ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет по предварительной заявке США 62/473,771, выданной 20 марта 2017 г., содержание которой включено в настоящее описание во всей полноте в виде ссылки.
УРОВЕНЬ ТЕХНИКИ
Малярия является трансмиссивным инфекционным заболеванием, вызываемым простейшими паразитами, и широко распространена в тропических и субтропических регионах, в том числе в некоторых частях Северной и Южной Америки, Азии и Африки. Из пяти видов паразитов Plasmodium, которые могут заразить человека (P. falciparum, P. vivax, P. ovale, P. malariae и P. knowlesi), наиболее серьезные формы заболевания вызываются P. falciparum и P. vivax. Кроме того, несколько видов паразитов Plasmodium заражают и других млекопитающих, помимо людей. Например, P. berghei, P. chabaudi, P. vinckei и P. yoelii могут вызывать малярию у некоторых грызунов.
Малярия ежегодно поражает примерно 515 миллионов человек, и от одного до трех миллионов из них умирают от этой болезни. Большинство современных противомалярийных препаратов нацелены на стадию бесполого размножения в крови, когда паразиты живут внутри эритроцитов. Несмотря на то, что паразиты на печеночной стадии и трансмиссивной стадии (передачи) не вызывают симптомов малярии, профилактика и препараты, блокирующие передачу, необходимы для профилактического предупреждения развития эпидемий этого заболевания и защиты уязвимых групп населения. Имеющиеся на сегодняшний день одобренные противомалярийные препараты, такие как хлорохин, атовакуон, пириметамин и сульфадоксин, ограничены лишь несколькими мишенями в цикле развития малярийного паразита у человека, и растущая широко распространенная устойчивость к современным препаратам побуждает к разработке новых противомалярийных средств, нацеленных на новые биологические мишени.
Криптоспоридиоз - еще одно паразитарное заболевание, вызываемое Cryptosporidium, родом простейших паразитов из типа Apicomplexa. Криптоспоридиоз чаще всего вызывается внутриклеточными паразитами, простейшими со сложным жизненным циклом, C. parvum и C. hominis. Это заболевание также может быть вызвано C. canis, C. felis, C. meleagridis и C. muris. Криптоспоридиоз поражает дистальные отделы тонкой кишки и может поражать дыхательные пути как у людей со здоровым иммунитетом, так и у людей с ослабленным иммунитетом. Криптоспоридиоз является одним из наиболее распространенных заболеваний, передаваемых через воду, и встречается во всем мире. Он также может передаваться другим животным, включая крупный рогатый скот, овец, свиней, лошадей, коз и гекконов. Нитазоксанид является современным стандартом лечения криптоспоридиоза, но этот препарат лишь частично эффективен у детей, а его эффективность у пациентов со СПИДом не выше, чем у плацебо.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, фармацевтическим композициям и способам лечения или профилактики паразитарных заболеваний, включая малярию и криптоспоридий, включающие/использующие соединение, описанные выше или в любом месте настоящего описания.
В некоторых вариантах осуществления эти фармацевтические композиции приготовлены в виде ветеринарных композиций для введения субъектам, которые не являются человеком.
Соединения могут иметь структуру, соответствующую формуле (I):
 (I)
(I)
где “пунктирная” связь может представлять собой одинарную или двойную связь;
R1 представляет собой необязательно замещенный арил или гетероарил;
R2 представляет собой необязательно замещенный алкокси, циклоалкокси или гетероциклил;
R3 представляет собой водород или -CH2-X;
R4 и R5 независимо представляют собой водород, -X или -CH2-X, R4 и R5 вместе могут формировать пяти- или шести-членное конденсированное кольцо, и по меньшей мере один из R4 и R5 не является водородом;
R6 и R7 независимо представляют собой водород или R; и
z1-z8 в каждом случае независимо выбирают из CH или N; где
-X в каждом случае независимо выбирают из -OH, -OR, -S(O)R, -S(O)2R, -N(R)-S(O)2R, -S(O)2-N(R)(R), -S(O)2-NHR, -N(R)-C(O)-R или -N(R)(R); и
R в каждом случае независимо представляет собой необязательно замещенный C1-C12 алкил;
или ее фармацевтически приемлемую соль. -X в каждом случае может быть независимо выбран из -OH -NH2 или -N(R)(R). В некоторых вариантах осуществления группы -X в R4 и/или R5 (включая группы -X, когда R4 и/или R5 представляет собой -CH2-X) могут быть выбраны из -OH, NH2 или -N(R)(R). R также в каждом случае может быть независимо выбран из C1-4 линейного или разветвленного углеводорода.
В некоторых вариантах осуществления, каждый из R6 и R7 представляет собой водород. В некоторых вариантах осуществления, R4 и R5 могут представлять собой одну и ту же функциональную группу, выбранную из -X или -CH2-X (например, R4 представляет собой -OH, и R5 представляет собой -OH, R4 представляет собой -NH2, и R5 представляет собой -NH2, R4 представляет собой -OCH3, и R5 представляет собой -OCH3 и т.д.). В других вариантах осуществления R5 представляет собой водород, а R4 представляет собой -X или -(CH2)-X. В других вариантах осуществления R4 представляет собой водород, а R5 представляет собой -X или -(CH2)-X. В некоторых вариантах осуществления, R4 и R5 независимо выбирают из -OH и -OR, и R4 и R5 вместе образуют 6-членное конденсированное кольцо. В некоторых вариантах осуществления R6 представляет собой C1-4 линейный или разветвленный углеводород. В других вариантах осуществления R6 представляет собой водород. R1 необязательно может быть замещен C6 арилом или гетероарилом (например, фенилом, фторфенилом, дифторфенилом, пиридилом и т.д.). В некоторых вариантах осуществления R2 представляет собой C1-4 линейный или разветвленный алкокси (например, метокси, этокси, пропокси, изопропокси и т.д.). В некоторых вариантах осуществления R2 представляет собой C1-4 линейный или разветвленный алкокси, замещенный одним или более F (например, -OCF3, -OCHF2 или -OCH2F). В других вариантах осуществления R2 представляет собой C3-6 гетероциклил (например, азиридинил, оксиранил, тииранил, оксетанил, азетидинил, триэтанил, диазетидинил, диоксетанил, дитриэтанил, пирролидинил, тетрагидрофуранил, имидазолил, пиразолил, оксазолил и т.д.). В предпочтительных вариантах осуществления R2 представляет собой оксетанил или азетидинил. В других вариантах осуществления R2 представляет собой C3-6 циклоалкокси (например, циклопропокси). В некоторых вариантах осуществления, R3 представляет собой -CH2-X, и -X представляет собой N(R)(R).
Каждый из z1-z8 может представлять собой CH. В некоторых вариантах осуществления один из z1-z4 представляет собой N, а остальные представляют собой CH. В некоторых вариантах осуществления один из z5-z8 представляет собой N, а остальные представляет собой CH. В некоторых вариантах осуществления каждый из z1 и z4 представляет собой N, и каждый из z3 и z2 представляет собой CH. В некоторых вариантах осуществления, каждый из z5 и z7 представляет собой N, и каждый из z6 и z8 представляет собой CH.
Любые стереоцентры в структуре, соответствующей формуле (I), могут иметь любую конфигурацию или присутствовать в виде рацемической смеси каждого стереоцентра (например, стреоизомеров, диастереомеров и т.д.). В некоторых вариантах осуществления соединения имеют структуру, соответствующую формуле (II):

В некоторых вариантах осуществления, соединения имеют структуру, соответствующую формуле (IIb):

В других вариантах осуществления, соединения имеют структуру, соответствующую формуле (IIa):
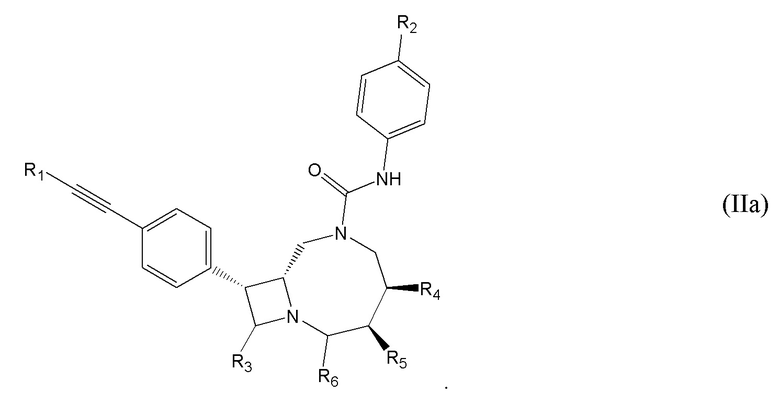
Соединения также могут иметь структуру, соответствующую формуле (III):
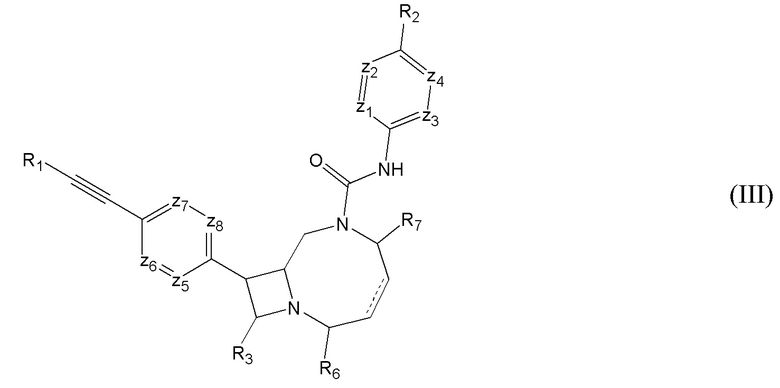
где “пунктирная” связь может представлять собой одинарную или двойную связь;
R1 представляет собой необязательно замещенный арил или гетероарил;
R2 представляет собой необязательно замещенный алкокси, циклоалкокси или гетероциклил;
R3 представляет собой водород или -CH2-X;
R6 и R7 независимо представляют собой водород или R; и
z1-z8 в каждом случае независимо выбирают из CH или N; где
-X в каждом случае независимо выбирают из -OH, -OR, -S(O)R, -S(O)2R, -N(R)-S(O)2R, -S(O)2-N(R)(R), -S(O)2-NHR, -N(R)-C(O)-R или -N(R)(R); и
R в каждом случае независимо представляет собой C1-C12 алкил;
при условии, что в случае, когда R6 представляет собой водород, R3 представляет собой -CH2-N(R)(R), и указанная “пунктирная” связь представляет собой двойную связь. В некоторых вариантах осуществления “пунктирная” связь представляет собой двойную связь. В других вариантах осуществления “пунктирная” связь представляет собой одинарную связь. -X в каждом случае может быть независимо выбран из -OH -NH2 или -N(R)(R). R также в каждом случае может быть независимо выбран из C1-4 линейного или разветвленного углеводорода.
Каждый из z1-z8 может представлять собой CH. В некоторых вариантах осуществления один из z1-z4 представляет собой N, остальные представляют собой CH. В некоторых вариантах осуществления один из z5-z8 представляет собой N, остальные представляют собой CH. В некоторых вариантах осуществления, каждый из z1 и z4 представляет собой N, и каждый из z3 и z2 представляет собой CH. В некоторых вариантах осуществления каждый из z5 и z7 представляет собой N, и каждый из z6 и z8 представляет собой CH.
В некоторых вариантах осуществления каждый из R6 и R7 представляет собой водород. В некоторых вариантах осуществления, R6 представляет собой низший алкил (например, метил, этил и т.д.). В некоторых вариантах осуществления R6 представляет собой C1-4 линейный или разветвленный углеводород. В других вариантах осуществления R6 представляет собой водород. R1 необязательно может быть замещен C6 арилом или гетероарилом (например, фенилом, фторфенилом, дифторфенилом, пиридилом и т.д.). В некоторых вариантах осуществления R2 представляет собой C1-4 линейный или разветвленный алкокси (например, метокси, этокси, пропокси, изопропокси и т.д.). В некоторых вариантах осуществления R2 представляет собой C1-4 линейный или разветвленный алкокси замещенный одним или более F (например, -OCF3, -OCHF2 или -OCH2F). В других вариантах осуществления R2 представляет собой C3-6 гетероциклил (например, азиридинил, оксиранил, тииранил, оксетанил, азетидинил, триэтанил, диазетидинил, диоксетанил, дитриэтанил, пирролидинил, тетрагидрофуранил, имидазолил, пиразолил, оксазолил и т.д.). В предпочтительных вариантах осуществления R2 представляет собой оксетанил или азетидинил. В других вариантах осуществления R2 представляет собой C3-6 циклоалкокси (например, циклопропокси).
В некоторых вариантах осуществления соединение может иметь структуру, соответствующую формуле (IV):

В некоторых вариантах осуществления соединение может представлять собой любое из соединений E1-E38 и их фармацевтически приемлемых солей.
В фармацевтических композициях можно использовать любое из описанных выше соединений. В некоторых вариантах осуществления фармацевтическая композиция может содержать фармацевтически приемлемый наполнитель и соединение, имеющее структуру, соответствующую формуле (I):
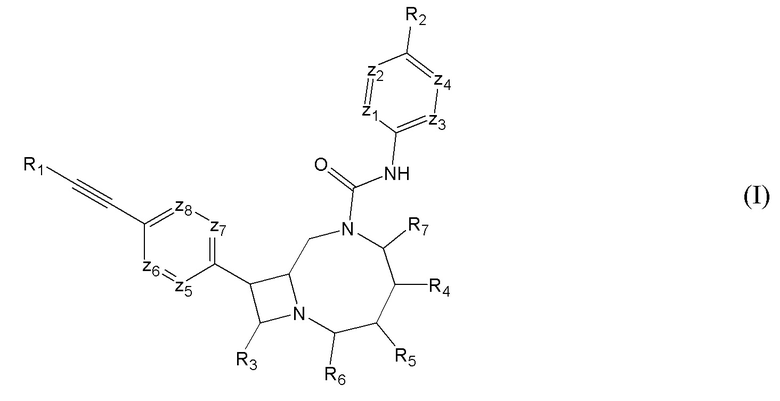
где “пунктирная” связь может представлять собой одинарную или двойную связь;
R1 представляет собой необязательно замещенный арил или гетероарил;
R2 представляет собой необязательно замещенный алкокси, циклоалкокси или гетероциклил;
R3 представляет собой водород или -CH2-X;
R4 и R5 независимо представляют собой водород, -X или -CH2-X, R4 и R5 вместе могут формировать пяти- или шести-членное конденсированное кольцо, и по меньшей мере один из R4 и R5 не является водородом;
R6 и R7 независимо представляют собой водород или R; и
z1-z8 в каждом случае независимо выбирают из CH или N; где
-X в каждом случае независимо выбирают из -OH, -OR, -S(O)R, -S(O)2R, -N(R)-S(O)2R, -S(O)2-N(R)(R), -S(O)2-NHR, -N(R)-C(O)-R или -N(R)(R); и
R в каждом случае независимо представляет собой необязательно замещенный C1-C12 алкил;
или его фармацевтически приемлемую соль. -X в каждом случае может быть независимо выбран из -OH -NH2 или -N(R)(R). В некоторых вариантах осуществления группы -X в R4 и/или R5 (включая группы -X, когда R4 и/или R5 представляет собой -CH2-X) могут быть выбраны из -OH, NH2 или -N(R)(R). Также, R в каждом случае может быть независимо выбран из C1-4 линейного или разветвленного углеводорода.
В некоторых вариантах осуществления фармацевтическая композиция может содержать фармацевтически приемлемый наполнитель и соединение, имеющее структуру, соответствующую формуле (III):

где “пунктирная” связь может представлять собой одинарную или двойную связь;
R1 представляет собой необязательно замещенный арил или гетероарил;
R2 представляет собой необязательно замещенный алкокси, циклоалкокси или гетероциклил;
R3 представляет собой водород или -CH2-X;
R6 и R7 независимо представляют собой водород или R; и
z1-z8 в каждом случае независимо выбирают из CH или N; где
-X в каждом случае независимо выбирают из -OH, -OR, -S(O)R, -S(O)2R, -N(R)-S(O)2R, -S(O)2-N(R)(R), -S(O)2-NHR, -N(R)-C(O)-R или -N(R)(R); и
R в каждом случае независимо представляет собой C1-C12 алкил;
при условии, что каждом случае, когда R6 представляет собой водород, R3 представляет собой -CH2-N(R)(R), и указанная “пунктирная” связь представляет собой двойную связь. В некоторых вариантах осуществления “пунктирная” связь представляет собой двойную связь. В других вариантах осуществления “пунктирная” связь представляет собой одинарную связь. -X в каждом случае может быть независимо выбран из -OH -NH2 или -N(R)(R). Также, R в каждом случае может быть независимо выбран из C1-4 линейного или разветвленного углеводорода. В некоторых вариантах осуществления соединение присутствует в фармацевтической композиции в эффективном количестве. Например, соединение может присутствовать в количестве, эффективном для лечения или профилактики малярии. В некоторых вариантах осуществления соединение может присутствовать в количестве, эффективном для лечения или профилактики заболевания, вызываемого паразитом из рода Cryptosporidium (например, криптоспоридиоза). В некоторых вариантах осуществления фармацевтическая композиция может быть приготовлена в виде лекарственной формы для лечения малярии и криптоспоридиоза.
Также раскрыты соответствующие способы лечения или профилактики заболевания у субъекта. В некоторых вариантах осуществления способ лечения или профилактики заболевания у субъекта содержит этап введения субъекту эффективного количества любого раскрытого в настоящем описании соединения. В некоторых вариантах осуществления эффективное количество соединения содержится в фармацевтической композиции (например, ветеринарной композиции и т.д.).
Паразитарное заболевание может представлять собой малярию. В некоторых вариантах осуществления малярия является фармакорезистентной малярией (например, малярией, устойчивой к хлорохину, хинину, пириметамину, сульфодоксину, мефлохину, артеметеру, люмефантрину, артесунату, амодиахину, дигидроартемизинину, пипераквину, прогуанилу, доксициклину, клиндамицину, артемизинину, атовахину или любой их комбинации и т.д.). В некоторых вариантах осуществления малярия представляет собой малярию на стадии развития в крови. В некоторых вариантах осуществления малярия представляет собой малярию на трансмиссивной стадии. В некоторых вариантах осуществления малярия представляет собой малярию на печеночной стадии. В некоторых вариантах осуществления субъект инфицирован вызывающим малярию паразитом, и указанное лечение предупреждает распространение указанной инфекции за пределы его печени. В некоторых вариантах осуществления малярия переносится видом москита, выбранным из P. falciparum, P. vivax, P. ovale, P. malariae, P. knowlesi, P. berghei, P. chabaudi, P. vinckei или P. yoelii. В предпочтительных вариантах осуществления вид москита представляет собой P. falciparum (в частности, когда субъектом является человек).
Паразитарное заболевание может представлять собой криптоспоридиоз. В некоторых вариантах осуществления криптоспоридиоз переносится C. parvum.
В некоторых вариантах осуществления субъектом является человек. В других вариантах осуществления субъект не является человеком (например, фармацевтическая композиция приготовлена в виде ветеринарной композиции). В некоторых вариантах осуществления, субъект представляет собой мышь, крысу, кролика, не являющегося человеком примата, ящерицу, геккона, корову, быка, овцу, ягненка, лошадь, жеребенка, свинью или поросенка.
Эти и другие аспекты изобретения станут очевидными специалисту в данной области из приведенного ниже описания, которое является всего лишь иллюстрацией различных вариантов осуществления изобретения. При реализации изобретение может включать дополнительные другие очевидные аспекты без отступления от сути и объема изобретения. Следовательно, описание, по сути, является иллюстративным, а не ограничивающим настоящее изобретение.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Следует понимать, что используемая в настоящем описании терминология предназначена для описания конкретных вариантов осуществления и не предназначена для ограничения. Кроме того, хотя на практике или при тестировании изобретения могут быть использованы любые способы и материалы, аналогичные или эквивалентные тем, которые описаны в настоящем документе, ниже описаны предпочтительные способы и материалы.
Термин “ацил”, используемый в настоящем описании, представляет собой водород или алкильную группу, определенную в настоящем описании, которая присоединена к исходной молекулярной группе через карбонильную группу, определенную в настоящем описании, и примером которого является формил (т.е. карбоксиальдегидная группа), ацетил, трифторацетил, пропионил и бутаноил. Типичные незамещенные ацильные группы включают от 1 до 6, от 1 до 11 или от 1 до 21 атомов углерода. В некоторых вариантах осуществления алкильная группа дополнительно замещена 1, 2, 3 или 4 заместителями, раскрытыми в настоящем описании.
Используемый в настоящем описании термин “алкил”, один или в комбинации с другими группами, относится к разветвленному или неразветвленному моновалентному насыщенному алифатическому углеводородному радикалу, содержащему от одного до двадцати атомов углерода (например, от одного до шестнадцати атомов углерода, от одного до двенадцати атомов углерода атомов, от одного до десяти атомов углерода или от одного до шести атомов углерода и т. д.).
Используемый в настоящем описании термин “алкилен” означает насыщенную двухвалентную углеводородную группу, полученную из насыщенного углеводорода с прямой или разветвленной цепью путем удаления двух атомов водорода, примером которого являются метилен, этилен и изопропилен. В некоторых вариантах осуществления алкилен может быть дополнительно замещен 1, 2, 3 или 4 заместителями, определенными в настоящем описании для алкильной группы.
Используемый в настоящем описании термин “алкенил”, один или в комбинации с другими группами, относится к углеводородному остатку с прямой или разветвленной цепью, имеющему олефиновую связь.
Используемый в настоящем описании термин “амино” представляет собой -N(RN1)2, где каждый RN1 независимо представляет собой Н, ОН, NO2, N(RN2)2, SO2ORN2, SO2RN2, SORN2, N-защитную группу, алкил, алкенил, алкинил, алкокси, арил, алкарил, циклоалкил, алкциклоалкил, карбоксиалкил (например, необязательно замещенный O-защитной группой, такой как необязательно замещенные арилалкоксикарбонильные группы или любые описанные в настоящем описании группы), сульфоалкил, ацил (например, ацетил, трифторацетил или другие описанные в настоящем описании группы), алкоксикарбонилалкил (например, необязательно замещенный O-защитной группой, такой как необязательно замещенные арилалкоксикарбонильные группы или любые описанные в настоящем описании группы), гетероциклил (например, гетероарил) или гетероциклилалкил (например, гетероарилалкил), где каждая из указанных групп RN1 необязательно может быть замещена, как определено в настоящем описании для каждой группы; или две группы RN1 объединены с образованием гетероциклила или N-защитной группы, и где каждая группа RN2 независимо представляет собой H, алкил или арил. Аминогруппы могут быть незамещенной аминогруппой (т.е., -NH2) или замещенной аминогруппой (т.е., -N(RN1)2). В предпочтительном варианте осуществления амино представляет собой -NH2 или -NHRN1, где RN1 независимо представляет собой OH, NO2, NH2, NRN22, SO2ORN2, SO2RN2, SORN2, алкил, карбоксиалкил, сульфоалкил, ацил (например, ацетил, трифторацетил или другие описанные в настоящем описании группы), алкоксикарбонилалкил (например, трет-бутоксикарбонилалкил) или арил, и каждая группа RN2 может представлять собой H, C1-20 алкил (например, C1-6 алкил) или C6-10 арил.
Термин “арил” относится к ароматическому моно- или полициклическому радикалу с 6-12 атомами углерода, имеющему по меньшей мере одно ароматическое кольцо. Примеры таких групп включают, без ограничения, фенил, нафтил, 1,2,3,4-тетрагидронафталил, 1,2-дигидронафталил, инданил и 1H-инденил.
“Арилалкильная” группа, используемая в настоящем описании, представляет собой арильную группу, определенную в настоящем описании, присоединенную к исходной молекулярной группе через алкиленовую группу, определенную в настоящем описании. Примеры незамещенных арилалкильных групп имеют от 7 до 30 атомов углерода (например, от 7 до 16 или от 7 до 20 атомов углерода, такие как C6-10 арил C1-6 алкил, C6-10 арил C1-10 алкил или C6-10 арил C1-20 алкил). В некоторых вариантах осуществления каждый из алкилена и арила могут быть дополнительно замещены 1, 2, 3 или 4 заместителями, определенными в настоящем описании для соответствующих групп.
Алкильные, карбоциклические и арильные группы могут быть замещенными или незамещенными. В случае замещения обычно присутствуют, например, от 1 до 4 заместителей. Эти заместители необязательно могут формировать кольцо с алкильной, карбоциклической или арильной группой, с которой они связаны. Заместители могут включать, например: углеродсодержащие группы, такие как алкил, арил, арилалкил (например, замещенный и незамещенный фенил, замещенный и незамещенный бензил); атомы галогена и галогенсодержащие группы, такие как галоалкил (например, трифторметил); кислородсодержащие группы, такие как спирты (например, гидроксил, гидроксиалкил, арил(гидроксил)алкил), простые эфиры (например, алкокси, циклоалкокси, арилокси, алкоксиалкил, арилоксиалкил, более предпочтительно, например, метокси, этокси, пропокси, изопропокси, циклопропокси и т.д.), альдегиды (например, карбоксальдегид), кетоны (например, алкилкарбонил, алкилкарбонилалкил, арилкарбонил, арилалкилкарбонил, арилкарбонилалкил), кислоты (например, карбокси, карбоксиалкил), производные кислот, такие как сложные эфиры (например, алкоксикарбонил, алкоксикарбонилалкил, алкилкарбонилокси, алкилкарбонилоксиалкил), амиды (например, аминокарбонил, моно- или ди-алкиламинокарбонил, аминокарбонилалкил, моно- или ди-алкиламинокарбонилалкил, ариламинокарбонил, карбаматы (например, алкоксикарбониламино, арилоксикарбониламино, аминокарбонилокси, моно- или ди-алкиламинокарбонилокси, ариламинокарбонилокси) или мочевины (например, моно- или ди-алкиламинокарбониламино или ариламинокарбониламино); азотсодержащие группы, такие как амины (например, амино, моно- или ди-алкиламино, аминоалкил, моно- или ди-алкиламиноалкил), азиды, нитрилы (например, циано, цианоалкил), нитро; серосодержащие группы, такие как тиолы, тиоэфиры, сульфоксиды и сульфоны (например, алкилтио, алкилсульфинил, алкилсульфонил, алкилтиоалкил, алкилсульфинилалкил, алкилсульфонилалкил, арилтио, арилсульфинил, арилсульфонил, арилтиоалкил, арилсульфинилалкил, арилсульфонилалкил); гетероциклил-гетероалкильные группы и гетероциклические группы, содержащие один или более гетероатомов (например, тиенил, фуранил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, оксадиазолил, тиадиазолил, азиридинил, азетидинил, пирролидинил, пирролинил, имидазолидинил, имидазолинил, пиразолидинил, тетрагидрофуранил, пиранил, пиронил, пиридил, пиразинил, пиридазинил, пиперидил, гексагидроазепинил, пиперазинил, морфолинил, тианафтил, бензофуранил, изобензофуранил, индолил, оксииндолил, изоиндолил, индазолил, индолинил, 7-азаиндолил, бензопиранил, кумаринил, изокумаринил, хинолинил, изохинолинил, нафтиридил, циннолинил, хиназолинил, пиридопиридил, бензоксазинил, хиноксалинил, хроменил, хроманил, изохроманил, фталазинил и карболинил).
Термин “азидо” представляет собой группу -N3, которая также может быть представлена как -N=N=N.
Термины “карбоциклический” и “карбоциклил”, используемые в настоящем описании, относятся к необязательно замещенной неароматической C3-12 моноциклической, бициклической или трициклической структуре, в которой кольца образованы атомами углерода. Карбоциклические структуры включают циклоалкильные, циклоалкенильные и циклоалкинильные группы.
Термин “циклоалкил” относится к моновалентному моно- или поликарбоциклическому радикалу, состоящему из от трех до десяти, предпочтительно от трех до шести атомов углерода. Примерами этого термина также являются радикалы, такие как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил, адамантил и инданил. В предпочтительном варианте осуществления “циклоалкильные” фрагменты необязательно могут быть замещены одним, двумя, тремя или четырьмя заместителями. Каждый заместитель независимо может представлять собой алкил, алкокси, галоген, амино, гидроксил или кислород, если специально не указано иное. Примеры циклоалкильных фрагментов включают, без ограничения, необязательно замещенный циклопропил, необязательно замещенный циклобутил, необязательно замещенный циклопентил, необязательно замещенный циклопентенил, необязательно замещенный циклогексил и необязательно замещенный циклогептил или фрагменты, в частности, приведенные в настоящем описании.
Термин “циано”, используемый в настоящем описании, представляет группу -CN.
Используемый в настоящем описании термин “гало” или “галоген” означает радикал фтор (фторо), хлор (хлоро), бром (бромо) или йод (йодо).
Термин “гетероалкил”, используемый в настоящем описании, относится к алкильной группе, определенной в настоящем описании, в которой каждый из одного или более составляющих ее атомов углерода заменен азотом, кислородом или серой. В некоторых вариантах осуществления гетероалкильная группа может быть дополнительно замещена 1, 2, 3 или 4 заместителями, описанными в настоящем описании для алкильных групп. Примерами гетероалкильных групп являются группа “алкокси”, которая в контексте настоящего описания относится к алкил-O-; и группа “алкоил”, которая в контексте настоящего описания относится к алкил-СО-. Алкокси-заместители или алкокси-содержащие заместители могут быть замещены, например, одной или более алкильными группами.
Термин “гетероарил” относится к ароматическому моно- или полициклическому радикалу, состоящему из 5-12 атомов, имеющему по меньшей мере одно ароматическое кольцо, содержащее один, два или три кольцевых гетероатома, выбранных из N, O и S, причем остальные атомы кольца являются C. Один или два кольцевых атома углерода гетероарильной группы могут быть замещены карбонильной группой. Примерами гетероарильных групп являются пиридил, бензоксазолил, бензоимидазолил и бензотиазолил.
Термин “гетероцикл” или “гетероциклил” обозначает моно- или полициклическое алкильное кольцо, где один, два или три атома углеродного кольца замещены гетероатомом, таким как N, O или S. Примеры гетероциклических групп включают, без ограничения, оксетанил, морфолинил, тиоморфолинил, пиперазинил, пиперидинил, пирролидинил, тетрагидропиранил, тетрагидрофуранил и 1,3-диоксанил. Гетероциклические группы могут быть незамещенными или замещенными и могут быть присоединены через углеродную структуру или через гетероатом(ы), при необходимости.
Термин “гетероциклил-гетероалкил” относится к гетероциклической группе, определенной в настоящем описании, присоединенной к исходной молекулярной группе через гетероалкильную группу (например, эфирную или алкокси группу). Примером гетероциклил-гетероалкильной группы является группа -OCH2CH2(морфолино).
Описанные выше гетероциклические и гетероарильные группы могут быть независимо замещены одним, двумя, тремя или более заместителями. Заместители могут включать, например: углеродсодержащие группы, такие как алкил, арил, арилалкил (например, замещенный и незамещенный фенил, замещенный и незамещенный бензил); атомы галогена и галогенсодержащие группы, такие как галоалкил (например, трифторметил); кислородсодержащие группы, такие как спирты (например, гидроксил, гидроксиалкил, арил(гидроксил)алкил), простые эфиры (например, алкокси, арилокси, алкоксиалкил, арилоксиалкил), альдегиды (например, карбоксальдегид), кетоны (например, алкилкарбонил, алкилкарбонилалкил, арилкарбонил, арилалкилкарбонил, арилкарбонилалкил), кислоты (например, карбокси, карбоксиалкил), производные кислот, такие как сложные эфиры (например, алкоксикарбонил, алкоксикарбонилалкил, алкилкарбонилокси, алкилкарбонилоксиалкил), амиды (например, аминокарбонил, моно- или ди-алкиламинокарбонил, аминокарбонилалкил, моно- или ди-алкиламинокарбонилалкил, ариламинокарбонил), карбаматы (например, алкоксикарбониламино, арилоксикарбониламино, аминокарбонилокси, моно- или ди-алкиламинокарбонилокси, ариламинокарбонилокси) и мочевины (например, моно- или ди-алкиламинокарбониламино или ариламинокарбониламино); азотсодержащие группы, такие как амины (например, амино, моно- или ди-алкиламино, аминоалкил, моно- или ди-алкиламиноалкил), азиды, нитрилы (например, циано, цианоалкил), нитро; серосодержащие группы, такие как тиолы, тиоэфиры, сульфоксиды и сульфоны (например, алкилтио, алкилсульфинил, алкилсульфонил, алкилтиоалкил, алкилсульфинилалкил, алкилсульфонилалкил, арилтио, арилсульфинил, арилсульфонил, арилтиоалкил, арилсульфинилалкил, арилсульфонилалкил); и гетероциклические группы, содержащие один или более гетероатомов (например, тиенил, фуранил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, оксадиазолил, тиадиазолил, азиридинил, азетидинил, пирролидинил, пирролинил, имидазолидинил, имидазолинил, пиразолидинил, тетрагидрофуранил, пиранил, пиронил, пиридил, пиразинил, пиридазинил, пиперидил, гексагидроазепинил, пиперазинил, морфолинил, тианафтил, бензофуранил, изобензофуранил, индолил, оксииндолил, изоиндолил, индазолил, индолинил, 7-азаиндолил, бензопиранил, кумаринил, изокумаринил, хинолинил, изохинолинил, нафтиридил, циннолинил, хиназолинил, пиридопиридил, бензоксазинил, хиноксалинил, хроменил, хроманил, изохроманил, фталазинил, бензотиазолил и карболинил).
Используемый в настоящем описании термин “гидроксил” означает группу -ОН. В некоторых вариантах осуществления гидроксильная группа может быть замещена О-защитной группой, определенной в настоящем описании.
Используемый в настоящем описании термин “N-защитная группа” означает группы, предназначенные для защиты аминогруппы от нежелательных реакций во время синтеза. Обычно используемые N-защитные группы раскрыты в работе Greene, “Protective Groups в Organic Synthesis”, 3-е изд. (John Wiley & Sons, New York, 1999), которая включена в настоящее описание в виде ссылки. Термин “O-защитная группа”, используемая в настоящем описании, означает группы, предназначенные для защиты кислородсодержащей (например, фенольной, гидроксильной или карбонильной) группы от нежелательных реакций во время синтеза. Обычно используемые O-защитные группы раскрыты в работе Greene, “Protective Groups в Organic Synthesis”, 3-е изд. (John Wiley & Sons, New York, 1999), которая включена в настоящее описание в виде ссылки. Термин “перфторалкил”, используемый в настоящем описании, представляет собой алкильную группу, определенную в настоящем описании, в которой каждый водородный радикал, связанный с алкильной группой, замещен фторидом. Примерами перфторалкильных групп являются трифторметил и пентафторэтил.
Используемый в настоящем описании термин “сульфонил” представляет группу -S(O)2-.
“Замещенный” углеводород может иметь в качестве заместителя один или более углеводородных радикалов, замещенных углеводородных радикалов или может содержать один или более гетероатомов. Примеры замещенных углеводородных радикалов включают, без ограничения, гетероциклы, такие как гетероарилы. Если не указано иное, углеводород, замещенный одним или более гетероатомами, может содержать от 1 до 20 гетероатомов. В других вариантах осуществления углеводород, замещенный одним или более гетероатомами, может содержать от 1 до 12 или от 1 до 8, или от 1 до 6, или от 1 до 4, или от 1 до 3, или от 1 до 2 гетероатомов. Примеры гетероатомов включают, без ограничения, кислород, азот, серу, фосфор, галоген (F, Cl, Br, I и т.д.), бор, кремний и т.д. В некоторых вариантах осуществления гетероатомы выбирают из группы, состоящей из кислорода, азота, серы, фосфора и галогена (F, Cl, Br, I и т.д.). В некоторых вариантах осуществления гетероатом или группа может замещать углерод. В некоторых вариантах осуществления гетероатом или группа может замещать водород. В некоторых вариантах осуществления замещенный углеводород может содержать один или более гетероатомов в каркасе или цепи молекулы (например, расположенной между двумя атомами углерода, как в случае “окса”). В некоторых вариантах осуществления замещенный углеводород может содержать один или более гетероатомов в виде боковой группы каркаса или цепи молекулы (например, ковалентно связанной с атомом углерода цепи или каркаса, как в случае “оксо”, замещение водорода в каркасе или цепи и т.д.).
Термин “заместитель” относится к группе, “замещенной” на, например, алкильную, галоалкильную, циклоалкильную, гетероциклическую, гетероциклоалкенильную, циклоалкенильную, арильную или гетероарильную группу на любом атоме этой группы, замещая на ней один или более атомов водорода. В одном из аспектов заместитель(и) в группе независимо представляет собой любой один или любую комбинацию двух или более допустимых атомов или групп атомов, указанных для этого заместителя. В другом аспекте заместитель сам может быть замещен любым из указанных выше заместителей. Кроме того, как используется в настоящем описании, фраза “необязательно замещенный” означает незамещенный (например, замещенный Н) или замещенный. Понятно, что замещение у данного атома ограничено валентностью. Обычные заместители включают галоген, C1-12 алкил с прямой или разветвленной цепью, C2-12 алкенил, C2-12 алкинил, C3-12 циклоалкил, C6-12 арил, C3-12 гетероарил, C3-12 гетероциклил, C1-12 алкилсульфонил, нитро, циано, -COOR, -C(O)NRR’, -OR, -SR, -NRR’ и оксо, такие как моно- или ди- или три-заместители с фрагментами, такими как трифторметокси, хлор, бром, фтор, метил, метокси, пиридил, фурил, триазил, пиперазинил, пиразолил, имидазолил и т.п., каждый из которых необязательно содержит один или более гетероатомов, таких как галоген, N, O, S и P. Радикалы R и R’ независимо представляют собой водород, C1-12 алкил, C1-12 галоалкил, C2-12 алкенил, C2-12 алкинил, C3-12 циклоалкил, C4-24 циклоалкилалкил, C6-12 арил, C7-24 аралкил, C3-12 гетероциклил, C3-24 гетероциклилалкил, C3-12 гетероарил или C4-24 гетероарилалкил. Если не указано иное, все группы, раскрытые в настоящем описании, необязательно могут содержать один или более общих заместителей в количестве согласно валентности. Далее, используемая в настоящем описании фраза “необязательно замещенный” означает незамещенный (например, замещенный атомом H) или замещенный. Используемый в настоящем описании термин “замещенный” означает, что атом водорода удален и замещен заместителем (например, обычным заместителем). Специалисту в области химии будет понятно, что заместитель при заданном атоме ограничен валентностью. Использование в виде префикса названий заместителей (радикалов), таких как алкил, без модификатора “необязательно замещенный” или “замещенный” означает, что конкретный заместитель является незамещенным. Однако использование термина “галоалкил”, без модификатора “необязательно замещенный” или “замещенный”, по-прежнему означает алкильную группу, в которой по меньшей мере один атом водорода замещен галогеном.
Следует понимать, что описание соединений в настоящем описании ограничено принципами химической связи, известными специалистам в данной области. Соответственно, когда группа может быть замещена одним или более из числа заместителей, такие замены выбирают таким образом, чтобы они соответствовали принципам химической связи в отношении валентностей и т.д., и давали соединения, которые по своей природе не являются нестабильными.
Представленные в настоящей заявке соединения могут иметь один или более асимметричных атомов углерода и могут существовать в форме оптически чистых энантиомеров, смесей энантиомеров, таких как рацематы, оптически чистых диастереоизомеров, смесей диастереоизомеров, диастереоизомерных рацематов или смесей диастереоизомерных рацематов. Оптически активные формы могут быть получены, например, путем разделения рацематов, асимметричным синтезом или асимметричной хроматографией (хроматография с хиральными адсорбентами или элюентом). Иными словами, некоторые из раскрытых соединений могут существовать в различных стереоизомерных формах. Стереоизомеры - это соединения, которые отличаются только пространственным расположением. Энантиомеры представляют собой пары стереоизомеров, чьи зеркальные отображения являются несовместимыми, чаще всего потому, что они содержат асимметрично замещенный атом углерода, который действует как хиральный центр. "Энантиомер" означает одну из пары молекул, которые являются зеркальным отображением друг друга и не могут быть совмещены. Диастереомеры представляют собой стереоизомеры, которые не относятся к зеркальным отображениям, чаще всего потому, что они содержат два или более асимметрично замещенных атома углерода и представляют конфигурацию заместителей вокруг одного или нескольких хиральных атомов углерода. Энантиомерные соединения могут быть получены, например, путем отделения энантиомера от рацемата с помощью одной или более хорошо известных техник и методов, таких как хиральная хроматография и методы разделения на их основе. Подходящие техники и/или методы отделения энантиомерного соединения, раскрытого в настоящем описании, от рацемической смеси могут быть легко определены специалистами в данной области. "Рацемат" или "рацемическая смесь" означает смесь, содержащую два энантиомера, причем такие смеси не проявляют оптической активности; т.е. они не вращают плоскость поляризованного света. "Геометрический изомер" означает изомеры, которые различаются ориентацией атомов-заместителей (например, в углерод-углеродной двойной связи, циклоалкильном кольце, мостиковой бициклической системе и т.д.). Атомы (кроме Н) на каждой стороне углерод-углеродной двойной связи могут находиться в конфигурации E (заместители находятся на противоположных сторонах двойной углерод-углеродной связи) или Z (заместители находятся на одной стороне). "R", "S", "S*", "R*", "E", "Z", "цис" и "транс" указывают конфигурацию относительно основной молекулы. Некоторые из раскрытых соединений могут существовать в атропоизомерных формах. Атропоизомеры представляют собой стереоизомеры, возникающие в результате затрудненного вращения вокруг одинарных связей, где стерический барьер является достаточно высоким для вращения, чтобы обеспечить возможность выделения конформеров. Раскрытые в настоящем описании соединения могут быть получены в виде отдельных изомеров любым изомер-специфическим синтезом, либо могут быть отделены от изомерной смеси. Обычные методы разделения включают образование соли свободного основания каждого изомера изомерной пары с использованием оптически активной кислоты (с последующей фракционной кристаллизацией и восстановлением свободного основания), образование соли кислотной формы каждого изомера изомерной пары с использованием оптически активного амина (с последующей фракционной кристаллизацией и восстановлением свободной кислоты) с образованием сложного эфира или амида каждого из изомеров изомерной пары с использованием оптически чистой кислоты, амина или спирта (с последующим хроматографическим разделением и удалением хирального вспомогательного вещества) или разделение изомерной смеси либо исходного материала, либо конечного продукта с помощью различных хорошо известных хроматографических методов. Когда стереохимия раскрытого соединения названа или изображена в виде структуры, названный или изображенный стереоизомер составляет по меньшей мере 60%, 70%, 80%, 90%, 99% или 99,9% по массе относительно других стереоизомеров. Когда один энантиомер назван или изображен в виде структуры, изображенный или названный энантиомер является по меньшей мере на 60%, 70%, 80%, 90%, 99% или 99,9% по массе оптически чистым. Когда один диастереомер назван или изображен в виде структуры, изображенный или названный диастереомер имеет чистоту по меньшей мере 60%, 70%, 80%, 90%, 99% или 99,9% по массе. Выраженная в процентах оптическая чистота - это отношение массы энантиомера к массе этого энантиомера плюс масса его оптического изомера. Диастереомерная чистота по массе представляет собой отношение массы одного диастереомера к массе всех диастереомеров. Когда стереохимия раскрытого соединения названа или изображена в виде структуры, названный или изображенный стереоизомер имеет по меньшей мере 60%, 70%, 80%, 90%, 99% или 99,9% чистоты по мольной доле относительно других стереоизомеров. Когда один энантиомер назван или изображен в виде структуры, изображенный или названный энантиомер имеет по меньшей мере 60, 70, 80, 90, 99 или 99,9% чистоты по мольной доле. Выраженная в процентах чистота в расчете на мольную долю представляет собой отношение молей энантиомера к молям этого энантиомера плюс моли его оптического изомера. Аналогично, выраженная в процентах чистота в расчете на мольную долю представляет собой отношение количества молей диастереомера к количеству молей этого диастереомера плюс количество молей его изомера. Когда раскрытое соединение названо или изображено в виде структуры без указания стереохимии, и соединение имеет по меньшей мере один хиральный центр, следует понимать, что название или структура включает любой энантиомер соединения, свободный от соответствующего оптического изомера, рацемическую смесь соединения или смеси, обогащенные одним энантиомером относительно его соответствующего оптического изомера. Когда раскрытое соединение названо или изображено в виде структуры без указания стереохимии и имеет два или более хиральных центра, следует понимать, что название или структура включает диастереомер, свободный от других диастереомеров, несколько диастереомеров, свободных от других диастереомерных пар, смесь диастереомеров, смесь диастереомерных пар, смесь диастереомеров, в которых один диастереомер обогащен относительно другого диастереомера(ов), или смесь диастереомеров, в которых один или более диастереомеров обогащены относительно других диастереомеров. Раскрытие охватывает все эти формы.
Используемый в настоящем описании термин “эффективное количество” или “терапевтически эффективное количество” агента представляет собой такое количество, которое достаточно для достижения полезных или требуемых результатов, таких как клинические результаты, и, как таковое, “эффективное количество” зависит от контекста, в котором оно применяется. Например, в контексте введения агента, который является противомалярийным агентом, эффективное количество агента представляет собой, например, количество, достаточное для достижения облегчения или улучшения, или для предотвращения или профилактики одного или более симптомов или состояний; уменьшения степени заболевания, расстройства или состояния; стабилизации (т.е. отсутствию ухудшения) состояния заболевания, расстройства или состояния; предотвращения распространения заболевания, расстройства или состояния (например, предотвращения распространения инфекции Plasmodium за пределы печени, предотвращения развития системного заболевания, предотвращения симптоматической стадии малярии, предотвращения возникновения инфекции Plasmodium и/или предотвращения дальнейшего распространения заболевания путем предотвращения передачи обратно к комару и т.д.); задержки или замедления развития заболевания, расстройства или состояния; улучшения или смягчения заболевания, расстройства или состояния; и ремиссии (частичной или полной), обнаруживаемой или не обнаруживаемой, по сравнению с ответом, полученным без введения агента.
Используемый в настоящем описании термин “фармацевтическая композиция” представляет собой композицию, содержащую соединение, раскрытое в настоящем описании, и фармацевтически приемлемый наполнитель. В некоторых вариантах осуществления фармацевтическую композицию изготавливают или продают в соответствии с разрешением государственного регулирующего органа как часть терапевтического режима лечения заболевания у млекопитающего. Фармацевтическая композиция может быть приготовлена, например, для перорального введения в виде стандартной лекарственной формы (например, таблетки, капсулы, каплетки, гелькапа или сиропа); для местного применения (например, в виде крема, геля, лосьона или мази); для внутривенного введения (например, в виде стерильного раствора, не содержащего частиц эмбола и в системе растворителей, подходящих для внутривенного применения); или в виде любой другой лекарственной формы, раскрытой в настоящем описании (см. ниже).
Пригодными фармацевтическими носителями для приготовления композиций по настоящему изобретению могут быть твердые вещества, жидкости или газы. Таким образом, композиции могут иметь форму таблеток, пилюль, капсул, суппозиториев, порошков, составов с энтеросолюбильным покрытием или защищенных иным образом (например, путем связывания с ионообменными смолами или упаковки в липид-белковые везикулы), составов с замедленным высвобождением, растворов, суспензий, эликсиров и аэрозолей. Носитель может быть выбран из различных масел, включая масла нефтяного, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло, минеральное масло и кунжутное масло. Вода, физиологический раствор, водная декстроза и гликоли являются предпочтительными жидкими носителями, особенно (когда они изотоничны плазме крови) для растворов для инъекций. Например, составы для внутривенного введения включают стерильные водные растворы активного ингредиента(ов), которые получают путем растворения твердого активного ингредиента(ов) в воде с получением водного раствора и обеспечения стерильности раствора. Подходящие фармацевтические наполнители включают крахмал, целлюлозу, тальк, глюкозу, лактозу, тальк, желатин, солод, рис, муку, мел, диоксид кремния, стеарат магния, стеарат натрия, моностеарат глицерина, хлорид натрия, сухое обезжиренное молоко, глицерин, пропиленгликоль, воду и этанол. Композиции могут содержать обычные фармацевтические добавки, такие как консерванты, стабилизирующие агенты, смачивающие или эмульгирующие агенты, соли для регулирования осмотического давления и буферы. Подходящие фармацевтические носители и их состав описаны в Remington's Pharmaceutical Sciences E. W. Martin. Такие композиции в любом случае будут содержать эффективное количество активного соединения вместе с носителем, подходящим для приготовления подходящей для введения реципиенту лекарственной формы.
Используемый в настоящем описании термин “фармацевтически приемлемая соль” относится к солям любого из раскрытых в настоящем описании соединений, которые в рамках здравого медицинского заключения подходят для использования в контакте с тканями людей и животных, не вызывая чрезмерной токсичности, раздражения, аллергии, и имеющие разумное соотношение польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области. Например, фармацевтически приемлемые соли описаны Berge et al., J. Pharmaceutical Sciences 66:1-19, 1977 и Pharmaceutical Salts: Properties, Selection, и Use, (Eds. P.H. Stahl и C.G. Wermuth), Wiley-VCH, 2008. Соли могут быть получены из фармацевтически приемлемых нетоксичных кислот и оснований, включая неорганические и органические кислоты и основания. Типичные кислотно-аддитивные соли включают такие соли, как ацетат, адипин, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентан пропионат, дихлорацетат, диглюконат, додецилсульфат, этансульфонат, фрмиат, фумарат, глюкогептонат, глутамат, глицерофосфат, гемисульфат, гептонат, гексаноат, гиппурат, гидробромид, гидрохлорид, гидроиодид, 2-гидроксиэтансульфонат, изетионат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, манделат, метансульфонат, мукат, 2-нафталинсульфонат, никотенат, нитрат, олеат, оксалат, пальмитат, памоат, пантотенат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, толуолсульфонат, ундеканоат и валерат. Типичные основные соли включают соли щелочных или щелочноземельных металлов, в том числе соли натрия, лития, калия, кальция и магния, алюминия, а также нетоксичные катионы аммония, четвертичного аммония и амина, включая, без ограничения, аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, кофеин и этиламин.
Используемый в настоящем описании термин “субъект” относится к любому организму, которому можно вводить композицию в соответствии с настоящим изобретением, например, для экспериментальных, диагностических, профилактических и/или терапевтических целей. Типичными субъектами являются любые животные (например, млекопитающие, такие как мыши, крысы, кролики, приматы, отличные от человека, и люди, ящерицы, гекконы и т.д.). Субъектом могут быть одомашненные животные (например, коровы, телята, овцы, ягнята, лошади, жеребята, свиньи, поросята и т.д.) или животные семейства Muridae (например, крысы, мыши и т.д.). Субъект может искать или нуждаться в лечении, требовать лечение, получать лечение, может получить лечение в будущем, или может быть человеком или животным, которое находится под наблюдением квалифицированного специалиста по конкретному заболеванию или состоянию.
Используемое в настоящем описании и хорошо понятное в данной области “лечить” состояние или “лечение” состояния (например, состояний, раскрытых в настоящем описании, таких как малярия), представляет собой подход для получения полезных или требуемых результатов, таких как клинические результаты. Полезные или требуемые результаты могут включать, без ограничения, облегчение или улучшение одного или более симптомов или состояний; уменьшение степени заболевания, расстройства или состояния; стабилизацию (т.е. отсутствие ухудшения) состояния заболевания, расстройства или состояния; предотвращение распространения заболевания, расстройства или состояния (например, предотвращение распространения инфекции Plasmodium за пределы печени или предотвращения передачи обратно к комару, предотвращения развития системного заболевания, предотвращения симптоматической стадии малярии и/или предотвращения возникновения инфекции Plasmodium); задержку или замедление развития заболевания, расстройства или состояния; улучшение или смягчение заболевания, расстройства или состояния; и ремиссию (частичную или полную), обнаруживаемую или не обнаруживаемую. “Паллиативное” заболевание, расстройство или состояние означает, что степень и/или нежелательные клинические проявления заболевания, расстройства или состояния уменьшаются и/или время прогрессирования замедляется или удлиняется по сравнению со степенью или временем прогрессирования при отсутствии лечения.
Термин “единичная лекарственная форма” относится к физически дискретной единице, подходящей в качестве стандартной дозы для людей и других млекопитающих, причем каждая единица содержит заранее определенное количество активного материала, рассчитанное для получения требуемого терапевтического эффекта, в сочетании с любым подходящим фармацевтическим наполнителем или наполнителями. Типичные неограничивающие единичные лекарственные формы включают таблетку (например, жевательную таблетку), каплету, капсулу (например, твердую капсулу или мягкую капсулу), пастилку, пленку, полоску, желатиновую капсулу и сироп (также см. ниже).
Другие признаки и преимущества изобретения описаны ниже в подробном описании и формуле изобретения.
СОЕДИНЕНИЯ
Настоящее изобретение относится к новым соединениям и фармацевтическим композициям, полезным для лечения малярии. Настоящее изобретение также относится к способам применения этих соединений и композиций.
В некоторых вариантах осуществления соединения могут представлять собой любые соединения, перечисленные в Таблице 1.


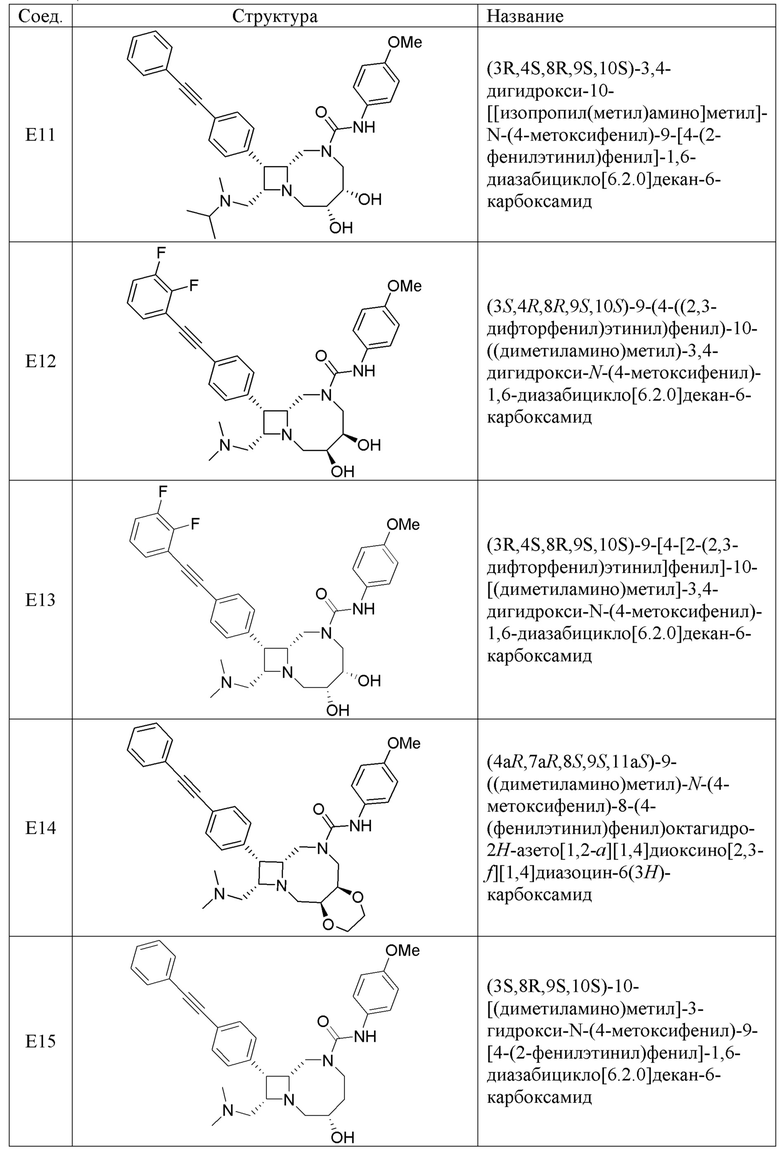
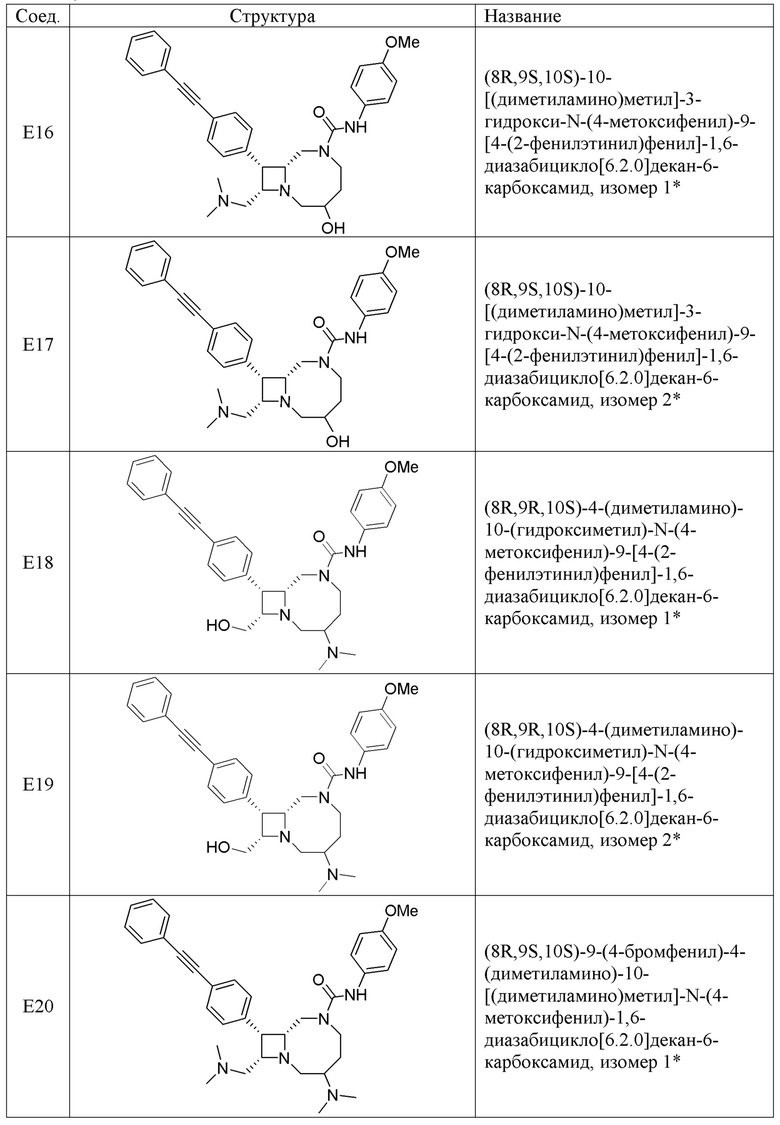




* Каждый стереоизомер выделяли, как описано в примере синтеза, и использовали во всех биологических экспериментах, описанных ниже. Однако стереохимия в положении C3 или C4 не может быть определена, хотя каждый диастереомер (т.е. изомер 1 или изомер 2) выделяли, как описано, и он не присутствовал в виде смеси.
Следует отметить, что в случае любого несоответствия между химическим названием и формулой как соединения с указанным химическим названием, так и соединения с указанной химической структурой следует рассматривать, как охваченные объемом изобретения.
Соединения по настоящему изобретению включают сами соединения, а также их соли и пролекарства, если это применимо. Соль, например, может быть образована между анионом и положительно заряженным заместителем (например, амином) в раскрытом в настоящем описании соединении. Подходящие анионы включают хлорид, бромид, йодид, сульфат, нитрат, фосфат, цитрат, метансульфонат, трифторацетат и ацетат. Аналогично, соль может быть образована между катионом и отрицательно заряженным заместителем (например, карбоксилатом) в раскрытом в настоящем описании соединении. Подходящие катионы включают ион натрия, ион калия, ион магния, ион кальция и катион аммония, такой как ион тетраметиламмония. Примеры пролекарств включают С1-6 алкиловые эфиры групп карбоновых кислот, которые при введении субъекту способны давать активные соединения.
Фармацевтически приемлемые соли соединений по настоящему изобретению включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Используемый в настоящем описании термин “фармацевтически приемлемая соль” относится к соли, образованной добавлением фармацевтически приемлемой кислоты или основания к соединению, раскрытому в настоящем описании. Используемое в настоящем описании выражение “фармацевтически приемлемый” относится к веществу, которое является приемлемым для использования в фармацевтических целях с токсикологической точки зрения и не взаимодействует с активным ингредиентом неблагоприятным образом.
Примеры подходящих солей кислот включают ацетат, адипинат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, гликолат, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, тартрат, тиоцианат, тозилат и ундеканоат. Другие кислоты, такие как щавелевая кислота, хотя сами по себе не являются фармацевтически приемлемыми, могут быть использованы при получении солей, полезных в качестве промежуточных соединений в процессе синтеза соединений по настоящему изобретению и их фармацевтически приемлемых кислотно-аддитивный солей. Соли, полученные из соответствующих оснований, включают соли щелочных металлов (например, натрия), щелочноземельных металлов (например, магния), соли аммония и N-(алкил)4+. Настоящее изобретение также предусматривает кватернизацию любых основных азотсодержащих групп, раскрытых в настоящем описании соединений. Такой кватернизацией могут быть получены растворимые или диспергируемые в воде или масле продукты. Солевые формы соединений любой из приведенных в настоящем описании формул могут представлять собой соли карбоксильных групп (например, соли L-аргинина, лизина, гистидина).
Списки подходящих солей можно найти в Remington's Pharmaceutical Sciences, 17-е изд., Mack Publishing Company, Easton, Pa., 1985, p. 1418; Journal of Pharmaceutical Science, 66, 2 (1977); и "Pharmaceutical Salts: Properties, Selection, и Use A Handbook; Wermuth, C. G. и Stahl, P. H. (eds.) Verlag Helvetica Chimica Acta, Zurich, 2002 [ISBN 3-906390-26-8], каждый из которых включен в настоящее описание во всей своей полноте в виде ссылки.
Нейтральные формы соединений могут быть восстановлены путем взаимодействия соли с основанием или кислотой и выделения исходного соединения обычным способом. Исходная форма соединения отличается от различных солевых форм определенными физическими свойствами, такими как растворимость в полярных растворителях, но в остальном для целей настоящего изобретения соли эквивалентны исходной форме соединения.
Дополнительно к солевым формам настоящее изобретение относится к соединениям, которые находятся в форме пролекарства. Пролекарствами раскрытых в настоящем описании соединений являются соединения, которые подвергаются химическим изменениям в физиологических условиях, давая в результате соединения по настоящему изобретению. Кроме того, пролекарства могут быть преобразованы в соединения по настоящему изобретению химическими или биохимическими методами в среде ex vivo. Например, пролекарства могут медленно превращаться в соединения по настоящему изобретению при помещении в резервуар для трансдермального пластыря с подходящим ферментом или химическим реагентом. Пролекарства часто полезны, потому что в некоторых ситуациях их легче вводить, чем исходный препарат. Они могут, например, быть более биодоступными при пероральном введении, чем исходное лекарственное средство. Пролекарство также может иметь улучшенную растворимость в фармакологических композициях по сравнению с исходным лекарственным средством. В данной области известно большое количество производных пролекарств, например в основе которых лежит гидролитическое расщепление или окислительная активация пролекарства. Примером пролекарства, без ограничения, может быть соединение по настоящему изобретению, которое вводят в виде эфира (“пролекарство”), который затем подвергается метаболическому гидролизу до карбоновой кислоты, активного вещества. Дополнительные примеры включают пептидильные производные соединения по настоящему изобретению.
Настоящее изобретение также включает различные гидратные и сольватные формы соединений.
Соединения по настоящему изобретению могут также содержать неестественные пропорции атомных изотопов одного или более атомов, которые составляют такие соединения. Например, соединения могут быть радиоактивно мечены радиоактивными изотопами, такими как, например, тритий (3H), йод-125 (125I) или углерод-14 (14C). Предполагается, что все изотопные варианты соединений по настоящему изобретению, радиоактивные или нет, входят в объем настоящего изобретения.
Способы
Раскрытые в настоящем описании соединения применяются в способах, представленных в настоящем документе, и, не будучи связанными какой-либо конкретной теорией, предполагается, что оказываемый ими требуемый эффект обеспечивается их способностью ингибировать рост или уничтожать простейших-паразитов, вызывающих малярию (например, P. Falciparum, P. vivax, P. ovale, P. malariae и/или P. knowlesi) и/или криптоспоридиоз (например, C. parvum, C. hominis, C. canis, C. felis, C. meleagridis и C. muris). В некоторых вариантах осуществления это лечение включает профилактику причины, такую как предотвращение распространения инфекции, вызываемой Plasmodium и/или Cryptosporidium, за пределы печени, предотвращение развития системного заболевания, предотвращение симптоматической стадии малярии, предотвращение возникновения инфекции и/или предотвращение дальнейшей передачи (например, комару). В некоторых вариантах осуществления лечение малярии относится к лечению, предназначенному для достижения выздоровления (например, P. vivax или P. malariae), например, полного выздоровления (т.е. очищения печени от гипнозоитов). В различных примерах способы включают предотвращение распространения инфицирования вызывающим малярию паразитом за пределы печени. По некоторым данным, лечение криптоспоридиоза включает профилактику причины, такую как предотвращение распространения криптоспоридиума за пределы инфицированных участков субъекта (например, печени, кишечника, дыхательных путей и т.д.).
Соединения могут быть полезными при лечении фармакорезистентной малярии, такой как малярии, резистентной к хлорохину, хинину, пириметамину, сульфодоксину, мефлохину, артеметеру, люмефантрину, артесунату, амодиахину, дигидроартемизинину, пипераквину, прогуанилу, доксициклину, клиндамицину, артемизинину, атовахину и любой их комбинации.
Фармацевтические композиции
1. Составы
Для использования в раскрытых в настоящем описании способах соединения могут быть приготовлены в виде фармацевтических или ветеринарных композиций. Выбранный состав может изменяться в зависимости от подлежащего лечению субъекта, способа введения и типа требуемого лечения (например, предотвращение, профилактика или терапия). Краткое описание методов составления лекарственных форм представлено в Remington: The Science и Practice of Pharmacy, 21st Edition, Lippincott Williams & Wilkins, (2005); и Enциклоpedia of Pharmaceutical Technology, eds. J. Swarbrick и J. C. Boylan, 1988-1999, Marcel Dekker, New York, каждый из которых включен в настоящее описание в виде ссылки. Примеры пути введения и составы описаны ниже.
При применении на практике раскрытых способов соединения (или их фармацевтически приемлемые соли) или композиции можно вводить любым из обычных и приемлемых путей и способов, известных в данной области. Таким образом, соединения или композиции могут быть введены, например, энтеральным или гастроинтестинальным путем (например, перорально или ректально), местно (например, нанесением на кожу или доступную слизистую оболочку (например, на поверхность в ротовой полости (например, подъязычно или буккально), интраназально, ректально или урогентильно), парентерально (например, путем нанесения или инъекции внутримышечной, внутривенной, подкожной, внутрисуставной, внутрипузырной, интратекальной, эпидуральной, окулярной или ушной), трансдермально или путем ингаляции (например, в виде аэрозоля).
Композиции могут быть в форме твердого вещества, жидкости или газа, как определено подходящими специалистами в данной области. Таким образом, в качестве общих примеров, фармацевтическая композиция может быть в форме таблеток, капсул, сиропов, пилюль, составов с энтеросолюбильным покрытием или другим защитным покрытием, составов с замедленным высвобождением, эликсиров, порошков, гранул, суспензий, эмульсий, растворов, гелей (например, гидрогелей), паст, мазей, кремов, пластырей, трансдермальных пластырей, каплей, суппозиториев, клизм, инъекционных препаратов, имплантатов, спреев или аэрозолей.
Композиции, как правило, содержит эффективное количество раскрытого в настоящем описании соединения и один или более фармацевтически приемлемых носителей или наполнителей, хорошо известных в данной области. Таким образом, композиции могут включать один или более разбавителей, буферов, консервантов, солей, углеводов, аминокислот, белков-носителей, жирных кислот, липидов и т.д. Раскрытые в настоящем описании соединения могут присутствовать в количестве, составляющем, например, 1-95% по массе от общей массы композиции.
Для инъекций составы могут быть приготовлены в обычных формах в виде жидких растворов или суспензий или в виде твердых форм, подходящих для растворения или суспендирования в жидкости перед инъекцией, или в виде эмульсий. Подходящие наполнители для этих составов включают, например, воду, физиологический раствор, декстрозу и глицерин. Такие композиции могут также содержать нетоксичные вспомогательные вещества, такие как смачивающие или эмульгирующие агенты, и рН-буферные агенты, такие как ацетат натрия, сорбитана монолаурат и т.д.
Составы для перорального применения включают таблетки, содержащие соединение в смеси с одним или более нетоксичными фармацевтически приемлемыми наполнителями. Этими наполнителями могут быть, например, инертные разбавители или наполнители (например, сахароза, сорбит, сахар, маннит, микрокристаллическая целлюлоза, крахмалы, включая картофельный крахмал, карбонат кальция, хлорид натрия, лактоза, фосфат кальция, сульфат кальция или фосфат натрия); гранулирующие и дезинтегрирующие агенты (например, производные целлюлозы, включая микрокристаллическую целлюлозу, крахмалы, включая картофельный крахмал, кроскармеллозу натрия, альгинаты или альгиновую кислоту); связующие агенты (например, сахароза, глюкоза, сорбит, акация, альгиновая кислота, альгинат натрия, желатин, крахмал, прежелатинизированный крахмал, микрокристаллическая целлюлоза, силикат магния алюминия, карбоксиметилцеллюлоза натрия, метилцеллюлоза, гидроксипропилметилцеллюлоза, этилцеллюлоза, поливинилпирролидон или этиленгликоль); и лубриканты, глиданты и антиадгезивы (например, стеарат магния, стеарат цинка, стеариновая кислота, диоксид кремния, гидрогенизированные растительные масла или тальк). Другие фармацевтически приемлемые наполнители могут представлять собой красители, ароматизаторы, пластификаторы, увлажнители и буферные агенты.
Составы для перорального введения также могут быть представлены в виде жевательных таблеток или в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем (например, картофельным крахмалом, лактозой, микрокристаллической целлюлозой, карбонатом кальция, фосфатом кальция или каолином), или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом. Порошки, гранулы и пеллеты могут быть приготовлены с использованием ингредиентов, упомянутых выше для таблеток и капсул, обычным способом, например, с использованием миксера, аппарата с псевдоожиженным слоем или оборудования для распылительной сушки.
Растворение или диффузия с контролируемым высвобождением может быть достигнута путем нанесения соответствующего покрытия на таблетку, капсулу, пеллету или гранулированный состав соединений или путем включения соединения в соответствующую матрицу. Покрытие с контролируемым высвобождением может включать одно или более, используемых для покрытия веществ, упомянутых выше, и/или, например, шеллак, пчелиный воск, гликовакс, касторовый воск, воск карнаубы, стеариловый спирт, глицерилмоностеарат, глицерилдистеарат, пальмитостеарат глицерина, этилцеллюлозу, акриловые смолы, dl-полимолочную кислоту, ацетатбутират целлюлозы, поливинилхлорид, поливинилацетат, винилпирролидон, полиэтилен, полиметакрилат, метилметакрилат, 2-гидроксиметакрилат, метакрилатные гидрогели, 1,3-бутиленгликоль, этиленгликольметакрилат и/или полиэтиленгликоль. В составе с матрицей с контролируемым высвобождением материал матрицы может также включать, например, гидратированную метилцеллюлозу, карнаубский воск и стеариловый спирт, карбопол 934, силикон, глицерилтристеарат, метилакрилат-метилметакрилат, поливинилхлорид, полиэтилен и/или галогенированный фторуглерод.
Жидкие формы для перорального введения, в которые могут быть введены соединения и композиции, включают водные растворы, подходящие ароматизированные сиропы, водные или масляные суспензии и ароматизированные эмульсии с пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и аналогичные фармацевтические несущие среды.
Фармацевтическая композиция также может быть приготовлена в виде ветеринарной композиции, предназначенной для использования субъектами, отличными от людей. Ветеринарные композиции в соответствии с настоящим изобретением могут быть в любых подходящих формах, соответствующих требуемым способам введения, например, для назального, перорального, внутрикожного, кожного или парентерального введения. В предпочтительном варианте осуществления композиция находится в форме, предназначенной для перорального введения и, например, может вводиться домашнему животному во время еды, либо путем смешивания с кормом, либо путем непосредственного введения в ротовую полость после приема пищи. Ветеринарные композиции по изобретению находятся в форме назальной, пероральной или инъецируемой жидкой суспензии или раствора или в твердой или полутвердой форме, в виде порошков, пеллет, капсул, гранул, таблеток, паст, имплантатов или гелей. В конкретном варианте осуществления композиции находятся в виде твердой формы для перорального введения, предпочтительно в виде таблеток. В некоторых случаях ветеринарные композиции могут содержать эффективное количество соединения для определенного вида животных (например, коровы, ягненка, козы, лошади и т.д.).
В предпочтительном варианте осуществления композиции по изобретению приготовлены в виде пеллет или таблеток для перорального введения. В соответствии с составом этого типа они включают моногидрат лактозы, микрокристаллическую целлюлозу, кросповидон/повидон, ароматизатор, прессованный сахар и стеарат магния в качестве наполнителей. Если композиции находятся в виде пеллет или таблеток, они представляют собой, например, 1 мг, 2 мг или 4 мг пеллеты или таблетки торасемида. Такие пеллеты или таблетки можно делить на части таким образом, чтобы они соответствовали дозировкам согласно изобретению, принимаемым за один или два приема в день. В следующем предпочтительном варианте осуществления композиции по изобретению приготовлены в виде растворов или суспензий для инъекций для парентерального введения. Инъецируемые композиции получают путем смешивания терапевтически эффективного количества торасемида с регулятором рН, буферным агентом, суспендирующим агентом, солюбилизирующим агентом, стабилизатором, агентом, регулирующим тоничность и/или консервантом, и путем превращения в смесь для внутривенной, подкожной, внутримышечной инъекции или перфузии в соответствии с общепринятым способом. Инъецируемые композиции могут быть лиофилизированы общепринятым способом. Примеры суспендирующих агентов включают метилцеллюлозу, полисорбат 80, гидроксиэтилцеллюлозу, ксантановую камедь, натриевую соль карбоксиметилцеллюлозы и полиэтоксилированный монолаурат сорбита. Примеры солюбилизирующего агента включают полиоксиэтилен-отвержденное касторовое масло, полисорбат 80, никотинамид, полиэтоксилированный монолаурат сорбита, макрогол и этиловый эфир жирной кислоты касторового масла. Кроме того, стабилизатор включает сульфит натрия, металлсульфит натрия и эфир, в то время как консервант включает метил п-гидроксибензоат, этил п-гидроксибензоат, сорбиновую кислоту, фенол, крезол и хлоркрезол. Примером тонизирующего агента является маннит. При приготовлении инъекционных суспензий или растворов желательно убедиться, что они являются изотоничными крови.
2. Наборы
Соединения и композиции могут быть упакованы в набор, необязательно, с одним или более другими фармацевтическими агентами (см. ниже). Неограничивающие примеры наборов включают те, которые содержат, например, две или более пилюли, пилюлю и порошок, суппозиторий и жидкость во флаконе или два крема для местного применения. Наборы могут включать необязательные компоненты, которые помогают при введении субъектам единичной дозы, такие как флаконы для восстановления порошковых форм, шприцы для инъекций, индивидуальные системы внутривенной доставки или ингаляторы. Кроме того, наборы единичных доз могут содержать инструкции по приготовлению и введению композиций. Наборы могут быть изготовлены в виде единичной дозы однократного применения для одного субъекта, многократного применения для конкретного субъекта (с постоянной дозой или в которой индивидуальные соединения могут различаться по эффективности в ходе терапии); или наборы могут содержать несколько доз, подходящих для введения нескольким субъектам (“объемная упаковка”). Компоненты набора могут находиться в картонных коробках, блистерных упаковках, бутылках и пробирках.
3. Дозирование
Доза соединения зависит от ряда факторов, таких как способ введения, возраст и масса тела субъекта, а также состояние субъекта, подлежащего лечению, и в конечном итоге будет определяться лечащим врачом или ветеринаром. Такое количество соединения, определенное лечащим врачом или ветеринаром, упоминается в настоящей заявке и в формуле изобретения как “терапевтически эффективное количество”. Например, доза соединения, раскрытого в настоящем описании, обычно находится в диапазоне от 1 до 1000 мг в сутки. Предпочтительно терапевтически эффективное количество находится в количестве от примерно 1 до примерно 500 мг в сутки.
Введение каждого лекарственного вещества, как описано в настоящем документе, может происходить независимо от одного до четырех раз в сутки в течение от одного дня до одного года и даже возможно в течение жизни субъекта. Хроническое, долгосрочное введение может быть указано.
4. Комбинированная терапия
Соединения и фармацевтические композиции могут быть приготовлены в виде лекарственной формы и использованы в комбинированной терапии, т.е. соединения и фармацевтические композиции могут быть приготовлены в виде лекарственной формы или вводиться одновременно, до или после применения одной или более других требуемых терапевтических или медицинских процедур. В конкретной комбинации методов лечения (терапевтических средств или процедур) для применения в комбинированном режиме учитываются совместимость требуемых терапевтических средств и/или процедур и требуемый терапевтический эффект, который должен быть достигнут. Также следует понимать, что применяемые виды терапии могут обеспечивать желаемый эффект при одном и том же расстройстве, или они могут обеспечивать разные эффекты (например, контроль любых побочных эффектов).
Примеры других лекарственных веществ для комбинирования с раскрытыми в настоящем описании соединениями включают лекарственные вещества для лечения малярии (например, хлорохин, хинин, пириметамин, сульфодоксин, мефлохин, артеметер, люмефантрин, артесунат, амодиахин, дигидроартемизинин, пипераквин, прогуанил, доксициклин, клиндамицин, артемизинин, атовахин, любое другое терапевтическое средство, одобренное для лечения малярии, и/или фармацевтические препараты для лечения криптоспоридиоза (например, нитазоксанид). Другие примеры лекарственных веществ для комбинирования с раскрытыми в настоящем описании соединениями включают фармацевтические препараты для лечения различных, но ассоциированных или родственных симптомов или показаний. Комбинированные способы могут включать применение двух (или более) агентов, составленных вместе или по отдельности, как определено специалистами в данной области техники. В одном из примеров два или более лекарственных вещества составлены вместе для одновременного или почти одновременного введения.
ПРИМЕРЫ
Приведенные ниже примеры иллюстрируют синтез репрезентативного количества соединений и применение этих соединений при лечении малярии. Соответственно, примеры предназначены для иллюстрации, а не для ограничения объема изобретения. Дополнительные соединения, специально не приведенные в качестве примеров, могут быть синтезированы обычными способами в комбинации со способами, раскрытыми в настоящем описании.
Синтез соединений
Материалы и способы
Все реакции выполняли в атмосфере N2 или аргона. Все реагенты и растворители приобретали у коммерческих поставщиков и использовали в том виде, в котором они были приобретены или синтезированы в соответствии с примечанием. Спектры ЯМР регистрировали на спектрометре Bruker 300 (300 МГц 1Н, 75 МГц, 13С) и Bruker 400 (400 МГц 1Н, 100 МГц, 13С). Химические сдвиги протонов приведены в миллионных долях (ppm) (6) относительно растворителя ЯМР. Данные представлены следующим образом: химические сдвиги, кратность (ш. = широкий, с=синглет, д=дублет, т=триплет, к=квартет, кв. = квинтет, м=мультиплет; константа(ы) связи в Гц; слияние). Если не указано иное, данные ЯМР собирали при 25°С. Флэш-хроматографию выполняли с использованием силикагеля 40-60 (60 Å меш) на Teledyne Isco Combiflash Rf. Для анализа чистоты во всех примерах чистоту измеряли по поглощению ультрафиолетового излучения при 210 нм, а идентичность определяли на масс-спектрометре SQ электрораспылительной ионизацией с регистрацией положительных ионов. Использовали следующий метод: тандемную жидкостную хромотографию/масс-спектрометрию (ЖХ/МС) выполняли на модуле разделения Waters 2795 и детекторе массы 3100. Подвижная фаза A состояла из 0,01% муравьиной кислоты в воде, тогда как подвижная фаза B состояла из 0,01% муравьиной кислоты в ацетонитриле. Градиент составлял от 5% до 95% подвижной фазы B в течение 2,5, 5 или 7,5 мин при скорости 1,75 мл/мин. Колонку Agilent Poroshell 120 EC-C18, 2,7 мМ, 3,0×30 мм использовали при температуре колонки, поддерживаемой на уровне 40°С. Вводили 2,1 мл раствора образца. Аналитическую ТСХ осуществляли на пластинах EM-Reagent 0,25 мм с силикагелем 60-F. Визуализацию осуществляли с помощью УФ-света и окрашивания водным раствором перманганата калия (KMnO4) с последующим нагреванием. Точные измерения массы получали на масс-спектрометре Agilent 6230 Time-of-Flight в виде (M+H)+. Чистоту и идентичность соединения также определяли с помощью СВЭЖХ-МС (UPLC-MS). Чистоту измеряли по УФ-поглощению при 210 нм. Идентичность определяли на масс-спектрометре SQ электрораспылительной ионизации с регистрацией положительных и отрицательных ионов. Подвижная фаза A состояла либо из 0,1% гидроксида аммония, либо 0,05% трифторуксусной кислоты в воде, тогда как подвижная фаза B состояла либо из 0,1% гидроксида аммония, либо 0,06% трифторуксусной кислоты в ацетонитриле. Градиент составлял от 5 до 95% подвижной фазы B в течение 2,65 мин при скорости 0,9 мл/мин. Использовали колонку Acquity BEH C18, 1,7 мкм, 2,1×50 мм с температурой колонки, поддерживаемой на уровне 65°C. Соединения растворяли в ДМСО при номинальной концентрации 1 мг/мл и вводили 1,0 мкл этого раствора. Хиральное разделение выполняли с помощью СФХ-МС (SFC-MS). Сверхкритический жидкостный хроматограф Berger G600 соединяли с одиночным квадрупольным масс-спектрометром Waters ZQ, работающим в режиме положительного APCI. С использованием сжиженного СО2, модифицированного 20% изопропанолом, проводили изократическое разделение в течение 5,0 минут при 4,0 мл/мин на колонке Chiralpak AD-H 4,6×100 мм, поддерживаемой при 40°С. Соединения растворяли в метаноле при номинальной концентрации 1 мг/мл и вводили 10 мкл этого раствора. Приведенные ниже примеры синтеза не имеют особых ограничений, и другие методы синтеза соединений по настоящему изобретению хорошо известны специалистам в данной области.
Сокращения
Во всех приведенных ниже примерах синтеза использованы различные сокращения, которые хорошо известны специалистам в данной области. В таблице 2 представлены различные примеры сокращений, использованных в примерах, и соответствующие ссылки.
Пример 1: (3S,4R,8R,9S,10S)-N-(4-циклопропоксифенил)-10-((диметиламино)метил)-3,4-дигидрокси-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E1”)




Промежуточное соединение 1: ((8R,9R,10S, Z)-9-(4-бромфенил)-6-((2-нитрофенил)сульфонил)-1,6-диазабицикло[6.2.0]дец-3-ен-10-ил)метанол
К раствору (8R,9R,10S, Z)-10-((бис(4-метоксифенил)(фенил)метокси)метил)-9-(4-бромфенил)-6-((2-нитрофенил)сульфонил)-1,6-диазабицикло[6.2.0]дец-3-ена (2,00 г, 2,66 ммоль) (Journal of Organic Chemistry (2012), 77(17), 7187-7211; WO 2015070204) в ДМСО (27,00 мл) добавляли TFA (3,03 г, 26,60 ммоль, 1,97 мл). Смесь перемешивали при 15°C в течение 20 часов. Реакционную смесь гасили добавлением раствора NaHCO3 (100 мл) и затем экстрагировали 180 мл DCM (3×60 мл). Комбинированные органические слои сушили над [NaSO4], фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир:DCM=1:1 до DCM:MeOH=10:1 с получением промежуточного соединения 1 (1,47 г, неочищенное) в виде твердого вещества черного цвета. Присутствие реакционного продукта подтверждали ЯМР и ЖХ/МС.
Промежуточное соединение 2: 2-(((8R,9S,10S, Z)-9-(4-бромфенил)-6-((2-нитрофенил)сульфонил)-1,6-диазабицикло[6.2.0]дец-3-ен-10-ил)метил)изоиндолин-1,3-дион
К смеси трифенилфосфина (1,52 г, 5,78 ммоль) в THF (5 мл), дегазированном и продутом N2, добавляли диизопропил азодикарбоксилат (1,17 г, 5,78 ммоль, 1,12 мл). К этой смеси, перемешиваемой при 0°C в атмосфере N2, добавляли смесь промежуточного соединения 1 (1,47 г, 2,89 ммоль, 1,00 экв.) и изоиндолин-1,3-дион (637,81 мг, 4,34 ммоль, 1,50 экв.) в THF (5 мл), дегазированном и продутом N2. Смесь перемешивали при 16°C в течение 18 часов в атмосфере N2. Реакционную смесь гасили добавлением H2O (20 мл) и затем экстрагировали DCM (3×20 мл). Комбинированные органические слои сушили над [Na2SO4], фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир:этилацетат=5:1 до 2:1), получая промежуточное соединение 2 (1,26 г, 1,98 ммоль, 68,39% выход) в виде твердого вещества коричневого цвета, которое подтверждали ЯМР и ЖХ/МС.
Промежуточное соединение 3: 2-(((8R,9S,10S)-9-(4-бромфенил)-3,4-дигидрокси-6-((2-нитрофенил)сульфонил)-1,6-диазабицикло[6.2.0]декан-10-ил)метил)изоиндолин-1,3-дион
К раствору промежуточного соединения 2 (1,26 г, 1,98 ммоль) в смеси ацетон/вода (15,00 мл) добавляли N-метилморфолина N-оксид (463,09 мг, 3,95 ммоль, 417,20 мкл) и затем OsO4 (5,02 мг, 19,76 мкмоль, 1,03 мкл). Смесь перемешивали при 15°C в течение 1,5 часов. Реакционную смесь сушили над [Na2SO4], фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=3/1 до 0:1) с получением промежуточного соединения 3 (1,11 г, 1,65 ммоль, 83,48% выход) в виде твердого вещества белого цыета, которое подтверждали ЖХ/МС и ЯМР.
Промежуточные соединения 4a и 4b: 2-(((3aR,6aR,7S,8R,10aS)-7-(4-бромфенил)-2,2-диметил-5-((2-нитрофенил)сульфонил)октагидро-3aH-азето[1,2-a][1,3]диоксоло[4,5-f][1,4]диазоцин-8-ил)метил)изоиндолин-1,3-дион (промежуточное соединение 4a) и 2-(((3aS,6aS,7S,8R,10aS)-7-(4-бромфенил)-2,2-диметил-5-((2-нитрофенил)сульфонил)октагидро-3aH-азето[1,2-a][1,3]диоксоло[4,5-f][1,4]диазоцин-8-ил)метил)изоиндолин-1,3-дион (промежуточное соединение 4b)
К раствору промежуточного соединения 3 (880,00 мг, 1,31 ммоль) и 2,2-диметоксипропана (272,97 мг, 2,62 ммоль, 321,14 мкл) в DCM (10,00 мл) добавляли TsOH (225,66 мг, 1,31 ммоль). Смесь перемешивали при 15°C. Через 20 часов реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1:1) с получением каждого из цис-диолов: промежуточное соединение 4a (менее полярное, 53,00 мг, 74,48 мкмоль, 5,68% выход) в виде твердого вещества белого цвета и промежуточное соединение 4b (более полярное, 260,00 мг, 365,38 мкмоль, 27,88% выход) в виде твердого вещества белого цвета, которые подтверждали ЯМР и ЖХ/МС.
Промежуточное соединение 5: ((3aR,6aR,7S,8S,10aS)-7-(4-бромфенил)-2,2-диметил-5-((2-нитрофенил)сульфонил)октагидро-3aH-азето[1,2-a][1,3]диоксоло[4,5-f][1,4]диазоцин-8-ил)метанамин
К раствору промежуточного соединения 4b (360,00 мг, 505,92 мкмоль) в этаноле (5,00 мл) добавляли гидрат гидразина (37,99 мг, 758,88 мкмоль, 36,88 мкл). После перемешивания при 70°C в течение 1,5 часов реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением промежуточного соединения 5 (239,00 мг, 411,02 мкмоль, 81,24% выход) в виде твердого вещества желтого цвета, которое подтверждали ЯМР и ЖХ/МС.
Промежуточное соединение 6: 1-((3aR,6aR,7S,8S,10aS)-7-(4-бромфенил)-2,2-диметил-5-((2-нитрофенил)сульфонил)октагидро-3aH-азето[1,2-a][1,3]диоксоло[4,5-f][1,4]диазоцин-8-ил)-N, N-диметилметанамин
К раствору промежуточного соединения 5 (239,00 мг, 411,02 мкмоль) и формальдегида (200,47 мг, 2,47 ммоль, 183,92 мкл) в DCM (500 мкл) добавляли MgSO4 (494,74 мг, 4,11 ммоль) и уксусную кислоту (49,36 мг, 822,04 мкмоль, 47,01 мкл). Через 20 мин добавляли триацетоксиборогидрид натрия (60,32 мг, 1,23 ммоль), и смесь перемешивали при 15°C в течение 23 часов. Реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1), получая промежуточное соединение 6 (210,00 мг, 344,53 мкмоль, 83,82% выход) в виде желтого масла, которое подтверждали ЖХ/МС.
Промежуточное соединение 7: 1-((3aR,6aR,7R,8S,10aS)-7-(4-бромфенил)-2,2-диметилоктагидро-3aH-азето[1,2-a][1,3]диоксоло[4,5-f][1,4]диазоцин-8-ил)-N, N-диметилметанамин
К раствору промежуточного соединения 6 (210,00 мг, 344,53 мкмоль) в CH3CN (2,00 мл) добавляли бензолтиол (17,08 мг, 155,04 мкмоль, 15,82 мкл) и затем добавляли Cs2CO3 (224,51 мг, 689,06 мкмоль). Смесь перемешивали при 40°C в течение 1,5 часов в атмосфере N2. Реакционную смесь гасили добавлением H2O (5 мл) и экстрагировали DCM 15 мл (3×5 мл). Комбинированные органические слои сушили над [Na2SO4], фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) получая промежуточное соединение 7 (72,00 мг, 169,66 мкмоль, 49,24% выход) в виде желтого масла, которое подтверждали ЯМР и ЖХ/МС.
Промежуточное соединение 8: (3aR,6aR,7S,8S,10aS)-7-(4-бромфенил)-N-(4-циклопропоксифенил)-8-((диметиламино)метил)-2,2-диметилгексагидро-3aH-азето[1,2-a][1,3]диоксоло[4,5-f][1,4]диазоцин-5(4H)-карбоксамид.
К раствору промежуточного соединения 7 (48,00 мг, 113,11 мкмоль) в DCM (2,00 мл) добавляли TEA (5,72 мг, 56,55 мкмоль, 7,84 мкл) и 1-(циклопропокси)-4-изоцианат-бензол (23,78 мг, 135,73 мкмоль) в атмосфере азота N2. После перемешивания при 15°C в течение 22 часов реакционную смесь гасили добавлением H2O (5 мл) и затем экстрагировали 15 мл DCM (3×5 мл). Объединенные органические слои концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1), получая промежуточное соединение 8 (29,40 мг, 49,04 мкмоль, 43,35% выход) в виде бледного масла, которое подтверждали ЖХ/МС.
Промежуточное соединение 9: (3aR,6aR,7S,8S,10aS)-N-(4-циклопропоксифенил)-8-((диметиламино)метил)-2,2-диметил-7-(4-(фенилэтинил)фенил)гексагидро-3aH-азето[1,2-a][1,3]диоксоло[4,5-f][1,4]диазоцин-5(4H)-карбоксамид
К раствору промежуточного соединения 8 (29,00 мг, 48,37 мкмоль) в CH3CN (1,00 мл) в атмосфере N2 добавляли TEA (97,89 мг, 967,38 мкмоль, 134,09 мкл) и этинилбензол (24,70 мг, 241,84 мкмоль, 26,56 мкл) и XPhos Pd G3 (6,14 мг, 7,26 мкмоль). После перемешивания при 70°C в течение 1 часа реакционную смесь гасили добавлением H2O (4 мл) и затем экстрагировали DCM 15 мл (3×5 мл). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 9 (30,00 мг) в виде бледного масла, которое подтверждали ЖХ/МС.
Синтез E1: (3S,4R,8R,9S,10S)-N-(4-циклопропоксифенил)-10-((диметиламино)метил)-3,4-дигидрокси-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 9 (20,00 мг, 32,22 мкмоль) в CH3CN (500,00 мкл) добавляли 1 M HCl (500,00 мкл). После перемешивания при 15°C в течение 18 часов реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением соединения E1 (8,20 мг, 14,12 мкмоль, 43,83% выход) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, хлороформ-d) δ ppm 0,73 (д, J=4,52 Гц, 4H) 1,26 (с, 1H) 2,32 (с, 6H) 2,60-2,70 (м, 2H) 2,86-2,92 (м, 1H) 2,96-3,05 (м, 2H) 3,22 (ш.д, J=15,56 Гц, 1H) 3,52-3,65 (м, 3H) 3,65-3,75 (м, 2H) 3,86 (ш.с, 2H) 3,97-4,05 (м, 1H) 4,13 (ш.с, 1H) 4,31 (ш.д, J=9,54 Гц, 1H) 6,95 (д, J=9,03 Гц, 2H) 7,24 (д, J=9,03 Гц, 2H) 7,32-7,46 (м, 6H) 7,49-7,57 (м, 4H) 8,37 (ш.с, 1H) 8,56 (ш.с, 1H).
Пример 2: (3S,4R,8R,9S,10S)-10-((диметиламино)метил)-3,4-диметокси-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E2”)
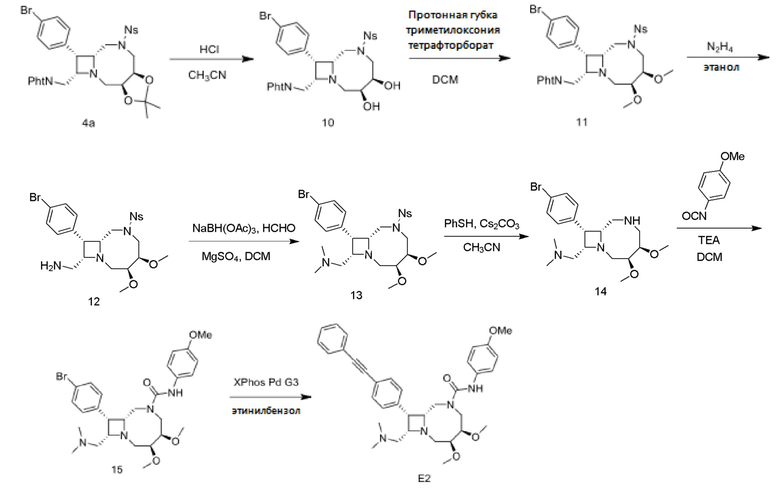
Промежуточное соединение 10: 2-(((3S,4R,8R,9S,10S)-9-(4-бромфенил)-3,4-дигидрокси-6-((2-нитрофенил)сульфонил)-1,6-диазабицикло[6.2.0]декан-10-ил)метил)изоиндолин-1,3-дион
Раствор промежуточного соединения 4a (170,00 мг, 238,90 мкмоль) в CH3CN (2,00 мл) и 1 M HCl (2,00 мл) перемешивали при 20°C в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM: MeOH=10:1) с получением промежуточного соединения 10 (140,00 мг, 208,49 мкмоль, 87,27% выход) в виде твердого вещества белого цвета, которое подтверждали ЯМР.
Промежуточное соединение 11: 2-(((3S,4R,8R,9S,10S)-9-(4-бромфенил)-3,4-диметокси-6-((2-нитрофенил)сульфонил)-1,6-диазабицикло[6.2.0]декан-10-ил)метил)изоиндолин-1,3-дион
К раствору промежуточного соединения 10 (125,00 мг, 186,15 мкмоль) в DCM (4,00 мл) добавляли триметилоксония тетрафторборат (137,67 мг, 930,75 мкмоль) и N1,N1,N8,N8-тетраметилнафтален-1,8-диамин (239,36 мг, 1,12 ммоль) при 0°C в атмосфере N2. После перемешивания при комнатной температуре (15°C) в течение 18 часов реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, этилацетат) с получением промежуточного соединения 11 (100,00 мг, 142,94 мкмоль, 76,79% выход) в виде твердого вещества белого цвета, которое подтверждали ЯМР.
Промежуточное соединение 12: ((3S,4R,8R,9S,10S)-9-(4-бромфенил)-3,4-диметокси-6-((2-нитрофенил)сульфонил)-1,6-диазабицикло[6.2.0]декан-10-ил)метанамин
К раствору промежуточного соединения 11 (100,00 мг, 142,94 мкмоль) в этаноле (3,00 мл) добавляли N2H4.H2O (10,73 мг, 214,42 мкмоль, 10,42 мкл). После перемешивания при 70°C в течение 1 часа реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного промежуточного соединения 12 (87,00 мг) в виде твердого вещества желтого цвета, которое подтверждали ЯМР.
Промежуточное соединение 13: 1-((3S,4R,8R,9S,10S)-9-(4-бромфенил)-3,4-диметокси-6-((2-нитрофенил)сульфонил)-1,6-диазабицикло[6.2.0]декан-10-ил)-N, N-диметилметанамин
К раствору промежуточного соединения 12 (87,00 мг, 152,77 мкмоль) в DCM (2,00 мл) добавляли HCHO (27,53 мг, 916,64 мкмоль, 25,25 мкл), MgSO4 (183,89 мг, 1,53 ммоль), NaBH(OAc)3 (97,14 мг, 458,32 мкмоль) и AcOH (18,35 мг, 305,55 мкмоль, 17,47 мкл). После перемешивания при 15°C в течение 3 часов реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM: MeOH=10:1) с получением промежуточного соединения 13 (46,00 мг, 76,98 мкмоль, 50,39% выход) в виде белого масла.
Промежуточное соединение 14: ((3S,4R,8R,9S,10S)-9-(4-бромфенил)-3,4-диметокси-1,6-диазабицикло[6.2.0]декан-10-ил)метанамин.
К раствору промежуточного соединения 13 (46,00 мг, 76,98 мкмоль) в CH3CN (2,00 мл) добавляли бензолтиол (12,72 мг, 115,48 мкмоль, 11,78 мкл) и Cs2CO3 (50,17 мг, 153,97 мкмоль). После перемешивания при 40°C в течение 1,5 часов в атмосфере N2 реакционную смесь гасили добавлением H2O (2 мл) и экстрагировали DCM 15 мл (3×5 мл). Комбинированные органические слои сушили над [Na2SO4], фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) с получением промежуточного соединения 14 (17,00 мг, 41,23 мкмоль, 53,55% выход) в виде белого масла, которое подтверждали ЖХ/МС.
Промежуточное соединение 15: (3S,4R,8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-3,4-диметокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 14 (17,00 мг, 41,23 мкмоль) в DCM (2,00 мл) добавляли TEA (2,09 мг, 20,61 мкмоль, 2,86 мкл) и 1-изоцианат-4-метокси-бензол (6,15 мг, 41,23 мкмоль, 5,30 мкл). После перемешивания в атмосфере N2 при 15°C в течение 2,5 часов реакционную смесь гасили добавлением H2O (5 мл) и затем экстрагировали DCM 15 мл (3×5 мл). Объединенные органические слои концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (DCM:MeOH=10:1) с получением промежуточного соединения 15 (33,00 мг, неочищенное) в виде прозрачного масла, которое подтверждали ЖХ/МС.
Синтез E2: (3S,4R,8R,9S,10S)-10-((диметиламино)метил)-3,4-диметокси-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 15 (33,00 мг, 58,77 мкмоль) в ацетонитриле (2,00 мл) добавляли TEA (118,94 мг, 1,18 ммоль, 162,93 мкл) и XPhos Pd G3 (7,46 мг, 8,82 мкмоль) в атмосфере N2. После перемешивания при 70°C в течение 1 часа реакционную смесь гасили добавлением H2O (2 мл) и затем экстрагировали DCM 15 мл (3×5 мл). Комбинированные органические слои сушили над [Na2SO4], фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1), а затем препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением 4,4 мг, 7,55 мкмоль, 12,85% выход в виде белого твердого вещества. Структуру подтверждали ЖХ/МС и ЯМР.
1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,06 (с, 6H) 2,39 (ш.с, 2H) 2,58 (ш.с, 2H) 3,03-3,14 (м, 1H) 3,30 (ш.с, 1H) 3,48 (с, 4H) 3,51-3,58 (м, 2H) 3,61-3,67 (м, 4H) 3,78 (с, 3H) 3,89-4,12 (м, 3H) 6,83 (д, J=8,91 Гц, 2H) 7,18 (д, J=8,78 Гц, 2H) 7,36 (ш.д, J=5,40 Гц, 3H) 7,43-7,47 (м, 2H) 7,48-7,51 (м, 2H) 7,52-7,56 (м, 2H) 7,99 (ш.с, 1H).
Пример 3: (4S,8R,9R,10S)-4-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E3”)


Промежуточное соединение 16: трет-бутил N-[(1S)-3-[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2-нитрофенил)сульфониламино]метил]-4-(тритилоксиметил)азетидин-1-ил]-1-[[трет-бутил(дифенил)силил]оксиметил]пропил]карбамат
К раствору N-(((2R,3R,4S)-3-(4-бромфенил)-4-((тритилокси)метил)азетидин-2-ил)метил)-2-нитробензолсульфонамида (WO 2015070204) (1,20 г, 1,72 ммоль) и (S)-трет-бутил(1-((трет-бутилдифенилсилил)окси)-4-оксобутан-2-ил)карбамата (US 20150266867) (911,52 мг, 2,06 ммоль) в DCM (40,00 мл) добавляли MgSO4 (2,07 г, 17,20 ммоль) и NaBH(OAc)3 (729,07 мг, 3,44 ммоль). После перемешивания при 15°C в течение 2 часов реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (15 мл) и экстрагировали DCM (2×5 мл). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=20:1 до 3:1) с получением промежуточного соединения 16 (2,00 г) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, хлороформ-d) δ=7,81-7,69 (м, 3H), 7,66-7,58 (м, 6H), 7,45-7,34 (м, 8H), 7,15-7,04 (м, 18H), 4,87-4,76 (м, 2H), 3,68-3,59 (м, 3H), 3,50-3,42 (м, 3H), 3,14 (ш.с, 1H), 3,03-2,97 (м, 1H), 2,95-2,82 (м, 2H), 2,46 (ш.с, 2H), 1,40 (с, 9H), 1,05 (с, 9H).
Промежуточное соединение 17: трет-бутил ((S)-4-((2R,3R,4S)-3-(4-бромфенил)-2-((2-нитрофенилсульфоноамидо)метил)-4-((тритилокси)метил)азетидин-1-ил)-1-гидроксибутан-2-ил)карбамат
К раствору промежуточного соединения 16 (500,00 мг, 444,74 мкмоль) в THF (6 мл) добавляли TBAF (174,42 мг, 667,11 мкмоль) при 15°C. После перемешивания при 15°C в течение 1 часа реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM:MeOH=100:1 до 20:1) с получением промежуточного соединения 17 (300,00 мг, 338,65 мкмоль, 76,15% выход) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, хлороформ-d) δ=7,83 (дд, J=7,8, 17,3 Гц, 2H), 7,76-7,63 (м, 2H), 7,22-7,12 (м, 15H), 7,01 (д, J=8,0 Гц, 2H), 5,05 (ш.с, 1H), 4,45 (ш.с, 1H), 3,78-3,70 (м, 1H), 3,59 (ш.с, 3H), 3,43 (д, J=9,5 Гц, 1H), 3,33-3,16 (м, 2H), 3,05 (д, J=6,0 Гц, 2H), 2,96-2,87 (м, 1H), 2,83-2,72 (м, 1H), 2,58-2,48 (м, 1H), 1,74 (дд, J=4,8, 14,8 Гц, 1H), 1,43 (с, 9H).
Промежуточное соединение 18: N-[[(2R,3R,4S)-1-[(3S)-3-амино-4-гидрокси-бутил]-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамид
К раствору промежуточного соединения 17 (800,00 мг, 903,08 мкмоль) в DCM (16,00 мл) добавляли TMSOTf (602,15 мг, 2,71 ммоль, 489,55 мкл) и 2,6-лютидин (387,06 мг, 3,61 ммоль, 420,72 мкл, 4 экв.). После перемешивания при 15°C в течение 16 часов реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM:MeOH=1:0 до 20:1) с получением промежуточного соединения 18 (700 мг, неочищенное) в виде твердого вещества белого цвета (содержащего TEA): 1H ЯМР (400 МГц, хлороформ-d) δ=7,85 (тд, J=2,3, 4,7 Гц, 1H), 7,76-7,72 (м, 1H), 7,70-7,63 (м, 2H), 7,25-7,16 (м, 15H), 6,98 (д, J=8,4 Гц, 2H), 3,72-3,60 (м, 3H), 3,52-3,42 (м, 3H), 3,27 (дд, J=6,6, 14,1 Гц, 1H), 3,14-3,00 (м, 3H), 2,83-2,68 (м, 2H), 1,85-1,62 (м, 2H).
Промежуточное соединение 19: N-[[(2R,3R,4S)-3-(4-бромфенил)-1-[(3S)-3-(диметиламино)-4-гидрокси-бутил]-4-(тритилоксиметил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамид
К раствору промежуточного соединения 18 (700,00 мг, 890,88 мкмоль) в DCM (15,00 мл) добавляли HCHO (86,77 мг, 1,07 ммоль, 79,61 мкл, 37% чистоты), MgSO4 (1,07 г, 8,91 ммоль), и затем порциями добавляли NaBH(OAc)3 (377,63 мг, 1,78 ммоль). После перемешивания при 15°C в течение 1 часа реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (100 мл) и экстрагировали DCM (2×30 мл). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM:MeOH=1:0 до 15:1) с получением промежуточного соединения 19 (500 мг, 614,40 мкмоль, 68,97% выход) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, хлороформ-d) δ=7,87 (дд, J=1,5, 7,5 Гц, 1H), 7,78-7,73 (м, 1H), 7,72-7,61 (м, 2H), 7,23-7,12 (м, 18H), 7,05 (д, J=8,0 Гц, 2H), 3,64-3,54 (м, 3H), 3,22-3,06 (м, 4H), 3,03-2,98 (м, 1H), 2,87-2,70 (м, 2H), 2,56 (тд, J=6,0, 12,0 Гц, 1H), 2,45 (ш.с, 1H), 2,22 (с, 6H), 1,57 (д, J=14,1 Гц, 1H), 1,41 (д, J=6,0 Гц, 1H).
Промежуточное соединение 20: (4S,8R,9R,10S)-9-(4-бромфенил)-N, N-диметил-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-амин
К раствору трифенилфосфин (1 г, 3,76 ммоль) в THF (19 мл) при 0°C медленно добавляли диизопропил азодикарбоксилат (0,72 мл, 3,76 ммоль). Затем эту смесь добавляли к раствору промежуточного соединения 19 (500 мг, 614,40 мкмоль) в THF (75 мл) при 0°C. После перемешивания при 15°C в течение 4 часов реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM:MeOH=20:1) с получением промежуточного соединения 20 (200,00 мг, 251,33 мкмоль, 40,91% выход) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, хлороформ-d) δ=8,14 (д, J=7,5 Гц, 1H), 7,65-7,55 (м, 3H), 7,25-7,19 (м, 19H), 3,62-3,50 (м, 4H), 3,42 (ш.с, 1H), 3,17-2,98 (м, 4H), 2,85 (дд, J=7,1, 9,3 Гц, 1H), 2,69-2,60 (м, 1H), 2,44-2,31 (м, 2H), 2,04 (ш.с, 6H), 1,89 (ш.с, 1H).
Промежуточное соединение 21: (4S,8R,9R,10S)-9-(4-бромфенил)-N, N-диметил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-амин
К раствору промежуточного соединения 20 (150,00 мг, 188,49 мкмоль) в ацетонитриле (3,00 мл) добавляли Cs2CO3 (368,49 мг, 1,13 ммоль) и бензолтиол (83,07 мг, 753,96 мкмоль, 76,92 мкл). После перемешивания при 40°C в течение 16 часов реакционную смесь гасили добавлением воды (15 мл) и экстрагировали этилацетатом (2×5 мл). Объединенные органические слои промывали солевым раствором (2×5 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) с получением промежуточного соединения 21 (90,00 мг, 147,39 мкмоль, 78,20% выход) в виде твердого вещества белого цвета, которое использовали без очистки.
Промежуточное соединение 22: (4S,8R,9R,10S)-9-(4-бромфенил)-4-(диметиламино)-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 21 (90,00 мг, 147,39 мкмоль) в DCM (3,00 мл) добавляли 1-изоцианат-4-метокси-бензол (26,38 мг, 176,87 мкмоль). После перемешивания при 15°C в течение 1 часа реакционную смесь гасили добавлением воды (15 мл) и экстрагировали DCM (2×5 мл). Объединенные органические слои промывали солевым раствором (2×10 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=20:1) с получением промежуточного соединения 22 (90,00 мг, 118,46 мкмоль, 80,37% выход) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, хлороформ-d) δ=7,28-7,18 (м, 21H), 6,82 (д, J=8,8 Гц, 2H), 4,15-4,06 (м, 1H), 3,77 (с, 3H), 3,64-3,47 (м, 4H), 3,18-2,98 (м, 3H), 2,92-2,82 (м, 2H), 2,70 (д, J=13,2 Гц, 1H), 2,43-2,33 (м, 7H), 2,10 (ш.с, 1H), 1,95 (ш.с, 1H).
Промежуточное соединение 23: (4S,8R,9R,10S)-4-(диметиламино)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 22 (64,00 мг, 84,24 мкмоль) и этинилбензола (25,81 мг, 252,71 мкмоль) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (7,13 мг, 8,42 мкмоль) и Cs2CO3 (54,89 мг, 168,47 мкмоль). После перемешивания при 70°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (этилацетат:метанол=10:1) с получением промежуточного соединения 23 (47,00 мг, 60,18 мкмоль, 71,44% выход) в виде твердого вещества коричневого цвета. 1H ЯМР (400 МГц, MeOD-d4) δ 7,54 (д, J=4,8 Гц, 2H), 7,38-7,35(м, 7H), 7,33-7,21 (м, 18H), 6,83 (д, J=8,8 Гц, 2H), 4,13 (с, 1H), 3,77 (с, 3H), 3,61-3,52 (м, 4H),3,19-3,17 (м, 2H), 2,94-2,90 (м, 2H), 2,73-2,44 (м, 1H), 2,17-1,97 (м, 1H).
Синтез E3: (4S,8R,9R,10S)-4-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 23 (40,00 мг, 51,22 мкмоль) в DCM (1 мл) добавляли TFA (58,40 мг, 512,17 мкмоль, 37,92 мкл). После перемешивания при 25°C в течение 12 часов реакционную смесь гасили насыщенным раствором NaHCO3 и экстрагировали DCM (3×5 мл) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением соединения E3 (21,70 мг, 40,28 мкмоль, 78,6% выход) в виде твердого вещества белого цвета. HRMS (ESI): вычисл. для C33H38N4O3 [M+H]+ 539,29, найденное 539,30. 1H ЯМР: (400 МГц, MeOD-d4) δ 7,60(д, J=4,8 Гц, 2H), 7,49-7,46 (м, 4H), 7,38-7,36 (м, 2H), 7,23-7,24 (д, J=8,8 Гц, 2H), 6,87 (д, J=8,8 Гц, 2H), 3,76 (с, 3H), 3,55-3,52 (м, 2H), 3,48-3,3,42 (м, 6H), 3,34-3,31 (м, 1H), 3,19 (с, 1H), 2,89 (с, 6H), 2,62 (с, 1H), 2,04 (м, 2H).
Пример 4: (3Z,8R,9S,10S)-10-[(диметиламино)метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамид (“E4”)


Промежуточное соединение 24: (3Z,8R,9R,10S)-9-(4-бромфенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]дец-3-ен
К раствору (8R,9R,10S, Z)-10-((бис(4-метоксифенил)(фенил)метокси)метил)-9-(4-бромфенил)-6-((2-нитрофенил)сульфонил)-1,6-диазабицикло[6.2.0]дец-3-ена и бензолтиола (132,09 мг, 1,20 ммоль, 122,31 мкл) в ацетонитриле (6 мл) добавляли Cs2CO3 (260,41 мг, 799,25 мкмоль) при 25°C. После перемешивания при 50°C в течение 12 часов реакционную смесь гасили H2O (5 мл) и экстрагировали DCM (10 мл) с получением органической фазы. Органическую фазу концентрировали и очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 24 (450 мг, 795,70 мкмоль, 99,56% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,26-7,16 (м, 19H), 5,90-5,84 (м, 1H), 5,67-5,60 (м, 1H), 3,61-3,53 (м, 4H), 3,61-3,52 (м, 4H), 3,11-3,09 (м, 2H), 2,86-2,84 (м, 2H), 2,48 (д, J=5,2 Гц, 1H).
Промежуточное соединение 25: (3Z,8R,9R,10S)-9-(4-бромфенил)-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамид
К раствору промежуточного соединения 24 (400 мг, 707,29 мкмоль) и Et3N (196,08 мкл, 1,41 ммоль) в DCM (2 мл) по каплям добавляли 1-изоцианат-4-метокси-бензол (116,04 мг, 778,02 мкмоль) при 0°C. После перемешивания при 25°C в течение 5 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 25 (400 мг, 559,68 мкмоль, 79,13% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,31-7,19 (м, 19H), 6,85-6,81 (м, 2H), 6,04 (с, 1H), 5,81-5,80 (м, 1H), 5,73-5,68 (м, 1H), 4,15 (д, J=16 Гц, 1H), 5,72 (д, J=16 Гц, 1H), 4,15-4,00 (м, 1H), 3,99-3,70 (м, 1H), 3,66-3,56 (м, 5H), 3,21-3,19 (м, 2H), 3,19-3,00 (м, 1H), 2,81-2,77 (м, 1H).
Промежуточное соединение 26: (3Z,8R,9R,10S)-9-(4-бромфенил)-10-(гидроксиметил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамид
К раствору промежуточного соединения 25 (360,00 мг, 503,71 мкмоль) в DCM (1 мл) по каплям добавляли TFA (3 мл, 5037 мкмоль) при 0°C. После перемешивания при 25°C в течение 1 часа реакционную смесь гасили насыщенным раствором NaHCO3 (3 мл) и экстрагировали DCM (5 мл) с получением органической фазы. Органическую фазу сушили над безводным Na2SO4, концентрировали и очищали препаративной ТСХ (петролейный эфир:этилацетат=1:2) с получением промежуточного соединения 26 (210 мг, 444,57 мкмоль, 88,26% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,48 (д, J=8,4 Гц, 2H), 7,38 (д, J=8,4 Гц, 2H), 7,21 (д, J=9,6 Гц, 2H), 6,83 (д, J=8,8 Гц, 2H), 6,06 (с, 2H), 5,85 (с, 1H), 5,74-5,71 (м, 1H), 4,26 (м, 1H), 4,03-3,95 (м, 1H), 3,77-3,59 (м, 6H), 3,28 (м, 1H), 2,90 (с, 1H).
Промежуточное соединение 27: (3Z,8R,9S,10S)-9-(4-бромфенил)-10-[(1,3-диоксоизоиндолин-2-ил)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамид
К раствору промежуточного соединения 26 (210,00 мг, 444,57 мкмоль), изоиндолин-1,3-диона (71,95 мг, 489,03 мкмоль) и PPh3 (233,21 мг, 889,13 мкмоль) в THF (2 мл) по каплям добавляли DIAD (179,79 мг, 889,13 мкмоль) при 25°C. После перемешивания при 25°C в течение 5 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 27 (200 мг, 332,51 мкмоль, 74,79% выход) в виде твердого вещества светло-коричневого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,80-7,67 (м, 17H), 7,54-7,67 (м, 22H), 7,25-7,20 (м, 2H), 6,80 (с, 2H), 6,06 (с, 2H), 5,85 (с, 1H), 5,74 (с, 1H), 4,22 (с, 1H) 4,02 (м, 1H), 3,77-3,75 (м, 4H), 3,70-3,66 (м, 3H), 3,50 (с, 3H), 3,12 (м, 1H), 2,88 (с, 1H).
Промежуточное соединение 28: (3Z,8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамид
К раствору промежуточного соединения 27 (60,00 мг, 99,75 мкмоль) в MeOH (1мл) по каплям добавляли 2-аминоэтанол (60,33 мкл, 997,52 мкмоль). После перемешивания при 25°C в течение 3 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=12:1) с получением промежуточного соединения 28 (33,00 мг, 70,01 мкмоль, 70,18% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,50 (д, J=13,6 Гц, 2H), 7,46 (м, 2H), 7,21 (д, J=5,5 Гц, 2H), 6,82 (д, J=9,6 Гц, 1H), 6,08 (с, 1H), 5,83 (с, 1H), 5,73(с, 1H), 4,22 (м, 2H), 4,02-4,00 (м, 1H), 3,77(с, 3H), 3,71-3,63 (м, 2H), 3,55-3,50 (м, 1H), 3,36-3,17 (м, 1H), 2,81-2,76 (м, 3H).
Промежуточное соединение 29: (3Z,8R,9S,10S)-9-(4-бромфенил)-10-[(диметиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 28 (23,00 мг, 48,79 мкмоль) в DCM (2 мл) добавляли NaBH(OAc)3 (51,70 мг, 243,96 мкмоль) при 25°C. После перемешивания при 25°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=12:1) с получением промежуточного соединения 29 (20 мг, 40,04 мкмоль, 82% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,53 (д, J=8,4 Гц, 2H), 7,31-7,26 (м, 2H), 7,19 (д, J=8,8 Гц, 3H), 6,82 (д, J=8,8 Гц, 3H), 6,14 (с, 1H), 5,83-5,86 (м, 2H), 4,12-4,02 (м, 3H), 3,90-3,59 (м, 9H), 3,19 (м, 1H), 3,01-2,80 (м, 2H), 2,38 (с, 6H).
Синтез E4: (3Z,8R,9S,10S)-10-[(диметиламино)метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамид
К раствору промежуточного соединения 29 (20,00 мг, 40,04 мкмоль) и этинилбензола (12,27 мг, 120,13 мкмоль, 13,19 мкл) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (3,39 мг, 4,00 мкмоль) и Cs2CO3 (26,09 мг, 80,09 мкмоль). После перемешивания при 70°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (0,04% система HCl/ацетонитрил/H2O) с получением (3Z,8R,9S,10S)-10-[(диметиламино)метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамида (2,00 мг, 3,84 мкмоль, 9,59% выход) в виде твердого вещества белого цвета. HRMS (ESI) вычисл. для C32H33N4O [M+H]+ 489,27, найденное 520,10; 1H ЯМР (400 МГц, MeOD-d4) δ7,74 (д, J=5,5 Гц, 2H), 7,67 (д, J=8,8 Гц, 2H), 7,53 (д, J=3,6 Гц, 2H), 7,20 (д, J=8,8 Гц, 2H), 7,18 (д, J=8,8 Гц, 2H), 6,13 (д, J=5,6 Гц, 1H), 5,78 (д, J=12,4 Гц, 1H), 4,33 (с, 2H), 4,30-4,21 (м, 9H), 3,90-3,75 (м, 5H), 3,50-3,31 (м, 3H), 2,77 (с, 6H).
Пример 5: (3S,4R,8R,9S,10S)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E5”)
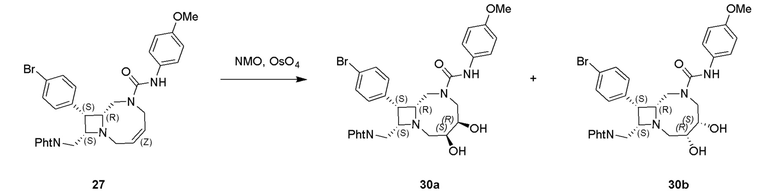

Промежуточные соединения 30a и 30b: (3S,4R,8R,9S,10S)-9-(4-бромфенил)-10-((1,3-диоксоизоиндолин-2-ил)метил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (30a) и (3R,4S,8R,9S,10S)-9-(4-бромфенил)-10-((1,3-диоксоизоиндолин-2-ил)метил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (30b)
К перемешиваемому раствору промежуточного соединения 27 (60 мг, 99,75 мкмоль) в смеси ацетон/H2O=10:1 (1 мл) добавляли NMO (17,53 мг, 149,62 мкмоль) и OsO4 (2,54 мг, 9,98 мкмоль) при комнатной температуре. После перемешивания при 25°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:5) с получением промежуточного соединения 30a (менее полярного) (25 мг, 39,34 мкмоль, 39,44% выход) в виде твердого вещества светло-коричневого цыета и промежуточного соединения 30b (более полярного) (25 мг, 39,34 мкмоль, 39,44% выход) в виде твердого вещества светло-коричневого цвета. Промежуточное соединение 30a: 1H ЯМР (400 МГц, CDCl3-d1) δ7,82 (д, J=2,8 Гц, 2H), 7,81 (д, J=2,0 Гц, 2H), 7,71-7,02 (м, 4H), 7,26 (м, 2H), 6,80 (д, J=1,2 Гц, 2H), 4,24-4,07 (м, 2H), 3,93-3,81 (м, 2H), 3,74(с, 3H), 3,62-3,54 (м, 3H), 3,28-2,88 (м, 1H), 2,65-2,49 (м, 2H).
Промежуточное соединение 30b: 1H ЯМР (400 МГц, CDCl3-d1) δ7,83 (д, J=2,8 Гц, 2H), 7,81 (д, J=2,0 Гц, 2H), 7,73-7,47 (м, 4H), 7,24 (м, 2H), 6,80 (д, J=8,8 Гц, 2H), 6,58 (с, 1H), 3,96-3,90 (м, 2H), 3,70-3,68 (м, 4H), 3,56-3,50 (м, 3H), 3,00 (с, 1H), 2,87-2,76 (м, 2H).
Промежуточное соединение 31a: (3S,4R,8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 30a (120 мг, 188,83 мкмоль) в MeOH (2 мл) добавляли 2-аминоэтанол (115,34 мг, 1,89 ммоль). После перемешивания при 25°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=8:1) с получением промежуточного соединения 31a (70 мг, 138,50 мкмоль, 73,35% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,47-7,43 (м, 3H), 7,42-7,40 (м, 1H), 7,35-7,20 (м, 3H), 7,18-6,79 (м, 2H), 4,31-4,27 (м, 1H), 3,89 (с, 1H), 3,89-3,79 (м, 5H), 3,76-3,23 (м, 2H), 2,91-2,86 (м, 2H), 2,72-2,61 (м, 2H).
Промежуточное соединение 32a: (3S,4R,8R,9S,10S)-9-(4-бромфенил)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору 31a (70,00 мг, 138,50 мкмоль) в DCM (2 мл) добавляли HCHO (112,41 мг, 1,39 ммоль, 103,13 мкл, 37% раствор) и MgSO4 (166,72 мг, 1,39 ммоль). После перемешивания при 25°C в течение 0,5 часа дополнительно добавляли NaBH(OAc)3 (146,77 мг, 692,52 мкмоль). Спустя еще 1,5 часа реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=5:1) с получением промежуточного соединения 32a (60 мг, 112,47 мкмоль, 81% выход) в виде светло-коричневого масла. 1H ЯМР (400 МГц, CDCl3-d1) δ7,48-7,43 (м, 4H), 7,42-7,40 (м, 1H), 7,27-7,20 (м, 2H), 6,81-6,78 (м, 2H), 4,26-4,03 (м, 3H), 3,64-3,54 (м, 5H), 3,27-3,24 (м, 1H).
Синтез E5: (3S,4R,8R,9S,10S)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 32a (20,00 мг, 40,04 мкмоль) и этинилбензола (12,27 мг, 120,13 мкмоль, 13,19 мкл) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (3,39 мг, 4,00 мкмоль, 0,10 экв.) и Cs2CO3 (26,09 мг, 80,09 мкмоль). После перемешивания при 70°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением (3S,4R,8R,9S,10S)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (2,00 мг, 3,84 мкмоль, 9,59% выход) в виде твердого вещества белого цвета. HRMS (ESI) вычисл. для C33H38N4O4 [M+H]+ 554,29, найденное 554,20. 1H ЯМР (400 МГц, MeOD-d4) δ7,59 (с, 4H), 7,51 (д, J=3,6 Гц, 2H), 7,38 (с, 3H), 7,16 (д, J=8,8 Гц, 2H), 6,85 (д, J=8,8 Гц, 2H), 4,20-4,11 (с, 3H), 3,85-3,82 (м, 4H), 3,75 (с, 3H), 3,57-3,44 (м, 3H), 3,12-2,97 (м, 4H), 2,97-2,56 (м, 6H).
Пример 6: (3R,4S,8R,9S,10S)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E6”)

Промежуточное соединение 31b: (3R,4S,8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 30b (30 мг, 47,21 мкмоль) в MeOH (2 мл) добавляли 2-аминоэтанол (28,84 мг, 472,10 мкмоль) при 25°C. После перемешивания при 25°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=8:1) с получением промежуточного соединения 31b (20 мг, 39,57 мкмоль, 83,82% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,51-7,48 (м, 3H), 7,46-7,43 (м, 4H), 6,83 (д, J=8,8 Гц, 2H), 7,77-3,73 (м, 3H), 3,65-3,51 (м, 2H), 3,23-3,15 (м, 1H), 2,80-2,73 (м, 3H).
Промежуточное соединение 32b: (3R,4S,8R,9S,10S)-9-(4-бромфенил)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 31a (10 мг, 19,79 мкмоль) в DCM (2 мл) добавляли HCHO (16,06 мг, 197,90 мкмоль, 14,73 мкл, 37% раствор) и MgSO4 (23,82 мг, 197,90 мкмоль) при 25°C. После перемешивания при 25°C в течение 0,5 часа дополнительно добавляли NaBH(OAc)3 (20,97 мг, 98,95 мкмоль). После перемешивания при 25°C в течение дополнительных 1,5 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=5:1) с получением промежуточного соединения 32b (10 мг, 18,75 мкмоль, 47,36% выход) в виде светло-коричневого масла. 1H ЯМР (400 МГц, CDCl3-d1) δ7,54-7,46 (м, 4H), 7,30-7,25 (м, 7H), 6,83-6,80 (д, J=8,8 Гц, 2H), 3,89-3,78 (м, 2H), 3,67-3,63 (м, 6H), 3,56-3,48 (м, 3H), 2,74-2,64 (м, 3H), 2,28 (с, 6H).
Синтез E6: (3R,4S,8R,9S,10S)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 32b (10 мг, 18,75 мкмоль) и этинилбензола (5,74 мг, 56,24 мкмоль, 6,18 мкл) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (1,59 мг, 1,87 мкмоль) и Cs2CO3 (12,22 мг, 37,49 мкмоль). После перемешивания при 70°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (0,04% система HCl/ACN/H2O) с получением (3R,4S,8R,9S,10S)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (2 мг, 3,61 мкмоль, 19,23% выход) в виде твердого вещества белого цвета. HRMS (ESI) вычисл. для C33H38N4O4 [M+H]+ 554,29, найденное 554,2; 1H ЯМР (400 МГц, MeOD-d4) δ7,62 (с, 4H), 7,53-7,51 (м, 2H), 7,40 (д, J=4,8 Гц, 3H), 7,24 (д, J=4,8 Гц, 2H), 6,87 (д, J=8,8 Гц, 2H), 4,18 (с, 3H), 4,08 (с, 1H), 3,94-3,93 (м, 3H), 3,76 (с, 4H), 3,56-3,52 (м, 2H), 3,42-3,40 (м, 2H), 3,23-3,21 (м, 2H), 2,99-2,95 (м, 1H), 3,70 (с, 6H).
Примеры 7-9
Приведенные ниже примеры соединений (E7-E9) получали согласно примеру 6 с перечисленными алкинами и конкретными альдегидами (где указано) в таблице 3, начиная либо с промежуточного соединения (“Пром.”) 32a, либо 32b.
(3S,4R,8R,9S,10S)-10-[(диметиламино)метил]-9-[4-[2-(2-фторфенил)этинил]фенил]-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E7”)
(3R,4S,8R,9S,10S)-10-[(диметиламино)метил]-9-[4-[2-(2-фторфенил)этинил]фенил]-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E8”)
(3S,4R,8R,9S,10S)-10-(диэтиламинометил)-3,4-дигидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E9”) BIMH2W60
Пример 10: (3S,4R,8R,9S,10S)-3,4-дигидрокси-10-[[изопропил(метил)амино]метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E10”)
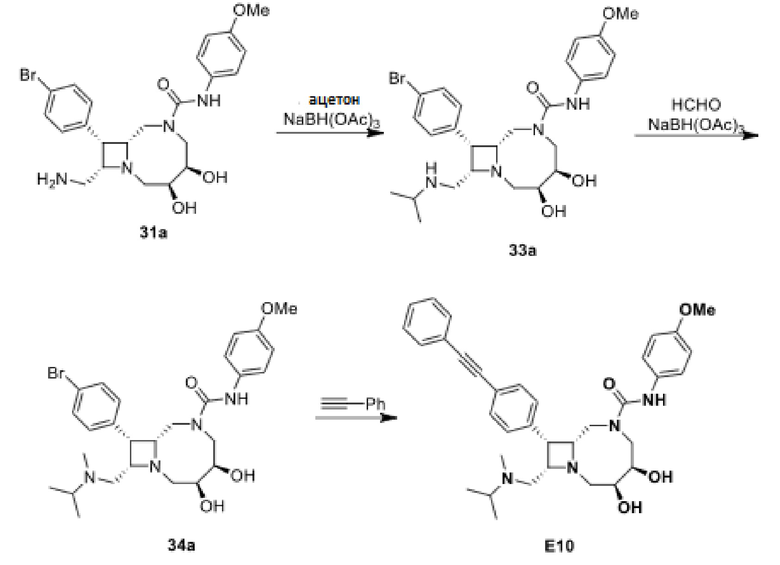
Промежуточное соединение 33a: (3S,4R,8R,9S,10S)-9-(4-бромфенил)-3,4-дигидрокси-10-[(изопропиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 31a (100,00 мг, 197,86 мкмоль), ацетона (68,95 мг, 1,19 ммоль, 87,28 мкл), AcOH (11,88 мг, 197,86 мкмоль, 11,31 мкл) и MgSO4 (238,16 мг, 1,98 ммоль) в DCM (1,00 мл) добавляли NaBH(OAc)3 (125,80 мг, 593,58 мкмоль). После перемешивания при 20°C в течение 12 часов реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного промежуточного соединения 33a (116,00 мг) в виде твердого вещества бледно-коричневого цвета.
Промежуточное соединение 34a: (3S,4R,8R,9S,10S)-9-(4-бромфенил)-3,4-дигидрокси-10-[[изопропил(метил)амино]метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 33a (116,00 мг, 211,88 мкмоль), формальдегида (103,08 мг, 1,27 ммоль, 94,57 мкл), MgSO4 (255,04 мг, 2,12 ммоль) и AcOH (12,72 мг, 211,88 мкмоль, 12,11 мкл) в DCM (1,00 мл) добавляли NaBH(OAc)3 (134,72 мг, 635,64 мкмоль). После перемешивания при 20°C в течение 2 часов реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного промежуточного соединения 34b (138,00 мг, неочищенное) в виде твердого вещества темно-желтого цвета.
Синтез E10: (3S,4R,8R,9S,10S)-3,4-дигидрокси-10-[[изопропил(метил)амино]метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 34a (30,00 мг, 53,43 мкмоль) и этинилбензола (10,91 мг, 106,86 мкмоль, 11,73 мкл) в CH3CN (1,00 мл) добавляли Et3N (16,22 мг, 160,29 мкмоль, 22,22 мкл) и XPhos Pd G3 (4,52 мг, 5,34 мкмоль). После перемешивания при 70°C в течение 2 часов реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением (3S,4R,8R,9S,10S)-3,4-дигидрокси-10-[[изопропил(метил)амино]метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (11,20 мг, 17,81 мкмоль, 33,34% выход, соль муравьиной кислоты) в виде твердого вещества светло-желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=8,65-8,37 (м, 1H), 7,57-7,52 (м, 2H), 7,49 (д, J=8,0 Гц, 2H), 7,42-7,34 (м, 5H), 7,25 (д, J=9,0 Гц, 2H), 6,81 (д, J=9,0 Гц, 2H), 4,36-4,21 (м, 1H), 4,13 (ш.с, 1H), 3,96 (д, J=14,1 Гц, 1H), 3,84-3,73 (м, 5H), 3,62-3,46 (м, 2H), 3,21 (д, J=15,6 Гц, 1H), 2,83 (д, J=14,1 Гц, 4H), 2,75-2,54 (м, 2H), 2,11 (ш.с, 3H), 0,99 (д, J=5,5 Гц, 3H), 0,71 (д, J=5,0 Гц, 3H).
Пример 11: (3R,4S,8R,9S,10S)-3,4-дигидрокси-10-[[изопропил(метил)амино]метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E11”)
Пример E11 синтезировали по схеме, описанной в примере 10, начиная с промежуточного соединения 31b. 1H ЯМР (400 МГц, хлороформ-d) δ=8,73-8,47 (м, 1H), 7,56-7,51 (м, 4H), 7,41-7,34 (м, 5H), 7,28 (д, J=8,9 Гц, 2H), 6,81 (д, J=8,9 Гц, 2H), 3,88 (д, J=5,4 Гц, 1H), 3,80-3,74 (м, 5H), 3,72-3,48 (м, 5H), 3,24 (дд, J=6,8, 14,1 Гц, 1H), 2,88 (т, J=11,9 Гц, 1H), 2,82-2,75 (м, 1H), 2,72 (д, J=14,2 Гц, 2H), 2,65-2,56 (м, 1H), 2,09 (с, 3H), 0,95 (д, J=6,5 Гц, 3H), 0,71 (д, J=6,4 Гц, 3H).
Пример 12: (3S,4R,8R,9S,10S)-9-[4-[2-(2,3-дифторфенил)этинил]фенил]-10-[(диметиламино)метил]-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E12”)

Промежуточное соединение 35a: (3S,4R,8R,9S,10S)-10-[(диметиламино)метил]-9-(4-этинилфенил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 32a (54,00 мг, 101,23 мкмоль) и этинилтриметилсилана (29,83 мг, 303,69 мкмоль, 42,01 мкл) в CH3CN (1,00 мл) добавляли Cs2CO3 (131,93 мг, 404,92 мкмоль) и XPhos Pd G3 (8,57 мг, 10,12 мкмоль). После перемешивания в атмосфере N2 при 70°C в течение двух часов реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением неочищенного продукта 2 (78,00 мг, неочищенный) в виде твердого вещества темно-желтого цвета.
Синтез E12: (3S,4R,8R,9S,10S)-9-[4-[2-(2,3-дифторфенил)этинил]фенил]-10-[(диметиламино)метил]-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору (3R,4S,8R,9S,10S)-10-[(диметиламино)метил]-9-(4-этинилфенил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (промежуточное соединение 35a) (21,60 мг, 45,13 мкмоль) и 1-бром-2,3-дифторбензола (8,71 мг, 45,13 мкмоль, 5,06 мкл) в CH3CN (500 мкл) добавляли Cs2CO3 (29,41 мг, 90,26 мкмоль) и XPhos Pd G3 (3,82 мг, 4,51 мкмоль). После перемешивания в атмосфере N2 при 40°C в течение 1 часа остаток очищали препаративной ТСХ (SiO2, DCM: MeOH=8:1) для удаления катализатора и затем очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением (3S,4R,8R,9S,10S)-9-[4-[2-(2,3-дифторфенил)этинил]фенил]-10-[(диметиламино)метил]-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (2,00 мг, 3,14 мкмоль, 6,96% выход, соль муравьиной кислоты) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=8,48-8,27 (м, 1H), 7,58 (д, J=7,9 Гц, 2H), 7,40 (д, J=7,9 Гц, 2H), 7,29 (д, J=8,4 Гц, 3H), 7,22-7,13 (м, 1H), 7,11-7,03 (м, 1H), 6,83 (д, J=8,8 Гц, 2H), 6,73 (ш.с, 1H), 3,89 (ш. с, 2H), 3,77 (с, 4H), 3,75-3,62 (м, 3H), 3,61-3,48 (м, 2H), 3,28 (дд, J=6,6, 14,1 Гц, 1H), 2,89 (д, J=12,8 Гц, 3H), 2,80 (д, J=14,6 Гц, 2H), 2,68 (д, J=7,5 Гц, 1H), 2,27 (с, 6H).
Пример 13: (3R,4S,8R,9S,10S)-9-(4-((2,3-дифторфенил)этинил)фенил)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E13”)


Промежуточное соединение 36b: (3R,4S,8R,9S,10S)-10-((диметиламино)метил)-9-(4-этинилфенил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору (3R,4S,8R,9S,10S)-9-(4-бромфенил)-10-[(диметиламино)метил]-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (промежуточное соединение 32b) (40,00 мг, 74,98 мкмоль) и этинил(триметил)силана (22,09 мг, 224,95 мкмоль, 31,12 мкл) в CH3CN (1,00 мл) добавляли Cs2CO3 (97,72 мг, 299,93 мкмоль) и XPhos Pd G3 (6,35 мг, 7,50 мкмоль). После перемешивания в атмосфере N2 при 70°C в течение 1 часа реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (DCM:MeOH=10:1, Rf=0,2) с получением промежуточного соединения 36b (30,00 мг, 62,69 мкмоль, 83,60% выход) в виде твердого вещества светло-желтого цвета.
Промежуточное соединение 37b: (3aS,6aR,7S,8S,10aR)-8-((диметиламино)метил)-7-(4-этинилфенил)-N-(4-метоксифенил)-2,2-диметилгексагидро-3aH-азето[1,2-a][1,3]диоксоло[4,5-f][1,4]диазоцин-5(4H)-карбоксамид
К раствору промежуточного соединения 36b (30,00 мг, 62,69 мкмоль) в 2,2-диметоксипропан (500 мкл) и DCM (500 мкл) добавляли TsOH (1,08 мг, 6,27 мкмоль). После перемешивания при 20°C в течение 1 часа реакционную смесь концентрировали и очищали препаративной ТСХ (DCM:MeOH=10:1) с получением промежуточного соединения 37b (20,00 мг, 38,56 мкмоль, 61,51% выход) в виде желтого масла.
Промежуточное соединение 38b: (3aS,6aR,7S,8S,10aR)-7-(4-((2,3-дифторфенил)этинил)фенил)-8-((диметиламино)метил)-N-(4-метоксифенил)-2,2-диметилгексагидро-3aH-азето[1,2-a][1,3]диоксоло[4,5-f][1,4]диазоцин-5(4H)-карбоксамид
К раствору промежуточного соединения 37b (20,00 мг, 38,56 мкмоль) и 1-бром-2,3-дифтор-бензола (22,33 мг, 115,68 мкмоль, 12,98 мкл) в CH3CN (500 мкл) добавляли Cs2CO3 (50,25 мг, 154,24 мкмоль) и XPhos Pd G3 (3,26 мг, 3,86 мкмоль). После перемешивания в атмосфере N2 при 70°C в течение одного часа реакционную смесь очищали препаративной ТСХ (DCM:MeOH=10:1) с получением промежуточного соединения 38b (15,00 мг, 23,78 мкмоль, 61,68% выход) в виде желтого масла.
Синтез E13: (3R,4S,8R,9S,10S)-9-(4-((2,3-дифторфенил)этинил)фенил)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К смеси промежуточного соединения 38b (15,00 мг, 23,78 мкмоль, 1,00 экв.) в CH3CN (500,00 мкл) добавляли 1 M HCl (500,00 мкл). После перемешивания при 15°C в течение 12 часов реакционную смесь концентрировали при пониженном давлении с получением остатка, который очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением (3S,4R,8R,9S,10S)-9-(4-((2,3-дифторфенил)этинил)фенил)-10-((диметиламино)метил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (11,00 мг, 17,28 мкмоль, 72,65% выход, соль муравьиной кислоты)) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, метанол-d4) δ=8,69-8,45 (м, 1H), 7,61 (с, 4H), 7,40-7,29 (м, 2H), 7,25-7,13 (м, 3H), 6,85 (д, J=9,0 Гц, 2H), 4,22 (дд, J=6,0, 15,6 Гц, 1H), 4,12 (д, J=6,0 Гц, 1H), 3,87-3,80 (м, 2H), 3,80-3,72 (м, 5H), 3,53-3,46 (м, 1H), 3,36 (ш.с, 1H), 3,00-2,88 (м, 3H), 2,84-2,73 (м, 2H), 2,41 (с, 6H).
Пример 14: (4aR,7aR,8S,9S,11aS)-9-((диметиламино)метил)-N-(4-метоксифенил)-8-(4-(фенилэтинил)фенил)октагидро-2H-азето[1,2-a][1,4]диоксино[2,3-f][1,4]диазоцин-6(3H)-карбоксамид (“E14”)

Промежуточное соединение 39a: (4aR,7aR,8S,9S,11aS)-8-(4-бромфенил)-9-((диметиламино)метил)-N-(4-метоксифенил)октагидро-2H-азето[1,2-a][1,4]диоксино[2,3-f][1,4]диазоцин-6(3H)-карбоксамид
Смесь промежуточного соединения 32a (30,00 мг, 56,24 мкмоль), NaOH (15,00 мг, 112,48 мкмоль, 180,30 мкл, 30% масс./об.) и TBAB (3,63 мг, 11,25 мкмоль) в DCE (1,00 мл) перемешивали при 55°C в течение 12 часов. Анализ ЖХ/МС показал, что осталось ~40% реагента 1. Детектировали один новый пик с требуемым промежуточным МС. Реакционную смесь разбавляли 10 мл DCM и промывали H2O (15 мл х 5). Затем промывали солевым раствором (3 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) с получением промежуточного соединения 39 (10,00 мг, 17,87 мкмоль, 31,78% выход) в виде бесцветного масла.
Синтез E14: (4aR,7aR,8S,9S,11aS)-9-((диметиламино)метил)-N-(4-метоксифенил)-8-(4-(фенилэтинил)фенил)октагидро-2H-азето[1,2-a][1,4]диоксино[2,3-f][1,4]диазоцин-6(3H)-карбоксамид
К раствору промежуточного соединения 39a (20,00 мг, 35,75 мкмоль) и этинилбензола (10,95 мг, 107,25 мкмоль, 11,77 мкл) в CH3CN (500 мкл) добавляли Cs2CO3 (46,59 мг, 143,00 мкмоль) и XPhos Pd G3 (3,03 мг, 3,58 мкмоль). После перемешивания в атмосфере N2 при 70°C в течение 1 часа реакционную смесь концентрировали, очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) для удаления катализатора и затем очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением (4aR,7aR,8S,9S,11aS)-9-((диметиламино)метил)-N-(4-метоксифенил)-8-(4-(фенилэтинил)фенил)октагидро-2H-азето[1,2-a][1,4]диоксино[2,3-f][1,4]диазоцин-6(3H)-карбоксамида (соль муравьиной кислоты), 6,00 мг, 10,33 мкмоль, 28,90% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=8,26 (ш.с, 1H), 7,55 (дд, J=2,0, 7,0 Гц, 2H), 7,51-7,42 (м, 4H), 7,40-7,33 (м, 3H), 7,20 (д, J=9,0 Гц, 2H), 6,83 (д, J=9,0 Гц, 2H), 4,21 (ш.д, J=15,6 Гц, 1H), 4,10 (ш.д, J=14,1 Гц, 1H), 4,06-3,94 (м, 3H), 3,86-3,76 (м, 5H), 3,66 (ш.с, 1H), 3,59-3,52 (м, 2H), 3,48-3,29 (м, 3H), 2,89 (ш.д, J=13,1 Гц, 1H), 2,60-2,33 (м, 3H), 2,03 (с, 6H), 1,83 (ш.с, 1H).
Пример 15: (3S,8R, 9S,10S)-10-[(диметиламино)метил]-3-гидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E15”)


Промежуточное соединение 40: N-[[(2R,3R,4S)-3-(4-бромфенил)-1-[(2S)-4-[трет-бутил(дифенил)силил]окси-2-гидрокси-бутил]-4-(тритилоксиметил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамид
К раствору N-[[(2R,3R,4S)-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамида (400,00 мг, 572,56 мкмоль) и LiClO4 (121,83 мг, 1,15 ммоль, 50,34 мкл) в CH3CN (4,00 мл) добавляли трет-бутил-[2-[(2R)-оксиран-2-ил]этокси]-дифенил-силан (Organic Letters (2016), 18(3), 468-471) (373,88 мг, 1,15 ммоль). После перемешивания при 80°C в течение 16 часов реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли H2O (20 мл) и экстрагировали DCM (3×20 мл). Объединенные органические слои промывали солевым раствором (3×10 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=2/1) с получением промежуточного соединения 40 (250,00 мг, 243,87 мкмоль, 42,59% выход) в виде твердого вещества светло-желтого цвета.
Промежуточное соединение 41: [(1S)-1-[[(2R,3R,4S)-2-[[ацетил-(2-нитрофенил)сульфонил-амино]метил]-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-1-ил]метил]-3-[трет-бутил(дифенил)силил]окси-пропил] ацетат
К раствору N-[[(2R,3R,4S)-3-(4-бромфенил)-1-[(2S)-4-[трет-бутил(дифенил)силил]окси-2-гидрокси-бутил]-4-(тритилоксиметил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамида (промежуточное соединение 40) (250,00 мг, 243,87 мкмоль), Et3N (24,68 мг, 243,87 мкмоль, 33,81 мкл) и DMAP (2,98 мг, 24,39 мкмоль) в DCM (3,00 мл) добавляли Ac2O (62,24 мг, 609,68 мкмоль, 57,10 мкл). После перемешивания при 15°C в течение 16 часов реакционную смесь гасили добавлением H2O (20 мл) и затем экстрагировали DCM (3×15 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=3/1, пластина 2) с получением промежуточного соединения 41 (270,00 мг, 243,42 мкмоль, 99,81% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 42: [(1S)-1-[[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2-нитрофенил)сульфониламино]метил]-4-(тритилоксиметил)азетидин-1-ил]метил]-3-[трет-бутил(дифенил)силил]окси-пропил] ацетат
К раствору [(1S)-1-[[(2R,3R,4S)-2-[[ацетил-(2-нитрофенил)сульфонил-амино]метил]-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-1-ил]метил]-3-[трет-бутил(дифенил)силил]окси-пропил]ацетата (промежуточное соединение 41) (270,00 мг, 243,42 мкмоль) в THF (2 мл) и H2O (2 мл) добавляли LiOH.H2O (20,43 мг, 486,84 мкмоль). После перемешивания при 15°C в течение 1 часа реакционную смесь разбавляли H2O (30 мл) и экстрагировали DCM (20 мл х 4). Объединенные органические слои промывали солевым раствором (20 мл х 3), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=3/1, пластина 1) с получением промежуточного соединения 42 (200,00 мг, 187,41 мкмоль, 76,99% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 43: [(1S)-1-[[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2-нитрофенил)сульфониламино]метил]-4-(тритилоксиметил)азетидин-1-ил]метил]-3-гидрокси-пропил] ацетат
К раствору [(1S)-1-[[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2-нитрофенил)сульфониламино]метил]-4-(тритилоксиметил)азетидин-1-ил]метил]-3-[трет-бутил(дифенил)силил]окси-пропил]ацетата (промежуточное соединение 42) (200,00 мг, 187,41 мкмоль) в THF (3 мл) добавляли TBAF (98,00 мг, 374,82 мкмоль). После перемешивания при 15°C в течение 2 часов реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли DCM (30 мл) и промывали H2O (15 мл х 5), затем солевым раствором (10 мл х 3), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1/1, пластина 1) с получением промежуточного соединения 43 (150,00 мг, 180,99 мкмоль, 96,57% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 44: [(3S,8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]ацетат
Смесь PPh3 (94,94 мг, 361,98 мкмоль, 2,00 экв.) и DIAD (73,20 мг, 361,98 мкмоль, 70,38 мкл, 2,00 экв.) в THF (2 мл) перемешивали при 0°C в атмосфере N2 с получением смеси молочно-белого цвета. К этой смеси добавляли раствор [(1S)-1-[[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2-нитрофенил)сульфониламино]метил]-4-(тритилоксиметил)азетидин-1-ил]метил]-3-гидрокси-пропил]ацетата (промежуточное соединение 43) (150,00 мг, 180,99 мкмоль) в THF (1 мл). После перемешивания при 15°C в течение 16 часов реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли DCM (30 мл) промывали H2O (10 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1/1, пластина 1) с получением промежуточного соединения 44 (150,00 мг, неочищенное) в виде твердого вещества белого цвета.
Промежуточное соединение 45: [(3S,8R,9R,10S)-9-(4-бромфенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил] ацетат
К раствору [(3S,8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]ацетата (промежуточное соединение 44) (150 мг, 185,01 мкмоль) в CH3CN (3 мл) добавляли Cs2CO3 (120,56 мг, 370,02 мкмоль) и бензолтиол (30,58 мг, 277,52 мкмоль, 28,31 мкл). После перемешивания при 40°C в течение 2 часов реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли H2O (30 мл) и экстрагировали DCM (20 мл х 5). Объединенные органические слои промывали солевым раствором (20 мл х 3), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1/1, пластина 1) с получением промежуточного соединения 45 (180,00 мг, неочищенное) в виде твердого вещества желтого цвета.
Промежуточное соединение 46: [(3S,8R,9R,10S)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]ацетат
К раствору [(3S,8R,9R,10S)-9-(4-бромфенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]ацетата (промежуточное соединение 45) (180 мг, 287,73 мкмоль) в DCM (3 мл) добавляли 1-изоцианат-4-метоксибензол (42,91 мг, 287,73 мкмоль, 36,99 мкл). После перемешивания при 15°C в течение 1 часа реакционную смесь гасили добавлением H2O (15 мл) и затем экстрагировали DCM (20 мл х 4). Объединенные органические слои промывали раствором NaHCO3 (15 мл), солевым раствором (15 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1/1, пластина 1) с получением промежуточного соединения 46 (200 мг, 258,15 мкмоль, 89,72% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 47: [(3S, 8R,9R,10S)-9-(4-бромфенил)-10-(гидроксиметил)-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил] ацетат
К раствору [(3S,8R,9R,10S)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]ацетата (промежуточное соединение 46) (200 мг, 258,15 мкмоль, 1,00 экв.) в DCM (3 мл) добавляли TFA (294,34 мг, 2,58 ммоль, 191,13 мкл). После перемешивания при 15°C в течение 2 часов реакционную смесь гасили добавлением раствора NaHCO3 (20 мл) и экстрагировали DCM (20 мл х 5). Объединенные органические слои промывали солевым раствором (10 мл х 3), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка который очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1/1, пластина 1) с получением промежуточного соединения 47 (130,00 мг, 244,16 мкмоль, 94,58% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 48: [(3S, 8R,9S,10S)-9-(4-бромфенил)-10-[(1,3-диоксоизоиндолин-2-ил)метил]-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил] ацетат
Смесь PPh3 (128,08 мг, 488,32 мкмоль, 2,00 экв.) и DIAD (98,74 мг, 488,32 мкмоль, 94,94 мкл, 2,00 экв.) в THF (1 мл) перемешивали при 0°C в атмосфере N2 с получением смеси молочно-белого цвета. К этой смеси молочно-белого цвета добавляли раствор (3S,8R,9R,10S)-9-(4-бромфенил)-10-(гидроксиметил)-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил]ацетата (промежуточное соединение 47) (130 мг, 244,16 мкмоль) и изоиндолин-1,3-дион (53,88 мг, 366,24 мкмоль) в THF (1 мл) при 0°C. После перемешивания при 15°C в течение 12 часов реакционную смесь концентрировали при пониженном давлении для удаления растворителя и затем очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1/1, пластина 1) с получением промежуточного соединения 48 (200 мг, неочищенное) в виде желтого масла.
Промежуточное соединение 49: (3S, 8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-3-гидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору [(3S,8R,9S,10S)-9-(4-бромфенил)-10-[(1,3-диоксоизоиндолин-2-ил)метил]-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил]ацетата (промежуточное соединение 48) (200 мг, 302,32 мкмоль) в EtOH (2 мл) добавляли N2H4.H2O (29,39 мкл, 604,64 мкмоль). После перемешивания при 70°C в течение 2 часов реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли H2O (10 мл) и экстрагировали DCM (10 мл х 5). Объединенные органические слои промывали солевым раствором (10 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1, пластина 1) с получением промежуточного соединения 49 (60 мг, 122,60 мкмоль, 40,55% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 50: (3S,8R,9S,10S)-9-(4-бромфенил)-10-[(диметиламино)метил]-3-гидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору (3S, 8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-3-гидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (промежуточное соединение 49) (60 мг, 122,60 мкмоль), формальдегида (54,77 мкл, 735,60 мкмоль), MgSO4 (147,57 мг, 1,23 ммоль) и AcOH (0,70 мкл, 12,26 мкмоль) в DCM (3 мл) добавляли NaBH(OAc)3 (77,95 мг, 367,80 мкмоль). После перемешивания при 15°C в течение 2 часов реакционную смесь фильтровали через целит и промывали раствором NaHCO3 (10 мл х 3). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения 50 (50 мг, 96,63 мкмоль, 78,81% выход) в виде твердого вещества белого цвета.
Синтез E15: (3S,8R,9S,10S)-10-[(диметиламино)метил]-3-гидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору (3S,8R,9S,10S)-9-(4-бромфенил)-10-[(диметиламино)метил]-3-гидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (промежуточное соединение 50) (50 мг, 96,63 мкмоль) и этинилбензола (29,61 мг, 289,89 мкмоль, 31,84 мкл) в CH3CN (1 мл) добавляли Cs2CO3 (125,94 мг, 386,52 мкмоль) и катализатор XPhos Pd G3 (8,18 мг, 9,66 мкмоль). После перемешивания в атмосфере N2 при 70°C в течение 1 часа реакционную смесь очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1, пластина 1) для удаления катализатора, получая неочищенный продукт, который затем очищали препаративной ВЭЖХ (колонка: Luna C18 150*25 5u; подвижная фаза: A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением продукта, фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (10 мг, 17,10 мкмоль, 17,7% выход, соль муравьиной кислоты)), в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=8,44 (ш.с, 1H), 7,54 (ш.д, J=7,0 Гц, 4H), 7,44-7,34 (м, 5H), 7,29-7,24 (м, 2H), 6,83 (ш.д, J=8,8 Гц, 2H), 6,29 (с, 1H), 5,71 (ш.с, 1H), 3,87 (ш.с, 2H), 3,77 (с, 3H), 3,75-3,62 (м, 4H), 3,52-3,38 (м, 1H), 3,18 (ш.дд, J=6,1, 13,6 Гц, 1H), 2,93-2,79 (м, 1H), 2,77-2,66 (м, 2H), 2,66-2,58 (м, 1H), 2,19 (с, 6H), 2,08-1,97 (м, 1H), 1,96-1,85 (м, 1H)
Примеры 16-17: Изомеры (8R,9S,10S)-10-[(диметиламино)метил]-3-гидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамида


Промежуточное соединение 51: (8R,9S,10S)-10-тритилоксиметил-3-гидрокси-N-(4-метоксифенил)-9-[4-бромфенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору (3Z,8R,9R,10S)-9-(4-бромфенил)-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамида (промежуточное соединение 25, 980,00 мг, 1,37 ммоль, 1,00 экв.) в THF (1,00 мл) добавляли BH3-Me2S (10 M, 686 мкл, 5,00 экв.) при 0°C, и реакционную смесь перемешивали при 25°C в течение 3 часов. Медленно добавляли H2O (1,24 г, 68,56 ммоль, 1,24 мл, 50,00 экв.), и реакционную смесь перемешивали при 25°C в течение 30 мин. Затем к реакционному раствору добавляли NaOH/H2O (18,27 г, 68,50 ммоль, 15% чистоты, 50,00 экв.) и H2O2 (6,66 г, 68,56 ммоль, 5,65 мл, 35% чистоты, 50,00 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 3 часов. Реакционную смесь гасили добавлением H2O (10 мл) и экстрагировали DCM (50 мл). Комбинированные органические слои сушили над безводным Na2SO4 и концентрировали с получением неочищенного продукта в виде твердого вещества белого цвета (смеси), который использовали на следующем этапе без дополнительной очистки.
Промежуточное соединение 52: [(8R,9R,10S)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил] ацетат
К раствору промежуточного соединения 51 (1,2 г, 1,64 ммоль), Et3N (497,85 мг, 4,92 ммоль) и DMAP (20,04 мг, 164 мкмоль) в DCM (2 мл) при 0°C добавляли Ac2O (334,86 мг, 3,28 ммоль). После перемешивания при 25°C в течение 3 часов реакционную смесь гасили добавлением H2O (10 мл) и экстрагировали DCM (20 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением двух изомеров [(8R,9R,10S)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]ацетата. Каждый изомер отделяли препаративной ТСХ с получением изомера 1 (126 мг, 9,92% выход, Rf=0,5) в виде твердого вещества белого цвета и изомера 2 (197 мг, 15,50% выход, Rf=0,4) в виде твердого вещества белого цвета, и смешанных фракций [(8R,9R,10S)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]ацетата (624 мг, 49,11% выход, Rf=0,3) в виде твердого вещества белого цвета. Несмотря на то, что каждый стереоизомер был выделен отдельно, невозможно было определить стереохимию положения 4 для каждого соединения.
Изомер 1: 1H ЯМР (400 МГц, CDCl3-d1) δ8,25 (с, 1H), 7,47 (д, J=8,8 Гц, 2H), 7,19-7,11 (м, 23H), 6,77 (д, J=8,8 Гц, 2H), 4,62 (с, 1H), 3,92-3,88 (м, 1H), 3,74-3,70 (м, 1H), 3,56-3,55 (м, 4H), 3,45-3,41 (м, 1H), 3,30-3,29 (м, 1H), 3,08-3,07 (м, 3H), 2,89-2,87 (м, 1H), 2,73-2,70 (м, 1H), 2,55-2,52 (м, 1H), 2,43 (с, 1H), 2,06 (с, 3H), 1,92-1,88 (м, 2H), 1,12-1,1,10 (м, 3H), 0,9-0,87 (м, 3H).
Изомер 2: 1H ЯМР (400 МГц, CDCl3-d1) δ7,34-7,22 (м, 23H), 7,09 (д, J=8 Гц, 2H), 6,83 (д, J=8,8 Гц, 2H), 5,07-5,04 (м, 1H), 3,92-3,88 (м, 1H), 3,77 (м, 5H), 3,65-3,58 (м, 3H), 3,25-3,27 (м, 2H), 3,03-3,00 (м, 1H), 3,02-2,99 (м, 1H), 2,61-2,57 (м, 1H), 2,45-2,44 (м, 1H), 2,16 (с, 3H), 2,08-2,05 (м, 1H), 1,74-1,71 (м, 2H), 1,00-0,95 (м, 1H).
Смешанные фракции 1H ЯМР (400 МГц, CDCl3-d1) δ7,52-7,17 (м, 21H), 6,84-6,82 (м, 3H), 4,81 (с, 1H), 4,29-4,16 (м, 1H), 3,99-3,77 (м, 1H), 3,67-3,66 (м, 1H), 3,65-3,63 (м, 6H), 3,63-3,56 (м, 5H), 3,40-3,39 (м, 3H), 3,16-3,12 (м, 2H), 2,96-2,94 (м, 1H), 2,63-,59 (м, 1H), 2,28-2,25 (м, 1H), 1,97 (с, 3H), 1,89-1,86 (м, 1H), 1,28-1,25 (м, 2H), 0,88-0,84 (м, 1).
Промежуточное соединение 53: (8R,9R,10S)-9-(4-бромфенил)-10-(гидроксиметил)-6-((4-метоксифенил)карбамоил)-1,6-диазабицикло[6.2.0]декан-3-ил ацетат
К раствору [(8R,9R,10S)-9-(4-бромфенил)-6-[(4-метокси фенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]ацетата (624 мг, 805,43 мкмоль) в DCM (2 мл) добавляли TFA (918,35 мг, 8,05 ммоль, 596,33 мкл). После перемешивания при 25°C в течение 3 часов реакционную смесь гасили добавлением NaHCO3 (10 мл) и экстрагировали DCM (20 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка, который очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 53 (265 мг, 61,80% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,46 (д, J=8 Гц, 2H), 7,38 (д, J=8 Гц, 2H), 7,26 (д, J=8 Гц, 2H), 6,83 (д, J=8 Гц, 2H), 6,47 (с, 1H), 4,80 (с, 1H), 3,81-3,77 (м, 4H), 3,64-3,57 (м, 5H), 3,52-3,47 (м, 3H), 3,25-3,22 (м, 1H), 3,21 (с, 1H), 2,97-2,84 (м, 1H), 22,33-2,23 (м, 2H), 2,24 (с, 2H), 2,15-1,96 (м, 2H).
Промежуточное соединение 54: [(8R,9S,10S)-9-(4-бромфенил)-10-[(1,3-диоксоизоиндолин-2-ил)метил]-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил]ацетат
К раствору промежуточного соединения 53 (275 мг, 516,5 мкмоль), изоиндолин-1,3-диона (83,59 мг, 568,15 мкмоль) и PPh3 (203,21 мг, 774,75 мкмоль) в THF (2 мл) по каплям добавляли DIAD (156,66 мг, 774,75 мкмоль) при 25°C. После перемешивания при 25°C в течение 5 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 54 (460 мг, неочищенное) в виде твердого вещества светло-желтого цвета. 1H ЯМР (4400 МГц, CDCl3-d1) δ7,79 (с, 2H), 7,70-7,65 (м, 20H), 7,55-7,53 (м, 20H), 7,48-7,45 (м, 30H), 6,86 (м, 3H), 6,62 (с, 1H), 4,76 (с, 2H), 3,86-3,83 (м, 6H), 3,79-7,77 (м, 3H), 3,65-3,64 (м, 3H), 3,54-3,49 (м, 1H), 3,09-3,04 (м, 1H), 2,85-2,81 (м, 1H), 2,56-2,53 (м, 1H), 3,37-3,14 (м, 1H), 2,21-2,14 (м, 1H).
Промежуточное соединение 55: [(8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил]ацетат.
К раствору промежуточного соединения 54 (460 мг, 695,35 мкмоль) в EtOH (1 мл) по каплям добавляли гидрат гидразина (53,28 мг, 1,04 ммоль, 51,73 мкл). После перемешивания при 80°C в течение 2 часов реакционную смесь фильтровали и концентрировали с получением промежуточного соединения 55 (500 мг, неочищенное) в виде коричневой жидкости, которую использовали на следующем этапе без очистки. HRMS (ESI): вычисл. для C24H28BrN4O3 [M+H]+ 531,15, найденное 531,1.
Промежуточное соединение 56: (8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-3-гидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 55 (300 мг, 564,50 мкмоль) в смеси EtOH:H2O=2:1 (1 мл) добавляли LiOH.H2O (47,37 мг, 1,13 ммоль). После перемешивания при 40°C в течение 2 часов реакционную смесь разбавляли H2O (3 мл) и экстрагировали DCM (10 мл) с получением органического слоя. Органический слой концентрировали и очищали препаративной ТСХ (дихлорметан:метанол=10:1) с получением промежуточного соединения 56 (90 мг, 32,58% выход) в виде белого твердого вещества. 1H ЯМР (4400 МГц, CDCl3-d1) δ7,52-7,47 (м, 2H), 7,34-7,32 (м, 18H), 7,27-7,22 (м, 3H), 6,84 (д, J=9,2 Гц, 2H), 6,17 (с, 1H), 3,87-3,84 (м, 1H), 3,78-3,74 (м, 3H), 3,70-3,66 (м, 3H), 3,57-3,55 (м, 2H), 3,13-3,10 (м, 1H), 2,77-2,71 (м, 1H), 2,04-1,88 (м, 5H).
Промежуточное соединение 57: (8R,9S,10S)-9-(4-бромфенил)-10-[(диметиламино)метил]-3-гидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 56 (90 мг, 183,9 мкмоль) в DCM (2 мл) добавляли HCHO (149,26 мг, 1,84 ммоль, 136,93 мкл, 37% раствор) и MgSO4 (442,72 мг, 3,68 ммоль). После перемешивания при 25°C в течение 0,5 часа добавляли NaBH(OAc)3 (194,88 мг, 919,49 мкмоль). После перемешивания при 25°C в течение 1,5 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=10:1) с получением промежуточного соединения 57 (58 мг, 60,95% выход) в виде светло-коричневого масла. 1H ЯМР (400 МГц, CDCl3-d1) δ7,51-7,44 (м, 2H), 7,30-7,23 (м, 5H), 6,85 (д, J=8,8 Гц, 2H), 6,15 (с, 1H), 3,89-3,87 (м, 2H), 3,83-3,78 (м, 3H), 3,73-3,67 (м, 4H), 3,20-3,15 (м, 1H), 2,72-2,69 (м, 2H), 2,60-2,51 (м, 2H), 2,14 (с, 5H), 2,05-2,026 (м, 4H), 2,00-1,91 (м, 1H).
Синтез E16 и E17: изомер 1 (E16) и изомер 2 (E17) (8R,9S,10S)-10-[(диметиламино)метил]-3-гидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамида
К раствору промежуточного соединения 57 (50 мг, 96,63 мкмоль) и этинилбензола (29,61 мг, 289,89 мкмоль) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (8,18 мг, 9,66 мкмоль) и Cs2CO3 (62,97 мг, 193,26 мкмоль). После перемешивания при 70°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением смеси изомеров (8R,9S,10S)-10-[(диметиламино)метил]-3-гидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамида. Каждый изомер отделяли, получая изомер 1 (5,1 мг, 9,80% выход) и изомер 2 (17,1 мг, 32,85% выход) в виде твердого вещества белого цвета.
E16: Изомер 1: HRMS (ESI): вычисл. для C32H35N4O2 [M+H]+ 539,29, найденное 529. 1H ЯМР (400 МГц, MeOD-d4) δ7,60-7,50 (м, 6H), 7,39-7,37 (м, 3H), 7,36-7,18 (м, 2H), 6,85 (д, J=8,8 Гц, 2H), 3,83-3,80 (м, 2H), 3,76 (с, 3H),3,69-3,68 (м, 2H), 3,55-3,52 (м, 3H), 3,15-3,09 (м, 2H), 3,01-3,00 (м, 2H), 3,41-3,34 (м, 2H), 2,26 (м, 6H), 1,75-1,72 (м, 1H).
E17: Изомер 2: HRMS (ESI): вычисл. для C32H35N4O2 [M+H]+ 539,29, найденное 529,40 1H ЯМР (400 МГц, MeOD-d4) δ7,61-7,54 (м, 6H), 7,50 (д, J=1,6 Гц, 3H), 7,38-7,36 (м, 2H), 6,85 (д, J=8,8 Гц, 2H), 3,84-3,80 (м, 3H), 3,75 (с, 3H), 3,67-3,61 (м, 2H), 3,55-3,51 (м, 1H), 3,08-3,05 (м, 2H), 2,83-2,80 (м, 1H), 2,74-2,68 (м, 3H), 2,26 (с, 6H), 2,03-1,94 (м, 2H).
Примеры 18-19: Изомер 1 (“E18”) и изомер 2 (“E19”): (8R,9R,10S)-4-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамида
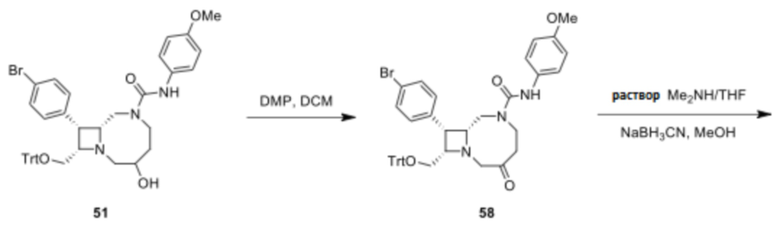

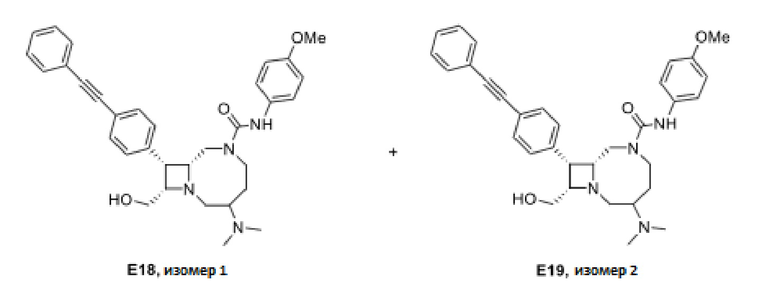
Промежуточное соединение 58: (8R,9R,10S)-9-(4-бромфенил)-N-(4-метоксифенил)-3-oxo-10-((тритилокси)метил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 51 (500,00 мг, 682,41 мкмоль, 1 экв.) в DCM (5 мл) добавляли реагент Десса-Мартина (347,32 мг, 818,89 мкмоль). После перемешивания при 25°C в течение 3 часов реакционную смесь гасили добавлением насыщенного Na2SO3 (5 мл) и экстрагировали DCM (10 мл) с получением органического слоя. Органический слой концентрировали и очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 58 (280 мг, 56,15% выход) в виде твердого вещества коричневого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,37-7,19 (м, 30H), 6,86-6,82 (м, 2H), 6,20 (с, 1H), 4,27-4,20 (м, 3H), 4,07-3,81 (м, 1H), 3,79-3,77 (м, 3H), 3,70-3,66 (м, 3H), 3,54-3,31 (м, 1H), 3,06-3,04 (м, 1H), 2,98-2,94 (м, 2H), 2,76-2,73 (м, 1H), 2,72-2,45 (м, 1H).
Промежуточное соединение 59: (8R,9R,10S)-9-(4-бромфенил)-3-(диметиламино)-N-(4-метоксифенил)-10-((тритилокси)метил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 58 (250 мг, 342,14 мкмоль) в MeOH (2 мл) добавляли N-метилметанамин (2 M, 1,71 мл) и CH3COOH (205,46 мг, 3,42 ммоль). После перемешивания при 25°C в течение 0,5 часа добавляли NaBH3CN (21,50 мг, 342,14 мкмоль). После перемешивания при 25°C в течение 1,5 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=15:1) с получением промежуточного соединения 59 (120 мг, 46,16% выход) в виде светло-коричневого масла. HRMS (ESI) вычисл. для C32H33N4O [M+H]+ 759,28, найденное 759,2.
Промежуточное соединение 60: (8R,9R,10S)-9-(4-бромфенил)-3-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 59 (115 мг, 151,36 мкмоль) в DCM (1 мл) добавляли TFA (172,58 мг, 1,51 ммоль). После перемешивания при 25°C в течение 12 часов реакционную смесь гасили добавлением насыщенного NaHCO3 (5 мл) и экстрагировали DCM (5 мл) с получением органического слоя. Органический слой собирали, концентрировали и затем очищали препаративной ТСХ (дихлорметан:метанол=10:1) с получением промежуточного соединения 60 (45,00 мг, 57,45% выход) в виде светло-коричневого масла. 1H ЯМР (400 МГц, CDCl3-d1) δ7,47-7,43 (м, 2H), 7,38-7,31 (м, 4H), 6,83 (д, J=8,8 Гц, 2H), 3,99 (с, 1H), 3,77 (с, 5H), 3,63-3,61 (м, 5H), 3,59-3,57 (м, 5H), 3,48-3,46 (м, 2H), 3,32-3,13 (м, 3H), 2,76 (с, 6H), 2,48-2,34 (м, 4H), 2,04-2,00 (м, 1H).
Синтез E18 и E19: изомер 1 (E18) и изомер 2 (E19) (8R,9R,10S)-4-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамида
К раствору промежуточного соединения 59 (30 мг, 57,98 мкмоль) и этинилбензола (17,76 мг, 173,94 мкмоль) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (4,91 мг, 5,80 мкмоль) и Cs2CO3 (37,78 мг, 115,96 мкмоль). После перемешивания при 70°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением каждого из стереоизомеров (8R,9R,10S)-4-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамида, выделенных как изомер 1 (E18) (2,60 мг, 8,32% выход) и изомер 2 (E19) (11,40 мг, 36,50% выход) в виде твердого вещества белого цвета.
E18: Изомер 1, HRMS (ESI) вычисл. для: C33H38N4O3 [M+H]+ 539,29, найденное 539,4; 1H ЯМР (400 МГц, MeOD-d4) δ8,34 (с, 1H), 7,56-7,51 (м, 2H), 7,50-7,48 (м, 4H), 7,37 (д, J=5,2 Гц, 2H), 7,26 (д, J=9,6 Гц, 2H), 6,85 (д, J=8,8 Гц, 1H), 4,09-4,05 (м, 1H), 3,75-3,72 (м, 3H), 3,66-3,65 (м, 1H), 3,52-3,46 (м, 4H), 3,26-3,20 (м, 2H), 2,64 (с, 6H), 2,55-2,52 (м, 1H), 1,97 (с, 2H).
E19: Изомер 2, HRMS (ESI) вычисл. для: C33H38N4O3 [M+H]+ 539,29, найденное 539,4; 1H ЯМР (400 МГц, MeOD-d4) δ8,55 (с, 1H), 7,61 (д, J=8 Гц, 2H), 7,51-7,49 (м, 4H), 7,38-7,36 (м, 3H), 7,23 (д, J=8,8 Гц, 2H), 6,86 (д, J=8,8 Гц, 2H), 4,21-4,11 (м, 1H), 3,76-3,73 (м, 5H), 3,76-3,73 (м, 2H), 3,62-3,40 (м, 3H), 3,31-3,13 (м, 3H), 2,81-2,77 (с, 6H), 2,64 (с, 1H), 2,46 (с, 1H), 2,06-2,03 (м, 1H).
Примеры 20-21: (8R,9S,10S)-9-(4-бромфенил)-4-(диметиламино)-10-[(диметиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида, изомер 1 (“E20”) и изомер 2 Пример 21 (“E21”)
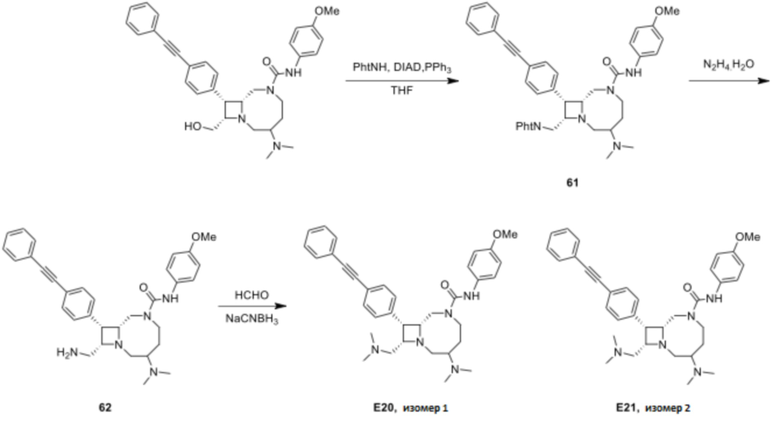
Промежуточное соединение 61: (8R,9S,10S)-3-(диметиламино)-10-((1,3-диоксоизоиндолин-2-ил)метил)-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору смеси примеров 18 и 19 (165 мг, 318,87 мкмоль, 1 экв.), изоиндолин-1,3-диона (51,61 мг, 350,75 мкмоль) и PPh3 (125,45 мг, 478,30 мкмоль) в THF (2 мл) добавляли по каплям DIAD (96,72 мг, 478,30 мкмоль). После перемешивания при 25°C в течение 12 часов остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=0:1) с получением промежуточного соединения 61 (184 мг, 89,25% выход) в виде твердого вещества светло-желтого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,83-7,81 (м, 1H), 7,73-7,72 (м, 2H), 7,55-7,26 (м, 5H), 6,82-6,79 (м, 2H), 3,94-3,74 (м, 2H), 3,72 (с, 3H), 3,67-3,56 (м, 5H), 3,54-3,55 (м, 2H), 3,35-3,06 (м, 1H), 2,77 (с, 6H), 1,26 (с, 1H).
Промежуточное соединение 62: (8R,9S,10S)-10-(аминометил)-3-(диметиламино)-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 61 (174 мг, 260,56 мкмоль) в EtOH (1 мл) по каплям добавляли гидрат гидразина (19,96 мг, 390,84 мкмоль). После перемешивания при 80°C в течение 2 часов реакционную смесь фильтровали и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=6:1) с получением промежуточного соединения 62 (50,00 мг, 35,69% выход) в виде коричневой жидкости. HRMS (ESI) вычисл. для C33H39N5O2 [M+H]+ 538,31, найденное 538,2.
Синтез E20 и E21: (4S,8R,9S,10S)-9-(4-бромфенил)-4-(диметиламино)-10-[(диметиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E20) и (4R,8R,9S,10S)-9-(4-бромфенил)-4-(диметиламино)-10-[(диметиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E21)
К перемешиваемому раствору промежуточного соединения 61 (50 мг, 96,81 мкмоль) в DCM (2,00 мл) добавляли HCHO (78,57 мг, 968,10 мкмоль, 72,08 мкл) и MgSO4 (116,53 мг, 968,10 мкмоль). После перемешивания при 25°C в течение 0,5 часа добавляли CH3COOH (5,81 мг, 96,81 мкмоль) и NaBH(OAc)3 (102,59 мг, 484,05 мкмоль). После перемешивания при 25°C в течение 1,5 часов реакционную смесь гасили добавлением H2O (5 мл) и экстрагировали DCM (20 мл х 3) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ с получением изомеров (8R,9S,10S)-9-(4-бромфенил)-4-(диметиламино)-10-[(диметиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида, которые разделяли в виде изомера 1 (E20) (2,90 мг, 5,33 мкмоль, 5,51% выход) в виде светло-коричневого масла (HRMS (ESI) вычисл. для: C35H43N5O2 [M+H]+ 566,34, найденное 566,4; 1H ЯМР (400 МГц, MeOD-d4) δ7,61-7,51 (м, 5H), 7,44-7,38 (м, 4H), 7,37-7,23 (м, 2H), 6,87-6,85 (м, 2H), 4,05-3,77 (м, 1H), 3,77 (с, 3H), 3,66-3,51 (м, 2H), 3,19-3,16 (м, 1H), 2,50-2,47 (м, 1H), 2,35-2,19 (м, 1H), 1,86-1,80 (м, 1H))) и изомера 2 (E21) (15,20 мг, 27,91 мкмоль, 28,83% выход) в виде светло-коричневого масла (1H ЯМР (400 МГц, MeOD-d4) δ7,56-7,54 (м, 6H), 7,52-7,51 (м, 3H), 7,23-7,22 (м, 2H), 6,83-6,84 (м, 2H), 4,14-4,12 (м, 1H), 3,87-3,81 (м, 1H), 3,76 (с, 3H), 3,67-3,48 (м, 2H), 3,24-3,22 (м, 2H), 2,77 (с, 6H), 2,71-2,60 (м, 4H), 2,32-2,31 (м, 2H), 2,31-2,30 (м, 1H)).
Пример 22: (3S,4R,8R,9R,10S)-3,4-дигидрокси-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(пиридин-3-илэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E22”)
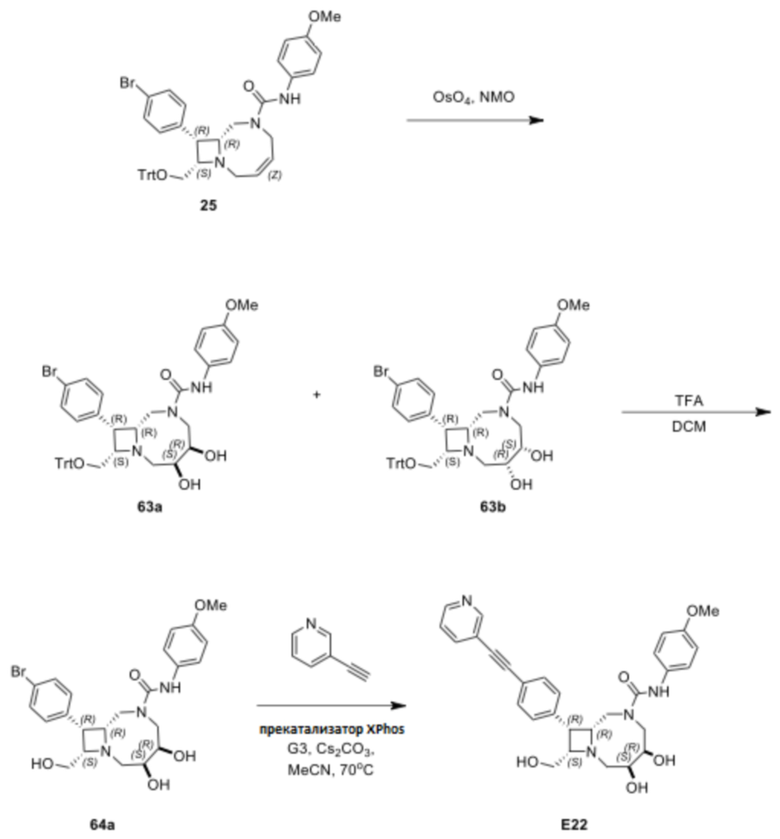
Промежуточные соединения 63a и 63b: (3S,4R,8R,9R,10S)-9-(4-бромфенил)-3,4-дигидрокси-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (63a) и (3R,4S,8R,9R,10S)-9-(4-бромфенил)-3,4-дигидрокси-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (63b)
К раствору промежуточного соединения 25 (220 мг, 307,83 мкмоль) и NMO (43,27 мг, 369,4 мкмоль) в смеси ацетон/H2O=10:1 (1 мл) добавляли OsO4 (7,83 мг, 30,78 мкмоль) при 25°C. После перемешивания при 25°C в течение 12 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:5) с получением промежуточного соединения 63a (120 мг, 52,07% выход) в виде твердого вещества белого цвета (1H ЯМР (400 МГц, CDCl3-d1) δ7,31-7,05 (м, 23H), 6,85-6,81 (м, 2H), 6,82 (д, J=8 Гц, 2H), 4,23-4,19 (м, 1H), 3,99 (с, 1H), 3,76-3,73 (м, 6H), 3,50-3,43 (м, 2H), 3,19-3,13 (м, 2H), 2,98-2,96 (м, 1H), 2,86-2,84 (м, 1H), 2,78-2,59 (м, 1H)); и промежуточного соединения 63b (134 мг, 58,14% выход) в виде твердого вещества белого цвета (1H ЯМР (400 МГц, CDCl3-d1) δ7,39-7,24 (м, 23H), 7,09-7,05 (м, 2H), 6,84 (д, J=8,8 Гц, 2H), 3,85-3,78 (м, 5H), 3,69-3,61 (м, 4H), 3,45-3,40 (м, 3H), 3,14-3,13 (м, 1H), 3,05-3,04 (м, 1H), 2,78-2,75 (м, 1H)).
Промежуточное соединение 64a: (3S,4R,8R,9R,10S)-9-(4-бромфенил)-3,4-дигидрокси-10-(гидроксиметил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 63a (60 мг, 80,14 мкмоль) в DCM (2 мл) добавляли TFA (91,37 мг, 801,39 мкмоль). После перемешивания при 25°C в течение 3 часов реакцию останавливали добавлением NaHCO3 (10 мл) и экстрагировали DCM (20 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат) с получением промежуточного соединения 64a (35 мг, 86,24% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,43 (д, J=7,6 Гц, 2H), 7,30 (д, J=8,4 Гц, 2H), 7,20 (д, J=8,8 Гц, 2H), 6,82 (д, J=8,8 Гц, 2H), 4,32-4,28 (м, 1H), 4,13-4,06 (м, 2H), 3,85 (м, 1H), 3,76 (с, 3H), 3,61 (с, 1H), 3,48 (с, 3H), 3,22 (м, 1H), 2,98-2,88 (м, 2H), 2,77-2,70 (м, 2H), 2,0 (с, 1H).
Синтез E22: (3S,4R,8R,9R,10S)-3,4-дигидрокси-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(пиридин-3-илэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 64a (20 мг, 39,5 мкмоль) и 3-этинилпиридина (8,15 мг, 78,99 мкмоль) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (3,34 мг, 3,95 мкмоль) и Cs2CO3 (25,74 мг, 78,99 мкмоль). После перемешивания при 70°C в течение 1 часа реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением (3S,4R,8R,9R,10S)-3,4-дигидрокси-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(пиридин-3-илэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (6,8 мг, 29,69% выход) в виде твердого вещества белого цвета (HRMS (ESI) вычисл. для C30H32N4O5 [M+H]+ 529,24, найденное 529,30; 1H ЯМР (400 МГц, MeOD-d4) δ8,69 (с, 1H), 8,51 (д, J=4 Гц, 1H), 7,98 (д, J=8 Гц, 1H), 7,60-7,52 (м, 4H), 7,48-7,45 (м, 1H), 7,16 (д, J=8,8 Гц, 2H), 6,84 (д, J=8,8 Гц, 2H), 4,16-4,11 (м, 2H), 3,84-3,82 (м, 2H), 3,75 (с, 3H), 3,64-3,58 (м, 4H), 3,39-3,60 (м, 2H), 2,88-2,79 (м, 3H)).
Пример 23: (3R,4S,8R,9R,10S)-3,4-дигидрокси-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(пиридин-3-илэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E23”)

Промежуточное соединение 64b: (3R,4S,8R,9R,10S)-9-(4-бромфенил)-3,4-дигидрокси-10-(гидроксиметил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 63b (130 мг, 173,63 мкмоль) в DCM (2 мл) добавляли TFA (197,98 мг, 1,74 ммоль). После перемешивания при 25°C в течение 3 часов реакционную смесь гасили добавлением NaHCO3 (10 мл) и экстрагировали DCM (20 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат) с получением промежуточного соединения 64b (75 мг, 85,30% выход) в виде твердого вещества белого цвета (1H ЯМР (400 МГц, CDCl3-d1) δ7,48 (д, J=7,6 Гц, 2H), 7,09-7,28 (м, 7H), 6,84 (д, J=8,8 Гц, 2H), 3,78 (д, J=10,8 Гц, 1H), 3,72-3,66 (м, 4H), 3,59-3,57 (м, 5H), 3,53-3,51 (м, 4H), 3,27-3,25 (м, 1H), 2,88-2,74 (м, 2H)).
Синтез E23: (3R,4S,8R,9R,10S)-3,4-дигидрокси-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(пиридин-3-илэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 64b (20 мг, 39,50 мкмоль) и 3-этинилпиридина (8,15 мг, 78,99 мкмоль) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (3,34 мг, 3,95 мкмоль) и Cs2CO3 (25,74 мг, 78,99 мкмоль). После перемешивания при 70°C в течение 2 часов реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением (3R,4S,8R,9R,10S)-3,4-дигидрокси-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(пиридин-3-илэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (7,4 мг, 29,69% выход) в виде твердого вещества белого цвета. HRMS (ESI) вычисл. для C30H32N4O5 [M+H]+ 529,24, найденное 529,30. 1H ЯМР (400 МГц, MeOD-d4) δ8,69 (с, 1H), 8,52 (д, J=5,2 Гц, 1H), 7,98 (д, J=8 Гц, 1H), 7,61-7,56 (м, 4H), 7,48-7,46 (м, 1H), 7,23 (д, J=8,8 Гц, 2H), 6,86 (д, J=9,2 Гц, 2H), 3,84-3,80 (м, 3H), 3,76 (с, 3H), 3,65-3,50 (м, 7H), 3,31-3,28 (м, 1H), 3,08-3,26 (м, 1H), 2,74-2,71 (м, 1H).
Синтез промежуточного соединения A5: [(1S)-1-[[трет-бутил(дифенил)силил]оксиметил]-3-oxo-пропил] ацетат

Промежуточное соединение A2: трет-бутил-[[(2S)-оксиран-2-ил]метокси]-дифенил-силан
К раствору (R)-оксиран-2-илметанола (3,00 г, 40,50 ммоль, 2,68 мл) и имидазола (5,51 г, 80,99 ммоль) в DCM (80 мл) при 0°C добавляли TBDPSCl (13,36 г, 48,60 ммоль, 12,48 мл). После перемешивания при 25°C в течение 12 часов реакционную смесь гасили добавлением H2O (20 мл) и экстрагировали DCM (60 мл х 2) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=10:1) с получением промежуточного соединения A2 (12,66 г, 100,00% выход) в виде бесцветной жидкости. 1H ЯМР (400 МГц, CDCl3-d1) δ7,69-7,68 (м, 4H), 7,46-7,40 (м, 5H), 8,87-3,85(м, 1H), 3,73-3,70 (м, 1H), 3,14-3,13 (м, 1H), 2,76-2,74 (м, 1H), 1,05 (с, 9H).
Промежуточное соединение A3: (2S)-1-[трет-бутил(дифенил)силил]оксипент-4-ен-2-ол
CuI (1,83 г, 9,60 ммоль, 1,5 экв.) помещали в трехгорлую колбу в атмосфере азота и затем добавляли безводный THF (20 мл). Полученную смесь охлаждали до -78° C, и затем по каплям добавляли винилмагния бромид (1 M, 22,40 мл, 3,5 экв.), при этом поддерживая внутреннюю температуру ниже -78°C. Гетерогенную смесь нагревали до -20°C и перемешивали при этой температуре в течение 30 мин. После охлаждения раствор снова охлаждали до -78°C, по каплям добавляли промежуточное соединение A2 (2,00 г, 6,40 ммоль, 1 экв.). Смесь перемешивали и оставляли постепенно нагреваться до 20°C в течение 12 часов. Реакционную смесь гасили добавлением H2O (20 мл) и экстрагировали DCM (60 мл х 2) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=7:1) с получением промежуточного соединения A3 (2,18 г, 100,00% выход) в виде бесцветной жидкости. 1H ЯМР (400 МГц, CDCl3-d1) δ7,67-7,65 (м, 4H), 7,44-7,37 (м, 7H), 5,82-5,76 (м, 1H), 5,10-4,90 (м, 2H), 3,81-3,78 (м, 1H), 3,68-3,65 (м, 1H), 3,57-3,54 (м, 1H), 2,46-2,44 (м, 1H), 2,25-2,22 (м, 1H), 1,05 (м, 9H).
Промежуточное соединение A4: [(1S)-1-[[трет-бутил(дифенил)силил]оксиметил]бут-3-енил]ацетат
К раствору промежуточного соединения A3 (2,18 г, 6,40 ммоль, 1 экв.), Et3N (1,94 г, 19,21 ммоль, 3 экв.) и DMAP (78,21 мг, 640,18 мкмоль, 0,1 экв.) в DCM (2 мл) при 0°C добавляли уксусный ангидрид (980,06 мг, 9,60 ммоль, 1,5 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 3 часов. Анализ ТСХ (петролейный эфир:этилацетат=10:1) показал завершение реакции, реакционную смесь гасили добавлением H2O (10 мл) и экстрагировали DCM (20 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=10:1) с получением промежуточного соединения A4 (2,30 г, 93,94% выход) в виде бесцветной жидкости. 1H ЯМР (400 МГц, CDCl3-d1) δ7,67-7,65 (м, 4H), 7,40-7,26 (м, 6H), 5,77-5,69 (м, 1H), 5,11-5,01 (м, 3H), 3,73-3,66 (м, 2H), 2,48-2,32 (м, 1H), 2,02 (м, 3H), 1,09 (м, 9H).
Промежуточное соединение A5: [(1S)-1-[[трет-бутил(дифенил)силил]оксиметил]-3-оксо-пропил] ацетат
Раствор промежуточного соединения A4 (1,70 г, 4,44 ммоль, 1 экв.) в DCM/MeOH=1:1 (30 мл) с температурой -78°C обрабатывали потоком O3 в O2 до тех пор, пока раствор не стал голубым. Затем через раствор пропускали газообразный N2 до момента исчезновения окраски. Полученный раствор обрабатывали Me2S (2,76 г, 44,44 ммоль, 10 экв.), нагревали до 20°C и перемешивали в течение 2 часов. Анализ ТСХ (петролейный эфир:этилацетат=5:1) показал завершение реакции, реакционную смесь разбавляли DCM (50 мл) и промывали H2O (10 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением промежуточного соединения A5 (1,70 г, неочищенного) в виде бесцветной жидкости без дополнительной очистки.
Пример 24: (4S,8R,9S,10S)-10-[(диметиламино)метил]-4-гидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E24”)
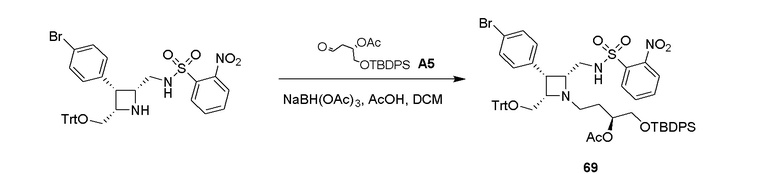


Промежуточное соединение 69: [(1S)-3-[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2-нитрофенил)сульфониламино]метил]-4-(тритилоксиметил)азетидин-1-ил]-1-[[трет-бутил(дифенил)силил]оксиметил]пропил] ацетат
К перемешиваемому раствору N-(((2R,3R,4S)-3-(4-бромфенил)-4-((тритилокси)метил)азетидин-2-ил)метил)-2-нитробензолсульфонамида (WO 2015070204) (1,20 г, 1,72 ммоль, 1 экв.) в DCM (12 мл) добавляли промежуточное соединение A5 (1,46 г, 3,78 ммоль, 2,2 экв.) при 25°C и CH3COOH (103,29 мг, 1,72 ммоль, 1 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 0,5 часа. К смеси добавляли NaBH(OAc)3 (1,09 г, 5,16 ммоль, 3 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 1,5 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (5 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 69 (1,67 г, неочищенное) в виде светло-коричневого масла. 1H ЯМР (400 МГц, CDCl3-d1) δ5,15-5,11 (м, `H), 4,96-4,95 (м, 1H), 4,10-4,07 (м, 1H), 3,62-3,44(м, 4H), 3,17-3,12 (м, 1H), 2,88-2,81 (м, 3H), 2,51-2,44 (м, 2H), 2,06-2,08 (м, 3H).
Промежуточное соединение 70: [(1S)-3-[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2-нитрофенил)сульфониламино]метил]-4-(тритилоксиметил)азетидин-1-ил]-1-(гидроксиметил)пропил]ацетат
К перемешиваемому раствору промежуточного соединения 69 (1,67 г, 1,56 ммоль, 1 экв.) в THF (16 мл) добавляли TBAF (1,22 г, 4,68 ммоль, 3 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 70 (920,00 мг, 71,16% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ 7,85-7,66 (м, 10), 7,26-6,97 (м, 20H), 5,02 (м, 1H), 4,85-4,82 (м, 1H), 4,05-3,76 (м, 1H), 3,70-3,52 (м, 3H), 3,12-2,86 (м, 3H), 2,03 (с, 5H), 1,52-1,46 (м, 2H).
Промежуточное соединение 71: [(4S,8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]ацетат
К перемешиваемому раствору промежуточного соединения 70 (660,00 мг, 796,36 мкмоль, 1 экв.) в THF (20 мл) добавляли смесь PPh3 (835,51 мг, 3,19 ммоль, 4 экв.) и DIAD (644,13 мг, 3,19 ммоль, 619,35 мкл, 4 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 71 (445,00 мг, неочищенное) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,92-7,90 (м, 1H), 7,69-7,61 (м, 3H), 7,26-7,16 (м, 20H), 4,42-4,40 (м, 2H), 4,14-4,10 (м, 1H), 3,57 (с, 3H), 3,56-3,45 (м, 1H), 3,10-3,03 (м, 3H), 2,85-2,84 (м, 1H), 2,37-2,31 (м, 1H), 2,13-1,98 (м, 1H), 1,87-1,54 (м, 1H),01,28-1,26 (м, 3H).
Промежуточное соединение 72: [(4S,8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]ацетат
К перемешиваемому раствору промежуточного соединения 71 (660,00 мг, 796,36 мкмоль, 1 экв.) в THF (20,00 мл) добавляли смесь PPh3 (835,51 мг, 3,19 ммоль, 4 экв.) и DIAD (644,13 мг, 3,19 ммоль, 619,35 мкл, 4 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 72 (445,00 мг, неочищенное) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,26-7,22 (м, 25H), 4,01-4,00 (м, 1H), 3,83-3,81 (м, 1H), 3,62-3,47 (м, 1H), 3,51-3,47 (м, 1H), 3,49-3,48 (м, 1H), 3,21-3,11 (м, 1H), 2,92-2,88 (м, 1H),2,49-2,47 (м, 2H), 2,05 (с, 3H), 1,85-1,084 (м, 1H), 1,60 (с, 3H).
Промежуточное соединение 73: [(4S,8R,9R,10S)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]ацетат
К перемешиваемому раствору промежуточного соединения 72 (154,00 мг, 246,17 мкмоль, 1 экв.) и Et3N (49,82 мг, 492,34 мкмоль, 68,25 мкл, 2 экв.) в DCM (2 мл) добавляли 1-изоцианат-4-метоксибензол (40,39 мг, 270,78 мкмоль, 34,82 мкл, 1,1 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (2 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 73 (169 мг, 88,61% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,33-7,23 (м, 20H), 6,83-6,82 (м, 2H), 4,59-4,57 (м, 1H), 4,30-4,27 (м, 1H), 3,78-3,75 (м, 4), 3,63-3,3,62 (м, 1H), 3,54-3,53 (м, 1H), 3,15-3,13 (м, 2H), 2,92-2,90 (м, 2H), 2,08-2,00 (м, 3H), 1,89-1,85 (с, 2H), 1,57 (м, 1H).
Промежуточное соединение 74: [(4S,8R,9R,10S)-9-(4-бромфенил)-10-(гидроксиметил)-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-4-ил]ацетат
К перемешиваемому раствору промежуточного соединения 73 (169,00 мг, 218,14 мкмоль, 1 экв.) в DCM (2 мл) добавляли TFA (248,72 мг, 2,18 ммоль, 10 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением NaHCO3 и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:2) с получением промежуточного соединения 74 (81,00 мг, 152,13 мкмоль, 69,74% выход) в виде твердого вещества белого цвета. HRMS (ESI): вычисл. для C25H30BrN3O5 [M+H]+ 532,14, найденное 534,1.
Промежуточное соединение 75: [(4S,8R,9S,10S)-9-(4-бромфенил)-10-[(1,3-диоксоизоиндолин-2-ил)метил]-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-4-ил]ацетат
К раствору промежуточного соединения 74 (81,00 мг, 104,55 мкмоль, 1 экв.), изоиндолин-1,3-диона (16,92 мг, 115,01 мкмоль, 1,1 экв.) и PPh3 (41,13 мг, 156,82 мкмоль, 1,5 экв.) в THF (2 мл) по каплям добавляли DIAD (31,71 мг, 156,82 мкмоль, 30,49 мкл, 1,5 экв.) при 25°C в атмосфере N2. Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 75 (58,00 мг, 83,86% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,83-7,81 (м, 2H), 7,73-7,71 (м, 2H), 7,51-7,49 (м, 4H), 7,25-7,23 (м, 2H), 6,89-6,84 (м, 2H), 6,56 (с, 1H), 4,59-4,55 (м, 1H), 4,28-4,27 (м, 1H), 3,82-3,78 (м, 4H), 3,58-3,55 (м, 3,50H), 3,50-3,25 (м, 2H), 2,99-2,98 (м, 1H), 2,37-2,12 (м, 1H), 2,08 (с, 5H).
Промежуточное соединение 76: (4S,8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-6-((4-метоксифенил)карбамоил)-1,6-диазабицикло[6.2.0]декан-4-илацетат
К раствору промежуточного соединения 75 (58,00 мг, 87,67 мкмоль, 1 экв.) в EtOH (1 мл) по каплям добавляли гидрат гидразина (6,72 мг, 131,51 мкмоль, 6,52 мкл, 98% чистоты, 1,5 экв.) при 25°C в атмосфере N2. Полученную реакционную смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка промежуточного соединения 76 без дополнительной очистки. HRMS (ESI): вычисл. для C25H31BrN4O4 [M+H]+ 531,15, найденное 531,1.
Промежуточное соединение 77: (4S,8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-4-гидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 76 (46,00 мг, 86,56 мкмоль, 1 экв.) в EtOH:H2O=1:1 (1 мл) добавляли LiOH.H2O (7,26 мг, 173,12 мкмоль, 2 экв.) при 25°C. Полученную реакционную смесь перемешивали при 25°C в течение 2 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (2 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением промежуточного соединения 77 (80,00 мг, неочищенное) в виде твердого вещества белого цвета. HRMS (ESI): ET6538-397-P1P вычисл. для C23H29BrN4O3 [M+H]+ 489,14, найденное 491,1.
Промежуточное соединение 78: (4S,8R,9S,10S)-9-(4-бромфенил)-10-((диметиламино)метил)-4-гидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 77 (757,00 мг, 1,01 ммоль, 1 экв.), изоиндолин-1,3-диона (164,07 мг, 1,12 ммоль, 1,1 экв.) и PPh3 (398,85 мг, 1,52 ммоль, 1,5 экв.) в THF (2 мл) по каплям добавляли DIAD (307,49 мг, 1,52 ммоль, 1,5 экв.) при 25°C в атмосфере N2. Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 78 (1,13 г, неочищенное) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,45-7,44 (м, 3H), 7,39-7,37 (м, 2H), 6,83-6,81 (м, 2H), 4,08-3,97 (м, 2H), 3,77-3,76 (м, 2H), 3,45-3,44 (м, 1H), 3,29-3,10 (м, 3H), 2,02 (с, 6H), 0,87 (с, 2H).
Синтез E24: (4S,8R,9S,10S)-10-[(диметиламино)метил]-4-гидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E24)
К раствору промежуточного соединения 78 (40,00 мг, 77,30 мкмоль, 1 экв.) и этинилбензола (23,68 мг, 231,90 мкмоль, 25,47 мкл, 3,00 экв.) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (6,54 мг, 7,73 мкмоль, 0,1 экв.) и Cs2CO3 (50,37 мг, 154,60 мкмоль, 2 экв.). Полученную реакционную смесь перемешивали при 70°C в течение 2 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением (4S,8R,9S,10S)-10-[(диметиламино)метил]-4-гидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамида (6,10 мг, 14,65% выход) в виде твердого вещества белого цвета. HRMS (ESI): вычисл. для C33H38N4O3 [M+H]+ 539,29, найденное 539,4 1H ЯМР (400 МГц, MeOD-d4) δ7,55-7,50 (м, 2H), 7,49-7,37 (м, 4H), 6,86-6,84 (м, 2H), 4,18-4,05 (м, 1H), 4,02-4,00 (м, 1H), 3,75 (с, 3H), 3,64-3,31 (м, 1H), 2,90-2,85 (м,3H), 2,43 (с, 6H), 2,35-2,34 (м, 1H), 2,34-2,06 (м, 1H).
Пример 25: (8R,9S,10S)-3,10-бис[(диметиламино)метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E25”)


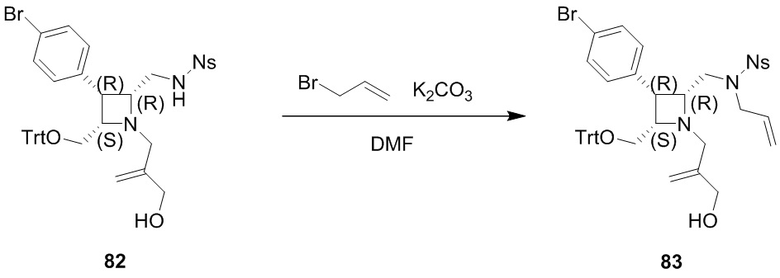

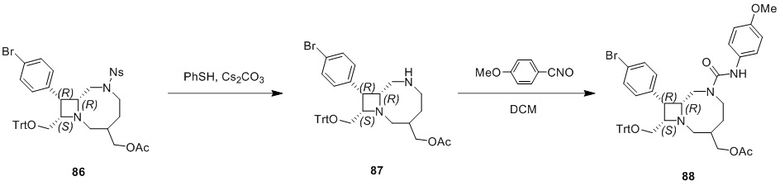





Промежуточное соединение 79: трет-бутил N-[[(2R,3R,4S)-1-аллил-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-2-ил]метил]-N- (2-нитрофенил)сульфонилкарбамат
К раствору N-(((2R,3R,4S)-3-(4-бромфенил)-4-((тритилокси)метил)азетидин-2-ил)метил)-2-нитробензолсульфонамида (WO 2015070204) (8,50 г, 11,51 ммоль, 1 экв.), DMAP (140,58 мг, 1,15 ммоль, 0,1 экв.) и Et3N (3,49 г, 34,52 ммоль, 4,79 мл, 3 экв.) в DCM (60 мл) добавляли BOC2O (2,76 г, 12,66 ммоль, 1,1 экв.), и реакционную смесь перемешивали при 25°C в течение 4 часов. Анализ ТСХ (петролейный эфир:DCM=3:1) показал завершение реакции, реакционную смесь гасили добавлением H2O (100 мл) и экстрагировали DCM (300 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали хроматографией на силикагеле (петролейный эфир:этилацетат=3:1) с получением промежуточного соединения 79 (8,19 г, 84,83% выход) в виде твердого вещества коричневого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ8,20 (д, J=4,4 Гц, 1H), 7,70 (с, 3H), 7,35-7,32 (м, 5H), 7,19-7,13 (м, 10H), 7,11 (м, 7H), 5,73-5,63 (м, 1H), 5,09-5,05 (м, 1H), 4,90-4,87 (м, 1H), 3,95-3,91 (м, 1H), 3,65-3,64 (м, 3H), 3,54-3,49 (м, 1H), 3,09-3,04 (м, 1H), 2,85-2,81 (м, 1H), 2,56-2,53 (м, 1H), 3,06-3,05 (м, 1H), 2,94-2,89 (м, 1H).
Промежуточное соединение 80: трет-бутил N-[[(2R,3R,4S)-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-2-ил]метил]-N-(2- нитрофенил)сульфонилкарбамат
К раствору промежуточного соединения 79 (7,70 г, 9,18 ммоль, 1 экв.) в EtOH (10 мл) добавляли 1,3-диметилгексагидропиримидин-2,4,6-трион (2,15 г, 13,77 ммоль, 1,5 экв.) и Pd(PPh3)4 (1,06 г, 917,98 мкмоль, 0,1 экв.), и реакционную смесь перемешивали при 40°C в течение 2 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением 10% NaOH (20 мл) и экстрагировали DCM (500 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=3:1) с получением промежуточного соединения 80 (6,44 г, 8,06 ммоль, 87,83% выход) в виде твердого вещества светло-коричневого вещества. 1H ЯМР (400 МГц, CDCl3-d1) δ8,24-8,22 (м, 1H), 7,72-7,65 (м, 3H), 7,33 (с, 4H), 7,22-7,19 (м, 16H), 7,42-7,34 (м, 2H), 3,95-3,89 (м, 1H), 3,06-3,04 (м, 1H), 3,02-2,95 (м, 1H), 2,05 (с, 1H), 1,03 (с, 9H).
Промежуточное соединение 81: трет-бутил N-[[(2R,3R,4S)-3-(4-бромфенил)-1-[2-(гидроксиметил)аллил]-4-(тритилоксиметил) азетидин-2-ил]метил]-N-(2-нитрофенил)сульфонилкарбамат
К раствору промежуточного соединения 80 (6,44 г, 8,06 ммоль, 1 экв.) и K2CO3 (3,34 г, 24,18 ммоль, 3 экв.) в DMF (60 мл) добавляли 2-(бромметил)проп-2-ен-1-ол (1,83 г, 12,09 ммоль, 1,5 экв.) при 25°C, и реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (50 мл) и экстрагировали DCM (100 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали хроматографией на силикагеле (петролейный эфир:этилацетат=2:1) с получением промежуточного соединения 81 (5,52 г, 78,83% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ8,20 (д, J=4,8 Гц, 2H), 8,02 (с, 1H), 7,72-7,67 (м, 3H), 7,42-7,40 (м, 2H), 7,30-7,26 (м, 2H), 7,18-7,16 (м, 10H), 7,08-7,07 (м, 7H), 4,80 (с, 1H), 4,65 (с, 1H), 4,06-3,98 (м, 3H), 3,84 (м, 1H), 3,65-3,62 (м, 2H), 3,55-3,51 (м, 2H), 3,03-3,00 (м, 2H), 1,30 (с, 9H).
Промежуточное соединение 82: N-[[(2R,3R,4S)-3-(4-бромфенил)-1-[2-(гидроксиметил)аллил]-4-(тритилоксиметил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамид
К раствору промежуточного соединения 81 (5,00 г, 5,75 ммоль, 1 экв.) в MeOH (50 мл) добавляли K2CO3 (3,97 г, 28,75 ммоль, 5 экв.), и реакционную смесь перемешивали при 80°C в течение 3 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (100 мл) и экстрагировали DCM (300 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали хроматографией на силикагеле (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 82 (2,50 г, 3,25 ммоль, 56,56% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ 7,82-7,78 (м, 2H), 7,71-7,69 (м, 1H), 7,18-7,13 (м, 18H), 5,10 (с, 1H), 5,01-4,97 (м, 2H), 4,12-4,07 (м, 2H), 3,70-3,58 (м, 3H), 3,34-3,23 (м, 1H), 3,07-3,06 (м, 1H), 2,93-2,89 (м, 2H).
Промежуточное соединение 83: N-аллил-N-[[(2R,3R,4S)-3-(4-бромфенил)-1-[2-(гидроксиметил)аллил]-4-(тритилоксиметил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамид
К раствору промежуточного соединения 82 (2,50 г, 3,25 ммоль, 1 экв.) в DMF (1,00 мл) добавляли 3-бромпроп-1-ен (590,18 мг, 4,88 ммоль, 1,5 экв.) и K2CO3 (1,35 г, 9,76 ммоль, 3 экв.) при 25°C, и реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (100 мл) и экстрагировали DCM (500 мл) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали хроматографией на силикагеле (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 83 (2,62 г, 99,68% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ8,02 (с, 1H), 7,66-7,53 (м, 1H), 7,37-7,35 (м, 3H), 7,15-7,11 (м, 19H), 5,43-5,36 (м, 1H), 5,00-4,98 (м, 1H), 4,88 (с, 1H), 4,80-4,76 (м, 2H), 4,04- (с, 2H), 3,82-3,79 (м, 2H), 3,52-3,32 (м, 3H), 3,16 (м, 1H), 3,06-3,02 (м, 2H), 2,96-2,88 (м, 2H).
Промежуточное соединение 84: [(3E,8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]дец-3-ен-3-ил]метанол
К раствору промежуточного соединения 83 (2,20 г, 2,72 ммоль, 1 экв.) в DCM (20 мл) добавляли [1,3-бис(2,4,6-триметилфенил)имидазолидин-2-илиден]-дихлор-[(2-изопропоксифенил)метилен]рутения (катализатор Ховейд-Граббса 2-го поколения) (426,12 мг, 680,04 мкмоль, 0,25 экв.), и реакционную смесь перемешивали при 40°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 84 (2,12 г, 100,00% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,89-7,87 (м, 1H), 7,70-7,61 (м, 3H), 7,26-7,22 (м, 17H), 7,11-7,08 (м, 2H), 5,68-5,64 (м, 1H), 4,15-4,05 (м, 1H), 3,99-3,64 (м, 2H), 3,53-3,52 (м, 1H), 3,31-3,22 (м, 2H), 3,12-3,09 (м, 2H), 2,85-2,84 (м, 1H), 2,17 (с, 1H).
Промежуточное соединение 85: [(8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]метанол
К раствору промежуточного соединения 84 (750,00 мг, 960,65 мкмоль, 1 экв.) и Et3N (1,94 г, 19,21 ммоль, 2,66 мл, 20 экв.) в THF (15 мл) добавляли 2-нитробензолсульфонгидразид (2,09 г, 9,61 ммоль, 10 экв.), и реакционную смесь перемешивали при 40°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (3 мл) и экстрагировали DCM (10 мл х 2) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 85 (1,50 г, 100,00% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,76-7,74 (д, J=5,6 Гц, 2H), 7,65-7,55 (м, 4H), 7,26-7,22 (м, 19H), 7,10-7,08 (д, J=8,4 Гц, 2H), 3,75-3,61 (м, 6H), 3,26-3,22 (м, 1H), 3,16-3,12 (м, 1H), 3,00-2,99 (м, 2H), 2,89-2,87 (м, 2H), 2,72-2,69 (м, 1H), 2,17 (с, 1H), 1,97-1,90 (м, 3H), 1,40-1,26 (м, 2H).
Промежуточное соединение 86: [(8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]метилацетат
К раствору промежуточного соединения 85 (1,49 г, 1,90 ммоль, 1 экв.), Et3N (576,78 мг, 5,70 ммоль, 3 экв.) и DMAP (23,21 мг, 190,00 мкмоль, 0,1 экв.) в DCM (15 мл) при 0°C добавляли уксусный ангидрид (387,94 мг, 3,80 ммоль, 355,91 мкл, 2 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 3 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (10 мл) и экстрагировали DCM (30 мл х 2) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 86 (1,51 г, 96,32% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,80-7,76 (м, 1H), 7,61-7,56 (м, 3H), 7,27-7,22 (м, 15H), 7,13-7,11 (д, J=8,4 Гц, 2H), 4,01-3,99 (м, 2H), 3,69-3,63 (м, 4H), 3,22-3,12 (м, 3H), 2,94-2,88 (м, 2H), 2,62-2,61 (м, 1H), 2,20-2,05 (м, 1H), 1,92 (с, 1H), 1,82 (с, 3H), 1,32-1,27 (м, 1H).
Промежуточное соединение 87: [(8R,9R,10S)-9-(4-бромфенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]метилацетат
К раствору промежуточного соединения 86 (1,51 г, 1,83 ммоль, 1. экв.) и бензолтиола (302,57 мг, 2,75 ммоль, 280,16 мкл, 1,5 экв.) в ацетонитриле (15 мл) по каплям добавляли Cs2CO3 (1,19 г, 3,66 ммоль, 2 экв.) при 25°C. Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (10 мл) и экстрагировали DCM (20 мл х 3) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1) с получением промежуточного соединения 87 (1,09 г, неочищенное) в виде твердого вещества белого цвета. HRMS (ESI): вычисл. для C37H39BrN2O3 [M+H]+ 639,21, найденное 639,1.
Промежуточное соединение 88: [(8R,9R,10S)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]метилацетат
К раствору промежуточного соединения 87 и Et3N (344,05 мг, 3,40 ммоль, 2 экв.) в DCM (10 мл) по каплям добавляли 1-изоцианат-4-метокси-бензол (278,91 мг, 1,87 ммоль, 1,1 экв.) при 0°C. Полученную реакционную смесь перемешивали при 25°C в течение 3 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (20 мл) и экстрагировали DCM (50 мл × 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 87 (1,10 г, 81,76% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,30-7,23 (м, 20H), 6,84-6,82 (м, 2H), 6,09 (с, 1H), 4,07-4,03 (м, 1H), 3,79-3,78 (м, 3H), 3,72 (м, 1H), 3,62-3,55 (м, 3H), 3,19-3,17 (м, 1H), 3,00-2,94 (м, 2H), 2,55-2,54 (м, 1H), 2,52-2,50 (м, 1H), 2,00 (с, 1H), 1,87 (с, 3H), 1,86-1,77 (м, 1H).
Промежуточное соединение 89: (8R,9R,10S)-9-(4-бромфенил)-3-(гидроксиметил)-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 88 (750,00 мг, 950,85 мкмоль, 1 экв.) в MeOH (7 мл) добавляли K2CO3 (394,25 мг, 2,85 ммоль, 3 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь фильтровали с получением фильтрата, который концентрировали, получая остаток. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 88 (757,00 мг, неочищенное) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,29-7,23 (м, 24H), 7,09-7,07 (м, 1H), 6,84-6,81 (м, 2H), 3,77 (с, 3H), 3,66-3,63 (м, 2H), 3,62 (м, 4H), 3,15-3,07 (м, 1H), 2,82-2,79 (м, 1H), 2,66-2,64 (м, 1H), 1,77 (с, 1H), 1,28-1,25 (с, 2H).
Промежуточное соединение 90: (8R,9R,10S)-9-(4-бромфенил)-3-[(1,3-диоксоизоиндолин-2-ил)метил]-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 89 (757,00 мг, 1,01 ммоль, 1 экв.), изоиндолин-1,3-диона (164,07 мг, 1,12 ммоль, 1,1 экв.) и PPh3 (398,85 мг, 1,52 ммоль, 1,5 экв.) в THF (2 мл) по каплям добавляли DIAD (307,49 мг, 1,52 ммоль, 1,5 экв.) при 25°C в атмосфере N2. Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 90 (1,13 г, неочищенное) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,70-7,68 (м, 3H), 7,68-7,65 (м, 8H), 7,49-7,47 (м, 3H), 7,22-7,17 (м, 3H), 6,74-6,72 (м, 2H), 5,99 (с, 1H), 3,84-3,77 (м, 2H), 3,75 (с, 3H), 3,64-3,60 (м, 4H), 3,54-3,50 (м, 3H), 3,16 (с, 1H), 3,05 (с, 1H), 2,94 (с, 1H), 2,64-2,62 (м, 1H), 1,88 (с, 1H).
Промежуточное соединение 91: (8R,9R,10S)-3-(аминометил)-9-(4-бромфенил)-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 90 (1,13 г, 1,29 ммоль, 1 экв.) в EtOH (2 мл) по каплям добавляли NH2NH2.H2O (98,86 мг, 1,94 ммоль, 98% чистоты, 1,5 экв.). Полученную реакционную смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (2 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=12:1) с получением промежуточного соединения 91 (586,00 мг, 60,91% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,21-7,14 (м, 20H), 7,12-7,09 (м, 3H), 6,81-6,78 (м, 2H), 3,76-3,70 (м, 5H), 3,59-3,51 (м, 5H), 3,17-3,14 (м, 1H), 3,00-2,99 (м, 1H), 2,77-2,72 (м, 1H), 2,68-2,46 (м, 3H), 1,91 (м, 2H), 1,69 (с, 1H), 1,63 (с, 1H).
Промежуточное соединение 92: (8R,9R,10S)-9-(4-бромфенил)-3-[(диметиламино)метил]-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 91 (586,00 мг, 785,80 мкмоль, 1 экв.) в DCM (6 мл) добавляли HCHO (637,77 мг, 7,86 ммоль, 585,11 мкл, 10 экв.) и MgSO4 (945,86 мг, 7,86 ммоль, 10 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 0,5 часа. К смеси добавляли CH3COOH (47,19 мг, 785,80 мкмоль, 1 экв.) и NaBH(OAc)3 (832,71 мг, 3,93 ммоль, 5 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 1,5 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (5 мл) и экстрагировали DCM (20 мл х 3) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=12:1) с получением промежуточного соединения 92 (370,00 мг, 60,85% выход) в виде светло-коричневого масла. 1H ЯМР (400 МГц, CDCl3-d1) δ7,34-7,29 (м, 2H), 7,76-7,22 (м, 10H), 7,14-7,12 (м, 2H), 6,83 (с, 1H), 6,83-6,79 (м, 2H), 3,76-3,67 (м, 4H), 3,63-3,60 (м, 4H), 3,15-3,14 (м, 1H), 2,97-2,96 (м, 1H), 2,73-2,64 (м, 3H), 2,73-2,65 (м, 1H), 2,02-1,88 (м, 3H).
Промежуточное соединение 93: (8R,9R,10S)-9-(4-бромфенил)-3-[(диметиламино)метил]-10-(гидроксиметил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 92 (370,00 мг, 478,16 мкмоль, 1 экв.) в DCM (1 мл) добавляли TFA (545,20 мг, 4,78 ммоль, 10 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением NaHCO3 (5 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан: метанол=8:1) с получением (172,00 мг, 67,68% выход) в виде твердого вещества светло-коричневого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,44 (д, J=8,0 Гц, 2H), 7,35 (д, J=8,0 Гц, 2H), 7,28-7,25 (м, 3H), 6,84 (д, J=8,8 Гц, 2H), 3,78-3,73 (м, 4H), 3,70-3,61 (м, 1H), 3,60-3,56 (м, 4H), 3,51-3,39 (м, 1H), 3,23-3,14 (м, 1H), 2,89-2,63 (м, 1H), 2,29 (м, 6H), 2,17-2,12 (м, 1H), 1,89 (с, 2H), 1,64-1,61 (м, 1H).
Промежуточное соединение 94: (8R,9S,10S)-9-(4-бромфенил)-3-[(диметиламино)метил]-10-[(1,3-диоксоизоиндолин-2-ил)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 93 (172,00 мг, 323,62 мкмоль, 1 экв.), изоиндолин-1,3-диона (52,38 мг, 355,98 мкмоль, 1,1 экв.) и PPh3 (127,32 мг, 485,43 мкмоль, 1,5 экв.) в THF (2 мл) по каплям добавляли DIAD (98,16 мг, 485,43 мкмоль, 1,5 экв.) при 25°C в атмосфере N2. Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=10:1) с получением промежуточного соединения 94 (152,00 мг, 71,10% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,81 (д, J=4,8 Гц, 2H), 7,70 (д, J=4,8 Гц, 2H), 7,51 (д, J=8,4 Гц, 2H), 7,44 (д, J=8,4 Гц, 2H), 7,26-7,23 (м, 3H), 6,82 (д, J=8,8 Гц, 2H), 3,92-3,89 (м, 1H), 3,76 (с, 3H), 3,70-3,68 (м, 2H), 3,66-3,64 (м, 2H), 3,57-3,53 (м, 4H), 2,79 (м, 1H), 2,67-2,61 (м, 1H), 2,39-2,38 (м, 1H), 2,36 (м, 3H), 2,16-1,99 (м, 1H), 1,26 (с, 1H).
Промежуточное соединение 95: (8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-3-[(диметиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 94 (152,00 мг, 230,09 мкмоль, 1 экв.) в EtOH (2 мл) по каплям добавляли NH2NH2.H2O (17,63 мг, 345,14 мкмоль, 17,12 мкл, 98% чистоты, 1,5 экв.). Полученную реакционную смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (2 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=5:1) с получением промежуточного соединения 95 (80,00 мг, 65,54% выход) в виде твердого вещества белого цвета. HRMS (ESI): вычисл. для C26H36BrN5O2 [M+H]+ 530,21, найденное 530,1.
Промежуточное соединение 96: (8R,9S,10S)-9-(4-бромфенил)-3,10-бис[(диметиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К перемешиваемому раствору промежуточного соединения 95 (120,00 мг, 226,20 мкмоль, 1 экв.) в DCM (2 мл) добавляли HCHO (183,43 мг, 2,26 ммоль, 168,28 мкл, 10 экв.) и MgSO4 (272,28 мг, 2,26 ммоль, 10 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 0,5 часа. К смеси добавляли CH3COOH (13,58 мг, 226,20 мкмоль, 1 экв.) и NaBH(OAc)3 (239,71 мг, 1,13 ммоль, 5 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 1,5 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (5 мл) и экстрагировали DCM (20 мл х 3) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=2:1) с получением (100,00 мг, 79,15% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,46 (д, J=8,4 Гц, 2H), 7,32 (д, J=8,4 Гц, 2H), 7,25 (д, J=12,8 Гц, 2H), 6,83 (д, J=8,4 Гц, 2H), 6,12 (с, 1H), 3,77 (с, 3H), 3,61-3,59 (м, 2H), 3,57-3,55 (м, 2H), 3,41-3,39 (м, 2H), 2,78-2,75 (м, 1H), 2,42-2,38 (м, 1H), 2,28-2,26 (м, 2H), 2,16 (с, 6H), 2,00 (м, 1H), 1,76 (м, 1H).
Синтез E25: (8R,9S,10S)-3,10-бис[(диметиламино)метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E25)
К раствору промежуточного соединения 96 (50,00 мг, 89,52 мкмоль, 1 экв.) и этинилбензола (27,43 мг, 268,55 мкмоль, 29,49 мкл, 3 экв.) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (7,58 мг, 8,95 мкмоль, 0,1 экв.) и Cs2CO3 (58,33 мг, 179,04 мкмоль, 2 экв.). Полученную реакционную смесь перемешивали при 70°C в течение 2 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (5 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением примера 25 (10,00 мг, 17,25 мкмоль, 19,27% выход) в виде твердого вещества белого цвета. HRMS (ESI): вычисл. для C36H45N5O2 [M+H]+ 580,36, найденное 580,5 1H ЯМР (400 МГц, MeOD-d4) δ7,56-7,51 (м, 6H), 7,38-7,37 (м, 2H), 7,37 (д, J=13,2 Гц, 2H), 6,85 (д, J=8,8 Гц, 2H), 4,02-3,98 (м, 2H), 3,853-3,82 (м, 2H), 3,75 (с, 3H), 3,54-3,51 (м, 1H), 2,99-2,91 (м,3H), 2,87 (м, 1H), 2,71 (с, 6H), 2,44 (с, 6H), 2,02 (м, 2H), 1,84-1,81 (м, 1H).
Пример 26: (8R,9S,10S)-10-[(диметиламино)метил]-3-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E26”)



Промежуточное соединение 97: [(8R,9R,10S)-9-(4-бромфенил)-10-(гидроксиметил)-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил]метилацетат
К перемешиваемому раствору промежуточного соединения 88 (180,00 мг, 228,20 мкмоль, 1 экв.) в DCM (3,6 мл) добавляли TFA (2,28 ммоль, 168,96 мкл, 10 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением насыщенного NaHCO3 (10 мл) и экстрагировали DCM (50 мл х 3) с получением органического слоя, и слой концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=12:1) с получением промежуточного соединения 97 (300,00 мг, неочищенное) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,47-7,45 (м, 1H), 7,38-7,32 (м, 2H), 7,26-7,22 (м, 3H), 6,84-6,82 (д, J=8,8 Гц, 2H), 6,08 (с, 1H), 4,24-4,20 (м, 1H), 4,11-4,07 (м, 1H), 3,80-3,77 (м, 4H), 3,65 (с, 4H), 3,55-3,54 (м, 2H), 3,48-3,43 (м,1H), 2,97 (с, 1H), 2,83 (с, 1H), 2,08-2,03 (м, 4H), 1,96-1,94 (м, 1H), 1,86-1,84 (м, 1H).
Промежуточное соединение 98: [(8R,9S,10S)-9-(4-бромфенил)-10-[(1,3-диоксоизоиндолин-2-ил)метил]-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил]метилацетат
К раствору промежуточного соединения 97 (300,00 мг, 549,00 мкмоль, 1 экв.), изоиндолин-1,3-диона (88,85 мг, 603,90 мкмоль, 1,1 экв.) и PPh3 (216,00 мг, 823,50 мкмоль, 1,5 экв.) в THF (2 мл) по каплям добавляли DIAD (166,52 мг, 823,50 мкмоль, 1,5 экв.) при 25°C. Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (петролейный эфир:этилацетат=1:1) с получением промежуточного соединения 98 (458,00 мг, неочищенное) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,80-7,70 (м, 2H), 7,68-7,65 (м, 12H), 7,49-7,46((м, 19H), 7,46-7,44 (м, 3H), 6,85-6,80 (м, 3H), 6,06 (с, 1H), 4,12-4,06 (м, 2H), 3,84-3,77 (м, 1H), 3,67-3,65 (м, 4H), 3,59-3,55 (м, 5H), 2,87-2,83 (м, 2H), 2,46-2,42 (м, 1H), 1,98 (с, 3H), 1,30-1,26 (м, 3H).
Промежуточное соединение 99: [(8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил]метилацетат
К раствору промежуточного соединения 98 (458,00 мг, 677,95 мкмоль, 1 экв.) в EtOH (2 мл) по каплям добавляли NH2NH2.H2O (51,95 мг, 1,02 ммоль, 98% чистоты, 1,5 экв.). Полученную реакционную смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (2 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=12:1) с получением промежуточного соединения 99 (180,00 мг, 48,67% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,47-7,45 (м, 3H), 7,38-7,34 (м, 2H), 7,24-7,22 (м, 2H), 6,84-6,81 (м, 2H), 6,07 (с, 1H), 4,20-4,18 (м, 1H), 4,10-4,09 (м, 1H), 3,77-3,62 (м, 5H), 3,60-3,57 (м, 3H), 3,45-3,33 (м, 2H), 3,35-3,32 (м, 2H), 2,95-2,92 (м, 1H), 2,77-2,76 (м, 2H), 2,55-2,54 (м, 1H), 2,03 (с, 3H), 1,94-1,74 (м, 2H), 1,27-1,24 (м, 2H).
Промежуточное соединение 100: [(8R,9S,10S)-9-(4-бромфенил)-10-[(диметиламино)метил]-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-3-ил]метилацетат
К перемешиваемому раствору промежуточного соединения 99 (180,00 мг, 329,99 мкмоль, 1 экв.) в DCM (2 мл) добавляли HCHO (267,83 мг, 3,30 ммоль, 245,71 мкл, 10 экв.) и MgSO4 (397,21 мг, 3,30 ммоль, 10 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 0,5 часа. К смеси добавляли CH3COOH (19,82 мг, 329,99 мкмоль, 1 экв.) и NaBH(OAc)3 (349,69 мг, 1,65 ммоль, 5 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 1,5 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (5 мл) и экстрагировали DCM (20 мл х 3) с получением органического слоя. Органический слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=12:1) с получением промежуточного соединения 100 (134,00 мг, 70,80% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3-d1) δ7,49-7,47 (м, 2H), 7,35-7,26 (м, 2H), 7,24-7,22 (м, 2H), 6,84-6,81 (м, 2H), 6,08 (с, 1H), 4,23-4,19 (м, 1H), 3,77 (с, 4H), 3,73-3,58 (м, 4H), 3,60-3,58 (м, 1H), 2,98-2,93(м, 1H), 2,95-2,93 (м, 1H), 2,76 (м, 2H), 2,56-2,51 (м, 2H), 2,05 (с, 6H), 1,92-1,88 (м, 2H).
Промежуточное соединение 101: [(8R,9S,10S)-10-[(диметиламино)метил]-6-[(4-метоксифенил)карбамоил]-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-3-ил]метилацетат
К раствору промежуточного соединения 100 (60,00 мг, 104,62 мкмоль, 1 экв.) и этинилбензола (32,05 мг, 313,85 мкмоль, 34,47 мкл, 3 экв.) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (8,86 мг, 10,46 мкмоль, 0,1 экв.) и Cs2CO3 (68,17 мг, 209,23 мкмоль, 2 экв.). Полученную реакционную смесь перемешивали при 70°C в течение 2 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=7:1) с получением промежуточного соединения 101 (42,00 мг, 67,50% выход) в виде твердого вещества коричневого цвета. HRMS (ESI): вычисл. для C36H42N4O4 [M+H]+ 595,32, найденное 565,2.
Синтез E26: (8R,9S,10S)-10-[(диметиламино)метил]-3-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E26)
К раствору промежуточного соединения 101 (42,00 мг, 70,62 мкмоль, 1 экв.) в MeOH (2 мл) добавляли K2CO3 (29,28 мг, 211,86 мкмоль, 3 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением H2O (5 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением соединения E26 (10,80 мг, 27,67% выход) в виде твердого вещества белого цвета. HRMS (ESI): вычисл. для C34H40N4O3 [M+H]+ 553,31, найденное 553,4. 1H ЯМР (400 МГц, MeOD-d4) δ7,57-7,51 (м, 6H), 7,39-7,38 (м, 3H), 6,85-6,83 (м, 2H), 3,91-3,82 (м, 1H), 3,76-3,71 (м, 1H), 3,71-3,69 (м, 4H), 3,76 (м, 2H), 3,03-2,94 (м, 3H), 2,59-2,47 (м, 4H), 1,95-1,92 (м, 1H), 1,91-1,82 (м, 2H).
Пример 27: (8R,9R,10S)-3-[(диметиламино)метил]-10-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E27”)

Промежуточное соединение 102: (8R,9R,10S)-3-[(диметиламино)метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 92 (30,00 мг, 38,77 мкмоль, 1 экв.) и этинилбензола (11,88 мг, 116,31 мкмоль, 12,77 мкл, 3 экв.) в ацетонитриле (1 мл) добавляли XPhos Pd G3 (3,28 мг, 3,88 мкмоль, 0,1 экв.) и Cs2CO3 (25,26 мг, 77,54 мкмоль, 2 экв.). Полученную реакционную смесь перемешивали при 70°C в течение 2 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (дихлорметан:метанол=7:1) с получением промежуточного соединения 102 (50,00 мг, неочищенное) в виде твердого вещества коричневого цвета. HRMS (ESI): вычисл. для C53H54N4O3 [M+H]+ 795,42, найденное 795,4
Синтез E27: (8R,9R,10S)-3-[(диметиламино)метил]-10-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E27)
К перемешиваемому раствору промежуточного соединения 102 (50,00 мг, 62,89 мкмоль, 1 экв.) в DCM (1 мл) добавляли TFA (71,71 мг, 628,91 мкмоль, 46,56 мкл, 10,00 экв.). Полученную реакционную смесь перемешивали при 25°C в течение 12 часов. Анализ ЖХ/МС показал завершение реакции, реакционную смесь гасили добавлением NaHCO3 (5 мл) и экстрагировали DCM (10 мл х 3) с получением органического слоя. Слой сушили над безводным Na2SO4 и концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением соединения E27 (4,70 мг, 13,52% выход) в виде твердого вещества белого цвета. HRMS (ESI): вычисл. для C33H37N4O2 [M+H]+ 553,29, найденное 553,4. 1H ЯМР (400 МГц, MeOD-d4) δ7,544-7,51 (м, 6H), 7,39-7,37 (м, 3H), 7,34-7,22 (м, 2H), 6,86-6,81 (м, 2H), 4,02-3,99 (м, 1H), 3,76-3,72 (м, 4H),3,70-3,66 (м, 1H), 3,41-3,40 (м, 1H), 3,38-3,35 (м, 1H), 3,12 (м, 1H), 2,85-2,75 (м, 1H), 2,16-2,09 (м, 2H), 1,81-1,77 (м, 1H).
Пример 28: (3R,8R,9R,10S)-3-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E28”)
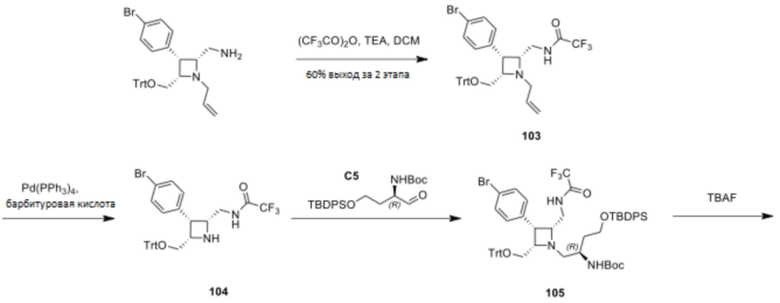




Промежуточное соединение 103: N-[[(2R,3R,4S)-1-аллил-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-2-ил]метил]-2,2,2-трифтор-ацетамид
К раствору [(2R,3R,4S)-1-аллил-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-2-ил]метанамин (J.O.C. (2012), 77(17), 7187-7211) (25,00 г, 31,62 ммоль, 1,00 экв.) в DCM (250,00 мл) добавляли TEA (25,59 г, 252,92 ммоль, 35,06 мл, 8,00 экв.), затем его охлаждали до 0°C. К реакционной смеси по каплям добавляли TFAA (26,56 г, 126,46 ммоль, 17,59 мл, 4,00 экв.). После перемешивания при 20°C в течение 16 часов реакционную смесь гасили добавлением раствора NaHCO3 (100 мл) и экстрагировали DCM (50 мл x 3), объединенные органические слои концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=20/1 до 1:1), получая промежуточное соединение 103 (11,00 г, 16,94 ммоль, 53,56% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,87-3,08 (м, 2 H) 3,09-3,24 (м, 2H) 3,30-3,51 (м, 3H) 3,57-3,69 (м, 2 H) 5,10 (ш.д, J=10,14 Гц, 1H) 5,18-5,33 (м, 1H) 5,80 (дд, J=17,28, 10,01, 7,44, 5,40 Гц, 1H) 6,11 (ш.с, 1H) 7,17-7,23 (м, 16H) 7,33 (д, J=8,38 Гц, 2H).
Промежуточное соединение 104: N-[[(2R,3R,4S)-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-2-ил]метил]-2,2,2-трифтор-ацетамид
К раствору промежуточного соединения 103 (11,00 г, 16,94 ммоль, 1,00 экв.) в EtOH (169,00 мл) добавляли 1,3-диметилбарбитуровую кислоту (3,97 г, 25,41 ммоль, 1,50 экв.) и Pd(PPh3)4 (1,96 г, 1,69 ммоль, 0,10 экв.) в атмосфере N2. Смесь перемешивали при 40°C в течение 16 часов. Реакционную смесь гасили добавлением раствора NaHCO3 (200 мл) и экстрагировали DCM (50 мл x 3), объединенный органический слой концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10/1 до 5:1), получая промежуточное соединение 104 (7,80 г, 12,80 ммоль, 75,55% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,26 (ш.с, 1H) 2,20-2,31 (м, 1H) 2,99-3,07 (м, 1H) 3,10-3,23 (м, 2H) 3,30-3,39 (м, 1H) 3,76 (ш.т, J=7,28 Гц, 1H) 4,27-4,49 (м, 2H) 6,10 (ш.с, 1H) 7,17-7,24 (м, 16H) 7,32-7,38 (м, 2H).
Промежуточное соединение 105: трет-бутил N-[(1R)-1-[[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2,2,2-трифторацетил)амино]метил]-4-(тритилоксиметил)азетидин-1-ил]метил]-3-[трет-бутил(дифенил)силил]окси-пропил]карбамат
К раствору промежуточного соединения 104 (4,00 г, 6,56 ммоль, 1,00 экв.), трет-бутил N-[(1R)-3-[трет-бутил(дифенил)силил]окси-1-формил-пропил]карбамата (синтезированный как описано в документе Bioorganic & Medicinal Chemistry (2006), 14(1), 214-236, включенном в настоящее описание во всей своей полноте в виде ссылки) (2,90 г, 6,56 ммоль, 1,00 экв.) в DCM (50,00 мл) добавляли MgSO4 (158,00 мг, 1,31 ммоль, 20,00 экв.) и NaBH(OAc)3 (139,10 мг, 656,30 мкмоль, 10,00 экв.). После перемешивания при 20°C в течение 16 часов, дополнительно добавляли трет-бутил N-[(1R)-3-[трет-бутил(дифенил)силил]окси-1-формил-пропил]карбамат (1 г). Спустя дополнительные 0,5 часа реакционную смесь гасили добавлением раствора NaHCO3 (200 мл) и экстрагировали DCM (50 мл x 3), объединенный органический слой концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=20/1 до 10:1) с получением промежуточного соединения 105 (4,20 г, 4,06 ммоль, 61,85% выход в виде смолы желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 1,01-1,05 (м, 9H) 1,41-1,47 (м, 10H) 1,63-1,78 (м, 1H) 1,82-1,92 (м, 1H) 2,65-2,76 (м, 1H) 2,82-3,01 (м, 2H) 3,13 (ш.дд, J=9,60, 6,09 Гц, 1H) 3,29-3,79 (м, 7H) 4,67 (ш.д, J=9,29 Гц, 1H) 7,11-7,25 (м, 17H) 7,31-7,50 (м, 8H) 7,63 (тд, J=7,37, 1,44 Гц, 4H) 8,40-8,73 (м, 1H).
Промежуточное соединение 106: трет-бутил N-[(1R)-1-[[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2,2,2-трифторацетил)амино]метил]-4-(тритилоксиметил)азетидин-1-ил]метил]-3-гидрокси-пропил]карбамат
К раствору промежуточного соединения 105 (4,20 г, 4,06 ммоль, 1,00 экв.) в THF (50,00 мл) добавляли TBAF (1,59 г, 6,09 ммоль, 1,50 экв.). После перемешивания при 20°C в течение 16 часов реакционную смесь концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10/1 до 2:1) с получением промежуточного соединения 106 (2,30 г, 2,89 ммоль, 71,23% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 1,41 (с, 3H) 1,45-1,53 (м, 9H) 1,58-1,77 (м, 2H) 2,49 (ш.дд, J=12,74, 4,33 Гц, 2H) 2,82 (т, J=12,17 Гц, 1H) 2,88-2,98 (м, 2H) 3,08-3,29 (м, 3H) 3,47 (тд, J=7,87, 3,20 Гц, 1H) 3,60-3,72 (м, 1H) 4,51 (ш.д, J=9,41 Гц, 1H) 7,12 (д, J=8,41 Гц, 1H) 7,09-7,14 (м, 1H) 7,16-7,26 (м, 17H) 7,34 (д, J=8,41 Гц, 1H) 8,13 (ш.с, 1H).
Промежуточное соединение 107: трет-бутил N-[(1R)-1-[[(2R,3R,4S)-3-(4-бромфенил)-2-[[(2,2,2-трифторацетил)амино]метил]-4-(тритилоксиметил)азетидин-1-ил]метил]-3-oxo-пропил]карбамат
К раствору промежуточного соединения 106 (2,30 г, 2,89 ммоль, 1,00 экв.) в DCM (30,00 мл) добавляли DMP (2,45 г, 5,78 ммоль, 1,79 мл, 2,00 экв.) при 0°C в атмосфере N2. Смесь перемешивали при 0°C в течение 1 часа. Анализ ЖХ/МС показал, что реагент 106 израсходован полностью, и основную массу составляет требуемый продукт, реакционную смесь гасили добавлением раствора NaHCO3 (2,5 г) и Na2SO3 (2,5 г) в H2O (20 мл) и экстрагировали DCM (20 мл x 3), и объединенный органический слой концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=2/1 до 0:1) с получением промежуточного соединения 107(1,80 г, 2,27 ммоль, 78,37% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 1,47 (с, 9H) 1,51-1,69 (м, 3H) 2,46-2,54 (м, 2H) 2,78-2,88 (м, 1H) 2,89-3,00 (м, 2H) 3,08-3,20 (м, 2H) 3,49-3,69 (м, 4H) 3,91-4,07 (м, 1H) 4,50-4,76 (м, 1H) 7,08-7,13 (м, 3H) 7,17-7,26 (м, 22H) 8,25 (ш.с, 1H) 9,63 (с, 1H).
Промежуточное соединение 108: трет-бутил N-[(3R,5Z,8R,9R,10S)-9-(4-бромфенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]дец-5-ен-3-ил]карбамат
К раствору промежуточного соединения 107 (1,70 г, 2,14 ммоль, 1,00 экв.) в MeOH (20,00 мл) и H2O (77,13 мг, 4,28 ммоль, 77,13 мкл, 2,00 экв.) добавляли K2CO3 (591,32 мг, 4,28 ммоль, 2,00 экв.). После перемешивания при 20°C в течение 16 часов, дополнительно добавляли K2CO3(200 мг). Спустя дополнительные 2 часа реакционную смесь разбавляли водой (30 мл), экстрагировали DCM (20 мл x 3), органический слой сушили над Na2SO4, затем концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=5/1 до 0:1) с получением промежуточного соединения 108 (750,00 мг, 771,30 мкмоль, 45,05% выход), 70% чистоты) в виде твердого вещества желтого цвета.
Промежуточное соединение 109: трет-бутил N-[(3R,8R,9R,10S)-9-(4-бромфенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]карбамат
К раствору промежуточного соединения 108 (750,00 мг, 1,10 ммоль, 1,00 экв.) в DCM (18,00 мл) добавляли AcOH (66,17 мг, 1,10 ммоль, 63,02 мкл, 1,00 экв.) и NaBH(OAc)3 (2,34 г, 11,02 ммоль, 10,00 экв.). Смесь перемешивали при 20°C в течение 0,5 часа. Анализ ЖХ/МС показал, что реагент 108 полностью израсходован, и основную массу составляет требуемый продукт, реакционную смесь гасили добавлением раствора NaHCO3 (20 мл) и экстрагировали DCM (10 мл x 3), объединенный органический слой концентрировали с получением промежуточного соединения 109 в виде твердого вещества желтого цвета (750 мг, неочищенное), которое использовали на следующем этапе без очистки.
Промежуточное соединение 110: трет-бутил N-[(3R,8R,9R,10S)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-3-ил]карбамат
К раствору промежуточного соединения 109 (750,00 мг, 769,02 мкмоль, 1,00 экв.) в DCM (10,00 мл) добавляли TEA (38,91 мг, 384,51 мкмоль, 53,30 мкл, 0,50 экв.) и 1-изоцианат-4-метокси-бензол (137,64 мг, 922,82 мкмоль, 118,66 мкл, 1,20 экв.). Смесь перемешивали при 20°C в течение 0,5 часа. Анализ ЖХ/МС показал, что реагент 109 полностью израсходован, и основную массу составляет требуемый продукт. Реакционную смесь концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат), дополнительно очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил). Промежуточное соединение 110 (150,00 мг, 180,33 мкмоль, 23,45% выход) получали в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 1,45 (ш.д, J=6,15 Гц, 9H) 2,18 (ш.с, 2H) 3,20-3,68 (м, 3H) 3,72-4,51 (м, 11H) 5,14-5,42 (м, 1H) 6,78 (ш.с, 1H) 6,88 (ш.т, J=7,97 Гц, 2H) 7,22-7,38 (м, 20H) 7,47-7,53 (м, 1H).
Промежуточное соединение 111: (3R,8R,9R,10S)-3-амино-9-(4-бромфенил)-10-(гидроксиметил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 110 (120,00 мг, 144,26 мкмоль, 1,00 экв.) в DCM (3,00 мл) добавляли TFA (1,54 г, 13,51 ммоль, 1,00 мл, 93,63 экв.) при 0°C. Смесь перемешивали при 20°C в течение 1 часа. Анализ ЖХ/МС показал, что 110 полностью израсходован, и основную массу составляет требуемый продукт, реакционную смесь гасили добавлением раствора NaHCO3 (20 мл) и затем промывали NH3.H2O (1 мл), экстрагировали DCM (10 мл x 3), и объединенный органический слой концентрировали с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) с получением промежуточного соединения 111 (60,00 мг, 122,60 мкмоль, 84,98% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 112: (3R,8R,9R,10S)-9-(4-бромфенил)-3-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 111 (60,00 мг, 122,60 мкмоль, 1,00 экв.), HCHO (99,50 мг, 1,23 ммоль, 91,28 мкл, 10,00 экв.) в DCM (3,00 мл) добавляли MgSO4 (147,57 мг, 1,23 ммоль, 10,00 экв.) и NaBH(OAc)3 (259,84 мг, 1,23 ммоль, 10,00 экв.) при 20°C. Смесь перемешивали при 20°C в течение 1 часа. Анализ ЖХ/МС показал, что реагент 111 полностью израсходован, и основную массу составляет требуемый продукт, реакционную смесь гасили добавлением раствора NaHCO3 (10 мл) и экстрагировали DCM (5 мл x 3), и объединенный органический слой концентрировали с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) с получением промежуточного соединения 112 (38,00 мг, 73,44 мкмоль, 59,90% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 1,84-1,99 (м, 1H) 2,49 (ш.с, 6H) 2,55 (ш.д, J=9,26 Гц, 2H) 2,88 (ш.д, J=11,69 Гц, 1H) 3,02-3,16 (м, 3H) 3,41 (ш.дд, J=11,58, 4,52 Гц, 1H) 3,51-3,61 (м, 3H) 3,62-3,86 (м, 6H) 3,98-4,17 (м, 1H) 4,73 (с, 1H) 6,78-6,91 (м, 2H) 7,23 (д, J=8,82 Гц, 2H) 7,34-7,53 (м, 4H).
Синтез E28: (3R,8R,9R,10S)-3-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E28)
К раствору промежуточного соединения 112 (25,00 мг, 48,31 мкмоль, 1,00 экв.), этинилбензола (14,80 мг, 144,93 мкмоль, 15,91 мкл, 3,00 экв.) в CH3CN (350,00 мкл) добавляли Cs2CO3 (47,22 мг, 144,93 мкмоль, 3,00 экв.) и XPhos Pd G3 (4,09 мг, 4,83 мкмоль, 0,10 экв.) в атмосфере N2. Смесь перемешивали при 70°C в течение 3 часов. Анализ ЖХ/МС показал, что реагент 1 полностью израсходован, и основную массу составляет требуемый продукт, реакционную смесь разбавляли водой (2 мл) и экстрагировали DCM (2 мл x 3), органический слой сушили над Na2SO4, затем концентрировали с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM: MeOH=10:1), затем дополнительно очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением соединения E28 (13,00 мг, 24,13 мкмоль, 49,95% выход в виде соли муравьиной кислоты) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 1,88 (ш.д, J=7,65 Гц, 1H) 2,38 (с, 7H) 2,49 (ш.дд, J=13,30, 9,29 Гц, 3H) 2,71 (ш.с, 1H) 2,79-2,96 (м, 1H) 3,09 (ш.д, J=15,94 Гц, 1H) 3,25 (ш.дд, J=13,30, 5,02 Гц, 1H) 3,41-3,73 (м, 6H) 3,78 (с, 3H) 3,90-4,03 (м, 1H) 6,84 (д, J=8,91 Гц, 2H) 7,21 (д, J=8,91 Гц, 2H) 7,32-7,40 (м, 3H) 7,46-7,57 (м, 6H).
Пример 29: (3S,8R,9R,10S)-3-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E29”)
Синтез выполняли аналогично описанному в примере 28 после синтеза промежуточного соединения 105 (промежуточное соединение 105), используя вместо промежуточного соединения 105 энантиомерный трет-бутил N-[(1S)-3-[трет-бутил(дифенил)силил]окси-1-формил-пропил]карбамат (Bioorganic & Medicinal Chemistry (2006)). 1H ЯМР (400 МГц, хлороформ-d) δ=7,64-7,46 (м, 6H), 7,44-7,34 (м, 3H), 7,33-7,29 (м, 1H), 7,29-7,25 (м, 2H), 6,85 (д, J=8,9 Гц, 2H), 6,29 (ш.с, 1H), 3,86 (ш.д, J=10,8 Гц, 1H), 3,80 (с, 3H), 3,78-3,72 (м, 2H), 3,72-3,65 (м, 1H), 3,65-3,54 (м, 4H), 3,21-3,11 (м, 1H), 2,99-2,87 (м, 1H), 2,72-2,60 (м, 2H), 2,37 (с, 6H), 2,15 (ш.с, 1H), 2,01 (ш.д, J=7,9 Гц, 1H).
Пример 30: (4R,8R,9R,10S)-4-(диметиламино)-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E30”)
Синтез выполняли как описано в примере 3, используя в качестве исходного материала энантиомерный (R)-трет-бутил (1-((трет-бутилдифенилсилил)окси)-4-оксобутан-2-ил)карбамат (US 20150266867). 1H ЯМР (400 МГц, хлороформ-d) δ=7,59-7,54 (м, 2H), 7,52-7,45 (м, 4H), 7,41-7,30 (м, 5H), 6,89-6,80 (м, 2H), 3,80 (с, 3H), 3,77-3,58 (м, 5H), 3,57 (ш.д, J=8,2 Гц, 1H), 3,51-3,43 (м, 2H), 3,29 (ш.д, J=13,6 Гц, 1H), 3,02 (ш.с, 1H), 2,67-2,44 (м, 2H), 2,34 (с, 6H), 1,81 (ш.с, 1H), 1,66 (ш.с, 1H).
Пример 31: (8R,9R,10S)-4-((диметиламино)метил)-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E31”)

Промежуточное соединение 113: N-[[(2R,3R,4S)-1-аллил-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-2-ил]метил]-N-[2-(гидроксиметил)аллил]-2-нитро-бензолсульфонамид
К раствору N-[[(2R,3R,4S)-1-аллил-3-(4-бромфенил)-4-(тритилоксиметил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамида (WO2015070204) (5,00 г, 6,77 ммоль, 1,00 экв.) в DMF (62,5 мл) добавляли K2CO3 (2,11 г, 15,23 ммоль, 2,25 экв.), и раствор охлаждали до 0°C, добавляя при этом 2-(бромметил)проп-2-ен-1-ола (2,04 г, 13,54 ммоль, 2,00экв.) в DMF (7,5 мл). Смесь перемешивали при 20°C в течение 16 часов. Анализ ЖХ/МС показал, что реагент 113 полностью израсходован, и детектировали требуемый МС. Реакционную смесь гасили добавлением воды (300 мл) и экстрагировали этилацетатом (100мл х 2). Объединенные органические слои промывали солевым раствором (100 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=20:1 до 2:1) с получением промежуточного соединения 113 (5,00 г, 6,18 ммоль, 91,32% выход) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, хлороформ-d) δ=8,00 (с, 1H), 7,68-7,62 (м, 1H), 7,60-7,46 (м, 3H), 7,30-7,23 (м, 3H), 7,19-7,12 (м, 10H), 7,07 (дд, J=2,9, 6,8 Гц, 6H), 5,57 (тдд, J=6,6, 10,4, 17,0 Гц, 1H), 5,10-5,00 (м, 2H), 4,89 (д, J=10,1 Гц, 1H), 4,69 (с, 1H), 3,91 (д, J=8,8 Гц, 3H), 3,70-3,61 (м, 1H), 3,54-3,38 (м, 3H), 3,26-3,14 (м, 3H), 2,98 (дд, J=4,4, 9,3 Гц, 1H), 2,85-2,73 (м, 2H).
Промежуточное соединение 114: [(3E,8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]дец-3-ен-4-ил]метанол
К раствору промежуточного соединения 113 (5,00 г, 6,18 ммоль, 1,00 экв.) в DCM (500,00 мл) добавляли катализатор Ховейд-Граббса 2-го поколения (774,77 мг, 1,24 ммоль, 0,20 экв.). Смесь перемешивали при 55°C в течение 2 часов. Анализ ТСХ (петролейный эфир:этилацетат=1:1) показал, что промежуточное соединение 113 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=20:1 до 1:1) с получением промежуточного соединения 114 (4,00 г, 5,12 ммоль, 82,90% выход) в виде твердого вещества коричневого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=7,88-7,83 (м, 1H), 7,64-7,51 (м, 3H), 7,18 (д, J=6,0 Гц, 2H), 7,13 (с, 14H), 7,06 (д, J=8,5 Гц, 2H), 5,81 (т, J=6,3 Гц, 1H), 4,07-4,02 (м, 3H), 3,96-3,88 (м, 1H), 3,59-3,32 (м, 6H), 3,19 (д, J=14,1 Гц, 1H), 3,09-2,93 (м, 3H), 2,83-2,79 (м, 1H).
Промежуточное соединение 115: [(8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]метанол
К раствору промежуточного соединения 114 (4,00 г, 5,12 ммоль, 1,00 экв.) в THF (51,00 мл) добавляли 2-нитробензолсульфонгидразид (5,56 г, 25,60 ммоль, 4,00экв.) и TEA (5,18 г, 51,23 ммоль, 7,10 мл, 10,00 экв.). После перемешивания при 40°C в течение 16 часов реакционную смесь гасили добавлением воды (100 мл) и экстрагировали этилацетатом (50 мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10/1 до 1:1) с получением промежуточного соединения 115 (3,60 г, неочищенное) в виде твердого вещества белого цвета.
Промежуточное соединение 116: [(8R,9R,10S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]метил ацетат
К раствору промежуточного соединения 115 (3,60 г, 4,60 ммоль, 1,00 экв.) в DCM (80,00 мл) добавляли DMAP (56,19 мг, 460,00 мкмоль, 0,1 экв.) и TEA (465,40 мг, 4,60 ммоль, 637,53 мкл, 1,00 экв.), затем Ac2O (939,07 мг, 9,20 ммоль, 861,53 мкл, 2,00 экв.) при 0°C. После перемешивания при 20°C в течение 2 часов реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (150 мл) и экстрагировали DCM (50мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=30/1 до 1:1) с получением промежуточного соединения 116 (3,00 г, 3,64 ммоль, 79,07% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 117: [(8R,9R,10S)-9-(4-бромфенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]метилацетат
К раствору промежуточного соединения 116 6 (3,00 г, 3,64 ммоль, 1,00 экв.) в ацетонитриле (80,00 мл) добавляли Cs2CO3 (1,42 г, 4,37 ммоль, 1,20 экв.) и бензолтиол (“PhSH”) (601,14 мг, 5,46 ммоль, 1,50 экв.) при 0°C. После перемешивания при 20°C в течение 16 часов реакционную смесь гасили добавлением воды (100 мл) и экстрагировали этилацетатом (30 мл х 2). Объединенные органические слои промывали солевым раствором (50 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10:1 до 1:1) с получением промежуточного соединения 117 (2,00 г, 3,13 ммоль, 85,90% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 118: [(8R,9R,10S)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-4-ил]метилацетат.
К раствору промежуточного соединения 117 (2,00 г, 3,13 ммоль, 1,00 экв.) в DCM (50,00 мл) добавляли 1-изоцианат-4-метокси-бензол (560,21 мг, 3,76 ммоль, 1,20 экв.) при 0°C. Смесь перемешивали при 15°C в течение 2 часов. Анализ ЖХ/МС показал, что промежуточное соединение 117 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь гасили добавлением воды (100 мл) и экстрагировали DCM (30мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM: MeOH=100:1 до 20:1) с получением промежуточного соединения 118 (2,20 г, 2,79 ммоль, 89,11% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=7,28-7,16 (м, 23H), 6,83 (д, J=9,0 Гц, 2H), 4,01-3,95 (м, 1H), 3,92-3,85 (м, 1H), 3,80-3,73 (м, 5H), 3,63-3,54 (м, 2H), 3,42 (т, J=8,0 Гц, 1H), 3,20-2,90 (м, 5H), 2,74 (дд, J=11,0, 14,1 Гц, 1H), 2,50-2,40 (м, 1H), 2,06 (с, 3H), 1,66 (дд, J=6,8, 13,8 Гц, 1H), 1,53-1,45 (м, 1H).
Промежуточное соединение 119: (8R,9R,10S)-9-(4-бромфенил)-4-(гидроксиметил)-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 118 (200,00 мг, 253,56 мкмоль, 1,00 экв.) в DCM (1,00 мл) и MeOH (1 мл) добавляли K2CO3 (175,22 мг, 1,27 ммоль, 5,00 экв.). Смесь перемешивали при 15°C в течение 4 часов. Анализ ЖХ/МС показал, что промежуточное соединение 118 полностью израсходовано, и детектировали один основной пик с требуемым МС. Реакционную смесь гасили добавлением воды (10 мл) и экстрагировали DCM (5 мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10:1 до 1:1) с получением промежуточного соединения 119 (150,00 мг, 200,88 мкмоль, 79,22% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=7,37 (д, J=8,5 Гц, 2H), 7,27 (с, 4H), 7,22 (с, 15H), 6,80 (д, J=8,5 Гц, 2H), 3,90 (д, J=16,1 Гц, 2H), 3,77 (с, 3H), 3,70-3,57 (м, 3H), 3,43-3,32 (м, 2H), 3,19-3,07 (м, 2H), 3,05-2,94 (м, 2H), 2,75 (д, J=11,5 Гц, 1H), 2,47 (т, J=12,0 Гц, 1H), 1,89 (ш.с, 1H), 1,52-1,38 (м, 2H).
Промежуточное соединение 120: (8R,9R,10S)-9-(4-бромфенил)-4-[(1,3-диоксоизоиндолин-2-ил)метил]-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид.
К раствору PPh3 (0,15 г) в 3 мл THF добавляли DIAD (0,11 мл) при 0°C. Через 5 минут эту смесь добавляли к раствору промежуточного соединения 119 (20,00 мг, 26,78 мкмоль, 1,00 экв.) и изоиндолин-1,3-диона (5,91 мг, 40,17 мкмоль, 1,50 экв.) в THF (1 мл) при 0°C. Смесь перемешивали при 15°C в течение 16 часов. Анализ ЖХ/МС показал, что промежуточное соединение 9 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1:1) с получением промежуточного соединения 120 (10,00 мг, 11,42 мкмоль, 42,63% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=7,87 (дд, J=3,0, 5,5 Гц, 1H), 7,75 (дд, J=3,3, 5,3 Гц, 2H), 7,32-7,28 (м, 7H), 7,27-7,20 (м, 15H), 6,80 (д, J=9,0 Гц, 2H), 6,17 (с, 1H), 3,83-3,75 (м, 3H), 3,74-3,56 (м, 5H), 3,52 (д, J=7,5 Гц, 1H), 3,24-3,12 (м, 3H), 3,03-2,96 (м, 1H), 2,83-2,74 (м, 1H), 2,45-2,30 (м, 2H), 1,83 (д, J=7,5 Гц, 1H).
Промежуточное соединение 121: (8R,9R,10S)-4-(аминометил)-9-(4-бромфенил)-N-(4-метоксифенил)-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 120 (500,00 мг, 570,87 мкмоль, 1,00 экв.) в EtOH (10,00 мл) добавляли N2H4.H2O (28,58 мг, 570,87 мкмоль, 27,75 мкл, 1,00 экв.). Смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал, что промежуточное соединение 120 израсходовано, и детектировали требуемый МС. Реакционную смесь гасили добавлением воды (30 мл) и экстрагировали DCM (10 мл х 2). Объединенные органические слои промывали солевым раствором (10 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM:MeOH=100:1 до 10:1) с получением промежуточного соединения 121 (150,00 мг, неочищенное) в виде твердого вещества желтого цвета, содержащего Ph3PO. 1H ЯМР (400 МГц, хлороформ-d) δ=7,64-7,56 (м, 4H), 7,51-7,45 (м, 3H), 7,39 (дт, J=2,8, 7,4 Гц, 5H), 7,14 (с, 13H), 6,71 (д, J=9,0 Гц, 2H), 3,79-3,62 (м, 4H), 3,56-3,46 (м, 2H), 3,31 (ш.с, 1H), 3,11-2,85 (м, 5H), 2,74 (д, J=12,5 Гц, 1H), 2,44-2,18 (м, 2H), 1,62-1,39 (м, 3H).
Промежуточное соединение 122: (8R,9R,10S)-9-(4-бромфенил)-4-((диметиламино)метил)-N-(4-метоксифенил)-10-((тритилокси)метил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 121 (150,00 мг, 201,14 мкмоль, 1,00 экв.) в DCM (5,00 мл) добавляли MgSO4 (290,53 мг, 2,41 ммоль, 12,00 экв.) и HCHO (98,21 мг, 1,21 ммоль, 90,10 мкл, 37% чистоты, 6,02 экв.) затем NaBH(OAc)3 (639,44 мг, 3,02 ммоль, 15,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Анализ ЖХ/МС показал, что промежуточное соединение 121 израсходовано, и детектировали один требуемый MS. Реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (20 мл) и экстрагировали DCM (5 мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=20:1) с получением промежуточного соединения 122 (60,00 мг, 77,54 мкмоль, 38,55% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=7,24-7,08 (м, 24H), 6,72 (д, J=9,0 Гц, 2H), 3,88 (д, J=14,1 Гц, 1H), 3,67 (с, 3H), 3,57-3,48 (м, 3H), 3,33-3,25 (м, 1H), 3,10-2,86 (м, 5H), 2,64 (дд, J=10,8, 14,3 Гц, 1H), 2,44-2,36 (м, 1H), 2,21 (с, 6H), 1,90 (д, J=12,0 Гц, 1H), 1,51 (ш.с, 2H), 1,36 (д, J=4,5 Гц, 2H).
Промежуточное соединение 123: (8R,9R,10S)-4-[(диметиламино)метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-10-(тритилоксиметил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 122 (60,00 мг, 77,54 мкмоль, 1,00 экв.) в ацетонитриле (1,00 мл) добавляли этинилбензол (23,76 мг, 232,62 мкмоль, 25,55 мкл, 3,00 экв.), Cs2CO3 (101,06 мг, 310,16 мкмоль, 4,00 экв.) и Xphos Pd G3 (6,56 мг, 7,75 мкмоль, 0,1 экв.). Смесь перемешивали при 70°C в течение 2 часов. Анализ ЖХ/МС показал, что реагент полностью израсходован, и детектировали требуемый МС. Реакционную смесь гасили добавлением воды (10 мл) и экстрагировали DCM (5 мл х 2). Объединенные органические слои промывали солевым раствором (5 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=20:1) с получением промежуточного соединения 123 (50,00 мг, 62,89 мкмоль, 81,11% выход) в виде твердого вещества желтого цвета.
Синтез E31: (8R,9R,10S)-4-((диметиламино)метил)-10-(гидроксиметил)-N-(4-метоксифенил)-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E31)
К раствору промежуточного соединения 123 (50,00 мг, 62,89 мкмоль, 1,00 экв.) в DCM (1,00 мл) добавляли TFA (71,71 мг, 628,90 мкмоль, 46,56 мкл, 10,00 экв.). Смесь перемешивали при 25°C в течение 1 часа. Анализ ЖХ/МС показал, что промежуточное соединение 123 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (10 мл) и экстрагировали DCM (5 мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) с получением соединения E31 (4,50 мг, 8,14 мкмоль, 12,95% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=7,54 (дд, J=2,0, 7,5 Гц, 2H), 7,48 (с, 4H), 7,37-7,29 (м, 5H), 6,82 (д, J=9,0 Гц, 2H), 4,05 (д, J=14,1 Гц, 1H), 3,78 (с, 3H), 3,73-3,62 (м, 4H), 3,60-3,52 (м, 2H), 3,46 (т, J=8,5 Гц, 1H), 3,15 (дд, J=5,3, 16,3 Гц, 2H), 2,86 (дд, J=10,5, 14,6 Гц, 1H), 2,58 (ш.с, 1H), 2,32 (с, 6H), 2,25 (т, J=12,3 Гц, 2H), 2,03 (д, J=12,5 Гц, 1H), 1,59 (ш.с, 2H), 1,53 (ш.с, 1H)
Пример 32: (8R,9S,10S)-10-[(диметиламино)метил]-4-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E32”)

Промежуточное соединение 124: (8R,9R,10S)-9-(4-бромфенил)-4-[(диметиламино)метил]-10-(гидроксиметил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 122 (400,00 мг, 516,93 мкмоль, 1,00 экв.) в DCM (10,00 мл) добавляли TFA (589,40 мг, 5,17 ммоль, 382,73 мкл, 10,00 экв.). Смесь перемешивали при 25°C в течение 1 часа. Анализ ЖХ/МС показал, что промежуточное соединение 122 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (10 мл) и экстрагировали DCM (5 мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM:MeOH=100:1 до 10:1) с получением промежуточного соединения 124 (130,00 мг, неочищенное) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=7,47 (д, J=8,8 Гц, 1H), 7,39-7,32 (м, 3H), 7,32-7,26 (м, 9H), 7,23 (д, J=9,3 Гц, 3H), 6,91 (д, J=8,4 Гц, 1H), 3,93 (д, J=14,1 Гц, 1H), 3,69 (с, 3H), 3,60-3,52 (м, 4H), 3,04 (д, J=14,1 Гц, 2H), 2,75-2,63 (м, 2H), 2,23 (с, 6H), 2,15 (д, J=11,9 Гц, 1H), 1,65-1,46 (м, 4H).
Промежуточное соединение 125: (8R,9S,10S)-9-(4-бромфенил)-4-[(диметиламино)метил]-10-[(1,3-диоксоизоиндолин-2-ил)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору PPh3 (0,15 г) в 3 мл THF добавляли DIAD (0,11 мл) при 0°C. Эту смесь (1,16 мл) добавляли к раствору промежуточного соединения 124 (30,00 мг, 56,45 мкмоль, 1,00 экв.) и изоиндолин-1,3-диона (12,46 мг, 84,67 мкмоль, 1,50 экв.) в THF (1,00 мл) при 0°C. Смесь перемешивали при 25°C в течение 16 часов. Анализ ЖХ/МС показал, что промежуточное соединение 124 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM: MeOH=20:1) с получением промежуточного соединения 125 (30,00 мг, неочищенное) в виде твердого вещества белого цвета, содержащего Ph3PO.
Промежуточное соединение 126: (8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-4-[(диметиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 125 (100,00 мг, 151,38 мкмоль, 1,00 экв.) в EtOH (2,00 мл) добавляли N2H4.H2O (7,58 мг, 151,38 мкмоль, 7,36 мкл, 1,00 экв.). Смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал, что промежуточное соединение 125 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь гасили добавлением воды (10 мл) и экстрагировали DCM (5 мл х 2). Объединенные органические слои промывали солевым раствором (5 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=8:1) с получением промежуточного соединения 126 (90,00 мг, неочищенное) в виде твердого вещества желтого цвета.
Промежуточное соединение 127: (8R,9S,10S)-9-(4-бромфенил)-4,10-бис[(диметиламино)метил]-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 126 (45,00 мг, 84,83 мкмоль, 1,00 экв.) в DCM (2,00 мл) добавляли HCHO (41,31 мг, 508,98 мкмоль, 37,90 мкл, 37% раствор, 6,00 экв.) и MgSO4 (122,53 мг, 1,02 ммоль, 12,00 экв.), после перемешивания в течение 30 мин добавляли NaBH(OAc)3 (269,68 мг, 1,27 ммоль, 15,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Анализ ЖХ/МС показал, что промежуточное соединение 126 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (10 мл) и экстрагировали DCM (3 мл х 2). Объединенные органические слои промывали солевым раствором (3 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) с получением промежуточного соединения 127 (20,00 мг, 35,81 мкмоль, 42,21% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=7,46-7,40 (м, 2H), 7,37-7,33 (м, 2H), 7,30 (д, J=9,0 Гц, 2H), 6,81 (д, J=9,0 Гц, 2H), 3,97 (д, J=14,1 Гц, 1H), 3,77 (с, 3H), 3,67 (д, J=15,6 Гц, 1H), 3,60-3,54 (м, 1H), 3,45-3,32 (м, 2H), 3,15-3,03 (м, 2H), 2,74 (дд, J=10,8, 14,3 Гц, 1H), 2,47-2,34 (м, 4H), 2,30 (с, 6H), 2,25-2,17 (м, 1H), 2,02 (с, 6H), 1,62-1,48 (м, 3H).
Синтез E32: (8R,9S,10S)-4,10-бис[(диметиламино)метил]-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E32)
К раствору промежуточного соединения 127 (15,00 мг, 26,86 мкмоль, 1,00 экв.) в ацетонитриле (1,00 мл) добавляли этинилбензол (10,97 мг, 107,42 мкмоль, 3,00экв.), Xphos Pd G3 (2,27 мг, 2,69 мкмоль, 0,10 экв.) и Cs2CO3 (35,00 мг, 107,42 мкмоль, 4,00 экв.). Смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал, что промежуточное соединение 127 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь гасили добавлением воды (10 мл) и экстрагировали этилацетатом (3 мл х 2). Объединенные органические слои промывали солевым раствором (3 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=8:1) с получением соединения E32 (4,30 мг, 7,42 мкмоль, 27,61% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=7,50-7,44 (м, 2H), 7,44-7,36 (м, 4H), 7,31-7,21 (м, 5H), 6,74 (д, J=8,5 Гц, 2H), 3,93 (д, J=14,6 Гц, 1H), 3,70 (с, 3H), 3,63-3,52 (м, 2H), 3,42-3,28 (м, 2H), 3,09-2,99 (м, 2H), 2,72 (дд, J=10,5, 14,1 Гц, 1H), 2,36 (д, J=10,0 Гц, 3H), 2,23 (с, 6H), 2,16 (т, J=12,5 Гц, 2H), 1,96 (ш.с, 6H), 1,54 (ш.с, 1H), 1,45 (ш.с, 2H).
Пример 33: (8R,9S,10S)-10-[(диметиламино)метил]-4-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E33”)

Промежуточное соединение 128: [(8R,9R,10S)-9-(4-бромфенил)-10-(гидроксиметил)-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-4-ил]метил ацетат
К раствору промежуточного соединения 118 (600,00 мг, 760,68 мкмоль, 1,00 экв.) в DCM (10,00 мл) добавляли TFA (867,33 мг, 7,61 ммоль, 563,20 мкл, 10,00 экв.). Смесь перемешивали при 20°C в течение 2 часов. Анализ ЖХ/МС показал, что промежуточное соединение 118 израсходовано, и детектировали требуемый МС. Реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (30 мл) и экстрагировали DCM (10 мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM/MeOH=1:0 до 20:1) с получением промежуточного соединения 128 (300,00 мг, 549,00 мкмоль, 72,17% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 129: [(8R,9S,10S)-9-(4-бромфенил)-10-[(1,3-диоксоизоиндолин-2-ил)метил]-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-4-ил]метилацетат
К раствору трифенилфосфин (1 г, 3,76 ммоль) в THF (19 мл) при 0°C медленно добавляли DIAD (0,73 мл, 3,76 ммоль). Полученную смесь (11 мл) добавляли к смеси промежуточного соединения 128 (300,00 мг, 549,00 мкмоль, 1,00 экв.) и изоиндолин-1,3-диона (121,16 мг, 823,50 мкмоль, 1,50 экв.) в THF (15,00 мл) при 0°C, и затем смесь перемешивали при 25°C в течение 16 часов в атмосфере N2. Анализ ЖХ/МС показал, что промежуточное соединение 128 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM:MeOH=1:0 до 10:1) с получением промежуточного соединения 129 (400,00 мг, неочищенное) в виде твердого вещества желтого цвета, содержащего Ph3PO.
Промежуточное соединение 130: [(8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-4-ил]метилацетат
К раствору промежуточного соединения 129 (400,00 мг, 592,09 мкмоль, 1,00 экв.) в EtOH (10,00 мл) добавляли N2H4.H2O (29,64 мг, 592,09 мкмоль, 28,78 мкл, 1,00 экв.). Смесь перемешивали при 70°C в течение 2 часов. Реакционную смесь гасили добавлением воды (30 мл) и экстрагировали DCM (30 мл х 2). Объединенные органические слои промывали солевым раствором (30 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM:MeOH=1:0 до 20:1) с получением промежуточного соединения 130 (280,00 мг, 513,32 мкмоль, 86,70% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 131: [(8R,9S,10S)-9-(4-бромфенил)-10-[(диметиламино)метил]-6-[(4-метоксифенил)карбамоил]-1,6-диазабицикло[6.2.0]декан-4-ил]метилацетат
К раствору промежуточного соединения 130 (140,00 мг, 256,66 мкмоль, 1,00 экв.) в DCM (5,00 мл) добавляли MgSO4 (370,73 мг, 3,08 ммоль, 12,00 экв.) и HCHO (124,99 мг, 1,54 ммоль, 114,67 мкл, 37% раствор, 6,00 экв.), затем частями добавляли NaBH(OAc)3 (815,95 мг, 3,85 ммоль, 15,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Анализ ЖХ/МС показал, что промежуточное соединение 130 израсходовано, и детектировали один требуемый МС. Реакционную смесь гасили добавлением насыщенного водного раствора NaHCO3 (20 мл) и экстрагировали DCM (5 мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=20:1) с получением промежуточного соединения 131 (73,00 мг, 127,28 мкмоль, 49,59% выход) в виде твердого вещества белого цвета.
Промежуточное соединение 132: [(8R,9S,10S)-10-[(диметиламино)метил]-6-[(4-метоксифенил)карбамоил]-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-4-ил]метилацетат
К раствору промежуточного соединения 131 (73,00 мг, 127,28 мкмоль, 1,00 экв.) в ацетонитриле (4,00 мл) добавляли этинилбензол (39 мг, 381,85 мкмоль, 3,00 экв.), Xphos Pd G3 (10,77 мг, 12,73 мкмоль, 0,10 экв.) и Cs2CO3 (165,89 мг, 509,14 мкмоль, 4,00 экв.). Смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал, что промежуточное соединение 131 полностью израсходовано, и детектировали требуемый МС. Реакционную смесь гасили добавлением воды (10 мл) и экстрагировали этилацетатом (3 мл х 2). Объединенные органические слои промывали солевым раствором (3 мл х 2), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, DCM:MeOH=10:1) с получением промежуточного соединения 132 (30,00 мг, 50,44 мкмоль, 39,63% выход) в виде твердого вещества коричневого цвета.
Синтез E33: (8R,9S,10S)-10-[(диметиламино)метил]-4-(гидроксиметил)-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E33)
К раствору промежуточного соединения 132 (30,00 мг, 50,44 мкмоль, 1,00 экв.) в MeOH (500,00 мкл)/DCM (0,5 мл) добавляли K2CO3 (34,86 мг, 252,20 мкмоль, 5,00 экв.). Смесь перемешивали при 15°C в течение 1 часа. Анализ ЖХ/МС показал, что промежуточное соединение 132 полностью израсходовано, и детектировали один основной пик с требуемым МС. Реакционную смесь гасили добавлением воды (10 мл) и экстрагировали DCM (5 мл х 2). Комбинированные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Luna C18 150*25 5u; подвижная фаза: [вода (0,225% муравьиная кислота)-ацетонитрил]; B%: 25%-45%, 12 мин) с получением соединения E33 (11,00 мг, 18,37 мкмоль, 36,42% выход, соль муравьиной кислоты)) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, метанол-d4) δ=7,58-7,49 (м, 6H), 7,38 (д, J=3,5 Гц, 3H), 7,29 (д, J=8,8 Гц, 2H), 6,81 (д, J=9,3 Гц, 2H), 3,92 (д, J=15,4 Гц, 1H), 3,82-3,71 (м, 5H), 3,59 (д, J=6,2 Гц, 2H), 3,45 (т, J=9,0 Гц, 1H), 3,20-3,05 (м, 2H), 3,01-2,91 (м, 2H), 2,88-2,81 (м, 1H), 2,48 (т, J=12,1 Гц, 1H), 2,40-2,29 (м, 6H), 1,83 (ш.с, 1H), 1,67-1,48 (м, 2H).
Пример 34: (8R,9S,10S)-10-((диметиламино)метил)-N-(4-метоксифенил)-2-метил-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E34”)



Промежуточное соединение 133: (2R,3R,4S)-1-аллил-3-(4-бромфенил)-4-(гидроксиметил)азетидин-2-карбонитрил
К раствору (2R,3R,4S)-1-аллил-3-(4-бромфенил)-4-((тритилокси)метил)азетидин-2-карбонитрила (J.O.C (2012), 77(17), 7187-7211) (16,00 г, 29,12 ммоль, 1,00 экв.) в DCM (291,00 мл) добавляли TFA (33,20 г, 291,20 ммоль, 21,56 мл, 10,00 экв.) при 25°C. Смесь перемешивали при 25°C в течение 16 часов. Анализ ТСХ показал, что реагент полностью израсходован, наблюдали наличие требуемого продукта, реакционную смесь гасили добавлением раствора NaHCO3 (500 мл) и экстрагировали DCM (200 мл x 3), сушили над Na2SO4, концентрировали с получением остатка (8,95 г, неочищенное) в виде твердого вещества желтого цвета, которое использовали на следующем этапе без очистки.
Промежуточное соединение 134: (2R,3R,4S)-3-(4-бромфенил)-4-(гидроксиметил)азетидин-2-карбонитрил
Смесь промежуточного соединения 133 (8,95 г, 29,14 ммоль, 1,00 экв.), 1,3-диметил барбитуровой кислоты (6,82 г, 43,71 ммоль, 1,50 экв.), Pd(PPh3)4 (3,37 г, 2,91 ммоль, 0,10 экв.) в EtOH (290,00 мл) дегазировали и продували N2 3 раза, и затем смесь перемешивали при 40°C в течение 16 часов в атмосфере N2. Анализ ЖХ/МС показал, что реагент 133 полностью израсходован, наблюдали требуемый МС, смесь концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (10 мМ NH4HCO3); B: ацетонитрил) с получением промежуточного соединения 134 (4,70 г, 17,60 ммоль, 60,38% выход) в виде твердого вещества оранжевого цвета.
Промежуточное соединение 135: (2R,3R,4S)-3-(4-бромфенил)-4-(гидроксиметил)-1-(1-метилаллил)азетидин-2-карбонитрил
К раствору промежуточного соединения 134 (1,60 г, 5,99 ммоль, 1,00 экв.) в THF (3,50 мл) добавляли P(OEt)3 (39,81 мг, 239,59 мкмоль, 41,04 мкл, 0,04 экв.), (E)-бут-2-ен-1-илацетат (724,70 мг, 6,35 ммоль, 1,06 экв.), аллил(хлор)палладия (21,92 мг, 59,90 мкмоль, 0,01 экв.) и DBU (911,89 мг, 5,99 ммоль, 902,86 мкл, 1,00 экв.), смесь дегазировали и продували N2 3 раза, и затем смесь перемешивали при 40°C в течение 16 часов в атмосфере N2. Реакционную смесь гасили добавлением воды (15 мл) и экстрагировали DCM (20 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10/1 до 0:1) с получением промежуточного соединения 135 (150,00 мг, 466,98 мкмоль, 7,80% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, метанол-d4) δ=1,20 (дд, J=11,04, 6,53 Гц, 3H) 3,11 (кв., J=7,03 Гц, 1H) 3,43-3,55 (м, 2H) 3,59-3,67 (м, 1H) 3,80-3,92 (м, 1H) 4,04-4,15 (м, 1H) 4,35 (д, J=8,53 Гц, 1H) 4,41 (д, J=8,53 Гц, 1H) 5,05-5,41 (м, 2H) 5,64-5,91 (м, 1H) 7,47-7,52 (м, 2H) 7,52-7,56 (м, 2H).
Промежуточное соединение 136: (2R,3R,4S)-3-(4-бромфенил)-4-[[трет-бутил(диметил)силил]оксиметил]-1-(1-метилаллил)азетидин-2-карбонитрил
К раствору промежуточного соединения 135 (1,23 г, 3,83 ммоль, 1,00 экв.) в DMF (6,00 мл) добавляли имидазол (1,56 г, 22,98 ммоль, 6,00 экв.) и TBSCl (2,31 г, 15,32 ммоль, 1,88 мл, 4,00 экв.). Смесь перемешивали при 25°C в течение 16 часов. Анализ ЖХ/МС показал, что реагент полностью израсходован, наблюдали требуемый МС, промывали водой (30 мл), экстрагировали этилацетатом (30 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=100/1 до 10:1) с получением промежуточного соединения 136 (1,60 г, 3,67 ммоль, 95,93% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, метанол-d4) δ ppm - 0,36- -0,13 (м, 7H) 0,69-0,77 (м, 9H) 1,19 (дд, J=18,57, 6,53 Гц, 3H) 3,04-3,17 (м, 1H) 3,39-3,47 (м, 1H) 3,53-3,59 (м, 1H) 3,61-3,72 (м, 1H) 3,82-3,92 (м, 1H) 4,33-4,44 (м, 1H) 5,07-5,35 (м, 2H) 5,61-5,85 (м, 1H) 7,46-7,50 (м, 2H) 7,52-7,56 (м, 2H).
Промежуточное соединение 137: [(2R,3R,4S)-3-(4-бромфенил)-4-[[трет-бутил(диметил)силил]оксиметил]-1-(1-метилаллил)азетидин-2-ил]метанамин
К раствору промежуточного соединения 136 (1,85 г, 4,25 ммоль, 1,00 экв.) в THF (200,00 мл) добавляли LiBHEt3 (1 M, 5,06 мл, 10,00 экв.) при 0°C в атмосфере N2. Смесь перемешивали при 25°C в течение 2 часов. Анализ ЖХ/МС показал, что реагент 136 полностью израсходован, наблюдали требуемый МС, реакцию гасили добавлением H2O (100 мл), экстрагировали этилацетатом (300 мл x 3), концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM/MeOH=1/0 до 10:1) с получением промежуточного соединения 137 (1,40 г, 3,19 ммоль, 74,95% выход) в виде масла.
Промежуточное соединение 138: N-[[(2R,3R,4S)-3-(4-бромфенил)-4-[[трет-бутил(диметил)силил]оксиметил]-1-(1-метилаллил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамид
К раствору промежуточного соединения 138 (1,36 г, 3,09 ммоль, 1,00 экв.) в THF (55,00 мл) добавляли TEA (9,91 г, 97,95 ммоль, 13,58 мл, 31,70 экв.) и 2-нитробензолсульфонил хлорид (2,05 г, 9,27 ммоль, 3,00 экв.) при 0°C. Смесь перемешивали при 25°C в течение 2 часов. Анализ ЖХ/МС показал, что реагент 1 полностью израсходован, наблюдали требуемый МС, смесь концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=20/1 до 5:1) с получением промежуточного соединения 138 (1,93 г, неочищенное) в виде желтого масла.
Промежуточное соединение 139: N-аллил-N-[[(2R,3R,4S)-3-(4-бромфенил)-4-[[трет-бутил(диметил)силил]оксиметил]-1-(1-метилаллил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамид
К раствору промежуточного соединения 138 (1,95 г, 3,12 ммоль, 1,00 экв.) в DMF (15,00 мл) добавляли K2CO3 (647,17 мг, 4,68 ммоль, 1,50 экв.), затем добавляли 3-бромпроп-1-ен (1,13 г, 9,36 ммоль, 808,84 мкл, 3,00 экв.). Смесь перемешивали при 25°C в течение 16 часов. Анализ ЖХ/МС показал, что реагент 138 полностью израсходован, наблюдали требуемый МС, смесь промывали солевым раствором (20 мл x 3), экстрагировали этилацетатом (20 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=20/1 до 5:1) с получением промежуточного соединения 139(1,90 г, 2,86 ммоль, 91,61% выход в виде смолы желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ =ppm - 0,34- -0,27 (м, 3H) -0,23- -0,18 (м, 3H) 0,70-0,75 (м, 9H) 1,14 (дд, J=16,76, 6,62 Гц, 3H) 3,11-3,42 (м, 5H) 3,15-3,79 (м, 6H) 4,69-4,82 (м, 1H) 4,93-5,04 (м, 1H) 5,05-5,22 (м, 2H) 5,28-5,50 (м, 1H) 5,67-5,81 (м, 1H) 7,32 (д, J=8,38 Гц, 2H) 7,43 (дд, J=8,38, 1,32 Гц, 2H) 7,52-7,62 (м, 3H) 7,62-7,70 (м, 1H).
Промежуточное соединение 140: [(3Z,8R,9R,10S)-9-(4-бромфенил)-2-метил-6-(2-нитрофенил)сульфонил-1,6-диазабицикло[6.2.0]дец-3-ен-10-ил]метокси-трет-бутил-диметил-силан
К раствору промежуточного соединения 139 (1,84 г, 2,77 ммоль, 1,00 экв.) в DCE (368,00 мл) добавляли катализатор Ховейд-Граббса 1-го поколения (569,51 мг, 692,50 мкмоль, 0,25 экв.). Смесь перемешивали при 90°C в течение 16 часов. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=5:1). Промежуточное соединение 140 (1,25 г, 1,96 ммоль, 70,88% выход) получали в виде легкого твердого вещества черного цвета.
Промежуточное соединение 141: [(8R,9R,10S)-9-(4-бромфенил)-2-метил-6-(2-нитрофенил)сульфонил-1,6-диазабицикло[6.2.0]декан-10-ил]метокси-трет-бутил-диметил-силан
К раствору промежуточного соединения 140 (1,00 г, 1,57 ммоль, 1,00 экв.) в THF (40,00 мл) добавляли TEA (1,43 г, 14,13 ммоль, 1,96 мл, 9,00 экв.) и 2-нитробензолсульфонгидразид (1,02 г, 4,71 ммоль, 3,00 экв.). Смесь перемешивали при 40°C в масляной бане в течение 16 часов. Анализ ЖХ/МС показал, что реагент 140 полностью израсходован, основную массу составлял требуемый продукт. Реакционную смесь гасили добавлением насыщенного раствора NaHCO3(20 мл) и экстрагировали три раза этилацетатом (30 мл x 3). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=20/1 до 5:1) с получением промежуточного соединения 141 (830,00 мг, 1,04 ммоль, 66,22% выход, 80% чистоты) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=ppm -0,29- -0,08 (м, 6H) 0,75 (д, J=14,05 Гц, 9H) 1,03-1,17 (м, 2H) 1,36-1,48 (м, 1H) 1,71-2,17 (м, 3H) 2,62 (ш.с, 1H) 2,75-2,94 (м, 1H) 3,00-3,29 (м, 2H) 3,37-3,48 (м, 1H) 3,49-3,67 (м, 3H) 3,69-3,87 (м, 2H) 7,28-7,46 (м, 4H) 7,53-7,66 (м, 3H) 7,78 (д, J=6,02 Гц, 1H).
Промежуточное соединение 142: [(8R,9R,10S)-9-(4-бромфенил)-2-метил-1,6-диазабицикло[6.2.0]декан-10-ил]метокси-трет-бутил-диметил-силан
К раствору промежуточного соединения 141 (830,00 мг, 1,30 ммоль, 1,00 экв.) в CH3CN (8,00 мл) добавляли Cs2CO3 (508,28 мг, 1,56 ммоль, 1,20 экв.) и бензолтиол (214,77 мг, 1,95 ммоль, 198,86 мкл, 1,50 экв.). После перемешивания при 20°C в течение 16 часов реакционную смесь разбавляли водой (15 мл), экстрагировали этилацетатом (15 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=5:1) с получением промежуточного соединения 142 (580,00 мг, 1,02 ммоль, 78,70% выход, 80% чистоты) в виде твердого вещества желтого цвета.
Промежуточное соединение 143: (8R,9R,10S)-9-(4-бромфенил)-10-[[трет-бутил(диметил)силил]оксиметил]-N-(4-метоксифенил)-2-метил-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 142 (550,00 мг, 1,21 ммоль, 1,00 экв.) в DCM (10,00 мл) добавляли TEA (61,36 мг, 606,35 мкмоль, 84,05 мкл, 0,50 экв.) и 1-изоцианат-4-метокси-бензол (180,88 мг, 1,21 ммоль, 155,93 мкл, 1,00 экв.) при 0°C. После перемешивания при 20°C в течение 3 часов реакционную смесь разбавляли водой (10 мл), экстрагировали DCM (20 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10/1 до 5:1) с получением промежуточного соединения 143 (700,00 мг, 1,16 ммоль, 95,99% выход) в виде твердого вещества желтого цвета.
Промежуточное соединение 144: (8R,9R,10S)-9-(4-бромфенил)-10-(гидроксиметил)-N-(4-метоксифенил)-2-метил-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 143 (690,00 мг, 1,14 ммоль, 1,00 экв.) в THF (10,00 мл) добавляли TBAF (596,13 мг, 2,28 ммоль, 2,00 экв.). Смесь перемешивали при 50°C в течение 16 часов. Анализ ТСХ показал, что реагент 1 полностью израсходован, наблюдали наличие требуемого продукта, промывали водой (10 мл x 3), экстрагировали этилацетатом (20 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM/MeOH=1/0 до 20:1) с получением промежуточного соединения 144 (440,00 мг, 900,86 мкмоль, 79,02% выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ= ppm 1,09 (д, J=6,53 Гц, 3H) 1,40 (д, J=16,56 Гц, 1H) 1,77-1,99 (м, 2H) 2,72-2,90 (м, 2H) 3,51 (д, J=6,02 Гц, 1H) 3,57-3,69 (м, 4H) 3,71-3,80 (м, 4H) 3,81-3,94 (м, 2H) 6,02-6,08 (м, 1H) 6,79-6,87 (м, 2H) 7,24 (д, J=9,03 Гц, 2H) 7,36-7,43 (м, 2H) 7,43-7,49 (м, 2H).
Промежуточное соединение 145: (8R,9S,10S)-9-(4-бромфенил)-10-[(1,3-диоксо-3a,7a-дигидроизоиндол-2-ил)метил]-N-(4-метоксифенил)-2-метил-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 144 (420,00 мг, 859,92 мкмоль, 1,00 экв.) в THF (18,00 мл) добавляли изоиндолин-1,3-дион (189,78 мг, 1,29 ммоль, 1,50 экв.), PPh3 (451,09 мг, 1,72 ммоль, 2,00 экв.) и DIAD (347,77 мг, 1,72 ммоль, 334,39 мкл, 2,00 экв.) при 0°C. Смесь перемешивали при 25°C в течение 16 часов. Анализ ЖХ/МС показал, что реагент 145 полностью израсходован, основную массу составлял требуемый продукт, который промывали водой (10 мл), экстрагировали этилацетатом (10 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=5/1 до 1:1) с получением промежуточного соединения 145 (800,00 мг, 645,63 мкмоль, 75,08% выход, 50% чистоты) в виде смолы коричневого цвета.
Промежуточное соединение 146: (8R,9S,10S)-10-(аминометил)-9-(4-бромфенил)-N-(4-метоксифенил)-2-метил-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору промежуточного соединения 145 (750,00 мг, 605,28 мкмоль, 1,00 экв.) в EtOH (14,00 мл) добавляли NH2NH2.H2O (30,30 мг, 605,28 мкмоль, 29,42 мкл, 1,00 экв.). Смесь перемешивали при 80°C в течение 2 часов. Анализ ЖХ/МС показал, что промежуточное соединение 145 полностью израсходовано, основную массу составлял требуемый продукт (промежуточное соединение 146), которое промывали водой (40 мл), экстрагировали этилацетатом (30 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, DCM/MEOH=0/1 до 10:1) с получением (10S,10aR)-9-амино-10-(4-бромфенил)-N-(4-метоксифенил)-6-метилоктагидропирроло[1,2-a][1,4]диазоцин-2(1H)-карбоксамида (“R146”) (190 мг) (1H ЯМР (400 МГц, хлороформ-d) δ=ppm 0,97 (д, J=6,02 Гц, 1H) 1,14 (д, J=8,03 Гц, 2H) 1,25-1,48 (м, 2 H) 1,87-2,09 (м, 2H) 2,72-2,84 (м, 1H) 3,01 (д, J=10,04 Гц, 1H) 3,15-3,23 (м, 1H) 3,28 (д, J=6,02 Гц, 1H) 3,45 (ш.с, 1H) 3,62-4,11 (м, 7H) 6,82 (д, J=10,04 Гц, 2H) 7,18-7,25 (м, 2H) 7,29 (д, J=8,03 Гц, 2 H) 7,49 (д, J=8,03 Гц, 2 H)) в виде смеси твердых веществ желтого цвета и промежуточного соединения 146 (40 мг) в виде твердого вещества желтого цвета (1H ЯМР (400 МГц, хлороформ-d) =ppm 0,99-1,16 (м, 3H) 1,44 (д, J=11,04 Гц, 1H) 1,87 (ш.с, 3H) 2,66 (ш.с, 1H) 2,80-2,91 (м, 1H) 2,92-3,00 (м, 1H) 3,01-3,15 (м, 1H) 3,38 (ш.с, 1H) 3,59 (д, J=12,55 Гц, 2H) 3,67-3,84 (м, 6H) 6,14 (с, 1H) 6,81 (д, J=8,53 Гц, 2H) 7,20 (д, J=8,53 Гц, 2H) 7,34 (д, J=8,03 Гц, 2H) 7,46 (д, J=8,03 Гц, 2Н)).
Промежуточное соединение 147: (8R,9S,10S)-9-(4-бромфенил)-10-((диметиламино)метил)-N-(4-метоксифенил)-2-метил-1,6-диазабицикло[6.2.0]декан-6-карбоксамид
К раствору соединения 146 (15,00 мг, 21,54 мкмоль, 1,00 экв.) в DCM (3,00 мл) добавляли HCHO (17,48 мг, 215,42 мкмоль, 16,04 мкл, 37% чистоты, 10,00 экв.) и MgSO4 (51,86 мг, 430,83 мкмоль, 20,00 экв.) с последующим добавлением NaBH(OAc)3 (45,66 мг, 215,42 мкмоль, 10,00 экв.). После перемешивания смеси при 20°C в течение 2 часов полученную смесь фильтровали, и фильтрат концентрировали. Остаток очищали препаративной ТСХ (SiO2, дихлорметан:метанол=20:1) с получением соединения 147 (5,00 мг, 7,76 мкмоль, 36,02% выход, 80% чистоты) в виде твердого вещества светло-желтого цвета.
Синтез E34: (8R,9S,10S)-10-((диметиламино)метил)-N-(4-метоксифенил)-2-метил-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E34)
К раствору промежуточного соединения 147 (5,00 мг, 9,70 мкмоль, 1,00 экв.) в CH3CN (500,00 мкл) добавляли этинилбензол (2,97 мг, 29,10 мкмоль, 3,19 мкл, 3,00 экв.), Cs2CO3 (12,64 мг, 38,80 мкмоль, 4,00 экв.), дегазировали с помощью N2 3 раза и затем добавляли XPhos Pd G3 (821,04 мкг, 0,97 мкмоль, 0,10 экв.). После перемешивания при 70°C в течение 3 часов реакционную смесь разбавляли водой (3 мл), экстрагировали этилацетатом (5 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1:1) с получением соединения E34 (5,00 мг, 9,32 мкмоль, 96,04% выход) в виде твердого вещества светло-желтого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=ppm 1,16 (д, J=6,17 Гц, 3H) 1,70-1,94 (м, 4H) 2,01 (ш.с, 6 H) 2,42-2,66 (м, 3H) 2,83-2,94 (м, 1H) 3,47 (ш.с, 2H) 3,53-3,62 (м, 1H) 3,65-3,82 (м, 6H) 6,04 (с, 1H) 6,83 (д, J=8,82 Гц, 2H) 7,24 (д, J=8,82 Гц, 2H) 7,33-7,39 (м, 3H) 7,39-7,44 (м, 2H) 7,50 (д, J=7,94 Гц, 2H) 7,53-7,57 (м, 2H).
Пример 35: (8R,9R,10S)-10-(гидроксиметил)-N-(4-метоксифенил)-2-метил-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E35”)

К раствору промежуточного соединения 144 (20,00 мг, 40,95 мкмоль, 1,00 экв.) в CH3CN (1,00 мл) добавляли этинилбензол (12,55 мг, 122,85 мкмоль, 13,49 мкл, 3,00 экв.), Cs2CO3 (53,37 мг, 163,80 мкмоль, 4,00 экв.), дегазировали с помощью N2 3 раза и затем добавляли XPhos Pd G3 (3,47 мг, 4,10 мкмоль, 0,10 экв.). Смесь перемешивали при 70°C в течение 16 часов. Анализ ЖХ/МС показал, что реагент 144 полностью израсходован, основную массу составлял требуемый продукт, который промывали водой (10 мл), экстрагировали этилацетатом (10 мл x 3), сушили над Na2SO4, концентрировали с получением остатка. Остаток очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (10mM NH4HCO3); B: ацетонитрил) с получением соединения E35 (8,30 мг, 16,29 мкмоль, 39,77% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ=1,10 (д, J=6,02 Гц, 3H) 1,40 (д, J=13,55 Гц, 1H) 1,76-2,01 (м, 3H) 2,72-3,03 (м, 1H) 2,76 (ш.с, 1H) 3,43-3,81 (м, 9H) 3,82-3,96 (м, 2H) 6,03-6,11 (м, 1H) 6,80-6,88 (м, 2H) 7,24 (д, J=9,03 Гц, 2H) 7,32-7,40 (м, 3H) 7,48-7,56 (м, 6H).
Пример 36: (3S,4R,8R,9S)-3,4-дигидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E36”)



Промежуточное соединение 150: (R)-2-(аллиламино)-1-(4-бромфенил)этан-1-ол
Во флакон, содержащий (R)-2-бромфенил)оксиран (8,0 г, 40,2 ммоль) добавляли аллиламин (12,03 мл, 161 ммоль). Смесь перемешивали при 55°C в течение 20 часов. После этого выполняли анализ ЖХ/МС, который показал, что реакция завершена, смесь охлаждали до комнатной температуры, и органический растворитель выпаривали при пониженном давлении. При добавлении к остатку гексана образовалось твердое вещество белого цвета, которое фильтровали и сушили под глубоким вакуумом в течение 2 часов с получением требуемого продукта, (R)-2-(аллиламино)-1-(4-бромфенил)этан-1-ола (9,66 г, 37,7 ммоль, 94% выход), в виде твердого вещества белого цвета. Реакционную смесь переносили на следующий этап без дополнительной очистки. 1H ЯМР (300 МГц, CDCl3) δ 7,53-7,36 (м, 2H), 7,29-7,13 (м, 2H), 5,96-5,72 (м, 1H), 5,28-5,02 (м, 2H), 4,67 (дд, J=9,0, 3,6 Гц, 1H), 3,41-3,18 (м, 2H), 2,87 (дд, J=12,2, 3,6 Гц, 1H), 2,66 (дд, J=12,2, 8,9 Гц, 1H).
Промежуточное соединение 151: (R)-2-(аллил(2-(4-бромфенил)-2-гидроксиэтил)амино)ацетонитрил
К раствору (R)-2-(аллиламино)-1-(4-бромфенил)этан-1-ола (9,66 г, 37,7 ммоль) в безводном ацетонитриле (60 мл) при комнатной температуре добавляли K2CO3 (7,82 г, 56,6 ммоль) и 2-бромацетонитрил (7,88 мл, 113 ммоль). Гетерогенную смесь перемешивали при 85°C в атмосфере азота в течение 18 часов. После этого анализ ЖХ/МС показал, что реакция завершена, и смесь охлаждали до комнатной температуры. Органический растворитель выпаривали при пониженном давлении, разбавляли водой (50 мл) и экстрагировали этилацетатом. Объединенные органические компоненты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (SiO2, этилацетат/гексан=20-30%) с получением (R)-2-(аллил(2-(4-бромфенил)-2-гидроксиэтил)амино)ацетонитрила (9 г, 30,5 ммоль, 81% выход) в виде твердого вещества бледно-желтого цвета. 1H ЯМР (300 МГц, CDCl3) δ 7,54-7,40 (м, 2H), 7,31-7,16 (м, 2H), 5,91-5,66 (м, 1H), 5,43-5,22 (м, 2H), 4,78-4,65 (м, 1H), 3,79-3,54 (м, 2H), 3,45-3,30 (м, 1H), 3,27-3,11 (м, 2H), 2,86-2,74 (м, 1H), 2,68-2,54 (м, 1H).
Промежуточное соединение 152: (R)-2-(аллил(2-(4-бромфенил)-2-хлорэтил)амино)ацетонитрил
К раствору пиридина (12,1 мл, 151 ммоль) в безводном DMF (100 мл) при 0°C по каплям добавляли тионилхлорид (4,38 мл, 60,3 ммоль). Смесь перемешивали при 0°C в течение 15 минут. К этому раствору добавляли (R)-2-(аллил(2-(4-бромфенил)-2-гидроксиэтил)амино)ацетонитрил (8,9 г, 30,2 ммоль) в DCM (10 мл) в течение 5 минут. Эту смесь перемешивали при 0°C в течение 30 минут, после чего анализ ТСХ показал, что реакция завершена. К реакционной смеси добавляли насыщенный водный раствор NaHCO3, и полученный раствор энергично перемешивали в течение 10 мин до его переноса в делительную воронку. Слои разделяли, и водную фазу экстрагировали DCM, и объединенные органические экстракты промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Остаток очищали колоночной хроматографией (SiO2, этилацетат/гексан=0-20%) с получением требуемого промежуточного соединения (5,00 г, 9,87 ммоль, 92% выход) в виде твердого вещества коричневого цвета. 1H ЯМР (300 МГц, CDCl3) δ ppm 7,57-7,42 (м, 2H), 7,32-7,23 (м, 2H), 5,84-5,58 (м, 1H), 5,37-5,14 (м, 2H), 4,91-4,71 (м, 1H), 3,65-3,38 (м, 2H), 3,26-2,94 (м, 4H).
Промежуточное соединение 153: (2R,3S)-1-аллил-3-(4-бромфенил)азетидин-2-карбонитрил
В 100 мл круглодонную колбу наливали (R)-2-(аллил(2-(4-бромфенил)-2-хлорэтил)амино)ацетонитрил (0,55 г, 1,754 ммоль) и безводный THF (10 мл) с получением бледно-желтого раствора. Колбу полностью погружали в баню сухой лед/ацетон и охлаждали до -50°C. К этому охлажденному раствору по каплям добавляли LiHMDS (1M, THF раствор). После добавления реакционную смесь перемешивали в течение 1 часа при -50°C. Анализы ЖХ/МС и ТСХ (20% EtOAc в гексанах, окрашивание KMnO4) показали полное исчезновение исходного материала. Реакционную смесь обрабатывали насыщенным водным раствором NH4Cl, и полученный раствор энергично перемешивали в течение 10 мин до его переноса в делительную воронку. Слои разделяли, водную фазу экстрагировали EtOAc, и объединенные органические экстракты промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Остаток очищали колоночной хроматографией (SiO2, этилацетат/гексан=0-20%) с получением двух основных наборов фракций. Первый набор фракций (0,16 г, 0,577 ммоль, 33% выход) дал побочный продукт, (2S,3S)-1-аллил-3-(4-бромфенил)азетидин-2-карбонитрил. Второй набор фракций (0,17 г, 0,613 ммоль, 35% выход) отверждался при отстаивании, давая требуемый продукт, (2R,3S)-1-аллил-3-(4-бромфенил)азетидин-2-карбонитрил. 1H ЯМР (300 МГц, CDCl3) δ 7,59-7,45 (м, 2H), 7,36-7,28 (м, 2H), 5,91-5,69 (м, 1H), 5,36-5,14 (м, 2H), 4,39-4,26 (м, 1H), 3,87-3,75 (м, 1H), 3,60-3,40 (м, 2H), 3,31-3,18 (м, 2H).
Промежуточное соединение 154: ((2R,3S)-1-аллил-3-(4-бромфенил)азетидин-2-ил)метанамин
К раствору (2R,3S)-1-аллил-3-(4-бромфенил)азетидин-2-карбонитрил (0,62 г, 2,25 ммоль) в DCM (23 мл), охлажденному до 0°C, при перемешивании медленно с помощью шприца добавляли DIBAL-H (2,41 мл, 13,51 ммоль). Через 15 минут реакционную смесь оставили нагреваться до комнатной температуры на 45 мин. После этого анализ ЖХ/МС показал, что реакция завершена, и реакционную смесь охлаждали до 0°C, и медленно добавляли метанол (1,366 мл, 33,8 ммоль). К этой смеси медленно (необходима осторожность, реакция экзотермическая) добавляли раствор Рошелля (Rochelle) (100 мл). Водную фазу экстрагировали DCM, и объединенные органические фазы сушили над MgSO4 и концентрировали при пониженном давлении с получением (2R,3S)-1-аллил-3-(4-бромфенил)азетидин-2-карбонитрила в виде желтого масла, которое переносили на следующий этап без очистки.
Промежуточное соединение 155: N-(((2R,3S)-1-аллил-3-(4-бромфенил)азетидин-2-ил)метил)-2-нитробензолсульфонамид
К раствору промежуточного соединения 154 (1,454 г, 5,171 ммоль) в DCM (26 мл), охлажденному до 0°C, добавляли 2,6-лютидин (1,797 мл, 15,51 ммоль) с последующим добавлением 2-нитробензол-1-сульфонил хлорида (1,261 г, 5,69 ммоль) в виде одной порции. Затем раствор оставляли нагреваться до комнатной температуры и перемешивали в течение 16 часов. К реакционной смеси добавляли воду, и водный слой экстрагировали DCM. Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Остаток очищали колоночной хроматографией (SiO2, этилацетат/гексан=0-20%) с получением промежуточного соединения 155 (2,4 г, 100% выход).
Промежуточное соединение 156: N-аллил-N-[[(2R,3S)-1-аллил-3-(4-бромфенил)азетидин-2-ил]метил]-2-нитро-бензолсульфонамид
К раствору промежуточного соединения 155 (5,00 г, 10,72 ммоль, 1,00 экв.) в DMF (10,00 мл) добавляли K2CO3 (2,22 г, 16,08 ммоль, 1,50 экв.) и 3-бромпроп-1-ен (1,95 г, 16,08 ммоль, 1,39 мл, 1,50 экв.). Смесь перемешивали при 25°C в течение 16 часов. Анализ ЖХ/МС показал, что реагент 156 полностью израсходован, основную массу составлял требуемый продукт. Реакционную смесь разбавляли водой (100 мл), экстрагировали DCM (20 мл x 3), органический слой сушили над Na2SO4, затем концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10/1 до 5:1) с получением промежуточного соединения 156 (5,00 г, 9,87 ммоль, 92,10% выход) в виде твердого вещества коричневого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,53 (с, 1H) 2,90 (дд, J=13,80, 6,65 Гц, 1H) 2,94-3,11 (м, 2H) 3,18-3,26 (м, 1H) 3,33 (дд, J=7,34, 2,20 Гц, 1H) 3,41 (дд, J=13,80, 5,14 Гц, 1H) 3,47-3,63 (м, 2H) 3,65-3,75 (м, 1H) 3,88 (дд, J=16,00, 5,96 Гц, 1H) 4,95 (дд, J=17,07, 1,13 Гц, 1H) 5,03-5,14 (м, 2H) 5,21 (дд, J=17,19, 1,51 Гц, 1H) 5,48 (ддт, J=16,89, 10,43, 6,23, 6,23 Гц, 1H) 5,64-5,87 (м, 1H) 7,35-7,40 (м, 2H) 7,42-7,49 (м, 2H) 7,55-7,62 (м, 2H) 7,64-7,72 (м, 2H).
Промежуточное соединение 157: (3Z,8R,9S)-9-(4-бромфенил)-6-(2-нитрофенил)сульфонил-1,6-диазабицикло[6.2.0]дец-3-ен
К раствору промежуточного соединения 156 (5,00 г, 9,87 ммоль, 1,00 экв.) в толуоле (500,00 мл) добавляли катализатор Ховейд-Граббса 1-го поколения (812 мг) в атмосфере N2. Смесь перемешивали при 60°C в водяной бане в течение 16 часов. Добавляли дополнительный катализатор Ховейд-Граббса 1-го поколения (1218 мг) и перемешивали в течение следующих 16 часов. Реакционную смесь концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=5/1 до 0:1) с получением промежуточного соединения 157 (1,20 г, 1,25 ммоль, 12,71% выход, 50% чистоты) в виде твердого вещества вместе с извлеченным SM (2,2 г). 1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,62 (ш.с, 1H) 3,15-3,31 (м, 2H) 3,38 (ш.с, 1H) 3,52 (ш.д, J=16,98 Гц, 1H) 3,60-3,79 (м, 3H) 3,82-3,95 (м, 1H) 4,03 (ш.дд, J=14,33, 6,62 Гц, 1H) 5,58-5,67 (м, 1H) 5,68-5,81 (м, 1H) 7,30 (ш.д, J=7,94 Гц, 2H) 7,45 (ш.д, J=8,16 Гц, 2H) 7,56 (ш.д, J=7,28 Гц, 1H) 7,61-7,67 (м, 2H) 7,83 (ш.д, J=7,50 Гц, 1H).
Промежуточное соединение 158: (3Z,8R,9S)-9-(4-бромфенил)-1,6-диазабицикло[6.2.0]дец-3-ен
К раствору промежуточного соединения 157 (1,70 г, 1,78 ммоль, 1,00 экв.) в CH3CN (17,00 мл) добавляли бензолтиол (293,67 мг, 2,67 ммоль, 271,92 мкл, 1,50 экв.) и Cs2CO3 (694,74 мг, 2,13 ммоль, 1,20 экв.). После перемешивания при 40°C в течение 16 часов реакционную смесь разбавляли в воде (100 мл), экстрагировали DCM (30 мл x 3), органический слой сушили над Na2SO4, затем концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=1/1 до дихлорметан:метанол=10:1) с получением промежуточного соединения 158 (650,00 мг, 1,55 ммоль, 87,18% выход, 70% чистоты) в виде черного масла. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,42-2,50 (м, 1H) 2,52-2,61 (м, 1H) 2,77-3,10 (м, 3H) 3,22 (ш.дд, J=16,22, 4,38 Гц, 1H) 3,36-3,50 (м, 3H) 3,53-3,69 (м, 3H) 3,77 (ш.т, J=8,99 Гц, 1H) 5,67-5,86 (м, 2H) 7,21-7,28 (м, 2H) 7,44 (д, J=8,33 Гц, 2H).
Промежуточное соединение 159: (3Z,8R,9S)-9-(4-бромфенил)-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]дец-3-ен-6-карбоксамид
К раствору промежуточного соединения 158 (650,00 мг, 1,55 ммоль, 1,00 экв.) в DCM (10,00 мл) добавляли TEA (78,52 мг, 775,92 мкмоль, 107,56 мкл, 0,50 экв.), и затем добавляли 1-изоцианат-4-метокси-бензол (277,75 мг, 1,86 ммоль, 239,44 мкл, 1,20 экв.) при 20°C в атмосфере N2. Смесь перемешивали при 20°C в течение 1 часа. Анализ ЖХ/МС показал, что реагент 158 полностью израсходован, основную массу составлял требуемый продукт. Реакционную смесь разбавляли водой (10 мл), экстрагировали DCM (5 мл x 3), органический слой сушили над Na2SO4, затем концентрировали с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=5/1 до 1:1) с получением промежуточного соединения 159 (560,00 мг, 1,27 ммоль, 81,84% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,37-2,56 (м, 1H) 3,24-3,33 (м, 1H) 3,34-3,41 (м, 1H) 3,42-3,53 (м, 1H) 3,54-3,64 (м, 2H) 3,68-3,74 (м, 2H) 3,77 (с, 3H) 3,86 (ш.дд, J=15,88, 7,28 Гц, 1H) 4,17 (ш.д, J=15,44 Гц, 1H) 5,64 (ш.д, J=11,69 Гц, 1H) 5,74-5,91 (м, 1H) 6,06 (с, 1H) 6,82 (д, J=8,82 Гц, 2H) 7,22 (д, J=9,04 Гц, 2H) 7,34 (д, J=8,16 Гц, 2H) 7,48 (д, J=8,38 Гц, 2H).
Промежуточные соединения 160a и 160b: (3S,4R,8R,9S)-9-(4-бромфенил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (160a);
(3R,4S,8R,9S)-9-(4-бромфенил)-3,4-дигидрокси-N-(4-метоксифенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (160b)
К раствору промежуточного соединения 159 (500,00 мг, 1,13 ммоль, 1,00 экв.) в ацетон (5,00 мл) и H2O (500,00 мкл) добавляли NMO (198,63 мг, 1,69 ммоль, 178,95 мкл, 1,50 экв.) и OsO4 (2,87 мг, 11,30 мкмоль, 0,59 мкл, 0,01 экв.) при -78°C. Затем полученную реакционную смесь перемешивали при 20°C в течение 0,5 часа. Анализ ТСХ показал, что реагент полностью израсходован, наблюдали наличие двух требуемых продуктов. Реакционную смесь концентрировали с получением остатка. Остаток очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат). Получали промежуточное соединение 160a (100,00 мг, 209,93 мкмоль, 61,92% выход) в виде твердого вещества белого цвета (1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,17-2,37 (м, 1H) 2,63 (ш.д, J=13,16 Гц, 1H) 2,90 (ш.дд, J=13,37, 8,55 Гц, 1H) 3,23 (ш.д, J=15,79 Гц, 1H) 3,39-3,45 (м, 1H) 3,46-3,52 (м, 2H) 3,58 (ш.д, J=5,26 Гц, 2H) 3,73-3,83 (м, 5H) 3,95 (ш.с, 1H) 4,11 (ш.дд, J=15,57, 5,04 Гц, 1H) 4,25 (ш.с, 1H) 6,81 (д, J=8,77 Гц, 2H) 7,16 (д, J=8,77 Гц, 2H) 7,32 (д, J=8,33 Гц, 2H) 7,44 (д, J=8,33 Гц, 2H) 7,92 (ш.с, 1H)) и промежуточное соединение 160b (275,00 мг, 577,29 мкмоль, 72,98% выход) в виде твердого вещества белого цвета (1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,27 (ш.с, 1H) 2,79 (ш.д, J=14,03 Гц, 1H) 2,93-3,05 (м, 1H) 3,27-3,52 (м, 1H) 3,45-3,52 (м, 3H) 3,54-3,70 (м, 5H) 3,73-3,82 (м, 4H) 6,79 (д, J=9,21 Гц, 2H) 7,21 (д, J=8,77 Гц, 2H) 7,25-7,30 (м, 1H) 7,27 (д, J=6,82 Гц, 1H) 7,28-7,31 (м, 1H) 7,47 (д, J=8,33 Гц, 2H)).
Синтез E36: (3S,4R,8R,9S)-3,4-дигидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E36)
К раствору промежуточного соединения 160a (10,00 мг, 20,99 мкмоль, 1,00 экв.), этинилбензола (6,43 мг, 62,97 мкмоль, 6,91 мкл, 3,00 экв.) в CH3CN (150,00 мкл) добавляли Cs2CO3 (27,36 мг, 83,96 мкмоль, 4,00 экв.) и Xphos Pd G3 (1,78 мг, 2,10 мкмоль, 0,10 экв.) в атмосфере N2. Смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал, что реагент 1 полностью израсходован, основную массу составлял требуемый продукт. Реакционную смесь фильтровали, фильтровальную лепешку промывали DCM (10 мл), фильтрат концентрировали с получением остатка. Остаток очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1:1) и затем очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением соединения E36 (4,00 мг, 8,04 мкмоль, 38,30% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,31-2,44 (м, 1H) 2,73 (ш.д, J=13,45 Гц, 1H) 2,92-2,99 (м, 1H) 3,33 (ш.д, J=15,66 Гц, 1H) 3,57 (ш.т, J=7,39 Гц, 2 H) 3,70 (ш.д, J=4,63 Гц, 1H) 3,75-3,85 (м, 4H) 3,87-3,94 (м, 1H) 4,00 (ш.с, 1H) 4,16 (ш.д, J=16,32 Гц, 1H) 6,83 (ш.д, J=8,82 Гц, 2H) 7,21 (ш.д, J=8,82 Гц, 2H) 7,35 (ш.с, 3H) 7,44 (ш.д, J=7,94 Гц, 2H) 7,49-7,59 (м, 4H) 7,82 (ш.с, 1H).
Пример 37: (3R,4S,8R,9S)-3,4-дигидрокси-N-(4-метоксифенил)-9-[4-(2-фенилэтинил)фенил]-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (“E37”)

К перемешиваемому раствору промежуточного соединения 160b (10,00 мг, 20,99 мкмоль, 1,00 экв.), этинилбензола (6,43 мг, 62,98 мкмоль, 6,92 мкл, 3,00 экв.) в CH3CN (150,00 мкл) добавляли Cs2CO3 (27,36 мг, 83,97 мкмоль, 4,00 экв.) и Xphos Pd G3 (1,78 мг, 2,10 мкмоль, 0,10 экв.) в атмосфере N2. Смесь перемешивали при 70°C в течение 1 часа. Анализ ЖХ/МС показал, что реагент 160b полностью израсходован, основную массу составлял требуемый продукт. Реакционную смесь фильтровали, фильтровальную лепешку промывали DCM (5 мл), фильтрат концентрировали с получением остатка. Остаток очищали препаративной ТСХ (SiO2, петролейный эфир/этилацетат=1:1), затем дополнительно очищали препаративной ВЭЖХ (колонка: Waters Xbridge Prep OBD C18 150*30 5u; A: вода (0,225% муравьиная кислота) B: ацетонитрил) с получением соединения E37 (5,00 мг, 10,05 мкмоль, 47,87% выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, хлороформ-d) δ ppm 2,32 (ш.с, 1H) 2,60 (ш.с, 2H) 2,87 (ш.д, J=14,11 Гц, 1H) 3,06 (дд, J=14,00, 6,95 Гц, 1H) 3,29-3,48 (м, 2H) 3,57-3,65 (м, 2H) 3,71 (ш.д, J=5,51 Гц, 4H) 3,77 (с, 3H) 3,80-3,84 (м, 1H) 6,66 (ш.с, 1H) 6,82 (д, J=8,82 Гц, 2H) 7,20-7,26 (м, 2H) 7,27 (с, 1H) 7,33-7,43 (м, 5H) 7,49-7,57 (м, 4H).
Пример 38: (3R,4S,8R,9S,10S)-N-(4-циклопропоксифенил)-10-((диметиламино)метил)-3,4-дигидрокси-9-(4-(фенилэтинил)фенил)-1,6-диазабицикло[6.2.0]декан-6-карбоксамид (E38)
Синтез соединения E38 выполняли аналогично синтезу соединения Е1, с тем отличием, что вместо промежуточного соединения 4a использовали промежуточное соединение 4b. Далее следовали такие же этапы реакций, как описаны выше.
Измерение биологической активности
Пример 39: In vitro культура P. falciparum на стадии развития в крови и анализ.
Линии P. falciparum (Dd2, 3D7, D6, K1, NF54, V1/3, HB3, 7G8, FCB и TM90C2B) получали из центра Malaria Research и Reference Reagent Resource Center (MR4). Изоляты P. falciparum содержали в O-положительной человеческой крови в атмосфере 93% N2, 4% CO2, 3% O2 при 37°C в полной культуральной среде (10,4 г/л RPMI 1640, 5,94 г/л HEPES, 5 г/л альбумакса II, 50 мг/л гипоксантина, 2,1 г/л бикарбоната натрия, 10% человеческой сыворотки и 43 мг/л гентамицина). Паразитов культивировали в среде до достижения уровня паразитемии 3-8%. Паразитемию определяли проверкой по меньшей мере 500 эритроцитов в мазке крови, окрашенном по Гимзе. Для скрининга соединений выполняли разведение средой до 2,0% паразитемии и 2,0% гематокрита. 25 мкл среды разливали в 384-луночные черные планшеты с прозрачным дном, и 100 нл каждого соединения в DMSO вносили в аналитические планшеты вместе с контрольным соединением (мефлохином). Затем, в аналитические планшеты вносили 25 мкл суспензии паразита в среде с получением конечной концентрации паразитемии 1% и конечной концентрации гематокрита 1%. Аналитические планшеты инкубировали в течение 72 часов при 37°C. В аналитические планшеты вносили 10 мкл детектирующего реагента, состоящего из 10×SYBR Green I (Invitrogen; поставляемого в 10000×концентрации) в буфере для лизиса (20 мМ Tris-HCl, 5 мМ ЭДТА, 0,16% (масс./об.) сапонина, 1,6% (об./об.) тритон X-100). Для оптимального окрашивания аналитические планшеты оставляли в темном месте при комнатной температуре на 24 часа. Аналитические планшеты считывали с помощью 505 дихроичных зеркал с установленной длиной волны возбуждения 485 нм и длиной волны излучения 530 нм на приборе Envision (PerkinElmer).
Следуя вышеописанному протоколу, измеряли 50% эффективную концентрацию (EC50) соединений E1-E38 в отношении P. falciparum Dd2, результаты измерений приведены ниже в Таблице 4, где +++ соответствует EC50 менее чем 50 нМ, ++ соответствует EC50 от 50 nM до 250 нМ, и+соответствует EC50 >250 нМ.
Пример 40: In vitro анализ печеночной стадии P. berghei на стадии личинки
Клетки HepG2 (ATCC) поддерживали в среде DMEM, дополненной 10% (об./об.) FBS (Sigma) и 1% (v/v) антибиотика-антимикотика в стандартном инкубаторе для тканевых культур (37°C, 5% CO2). Москитов A. Stephensi, инфицированных P. berghei (ANKA GFP-luc), получали из инсектария медицинского центра Лангоне при Нью-Йоркском университете (the New York University Langone Medical Center Insectary). Для анализа в 384-луночный микротитровальный планшет в двух экземплярах добавляли ∼17500 HepG2 клеток на лунку. Через 18-24 часов при 37°C среду заменяли, и добавляли соединения. Через 1 час в планшеты добавляли паразитов, полученных из только что рассеченных москитов (4000 паразитов на лунку), планшеты вращали в течение 10 мин со скоростью 1000 об./мин и затем инкубировали при 37°C. Конечный анализируемый объем был равен 30 мкл. Через 48 часов инкубации при 37°C в планшет с паразитами добавляли Bright-Glo (Promega) для измерения относительной люминесценции. Относительную интенсивность сигнала каждого планшета оценивали с помощью системы EnVision (PerkinElmer).
Пример 41: Культивирование на микропатерннированной поверхности (micropatterned co-culture, MPCC) представляет собой систему совместного культивирования in vitro первичных гепатоцитов человека, организованных в колонии и окруженных поддерживающими стромальными клетками. Гепатоциты в этом формате сохраняют функциональный фенотип до 4-6 недель без пролиферации, что оценивается по основным функциям, характерным для печени, и экспрессии генов. Вкратце, 96-луночные планшеты равномерно покрывали коллагеном типа I из крысиного хвоста (50 мкг/мл) и с помощью методами легкой литографии структурировали коллаген в микродомены размером 500 мкм, которые обеспечивают селективную адгезию гепатоцитов. Для создания MPCC криоконсервированные первичные гепатоциты человека (BioreclamationIVT) осаждали центрифугированием при 100g в течение 6 минут при 4°C, оценивали их жизнеспособность путем исключения с помощью трипанового синего (обычно 70-90%) и высевали на чашки с микропаттерном, образованным коллагеном, (каждая лунка содержала примерно 10000 гепатоцитов, организованных в колонии по 500 мкМ) в бессывороточной среде DMEM с 1% пенициллин-стрептомицином. Клетки промывали бессывороточной DMEM с 1% пенициллин-стрептомицином через 2-3 ч и заменяли культуральной средой 48 для гепатоцитов человека. Мышиные фибробласты 3T3-J2 высевали (7000 клеток на лунку) через 24 часа после посева гепатоцитов.
MPCC инфицировали 75000 спорозоитами (NF54) (Университет Джона Хопкинса) через 1 день после посева гепатоцитов. После инкубации при 37°С и 5% СО2 в течение 3 ч лунки промывали один раз PBS и добавляли соответствующие соединения. Культуры дозировали ежедневно. Образцы фиксировали на день 3,5 после заражения. Для иммунофлуоресцентного окрашивания MPCC фиксировали с помощью охлажденного до -20°C метанола в течение 10 мин при 4°C, дважды промывали PBS, блокировали 2% BSA в PBS и инкубировали с мышиными антителами к Hsp70 P.falciparum (клон 4C9, 2 мкг/мл) в течение 1 ч при комнатной температуре. Образцы промывали PBS, затем инкубировали с Alexa 488-конъюгированным вторичным козьим антимышиным антителом в течение 1 ч при комнатной температуре. Образцы промывали PBS, выполняли контрастную окраску с ДНК-красителем Hoechst 33258 (Invitrogen; 1:1000) и фиксировали на предметных стеклах с помощью раствора Fluoromount G (Southern Biotech). Изображения получали на флуоресцентном микроскопе Nikon Eclipse Ti. Диаметры паразитов развивающихся на печеночной стадии измеряли и использовали для вычисления соответствующей площади.
Пример 42: Биохимический анализ цитоплазматической PheRS P. falciparum
Белковые последовательности как субъединицы α-(PF3D7_0109800), так и β-(PF3D7_1104000) цитоплазматического PheRS P. falciparum получали из PlasmoDB (http://plasmodb.org/lasmo/). Полноразмерные гены α- и β-субъединиц, оптимизированные для экспрессии в E.coli, клонировали в векторы экспрессии pETM11 (устойчивость к канамицину) и pETM20 (устойчивость к ампициллину) с помощью сайтов Nco1 и Kpn1 и ко-трансформировали в клетки B834 E. coli. Экспрессию белка индуцировали добавлением 0,5 мМ изопропил β-d-1-тиогалактопиранозида (IPTG), и клетки выращивали до достижения плотности OD600 0,6-0,8 при 37°C. Затем, после индукции, их оставляли расти при 18°С в течение 20 часов. Клетки отделяли центрифугированием при 5000g в течение 20 мин, и бактериальные осадки суспендировали в буфере, состоящем из 50 мМ Трис-HCl (pH 7,5), 200 мМ NaCl, 4 мМ β-меркаптоэтанол, 15% (об./об.) глицерин, 0,1 мг/мл лизоцима и 1 мМ фенилметилсульфонил фторид (PMSF). Клетки лизировали ультразвуком и очищали центрифугированием при 20000g в течение 1 часа. Надосадочную жидкость наносили на предварительно упакованную колонку NiNTA (GE Healthcare), и связанные белки элюировали градиентом, смешивая с элюирующим буфером (50 мМ Трис-HCl (pH 7,5), 80 мМ NaCl, 4 мМ β-меркаптоэтанол, 15% (об./об.) глицерин, 1 М имидазол). Чистые фракции объединяли и загружали в гепариновую колонку для дальнейшей очистки. И в этом случае, связанные белки элюировали градиентом с использованием элюирующего буфера с гепарином (50 мМ Трис-HCl (pH 7,5), 1 М NaCl, 4 мМ β-меркаптоэтанол, 15% (об./об.) глицерин). Чистые фракции снова объединяли и диализовали в течение ночи в буфере, содержащем 50 мМ Трис-HCl (рН 7,5), 200 мМ NaCl, 4 мМ β-меркаптоэтанол, 1 мМ DTT и 0,5 мМ ЭДТА. К образцу белка добавляли TEV протеазу (соотношение протеаза:белок=1:50) и инкубировали при 20°C в течение 24 часов для удаления полигистидиновой метки. Далее белок очищали с помощью гель-фильтрационной хроматографии на колонке GE HiLoad 60/600 Superdex в 50 мМ Трис-HCl (pH 7,5), 200 мМ NaCl, 4 мМ β-меркаптоэтанол, 1 мМ MgCl2. Элюированный белок (гетеро-димер cpheRS P. falciparum) собирали, оценивали его чистоту с помощью SDS-PAGE и хранили при -80°C.
Кодируемую в ядре tRNAPhe P. falciparum синтезировали методом транскрипции in vitro, как описано в Nature 538, 344-349 (20 октября 2016 г.) doi:10,1038/nature19804. Оценку аминоацилирования и ферментативного ингибирования цитозольных PheRS P. falciparum выполняли, как описано в Biochem. J. 465, 459-469 (2015). Ферментативные анализы выполняли в буфере, содержащем 30 мМ HEPES (рН 7,5), 150 мМ NaCl, 30 мМ KCl, 50 мМ MgCl2, 1 мМ DTT, 100 мкМ АТФ, 100 мкМ L-фенилаланин, 15 мкМ tRNAPhe P. falciparum, 2 ед/мл неорганической пирофосфатазы E.coli (NEB) и 500 нМ рекомбинантного PheRS P. falciparum, при 3°C. Реакции в разные моменты времени останавливали добавлением 40 мМ ЭДТА и последующим переносом на лед. Рекомбинантный мальтозосвязывающий белок использовали в качестве отрицательного контроля. Анализы ингибирования cPheRS выполняли с использованием следующих концентраций ингибитора: 0,01 нМ, 0,1 нМ, 1 нМ, 10 нМ, 100 нМ, 1 мкМ, 5 мкМ и 10 мкМ для сильных связующих веществ, и 1 нМ, 10 нМ, 100 нМ, 1 мкМ, 10 мкМ, 100 мкМ и 500 мкМ для более слабых связующих веществ в аналитическом буфере. Ферментативные и ингибирующие эксперименты выполняли по два раза в трех экземплярах.
Пример 43: Оценка цитотоксичности в клетках млекопитающих
Клетки млекопитающих (HepG2, A549 и HEK293) получали из АТСС и культивировали обычным способом в среде DMEM с 10% FBS и 1% (об./об.) антибиотика-антимикотика. Для оценки цитотоксичности высевали 1×106 клеток в 384-луночные планшеты за 1 день до обработки соединением. Клетки обрабатывали увеличивающимися дозами соединения в течение 72 ч, а жизнеспособность измеряли с помощью Cell-Titer Glo (Promega). Все клеточные линии проверяли на загрязнение микоплазмой с помощью универсального набора для обнаружения микоплазмы (ATCC).
Пример 44: In vitro оценка ADME/PK и безопасности.
In vitro оценку некоторых характеристик (связывание белков, микросомальная стабильность, стабильность гепатоцитов, ингибирование цитохрома P450 (CYP) и растворимость в воде) выполняли стандартными для данной отрасли методами. Изучение ингибирования ионных каналов выполняли с помощью системы Q-Patch стандартными методами.
Пример 45: In vivo оценка стадии развития в крови P. berghei
Мышам CD-1 (n=4 на экспериментальную группу; самки в возрасте 6-7 недель; 20-24 г, Charles River) внутривенно инокулировали примерно 1×105 паразита P. berghei (ANKA GFP-luc), находящегося на стадии развития в крови, за 24 ч до лечения, и перорально вводили (в 0 ч) соединения. Паразитемию контролировали с помощью системы in vivo визуализации (IVIS SpectrumCT, PerkinElmer) с получением биолюминесцентного сигнала (150 мг/кг люциферина вводили внутрибрюшинно за 10 мин до визуализации). Кроме того, у каждой мыши периодически брали образцы мазков крови, окрашивали по Гимзе и изучали под микроскопом с целью визуального обнаружения паразитемии крови. Животных с паразитемией, превышающей 25%, гуманно умерщвляли.
Пример 46: In vivo оценка этиотропной профилактики инфекции, вызываемой P. berghei
Мышам CD-1 (n=4 на экспериментальную группу; самки в возрасте 6-7 недель; 20-24 г, Charles River) внутривенно инокулировали примерно 1×105 спорозоитов P. berghei (ANKA GFP-luc) из предварительно рассеченных москитов A. stephensi. Сразу после заражения мышам вводили единичные пероральные дозы соединения; развитие инфекции отслеживали, как описано для анализа эритроцитарной стадии P. berghei. В экспериментах по изучению временной зависимости лечения соединением (единичная пероральная доза 10 мг/кг) изменялось от 5 дней до заражения до 2 дней после заражения.
Пример 47: In vivo оценка трансмиссивной стадии P. berghei
Мышей CD-1 (n=3 на экспериментальную группу; самки в возрасте 6-7 недель; 21-24 г, Charles River) инфицировали P. berghei (ANKA GFP-luc) за 96 ч до обработки несущей средой или соединением (день 0). На второй день самкам москитов A. stephensi давали возможность напитаться кровью мышей в течение 20 минут. Через 1 неделю (день 9) средние кишки комаров иссекали и под микроскопом (увеличение 12,5×) подсчитывали количество ооцистов.
Пример 48: In vivo оценка стадии развития в крови P. falciparum
Выбирали in vivo адаптированный P. falciparum (3D7HLH/BRD), как описано в PLoS One 3, e2252 (2008). Вкратце, мышам NSG (n=2 на экспериментальную группу; самки в возрасте 4-5 недель; 19-21 г; лаборатория Джексона) ежедневно внутрибрюшинно инъецировали 1 мл эритроцитов человека (O-положительные, 50% гематокрит, 50% RPMI 1640 с 5% альбумаксом) для получения мышей с гуманизированными циркулирующими эритроцитами (huRBC NSG). Примерно 2×107 P. falciparum 3D7HLH/BRD, находящихся на стадии развития в крови (FASEB J. 25, 3583-3593 (2011)), внутривенно вводили мышам huRBC NSG, и через 5 недель после заражения паразитемия достигала >1%. После трех пассажей in vivo паразитов замороживали и использовали в экспериментах. Примерно через 48 ч после заражения 1×107 P. falciparum 3D7HLH/BRD, находящимися на стадии развития в крови, средний уровень паразитемии составил примерно 0,4%. Мышей huRBC NSG перорально обрабатывали однократной дозой соединения, и в течение 30 дней отслеживали паразитемию с помощью IVIS по биолюминесцентному сигналу (150 мг/кг люциферина вводили внутрибрюшинно примерно за 10 минут до визуализации).
Пример 49: In vivo оценка трансмиссивной стадии P. falciparum
Мышей huRBCNSG (n=2 на экспериментальную группу; самки в возрасте 4-5-недель; 18-20 г; лаборатория Джексона) инфицировали P. falciparum 3D7HLH/BRD, находящимися на стадии развития в крови, в течение 2 недель для развития зрелых гаметоцитов. Затем мышей лечили однократной пероральной дозой соединения. Образцы крови собирали в течение 11 дней. Для детектирования на стадий паразита молекулярном уровне у контрольных и обработанных мышей брали по 40 мкл крови. Вкратце, общую РНК выделяли из образцов крови с помощью набора RNeasy Plus Kit, используя колонки для удаления геномной ДНК (Qiagen). Синтез первой цепи кДНК выполняли из экстрагированной РНК с помощью системы для синтеза первой цепи SuperScript III (Life Technologies). Выполняли количественное определение стадии паразитов с помощью специфичного в отношении стадии метода количественной ПЦР в реальном времени (qRT), как описано в Sci. Transl. Med. 6, 244re5 (2014). Для измерения уровней транскриптов PF3D7_0501300 (паразитов на кольцевой стадии), PF3D7_1477700 (незрелых гаметоцитов) и PF3D7_1031000 (зрелых гаметоцитов) разрабатывали праймеры. Праймеры для транскрипта PF3D7_1120200 (P. falciparum UCE) использовали в качестве конститутивно экспрессируемого маркера паразита. Анализ выполняли с помощью кДНК с общим реакционным объемом 20 мкл, содержащим праймеры для каждого гена с конечной концентрацией 250 нМ. Амплификацию выполняли на аппарате Viia7 qRT-PCR (Life Technologies) с использованием SYBR Green Master Mix (Applied Biosystems) со следующими условиями реакции: 1 цикл × 10 минут при 95°C и 40 циклов × 1 с при 95°C и 20 с при 60°С. Каждый образец кДНК запускали в трех экземплярах, и для анализа использовали среднее значение Ct. Значения Ct, превышающие пороговое значение (отрицательный контроль) для каждого маркера, считали отрицательными на наличие специфических транскриптов. Образцы крови каждой мыши перед инокуляцией паразитов также тестировали на “фоновый шум” с использованием тех же наборов праймеров. Амплификация не была обнаружена ни в одном из образцов.
Пример 50: In vivo оценка печеночной стадии P. falciparum
Мышам C57BL/6, нокаутным по гену FRG (репопулированные человеческими гепатоцитами >70%) (нокаут FRG huHep; n=2 на экспериментальную группу; самки в возрасте 5,5-6 месяцев; 19-21 г; Yecuris), инокулировали внутривенно примерно 1×105 спорозоитов P. falciparum (NF54HT-GFP-luc) и вводили соединение в виде единичной пероральной дозы 10 мг/кг через день после инокуляции. Инфекцию ежедневно отслеживали с помощью IVIS. Ежедневное приживление эритроцитов человека (0,4 мл, O-положительные, 50% гематокрит, 50% RPMI 1640 с 5% альбумаксом) начинали через 5 дней после инокуляции. Для анализа кПЦР собирали образцы крови (40 мкл) через 7 дней после инокуляции. Для детектирования на молекулярном уровне паразита, находящегося на стадии развития крови, у контрольных и обработанных мышей брали 40 мкл крови. Вкратце, общую РНК выделяли из образцов крови с помощью набора RNeasy Plus Kit с колонками для удаления геномной ДНК (Qiagen). Синтез первой цепи кДНК выполняли из экстрагированной РНК с помощью системы синтеза первой цепи SuperScript III (Life Technologies). Количественное определение наличия паразитов на стадии развития в крови выполняли с помощью высоко-специфичного количественного ПЦР в реальном времени, как описано в PLOS Comput. Biol. 9, e1003392 (2013). Для измерения уровней транскриптов PF3D7_1120200 (P. falciparum UCE) разрабатывали праймеры. Анализ выполняли с использованием кДНК в общем реакционном объеме 20 мкл, содержащем праймеры для каждого гена с конечной концентрацией 250 нМ. Амплификацию выполняли на аппарате Viia7 qRT-PCR (Life Technologies) с использованием SYBR Green Master Mix (Applied Biosystems) со следующими условиями реакции: 1 цикл × 10 мин при 95°C и 40 циклов × 1 с при 95°C и 20 с при 60°C. Каждый образец кДНК запускали в трех экземплярах, и для анализа использовали среднее значение Ct. Значения Ct, превышающие порог (отрицательный контроль) для каждого маркера, считали отрицательными на наличие специфических транскриптов. Перед инокуляцией паразита образцы крови от каждой мыши также тестировали на фоновый шум с использованием тех же наборов праймеров. Амплификация не была обнаружено ни в одном из образцов.
Пример 51: In vivo оценка C. parvum
Для оценки эффективности соединения in vivo использовали модель хронической бессимптомной инфекции C. parvum у NOD SCID гамма мышей. NOD SCID гамма мышей инфицировали ооцистами ~ 1×105 C. parvum через желудочный зонд через 5-7 дней после отъема. Инфицированные животные начинают выделять ооцисты через 1 неделю после заражения, которые измеряют количественной ПЦР (кПЦР). Основываясь на опыте с соединением, использованным в качестве положительного контроля, паромомицином, для достижения 80% мощности для выявления уменьшения паразитов на 80% через четыре дня приема лекарственного вещества экспериментальная группа должна состоять из четырех мышей. В дополнение к экспериментальным группам, получающим лечение, в каждый эксперимент включали дополнительные группы отрицательного (желудочный зонд с носителем ДМСО/метилцеллюлоза) и положительного (2000 мг/кг паромомицина один раз в сутки) контроля. Мышей инфицировали через 5-7 дней после отъема (день -6), инфекцию подтверждали через 1 неделю (день 0), а экспериментальные соединения вводили через желудочный зонд в дни 1-4. Дозировка была такой же, как указано. Эффективность лечения оценивали путем измерения ооцист, выделенных с фекалиями, методом кПЦР на 5-й день.
Пример 52: In vitro оценка EC50 C. parvum.
Клетки человеческой илеоцекальной аденокарциномы (HCT-8) получали из ATCC и хранили в колбах T-75 для тканевой культуры со средой RPMI 1640 с HEPES, пируватом натрия (1 мМ) и L-глютамином (ATCC) с добавлением 10% лошадиной сыворотки (ATCC) и 120 ед./мл пенициллина и 120 мкг/мл стрептомицина. Клетки высевали в 384-луночные, обработанные тканевой культурой, микролуночные планшеты с черными стенками и прозрачным дном (BD Falcon) с плотностью 8850 клеток/лунка и оставляли расти до смыкания монослоя. Затем в них нокулировали 5,5х103 праймированных ооцист C. parvum (Bunchgrass Farms, Deary, ID), суспендированных в среде для инокуляции (RPMI 1640 без лошадиной сыворотки). Ооцисты были праймированы для эксцистирования согласно ранее описанному протоколу (J. Eukaryot. Microbiol. 46:56S-57S). Вкратце, ооцисты обрабатывали в течение 10 минут 10 мМ HCl при 37°С, центрифугировали и обрабатывали 2 мМ раствором таурохолата натрия (Sigma-Aldrich) в фосфатно-солевом буфере (PBS) с Ca2+ и Mg2+. Суспензию инкубировали в течение 10 мин при 16°С и затем разбавляли средой для инокуляции и добавляли в каждую лунку. Инфицированные клетки инкубировали при 37°С в течение 3 ч, после чего оценивали эквивалентный объем ростовой среды, содержащей 20% лошадиной сыворотки (общая концентрация сыворотки 10%). Соединения разбавляли и анализировали, используя фиксированные дозы: 0,12, 0,37, 1,1, 3,3 и 10 мкМ (каждая концентрация, n=14) для получения кривых EC50. В случае окончательных кривых ЕС50 три лунки оставляли незараженными, но обрабатывали каждой из соответствующих концентраций соединения для оценки фонового окрашивания. Все кривые получали в виде кривой зависимости log[ингибитор]-ответ с переменным угловым коэффициентом, используя программное обеспечение GraphPad Prism (уравнение 3), с установленным на 0 нижним пределом.
Следуя вышеописанному протоколу, для нескольких соединений со структурой, соответствующей формуле (I), измеряли 50% эффективную концентрацию (ЕС50) в отношении C. parvum; результаты представлены в таблице 5 ниже.
Другие варианты осуществления
Раскрытие каждого патента, патентной заявки и публикации, упомянутых в настоящей заявке, включено в настоящее описание во всей воей полноте в виде ссылки. Также в виде ссылки включено раскрытие публикации США № 2016/0289235. В конкретных вариантах осуществления соединение по настоящему изобретению не является соединением, описанным в таблице 1 публикации США № 2016/0289235.
Хотя в настоящем изобретении предоставлены конкретные варианты осуществления, следует понимать, что оно может быть дополнительно модифицировано, и настоящая заявка охватывает любые изменения, применения или адаптации следующего: в общем случае, принципы, раскрытые в настоящем описании и включающие такие отклонения от настоящего раскрытия, относятся к известной или обычной практике в области техники, к которой относится настоящее изобретение, и могут применяться к существенным признакам, изложенным выше.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ ЦНС | 2015 |
|
RU2733756C2 |
| ПРОИЗВОДНОЕ РЕЗОРЦИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 | 2016 |
|
RU2697703C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ УСИЛЕНИЯ ДЕГРАДАЦИИ БЕЛКОВ-МИШЕНЕЙ И ДРУГИХ ПОЛИПЕПТИДОВ С ПОМОЩЬЮ Е3 УБИКВИТИН ЛИГАЗЫ | 2013 |
|
RU2666530C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ УСИЛЕНИЯ ДЕГРАДАЦИИ БЕЛКОВ-МИШЕНЕЙ И ДРУГИХ ПОЛИПЕПТИДОВ С ПОМОЩЬЮ E3 УБИКВИТИН ЛИГАЗЫ | 2013 |
|
RU2781452C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2567238C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2675857C2 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДИНКАРБОКСАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2017 |
|
RU2778478C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2809023C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
Изобретение относится к соединению, имеющему структуру, соответствующую формуле (I), или к его фармацевтически приемлемой соли, где R1 представляет собой фенил, необязательно замещенный одним или двумя F, или пиридинил; R2 представляет собой С1-С6 алкокси или С3-С6 циклоалкокси; R3 представляет собой водород или –CH2–X; R4 и R5 независимо представляют собой водород, –OH, –OR, –N(R)(R), –CH2OH, – CH2OR или –CH2N(R)(R), или R4 и R5 вместе могут формировать диоксин и по меньшей мере один из R4 и R5 не является водородом; R6 и R7 независимо представляют собой водород; и z1–z8 независимо представляют собой CH; где –X в каждом случае независимо выбирают из –OH или –N(R)(R); и R в каждом случае независимо представляет собой необязательно замещенный C1–C12 алкил. Изобретение также относится к фармацевтической композиции на основе соединения формулы (I). Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения и профилактики паразитарного заболевания, представляющего собой малярию или криптоспоридиоз. 5 н. и 56 з.п. ф-лы, 5 табл., 52 пр.

1. Соединение, имеющее структуру, соответствующую формуле (I):
 , (I)
, (I)
где R1 представляет собой фенил, необязательно замещенный одним или двумя F, или пиридинил;
R2 представляет собой С1-С6 алкокси или С3-С6 циклоалкокси;
R3 представляет собой водород или –CH2–X;
R4 и R5 независимо представляют собой водород, –OH, –OR, –N(R)(R), –CH2OH, – CH2OR или –CH2N(R)(R), или R4 и R5 вместе могут формировать диоксин и по меньшей мере один из R4 и R5 не является водородом;
R6 и R7 независимо представляют собой водород; и
z1–z8 независимо представляют собой CH; где
–X в каждом случае независимо выбирают из –OH или –N(R)(R); и
R в каждом случае независимо представляет собой необязательно замещенный C1-C12 алкил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, имеющее структуру, соответствующую формуле (II):
 (II).
(II).
3. Соединение по п.1, имеющее структуру, соответствующую формуле (IIb):
 (IIb).
(IIb).
4. Соединение по п.1, имеющее структуру, соответствующую формуле (IIa):
 (IIa).
(IIa).
5. Соединение по любому из пп.1-4, где R4 и R5 представляют собой одинаковые функциональные группы, выбранные из –OH, –N(R)(R), –(CH2)–OH или –(CH2)–N(R)(R).
6. Соединение по любому из пп.1-4, где R5 представляет собой водород, и R4 представляет собой –OH, –N(R)(R), –(CH2)–OH или –(CH2)–N(R)(R).
7. Соединение по любому из пп.1-4, где R4 представляет собой водород, и R5 представляет собой OH, –N(R)(R), –(CH2)–OH или –(CH2)–N(R)(R).
8. Соединение по любому из пп.1-4, где R4 и R5 независимо выбирают из –OH и –OR.
9. Соединение по любому из пп.1-8, где R в каждом случае независимо представляет собой C1-4 алкил.
10. Соединение по любому из пп.1-9, где R2 представляет собой C1-4 линейный или разветвленный алкокси.
11. Соединение по любому из пп.1-10, где R2 представляет собой метокси, этокси, пропокси или изопропокси.
12. Соединение по любому из пп.1-9, где R2 представляет собой C3-6 циклоалкокси.
13. Соединение по п.12, где R2 представляет собой циклопропокси.
14. Соединение, имеющее структуру, соответствующую формуле (III):
 (III),
(III),
где «пунктирная» связь может представлять собой одинарную или двойную связь;
R1 представляет собой фенил;
R2 представляет собой С1-С6 алкокси;
R3 представляет собой водород или –CH2–X;
R6 и R7 независимо представляют собой водород или R; и
z1–z8 в каждом случае независимо выбирают из CH; где
–X в каждом случае независимо выбирают из –OH или –N(R)(R); и
R в каждом случае независимо представляют собой C1-C6 алкил;
при условии, что в каждом случае, когда R6 представляет собой водород, R3 представляет собой –CH2–N(R)(R) и указанная «пунктирная» связь представляет собой двойную связь,
или его фармацевтически приемлемая соль.
15. Соединение по п.14, имеющее структуру, соответствующую формуле (IV):
 (IV).
(IV).
16. Соединение по п.14 или 15, где указанная «пунктирная» связь представляет собой двойную связь.
17. Соединение по п.14 или 15, где R6 представляет собой C1-4 алкил.
18. Соединение по любому из пп.14-17, где R2 представляет собой метокси, этокси, пропокси или изопропокси.
19. Соединение по п.1 или 14, где указанное соединение выбирают из группы, состоящей из соединений
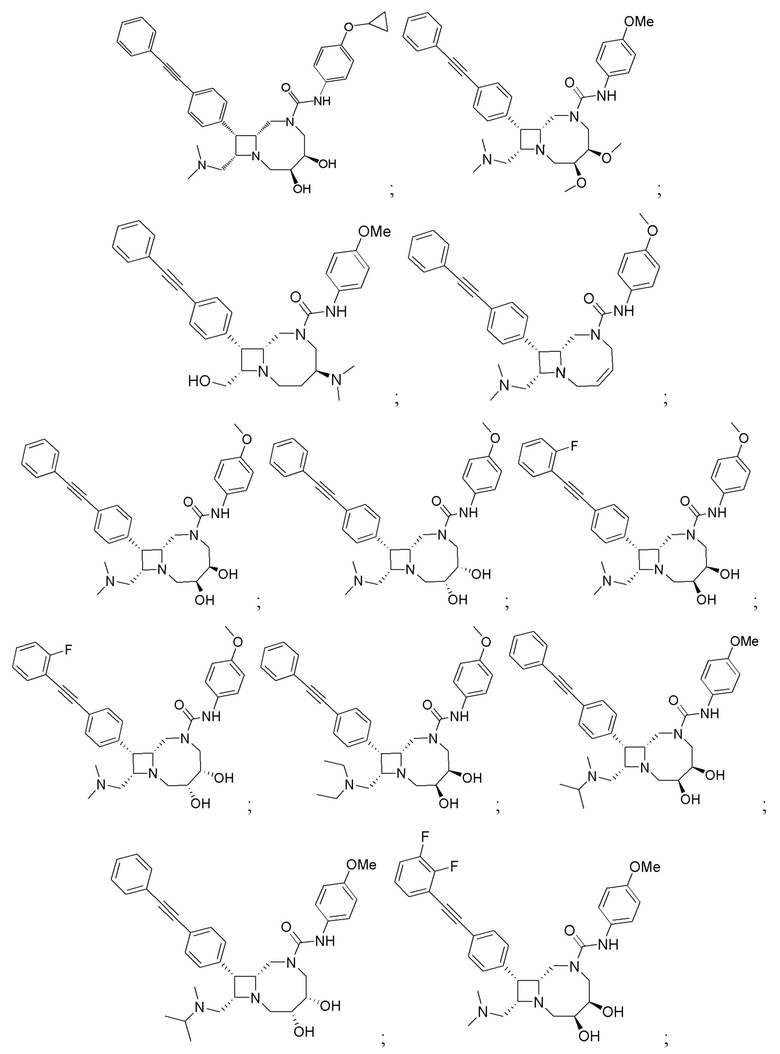

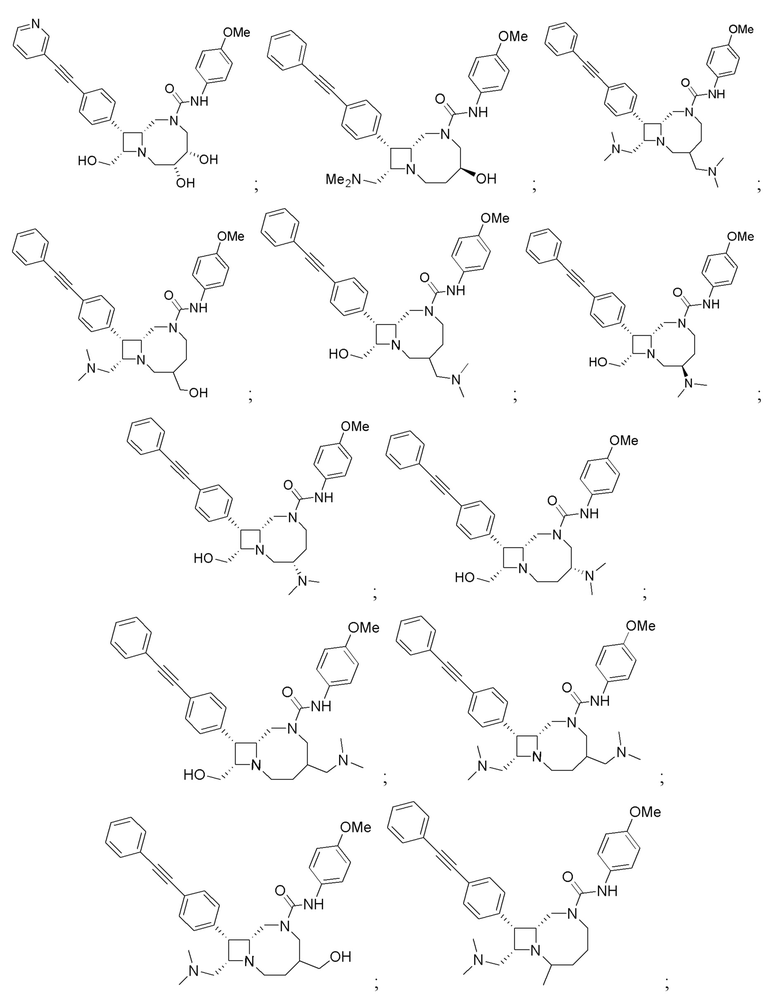

или их фармацевтически приемлемых солей.
20. Фармацевтическая композиция для лечения или профилактики паразитарного заболевания, выбранного из малярии и криптоспоридиоза, содержащая фармацевтически приемлемый наполнитель и эффективное количество соединения по любому из пп.1-19 или его фармацевтически приемлемую соль.
21. Фармацевтическая композиция по п.20, приготовленная в виде ветеринарной композиции.
22. Фармацевтическая композиция по любому из пп.20-21, в которой соединение находится в количестве, эффективном для лечения малярии.
23. Фармацевтическая композиция по п.20, где указанное соединение присутствует в эффективном количестве для лечения криптоспоридиоза.
24. Способ лечения или профилактики паразитарного заболевания, выбранного из малярии и криптоспоридиоза, у субъекта, содержащий введение субъекту эффективного количества соединения по любому из пп.1-19 или его фармацевтически приемлемой соли.
25. Способ по п.24, где указанное паразитарное заболевание представляет собой малярию.
26. Способ по п.24, где указанная малярия является фармакорезистентной малярией.
27. Способ по п.26, где фармакорезистентная малярия является резистентной к хлорохину, хинину, пириметамину, сульфадоксину, мефлохину, артеметеру, люмефантрину, артесунату, амодиахину, дигидроартемизинину, пипераквину, прогуанилу, доксициклину, клиндамицину, артемизинину, атовахину или любой их комбинации.
28. Способ по п.24, где указанная малярия представляет собой малярию печеночной стадии.
29. Способ по п.28, где печень указанного субъекта инфицирована вызывающим малярию паразитом и указанное лечение предотвращает распространение указанной инфекции за пределы печени.
30. Способ по п.24, где указанная малярия представляет собой малярию на стадии развития в крови.
31. Способ по п.24, где указанная малярия представляет собой малярию на трансмиссивной стадии.
32. Способ по п.24, где указанная малярия переносится видом москита, выбранным из P. falciparum, P. vivax, P. ovale, P. malariae, P. knowlesi, P. berghei, P. chabaudi, P. vinckei или P. yoelii.
33. Способ по п.30, где указанный вид москита представляет собой P. falciparum.
34. Способ по п.24, где указанное паразитарное заболевание представляет собой криптоспоридиоз.
35. Способ по п.34, где указанный криптоспоридиоз переносится C. parvum.
36. Способ по п.24, где указанный субъект представляет собой человека.
37. Способ по п.24, где указанный субъект не является человеком.
38. Способ по п.37, где указанный субъект представляет собой мышь, крысу, кролика, не относящегося к человеку примата, ящериц, гекконов, сову, теленка, овцу, ягненка, лошадь, жеребенка, свинью или поросенка.
39. Способ лечения или профилактики паразитарного заболевания, выбранного из малярии и криптоспоридиоза, у субъекта, включающий введение субъекту фармацевтической композиции по любому из пп.20-23.
40. Способ по п.39, где указанное паразитарное заболевание представляет собой малярию.
41. Способ по п.39, где указанная малярия является фармакорезистентной малярией.
42. Способ по п.41, где фармакорезистентная малярия является резистентной к хлорохину, хинину, пириметамину, сульфадоксину, мефлохину, артеметеру, люмефантрину, артесунату, амодиахину, дигидроартемизинину, пипераквину, прогуанилу, доксициклину, клиндамицину, артемизинину, атовахину или любой их комбинации.
43. Способ по п.39, где указанная малярия представляет собой малярию печеночной стадии.
44. Способ по п.43, где печень указанного субъекта инфицирована вызывающим малярию паразитом и указанное лечение предотвращает распространение указанной инфекции за пределы печени.
45. Способ по п.39, где указанная малярия представляет собой малярию на стадии развития в крови.
46. Способ по п.39, где указанная малярия представляет собой малярию на трансмиссивной стадии.
47. Способ по п.39, где указанная малярия переносится видом москита, выбранным из P. falciparum, P. vivax, P. ovale, P. malariae, P. knowlesi, P. berghei, P. chabaudi, P. vinckei или P. yoelii.
48. Способ по п.39, где указанный вид москита представляет собой P. falciparum.
49. Способ по п.39, где указанное паразитарное заболевание представляет собой криптоспоридиоз.
50. Способ по п.49, где указанный криптоспоридиоз переносится C. parvum.
51. Способ по п.39, где указанный субъект представляет собой человека.
52. Способ по п.39, где указанный субъект не является человеком.
53. Способ по п.52, где указанный субъект представляет собой мышь, крысу, кролика, не относящегося к человеку примата, ящериц, гекконов, сову, теленка, овцу, ягненка, лошадь, жеребенка, свинью или поросенка.
54. Соединение по п.1, где указанное соединение имеет структуру
 ;
;
или его фармацевтически приемлемая соль.
55. Соединение по п.54, где указанное соединение имеет структуру

56. Фармацевтическая композиция по п.20, где указанная фармацевтическая композиция содержит соединение, имеющее структуру
 ;
;
или его фармацевтически приемлемую соль.
57. Фармацевтическая композиция по п.56, где указанная фармацевтическая композиция содержит соединение, имеющее структуру
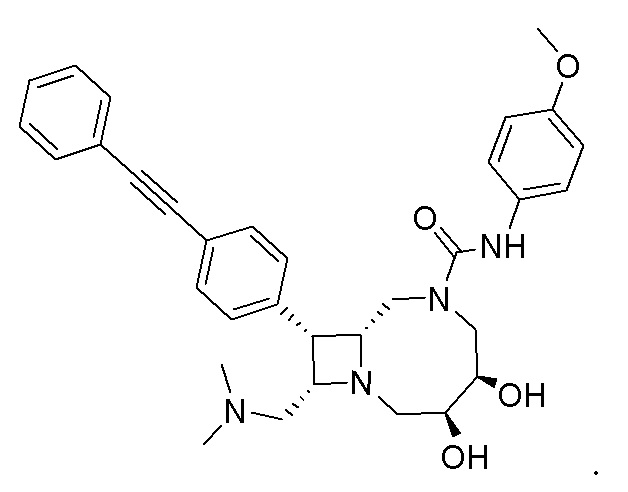
58. Способ по п.24, где указанное соединение имеет структуру
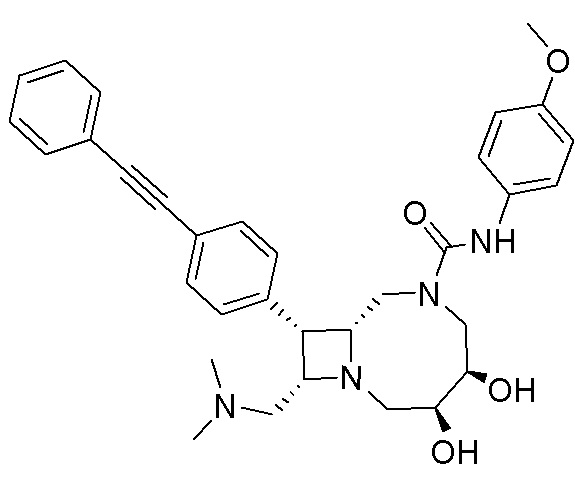 ;
;
или его фармацевтически приемлемая соль.
59. Способ по п.58, где указанное соединение имеет структуру

60. Способ по п.58, где указанная фармацевтическая композиция содержит соединение, имеющее структуру
 ;
;
или его фармацевтически приемлемую соль.
61. Способ по п.60, где указанная фармацевтическая композиция содержит соединение, имеющее структуру

| US 20160289235 A1, 06.10.2016 | |||
| NOBUTAKA KATO et al | |||
| Nature, vol | |||
| Радиоприемное устройство | 1922 |
|
SU538A1 |
| Замок с запорным засовом и ручкой | 1927 |
|
SU7625A1 |
| Способ получения жидкой протравы для основных красителей | 1923 |
|
SU344A1 |
| US 3176017 A1, 30.03.1965 | |||
| ПРОИЗВОДНЫЕ ДИАЗАБИЦИКЛИЧЕСКИХ АЛКАНОВ С NK-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2003 |
|
RU2310656C2 |
| ПРОИЗВОДНЫЕ АРТЕМИЗИНИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1999 |
|
RU2236413C2 |

