Настоящее изобретение относится к фармацевтической композиции с замедленным высвобождением действующего вещества, содержащей 3-(3-диметиламино-1-этил-2-метилпропил)фенол или одну из его фармацевтически приемлемых солей в соответствующей матрице.
3-(3-диметиламино-1-этил-2-метилпропил)фенол известен из заявки ЕР 0693475 В1 в качестве обладающего анальгетическим действием лекарственного средства и может назначаться для перорального применения. При использовании обычных композиций, предназначенных для перорального введения 3-(3-диметиламино-1-этил-2-метилпропил)фенола, это действующее вещество быстро высвобождается в желудочно-кишечном тракте, вследствие чего практически сразу же проявляется его анальгетическая эффективность. Вместе с тем наблюдается такое же быстрое прекращение подобного действия. Тем самым до настоящего времени лечение сильных хронических болей с помощью 3-(3-диметиламино-1-этил-2-метилпропил)фенола предусматривает необходимость вводить это лекарственное средство в организм через относительно короткие промежутки времени, например до четырех-шести раз в день с целью обеспечить таким путем достаточную концентрацию действующего вещества в плазме крови пациента. Однако подобная необходимость прибегать к частой дозировке легко приводит к ошибкам при приеме препарата, а также к нежелательным колебаниям в концентрации действующего вещества в крови, что, в свою очередь, отрицательно сказывается на соблюдении пациентом режима и схемы лечения и на терапевтическом эффекте, прежде всего при лечении хронических болей. Из сказанного ясно, что существует настоятельная потребность в создании фармацевтической, предназначенной для перорального применения лекарственной формы с замедленным, постепенным высвобождением (ретард-формы, т.е. формы пролонгированного действия) действующего вещества 3-(3-диметиламино-1-этил-2-метилпропил)фенол.
Из уровня техники широко известны ретард-композиции, состав которых включает множество разнообразных действующих веществ. Обычными ретард-формами являются, в частности, формы с покрытием, обеспечивающим замедленное высвобождение действующего вещества, и формы с матрицей, также обеспечивающей замедленное высвобождение действующего вещества.
В первом случае (ретард-формы с покрытием, описанные, например, в заявке DE 3625458 А1) на содержащую действующее вещество сердцевину фармацевтической композиции наносят обеспечивающее замедленное высвобождение действующего вещества покрытие из одного либо нескольких гидрофильных и/или гидрофобных полимеров.
Во втором случае (ретард-формы с матрицей) действующее вещество заключено в образованную из одного либо нескольких носителей матрицу, которая регулирует высвобождение действующего вещества. Так, например, в заявке DE 3309516 А1 описан способ получения композиций с матрицей с использованием в качестве носителя гидроксипропилметилцеллюлозы (ГПМЦ) и с частично замедленным высвобождением действующего вещества, при этом на долю носителя приходится не более одной трети массы композиции, и он состоит по меньшей мере из какого-либо одного типа гидроксипропилметилцеллюлозы, которая содержит 16-24 мас.% метоксигрупп и 4-32 мас.% гидроксипропила и среднечисленная молекулярная масса которой составляет по меньшей мере 50000. В указанной заявке DE 3309516 А1 описаны композиции, содержащие ГПМЦ с вязкостью (в 2 мас.%-ном водном растворе при 20°С) от 15 до 30000 сП (от 15 до 30000 мПа·с). Однако в этой публикации ничего не говорится о специфике высвобождения действующего вещества вне зависимости от значения рН среды растворения.
Исходя из вышеизложенного, в основу настоящего изобретения была положена задача получить и предложить содержащую 3-(3-диметиламино-1-этил-2-метилпропил)фенол фармацевтическую композицию с замедленным высвобождением действующего вещества.
Эта задача решается с помощью фармацевтической композиции с замедленным высвобождением действующего вещества, которая содержит 3-(3-диметиламино-1-этил-2-метилпропил)фенол или одну из его фармацевтически приемлемых солей в матрице, которая обеспечивает замедленное высвобождение действующего вещества и содержит от 1 до 80 мас.%, предпочтительно от 5 до 80 мас.%, одного либо нескольких гидрофильных или гидрофобных полимеров в качестве образующих ее фармацевтически приемлемых веществ и которая характеризуется следующей скоростью высвобождения действующего вещества in vitro (измерения рекомендуемым согласно Европ. Фармакопее методом с использованием мешалки при частоте ее вращения 75 об/мин в буферном растворе (также согласно Европ. Фармакопее) при значении рН 6,8 при температуре 37°С и обнаружение с помощью УФ-спектрометрии):
3-35 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 0,5 часа,
5-50 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 1 часа,
10-75 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 2 часов,
15-82 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 3 часов,
30-97 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 6 часов,
более 50 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 12 часов,
более 70 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 18 часов,
более 80 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 24 часов.
Как неожиданно было установлено, действующее вещество 3-(3-диметиламино-1-этил-2-метилпропил)фенол высвобождается из предлагаемой в изобретении композиции при пероральном применении постепенно, с замедлением, что позволяет вводить эту композицию в организм в интервалы времени по меньшей мере в 12 часов. Тем самым предлагаемая в изобретении композиция может применяться для терапии болей, при которой частоту введения анальгетика - 3-(3-диметиламино-1-этил-2-метилпропил)фенола - с целью обеспечить достаточную концентрацию этого действующего вещества в плазме можно сократить до одноразового приема в сутки, например с интервалом в 24 часа, или же до двухразового приема в сутки, предпочтительно с интервалами в 12 часов. Продолжительность действия анальгетика и поддержание требуемого его уровня в плазме крови подтверждаются данными проводившихся одновременно анализов и экспериментальными исследованиями.
Особенно неожиданным при этом оказался тот факт, что предлагаемая в изобретении композиция не только благодаря замедленному высвобождению активного вещества обеспечивает продолжительное терапевтическое действие в течение относительно длительного промежутка времени (по меньшей мере в течение 12 часов), но и позволяет одновременно при первом же приеме лекарственного средства быстро достичь необходимой концентрации активного вещества в плазме, что сразу же приводит к ослаблению боли у пациента ("быстрая атака"). Тем самым при введении в организм страдающего от боли пациента предлагаемой в изобретении композиции удается быстро смягчить испытываемую им боль, сохраняя при этом дальнейшую анальгетическую эффективность. Таким образом, предлагаемая в изобретении композиция объединяет в себе свойства композиции с незамедлительным высвобождением действующего вещества - быстрое смягчение боли благодаря достижению достаточно высокой концентрации действующего вещества вскоре после приема лекарственного средства - и свойства композиции с замедленным высвобождением активного вещества - продолжительное анальгетическое действие благодаря сохранению достаточно высокого уровня активного вещества в течение длительного промежутка времени. Тем самым пациент уже при первом приеме анальгетика, содержащегося в предлагаемой согласно изобретению композиции, имеет возможность сразу же эффективно бороться со своей болью и одновременно без каких-либо других мер, а только за счет регулярного приема через каждые 12 часов (или 24 часа) успешно продолжать эту борьбу в течение определенного промежутка времени.
Действующее вещество в предлагаемой согласно изобретению композиции заключено, как указывалось выше, в матрицу, обеспечивающую замедленное высвобождение действующего вещества. Вместе с тем возможен и иной подход, а именно, действующее вещество заключено в матрицу с обычным профилем его высвобождения, а обеспечить замедленное высвобождение можно за счет нанесения соответствующего ретардирующего покрытия.
Другая возможность добиться замедленного высвобождения действующего вещества заключается в применении системы высвобождения, основанной на принципе осмоса.
В том случае, когда предлагаемая в изобретении композиция содержит матрицу с замедленным высвобождением действующего вещества, состав этой матрицы включает от 1 до 80 мас.% одного либо нескольких гидрофильных или гидрофобных полимеров в качестве фармацевтически приемлемых матрицеобразователей, таких, например, как камеди, простые и сложные эфиры целлюлозы, акриловые смолы, производные протеинов, жиры, воски, жирные спирты или эфиры жирных кислот. При использовании в качестве матрицеобразователей гидрофильных полимеров предпочтительно предусмотреть, чтобы их содержание в матрице составляло от 5 до 80 мас.%.
Еще одним объектом настоящего изобретения является фармацевтическая композиция, которая содержит 3-(3-диметиламино-1-этил-2-метилпропил)фенол или одну из его фармацевтически приемлемых солей в матрице, обеспечивающей замедленное высвобождение действующего вещества и содержащей от 1 до 80 мас.%, предпочтительно от 5 до 80 мас.%, одного либо нескольких гидрофильных или гидрофобных полимеров в качестве образующих ее фармацевтически приемлемых веществ, и которая отличается тем, что в качестве фармацевтически приемлемых матрицеобразователей она содержит простые и/или сложные эфиры целлюлозы с вязкостью в 2 мас.%-ном водном растворе при 20°С от 3000 до 150000 мПа·с (Вязкость при этом определяют посредством капиллярной вискозиметрии согласно Европ. Фармакопее). Предлагаемые композиции характеризуются вышеуказанным в соответствии с изобретением профилем высвобождения действующего вещества.
В качестве фармацевтически приемлемых матрицеобразователей предпочтительно применять простые и/или сложные эфиры целлюлозы, вязкость которых в 2 мас.%-ном водном растворе при 20°С составляет от 10000, предпочтительно от 50000 до 150000 мПа·с.
Особенно пригодные в качестве фармацевтически приемлемых матрицеобразователей выбраны из группы, включающей различные типы гидроксипропилметилцеллюлозы (ГПМЦ), гидроксиэтилцеллюлозы, гидроксипропилцеллюлозы (ГПЦ), метилцеллюлозы, этилцеллюлозы и карбоксиметилцеллюлозы, и прежде всего из группы, включающей различные типы ГПМЦ, гидроксиэтилцеллюлозы и ГПЦ. Наиболее предпочтительной из них является группа ГПМЦ с вязкостью порядка 100000 мПа·с, измеренной в 2 мас.%-ном водном растворе при 20°С.
Действующее вещество 3-(3-диметиламино-1-этил-2-метилпропил)фенол может быть представлен как таковой, т.е. в виде свободного основания, а также в виде одной из его фармацевтически приемлемых солей, например в виде гидрохлорида. Получение свободного основания известно из заявки ЕР 0693475 А1. Поскольку, однако, в данной публикации ничего не говорится о получении фармацевтически приемлемых солей, таких как гидрохлорид, следует отметить, что эти соли можно получать с помощью общеизвестных из уровня техники методов исходя из свободного основания.
3-(3-диметиламино-1-этил-2-метилпропил)фенол имеет два асимметрических центра, вследствие чего это соединение может быть представлено в форме четырех различных стереоизомеров. В предлагаемой согласно изобретению композиции 3-(3-диметиламино-1-этил-2-метилпропил)фенол может быть представлен в виде смеси всех четырех диастереомеров в любом их соотношении в смеси, вместе с тем он может быть представлен также в виде смеси двух либо трех из четырех возможных стереоизомеров или же в форме чистых стереоизомеров. Предпочтительными стереоизомерами при этом являются (+)-(1S,2S)-3-(3-диметиламино-1-этил-2-метилпропил)фенол и (-)-(1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенол, которые в предлагаемой согласно изобретению композиции могут быть представлены в виде смеси, прежде всего в виде смеси при их соотношении 1:1 (рацемат) или, что особенно предпочтительно, в форме чистых изомеров. В соответствии с этим под понятием "действующее (активное) вещество" в контексте настоящего описания имеется в виду, что 3-(3-диметиламино-1-этил-2-метилпропил)фенол может быть представлен в виде смеси его различных стереоизомеров или в виде одного из его чистых стереоизомеров, соответственно в виде свободного основания либо в виде одной из его фармацевтически приемлемых солей.
В предлагаемых согласно изобретению лекарственных средствах высвобождаемое с замедлением действующее вещество содержится предпочтительно в количестве от 0,5 до 85 мас.%, а содержание фармацевтически приемлемых матрицеобразователей составляет от 8 до 40 мас.%. К особенно предпочтительным относятся лекарственные средства, содержащие высвобождаемое с замедлением действующее вещество в количестве от 3 до 70 мас.%, прежде всего от 8 до 66 мас.%, и содержащие фармацевтически приемлемые матрицеобразователи в количестве от 10 до 35 мас.%, прежде всего от 10 до 30 мас.%. Если в качестве действующего вещества используют энантиомерочистый (т.е. в виде чистого энантиомера) (+)-(1S,2S)-3-(3-диметиламино-1-этил-2-метилпропил)фенол (или смесь (+)- и (-)-энантиомеров с большим избытком (+)-энантиомера), то особенно предпочтительно, чтобы содержание действующего вещества не превышало нижнего предела, т.е. составляло от 0,5 до 25 мас.% (в пересчете на общую массу). Если же в качестве действующего вещества используют энантиомерочистый (-)-(1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенол (или смесь (+)- и (-)-энантиомеров с большим избытком (-)-энантиомера), то особенно предпочтительно, чтобы содержание действующего вещества составляло от 16 до 66 мас.%.
В качестве других компонентов матрицы в предлагаемой согласно изобретению композиции возможно использование хорошо усваиваемых организмом (перевариваемых) длинноцепочечных (т.е. с 8-50 С-атомами, предпочтительно с 12-40 С-атомами) незамещенных либо замещенных углеводородов, таких, например, как жирные спирты, глицериловые эфиры жирных кислот, минеральные и растительные масла, а также воски, предпочтительны при этом углеводороды с температурой кипения от 25 до 90°С. Прежде всего предпочтительны жирные спирты, наиболее предпочтительными из которых являются лауриловый спирт, миристиловый спирт, стеариловый спирт, цетиловый спирт и цетилстеариловый спирт. Их содержание в матрице составляет от 0 до 60 мас.%. Вместо указанных компонентов или в дополнение к ним могут использоваться также полиэтиленгликоли, содержание которых в матрице может составлять от 0 до 60 мас.%.
В качестве других компонентов в предлагаемых согласно изобретению композициях могут содержаться далее обычно используемые в фармацевтике вспомогательные вещества, такие как наполнители, например лактоза, микрокристаллическая целлюлоза (МКЦ) или гидрофосфат кальция, а также вещества, придающие скользкость, смазывающие вещества и регуляторы текучести, например тальк, стеарат магния, стеариновая кислота и/или высокодисперсный диоксид кремния, общее содержание которых в таблетке составляет от 0 до 80 мас.%, предпочтительно от 5 до 65 мас.%.
Во многих случаях скорость высвобождения действующего вещества из готовой лекарственной формы зависит от значения рН среды, где происходит такое высвобождение. При прохождении лекарственного средства через желудочно-кишечный тракт это значение рН может колебаться в пределах от менее 1 до порядка 8. Эти колебания могут зависеть от индивидуальных особенностей человека, принимающего лекарственное средство. Более того, даже у одного и того же человека значение рН в зависимости от времени прохождения через желудочно-кишечный тракт может изменяться при каждом следующем приеме препарата. Поскольку скорость высвобождения действующего вещества из лекарственного средства зависит от значения рН, это может обусловить различную скорость такого высвобождения in vivo и тем самым различную степень биосовместимости. Следует, однако, особо подчеркнуть, что профиль высвобождения действующего вещества (в виде основания или в виде одной из его фармацевтически приемлемых солей) из предлагаемой в изобретении фармацевтической композиции неожиданно не зависит от значения рН, которое обычно подвержено воздействию определенных физиологических факторов при прохождении через желудочно-кишечный тракт. Профиль высвобождения при значениях рН окружающей среды, равных 1,2, 4,0 и 6,8, остается идентичным во всех случаях, равно как и при сравнении с высвобождением при зависящих от времени значениях рН, изменяющихся от 1,2 до 7,2 при промежуточных значениях, равных 2,3 и 6,8.
Как было установлено, для достижения замедленного высвобождения действующего вещества из предпочтительно представленной в форме таблеток предлагаемой в изобретении композиции, вне зависимости от сохранения в остальном неизменными размера и состава таблеток касательно действующего вещества, матрицеобразователя и возможных других компонентов, не играет принципиальной роли, используют ли в качестве наполнителя водорастворимый наполнитель, например лактозу, нерастворимый, не набухающий в водной среде наполнитель, например гидрофосфат кальция, или же нерастворимый, набухающий в водной среде наполнитель, например микрокристаллическую целлюлозу. Для всех подобных лекарственных средств характерен в целом аналогичный профиль высвобождения действующего вещества.
Неожиданным является далее тот факт, что при определенном, заданном количестве действующего вещества в предлагаемых согласно изобретению композициях содержание в них матрицеобразователей и возможных других компонентов может варьироваться в каждом случае в относительно широких пределах при сохранении при этом терапевтической эффективности по меньшей мере в течение 12 ч, соответственно при двукратном суточном приеме препарата (при условии, что выдерживаются указанные выше количественные пределы в отношении действующего вещества, матрицеобразователей и других возможных компонентов). Обеспечить требуемую эффективность в течение по меньшей мере 12 ч удается, например, при содержании действующего вещества порядка 32,25 мас.% (в пересчете на массу всей композиции) как в композиции из примерно 12,9 мас.%, используемой в качестве матрицеобразователя ГПМЦ с вязкостью 100000 мПа·с, и примерно 52,6 мас.%, используемой, например, в качестве наполнителя МКЦ, так и в композиции из примерно 25,8 мас.% той же ГПМЦ и примерно 39,7 мас.% МКЦ (или моногидрата лактозы), при условии, что в остальном используются одинаковые количества средств, придающих скользкость, смазывающих средств и регуляторов текучести. Подобное справедливо также для предлагаемых в изобретении композиций с более высоким или более низким содержанием действующего вещества в указанных выше пределах.
Совершенно неожиданным оказался и тот факт, что при введении пробандам предлагаемых в изобретении фармацевтических композиций с замедленным высвобождением действующего вещества, несмотря на высокий, так называемый "эффект первого прохождения" действующего вещества, вопреки ожиданиям удается достичь не изменяющейся по сравнению с композициями с быстрым высвобождением действующего вещества биодоступности.
К предпочтительным относятся далее те из предлагаемых в изобретении композиций, показатель tмакс которых на графике зависимости концентрации в плазме от времени in vivo после перорального введения соответствующей композиции составляет от 2 до 10 ч, прежде всего от 3,5 до 6 ч и наиболее предпочтительно от 4 до 5,5 ч, т.е. концентрация действующего вещества в плазме достигает максимального уровня в тот же промежуток времени.
Предлагаемая в изобретении композиция содержит действующее вещество 3-(3-диметиламино-1-этил-2-метилпропил)фенол как таковой и/или в виде фармацевтически приемлемой соли в количестве от обычно 2,5 до 800 мг, прежде всего от 5 до 400 мг, особенно предпочтительно от 10 до 250 мг (масса действующего вещества 3-(3-диметиламино-1-этил-2-метилпропил)фенола в виде гидрохлорида) из расчета на одну унифицированную дозу, при этом на профиль высвобождения действующего вещества из композиции по изобретению его точно подобранное количество не оказывает какого-либо влияния, при условии, что соблюдаются вышеуказанные пределы его содержания. Поскольку оба особенно предпочтительных энантиомера (+)-(1S,2S)-3-(3-диметиламино-1-этил-2-метилпропил)фенол и (-)-(1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенол обладают действием различной степени эффективности, целесообразно более сильный (+)-(1S,2S)-3-(3-диметиламино-1-этил-2-метилпропил)фенол использовать для предлагаемых в изобретении композиций в количестве от 2,5 до 80 мг, прежде всего от 5 до 40 мг и особенно предпочтительно в количестве от 10 до 25 мг (в пересчете на гидрохлорид), тогда как другой из них (-)-(1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенол должен быть представлен в предлагаемых композициях в количестве от 25 до 800 мг, прежде всего от 50 до 400 мг и наиболее предпочтительно в количестве от 100 до 250 мг (в пересчете на гидрохлорид), при одном условии: соблюдении вышеуказанных пределов содержания.
Под фармацевтически приемлемыми солями действующего вещества в контексте настоящего описания подразумеваются такие соли действующего вещества, которые при их фармацевтическом применении, прежде всего при введении в организм млекопитающего и/или человека, обладают физиологической совместимостью. Подобные фармацевтически приемлемые соли можно образовывать, например, с помощью неорганических или органических кислот.
Предлагаемые в изобретении фармацевтические композиции могут быть представлены как в виде простых таблеток, так и в виде таблеток с покрытием (покрытых оболочкой), например в виде филмтаблеток (таблеток с пленочным или лаковым покрытием) или драже. Обычно таблетки изготавливают круглыми и двояковыпуклыми, при этом таблетки могут иметь также продолговатую форму, позволяющую делить их на определенные части. Кроме того, композиции могут быть представлены в виде гранул, шариков, пеллет или микрокапсул, которые расфасовывают в сашé либо капсулы или из которых можно прессовать распадающиеся таблетки.
При изготовлении таблеток с покрытием это покрытие можно наносить в виде одного или нескольких слоев. Материалом для таких целей могут служить известные типы гидроксипропилметилцеллюлозы с низкой вязкостью от порядка 1 до 100 мПа·с и низкой молекулярной массой менее 10000 (например Pharmacoat 606 с вязкостью 6 мПа·с в 2 мас.%-ном водном растворе при 20°С), которые если и влияют каким-либо образом на профиль высвобождения действующего вещества из предлагаемых в изобретении лекарственных средств, то лишь в минимальной степени. Известные специалистам диффузионные покрытия, например на основе набухающих, но не растворимых в воде поли(мет)акрилатов, могут обусловить некоторые изменения в профиле замедленного высвобождения действующего вещества из фармацевтических композиций по изобретению. С учетом этого содержащую действующее вещество, высвобождающую его с замедлением сердцевину таблетки с содержанием действующего вещества предпочтительно в количестве от 0,5 до 85 мас.%, особенно предпочтительно от 3 до 70 мас.% и наиболее предпочтительно от 8 до 66 мас.%, можно дополнительно покрывать оболочкой из высвобождающегося без замедления действующего вещества в качестве начальной дозы, которую (оболочку) наносят различными, известными специалистам методами, например дражированием, напылением из растворов либо суспензий или распылением порошка, не принимая при этом никаких особых мер по достижению главной цели: обеспечить требуемое замедленное высвобождение действующего вещества при одновременном быстром его накоплении для ускоренного ослабления боли при первом приеме предлагаемой в изобретении фармацевтической композиции.
Согласно другим вариантам осуществления изобретения изготавливают многослойные таблетки и таблетки с оболочкой, из которых 3-(3-диметиламино-1-этил-2-метилпропил)фенол или одна из его фармацевтически приемлемых солей, содержащиеся в одном либо нескольких слоях многослойной таблетки в количестве предпочтительно от 0,5 до 85 мас.%, особенно предпочтительно от 3 до 70 мас.% и наиболее предпочтительно от 8 до 66 мас.%, соответственно содержащиеся в сердцевине таблетки с оболочкой в количестве предпочтительно от 0,5 до 85 мас.%, особенно предпочтительно от 3 до 70 мас.% и наиболее предпочтительно от 8 до 66 мас.%, высвобождаются с замедлением благодаря использованию фармацевтически приемлемого матрицеобразователя, а высвобождение действующего вещества из одного либо нескольких других слоев многослойной таблетки, соответственно из покровного слоя таблетки с оболочкой происходит без замедления. Многослойные таблетки и таблетки с оболочкой могут иметь один или несколько слоев, не содержащих действующее вещество.
Вместо обеспечивающей высвобождение действующего вещества с замедлением матрицы в фармацевтической композиции с замедленным высвобождением можно использовать матрицу с нормальным режимом высвобождения действующего вещества, снабдив ее ретардирующим покрытием, т.е. покрытием, обеспечивающим замедленное высвобождение. При этом действующее вещество может, например, содержаться в обычной матрице из микрокристаллической целлюлозы и других (в случае их использования) фармацевтических вспомогательных веществ, таких, в частности, как связующие, наполнители, вещества, придающие скользкость, смазывающие вещества и регуляторы текучести, с нанесенным на них покрытием из материала, позволяющего регулировать замедленное высвобождение действующего вещества в водной среде. Пригодными для указанных целей средствами покрытия являются среди прочих не растворимые в воде воски и полимеры, такие как полиметакрилаты (Eudragit и т.п.), или не растворимая в воде целлюлоза, прежде всего этилцеллюлоза. При необходимости в состав материала покрытия могут входить также водорастворимые полимеры, такие как поливинилпирролидон, водорастворимая целлюлоза, такая как гидроксипропилметилцеллюлоза или гидроксипропилцеллюлоза, другие водорастворимые средства, такие как полисорбат 80, или гидрофильные порообразователи, такие как полиэтиленгликоль, лактоза или маннит.
Наряду или в дополнение к возможностям использования в фармацевтической композиции с замедленным высвобождением действующего вещества матрицы, обеспечивающей такое замедленное высвобождение, или матрицы с нормальным режимом высвобождения действующего вещества, но снабженной ретардирующим его высвобождение покрытием, с целью достичь замедленное высвобождение действующего вещества можно предусмотреть соответствующую систему его высвобождения, основанную на принципе осмоса. В такой системе высвобождения, предпочтительно при пероральном применении, по меньшей мере одна ее поверхность, предпочтительно все ее поверхности, предпочтительно та(те) из них, которая(-ые) контактирует(-ют) или может(могут) контактировать со средой высвобождения, является(-ются) полупроницаемой(-ыми), предпочтительно снабжена(-ны) полупроницаемым покрытием, благодаря чему поверхность(-ти) способна(-ны) пропускать среду высвобождения, тогда как для действующего вещества она(они) в основном, предпочтительно полностью, остается(-ются) непроницаемой(-ыми), при этом поверхность(-ти) и/или покрытие (если таковое предусмотрено) имеет(-ют) по меньшей мере одно отверстие для высвобождения действующего вещества. Действующее вещество 3-(3-диметиламино-1-этил-2-метилпропил)фенол или его фармацевтически приемлемая соль, предпочтительно (+)-(1S,2S)-3-(3-диметиламино-1-этил-2-метилпропил)фенол или его фармацевтически приемлемая соль и/или (-)-(1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенол или его фармацевтически приемлемая соль, соответственно их смесь, может при этом - но не обязательно - находиться в матрице. Предпочтительно под вышеназванной системой подразумевается система в форме таблетки с отверстием для выделения, осмотической сердцевиной лекарственного средства, полупроницаемой мембраной и полимерной частью, обеспечивающей требуемое давление. Наглядным и предпочтительным примером подобной системы может служить система OROS® фирмы ALZA Corporation, США, более подробную информацию о которой можно найти на сайте фирмы в сети Интернет. Система OROS® включает группу таких систем, как OROS® Push-Pull™, OROS® Delayed Push-Pull™, OROS® Multi-Layer Push-Pull™, OROS® Push-Stick System, а также частично L-OROS™. Варианты и примеры конкретного получения систем высвобождения, основанных на принципе осмоса, описаны в патентах US 4765989, US 4783337 и US 4612008, которые в полном объеме включены в настоящее описание в качестве ссылки.
Предлагаемые в изобретении композиции можно получать, например, с помощью следующего общего способа.
После последовательного взвешивания требуемых количеств каждого из компонентов композиции (действующего вещества, матрицеобразователя и других возможных компонентов) их просеивают с помощью обычного грохота. В этих целях можно использовать грохот типа Quadro Comil U10, наиболее распространенный размер отверстий сита которого составляет примерно 0,813 мм. Затем просеянный материал смешивают в контейнерном смесителе, например смесителе такого типа фирмы Bohle; типичные рабочие условия: продолжительность порядка 15 мин ± 45 с при частоте вращения 20±1 об/мин. После этого из полученной порошковой смеси с помощью таблетировочного пресса прессуют таблетку. В этих целях можно использовать, например, таблетировочный пресс типа Korsch EKO, оснащенный круглым пуансоном диаметром 10 мм с вогнутой по форме драже рабочей поверхностью. Возможен и иной подход, а именно: сначала порошковую смесь уплотняют, затем уплотненный материал просеивают (последовательно через сито терочного типа Comill с размером отверстий 3 мм и через сито с круглыми отверстиями размером 1,2 мм) и после этого образующийся таким путем гранулят при добавлении смазывающих веществ (например, стеарата магния) прессуют описанным выше образом, например с помощью таблетировочного пресса типа Korsch EKO, оснащенного круглыми пуансонами диаметром 10 мм. Гранулирование можно осуществлять методом мокрой грануляции на основе водных или органических растворителей; предпочтительны при этом водные растворители с соответствующими связующими либо без таковых. Описанный способ получения можно без проблем адаптировать к конкретным требованиям с учетом особенностей получаемой лекарственной формы, используя в этих целях общеизвестные из уровня техники методы.
Получение предлагаемых в изобретении фармацевтических композиций отличается высокой воспроизводимостью характеристик высвобождения действующего вещества из получаемых составов, содержащих 3-(3-диметиламино-1-этил-2-метилпропил)фенол или одну из его фармацевтически приемлемых солей. На протяжении всего периода хранения в течение по меньшей мере одного года в нормальных условиях согласно нормам ICH Q1AR-Stability-Testing-Guideline профиль высвобождения действующего вещества из предлагаемых в изобретении лекарственных средств сохраняет, как было установлено, стабильность.
В результате ежедневного одно- или двухразового приема пациентом, страдающим продолжительными сильными болями, предлагаемой в изобретении фармацевтической композиции удается с большой степенью надежности достичь высокого терапевтического эффекта.
Примеры
Представленные ниже примеры служат иллюстрацией настоящего изобретения и предпочтительных вариантов его осуществления, не ограничивая при этом объем изобретения.
Пример 1
Состав таблеток с матрицей из расчета на одну таблетку
Указанные таблетки получали в виде партии численностью 1000 штук по следующей технологии.
Все компоненты взвешивали в требуемых количествах, затем просеивали на грохоте типа Quadro Comil U10 с размером отверстий сита 0,813 мм, смешивали с помощью контейнерного смесителя (тип Bohle LM 40) в течение 15 мин ± 15 с при частоте вращения 20±1 об/мин и из полученной смеси с помощью эксцентрикового пресса типа Korsch EKO прессовали выпуклые по типу драже таблетки диаметром 10 мм, с радиусом кривизны 8 мм и со средней массой одной таблетки 310 мг.
Высвобождение действующего вещества in vitro определяли рекомендуемым согласно Европ. Фармакопее методом с использованием мешалки при частоте ее вращения 75 об/мин в 900 мл буферного раствора при рН 6,8 (также согласно Европ. Фармакопее) при температуре 37°С и обнаружении с помощью УФ-спектрометрии. Полученные данные приведены в нижеследующей таблице.
Пример 2
Способом, аналогично описанному в примере 1, получали 3000 таблеток с матрицей следующего состава из расчета на одну таблетку:
Высвобождение действующего вещества in vitro определяли аналогично примеру 1.
Пример 3
Состав таблеток с матрицей из расчета на одну таблетку
Указанные таблетки получали в виде партии численностью 3000 штук способом, аналогично описанному в примере 1. Высвобождение действующего вещества in vitro определяли также аналогично примеру 1, но дополнительно, при прочих равных условиях, выявляли высвобождение действующего вещества при частоте вращения мешалки 50 об/мин и 100 об/мин.
Пример 4
Состав таблеток с матрицей из расчета на одну таблетку
Указанные таблетки получали в виде партии численностью 200 штук способом, аналогично описанному в примере 1. Высвобождение действующего вещества in vitro определяли также аналогично примеру 1.
Пример 5
Состав таблеток с матрицей из расчета на одну таблетку
Указанные таблетки получали в виде партии численностью 100 штук способом, аналогично описанному в примере 1. Высвобождение действующего вещества in vitro определяли также аналогично примеру 1.
Пример 6
Состав таблеток с матрицей из расчета на одну таблетку
Указанные таблетки получали в виде партии численностью 100 штук способом, аналогично описанному в примере 1. Высвобождение действующего вещества in vitro определяли также аналогично примеру 1.
Пример 7
Состав таблеток с матрицей из расчета на одну таблетку
Указанные таблетки получали в виде партии численностью 200 штук способом, аналогично описанному в примере 1. Высвобождение действующего вещества in vitro определяли также аналогично примеру 1.
Пример 8
Состав таблеток с матрицей из расчета на одну таблетку
Указанные таблетки получали в виде партии численностью 200 таблеток следующим образом:
Действующее вещество, Cellactose, Natrosol и Cutina смешивали между собой, затем нагревали в сушильном шкафу до 80°С и гранулировали в бытовом миксере типа Kenwood Chef. Охлажденный гранулят пропускали через сито с размером отверстий 1 мм. После смешения со стеаратом магния и тальком из гранулята с помощью эксцентрикового пресса типа Korsch EKO прессовали таблетки продолговатой формы размером 6×15 мм с насечкой.
Высвобождение действующего вещества in vitro определяли также аналогично примеру 1.
Пример 9
Состав таблеток с матрицей из расчета на одну таблетку
Указанные таблетки получали в виде партии численностью 100 штук способом, аналогично описанному в примере 1. Высвобождение действующего вещества in vitro определяли также аналогично примеру 1.
Пример 10
Состав таблеток с матрицей из расчета на одну таблетку
Указанные таблетки получали в виде партии численностью 100 штук способом, аналогично описанному в примере 1.
Высвобождение действующего вещества in vitro определяли при следующих условиях:
(А) Применение рекомендуемого согласно Европ. Фармакопее метода с использованием мешалки при частоте ее вращения 75 об/мин в 900 мл буферного раствора при значении рН 7,2 согласно Фармакопее США 22 при температуре 37°С и обнаружении с помощью УФ-спектроскопии.
(Б) Применение рекомендуемого согласно Европ. Фармакопее метода с использованием мешалки при частоте ее вращения 75 об/мин, при этом в промежуток времени с 0-й по 30-тую мин значение рН составляло 1,2, с 30-й по 120-тую мин - 2,3, со 120-й по 180-тую мин - 6,5, а в течение остального времени эксперимента значение рН составляло 7,2. В нижеследующей таблице приведены результаты эксперимента, проведенного при обоих условиях.
Результаты эксперимента показывают, что высвобождение действующего вещества из предлагаемых в изобретении композиций практически не зависит от значения рН среды высвобождения.
Пример 11
Состав сферических гранул (пеллет) из расчета на одну пеллету
Указанные пеллеты получали следующим образом. Действующее вещество, Avicel и L-HPC в течение 10 мин смешивали в планетарном смесителе (смеситель фирмы Kenwood), после чего гранулировали с использованием воды. Влажный гранулят экструдировали в экструдере типа Nica с матрицей размером 0,8×0,8 мм, а затем в течение 10 мин гранулам с помощью сфероидизатора типа Nica при частоте вращения 500 об/мин придавали сферическую форму (загрузка 1 кг). Пеллеты в течение ночи сушили в камерной сушилке при 50°С и после этого разделяли на фракции по крупности.
На пеллеты размером 0,6-1,0 мм (выход порядка 95%) в установке WSG (Glatt GPCG1 с насадкой фирмы Wurster) при температуре приточного воздуха 60°С (температура продукта 40°С) с использованием водной дисперсии из Aquacoat и ДБС (20%, в пересчете на содержание Aquacoat в твердом виде) наносили покрытие, в результате чего масса пеллет увеличивалась на 9,8% (в пересчете на исходную массу). Указанную покрывную дисперсию получали в соответствии с рекомендациями изготовителя (фирма FMC), при этом ДБС совместно с твином 80 гомогенизировали в частичном количестве воды, а затем добавляли к разбавленной дисперсии Aquacoat. Содержание твердого вещества в готовой дисперсии составляло 20 мас.% и ее перемешивали по меньшей мере в течение 3 ч. Пеллеты с нанесенным на них покрытием сушили в установке WSG и в течение 2 ч выдерживали в камерной сушилке при 60°С. Высвобождение действующего вещества определяли аналогично примеру 1, с тем, однако, отличием, что при этом использовали смеситель корзиночного типа с частотой его вращения 100 об/мин.
Клинические исследования
Для выявления фармакокинетических характеристик проводили перекрестное моноцентрическое, открытое, рандомизированное исследование с использованием 4-х лекарственных форм в разовой дозе, в ходе которого 16 здоровым, белым мужчинам - пробандам в возрасте от 18 до 45 лет вводили различные лекарственные формы, содержавшие в качестве действующего вещества (-)-(1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенол в виде гидрохлорида. В экспериментах по выявлению указанных характеристик использовали методы ЖХВР (жидкостная хроматография высокого разрешения).
При введении применяли следующие лекарственные формы:
"Капсула 100 мг": капсулы с быстрым высвобождением действующего вещества и массой действующего вещества 100 мг.
"Капсула 25 мг": капсулы с быстрым высвобождением действующего вещества и массой действующего вещества 25 мг.
"Таблетка 100 мг": таблетка согласно примеру 1 (масса действующего вещества 100 мг).
"Таблетка 200 мг": таблетка согласно примеру 2 (масса действующего вещества 200 мг)
(Вышеуказанные капсулы представляли собой белые непрозрачные твердожелатиновые капсулы размером 0 EL с общей массой 360 мг, имевшие следующий состав:
"Капсула 100 мг": 100 мг (-)-(1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенола, гидрохлорида, 152 мг микрокристаллической целлюлозы, 8 мг аэросила, 20 мг стеарата магния и 80 мг Primojel (натрийкарбоксиметилкрахмал тип А, продукт фирмы Avebe);
"Капсула 25 мг": 25 мг (-)-(1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенола, гидрохлорида, 227 мг микрокристаллической целлюлозы, 8 мг аэросила, 20 мг стеарата магния и 80 мг Primojel (натрийкарбоксиметилкрахмал тип А, продукт фирмы Avebe)).
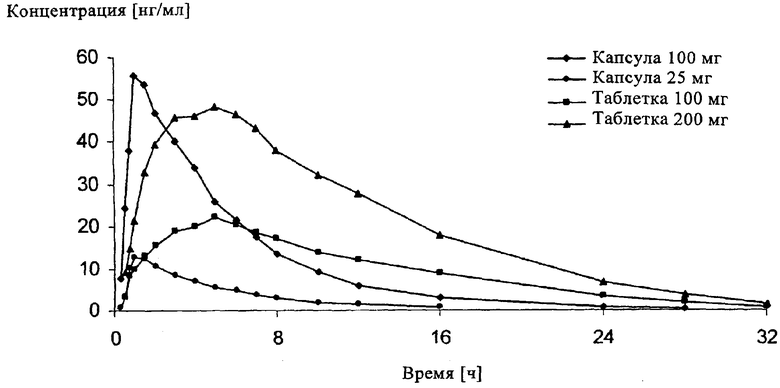
Наиболее важные фармакокинетические характеристики приведены в нижеследующей таблице, а полученные экспериментальным путем данные о средней концентрации действующего вещества в сыворотке представлены в графическом виде на прилагаемом к описанию чертеже.
AUC обозначает площадь под кривой (от англ. "area under curve"),
MRT обозначает среднюю продолжительность нахождения в организме (от англ. "mean residence time"),
HVD обозначает период времени, в течение которого концентрация действующего вещества в плазме сохраняется на уровне не ниже половины от максимальной (от англ. "half-value duration").
С одной стороны, в первую очередь сравнение "Капсулы 100 мг" и "Таблетки 100 мг" показывает, что предлагаемые в изобретении композиции позволяют высокоэффективно решить задачу по получению содержащей 3-(3-диметиламино-1-этил-2-метилпропил)фенол фармацевтической композиции с замедленным высвобождением действующего вещества. Вместе с тем при сравнении "Таблетки 100 мг" и "Таблетки 200 мг" можно отметить и такой положительный факт, как исключительно удачно выбранная дозировка действующего вещества, обеспечивающая требуемый профиль его высвобождения. Кроме того, при сравнении обеих предлагаемых в изобретении композиций "Таблетка 100 мг" и "Таблетка 200 мг" можно установить, что вначале они высвобождают действующее вещество в довольно заметном количестве, однако медленнее, чем обе композиции с быстрым высвобождением действующего вещества; тем не менее уровень его содержания в плазме при использовании обеих ретард-композиций уже по истечении 1 ч составляет более 10 нг/мл и по истечении 16 ч все еще остается достаточно высоким для обеспечения анальгетического эффекта. Помимо этого одновременные исследования действия только "Таблетки 100 мг" показывают, что при повторном введении этого лекарственного средства с интервалом в 12 ч удается достичь уровня его содержания в сыворотке не ниже 20 нг/мл, что уже при 2-разовом суточном приеме обеспечивает высокий анальгетический эффект. Это означает существенный прогресс в лечении прежде всего хронических болей и позволяет значительно улучшить соблюдение пациентом режима и схемы лечения.
Настоящее изобретение относится к области лекарственных средств, в частности к фармацевтическим композициям, обладающим анальгетическим действием с замедленным высвобождением действующего вещества, которая содержит 3-(3-диметиламино-1-этил-2-метилпропил)фенол или одну из его фармацевтически приемлемых солей в матрице, а также к таблеткам для двухразового суточного приема внутрь, содержащих указанные композиции. Техническим результатом является обеспечение замедленного высвобождения активного компонента из указанных композиций и таблеток. 4 н. и 9 з.п. ф-лы, 1 ил.
3-35 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 0,5 ч,
5-50 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 1 ч,
10-75 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются высвобождались по истечении 2 ч,
15-82 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 3 ч,
30-97 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 6 ч,
более 50 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 12 ч,
более 70 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 18 ч,
более 80 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 24 ч.
3-35 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола (в пересчете на 100 мас.% действующего вещества) высвобождаются по истечении 0,5 ч,
5-50 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 1 ч,
10-75 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются высвобождались по истечении 2 ч,
15-82 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 3 ч,
30-97 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 6 ч,
более 50 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 12 ч,
более 70 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола высвобождаются по истечении 18 ч,
более 80 мас.% 3-(3-диметиламино-1-этил-2-метилпропил)фенола 30 высвобождаются по истечении 24 ч.
| Зонд для измерения параметров электронных пучков электроннолучевых приборов | 1977 |
|
SU693475A1 |
| US 4389393 A, 01.12.1983 | |||
| DE 3625458 A1, 12.02.1987. | |||

