ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому способу получения соединения, представленного следующей формулой (2)
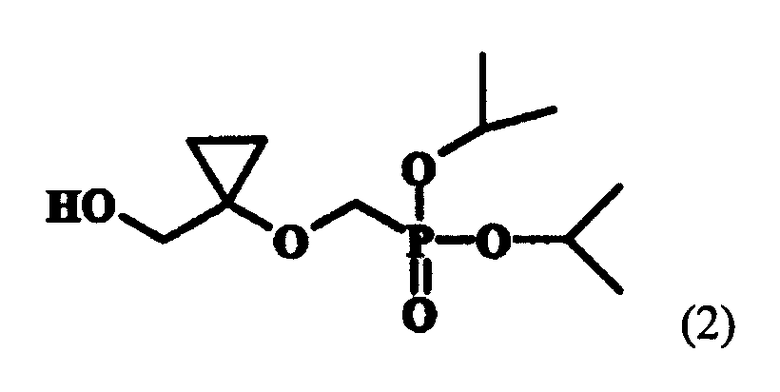
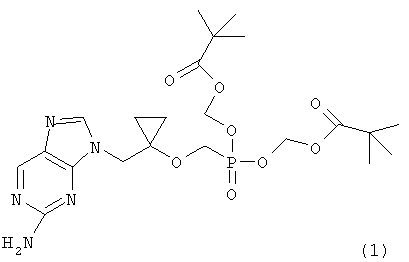
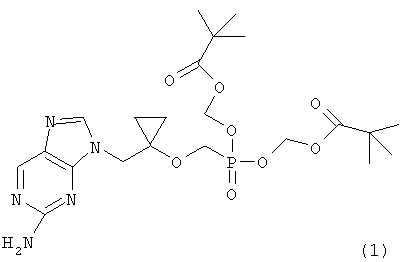
которое представляет собой ключевое промежуточное соединение для синтеза аналога антивирусного (особенно против вируса гепатита В) нуклеозида, представленного следующей формулой (1):
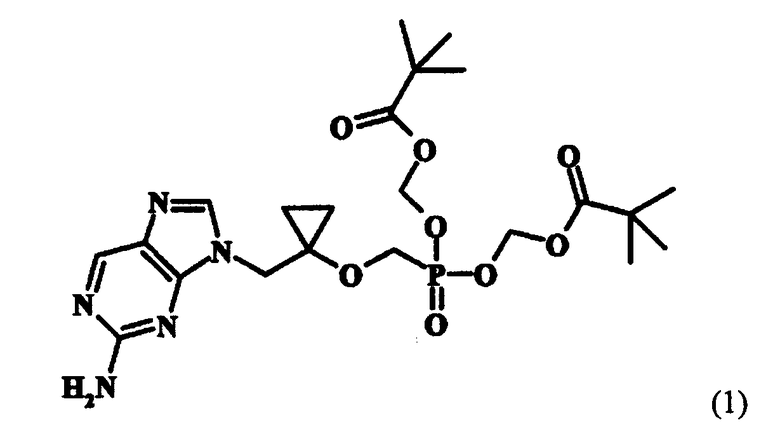
Настоящее изобретение также относится к новым промежуточным соединениям и способу получения соединения формулы (1) [соединение (1), ниже] из соединения формулы (2) [соединение (2), ниже], полученного согласно настоящему изобретению.
УРОВЕНЬ ТЕХНИКИ
Соединение (1) представляет собой терапевтический агент против гепатита B (корейская заявка на патент № 2002-0003051, WO 02/057288), и соединение (2) может применяться как важный реагент для получения соединения (1), совместно с соединением следующей формулы (3):
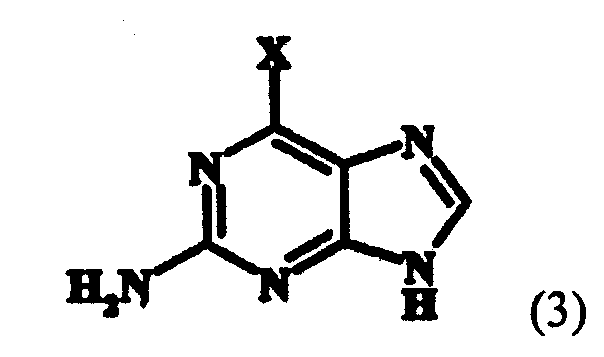
в которой X представляет собой атом фтора, атом хлора, атом брома или атом иода.
Известно, что такие производные пурина, как соединение (1), обладают противораковой и антивирусной активностями, и десять (10) или более видов таких производных, включая AZT, 3TC, ACV и так далее, уже являются коммерчески доступными.
Соединение (2), применяемое как важное промежуточное соединение для получения соединения (1), было получено способом, представленным на следующей схеме реакции 1.
Схема реакции 1:
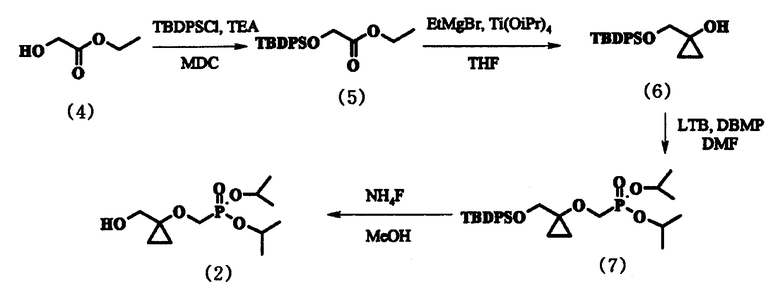
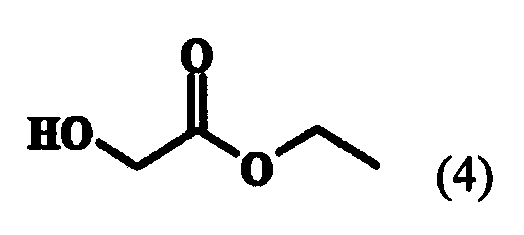
В указанном способе схемы 1 в качестве исходного вещества используют этилгликолят следующей формулы (4):
В способе по схеме 1, этилгликолят формулы (4) вводили в реакцию с трет-бутил(дифенил)силилхлоридом с образованием соединения формулы (5), которое вводили в реакцию с этилмагнийбромидом и тетраизопропоксидом титана согласно способу, известному из уровня техники (Syn. Lett, 07, 1053-1054, 1999), что приводит к циклопропиловому спирту формулы (6), который затем выделяли как твердое вещество из гептана. Полученное таким образом соединение формулы (6) растворяли в диметилформамиде и вводили в реакцию с трет-бутоксидом лития и диизопропилбромметил фосфонатом, получая соединение формулы (7). Это соединение формулы (7) кипятили с фторидом аммония в метаноле, что приводило к соединению (2).
Вышеупомянутый способ, однако, имеет тот недостаток, что образуется загрязненная форма соединения (2), поскольку диизопропилбромметилфосфонат, применяемый на стадии синтеза соединения формулы (7), остается в реакционном растворе и таким образом всегда содержится в конечном полученном соединении (2) в количестве 7-15%. Далее, плохая стабильность соединения формулы (6) к трет-бутоксиду лития на стадии синтеза соединения формулы (7) ответственна за непостоянство выхода и трудности выделения.
Поэтому для специалистов в данной области желательно повысить чистоту соединения (2) и улучшить стабильность соединения формулы (6) во время синтеза соединения формулы (7).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Таким образом, авторы настоящего изобретения провели масштабные исследования способа получения соединения (2) с целью его улучшения и в результате выяснили, что упомянутые недостатки ранее предложенного способа могут быть преодолены путем применения в качестве реагента тритилхлорида вместо трет- бутил(дифенил)силилхлорида, что приводит к соединению (2) высокой чистоты. Авторы изобретения также нашли, что соединение (1) может быть получено с высоким выходом путем применения высокочистого соединения (2), полученного таким образом, и тем самым завершили настоящее изобретение.
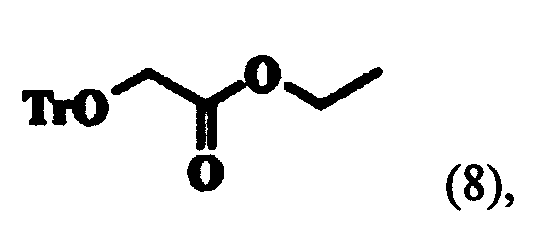
Поэтому целью настоящего изобретения является создание нового способа получения соединения (2). Другой целью настоящего изобретения являются новые промежуточные соединения, полученные при проведении способа получения соединения (2). Дальнейшей целью настоящего изобретения является способ получения аналога нуклеозида формулы (1), который может быть применен в качестве антивирусного агента, с применением соединения (2), полученного по способу согласно настоящему изобретению. Настоящее изобретение представляет способ получения соединения (2), включающий в себя стадии реагирования соединения формулы (4) с тритилхлоридом для получения этилового эфира тритилоксиуксусной кислоты следующей формулы (8):
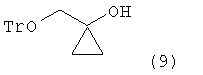
реагирования соединения формулы (8) с этилмагнийгалогенидом для получения 1-тритилоксиметилциклопропанола следующей формулы (9):
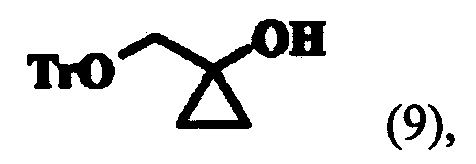
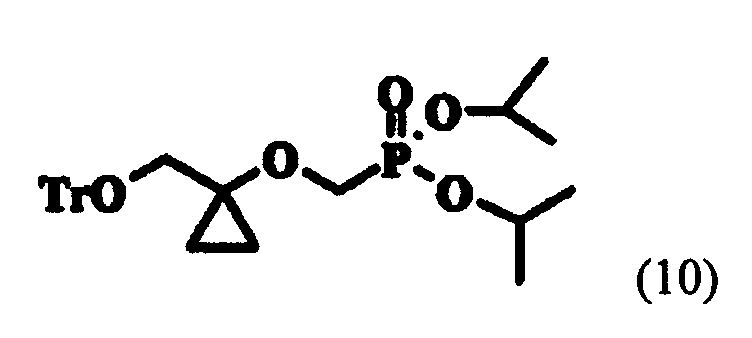
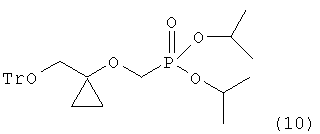
реагирования 1-тритилоксиметилциклопропанола формулы (9) с диизопропилбром-метилфосфонатом в растворителе в присутствии основания для получения диизопропилового эфира (l-тритилоксиметил-циклопропоксиметил-фосфоновой кислоты следующей формулы (10) в твердой форме:
и превращения тритильной группы соединения формулы (10) в гидроксильную группу.
При проведении способа настоящего изобретения N-метилпирролидон (NMP) может быть выбран более предпочтительно в качестве растворителя для стадии сочетания циклопропилоксигруппы формулы (9) с фосфонатной группой с образованием соединения (2) с лучшим выходом и чистотой.
Способ настоящего изобретения может быть изображен следующей схемой реакции 2, на которой X представляет собой атом фтора, атом хлора, атом брома или атом иода.
Схема реакции 2:
Предпочтительные количества каждого реагента и условия реакции, включая температуру реакции и способ очистки, могут быть конкретно объяснены ниже.
Во-первых, определены следующие сокращения, примененные в настоящем описании:
На первой стадии реакции этилгликолята формулы (4) с тритилхлоридом, 1,0 эквивалента тритилхлорида (TrCl) и 1-1,3 эквивалента пиридина добавляли к 1,0-1,5 эквиваленту соединения формулы (4) и смесь перемешивали в присутствии EDC при температуре около 30-60°C. После перемешивания кислотно-основная обработка смеси приводила к соединению формулы (8), которое затем обрабатывали гексаном для перевода в твердую форму или применяли в следующей реакции без дальнейшей очистки. К соединению формулы (8) прибавляли 2,1-3,1 эквивалента этилмагнийгалогенида, предпочтительно этилмагний хлорида или этилмагний бромида, и 0,2-0,6 эквивалента тетраизопропоксида титана и проводили реакцию Кулинковича при 5-15°C (J. Am. Chem. Soc, 1995, 117, 9919-9920). Затем туда добавляли водный раствор лимонной кислоты, реакционную смесь перемешивали и экстрагировали, что дало соединение формулы (9). Соединение формулы (9) растворяли в растворителе, особенно предпочтительно в NMP, туда добавляли 1,3-1,7 эквивалента DBMP и 1,5-2,0 эквивалента LTB и смесь перемешивали более 6-19 часов при условии не превышения 45°C; и подвергали кислотно-основной обработке, что дало соединение формулы (10). Соединение формулы (10), полученное таким образом, переводили в твердую форму обработкой гептаном при низкой температуре. К соединению формулы (10) добавляли 1,5-2,5 эквивалента TFA и 0,1-0,5 мл/г воды и смесь перемешивали при комнатной температуре. После перемешивания, кислотно-основной обработки и фильтрации полученного таким образом твердого вещества и экстракции получали соединение (2). Здесь кислотно-основная обработка предпочтительно проводилась гидроксидом натрия, а экстракция - метиленхлоридом.
Способ настоящего изобретения приводит к соединению (2) высокой чистоты от 98 до 100%.
Соединения формул (9) и (10), полученные как промежуточные соединения для способа настоящего изобретения, сами по себе также являются новыми соединениями. Поэтому настоящее изобретение далее представляет эти новые промежуточные соединения.
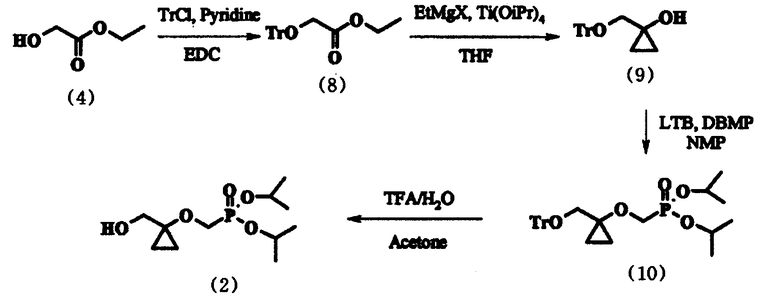
Соединение (2), полученное по упомянутому способу, представляет собой ключевое промежуточное соединение для синтеза аналога нуклеозида формулы (1), как упомянуто ранее. Конкретно, соединение (1) может быть получено по способу, включающему стадии введения уходящей группы в соединение (2) для получения соединения следующей формулы (11):
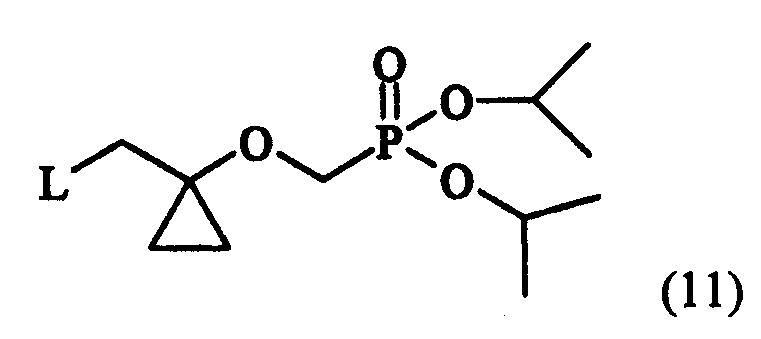
в которой L представляет собой уходящую группу, предпочтительно метансульфонилокси-, пара-толуолсульфонилокси-группу, или атом галогена; сочетания соединения формулы (11) с соединением формулы (3) для получения соединения следующей формулы (12):
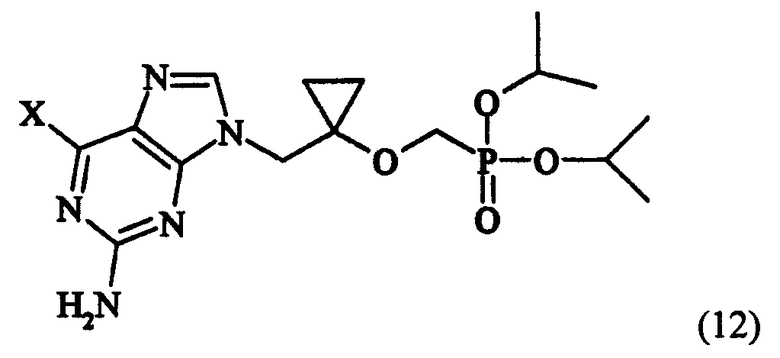
в которой X определен как ранее, и гидролиза соединения формулы (12) для получения соединения следующей формулы (13):
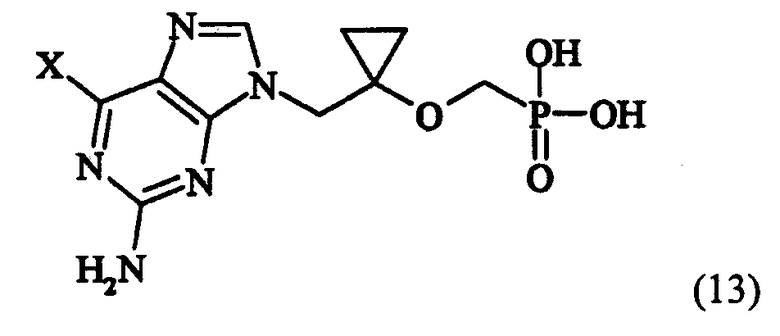
в которой X определен как ранее, удаления группы X из соединения формулы (13) и одновременно введения трет-бутилкарбонилоксиметильной группы в фрагмент фосфоновой кислоты. Подробные условия реакции вышеупомянутого способа описаны в предшествующей заявке авторов (корейская заявка на патент № 2002-0003051, WO 02/057288). Поэтому настоящее изобретение представляет способ получения соединения (1) из соединения (2), полученного по способу, представленному на схеме реакции 2.
Настоящее изобретение будет более конкретно объяснено на следующих примерах. Однако должно быть понятно, что эти примеры предназначены для иллюстрации настоящего изобретения, но не для ограничения, любым способом, объема настоящего изобретения.
В примерах ниже, условия высокоэффективной жидкостной хроматографии (ВЭЖХ) для определения завершения реакции представляют собой следующие:
[условия ВЭЖХ]
Примеры
Пример 1.
Получение этилового эфира тритилоксиуксусной кислоты (8)
Тритилхлорид (279 г, 1,0 моль) растворяли в EDC (680 мл, 5 мл/г по отношению к этилгликоляту) и добавляли к смеси этилгликолят (135 г, 1,3 моль). К ним добавляли пиридин (99 г, 1,25 моль) и смесь перемешивали в течение 19 часов при 40°C. После подтверждения методом ВЭЖХ завершения реакции добавляли 0,5 N раствор водной соляной кислоты (270 мл, 2 мл/г по отношению к этилгликоляту) для того, чтобы сделать реакционный раствор двухфазным, затем экстрагировали. После повторного проведения экстракции EDC отгоняли при уменьшенном давлении. Для того чтобы получить соединение формулы (8) в твердой форме, к концентрированному соединению добавляли гексан (680 мл), температуру понижали до 0°C, смесь перемешивали в течение около 3 часов и фильтровали.
[ВЭЖХ]
1H ЯМР (400 МГц, CDCl3 ) д 1,23 (т, 3H, J=8 Гц), 3,78 (с, 2H), 4,14 (кв, 2H, J=8 Гц), 7,26-7,22 (м, 3H), 7,33-7,29 (м, 6H), 7,50-7,47 (м, 6H);
13C ЯМР (400 МГц, CDCl3 ) д 10,8, 57,3, 59,3, 84,0, 123,9, 124,6, 125,3, 140,0, 166,7.
Пример 2.
Получение l-тритилоксиметил-циклопропанола (9)
При допущении, что выход в примере 1 составляет 100%, к соединению формулы (8), полученному в примере 1, добавляли тетрагидрофуран (1040 мл, 3 мл/г по отношению к соединению формулы (8)) и смесь охлаждали до 0°C. Туда добавляли тетраизопропоксид титана (113,8 г, 0,4 моль) и затем по каплям добавляли этилмагнийбромид (1500 мл, 3,0 моль, 1M концентрация) при 5-15°C в течение 3-6 часов. После подтверждения методом ВЭЖХ завершения реакции добавляли 20% водный раствор лимонной кислоты [1790 мл, 5 мл/г по отношению к соединению формулы (8)] к реакционному раствору при условии не превышения 35°C, который перемешивали в течение около 1 часа. После перемешивания тетрагидрофуран отгоняли при уменьшенном давлении и остаток дважды экстрагировали, первый раз 1390 мл этилацетата [4 мл/г по отношению к соединению формулы (8)] и второй раз 690 мл того же растворителя [2 мл/г по отношению к соединению формулы (8)]. Таким образом, полученный органический слой промывали насыщенным водным раствором NaHCO3 [690 мл, 2 мл/г по отношению к соединению формулы (8)] и концентрировали при уменьшенном давлении, что дало соединение формулы (9), обозначенное в заголовке примера.
[ВЭЖХ]
1H ЯМР (400 МГц, CDCl3 ) д 0,45 (дд, 2H, J=8 Гц), 0,80 (дд, 2H, J=8 Гц), 2,59 (с, 1H), 3,18 (с, 2H), 7,23-7,32 (м, 9H), 7,45-7,47 (м, 6H);
13C ЯМР (400 МГц, CDCl3) д 8,4, 51,9, 63,9, 83,0, 123,7, 124,6, 125,3, 140,5.
Пример 3.
Получение диизопропилового эфира (l-тритилоксиметил-циклопропоксиметил-фосфоновой кислоты (10)
К соединению формулы (9) (261,25 г, 0,79 моль), полученному концентрированием при уменьшенном давлении, в примере 2 (предполагается, что соединение формулы (9) получали с выходом 79%, начиная от примера 1, так как величина пика в ВЭЖХ составляла 78,54%), добавляли NMP [1050 мл, 4 мл/г по отношению к соединению формулы (9)] и DBMP (307 г, 1,2 моль). К реакционной смеси добавляли LTB (107 г, 1,3 моль), которую затем перемешивали при условии не превышения 45°C. После около 6-19 часов завершение реакции подтверждали методом ВЭЖХ и добавляли туда 14% водный раствор NH4Cl [1830 мл, 7 мл/г по отношению к соединению формулы (9)] для того, чтобы остановить реакцию. Туда добавляли дважды MTBE [первый раз: 1050 мл, 4 мл/г по отношению к соединению формулы (9); второй раз: 520 мл, 2 мл/г по отношению к соединению формулы (9)] для разделения фаз. Таким образом полученные органические слои объединяли и промывали 21% водным раствором NaCl [1650 мл, 6,3 мл/г по отношению к соединению формулы (9)]. Остающийся органический слой концентрировали при уменьшенном давлении и туда добавляли гептан [1300 мл, 5 мл/г по отношению к соединению формулы (8)]. Смесь охлаждали до -10°C и фильтровали после приблизительно 3 часов, что дало соединение формулы (10), обозначенное в заголовке примера (586 г, чистота 96,23%, выход 71,3%) в виде твердого вещества.
[ВЭЖХ]
ЯМР: Не наблюдали других пиков, кроме пиков соединения формулы (10).
1H ЯМР (400 МГц, CDCl3) д 0,54 (дд, 2H, J=8 Гц), 0,80 (дд, 2H, J=8 Гц), 1,33-1,29 (м, 12H), 3,22 (с, 2H), 3,92 (д, 2H, J=8 Гц), 4,67-4,76 (м, 2H), 7,21-7,31 (м, 9H), 7,43-7,46 (м, 6H)
13C ЯМР (400 МГц, CDCl3) д 11,9, 24,4, 24,4, 24,5, 24,6, 63,5, 63,8, 64,0, 65,1, 67,3, 71,3, 71,4, 86,9, 127,4, 128,3, 129,1, 144,3
Пример 4.
Получение диизопропил {[l-(гидроксиметил)-циклопропил] окси}метилфосфоната (2)
Соединение формулы (10), полученное в примере 3 (59,15 г, 116,3 ммоль), растворяли в ацетоне [59,2 мл, 1 мл/г по отношению к соединению формулы (10)], добавляли воду (5,9 мл, 327,8 ммоль) и TFA (26,52g, 232,6 ммоль) и смесь перемешивали при комнатной температуре. После того как методом ВЭЖХ было установлено, что содержание соединения формулы (10) составило 7% или менее, туда добавляли 3N водный раствор NaOH [75 мл, 2,6 мл/г по отношению к соединению формулы (10)], и ацетон удаляли из смеси отгонкой при уменьшенном давлении. Фильтровали твердое вещество, полученное в ходе реакции, и фильтрат экстрагировали дважды метиленхлоридом [118,3 мл ×2; 2 мл/г по отношению к соединению формулы (10)]. Таким образом, полученный органический слой концентрировали при уменьшенном давлении, что дало соединение, обозначенное в заголовке примера (2) [31,71 г, чистота 98%, выход по отношению к соединению формулы (10) 102,4%].
[ВЭЖХ]
[ЯМР] Кроме пика для соединения (2) наблюдался только пик метиленхлорида (растворитель).
1H ЯМР (400 МГц, DMSO) д 0,55 (дд, 2H, J=8 Гц), 0,72 (дд, 2H, J=8 Гц), 1,22-1,24 (м, 12H), 3,32 (с, 2H), 3,53 (д, 2H, J=4 Гц), 3,81 (д, 2H, J=8 Гц), 4,53-4,72 (м, 2H), 4,73 (т, 1H)
13C ЯМР (400 МГц, DMSO) д 7,44, 20,7, 20,75, 20,85, 20,88, 58,81, 60,47, 60,69, 61,62, 61,77, 67,12, 67,18
Как объяснено выше, когда тритилхлорид применяли согласно настоящему изобретению, промежуточное соединение формулы (10) может быть получено как твердое вещество, что решает проблему предшествующего уровня техники, состоящую в том, что диизопропилбромметилфосфонат не удаляется, но остается в соединении (2), что уменьшает его чистоту. Особенно, если N- метилпирролидон выбирали взамен диметилформамида в качестве растворителя для получения соединения формулы (10), стабильность соединения формулы (9) к трет-бутоксиду лития значительно улучшается, что действует благоприятным образом на чистоту и выход соединения (2). Кроме того, соединение (1), которое может быть применено как антивирусный агент, может быть получено, преимущественно с высоким выходом, с применением высокочистого соединения (2), полученного согласно настоящему изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ FXa С ЦИКЛИЧЕСКИМ АМИДОКСИМОМ ИЛИ ЦИКЛИЧЕСКИМ АМИДРАЗОНОМ В КАЧЕСТВЕ P4 СУБЪЕДИНИЦЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПРОИЗВОДНЫЕ | 2009 |
|
RU2468024C2 |
| НОВОЕ ПРОИЗВОДНОЕ ГЛЮЦИТОЛА, ЕГО ПРОЛЕКАРСТВО И ЕГО СОЛЬ И СОДЕРЖАЩИЙ ИХ ТЕРАПЕВТИЧЕСКИЙ АГЕНТ ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА | 2005 |
|
RU2386631C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2016 |
|
RU2740019C1 |
| АГОНИСТЫ РЕЦЕПТОРА МЕЛАНОКОРТИНА | 2007 |
|
RU2411240C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ ПАРАЗИТАРНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2793122C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА RIP-1 КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2800652C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ВЫПОЛНЯЮЩЕЕ ФУНКЦИИ ИНГИБИТОРА WEE1, И ВАРИАНТЫ ЕГО ПРИМЕНЕНИЯ | 2018 |
|
RU2783243C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛОСАРТАНА | 2006 |
|
RU2412940C2 |
| АЗААДАМАНТАНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2450002C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ДЕЗОКСИ-2', 2'-ДИФТОРЦИТИДИНА | 2005 |
|
RU2360919C2 |
Настоящее изобретение относится к способу получения диизопропил {[1-(гидроксиметил)-циклопропил]окси}метилфосфоната формулы (2), который является ключевым промежуточным соединением в синтезе аналога антивирусного нуклеозида. Настоящее изобретение также относится к новым промежуточным соединениям формул (9) и (10) и способу получения из соединения (2), полученного согласно настоящему изобретению, аналога антивирусного нуклеозида (особенно против вируса гепатита В) формулы (1)
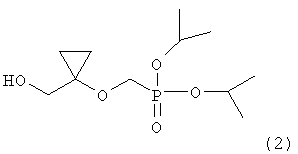
 ,
,
Технический результат - получение соединений формул (1) и (2) высокой степени чистоты с высоким выходом.
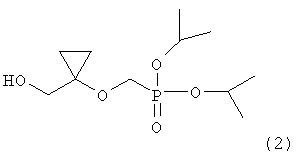 ,
,
включающий стадии реагирования соединения следующей формулы (4):
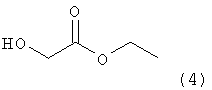 ,
,
с тритилхлоридом с получением этилового эфира тритилоксиуксусной кислоты следующей формулы (8):
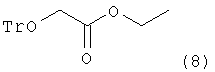 ,
,
реагирование соединения формулы (8) с этилмагнийгалогенидом с получением 1-тритилоксиметил-циклопропанола следующей формулы (9):
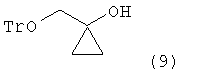 ,
,
сочетание 1-тритилоксиметил-циклопропанола формулы (9) с диизопропилбром-метилфосфонатом в растворителе в присутствии основания для получения диизопропилового эфира (1-тритилоксиметил-циклопропоксиметил-фосфоновой кислоты следующей формулы (10) в твердом виде:
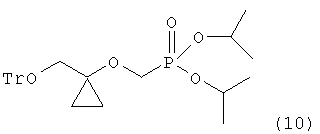
и превращения тритильной группы соединения формулы (10) в гидроксильную группу.
,
включающий в себя стадии введения уходящей группы в соединение формулы (2), полученного по способу по п.1, для получения соединения следующей формулы (11):
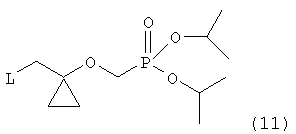
в которой L представляет собой уходящую группу;
сочетания соединения формулы (11) с соединением следующей формулы (3):
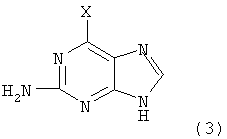
в который Х представляет собой атом фтора, атом хлора, атом брома, или атом йода для получения соединения следующей формулы (12):
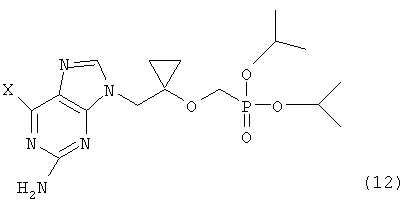
в которой Х определен как ранее; гидролиза соединения формулы (12) для получения соединения следующей формулы (13):
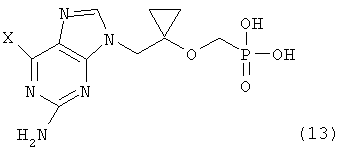
в которой Х определен как ранее; удаления группы Х из соединения формулы (13), и одновременно введения трет-бутилкарбонилоксиметильной группы в фрагмент фосфоновой кислоты.
| Приспособление для разматывания лент с семенами при укладке их в почву | 1922 |
|
SU56A1 |
| US 5817647, 06.10.1998 | |||
| US 5688778, 18.11.1997 | |||
| WO 9307157, 15.04.1993 | |||
| RU 98110564, 27.03.2000. | |||

