Область техники, к которой относится изобретение
Настоящее изобретение касается соединения, имеющего формулу (I), или его изомера или фармацевтически приемлемой соли, и их применения в производстве лекарственного средства для лечения заболеваний, связанных с RIP-1 (белок, взаимодействующий с рецептором) киназой.
Предшествующий уровень техники
RIP1 представляет собой важную киназу, управляющую некрозом и апоптозом клеток. RIP1 также участвует в различных сигнальных путях воспалительного ответа и может стимулироваться лигандами из TNF и TLR семейств, запуская воспалительный ответ (Weinlich, R. Nat. Rev. Mol. Cell. Biol. 2017, 18, 127; Pasparakis M. et al., Nature 2015, 517, 311; Berger, S.B. Cell Death Discovery, 2016, 2, e16056). RIP1 запускает ряд сигнальных путей воспалительного ответа [Fas лиганд, апоптоз-индуцирующий лиганд семейства TNF (TRAIL), TLR3 и TLR4] через TNF рецептор 1 (Lukens J. R., et al. Nature, 2013, 498, 224). Ингибирование активности RIP1 может применяться для лечения многих воспалительных заболеваний.
Краткое описание изобретения
В настоящем изобретении описано соединение, имеющее формулу (I), или его изомер или фармацевтически приемлемая соль,
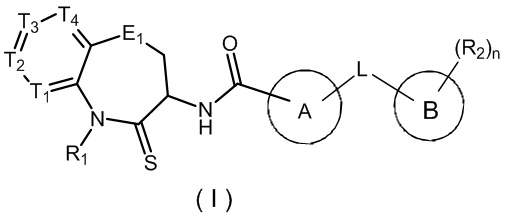
где:
T1 выбран из N и CR1t;
T2 выбран из N и CR2t;
T3 выбран из N и CR3t;
T4 выбран из N и CR4t;
E1 выбран из C(R1e)2, O, C(=O), S и NR2e;
Цикл A выбран из 1,2,4-триазолила, 1,2,3-триазолила, имидазолила, изоксазолила, 1,3,4-оксадиазолила, 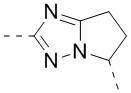 , тетразолила, пиридила и
, тетразолила, пиридила и 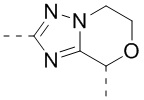 , где 1,2,4-триазолил, 1,2,3-триазолил, имидазолил, изоксазолил, 1,3,4-оксадиазолил,
, где 1,2,4-триазолил, 1,2,3-триазолил, имидазолил, изоксазолил, 1,3,4-оксадиазолил, 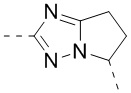 , тетразолил, пиридил и необязательно замещены 1, 2 или 3 галогенами или C1-3 алкилами;
, тетразолил, пиридил и необязательно замещены 1, 2 или 3 галогенами или C1-3 алкилами;
Цикл B выбран из фенила и ;
;
L выбран из простой связи, O, C(=O), S, NH и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Ra;
R1 выбран из H и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rb;
R1t, R2t, R3t и R4t каждый независимо выбраны из H, F, Cl, Br, I, OH, CN, NH2, C1-3 алкила, COOH и -C(=O)NH2, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rc;
R1e каждый независимо выбран из H, F, Cl, Br, I, OH, CN, NH2 и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rd;
R2e выбран из H и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Re;
R2 каждый независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, C1-3 алкила и C1-3 алкокси-группы, где C1-3 алкил и C1-3 алкокси-группа необязательно замещены 1, 2 или 3 заместителями Rf;
n равен 1, 2, 3, 4 или 5;
Ra, Rb, Rc, Rd, Re и Rf каждый независимо выбраны из F, Cl, Br, I, OH, CN, NH2 и D.
В настоящем изобретении описано соединение, имеющее формулу (I), или его изомер или фармацевтически приемлемая соль,
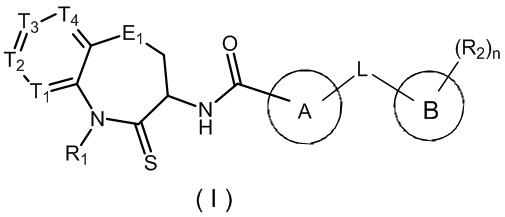 ,
,
где
T1 выбран из N и CR1t;
T2 выбран из N и CR2t;
T3 выбран из N и CR3t;
T4 выбран из N и CR4t;
E1 выбран из C(R1e)2, O, C(=O), S и NR2e;
Цикл A выбран из 1,2,4-триазолила, 1,2,3-триазолила, имидазолила, изоксазолила, 1,3,4-оксадиазолила и, где 1,2,4-триазолил, 1,2,3-триазолил, имидазолил, изоксазолил, 1,3,4-оксадиазолил и необязательно замещены 1, 2 или 3 галогенами или C1-3 алкилами;
Цикл B представляет собой фенил;
L выбран из простой связи, O, C(=O), S, NH и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Ra;
R1 выбран из H и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rb;
R1t, R2t, R3t и R4t каждый независимо выбраны из H, F, Cl, Br, I, OH, CN, NH2 и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rc;
R1e каждый независимо выбран из H, F, Cl, Br, I, OH, CN, NH2 и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rd;
R2e выбран из H и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Re;
R2 каждый независимо выбран из H, F, Cl, Br, I, OH, NH2, CN и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rf;
n равен 1, 2, 3, 4 или 5;
Ra, Rb, Rc, Rd, Re и Rf каждый независимо выбраны из F, Cl, Br, I, OH, CN, NH2 и D.
В настоящем изобретении описано соединение, имеющее формулу (I), или его изомер или фармацевтически приемлемая соль,
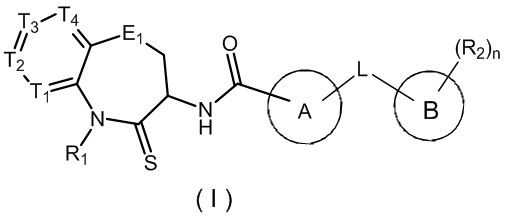 ,
,
где
T1 представляет собой N или CR1t;
T2 представляет собой N или CR2t;
T3 представляет собой N или CR3t;
T4 представляет собой N или CR4t;
E1 is C(R1e)2, O, C(=O), S, или NR2e;
Цикл A представляет собой 5-членный гетероарил, необязательно замещенный C1-3 алкилом;
Цикл B представляет собой фенил или 6-членный гетероарил;
L представляет собой O, C(=O), S, NH или C1-3 алкил, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Ra;
R1 выбран из H и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rb;
R1t, R2t, R3t и R4t каждый независимо выбраны из H, F, Cl, Br, I, OH, CN, NH2 и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rc;
R1e каждый независимо выбран из H, F, Cl, Br, I, OH, CN, NH2 и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rd;
R2e выбран из H и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Re;
R2 каждый независимо выбран из H, F, Cl, Br, I, OH, NH2, CN и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rf;
n равен 1, 2, 3, 4 или 5;
Ra, Rb, Rc, Rd, Re и Rf каждый независимо выбраны из F, Cl, Br, I, OH, CN и NH2.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R1 выбран из H и CH3, где CH3 необязательно замещен 1, 2 или 3 заместителями Rb, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R1 выбран из H, CH3 и CD3, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R1 выбран из H и CH3, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R1t, R2t, R3t и R4t каждый независимо выбраны из H, F, Cl, Br, I, OH, CN, NH2, CH3, COOH и -C(=O)NH2, где CH3 необязательно замещен 1, 2 или 3 заместителями Rc, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R1t, R2t, R3t и R4t каждый независимо выбраны из H, F, Cl, Br, I, OH, CN, NH2 и CH3, где CH3 необязательно замещен 1, 2 или 3 заместителями Rc, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R1t, R2t, R3t и R4t каждый независимо выбраны из H, F, Cl, Br, I, OH, CN, NH2 и CH3, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R1e каждый независимо выбран из H, F, Cl, Br, I, OH, CN, NH2 и CH3, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R2e выбран из H и CH3, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше E1 представляет собой CH2, O, C(=O), S или NH, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R2 каждый независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, CH3 и OCH3, где CH3 и OCH3 необязательно замещены 1, 2 или 3 заместителями Rf, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R2 каждый независимо выбран из H, F, Cl, Br, I, OH, NH2, CN, CH3, OCH3, CF3 и OCF3, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше R2 каждый независимо выбран из H, F, Cl, Br, I, OH, NH2 и CN, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше Цикл A выбран из  ,
,  ,
, 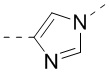 ,
, 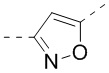 ,
, 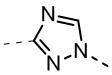 ,
, 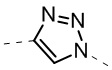 ,
, 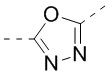 ,
, 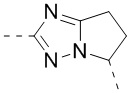 ,
, 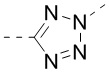 ,
, 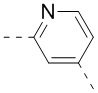 , и , где , , , , , , , , , и необязательно замещены 1, 2 или 3 галогенами или C1-3 алкилами, и другие переменные имеют значения, указанные в настоящем тексте.
, и , где , , , , , , , , , и необязательно замещены 1, 2 или 3 галогенами или C1-3 алкилами, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше Цикл A выбран из , , , , , , и .
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше Цикл A представляет собой пирролил, имидазолил, пиразолил, фурил, тиенил, оксазолил, изоксазолил, тиазолил, 1,2,3-триазолил, 1,2,4-триазолил или 1,3,4-тиадиазолил, где пирролил, имидазолил, пиразолил, фурил, тиенил, оксазолил, изоксазолил, тиазолил, 1,2,3-триазолил, 1,2,4-триазолил или 1,3,4-тиадиазолил необязательно замещен C1-3 алкилом, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше L выбран из простой связи, -CH2- и -O-, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше L выбран из простой связи и -CH2-, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше Цикл A представляет собой  , и другие переменные имеют значения, указанные в настоящем тексте.
, и другие переменные имеют значения, указанные в настоящем тексте.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанный выше Цикл B представляет собой фенил или пиридил, и другие переменные имеют значения, указанные в настоящем тексте.
Настоящее изобретение включает варианты осуществления, реализующиеся при комбинировании любых указанных выше переменных.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанное выше соединение выбрано из
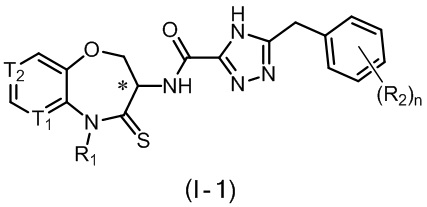
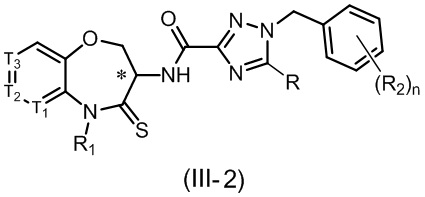
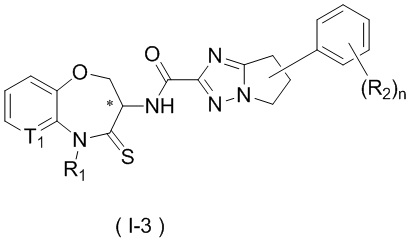 и
и 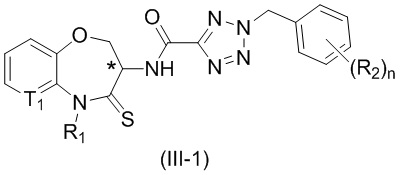
где
n, T1, T2, T3, R1, R2 и R имеют указанные в настоящем тексте значения;
атом углерода с “*” представляет собой хиральный атом углерода, который существует в форме индивидуального (R) или (S) энантиомера или смеси, обогащенной одним энантиомером;
или его изомер или фармацевтически приемлемая соль.
В некоторых вариантах осуществления, описанных в настоящем тексте, указанное выше соединение выбрано из
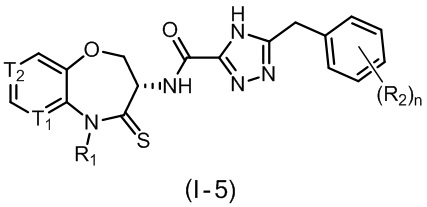
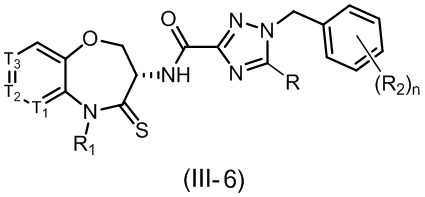
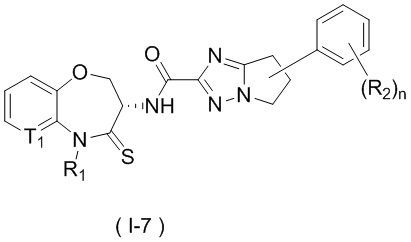 и
и 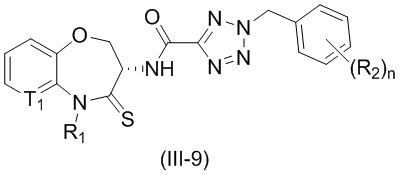
где
n, T1, T2, T3, R1, R2 и R имеют указанные в настоящем тексте значения;
или его изомер или фармацевтически приемлемая соль.
В настоящем изобретении описаны также соединения, имеющие приведенные ниже формулы, или их изомеры или фармацевтически приемлемые соли:
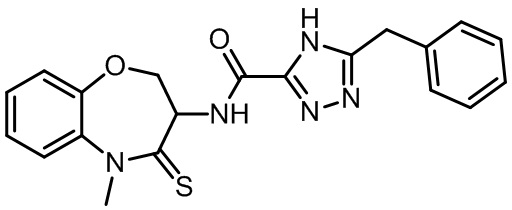
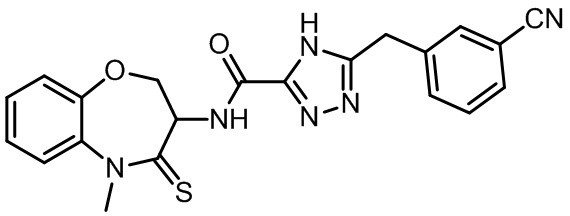
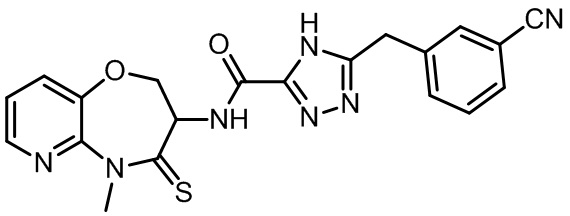
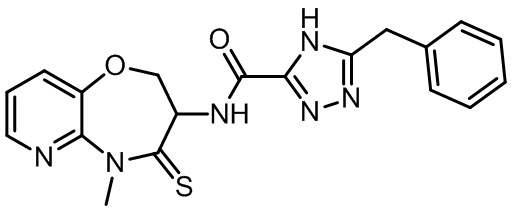
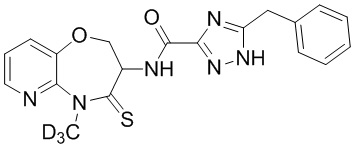
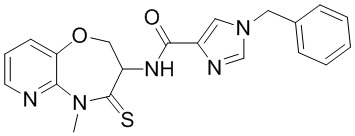
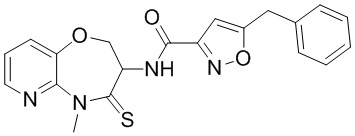
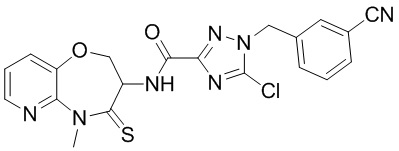
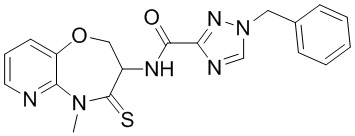
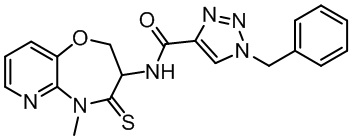
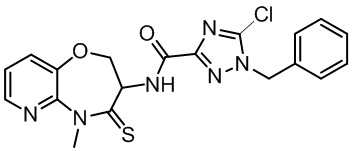
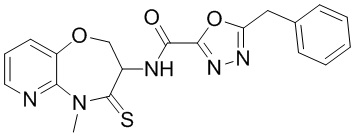
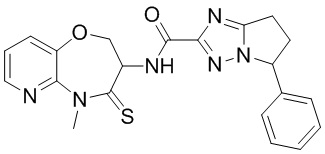
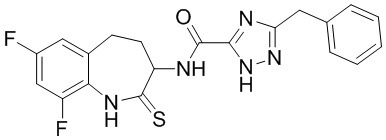
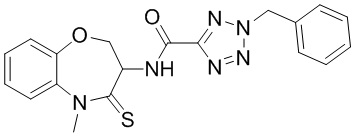
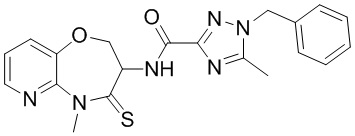
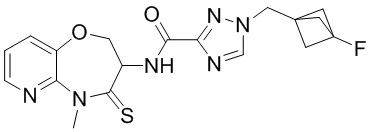
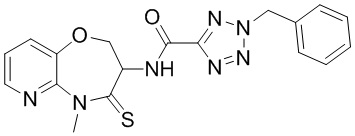
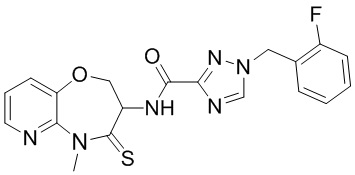
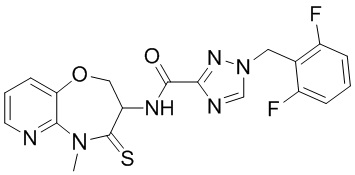
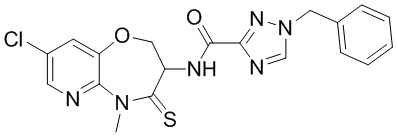
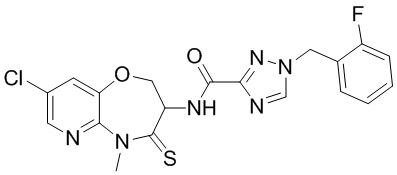
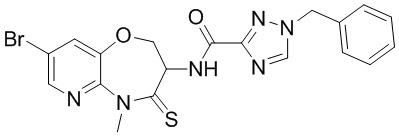
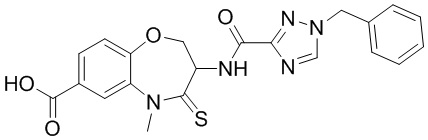
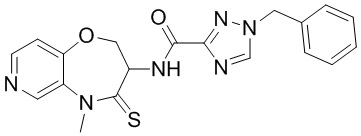
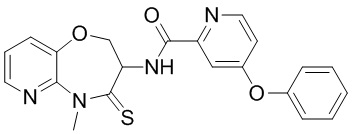
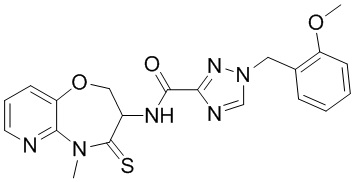
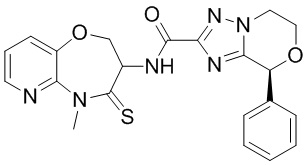
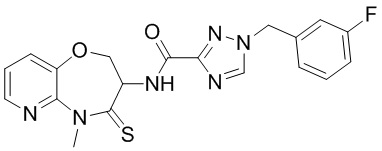
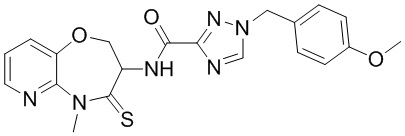
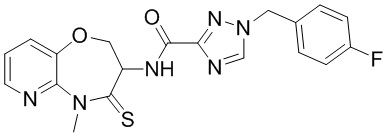
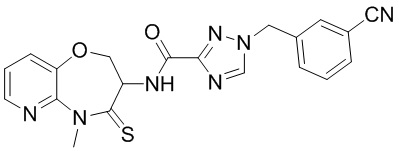
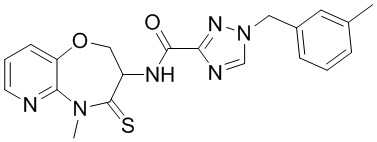
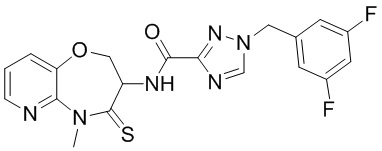
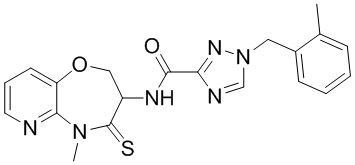
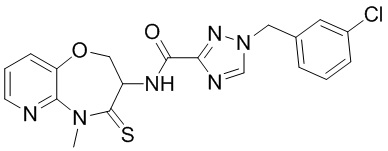
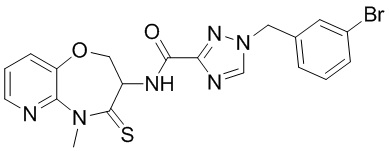
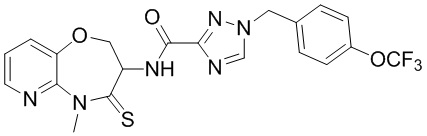
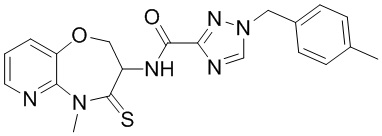
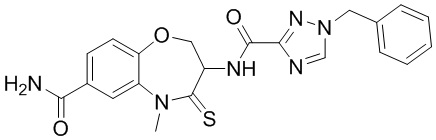
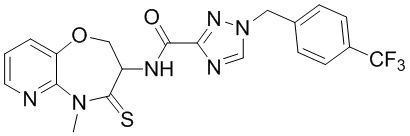
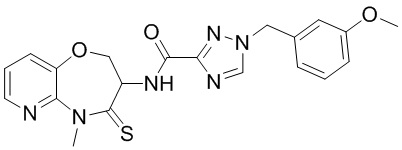
В некоторых вариантах осуществления, описанных в настоящем тексте, указанное выше соединение выбрано из
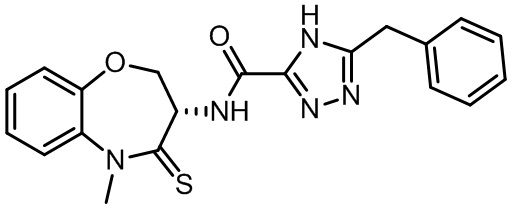
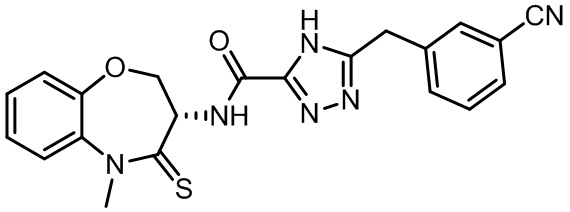

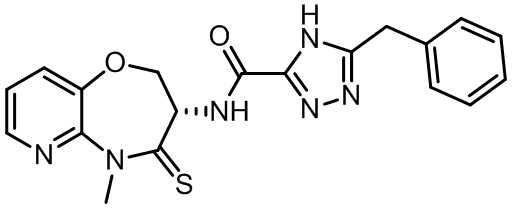
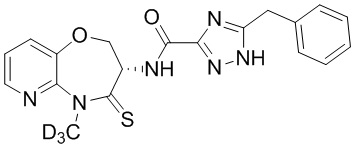
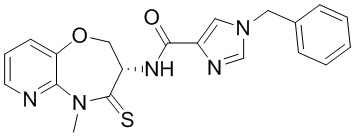
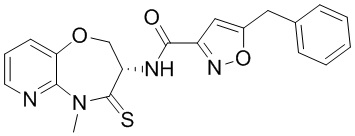
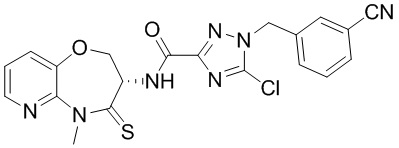
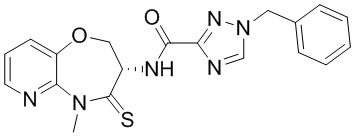
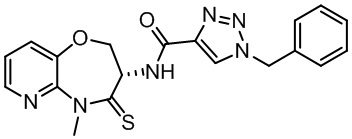
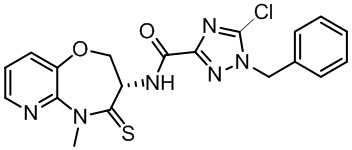
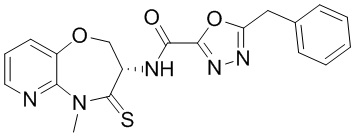
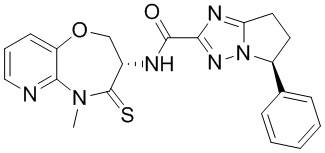
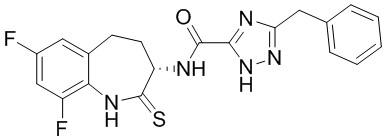
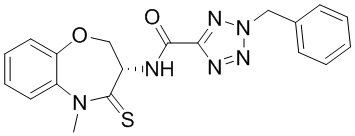
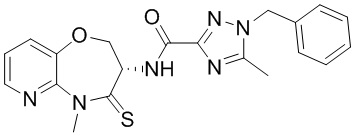
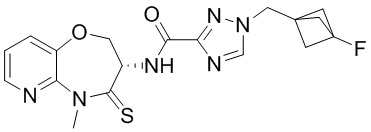
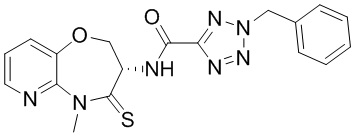
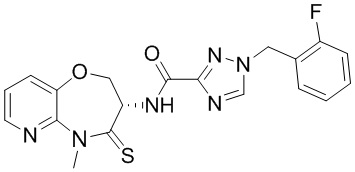
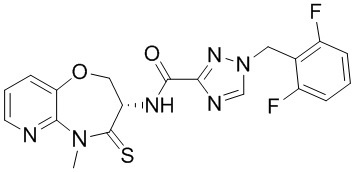
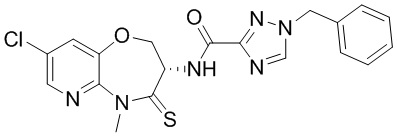
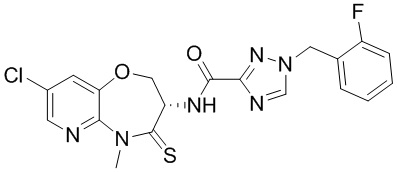
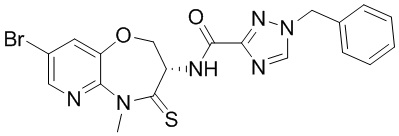
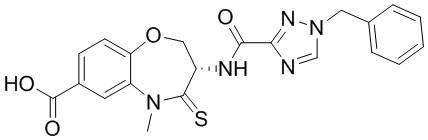
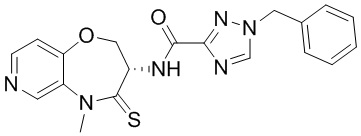
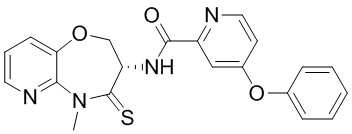
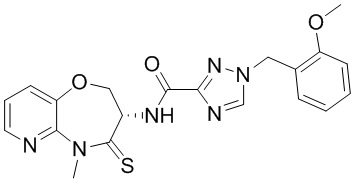
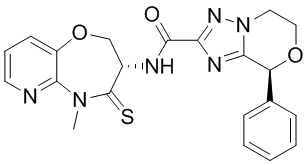
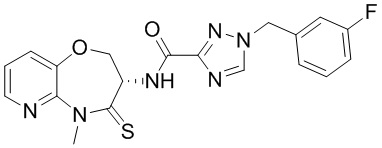
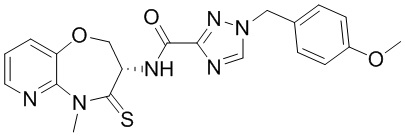
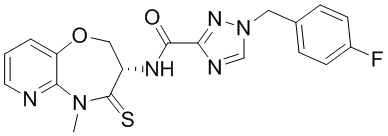
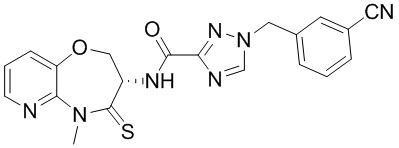
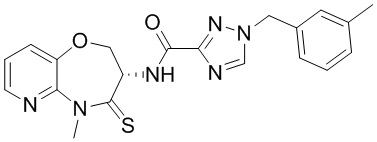
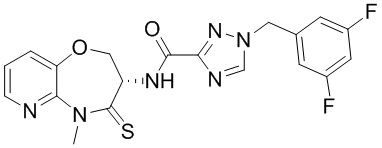
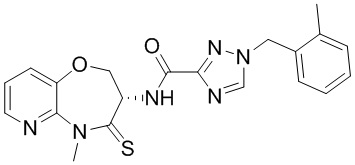
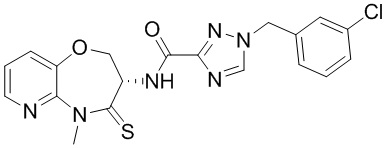
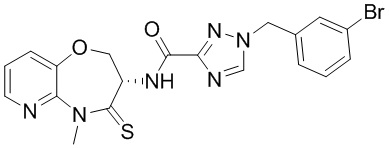
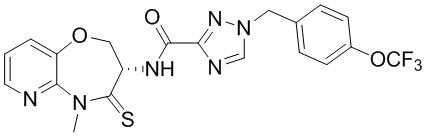
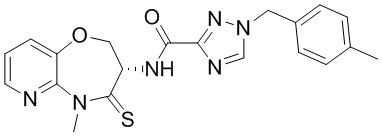
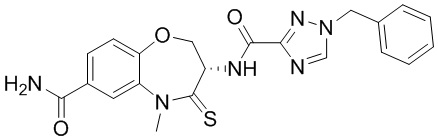
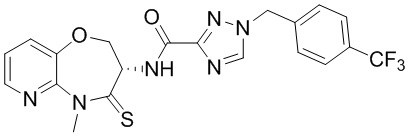 и
и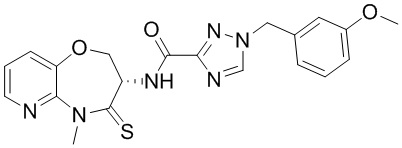
или их изомеров или фармацевтически приемлемых солей.
В настоящем изобретении описано также применение указанного выше соединения, или его изомера или фармацевтически приемлемой соли, в производстве лекарственного средства для лечения заболеваний, связанных с RIP-1 киназой.
Технический эффект изобретения
В качестве нового ингибитора RIP-1 киназы, соединение по настоящему изобретению оказывает активное ингибирующее действие на запрограммированный некроз (некроптоз) U937 клеток, вызываемый TNFα/QVD-OPh, и оказывает хороший ингибирующий эффект, проявляющийся в снижении температуры тела у мышей в модели TNF-запускаемого синдрома системной воспалительной реакции.
Определения и термины
Если не указано иное, перечисленные ниже термины и обороты, использующиеся в настоящем тексте, имеют указанные далее значения. Частные термины или обороты не должны считаться неопределенными или неясными в отсутствие конкретизирующего определения, а должны пониматься в своем обычном значении. Когда в настоящем тексте используется торговое название, оно означает соответствующий ему материал или активный ингредиент.
Термин "фармацевтически приемлемый" используется в настоящем тексте в отношении тех соединений, материалов, композиций и/или дозированных форм, которые подходят для применения в контакте с тканями человека или животных в рамках современных медицинских знаний, не обладают избыточной токсичностью, раздражающим действием, не вызывают аллергических реакций или других проблем или осложнений, и имеют разумное соотношение польза/риск.
Термин "фармацевтически приемлемая соль" означает соль соединения по настоящему изобретению, которую получают реакцией соединения, содержащего определенный раскрытый в настоящем тексте заместитель, с относительно нетоксичной кислотой или основанием. Когда соединения по настоящему изобретению содержат относительно кислую функциональную группу, можно получить соль с основанием путем контакта нейтральной формы соединения с достаточным количеством основания, без растворителя или в подходящем нейтральном растворителе. Фармацевтически приемлемая соль с основанием включает соль натрия, калия, кальция, аммония, органического амина или магния, или похожие соли. Когда соединения по настоящему изобретению содержат относительно основную функциональную группу, можно получить соль с кислотой путем контакта нейтральной формы соединения с достаточным количеством кислоты, без растворителя или в подходящем нейтральном растворителе. Фармацевтически приемлемая соль с кислотой включает соль с неорганической кислотой, где неорганическая кислота включает, например, соляную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, иодоводородную кислоту, фосфорную кислоту и т.п.; и соль с органической кислотой, где органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфокислоту, п-толуолсульфокислоту, лимонную кислоту, винную кислоту и метансульфокислоту и т.п.; и соль с аминокислотой (такой как аргинин и т.п.), и соль с органической кислотой, такой как глюкуроновая кислота и т.п. Некоторые соединения по настоящему изобретению содержат одновременно основную и кислую функциональные группы и могут быть превращены как в соль с кислотой, так и в соль с основанием.
Фармацевтически приемлемая соль по настоящему изобретению может быть получена из материнского соединения, содержащего кислый или основный фрагмент, стандартными химическими методами. В целом, такую соль можно получить путем реакции свободной кислотной или основной формы соединения со стехиометрическим количеством подходящего основания или кислоты, в воде или в органическом растворителе или в их смеси.
Если не указано иное, термин "изомер" включает геометрические изомеры, цис- или транс-изомеры, стереоизомеры, энантиомеры, оптические изомеры, диастереомеры и таутомеры.
Соединения по настоящему изобретению могут присутствовать в определенной геометрической или стереоизомерной форме. Настоящее изобретение охватывает все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереоизомеры, (D)-изомеры, (L)-изомеры и рацемические смеси и другие смеси, например, смесь, обогащенную энантиомером или диастереомером, и все они входят в объем настоящего изобретения. Заместитель, такой как алкил, может содержать дополнительный асимметрический атом углерода. Все такие изомеры и их смеси входят в объем настоящего изобретения.
Если не указано иное, термин "энантиомер" или "оптический изомер" означает стереоизомеры, являющиеся зеркальным отражением друг друга.
Если не указано иное, термин "цис-транс изомер" или "геометрический изомер" отражает отсутствие возможности свободного вращения вокруг двойной связи или простой связи между атомами углерода, находящимися в составе цикла.
Если не указано иное, термин "диастереомер" означает стереоизомеры, в молекуле которых содержатся два или больше хиральных центра, и которые не являются зеркальным отражением друг друга.
Если не указано иное, "(D)" или "(+)" означает декстроизомер, "(L)" или "(-)" означает левовращающий изомер, а "(DL)" или "(±)" означает рацемат.
Если не указано иное, сплошная клиновидная связь ( ) и пунктирная клиновидная связь (
) и пунктирная клиновидная связь ( ) означают абсолютную конфигурацию стереоцентра; сплошная прямая связь (
) означают абсолютную конфигурацию стереоцентра; сплошная прямая связь ( ) и пунктирная прямая связь (
) и пунктирная прямая связь ( ) означают относительную конфигурацию стереоцентра; волнистая линия (
) означают относительную конфигурацию стереоцентра; волнистая линия ( ) означает сплошную клиновидную связь () или пунктирную клиновидную связь (); или волнистая линия () означает сплошную прямую связь () и пунктирную прямую связь ().
) означает сплошную клиновидную связь () или пунктирную клиновидную связь (); или волнистая линия () означает сплошную прямую связь () и пунктирную прямую связь ().
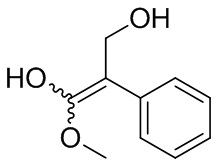
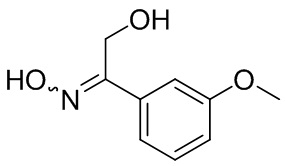
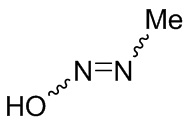
Если не указано иное, когда в соединении присутствует двойная связь, такая как двойная связь углерод-углерод, двойная связь углерод-азот и двойная связь азот-азот, и к каждому атому у двойной связи присоединены два разных заместителя (в двойной связи с участием атома азота свободная пара неподеленных электронов у атома азота считается одним из заместителей), соединение представляет собой (Z) изомер, (E) изомер или смесь этих двух изомеров соединения, если атомы у двойной связи в соединении связаны с заместителями волнистой линией ( ). Например, соединение, имеющее изображенную ниже формулу (A), означает, что это соединение представляет собой единственный изомер формулы (A-1) или формулы (A-2) или смесь двух изомеров формулы (A-1) и формулы (A-2); а соединение, имеющее изображенную ниже формулу (B), означает, что это соединение представляет собой единственный изомер формулы (B-1) или формулы (B-2) или смесь двух изомеров формулы (B-1) и формулы (B-2). Соединение, имеющее изображенную ниже формулу (C), означает, что это соединение представляет собой единственный изомер формулы (C-1) или формулы (C-2), или смесь двух изомеров формулы (C-1) и формулы (C-2).
). Например, соединение, имеющее изображенную ниже формулу (A), означает, что это соединение представляет собой единственный изомер формулы (A-1) или формулы (A-2) или смесь двух изомеров формулы (A-1) и формулы (A-2); а соединение, имеющее изображенную ниже формулу (B), означает, что это соединение представляет собой единственный изомер формулы (B-1) или формулы (B-2) или смесь двух изомеров формулы (B-1) и формулы (B-2). Соединение, имеющее изображенную ниже формулу (C), означает, что это соединение представляет собой единственный изомер формулы (C-1) или формулы (C-2), или смесь двух изомеров формулы (C-1) и формулы (C-2).
 (A)
(A) 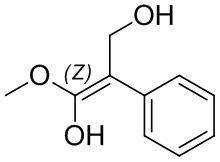 (A-1)
(A-1) 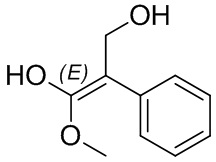 (A-2)
(A-2)
 (B)
(B) 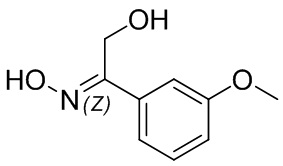 (B-1)
(B-1) 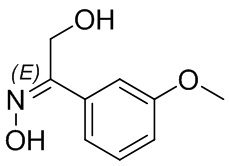 (B-2)
(B-2)
 (C)
(C) 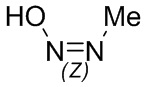 (C-1)
(C-1) 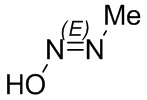 (C-2)
(C-2)
Соединения по настоящему изобретению могут присутствовать в определенной форме. Если не указано иное, термины "таутомер" или "таутомерная форма" означают, что разные функциональные группы находятся в динамическом равновесии при комнатной температуре и могут быстро превращаться друг в друга. Если таутомеры возможны (как в растворе, например), может устанавливаться таутомерное равновесие. Например, протонные таутомеры (известные также как прототропные таутомеры) включают взаимопревращение путем переноса протона, такое как кето-енольная изомеризация и имин-енаминная изомеризация. Валентные таутомеры включают взаимные превращения путем перегруппировки некоторых связывающих электронов. Частным примером кето-енольной таутомеризации является взаимное превращение между двумя таутомерами - пентан-2,4-дионом и 4-гидроксипент-3-ен-2-оном.
Если не указано иное, термин "обогащенный одним изомером", "изомерно обогащенный", "обогащенный одним энантиомером" или "энантиомерно обогащенный" означает, что содержание одного изомера или энантиомера составляет менее 100%, и содержание одного изомера или энантиомера составляет 60% или больше, или 70% или больше, или 80% или больше, или 90% или больше, или 95% или больше, или 96% или больше, или 97% или больше, 98% или больше, 99% или больше, 99.5% или больше, 99.6% или больше, 99.7% или больше, 99.8% или больше, или 99.9% или больше.
Если не указано иное, термин "изомерный избыток" или "энантиомерный избыток" означает разницу между относительными процентными содержаниями двух изомеров или двух энантиомеров. Например, если один изомер или энантиомер присутствует в количестве 90%, а второй изомер или энантиомер присутствует в количестве 10%, изомерный или энантиомерный избыток (значение ee) составляет 80%.
Оптически активный (R)- и (S)-изомер, или D и L изомер можно получить с использованием методик хирального синтеза или хиральных реагентов или других общеизвестных методов. Если необходимо получить один вид энантиомера определенного соединения по настоящему изобретению, чистый целевой энантиомер можно получить асимметрическим синтезом или с помощью дериватизующего хирального агента с последующим разделением полученной диастереомерной смеси и отщеплением дериватизующей группы. Альтернативно, когда молекула содержит основную функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), соединение реагирует с подходящей оптически активной кислотой или основанием с образованием диастереомерной соли, которую затем разделяют на индивидуальные диастереомеры известными в данной области способами, получая чистый эннатиомер. Кроме того, энантиомер и диастереомер можно выделить с использованием хроматографии на хиральной неподвижной фазе, опционально с применением методов химической дериватизации (например, с получением карбамата из амина).
Соединения по настоящему изобретению могут иметь неприродное соотношение изотопов атомов по одному или больше атомам, составляющим данное соединение. Например, соединение может быть помечено радиоактивным изотопом, таким как тритий (3H), иод-125 (125I) или C-14 (14C). В другом примере, водород может быть заменен на более тяжелый изотоп водорода, формируя дейтерированное лекарственное средство. Связь между дейтерием и углеродом сильнее, чем между обычным водородом и углеродом. По сравнению с недейтерированными лекарственными средствами, дейтерированные лекарственные средства обладают преимуществом, заключающимся в уменьшении побочных токсических эффектов, повышении стабильности лекарственного средства, повышенной эффективности и увеличенном времени полужизни лекарственного средства. Все изменения в изотопном составе соединений по настоящему изобретению, вне зависимости от радиоактивности изотопов, включены в объем настоящего изобретения.
Термин "опциональный" или "необязательный" означает, что описанное далее событие или условие может иметь место, но не является обязательным, и что данный термин включает случаи когда указанное событие или условие выполняется, и случаи когда указанное событие или условие не выполняется.
Термин "замещенный" означает, что один или больше атомов водорода у определенного атома заменены на заместитель, включая дейтерий и варианты водорода, при условии соблюдения валентности данного атома и стабильности образующегося замещенного соединения. Когда заместитель представляет собой оксо-группу (т.е. =O), это означает, что заменены два атома водорода. Положения в составе ароматического кольца не могут быть заменены на оксо-группу. Термин "необязательно замещен" означает, что атом может иметь или не иметь заместитель(-ли), если не указано иное, вид и число заместителей могут быть разными, при условии их химической возможности.
Когда переменная (такая как R) встречается в конструкции или в структуре соединения больше одного раза, значение этой переменной в каждом отдельном случае не зависит от других. Так, например, если группа имеет от 0 до 2 заместителей R, данная группа необязательно может иметь до двух заместителей R, где значение R в каждом случае является независимым. Кроме того, комбинация заместителей и/или их вариантов является разрешенной только в том случае, когда эта комбинация приводит к устойчивому соединению.
Когда число соединяющих групп равно 0, например в -(CRR)0-, это означает, что эта соединяющая группа представляет собой простую связь.
Когда одна из переменных представляет собой простую связь, это означает, что две группы, соединенные этой простой связью, связаны напрямую. Например, когда L в A-L-Z представляет собой простую связь, структура A-L-Z фактически представляет собой A-Z.
Когда заместитель является вакантным, это означает, что заместителя нет. Например, когда X вакантный в A-X, структура A-X фактически представляет собой А.
Когда для заместителя не указан атом, через который этот заместитель связан с замещаемой группой, такой заместитель может быть связан через любой из своих атомов. Например, пиридильная группа как заместитель может быть связана с замещаемой группой через любой из атомов углерода пиридинового кольца.
Когда для соединяющей группы не указано направление связывания, ее направление связывания может быть любым. Например, когда соединяющая группа L в 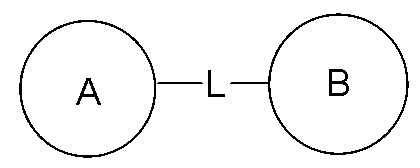 представляет собой -M-W-, то -M-W- может быть связана с циклом A и циклом B в порядке прочтения слева направо и формировать комбинацию
представляет собой -M-W-, то -M-W- может быть связана с циклом A и циклом B в порядке прочтения слева направо и формировать комбинацию 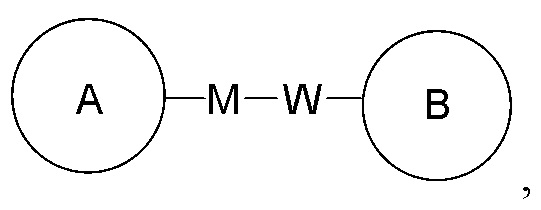 или она может быть соединена с циклом A и циклом B в обратном направлении, формируя комбинацию
или она может быть соединена с циклом A и циклом B в обратном направлении, формируя комбинацию 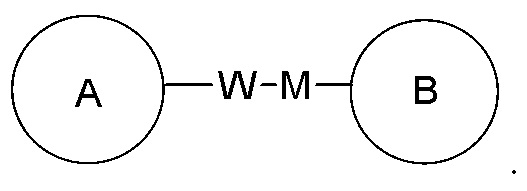 Комбинация соединяющих групп, заместителей и/или их вариантов является разрешенной только в том случае, когда такая комбинация приводит к формированию устойчивого соединения.
Комбинация соединяющих групп, заместителей и/или их вариантов является разрешенной только в том случае, когда такая комбинация приводит к формированию устойчивого соединения.
Если не указано иное, термин "галоген" сам по себе или как часть другого заместителя означает атом фтора, хлора, брома или иода.
Если не указано иное, термин "C1-3 алкокси-группа" означает алкильную группу, содержащую от 1 до 3 атомов углерода и соединенную с остальной частью молекулы через атом кислорода. C1-3 алкокси-группа включает C1-2, C2-3, C3, C2 алкокси-группы и т.п. Примеры C1-3 алкокси-групп включают (но не ограничиваются только ими) метокси, этокси, пропокси (включая н-пропокси и изопропокси) и т.п.
Если не указано иное, термин "C1-3 алкил" используется для обозначения линейной или разветвленной насыщенной углеводородной группы, состоящей из 1-3 атомов углерода. C1-3 алкильная группа включает C1-2 и C2-3 алкильные группы и т.п. Она может быть одновалентной (например, метил), двухвалентной (например, метилен) или многовалентной (например, метенил). Примеры C1-3 алкильных групп включают (но не ограничиваются только ими) метил (Me), этил (Et), пропил (включая н-пропил и изопропил) и т.п.
Если не указано иное, термины "5-6-членное гетероароматическое кольцо" и "5-6-членный гетероарил" могут использоваться взаимозаменяемо. Термин "5-6-членный гетероарил" означает одновалентную группу, содержащую сопряженную π-электронную систему и состоящую из 5 или 6 атомов в кольце, из которых 1, 2, 3 или 4 атомов в кольце представляют собой гетероатомы, независимо выбранные из O, S и N, и остальные атомы представляют собой атомы углерода, где атом азота необязательно является кватернизованным, и атомы азота и серы необязательно окислены (например, NO и S(O)p, p равен 1 или 2). 5-6-членная гетероарильная группа может быть присоединена к остальной части молекулы через гетероатом или через атом углерода. 5-6-членная гетероарильная группа включает 5-членные и 6-членные гетероарильные группы. Примеры 5-6-членных гетероарильных групп включают (но не ограничиваются только ими) пирролил (включая N-пирролил, 2-пирролил, 3-пирролил и т.п.), пиразолил (включая 2-пиразолил и 3-пиразолил и т.п.), имидазолил (включая N-имидазолил, 2-имидазолил, 4-имидазолил и 5-имидазолил и т.п.), оксазолил (включая 2-оксазолил, 4-оксазолил и 5-оксазолил и т.п.), триазолил (1H-1,2,3-триазолил, 2H-1,2,3-триазолил, 1H-1,2,4-триазолил и 4H-1,2,4-триазолил и т.п.), тетразолил, изоксазолил (3-изоксазолил, 4-изоксазолил и 5-изоксазолил и т.п.), тиазолил (включая 2-тиазолил, 4-тиазолил и 5-тиазолил и т.п.), фурил (включая 2-фурил и 3-фурил и т.п.), тиенил (включая 2-тиенил и 3-тиенил и т.п.), пиридил (включая 2-пиридил, 3-пиридил и 4-пиридил и т.п.), пиразинил или пиримидинил (включая 2-пиримидинил и 4-пиримидинил и т.п.).
Если не указано иное, Cn-n+m или Cn-Cn+m включает любое число атомов углерода от n до n+m. Например, C1-12 включает C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12. Cn-n+m или Cn-Cn+m также включает любой диапазон от n до n+m. Например, C1-12 включает C1-3, C1-6, C1-9, C3-6, C3-9, C3-12, C6-9, C6-12, C9-12 и т.п. Аналогично, кольцо от n-членного до n+m-членного означает, что число атомов в кольце составляет от n до n+m. Например, 3-12-членное кольцо включает 3-членное кольцо, 4-членное кольцо, 5-членное кольцо, 6-членное кольцо, 7-членное кольцо, 8-членное кольцо, 9-членное кольцо, 10-членное кольцо, 11-членное кольцо и 12-членное кольцо. Кольцо от n-членного до n+m-членного означает, что число атомов в кольце включает любой диапазон от n до n+m. Например, 3-12-членное кольцо включает 3-6-членное кольцо, 3-9-членное кольцо, 5-6-членное кольцо, 5-7-членное кольцо, 6-7-членное кольцо, 6-8-членное кольцо и 6-10-членное кольцо и т.п.
Термин "уходящая группа" означает функциональную группу или атом, которые могут замещаться другой функциональной группой или атомом в ходе реакции замещения. Например, репрезентативные уходящие группы включают трифлат; хлор, бром и иод; сульфонатную группу, такую как мезилат, тозилат, п-бромбензолсульфонат, п-толуолсульфонат и т.п.; ацилокси-группу, такую как ацетокси, трифторацетокси и т.п.
Термин "защитная группа" включает (но не ограничивается только ими) "амино-защитную группу", "гидрокси-защитную группу" или "тио-защитную группу". Термин "амино-защитная группа" означает защитную группу, подходящую для блокирования побочных реакций по атому азота аминогруппы. Репрезентативные амино-защитные группы включают (но не ограничиваются только ими): формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкокси-карбонил, такой как трет-бутоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и т.п. Термин "гидрокси-защитная группа" означает защитную группу, подходящую для блокирования побочных реакций по гидрокси-группе. Репрезентативные гидрокси-защитные группы включают (но не ограничиваются только ими): алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и т.п.
Структуры соединений по настоящему изобретению могут быть подтверждены стандартными методами, известными квалифицированным специалистам в данной области техники. Если настоящее описание касается абсолютной конфигурации соединения, то абсолютную конфигурацию можно подтвердить такими общеизвестными в данной области методами, как рентгеноструктурный анализ монокристалла. В методе рентгеноструктурного анализа собираются данные интенсивности дифракции на выращенном монокристалле с помощью дифрактометра Bruker D8 с источником излучения CuKα в сканирующем режиме ϕ/ωскан; после сбора этой информации структуру кристалла далее анализируют прямым методом (Shelxs97) для определения абсолютной конфигурации.
Соединения по настоящему изобретению могут быть получены различными методами синтеза, хорошо известными квалифицированным специалистам в данной области, включая описанные ниже частные варианты осуществления, варианты, образующиеся при комбинации описанных ниже частных вариантов осуществления с другими методами химического синтеза, и эквивалентные замены, хорошо известные квалифицированным специалистам в данной области. Альтернативные варианты осуществления включают (но не ограничиваются только ими) описанные в настоящем тексте варианты осуществления.
Растворители, использующиеся по настоящему изобретению, являются коммерчески доступными.
В настоящем тексте используются следующие сокращения и аббревиатуры: водн. = водный; NaNO2 означает нитрит натрия; H2SO4 означает серную кислоту; CuCl2 означает хлорид меди; LiOH⋅H2O означает гидроксид лития моногидрат; NaBH3CN означает цианоборгидрид натрия; HCl/EtOAc означает раствор хлороводорода в этилацетате; HCl означает соляную кислоту; CO2 означает диоксид углерода; ACN означает ацетонитрил; FA означает муравьиную кислоту; H2O означает воду; NH3H2O означает аммиак; Na2SO4 означает сульфат натрия; MgSO4 означает сульфат магния; NCS означает N-хлорсукцинимид; NH4HCO3 означает бикарбонат аммония; Pd/C означает палладий на угле; фунт/кв.дюйм это единица давления; MeCN означает ацетонитрил; TsOH⋅H2O означает моногидрат п-толуолсульфокислоты; Pd(dppf)Cl2 означает [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорид; Neu означает нейтральный; ВЭЖХ означает Высокоэффективную жидкостную хроматографию; HATU означает O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; K2CO3 означает карбонат калия; EDC означает N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорид; m-CPBA означает 3-хлорпербензойную кислоту; экв. означает эквивалент или эквивалентность; T3P означает пропилфосфоновый ангидрид; CDI означает карбонил диимидазол; ДХМ означает дихлорметан; ПЭ означает петролейный эфир; DIAD означает диизопропил азодикарбоксилат; ДМФА означает N,N-диметилформамид; ДМСО означает диметилсульфоксид; EtOAc означает этилацетат; EtOH означает этанол; MeOH означает метанол; CBz означает бензилоксикарбонил, который представляет собой амино-защитную группу; BOC означает трет-бутоксикарбонил, который представляет собой амино-защитную группу; HOAc означает уксусную кислоту; NaCNBH3 означает цианоборгидрид натрия; r.t. или rt означает комнатную температуру; O/N = в течение ночи; ТГФ означает тетрагидрофуран; Boc2O означает ди-трет-бутил-дикарбонат; ТФУК означает трифторуксусную кислоту; SOCl2 означает тионилхлорид; CS2 означает дисульфид углерода; TsOH означает п-толуолсульфокислоту; NFSI означает N-фтор-N-(фенилсульфонил)бензолсульфонамид; н-Bu4NF означает фторид тетрабутиламмония; iPrOH означает 2-пропанол; т.пл. означает температуру плавления; LDA означает диизопропиламид лития; IPAm означает изопропиламин; DIPEA означает N,N-диизопропилэтиламин; Xantphos означает 4,5-бис(дифенилфосфино)-9,9- диметилксантан; t-Bu Xphos означает 2-ди-трет-бутилфосфино-2',4',6'- триизопропилбифенил; DPPP означает 1,3-бис(дифенилфосфино)пропан; Et3SiH означает триэтилсилан; DMAP означает 4-диметиламинопиридин; Py означает пиридин; DPBS означает фосфатный буфер Дульбекко; TNF-α означает фактор некроза опухоли; IC50 означает концентрацию полумаксимального ингибирования; мг означает миллиграммы, кг означает килограммы; и HEPES означает N-2-гидроксиэтилпиперазин-N-2- этансульфокислотный буфер.
Соединениям даны названия согласно общим принципам составления названий или с помощью программы ChemDraw®, а коммерчески доступные соединения названы согласно названиям от их поставщиков.
Подробное описание изобретения
Настоящее изобретение подробнее описано ниже с помощью примеров. Однако приведенные примеры никоим образом не ограничивают объем настоящего изобретения. Настоящее изобретение подробно описано в настоящем тексте, и описаны также варианты его осуществления. Квалифицированным специалистам в данной области будет очевидно, что в вариантах осуществления, описанных в настоящем тексте, могут быть сделаны различные изменения и модификации без выхода за рамки сути и объема настоящего изобретения.
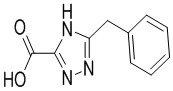
Сравнительный пример 1: Фрагмент BB-1
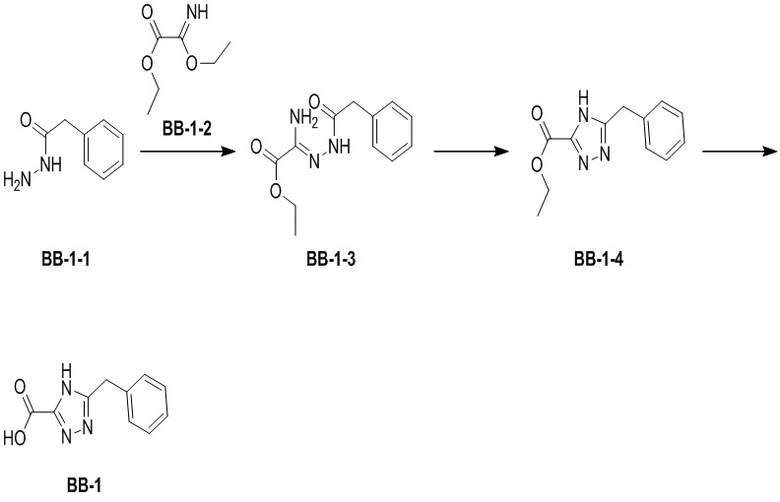
Путь синтеза:
Стадия 1: синтез соединения BB-1-3
В предварительно высушенную 100-миллилитровую колбу помещали BB-1-1 (2.60 г, 17.31 ммоль, 1.00 экв.) и растворитель EtOH (11.25 мл) и изопропиловый эфир (37.50 мл). BB-1-2 (2.59 г, 17.83 ммоль, 1.03 экв.) медленно добавляли при перемешивании, и реакционный раствор перемешивали при 20°C в течение 12 часов. Реакционный раствор фильтровали, и осадок на фильтре промывали изопропиловым эфиром (30 мл * 3), получая соединение BB-1-3. 1H ЯМР (400 МГц, ДМСО-d6) δ = 9.95 (с, 1H), 9.90 (с, 1H), 7.39 - 7.08 (м, 6H), 4.21 - 4.19 (м, 2H), 3.82 (с, 2H), 1.27 - 1.23 (м, 3H); LCMS m/z = 250.1 [M+H]+.
Стадия 2: синтез соединения BB-1-4
В предварительно высушенную 250-миллилитровую колбу помещали соединение BB-1-3 (7.00 г, 28.08 ммоль, 1.00 экв.) и ксилол (100.00 мл). Устанавливали насадку Дина-Старка. Реакционную смесь нагревали при 180°C в течение 10 часов. Добавляли в реакционную систему 100 мл изопропилового эфира, и смесь перемешивали 30 минут в ледяной бане. Реакционную смесь фильтровали, и осадок на фильтре промывали 50 мл н-гексана, получая соединение BB-1-4. 1H ЯМР (400 МГц, ДМСО-d6) δ = 14.44 (ушир.с, 1H), 7.42 - 7.14 (м, 4H), 4.29 (кв, J = 6.5 Гц, 2H), 4.13 (ушир.с, 2H), 1.28 (ушир.т, J = 7.0 Гц, 3H); LCMS m/z = 232.2 [M+H]+.
Стадия 3: синтез соединения BB-1
В колбу помещали соединение BB-1-4 (4.10 г, 17.73 ммоль, 1.00 экв.), тетрагидрофуран (1.00 мл) и воду (200.00 мкл), затем добавляли моногидрат гидроксида лития (1.74 г, 41.49 ммоль, 2.34 экв.). Раствор перемешивали при 20°C в течение 12 часов. Растворитель удаляли при пониженном давлении. Добавляли по каплям 25 мл 1н. водного раствора соляной кислоты при перемешивании до значения pH 2. Соединение BB-1 получали после фильтрования при отсасывании. LCMS m/z = 202.1 [M-H]-.
Сравнительный пример 2: Фрагмент BB-2
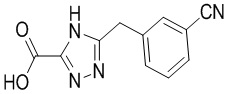
Путь синтеза:
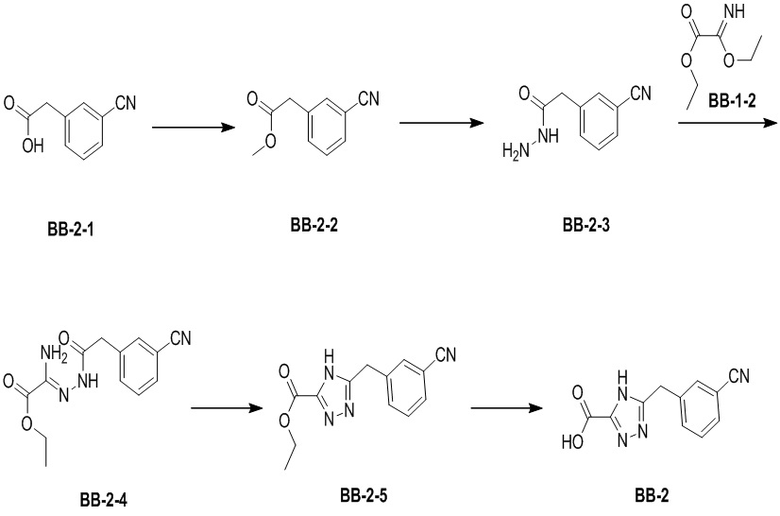
Стадия 1: синтез соединения BB-2-2
В раствор BB-2-1 (2.5 г, 15.51 ммоль, 2.94 мл, 1 экв.) в дихлорметане (40 мл) добавляли метанол (4.97 г, 155.13 ммоль, 6.28 мл, 10 экв.) при 23°C. Затем добавляли TMSCHN2 (2 M, 9.31 мл, 1.2 экв.), и смесь перемешивали еще 2 часа. Исходные соединения полностью исчезали, согласно данным ТСХ-мониторинга (петролейный эфир:этилацетат = 5:1). Добавляли в реакционный раствор 0.1 мл ледяной уксусной кислоты, и раствор упаривали при пониженном давлении, получая BB-2-2.
Стадия 2: синтез соединения BB-2-3
В раствор BB-2-2 (3 г, 17.12 ммоль, 1 экв.) в метаноле (30 мл) добавляли гидразин гидрат (3.21 г, 51.37 ммоль, 3.12 мл, 80% чистота, 3 экв.), и смесь перемешивали на масляной бане при 70°C в течение 2 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор упаривали при пониженном давлении для удаления растворителя и добавляли 50 мл этилацетата. Смесь промывали 30 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая BB-2-3. 1H ЯМР (400 МГц, ДМСО-d6) δ = 9.25 (ушир.с, 1 H), 7.66 - 7.74 (м, 2 H), 7.45 - 7.65 (м, 2 H), 4.24 (с, 2 H), 3.43 (с, 2 H); LCMS m/z = 176.1 [M+H]+.
Стадия 3: синтез соединения BB-2-4
В предварительно высушенную трехгорлую колбу помещали BB-1-2 (2.07 г, 14.27 ммоль, 1 экв.), этанол (10 мл) и изопропиловый эфир (20 мл), затем добавляли BB-2-3 (2.5 г, 14.27 ммоль, 1 экв.). Смесь оставляли для прохождения реакции при 25°C на 5 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор фильтровали, и осадок на фильтре промывали 50 мл петролейного эфира, получая BB-2-4. LCMS m/z = 275.2 [M+H]+.
Стадия 4: синтез соединения BB-2-5
В виалу для проведения реакций в микроволновой печи помещали BB-2-4 (0.5 г, 1.82 ммоль, 1 экв.), молекулярные сита (4Å, 0.05 г) и ксилол (15 мл). Виалу для проведения реакций в микроволновой печи герметично закрывали, и реакционную смесь перемешивали в микроволновом реакторе при 150°C в течение 4 часов. Мониторинг методом LCMS показывал появление сигнала целевого продукта. Добавляли в реакционный раствор 30 мл ацетонитрила, и смесь фильтровали. Фильтрат упаривали при пониженном давлении, получая BB-2-5. LCMS m/z = 257.1 [M+H]+.
Стадия 5: синтез соединения BB-2
В раствор BB-2-5 (1 г, 3.90 ммоль, 1 экв.) в тетрагидрофуране (50 мл) добавляли раствор моногидрата гидроксида лития (654.96 мг, 15.61 ммоль, 4 экв.) в воде (15 мл) при 21°C, и смесь перемешивали 12 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Добавляли в реакционный раствор 15 мл воды, и смесь экстрагировали этилацетатом (30 мл). Результирующую водную фазу доводили до значения pH около 3 добавлением 2н. водного раствора соляной кислоты и лиофилизовывали, получая BB-2. LCMS m/z = 227.0 [M-H]-.
Сравнительный пример 3: Фрагмент BB-3
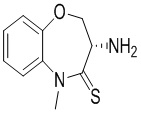
Путь синтеза:
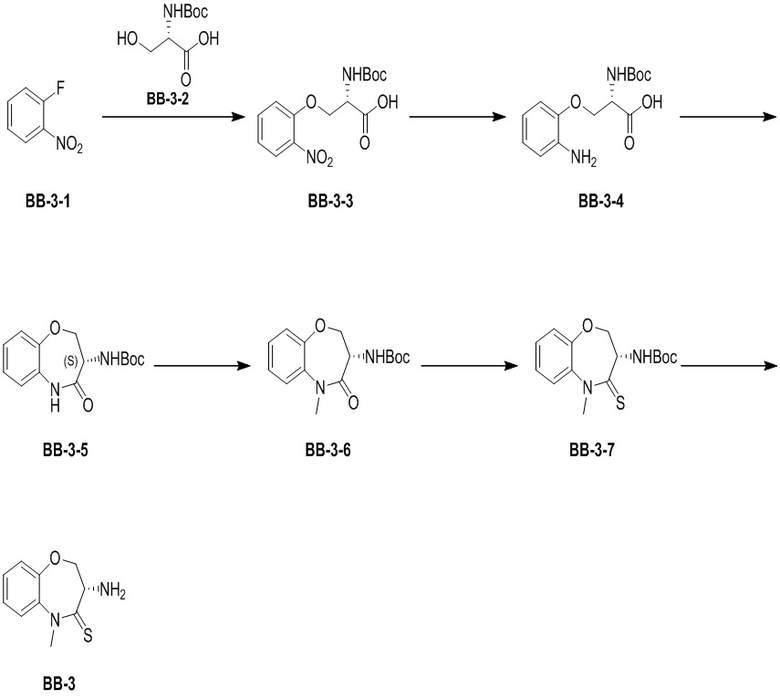
Стадия 1: синтез соединения BB-3-3
BB-3-2 (20.00 г, 97.46 ммоль, 1.00 экв.) растворяли в ДМФА (50 мл), и полученный раствор добавляли по каплям в суспензию NaH (8.19 г, 204.67 ммоль, 60% чистота, 2.10 экв.) в ДМФА (150 мл). Затем добавляли по каплям соединение BB-3-1 (13.75 г, 97.46 ммоль, 10.26 мл, 1.00 экв.). Реакционный раствор перемешивали при 20°C в течение 16 часов. Затем добавляли в реакционный раствор этилацетат (200 мл) и воду (200 мл), и водную фазу доводили до значения pH около 1 добавлением соляной кислоты. Смесь экстрагировали этилацетатом (200 мл). Органическую фазу сушили над безводным сульфатом магния и фильтровали, фильтрат упаривали при пониженном давлении, получая соединение BB-3-3. LCMS m/z = 227.1 [M-100+H]+.
Стадия 2: синтез соединения BB-3-4
Раствор соединения BB-3-3 (20.00 г, 61.29 ммоль, 1.00 экв.) и влажного Pd/C (5.00 г, 5% чистота) в метаноле (200.00 мл) перемешивали при 20°C в атмосфере водорода (15 фунт/кв. дюйм) 5 часов. После окончания реакции раствор фильтровали, и фильтрат упаривали при пониженном давлении, получая соединение BB-3-4. LCMS m/z = 297.1 [M+H]+.
Стадия 3: синтез соединения BB-3-5
В раствор соединения BB-3-4 (10.00 г, 33.75 ммоль, 1.00 экв.) и N,N-диизопропилэтиламина (4.36 г, 33.75 ммоль, 5.89 мл, 1.00 экв.) в ДМСО (100.00 мл) добавляли HATU (12.83 г, 33.75 ммоль, 1.00 экв.). Реакционную смесь перемешивали при 20°C в течение 2 часов. После окончания реакции добавляли в раствор воду (200 мл) и этилацетат (200 мл), и слои разделяли. Органическую фазу упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 10:1 ~ 1:1), получая соединение BB-3-5. LCMS m/z = 223.1 [M-56+H]+.
Стадия 4: синтез соединения BB-3-6
В высушенную 250-миллилитровую трехгорлую колбу помещали соединение BB-3-5 (6.00 г, 21.56 ммоль, 1.00 экв.), ДМФА (60.00 мл) и карбонат цезия (10.54 г, 32.34 ммоль, 1.50 экв.), затем добавляли иодметан (3.06 г, 21.56 ммоль, 1.34 мл, 1.00 экв.) по каплям. Контролировали температуру в реакционной смеси таким образом, чтобы она не поднималась выше 20°C. Реакционную смесь перемешивали при 16°C в течение 16 часов. После окончания реакции добавляли воду (100 мл) и этилацетат (100 мл), и слои разделяли. Органическую фазу промывали насыщенным водным раствором хлорида натрия (100 мл * 2), сушили над сульфатом магния и фильтровали. Фильтрат упаривали при пониженном давлении, получая соединение BB-3-6. LCMS m/z = 237.1 [M-56+H]+.
Стадия 5: синтез соединения BB-3-7
В раствор пентасульфида фосфора (760.35 мг, 3.42 ммоль, 363.80 мкл, 5 экв.) в ТГФ (10 мл) добавляли карбонат натрия (362.57 мг, 3.42 ммоль, 5 экв.) при 20°C, и смесь перемешивали еще 0.5 часа. Затем добавляли BB-3-6 (0.2 г, 684.16 мкмоль, 1 экв.), и реакционный раствор нагревали на масляной бане при 70°C и перемешивании в течение 9.5 часов. Мониторинг методом LCMS показывал появление сигнала целевого продукта. Добавляли в реакционный раствор 20 мл этилацетата, и смесь фильтровали. Полученный фильтрат упаривали при пониженном давлении, получая соединение BB-3-7. LCMS m/z = 253.1 [M-56+H]+.
Стадия 6: синтез соединения BB-3
К BB-3-7 (630.84 мг, 2.05 ммоль, 1 экв.) добавляли раствор хлороводорода в этилацетате (4M, 20.00 мл, 39.11 экв.), и смесь перемешивали при 23°C в течение 1 часа. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор упаривали при пониженном давлении, получая сырой продукт, затем добавляли 30 мл насыщенного водного раствора бикарбоната натрия. Смесь экстрагировали этилацетатом (15 мл * 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая соединение BB-3. 1H ЯМР (400 МГц, CDCl3) δ = 7.21 - 7.32 (м, 3 H), 7.15 - 7.19 (м, 1 H), 4.40 (дд, J = 9.92, 6.62 Гц, 1 H), 4.10 (дд, J = 11.36, 9.81 Гц, 1 H), 3.77 - 3.94 (м, 4 H); LCMS m/z = 209.1 [M+H]+.
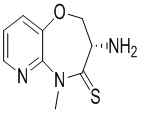
Сравнительный пример 4: Фрагмент BB-4
Путь синтеза:
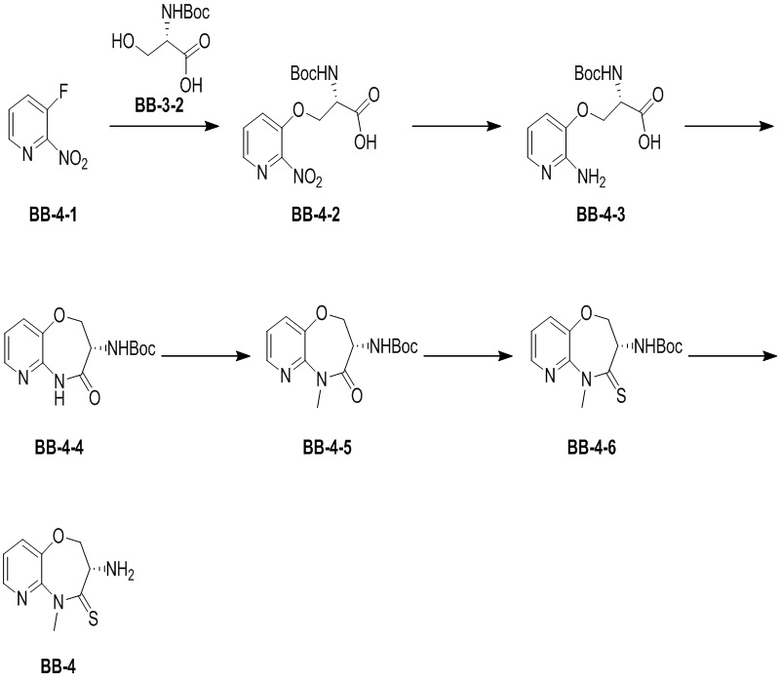
Стадия 1: синтез соединения BB-4-2
В раствор BB-3-2 (13.13 г, 63.98 ммоль, 1 экв.) в ДМФА (100 мл) добавляли NaH (5.12 г, 127.96 ммоль, 60% чистота, 2 экв.) при 0°C, и смесь перемешивали 1 час. Затем добавляли BB-4-1 (10 г, 70.38 ммоль, 1.1 экв.), и смесь перемешивали при 28°C в течение 9 часов. Мониторинг методом LCMS показывал появление сигнала целевого продукта. Реакционный раствор добавляли в 1000 мл воды, и смесь экстрагировали этилацетатом (300 мл * 4). Водную фазу доводили до значения pH около 5 добавлением 2н. раствора соляной кислоты и затем экстрагировали этилацетатом (300 мл * 5). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая соединение BB-4-2. LCMS m/z = 326.0 [M-H]-.
Стадия 2: синтез соединения BB-4-3
В раствор BB-4-2 (20.00 г, 61.11 ммоль, 1 экв.) в метаноле (200 мл) добавляли влажный Pd/C (10 г, 30.55 ммоль, 5 % чистота) при 23°C. Смесь перемешивали в атмосфере водорода (15 фунт/кв. дюйм) 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор фильтровали, и полученный фильтрат упаривали при пониженном давлении, получая соединение BB-4-3. LCMS m/z = 298.1 [M+H]+.
Стадия 3: синтез соединения BB-4-4
В раствор BB-4-3 (16.00 г, 53.82 ммоль, 1 экв.) в этилацетате (400 мл) добавляли N,N-диизопропилэтиламин (20.87 г, 161.45 ммоль, 28.12 мл, 3 экв.) и 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан-2,4,6-триоксид (68.49 г, 107.63 ммоль, 64.01 мл, 50%-ный раствор в этилацетате, 2 экв.) при 23°C. Смесь перемешивали еще 2 часа. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор промывали насыщенным водным раствором хлорида натрия (200 мл * 2). Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 10:1 ~ 1:1), получая соединение BB-4-4. LCMS m/z = 224.1 [M-56+H]+.
Стадия 4: синтез соединения BB-4-5
В раствор BB-4-4 (1 г, 3.58 ммоль, 1 экв.) в ДМФА (30 мл) добавляли карбонат цезия (1.40 г, 4.30 ммоль, 1.2 экв.) и иодметан (609.86 мг, 4.30 ммоль, 267.48 мкл, 1.2 экв.) при 20°C. Смесь перемешивали 1 час. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор выливали в 100 мл воды, и смесь экстрагировали этилацетатом (30 мл * 5). Органические фазы объединяли и промывали 50 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая соединение BB-4-5. LCMS m/z = 238.1 [M-56+H]+.
Стадия 5: синтез соединения BB-4-6
В раствор пентасульфида фосфора (3.79 г, 17.05 ммоль, 10 экв.) в ТГФ (30 мл) добавляли карбонат натрия (1.81 г, 17.05 ммоль, 10 экв.) при 20°C, и смесь перемешивали 0.5 часа. Затем добавляли BB-4-5 (0.5 г, 1.70 ммоль, 1 экв.), и реакционный раствор нагревали на масляной бане при 70°C и перемешивании в течение 9.5 часов. По данным LCMS мониторинга, исходные соединения практически полностью исчезали, и наблюдался сигнал целевого продукта. Реакционный раствор фильтровали, получая фильтрат. Добавляли в фильтрат 50 мл этилацетата, и смесь промывали 30 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 10:1 ~ 1:1), получая соединение BB-4-6. LCMS m/z = 310.3 [M+H]+.
Стадия 6: синтез соединения BB-4.
К BB-4-6 (0.6 г, 1.94 ммоль, 1 экв.) добавляли раствор хлороводорода в этилацетате (4 M, 20 мл, 41.25 экв.), и смесь перемешивали при 23°C в течение 1 часа. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор упаривали при пониженном давлении, получая сырой продукт, и добавляли 30 мл насыщенного водного раствора бикарбоната натрия. Смесь экстрагировали этилацетатом (20 мл * 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая соединение BB-4. LCMS m/z = 210.1 [M+H]+.
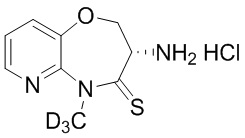
Сравнительный пример 5: Фрагмент BB-5
Путь синтеза:
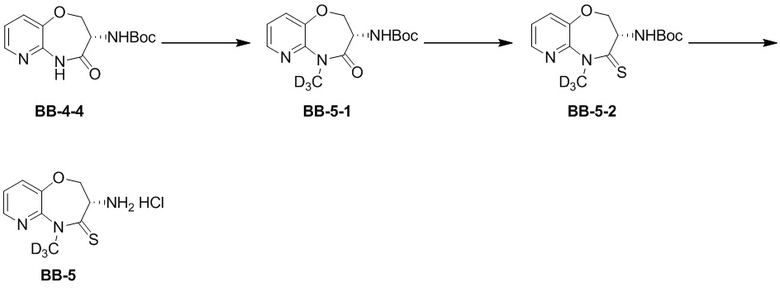
Стадия 1: синтез соединения BB-5-1
В раствор BB-4-4 (0.1 г, 358.05 мкмоль, 1 экв.) в тетрагидрофуране (5 мл) добавляли дейтерированный иодметан (101.64 мг, 716.10 мкмоль, 43.62 мкл, 2 экв.) и карбонат цезия (349.98 мг, 1.07 ммоль, 3 экв.) при 25°C, и смесь перемешивали еще 2 часа. Этилацетат (20 мл) добавляли в реакционный раствор, затем смесь промывали насыщенным водным раствором хлорида натрия (10 мл) и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали суспендированием в метил-третбутиловом эфире (5 мл), получая соединение BB-5-1. LCMS m/z = 297.2 [M+H]+.
Стадия 2: синтез соединения BB-5-2
В раствор пентасульфида фосфора (2.25 г, 10.12 ммоль, 1.08 мл, 10 экв.) в ТГФ (10 мл) добавляли карбонат натрия (1.07 г, 10.12 ммоль, 10 экв.) при 30°C, и смесь перемешивали 0.5 часа. Затем добавляли BB-5-1(0.3 г, 1.01 ммоль, 1 экв.), и реакционный раствор перемешивали на масляной бане при 70°C в течение 10 часов. Этилацетат (20 мл) добавляли в реакционный раствор. Смесь фильтровали через целит, и фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 10:1 ~ 1:1), получая соединение BB-5-2. LCMS m/z = 312.9 [M+H]+.
Стадия 3: синтез соединения BB-5
HCl/EtOAc (4M, 5.69 мл, 59.27 экв.) добавляли к BB-5-2 (0.12 г, 384.12 мкмоль, 1 экв.) при 28°C, и смесь перемешивали в течение 1 часа. Реакционный раствор упаривали при пониженном давлении, получая соединение BB-5. LCMS m/z = 213.1 [M+H]+.
Сравнительный пример 6: Фрагмент BB-6
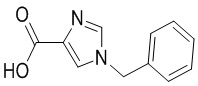
Путь синтеза:
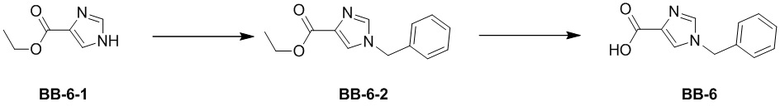
Стадия 1: синтез соединения BB-6-2
В раствор BB-6-1 (2 г, 14.27 ммоль, 1 экв.) и Cs2CO3 (5.11 г, 15.70 ммоль, 1.1 экв.) в ДМФА (20 мл) добавляли бензилбромид (2.44 г, 14.27 ммоль, 1.70 мл, 1 экв.) при 25°C, и смесь перемешивали 3 часа. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и появлялся сигнал с целевой массой. Реакционный раствор выливали в воду (100 мл), и смесь экстрагировали этилацетатом (30 мл*5). Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении. Сырой продукт очищали методом колоночной флэш-хроматографии на силикагеле (петролейный эфир:этилацетат = 10:1 ~ 1:1), получая BB-6-2. 1H ЯМР (400 МГц, CDCl3) δ = 7.58 (д, J = 1.00 Гц, 1 H), 7.55 (с, 1 H), 7.33 - 7.40 (м, 3 H), 7.14 - 7.20 (м, 2 H), 5.13 (с, 2 H), 4.34 (кв, J = 7.20 Гц, 2 H), 1.36 (т, J = 7.20 Гц, 3 H); LCMS m/z = 231.1 [M+1]+.
Стадия 2: синтез соединения BB-6
В раствор BB-6-2 (1.50 г, 6.51 ммоль, 1 экв.) в тетрагидрофуране (26 мл) добавляли раствор моногидрата гидроксида лития (2.19 г, 52.11 ммоль, 8 экв.) в воде (26 мл) при 28°C, и смесь перемешивали еще 12 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и появлялся сигнал с целевой массой. Реакционный раствор упаривали при пониженном давлении. После удаления растворителя добавляли воду (20 мл), и смесь доводили до значения pH около 2 добавлением 2н. раствора соляной кислоты. Выпавший осадок отфильтровывали, получая BB-6. 1H ЯМР (400 МГц, ДМСО-d6) δ = 7.87 (д, J = 2.20 Гц, 2 H), 7.27 - 7.43 (м, 5 H), 5.24 (с, 2 H); LCMS m/z = 201.1 [M-1]-.
Сравнительный пример 7: Фрагмент BB-7
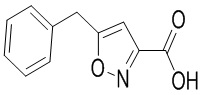
Путь синтеза:
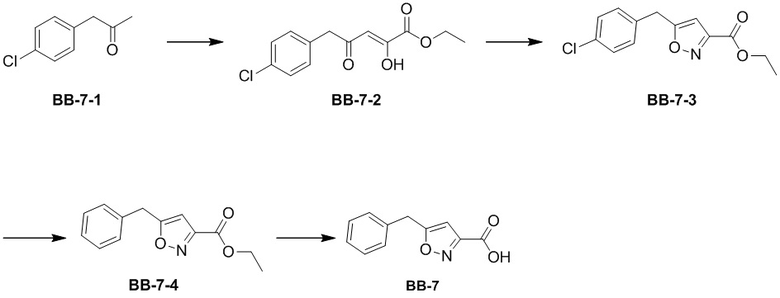
Стадия 1: синтез соединения BB-7-2
В раствор BB-7-1 (5 г, 29.65 ммоль, 1 экв.) в толуоле (70 мл) добавляли диэтилоксалат (4.77 г, 32.62 ммоль, 4.45 мл, 1.1 экв.) и этилат натрия (2.62 г, 38.55 ммоль, 1.3 экв.) при 0°C, и смесь нагревали при 20°C в течение 12 часов. Согласно данным LCMS анализа, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор упаривали при пониженном давлении, разбавляли остаток этилацетатом (200 мл) и промывали насыщенным водным раствором хлорида натрия (200 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая сырой продукт, который напрямую использовали в следующей стадии без очистки. Получали соединение BB-7-2. 1H ЯМР (400 МГц, CDCl3) δ = 6.87 - 7.19 (м, 4 H), 5.78 (ушир.с, 1 H), 4.04 (ушир.с, 2 H), 2.92 - 3.44 (м, 2 H), 1.18 (ушир.с, 3 H); LCMS m/z = 269.1[M+1]+.
Стадия 2: синтез соединения BB-7-3
В раствор BB-7-2 (7 г, 26.05 ммоль, 1 экв.) в этаноле (200 мл) добавляли гидроксиламин гидрохлорид (1.81 г, 26.05 ммоль, 1 экв.), и смесь нагревали при 80°C в течение 12 часов. Согласно данным LCMS анализа, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор упаривали при пониженном давлении, получая BB-7-3. 1H ЯМР (400 МГц, CDCl3) δ = 7.33 (м, 2 H); 7.20 (м, 2 H), 6.35 (с, 1 H), 4.41 - 4.47 (м, 2 H), 4.13 (кв, J = 7.20 Гц, 2 H), 1.30 - 1.33 (т, 3 H); LCMS m/z = 266.1[M+1]+.
Стадия 3: синтез соединения BB-7-4
В раствор BB-7-3 (4 г, 15.06 ммоль, 1 экв.) в этаноле (100 мл) добавляли Pd/C (4 г, 5% чистота), и смесь нагревали при 30°C в атмосфере водорода (30 фунт/кв.дюйм) 12 часов. Согласно данным LCMS анализа, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор фильтровали через целит, и фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (дихлорметан:метанол = 10:1), получая соединение BB-7-4. LCMS m/z = 232.1 [M+1]+.
Стадия 4: синтез соединения BB-7
В раствор BB-7-4 (0.5 г, 2.16 ммоль, 1 экв.) в тетрагидрофуране (15 мл) и воде (5 мл) добавляли моногидрат гидроксида лития (453.17 мг, 10.80 ммоль, 5 экв.), и смесь нагревали при 30°C в течение 2 часов. Исходные соединения полностью прореагировали, и наблюдалось новое пятно в ТСХ (петролейный эфир:этилацетат = 5:1). Реакционный раствор упаривали при пониженном давлении для удаления органического растворителя, и добавляли 2М раствор соляной кислоты, доводя значение pH примерно до 2. Выпадал белый осадок. Смесь фильтровали, и осадок на фильтре собирали, получая BB-7. 1H ЯМР (400 МГц, ДМСО-d6) δ = 7.24 - 7.41 (м, 5 H), 6.57 (с, 1 H), 4.22 (с, 2 H).
Сравнительный пример 8: Фрагмент BB-8
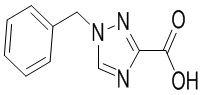
Путь синтеза:
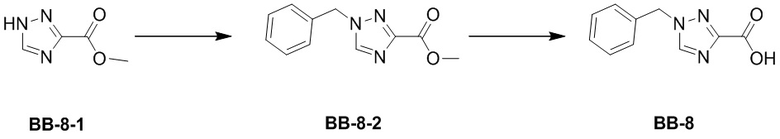
Стадия 1: синтез соединения BB-8-2
В раствор BB-8-1(2 г, 15.74 ммоль, 1 экв.) в ДМФА (32 мл) добавляли карбонат цезия (6.67 г, 20.46 ммоль, 1.3 экв.) и бензилбромид (3.23 г, 18.88 ммоль, 2.24 мл, 1.2 экв.), и смесь перемешивали при 20°C в течение 2 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Добавляли в реакционный раствор воду (30 мл) и этилацетат (30 мл), и слои разделяли. Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной флэш-хроматографии на силикагеле (петролейный эфир:этилацетат = 10:1-3:1), получая BB-8-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.11 (с, 1 H), 7.34 - 7.43 (м, 3 H), 7.27 - 7.33 (м, 2 H), 5.43 (с, 2 H), 4.00 (с, 3 H); LCMS m/z = 218.1 [M+1]+.
Стадия 2: синтез соединения BB-8
BB-8-2 (0.3 г, 1.38 ммоль, 1 экв.) растворяли в тетрагидрофуране (12 мл) и воде (4 мл) и добавляли моногидрат гидроксида лития (173.85 мг, 4.14 ммоль, 3 экв.). Реакционную смесь перемешивали при 25°C в течение 12 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и появлялся пик целевого продукта. Этилацетат (10 мл) добавляли в реакционный раствор, и водную фазу доводили до значения pH 3-4 добавлением 1М раствора соляной кислоты. Выпадал осадок. Смесь фильтровали, получая BB-8. 1H ЯМР (400 МГц, ДМСО-d6) δ =13.25 (ушир.с, 1 H), 8.78 (с, 1 H), 7.27 - 7.40 (м, 5 H), 5.47 (с, 2 H); LCMS m/z = 204.1 [M+1]+.
Сравнительный пример 9: Фрагмент BB-9
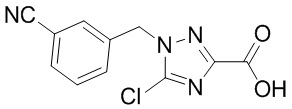
Путь синтеза:
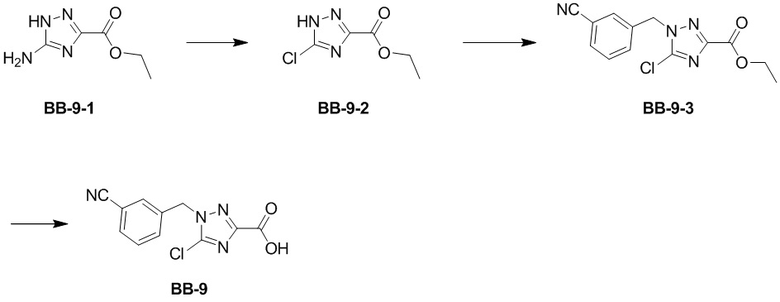
Стадия 1: синтез соединения BB-9-2
NaNO2 (662.86 мг, 9.60 ммоль, 1.5 экв.) добавляли в раствор BB-9-1 (1 г, 6.40 ммоль, 1 экв.) в H2SO4 (1 M, 12.80 мл, 2 экв.) и воде (10 мл) при 0°C, и смесь перемешивали при 0°C в течение 0.5 часа. CuCl2 (1.72 г, 12.80 ммоль, 2 экв.) растворяли в концентрированной соляной кислоте (15 мл), и полученный раствор добавляли в описанную выше смесь. Полученную смесь перемешивали при 0°C в течение 1.5 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и появлялся пик целевого продукта. Этилацетат (10 мл) добавляли в реакционный раствор, и водную фазу экстрагировали этилацетатом (15 мл*2). Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали методом колоночной флэш-хроматографии на силикагеле (петролейный эфир:этилацетат = 10:1 ~ 1:2), получая BB-9-2. 1H ЯМР (400 МГц, CDCl3) = 4.46 (кв, J = 21.60 Гц, 2 H), 1.38 (т, J = 14.40 Гц, 3 H); LCMS m/z = 176.0 [M+1]+.
Стадия 2: синтез соединения BB-9-3
В раствор BB-9-2 (760.78 мг, 4.33 ммоль, 1 экв.) в ДМФА (5 мл) добавляли карбонат цезия (1.84 г, 5.63 ммоль, 1.3 экв.) и 3-цианобензилбромид (889.31 мг, 5.20 ммоль, 617.58 мкл, 1.2 экв.), и реакционную смесь перемешивали при 20°C в течение 2 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и появлялся пик целевого продукта. Воду (10 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (10 мл*2). Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, и сырой продукт очищали методом колоночной флэш-хроматографии на силикагеле (петролейный эфир:этилацетат = 20:1 ~ 2:1), получая BB-9-3. LCMS m/z = 291.0 [M+1]+.
Стадия 3: синтез соединения BB-9
BB-9-3 (0.08 г, 301.10 мкмоль, 1 экв.) растворяли в тетрагидрофуране (6 мл) и воде (2 мл), и добавляли гидроксид лития моногидрат (37.90 мг, 903.29 мкмоль, 3 экв.). Смесь нагревали при 2°C в течение 12 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и появлялся пик целевого продукта. Этилацетат (5 мл) добавляли в реакционный раствор. Водную фазу доводили до значения pH около 6 добавлением 1М раствора соляной кислоты и лиофилизовывали, получая BB-9. LCMS m/z = 263.1 [M+1]+.
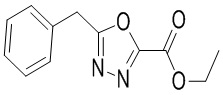
Сравнительный пример 10: Фрагмент BB-10
Путь синтеза:
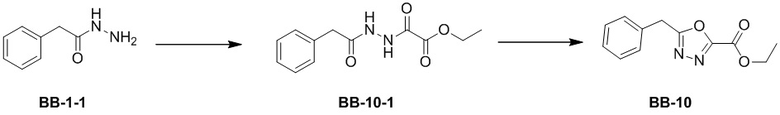
Стадия 1: синтез соединения BB-10-1
В раствор BB-1-1 (3 г, 19.98 ммоль, 1 экв.) в дихлорметане (90 мл) добавляли триэтиламин (6.06 г, 59.93 ммоль, 8.34 мл, 3 экв.) добавляли этил хлороксоацетат (3.55 г, 25.97 ммоль, 2.91 мл, 1.3 экв.), и смесь перемешивали при 25°C в течение 5 часов. Согласно данным LCMS анализа, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакцию гасили добавлением воды (60 мл) и экстрагировали дихлорметаном (60 мл*3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении. Сырой продукт очищали методом колоночной флэш-хроматографии на силикагеле (петролейный эфир:этилацетат = 5:1-1:1), получая BB-10-1. 1H ЯМР (400 МГц, ДМСО-d6) δ = 10.73 (ушир.с, 1 H), 10.30 (ушир.с, 1 H), 7.22 - 7.34 (м, 5 H), 4.26 (кв, J = 7.00 Гц, 2 H), 3.51 (с, 2 H), 1.27 (т, J = 7.10 Гц, 3 H); LCMS m/z = 251.1[M+1]+.
Стадия 2: синтез соединения BB-10
В раствор BB-10-1 (529.69 мг, 2.12 ммоль, 1 экв.) в ацетонитриле (15 мл) добавляли оксихлорид фосфора (486.82 мг, 3.17 ммоль, 295.04 мкл, 1.5 экв.). Реакционную смесь перемешивали при 90°C в течение 2 часов. Реакционный раствор выливали в насыщенный раствор бикарбоната натрия (20 мл), и смесь интенсивно перемешивали 5 минут. Смесь экстрагировали два раза этилацетатом (20 мл). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая BB-10. 1H ЯМР (400 МГц, CDCl3) δ = 7.23 - 7.10 (м, 5H), 4.36 - 4.29 (м, 2H), 4.12 (с, 2H), 1.34 - 1.23 (м, 3H); LCMS m/z = 233.1 [M+1]+.
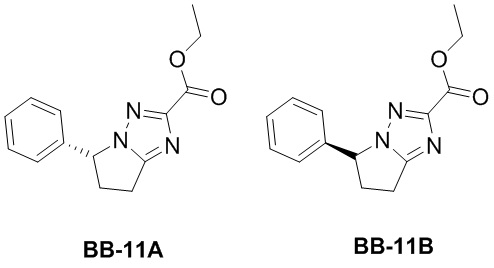
Сравнительный пример 11: Фрагмент BB-11A, BB-11B
Путь синтеза:
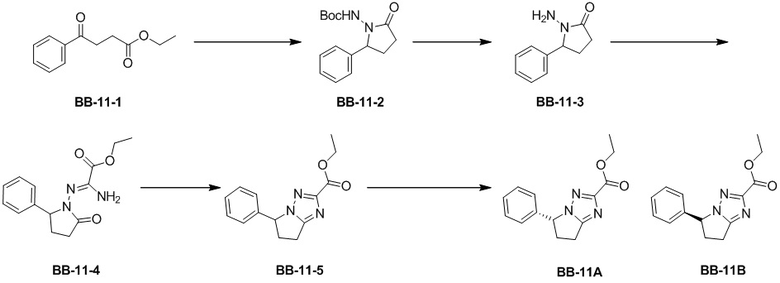
Стадия 1: синтез соединения BB-11-2
В раствор BB-11-1 (5 г, 26.01 ммоль, 1 экв.) в ледяной уксусной кислоте (39 мл) и тетрагидрофуране (150 мл) добавляли трет-бутилкарбазат (17.19 г, 130.07 ммоль, 5 экв.), и смесь нагревали при 55°C в течение 12 часов. Смесь охлаждали до комнатной температуры (25°C) и порциями добавляли NaBH3CN (8.17 г, 130.07 ммоль, 5 экв.). Смесь нагревали до 55°C и оставляли для прохождения реакции на 12 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли насыщенный раствор бикарбоната натрия для доведения значения pH до 7~8. Смесь экстрагировали этилацетатом (150 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая BB-11-2. LCMS m/z = 221.1 [M-56+1]+.
Стадия 2: синтез соединения BB-11-3
ВВ-11-2 (7 г, 25.33 ммоль, 1 экв.) добавляли в раствор HCl/EtOAc (4M, 60 мл), и полученную смесь нагревали при 25°C в течение 2 часов. Реакционный раствор упаривали при пониженном давлении, получая сырой продукт. В сырой продукт добавляли насыщенный раствор бикарбоната натрия, доводя значение pH до 7~8. Смесь экстрагировали этилацетатом (50 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт BB-11-3. LCMS m/z = 177.1 [M+1]+.
Стадия 3: синтез соединения BB-11-4.
В раствор BB-11-3 (4 г, 22.70 ммоль, 1 экв.) в этаноле (50 мл) добавляли этил 2-этокси-2-иминоацетат (16.47 г, 113.50 ммоль, 5 экв.), и смесь нагревали при 85°C в течение 12 часов. Реакционный раствор упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (дихлорметан:метанол = от 10:0 до 5:1), получая BB-11-4. LCMS m/z = 276.2 [M+1]+.
Стадия 4: синтез соединения BB-11-5
BB-11-4 (1.3 г, 4.72 ммоль, 1 экв.) добавляли в оксихлорид фосфора (30 мл) при 25°C, и смесь нагревали при 120°C в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и затем медленно выливали в теплую воду (150 мл). Смесь доводили до значения pH 7~8 насыщенным водным раствором бикарбоната натрия и затем экстрагировали этилацетатом (100 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (дихлорметан:метанол = от 10:0 до 10:1), получая BB-11-5. 1H ЯМР (400 МГц, ДМСО-d6) δ = 7.34 - 7.43 (м, 3 H), 7.21 - 7.28 (м, 2 H), 5.58 (ушир.т, J = 7.12 Гц, 1 H), 4.27 (ушир. дд, J = 6.80, 5.04 Гц, 2 H), 2.88 - 3.28 (м, 4 H), 1.26 (ушир.т, J = 7.12 Гц, 3 H). LCMS m/z = 258.2 [M+1]+.
Стадия 5: синтез соединения BB-11A и BB-11B
Соединение BB-11 анализировали методом сверхкритической жидкостной хроматографии (Chiralcel AD-3 3мкм, 0.46см id x 15мл; Подвижная фаза: A = SFC CO2 и B = MeOH (0.05% IPAm); Градиент: B в A от 10% до 40% за 6 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), и было показано, что времена удерживания BB-11A и BB-11B составляли 2.217 мин и 2.427 мин, соответственно. BB-11A и BB-11B получали посредством разделения методом сверхкритической жидкостной хроматографии (Колонка: DAICEL CHIRALPAK AD (250мм*50мм, 10 мкм); Подвижная фаза: [0.1% NH3H2O MEOH]; MeOH%: 30%-30%, 2.56 мин). Соединение BB-11A анализировали методом сверхкритической жидкостной хроматографии (Chiralcel AD-3 3мкм, 0.46 см id × 15 мл; Подвижная фаза: A = сверхкритический диоксид углерода, B = метанол (содержит 0.05% IPAm); Градиент: B от 10% до 40% за 6 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), и было показано, что время удерживания BB-11A составляет 2.235 мин, а время удерживания BB-11B составляет 2.439 мин.
Сравнительный пример 12: Фрагмент BB-12
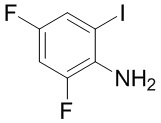
Путь синтеза:
Стадия 1: синтез соединения BB-12.
В раствор BB-12-1 в этаноле (100 мл) добавляли I2 (11.40 г, 44.92 ммоль, 9.05 мл, 1 экв.) и Ag2SO4 (14.01 г, 44.92 ммоль, 7.61 мл, 1 экв.) при 25°C, и смесь перемешивали 12 часов. Реакционный раствор фильтровали через целит. Добавляли в фильтрат воду (150 мл) и этилацетат (150 мл), и слои разделяли. Водную фазу экстрагировали этилацетатом (50 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт растворяли в этилацетате и добавляли раствор хлороводород/этилацетат (4 M). Смесь перемешивали при комнатной температуре в течение 0.5 часа и фильтровали. Осадок на фильтре отделяли. Добавляли к осадку этилацетат (50 мл) и добавляли насыщенный раствор бикарбоната натрия, доводя значение pH до 8~9. Слои разделяли. Водную фазу экстрагировали этилацетатом (25 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении (водоструйный насос, 45°C), получая сырой продукт, и полученный сырой продукт очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 10:1 ~ 1:1), получая BB-12. 1H ЯМР (400 МГц, CDCl3) δ = 7.22 (дт, J = 7.68, 2.37 Гц, 1 H), 6.83 (ддд, J = 10.85, 8.22, 2.76 Гц, 1 H), 3.97 (ушир.с, 2 H).
Сравнительный пример 13: Фрагмент BB-13
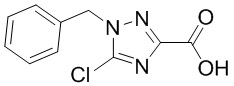
Путь синтеза:
Стадия 1: синтез соединения BB-13-1
В раствор BB-9-2 (760.78 мг, 4.33 ммоль, 1 экв.) в ДМФА (5 мл) добавляли карбонат цезия (1.84 г, 5.63 ммоль, 1.3 экв.) и бензилбромид (889.31 мг, 5.20 ммоль, 617.58 мкл, 1.2 экв.), и реакционную смесь перемешивали при 20°C в течение 2 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и появлялся пик целевого продукта. Добавляли в реакционный раствор воду (10 мл), и смесь экстрагировали этилацетатом (10 мл*2). Органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали методом колоночной флэш-хроматографии на силикагеле (петролейный эфир:этилацетат = 20:1~2:1), получая BB-13-1. 1H ЯМР (400 МГц, CDCl3) δ =7.34 - 7.41 (м, 3 H), 7.29 - 7.34 (м, 2 H), 5.42 (с, 2 H), 4.47 (кв, J = 22.40 Гц, 2 H), 1.43 (т, J = 15.60 Гц, 3 H); LCMS m/z = 266.0 [M+1]+.
Стадия 2: синтез соединения BB-13
BB-13-1 (0.08 г, 301.10 мкмоль, 1 экв.) растворяли в тетрагидрофуране (6 мл) и воде (2 мл), и добавляли гидроксид лития моногидрат (37.90 мг, 903.29 мкмоль, 3 экв.). Смесь нагревали при 25°C в течение 12 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и появлялся пик целевого продукта. Добавляли в реакционный раствор 5 мл этилацетата. Водную фазу доводили до значения pH около 6 добавлением 1М раствора соляной кислоты и лиофилизовывали, получая BB-13. 1H ЯМР (400 МГц, ДМСО-d6) δ = 7.32 - 7.42 (м, 3 H), 7.25 - 7.30 (м, 2 H), 5.48 (с, 2 H); LCMS m/z = 238.1 [M+1]+.
Пример 1. WX001
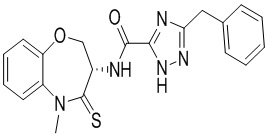
Путь синтеза:
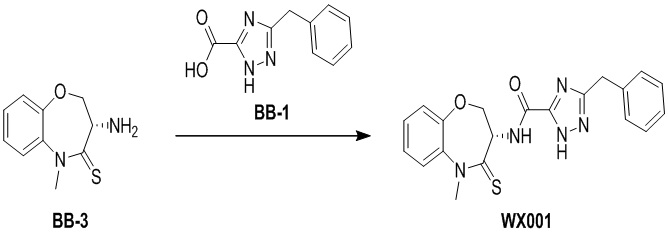
Стадия 1: синтез соединения WX001
Соединение BB-3 (0.58 г, 2.78 ммоль, 1 экв.), соединение BB-1 (849 мг, 4.18 ммоль, 1.5 экв.), N,N-диизопропилэтиламин (1.26 г, 9.75 ммоль, 1.70 мл, 3.5 экв.) растворяли в этилацетате (10 мл) и затем добавляли по каплям 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан-2,4,6-триоксид (4.43 г, 6.96 ммоль, 4.14 мл, 50%-ный раствор в этилацетате, 2.5 экв.). После окончания добавления смесь перемешивали при 25°C в течение 12 часов. Добавляли в реакционный раствор 20 мл воды, и смесь экстрагировали этилацетатом (5 мл*2). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия (10 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле и затем методом сверхкритической жидкостной хроматографии (Chiralcel OJ-3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A = сверхкритический диоксид углерода, B = метанол (содержит 0.05% IPAm); Градиент: B от 10% до 40% за 5 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), получая соединение WX001. 1H ЯМР (400 МГц, CDCl3) δ = 8.71 (д, J = 7.53 Гц, 1H), 7.20-7.38 (м, 9H), 5.15 - 5.25 (м, 1H), 4.67 (дд, J = 6.53, 9.54 Гц, 1H), 4.29 (дд, J = 9.79, 10.79 Гц, 1H), 4.21 (с, 2H), 3.89 (с, 3H); LCMS m/z = 394.1 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 96.36%.
Пример 2. WX002
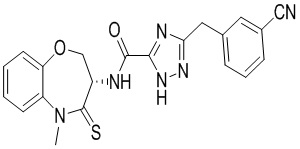
Путь синтеза:
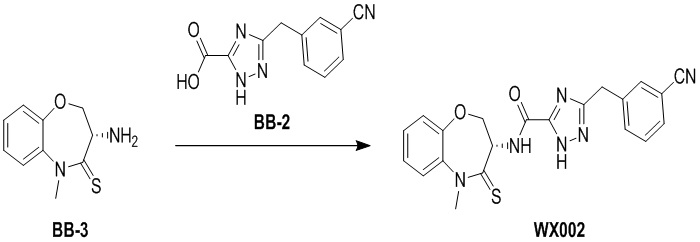
Стадия 1: синтез соединения WX002
В раствор BB-3 (0.06 г, 288.07 мкмоль, 1 экв.) и BB-2 (131.48 мг, 576.15 мкмоль, 2 экв.) в ДМФА (10 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан- 2,4,6-триоксид (458.30 мг, 720.19 мкмоль, 428.32 мкл, 50% чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (111.69 мг, 864.22 мкмоль, 150.53 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Добавляли в реакционный раствор 20 мл этилацетата, и смесь промывали 15 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 150 мм*25 мм*5 мкм; Подвижная фаза: [вода (10 мM NH4HCO3)-ACN]; ацетонитрил%: 35%-65%, 10.5 мин), получая соединение WX002. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.60 (ушир.д, J = 8.16 Гц, 1 H), 7.69 - 7.81 (м, 2 H), 7.51 - 7.65 (м, 3 H), 7.36 - 7.46 (м, 2 H), 7.25 - 7.33 (м, 1 H), 4.97 (дт, J = 10.58, 7.72 Гц, 1 H), 4.35 - 4.52 (м, 2 H), 4.21 (с, 2 H), 3.79 (с, 3 H); LCMS m/z = 419.1 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD-3, 3 мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 100%.
Пример 3. WX003
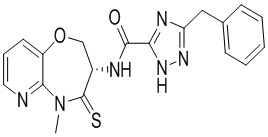
Путь синтеза:
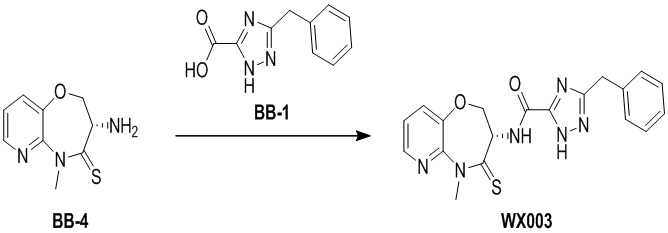
Стадия 1: синтез соединения WX003
В раствор BB-4 (60 мг, 286.71 мкмоль, 1 экв.) и BB-1 (87.39 мг, 430.07 мкмоль, 1.5 экв.) в ДМФА (10 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан- 2,4,6-триоксид (456.13 мг, 716.78 мкмоль, 426.29 мкл, 50% чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (111.16 мг, 860.13 мкмоль, 149.82 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Добавляли в реакционный раствор 30 мл этилацетата, и смесь промывали 20 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 150 мм*25мм*5 мкм; Подвижная фаза: [вода (10 мM NH4HCO3)-ACN]; ацетонитрил%: 35%-65%, 10.5 мин), получая соединение WX003. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.63 (ушир.с, 1 H), 8.46 (дд, J = 4.63, 1.54 Гц, 1 H), 7.79 (д, J = 7.94 Гц, 1 H), 7.48 (дд, J = 8.05, 4.74 Гц, 1 H), 7.23 - 7.37 (м, 5 H), 5.01 (дт, J = 10.80, 7.28 Гц, 1 H), 4.43 - 4.72 (м, 2 H), 4.14 (ушир.с, 2 H), 3.80 (с, 3 H); LCMS m/z = 395.2 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 96.3%.
Пример 4. WX004
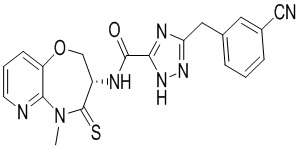
Путь синтеза:
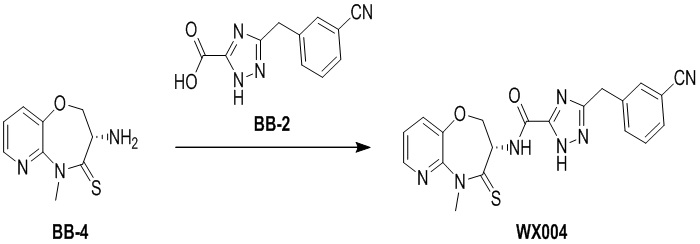
Стадия 1: синтез соединения WX004
В раствор BB-4 (60 мг, 286.71 мкмоль, 1 экв.) и BB-2 (130.86 мг, 573.43 мкмоль, 2 экв.) в ДМФА (10 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан- 2,4,6-триоксид (456.13 мг, 716.78 мкмоль, 426.29 мкл, 50 % чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (111.16 мг, 860.14 мкмоль, 149.82 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Добавляли в реакционный раствор 30 мл этилацетата, и смесь промывали 20 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 15 мм*25 мм*5 мкм; Подвижная фаза: [вода (10 мM NH4HCO3)-ACN]; ацетонитрил%: 35%-65%, 10.5 мин), получая соединение WX004. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.73 (ушир.с, 1 H), 8.46 (д, J = 4.85 Гц, 1 H), 7.69 - 7.86 (м, 3 H), 7.44 - 7.65 (м, 3 H), 5.01 (дт, J = 11.08, 7.14 Гц, 1 H), 4.48 - 4.68 (м, 2 H), 4.22 (с, 2 H), 3.80 (с, 3 H); LCMS m/z = 420.1 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 100 %.
Пример 5. WX005
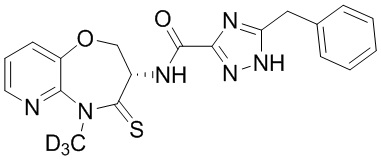
Путь синтеза:
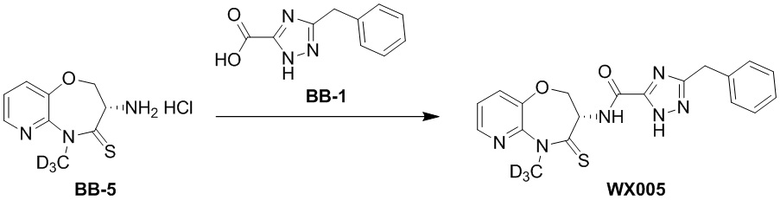
Стадия 1: синтез соединения WX005
В раствор BB-5 (100 мг, 471.06 мкмоль, 1 экв.) и BB-1 (130.86 мг, 573.43 мкмоль, 1.2 экв.) в ДМФА (10 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан- 2,4,6-триоксид (599.53 мг, 942.12 мкмоль, 560.31 мкл, 50 % чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (182.64 мг, 1.41 ммоль, 246.15 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Этилацетат (30 мл) добавляли в реакционный раствор, и смесь промывали насыщенным водным раствором хлорида натрия (20 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX005. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.62 (ушир.с, 1H), 8.46 (дд, J = 1.75, 4.82 Гц, 1H), 7.79 (дд, J = 1.32, 7.89 Гц, 1H), 7.48 (дд, J = 4.82, 7.89 Гц, 1H), 7.20-7.37 (м, 5H), 5.01 (тд, J = 7.34, 11.18 Гц, 1H), 4.60 (ушир.с, 1H), 4.49-4.55 (м, 1H), 4.14 (ушир.с, 2H); LCMS m/z = 398.2 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 100%.
Пример 6. WX006
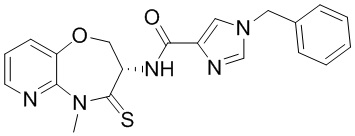
Путь синтеза:
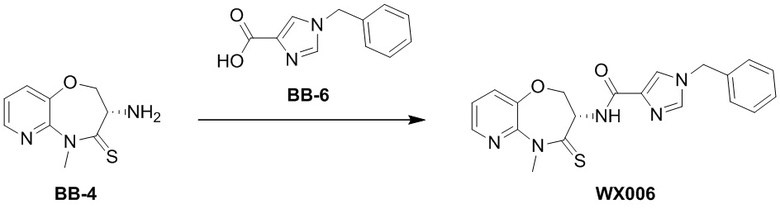
Стадия 1: синтез соединения WX006
В раствор BB-4 (10 мг, 47.79 мкмоль, 1 экв.) и BB-1 (11.60 мг, 57.34 мкмоль, 1.2 экв.) в ДМФА (1 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан-2,4,6-триоксид (60.82 мг, 95.57 мкмоль, 56.84 мкл, 50 % чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (18.53 мг, 143.36 мкмоль, 24.97 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор напрямую очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX006. 1H ЯМР (400 МГц, CDCl3) δ = 9.01 (ушир.с, 1H), 8.34-8.46 (м, 2H), 7.51-7.64 (м, 2H), 7.43 (ушир.с, 3H), 7.28-7.31 (м, 2H), 7.22-7.26 (м, 1H), 5.29 (ушир.с, 2H), 5.20 (ушир.д, J = 7.02 Гц, 1H), 4.58-4.69 (м, 2H), 3.91 (с, 3H); LCMS m/z = 394.2 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 100%.
Пример 7. WX007
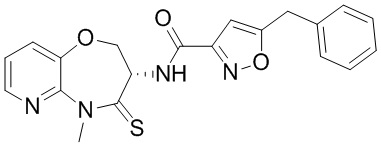
Путь синтеза:
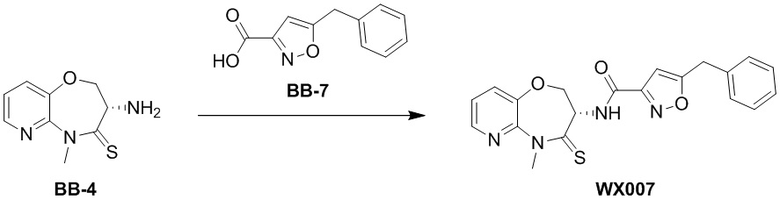
Стадия 1: синтез соединения WX007
В раствор BB-4 (10 мг, 47.79 мкмоль, 1 экв.) и BB-7 (11.65 мг, 57.35 мкмоль, 1.2 экв.) в ДМФА (1 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан-2,4,6-триоксид (60.82 мг, 95.57 мкмоль, 56.84 мкл, 50% чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (18.53 мг, 143.36 мкмоль, 24.97 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор напрямую очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX007. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.98 (д, J = 8.33 Гц, 1H), 8.46 (дд, J = 1.32, 4.82 Гц, 1H), 7.77 (дд, J = 1.75, 7.89 Гц, 1H), 7.47 (дд, J = 4.60, 8.11 Гц, 1H), 7.22-7.40 (м, 5H), 6.54 (с, 1H), 5.00 (тд, J = 7.45, 11.40 Гц, 1H), 4.59-4.70 (м, 1H), 4.48-4.56 (м, 1H), 4.22 (с, 2H), 3.79 (с, 3H); LCMS m/z = 395.1 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 100 %.
Пример 8. WX008
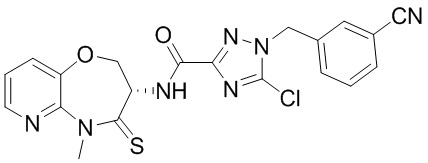
Путь синтеза:
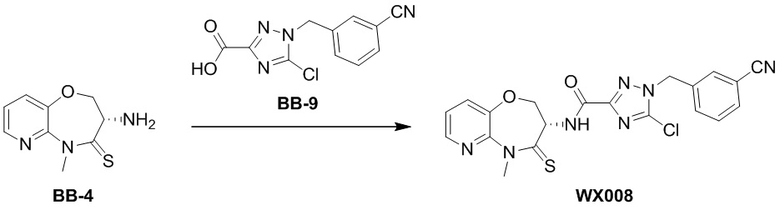
Стадия 1: синтез соединения WX008
В раствор BB-4 (10 мг, 47.79 мкмоль, 1 экв.) и BB-8 (15.06 мг, 57.35 мкмоль, 1.2 экв.) в ДМФА (1 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан-2,4,6-триоксид (60.82 мг, 95.57 мкмоль, 56.84 мкл, 50% чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (18.53 мг, 143.36 мкмоль, 24.97 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор напрямую очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX008. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.85 (д, J = 7.89 Гц, 1H), 8.46 (д, J = 3.07 Гц, 1H), 7.75-7.89 (м, 3H), 7.61 (д, J = 4.82 Гц, 2H), 7.48 (дд, J = 4.82, 7.89 Гц, 1H), 5.58 (с, 2H), 4.94-5.06 (м, 1H), 4.65 (т, J = 10.74 Гц, 1H), 4.45-4.57 (м, 1H), 3.80 (с, 3H); LCMS m/z = 454.1 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 100 %.
Пример 9. WX009
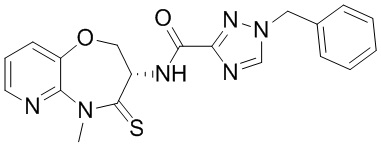
Путь синтеза:
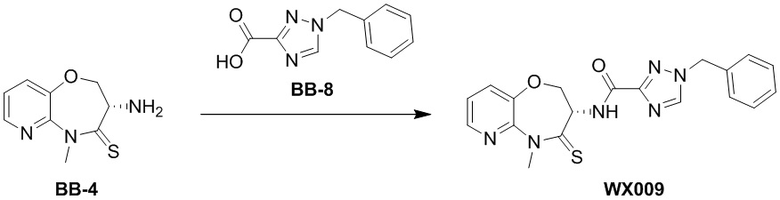
Стадия 1: синтез соединения WX009
В раствор BB-4 (10 мг, 47.79 мкмоль, 1 экв.) и BB-9 (11.65 мг, 57.35 мкмоль, 1.2 экв.) в ДМФА (1 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан-2,4,6-триоксид (60.82 мг, 95.57 мкмоль, 56.84 мкл, 50% чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (18.53 мг, 143.36 мкмоль, 24.97 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор напрямую очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX009. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.84 (с, 1H), 8.71 (д, J = 7.89 Гц, 1H), 8.46 (дд, J = 1.53, 4.60 Гц, 1H), 7.79 (дд, J = 1.53, 8.11 Гц, 1H), 7.48 (дд, J = 4.82, 7.89 Гц, 1H), 7.26-7.42 (м, 5H), 5.49 (с, 2H), 4.94-5.08 (м, 1H), 4.48-4.63 (м, 2H), 3.80 (с, 3H); LCMS m/z = 395.1 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 100 %.
Пример 10. WX010
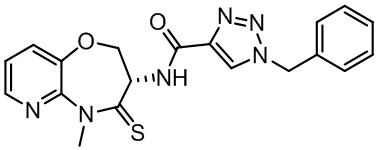
Путь синтеза:
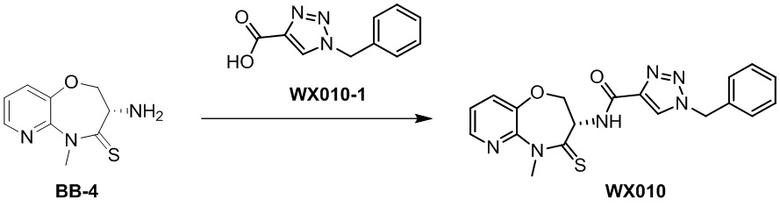
Стадия 1: синтез соединения WX010
В раствор BB-4 (10 мг, 47.79 мкмоль, 1 экв.) и WX010-1 (11.65 мг, 57.35 мкмоль, 1.2 экв.) в ДМФА (1 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан- 2,4,6-триоксид (60.82 мг, 95.57 мкмоль, 56.84 мкл, 50% чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (18.53 мг, 143.36 мкмоль, 24.97 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор напрямую очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX010. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.75 (д, J = 8.33 Гц, 1H), 8.69 (с, 1H), 8.46 (дд, J = 1.53, 4.60 Гц, 1H), 7.79 (дд, J = 1.53, 8.11 Гц, 1H), 7.48 (дд, J = 4.82, 7.89 Гц, 1H), 7.30-7.41 (м, 5H), 5.65 (с, 2H), 4.97-5.10 (м, 1H), 4.58-4.68 (м, 1H), 4.47-4.57 (м, 1H), 3.80 (с, 3H); LCMS m/z = 395.1 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 100 %.
Пример 11. WX011
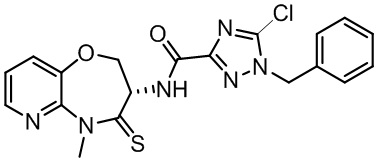
Путь синтеза:
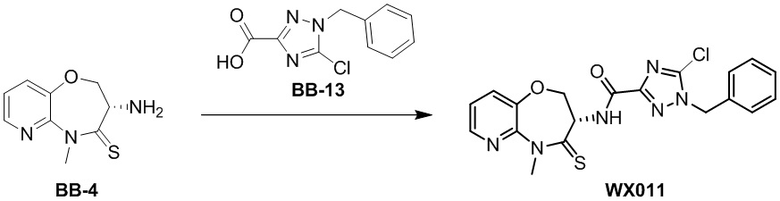
Стадия 1: синтез соединения WX011
В раствор BB-4 (10 мг, 47.79 мкмоль, 1 экв.) и BB-13 (14.76 мг, 62.13 мкмоль, 1.2 экв.) в ДМФА (1 мл) добавляли 2,4,6-трибутил-1,3,5,2,4,6-триоксатрифосфинан- 2,4,6-триоксид (60.82 мг, 95.57 мкмоль, 56.84 мкл, 50 % чистота в этилацетате, 2.5 экв.) и N,N-диизопропилэтиламин (18.53 мг, 143.36 мкмоль, 24.97 мкл, 3 экв.), и смесь перемешивали при 25°C в течение 10 часов. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор напрямую очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX011. 1H ЯМР (400 МГц, ДМСО- d6) δ = 8.83 (д, J = 7.89 Гц, 1H), 8.41-8.49 (м, 1H), 7.77 (дд, J = 1.32, 7.89 Гц, 1H), 7.46 (дд, J = 4.82, 7.89 Гц, 1H), 7.30-7.42 (м, 3H), 7.26 (д, J = 7.02 Гц, 2H), 5.48 (с, 2H), 4.98 (тд, J = 7.13, 11.62 Гц, 1H), 4.59-4.67 (м, 1H), 4.49 (дд, J = 6.80, 9.87 Гц, 1H), 3.78 (с, 3H); LCMS m/z = 429.0 [M+H]+. Согласно данным анализа методом сверхкритической жидкостной хроматографии (Хиральная колонка: Колонка: Chiralpak OD -3, 3мкм, 0.46 см id × 5 см L; Подвижная фаза: A: CO2, B: MeOH (0.05% IPAm); Градиент: B/A = 10% - 40% в течение 3 минут; Скорость потока: 4.0 мл/мин; Длина волны: 220 нм), ee % составлял 100%.
Пример 12. WX012
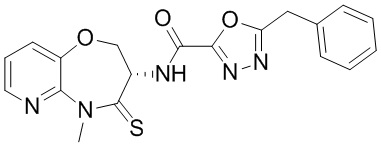
Путь синтеза:
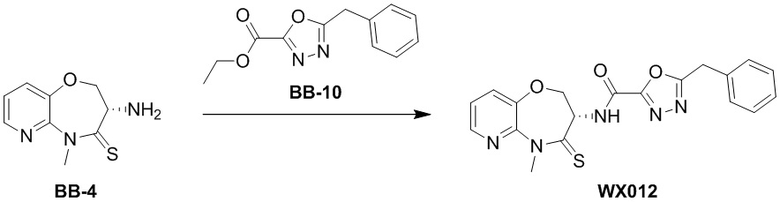
Стадия 1: синтез соединения WX012
В раствор BB-4 (15 мг, 71.68 мкмоль, 1 экв.) и BB-10 (33.29 мг, 143.36 мкмоль, 2 экв.) в толуоле (3 мл) добавляли триметилалюминий (2M, 53.76 мкл, 1.5 экв.), и смесь перемешивали при 25°C в течение 1 часа. По данным LCMS мониторинга, исходные соединения полностью прореагировали, и наблюдался сигнал целевого продукта. Реакционный раствор напрямую очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX012. LCMS m/z = 396.2 [M+H]+.
Пример 13. WX013
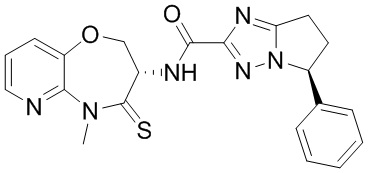
Путь синтеза:
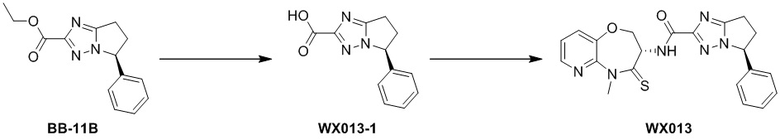
Стадия 1: синтез соединения WX013-1
LiOH⋅H2O (245.59 мг, 5.85 ммоль, 3 экв.) добавляли в раствор BB-11B (501.93 мг, 1.95 ммоль, 1 экв.) в тетрагидрофуране (9 мл) и воде (3 мл), и смесь нагревали при 25°C в течение 2 часов. Воду (10 мл) и этилацетат (10 мл) добавляли в реакционный раствор, и слои разделяли. Водную фазу экстрагировали этилацетатом (10 мл*2). Результирующую водную фазу доводили до значения pH 5-6 добавлением 1M раствора HCl и экстрагировали этилацетатом (10 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении (водоструйный насос, 45°C), получая WX013-1. LCMS m/z = 230.2 [M+1]+.1H ЯМР (400 МГц, ДМСО-d6) δ = 12.75 - 13.57 (м, 1 H), 7.33 - 7.44 (м, 3 H), 7.18 - 7.28 (м, 2 H), 5.57 (дд, J = 7.97, 6.21 Гц, 1 H), 3.06 - 3.22 (м, 2 H), 2.94 - 3.03 (м, 1 H), 2.53 - 2.61 (м, 1 H).
Стадия 2: синтез соединения WX013
N,N-диизопропилэтиламин (23.16 мг, 179.20 мкмоль, 31.21 мкл, 2.5 экв.) и WX013-1 (24.65 мг, 107.52 мкмоль, 1.5 экв.) добавляли в раствор BB-1 (15 мг, 71.68 мкмоль, 1 экв.) в ДМФА (1.5 мл), и затем добавляли пропилфосфоновый ангидрид (T3P, 68.42 мг, 107.52 мкмоль, 63.94 мкл, 50% чистота, 1.5 экв.). Смесь нагревали при 25°C в течение 1 часа. Реакционный раствор фильтровали, и фильтрат собирали. Фильтрат очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая WX013. LCMS m/z = 421.1 [M+1]+.1H ЯМР (400 МГц, ДМСО-d6) δ = 8.68 (д, J = 8.04 Гц, 1 H), 8.46 (дд, J = 4.72, 1.32 Гц, 1 H), 7.78 (дд, J = 8.04, 1.38 Гц, 1 H), 7.47 (дд, J = 8.04, 4.64 Гц, 1 H), 7.32 - 7.43 (м, 3 H), 7.24 (ушир.д, J = 6.65 Гц, 2 H), 5.44 - 5.68 (м, 1 H), 5.00 (дт, J = 11.40, 7.40 Гц, 1 H), 4.42 - 4.68 (м, 2 H), 3.79 (с, 3 H), 2.90 - 3.23 (м, 4 H).
Пример 14. WX014
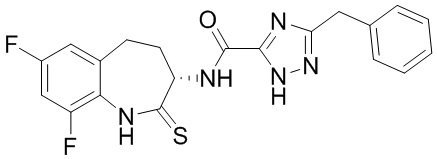
Путь синтеза:
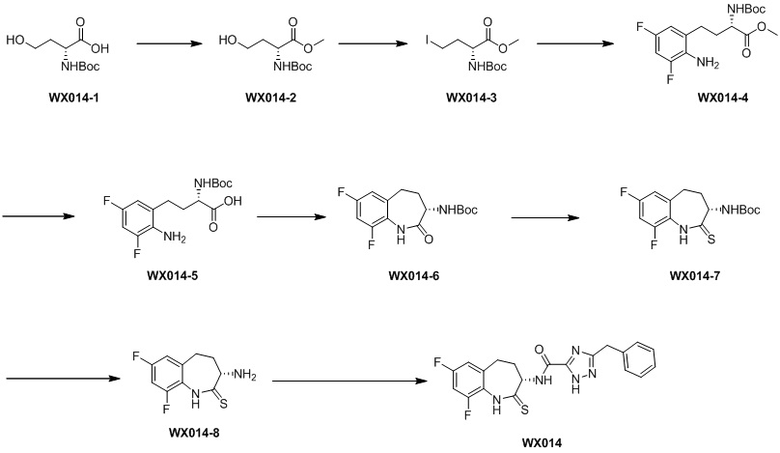
Стадия 1: синтез соединения WX014-2
WX014-1 растворяли в ТГФ (100 мл) и MeOH (100 мл), и реакционную колбу помещали в ледяную баню (0°C). Добавляли в колбу триметилсилил диазометан (2M, 47.89 мл, 2.1 экв.), смесь нагревали до 25°C и перемешивали в течение 2 часов. Ледяную уксусную кислоту добавляли по каплям до тех пор, пока раствор не стал бледно-желтым. Реакционный раствор упаривали при пониженном давлении, получая соединение WX014-2.
Стадия 2: синтез соединения WX014-3
Трифенилфосфин (14.30 г, 54.53 ммоль, 1.2 экв.) и имидазол (3.71 г, 54.53 ммоль, 1.2 экв.) растворяли в дихлорметане (120 мл) при 25°C и медленно добавляли I2 (13.84 г, 54.53 ммоль, 10.98 мл, 1.2 экв.) в полученную смесь. Смесь перемешивали 10 минут и добавляли в нее раствор WX014-2 в ДХМ (80 мл). Смесь нагревали при 25°C в течение 12 часов. Реакционный раствор фильтровали, и фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 10:1 ~ 1:1), получая WX014-3. LCMS m/z = 244.1 [M-100+1]+. 1H ЯМР (400 МГц, CDCl3) δ = 5.10 (ушир.д, J = 4.82 Гц, 1 H), 4.35 (ушир.д, J = 3.96 Гц, 1 H), 3.77 (с, 3 H), 3.18 (т, J = 7.46 Гц, 2 H), 2.30 - 2.49 (м, 1 H), 2.18 (д.кв, J = 14.32, 7.22 Гц, 1 H), 1.43 - 1.47 (м, 9 H).
Стадия 3: синтез соединения WX014-4
1,2-Дибромэтан (82.12 мг, 437.12 мкмоль, 32.98 мкл, 0.3 экв.) добавляли к цинковому порошку (285.83 мг, 4.37 ммоль, 3 экв.) в ДМФА (2 мл). Смесь перемешивали при 60°C в течение 30 минут, и затем охлаждали до комнатной температуры (25°C). Добавляли в реакционную колбу триметилхлорсилан (9.50 мг, 87.42 мкмоль, 11.10 мкл, 0.06 экв.), и смесь перемешивали при 25°C в течение 30 минут. Раствор WX014-2 в ДМФА (2 мл) добавляли в полученную смесь, и смесь нагревали при 25°C в течение 30 минут, затем оставляли на 30 минут. Верхний эмульсионный слой отделяли и медленно добавляли в раствор трис(дибензилиденацетон)дипалладия (44.08 мг, 48.14 мкмоль, 0.05 экв.), три-о-метилфенилфосфина (58.61 мг, 192.55 мкмоль, 0.2 экв.) и BB-12 в ДМФА (2 мл), и смесь нагревали при 20°C в течение 1 часа. Добавляли в реакционный раствор воду (5 мл) и метил-трет-бутиловый эфир (5 мл), и слои разделяли. Водную фазу экстрагировали метил-трет-бутиловым эфиром (5 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая WX014-4. LCMS m/z = 289.1 [M-56+1]+.
Стадия 4: синтез соединения WX014-5
LiOH⋅H2O (219.33 мг, 5.23 ммоль, 3 экв.) добавляли в раствор WX014-4 в тетрагидрофуране (15 мл) и воде (15 мл), и смесь нагревали при 25°C в течение 2 часов. добавляли в реакционный раствор 10 мл воды и 10 мл этилацетата, и слои разделяли. Водную фазу экстрагировали этилацетатом (10 мл*2). Результирующую водную фазу доводили до значения pH 5~6 добавлением 1M раствора HCl и экстрагировали этилацетатом (10 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая WX014-5. LCMS m/z = 275.1 [M-56+1]+.
Стадия 5: синтез соединения WX014-6
Пропилфосфоновый ангидрид (895.80 мг, 1.41 ммоль, 837.20 мкл, 50% чистота, 1.5 экв.) добавляли в раствор N,N-диизопропилэтиламина (303.22 мг, 2.35 ммоль, 408.65 мкл, 2.5 экв.) и WX014-5 в ДМФА (20 мл), и смесь нагревали при 25°C в течение 2 часов. Добавляли в реакционный раствор воду (15 мл) и этилацетат (15 мл), и слои разделяли. Водную фазу экстрагировали этилацетатом (15 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 10:1 ~ 1:1), получая WX014-6. 1H ЯМР (400 МГц, CDCl3) δ = 7.40 (ушир.с, 1 H), 6.76 - 6.87 (м, 2 H), 5.46 (ушир.д, J = 7.46 Гц, 1 H), 4.22 - 4.37 (м, 2 H), 2.88 - 3.03 (м, 1 H), 2.63 - 2.73 (м, 2 H), 1.42 (с, 9 H).
Стадия 6: синтез соединения WX014-7
Реагент Лоуссона (77.70 мг, 192.12 мкмоль, 1.5 экв.) добавляли в раствор WX014-6 (40 мг, 128.08 мкмоль, 1 экв.) в толуоле (5 мл) при 20°C, и смесь нагревали при 70°C в течение 12 часов. Реакционный раствор упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 10:1 ~ 1:1), получая WX014-7. LCMS m/z = 273.1 [M-56+1]+.
Стадия 7: синтез соединения WX014-8
HCl/EtOAc (4M, 2 мл) добавляли в раствор WX014-7 в этилацетате (3 мл), и смесь нагревали при 25°C в течение 2 часов. Реакционный раствор упаривали при пониженном давлении (водоструйный насос, 40°C), получая сырой продукт WX014-8. LCMS m/z = 229.2 [M+1]+.
Стадия 8: синтез соединения WX014
DIPEA (18.31 мг, 141.66 мкмоль, 24.67 мкл, 2.5 экв.) и T3P (54.09 мг, 84.99 мкмоль, 50.55 мкл, 50% чистота, 1.5 экв.) добавляли в раствор WX014-8 (15 мг, 56.66 мкмоль, 1 экв., HCl) и BB-1 (14.97 мг, 73.66 мкмоль, 1.3 экв.) в ДМФА (2 мл), и смесь нагревали при 25°C в течение 2 часов. Добавляли в реакционный раствор воду (5 мл) и этилацетат (5 мл), и слои разделяли. Водную фазу экстрагировали этилацетатом (5 мл*2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150мм*30мм*5мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая WX014. LCMS m/z = 414.2 [M+1]+.1H ЯМР (400 МГц, CD3OD) δ = 7.21 - 7.37 (м, 5 H), 6.97 - 7.13 (м, 2 H), 4.71 (ушир.д, J = 4.14 Гц, 1 H), 4.18 (ушир.с, 2 H), 2.84 - 2.96 (м, 1 H), 2.64 - 2.83 (м, 2 H), 2.17 (тд, J = 11.67, 7.78 Гц, 1 H).
Пример 15. WX015
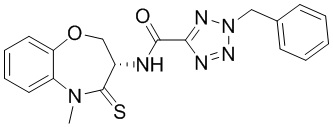
Путь синтеза:
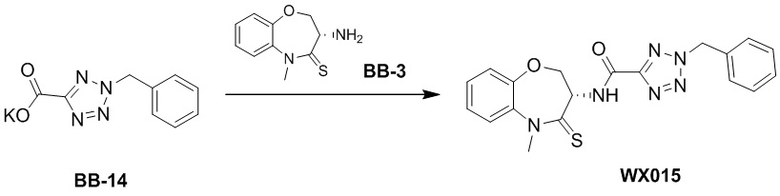
Стадия 1: синтез соединения WX015
В круглодонную колбу, содержащую N,N-диметилформамид (10 мл), добавляли BB-3 (20 мг, 96.02 мкмоль, 1.1 экв.) и DIPEA (23.69 мг, 183.32 мкмоль, 31.93 мкл, 2.1 экв.). После растворения добавляли BB-14 (21.24 мг, 87.30 мкмоль, 1 экв.) и T3P (55.55 мг, 87.30 мкмоль, 51.92 мкл, 50% чистота, 1 экв.), и затем смесь оставляли для прохождения реакции при перемешивании при 25°C на 12 часов. LCMS анализ показал практически полное исчезновение исходных веществ. Реакционный раствор упаривали при пониженном давлении до объема примерно 5 мл. ВЭЖХ (Колонка: Welch Xtimate C18 150 мм*25 мм*5 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин) использовали для очистки и получали продукт WX015. 1H ЯМР (400 МГц, CDCl3) δ = 8.78 (ушир.д, J = 7.5 Гц, 1H), 7.43 - 7.35 (м, 5H), 7.35 - 7.26 (м, 4H), 5.82 (с, 2H), 5.21 (тд, J = 6.9, 11.0 Гц, 1H), 4.71 (дд, J = 6.5, 9.5 Гц, 1H), 4.33 - 4.21 (м, 1H), 3.87 (с, 3H); LCMS m/z = 395.1 [M+1]+.
Пример 16. WX016
Путь синтеза:
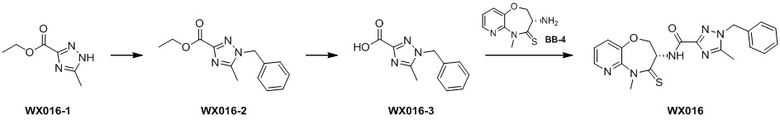
Стадия 1: синтез соединения WX016-2
В круглодонную колбу помещали ДМСО (10 мл) и затем медленно добавляли WX016-1 (1.31 г, 8.44 ммоль, 1 экв.), K2CO3 (1.40 г, 10.13 ммоль, 1.2 экв.) и бензилбромид (1.59 г, 9.29 ммоль, 1.10 мл, 1.1 экв.). Смесь непрерывно перемешивали при 25°C в течение 12 часов. После окончания реакции добавляли в реакционный раствор воду (50 мл), и смесь экстрагировали два раза этилацетатом (30 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (5 мл × 2), и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 100:1~1:1), получая WX016-2. 1H ЯМР (400 МГц, CDCl3) δ = 7.39 - 7.30 (м, 3H), 7.19 (дд, J = 1.8, 7.6 Гц, 2H), 5.39 (с, 2H), 4.49 (кв, J = 7.1 Гц, 2H), 2.43 (с, 3H), 1.44 (т, J = 7.1 Гц, 3H); LCMS m/z = 246.1 [M+1]+.
Стадия 2: синтез соединения WX016-3
В круглодонную колбу помещали ТГФ (7.5 мл) и H2O (2.5 мл) и затем медленно добавляли WX016-2 (1 г, 4.08 ммоль, 1 экв.) и LiOH⋅H2O (513.26 мг, 12.23 ммоль, 3 экв.). Смесь оставляли для прохождения реакции при перемешивании при 25°C на 2 часа. Реакционный раствор упаривали при пониженном давлении и добавляли по каплям 2н. раствор соляной кислоты, доводя pH до кислого. Смесь фильтровали, получая осадок на фильтре, который представлял собой целевое соединение WX016-3. 1H ЯМР (400 МГц, CDCl3) δ = 7.42 - 7.28 (м, 3H), 7.25 - 7.19 (м, 2H), 5.40 (с, 2H), 2.56 (с, 3H); LCMS m/z = 217.9 [M+1]+.
Стадия 3: синтез соединения WX016
В круглодонную колбу помещали N,N-диметилформамид (1 мл), WX016-3 (80 мг, 368.28 мкмоль, 1 экв.) и BB-4 (77.07 мг, 368.28 мкмоль, 1 экв.), затем добавляли T3P (351.54 мг, 552.43 мкмоль, 328.55 мкл, 50% чистота, 1.5 экв.) и DIPEA (142.79 мг, 1.10 ммоль, 192.45 мкл, 3 экв.). Смесь непрерывно перемешивали при 25°C в течение 5 часов, затем упаривали, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Welch Xtimate C18 150 мм*25 мм*5 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин), получая соединение WX016. 1H ЯМР (400 МГц, CDCl3) δ = 8.72 (ушир.д, J = 7.3 Гц, 1H), 8.37 (дд, J = 1.4, 4.6 Гц, 1H), 7.56 (дд, J = 1.3, 8.0 Гц, 1H), 7.38 - 7.27 (м, 4H), 7.18 (ушир.д, J = 6.0 Гц, 2H), 5.36 (с, 2H), 5.23 (тд, J = 6.8, 11.0 Гц, 1H), 4.78 (дд, J = 6.5, 9.5 Гц, 1H), 4.32 (дд, J = 9.8, 11.0 Гц, 1H), 3.93 (с, 3H), 2.41 (с, 3H); LCMS m/z = 409.1 [M+1]+.
Пример 17. WX017
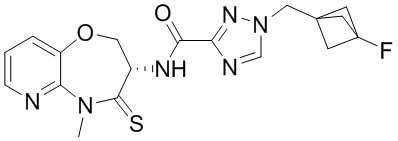
Путь синтеза:
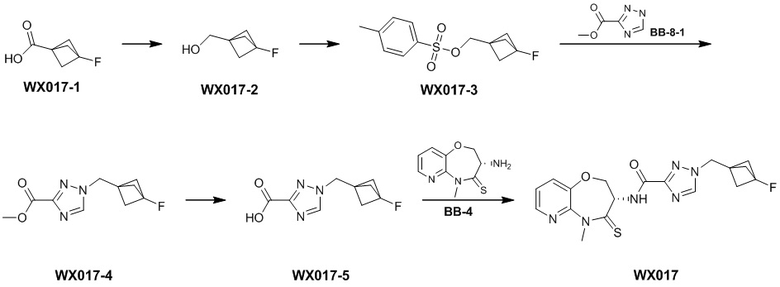
Стадия 1: синтез соединения WX017-2
В реакционную колбу помещали литийалюминий гидрид (629.99 мг, 16.60 ммоль, 2 экв.) и ТГФ (5 мл) при 25°C, затем медленно добавляли по каплям раствор WX017-1 (1.08 г, 8.30 ммоль, 1 экв.) в ТГФ (5 мл). После окончания добавления смесь нагревали до 35°C и непрерывно перемешивали в течение 1 часа. Реакцию гасили добавлением 6 мл насыщенного водного раствора Na2SO4. Смесь фильтровали и промывали этилацетатом (10 мл × 3). Органические фазы объединяли, сушили над безводным MgSO4 и фильтровали. Фильтрат упаривали при пониженном давлении. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~5:1), получая соединение WX017-2. 1H ЯМР (400 МГц, CDCl3) δ = 3.84 (с, 2H), 2.01 (д, J = 2.5 Гц, 6H).
Стадия 2: синтез соединения WX017-3
В реакционную колбу помещали WX017-2, ДХМ (10 мл) и пиридин (1.40 г, 17.70 ммоль, 1.43 мл, 4.11 экв.), затем медленно добавляли п-толуолсульфонилхлорид (1.23 г, 6.46 ммоль, 1.5 экв.). Смесь нагревали до 40°C и оставляли для прохождения реакции на 24 часа. Новое пятно (Rf = 0.7) появлялось в ТСХ (петролейный эфир:этилацетат = 5:1). Реакционный раствор сразу упаривали при пониженном давлении, и сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~30:1), получая соединение WX017-3. 1H ЯМР (400 МГц, CDCl3) δ = 7.78 (д, J = 8.3 Гц, 2H), 7.35 (д, J = 8.0 Гц, 2H), 4.24 (с, 2H), 2.46 (с, 3H), 1.99 (д, J = 2.3 Гц, 6H).
Стадия 3: синтез соединения WX017-4
В грушевидную колбу помещали ацетонитрил (10 мл), WX017-3 (500 мг, 1.85 ммоль, 1.1 экв.) и BB-8-1 (213.72 мг, 1.68 ммоль, 1 экв.) при 25°C, затем добавляли K2CO3 (278.88 мг, 2.02 ммоль, 1.2 экв.). Смесь нагревали до 70°C и оставляли для прохождения реакции на 48 часов. Реакционный раствор упаривали, и сырой продукт очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир:этилацетат = 100:1~1:2), получая соединение WX017-4. 1H ЯМР (400 МГц, CDCl3) δ = 8.12 (с, 1H), 4.57 (с, 2H), 4.01 (с, 3H), 2.05 (д, J = 2.5 Гц, 6H).
Стадия 4: синтез соединения WX017-5
В круглодонную колбу, содержащую ТГФ (2 мл), добавляли WX017-4 (100 мг, 444.01 мкмоль, 1 экв.) при 25°C. После растворения добавляли раствор LiOH⋅H2O (55.90 мг, 1.33 ммоль, 3 экв.) в MeOH (1 мл) по каплям, и смесь нагревали при 25°C в течение 5 часов. Смесь упаривали при пониженном давлении, и остаток растворяли в тетрагидрофуране (5 мл). Добавляли 1н. раствор соляной кислоты, доводя pH до 4, затем добавляли толуол (5 мл). Смесь хорошо перемешивали и упаривали досуха растворитель при пониженном давлении, получая сырой продукт WX017-5, который напрямую использовали в следующей стадии без очистки.
Стадия 5: синтез соединения WX017
В реакционную колбу помещали этилацетат (10 мл), WX017-5 (207 мг, 588.09 мкмоль, 1 экв.) и BB-4 (143.52 мг, 588.09 мкмоль, 1 экв.) при 25°C. После растворения добавляли T3P (1.12 г, 1.76 ммоль, 1.05 мл, 50% чистота, 3 экв.) и DIPEA (228.02 мг, 1.76 ммоль, 307.30 мкл, 3 экв.), и смесь нагревали при 25°C в течение 8 часов. После окончания реакции добавляли этилацетат (10 мл). Органическую фазу промывали водой (10 мл × 3), затем насыщенным водным раствором хлорида натрия (10 мл × 1) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Welch Xtimate C18 150 мм*25 мм*5 мкм; Подвижная фаза: [вода (0.05% NH3H2O)-ACN]; ацетонитрил%: 35%-65%, 8 мин), получая соединение WX017. 1H ЯМР (400 МГц, CDCl3) δ = 8.83 (ушир.д, J = 6.8 Гц, 1H), 8.37 (дд, J = 1.4, 4.6 Гц, 1H), 8.08 (с, 1H), 7.57 (дд, J = 1.3, 8.0 Гц, 1H), 7.29 (дд, J = 4.8, 8.0 Гц, 1H), 5.20 (тд, J = 6.7, 11.2 Гц, 1H), 4.80 (дд, J = 6.4, 9.4 Гц, 1H), 4.54 (с, 2H), 4.33 (дд, J = 9.8, 11.0 Гц, 1H), 3.94 (с, 3H), 2.05 (д, J = 2.3 Гц, 6H); LCMS m/z = 403.2 [M+1]+.
Пример 18. WX018
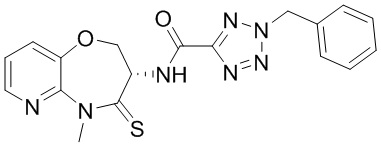
Путь синтеза:
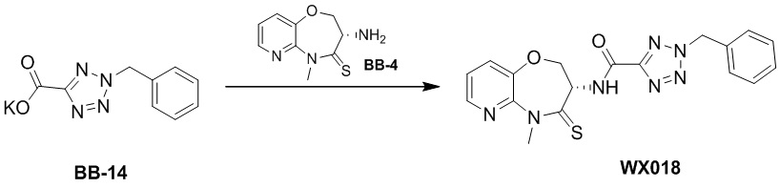
Синтез соединения WX018
В круглодонную колбу, содержащую N,N-диметилформамид (10 мл), добавляли BB-4 (200 мг, 955.71 мкмоль, 1.1 экв.) и DIPEA (235.80 мг, 1.82 ммоль, 317.80 мкл, 2.1 экв.). После растворения добавляли BB-14 (210.50 мг, 868.83 мкмоль, 1 экв.) и T3P (552.89 мг, 868.83 мкмоль, 516.72 мкл, 50% чистота, 1 экв.), и смесь перемешивали при 25°C в течение 12 часов. Реакционный раствор упаривали при пониженном давлении до объема примерно 5 мл. ВЭЖХ (Колонка: Welch Xtimate C18 150 мм*25 мм*5 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин) использовали для очистки и получали соединение WX018. 1H ЯМР (400 МГц, CDCl3) δ = 8.86 (ушир.д, J = 6.8 Гц, 1H), 8.38 (дд, J = 1.6, 4.6 Гц, 1H), 7.69 - 7.27 (м, 7H), 5.83 (с, 2H), 5.20 (тд, J = 6.7, 11.2 Гц, 1H), 4.78 (дд, J = 6.3, 9.5 Гц, 1H), 4.33 (дд, J = 9.7, 11.2 Гц, 1H), 3.93 (с, 3H); LCMS m/z = 396.1 [M+1]+.
Пример 19. WX019
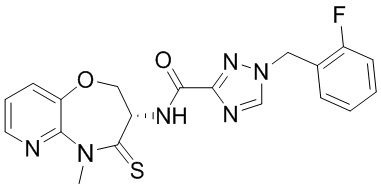
Путь синтеза:
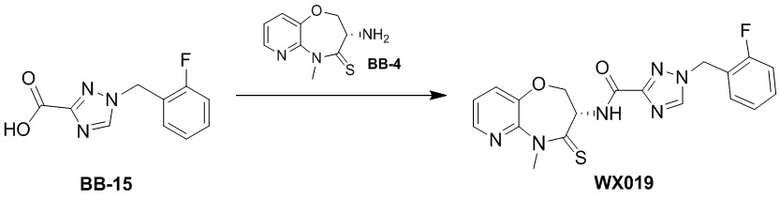
Стадия 1: синтез соединения WX019
В круглодонную колбу, содержащую N,N-диметилформамид (10 мл), добавляли BB-15 (156.11мг, 745.97 мкмоль, 1.1 экв.), BB-4 (150 мг, 678.16 мкмоль, 1 экв.), T3P (431.55 мг, 678.16 мкмоль, 403.32 мкл, 50% чистота, 1 экв.) и DIPEA (184.06 мг, 1.42 ммоль, 248.06 мкл, 2.1 экв.), затем начинали перемешивание. Смесь оставляли для прохождения реакции при перемешивании при 25°C в течение 12 часов. После окончания реакции раствор упаривали при пониженном давлении до объема примерно 5 мл. ВЭЖХ (Колонка: Welch Xtimate C18 150 мм*25 мм*5 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)- ACN]; ацетонитрил%: 35%-65%, 8 мин) использовали для очистки и получали продукт WX019. 1H ЯМР (400 МГц, CDCl3) δ = 8.81 (ушир.д, J = 7.3 Гц, 1H), 8.37(дд, J = 1.5, 4.8 Гц, 1H), 8.14 (с, 1H), 7.56 (дд, J = 1.5, 8.0 Гц, 1H), 7.41 - 7.27 (м, 3H), 7.21 - 7.06 (м, 2H), 5.45 (с, 2H), 5.20 (тд, J = 6.7, 11.2 Гц, 1H), 4.78 (дд, J = 6.5, 9.5 Гц, 1H), 4.31 (дд, J = 9.5, 11.0 Гц, 1H), 3.93 (с, 3H); 19F ЯМР (376 МГц, CDCl3) δ = -109.59 ~ -130.05 (м, 1F); LCMS m/z = 413.1 [M+1]+.
Пример 20. WX020
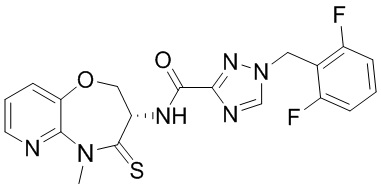
Путь синтеза:
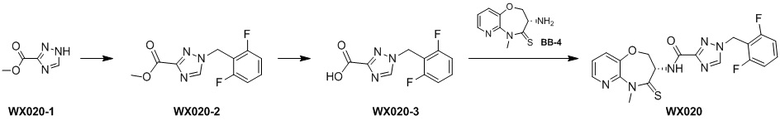
Стадия 1: синтез соединения WX020-2
В ацетонитрил (15 мл) добавляли WX020-1 (3 г, 23.60 ммоль, 1 экв.), 2,6-дифторбензилбромид (5.37 г, 25.96 ммоль, 1.1 экв.) и K2CO3 (3.91 г, 28.32 ммоль, 1.2 экв.), затем наичнали перемешивание. Смесь нагревали при 50°C в течение 12 часов. После окончания реакции по данным ТСХ-мониторинга, реакционный раствор фильтровали для удаления K2CO3, и осадок на фильтре промывали три раза этилацетатом (15 мл). Добавляли в фильтрат насыщенный водный раствор хлорида натрия (50 мл) и отделяли органическую фазу после экстракции. Водную фазу экстрагировали этилацетатом (30 мл) снова. Органические фазы объединяли и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир:этилацетат = 100:1~100:100), получая соединение WX020-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.17 (с, 1H), 7.46 - 7.33 (м, 1H), 7.04- 6.91 (м, 2H), 5.51 (с, 2H), 4.13 - 3.85 (м, 3H).
Стадия 2: синтез соединения WX020-3
В круглодонную колбу, содержащую ТГФ (30 мл), добавляли WX020-2 (1 г, 3.95 ммоль, 1 экв.). После растворения добавляли раствор LiOH⋅H2O (331.43 мг, 7.90 ммоль, 2 экв.) в H2O (5 мл), и смесь оставляли для прохождения реакции при перемешивании при 20°C в течение 2 часов. Реакционный раствор доводили до значения pH 6 добавлением 1М раствора соляной кислоты, и смесь упаривали при пониженном давлении для удаления растворителя, получая сырой продукт WX020-3; LCMS m/z = 239.9 [M+1]+.
Стадия 3: синтез соединения WX020
В круглодонную колбу, содержащую N,N-диметилформамид (10 мл), добавляли BB-4 (192.49 мг, 919.82 мкмоль, 1.1 экв.), WX020-3 (200 мг, 836.20 мкмоль, 1 экв.), T3P(532.12 мг, 836.20 мкмоль, 497.31 мкл, 50% чистота, 1 экв.) и DIPEA (226.95 мг, 1.76 ммоль, 305.86 мкл, 2.1 экв.), и смесь оставляли для прохождения реакции при перемешивании при 25°C в течение 12 часов. Реакционный раствор упаривали при пониженном давлении до объема примерно 5 мл, и остаток очищали методом препаративной ВЭЖХ (Колонка: Welch Xtimate C18 150 мм*25 мм*5 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин), получая соединение WX020. 1H ЯМР (400 МГц, CDCl3) δ = 8.79 (ушир.д, J = 7.0 Гц, 1H), 8.36 (дд, J = 1.5, 4.8 Гц, 1H), 8.14 (с, 1H), 7.55 (дд, J = 1.5, 8.0 Гц, 1H), 7.43 - 7.26 (м, 2H), 6.97 (т, J = 7.9 Гц, 2H), 5.49 (с, 2H), 5.19 (тд, J = 6.7, 11.1 Гц, 1H), 4.77 (дд, J = 6.3, 9.5 Гц, 1H), 4.30 (дд, J = 9.7, 10.9 Гц, 1H), 3.92 (с, 3H); 19F ЯМР (376 МГц, CDCl3) δ = -104.74 ~ -120.36 (м, 1F); LCMS m/z =431.1 [M+1]+.
Пример 21. WX021
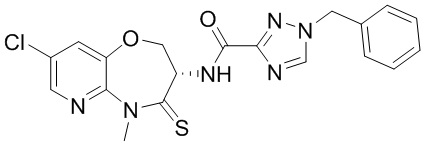
Путь синтеза:
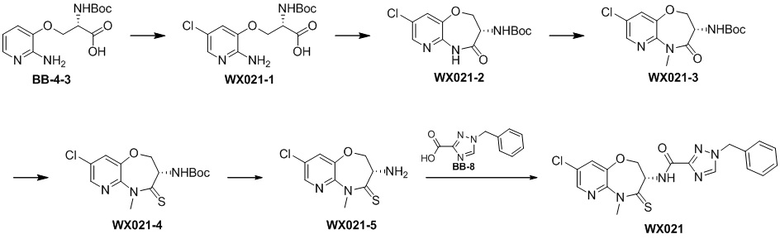
Стадия 1: синтез соединения WX021-1
В круглодонную колбу, содержащую ДХМ (20 мл), добавляли BB-3-4 (2 г, 6.73 ммоль, 1 экв.), и смесь перемешивали при 40°C. Медленно добавляли NCS (988.11 мг, 7.40 ммоль, 1.1 экв.), затем реакционную смесь непрерывно перемешивали при 40°C в течение 5 часов. Реакционный раствор упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (элюент: ДХМ:MeOH = 100:1~100:10), получая соединение WX021-1. 1H ЯМР (400 МГц, CD3OD) δ = 7.54 (д, J = 1.5 Гц, 1H), 7.34 (д, J = 1.8 Гц, 1H), 6.95 - 6.77 (м, 1H), 5.49 (с, 2H), 4.70 - 4.60 (м, 2H), 4.53 (ушир. дд, J = 4.3, 9.5 Гц, 1H), 4.39 - 4.26 (м, 1H), 1.46 (с, 9H). LCMS m/z = 331.9 [M+1]+.
Стадия 2: синтез соединения WX021-2
В круглодонную колбу, содержащую ДХМ (10 мл), добавляли WX021-1 (1.60 г, 4.82 ммоль, 1 экв.), T3P (6.14 г, 9.65 ммоль, 5.74 мл, 50% чистота, 2 экв.) и DIPEA (1.87 г, 14.47 ммоль, 2.52 мл, 3 экв.), и смесь нагревали при 20°C в течение 3 часов. Реакционный раствор выливали в насыщенный водный раствор хлорида натрия (300 мл) и слои разделяли, отделяя органическую фазу. Водную фазу экстрагировали два раза этилацетатом (100 мл). Органические фазы объединяли и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:20~100:100), получая соединение WX021-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.22 (ушир.с, 1H), 8.04 (д, J = 2.0 Гц, 1H), 7.41 (д, J = 2.3 Гц, 1H), 5.59 (ушир.с, 1H), 4.71 - 4.46 (м, 2H), 4.23 - 4.05 (м, 1H), 1.46 (с, 9H); LCMS m/z = 257.8 [M-56+1]+.
Стадия 3: синтез соединения WX021-3
В круглодонную колбу, содержащую N,N-диметилацетамид (10 мл), добавляли WX021-2 (200 мг, 637.48 мкмоль, 1 экв.), иодметан (1.17 г, 8.24 ммоль, 513.16 мкл, 12.93 экв.) и карбонат цезия (415.41 мг, 1.27 ммоль, 2 экв.). Смесь оставляли для прохождения реакции при перемешивании при 25°C в атмосфере азота на 1 час. После окончания реакции раствор выливали в насыщенный водный раствор хлорида натрия (20 мл), и смесь экстрагировали три раза этилацетатом (15 мл). Органические фазы объединяли и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт растворяли в этилацетате (2 мл) и очищали методом препаративной ТСХ (петролейный эфир:этилацетат = 3:1), получая продукт WX021-3. 1H ЯМР (400 МГц, CDCl3) δ = 8.23 (д, J = 2.3 Гц, 1H), 7.47 (д, J = 2.0 Гц, 1H), 5.55 (ушир.с, 1H), 4.82 - 4.49 (м, 2H), 4.35 - 4.14 (м, 1H), 3.47 (с, 3H), 1.41 (с, 9H).
Стадия 4: синтез соединения WX021-4
В круглодонную колбу, содержащую толуол (10 мл), добавляли WX021-3 (177 мг, 540.02 мкмоль, 1 экв.) и реагент Лоуссона (327.63 мг, 810.04 мкмоль, 1.5 экв.) при 25°C, смесь медленно нагревали при 110°C и оставляли для прохождения реакции на 12 часов. Реакционный раствор выливали в насыщенный водный раствор хлорида натрия (100 мл), и смесь экстрагировали три раза этилацетатом (30 мл). Органические фазы объединяли и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ТСХ (петролейный эфир:этилацетат = 3:1), получая соединение WX021-4. LCMS m/z = 287.8 [M-56+1]+.
Стадия 5: синтез соединения WX021-5
В круглодонную колбу, содержащую дихлорметан (5 мл), добавляли WX021-4 (35 мг, 101.80 мкмоль, 1 экв.) при 25°C. Добавляли по каплям трифторуксусную кислоту (174.10 мг, 1.53 ммоль, 113.06 мкл, 15 экв.) при перемешивании, и смесь оставляли для прохождения реакции на 2 часа. Реакционный раствор упаривали досуха при пониженном давлении, получая сырой продукт WX021-5, который напрямую использовали в следующей стадии.
Стадия 6: синтез соединения WX021
В круглодонную колбу, содержащую N,N-диметилацетамид (5 мл), добавляли WX021-5 (20 мг, 82.06 мкмоль, 1 экв.), BB-8 (16.68 мг, 82.06 мкмоль, 1 экв.), T3P (52.22 мг, 82.06 мкмоль, 48.81 мкл, 50% чистота, 1 экв.) и DIPEA (22.27 мг, 172.33 мкмоль, 30.02 мкл, 2.1 экв.), и смесь нагревали при 25°C в течение 5 часов. Добавляли насыщенный водный раствор хлорида натрия (30 мл), и смесь экстрагировали этилацетатом (30 мл × 3). Органическую фазу упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом ВЭЖХ (система с муравьиной кислотой) (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 40%-70%, 8 мин), получая соединение WX021. 1H ЯМР (400 МГц, CDCl3) δ = 8.77 (ушир.д, J = 7.3 Гц, 1H), 8.32 (д, J = 2.3 Гц, 1H), 8.04 (с, 1H), 7.57 (д, J = 2.0 Гц, 1H), 7.44 - 7.34 (м, 3H), 7.32 - 7.27 (м, 2H), 5.40 (с, 2H), 5.26 - 5.16 (м, 1H), 4.77 (дд, J = 6.3, 9.5 Гц, 1H), 4.39 - 4.27 (м, 1H), 3.90 (с, 3H); LCMS m/z = 429.0 [M+1]+.
Пример 22. WX022
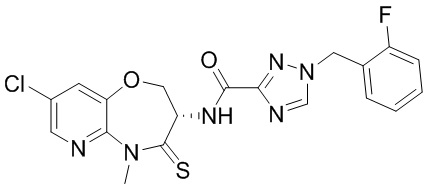
Путь синтеза:
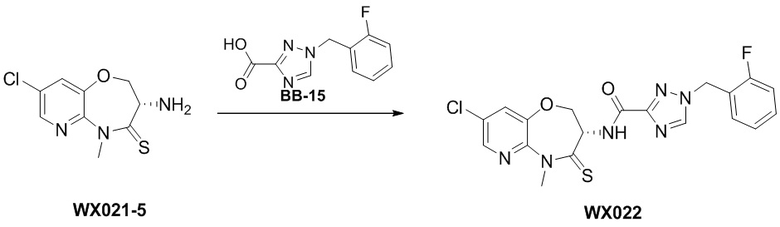
Стадия 1: синтез соединения WX022
В N,N-диметилацетамид (5 мл) добавляли WX021-5 (20 мг, 82.06 мкмоль, 1 экв.), BB-15 (18.15 мг, 82.06 мкмоль, 1 экв.), T3P (52.22 мг, 82.06 мкмоль, 48.81 мкл, 50% чистота, 1 экв.) и DIPEA (22.27 мг, 172.33 мкмоль, 30.02 мкл, 2.1 экв.), и смесь нагревали при 25°C в течение 5 часов. Добавляли 30 мл насыщенного водного раствора хлорида натрия, и смесь экстрагировали этилацетатом (30 мл × 3), получая органическую фазу. Органическую фазу упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 42%-72%, 8 мин), получая соединение WX022. 1H ЯМР (400 МГц, CDCl3) δ = 8.77 (ушир.д, J = 6.5 Гц, 1H), 8.32 (д, J = 2.0 Гц, 1H), 8.14 (с, 1H), 7.57 (д, J = 2.3 Гц, 1H), 7.41 - 7.31 (м, 2H), 7.20 - 7.07 (м, 2H), 5.45 (с, 2H), 5.30 - 5.08 (м, 1H), 4.76 (дд, J = 6.3, 9.5 Гц, 1H), 4.32 (т, J = 10.3 Гц, 1H), 3.93 - 3.87 (м, 1H); 19F ЯМР (376 МГц, CDCl3) δ = -110.66 ~ -127.90 (м, 1F); LCMS m/z = 447.1[M+1]+.
Пример 23. WX023
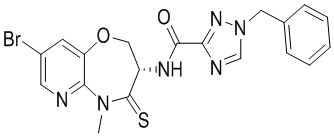
Путь синтеза:
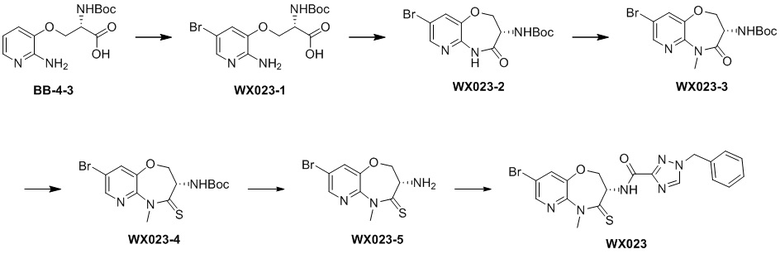
Стадия 1: синтез соединения WX023-1
BB-4-3 (3.00 г, 10.09 ммоль, 1 экв.) и N-бромсукцинимид (1.98 г, 11.10 ммоль, 1.1 экв.) медленно добавляли в ДХМ (30 мл) при 25°C, и реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 12 часов. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали дихлорметаном (100 мл × 2). Органические фазы объединяли и последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), получая органическую фазу. Органическую фазу упаривали при пониженном давлении. Сырой продукт очищали методом колоночной хроматографии на силикагеле (дихлорметан:метанол = 100:1~100:5), получая соединение WX023-1.
Стадия 2: синтез соединения WX023-2
В круглодонную колбу помещали этилацетат (100 мл) при 25°C. Затем медленно добавляли WX023-1 (5.60 г, 14.89 ммоль, 1 экв.), T3P (14.21 г, 22.33 ммоль, 13.28 мл, 50% чистота, 1.5 экв.) и DIPEA (3.85 г, 29.77 ммоль, 5.19 мл, 2 экв.). Реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 1 час. Реакционный раствор медленно выливали в воду (200 мл), и смесь экстрагировали этилацетатом (200 мл × 2). Органические фазы объединяли и последовательно промывали водой (200 мл) и насыщенным водным раствором хлорида натрия (200 мл), получая органическую фазу. Органическую фазу упаривали при пониженном давлении. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX023-2.
Стадия 3: синтез соединения WX023-3
В круглодонную колбу помещали N,N-диметилформамид (10 мл) при 25°C. Затем медленно добавляли WX023-2 (1.80 г, 5.03 ммоль, 1 экв.), иодметан (5.71 г, 40.23 ммоль, 2.50 мл, 8.01 экв.) и карбонат цезия (3.60 г, 11.06 ммоль, 2.2 экв.). Реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 1 час. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли и последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл). Органическую фазу упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~3:1), получая соединение WX023-3. 1H ЯМР (400 МГц, CDCl3) δ = 8.32 (д, J = 2.0 Гц, 1H), 7.61 (д, J = 2.3 Гц, 1H), 5.55 (ушир.с, J = 4.8 Гц, 1H), 4.79 - 4.44 (м, 2H), 4.34 - 4.11 (м, 1H), 3.46 (с, 3H), 1.40 (с, 9H).
Стадия 4: синтез соединения WX023-4
В круглодонную колбу помещали толуол (50 мл) при 25°C, затем медленно добавляли WX023-3 (1.35 г, 3.63 ммоль, 1 экв.) и реагент Лоуссона (2.20 г, 5.44 ммоль, 1.5 экв.). Реакционный раствор непрерывно перемешивали при 110°C в атмосфере азота 11 часов. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли и последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл). Органическую фазу упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX023-4. 1H ЯМР (400 МГц, CDCl3) δ = 8.38 (д, J = 2.0 Гц, 1H), 7.65 (д, J = 2.3 Гц, 1H), 6.10 (ушир.д, J = 7.3 Гц, 1H), 4.81 - 4.67 (м, 1H), 4.58 (дд, J = 6.1, 9.4 Гц, 1H), 4.23 (дд, J = 9.5, 11.3 Гц, 1H), 3.87 (с, 3H), 1.41 (с, 9H).
Стадия 5: синтез соединения WX023-5
В круглодонную колбу помещали дихлорметан (40 мл) при 25°C, затем медленно добавляли WX023-4 (980 мг, 2.52 ммоль, 1 экв.) и трифторуксусную кислоту (6.16 г, 54.02 ммоль, 4, 21.40 экв.). Реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 11 часов. Реакционный раствор медленно выливали в насыщенный водный раствор бикарбоната натрия (100 мл), и смесь экстрагировали два раза дихлорметаном (100 мл × 2). Органические фазы объединяли и последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл). Органическую фазу упаривали при пониженном давлении, получая сырое соединение WX023-5, которое напрямую использовали в следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ = 8.32 (д, J = 2.0 Гц, 1H), 7.59 (д, J = 2.3 Гц, 1H), 4.40 (дд, J = 6.3, 10.0 Гц, 1H), 4.10 (т, J = 10.8 Гц, 1H), 3.88 - 3.71 (м, 4H).
Стадия 6: синтез соединения WX023
В круглодонную колбу помещали N,N-диметилацетамид (5 мл) при 25°C. Затем медленно добавляли WX023-5 (288 мг, 999.43 мкмоль, 1 экв.), T3P (954.00 мг, 1.50 ммоль, 891.59 мкл, 50% чистота, 1.5 экв.), BB-8 (406.16 мг, 2.00 ммоль, 2 экв.) и DIPEA (387.50 мг, 3.00 ммоль, 522.24 мкл, 3 экв.). Реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 2 часа. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли и последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл). Органическую фазу упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 42%-72%, 8 мин), получая соединение WX023. 1H ЯМР (400 МГц, CDCl3) δ = 8.77 (ушир.д, J = 7.0 Гц, 1H), 8.42 (д, J = 2.0 Гц, 1H), 8.04 (с, 1H), 7.71 (д, J = 2.0 Гц, 1H), 7.45 - 7.33 (м, 3H), 7.33 - 7.27 (м, 2H), 5.40 (с, 2H), 5.21 (тд, J = 6.7, 11.3 Гц, 1H), 4.77 (дд, J = 6.3, 9.5 Гц, 1H), 4.33 (дд, J = 9.8, 11.0 Гц, 1H), 3.90 (с, 3H); LCMS m/z =475.0 [M+1]+.
Пример 24. WX024
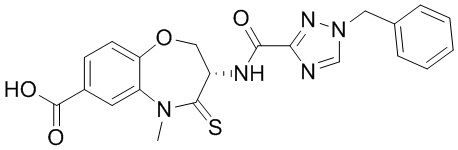
Путь синтеза:
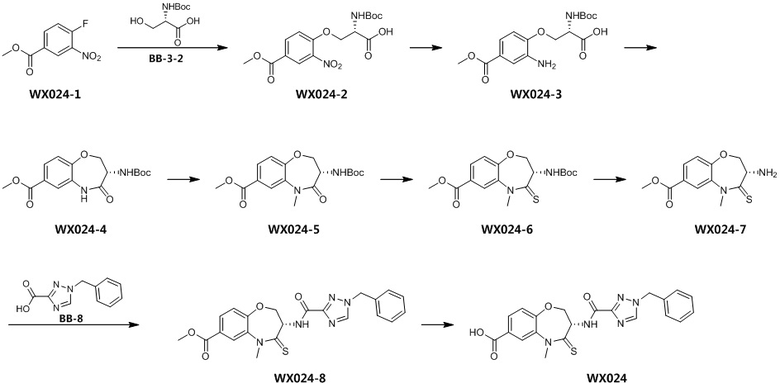
Стадия 1: синтез соединения WX024-2
В круглодонную колбу помещали ТГФ (100 мл). Затем медленно добавляли WX024-1 (10 г, 50.22 ммоль, 1 экв.), BB-3-2 (11.34 г, 55.24 ммоль, 1.1 экв.) и карбонат цезия (29.45 г, 90.39 ммоль, 1.8 экв.). Смесь непрерывно перемешивали при 65°C в течение 16 часов. Реакционный раствор выливали в воду (300 мл), затем добавляли 2М раствор соляной кислоты, доводя pH до 3. Смесь экстрагировали этилацетатом (500 мл × 2). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия (50 мл × 2). Органическую фазу упаривали при пониженном давлении, получая соединение WX024-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.46 (д, J = 1.8 Гц, 1H), 8.20 (дд, J = 2.0, 8.8 Гц, 1H), 7.12 (д, J = 8.8 Гц, 1H), 5.62 (ушир.д, J = 8.0 Гц, 1H), 4.83 - 4.61 (м, 2H), 4.45 (дд, J = 3.0, 9.3 Гц, 1H), 3.93 (с, 3H), 1.45 (с, 10H).
Стадия 2: синтез соединения WX024-3
В колбу для гидрирования добавляли MeOH (300 мл). Затем медленно добавляли WX024-2 (25.8 г, 67.13 ммоль, 1 экв.) и Pd/C (2.6 г, 26.02 ммоль, 10% чистота). Три раза заменяли атмосферу в колбе на аргон, и смесь непрерывно перемешивали при 30°C в атмосфере водорода (40 Фунт/кв. дюйм) 24 часа. Новое пятно появлялось в ТСХ (петролейный эфир:этилацетат:уксусная кислота = 1:1:0.1). Реакционный раствор фильтровали через целит, и фильтрат упаривали при пониженном давлении, получая соединение WX024-3. 1H ЯМР (400 МГц, CDCl3) δ = 7.50 - 7.44 (м, 1H), 7.39 (ушир.с, 1H), 6.70 (д, J = 8.5 Гц, 1H), 6.53 (ушир.с, 2H), 6.00 (ушир.д, J = 7.5 Гц, 1H), 4.73 (ушир.д, J = 7.0 Гц, 1H), 4.43 (ушир.д, J = 8.5 Гц, 1H), 4.31 - 4.24 (м, 1H), 3.85 (с, 3H), 1.41 (с, 9H).
Стадия 3: синтез соединения WX024-4
В круглодонную колбу помещали этилацетат (200 мл). Затем медленно добавляли WX024-3, T3P (33.67 г, 52.91 ммоль, 31.47 мл, 50% чистота, 1.5 экв.) и DIEA (13.68 г, 105.83 ммоль, 18.43 мл, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор экстрагировали водой (200 мл), и слои разделяли. Водную фазу экстрагировали этилацетатом (200 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (100 мл × 2) и упаривали при пониженном давлении. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX024-4. 1H ЯМР (400 МГц, CDCl3) δ = 8.15 (ушир.с, 1H), 7.80 (дд, J = 2.0, 8.5 Гц, 1H), 7.73 (д, J = 2.0 Гц, 1H), 7.13 (д, J = 8.3 Гц, 1H), 5.62 (ушир.д, J = 5.3 Гц, 1H), 4.71 - 4.59 (м, 2H), 4.33 - 4.23 (м, 1H), 3.95 - 3.89 (м, 3H), 1.43 (с, 9H).
Стадия 4: синтез соединения WX024-5
В круглодонную колбу помещали N,N-диметилформамид (200 мл). Затем медленно добавляли WX024-4 (17 г, 50.54 ммоль, 1 экв.), карбонат цезия (49.40 г, 151.63 ммоль, 3 экв.) и иодметан (21.52 г, 151.63 ммоль, 9.44 мл, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Воду (300 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (300 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (100 мл × 2) и упаривали при пониженном давлении. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX024-5. 1H ЯМР (400 МГц, CDCl3) δ = 7.92 - 7.83 (м, 2H), 7.18 (д, J = 8.4 Гц, 1H), 5.50 (ушир.д, J = 6.1 Гц, 1H), 4.72 - 4.52 (м, 2H), 4.28 - 4.17 (м, 1H), 3.92 (с, 3H), 3.42 (с, 3H), 1.38 (с, 9H).
Стадия 5: синтез соединения WX024-6
В круглодонную колбу помещали толуол (40 мл). Затем медленно добавляли WX024-5 (4.01 г, 11.45 ммоль, 1 экв.), реагент Лоуссона (5.09 г, 12.59 ммоль, 1.1 экв.) и Boc2O (10.60 г, 48.55 ммоль, 11.15 мл, 4.24 экв.). Смесь нагревали до 110°C и перемешивали в течение 12 часов. Реакционный раствор медленно выливали в насыщенный водный раствор хлорида натрия (30 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (30 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX024-6. 1H ЯМР (400 МГц, CDCl3) δ = 7.99 - 7.92 (м, 2H), 7.21 (д, J = 8.3 Гц, 1H), 6.02 (ушир.д, J = 7.8 Гц, 1H), 4.80 - 4.69 (м, 1H), 4.53 (дд, J = 6.3, 9.5 Гц, 1H), 4.22 (дд, J = 9.5, 11.3 Гц, 1H), 3.94 (с, 3H), 3.86 (с, 3H), 1.46 - 1.33 (м, 9H).
Стадия 6: синтез соединения WX024-7
В круглодонную колбу помещали этилацетат (10 мл). Затем медленно добавляли WX024-6 (500 мг, 1.36 ммоль, 1 экв.) и HCl/этилацетат (4M, 10 мл, 29.31 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор упаривали при пониженном давлении, получая соединение WX024-7. 1H ЯМР (400 МГц, CDCl3) δ = 8.04 - 7.89 (м, 2H), 7.28 (ушир.с, 1H), 4.93 (ушир.с, 1H), 4.63 (ушир.с, 1H), 4.44 (ушир.с, 1H), 3.93 (с, 3H), 3.88 - 3.85 (м, 1H), 3.81 (с, 3H).
Стадия 7: синтез соединения WX024-8
В круглодонную колбу помещали N,N-диметилформамид (5 мл). Затем медленно добавляли WX024-7 (245 мг, 809.18 мкмоль, 1 экв., HCl), BB-8 (164.42 мг, 809.18 мкмоль, 1 экв.), HATU (461.51 мг, 1.21 ммоль, 1.5 экв.) и DIPEA (313.74 мг, 2.43 ммоль, 422.83 мкл, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор выливали в воду (20 мл), и смесь экстрагировали этилацетатом (20 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (5 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:100), получая соединение WX024-8. 1H ЯМР (400 МГц, CDCl3) δ = 8.70 (ушир.д, J = 7.3 Гц, 1H), 8.02 - 7.94 (м, 3H), 7.42 - 7.34 (м, 3H), 7.31 - 7.28 (м, 2H), 5.39 (с, 2H), 5.20 (тд, J = 6.8, 11.0 Гц, 1H), 4.72 (дд, J = 6.5, 9.4 Гц, 1H), 4.31 (т, J = 10.3 Гц, 1H), 3.95 (с, 3H), 3.89 (с, 3H).
Стадия 8: синтез соединения WX024
В круглодонную колбу помещали ТГФ (4 мл) и H2O (1 мл). Затем медленно добавляли WX024-8 (50 мг, 110.74 мкмоль, 1 экв.) и LiOH·H2O (4.65 мг, 110.74 мкмоль, 12.79 мкл, 1 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор очищали методом ВЭЖХ (Колонка: Phenomenex Gemini-NX 80 мм*40 мм*3 мкм; Подвижная фаза: [вода (0.05% NH3⋅H2O + 10 мM NH4HCO3) -ACN]; ацетонитрил%: 11%-31%, 8 мин), получая соединение WX024. 1H ЯМР (400 МГц, CDCl3) δ = 8.78 (ушир.с, 1H), 8.18 - 7.82 (м, 3H), 7.36 (ушир.с, 5H), 5.64 - 5.32 (м, 2H), 5.26 (ушир.с, 1H), 4.72 (ушир.с, 1H), 4.32 (ушир.с, 1H), 3.86 (ушир.с, 3H); LCMS m/z = 438.1 [M+1]+.
Пример 25. WX025
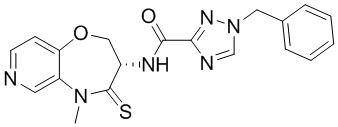
Путь синтеза:
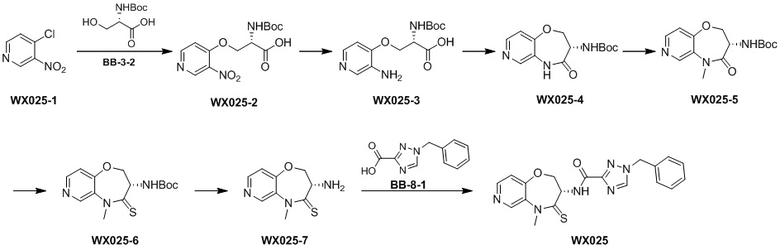
Стадия 1: синтез соединения WX025-2
NaH (2.78 г, 69.38 ммоль, 60% чистота, 2.2 экв.) добавляли в раствор BB-3-2 (7.12 г, 34.69 ммоль, 1.1 экв.) в N,N-диметилформамиде (80 мл) при 0°C в атмосфере азота, и смесь оставляли для прохождения реакции на 2 часов. Реакционный раствор помещали в ледяную баню и добавляли WX025-1 (5 г, 31.54 ммоль, 1 экв.). Реакционный раствор нагревали до 25°C и оставляли для прохождения реакции на 12 часов. Смесь медленно выливали в ледяную воду (100 мл) и экстрагировали этилацетатом (150 мл × 2). Водную фазу доводили до значения pH 5~6 добавлением 1M раствора HCl и экстрагировали этилацетатом (150 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая соединение WX025-2. LCMS m/z = 328.1 [M+1]+.
Стадия 2: синтез соединения WX025-3
Pd/C (3 г, 5% чистота) добавляли в раствор WX025-2 (9 г, 27.50 ммоль, 1 экв.) в этилацетате (60 мл) и метаноле (30 мл). Атмосферу в колбе три раза заменяли на H2, и смесь нагревали при 25°C в атмосфере водорода (15 Фунт/кв. дюйм) в течение 12 часов. Реакционный раствор фильтровали через целит, и фильтрат упаривали при пониженном давлении, получая соединение WX025-3. LCMS m/z = 298.1 [M+1]+.
Стадия 3: синтез соединения WX025-4
T3P (20.87 г, 32.79 ммоль, 19.50 мл, 50% чистота, 1.5 экв.) добавляли в раствор DIPEA (7.06 г, 54.66 ммоль, 9.52 мл, 2.5 экв.) и WX025-3 (6.5 г, 21.86 ммоль, 1 экв.) в N,N-диметилформамиде (200 мл), и смесь нагревали при 25°C в течение 3 часов. Воду (200 мл) добавляли в реакционный раствор на ледяной бане, и смесь экстрагировали этилацетатом (200 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 10:1~1:2), получая соединение WX025-4. 1H ЯМР (400 МГц, CDCl3) δ = 8.37 (ушир.с, 1 H), 8.20 - 8.31 (м, 2 H), 6.97 (д, J = 5.70 Гц, 1 H), 5.69 (ушир.д, J = 4.38 Гц, 1 H), 4.54 - 4.71 (м, 2 H), 4.22 - 4.35 (м, 1 H), 1.45 - 1.52 (м, 9 H); LCMS m/z = 280.1[M+1]+.
Стадия 4: синтез соединения WX025-5
Иодметан (384.21 мг, 2.71 ммоль, 168.51 мкл, 1.05 экв.) добавляли в раствор WX025-4 (0.72 г, 2.58 ммоль, 1 экв.) и карбоната цезия (1.01 г, 3.09 ммоль, 1.2 экв.) в тетрагидрофуране (14 мл), и смесь нагревали при 20°C в течение 12 часов. Добавляли в реакционный раствор воду (25 мл) и этилацетат (25 мл), и слои разделяли. Водную фазу экстрагировали этилацетатом (25 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (дихлорметан:метанол = 10:0~10:1), получая соединение WX025-5. 1H ЯМР (400 МГц, CDCl3) δ = 8.50 (с, 1 H), 8.39 (д, J = 5.27 Гц, 1 H), 7.06 (д, J = 5.40 Гц, 1 H), 5.54 (ушир.д, J = 6.02 Гц, 1 H), 4.56 - 4.73 (м, 2 H), 4.25 - 4.37 (м, 1 H), 3.47 (с, 3 H), 1.41 (с, 9 H); LCMS m/z = 294.1 [M+1]+.
Стадия 5: синтез соединения WX025-6
Реагент Лоуссона (110.32 мг, 272.74 мкмоль, 0.8 экв.) добавляли в раствор WX025-5 (0.1 г, 340.93 мкмоль, 1 экв.) в толуоле (2 мл) при 20°C, и смесь нагревали при 90°C в течение 12 часов. Этилацетат (5 мл) добавляли в реакционный раствор. Смесь фильтровали, и фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии (петролейный эфир:этилацетат = 10:1~1:2), получая соединение WX025-6. LCMS m/z = 310.1 [M+1]+.
Стадия 6: синтез соединения WX025-7
HCl/этилацетат (3 мл) добавляли в раствор WX025-6 (53 мг, 171.31 мкмоль, 1 экв.) в этилацетате (3 мл), и смесь нагревали при 25°C в течение 2 часов. Реакционный раствор упаривали при пониженном давлении, получая соединение WX025-7. LCMS m/z = 210.1 [M+1]+.
Стадия 7: синтез соединения WX025
DIPEA (77.82 мг, 602.10 мкмоль, 104.87 мкл, 3 экв.) и T3P (127.72 мг, 401.40 мкмоль, 119.36 мкл, 2 экв.) добавляли в раствор WX025-7 (42 мг, 200.70 мкмоль, 1 экв.) и BB-8 (61.17 мг, 301.05 мкмоль, 1.5 экв.) в N,N-диметилформамиде (3 мл), и смесь нагревали при 25°C в течение 2 часов. Добавляли в реакционный раствор воду (5 мл) и этилацетат (5 мл), и слои разделяли. Водную фазу экстрагировали этилацетатом (5 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX025. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.83 (д, J = 8.16 Гц, 2 H), 8.64 - 8.65 (м, 1 H), 8.67 (д, J = 7.72 Гц, 1 H), 8.54 (д, J = 5.28 Гц, 1 H), 7.26 - 7.42 (м, 6 H), 5.49 (с, 2 H), 5.04 (дт, J = 11.24, 6.84 Гц, 1 H), 4.44 - 4.64 (м, 2 H), 3.83 (с, 3 H); LCMS m/z = 395.2 [M+1]+.
Пример 26. WX026
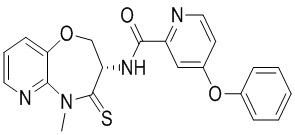
Путь синтеза:
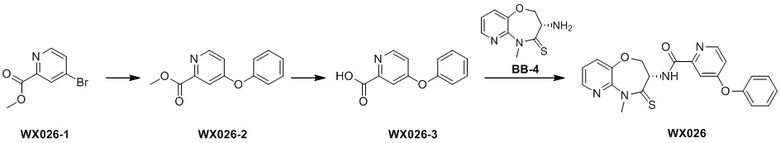
Стадия 1: синтез соединения WX026-2
В раствор WX026-1 (1 г, 4.63 ммоль, 1 экв.) и фенола (871.27 мг, 9.26 ммоль, 814.27 мкл, 2 экв.) в N,N-диметилформамиде (10 мл) добавляли карбонат калия (1.28 г, 9.26 ммоль, 2 экв.), бромид меди (66.40 мг, 462.89 мкмоль, 14.10 мкл, 0.1 экв.) и ацетилацетон (23.17 мг, 231.45 мкмоль, 23.77 мкл, 0.05 экв.), и смесь перемешивали при 90°C в течение 10 часов. Воду (100 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали, фильтрат упаривали при пониженном давлении, получая соединение WX026-2. LCMS m/z = 230.2 [M+1]+.
Стадия 2: синтез соединения WX026-3
В раствор WX026-2 (0.45 г, 1.96 ммоль, 1 экв.) в тетрагидрофуране (5 мл) добавляли раствор LiOH·H2O (164.76 мг, 3.93 ммоль, 2 экв.) в воде (1 мл), и смесь перемешивали при 18°C в течение 1 часа. Этилацетат (20 мл) добавляли в реакционный раствор, и смесь доводили до значения pH 2 добавлением 2н. раствора соляной кислоты. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении, получая соединение WX026-3. LCMS m/z = 216.1 [M+1]+.
Стадия 3: синтез соединения WX026
В раствор BB-4 (0.3 г, 679.40 мкмоль, 1 экв.) в N,N-диметилформамиде (2 мл) добавляли WX026-3 (153.52 мг, 713.37 мкмоль, 1.05 экв.), DIPEA (263.42 мг, 2.04 ммоль, 355.01 мкл, 3 экв.) и T3P (648.52 мг, 1.02 ммоль, 606.09 мкл, 50% чистота, 1.5 экв.) при 18°C, и смесь перемешивали в течение 1 часа. Реакционный раствор очищали методом ВЭЖХ (Колонка: Phenomenex Luna C18 150 мм*30 мм*5 мкм; Подвижная фаза: [вода (0.05% HCl)-ACN]; ацетонитрил%: 20%-50%, 12 мин), получая соединение WX026. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.84 (с, 1H), 8.60 (д, J = 7.89 Гц, 1H), 8.36 (дд, J = 1.32, 4.82 Гц, 1H), 7.70 (дд, J = 1.53, 8.11 Гц, 1H), 7.25-7.46 (м, 5H), 5.49 (с, 2H), 4.85 (тд, J = 7.67, 11.40 Гц, 1H), 4.63-4.75 (м, 1H), 4.51 (дд, J = 7.45, 9.65 Гц, 1H), 4.03 (ушир.с, 3H); LCMS m/z = 407.1 [M+1]+.
Пример 27. WX027
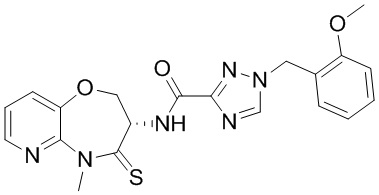
Путь синтеза:
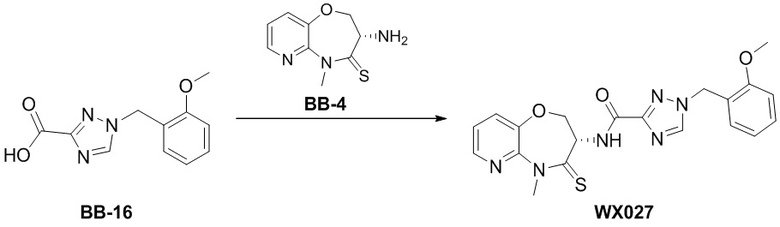
Стадия 1: синтез соединения WX027
В круглодонную колбу помещали N,N-диметилацетамид (5 мл) при 25°C. Затем медленно добавляли BB-4 (20 мг, 45.29 мкмоль, 1 экв.), T3P (43.23 мг, 67.94 мкмоль, 40.41 мкл, 50% чистота, 1.5 экв.), BB-16 (15.85 мг, 67.94 мкмоль, 1.5 экв.) и DIPEA (17.56 мг, 135.88 мкмоль, 23.67 мкл, 3 экв.), и реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 2 часа. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 42%-72%, 8 мин), получая соединение WX027. 1H ЯМР (400 МГц, CDCl3) δ = 8.78 (ушир.д, J = 7.0 Гц, 1H), 8.47 - 8.25 (м, 1H), 8.08 (с, 1H), 7.60 - 7.50 (м, 1H), 7.35 (т, J = 7.9 Гц, 1H), 7.32 - 7.26 (м, 2H), 6.96 (т, J = 7.4 Гц, 1H), 6.91 (д, J = 8.0 Гц, 1H), 5.38 (с, 2H), 5.20 (тд, J = 6.7, 11.4 Гц, 1H), 4.77 (дд, J = 6.3, 9.5 Гц, 1H), 4.38 - 4.23 (м, 1H), 3.92 (с, 3H), 3.84 (с, 3H); LCMS m/z =425.1 [M+1]+.
Пример 28. WX028
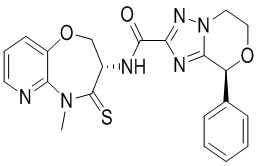
Путь синтеза:
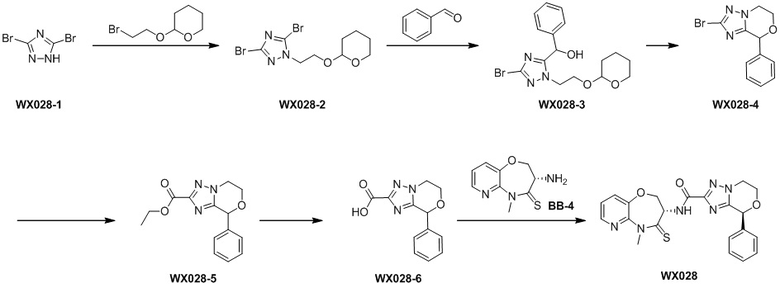
Стадия 1: синтез соединения WX028-2
В круглодонную колбу помещали MeCN (80 мл) и затем медленно добавляли WX028-1 (4 г, 17.63 ммоль, 1 экв.). После растворения добавляли 2-(2-бромэтокси)тетрагидропиран (4.42 г, 21.16 ммоль, 3.21 мл, 1.2 экв.) и DIPEA (2.51 г, 19.40 ммоль, 3.38 мл, 1.1 экв.), смесь нагревали до 90°C и непрерывно перемешивали в течение 3 часов. Воду (100 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (30 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX028-2. 1H ЯМР (400 МГц, CDCl3) δ = 4.55 (ушир.с, 1H), 4.34 (т, J = 5.3 Гц, 2H), 4.12 - 4.06 (м, 1H), 3.79 (тд, J = 5.4, 10.8 Гц, 1H), 3.64 - 3.53 (м, 1H), 3.50 - 3.41 (м, 1H), 1.76 - 1.58 (м, 2H), 1.57 - 1.43 (м, 4H).
Стадия 2: синтез соединения WX028-3
В круглодонную колбу помещали ТГФ (50 мл) и затем медленно добавляли WX028-2 (4.99 г, 14.06 ммоль, 1 экв.). Смесь охлаждали до -78°C и добавляли н-бутиллитий (2.5 M, 6.75 мл, 1.2 экв.). Смесь перемешивали в течение 1 часа и медленно добавляли бензальдегид (2.98 г, 28.11 ммоль, 2.84 мл, 2 экв.). Смесь перемешивали при -78°C в течение 0.5 часа, затем нагревали до -50°C и перемешивали 0.5 часа. После окончания реакции смесь выливали в смесь насыщенного водного раствора хлорида аммония (200 мл) и этилацетата (200 мл), и слои разделяли. Водную фазу экстрагировали этилацетатом (200 мл). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (50 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX028-3. 1H ЯМР (400 МГц, CDCl3) δ = 7.45 - 7.29 (м, 5H), 6.09 (д, J = 5.8 Гц, 1H), 4.51 (ушир.с, 1H), 4.34 (ддкв., J = 3.5, 5.5, 7.2 Гц, 1H), 4.26 - 4.15 (м, 1H), 4.07 - 3.97 (м, 1H), 3.78 - 3.56 (м, 2H), 3.47 (дт, J = 5.9, 10.7 Гц, 1H), 1.85 - 1.64 (м, 2H), 1.52 (ушир.д, J = 6.3 Гц, 4H).
Стадия 3: синтез соединения WX028-4
В круглодонную колбу помещали толуол (50 мл). Затем медленно добавляли WX028-3 (4.7 г, 12.30 ммоль, 1 экв.) и TsOH⋅H2O (3.04 г, 15.98 ммоль, 1.3 экв.), и смесь непрерывно перемешивали при 110°C в течение 5 часов. Воду (50 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (20 мл × 2), и упаривали при пониженном давлении. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX028-4. 1H ЯМР (400 МГц, CDCl3) δ = 7.45 - 7.34 (м, 5H), 5.87 (с, 1H), 4.33 - 4.27 (м, 2H), 4.19 - 4.06 (м, 2H).
Стадия 4: синтез соединения WX028-5
В автоклав помещали EtOH (20 мл). Затем медленно добавляли WX028-4 (500 мг, 1.78 ммоль, 1 экв.), Pd(dppf)Cl2 (261.21 мг, 356.99 мкмоль, 0.2 экв.) и TEA (1.81 г, 17.85 ммоль, 2.48 мл, 10 экв.). Три раза заменяли атмосферу в автоклаве на аргон, и смесь непрерывно перемешивали при 100°C в атмосфере CO (1 МПа) 24 часа. Воду (50 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (10 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 42%-72%, 8 мин), получая соединение WX028-5. 1H ЯМР (400 МГц, CDCl3) δ = 7.47 - 7.31 (м, 5H), 6.02 - 5.93 (м, 1H), 4.54 - 4.38 (м, 4H), 4.34 - 4.25 (м, 1H), 4.19 - 4.10 (м, 1H), 1.42 (т, J = 7.1 Гц, 3H).
Стадия 5: синтез соединения WX028-6
В круглодонную колбу помещали ТГФ (4 мл) и H2O(1 мл). Затем медленно добавляли WX028-5 (260 мг, 951.38 мкмоль, 1 экв.) и LiOH⋅H2O (119.77 мг, 2.85 ммоль, 3 экв.). Смесь непрерывно перемешивали при 25°C в течение 1 часа. Реакционный раствор упаривали досуха на роторном испарителе при пониженном давлении для удаления ТГФ. Добавляли по каплям 1M водный раствор HCl, доводя pH до 3, и смесь фильтровали, получая продукт WX028-6. 1H ЯМР (400 МГц, ДМСО-d6) δ = 7.40 (с, 5H), 6.00 (с, 1H), 4.50 - 4.40 (м, 1H), 4.40 - 4.29 (м, 2H), 4.26 - 4.11 (м, 1H).
Стадия 6: синтез соединения WX028-7
В круглодонную колбу помещали N,N-диметилформамид (1 мл). Затем медленно добавляли WX028-6 (30 мг, 122.33 мкмоль, 1 экв.), BB-4 (54.02 мг, 122.33 мкмоль, 1 экв.), T3P (116.77 мг, 183.50 мкмоль, 109.13 мкл, 50% чистота, 1.5 экв.) и DIPEA (47.43 мг, 366.99 мкмоль, 63.92 мкл, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин), получая рацемат соединения WX028, который затем разделяли методом хиральной сверхкритической жидкостной хроматографии (Колонка: DAICEL CHIRALCEL OD (250мм*30мм, 10мкм); Подвижная фаза: [Neu-ETOH]; этанол %: 45%-45%, мин), получая активный изомер WX028. 1H ЯМР (400 МГц, CDCl3) δ = 8.65 (ушир.д, J = 7.1 Гц, 1H), 8.37 (ушир.д, J = 3.3 Гц, 1H), 7.56 (ушир.д, J = 7.8 Гц, 1H), 7.48 - 7.36 (м, 5H), 7.32 - 7.27 (м, 1H), 5.99 (с, 1H), 5.33 - 5.16 (м, 1H), 4.74 (дд, J = 6.4, 9.5 Гц, 1H), 4.40 (ушир.д, J = 4.3 Гц, 2H), 4.36 - 4.24 (м, 2H), 4.19 - 4.07 (м, 1H), 3.92 (с, 3H); LCMS m/z = 437.1 [M+1]+.
Пример 29. WX029
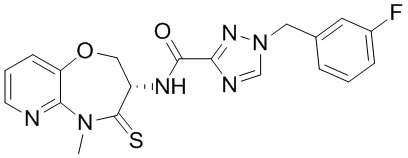
Путь синтеза:
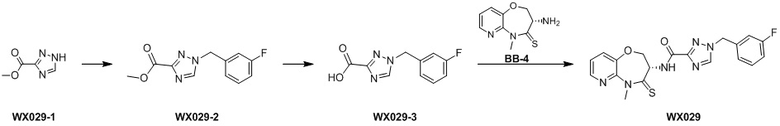
Стадия 1: синтез соединения WX029-2
В круглодонную колбу помещали MeCN (20 мл) при 25°C, затем медленно добавляли WX029-1 (2.02 г, 15.89 ммоль, 1 экв.), K2CO3 (4.39 г, 31.79 ммоль, 2 экв.) и 3-фторбензилхлорид (2.76 г, 19.07 ммоль, 2.28 мл, 1.2 экв.). Реакционный раствор непрерывно перемешивали при 60°C в атмосфере азота 12 часов. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:20~100:75), получая соединение WX029-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.14 (с, 1H), 7.36 (дт, J = 5.9, 8.0 Гц, 1H), 7.11 - 7.03 (м, 2H), 6.99 (ушир.д, J = 9.0 Гц, 1H), 5.42 (с, 2H), 4.14 - 3.89 (м, 3H).
Стадия 2: синтез соединения WX029-3
В круглодонную колбу помещали ТГФ (14 мл) и H2O (2 мл) при 25°C, и затем медленно добавляли WX029-2 и LiOH⋅H2O (642.21 мг, 15.31 ммоль, 3 экв.). Реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 1 час. Реакционный раствор доводили до значения pH 6~7 разбавленной соляной кислотой (1M), выпавший осадок отфильтровывали и сышили, получая соединение WX029-3.
Стадия 3: синтез соединения WX029
В круглодонную колбу помещали N,N-диметилацетамид (5 мл) при 25°C, и затем медленно добавляли WX029-3 (15.03 мг, 67.94 мкмоль, 1.5 экв.), T3P (43.23 мг, 67.94 мкмоль, 40.41 мкл, 50% чистота, 1.5 экв.), BB-4 (15.03 мг, 67.94 мкмоль, 1.5 экв.) и DIPEA (17.56 мг, 135.87 мкмоль, 23.67 мкл, 3 экв.). Реакционный раствор непрерывно перемешивали в атмосфере азота 2 часа. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин), получая продукт WX029. 1H ЯМР (400 МГц, CDCl3) δ = 8.83 (ушир.д, J = 7.0 Гц, 1H), 8.37 (д, J = 3.8 Гц, 1H), 8.10 (с, 1H), 7.57 (д, J = 8.0 Гц, 1H), 7.40 - 7.27 (м, 2H), 7.15 - 6.87 (м, 3H), 5.40 (с, 2H), 5.29 - 5.07 (м, 1H), 4.79 (дд, J = 6.3, 9.3 Гц, 1H), 4.32 (т, J = 10.3 Гц, 1H), 3.93 (с, 3H); 19F ЯМР (376 МГц, CDCl3) δ = -111.33 (с, 1F); LCMS m/z =413.0 [M+1]+.
Пример 30. WX030
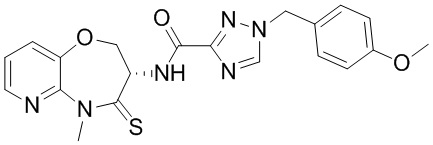
Путь синтеза:
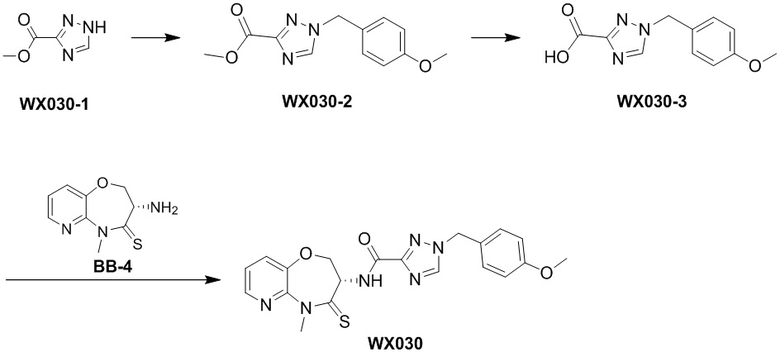
Стадия 1: синтез соединения WX030-2
В круглодонную колбу помещали MeCN (20 мл) при 25°C и затем медленно добавляли WX030-1 (2.04 г, 16.05 ммоль, 1экв.), K2CO3 (4.44 г, 32.10 ммоль, 2 экв.) и 4-метоксибензилхлорид (3.02 г, 19.26 ммоль, 2.62 мл, 1.2 экв.). Реакционный раствор непрерывно перемешивали при 60°C в атмосфере азота 12 часов. Смесь медленно выливали в воду (100 мл) и экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:20~100:75), получая соединение WX030-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.05 (с, 1H), 7.27 (д, J = 8.8 Гц, 2H), 6.98 - 6.87 (м, 2H), 5.43 - 5.26 (м, 2H), 4.03 - 3.97 (м, 3H), 3.82 (с, 3H).
Стадия 2: синтез соединения WX030-3
В круглодонную колбу помещали ТГФ (14 мл) и H2O (2 мл) при 25°C, и WX030-2 (1.5 г, 6.07 ммоль, 1 экв.) и LiOH⋅H2O (763.68 мг, 18.20 ммоль, 3 экв.). Смесь непрерывно перемешивали при 25°C в атмосфере азота 1 час. Реакционный раствор доводили до значения pH 6~7 разбавленной соляной кислотой (1 M), выпавший осадок отфильтровывали и сушили, получая соединение WX030-3.
Стадия 3: синтез соединения WX030
В круглодонную колбу помещали N,N-диметилацетамид (5 мл) при 25°C, затем медленно добавляли WX030-3 (15.84 мг, 67.94 мкмоль, 1.5 экв.), T3P (43.23 мг, 67.94 мкмоль, 40.41 мкл, 50% чистота, 1.5 экв.), BB-4 (15.84 мг, 67.94 мкмоль, 1.5 экв.) и DIPEA (17.56 мг, 135.87 мкмоль, 23.67 мкл, 3 экв.), и раствор непрерывно перемешивали при 25°C в атмосфере азота 2 часа. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин), получая продукт WX030. 1H ЯМР (400 МГц, CDCl3) δ = 8.80 (ушир.с, 1H), 8.37 (ушир.с, 1H), 7.98 (ушир.с, 1H), 7.56 (ушир.с, 1H), 7.38 - 7.28 (м, 1H), 7.26 (ушир.с, 2H), 6.91 (ушир.с, 2H), 5.41 - 5.10 (м, 3H), 4.78 (ушир.с, 1H), 4.31(ушир.с, 1H), 3.98 - 3.73 (м, 6H); LCMS m/z =425.1 [M+1]+.
Пример 31. WX031
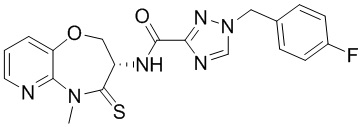
Путь синтеза:
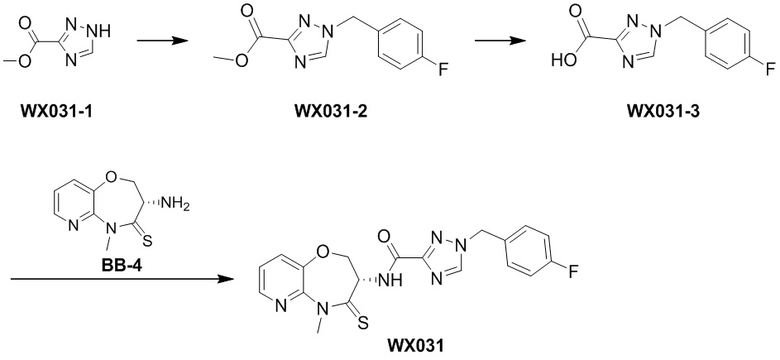
Стадия 1: синтез соединения WX031-2
В круглодонную колбу помещали MeCN (20 мл). Затем добавляли WX031-1 (1.5 г, 11.80 ммоль, 1 экв.), K2CO3 (2.45 г, 17.70 ммоль, 1.5 экв.) и 4-фторбензилхлорид (1.88 г, 12.98 ммоль, 1.55 мл, 1.1 экв.), и смесь непрерывно перемешивали при 50°C в течение 12 часов. Реакционный раствор выливали в воду (50 мл), и смесь экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (10 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:100), получая соединение WX031-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.09 (с, 1H), 7.30 (дд, J = 5.3, 8.3 Гц, 2H), 7.08 (т, J = 8.5 Гц, 2H), 5.39 (с, 2H), 4.00 (с, 3H).
Стадия 2: синтез соединения WX031-3
В круглодонную колбу помещали ТГФ (8 мл) и H2O (2 мл). Затем добавляли WX031-2 (1.06 г, 4.52 ммоль, 1 экв.) и LiOH⋅H2O (569.16 мг, 13.56 ммоль, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор упаривали при пониженном давлении для удаления ТГФ. 1М раствор соляной кислоты добавляли по каплям до pH меньше 3, и смесь фильтровали, получая соединение WX031-3. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.79 (с, 1H), 7.39 (дд, J = 5.5, 8.8 Гц, 2H), 7.25 - 7.17 (м, 2H), 5.47 (с, 2H).
Стадия 3: синтез соединения WX031
В круглодонную колбу помещали N,N-диметилформамид (1 мл). Затем медленно добавляли WX031-3 (18.03 мг, 81.53 мкмоль, 1.2 экв.), BB-4 (30 мг, 67.94 мкмоль, 1 экв.), DIPEA (26.34 мг, 203.82 мкмоль, 35.50 мкл, 3 экв.) и T3P (64.85 мг, 101.91 мкмоль, 60.61 мкл, 50% чистота, 1.5 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор напрямую очищали методом ВЭЖХ (Колонка: Xtimate C18 100*30мм*3мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 40%-70%, 8 мин), получая соединение WX031. 1H ЯМР (400 МГц, CDCl3) δ = 8.81 (ушир.д, J = 7.3 Гц, 1H), 8.37 (дд, J = 1.4, 4.6 Гц, 1H), 8.06 (с, 1H), 7.56 (дд, J = 1.4, 7.9 Гц, 1H), 7.33 - 7.27 (м, 3H), 7.07 (т, J = 8.5 Гц, 2H), 5.37 (с, 2H), 5.20 (тд, J = 6.7, 11.2 Гц, 1H), 4.78 (дд, J = 6.4, 9.4 Гц, 1H), 4.32 (дд, J = 9.7, 10.9 Гц, 1H), 3.93 (с, 3H); LCMS m/z = 413.1 [M+1]+.
Пример 32. WX032
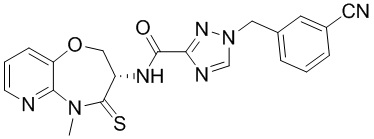
Путь синтеза:
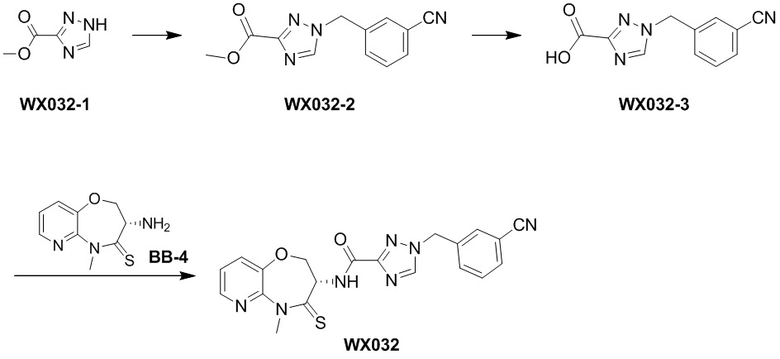
Стадия 1: синтез соединения WX032-2
В MeCN (20 мл) добавляли WX032-1 (1.5 г, 11.80 ммоль, 1 экв.), 3-хлорметилбензонитрил (1.97 г, 12.98 ммоль, 1.72 мл, 1.1 экв.) и K2CO3 (2.45 г, 17.70 ммоль, 1.5 экв.), и реакционный раствор непрерывно перемешивали при 50°C в течение 12 часов. Водуа (50 мл) добавляли в раствор, и смесь экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (20 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:100), получая соединение WX032-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.21 (с, 1H), 7.67 (т, J = 4.3 Гц, 1H), 7.58 (с, 1H), 7.52 (д, J = 4.8 Гц, 2H), 5.47 (с, 2H), 4.01 (с, 3H).
Стадия 2: синтез соединения WX032-3
В круглодонную колбу помещали ТГФ (8 мл) и H2O (2 мл). Затем добавляли WX032-2 (1.4 г, 5.78 ммоль, 1 экв.) и LiOH⋅H2O (727.53 мг, 17.34 ммоль, 3 экв.), и реакционный раствор непрерывно перемешивали при 25°C в течение 2 часов. Раствор упаривали при пониженном давлении для удаления ТГФ. 1М раствор соляной кислоты добавляли по каплям до pH меньше 3, и смесь фильтровали, получая целевое соединение WX032-3. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.83 - 8.80 (м, 1H), 7.86 - 7.80 (м, 2H), 7.68 - 7.57 (м, 2H), 5.59 - 5.52 (м, 2H).
Стадия 3: синтез соединения WX032
В круглодонную колбу помещали N,N-диметилформамид (1 мл). Затем медленно добавляли WX032-3 (15.50 мг, 67.94 мкмоль, 1 экв.), BB-4 (30 мг, 67.94 мкмоль, 1 экв.), DIPEA (26.34 мг, 203.82 мкмоль, 35.50 мкл, 3 экв.) и T3P (64.85 мг, 101.91 мкмоль, 60.61 мкл, 50% чистота, 1.5 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор напрямую очищали методом ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин), получая соединение WX032. 1H ЯМР (400 МГц, CDCl3) δ = 8.85 (ушир.д, J = 7.3 Гц, 1H), 8.37 (д, J = 3.3 Гц, 1H), 8.17 (с, 1H), 7.65 (ушир.д, J = 2.8 Гц, 1H), 7.60 - 7.55 (м, 2H), 7.53 - 7.47 (м, 2H), 7.29 (дд, J = 4.6, 7.9 Гц, 1H), 5.45 (с, 2H), 5.26 - 5.15 (м, 1H), 4.79 (дд, J = 6.3, 9.5 Гц, 1H), 4.38 - 4.28 (м, 1H), 3.93 (с, 3H); LCMS m/z = 420.1 [M+1]+.
Пример 33. WX033
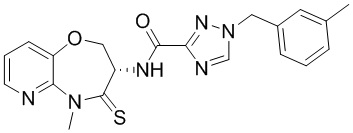
Путь синтеза:
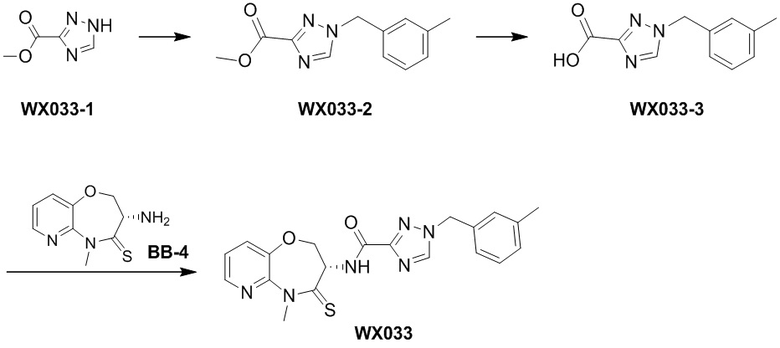
Стадия 1: синтез соединения WX033-2
В круглодонную колбу помещали MeCN (20 мл). Добавляли WX033-1(1.5 г, 11.80 ммоль, 1 экв.), 3-метилбензилхлорид (1.83 г, 12.98 ммоль, 1.72 мл, 1.1 экв.) и K2CO3 (2.45 г, 17.70 ммоль, 1.5 экв.), и смесь непрерывно перемешивали при 50°C в течение 12 часов. Вода (50 мл) добавляли в раствор, и смесь экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (10 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:100), получая соединение WX033-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.08 (с, 1H), 7.33 - 7.27 (м, 1H), 7.18 (д, J = 7.6 Гц, 1H), 7.10 (ушир.д, J = 1.8 Гц, 2H), 5.38 (с, 2H), 4.05 - 3.97 (м, 3H), 2.38 - 2.31 (м, 3H).
Стадия 2: синтез соединения WX033-3
В круглодонную колбу помещали ТГФ (8 мл) и H2O (2 мл). Затем добавляли WX033-2 (1.6 г, 6.92 ммоль, 1 экв.) и LiOH⋅H2O (870.95 мг, 20.76 ммоль, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор упаривали при пониженном давлении для удаления ТГФ. 1М раствор соляной кислоты добавляли по каплям до pH меньше 3, и смесь фильтровали, получая соединение WX033-3. 1H ЯМР (400 МГц, ДМСО-d6) δ = 13.28 (ушир.с, 1H), 8.79 (с, 1H), 7.30 - 7.23 (м, 1H), 7.17 - 7.07 (м, 3H), 5.43 (с, 2H), 2.29 (с, 3H).
Стадия 3: синтез соединения WX033
В круглодонную колбу помещали N,N-диметилформамид (1 мл). Затем медленно добавляли WX033-3 (17.71 мг, 81.53 мкмоль, 1.2 экв.), BB-4 (30 мг, 67.94 мкмоль, 1 экв.), DIPEA (26.34 мг, 203.82 мкмоль, 35.50 мкл, 3 экв.) и T3P (64.85 мг, 101.91 мкмоль, 60.61 мкл, 50% чистота, 1.5 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор напрямую очищали методом ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 37%-67%, 8 мин), получая соединение WX033. 1H ЯМР (400 МГц, CDCl3) δ = 8.81 (ушир.д, J = 7.0 Гц, 1H), 8.37 (д, J = 3.3 Гц, 1H), 8.03 (с, 1H), 7.57 (д, J = 6.8 Гц, 1H), 7.31 - 7.27 (м, 2H), 7.20 - 7.15 (м, 1H), 7.11 - 7.07 (м, 2H), 5.36 (с, 2H), 5.22 (тд, J = 6.8, 11.5 Гц, 1H), 4.79 (дд, J = 6.4, 9.4 Гц, 1H), 4.36 - 4.27 (м, 1H), 3.93 (с, 3H), 2.34 (с, 3H); LCMS m/z = 409.1 [M+1]+.
Пример 34. WX034
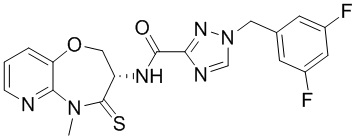
Путь синтеза:
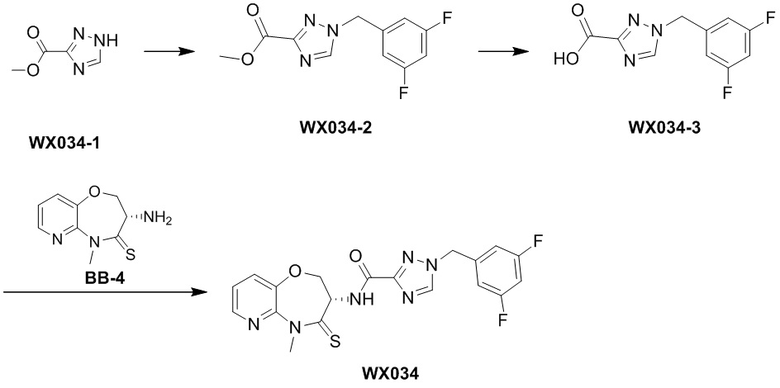
Стадия 1: синтез соединения WX034-2.
В одногорлую колбу помещали MeCN (20 мл). Затем добавляли 1-хлорметил-3,5-дифторбензол (2.11 г, 12.98 ммоль, 573.45 мкл, 1.1 экв.), WX034-1 (1.5 г, 11.80 ммоль, 1 экв.) и K2CO3 (2.45 г, 17.70 ммоль, 1.5 экв.), и смесь непрерывно перемешивали при 50°C в течение 12 часов. Новое пятно появлялось в ТСХ (петролейный эфир:этилацетат = 1:2). Воду (50 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (20 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:100), получая соединение WX034-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.22 (с, 1H), 6.92 - 6.77 (м, 3H), 5.50 - 5.38 (м, 2H), 4.11 - 3.99 (м, 3H).
Стадия 2: синтез соединения WX034-3
В круглодонную колбу помещали ТГФ (8 мл) и H2O (2 мл). Затем добавляли WX034-2 (1 г, 3.95 ммоль, 1 экв.) и LiOH⋅H2O (497.15 мг, 11.85 ммоль, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор упаривали при пониженном давлении для удаления ТГФ. 1М раствор соляной кислоты добавляли по каплям до pH меньше 3, и смесь фильтровали, получая соединение WX034-3. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.80 (с, 1H), 7.24 (ушир.т, J = 9.4 Гц, 1H), 7.07 (ушир.д, J = 6.3 Гц, 2H), 5.52 (с, 2H).
Стадия 3: синтез соединения WX034.
В круглодонную колбу помещали N,N-диметилформамид (1 мл). Затем медленно добавляли WX034-3 (19.50 мг, 81.53 мкмоль, 1.2 экв.), BB-4 (30 мг, 67.94 мкмоль, 1 экв.), DIPEA (26.34 мг, 203.82 мкмоль, 35.50 мкл, 3 экв.) и T3P (64.85 мг, 101.91 мкмоль, 60.61 мкл, 50% чистота, 1.5 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор напрямую очищали методом ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 37%-67%, 8 мин), получая соединение WX034. 1H ЯМР (400 МГц, CDCl3) δ = 8.85 (ушир.д, J = 7.0 Гц, 1H), 8.37 (дд, J = 1.5, 4.6 Гц, 1H), 8.16 (с, 1H), 7.57 (дд, J = 1.5, 8.0 Гц, 1H), 7.29 (дд, J = 4.7, 7.9 Гц, 1H), 6.85 - 6.73 (м, 3H), 5.38 (с, 2H), 5.21 (тд, J = 6.6, 11.2 Гц, 1H), 4.79 (дд, J = 6.4, 9.5 Гц, 1H), 4.33 (дд, J = 9.6, 11.0 Гц, 1H), 3.93 (с, 3H); LCMS m/z = 431.1 [M+1]+.
Пример 35. WX035
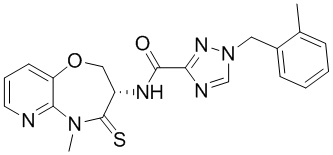
Путь синтеза:
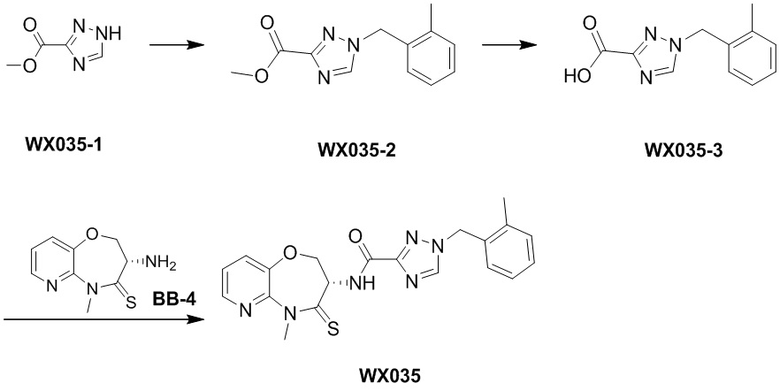
Стадия 1: синтез соединения WX035-2
В MeCN (20 мл) добавляли WX035-1 (1.5 г, 11.80 ммоль, 1 экв.), 1-хлорметил-2-толуол (1.83 г, 12.98 ммоль, 1.72 мл, 1.1 экв.) и K2CO3 (2.45 г, 17.70 ммоль, 1.5 экв.), и смесь непрерывно перемешивали при 50°C в течение 12 часов. Воду (50 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (20 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:100), получая соединение WX035-2. 1H ЯМР (400 МГц, CDCl3) δ = 7.89 (с, 1H), 7.36 - 7.29 (м, 1H), 7.26 - 7.16 (м, 3H), 5.42 (с, 2H), 4.00 (с, 3H), 2.27 (с, 3H).
Стадия 2: синтез соединения WX035-3
В круглодонную колбу помещали ТГФ (8 мл) и H2O (2 мл). Затем добавляли WX035-2 (1.6 г, 6.92 ммоль, 1 экв.) и LiOH·H2O (870.95 мг, 20.76 ммоль, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор упаривали при пониженном давлении для удаления ТГФ. 1М раствор соляной кислоты добавляли по каплям до pH меньше 3, и смесь фильтровали, получая WX035-3. 1H ЯМР (400 МГц, ДМСО-d6) δ = 13.28 (ушир.с, 1H), 8.79 (с, 1H), 7.30 - 7.23 (м, 1H), 7.17 - 7.07 (м, 3H), 5.43 (с, 2H), 2.29 (с, 3H).
Стадия 3: синтез соединения WX035
В круглодонную колбу помещали N,N-диметилформамид (1 мл). Затем медленно добавляли WX035-3 (17.71 мг, 81.53 мкмоль, 1.2 экв.), BB-4 (30 мг, 67.94 мкмоль, 1 экв.), DIPEA (26.34 мг, 203.82 мкмоль, 35.50 мкл, 3 экв.) и T3P (64.85 мг, 101.91 мкмоль, 60.61 мкл, 50% чистота, 1.5 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор напрямую очищали методом ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 37%-67%, 8 мин), получая соединение WX035. 1H ЯМР (400 МГц, CDCl3) δ = 8.81 (ушир.д, J = 7.0 Гц, 1H), 8.37 (д, J = 4.5 Гц, 1H), 7.86 (с, 1H), 7.57 (д, J = 7.8 Гц, 1H), 7.34 - 7.27 (м, 2H), 7.26 - 7.15 (м, 3H), 5.40 (с, 2H), 5.22 (тд, J = 6.7, 11.2 Гц, 1H), 4.79 (дд, J = 6.4, 9.4 Гц, 1H), 4.38 - 4.26 (м, 1H), 3.93 (с, 3H), 2.29 (с, 3H); LCMS m/z = 409.1 [M+1]+.
Пример 36. WX036
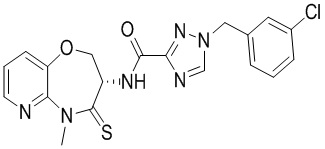
Путь синтеза:
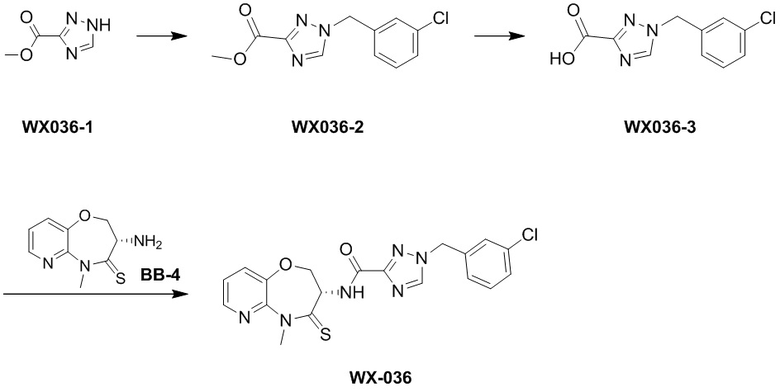
Стадия 1: синтез соединения WX036-2
В круглодонную колбу помещали MeCN (20 мл) при 25°C. Затем медленно добавляли WX036-1 (2.02 г, 15.89 ммоль, 1 экв.), K2CO3 (4.39 г, 31.79 ммоль, 2 экв.) и 1-хлор-3-хлорметилбензол (3.07 г, 19.07 ммоль, 2.42 мл, 1.2 экв.). Реакционный раствор непрерывно перемешивали при 60°C в атмосфере азота 12 часов. Смесь медленно выливали в воду (100 мл) и экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:20~100:75), получая соединение WX036-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.15 (с, 1H), 7.35 - 7.26 (м, 3H), 7.17 (д, J = 6.8 Гц, 1H), 5.40 (с, 2H), 4.04 - 3.93 (м, 3H).
Стадия 2: синтез соединения WX036-3
В круглодонную колбу помещали ТГФ (14 мл) и H2O (2 мл) при 25°C. Затем медленно добавляли WX036-2 (1.80 г, 7.15 ммоль, 1 экв.) и LiOH⋅H2O (900.33 мг, 21.46 ммоль, 3 экв.). Реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 1 час. Реакционный раствор доводили до значения pH 6~7 разбавленной соляной кислотой (1 M), и осадок отфильтровывали, получая соединение WX036-3.
Стадия 3: синтез соединения WX036.
В круглодонную колбу помещали N,N-диметилацетамид (5 мл) при 25°C. Затем медленно добавляли BB-4 (20 мг, 45.29 мкмоль, 1 экв.), T3P (43.23 мг, 67.94 мкмоль, 40.41 мкл, 50% чистота, 1.5 экв.), WX036-3 (16.14 мг, 67.94 мкмоль, 1.5 экв.) и DIPEA (17.56 мг, 135.87 мкмоль, 23.67 мкл, 3 экв.), и реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 2 часа. Реакционную смесь медленно выливали в воду (100 мл) и экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт выделяли методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин), получая соединение WX036. 1H ЯМР (400 МГц, CDCl3) δ = 8.95 - 8.69 (м, 1H), 8.37 (ушир.д, J = 3.1 Гц, 1H), 8.10 (с, 1H), 7.68 - 7.47 (м, 1H), 7.38 - 7.27 (м, 3H), 7.26 - 7.20 (м, 1H), 7.16 (ушир.д, J = 7.0 Гц, 1H), 5.38 (с, 2H), 5.26 - 5.10 (м, 1H), 4.79 (дд, J = 6.5, 9.4 Гц, 1H), 4.41 - 4.20 (м, 1H), 3.93 (с, 3H); LCMS m/z =429.0 [M+1]+.
Пример 37. WX037
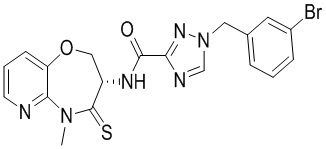
Путь синтеза:
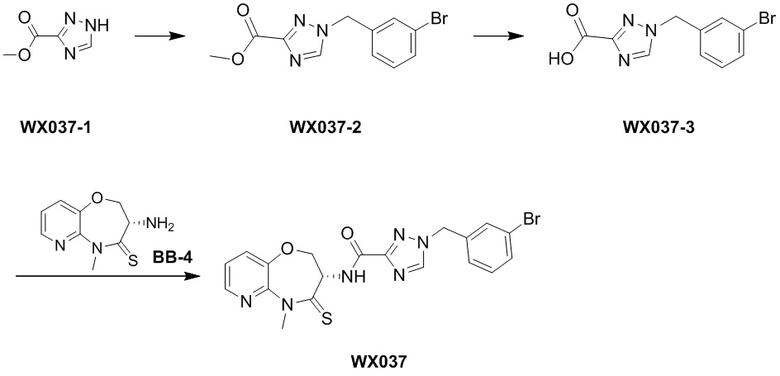
Стадия 1: синтез соединения WX037-2
В круглодонную колбу помещали MeCN (20 мл) при 25°C. Затем медленно добавляли WX037-1 (2.03 г, 15.97 ммоль, 1 экв.), K2CO3 (4.41 г, 31.94 ммоль, 2 экв.) и 1-бром-3-хлорметилбензол (3.94 г, 19.17 ммоль, 241.82 мкл, 1.2 экв.). Реакционный раствор непрерывно перемешивали при 60°C в атмосфере азота 12 часов. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:20~100:75), получая соединение WX037-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.14 (с, 1H), 7.59 - 7.38 (м, 2H), 7.30 - 7.26 (м, 1H), 7.25 - 7.20 (м, 1H), 5.39 (с, 2H), 4.01 (с, 3H).
Стадия 2: синтез соединения WX037-3
В круглодонную колбу помещали ТГФ (14 мл) и H2O (2 мл) при 25°C. Затем медленно добавляли WX037-2 (2.0 г, 6.75 ммоль, 1 экв.) и LiOH⋅H2O (850.20 мг, 20.26 ммоль, 3 экв.). Реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 1 час. Реакционный раствор доводили до значения pH 6~7 разбавленной соляной кислотой (1 M), и осадок отфильтровывали, получая соединение WX037-3.
Стадия 3: синтез соединения WX037
В круглодонную колбу помещали N,N-диметилацетамид (5 мл) при 25°C. Затем медленно добавляли BB-4 (20 мг, 45.29 мкмоль, 1 экв.), T3P (43.23 мг, 67.94 мкмоль, 40.41 мкл, 50% чистота, 1.5 экв.), WX037-3 (19.16 мг, 67.94 мкмоль, 1.5 экв.) и DIPEA (17.56 мг, 135.87 мкмоль, 23.67 мкл, 3 экв.), и реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 2 часа. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт выделяли методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 35%-65%, 8 мин), получая соединение WX037. 1H ЯМР (400 МГц, CDCl3) δ = 8.83 (ушир.д, J = 6.4 Гц, 1H), 8.37 (ушир.д, J = 4.5 Гц, 1H), 8.10 (с, 1H), 7.66 - 7.27 (м, 4H), 7.25 - 7.15 (м, 2H), 5.37 (с, 2H), 5.26 - 5.06 (м, 1H), 4.79 (ушир. дд, J = 6.5, 9.1 Гц, 1H), 4.33 (ушир.т, J = 10.3 Гц, 1H), 3.93 (с, 3H); LCMS m/z =474.8 [M+1]+.
Пример 38. WX038
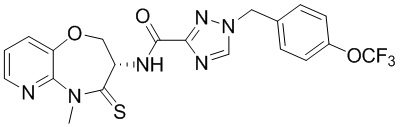
Путь синтеза:
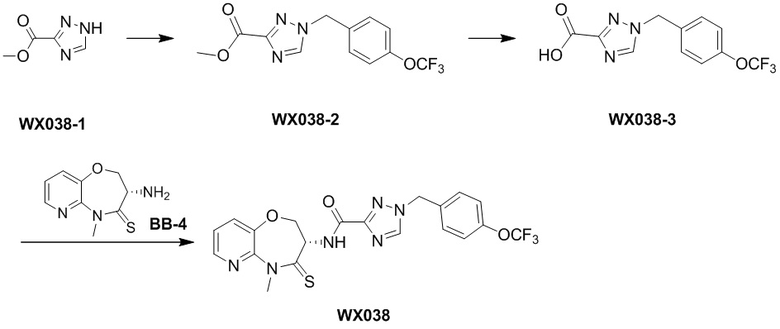
Стадия 1: синтез соединения WX038-2
В круглодонную колбу помещали MeCN (20 мл). Затем добавляли 1-хлорметил-4-трифторметоксибензол (2.73 г, 12.98 ммоль, 517.03 мкл, 1.1 экв.), WX038-1 (1.5 г, 11.80 ммоль, 1 экв.) и K2CO3 (2.45 г, 17.70 ммоль, 1.5 экв.), и смесь непрерывно перемешивали при 50°C в течение 12 часов. Воду (50 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (20 мл × 2) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:100), получая соединение WX038-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.17 (с, 1H), 7.36 (д, J = 8.5 Гц, 2H), 7.27 - 7.23 (м, 2H), 5.50 - 5.40 (м, 2H), 4.08 - 3.96 (м, 3H).
Стадия 2: синтез соединения WX038-3
В круглодонную колбу помещали ТГФ (16 мл) и H2O (4 мл). Затем добавляли WX038-2 (1.9 г, 6.31 ммоль, 1 экв.) и LiOH⋅H2O (794.01 мг, 18.92 ммоль, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор упаривали при пониженном давлении для удаления ТГФ. 1М раствор соляной кислоты добавляли по каплям до pH меньше 3, и осадок отфильтровывали, получая соединение WX038-3. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.81 (с, 1H), 7.51 - 7.33 (м, 4H), 5.53 (с, 2H).
Стадия 3: синтез соединения WX038
В круглодонную колбу помещали N,N-диметилформамид (1 мл). Затем медленно добавляли WX038-3 (23.41 мг, 81.53 мкмоль, 1.2 экв.), BB-4 (30 мг, 67.94 мкмоль, 1 экв.), DIPEA (26.34 мг, 203.82 мкмоль, 35.50 мкл, 3 экв.) и T3P (64.85 мг, 101.91 мкмоль, 60.61 мкл, 50% чистота, 1.5 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор напрямую очищали методом ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 40%-70%, 8 мин), получая соединение WX038. 1H ЯМР (400 МГц, CDCl3) δ = 8.84 (ушир.д, J = 7.3 Гц, 1H), 8.38 (дд, J = 1.5, 4.5 Гц, 1H), 8.11 (с, 1H), 7.58 (дд, J = 1.5, 8.0 Гц, 1H), 7.34 (д, J = 8.8 Гц, 2H), 7.32 - 7.29 (м, 1H), 7.27 (с, 5H), 7.26 - 7.22 (м, 2H), 5.42 (с, 2H), 5.28 - 5.15 (м, 1H), 4.80 (дд, J = 6.5, 9.5 Гц, 1H), 4.37 - 4.29 (м, 1H), 3.94 (с, 3H); LCMS m/z = 479.1 [M+1]+.
Пример 39. WX039
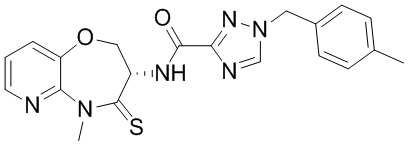
Путь синтеза:
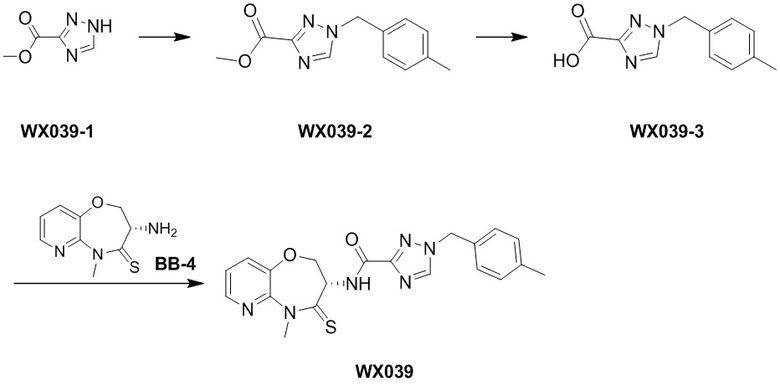
Стадия 1: синтез соединения WX039-2
В круглодонную колбу помещали MeCN (20 мл) при 25°C. Затем медленно добавляли WX039-1 (2.0 г, 15.74 ммоль, 1 экв.), K2CO3 (4.35 г, 31.48 ммоль, 2 экв.) и 1-хлорметил-4-метилбензол (2.66 г, 18.89 ммоль, 2.48 мл, 1.2 экв.). Реакционный раствор непрерывно перемешивали при 60°C в атмосфере азота 12 часов. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:20~100:75), получая соединение WX039-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.04 (с, 1H), 7.23 - 7.17 (м, 4H), 5.37 (с, 2H), 4.00 (с, 3H), 2.47 - 2.25 (м, 3H).
Стадия 2: синтез соединения WX039-3
В круглодонную колбу помещали ТГФ (14 мл) и H2O (2 мл) при 25°C. Затем медленно добавляли WX039-2 (1.5 г, 6.49 ммоль, 1 экв.) и LiOH⋅H2O (816.52 мг, 19.46 ммоль, 3 экв.). Реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 1 час. Реакционный раствор доводили до значения pH 6~7 разбавленной соляной кислотой (1 M), и осадок отделяли фильтрованием, получая соединение WX039-3.
Стадия 3: синтез соединения WX039
В N,N-диметилацетамид (5 мл) добавляли BB-4 (20 мг, 45.29 мкмоль, 1 экв.), T3P (43.23 мг, 67.94 мкмоль, 40.41 мкл, 50% чистота, 1.5 экв.), WX039-3 (14.76 мг, 67.94 мкмоль, 1.5 экв.) и DIPEA (17.56 мг, 135.87 мкмоль, 23.67 мкл, 3 экв.) при 25°C, и реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 2 часа. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли, последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл) и упаривали при пониженном давлении, получая сырой продукт. Сырой продукт выделяли методом препаративной ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 40%-70%, 8 мин), получая продукт WX039. 1H ЯМР (400 МГц, CDCl3) δ = 8.80 (ушир.д, J = 7.3 Гц, 1H), 8.37 (дд, J = 1.5, 4.5 Гц, 1H), 8.00 (с, 1H), 7.56 (дд, J = 1.5, 7.8 Гц, 1H), 7.28 (дд, J = 4.8, 8.0 Гц, 1H), 7.19 (с, 4H), 5.35 (с, 2H), 5.21 (тд, J = 6.8, 11.0 Гц, 1H), 4.78 (дд, J = 6.4, 9.4 Гц, 1H), 4.32 (дд, J = 9.7, 11.2 Гц, 1H), 3.93 (с, 3H), 2.63 - 2.13 (м, 3H); LCMS m/z =409.1 [M+1]+.
Пример 40. WX040
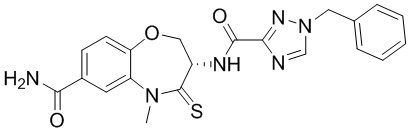
Путь синтеза:
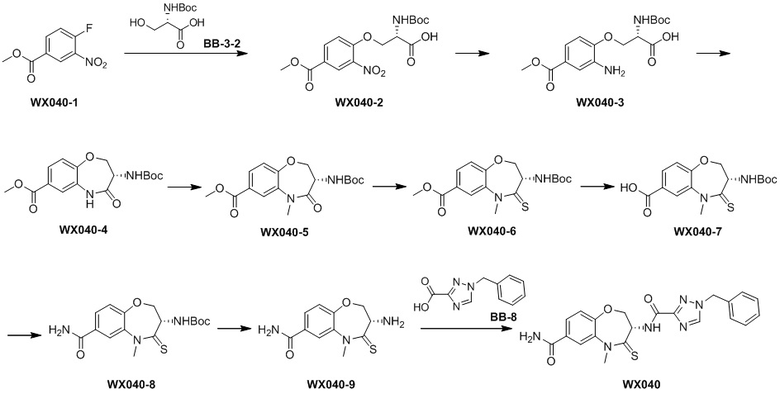
Стадия 1: синтез соединения WX040-2
В круглодонную колбу помещали ТГФ (100 мл). Затем медленно добавляли WX040-1 (10 г, 50.22 ммоль, 1 экв.), BB-3-2 (11.34 г, 55.24 ммоль, 1.1 экв.) и карбонат цезия (29.45 г, 90.39 ммоль, 1.8 экв.), и смесь непрерывно перемешивали при 65°C в течение 16 часов. ТСХ-мониторинг (петролейный эфир:этилацетат:уксусная кислота = 1:1:0.1) показал, что исходные соединения полностью прореагировали. Реакционный раствор выливали в воду (300 мл), затем добавляли 2М раствор соляной кислоты, доводя pH до 3. Смесь экстрагировали этилацетатом (500 мл × 2). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия (50 мл × 2). Органическую фазу затем упаривали при пониженном давлении, получая WX040-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.46 (д, J = 1.8 Гц, 1H), 8.20 (дд, J = 2.0, 8.8 Гц, 1H), 7.12 (д, J = 8.8 Гц, 1H), 5.62 (ушир.д, J = 8.0 Гц, 1H), 4.83 - 4.61 (м, 2H), 4.45 (дд, J = 3.0, 9.3 Гц, 1H), 3.93 (с, 3H), 1.45 (с, 10H).
Стадия 2: синтез соединения WX040-3
В колбу для гидрирования помещали MeOH (300 мл). Затем медленно добавляли WX040-2 (25.8 г, 67.13 ммоль, 1 экв.) и Pd/C (2.6 г, 26.02 ммоль, 10% чистота). Три раза заменяли атмосферу в колбе на аргон, и смесь непрерывно перемешивали при 30°C в атмосфере водорода (40 фунт/кв.дюйм) 24 часа. Новое пятно появлялось в ТСХ (петролейный эфир:этилацетат:уксусная кислота = 1:1:0.1). Реакционный раствор фильтровали через целит, и фильтрат упаривали при пониженном давлении, получая соединение WX040-3. 1H ЯМР (400 МГц, CDCl3) δ = 7.50 - 7.44 (м, 1H), 7.39 (ушир.с, 1H), 6.70 (д, J = 8.5 Гц, 1H), 6.53 (ушир.с, 2H), 6.00 (ушир.д, J = 7.5 Гц, 1H), 4.73 (ушир.д, J = 7.0 Гц, 1H), 4.43 (ушир.д, J = 8.5 Гц, 1H), 4.31 - 4.24 (м, 1H), 3.85 (с, 3H), 1.41 (с, 9H).
Стадия 3: синтез соединения WX040-4
В круглодонную колбу помещали этилацетат (200 мл). Затем медленно добавляли WX040-3 (12.5 г, 35.28 ммоль, 1 экв.), T3P (33.67 г, 52.91 ммоль, 31.47 мл, 50% чистота, 1.5 экв.) и DIPEA (13.68 г, 105.83 ммоль, 18.43 мл, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Воду (200 мл) добавляли в реакционный раствор, слои разделяли. Водную фазу экстрагировали этилацетатом (200 мл × 2). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия (100 мл × 2), затем органическую фазу упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX040-4. 1H ЯМР (400 МГц, CDCl3) δ = 8.15 (ушир.с, 1H), 7.80 (дд, J = 2.0, 8.5 Гц, 1H), 7.73 (д, J = 2.0 Гц, 1H), 7.13 (д, J = 8.3 Гц, 1H), 5.62 (ушир.д, J = 5.3 Гц, 1H), 4.71 - 4.59 (м, 2H), 4.33 - 4.23 (м, 1H), 3.95 - 3.89 (м, 3H), 1.43 (с, 9H).
Стадия 4: синтез соединения WX040-5
В круглодонную колбу помещали N,N-диметилформамид (200 мл). затем медленно добавляли WX040-4 (17 г, 50.54 ммоль, 1 экв.), карбонат цезия (49.40 г, 151.63 ммоль, 3 экв.) и иодметан (21.52 г, 151.63 ммоль, 9.44 мл, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Воду (300 мл) добавляли в реакционный раствор, и смесь экстрагировали этилацетатом (300 мл × 2). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия (100 мл × 2), органическую фазу затем упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX040-5. 1H ЯМР (400 МГц, CDCl3) δ = 7.92 - 7.83 (м, 2H), 7.18 (д, J = 8.4 Гц, 1H), 5.50 (ушир.д, J = 6.1 Гц, 1H), 4.72 - 4.52 (м, 2H), 4.28 - 4.17 (м, 1H), 3.92 (с, 3H), 3.42 (с, 3H), 1.38 (с, 9H).
Стадия 5: синтез соединения WX040-6
В толуол (40 мл) медленно добавляли WX040-5 (4.01 г, 11.45 ммоль, 1 экв.), реагент Лоуссона (5.09 г, 12.59 ммоль, 1.1 экв.) и Boc2O (10.60 г, 48.55 ммоль, 11.15 мл, 4.24 экв.), и смесь нагревали до 110°C и непрерывно перемешивали в течение 12 часов. Реакционный раствор медленно выливали в насыщенный водный раствор хлорида натрия (30 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия (30 мл × 2), органическую фазу затем упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1~1:1), получая соединение WX040-6. 1H ЯМР (400 МГц, CDCl3) δ = 7.99 - 7.92 (м, 2H), 7.21 (д, J = 8.3 Гц, 1H), 6.02 (ушир.д, J = 7.8 Гц, 1H), 4.80 - 4.69 (м, 1H), 4.53 (дд, J = 6.3, 9.5 Гц, 1H), 4.22 (дд, J = 9.5, 11.3 Гц, 1H), 3.94 (с, 3H), 3.86 (с, 3H), 1.46 - 1.33 (м, 9H).
Стадия 6: синтез соединения WX040-7
В круглодонную колбу помещали ТГФ (8 мл) и H2O (2 мл). Затем медленно добавляли WX040-6 (800 мг, 2.18 ммоль, 1 экв.) и LiOH⋅H2O (183.23 мг, 4.37 ммоль, 2 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Разбавленную соляную кислоту добавляли по каплям в реакционный раствор до pH<3, и осадок отфильтровывали, получая соединение WX040-7. 1H ЯМР (400 МГц, CDCl3) δ = 7.89 - 7.79 (м, 1H), 7.25 - 7.20 (м, 1H), 7.02 - 6.95 (м, 1H), 6.17 - 6.02 (м, 1H), 4.88 - 4.74 (м, 1H), 4.56 (дд, J = 6.4, 9.7 Гц, 1H), 4.31 - 4.17 (м, 1H), 3.85 (с, 3H), 1.41 (с, 9H).
Стадия 7: синтез соединения WX040-8
В круглодонную колбу помещали N,N-диметилформамид (5 мл). Затем медленно добавляли WX040-7 (200 мг, 567.53 мкмоль, 1 экв.), NH4Cl (91.07 мг, 1.70 ммоль, 3 экв.), HATU (647.37 мг, 1.70 ммоль, 253.15 мкл, 3.00 экв.) и DIPEA (366.75 мг, 2.84 ммоль, 494.27 мкл, 5 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор напрямую очищали методом ВЭЖХ (Колонка: Xtimate C18 100 мм*30 мм*3 мкм; Подвижная фаза: [вода (0.225% муравьиная кислота)-ACN]; ацетонитрил%: 30%-60%, 8 мин), получая соединение WX040-8. 1H ЯМР (400 МГц, CDCl3) δ = 7.82 (с, 1H), 7.64 (ушир.д, J = 8.3 Гц, 1H), 7.22 (д, J = 8.3 Гц, 1H), 6.98 (с, 2H), 5.01 (ушир.с, 1H), 4.86 - 4.66 (м, 1H), 4.52 (дд, J = 6.3, 9.5 Гц, 1H), 4.30 - 4.11 (м, 1H), 3.86 (с, 3H), 1.40 (с, 9H).
Стадия 8: синтез соединения WX040-9
В круглодонную колбу помещали этилацетат (10 мл). Затем медленно добавляли WX040-8 (40 мг, 113.82 мкмоль, 1 экв.) и HCl/этилацетат (4M, 10.00 мл, 351.42 экв.), и смесь непрерывно перемешивали при 25°C в течение 1 часа. Реакционный раствор упаривали при пониженном давлении, получая соединение WX040-9. 1H ЯМР (400 МГц, ДМСО-d6) δ = 8.16 - 8.04 (м, 2H), 7.91 (д, J = 8.5 Гц, 1H), 7.57 (с, 1H), 7.37 (д, J = 8.3 Гц, 1H), 4.62 - 4.41 (м, 3H), 3.82 (с, 3H).
Стадия 9: синтез соединения WX040
В ДМСО (1 мл) медленно добавляли WX040-9 (30 мг, 119.38 мкмоль, 1 экв., HCl), BB-8 (24.26 мг, 119.38 мкмоль, 1 экв.), HATU (68.09 мг, 179.07 мкмоль, 1.5 экв.) и DIEA (46.28 мг, 358.13 мкмоль, 62.38 мкл, 3 экв.), и смесь непрерывно перемешивали при 25°C в течение 2 часов. Реакционный раствор напрямую очищали методом ВЭЖХ (Колонка: Phenomenex Gemini-NX 80 мм*30 мм*3 мкм; Подвижная фаза: [вода (10 мM NH4HCO3)-ACN]; ацетонитрил%: 28%-58%, 9 мин), получая соединение WX040. 1H ЯМР (400 МГц, CDCl3) δ =8.70 (ушир.д, J = 7.3 Гц, 1H), 8.04 (с, 1H), 7.69 (с, 1H), 7.39 - 7.36 (м, 1H), 7.31 - 7.28 (м, 7H), 5.39 (с, 2H), 5.20 (тд, J = 6.8, 11.0 Гц, 1H), 4.72 (дд, J = 6.5, 9.4 Гц, 1H), 4.31 (т, J = 10.3 Гц, 1H), 3.89 (с, 3H); LCMS m/z = 437.1 [M+1]+.
Пример 41. WX041
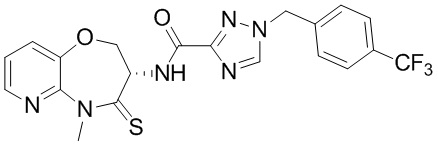
Путь синтеза:
Стадия 1: синтез соединения WX041-2
В круглодонную колбу помещали MeCN (20 мл) при 25°C. Затем медленно добавляли WX041-1 (2.68 г, 21.09 ммоль, 1 экв.), K2CO3 (5.83 г, 42.17 ммоль, 2 экв.) и 4-(хлорметил)-трифторметилбензол (4.92 г, 25.30 ммоль, 3.73 мл, 1.2 экв.). Реакционный раствор непрерывно перемешивали при 60°C в атмосфере азота 12 часов. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли и последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), органическую фазу затем упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:20-100:75), получая соединение WX041-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.17 (с, 1H), 7.65 (д, J = 8.0 Гц, 2H), 7.40 (д, J = 8.0 Гц, 2H), 5.49 (с, 2H), 4.08 - 3.91 (м, 3H).
Стадия 2: синтез соединения WX041-3
В круглодонную колбу помещали ТГФ (20 мл) и H2O (4 мл) при 25°C. Медленно добавляли WX041-2 (2.40 г, 8.41 ммоль, 1 экв.) и LiOH⋅H2O (1.06 г, 25.24 ммоль, 3 экв.). Реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 1 час. Реакционный раствор доводили до значения pH 6~7 разбавленной соляной кислотой (1 M), и осадок отфильтровывали, получая соединение WX041-3.
Стадия 3: синтез соединения WX041
В круглодонную колбу помещали N,N-диметилацетамид (5 мл) при 25°C. Медленно добавляли BB-4 (20 мг, 45.29 мкмоль, 1 экв.), T3P (43.23 мг, 67.94 мкмоль, 40.41 мкл, 50% чистота, 1.5 экв.), WX041-3 (18.42 мг, 67.94 мкмоль, 1.5 экв.) и DIPEA (17.56 мг, 135.87 мкмоль, 23.67 мкл, 3 экв.), и реакционный раствор непрерывно перемешивали при 25°C в атмосфере азота 2 часа. Реакционный раствор медленно выливали в воду (100 мл), и смесь экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли и последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), затем органическую фазу упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом препаративной ВЭЖХ (Колонка: Phenomenex Gemini-NX 80 мм*30 мм*3 мкм; Подвижная фаза: [вода (10 мM NH4HCO3)-ACN], получая соединение WX041. 1H ЯМР (400 МГц, CDCl3) δ = 8.77 (ушир.д, J = 7.5 Гц, 1H), 8.31 (ушир.д, J = 3.6 Гц, 1H), 8.07 (с, 1H), 7.65 - 7.45 (м, 3H), 7.33 (ушир.д, J = 8.0 Гц, 2H), 7.22 (дд, J = 4.8, 7.9 Гц, 1H), 5.40 (с, 2H), 5.23 - 4.97 (м, 1H), 4.72 (дд, J = 6.5, 9.4 Гц, 1H), 4.25 (т, J = 10.3 Гц, 1H), 3.87 (с, 3H); 19F ЯМР (377 МГц, CDCl3) δ = -43.60 (с, 1F); LCMS m/z =463.1 [M+1]+.
Пример 42. WX042
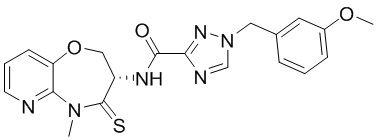
Путь синтеза:
Стадия 1: синтез соединения WX042-2
В MeCN (20 мл) добавляли WX042-1 (2 г, 15.74 ммоль, 1 экв.), 3-хлорметиланизол (2.46 г, 15.74 ммоль, 2.14 мл, 1 экв.) и K2CO3 (2.61 г, 18.88 ммоль, 1.2 экв.), и смесь нагревали при 50°C в течение 12 часов. Реакционный раствор фильтровали для удаления K2CO3. Осадок на фильтре промывали этилацетатом (15 мл × 3), и фильтрат промывали насыщенным водным раствором хлорида натрия (50 мл). Органическую фазу упаривали при пониженном давлении, получая сырой продукт. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир:этилацетат = 100:1-100:100), получая соединение WX042-2. 1H ЯМР (400 МГц, CDCl3) δ = 8.10 - 8.01 (м, 1H), 7.30 - 7.21 (м, 1H), 6.91 - 6.80 (м, 2H), 6.77 (с, 1H), 5.34 (с, 2H), 3.96 (с, 3H), 3.75 (с, 3H).
Стадия 2: синтез соединения WX042-3
В ТГФ (30 мл) добавляли WX042-2 (2.5 г, 10.11 ммоль, 1 экв.). Затем добавляли раствор LiOH⋅H2O (509.17 мг, 12.13 ммоль, 1.2 экв.) в H2O (3 мл). Реакционную смесь перемешивали при 25°C в течение 3 часов. ТСХ-мониторинг (петролейный эфир:этилацетат = 1:1) показал, что исходные соединения полностью прореагировали. Реакционный раствор доводили до значения pH 6 добавлением 1M раствора соляной кислоты, и смесь упаривали при пониженном давлении для удаления растворителя. Добавляли толуол (10 мл), и смесь упаривали при пониженном давлении еще раз для удаления толуола, получая соединение WX042-3. 1H ЯМР (400 МГц, CDCl3) δ = 7.19 (с, 4H), 7.21 - 7.17 (м, 1H), 4.65 (с, 2H), 3.79 (с, 3H); LCMS m/z = 233.9 [M+1]+.
Стадия 3: синтез соединения WX042
В N,N-диметилформамид (5 мл) добавляли BB-4 (94.67 мг, 214.39 мкмоль, 1 экв.), WX042-3 (50 мг, 214.39 мкмоль, 1 экв.), T3P (163.71 мг, 257.26 мкмоль, 153.00 мкл, 50% чистота, 1.2 экв.) и DIPEA (41.56 мг, 321.58 мкмоль, 56.01 мкл, 1.5 экв.), и смесь нагревали при 25°C в течение 2 часов. LCMS анализ показал, что исходные соединения полностью прореагировали. Реакционный раствор упаривали при пониженном давлении до объема примерно 5 мл. Остаток очищали методом ВЭЖХ (система с муравьиной кислотой), получая соединение WX042. 1H ЯМР (400 МГц, CDCl3) δ = 8.81 (ушир.д, J = 7.0 Гц, 1H), 8.37 (дд, J = 1.6, 4.6 Гц, 1H), 8.04 (с, 1H), 7.57 (дд, J = 1.6, 7.9 Гц, 1H), 7.33 - 7.27 (м, 2H), 7.00 - 6.69 (м, 3H), 5.36 (с, 2H), 5.22 (тд, J = 6.7, 11.1 Гц, 1H), 4.79 (дд, J = 6.3, 9.5 Гц, 1H), 4.32 (дд, J = 9.5, 11.0 Гц, 1H), 3.93 (с, 3H), 3.79 (с, 3H).
Пример теста 1: In vitro тестирование
Стадии тестирования:
1.1 Реагенты
1.2 Приборы и материалы
CULTURPLATE-384 +LID /50W Brand PerkinElmer cat#6007680
Планшет-ридер Envision (Perkin Elmer, Cat#2104-0010)
1.3 Приготовление реагентов
(1) 1 × DPSB: В 100 мл 10×DPBS добавляли 900 мл деионизованной воды.
(2) 1640 полная среда: В 89 мл среды RPMI-1640 добавляли 10 мл FBS и 1 мл смеси пенициллин/стрептомицин
2. Стадии тестирования
1. Клетки U937 центрифугировали, и затем среду отбрасывали. Клетки ресуспендировали в полной смеси 1640 и проводили подсчет. Концентрацию клеток доводили до 5×105 клеток/мл для использования, и клетки распределяли в соответствии с тестируемыми соединениями.
2. В полученную суспензию клеток U937 добавляли QVD-OPh в финальной концентрации 25 мкM и TNF-α в финальной концентрации 100 нг/мл, после этого смесь осторожно перемешивали.
3. В 384-луночный планшет добавляли 30 мкл/лунку клеток U937, содержащих 25 мкM QVD-OPh и TNF-α в финальной концентрации 100 нг/мл, и готовили лунки пустого контроля без QVD-OPh и TNF-α.
4. В группу с добавлением соединения добавляли 10 мкл тестируемого соединения в 4× концентрации относительно финальной, а в лунки пустого контроля добавляли ДМСО в соответствующей концентрации.
5. 384-луночный планшет помещали в инкубатор с 37°C и 5% CO2 на 24 часа.
6. Через 24 часа 384-луночный планшет вынимали и уравновешивали при комнатной температуре 20 минут.
7. В каждую лунку добавляли 40 мкл CTG, и затем планшет встряхивали на шейкере в темноте в течение 2 минут.
8. Планшет инкубировали при комнатной температуре в темноте 10 минут и затем считывали на планшет-ридере Envision.
Результаты тестирования:
Результаты тестирования соединений по настоящему изобретению представлены в таблице 1.
Заключение по результатам тестирования
Приведенные выше в таблице результаты показывают, что соединения по настоящему изобретению демонстрируют хорошую ингибирующую активность в клеточном анализе активности ингибирования TNFα/QVD-OPh-индуцированного запрограммированного некроза клеток (некроптоз).
Пример теста 2: Тестирование активности in vitro на мышином RIP1
Линия клеток
L929 (приобретено в ATCC, Item No.: CCL-1)
Реагент
Прибор
Планшет-ридер (Perkin Elmer, Envision)
Приготовление реагентов
Полная среда для клеток L929: EMEM + 10% лошадиной сыворотки (соотношение по объему) + 1% двойной смеси антибиотиков (соотношение по объему)
Стадии тестирования
1. Выращенные клетки L929 обрабатывали трипсином, центрифугировали, ресуспендировали в полной среде для выращивания и затем проводили подсчет. Концентрацию клеток доводили до 1×105/мл путем разведения полной средой для выращивания.
2. Суспензию клеток добавляли в 96-луночный планшет в концентрации 100 мкл на лунку и инкубировали при 37°C и 5% CO2 в течение 24 часов.
3. Разные концентрации тестируемых соединений добавляли в каждую лунку, а в контрольную группу добавляли диметилсульфоксид. Планшет инкубировали при 37°C и 5% CO2 в течение 1 часа.
4. В каждую лунку добавляли TNF-α в финальной концентрации 50 нг/мл и Q-VD в финальной концентрации 50 мкM, и планшет инкубировали при 37°C и 5% CO2 в течение 24 часов.
5. В каждую лунку добавляли 50 мкл CellTiter Glo флуоресцентных клеток, и планшет встряхивали в темноте 10 мин.
6. Планшет считывали на планшет-ридере.
Результаты тестирования
Результаты тестирования соединений по настоящему изобретению представлены в таблице 2.
Заключение по результатам тестирования
Приведенные выше в таблице результаты показывают, что соединения по настоящему изобретению демонстрируют хорошую ингибирующую активность в клеточном анализе активности ингибирования TNFα/QVD-OPh-индуцированного запрограммированного некроза клеток (некроптоз) на мышиных клетках L929.
Пример теста 3: Исследование фармакокинетики соединения
Стадии тестирования
Отбирали восемь здоровых взрослых мышей C57BL/6 (возраст 6-9 недель, приобретены у Shanghai Lingchang Biotechnology Co., Ltd.) и случайным образом распределяли их в две группы по 4 мыши в каждой группе (n = 2). Одной группе вводили 0.5 мг/кг тестируемого соединения посредством внутривенной инъекции, а другой группе вводили 1 мг/кг тестируемого соединения через зонд для принудительного кормления. Брали пробы крови соответственно у животных из «внутривенной» группы и из «пероральной» группы через 0.083, 0.25, 0.5, 1.0, 2.0, 4.0, 6.0, 8.0, 24 часа после введения. Концентрацию соединения в крови определяли с помощью метода LC-MS/MS. Фармакокинетические параметры вычисляли с помощью программы WinNonlin™ Version 6.3 (Pharsight, Mountain View, CA), используя некомпартментный метод и линейный логарифмический метод трапеций. AUC0-last - это площадь под кривой зависимости концентрации в плазме крови от времени, с точки 0 до момента времени последней детектируемой концентрации; Cmax - это пиковая концентрация; Tmax - это время достижения пика; T1/2 - это время полураспада; CL - это скорость выведения; Tlast - это время, соответствующее последней поддающейся количественному определению концентрации; p.o. означает «перорально»; i.v. означает внутривенную инъекцию; и F% - это пероральная бодоступность.
Результаты тестирования
Результаты тестирования соединения по настоящему изобретению представлены в таблице 3.
Заключение по результатам тестирования
Соединение по настоящему изобретению демонстрирует хорошие фармакокинетические характеристики, концентрацию в плазме in vivo и биодоступность.
Пример теста 4: Исследование in vivo эффективности в мышиной модели синдрома системной воспалительной реакции (SIRS), запускаемой фактором некроза опухоли (TNF-α)
Реагент
Приборы и материалы
1) Электронные весы: бренд Changzhou Tianzhiping Instrument Equipment Co., Ltd., модель JY20002
2) Анальный термометр: бренд PHYSITEMP, модель BAT-12
Приготовление реагентов
1) TNF-α: TNF-α недолго центрифугировали (10000 об/мин, 30с) перед тем как открыть крышку. Один флакон TNF-α (1 мг) растворяли в 1 мл стерильной воды, получая исходный раствор с концентрацией 1 мг/мл. Исходный раствор хранили при 4°C, и он может храниться в течение недели. 1000 мкл исходного раствора отвешивали и добавляли в 5667 мкл стерильной воды, затем добавляли и растворяли 333 мг трегалозы, получая рабочий раствор.
2) zVAD: 20 мг zVAD отвешивали и добавляли в 480 мкл ДМСО, и смесь перемешивали на вихревой мешалке, обрабатывали ультразвуком и нагревали при 37°C, затем добавляли 15.52 мл физиологического солевого раствора. Получали рабочий раствор с концентрацией 1.25 мг/мл.
Стадии тестирования
1) Мышей адаптировали к условиям в течение 3 дней, затем случайным образом группировали по клеткам, по 5 мышей в каждой клетке. Прикрепляли бирки на ухо.
2) Сначала мышам вводили тестируемое соединение через желудочный зонд в соответствии с их весом тела, и затем посредством интраперитонеальной инъекции вводили zVAD в дозировке 250 мкг на мышь. После интраперитонеальной инъекции zVAD немедленно измеряли анальную температуру (0 час) и фиксировали время. Время, когда мышам был введен zVAD, принимали за нулевой час (0 час).
3) Через 15 минут инъекционно вводили TNF-α в хвостовую вену в дозировке 200 мкл на мышь, и через 15 минут измеряли анальную температуру (0.5 час).
4) Через 1 час посредством интраперитонеальной инъекции вводили zVAD в дозировке 125 мкг на мышь, и немедленно измеряли анальную температуру (1 час). Анальную температуру снова измеряли через 2 часа, 3 часа и 4 часа.
Результаты тестирования
Результаты тестирования соединения по настоящему изобретению представлены в таблице 4.
WX009 (10 мг/кг)
Заключение по результатам тестирования
Соединение по настоящему изобретению показало очень хорошее защитное действие в мышиной модели ингибирования синдрома системной воспалительной реакции (SIRS), запускаемой TNF-α/zVAD, где снижение температуры тела у мышей значительно уменьшалось, и коэффициент выживаемости животных вырастал с 0% до 100%.
Пример теста 5: Тест проницаемости in vitro на клетках Caco-2
Культура клеток
Для инкубирования клеток использовали MEM (минимальную питательную среду) с добавлением 2 мM L-глутамина, 10% фетальной бычьей сыворотки (FBS), 100 Ед/мл пенициллин-G и 100 мкг/мл стрептомицина. Условия для инкубирования клеток: 37±1°C, 5% CO2 и насыщенная влажность. Когда клетки вырастали до 80-90% конфлюентности, добавляли трипсин (0.05%, вес./об.)/ЭДТА (0.02%, вес./об.). Клетки высевали в BD Falcon's Transwell-96-луночный планшет с плотностью 1×105 клеток/см2. Клетки инкубировали в инкубаторе с диоксидом углерода 22 дня и затем использовали для тестирования транспорта.
Тестирование транспорта
Сбалансированный солевой раствор Хенкса, содержащий 10 мM HEPES (pH 7.40±0.05) использовали в данном тесте в качестве буфера для тестирования транспорта. Исследовали двухсторонний транспорт тестируемого соединения и лекарственного средства дигоксин в концентрации 2 мкM, каждый из них в двух повторностях. Исследовали транспорт фенотерола и пропранолола от апикальной стороны к базолатеральной стороне (A-B). Концентрацию ДМСО в системе для инкубирования поддерживали ниже 1%. После добавления образца планшет с клетками инкубировали при 37±1°C, 5% CO2 и насыщенной влажности в течение 120 минут. Все образцы перемешивали на вихревой мешалке и затем центрифугировали при 3220 об/мин и 20°C в течение 20 минут. Контрольные и тестируемые образцы разводили 1:1 (об.:об.) ультрачистой водой и затем хранили при 4°C. Для анализа и тестирования применяли метод жидкостной хроматографии с тандемной масс-спектрометрией (LC/MS/MS).
Анализ образцов
В этом тестировании применяли метод жидкостной хроматографии с тандемной масс-спектрометрией (LC/MS/MS) для полуколичественного анализа соотношения площадей пиков тестируемого соединения и контрольных веществ фенотерола, пропранолола и дигоксина относительно внутреннего стандарта в исходном растворе, в растворе акцепторного отсека и в супернатанте дозировочной лунки.
Обсчет результатов
Кажущийся коэффициент проницаемости (Papp, см/с), коэффициент эффлюкса и коэффициент выделения вычисляли по приведенным ниже формулам.
Кажущийся коэффициент проницаемости (Papp, см/с) вычисляли по следующей формуле:
Papp = (dCr/dt) × Vr / (A × C0)
где dCr/dt это кумулятивная концентрация соединения с акцепторной стороны на единицу времени (мкM/с); Vr это объем раствора с акцепторной стороны (объемы растворов с апикальной стороны и с базолатеральной стороны составляли 0.075 мл и 0.250 мл, соответственно); A - это относительная площадь поверхности монослоя клеток (0.0804 см2); C0 - это соотношение площадей пиков начальной концентрации (нM) тестируемого соединения и контрольных соединений с донорной стороны.
Коэффициент эффлюкса вычисляли по следующей формуле:
Коэффициент эффлюкса = Papp (BA) / Papp (AB)
Коэффициент выделения вычисляли по следующей формуле:
% коэффициент выделения = 100 × [(Vr × Cr) + (Vd × Cd)] / (Vd × C0),
где C0 это начальная концентрация (нM) тестируемого соединения с донорной стороны или соотношение площадей пиков контрольных соединений; Vd - это объем с донорной стороны (с апикальной стороны это 0.075 мл, а с базолатеральной стороны это 0.250 мл); Cd и Cr - это соотношение площадей пиков финальных концентраций (нM) тестируемого соединения или контрольных соединений с донорной стороны и акцепторной стороны, соответственно.
Результаты тестирования
Результаты тестирования соединения по настоящему изобретению представлены в таблице 5.
Заключение по результатам тестирования
Соединение по настоящему изобретению представляет собой соединение с высокой проницаемостью и низким коэффициентом эффлюкса.
Пример теста 6. Определение коэффициента связывания с белками плазмы (PPB)
Взвешивали 796 мкл бланковой плазмы крови человека, SD-крыс и CD-1 мышей (плазму приобретали у Bioreclamation IVT), и добавляли рабочий раствор тестируемого соединения или рабочий раствор варфарина, так чтобы финальная концентрация тестируемого соединения и варфарина в образце плазмы составляла 2 мкM. Образцы тщательно перемешивали. Финальная концентрация ДМСО в органической фазе составляла 0.5%. 50 мкл образцов плазмы с тестируемым соединением и варфарином переносили пипеткой в лунки планшета, и немедленно добавляли соответствующий объем бланковой плазмы или буфера, так чтобы финальный объем в каждой лунке с образцом составлял 100 мкл, где соотношение по объему плазма:буфер для диализа составляло 1:1. Затем добавляли в эти образцы стоп-реагент. Полученные образцы использовали как образцы T0 для определения выделения и стабильности. Образцы плазмы с тестируемым соединением и варфарином помещали в верхний отдел каждой диализной ячейки, а бланковый буфер для диализа помещали в соответствующий нижний отдел диализной ячейки. Диализный планшет затем герметизировали воздухопроницаемой мембраной, помещали в увлажненный инкубатор с 5% CO2 и инкубировали при встряхивании при 100 об/мин при 37°C в течение 4 часов. После окончания диализа 50 мкл образца диализного буфера и образца диализной плазмы переносили пипеткой в новый планшет для образцов. Добавляли в образцы соответствующий объем соответствующей бланковой плазмы или буферного раствора, так чтобы финальный объем в каждой лунке с образцом составлял 100 мкл, где соотношение по объему плазма:буфер для диализа составляло 1:1. Во всех образцах проводили осаждение белка и затем анализировали методом LC/MS/MS. Процент несвязанного (% несвязанного), процент связанного (% связанного), и процент выделения (% выделения) для тестируемого соединения вычисляли с помощью следующих формул:
% несвязанного = 100 * FC / TC,
% связанного = 100 - % несвязанного, и
% выделения = 100 * (FC + TC) / T0,
где FC это концентрация соединения в буферном конце диализной ячейки; TC это концентрация соединения в плазменном конце диализной ячейки; и T0 это концентрация соединения в образце плазмы в нулевой момент времени.
Результаты тестирования
Результаты тестирования соединения по настоящему изобретению представлены в Таблице 6.
Заключение по результатам тестирования
Соединение по настоящему изобретению имеет приемлемый коэффициент связывания с белками плазмы человека, крысы и мыши, и процент содержания данного лекарственного соединения в несвязанном состоянии в плазме крови человека составляет 5.7%.
Пример теста 7: Определение фармакокинетических параметров на собаках
Стадии тестирования
Отбирали четыре здоровых взрослых собаки породы бигль и случайным образом распределяли их на две группы. Одной группе вводили исследуемое соединение посредством внутривенной инъекции, а другой группе вводили исследуемое соединение через желудочный зонд. Брали пробы крови соответственно у животных из «внутривенной» группы и из «пероральной» группы через 0.083, 0.25, 0.5, 1.0, 2.0, 4.0, 6.0, 8.0, 24 часов после введения. Концентрацию в крови определяли методом LC-MS/MS. Фармакокинетические параметры вычисляли с помощью программы WinNonlin™ Version 6.3 (Pharsight, Mountain View, CA), используя некомпартментный метод и линейный логарифмический метод трапеций. AUC0-last - это площадь под кривой зависимости концентрации в плазме крови от времени, с точки 0 до момента времени последней детектируемой концентрации; Cmax - это пиковая концентрация; Tmax - это время достижения пика; T1/2 - это время полураспада; CL - это скорость выведения; Vdss это кажущийся объем распределения; p.o. означает «перорально»; i.v. означает внутривенную инъекцию; и F% - это пероральная бодоступность.
Результаты тестирования
Результаты тестирования соединений по настоящему изобретению представлены в таблице 7.
Заключение по результатам тестирования
Соединения по настоящему изобретению показали хорошие фармакокинетические параметры и высокую концентрацию in vivo в тестах на собаках. В частности, время полураспада соединений по настоящему изобретению достигает примерно 5 часов, что относительно идеально.
Пример теста 8: Исследование киназной селективности у соединения по настоящему изобретению
Целью данного исследования было определить in vitro ингибирующую активность WX009 в отношении разных киназ. Исследованные в этом тесте киназы включали 413 киназ, и метод определения активности был предоставлен компанией Eurofins Pharma Discovery Service. В данном тесте использовали метод определения активности киназ с помощью 33P-радиоактивной метки. Концентрация WX009 составляла 1 мкM, а концентрация АТФ составляла 10 мкM. Результаты исследования представлены в Таблице 8.
Заключение по результатам тестирования: Соединение по настоящему изобретению показывает высокоспецифичную киназную селективность и оказывает высокоактивное ингибирующее действие почти исключительно на RIP1. Поэтому соединение по настоящему изобретению имеет низкий риск воздействия на другие киназные мишени, не являющиеся целевыми.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ FXIA, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2020 |
|
RU2802878C1 |
| СПОСОБ СИНТЕЗА БИЛИРУБИНА | 2022 |
|
RU2838556C2 |
| ПРОИЗВОДНОЕ НИТРОИМИДАЗОЛА ПРОТИВ ТУБЕРКУЛЕЗА ЛЕГКИХ | 2016 |
|
RU2675622C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2016 |
|
RU2740019C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| ХИНОЛИНИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2803116C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2567238C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2675857C2 |
| ПРОИЗВОДНОЕ С КОНДЕНСИРОВАННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРА A | 2018 |
|
RU2748993C1 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
Изобретение относится к соединению, имеющему формулу (I), или его стереоизомеру, или фармацевтически приемлемой соли, которые обладают ингибирующей активностью в отношении RIP-1 киназы. В формуле (I) T1 выбран из N и CR1t; Т2 выбран из N и CR2t; Т3 представляет собой CR3t; Т4 представляет собой CR4t; E1 выбран из C(R1e)2 и О; цикл А выбран из 1,2,4-триазолила,  где 1,2,4-триазолил,
где 1,2,4-триазолил,  необязательно замещены 1, 2 или 3 галогенами или С1-3 алкилами; цикл В выбран из фенила и
необязательно замещены 1, 2 или 3 галогенами или С1-3 алкилами; цикл В выбран из фенила и  L выбран из простой связи, О и C1-3 алкилена; R1 выбран из Н и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rb; R1t, R2t, R3t и R4t каждый независимо выбраны из Н, F, Cl, Br, I, CN, C1-3 алкила, СООН и -C(=O)NH2; R1e каждый независимо выбран из Н и C1-3 алкила; R2 каждый независимо выбран из Н, F, Cl, Br, I, CN, C1-3 алкила и C1-3 алкокси-группы, где C1-3 алкил и C1-3 алкокси-группа необязательно замещены 1, 2 или 3 заместителями Rf; n равен 1, 2, 3, 4 или 5; Rb и Rf каждый независимо выбран из F, Cl, Br, I и D. Изобретение относится также к конкретным соединениям и к применению указанных соединений в производстве лекарственного средства для лечения заболевания, связанного с RIP-1 киназой. 3 н. и 13 з.п. ф-лы, 8 табл., 42 пр.
L выбран из простой связи, О и C1-3 алкилена; R1 выбран из Н и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rb; R1t, R2t, R3t и R4t каждый независимо выбраны из Н, F, Cl, Br, I, CN, C1-3 алкила, СООН и -C(=O)NH2; R1e каждый независимо выбран из Н и C1-3 алкила; R2 каждый независимо выбран из Н, F, Cl, Br, I, CN, C1-3 алкила и C1-3 алкокси-группы, где C1-3 алкил и C1-3 алкокси-группа необязательно замещены 1, 2 или 3 заместителями Rf; n равен 1, 2, 3, 4 или 5; Rb и Rf каждый независимо выбран из F, Cl, Br, I и D. Изобретение относится также к конкретным соединениям и к применению указанных соединений в производстве лекарственного средства для лечения заболевания, связанного с RIP-1 киназой. 3 н. и 13 з.п. ф-лы, 8 табл., 42 пр.
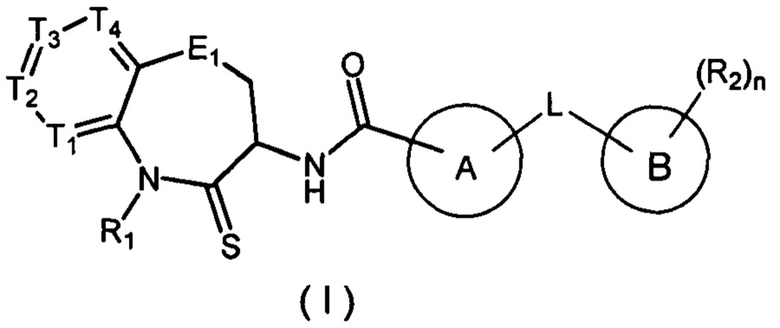
1. Соединение, имеющее формулу (I), или его стереоизомер, или фармацевтически приемлемая соль
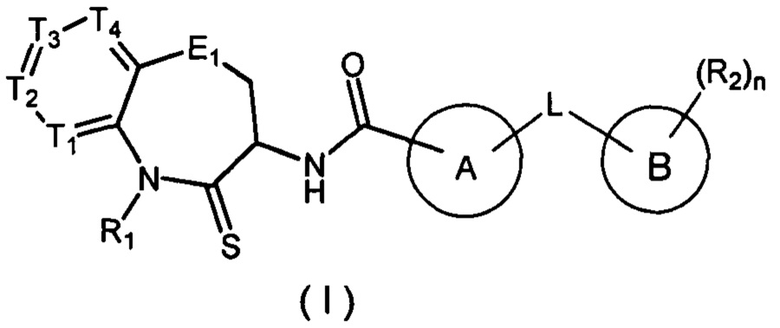 ,
,
где
T1 выбран из N и CR1t;
Т2 выбран из N и CR2t;
Т3 представляет собой CR3t;
Т4 представляет собой CR4t;
E1 выбран из C(R1e)2 и О;
цикл А выбран из 1,2,4-триазолила, 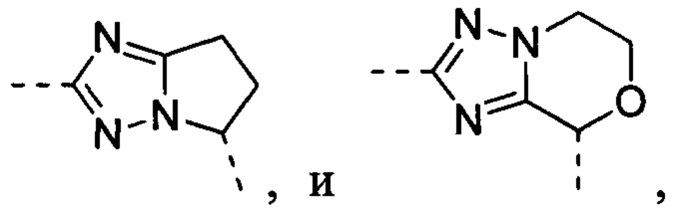 где 1,2,4-триазолил,
где 1,2,4-триазолил, 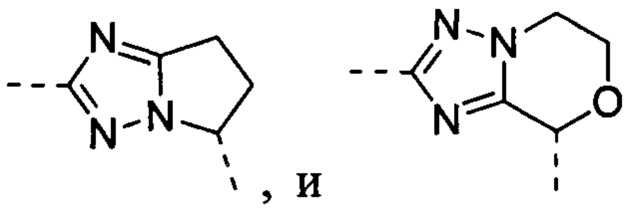 необязательно замещены 1, 2 или 3 галогенами или С1-3 алкилами;
необязательно замещены 1, 2 или 3 галогенами или С1-3 алкилами;
цикл В выбран из фенила и 
L выбран из простой связи, О и C1-3 алкилена;
R1 выбран из Н и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 заместителями Rb;
R1t, R2t, R3t и R4t каждый независимо выбраны из Н, F, Cl, Br, I, CN, C1-3 алкила, СООН и -C(=O)NH2;
R1e каждый независимо выбран из Н и C1-3 алкила;
R2 каждый независимо выбран из Н, F, Cl, Br, I, CN, C1-3 алкила и C1-3 алкокси-группы, где C1-3 алкил и C1-3 алкокси-группа необязательно замещены 1, 2 или 3 заместителями Rf;
n равен 1, 2, 3, 4 или 5;
Rb и Rf каждый независимо выбран из F, Cl, Br, I и D.
2. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 1, где R1 выбран из Н и СН3, где СН3 необязательно замещен 1, 2 или 3 заместителями Rb.
3. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 2, где R1 выбран из Н, СН3 и CD3.
4. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 1, где R1t, R2t, R3t и R4t каждый независимо выбраны из Н, F, Cl, Br, I, CN, СН3, СООН и -C(=O)NH2.
5. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 4, где R1t, R2t, R3t и R4t каждый независимо выбраны из Н, F, Cl, Br, I, CN и СН3.
6. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 1, где R1e каждый независимо выбран из Н и СН3.
7. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 1 или 6, где E1 выбран из СН2 и О.
8. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 1, где R2 каждый независимо выбран из Н, F, Cl, Br, I, CN, СН3 и ОСН3, где СН3 и ОСН3 необязательно замещены 1, 2 или 3 заместителями Rf.
9. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 8, где R2 каждый независимо выбран из Н, F, Cl, Br, I, CN, СН3, ОСН3, CF3 и OCF3.
10. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 1, где цикл А выбран из 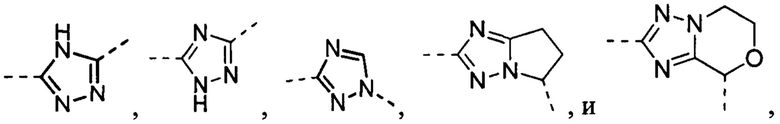 где
где 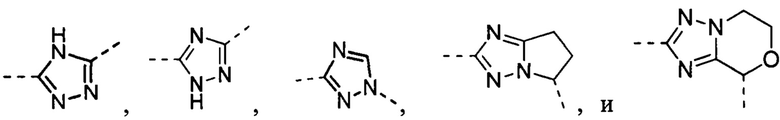 необязательно замещены 1, 2 или 3 галогенами или С1-3 алкилами.
необязательно замещены 1, 2 или 3 галогенами или С1-3 алкилами.
11. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 1, где L выбран из простой связи, -СН2- и -О-.
12. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому из пп. 1-3, 8 и 9, где соединение выбрано из
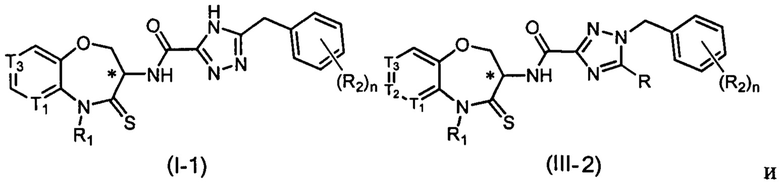
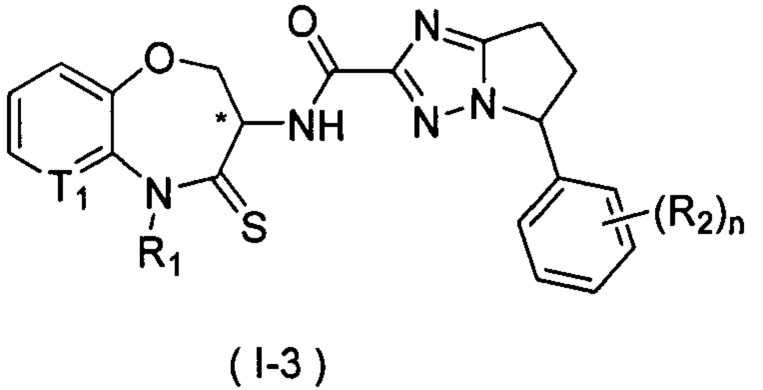 ,
,
где
n, T1, Т2 и Т3 имеют значения, указанные в п. 1;
R1 имеет значение, указанное в п. 1, 2 или 3;
R2 имеет значение, указанное в п. 1, 8 или 9;
R выбран из Н и галогена;
атом углерода с "*" представляет собой хиральный атом углерода, который существует в форме индивидуального (R) или (S) энантиомера или смеси, обогащенной одним энантиомером.
13. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 12, где соединение выбрано из
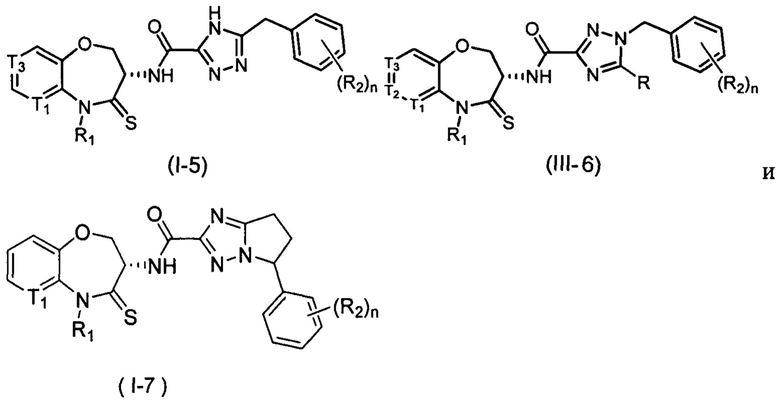 ,
,
где
n, T1, Т2 и Т3 имеют значения, указанные в п. 1;
R1 имеет значение, указанное в п. 1, 2 или 3;
R2 имеет значение, указанное в п. 1, 8 или 9;
R выбран из Н и галогена.
14. Соединение, имеющее формулу:
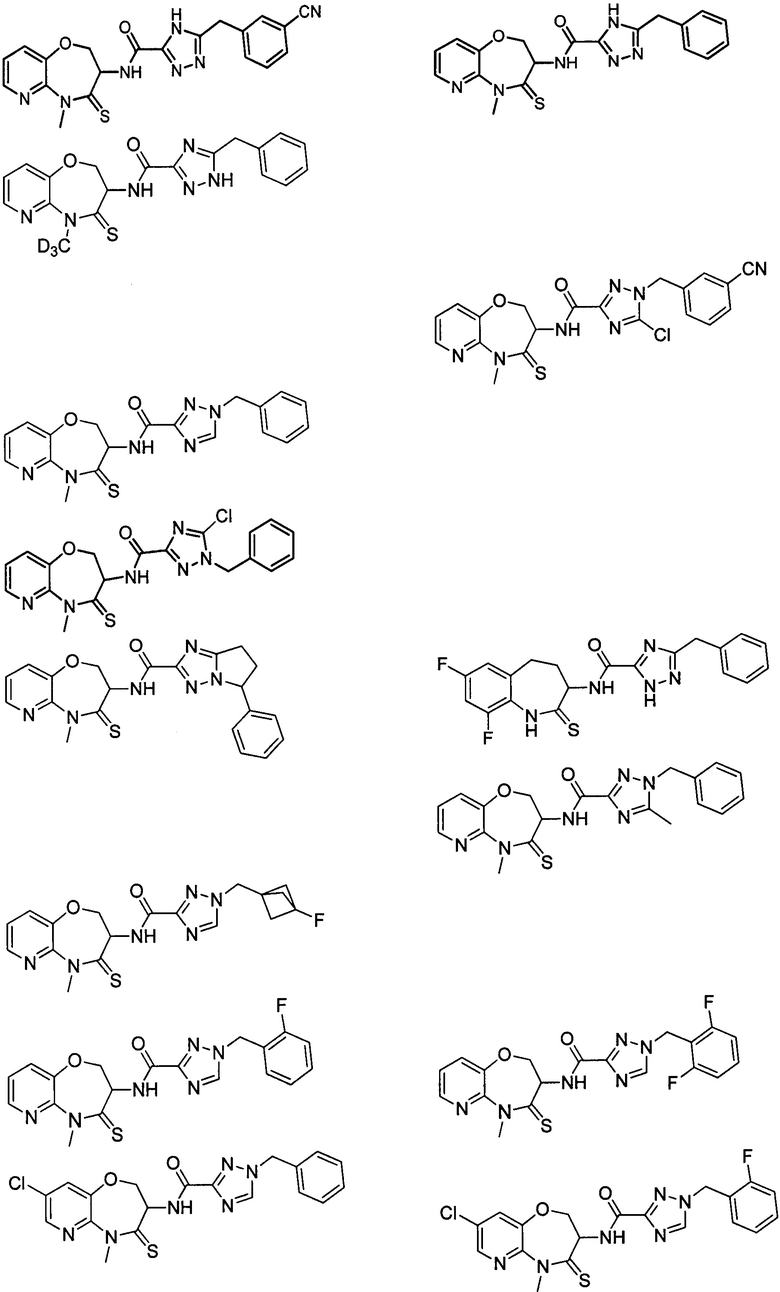
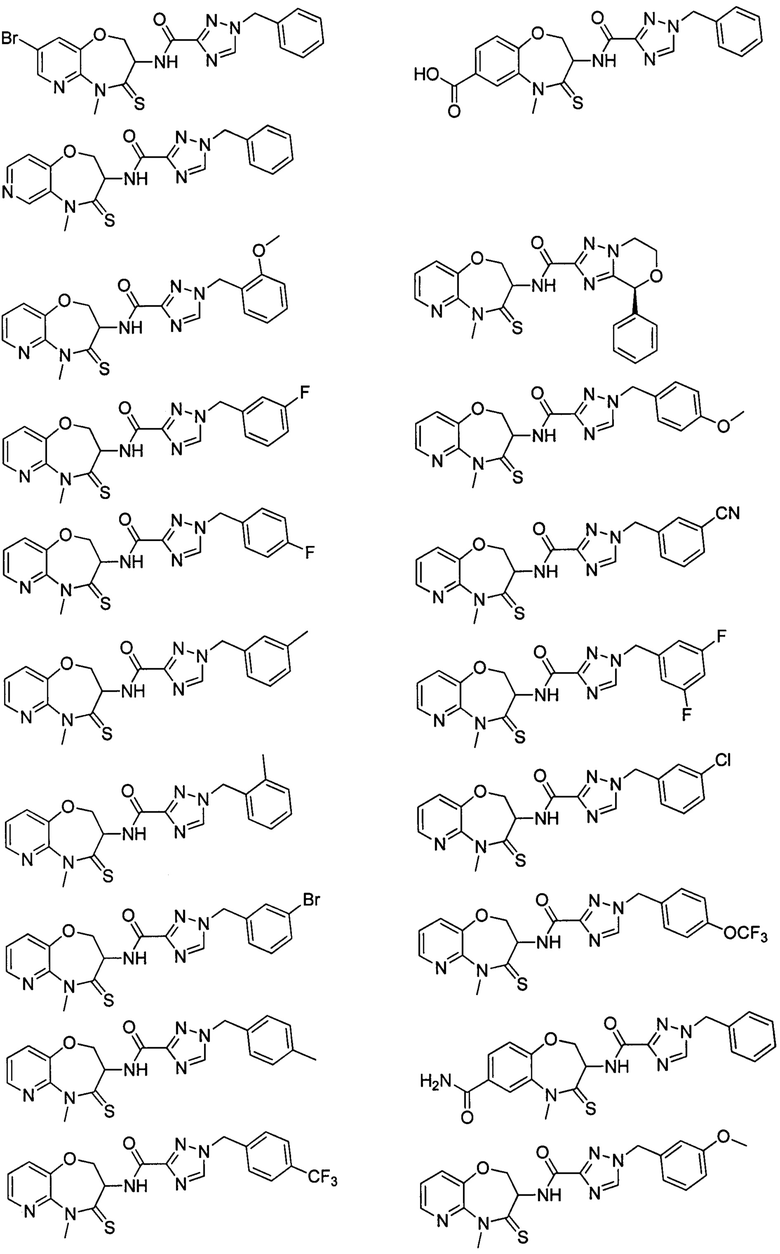
или его стереоизомер, или фармацевтически приемлемая соль.
15. Соединение или его стереоизомер, или фармацевтически приемлемая соль по п. 14, где соединение выбрано из следующих:
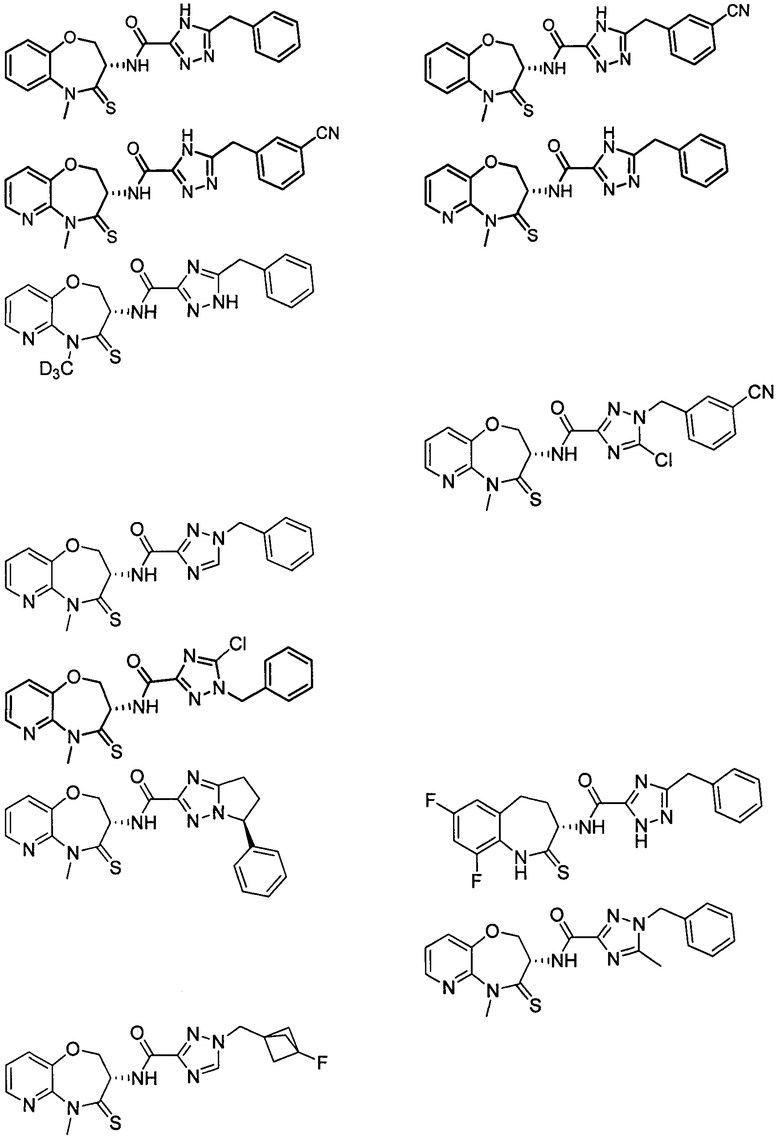
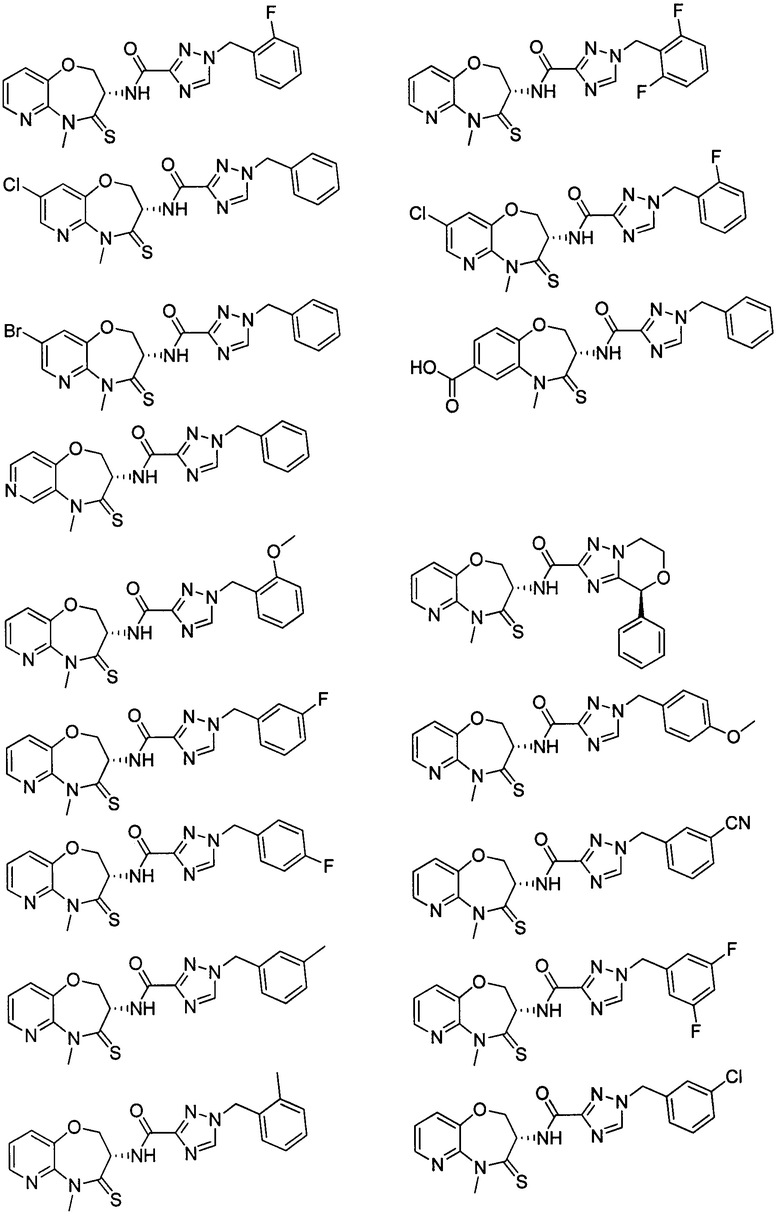
 .
.
16. Применение соединения или его стереоизомера, или фармацевтически приемлемой соли по любому из пп. 1-15 в производстве лекарственного средства для лечения заболевания, связанного с RIP-1 киназой.
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Механический подаватель стеблей к декортикаторам | 1932 |
|
SU28991A1 |
| CN 104418866 А, 18.03.2015 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |

