Перекрестная ссылка на родственные заявки
CN201711058653.5, дата подачи: 01 ноября 2017 г.
Область техники, к которой относится изобретение
Настоящее изобретение относится к макроциклическому соединению и к его применению в изготовлении лекарственного препарата для лечения заболеваний, связанных с Wee1, конкретно относится к соединению, представленному формулой (II), его изомеру или его фармацевтически приемлемой соли.
Уровень техники изобретения
Ход клеточного цикла является сложным процессом, контролируемым рядом регуляторных систем клеточного цикла. Основным компонентом регуляторных систем клеточного цикла являются комплексы CKD/циклинов, образованные комбинацией циклин-зависимых киназ (CDK) и циклинов, которые могут способствовать вхождению клетки в цикл пролиферации, в котором комплекс CDK1 (человеческого гомолога, также известного как CDC2)/циклина B играет ключевую роль в контроле вхождения клетки в M-фазу.
Перед вхождением клетки в M-фазу репликация ДНК должна завершиться. Вследствие воздействия различных эндогенных и экзогенных факторов зачастую ДНК будет мутирована или повреждена. Необходима репарация таких аномальных ДНК, в противном случае они будут обуславливать митотическую катастрофу и гибель клеток. Основной функцией контрольных точек клеточного цикла является задержка клеточного цикла и обеспечение завершения клеткой репарации ДНК перед вхождением в M-фазу. Контрольная точка G1/S в конце G1-фазы и контрольная точка G2/M в G2-фазе представляют собой две основные контрольные точки клеточного цикла, которые ответственны за идентификацию и репарацию повреждения ДНК. В нормальных клетках может применяться контрольная точка G1/S для завершения репарации ДНК на G1-фазе. Однако около 50% раковых клеток имеют дефект гена-супрессора опухолей p53, который обуславливает у них отсутствие функции контрольной точки G1/S, поэтому они должны больше полагаться на контрольную точку G2/M для завершения репарации ДНК. Контрольная точка G2/M редко поддается мутации. По этой причине раковые клетки могут избежать обработки средствами для повреждения ДНК и облучения.
Протеинкиназа Wee1 является регулятором клеточного цикла, который принадлежит к семейству серин- и треонин-протеинкиназ в ядре, и является ключевой киназой в контрольной точке G2/M. Семейство протеинкиназ «Wee1» у человека в основном включает Wee1 и Myt1, которые могут фосфорилировать сайт Tyr15 CDC2, подавлять активацию комплекса CDC2/циклина B и предотвращать вступление клеток в M-фазу до завершения репарации ДНК. Myt1 также может фосфорилировать сайт Thr14 на CDC2, что также обеспечивает негативную регуляцию активности CDC2. Киназа Wee1 высоко экспрессируется во множестве раковых клеток. Посредством ингибирования киназы Wee1 опухолевые клетки могут непосредственно пропустить репарацию ДНК на G2-фазе и вступать в митоз заблаговременно, что приводит к смерти опухолевых клеток и обеспечивает достижение цели лечения рака.
В настоящее время ингибитор Wee1, разработанный AstraZeneca, AZD1775, достиг фазы II клинических испытаний, и более 30 клинических испытаний находятся на стадии разработки, в которых были показаны хорошие терапевтические эффекты. AZD1775 впервые был разработан Merck, поэтому он также известен как MK-1775. В сентябре 2013 г. Merck передала соединение AstraZeneca Worldwide, и связанные патенты главным образом включают US20070254892, WO2007126122, EP2213673, WO2008133866, WO2011034743 и т. д. Abbott и Abbvie также провели изучение ингибиторов Wee1, и связанные с этим патенты главным образом включают US2012220572, WO2013126656, WO2013012681, WO2013059485, WO2013013031, WO2013126656 и т. д. Патенты Almac в отношении ингибиторов Wee1 включают WO2014167347, WO2015019037 и WO2015092431.
В WO2008133866 раскрыто соединение AZD1775, структура которого является следующей:
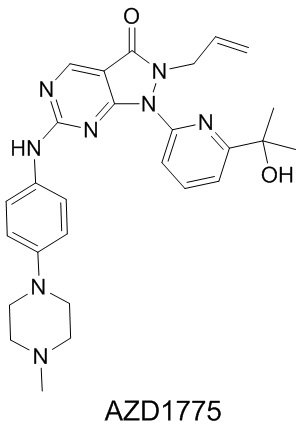 .
.
Подробное описание изобретения
Раскрывается соединение, представленное формулой (II), его изомер или его фармацевтически приемлемая соль,
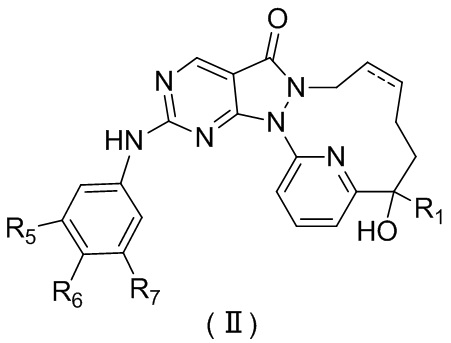 ,
,
где
 представляет собой одинарную связь или двойную связь;
представляет собой одинарную связь или двойную связь;
R1 выбран из H и C1-3алкила, где C1-3алкил необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R5 выбран из H и C1-3алкила, где C1-3алкил необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R6 выбран из R61,  ,
,  и
и 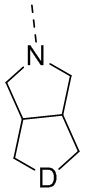 ;
;
r равняется 1 или 2;
m равняется 1 или 2;
D выбран из -N(R2)-, -N+(O-)(R2)- и -C(R3)(R4)-;
R2 выбран из H и C1-3алкила, где C1-3алкил необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R3 и R4 независимо выбраны из H, F, Cl, Br, I, OH, NH2 и C1-3алкила, где NH2 и C1-3алкил необязательно замещены R, и при этом число R равняется 1, 2 или 3;
в качестве альтернативы R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 5-7-членный циклоалкил или 5-7-членный гетероциклоалкил, где 5-7-членный циклоалкил и 5-7-членный гетероциклоалкил необязательно замещены R, и при этом число R равняется 1, 2 или 3;
R61 выбран из H, F, Cl, Br, I, OH, NH2, C1-3алкокси и -O-C3-6циклоалкила, где C1-3алкокси и -O-C3-6циклоалкил необязательно замещены R, и при этом число R равняется 1, 2 или 3;
R7 выбран из H, F, Cl, Br, I, OH, NH2, C1-3алкила, C1-3алкокси и 5-6-членного гетероциклоалкила, где C1-3алкил, C1-3алкокси и 5-6-членный гетероциклоалкил необязательно замещены R, и при этом число R равняется 1, 2 или 3;
в качестве альтернативы R6 и R7 вместе с атомами кольца, к которым они присоединены, образуют кольцо A, и при этом кольцо A выбрано из 5-7-членного гетероциклоалкила, который необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R независимо выбран из F, Cl, Br, I, OH, NH2, C1-3алкила, C1-3алкокси и C1-3алкиламино;
5-7-членный гетероциклоалкил содержит 1, 2, 3 или 4 гетероатома или гетероатомные группы, независимо выбранные из –NH-, -S- и N.
В некоторых вариантах осуществления настоящего изобретения R независимо выбран из F, Cl, Br, I, OH, NH2, CH3, Et, -OCH3 и 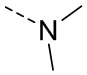 , и другие переменные определены в настоящем изобретении.
, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R1 выбран из H, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R2 выбран из H, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R3 и R4 независимо выбраны из H, F, Cl, Br, I, OH, NH2, -NH(CH3), -N(CH3)2, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R3 выбран из H, F, Cl, Br, I, OH, NH2, -NH(CH3), -N(CH3)2, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R4 выбран из H, F, Cl, Br, I, OH, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R5 выбран из H, CH3 и Et, где CH3 и Et необязательно замещены R, и при этом число R равняется 1, 2 или 3; и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R5 выбран из H, CH3 и –CH2OH, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R61 выбран из H, F, Cl, Br, I, OH, NH2, -OCH3, 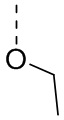 и
и 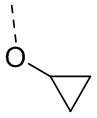 , где -OCH3, и необязательно замещены R, и при этом число R равняется 1, 2 или 3; и другие переменные определены в настоящем изобретении.
, где -OCH3, и необязательно замещены R, и при этом число R равняется 1, 2 или 3; и другие переменные определены в настоящем изобретении.
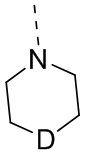
В некоторых вариантах осуществления настоящего изобретения R61 выбран из H, F, Cl, Br, I, OH, NH2, -OCH3, 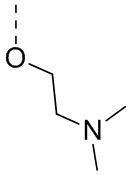 и , и другие переменные определены в настоящем изобретении.
и , и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R6 выбран из R61, 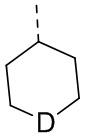 , ,
, ,  и
и  , и другие переменные определены в настоящем изобретении.
, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R6 выбран из R61, 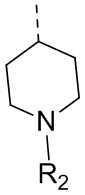 ,
,  ,
, 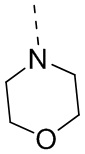 ,
,  ,
, 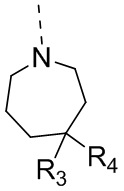 ,
, 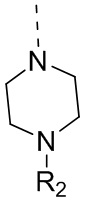 ,
, 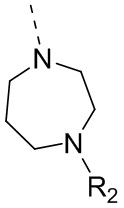 и
и  , и другие переменные определены в настоящем изобретении.
, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 5-7-членный циклоалкил или 5-7-членный гетероциклоалкил, при этом 5-7-членный циклоалкил или 5-7-членный гетероциклоалкил необязательно замещен R, и при этом число R равняется 1, 2 или 3; где фрагмент  представляет собой
представляет собой 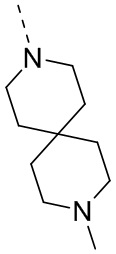 , и другие переменные определены в настоящем изобретении.
, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R6 выбран из H, F, Cl, Br, I, OH, NH2, -OCH3, , , 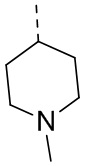 ,
,  ,
,  и другие переменные определены в настоящем изобретении.
и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R7 выбран из H, F, Cl, Br, I, OH, NH2, CH3, -OCH3, и  , где CH3, -OCH3, и необязательно замещены R, и при этом число R равняется 1, 2 или 3; и другие переменные определены в настоящем изобретении.
, где CH3, -OCH3, и необязательно замещены R, и при этом число R равняется 1, 2 или 3; и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R7 выбран из H, F, Cl, Br, I, OH, NH2, -OCH3,  , и
, и  , и другие переменные определены в настоящем изобретении.
, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения кольцо A выбрано из пиперидила, который необязательно замещен R, и при этом число R равняется 1, 2 или 3, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения фрагмент 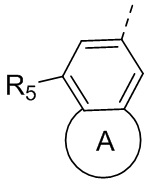 представляет собой
представляет собой  , и другие переменные определены в настоящем изобретении.
, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения фрагмент 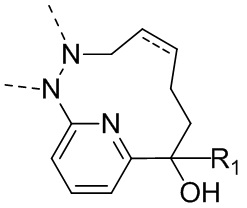 выбран из
выбран из 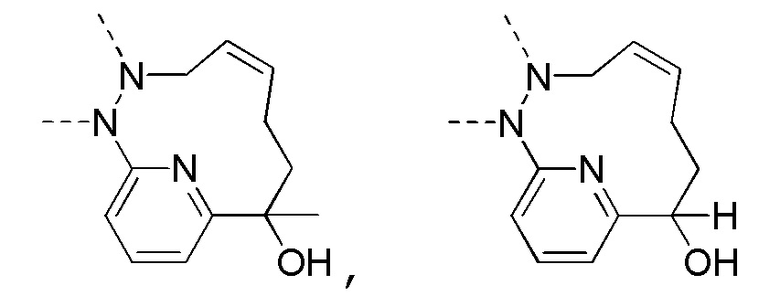 , и
, и 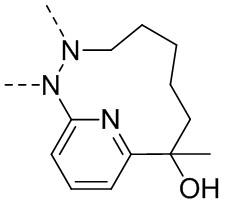 , и другие переменные определены в настоящем изобретении.
, и другие переменные определены в настоящем изобретении.
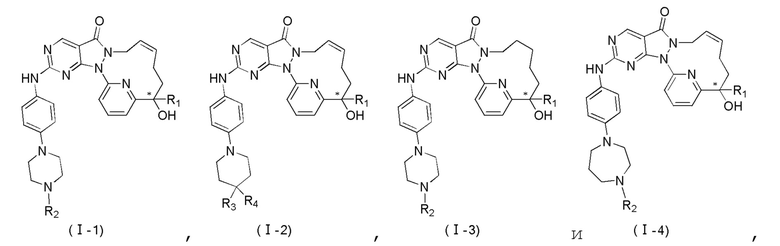
В некоторых вариантах осуществления настоящего изобретения соединение, его изомер или его фармацевтически приемлемая соль выбраны из
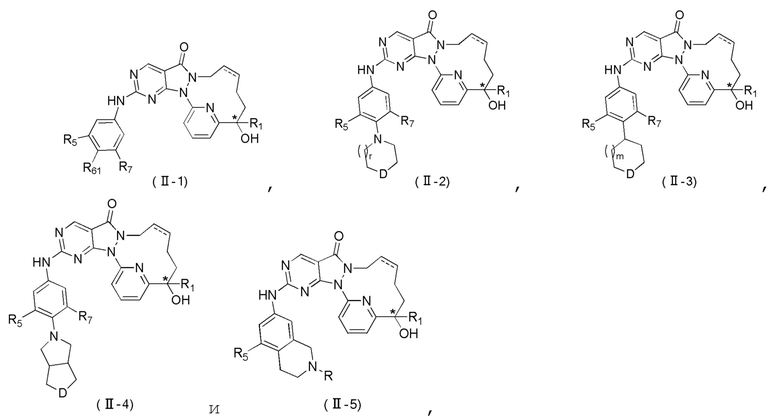
где
D выбран из -N(R2)-, -N+(O-)(R2)- и -C(R3)(R4)-;
r, m, R1, R2, R3, R4, R5, R61 и R7 определены в настоящем изобретении;
атом углерода, обозначенный с помощью «*», представляет собой хиральный атом углерода, и находятся в форме одного (R)- или (S)-энантиомера или обогащены одним энантиомером.
В некоторых вариантах осуществления настоящего изобретения соединение, его изомер или его фармацевтически приемлемая соль выбраны из
 ,
,
где
D выбран из -N(R2)-, -N+(O-)(R2)- и -C(R3)(R4)-;
r, R1, R2, R3, R4 определены в настоящем изобретении;
атом углерода, обозначенный с помощью «*», представляет собой хиральный атом углерода, и находятся в форме одного (R)- или (S)-энантиомера или обогащены одним энантиомером.
В некоторых вариантах осуществления настоящего изобретения фрагмент 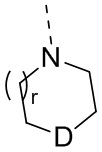 выбран из
выбран из  и
и 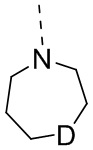 , и другие переменные определены в настоящем изобретении.
, и другие переменные определены в настоящем изобретении.
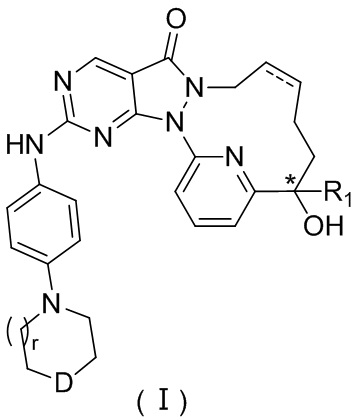
В настоящем изобретении предусмотрены соединение, представленное формулой (I), его изомер или его фармацевтически приемлемая соль,
 ,
,
где
представляет собой одинарную связь или двойную связь;
r равняется 1 или 2;
D выбран из -N(R2)- и -C(R3)(R4)-;
R1 выбран из H и C1-3алкила, где C1-3алкил необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R2 выбран из H и C1-3алкила, где C1-3алкил необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R3 и R4 независимо выбраны из H, F, Cl, Br, I, OH, NH2 и C1-3алкила, где NH2 или C1-3алкил необязательно замещен R, и при этом число R равняется 1, 2 или 3;
в качестве альтернативы R3 и R4 соединены с образованием 5-7-членного циклоалкила или гетероциклоалкила, при этом 5-7-членный циклоалкил и гетероциклоалкил необязательно замещены R, и при этом число R равняется 1, 2 или 3;
R независимо выбран из F, Cl, Br, I, OH, NH2 и C1-3алкила;
5-7-членный гетероциклоалкил содержит 1, 2, 3 или 4 гетероатома или гетероатомные группы, независимо выбранные из –NH-, -S- и N;
атом углерода, обозначенный с помощью «*», представляет собой хиральный атом углерода, и при этом они находятся в форме одного (R)- или (S)-энантиомера или обогащены одним энантиомером.
В некоторых вариантах осуществления настоящего изобретения R независимо выбран из F, Cl, Br, I, OH, NH2, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R1 выбран из H, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R2 выбран из H, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R3 и R4 независимо выбраны из H, F, Cl, Br, I, OH, NH2, -NH(CH3), -N(CH3)2, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R3 выбран из H, F, Cl, Br, I, OH, NH2, -NH(CH3), -N(CH3)2, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R4 выбран из H, F, Cl, Br, I, OH, CH3 и Et, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения фрагмент 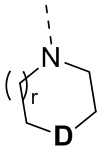 выбран из
выбран из  и
и  , и другие переменные определены в настоящем изобретении.
, и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения фрагмент выбран из  ,
,  , и , и другие переменные определены в настоящем изобретении.
, и , и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения R3 и R4 соединены с образованием 5-7-членного циклоалкила или гетероциклоалкила, при этом 5-7-членный циклоалкил или гетероциклоалкил необязательно замещен R, и при этом число R равняется 1, 2 или 3; где фрагмент представляет собой , и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения фрагмент выбран из  и другие переменные определены в настоящем изобретении.
и другие переменные определены в настоящем изобретении.
В некоторых вариантах осуществления настоящего изобретения фрагмент выбран из  , и , и другие переменные определены в настоящем изобретении.
, и , и другие переменные определены в настоящем изобретении.
Некоторые варианты осуществления настоящего изобретения получены за счет любой комбинации указанных выше переменных.
В некоторых вариантах осуществления настоящего изобретения соединение, его изомер или его фармацевтически приемлемая соль выбраны из
где
R1, R2, R3, R4 определены в настоящем изобретении.
В настоящем изобретении также предусмотрены соединение, показанное ниже, его изомер или его фармацевтически приемлемая соль, которое выбрано из
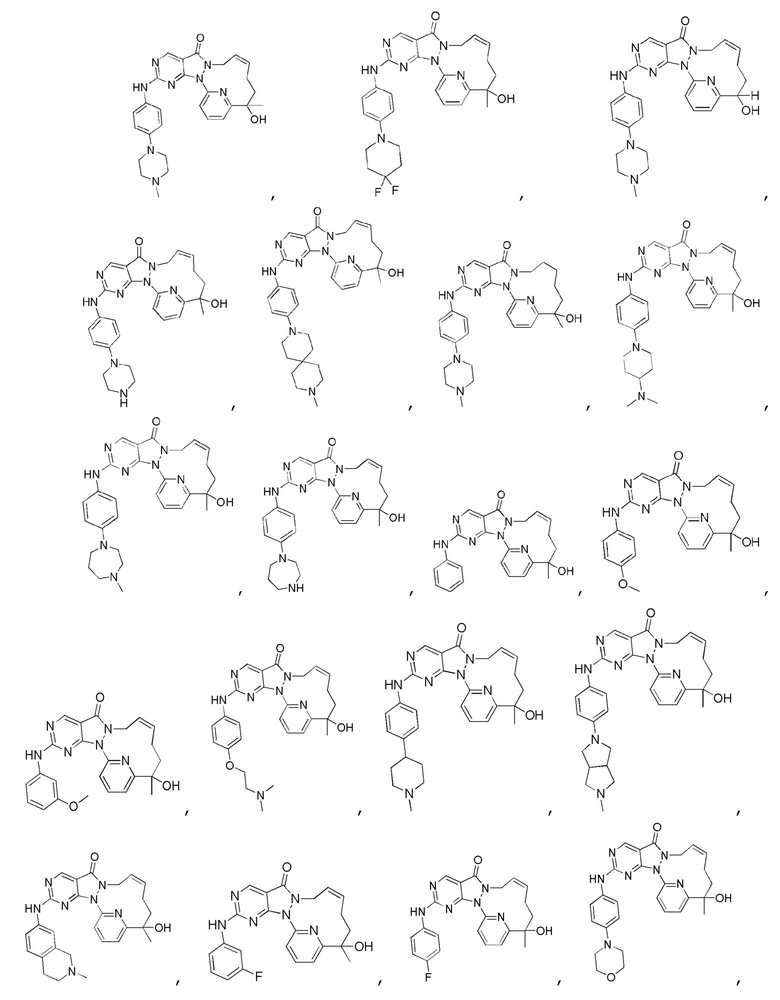

В некоторых вариантах осуществления настоящего изобретения соединение, его изомер или фармацевтически приемлемая соль выбраны из

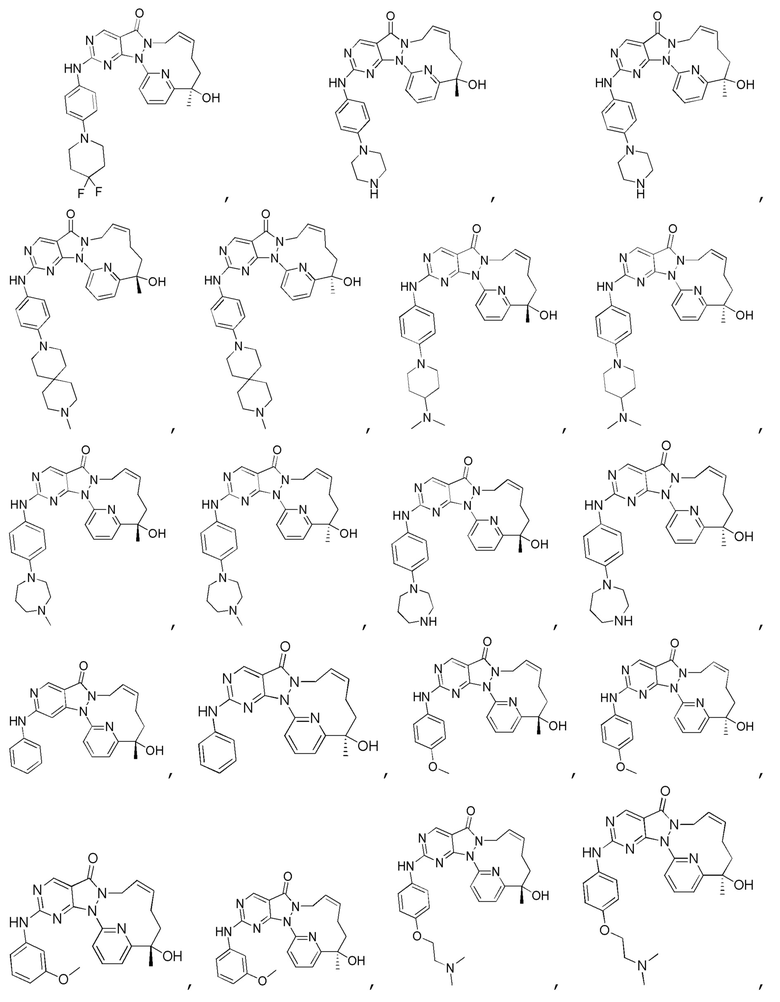
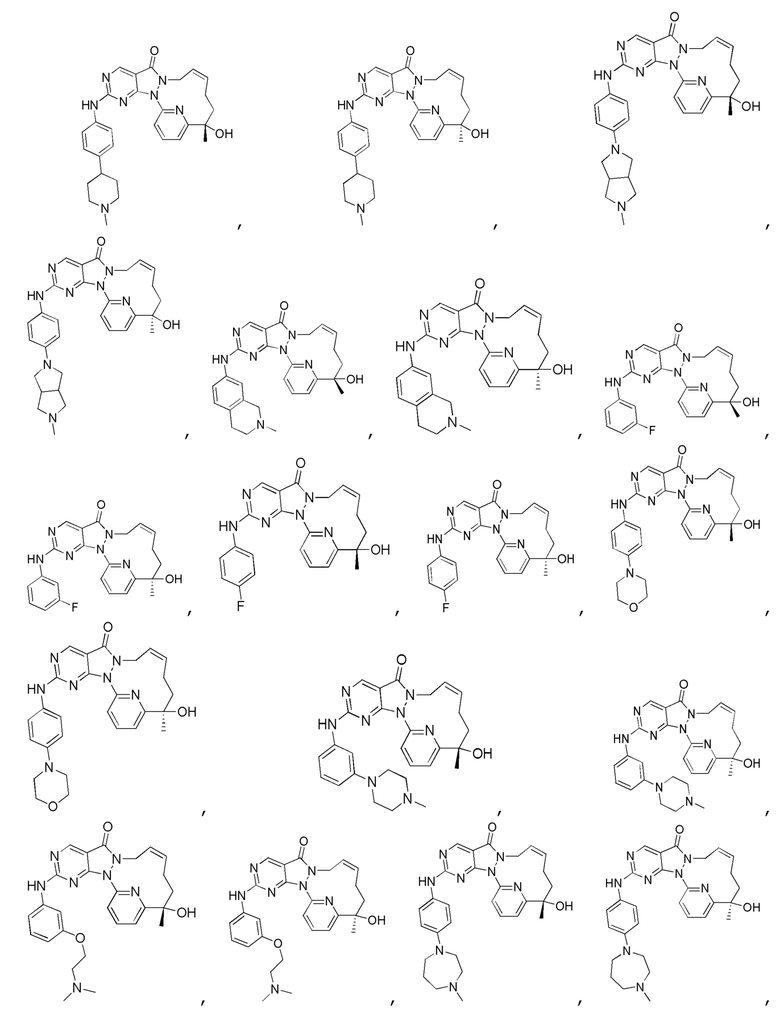

В настоящем изобретении также предусмотрено применение соединения или его фармацевтически приемлемой соли в изготовлении лекарственного препарата, предназначенного для лечения заболеваний, связанных с Wee1.
В некоторых вариантах осуществления настоящего изобретения в указанном выше применении лекарственный препарат применяется для лечения солидных опухолей.
Технические эффекты
В качестве нового ингибитора Wee1 соединение по настоящему изобретению обладает хорошим ингибирующим эффектом в отношении киназы Wee1 и характеризуется хорошей проницаемостью. В отношении фармакокинетики оно характеризуется несколькими в значительной степени улучшенными фармакокинетическими показателями, включающими скорость выведения in vivo, период полувыведения, интегральную концентрацию in vivo и биодоступность. С точки зрения эффективности лекарственного средства in vivo соединение по настоящему изобретению характеризуется в значительной степени улучшенным эффектом в отношении подавления опухоли, и при этом хиральность соединения оказывает неожиданный эффект в отношении эффективности лекарственного средства in vivo.
Определения и описание
Если не указано иное, при использовании в описании и формуле настоящего изобретения следующие термины имеют следующие значения. Конкретный термин или выражение при отсутствии точного определения не следует считать неопределенным или неясным, а следует понимать в соответствии с общепринятым значением. Если в данном документе встречается торговое название, то предполагается, что оно относится к соответствующему продукту или его активному ингредиенту. Термин «фармацевтически приемлемый» используется в данном документе применительно к тем соединениям, материалам, композициям и/или лекарственным формам, которые в рамках тщательной медицинской оценки являются подходящими для применения в контакте с тканями человека и животного без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений в соответствии с обоснованным соотношением польза/риск.
Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которую получают путем осуществления реакции соединения, содержащего конкретный заместитель по настоящему изобретению, с относительно нетоксичными кислотой или основанием. Если соединение по настоящему изобретению содержит относительно кислотную функциональную группу, то соль присоединения основания может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения основания включает соль натрия, калия, кальция, аммония, органического амина или магния, или подобные соли. Если соединение по настоящему изобретению содержит относительно основную функциональную группу, то соль присоединения кислоты может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, где неорганическая кислота включает, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту и т. п.; и соль органической кислоты, где органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту, метансульфоновую кислоту и т. п.; и соль аминокислоты (такой как аргинин и т. п.), и соль органической кислоты, такой как глюкуроновая кислота и т. п. Некоторые конкретные соединения по настоящему изобретению, которые содержат как основные, так и кислотные функциональные группы, могут быть превращены в любую соль присоединения основания или кислоты.
Фармацевтически приемлемая соль по настоящему изобретению может быть получена из исходного соединения, которое содержит кислотный или основный фрагмент, с помощью традиционного химического способа. Как правило, такая соль может быть получена путем осуществления реакции свободной кислотной или основной формы соединения со стехиометрическим количеством соответствующих основания или кислоты в воде или органическом растворителе или в их смеси.
Соединение по настоящему изобретению может характеризоваться конкретной геометрической или стереоизомерной формой. В настоящем изобретении подразумеваются все такие соединения, в том числе цис- и транс-изомер, (−)- и (+)-энантиомер, (R)- и (S)-энантиомер, диастереоизомер, (D)-изомер, (L)-изомер, а также рацемическая смесь и другие смеси, например, энантиомерно или диастереоизомерно обогащенная смесь, все из которых охватываются объемом настоящего изобретения. Заместитель, такой как алкил, может содержать дополнительный асимметричный атом углерода. Все данные изомеры и их смеси охватываются объемом настоящего изобретения.
Если не указано иное, термины «энантиомер» или «оптический изомер» относятся к стереоизомерам, которые представляют собой зеркальные отражения друг друга.
Если не указано иное, термины «цис-транс-изомер» или «геометрический изомер» определяются неспособностью к свободному вращению вокруг двойной связи или одинарной связи между атомами углерода в кольце.
Если не указано иное, термин «диастереомер» относится к стереоизомерам, молекулы которых имеют два или более хиральных центров и не являются зеркальными отражениями друг друга.
Если не указано иное, «(D)» или «(+)» обозначает правостороннее вращение, «(L)» или «(−)» обозначает левостороннее вращение, «(DL)» или «(±)» обозначает рацемизацию.
Если не указано иное, абсолютная конфигурация стереогенного центра представлена связью, обозначенной клиновидной сплошной линией ( ), и связью, обозначенной клиновидной пунктирной линией (
), и связью, обозначенной клиновидной пунктирной линией ( ), а относительная конфигурация стереогенного центра представлена связью, обозначенной прямой сплошной линией (
), а относительная конфигурация стереогенного центра представлена связью, обозначенной прямой сплошной линией ( ), и связью, обозначенной прямой пунктирной линией (
), и связью, обозначенной прямой пунктирной линией ( ). Волнистая линия (
). Волнистая линия (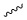 ) представляет связь, обозначенную клиновидной сплошной линией (), или связь, обозначенную клиновидной пунктирной линией (), или представляет связь, обозначенную прямой сплошной линией (), или связь, обозначенную прямой пунктирной линией ().
) представляет связь, обозначенную клиновидной сплошной линией (), или связь, обозначенную клиновидной пунктирной линией (), или представляет связь, обозначенную прямой сплошной линией (), или связь, обозначенную прямой пунктирной линией ().
Соединение по настоящему изобретению может находиться в конкретной форме. Если не указано иное, термины «таутомер» или «таутомерная форма» относятся к тому факту, что различные функциональные изомеры находятся в состоянии динамического равновесия при комнатной температуре и могут быстро превращаться друг в друга. Если возможно наличие таутомеров (как, например, в растворе), то таутомеры могут достигать состояния химического равновесия. Например, протонные таутомеры (также известные как прототропные таутомеры) предусматривают взаимопревращения посредством миграции протона, такие как кето-енольная изомеризация и имино-енаминовая изомеризация. Валентный таутомер предусматривает взаимное преобразование с участием некоторых связывающих электронов. Конкретным примером кето-енольной таутомеризации является взаимопревращение двух таутомеров – пентан-2,4-диона и 4-гидроксипент-3-ен-2-она.
Если не указано иное, термины «обогащенный одним изомером», «обогащенный изомером», «обогащенный одним энантиомером» или «обогащенный энантиомером» относятся к содержанию одного из изомеров или энантиомеров, которое составляет менее 100%, и при этом содержание изомера или энантиомера составляет 60% или больше, или 70% или больше, или 80% или больше, или 90% или больше, или 95% или больше, или 96% или больше, или 97% или больше, или 98% или больше, или 99% или больше, или 99,5% или больше, или 99,6% или больше, или 99,7% или больше, или 99,8% или больше, или 99,9% или больше.
Если не указано иное, термины «избыток изомера» или «избыток энантиомера» относятся к разнице между значениями относительного процентного содержания двух изомеров или энантиомеров. Например, если содержание одного из изомеров или энантиомеров составляет 90%, а другого – 10%, то избыток изомера или энантиомера (значение ee) составляет 80%.
Оптически активный (R)- и (S)-изомер или D- и L-изомер можно получить с применением хирального синтеза, или хиральных реагентов, или других традиционных методик. Если требуется получение одного типа энантиомера конкретного соединения по настоящему изобретению, чистый необходимый энантиомер может быть получен путем асимметрического синтеза или дериватизации с помощью хирального вспомогательного вещества с последующим разделением полученной диастереомерной смеси и отщеплением вспомогательной группы. В качестве альтернативы, если молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксильная), соединение реагирует с соответствующими оптически активными кислотой или основанием с образованием соли диастереомерного изомера, которую затем подвергают диастереомерному разделению посредством традиционного способа, известного из уровня техники, с получением чистого энантиомера. Кроме того, энантиомер и диастереоизомер обычно выделяют посредством хроматографии, в которой используется хиральная неподвижная фаза, при этом необязательно в комбинации со способом химической дериватизации (например, карбамат, полученный из амина). Соединение по настоящему изобретению может содержать неприродное соотношение атомных изотопов при одном или более атомах, которые составляют соединение. Например, соединение может быть мечено радиоактивным изотопом, таким как тритий (3H), йод-125 (125I) или C-14 (14C). В качестве другого примера водород может быть заменен тяжелым водородом с образованием дейтерированного лекарственного средства, и при этом связь, образуемая между дейтерием и углеродом, является более прочной, чем связь, образуемая между обычным водородом и углеродом. По сравнению с недейтерированными лекарственными средствами дейтерированные лекарственные средства характеризуются менее выраженными побочными эффектами и более высокой стабильностью лекарственного средства с усилением эффективности и продлением биологического периода полувыведения лекарственного средства. Все изотопные варианты соединения по настоящему изобретению, вне зависимости от того, являются ли они радиоактивными, охватываются объемом настоящего изобретения. Термин «фармацевтически приемлемый носитель» относится к любому средству или несущей среде, которые способны доставлять эффективное количество активного вещества по настоящему изобретению, не оказывают отрицательного воздействия на биологическую активность активного вещества и не вызывают какого-либо токсичного побочного эффекта у хозяина или пациента. Иллюстративный носитель включает воду, растительное и минеральное масло, основу для крема, основу для лосьона, основу для мази и т. п. Основа содержит суспендирующее средство, загуститель, вещество, способствующее проникновению, и т. п. Их составы хорошо известны специалистам в области косметических средств или в области фармацевтических препаратов для местного применения.
Применительно к лекарственному препарату или фармакологически активному средству термин «эффективное количество» или «терапевтически эффективное количество» относится к нетоксичному, но достаточному количеству для достижения необходимого эффекта лекарственного препарата или средства. В отношении лекарственной формы по настоящему изобретению для перорального применения «эффективное количество» активного вещества в композиции относится к количеству, необходимому для достижения необходимого эффекта при объединении с другим активным веществом в композиции. Эффективное количество отличается для каждого человека и определяется в зависимости от возраста и общего состояния реципиента, а также от конкретного активного вещества. Соответствующее эффективное количество в каждом отдельном случае может быть определено специалистом в данной области на основе обычного эксперимента.
Термины «активный ингредиент», «терапевтическое средство», «активное вещество» или «активное средство» относятся к химическому соединению, с помощью которого можно эффективно лечить целевое нарушение, заболевание или состояние.
«Необязательный» или «необязательно» означает, что последующее событие или условие может реализовываться, но не является необходимым, и что термин включает случаи, в которых событие или условие реализуется, и случаи, в которых событие или условие не реализуется.
Термин «замещенный» означает, что один или более атомов водорода при конкретном атоме замещены заместителем, в том числе дейтерием и вариантами водорода, при условии, что валентность конкретного атома является нормальной, и замещенное соединение является стабильным. Если заместитель представляет собой атом кислорода (т. е. =O), то это означает, что два атома водорода являются замещенными. Положения в ароматическом кольце не могут быть замещены кетоном. Термин «необязательно замещенный» означает, что атом может быть замещен или не замещен заместителем, если не указано иное, причем тип и число заместителей могут быть произвольными при условии, что это химически достижимо.
Если любая переменная (такая как R) встречается более одного раза в составе или структуре соединения, то определение переменной в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, то данная группа может быть необязательно замещена не более чем двумя R, при этом определение R в каждом случае является независимым. Более того, комбинация заместителя и/или его варианта является допустимой, только если такая комбинация приводит к образованию стабильного соединения.
Если число линкерных групп равно 0, например -(CRR)0-, это означает, что линкерная группа представляет собой одинарную связь.
Если одна из переменных выбрана из одинарной связи, это означает, что две группы, соединенные одинарной связью, соединены непосредственно. Например, если L в A-L-Z представляет собой одинарную связь, то структура A-L-Z фактически представляет собой A-Z.
Если в перечисленном заместителе не указано, посредством какого атома он присоединен к замещаемой группе, такой заместитель может быть связан посредством любого из его атомов. Например, пиридил как заместитель может быть присоединен к замещаемой группе посредством любого атома углерода на пиридиновом кольце. Если в перечисленной линкерной группе не указано направление связывания, то направление связывания является произвольным; например, если линкерная группа L, содержащаяся в 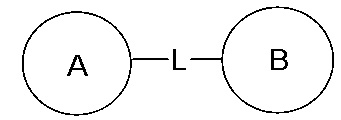 , представляет собой –M-W, то –M-W- может связывать кольцо A и кольцо B с образованием
, представляет собой –M-W, то –M-W- может связывать кольцо A и кольцо B с образованием 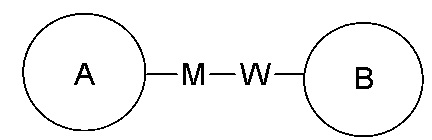 в направлении, соответствующем порядку чтения слева направо, и с образованием
в направлении, соответствующем порядку чтения слева направо, и с образованием  в направлении, противоположном порядку чтения слева направо. Комбинации линкерных групп, заместителей и/или их вариантов являются допустимыми, только если такие комбинации дают в результате стабильные соединения.
в направлении, противоположном порядку чтения слева направо. Комбинации линкерных групп, заместителей и/или их вариантов являются допустимыми, только если такие комбинации дают в результате стабильные соединения.
Если не указано иное, термин «гетеро» представляет гетероатом или гетероатомную группу (например, группу атомов, содержащую гетероатом), в том числе атом, отличный от атома углерода (C) и водорода (H), и группу атомов, содержащую вышеуказанный гетероатом, например, в том числе атом кислорода (O), азота (N), серы (S), кремния (Si), германия (Ge), алюминия (Al), бора (B), -O-, -S-, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2- и группу, состоящую из -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- и -S(=O)N(H)-, каждое из которых необязательно замещено.
Если не указано иное, термин «кольцо» относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Так называемое кольцо включает одинарное кольцо, двойное кольцо, спирокольцо, конденсированное кольцо или кольцо с мостиковой связью. Число атомов в кольце обычно определяется как число членов в кольце, например, «5-7-членное кольцо» означает, что от 5 до 7 атомов расположены в виде кольца. Если не указано иное, кольцо необязательно содержит от 1 до 3 гетероатомов. Следовательно, «5-7-членное кольцо» включает, например, фенил, пиридинил и пиперидинил; с другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает пиридил и пиперидинил, но не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую по меньшей мере одно кольцо, где каждое «кольцо» независимо соответствует вышеуказанному определению.
Если не указано иное, термин «гетероцикл» или «гетероцикло» относится к стабильному моноциклическому, бициклическому или трициклическому кольцу, содержащему гетероатом или гетероатомную группу, которое может быть насыщенным, частично ненасыщенным или ненасыщенным (ароматическим) и может содержать атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, независимо выбранных из N, O и S, где любой из вышеуказанного гетероцикла может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Гетероатомы, представляющие собой азот и серу, необязательно могут быть окислены (т. е., NO и S(O)p, p равняется 1 или 2). Атом азота может быть замещенным или незамещенным (т. е. N или NR, где R представляет собой H или другие заместители, уже определенные в данном документе). Гетероцикл может быть присоединен к боковой группе посредством любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является стабильным, гетероцикл, описанный в данном документе, может подвергаться замещению в положении, соответствующему атому углерода или азота. Атом азота в гетероцикле необязательно кватернизирован. В предпочтительном варианте осуществления, если общее число атомов S и O в гетероцикле превышает 1, то гетероатомы не являются смежными друг с другом. В другом предпочтительном варианте осуществления общее число атомов S и O в гетероцикле не превышает 1. Используемый в данном документе термин «ароматическая гетероциклическая группа» или «гетероарил» относится к стабильному 5-, 6- или 7-членному моноциклическому или бициклическому или 7-, 8-, 9- или 10-членному бициклическому гетероциклическому ароматическому кольцу, которое содержит атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, независимо выбранных из N, O и S. Атом азота может быть замещенным или незамещенным (т. е. N или NR, где R представляет собой H или другие заместители, уже определенные в данном документе). Гетероатомы, представляющие собой азот и серу, необязательно могут быть окислены (т. е. NO и S(O)p, p равняется 1 или 2). Следует отметить, что общее число атомов S и O в ароматическом гетероцикле не превышает один. Кольцо с мостиковой связью также включено в определение гетероцикла. Кольцо с мостиковой связью образуется, если один или более атомов (т. е. C, O, N или S) соединяют два несмежных атома углерода или азота. Предпочтительное кольцо с мостиковой связью включает без ограничения один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Следует отметить, что мостиковая связь всегда превращает моноциклическое кольцо в трициклическое кольцо. В кольце с мостиковой связью заместитель в кольце также может присутствовать при мостиковой связи.
Примеры гетероциклического соединения включают без ограничения акридинил, азоцинил, бензимидазолил, бензофуранил, бензoмеркаптофуранил, бензoмеркаптофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензoтетразолил, бензoизоксазолил, бензoизотиазолил, бензoимидазолинил, карбазолил, 4aH-карбазолил, карболинил, бензoдигидропиранил, хромен, циннолинил декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изобензoфуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксииндолил, пиримидинил, фенантридинил, фенантролинил, феназин, фенотиазин, бензоксантинил, фенолоксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазолил, пиридоимидазолил, пиридотиазолил, пиридинил, пирролидинил, пирролинил, 2H-пирролил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1H-1,2,3-триазолил, 2H-1,2,3-триазолил, 1H-1,2,4-триазолил, 4H-1,2,4-триазолил и ксантенил. Также включены соединения с конденсированными кольцами и спиросоединения.
Если не указано иное, термин «гидрокарбил» или его гипонимы (например, алкил, алкенил, алкинил, арил и т. д.), сами по себе или в качестве части другого заместителя, относятся к линейному, имеющему разветвленную цепь или циклическому углеводородному радикалу или любой их комбинации, при этом они могут быть полностью насыщенными (например, алкил), моно- или полиненасыщенными (например, алкенил, алкинил и арил), могут быть моно-, ди- или полизамещенными, могут быть одновалентными (например, метил), двухвалентными (например, метилен) или многовалентными (например, метенил), могут также включать двухвалентную или многовалентную группу, иметь определенное число атомов углерода (например, C1-C12 означает 1-12 атомов углерода, при этом C1-12 выбран из C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12; C3-12 выбран из C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12). Термин «гидрокарбил» включает без ограничения алифатический гидрокарбил и ароматический гидрокарбил, при этом алифатический гидрокарбил включает линейный и циклический гидрокарбил, в частности включает без ограничения алкил, алкенил и алкинил. Ароматический гидрокарбил включает без ограничения 6-12-членный ароматический гидрокарбил, такой как фенил, нафтил и т. п. В некоторых вариантах осуществления термин «гидрокарбил» относится к линейной или разветвленной группе или их комбинации, которая может быть полностью насыщенной, моно- или полиненасыщенной и может включать двухвалентную или многовалентную группу. Примеры насыщенной гидрокарбильной группы включают без ограничения метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил и гомолог или изомер н-амила, н-гексила, н-гептила, н-октила и других групп атомов. Ненасыщенный гидрокарбил содержит одну или более двойных или тройных связей. Примеры ненасыщенного алкила включают без ограничения винил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и гомологи и изомеры более высокого порядка.
Если не указано иное, термин «гетерогидрокарбил» или его гипонимы (такие как гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т. д.), сами по себе или как часть другого заместителя, относятся к стабильной линейной, разветвленной или циклической углеводородной группе или любой их комбинации, которая содержит конкретное число атомов углерода и по меньшей мере один гетероатом. В некоторых вариантах осуществления термин «гетероалкил», сам по себе или в комбинации с другим термином, относится к стабильной линейной цепи, разветвленной алкильной группе или их комбинации, которые содержат конкретное число атомов углерода и по меньшей мере один гетероатом. В конкретном варианте осуществления гетероатом выбран из B, O, N и S, где атомы азота и серы необязательно окислены, и гетероатом, представляющий собой атом азота, необязательно кватернизирован. Гетероатом или гетероатомная группа могут находиться в любом внутреннем положении гетерогидрокарбила, в том числе в положении, где гидрокарбил присоединяется к остальной части молекулы. Но термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкил) используются в их общепринятом значении и означают алкильную группу, соединенную с остальной частью молекулы посредством атома кислорода, аминогруппы или атома серы соответственно. Примеры включают без ограничения -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. Могут присутствовать не более двух последовательно расположенных гетероатомов, например -CH2-NH-OCH3.
Если не указано иное, термины «циклогидрокарбил», «гетероциклогидрокарбил» или их гипонимы (такие как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т. д.), применяемые сами по себе или в комбинации с другим термином, относятся к циклизированному «гидрокарбилу» или «гетерогидрокарбилу». Кроме того, в случае гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкил и гетероциклоалкил) один гетероатом может занимать положение, в котором гетероцикл присоединяется к остальной части молекулы. Примеры циклогидрокарбила включают без ограничения циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т. п. Неограничивающие примеры гетероциклогидрокарбила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофураниндол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
Если не указано иное, термин «гетероциклоалкил», сам по себе или в комбинации с другими терминами, означает циклизированный «гетероалкил». Кроме того, в случае «гетероциклоалкила» гетероатомы могут занимать положение присоединения гетероциклоалкила к остальной части молекулы. В некоторых вариантах осуществления гетероциклоалкил представляет собой 4-6-членный гетероциклоалкил; в других вариантах осуществления гетероциклоалкил представляет собой 5-6-членный гетероциклоалкил. Примеры гетероциклоалкила включают без ограничения азетидинил, оксетанил, тиетанил, пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил, тетрагидрофуранил, тетрагидропиранил, пиперидинил, пиперазинил, морфолинил, диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил, гомопиперидинил или оксетанил.
Если не указано иное, термин «алкил» относится к линейной цепи или разветвленной насыщенной углеводородной группе, которая может быть монозамещенной (например, -CH2F) или полизамещенной (например, -CF3), может быть одновалентной (например, метил), двухвалентной (например, метилен) или многовалентной (например, метенил). Примеры алкила включают метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил) и т. п.
Если не указано иное, термин «алкенил» относится к алкильной группе, содержащей одну или более углерод-углеродных двойных связей в любом положении в цепи, которая может быть монозамещенной или полизамещенной и может быть одновалентной, двухвалентной или многовалентной. Примеры алкенила включают этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил и т. п.
Если не указано иное, термин «алкинил» относится к алкильной группе, содержащей одну или более углерод-углеродных тройных связей в любом положении в цепи, которая может быть монозамещенной или полизамещенной и может быть одновалентной, двухвалентной или многовалентной. Примеры алкинила включают этинил, пропинил, бутинил, пентинил и т. п.
Если не указано иное, циклоалкил включает любой стабильный циклический или полициклический гидрокарбил и любой атом углерода, который является насыщенным, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или многовалентным. Примеры циклоалкила включают без ограничения циклопропил, норборнанил, [2.2.2]бициклооктан, [4.4.0]бициклодеканил и т. п.
Если не указано иное, циклоалкенил включает любой стабильный циклический или полициклический гидрокарбил, содержащий одну или более ненасыщенных углерод-углеродных двойных связей в любом положении в кольце, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или многовалентным. Примеры циклоалкенила включают без ограничения циклопентенил, циклогексенил и т. п.
Если не указано иное, циклоалкинил включает любой стабильный циклический или полициклический гидрокарбил, содержащий одну или более углерод-углеродных тройных связей в любом положении в кольце, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или многовалентным.
Если не указано иное, термины «галогено» или «галоген», сами по себе или как часть другого заместителя, относятся к атому фтора, хлора, брома или йода. Кроме того, подразумевается, что термин «галогеналкил» включает моногалогеналкил и полигалогеналкил. Например, подразумевается, что термин «галоген(C1-C4)алкил» включает без ограничения трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т. п. Примеры галогеналкила включают без ограничения трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
Термин «алкокси» означает алкил, определенный выше, содержащий конкретное число атомов углерода, присоединенных посредством кислородного мостика. Если не указано иное, C1-6алкокси включает C1-, C2-, C3-, C4-, C5- и C6алкокси. Примеры алкокси включают без ограничения метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентокси.
Если не указано иное, термин «арил» относится к полиненасыщенному ароматическому заместителю, который может быть моно- или полизамещенным, может быть одновалентным, двухвалентным или многовалентным, может представлять собой одно кольцо или несколько колец (например, от одного до трех колец; где по меньшей мере одно кольцо является ароматическим), которые являются конденсированными совместно или соединенными ковалентно. Термин «гетероарил» относится к арилу (или кольцу), содержащему от одного до четырех гетероатомов. В иллюстративном примере гетероатом выбран из B, O, N и S, где атомы азота и серы необязательно окислены, и атом азота необязательно кватернизирован. Гетероарил может быть присоединен к остальной части молекулы посредством гетероатома. Неограничивающие примеры арила или гетероарила включают фенил, нафтил, бифенил, пирролил, пиразолил, имидазолил, пиразинил, оксазолил, фенилоксазолил, изоксазолил, тиазолил, фуранил, тиенил, пиридил, пиримидинил, бензотиазолил, пуринил, бензимидазолил, индолил, изохинолил, хиноксалинил, хинолил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместитель в любой из указанных выше арильных и гетероарильных кольцевых систем выбран из приемлемых заместителей, описанных ниже.
Если не указано иное, при использовании арила в сочетании с другими терминами (например, арилокси, арилтио, арилалкил) арил включает арильное и гетероарильное кольцо, как определено выше. Таким образом, подразумевается, что термин «арилалкил» включает группы (например, бензил, фенэтил, пиридилметил и т. д.), в которых арил присоединен к алкилу, в том числе к алкилу, где атом углерода (например, метиленовый) был заменен таким атомом, как атом кислорода, например, феноксиметил, 2-пиридилоксиметил, 3-(1-нафтилокси)пропил и т. п.
Термин «уходящая группа» относится к функциональной группе или атому, которые могут быть заменены другими функциональной группой или атомом посредством реакции замещения (такой как реакция замещения по аффинности). Например, иллюстративные уходящие группы включают трифлат; хлор, бром и йод; сульфонатную группу, например, мезилат, тозилат, п-бромбензолсульфонат, п-толуолсульфонаты и т. п.; ацилокси, например, ацетокси, трифторацетокси и т. п.
Термин «защитная группа» включает без ограничения «защитную группу для аминогруппы», «защитную группу для гидроксигруппы» или «защитную группу для тиогруппы». Термин «защитная группа для аминогруппы» относится к защитной группе, подходящей для блокирования побочной реакции с участием атома азота аминогруппы. Иллюстративные защитные группы для аминогруппы включают без ограничения формил, ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т. п. Термин «защитная группа для гидроксигруппы» относится к защитной группе, подходящей для блокирования побочной реакции с участием гидроксигруппы. Иллюстративные защитные группы для гидроксигруппы включают без ограничения алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т. п.
Соединение по настоящему изобретению может быть получено посредством различных способов синтеза, хорошо известных специалистам в данной области техники, в том числе посредством следующего перечисленного варианта осуществления, варианта осуществления, образованного комбинацией следующего перечисленного варианта осуществления и других способов химического синтеза, и эквивалентных замен, хорошо известных специалистам в данной области техники. Предпочтительный вариант осуществления включает без ограничения вариант осуществления настоящего изобретения.
Используемый в настоящем изобретении растворитель является коммерчески доступным. В настоящем изобретении используются следующие сокращения: вод. обозначает воду; HATU обозначает O-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; EDC обозначает N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид; m-CPBA обозначает 3-хлорпероксибензойную кислоту; экв. обозначает эквивалент; CDI обозначает карбонилдиимидазол; DCM обозначает дихлорметан; PE обозначает петролейный эфир; DIAD обозначает диизопропилазодикарбоксилат; DMF обозначает N,N-диметилформамид; DMSO обозначает диметилсульфоксид; EtOAc обозначает этилацетат; EtOH обозначает этанол; MeOH обозначает метанол; CBz обозначает бензилоксикарбонил, который является защитной группой для аминогруппы; BOC обозначает трет-бутоксикарбонил, который является защитной группой для аминогруппы; HOAc обозначает уксусную кислоту; NaCNBH3 обозначает цианоборогидрид натрия; к. т. обозначает комнатную температуру; O/N обозначает в течение ночи; THF обозначает тетрагидрофуран; Boc2O обозначает ди-трет-бутилдикарбонат; TFA обозначает трифторуксусную кислоту; DIPEA обозначает диизопропилэтиламин; SOCl2 обозначает тионилхлорид; CS2 обозначает дисульфид углерода; TsOH обозначает п-толуолсульфоновую кислоту; NFSI обозначает N-фтор-N-(фенилсульфонил)бензолсульфонамид; NCS обозначает N-хлорсукцинимид; n-Bu4NF обозначает фторид тетрабутиламмония; iPrOH обозначает 2-пропанол; т. пл. обозначает точку плавления; LDA обозначает диизопропиламинолитий; EA обозначает этилацетат; NH3H2O обозначает аммиак; DEA обозначает диэтаноламин; m-CPBA обозначает м-хлорпероксибензойную кислоту; ACN обозначает ацетонитрил; Tris-HCl обозначает трисгидроксиметиламинометана гидрохлорид; EDTA обозначает этилендиаминтетрауксусную кислоту; IPA обозначает изопропиловый спирт; NEAA обозначает заменимые аминокислоты.
Названия соединениям давали самостоятельно или с помощью программного обеспечения ChemDraw®, а для коммерчески доступных соединений используются их названия в соответствии с каталогом поставщика.
Подробное описание предпочтительного варианта осуществления
Следующие примеры дополнительно иллюстрируют настоящее изобретение, однако настоящее изобретение ими не ограничивается. Настоящее изобретение было подробно описано в описании и его конкретные варианты осуществления также были раскрыты, для специалиста в данной области техники будет очевидно, что модификации и улучшения вариантов осуществления настоящего изобретения находятся в пределах сущности и объема настоящего изобретения.
Промежуточное соединение 1
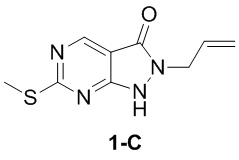
Его получали со ссылкой на способ синтеза, описанный в WO2007126122.
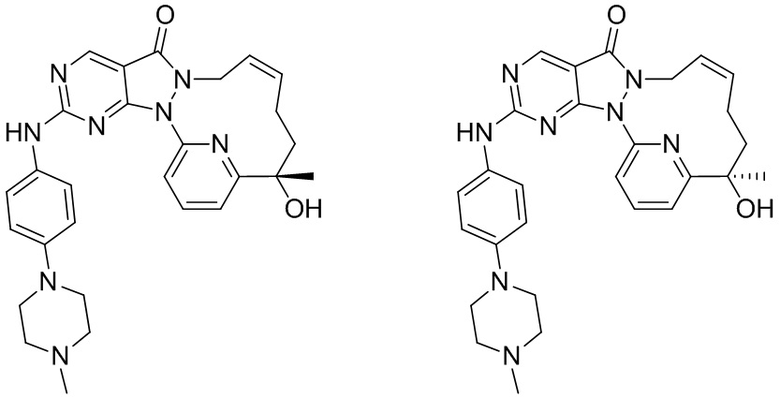
Пример 1. Соединение 1 и соединение 2
Путь синтеза:
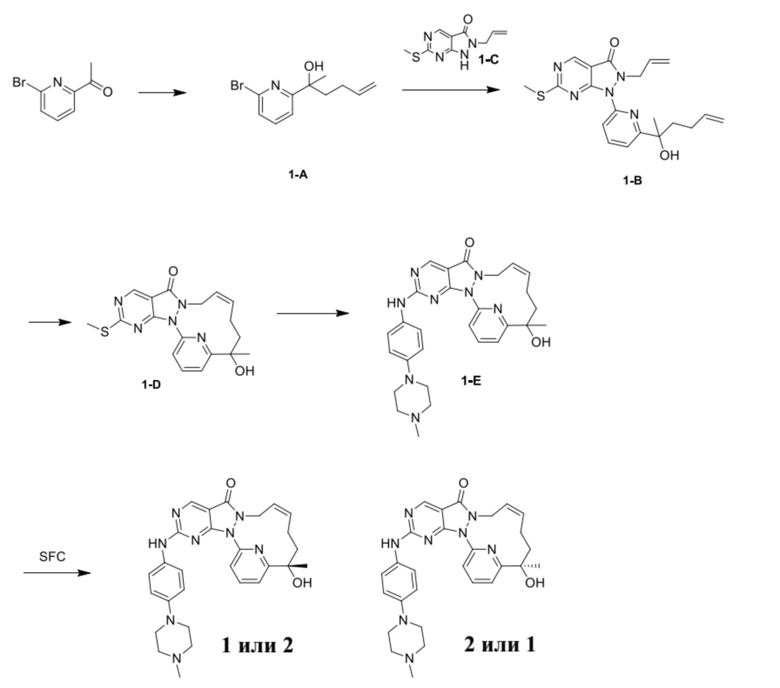
Стадия 1. Синтез соединения 1-A
К раствору 2-ацетил-6-бромпиридина (7,35 г, 36,74 ммоль) в THF (150 мл) добавляли по каплям 3-бутенилмагния бромид (1 M, 55,12 мл) при 0-15oC в защитной атмосфере азота и затем реакционный раствор перемешивали при 10-20oC в течение 3 часов. Добавляли 100 мл насыщенного раствора хлорида аммония для гашения реакции. Отделяли органический слой и промывали с помощью 50 мл насыщенного раствора хлорида натрия, высушивали над безводным сульфатом натрия и концентрировали до сухого состояния с получением коричневого масла. Данное коричневое масло очищали посредством колоночной хроматографии на силикагеле (PE/EA = 7/1) с получением 1-A. 1H ЯМР (400 МГц, DMSO-d6) δ 7,73 (t, J=8,0 Гц, 1H), 7,64 (d, J=7,2 Гц, 1H), 7,46 (d, J=7,2 Гц, 1H), 5,78-5,7 (m, 1H), 4,94-4,85 (m, 2H), 2,07-2,01 (m, 1 H), 1,90-1,71 (m, 3H), 1,41 (s, 3H).
Стадия 2. Синтез соединения 1-B
К смеси 1-A (3,47 г, 13,55 ммоль) и 1-C (3,01 г, 13,55 ммоль) в диоксане (150 мл) добавляли N,N'-диметилэтилендиамин (1,31 г, 14,90 ммоль, 1,60 мл), йодид меди (2,58 г, 13,55 ммоль) и карбонат калия (2,62 г, 18,97 ммоль) с последующей заменой азота три раза. Затем смесь перемешивали при 95oC в защитной атмосфере азота в течение 1,5 часа и добавляли 200 мл гидроксида аммония (28%), затем экстрагировали этилацетатом (300 мл × 2), органические слои объединяли, промывали с помощью 200 мл насыщенного солевого раствора, высушивали над безводным сульфатом натрия и концентрировали до сухого состояния при пониженном давлении. Смесь очищали посредством колоночной хроматографии на силикагеле (PE/EA = 3/1) с получением 1-B. 1H ЯМР (400 МГц, DMSO-d6) δ 9,02 (s,1H), 8,04 (t, J=8,0 Гц, 1H), 7,76 (d, J=7,2 Гц, 1H), 7,64 (d, J=7,6 Гц, 1H), 5,77-5,67 (m, 2H), 5,01-4,79 (m,6H), 2,56 (s, 3H), 2,15-2,11 (m, 1H), 1,85-1,75 (m, 2H), 1,70-1,60 (m, 1H), 1,46 (s, 3H).
Стадия 3. Синтез соединения 1-D
К раствору 1-B (2,06 г, 5,18 ммоль) в толуоле (700 мл) добавляли катализатор Граббса второго поколения (1,32 г, 1,55 ммоль) и смесь перемешивали при 80oC в атмосфере азота в течение 6 часов. Добавляли дополнительное количество катализатора Граббса второго поколения (0,65 г, 0,775 ммоль) и смесь перемешивали при 80oC в атмосфере азота в течение 3 часов. Затем реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат концентрировали до сухого состояния с получением коричневого остатка, который очищали посредством колоночной хроматографии на силикагеле (PE/EA = 1/1) с получением 1-D. 1H ЯМР (400 МГц, DMSO-d6) δ 9,07 (s,1H), 8,06 (t, J=8,0 Гц, 1H), 7,79 (d, J=8,2 Гц, 1H), 7,70 (d, J=7,6 Гц, 1H), 5,39-5,25 (m, 3H), 4,66 (d, J=5,2 Гц, 2H), 2,62 (s, 3H), 2,40-1,95 (m, 3H), 1,85-1,65 (m, 1H) 1,64 (s, 3H).
Стадия 4. Синтез соединения 1-E
К раствору соединения 1-D (360 мг, 974,45 мкмоль) в толуоле (35 мл) добавляли м-хлорпероксибензойную кислоту (265,09 мг, 1,31 ммоль, чистота 85%) и реакционную смесь перемешивали при 25oC в течение 2 часов. К реакционному раствору добавляли 4-(4-метилпиперазин)анилин (242,30 мг, 1,27 ммоль) и N,N-диизопропилэтиламин (503,76 мг, 3,90 ммоль). Реакционный раствор перемешивали при 25oC в течение 12 часов. К реакционному раствору добавляли 25 мл воды и перемешивали и водную фазу экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, однократно промывали насыщенным раствором бикарбоната натрия (30 мл) и насыщенным солевым раствором (30 мл) в указанном порядке и высушивали над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением неочищенного продукта, который отделяли посредством препаративной жидкостной (нейтральной) хроматографии с получением 1-E. 1H ЯМР (400 МГц, CDCl3) δ ppm 1,70 (s, 3 H) 1,78 (br d, J=13,54 Гц, 2 H), 2,04 (br d, J=6,54 Гц, 1 H), 2,08-2,23 (m, 2 H), 2,39 (s, 3 H), 2,62-2,64 (m, 4 H), 3,21-3,24 (m, 4 H), 4,24 (br s, 1 H), 4,51 (br d, J=13,54 Гц, 1 H), 5,61-5,88 (m, 2 H), 6,95 (d, J=9,04 Гц, 2 H), 7,49 (d, J=9,04 Гц, 3 H), 7,81-7,90 (m, 2 H), 8,87 (s, 1 H); MS масса/заряд: 513,1 [M+H]+.
Стадия 5. Синтез соединений 1 и 2
Соединение 1-E (100 мг, 195,08 мкмоль) подвергали хиральному разделению посредством SFC (хроматографическая колонка: A.D. 250 × 50 мм, внутренний диаметр 10 мкм, подвижная фаза: A: CO2 в сверхкритическом состоянии, B: EtOH (0,1% NH3H2O), A:B = 55:45 при 200 мл/мин.) с получением 1, время удерживания: 21 мин. 1H ЯМР (400 МГц, CDCl3) δ ppm 1,60 (s, 3 H), 1,68 (br s, 2 H), 1,94 (br d, J=7,04 Гц, 1 H), 2,00-2,20 (m, 2 H), 2,30 (s, 3 H), 2,52-2,55 (m, 4 H), 3,12-3,15 (m, 4 H), 4,24 (br s, 1 H), 4,42 (br d, J=9,54 Гц, 1 H), 5,65 (br s, 2 H), 6,85 (d, J=9,04 Гц, 2 H), 7,17 (s, 1 H), 7,40 (d, J=9,04 Гц, 2 H), 7,69-7,81 (m, 2 H), 8,77 (s, 1 H). MS масса/заряд: 513,1 [M+H]+. И 2, время удерживания: 42 мин. 1H ЯМР (400 МГц, CDCl3) δ ppm 1,61 (s, 3 H), 1,70 (br d, J=5,02 Гц, 2 H), 1,94 (br s, 1 H), 2,03-2,18 (m, 2 H), 2,30 (s, 3 H), 2,52-2,55 (m, 4 H), 3,12-3,15 (m, 4 H), 4,16 (br s, 1 H), 4,42 (br d, J=11,04 Гц, 1 H), 5,66 (br s, 2 H), 6,85 (d, J=9,04 Гц, 2 H), 7,18 (s, 1 H), 7,40 (d, J=9,04 Гц, 2 H), 7,71-7,81 (m, 2 H), 8,77 (s, 1 H). MS масса/заряд: 513,1 [M+H]+.
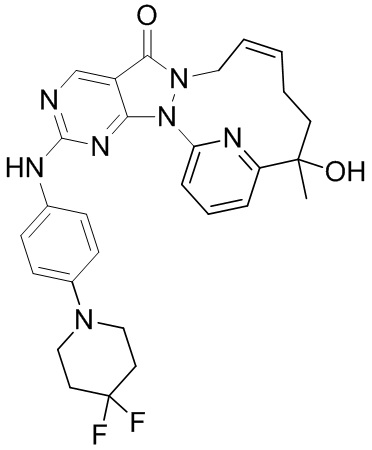
Пример 2. Соединение 3
Путь синтеза:
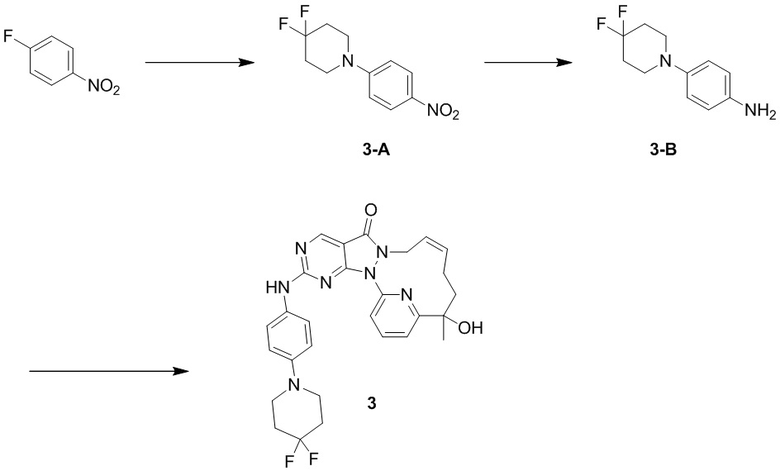
Стадия 1. Синтез соединения 3-A
Соединение 4-фторнитробензол (2,46 г, 17,45 ммоль, 1,85 мл) добавляли к DMF (30 мл) с последующим добавлением K2CO3 (5,48 г, 39,66 ммоль) и 4,4-дифторпиперидина гидрохлорида (2,5 г, 15,86 ммоль), обеспечивали осуществление реакции в реакционной смеси при 60oC в течение 6 часов. К реакционному раствору добавляли H2O (20 мл) для разбавления, а затем добавляли EtOAc (20 мл) для экстракции, органическую фазу высушивали, фильтровали и концентрировали с получением неочищенного продукта, который очищали посредством колоночной хроматографии (SiO2, PE/EA = от 100/1 до 20/1) с получением соединения 3-A. 1H ЯМР (400 МГц, CDCl3): 8,05-8,21 (m, 2 H), 6,78-6,93 (m, 2 H), 3,53-3,65 (m, 4 H), 2,09 (ddd, J=19,46, 13,51, 5,84 Гц, 4 H); MS масса/заряд: 242,09 [M+H]+.
Стадия 2. Синтез соединения 3-B
Соединение 3-A (1,5 г, 6,19 ммоль) добавляли к THF (15 мл), затем добавляли Pd/C (0,2 г, чистота 10%) и реакционный раствор перемешивали при 25oC в течение 2 часов в атмосфере H2 (15 фунтов/кв. дюйм). Реакционный раствор фильтровали для удаления Pd/C и органическую фазу высушивали путем выпаривания на роторном испарителе с получением 3-B. 1H ЯМР (400 МГц, CDCl3): 6,69-6,84 (m, 2 H), 6,53-6,64 (m, 2 H), 3,02-3,17 (m, 4 H), 1,97-2,13 (m, 4 H). MS масса/заряд: 212,11 [M+H]+.
Стадия 3. Синтез соединения 3
К толуолу (2 мл) добавляли 1-D (0,2, 541,36 мкмоль), затем добавляли m-CPBA (153,87 мг, 757,90 мкмоль, чистота 85%), реакционную смесь перемешивали при 25oC в течение 1 часа, затем добавляли 3-B (126,39 мг, 595,50 мкмоль), реакционную смесь перемешивали при 25oC в течение 2 часов, к реакционному раствору добавляли насыщенный раствор Na2SO3 (10 мл) с последующей экстракцией с помощью EtOAc (10 мл × 3), органическую фазу высушивали, фильтровали и концентрировали с получением коричневого масла, которое очищали посредством препаративной TLC (SiO2, PE/EA = 1:1) и препаративной высокоэффективной жидкостной хроматографии с получением 3. 1H ЯМР (400 МГц, CDCl3): 8,86 (s, 1 H), 7,84-7,90 (m, 1 H), 7,77-7,83 (m, 1 H), 7,45-7,57 (m, 3 H), 6,95 (d, J=9,04 Гц, 2 H), 5,72 (br s, 2 H), 4,22 (s, 1 H), 3,28-3,38 (m, 4 H), 2,04-2,24 (m, 6 H), 1,61-1,72 (m, 7 H). MS масса/заряд: 533,24 [M+H]+.
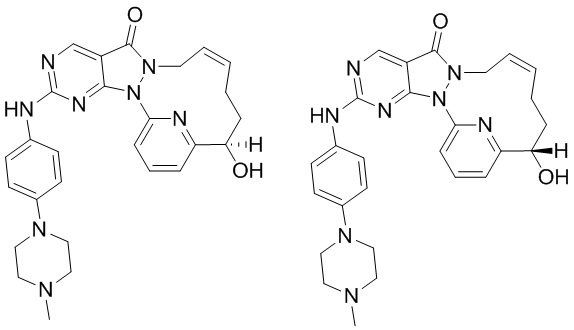
Пример 3. Соединение 4 и соединение 5
Путь синтеза
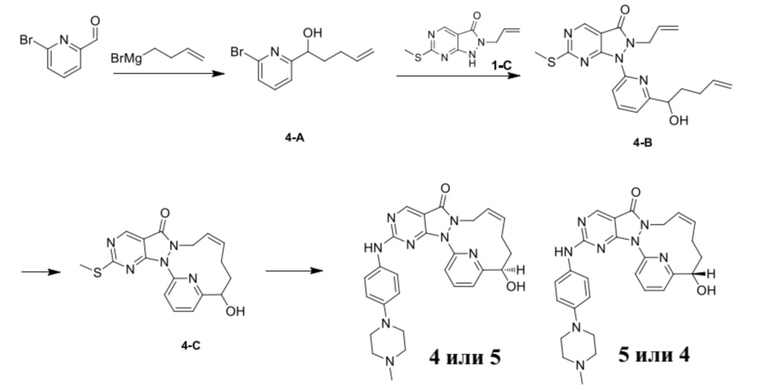
Стадия 1. Синтез соединения 4-A
К раствору соединения 6-бромпиридин-2-карбальдегида (15 г, 80,64 ммоль) в THF (150 мл) добавляли по каплям 3-бутенилмагния бромид (0,5 M, 725,78 мл) при 0-15oC в защитной атмосфере азота. А затем реакционный раствор перемешивали при 10-20oC в течение 12 часов. Добавляли 50 мл насыщенного раствора хлорида аммония. Реакционный раствор экстрагировали с помощью 200 мл этилацетата. Органическую фазу промывали с помощью 100 мл насыщенного солевого раствора, высушивали над безводным сульфатом натрия, фильтровали и высушивали путем выпаривания на роторном испарителе с получением темно-коричневого масла, которое очищали посредством колоночной хроматографии на силикагеле (PE/EA = 5/1) с получением 4-A. MS масса/заряд: 242,0 [M+H]+.
Стадия 2. Синтез соединения 4-B
В реакционную колбу добавляли соединение 4-A (1,29 г, 5,34 ммоль), промежуточное соединение 1-C (1,13 г, 5,08 ммоль), CuI (1,02 г, 5,34 ммоль), K2CO3 (1,03 г, 7,43 ммоль), N,N'-диметилэтилендиамин (518,25 мг, 5,88 ммоль, 632,78 мкл), 1,4-диоксан (20 мл) с последующей заменой азота три раза. Затем реакционный раствор перемешивали при 95oC в течение 13 часов в атмосфере азота. Добавляли 50 мл гидроксида аммония. Реакционный раствор экстрагировали с помощью 100 мл этилацетата и органическую фазу промывали с помощью 50 мл насыщенного солевого раствора, высушивали над безводным сульфатом натрия, фильтровали и высушивали путем выпаривания на роторном испарителе с получением 4-B. MS масса/заряд: 384,2 [M+H]+.
Стадия 3. Синтез соединения 4-C
В реакционную колбу добавляли соединение 4-B (1,3 г, 3,39 ммоль), катализатор Граббса второго поколения (863,44 мг, 1,02 ммоль) и толуол (455 мл) с последующей заменой азота три раза. Затем данный реакционный раствор перемешивали при 80oC в течение 6 часов в атмосфере азота. Реакционный раствор фильтровали и фильтрат высушивали путем выпаривания на роторном испарителе и очищали посредством колоночной хроматографии (диоксид кремния, петролейный эфир:этилацетат = 10:1-1:1) с получением неочищенного 4-C. MS масса/заряд: 355,9 [M+H]+.
Стадия 4. Синтез соединения 4 и соединения 5
В реакционную колбу добавляли соединение 4-C (0,159 г, 447,37 мкмоль), m-CPBA (124,43 мг, 612,89 мкмоль) и толуол (2 мл) и реакционный раствор перемешивали при 25oC в течение 1 часа. К реакционному раствору добавляли 4-(4-метилпиперазин)анилин (119,80 мг, 626,32 мкмоль), диизопропилэтилендиамин (289,09 мг, 2,24 ммоль, 389,61 мкл) и реакционный раствор перемешивали при 25oC в течение 12 часов. К реакционному раствору добавляли 5 мл насыщенного водного раствора сульфита натрия и добавляли 10 мл DCM для экстракции. Органическую фазу высушивали над безводным сульфатом натрия, фильтровали и высушивали путем выпаривания на роторном испарителе с получением неочищенного продукта. Неочищенный продукт растворяли в 2 мл метанола и разделяли с помощью препаративной жидкостной хроматографии (хроматографическая колонка: Luna C18 100 * 30 мм, 5 мкм; подвижная фаза: [вода (0,1% TFA) – ACN]; B (ацетонитрил), %: 15% - 35%, 10 мин.), и выделяли с помощью основной смолы Albert-21 (pH регулировали до 7), фильтровали и концентрировали с получением остатка. Вышеописанную операцию повторяли с получением другой партии остатков и две партии остатков разделяли посредством SFC (хроматографическая колонка: Chiralpak AD-H 250 * 30 мм, внутренний диаметр 5 мкм, подвижная фаза: A: CO2, B: IPA (0,1% NH3H2O), градиент: B, % = 45%, скорость потока: 75 г/мин.) с получением 4, время удерживания: 10,5 мин. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,85 (s, 1 H), 7,82-7,89 (m, 2 H), 7,45-7,54 (m, 2 H), 7,18-7,24 (m, 1 H), 6,91-6,96 (m, 2 H), 5,71-5,73 (m, 2 H), 4,92-4,97 (m, 1 H), 4,41-4,47 (m, 1 H), 3,19-3,27 (m, 4 H), 2,60-2,72 (m, 4 H), 2,41 (s, 3 H), 1,5-2,2 (m, 6 H); MS масса/заряд: 499,2 [M+H]+.
И 5, время удерживания: 16 мин.
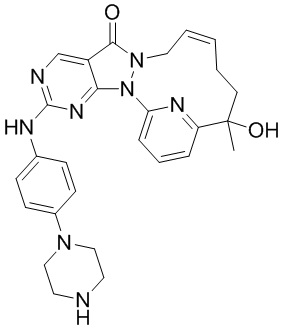
Пример 4. Соединение 6
Путь синтеза
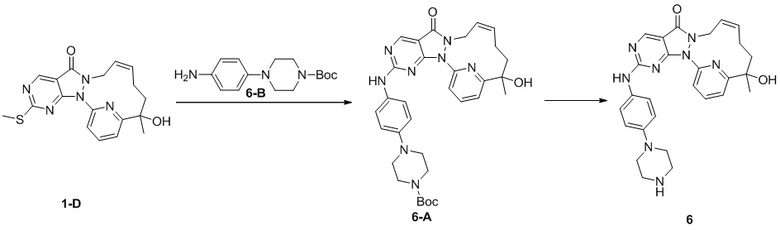
Стадия 1. Синтез соединения 6-A
К раствору соединения 1-D (128 мг, 346,47 мкмоль) в толуоле (30 мл) добавляли м-хлорпероксибензойную кислоту (92,85 мг, 457,34 мкмоль, чистота 85%) и реакционную смесь перемешивали при 25oC в течение 2 часов. К реакционному раствору добавляли 6-B (134,54 мг, 485,06 мкмоль) и N,N-диизопропилэтиламин (223,89 мг, 1,73 ммоль). Реакционный раствор перемешивали при 25oC в течение 12 часов. К реакционному раствору добавляли 10 мл воды и перемешивали и водную фазу экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли, однократно промывали насыщенным раствором бикарбоната натрия (20 мл) и насыщенным солевым раствором (20 мл) в указанном порядке и высушивали над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением неочищенного продукта 6-A. MS масса/заряд: 599,1 [M+H]+.
Стадия 2. Синтез соединения 6
К раствору соединения 6-A (265 мг, 442,63 мкмоль) в дихлорметане (15 мл) добавляли трифторуксусную кислоту (7,70 г, 67,53 ммоль) и реакционную смесь перемешивали при 20oC в течение 30 минут. Реакционный раствор выпаривали на роторном испарителе с получением неочищенного продукта, к которому добавляли насыщенный раствор бикарбоната натрия для регулирования pH до 7-8, и водную фазу экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, однократно промывали насыщенным солевым раствором (20 мл) и высушивали над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением неочищенного продукта, который разделяли посредством препаративной жидкостной (нейтральной) хроматографии с получением 6. 1H ЯМР (400 МГц, CDCl3) δ ppm 1,61 (s, 3 H), 1,70 (br d, J=5,02 Гц, 2 H), 1,86-1,97 (m, 1 H), 2,00-2,14 (m, 2 H), 2,97-3,01 (m, 4 H), 3,05-3,09 (m, 4 H), 4,23 (br s, 1 H), 4,42 (br d, J=13,04 Гц, 1 H), 5,65 (br s, 2 H), 6,86 (d, J=9,04 Гц, 2 H), 7,40 (br d, J=9,04 Гц, 3 H), 7,71-7,82 (m, 2 H), 8,78 (s, 1 H). MS масса/заряд: 499,0 [M+H]+.
Пример 5. Соединение 7

Путь синтеза
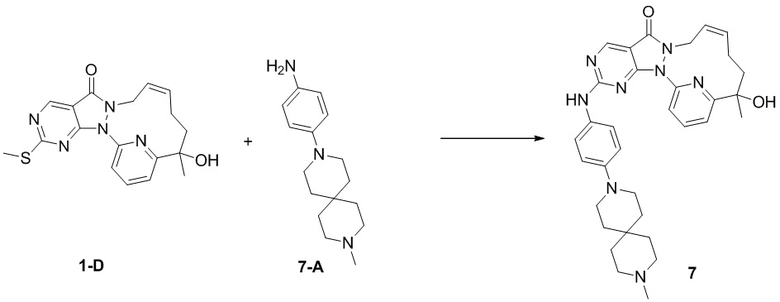
Стадия 1. Синтез соединения 7
К раствору соединения 1-D (0,35 г, 947,38 мкмоль) в толуоле (30 мл) добавляли м-хлорпероксибензойную кислоту (257,73 мг, 1,27 ммоль, чистота 85%) и реакционную смесь перемешивали при 25°C в течение 2,5 часа. К реакционному раствору добавляли 7-A (319,46 мг, 1,23 ммоль) и N,N-диизопропилэтиламин (612,21 мг, 4,74 ммоль). Реакционный раствор перемешивали при 25°C в течение 12 часов. К реакционному раствору добавляли 10 мл воды и перемешивали и водную фазу экстрагировали этилацетатом (15 мл × 3). Органические фазы объединяли, однократно промывали насыщенным раствором бикарбоната натрия (20 мл) и насыщенным солевым раствором (20 мл) в указанном порядке и высушивали над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением неочищенного продукта, который разделяли посредством препаративной жидкостной (нейтральной) хроматографии и препаративной TLC (дихлорметан/метанол = 15/1) с получением 7.
1H ЯМР (400 МГц, CDCl3) δ ppm 1,60 (br s, 11 H), 1,69 (br d, J=14,82 Гц, 2 H), 1,94 (br s, 1 H), 2,06 (br s, 2 H), 2,33 (s, 3 H), 2,48 (br s, 4 H), 3,05-3,10 (m, 4 H), 4,24 (br s, 1 H), 4,41 (br d, J=11,80 Гц, 1 H), 5,65 (br s, 2 H), 6,86 (br d, J=8,54 Гц, 2 H), 7,18 (br s, 1 H), 7,39 (br d, J=8,78 Гц, 2 H), 7,71-7,82 (m, 2 H), 8,77 (s, 1 H). MS масса/заряд: 581,1 [M+H]+.
Пример 6. Соединение 8

Путь синтеза
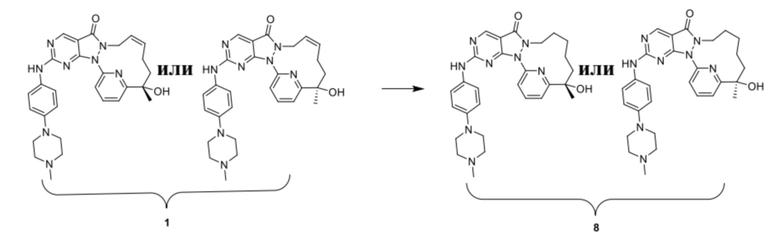
Стадия 1. Синтез соединения 8
К безводному метанолу (20 мл) добавляли палладий на угле (0,04 г, чистота 10%), затем добавляли соединение 1 (80 мг, 156,07 мкмоль) с последующими заменой водорода три раза и повышением давления до 15 фунтов/кв. дюйм. Обеспечивали проведение реакции в реакционном растворе при 25°C в течение 10 часов. Реакционный раствор фильтровали через воронку с пятью отверстиями с засыпанной диатомовой землей. Осадок на фильтре промывали метанолом (2 × 20 мл). Фильтрат высушивали путем выпаривания на роторном испарителе под действием давления с получением остатка. Остаток растворяли в 2 мл метанола и разделяли посредством HPLC (хроматографическая колонка: Waters Xbridge 150 * 25 мм, 5 мкм; подвижная фаза: [вода (0,05% HCl) – ACN]; B, %: 15% - 35%, 12 мин.) с получением 8. 1H ЯМР (400 МГц, CD3OD) δ = 8,71 (s, 1H), 7,99-7,93 (m, 1H), 7,99-7,93 (m, 1H), 7,85 (br d, J=8,2 Гц, 1H), 7,62 (d, J=7,5 Гц, 1H), 7,52 (br d, J=8,8 Гц, 2H), 6,92 (d, J=9,0 Гц, 2H), 3,83-3,74 (m, 2H), 3,65-3,61 (m, 1H), 3,48-3,39 (m, 1H), 3,31-3,29 (m, 2H), 3,25-3,20 (m, 3H), 2,85-2,79 (m, 3H), 2,68 (s, 2H), 2,50 (s, 3H), 2,38-2,21 (m, 3H), 1,90 (br dd, J=7,3, 13,7 Гц, 1H), 1,48 (s, 3H); MS масса/заряд: 514,62 [M+H]+.
Пример 7. Соединение 9
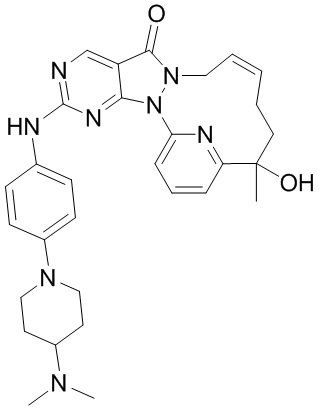
Путь синтеза

Стадия 1. Синтез соединения 9
За исключением применения соответствующих исходных материалов, 9 получали таким же способом, как и проводили синтез соединения 1-E в примере 1.
1H ЯМР (400 МГц, CDCl3) δ ppm 1,69 (s, 3 H) 1,80 (br s, 4 H), 1,95-2,05 (m, 3 H), 2,11-2,25 (m, 2 H), 2,35 (s, 6 H), 2,74 (br t, J=12,18 Гц, 2 H), 3,72 (br d, J=11,54 Гц, 2 H), 4,26 (br s, 1 H), 4,50 (br d, J=13,30 Гц, 1 H), 5,74 (br s, 2 H), 6,95 (d, J=9,04 Гц, 2 H), 7,26 (s, 1 H), 7,47 (d, J=9,04 Гц, 2 H), 7,80-7,91 (m, 2 H), 8,86 (s, 1 H); MS масса/заряд: 541,1 [M+H]+.
Пример 8. Соединение 10

Путь синтеза
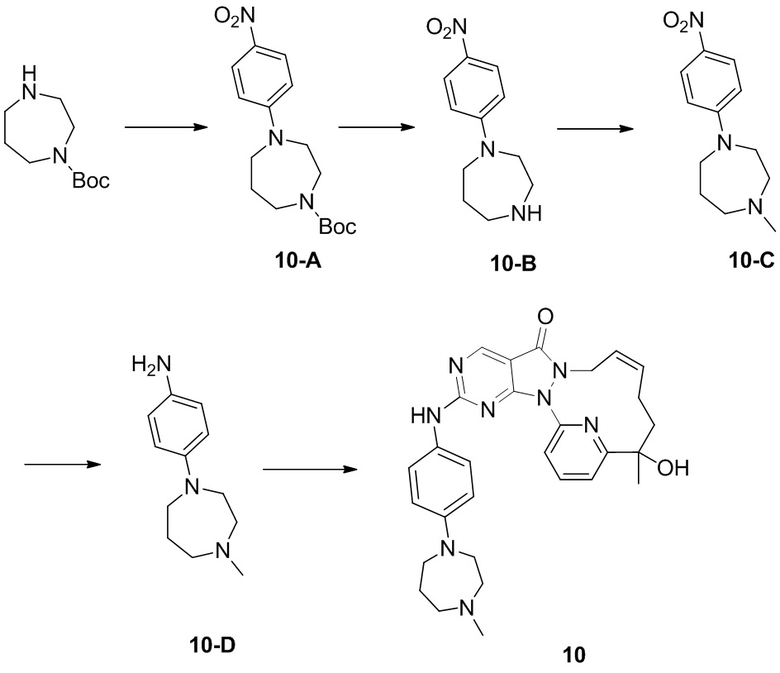
Стадия 1. Синтез соединения 10-A
К раствору соединения 1-Boc-гомопиперазина (3 г, 14,98 ммоль) в диметилсульфоксиде (40 мл) добавляли п-нитрофторбензол (2,52 г, 14,98 ммоль) и карбонат калия (3,00 г, 21,72 ммоль). Реакционный раствор перемешивали при 100°C в течение 12 часов. К реакционному раствору добавляли 120 мл воды и перемешивали, фильтровали и осадок на фильтре промывали с помощью 20 мл воды, а затем высушивали на роторном испарителе с получением неочищенного продукта 10-A.
1H ЯМР (400 МГц, CDCl3) δ ppm 1,36-1,46 (m, 9 H), 2,00 (br d, J=5,78 Гц, 2 H), 3,24-3,42 (m, 2 H), 3,61-3,73 (m, 6 H), 6,68 (br d, J=9,28 Гц, 2 H), 8,14 (d, J=9,28 Гц, 2 H).
Стадия 2. Синтез соединения 10-B
К раствору соединения 10-A (3 г, 9,34 ммоль) в дихлорметане (18 мл) добавляли трифторуксусную кислоту (9,24 г, 81,04 ммоль) и реакционный раствор перемешивали при 30°C в течение 1 часа. Реакционный раствор концентрировали до сухого состояния, разбавляли с помощью 20 мл воды, водную фазу экстрагировали с помощью 30 мл дихлорметана, органическую фазу удаляли, pH водной фазы регулировали до 11-12 с помощью 10% раствора гидроксида натрия и водную фазу экстрагировали дихлорметаном (40 мл × 3), органическую фазу промывали с помощью 40 мл насыщенного солевого раствора, высушивали над безводным сульфатом натрия и фильтровали и реакционный раствор концентрировали до сухого состояния с получением неочищенного соединения 10-B. MS масса/заряд: 220,0 [M+H]+.
Стадия 3. Синтез соединения 10-C
К раствору соединения 10-B (1,87 г, 8,45 ммоль) в метаноле (18,00 мл) добавляли формальдегид (6,72 г, 82,83 ммоль), ацетат боргидрида натрия (3,58 г, 16,90 ммоль) и уксусную кислоту (507,54 мг, 8,45 ммоль) и перемешивали при 30°C в течение 1 часа. Регулировали pH реакционного раствора до 5 с помощью 2 н. хлористоводородной кислоты. Реакционный раствор концентрировали, а затем его pH регулировали до 11 с помощью 10% раствора гидроксида натрия, экстрагировали дихлорметаном (50 мл × 3), органическую фазу промывали с помощью 30 мл насыщенного солевого раствора и высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного соединения 10-C. 1H ЯМР (400 МГц, CDCl3) δ ppm 1,96 (квин, J=5,90 Гц, 2 H), 2,29-2,36 (m, 3 H), 2,46-2,53 (m, 2 H), 2,63-2,68 (m, 2 H), 3,52 (t, J=6,26 Гц, 2 H), 3,57-3,61 (m, 2 H), 6,56 (d, J=9,54 Гц, 2 H), 8,03 (d, J=9,54 Гц, 2 H). MS масса/заряд: 235,9 [M+H]+.
Стадия 4. Синтез соединения 10-D
К раствору соединения 10-C (1,5 г, 6,38 ммоль) в тетрагидрофуране (20 мл) добавляли палладий на угле (760 мг, 644,07 мкмоль, чистота 10%) и смесь перемешивали при 30°C и давлении водорода (15 фунтов/кв. дюйм) в течение 12 часов. После завершения реакции реакционный раствор фильтровали через диатомовую землю и концентрировали с получением неочищенного продукта 10-D.
1H ЯМР (400 МГц, CDCl3) δ ppm 1,91 (dt, J=11,42, 6,08 Гц, 2 H), 2,27-2,37 (m, 3 H), 2,45-2,54 (m, 2 H), 2,58-2,64 (m, 2 H), 3,34 (t, J=6,54 Гц, 2 H), 3,38-3,46 (m, 2 H), 6,48-6,53 (m, 2 H), 6,55-6,61 (m, 2 H). MS масса/заряд: 206,1 [M+H]+.
Стадия 5. Синтез соединения 10
За исключением применения соответствующих исходных материалов, 10 получали таким же способом, как и проводили синтез соединения 1-E в примере 1.
1H ЯМР (400 МГц, CDCl3) δ ppm 1,60 (s, 3 H) 1,68 (br d, J=3,76 Гц, 2 H), 1,94-1,99 (m, 2 H), 2,00-2,17 (m, 2 H), 2,33 (s, 3 H), 2,47-2,59 (m, 2 H), 2,63-2,70 (m, 2 H), 3,43 (t, J=6,26 Гц, 2 H), 3,50-3,54 (m, 2 H), 4,17 (br s, 1 H), 4,41 (br d, J=11,04 Гц, 1 H), 5,65 (br s, 2 H), 6,60 (d, J=8,78 Гц, 2 H), 7,17 (br d, J=7,54 Гц, 1 H), 7,31 (br d, J=8,54 Гц, 2 H), 7,70-7,80 (m, 2 H), 8,75 (s, 1 H). MS масса/заряд: 527,1 [M+H]+.
Пример 9. Соединение 11
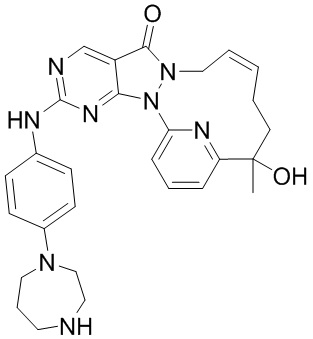
Путь синтеза
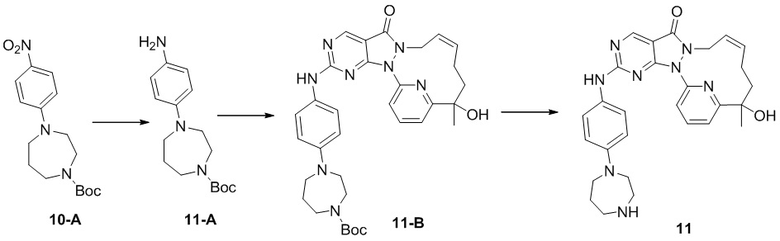
Стадия 1. Синтез соединения 11-A
За исключением применения соответствующих исходных материалов, неочищенный продукт 11-A получали таким же способом, как и проводили синтез соединения 10-D в примере 8. MS масса/заряд: 292,1 [M+H]+.
Стадия 2. Синтез соединения 11-B
За исключением применения соответствующих исходных материалов, неочищенный продукт 11-B получали таким же способом, как и проводили синтез соединения 1-E в примере 1. MS масса/заряд: 613,3 [M+H]+.
Стадия 3. Синтез соединения 11
За исключением применения соответствующих исходных материалов, 11 получали таким же способом, как и проводили синтез соединения 6 в примере 4.
1H ЯМР (400 МГц, CDCl3) δ ppm 1,60 (s, 3 H), 1,91-1,95 (m, 2 H), 2,05-2,20 (m, 4 H), 2,83-2,89 (m, 2 H), 3,02-3,07 (m, 2 H), 3,51-3,56 (m, 4 H), 4,23 (br s, 1 H) 4,41 (br d, J=13,56 Гц, 1 H), 5,66 (br s, 2 H), 6,61 (d, J=9,04 Гц, 2 H), 7,18 (br d, J=7,54 Гц, 1 H), 7,32 (br d, J=8,54 Гц, 2 H), 7,71-7,79 (m, 2 H), 8,75 (s, 1 H). MS масса/заряд: 613,3 [M+H]+.
Пример 10. Соединение 12-A и соединение 12-B
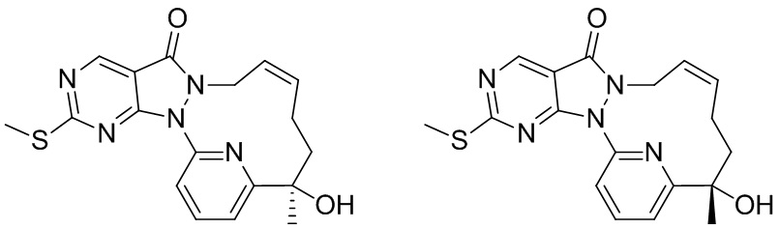
Путь синтеза
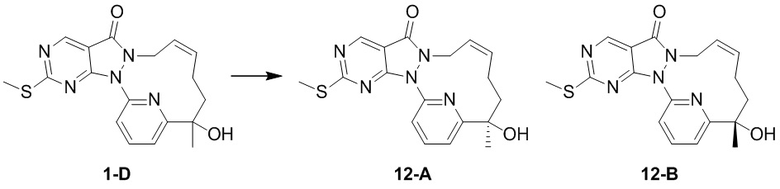
Стадия 1. Синтез соединения 12-A и соединения 12-B
Соединение 1-D (3,9 г, 10,56 мкмоль) подвергали хиральному разделению посредством SFC (хроматографическая колонка: DAICEL CHIRALPAK AS 250 мм * 50 мм, внутренний диаметр 10 мкм; подвижная фаза: A: CO2 в сверхкритическом состоянии, B: MeOH (0,1% NH3H2O), A:B = 75:25 при 200 мл/мин.) с получением 12-A, время удерживания: 13,1 мин.
1H ЯМР (400 МГц, CDCl3) δ = 8,97 (s, 1H), 7,94-7,89 (t, 1H), 7,81 (d, 1H), 7,32 (d, J=7,3 Гц, 1H), 5,93-5,46 (br s, 2H), 4,55( m, 1H), 4,50-4,34 (br s, 1H), 4,17 (s, 1H), 2,60 (s, 3H), 2,15 (m, J=14,1 Гц, 2H), 2,00 (m, 1H), 1,81-1,72 (m, 1H), 1,69 (s, 3H). MS масса/заряд: 370,2 [M+H]+.
И 12-B, время удерживания: 16,7 мин.
1H ЯМР (400 МГц, CDCl3) δ = 8,97 (s, 1H), 7,95-7,88 (t, 1H), 7,81 (d, 1H), 7,32 (d, J=7,8 Гц, 1H), 6,04-5,43 (br s, 2H), 4,61-4,52 (m, 1H), 4,49-4,31 (br s, 1H), 4,17 (s, 1H), 2,60 (s, 3H), 2,15 (m, J=14,1 Гц, 2H), 2,05-1,95 (m, 1H), 1,81-1,72 (m, 1H), 1,69 (s, 3H). MS масса/заряд: 370,1 [M+H]+.
Пример 11. Соединение 12
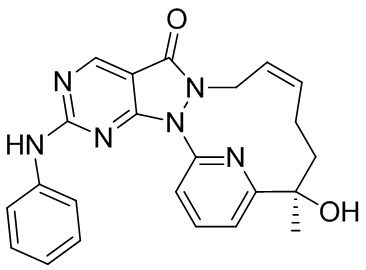
Путь синтеза

Стадия 1. Синтез соединения 12
Соединение 12-A (0,1 г, 270,68 мкмоль) растворяли в толуоле (7 мл), добавляли м-хлорпероксибензойную кислоту (70,07 мг, 324,82 мкмоль, чистота 80%) и реакционную смесь перемешивали при 30°C в течение 1 часа. К реакционному раствору добавляли анилин (27,73 мг, 297,75 мкмоль, 27,18 мкл), N,N-диизопропилэтилендиамин (87,46 мг, 676,70 мкмоль, 117,87 мкл) и реакционный раствор перемешивали при 30°C в течение 12 часов. К реакционному раствору добавляли 5 мл насыщенного водного раствора сульфита натрия и добавляли 10 мл DCM для экстракции. Органическую фазу высушивали над безводным сульфатом натрия, фильтровали и высушивали путем выпаривания на роторном испарителе с получением неочищенного продукта. Неочищенный продукт разделяли посредством препаративной жидкостной хроматографии (хроматографическая колонка: Waters Xbridge 150 * 25 мм, 5 мкм; подвижная фаза: [H2O (10 мМ NH4HCO3) – ACN]; B, %: 33% - 63%, 7 мин.) с получением 12.
1H ЯМР (400 МГц, CDCl3) δ = 8,90 (s, 1H), 7,92-7,86 (t, 1H), 7,85-7,80 (d, 1H), 7,62 (d, J=7,9 Гц, 2H), 7,57-7,50 (br s, 1H), 7,37 (t, J=7,9 Гц, 2H), 7,30 (d, 1H), 7,13 (t, J=7,2 Гц, 1H), 5,73 (br s, 2H), 4,52 (m, J=12,6 Гц, 1H), 4,43-4,27 (br s, 1H), 4,25-4,21 (s, 1H), 2,24-2,07 (m, 2H), 2,05-1,93 (m, 1H), 1,82-1,72 (m, 1H), 1,69 (s, 3H). MS масса/заряд: 415,2 [M+H]+.
Пример 12. Соединение 13
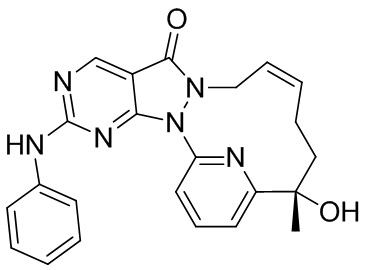
Путь синтеза

Стадия 1. Синтез соединения 13
За исключением применения соответствующих исходных материалов, 13 получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,88 (s, 1H), 7,98 (br s, 1H), 7,92-7,84 (t, 1H), 7,84-7,77 (d, 1H), 7,66-7,56 (d, 2H), 7,35 (t, J=7,3 Гц, 2H), 7,29 (d, 1H), 7,17-7,08 (t, 1H), 5,72 (br s, 2H), 4,61-4,45 (m, 1H), 4,43-4,31 (br s, 1H), 4,31-4,21 (s, 1H), 2,14 (m, J=12,3 Гц, 2H), 2,05-1,95 (m, 1H), 1,81-1,73 (m, 1H), 1,68 (s, 3H). MS масса/заряд: 415,2 [M+H]+.
Пример 13. Соединение 14
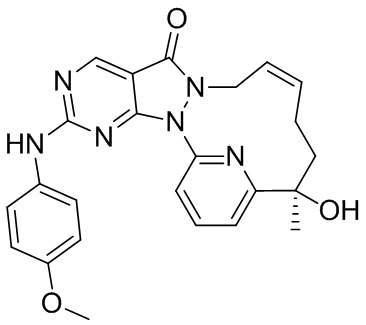
Путь синтеза
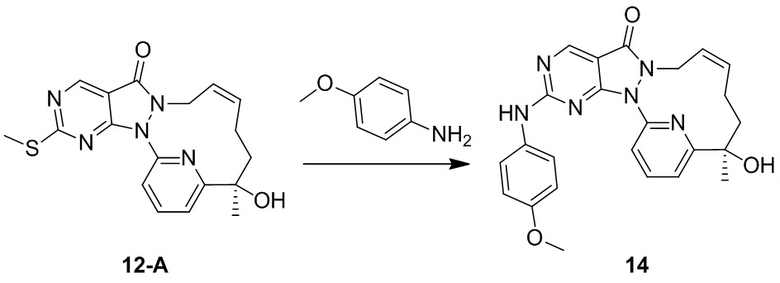
Стадия 1. Синтез соединения 14
За исключением применения соответствующих исходных материалов, 14 получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,86 (s, 1H), 7,89-7,83 (t, 1H), 7,82-7,76 (d, 1H), 7,49 (d, J=9,0 Гц, 2H), 7,26-7,24 (d, 1H), 6,91 (d, J=9,0 Гц, 2H), 5,74 (br s, 2H), 4,49 (m, J=12,8 Гц, 1H), 4,41-4,28 (br s, 1H), 4,23 (s, 1H), 3,83 (s, 3H), 2,23-2,07 (m, 2H), 2,05-1,93 (m, 1H), 1,81-1,72 (m, 1H), 1,68 (s, 3H). MS масса/заряд: 445,2 [M+H]+.
Пример 14. Соединение 15

Путь синтеза

Стадия 1. Синтез соединения 15
За исключением применения соответствующих исходных материалов, 15 получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,86 (s, 1H), 7,89-7,83 (t, 1H), 7,82-7,77 (d, 1H), 7,49 (d, J=8,8 Гц, 2H), 7,36 (br s, 1H), 7,26 (d, 1H), 6,91 (d, J=9,0 Гц, 2H), 5,75 (br s, 2H), 4,48 (m, 1H), 4,30 (br s, 1H), 4,23 (s, 1H), 3,84 (s, 3H), 2,15 (m, J=14,1 Гц, 2H), 2,05-1,94 (m, 1H), 1,77 (m, J=13,8 Гц, 1H), 1,68 (s, 3H). MS масса/заряд: 445,2 [M+H]+.
Пример 15. Соединение 16
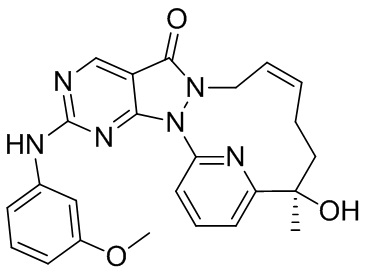
Путь синтеза
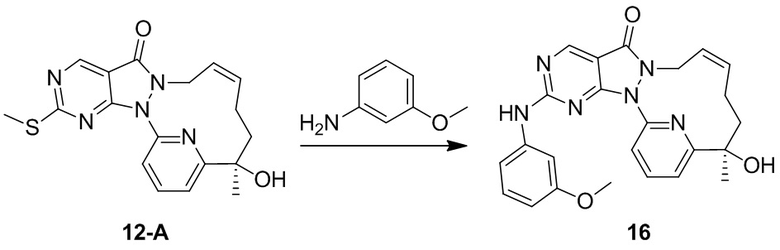
Стадия 1. Синтез соединения 15
За исключением применения соответствующих исходных материалов, 16 получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,90 (s, 1H), 7,92-7,87 (t, 1H), 7,87-7,83 (d, 1H), 7,50 (br s, 1H), 7,41 (t, J=2,1 Гц, 1H), 7,31-7,28 (d, 1H),7,26-7,23 (d, 1H), 7,08 (dd, J=7,6 Гц, 1H), 6,68 (dd, J=2,0, 8,3 Гц, 1H), 5,74 (br s, 2H), 4,52 (m, J=9,0 Гц, 1H), 4,35 (br s, 1H), 4,23 (s, 1H), 3,81 (s, 3H), 2,21-2,09 (m, 2H), 2,06-1,95 (m, 1H), 1,77 (m, J=13,3 Гц, 1H), 1,69 (s, 3H). MS масса/заряд: 445,2 [M+H]+.
Пример 16. Соединение 17
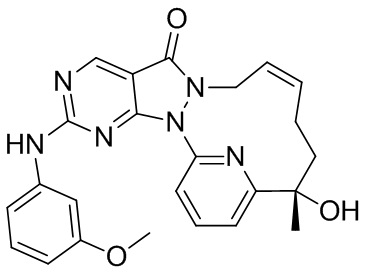
Путь синтеза
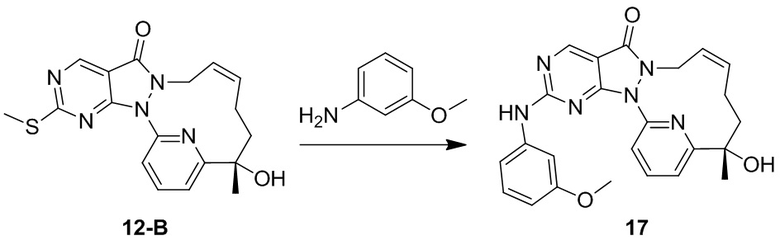
Стадия 1. Синтез соединения 17
За исключением применения соответствующих исходных материалов, 17 получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,90 (s, 1H), 7,92-7,87 (t, 1H), 7,87-7,82 (d, 1H), 7,49 (br s, 1H), 7,41 (t, J=2,1 Гц, 1H), 7,30-7,28 (d, 1H), 7,26-7,23 (d, 1H), 7,07 (dd, J=7,5 Гц, 1H), 6,68 (dd, J=1,8, 8,3 Гц, 1H), 5,74 (br s, 2H), 4,52 (m, J=14,6 Гц, 1H), 4,35 (br s, 1H), 4,22 (s, 1H), 3,81 (s, 3H), 2,22-2,08 (m, 2H), 2,06-1,93 (m, 1H), 1,82-1,73 (m, 1H), 1,69 (s, 3H). MS масса/заряд: 445,2 [M+H]+.
Пример 17. Соединение 18
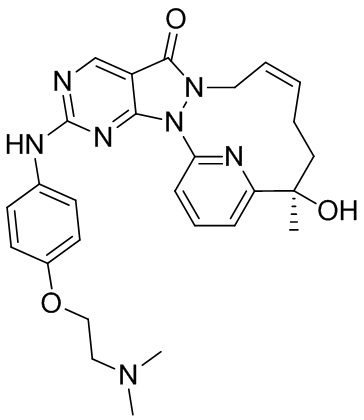
Путь синтеза
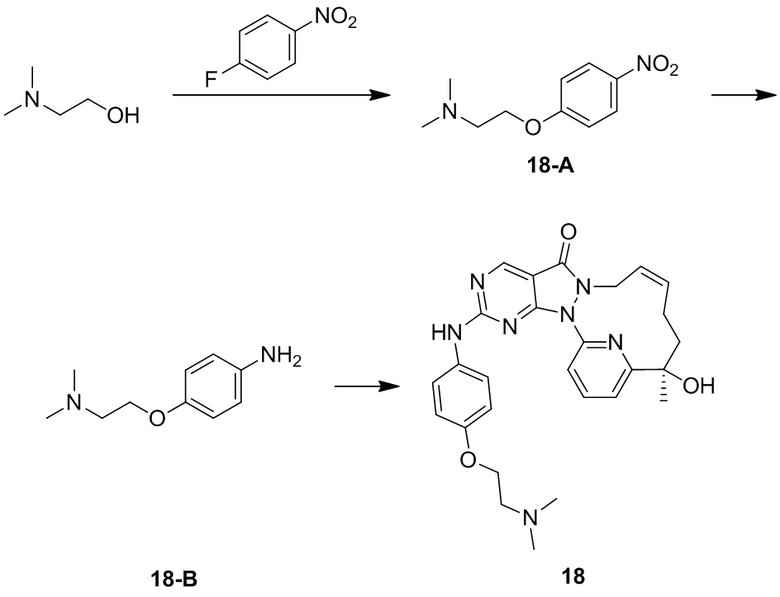
Стадия 1. Синтез соединения 18-A
Соединение гидрид натрия (1,68 г, 42,07 ммоль, чистота 60%) растворяли в DMF (20 мл) и медленно добавляли N,N-диметилэтаноламин (1,5 г, 16,83 ммоль, 1,69 мл) при 0oC в атмосфере азота. После перемешивание в течение 1 часа к реакционному раствору добавляли смесь п-фторнитробензола (2,37 г, 16,83 ммоль) в DMF (2 мл) и перемешивали при 15oC в атмосфере азота в течение 16 часов. Для гашения реакции добавляли насыщенный раствор хлорида аммония (20 мл), проводили экстрагирование этилацетатом (20 мл × 3), органические фазы объединяли, однократно промывали насыщенным солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и высушивали путем выпаривания на роторном испарителе с получением неочищенного продукта. Неочищенный продукт разделяли посредством колоночной хроматографии (дихлорметан/метанол = 10/1, TLC (дихлорметан/метанол = 10/1, Rf = 0,6)) с получением 18-A. 1H ЯМР (400 МГц, DMSO-d6) δ = 8,22-8,17 (d, 2H), 7,15 (d, J=8,1 Гц, 2H), 4,19 (t, J=5,8 Гц, 2H), 2,64 (t, J=5,6 Гц, 2H), 2,21 (s, 6H). MS масса/заряд: 211,0 [M+H]+.
Стадия 2. Синтез соединения 18-B
Соединение 18-A (1 г, 4,76 ммоль) растворяли в THF (20 мл), добавляли Pd/C (650 мг, 548,85 мкмоль, чистота 10%) с последующими заменой водорода три раза и повышением давления до 15 фунтов/кв. дюйм. Обеспечивали проведение реакции в реакционном растворе при 20°C в течение 16 часов. Реакционный раствор фильтровали через воронку с пятью отверстиями с засыпанной диатомовой землей, осадок на фильтре промывали метанолом (2 × 20 мл) и фильтрат высушивали путем выпаривания на роторном испарителе с получением 18-B.
1H ЯМР (400 МГц, CDCl3) δ = 6,79-6,74 (d, 2H), 6,65-6,60 (d, 2H), 3,99 (t, J=5,8 Гц, 2H), 2,69 (t, J=5,8 Гц, 2H), 2,32 (s, 6H). MS масса/заряд: 181,1 [M+H]+.
Стадия 3. Синтез соединения 18
За исключением применения соответствующих исходных материалов, 18 получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,86 (s, 1H), 7,90-7,83 (t, 1H), 7,82-7,76 (d, 1H), 7,48 (d, J=9,0 Гц, 2H), 7,37 (br s, 1H), 7,25 (d, 1H), 6,93 (d, J=8,9 Гц, 2H), 5,75 (br s, 2H), 4,49 m, J=13,3 Гц, 1H), 4,32 (br s, 1H), 4,26-4,19 (s, 1H), 4,09 (t, J=5,7 Гц, 2H), 2,76 (t, J=5,7 Гц, 2H), 2,37 (s, 6H), 2,25-2,08 (m, 2H), 2,06-1,93 (m, 1H), 1,81-1,73 (m, 1H), 1,68 (s, 3H). MS масса/заряд: 502,3 [M+H]+.
Пример 18. Соединение 19

Путь синтеза
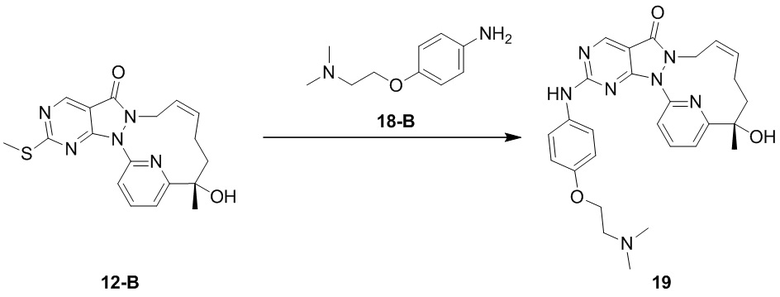
Стадия 1. Синтез соединения 19
За исключением применения соответствующих исходных материалов, 19 получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,85 (s, 1H), 7,89-7,83 (t, 1H), 7,82-7,75 (d, 1H), 7,65-7,52 (br s, 1H), 7,51-7,45 (d, 2H), 7,25 (d, 1H), 6,92 (d, J=9,0 Гц, 2H), 5,74 (br s, 2H), 4,49 (m, J=12,8 Гц, 1H), 4,39-4,29 (br s, 1H), 4,28-4,16 (s, 1H), 4,08 (t, J=5,7 Гц, 2H), 2,75 (t, J=5,7 Гц, 2H), 2,36 (s, 6H), 2,22-2,08 (m, 2H), 2,05-1,93 (m, 1H), 1,77 (m, J=4,6 Гц, 1H), 1,68 (s, 3H). MS масса/заряд: 502,3 [M+H]+.
Пример 19. Соединение 20
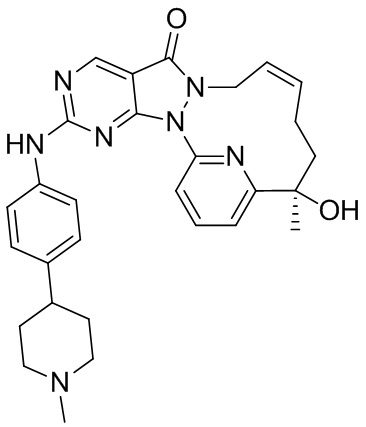
Путь синтеза

Стадия 1. Синтез соединения 20
За исключением применения соответствующих исходных материалов, 20 получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,88 (s, 1H), 7,90-7,85 (t, 1H), 7,84-7,79 (d, 1H), 7,54 (d, J=8,5 Гц, 2H), 7,52-7,45 (br s, 1H), 7,31-7,27 (d, 1H), 7,22 (d, J=8,5 Гц, 2H), 5,75 (br s, 2H), 4,51 (m, J=13,8 Гц, 1H), 4,41-4,28 (br s, 1H), 4,27-4,20 (s, 1H), 3,00 (d, J=11,5 Гц, 2H), 2,48 (m, J=5,6, 10,6 Гц, 1H), 2,34 (s, 3H), 2,23-2,12 (m, 2H), 2,11-2,03 (m, 3H), 1,99 (m, J=10,5 Гц, 1H), 1,84 (m, J=3,5, 9,8 Гц, 3H), 1,78-1,72 (m, 1H), 1,69 (s, 3H). MS масса/заряд: 512,5 [M+H]+.
Пример 20. Соединение 21

Путь синтеза
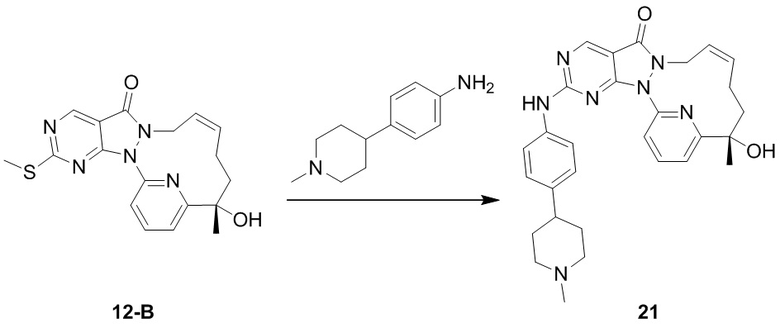
Стадия 1. Синтез соединения 21
За исключением применения соответствующих исходных материалов, 21 получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,88 (s, 1H), 7,90-7,85 (t, 1H), 7,84-7,77 (d, 1H), 7,61 (br s, 1H), 7,57-7,51 (d, 2H), 7,29 (d, 1H), 7,24-7,19 (d, 2H), 5,73 (br s, 2H), 4,51 (m, J=12,9 Гц, 1H), 4,43-4,29 (br s, 1H), 4,28-4,18 (m, 1H), 3,00 (d, J=11,4 Гц, 2H), 2,49 (m, J=5,2, 10,7 Гц, 1H), 2,34 (s, 3H), 2,23-2,12 (m, 2H), 2,12-2,03 (m, 3H), 2,02-1,93 (m, 1H), 1,84 (m, J=3,5, 9,6 Гц, 3H), 1,76-1,73 (m, 1H), 1,69 (s, 3H). MS масса/заряд: 512,4 [M+H]+.
Пример 21. Соединение 22
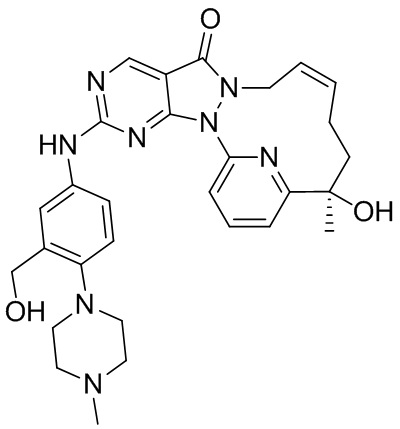
Путь синтеза
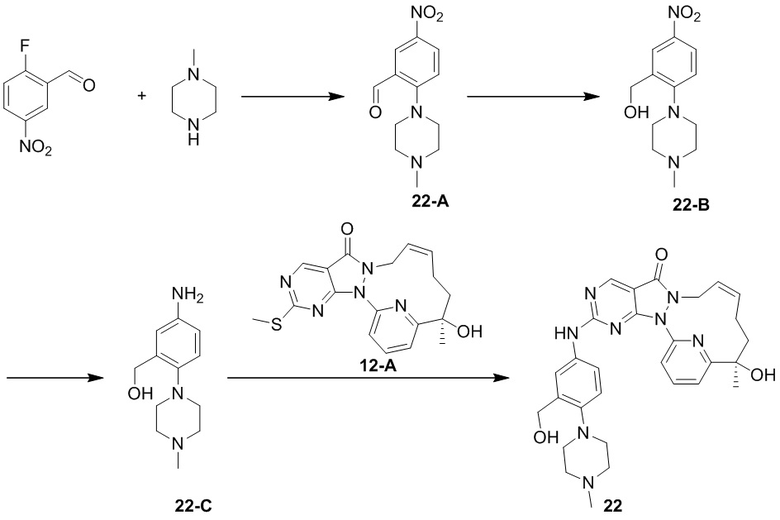
Стадия 1. Синтез соединения 22-A
В реакционную колбу добавляли 2-фтор-5-нитробензальдегид (8 г, 47,31 ммоль), 1-метилпиперазин (9,48 г, 94,61 ммоль, 10,49 мл), карбонат калия (13,08 г, 94,61 ммоль) и N,N-диметилформамид (20 мл). Затем обеспечивали осуществление реакции в смеси при 90°C в течение 2 часов. Реакционную систему охлаждали до комнатной температуры, добавляли воду (40 мл), экстрагировали этилацетатом (40 мл), промывали водой (3 × 30 мл) и насыщенным солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт разделяли посредством колоночной флэш-хроматографии (число меш силикагеля: 200 меш; петролейный эфир:этилацетат = от 1:1 до 0:1) и очищали с получением соединения 22-A.
1H ЯМР (400 МГц, CDCl3) δ= 10,11 (s, 1H), 8,64 (d, J=2,9 Гц, 1H), 8,31 (dd, J=2,9, 9,0 Гц, 1H), 7,10 (d, J=9,0 Гц, 1H), 3,40-3,31 (m, 4H), 2,70-2,62 (m, 4H), 2,40 (s, 3H).
Стадия 2. Синтез соединения 22-B
В реакционную колбу добавляли 22-A (2,4 г, 9,63 ммоль) и THF (20 мл), затем систему охлаждали до 0°C на ледяной бане с последующим добавлением боргидрида натрия (910,59 мг, 24,07 ммоль) и метанола (6 мл), реакционную систему перемешивали и обеспечивали осуществление реакции при 25°C в течение 2 часов. Реакционную систему охлаждали до 0°C на бане с ледяной водой, и медленно добавляли воду (50 мл), и перемешивали в течение 10 мин. для гашения реакции и реакционный раствор экстрагировали дихлорметаном (3 × 50 мл). Органические фазы объединяли, промывали насыщенным солевым раствором (2 × 50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который разделяли посредством колоночной флэш-хроматографии (число меш силикагеля: 200 меш; петролейный эфир:этилацетат = от 1:1 до 0:1), очищали с получением соединения 22-B. LCMS (ESI) (5-95AB): масса/заряд: 251,28 [M+1]; 1H ЯМР (400 МГц, CDCl3) δ = 8,28 (d, J=2,8 Гц, 1H), 8,15 (dd, J=2,8, 8,8 Гц, 1H), 7,16 (d, J=8,8 Гц, 1H), 4,81 (s, 2H), 3,09 (t, J=4,8 Гц, 4H), 2,63 (br s, 4H), 2,39 (s, 3H).
Стадия 3. Синтез соединения 22-C
В предварительно высушенный одногорлый сосуд добавляли соединение 22-B (0,5 г, 1,99 ммоль) и метанол (20 мл), проводили насыщение азотом и затем добавляли палладий на угле (0,08 г, чистота 10%) с последующими заменой азота три раза, затем заменой водорода три раза и повышением давления до 15 фунтов/кв. дюйм. Реакционный раствор перемешивали при 25°C в течение 2 часов. В реакционную систему добавляли 10 мл тетрагидрофурана для разбавления, пропускали реакционный раствор через воронку с пятью отверстиями с засыпанной диатомовой землей и осадок на фильтре промывали тетрагидрофураном (5 × 10 мл). Фильтраты объединяли и концентрировали при пониженном давлении с получением соединения 22-C. LCMS (ESI): масса/заряд: 221,30 [M+1]; 1H ЯМР (400 МГц, CDCl3) δ = 7,07 (d, J=8,4 Гц, 1H), 6,58 (dd, J=2,6, 8,4 Гц, 1H), 6,46 (d, J=2,6 Гц, 1H), 4,71 (s, 2H), 3,58-3,57 (m, 1H), 3,59 (br s, 1H), 2,96 (t, J=4,8 Гц, 4H), 2,72-2,48 (m, 4H), 2,36 (s, 3H), 1,30-1,22 (m, 1H).
Стадия 4. Синтез соединения 22
В реакционную колбу добавляли промежуточное соединение 12-A (150 мг, 406,02 мкмоль), толуол (8 мл) и м-хлорпероксибензойную кислоту (203,61 мг, 1,00 ммоль, чистота 85%) и реакционный раствор перемешивали и обеспечивали осуществление реакции при 25°C в течение 1,5 часа, затем добавляли соединение 22-C (89,9 мг, 406,02 мкмоль) и N,N-диизопропилэтиламин (131,19 мг, 1,02 ммоль, 176,80 мкл), реакционный раствор перемешивали и обеспечивали осуществление реакции в течение 10 часов. В реакционную систему добавляли 10 мл насыщенного раствора сульфита натрия и ее экстрагировали с помощью 10 мл этилацетата, органическую фазу промывали с помощью 10 мл насыщенного солевого раствора, высушивали над безводным сульфатом натрия, концентрировали под давлением и высушивали путем выпаривания на роторном испарителе с получением неочищенного продукта, который разделяли посредством HPLC (хроматографическая колонка: Waters Xbridge 150 мм * 25 мм, 5 мкм; подвижная фаза: [вода (0,05% HCl) – ацетонитрил]; B, %: 10% - 30%, 12 мин.), затем добавляли насыщенный раствор бикарбоната натрия для доведения pH до 7 с последующим концентрированием при пониженном давлении для удаления ацетонитрила. Раствор экстрагировали дихлорметаном (3 × 10 мл), органическую фазу высушивали над безводным сульфатом натрия и концентрировали путем выпаривания на роторном испарителе при пониженном давлении с получением продукта, который очищали с помощью пластины с силикагелем для тонкослойной хроматографии (дихлорметан:метанол = 10:1) с получением соединения 22. LCMS (ESI): масса/заряд: 542,63 [M+1]; 1H ЯМР (400 МГц, CDCl3) δ = 8,87 (s, 1H), 7,98-7,89 (m, 1H), 7,83 (br d, J=7,9 Гц, 1H), 7,71-7,57 (m, 2H), 7,39 (br d, J=8,6 Гц, 1H), 7,30 (br d, J=7,7 Гц, 1H), 7,22 (d, J=8,6 Гц, 1H), 5,74 (br s, 2H), 4,78 (d, J=5,1 Гц, 2H), 4,51 (br d, J=12,8 Гц, 1H), 4,35 (br s, 1H), 3,07 (br s, 4H), 2,73 (br s, 4H), 2,45 (s, 3H), 2,22-2,10 (m, 2H), 2,06-1,93 (m, 1H), 1,77 (br d, J=13,5 Гц, 1H), 1,69 (s, 3H), 1,26 (s, 1H).
Пример 22. Соединение 23

Путь синтеза
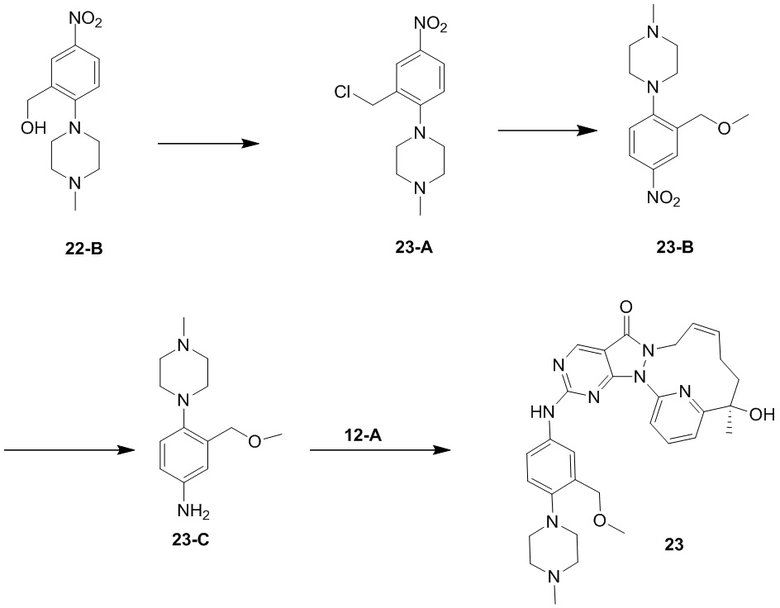
Стадия 1. Синтез соединения 23-A
Соединение 22-B (450 мг, 1,79 ммоль) и сульфоксидхлорид (426,11 мг, 3,58 ммоль, 259,82 мкл) растворяли в хлороформе (10 мл) и реакционный раствор перемешивали и обеспечивали осуществление реакции при 25°C в течение 0,5 часа. Реакционную систему помещали на баню с ледяной водой и медленно добавляли по каплям насыщенный водный раствор карбоната натрия для регулирования pH до 7-8. Реакционную систему экстрагировали этилацетатом (20 мл), промывали водой (3 × 10 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 23-A. LCMS (ESI): масса/заряд: 269,73[M+1].
Стадия 2. Синтез соединения 23-B
Соединение 23-A (0,465 г, 1,72 ммоль) растворяли в безводном MeOH (5 мл), после растворения добавляли метоксид натрия (372,51 мг, 6,90 ммоль) и затем реакционную систему помещали на масляную баню и нагревали до 60°C, перемешивали и обеспечивали осуществление реакции в течение 2 часов. Реакционную систему охлаждали до комнатной температуры, добавляли воду (10 мл), путем выпаривания на роторном испарителе при пониженном давлении удаляли метанол и затем реакционную систему экстрагировали этилацетатом (3 × 10 мл), органические фазы объединяли и промывали насыщенным солевым раствором (2 × 10 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью пластины с силикагелем для тонкослойной хроматографии с получением соединения 23-B. LCMS (ESI): масса/заряд: 265,31[M+1].
Стадия 3. Синтез соединения 23-C
Соединение 23-B (0,107 г, 403,31 мкмоль) растворяли в безводном тетрагидрофуране (4 мл), проводили насыщение азотом и добавляли палладий на угле (0,05 г, чистота 10%) с последующими заменой азота три раза, затем заменой водорода три раза и повышением давления до 15 фунтов/кв. дюйм. Реакционный раствор перемешивали и обеспечивали осуществление реакции при 25°C в течение 0,5 часа. В реакционную систему добавляли 10 мл безводного тетрагидрофурана для разбавления, пропускали реакционный раствор через воронку с пятью отверстиями с засыпанной диатомовой землей и осадок на фильтре промывали безводным тетрагидрофураном (3 × 10 мл). Фильтраты объединяли и концентрировали при пониженном давлении с получением соединения 23-C. LCMS (ESI): масса/заряд: 235,32[M+1].
Стадия 4. Синтез соединения 23
К промежуточному соединению 12-A (113,03 мг, 305,96 мкмоль) добавляли толуол (5 мл) и м-хлорпероксибензойную кислоту (124,23 мг, 611,92 мкмоль, чистота 85%). После перемешивания и осуществления реакции при 25°C в течение 2 часов, добавляли соединение 23-C (72 мг, 305,96 мкмоль) и N,N-диизопропилэтиламин (98,86 мг, 764,90 мкмоль, 133,23 мкл), а затем реакционный раствор перемешивали и обеспечивали осуществление реакции в течение 10 часов. Реакционную систему добавляли 10 мл насыщенного раствора сульфита натрия, экстрагировали с помощью 10 мл этилацетата и органическую фазу промывали с помощью 10 мл насыщенного солевого раствора, высушивали над безводным сульфатом натрия, концентрировали под давлением и высушивали путем выпаривания на роторном испарителе с получением неочищенного продукта. Неочищенный продукт растворяли в 1 мл метанола и разделяли посредством HPLC (хроматографическая колонка: Waters Xbridge 150 * 25, 5 мкм; подвижная фаза: [вода (0,05% HCl) – ацетонитрил]; B, %: 15% - 30%, 12 мин.), добавляли насыщенный раствор бикарбоната натрия для доведения pH до 7, путем выпаривания на роторном испарителе при пониженном давлении удаляли ацетонитрил, раствор экстрагировали дихлорметаном (3 × 10 мл), органическую фазу высушивали над безводным сульфатом натрия и концентрировали путем выпаривания на роторном испарителе при пониженном давлении с получением соединения 23. LCMS (ESI): масса/заряд: 556,66[M+1]. 1H ЯМР (400 МГц, CDCl3) δ = 8,79 (s, 1H), 7,87-7,65 (m, 3H), 7,38 (br d, J=6,8 Гц, 1H), 7,19 (br d, J=2,6 Гц, 1H), 7,01 (d, J=8,5 Гц, 1H), 5,65 (br s, 2H), 4,46 (s, 2H), 3,41-3,41 (m, 1H), 3,38 (d, J=17,8 Гц, 3H), 3,40-3,32 (m, 1H), 2,89 (br t, J=4,6 Гц, 4H), 2,56 (br s, 4H), 2,33 (s, 3H), 2,11-2,02 (m, 2H), 1,99-1,85 (m, 2H), 1,60 (s, 3H).
Пример 23. Соединение 24

Путь синтеза
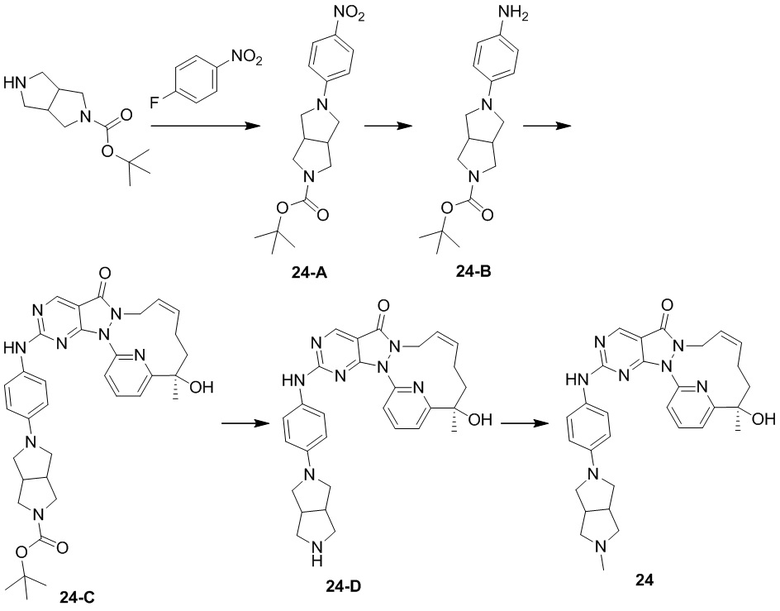
Стадия 1. Синтез соединения 24-A
Соединение 2-BOC-гексагидропирроло[3,4-c]пиррол (650 мг, 3,06 ммоль) и п-фторнитробензол (475,23 мг, 3,37 ммоль) растворяли в DMSO (15 мл) и добавляли K2CO3 (846,34 мг, 6,12 ммоль). Затем смесь перемешивали при 60°C в течение 12 часов. Медленно добавляли 50 мл воды и перемешивали в течение 10 минут, фильтровали и осадок на фильтре высушивали путем выпаривания на роторном испарителе с получением 24-A.
1H ЯМР (400 МГц, DMSO-d6) δ = 8,06 (d, J=9,0 Гц, 2H), 6,62 (d, J=9,3 Гц, 2H), 3,62 (m, 2H), 3,58-3,48 (m, 2H), 3,32-3,26 (m, 2H), 3,17 (m, 2H), 3,09-2,96 (m, 2H), 1,39 (s, 9H). MS масса/заряд: 278,1 [M+H-56]+.
Стадия 2. Синтез соединения 24-B
За исключением применения соответствующих исходных материалов, 24-B получали таким же способом, как и проводили синтез соединения 18-B в примере 17.
1H ЯМР (400 МГц, DMSO-d6) δ = 6,48 (d, J=8,8 Гц, 2H), 6,37 (d, J=8,8 Гц, 2H), 4,32 (s, 2H), 3,51 (m, 2H), 3,20 (m, J=7,5 Гц, 2H), 3,18-3,10 (m, 2H), 3,01 (m, J=10,5 Гц, 2H), 2,96-2,86 (m, 2H), 1,38 (s, 9H). MS масса/заряд: 304,2 [M+H]+.
Стадия 3. Синтез соединения 24-C
За исключением применения соответствующих исходных материалов, 24-C получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,83 (s, 1H), 7,89-7,84 (t, 1H), 7,83-7,79 (d, 1H), 7,41 (d, J=8,0 Гц, 2H), 7,24 (d, 1H), 6,55 (d, J=8,8 Гц, 2H), 5,74 (br s, 2H), 4,47 (m, 1H), 4,40-4,27 (br s, 1H), 4,26-4,21 (s, 1H), 3,67 (m, 2H), 3,55 (m, 2H), 3,42 (m, 1H), 3,24 (m, J=9,8 Гц, 3H), 3,03 (m, 2H), 2,15 (m, J=14,0 Гц, 2H), 2,06-1,97 (m, 1H), 1,77 (m, J=14,1 Гц, 1H), 1,68 (s, 3H), 1,47 (s, 9H). MS масса/заряд: 625,4 [M+H]+.
Стадия 4. Синтез соединения 24-D
За исключением применения соответствующих исходных материалов, 24-D получали таким же способом, как и проводили синтез соединения 22-D в примере 21. MS масса/заряд: 525,2 [M+H]+.
Стадия 5. Синтез соединения 24
За исключением применения соответствующих исходных материалов, 24 получали таким же способом, как и проводили синтез соединения 22 в примере 21.
1H ЯМР (400 МГц, CDCl3) δ = 8,82 (s, 1H), 7,88-7,83 (t, 1H), 7,83-7,76 (d, 1H), 7,65 (br s, 1H), 7,40 (d, J=8,6 Гц, 2H), 7,24 (d, J=7,4 Гц, 1H), 6,65 (d, J=8,9 Гц, 2H), 5,73 (br s, 2H), 4,48 (m, J=10,9 Гц, 1H), 4,40-4,09 (m, 2H), 3,35 (m, J=5,5 Гц, 2H), 3,27-3,20 (m, 2H), 3,05-2,96 (m, 2H), 2,88-2,75 (m, 2H), 2,46 (m, J=7,8 Гц, 2H), 2,37 (s, 3H), 2,14 (m, J=14,0 Гц, 2H), 2,00 (m, J=9,1 Гц, 1H), 1,80-1,72 (m, 1H), 1,67 (s, 3H). MS масса/заряд: 539,5 [M+H]+.
Пример 24. Соединение 25
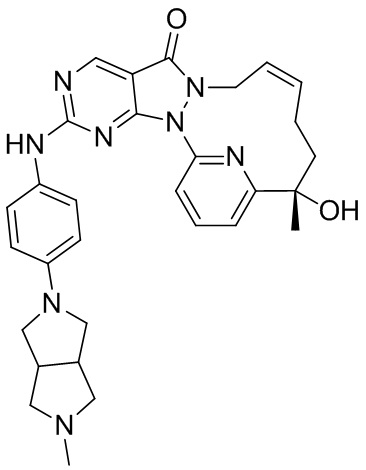
Путь синтеза
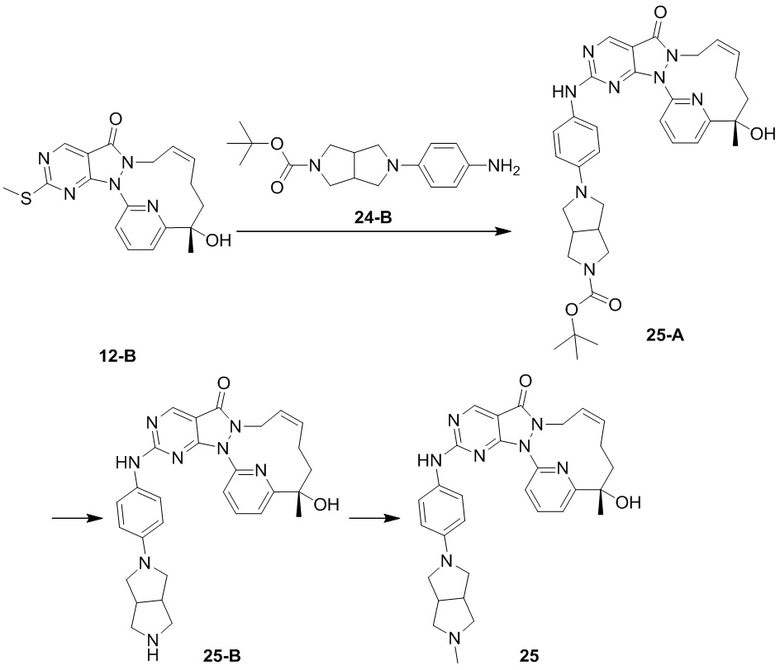
Стадия 1. Синтез соединения 25-A
За исключением применения соответствующих исходных материалов, 25-A получали таким же способом, как и проводили синтез соединения 12 в примере 11.
1H ЯМР (400 МГц, CDCl3) δ = 8,83 (s, 1H), 7,89-7,84 (t, 1H), 7,84-7,79 (d, 1H), 7,41 (d, J=8,1 Гц, 2H), 7,24 (d, 1H), 6,55 (d, J=8,9 Гц, 2H), 5,91-5,50 (br s, 2H), 4,46 (m, 1H), 4,39-4,27 (m, 1H), 4,26-4,22 (s, 1H), 3,67 (m, 2H), 3,54 (m, 2H), 3,42 (m, 1H), 3,24 (m, J=9,4 Гц, 3H), 3,02 (m, 2H), 2,22-2,09 (m, 2H), 2,07-1,96 (m, 1H), 1,77 (m, J=13,4 Гц, 1H), 1,68 (s, 3H), 1,47 (s, 9H). MS масса/заряд: 625,4 [M+H]+.
Стадия 2. Синтез соединения 25-B
За исключением применения соответствующих исходных материалов, 25-B получали таким же способом, как и проводили синтез соединения 22-D в примере 21. MS масса/заряд: 525,3 [M+H]+.
Стадия 3. Синтез соединения 25
За исключением применения соответствующих исходных материалов, 25 получали таким же способом, как и проводили синтез соединения 22 в примере 21.
1H ЯМР (400 МГц, CDCl3) δ = 8,82 (s, 1H), 7,88-7,83 (t, 1H), 7,82-7,76 (d, 1H), 7,40 (d, J=8,6 Гц, 2H), 7,24 (d, J=7,3 Гц, 1H), 6,65 (d, J=8,8 Гц, 2H), 5,73 (br s, 2H), 4,47 (m, J=12,9 Гц, 1H), 4,38-4,08 (m, 2H), 3,41-3,30 (m, 2H), 3,26-3,18 (m, 2H), 2,99 (m, 2H), 2,85-2,76 (m, 2H), 2,45 (m, J=7,9 Гц, 2H), 2,36 (s, 3H), 2,20-2,11 (m, 2H), 2,05-2,00 (m, 1H), 1,79-1,73 (m, 1H), 1,67 (s, 3H). MS масса/заряд: 539,5 [M+H]+.
Пример 25. Соединение 26
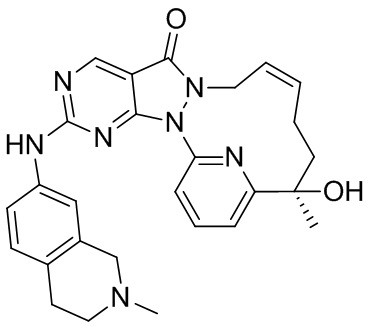
Путь синтеза

Стадия 1. Синтез соединения 26-B
За исключением применения соответствующих исходных материалов, неочищенный продукт 26-B получали таким же способом, как и проводили синтез соединения 10-C в примере 8. 1H ЯМР (400 МГц, CDCl3) δ ppm 7,92 (dd, J=8,4, 2,4 Гц, 1 H) 7,85 (d, J=2,6 Гц, 1 H) 7,19 (s, 1 H) 3,58 (s, 2 H) 2,94 (t, J=5,6 Гц, 2 H) 2,66 (t, J=6,0 Гц, 2 H) 2,40-2,45 (m, 3 H). MS масса/заряд: 193,0 [M+H]+.
Стадия 2. Синтез соединения 26-C
За исключением применения соответствующих исходных материалов, 26-C получали таким же способом, как и проводили синтез соединения 10-D в примере 8. MS масса/заряд: 163,3 [M+H]+.
Стадия 3. Синтез соединения 26
К раствору соединения 12-A (100 мг, 270,68 мкмоль) в толуоле (7 мл) и дихлорметане (1 мл) добавляли м-хлорпероксибензойную кислоту (70,07 мг, 324,82 мкмоль, чистота 80%). Реакционный раствор перемешивали при 20°C в течение 1 часа. К реакционному раствору добавляли N,N-диизопропилэтиламин (104,95 мг, 812,04 мкмоль) и 26-B (52,70 мг, 324,82 мкмоль) и реакционный раствор перемешивали при 20°C в течение 12 часов. К реакционному раствору добавляли воду (10 мл), водную фазу экстрагировали этилацетатом (15 мл × 3), органические фазы объединяли и промывали насыщенным раствором бикарбоната натрия (20 мл), разделяли и органическую фазу промывали насыщенным солевым раствором (20 мл) и отделяли. Органическую фазу высушивали над безводным сульфатом натрия, фильтровали и растворитель удаляли путем выпаривания на роторном испарителе с получением неочищенного продукта. Неочищенный продукт разделяли посредством препаративной жидкостной хроматографии с получением 26. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,79 (s, 1 H), 7,73-7,83 (m, 2 H), 7,45 (s, 1 H), 7,34 (s, 1 H), 7,21 (s, 1 H), 7,02 (d, J=8,0 Гц, 1 H), 5,67 (br s, 2 H), 4,41- 4,44 (d, J=12,8 Гц, 1 H), 4,16 (s, 1 H), 3,50-3,51 (m, 2 H), 2,82-2,87 (m, 2 H), 2,53-2,72 (m, 2 H), 2,42 (s, 3 H), 2,00-2,16 (m, 2 H), 1,88-1,98 (m, 1 H), 1,69-1,72 (m, 1 H), 1,61 (s, 3 H); MS масса/заряд: 484,3 [M+H]+.
Пример 26. Соединение 27
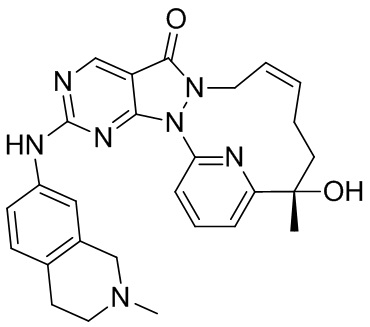
Путь синтеза

Стадия 1. Синтез соединения 27
За исключением применения соответствующих исходных материалов, 27 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,88 (s, 1 H), 7,82-7,92 (m, 2 H), 7,54 (s, 1 H), 7,43 (s, 1 H), 7,30 (s, 1 H), 7,10 (d, J=8,2 Гц, 1 H), 5,77 (br s, 2 H), 4,49-4,53 (d, J=13,6 Гц, 1 H), 4,26 (s, 1 H), 3,53-3,66 (m, 2 H), 2,91-2,94 (m, 2 H), 2,72-2,74 (m, 2 H), 2,50 (s, 3 H), 2,15-2,21 (m, 2 H), 1,99-2,04 (m, 1 H), 1,79-1,81 (m, 1 H), 1,70 (s, 3 H). MS масса/заряд: 484,3 [M+H]+.
Пример 27. Соединение 28

Путь синтеза
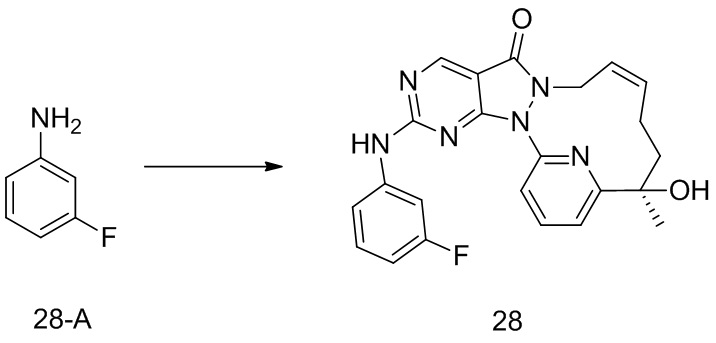
Стадия 1. Синтез соединения 28
За исключением применения соответствующих исходных материалов, 28 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,84 (m, 1 H), 7,83-7,90 (m, 1 H), 7,72-7,79 (m, 2 H), 7,20-7,25 (m, 2 H), 7,03 (d, J=7,6 Гц, 1 H), 6,74 (t, J=8,2 Гц 1 H), 5,67 (br s, 2 H), 4,46 (d, J=13,2 Гц, 1 H), 4,15 (s, 1 H), 1,98-2,17 (m, 2 H), 1,85-1,97 (m, 1 H), 1,70 (d, J=14,4 Гц, 1 H), 1,62 (s, 3 H); MS масса/заряд: 433,0 [M+H]+.
Пример 28. Соединение 29

Путь синтеза
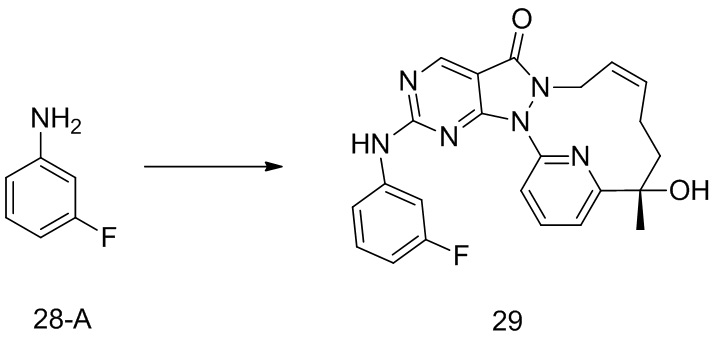
Стадия 1. Синтез соединения 29
За исключением применения соответствующих исходных материалов, 29 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,83 (s, 1 H), 7,82-7,90 (m, 1 H), 7,67-7,79 (m, 2 H), 7,22-7,24 (m, 2 H), 7,03 (d, J=7,6 Гц, 1 H), 6,73 (t, J=7,2 Гц, 1 H), 5,67 (br s, 2 H), 4,46 (d, J=10,8 Гц, 1 H), 4,15 (s, 1 H), 2,00-2,17 (m, 2 H), 1,85-1,97 (m, 1 H), 1,70 (d, J=14,4 Гц, 1 H), 1,62 (s, 3 H); MS масса/заряд: 433,0 [M+H]+.
Пример 29. Соединение 30
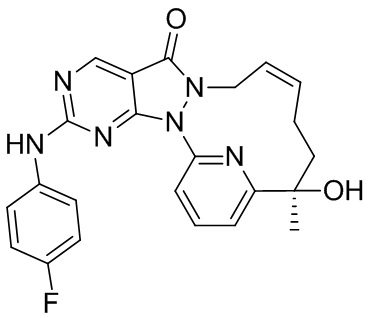
Путь синтеза
Стадия 1. Синтез соединения 30
За исключением применения соответствующих исходных материалов, 30 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,80 (s, 1 H), 7,76-7,82 (m, 1 H), 7,65-7,70 (d, J=8,0 Гц, 1 H), 7,45-7,51 (m, 2 H), 7,18-7,22 (d, 1 H), 6,93-7,01 (t, J=8,4 Гц, 2 H), 5,64 (br s, 2 H), 4,38-4,46 (m, 1 H), 2,01-2,17 (m, 2 H), 4,18 (s, 1 H), 1,86-1,98 (m, 1 H), 1,65-1,74 (m, 1 H), 1,61 (m, 3 H). MS масса/заряд: 433,0 [M+H]+.
Пример 30. Соединение 31
Путь синтеза

Стадия 1. Синтез соединения 31
За исключением применения соответствующих исходных материалов, 31 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,80 (s, 1 H), 7,75-7,84 (m, 1 H), 7,65-7,71 (d, J=8,0 Гц, 1 H), 7,44-7,53 (m, 2 H), 7,20-7,22 (d, 1 H), 6,94-7,01 (t, J=8,8 Гц, 2 H), 5,64 (br s, 2 H), 4,38-4,48 (m, 1 H), 4,17 (s, 1 H), 2,00-2,18 (m, 2 H), 1,88-1,95 (m, 1 H), 1,65-1,74 (m, 1 H), 1,61 (s, 3 H). MS масса/заряд: 433,0 [M+H]+.
Пример 31. Соединение 32
Путь синтеза

Стадия 1. Синтез соединения 32
За исключением применения соответствующих исходных материалов, 32 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,77 (s, 1 H), 7,69-7,82 (m, 2 H), 7,36-7,45 (d, J=8,8 Гц, 2 H), 7,16-7,20 (d, 1 H), 6,80-6,86 (d, J=8,78 Гц, 2 H), 5,64 (br s, 2 H), 4,39-4,44 (d, J=10,28 Гц, 1 H), 4,20 (s, 1 H), 3,75-3,87 (m, 4 H), 3,01-3,15 (m, 4 H), 1,99-2,18 (m, 2 H), 1,88-1,99 (m, 1 H), 1,65-1,71 (m, 1 H), 1,61 (m, 3 H); MS масса/заряд: 500,3 [M+H]+.
Пример 32. Соединение 33

Путь синтеза
Стадия 1. Синтез соединения 33 222
За исключением применения соответствующих исходных материалов, 33 получали таким же способом, как и проводили синтез соединения 26 в примере 25.
1H ЯМР (400 МГц, CDCl3) δ ppm 8,84 (s, 1 H) 7,77-7,89 (m, 2 H) 7,46 – 7,51 (d, J=9,2 Гц, 2 H), 7,26 (s, 1 H), 6,86-6,92 (d, J=8,8 Гц, 2 H), 5,71 (br s, 2 H), 4,44-4,49 (d, J=13,0 Гц, 1 H), 4,25 (s, 1 H), 3,81-3,94 (m, 4 H), 3,08-3,21 (m, 4 H), 2,09-2,24 (m, 2 H), 1,94-2,04 (m, 1 H), 1,74-1,79 (m, 1 H), 1,67 (s, 3 H); MS масса/заряд: 500,0 [M+H]+.
Пример 33. Соединение 34

Путь синтеза
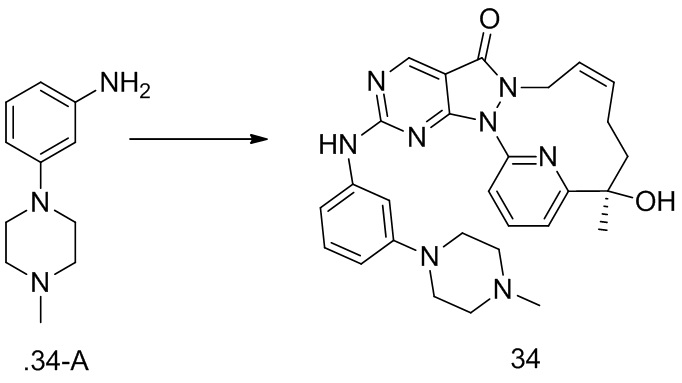
Стадия 1. Синтез соединения 34
За исключением применения соответствующих исходных материалов, 34 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,79 (s, 1 H), 7,73-7,87 (m, 2 H), 7,69 (br s, 1 H), 7,10-7,19 (m, 2 H), 6,90-6,97 (d, J=7,2 Гц, 1 H), 6,56-6,63 (d, J=7,6 Гц, 1 H), 5,55 (br s, 2 H), 4,45 (br s, 1 H), 4,32 (br s, 1 H), 3,07 (br s, 4 H), 2,44 (br s, 4 H), 2,27 (s, 3 H), 2,02-2,13 (m, 2 H), 1,85-1,95 (d, J=8,2 Гц, 1 H), 1,65-1,72 (d, J=6,2 Гц, 1 H), 1,62 (s, 3 H); MS масса/заряд: 513,3 [M+H]+.
Пример 34. Соединение 35
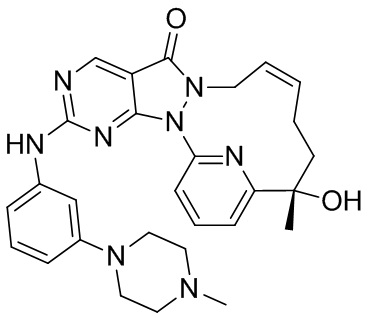
Путь синтеза
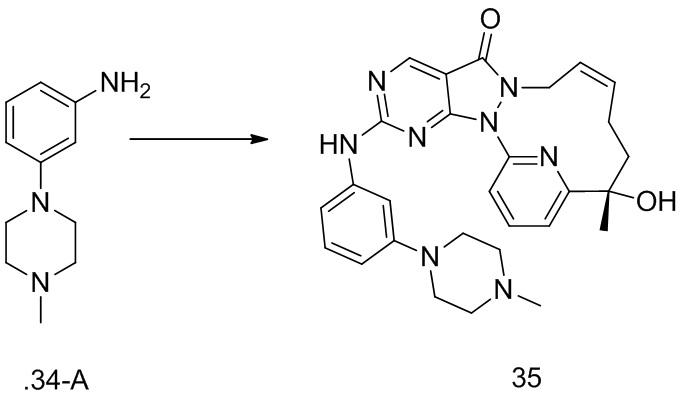
Стадия 1. Синтез соединения 35
За исключением применения соответствующих исходных материалов, 35 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,79 (s, 1 H), 7,73-7,95 (m, 2 H), 7,69 (br s, 1 H), 7,09-7,19 (m, 2 H), 6,89-6,95 (d, J=7,0 Гц, 1 H), 6,52-6,63 (d, J=7,6 Гц, 1 H), 5,54 (br s, 2 H), 4,45 (br s, 1 H), 4,33 (br s, 1 H), 3,07 (br s, 4 H), 2,44 (br s, 4 H), 2,27 (s, 3 H), 2,04-2,12 (m, 2 H), 1,85-1,95 (d, J=8,4 Гц, 1 H), 1,64-1,72 (d, J=7,2 Гц, 1 H), 1,62 (s, 3 H); MS масса/заряд: 513,3 [M+H]+.
Пример 35. Соединение 36
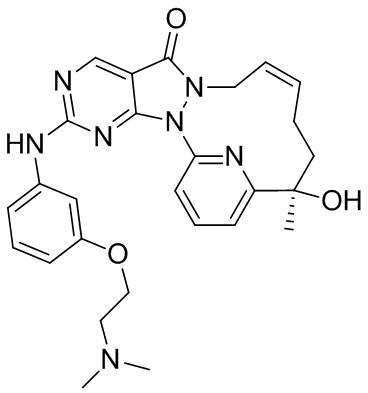
Путь синтеза

Стадия 1. Синтез соединения 36-B
К раствору соединения 36-A (1,5 г, 10,78 ммоль) в N,N-диметилформамиде (25 мл) добавляли карбонат калия (4,47 г, 32,35 ммоль) и 2-хлорэтилдиметиламина гидрохлорид (2,33 г, 16,17 ммоль). Реакционный раствор перемешивали при 85°C в течение 2 часов. К реакционному раствору добавляли воду (10 мл) и водную фазу экстрагировали этилацетатом (15 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором (15 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат высушивали путем выпаривания на роторном испарителе с получением неочищенного продукта. Неочищенный продукт разделяли посредством колоночной хроматографии (дихлорметан/метанол = 10/1) с получением 36-B. MS масса/заряд: 211,3 [M+H]+.
Стадия 2. Синтез соединения 36-C
К раствору соединения 36-B (717 мг, 3,41 ммоль) в этаноле (20 мл) добавляли влажный палладий на угле (300 мг, 253,31 мкмоль) и реакционный раствор перемешивали при 25°C в течение 12 часов в атмосфере водорода (15 фунтов/кв. дюйм). Реакционный раствор фильтровали и фильтрат высушивали путем выпаривания на роторном испарителе с получением 36-C. 1H ЯМР (400 МГц, CDCl3) δ ppm 7,07 (t, J=7,9 Гц, 1 H), 6,26-6,38 (m, 3 H), 4,05 (t, J=5,8 Гц, 2 H), 2,72 (t, J=5,8 Гц, 2 H), 2,29-2,41 (m, 6 H); MS масса/заряд: 181,2 [M+H]+.
Стадия 3. Синтез соединения 36
За исключением применения соответствующих исходных материалов, 36 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,81 (s, 1 H), 7,82-7,88 (m, 1 H), 7,70-7,81 (m, 1 H), 7,60 (s, 1 H), 7,34 (s, 1 H), 7,12-7,19 (m, 1 H), 6,98-7,02 (d, J=7,4 Гц, 1 H), 6,58-6,62 (d, J=8,2, 1 H), 5,64 (br s, 2 H), 4,40-4,52 (d, J=10,2 Гц, 1 H), 4,21 (br s, 1 H), 3,93-4,01 (t, J=5,4 Гц, 2 H), 2,61-2,68 (t, J=5,4 Гц, 2 H), 2,27 (s, 6 H), 2,04-2,14 (m, 2 H), 1,88-1,94 (d, J=11,0 Гц, 1 H), 1,65-1,70 (m, 1 H), 1,61 (s, 3 H); MS масса/заряд: 502,2 [M+H]+.
Пример 36. Соединение 37
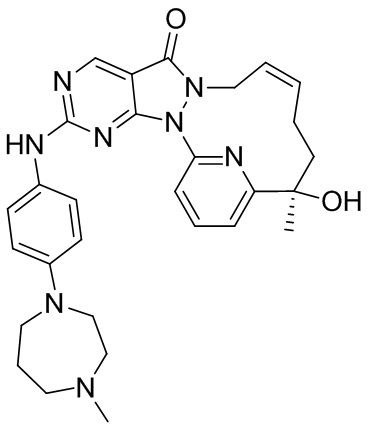
Путь синтеза
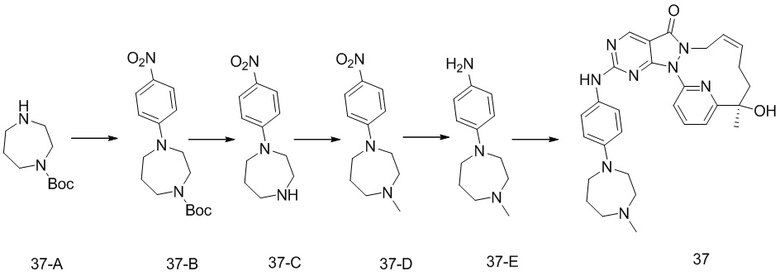
Стадия 1. Синтез соединения 37-B
К раствору 4-фторнитробензола (2,52 г, 14,98 ммоль) в диметилсульфоксиде (40 мл) добавляли карбонат калия (3,00 г, 21,72 ммоль) и 37-A (3 г, 14,98 ммоль), реакционный раствор перемешивали при 100°C в течение 12 часов. К реакционному раствору добавляли 120 мл воды и перемешивали, суспензию фильтровали и осадок на фильтре однократно промывали с помощью 20 мл воды с получением 37-B. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,14 (d, J=9,2 Гц, 2 H), 6,68 (d, J=9,2 Гц, 2 H), 3,61-3,73 (m, 6 H), 3,24-3,42 (m, 2 H), 2,00 (d, J=5,8 Гц, 2 H), 1,36-1,46 (m, 9 H).
Стадия 2. Синтез соединения 37-C
К раствору 37-B (3 г, 9,34 ммоль) в дихлорметане (18 мл) добавляли трифторуксусную кислоту (9,24 г, 81,04 ммоль) и реакционную смесь перемешивали при 30°C в течение 1 часа. Реакционный раствор концентрировали до сухого состояния, к неочищенному продукту добавляли 20 мл воды, водную фазу экстрагировали дихлорметаном (30 мл) и органическую фазу удаляли. С помощью 10% водного раствора гидроксида натрия регулировали pH водной фазы до 11-12, водную фазу экстрагировали дихлорметаном (40 мл × 3), органические фазы объединяли и промывали насыщенным солевым раствором (40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат высушивали путем выпаривания на роторном испарителе с получением 37-C. MS масса/заряд: 222,0 [M+H]+.
Стадия 3. Синтез соединения 37-D
К раствору 37-C (1,87 г, 8,45 ммоль) в метаноле (18 мл) добавляли формальдегид (6,72 г, 82,83 ммоль), ацетат боргидрида натрия (3,58 г, 16,90 ммоль) и уксусную кислоту (507,54 мг, 8,45 ммоль), реакционный раствор перемешивали при 30°C в течение 1 часа. Регулировали pH реакционного раствора до 5 путем добавления разбавленной 2 моль/л хлористоводородной кислоты, затем органическую фазу отгоняли, pH водной фазы регулировали до 11 с помощью 10% водного раствора гидроксида натрия, водную фазу экстрагировали дихлорметаном (50 мл × 3), органические фазы объединяли и промывали насыщенным солевым раствором (30 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат высушивали путем выпаривания на роторном испарителе с получением 37-D. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,03 (d, J=9,6 Гц, 2 H), 6,56 (d, J=9,6 Гц, 2 H), 3,57-3,61 (m, 2 H), 3,52 (t, J=6,2 Гц, 2 H), 2,63-2,68 (m, 2 H), 2,46-2,53 (m, 2 H), 2,29-2,36 (m, 3 H), 1,96 (d, J=5,8 Гц, 2 H); MS масса/заряд: 235,9 [M+H]+.
Стадия 4. Синтез соединения 37-E
К раствору 37-D (1,5 г, 6,38 ммоль) в тетрагидрофуране (20 мл) добавляли палладий на угле (760 мг, 644,07 мкмоль) и реакционный раствор перемешивали при 30°C в течение 12 часов в атмосфере водорода (15 фунтов/кв. дюйм). Реакционный раствор фильтровали и фильтрат высушивали путем выпаривания на роторном испарителе с получением 37-E. 1H ЯМР (400 МГц, CDCl3) δ ppm 6,55-6,61 (m, 2 H), 6,48-6,53 (m, 2 H), 3,38-3,46 (m, 2 H), 3,34 (t, J=6,6 Гц, 2 H), 2,58-2,64 (m, 2 H), 2,45-2,54 (m, 2 H), 2,27-2,37 (m, 3 H), 1,91 (d, J=11,4, 2 H); MS масса/заряд: 206,1 [M+H]+.
Стадия 5. Синтез соединения 37
К раствору 12-A (200 мг, 541,36 мкмоль) в толуоле (10 мл) добавляли м-хлорпероксибензойную кислоту (140,13 мг, 649,63 мкмоль) и реакционный раствор перемешивали при 15°C в течение 4 часов, к реакционному раствору добавляли 37-E (133,37 мг, 649,63 мкмоль) и N,N-диизопропилэтиламин (209,90 мг, 1,62 ммоль) и реакционный раствор перемешивали при 15°C в течение 12 часов. К реакционному раствору добавляли 15 мл воды, водную фазу экстрагировали этилацетатом (15 мл * 3), органические фазы объединяли и промывали сульфитом натрия (20 мл), а затем дополнительно промывали бикарбонатом натрия (20 мл) и насыщенным солевым раствором (20 мл), высушивали над безводным сульфатом натрия, фильтровали и фильтрат высушивали путем выпаривания на роторном испарителе с получением неочищенного продукта. Неочищенный продукт разделяли посредством колоночной хроматографии (дихлорметан/метанол = 10/1, 0,5% NH3.H2O) и препаративной жидкостной (нейтральной) хроматографии с получением 37. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,75 (s, 1 H), 7,72-7,79 (m, 2 H), 7,31 (d, J=8,0 Гц, 2 H), 7,17 (d, J=7,6 Гц, 1 H), 6,60 (d, J=8,8 Гц, 2 H), 5,65 (s, 2 H), 4,41 (d, J=9,4 Гц, 1 H), 4,18 (s, 1 H), 3,48-3,54 (m, 2 H), 3,43 (t, J=6,2 Гц, 2 H), 2,62-2,69 (m, 2 H), 2,49-2,55 (m, 2 H), 2,32 (s, 3 H), 2,01-2,15 (m, 2 H), 1,94-1,98 (m, 2 H), 1,69 (d, J=13,8 Гц, 2 H), 1,60 (s, 3 H); MS масса/заряд: 527,1 [M+H]+.
Пример 37. Соединение 38
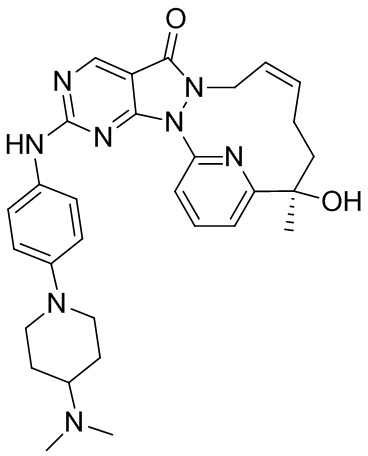
Путь синтеза
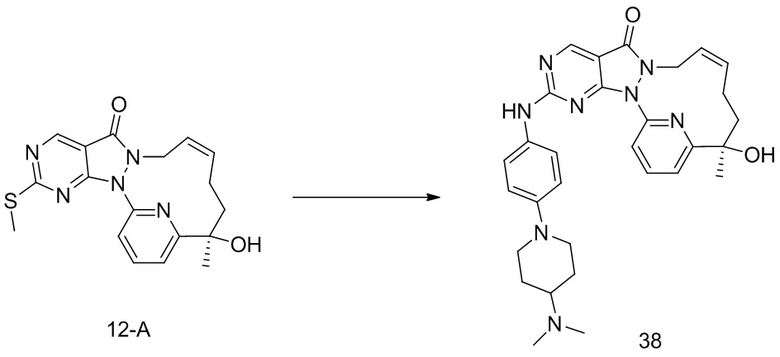
Стадия 1. Синтез соединения 38
За исключением применения соответствующих исходных материалов, 38 получали таким же способом, как и проводили синтез соединения 37 в примере 36. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,78 (s, 1 H), 7,71-7,82 (m, 2 H), 7,39 (d, J=8,8 Гц, 2 H), 7,18 (s, 1 H), 6,86 (d, J=8,8 Гц, 2 H), 5,65 (s, 2 H), 4,42 (d, J=12,4 Гц, 1 H), 4,18 (s, 1 H), 3,63 (d, J=11,6 Гц, 2 H), 2,65 (t, J=12,4 Гц, 2 H), 2,28 (s, 6 H), 2,04-2,17 (m, 2 H), 1,89 (d, J=12,4 Гц, 3 H), 1,68-1,83 (m, 4 H), 1,61 (s, 3 H); MS масса/заряд: 541,1 [M+H]+.
Пример 38. Соединение 39
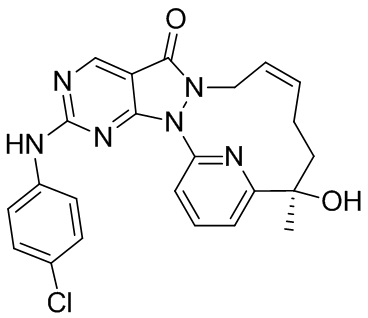
Путь синтеза

Стадия 1. Синтез соединения 39
К раствору 12-A (100 мг, 270,68 мкмоль) в толуоле (7 мл) добавляли м-хлорпероксибензойную кислоту (70,07 мг, 324,82 мкмоль), реакционный раствор перемешивали при 25°C в течение 1 часа, к реакционному раствору добавляли 4-хлоранилин (37,98 мг, 297,75 мкмоль) и N,N-диизопропилэтиламин (139,93 мг, 1,08 ммоль) и реакционный раствор перемешивали при 25°C в течение 12 часов. К реакционному раствору добавляли 10 мл воды, водную фазу экстрагировали этилацетатом (15 мл × 3), органические фазы объединяли, затем промывали бикарбонатом натрия (20 мл) и насыщенным солевым раствором (20 мл), и высушивали над сульфатом натрия, и фильтровали, фильтрат высушивали путем выпаривания на роторном испарителе с получением неочищенного продукта. Неочищенный продукт разделяли посредством препаративной жидкостной (нейтральной) хроматографии с получением 39. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,82 (s, 1 H), 7,80-7,85 (m, 1 H), 7,69 (d, J=7,8 Гц, 1 H), 7,60 (s, 1 H), 7,47-7,52 (m, 2 H), 7,22-7,25 (m, 2 H), 5,64 (s, 2 H), 4,45 (d, J=13,2 Гц, 1 H), 4,18 (br s, 1 H), 2,05-2,14 (m, 1 H), 1,85-1,96 (m, 1 H), 1,69 (d, J=17,6 Гц, 2 H), 1,62 (s, 3 H); MS масса/заряд: 449,0 [M+H]+.
Пример 39. Соединение 40

Путь синтеза

Стадия 1. Синтез соединения 40-A
К раствору гидрида натрия в DMF (20 мл) добавляли йодид калия (142,91 мг, 860,89 мкмоль), реакционный раствор перемешивали при 5-10°C в защитной атмосфере азота и к реакционному раствору медленно добавляли раствор циклопропанола (500 мг, 8,61 ммоль) в DMF (0,5 мл) и перемешивали при 5-10°C в течение 1 часа. Затем к реакционному раствору добавляли раствор п-фторнитробензола (1,34 г, 9,47 ммоль) в DMF (0,5 мл) и реакционный раствор перемешивали при 25°C в течение 12 часов. К реакционному раствору добавляли насыщенный раствор хлорида аммония (15 мл), водную фазу экстрагировали этилацетатом (20 мл × 3), органические фазы объединяли, и однократно промывали насыщенным солевым раствором (20 мл), и высушивали над безводным сульфатом натрия, фильтровали и фильтрат высушивали путем выпаривания на роторном испарителе с получением неочищенного продукта, который разделяли посредством колоночной хроматографии (петролейный эфир/этилацетат = 1-10/1) с получением 40-A.
1H ЯМР (400 МГц, CDCl3) δ = 8,10-8,16 (m, 2 H), 7,05 (d, J=8,06 Гц, 2 H), 3,74-3,79 (m, 1 H), 0,72-0,84 (m, 4 H).
Стадия 2. Синтез соединения 40-B
За исключением применения соответствующих исходных материалов, 40-B получали таким же способом, как и проводили синтез соединения 18-B в примере 17.
1H ЯМР (400 МГц, CDCl3) δ = 6,87-6,93 (m, 2 H), 6,64-6,70 (m, 2 H), 3,65-3,71 (m, 1 H), 0,71-0,80 (m, 4 H). MS масса/заряд: 150,0 [M+H]+.
Стадия 3. Синтез соединения 40
За исключением применения соответствующих исходных материалов, 40 получали таким же способом, как и проводили синтез соединения 22 в примере 21.
1H ЯМР (400 МГц, CDCl3) δ = 8,86 (s, 1H), 7,89-7,83 (t, 1H), 7,82-7,77 (d, 1H), 7,49 (d, J=9,0 Гц, 2H), 7,42 (br s, 1H), 7,25 (d, 1H), 7,05 (d, J=8,8 Гц, 2H), 5,75 (br s, 2H), 4,50 (m, J=12,3 Гц, 1H), 4,33 (br s, 1H), 4,25-4,20 (s, 1H), 3,78-3,73 (m, 1H), 2,22-2,08 (m, 2H), 2,01 (m, J=10,5 Гц, 1H), 1,77 (m, J=13,8 Гц, 1H), 1,68 (s, 3H), 0,82-0,78 (m, 4H). MS масса/заряд: 471,3 [M+H]+.
Пример 40. Соединение 41
Путь синтеза

Стадия 1. Синтез соединения 41
За исключением применения соответствующих исходных материалов, 40 получали таким же способом, как и проводили синтез соединения 22 в примере 21.
1H ЯМР (400 МГц, CDCl3) δ = 8,85 (s, 1H), 7,90-7,83 (t, 1H), 7,82-7,76 (d, 1H), 7,49 (d, J=8,8 Гц, 2H), 7,29-7,24 (d, 1H), 6,94 (d, J=8,8 Гц, 2H), 5,72 (br s, 2H), 4,50 (m, J=12,5 Гц, 1H), 4,42-4,29 (br s, 1H), 4,29-4,21 (s, 1H), 3,40-3,26 (m, 4H), 2,20-2,05 (m, 6H), 2,05-1,95 (m, 1H), 1,79-1,74 (m, 1H), 1,68 (s, 3H). MS масса/заряд: 534,1 [M+H]+.
Пример 41. Соединение 42

Путь синтеза

Стадия 1. Синтез соединения 37
За исключением применения соответствующих исходных материалов, 37 получали таким же способом, как и проводили синтез соединения 26 в примере 25. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,82 (s, 1 H) 7,82-7,88 (m, 1 H) 7,70-7,81 (m, 1 H) 7,58 (br s, 1 H) 7,35 (s, 1 H) 7,12-7,20 (d, J=8,2 Гц, 1 H) 6,58-6,62 (d, J=8,2, 1 H) 5,64 (br s, 2 H) 4,42-4,51 (d, J=12,0 Гц, 1 H) 4,19 (br s, 1 H) 3,94-4,01 (t, J=5,6 Гц, 2 H) 2,62-2,68 (t, J=5,4 Гц, 2 H) 2,27 (s, 6 H) 2,03-2,14 (m, 2 H) 1,86-1,96 (d, J=11,2 Гц, 1 H) 1,69-1,73 (m, 1 H) 1,61 (s, 3 H); MS масса/заряд: 502,2 [M+H]+.
Экспериментальный пример 1. Ферментативная ингибирующая активность in vitro соединений по настоящему изобретению
Экспериментальные тесты проводили в Eurofins, и результаты были предоставлены компанией.
В тестовой системе присутствовали 20 мМ Tris-HCl, pH 8,5, 0,2 мМ EDTA, 500 мкМ полипептидный субстрат (LSNLYHQGKFLQTFCGSPLYRRR), 10 мМ ацетат магния и 10 мкМ [γ-33P]-ATP (интенсивность которого составляла приблизительно 500 имп. в мин./пмоль). После добавления смешанного раствора Mg2+ и ATP начиналась реакция, и реакционный раствор инкубировали при комнатной температуре в течение 40 мин. Для завершения реакции добавляли 3% фосфатный буфер. Фильтровали 10 мкл реакционного раствора на фильтре непрерывного действия P30, три раза промывали с помощью 75 мМ фосфатного буфера и один раз метанолом, при этом каждый раз в течение 5 минут. После высушивания считывали значение с помощью сцинтилляционного способа подсчета.
Таблица 1. Результаты определения ферментативной активности in vitro соединений по настоящему изобретению (IC50)
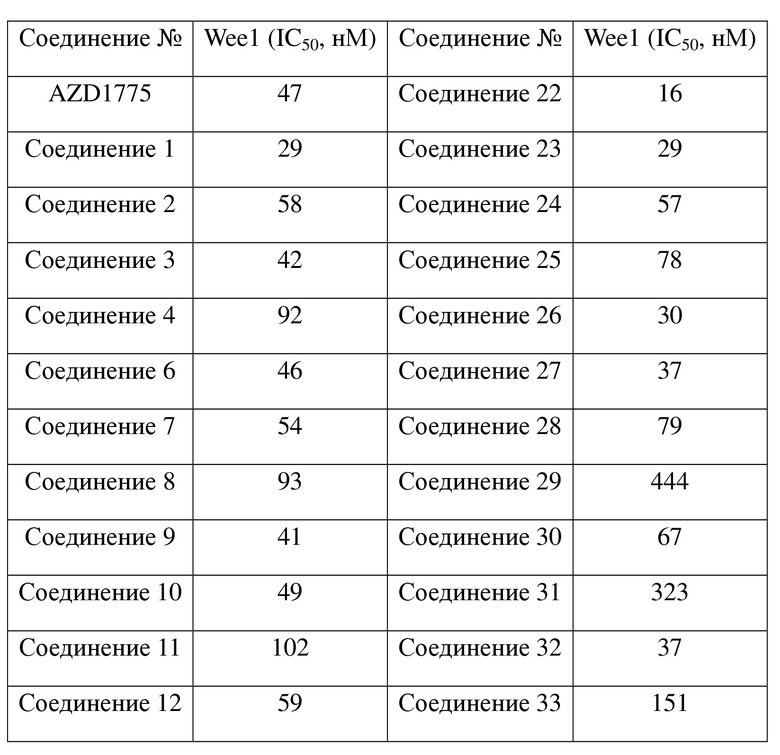

Заключение эксперимента.
Соединения по настоящему изобретению обладают хорошим ингибирующим эффектом в отношении киназы Wee1.
Экспериментальный пример 2. Тест на проницаемость in vitro соединений по настоящему изобретению
В исследовании применяли клетки MDR1-MDCK II, утвержденные лабораторией Пита Борста Института рака Нидерландов, которые представляют собой тип клеток Мадин-Дарби почек собак, трансфицированных геном человека, обуславливающим множественную лекарственную устойчивость (MDR1). Клетки могут стабильно экспрессировать эффлюксный переносчик P-гликопротеин (P-gp), поэтому они являются подходящими для скрининга субстратов или ингибиторов P-gp, и с их помощью можно предсказать, обладают ли соединения высокой проницаемостью через барьер выведения, такой как двенадцатиперстная кишка, гематоэнцефалический барьер, ядро гепатоцитов и нефрон. Авторы настоящего изобретения применяли клетки MDR1-MDCK II от 5-го до 35-го поколения для исследования проницаемости.
Клетки MDR1-MDCK II культивировали в среде α-MEM (α-минимальная эссенциальная среда) в условиях 37±1°C, 5% CO2 и насыщенной относительной влажности. После этого клетки инокулировали в 96-луночном планшете Transwell от BD при плотности инокуляции 2,3 × 105 клеток/см2, а затем клетки помещали в инкубатор с диоксидом углерода на 4-7 дней и применяли для экспериментов на перенос. Способ получения среды α-MEM был следующим: жидкую питательную основу получали путем растворения порошка (порошок α-MEM от Gibco, № по кат. 11900) в чистой воде и добавления L-глутамина и NaHCO3. Для получения полной среды в нее добавляли 10% FBS (эмбриональная бычья сыворотка), 1% PS (содержатся два антибиотика) и 1% NEAA при использовании. Ингредиенты среды α-MEM показаны в таблице 2.
Таблица 2. Список ингредиентов α-MEM (1 л ×)

Вводили AZD1775 (или соединения по настоящему изобретению) и дигоксин в концентрации 2 мкМ, двунаправленно (направления A B и B A), и обеспечивали две повторности в двух лунках. Фенотерол и пропранолол тестировали в концентрации 2 мкМ, вводили однонаправленно (направление A-B), и обеспечивали две повторности в двух лунках.
Раствор, подлежащий применению, предварительно инкубировали на водной бане при 37±1°C в течение 30 минут. Содержащий дозу раствор и принимающий раствор добавляли в соответствующие положения лунок планшета для клеток (75 и 250 мкл для каждой верхней и нижней лунки соответственно) для начала эксперимента по двунаправленному переносу. После загрузки планшеты для клеток помещали в инкубатор при 37±1°C, 5% CO2 и относительной влажности при насыщении в течение 150 минут. Информация о сборе образцов показана в таблице 3.
Таблица 3. Информация о сборе образцов

Примечание: T0 представляет собой исходный образец содержащего дозу раствора.
После перемешивания вихревым способом все образцы центрифугировали при 3220 g в течение 10 минут. Соответствующий объем надосадочной жидкости переносили в планшет для анализа образца и анализировали с помощью LC/MS/MS. Если образцы не анализировали непосредственно после закрытия, их хранили при 2-8°C.
После завершения эксперимента по переносу проводили тестирование на целостность клеток MDR1-MDCK II с применением анализа отторжения Люцифера желтого. После инкубирования в течение 30 минут раствора, содержащего флуоресцентный желтый краситель, собирали образцы с флуоресцентным желтым красителем и определяли относительную единицу флуоресценции (RFU) флуоресцентного желтого красителя в образце при 425/528 нм (возбуждение/испускание) с помощью планшет-ридера 2e.
Применяли полуколичественный анализ для тестируемых продуктов AZD1775 (или соединения по настоящему изобретению), контрольных продуктов: фенотерола, пропранолола и дигоксина. Применяли соотношение площади пика аналита и внутреннего стандарта как концентрацию эталонного вещества. Экспериментальные результаты показаны в таблице 4.
Таблица 4. Скорость проникновения (10-6 см/с)

Заключение эксперимента.
Свойства в отношении проницаемости соединений по настоящему изобретению значительно улучшены по сравнению с AZD1775, что является преимущественным для использования лекарственных средств в отношении организмов.
Экспериментальный пример 3. Оценивание фармакокинетических характеристик соединений
Целью данного эксперимента являлось исследование фармакокинетики AZD1775 (или соединений по настоящему изобретению) в плазме крови самок голых мышей Balb/c после однократного внутривенного, однократного перорального введения.
Двенадцать мышей (предоставленных Lingchang Biotech) произвольным образом разделяли на две группы, по 6 самок в каждой группе, и образцы собирали посредством перекрестного забора крови. Всем животным в группе с внутривенным введением назначали 1 мг/кг AZD1775 (или соединений по настоящему изобретению) посредством внутривенной инъекции. Состав представлял собой прозрачный раствор 5% HP-betaCD (Kunshan Ruisk Chemical Materials Co., Ltd.), содержащий 0,2 мг/мл AZD1775 (или соединений по настоящему изобретению). Животным в группе с пероральным введением назначали AZD1775 (или соединения по настоящему изобретению) в дозе 10 мг/кг посредством зонда. Состав представлял собой однородную суспензию 0,5% метилцеллюлозы, содержащую 1 мг/мл AZD1775 (или соединений по настоящему изобретению). В группе с внутривенным введением образцы плазмы крови собирали в 9 моментов времени: через 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения; в группе с пероральным введением образцы плазмы крови собирали в 8 моментов времени: через 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения. Образцы анализировали с помощью LC-MS/MS с получением данных о концентрации в плазме крови AZD1775 (или соединений по настоящему изобретению) и рассчитывали фармакокинетические параметры, такие как пиковая концентрация, пиковое время, скорость выведения, период полувыведения, площадь под кривой зависимости концентрации от времени, биодоступность и т. д. Экспериментальные результаты показаны в таблице 5.
Таблица 5. Результаты тестирования фармакокинетических характеристик

Заключение. По сравнению с AZD1775 соединения по настоящему изобретению характеризуются несколькими в значительной степени улучшенными показателями фармакокинетики у мышей, среди которых скорость выведения in vivo, период полувыведения, интегральная концентрация in vivo и биодоступность обеспечивают очевидные преимущества.
Экспериментальный пример 4. Исследование in vivo
(1) Фармакодинамическое исследование in vivo соединений на модели рака толстой кишки человека, полученной путем подкожной ксенотрансплантации опухоли из клеток LoVo голым мышам BALB/c
Экспериментальный способ. Выбранное экспериментальное животное (предоставленное Shanghai Xipu'er-bikai Experimental Animal Co., Ltd.) представляло собой голых мышей BALB/c, возрастом 6-8 недель, весом 18-22 грамм.
Клетки LoVo рака толстой кишки человека культивировали в монослое in vitro. Условия культивирования были следующими: среда Хэма F-12, содержащая 10% эмбриональной бычьей сыворотки, 100 Ед./мл пенициллина, 100 мкг/мл стрептомицина и 2 мМ глутамина, культивирование при 37ºC, 5% CO2. Проводили традиционную обработку для открепления клеток с помощью трипсин-EDTA два раза в неделю для пассирования. Если насыщение клеток составляло 80%-90%, клетки собирали, подсчитывали и инокулировали. В правый бок каждой голой мыши подкожно инокулировали 0,1 мл (10 × 106) клеток LoVo. Когда средний объем опухоли достигал 213 мм3, их разделяли на группы и начинали введение лекарственного средства. Дозировка: 40 мг/кг два раза в сутки. Экспериментальный показатель служил для исследования того, происходило ли подавление, замедление или устранение роста опухоли. Диаметр опухоли измеряли с помощью штангенциркуля с нониусом два раза в неделю. Формула для расчета объема опухоли представляла собой: V = 0,5a × b2, a и b представляют собой длинный и короткий диаметры опухоли соответственно.
Противоопухолевую эффективность соединения оценивали с помощью TGI (%) или относительной скорости разрастания опухоли T/C (%). Значение TGI (%) отражает степень подавления роста опухоли. Расчет TGI (%): TGI (%) = [(1 – (средний объем опухоли в конце введения в определенной группе обработки – средний объем опухоли в начале введения в данной группе обработки))/(средний объем опухоли в контрольной группе, получающей растворитель, в конце обработки – средний объем опухоли в контрольной группе, получающей растворитель, в начале обработки)] × 100%.
После последнего введения через 16 дней экспериментальные результаты являлись следующими.
Таблица 6. Результаты касательно эффективности in vivo лекарственного средства в отношении опухолей у мышей
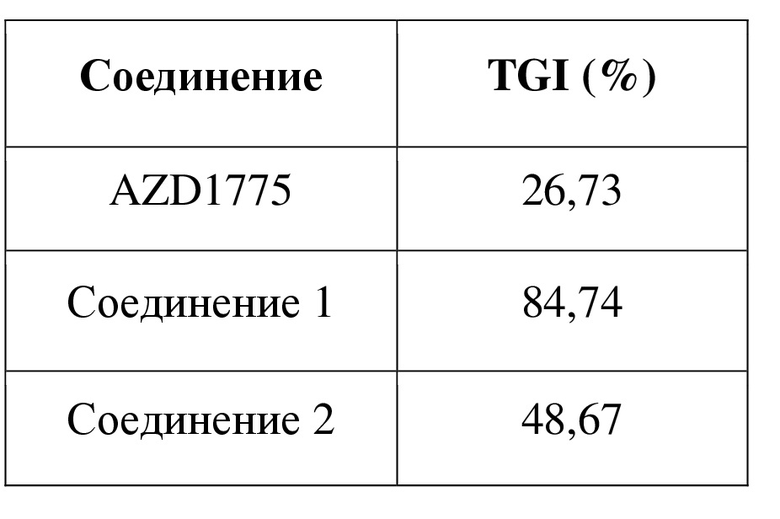
Заключение. По сравнению с AZD1775 соединения по настоящему изобретению обладают в значительной степени увеличенным ингибирующим эффектом в отношении опухолей у мышей, и хиральность соединения оказывает неожиданный эффект на эффективность in vivo лекарственного средства.
(2) Фармакодинамическое исследование in vivo соединения на модели рака поджелудочной железы человека, полученной путем подкожной трансплантации опухоли из клеток BxPC-3 голым мышам BALB/c.
Экспериментальный способ. Выбранное экспериментальное животное представляло собой голых мышей BALB/c, возрастом 6-8 недель, весом 18-22 грамма.
Клетки BxPC-3 10-го поколения культивировали в монослое in vitro. Условия культивирования являлись следующими: среда RPMI 1640 (изготовитель: gibco, номер изделия: 22400-089), содержащая 10% эмбриональной бычьей сыворотки, 100 Ед./мл пенициллина и 100 мкг/мл стрептомицина, культивирование при 37°C, 5% CO2, пассирование выполняли 4 раза. Способ пассирования представлял собой традиционную обработку для открепления клеток с помощью трипсин-EDTA два раза в неделю. Когда насыщение клеток достигало 80%-90%, клетки открепляли с помощью трипсин-EDTA, подсчитывали и повторно суспендировали в PBS при плотности 5 x 107 клеток/мл. Каждому животному инокулировали 0,1 мл (5 × 106) клеток BxPC-3 в правый бок. Когда средний объем опухоли достигал 153 мм3, их произвольным образом разделяли на группы в соответствии с объемом опухоли и начинали введение лекарственного средства. Дозировка: 25 мг/кг, один раз в сутки.
Экспериментальный показатель служил для исследования того, происходило ли подавление, замедление или устранение роста опухоли. Диаметр опухоли измеряли с помощью штангенциркуля с нониусом два раза в неделю. Противоопухолевую эффективность соединения оценивали с помощью TGI (%) или относительной скорости разрастания опухоли T/C (%). Значение TGI (%) отражает степень подавления роста опухоли. Расчет TGI (%): TGI (%) = [(1 – (средний объем опухоли в конце введения в определенной группе обработки – средний объем опухоли в начале введения в данной группе обработки))/(средний объем опухоли в контрольной группе, получающей растворитель, в конце обработки – средний объем опухоли в контрольной группе, получающей растворитель, в начале обработки)] × 100%.
После последнего введения через 27 дней экспериментальные результаты являлись следующими.
Таблица 7. Результаты касательно эффективности in vivo лекарственного средства в отношении опухолей у мышей
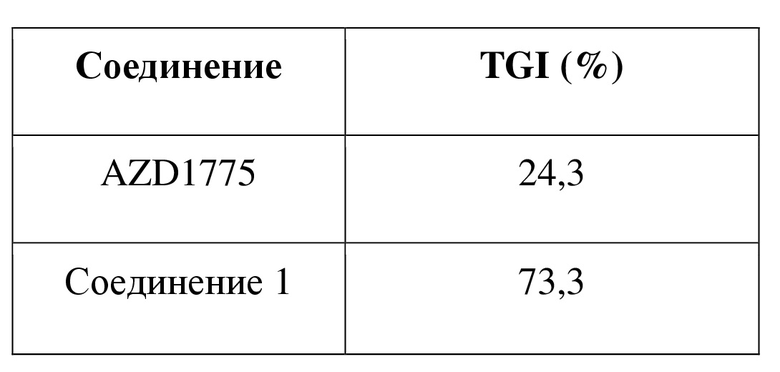
Заключение. Из таблицы 7 можно увидеть, что соединение по настоящему изобретению обладает в значительной степени увеличенным ингибирующим эффектом в отношении опухолей у мышей по сравнению с AZD1775.
(3) Противоопухолевая эффективность in vivo соединений на животной модели рака толстой кишки, полученной путем трансплантации опухоли из клеток CT26 мышам.
Экспериментальный способ. Выбранное экспериментальное животное представляло собой голых мышей BALB/c, возрастом 7 недель, весом 16-20 грамм, самки.
Клетки: клетки CT26 рака толстой кишки мыши (клеточный банк Комитета по хранению типовых культур Китайской академии наук) культивировали в монослое in vitro. Условия культивирования были следующими: среда RPMI-1640, содержащая 10% эмбриональной бычьей сыворотки, и инкубатор с условиями 37°C, 5% CO2. Проводили традиционную обработку для открепления клеток с помощью трипсин-EDTA два раза в неделю для пассирования. Когда клетки находились в экспоненциальной фазе роста и насыщение составляло 80% - 90%, клетки собирали, подсчитывали и инокулировали. В правый бок каждой мыши подкожно инокулировали 0,1 мл DPBS (содержащего 3 × 105 клеток CT26). Когда средний объем опухоли достигал 50-70 мм3, выполняли рандомизированное введение в соответствии с объемом опухоли. Дозировка: 30 мг/кг два раза в сутки.
Экспериментальный показатель служил для исследования того, происходило ли подавление, замедление или устранение роста опухоли. Диаметр опухоли измеряли с помощью штангенциркуля с нониусом два раза в неделю. Противоопухолевую эффективность соединения оценивали с помощью TGI (%) или относительной скорости разрастания опухоли T/C (%). Значение TGI (%) отражает степень подавления роста опухоли. Расчет TGI (%): TGI (%) = [(1 – (средний объем опухоли в конце введения в определенной группе обработки – средний объем опухоли в начале введения в данной группе обработки))/(средний объем опухоли в контрольной группе, получающей растворитель, в конце обработки – средний объем опухоли в контрольной группе, получающей растворитель, в начале обработки)] × 100%.
После последнего введения через 18 дней экспериментальные результаты являлись следующими.
Таблица 8. Результаты касательно эффективности in vivo лекарственного средства в отношении опухолей у мышей
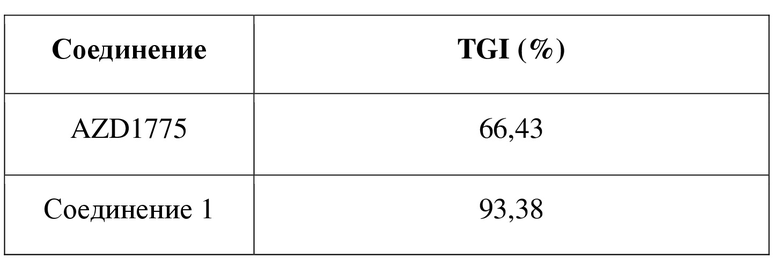
Заключение. Из таблицы 8 можно увидеть, что соединение по настоящему изобретению обладает в значительной степени увеличенным ингибирующим эффектом в отношении опухолей у мышей по сравнению с AZD1775.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| ИНГИБИТОР PDE4 | 2017 |
|
RU2743126C2 |
| ПИРИМИДОИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ДНК-PK | 2020 |
|
RU2796163C1 |
| ИНГИБИТОР LSD1, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2763898C2 |
| ГЛУТАРИМИДНОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ КОЛЬЦОМ, КОНДЕНСИРОВАННЫМ С ФУРАНОМ | 2022 |
|
RU2836921C1 |
| ПИРИДОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, ВЫПОЛНЯЮЩИЕ ФУНКЦИЮ ДВОЙНЫХ ИНГИБИТОРОВ mTORC 1/2 | 2018 |
|
RU2771201C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ 7-ЗАМЕЩЕННОГО ПИРРОЛОТРИАЗИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2018 |
|
RU2745548C1 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ДЕЙСТВУЮЩИЕ НА БЕЛКИ CRBN | 2019 |
|
RU2801068C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| АГОНИСТ S1P1 И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2754845C2 |
Группа изобретений относится к фармацевтической химии и включает соединение формулы (II), конкретные макроциклические соединения, указанные в формуле изобретения, их стереоизомеры и фармацевтически приемлемые соли, а также их применение в изготовлении лекарственного средства. В формуле (II) R1 выбран из H и C1-3алкила; R5 выбран из H и C1-3алкила, где C1-3алкил необязательно замещен R, R6 является таким, как указано в формуле изобретения; r и m равняются 1 или 2; D выбран из -N(R2)- и -C(R3)(R4)-; R2 выбран из H и C1-3алкила; R3 и R4 независимо выбраны из H, F, Cl, Br, I, NH2 и C1-3алкила, где NH2 необязательно замещен R, и при этом число R равняется 1, 2 или 3; в качестве альтернативы R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 5-6-членный гетероциклоалкил, который содержит N в качестве гетероатома, число гетероатомов равняется одному или двум, где 5-6-членный гетероциклоалкил необязательно замещен R, и при этом число R равняется 1, 2 или 3; R61 выбран из H, F, Cl, Br, I, C1-3алкокси и -O-C3-6циклоалкила, где C1-3алкокси необязательно замещен R, и при этом число R равняется 1, 2 или 3; R7 выбран из H, F, Cl, Br, I, C1-3алкила, C1-3алкокси и 5-6-членного гетероциклоалкила, который содержит N в качестве гетероатома, число гетероатомов равняется одному или двум, где C1-3алкил, C1-3алкокси и 5-6-членный гетероциклоалкил необязательно замещены R, и при этом число R равняется 1, 2 или 3; в качестве альтернативы R6 и R7 вместе с атомами кольца, к которым они присоединены, образуют кольцо A, и при этом кольцо A представляет собой 5-6-членный гетероциклоалкил, который содержит N в качестве гетероатома, число гетероатомов равняется одному или двум, который необязательно замещен R, и при этом число R равняется 1, 2 или 3; R независимо выбран из OH, C1-3алкила, C1-3алкокси и C1-3диалкиламино. Технический результат - макроциклические соединения, проявляющие ингибирующую активность в отношении киназы Wee1. 3 н. и 17 з.п. ф-лы, 8 табл., 42 пр.
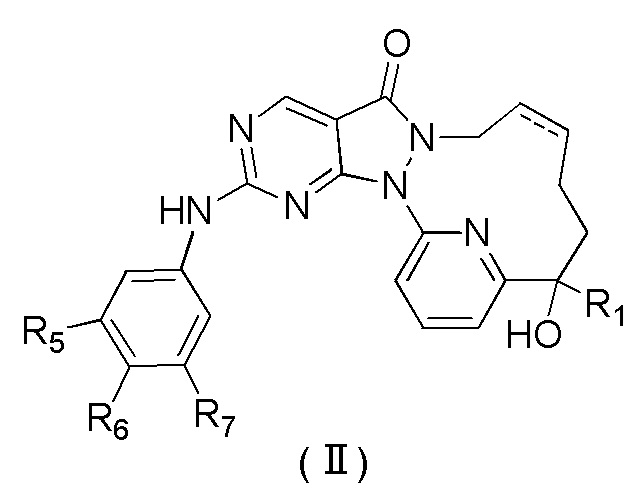
1. Соединение, представленное формулой (II), его стереоизомер или его фармацевтически приемлемая соль,
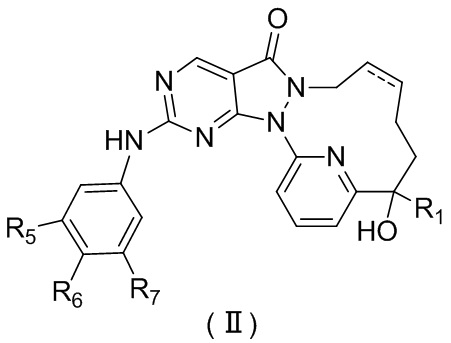 ,
,
где
 представляет собой одинарную связь или двойную связь;
представляет собой одинарную связь или двойную связь;
R1 выбран из H и C1-3алкила;
R5 выбран из H и C1-3алкила, где C1-3алкил необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R6 выбран из R61, 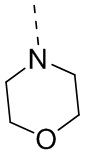 ,
, 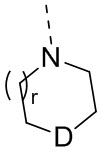 ,
, 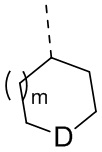 и
и  ;
;
r равняется 1 или 2;
m равняется 1 или 2;
D выбран из -N(R2)- и -C(R3)(R4)-;
R2 выбран из H и C1-3алкила;
R3 и R4 независимо выбраны из H, F, Cl, Br, I, NH2 и C1-3алкила, где NH2 необязательно замещен R, и при этом число R равняется 1, 2 или 3;
в качестве альтернативы R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 5-6-членный гетероциклоалкил, который содержит N в качестве гетероатома, число гетероатомов равняется одному или двум, где 5-6-членный гетероциклоалкил необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R61 выбран из H, F, Cl, Br, I, C1-3алкокси и -O-C3-6циклоалкила, где C1-3алкокси необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R7 выбран из H, F, Cl, Br, I, C1-3алкила, C1-3алкокси и 5-6-членного гетероциклоалкила, который содержит N в качестве гетероатома, число гетероатомов равняется одному или двум, где C1-3алкил, C1-3алкокси и 5-6-членный гетероциклоалкил необязательно замещены R, и при этом число R равняется 1, 2 или 3;
в качестве альтернативы R6 и R7 вместе с атомами кольца, к которым они присоединены, образуют кольцо A, и при этом кольцо A представляет собой 5-6-членный гетероциклоалкил, который содержит N в качестве гетероатома, число гетероатомов равняется одному или двум, который необязательно замещен R, и при этом число R равняется 1, 2 или 3;
R независимо выбран из OH, C1-3алкила, C1-3алкокси и C1-3диалкиламино.
2. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где R независимо выбран из OH, CH3, Et, -OCH3 и  .
.
3. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1 или 2, где R1 выбран из H, CH3 и Et;
или R2 выбран из H, CH3 и Et;
или R3 и R4 независимо выбраны из H, F, Cl, Br, I, NH2, -NH(CH3), -N(CH3)2, CH3 и Et;
или R5 выбран из H, CH3 и Et, где CH3 и Et необязательно замещены R, и при этом число R равняется 1, 2 или 3;
или R61 выбран из H, F, Cl, Br, I, -OCH3,  и
и  , где -OCH3 и необязательно замещены R, и при этом число R равняется 1, 2 или 3;
, где -OCH3 и необязательно замещены R, и при этом число R равняется 1, 2 или 3;
или R6 выбран из R61, ,  , ,
, ,  и
и  ;
;
или R7 выбран из H, F, Cl, Br, I, CH3, -OCH3, и  , где CH3, -OCH3, и необязательно замещены R, и при этом число R равняется 1, 2 или 3;
, где CH3, -OCH3, и необязательно замещены R, и при этом число R равняется 1, 2 или 3;
или кольцо A выбрано из пиперидила, который необязательно замещен R, и при этом число R равняется 1, 2 или 3;
или фрагмент 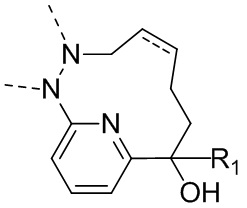 выбран из
выбран из 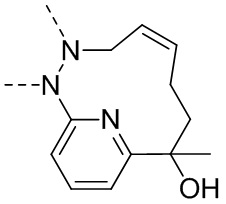 ,
,  и
и  .
.
4. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, где R3 выбран из H, F, Cl, Br, I, NH2, -NH(CH3), -N(CH3)2, CH3 и Et.
5. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, где R4 выбран из H, F, Cl, Br, I, CH3 и Et.
6. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, где R5 выбран из H, CH3 и -CH2OH.
7. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, где R61 выбран из H, F, Cl, Br, I, -OCH3,  и .
и .
8. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, где R6 выбран из R61,  ,
, 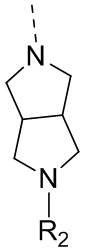 , ,
, , 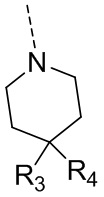 ,
, 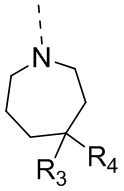 ,
, 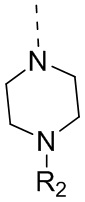 и
и  .
.
9. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где фрагмент  представляет собой
представляет собой  .
.
10. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1, 7, 8 или 9, где R6 выбран из H, F, Cl, Br, I, -OCH3, , ,  ,
,  , ,
, ,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и  .
.
11. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, где R7 выбран из H, F, Cl, Br, I, -OCH3,  , и
, и  .
.
12. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 3, где фрагмент 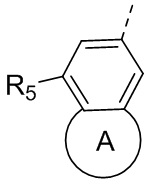 представляет собой
представляет собой  .
.
13. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1-7 или 11, которые выбраны из
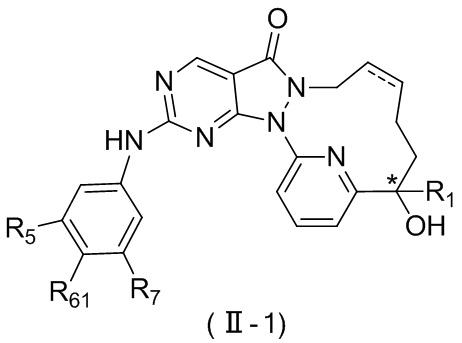 ,
, 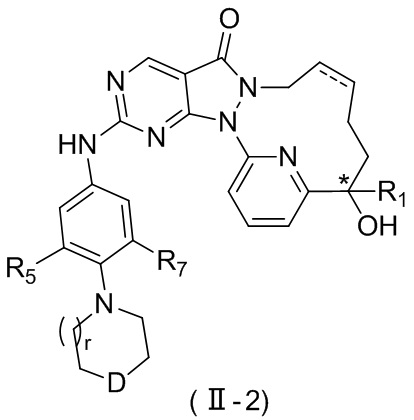 ,
,  ,
, 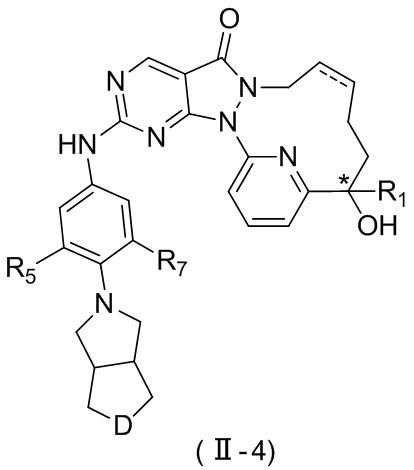 и
и  ,
,
где
D выбран из -N(R2)- и -C(R3)(R4)-;
r, m, R1, R2, R3, R4, R5, R61 и R7 определены в любом из пп. 1-7 или 11;
атом углерода, обозначенный с помощью «*», представляет собой хиральный атом углерода, и находятся в форме одного (R)- или (S)-энантиомера.
14. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 13, которые выбраны из
 ,
,
где
D выбран из -N(R2)-, и -C(R3)(R4)-;
r, R1, R2, R3, R4 определены в любом из пп. 1-5;
атом углерода, обозначенный с помощью «*», представляет собой хиральный атом углерода, и находятся в форме одного (R)- или (S)-энантиомера.
15. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 14, где фрагмент  выбран из
выбран из  и
и  .
.
16. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 14, которые выбраны из

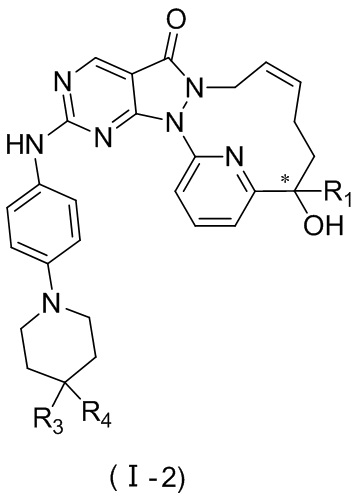
 и
и  ,
,
где R1, R2, R3, R4 определены в любом из пп. 1-5;
атом углерода, обозначенный с помощью «*», представляет собой хиральный атом углерода, и находятся в форме одного (R)- или (S)-энантиомера.
17. Соединение, его стереоизомер или его фармацевтически приемлемая соль, которое выбрано из
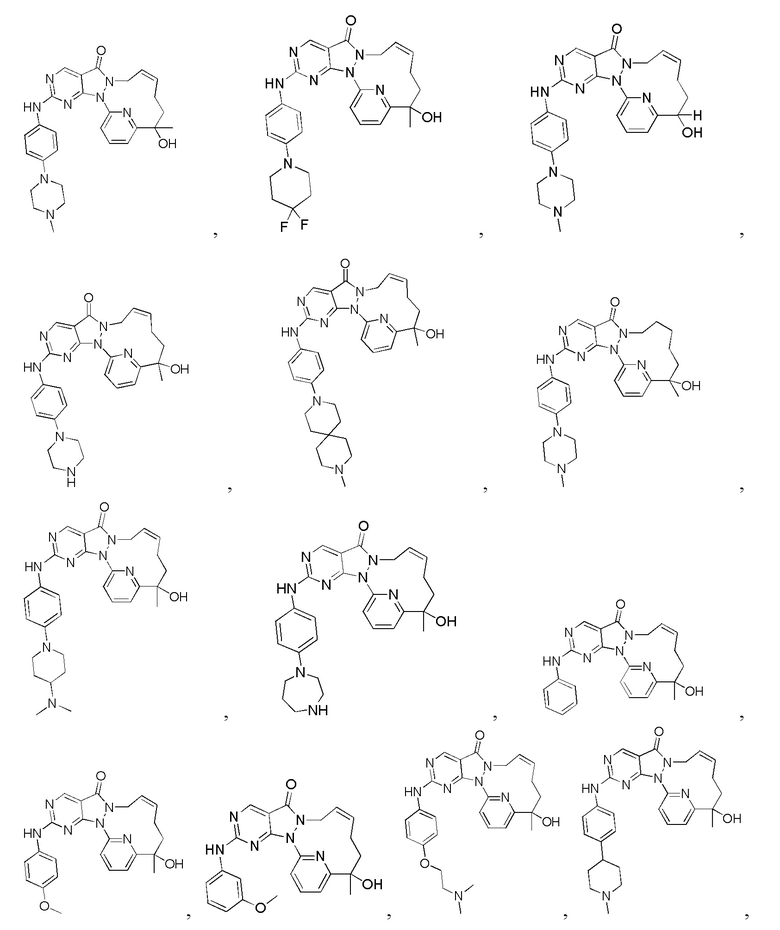
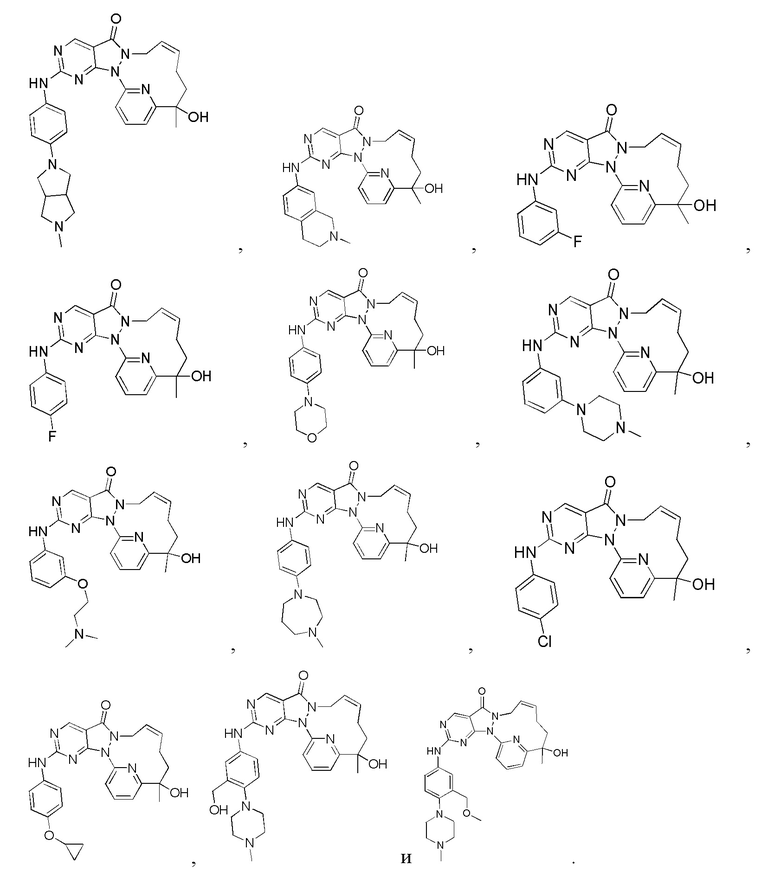
18. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 17, которые выбраны из

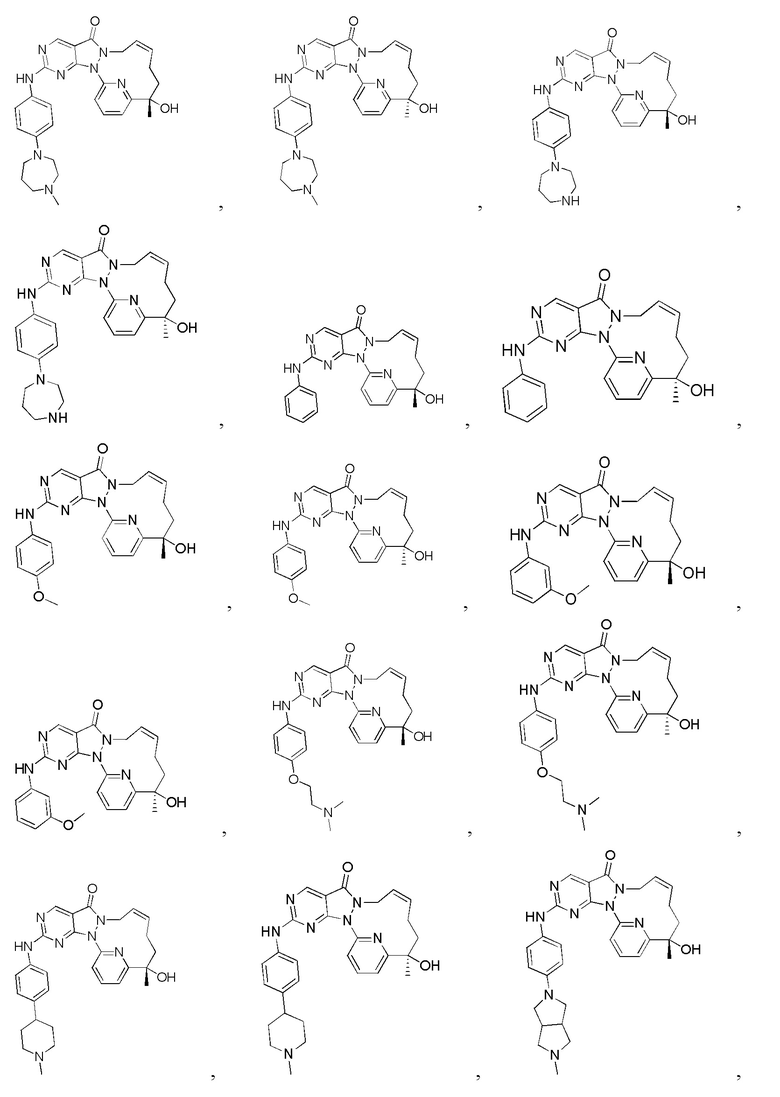


19. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-18 в изготовлении лекарственного средства, предназначенного для лечения заболеваний, связанных с Wee1.
20. Применение по п. 19, где лекарственное средство применяется для лечения солидных опухолей.
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ TRK | 2011 |
|
RU2594742C2 |

