
Препарат готовили путем растворения Соединения А в небольшом объеме двойного эквимолярного количества HCl с последующим осторожным перемешиванием и разбавлением до 1 мл. рН конечного раствора доводили до 3,6. Растворимость Соединения А в этом носителе по меньшей мере в 20 раз выше, чем в одной воде.
Пример 25
Препарат готовили путем растворения Соединения А в воде и добавляли HCl для получения рН 1, после этого раствор осторожно перемешивали. рН конечного раствора доводили до 3,0 при помощи NaOH. Растворимость Соединения А в этом носителе по меньшей мере в 40 раз выше, чем в одной воде. Этот препарат перорально вводили крысам при сравнительном исследовании кинетики.
Пример 26
Препарат готовили путем растворения Соединения А в 1 мл смеси DMA/миглиол с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 4000 раз выше, чем в одной воде.
Пример 27
Препарат готовили путем растворения Соединения А в 1 мл смеси этанол/миглиол с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 4000 раз выше, чем в одной воде.
Пример 28
Препарат готовили путем растворения Соединения А в 1 мл этанола с последующим осторожным перемешиванием. Вещество стабильно в этом препарате в течение более чем 1 недели.
Пример 29
Для получения наночастиц использовали исходный раствор приблизительно 100 мМ Соединения А в этаноле. Также включали 25%-ный (мас./мас.) миглиол в пересчете на количество вещества. Растворы разбавляли 1/10 раствором стабилизатора, содержащего 0,2 (мас./мас.)% PVP (поливинилпирролидона) и 0,25 мМ SDS (додецилсульфата натрия) в воде. Смешивание, которое считается критическим параметром при получении наночастиц, было быстрым и немедленным. Раствор лекарственного средства быстро вводили в раствор стабилизатора во время обработки ультразвуком. После разбавления 1/10 в водном растворе получали наночастицы размером приблизительно 150 нм. Через 6 часов при комнатной температуре размеры частиц не изменялись.
Пример 30
Препарат готовили путем растворения Соединения А в смеси физиологический раствор/этанол/solutol 90/5/5 (мас./мас.)% с последующим осторожным перемешиванием. Раствор перорально вводили крысам, и концентрация в плазме Соединения Г через 1 час составляла 0,56 мкмоль/л. Раствор подкожно вводили крысам, и концентрации g плазме Соединений Г и А через 1 час составляли 0,24 мкмоль/л и 0,6 мкмоль/л соответственно.
Пример 31
Препарат готовили путем растворения Соединения Б в смеси физиологический раствор/этанол/solutoi 90/5/5 (мас./мас.)% с последующим осторожным перемешиванием. Раствор перорально вводили крысам, и концентрации в плазме Соединения Б и Соединения Д составляли соответственно 0,07 мкмоль/л и 0,65 мкмоль/л спустя 1 час. Раствор подкожно вводили крысам, и концентрации в плазме Соединений Б и Д составляли через 1 час 0,4 мкмоль/л и 0,3 мкмоль/л соответственно.
Пример 32
Препарат готовили путем растворения Соединения В в смеси физиологический раствор/этанол/solutol 90/5/5 (мас./мас.)% с последующим осторожным перемешиванием. Раствор перорально вводили крысам, и концентрации в плазме Соединения В и Соединения Е составляли через 1 час соответственно 0,2 мкмоль/л и 0,5 мкмоль/л. Раствор подкожно вводили крысам, и концентрации в плазме Соединений В и Е через 1 час составляли 0,35 мкмоль/л и 0,5 мкмоль/л соответственно.
Пример 33
Препарат готовили путем растворения соли Соединения Г в 1 мл физиологического раствора с последующим осторожным перемешиванием.
Пример 34
Препарат готовили путем растворения соли Соединения Г в 1 мл смеси физиологический раствор/этанол с последующим осторожным перемешиванием.
Пример 35
Препарат готовили путем растворения соли Соединения Г в 1 мл смеси физиологический раствор/этанол с последующим осторожным перемешиванием. Раствор подкожно вводили крысам, и через 1 час концентрация в плазме Соединения Г составляла 0,55 мкмоль/л.
Пример 36
Препарат готовили путем растворения соли Соединения Д в 1 мл смеси физиологический раствор/этанол с последующим осторожным перемешиванием. Раствор подкожно вводили крысам, и через 1 час концентрация в плазме Соединения Д составляла 0,75 мкмоль/л.
Пример 37
Препарат готовили путем растворения соли Соединения Е в 1 мл смеси физиологический раствор/этанол с последующим осторожным перемешиванием. Раствор подкожно вводили крысам, и через 1 час концентрация в плазме Соединения Е составляла 0,92 мкмоль/л.
Пример 38
Препарат готовили путем растворения соли Соединения Д в 1 мл
физиологического раствора с последующим осторожным перемешиванием.
Пример 39
Препарат готовили путем растворения соли Соединения Е в 1 мл
физиологического раствора с последующим осторожным перемешиванием.
Пример 40
Раствор готовили путем растворения избытка Соединения А в виде эзилата в 3 мл воды с последующим осторожным перемешиванием в течение ночи. После фильтрации наблюдалась конечная концентрация раствора 14 мг/мл при рН 2,7.
Пример 41
Раствор готовили путем растворения 112 мг Соединения А в виде эзилата в 3 мл буферного раствора фосфата натрия с последующим осторожным перемешиванием в течение ночи. После фильтрации наблюдалась конечная концентрация раствора 33 мг/мл при рН 2,7.
Пример 42
Раствор готовили путем растворения 20 мг Соединения А в виде эзилата в 3 мл буферного раствора фосфата натрия с последующим осторожным перемешиванием в течение ночи. После фильтрации наблюдалась конечная концентрация раствора 1,6 мг/мл при рН 6,5.
Пример 43
Следующие лиофильно высушенные препараты могут быть приготовлены в соответствии с методиками, описанными в одном или более приведенном выше Примере 1-29:
а)
б)
в)
г)
д)
е)
ж)
з)
Растворы, возможно, стерильно фильтровали, например, через мембранный фильтр с диаметром пор 0,22 мкм. Растворы (стерильные или иные) разливали в подходящие сосуды (например, ампулы) и препараты лиофильно высушивали, используя стандартное оборудование. Ампулы могут быть запаяны в установке для лиофильной сушки в атмосфере азота.
Пример 44
Эксципиенты и лекарственное средство смешивали и гранулировали с поливинилпирролидоном К90, растворенным в воде. Затем гранулы сушили в сушильном шкафу. Гранулят делали скользким при помощи стеарилфумарата натрия и прессовали в таблетки, используя эксцентриковый пресс.
Три отдельные таблетки тестировали на высвобождение лекарственного средства в 900 мл среды, используя аппарат 2 для растворения по USP (Фармакопея США) (мешалка+корзина1) при 50 об/мин и 37°С. Используемые среды для растворения представляли собой 0,1 М соляную кислоту (рН 1) и 0,1 М буферный раствор фосфата натрия (рН 6,8). Оперативный количественный анализ проводили, используя С Technologies оптиковолоконную систему с аналитической длиной волны 220 нм, когда в качестве среды для растворения использовали 0,1 М HCl, и 260 нм, когда в качестве среды для растворения использовали фосфатный буфер с рН 6,8. В качестве эталонной длины волны с обеими средами использовали 350 нм. В течение первых двух часов анализа величину высвобождения измеряли каждые 15 минут, а затем каждый час на протяжении оставшегося времени анализа. Результаты представлены в таблице ниже.
[1 Специально сделанная квадратная корзина из сетчатой проволоки, припаянной одной из верхних узких сторон к концу стального стержня. Стержень пропущен через крышку сосуда для растворения и зафиксирован посредством двух тефлоновых гаек на расстоянии 3,2 см от центра сосуда. Нижний край днища корзины установлен на расстоянии 1 см выше мешалки. Корзина с тестируемой таблеткой, установленной на ее краю, ориентирована вдоль течения потока].
Пример 45
Эксципиенты и лекарственное средство смешивали и гранулировали с поливинилпирролидоном К90, растворенным в воде. Затем гранулы сушили в сушильном шкафу. Гранулят делали скользким стеарилфумаратом натрия и прессовали в таблетки, используя эксцентриковый пресс.
Пример 46
Эксципиенты и лекарственное средство смешивали и гранулировали с поливинилпирролидоном К90, растворенным в воде. Затем гранулы сушили в сушильном шкафу. Гранулят делали скользким стеарилфумаратом натрия и прессовали в таблетки, используя эксцентриковый пресс.
Пример 47
Эксципиенты и лекарственное средство смешивали и гранулировали с поливинилпирролидоном К90, растворенным в воде. Затем гранулы сушили в сушильном шкафу. Гранулят делали скользким стеарилфумаратом натрия и прессовали в таблетки, используя эксцентриковый пресс.
Пример 48
Препарат готовили путем растворения Соединения А в подкисленном PEG 414 с последующим осторожным перемешиванием.
Пример 49
Препарат готовили путем растворения Соединения А в подкисленном PEG 300 с последующим осторожным перемешиванием.
Пример 50
Препарат готовили путем растворения Соединения А в подкисленном PEG 200 с последующим осторожным перемешиванием.
Пример 51
Препарат готовили путем растворения Соединения Ж в смеси физиологический раствор/этанол/solutol 90/5/5 (мас./мас.)% с последующим осторожным перемешиванием.
Пример 52
Препарат готовили путем растворения Соединения К в смеси физиологический раствор/этанол/solutol 90/5/5 (мас./мас.)% с последующим осторожным перемешиванием.
Пример 53
Препарат готовили путем растворения Соединения 3 в смеси физиологический раствор/этанол/solutol 90/5/5 (мас./мас.)% с последующим осторожным перемешиванием.
Пример 54
Препарат может быть приготовлен в соответствии с приведенным выше Примером 47.
Пример 55
Препарат может быть изготовлен в соответствии с приведенным выше Примером 47.
Пример 56
Препарат может быть изготовлен в соответствии с приведенным выше Примером 47.
Пример 57
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 25/10/65 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 100 раз выше, чем в одной воде. Препарат стабилен в морозильной камере в течение по меньшей мере 2 месяцев.
Пример 58
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 50/10/40 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 2000 раз выше, чем в одной воде.
Пример 59
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 40/10/50 (мас./мас.)% с последующим осторожным перемешиванием. Растворимость Соединения А в этом носителе по меньшей мере в 1500 раз выше, чем в одной воде.
Пример 60
Препарат готовили путем растворения Соединения А в этаноле с последующим осторожным перемешиванием, после этого добавляли лимонную кислоту и воду до конечного объема, а рН доводили до 3,2. Растворимость Соединения А в этом носителе по меньшей мере в 100 раз выше, чем в одной воде. Препарат стабилен в морозильной камере по в течение меньшей мере 1 месяца.
Пример 61
Препарат готовили путем растворения Соединения А и лимонной кислоты в физиологическом растворе с последующим осторожным перемешиванием. рН доводили до 3,6. Препарат стабилен в морозильной камере в течение по меньшей мере 3 месяцев.
Пример 62
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 40/5/55 (мас./мас.)%, содержащей лимонную кислоту, с последующим осторожным перемешиванием и доведением рН до 3,6. Препарат стабилен в морозильной камере в течение по меньшей мере 1 месяца.
Пример 63
Препарат готовили путем растворения Соединения А в смеси PEG 400/этанол/вода 20/5/75 (мас./мас.)%, содержащей лимонную кислоту, с последующим осторожным перемешиванием и доведением рН до 3,2.
Пример 64
Винная кислота: количество, эквимолярное компоненту А (уксуснокислой соли Соединения Г) плюс избыток 5 мМ
Препарат готовили путем растворения Соединения Г в подкисленной смеси PEG 400/этанол/вода 40/5/55 (мас./мас.)% с последующим осторожным перемешиванием. рН этого раствора доводили до 3,6 путем добавления HCl. Препараты Соединения Г в этом носителе стабильны в течение по меньшей мере 2 месяцев при <-15°С.
Пример 65
НРМС суспендировали в горячей воде и добавляли расплавленный Solutol при сильном перемешивании. Этот раствор охлаждали и добавляли Соединение А при сильном перемешивании с получением хорошо диспергированной суспензии.
Пример 66
НРМС суспендировали в горячей воде и добавляли расплавленный Solutol при сильном перемешивании. Этот раствор охлаждали и добавляли Соединение А (безилат) при сильном перемешивании с получением хорошо диспергированной суспензии.
Пример 67
Препарат готовили путем растворения Соединения А и лимонной кислоты в физиологическом растворе и осторожного перемешивания. рН доводили до 3,6. Препарат стабилен в морозильной камере в течение по меньшей мере 3 месяцев.
Пример 68
Для получения наночастиц использовали исходный раствор приблизительно 100 мМ Соединения Б в этаноле. Включали также 25%-ный (мас./мас.) миглиол в пересчете на количество вещества. Растворы разбавляли 1/10 раствором стабилизатора, содержащего 0,2 (мас./мас.)% PVP и 0,25 мМ SDS в воде. Критическая стадия смешивания была быстрой и немедленной. Раствор лекарственного средства быстро вводили в раствор стабилизатора во время обработки ультразвуком. После разбавления 1/10 в водном растворе получали наночастицы размером приблизительно 110 нм. Через 6 часов при комнатной температуре размеры частиц не изменялись.
Вместо этанола возможно может быть использован DMA (N,N-диметилацетамид), миглиол может быть исключен, и разбавление может быть больше (1/20). Посредством различных сочетаний могут быть получены частицы размером в интервале 100-300 нм.
Пример 69
Препарат готовили путем растворения Соединения Б в смеси PEG 400/этанол/вода 50/5/45 (мас./мас.)% с последующим осторожным перемешиванием. Препараты Б (при 0,5 мг/мл) в этом носителе стабильны в течение по меньшей мере 1 месяца при <-15°С.
Пример 70
Препарат готовили путем растворения Соединения Б в смеси PEG 400/этанол/вода 60/5/35 (мас./мас.)% с последующим осторожным перемешиванием.
Пример 71
НРМС суспендировали в горячей воде и добавляли расплавленный Solutol при сильном перемешивании. Этот раствор охлаждали и добавляли Соединение Б при сильном перемешивании с получением хорошо диспергированной суспензии.
Пример 72
Препарат готовили путем растворения Соединения Д в 9 мг/мл NaCl путем осторожного перемешивания. Полученное в этом препарате значение рН составляет 8-9.
Пример 73
Препарат готовили путем растворения Соединения В в смеси PEG 400/этанол/вода 50/5/45 (мас./мас.)% с последующим осторожным перемешиванием. Препараты В (при 0,5 мг/мл) в этом носителе стабильны в течение по меньшей мере 1 месяца при комнатной температуре и ниже.
Пример 74
Препарат готовили путем растворения Соединения В в смеси гидроксипропил-β-циклодекстрин/вода 20/80 (мас./мас.)% с последующим осторожным перемешиванием. Препараты В в этом носителе стабильны в течение по меньшей мере 2 недель при <8°С.
Пример 75
Препарат готовили путем растворения Соединения Е в 9 мг/мл NaCI путем осторожного перемешивания. Полученное в этом препарате значение рН составляет 3-4. Препараты Соединения Е в этом носителе стабильны в течение по меньшей мере 2 недель при комнатной температуре и ниже,
Пример 76
Таблетку готовили в соответствии с общим способом Примера 44.
Данные по высвобождению
Данные получали в соответствии с общим способом Примера 44, но используя 500 мл среды и 75 об/мин.
Пример 77
Таблетку готовили в соответствии с общим способом Примера 44.
Могут быть изготовлены другие препараты, в которых количество безилата Соединения А находится в интервале 50-300 мг; при этом соотношение других компонентов аналогично соотношениям в Примере 77.
Пример 78
Таблетку готовили в соответствии с общим способом Примера 44.
Могут быть изготовлены другие препараты, в которых используют 100 мг или 200 мг соли геми-нафталин-1,5-дисульфоновой кислоты Соединения Б; при этом соотношение других компонентов аналогично соотношениям в Примере 78.
Данные по биологической активности соединений
Ингибирующая активность соединений по изобретению в отношении тромбина была подтверждена в следующих тестах.
Тест А
Определение тромбинового времени (ТТ)
Раствор ингибитора (25 мкл) инкубируют с плазмой (25 мкл) в течение 3 минут. Затем добавляют тромбин человека (Т 6769, Sigma Chem. Co или Hematologic Technologies) в буферном растворе рН 7,4 (25 мкл, 4,0 NIH единицы/мл) и измеряют время свертывания на автоматическом приборе (КС 10, Amelung).
Тромбиновое время (ТТ) выражают в абсолютных значениях (секундах), равно как и в виде отношения ТТ без ингибитора (ТТ0) к ТТ с ингибитором (TT1). Строят кривые зависимости последних отношений (интервал 1-0) от концентрации ингибитора (в log-преобразовании) и осуществляют подгонку к сигмоидальным кривым дозового ответа согласно уравнению:
y=а/[1+(х/lC50)s]
где: а = максимальный интервал, то есть 1; s = наклон кривой дозового ответа; и IC50 = концентрация ингибитора, при которой происходит удвоение времени свертывания. Вычисления выполняются на ПК с использованием программного обеспечения GraFit Version 3 с установочными параметрами уравнения, равными: старт от 0, окончание = 1 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, Лондон, Соединенное Королевство).
Тест В
Определение частичного активированного тромбопластинового времени (APTT)
АРТТ определяют в совокупной нормальной цитратной плазме человека с помощью реагента РТТ Automated 5, производимого Stago. Ингибиторы добавляют к плазме (10 мкл раствора ингибитора к 90 мкл плазмы), инкубируют с АРТТ-реагентом в течение 3 минут с последующим добавлением 100 мкл раствора хлорида кальция (0,025М) и определяют АРТТ, используя анализатор коагуляции KC10 (Amelung) в соответствии с инструкциями производителя реагента.
Время свертывания выражают в абсолютных значениях (секундах), равно как и в виде отношения АРТТ без ингибитора (АРТТ0) к АРТТ с ингибитором (APTTi). Строят кривые зависимости последних отношений (интервал 1-0) от концентрации ингибитора (в log-преобразовании) и осуществляют подгонку к сигмоидальным кривым дозового ответа согласно уравнению:
y=а/[1+(х/lC50)s]
где: а = максимальный интервал, то есть 1; s = наклон кривой дозового ответа; и IC50 = концентрация ингибитора, при которой происходит удвоение времени свертывания. Вычисления выполняются на ПК с использованием программного обеспечения GraFit Version 3 с установочными параметрами уравнения, равными: старт от 0, окончание = 1 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, Лондон, Соединенное Королевство).
IC50АРТТ определяют как концентрацию ингибитора в плазме человека, увеличивающую в два раза частичное активированное тромбопластиновое время.
Тест С
Определение тромбинового времени ex vivo
Ингибирование тромбина после перорального введения соединений по изобретению, растворенных в смеси этанол/Солютол K/вода (5:5:90) исследуют на находящихся в сознании крысах, которых за один или два дня до эксперимента снабжают катетером для отбора крови из сонной артерии. В день эксперимента в фиксированное время после введения соединения отбирают образцы крови в пластиковые пробирки из расчета 1 часть раствора цитрата натрия (0,13 моль в л) и 9 частей крови. Пробирки центрифугируют с целью получения обедненной по тромбоцитам плазмы. Образцы плазмы в объеме 50 мкл осаждают 100 мкл охлажденного ацетонитрила. Образцы центрифугируют в течение 10 минут при 40000 об/мин. 75 мкл супернатанта разбавляют 75 мкл 0,2%-ной муравьиной кислоты. Получающиеся растворы в объеме 10 мкл анализируют с помощью LC-MS/MS и, используя стандартные кривые, определяют концентрации ингибитора тромбина.
Результаты биологических тестов
Соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab, соединение Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab в форме трифторуксуснокислой соли и соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF) демонстрировали значения IC50TT в тесте А меньше 0,02 мкМ.
Соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab и соединение Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab в форме трифторуксуснокислой соли демонстрировали значения IC50APTT в Тесте В меньше 1 мкМ.
Анализ в соответствии с вышеприведенным Тестом С подтвердил, что соединения являются биодоступными при пероральном введении крысам.
Предложены следующие конкретные аспекты изобретения:
1. Фармацевтический препарат с немедленным высвобождением, содержащий в качестве активного ингредиента соединение формулы (I):
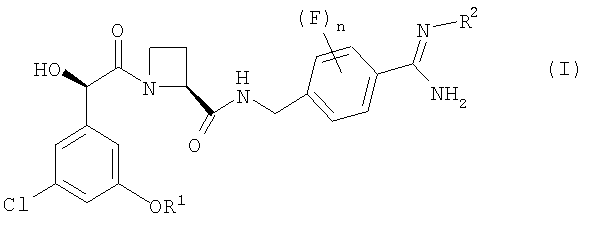
где
R1 представляет собой С1-2алкил, замещенный одним или более чем одним заместителем фторо;
R2 представляет собой водород, гидрокси, метокси или этокси; и
n равно 0, 1 или 2;
или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель;
при условии, что препарат не содержит исключительно:
- раствор одного активного ингредиента и воду;
- раствор одного активного ингредиента и диметилсульфоксид; или
- раствор одного активного ингредиента в смеси этанол:12-гидроксистеарат PEG 660:вода в соотношении 5:5:90.
2. Фармацевтический препарат с немедленным высвобождением, как он определен в аспекте 1, в котором активный ингредиент представляет собой:
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OMe);
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab;
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OH);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OH);
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab; или
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OH).
3. Твердый фармацевтический препарат с немедленным высвобождением, как он определен в аспекте 1, в котором активный ингредиент представляет собой:
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe);
РЬ(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-(S)Aze-Pab(2,6-диF)(ОМе); или
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe),
или их фармацевтически приемлемую соль.
4. Твердый фармацевтический препарат с немедленным высвобождением, как он определен в аспекте 1, в котором активный ингредиент представляет собой Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe) или его соль С1-6алкансульфоновой кислоты или, возможно, замещенной арилсульфоновой кислоты.
5. Инъекционный фармацевтический препарат с немедленным высвобождением, как он определен в аспекте 1, где активный ингредиент представляет собой:
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab;
Ph(3-Cl)(5-OCHF2)-(Р)СН(ОН)С(O)-(S)Aze-Pab(2,6-диF); или
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab.
6. Применение препарата, как он описан в аспекте 1, в качестве лекарства.
7. Применение препарата, как он описан в аспекте 1, в изготовлении лекарства для лечения сердечно-сосудистого расстройства.
8. Способ лечения сердечно-сосудистого расстройства у пациента, страдающего указанным расстройством или имеющего риск указанного расстройства, который включает введение пациенту терапевтически эффективного количества фармацевтического препарата, как он описан в аспекте 1.
9. Способ изготовления фармацевтического препарата с немедленным высвобождением, как он описан в аспекте 1.
10. Соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(ОН).
Также предложен препарат, который можно получить посредством любого из Методов и/или Примеров, описанных здесь.
Изобретение относится к области медицины и фармацевтики и касается пероральной таблеточной композиции с немедленным высвобождением, содержащей в качестве активного ингредиента соединение формулы (1) и фармацевтически приемлемый разбавитель или носитель. Изобретение обеспечивает повышенную скорость высвобождения соединения формулы (1). 8 з.п. ф-лы, 23 табл.
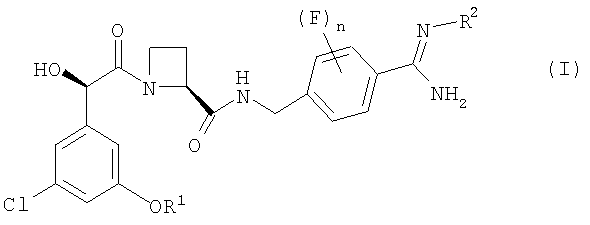
1. Пероральная фармацевтическая таблеточная композиция с немедленным высвобождением для антикоагулянтной терапии, содержащая в качестве активного ингредиента соединение формулы (I)
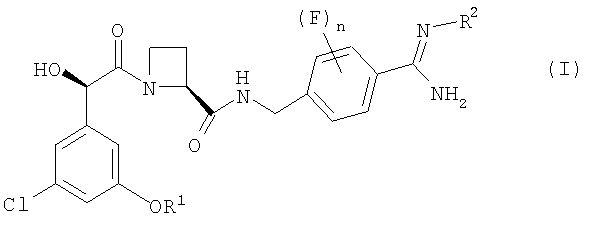
где R1 представляет собой -CHF2 или -CH2CH2F;
R2 представляет собой водород, гидрокси, метокси или этокси; и
n равно 0 или 2; или его фармацевтически приемлемую соль; и по меньшей мере один фармацевтически приемлемый разбавитель или носитель в количестве вплоть до 79% (мас./мас.) от конечной композиции, выбранный из одноосновного фосфата кальция, двухосновного фосфата кальция (включая дигидрат двухосновного фосфата кальция и ангидрат двухосновного фосфата кальция), трехосновного фосфата кальция, лактозы, микрокристаллической целлюлозы, силикатированной микрокристаллической целлюлозы, маннита, сорбита, крахмала (такого как кукурузный, картофельный или рисовый), глюкозы, лактата кальция и карбоната кальция; где активный ингредиент и другие возможные разбавители или эксципиенты дополняют композицию до 100% (мас./мас.).
2. Фармацевтическая композиция с немедленным высвобождением по п.1, где фармацевтически приемлемая соль соединения формулы (I) представляет собой соль присоединения кислоты.
3. Фармацевтическая композиция с немедленным высвобождением по п.1, где активный ингредиент выбран из
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OMe);
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab;
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OH);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OH);
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab; или
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OH),
или фармацевтически приемлемой соли любого из них.
4. Композиция по п.1, где активный ингредиент представляет собой кристаллическую соль соединений
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-(S)Aze-Pab(2,6-диF)(ОМе); или
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe).
5. Композиция по п.1, где активный ингредиент представляет собой соль присоединения этансульфоновой кислоты, н-пропансульфоновой кислоты, бензолсульфоновой кислоты, 1,5-нафталиндисульфоновой кислоты или н-бутансульфоновой кислоты Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe) или Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-(S)Aze-Pab(2,6-диF)(ОМе).
6. Композиция по любому из пп.1-5, где активный ингредиент представляет собой соль бензолсульфоновой кислоты Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe).
7. Композиция по любому из пп.1-5, где активный ингредиент представляет собой соль бензолсульфоновой кислоты Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe), характеризующуюся картиной дифракции рентгеновских лучей на порошке, характеризующейся пиками со значениями d равными 5,9, 4,73, 4,09 и 4,08 Å.
8. Композиция по любому из пп.1-5, где активный ингредиент представляет собой соль геми-1,5-нафталиндисульфоновой кислоты Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OMe).
9. Композиция по любому из пп.1-5, где активный ингредиент представляет собой соль геми-1,5-нафталиндисульфоновой кислоты Ph(3-CI)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OMe), характеризующуюся картиной дифракции рентгеновских лучей на порошке, характеризующейся пиками со значениями d равными 18,3, 9,1, 5,6, 5,5, 4,13, 4,02, 3,86, 3,69 и 3,63 Å.
| Патрон для нарезки резьбы на сверлильном станке | 1929 |
|
SU13671A1 |
| Способ осаждения мути из сернокислотной вытяжки титаножелезных руд | 1934 |
|
SU42059A1 |
| ИНГИБИТОРЫ ТРОМБИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2172741C2 |

