Изобретение относится к новому способу получения лосартана, производного имидазола с химическим названием 2-н-бутил-4-хлор-5-гидрoкcимeтил-1-{[2'-(1H-тeтpaзoл-5-ил)бифeнил-4-]мeтил}имидaзoл, а также его фармакологически эффективным солям. Кроме того, изобретение относится к новым промежуточным продуктам, которые подходят для получения лосартана, а также к новым способам получения промежуточных соединений, которые подходят для получения лосартана.
Лосартан и эффективные и экономические способы его получения представляют большой интерес, так как лосартан оказался потенциальным эффективным веществом для борьбы с высоким кровяным давлением у млекопитающих, включая человека, и появляющимися в результате этого заболеваниями.
Лосартан и его получение были описаны впервые в ЕР-А-253310. Синтез включал в качестве существенной стадии N-алкилирование, реакцию имидазола, например, с производным бромметил-бифенила (ЕР 253310 В1, стр.213, п.6).
В ЕР-А-291969 описаны тритил-защищенные производные тетразола, которые подходят для получения лосартана.
В WO 03/093262 описано получение лосартана из тритил-защищенных производных тетразола путем отщепления защитных групп.
Получение лосартан-калия, его обычной формы, имеющейся в продаже, из лосартана описывалось неоднократно (ср., например, ЕР 324377 А, стр.191, пример 316, ч.D и WO 95/17396, стр.18, пример 4 и стр.24, пример 9, стадия С).
Названные выше пути синтеза можно, однако, улучшить для получения лосартана в промышленных масштабах, так как общий выход оставляет желать лучшего.
Все этапы синтеза являются общими, сначала получают производное 1-Н-имидазола, которое затем алкилируют в 1 положении. При такой реакции имеется возможность образования двух изомеров в зависимости от того, какой из двух атомов азота алкилируется.
Из J. Org. Chem. 1997 62(24), 8449-8454 (см. таблицу 1) известно целевое строение производного имидазола, алкилированного в 1 положении, из N-однозамещенного амидина. О получении подходящих предварительных стадий для синтеза лосартана, однако, не говорилось.
Поэтому задачей было найти новые способы синтеза и приготовить промежуточные продукты для получения лосартана и его фармакологически эффективных солей. В частности, задачей изобретения являются новые способы синтеза и приготовление промежуточных продуктов для получения лосартана и его фармакологически эффективных солей, с помощью которых лосартан можно получить с высоким общим выходом продукта.
Кроме того, задачей изобретения являются новые способы синтеза и приготовление промежуточных продуктов для получения лосартана и его фармакологически эффективных солей, которые можно получить в технических масштабах, используя незначительную аппаратуру. Кроме того, должны использоваться промышленно хорошо подходящие исходные вещества, а токсичные вещества или вещества с обязательной маркировкой не должны применяться.
В связи с этим были найдены описанные объекты.
Центральным аспектом изобретения является получение соединения общей формулы I

где R1 обозначает радикал R1a или радикал R1b.
В отношении R1a речь идет о радикале общей формулы II,
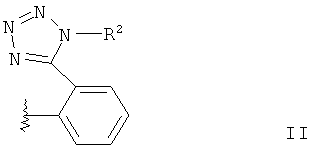
где R2 обозначает защитную группу тетразола.
В общей формуле II «линия в виде змейки» обозначает точку присоединения, например к соединению согласно общей формуле I.
Подходящие защитные группы тетразола в радикале приведенной выше общей формулы II известны из ЕР-А-291969 и WO 03/093262 (ср., там заместитель триарилметил в соединении общей формулы (II)). Подходящими защитными группами тетразола являются, в частности, трифенилметил или трет-бутил.
Что касается радикала R1b общей формулы I, то речь идет о радикале, который находится в состоянии связать группу фенилена соединения общей формулы I при помощи С-С-связи с другой группой арила.
Что касается радикала R1b общей формулы I, то речь идет о радикале, который в состоянии связать группу фенилена соединения общей формулы I путем реакции с дополнительным радикалом R3, компонент соединения общей формулы III, содержащего другую единицу фенилена,

где R4 обозначает радикал общей формулы II,
при образовании С-С-связи между группой фенилена соединения общей формулы I и группой фенилена соединения общей формулы III. С-С-связь возникает при этом обычно при отщеплении радикалов R1b и R3.
Соединение общей формулы I получают в результате реакции обмена соединения общей формулы IV

где R5
- в случае, если R1 в формуле I является радикалом R1a, обозначает радикал общей формулы II, и
- в случае, если R1 в формуле является радикалом R1b, имеет то же значение, что и радикал R1b в формуле I,
с соединением общей формулы V,
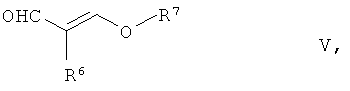
где R6 является галогеном из группы Сl, Вr, I, предпочтительно Br, a R7 обозначает разветвленную или неразветвленную группу C1-C6-алкила, предпочтительно группу изопропила.
Описанную выше реакцию (реакция взаимодействия соединения общей формулы IV с соединением общей формулы V) проводят предпочтительно в присутствии основания Бренстеда, в частности слабого основания Бренстеда. Подходящими основаниями Бренстеда являются щелочные карбонаты или щелочные гидрогенкарбонаты, например карбонат натрия, карбонат калия или бикарбонат натрия. Предпочтительным является карбонат калия.
Реакция проходит предпочтительно в двухфазной системе, при которой одна фаза образуется из водного раствора, а другая фаза образуется из раствора, который включает в себя органический растворитель, смешиваемый с водой не бесконечно. Примерами подходящих растворителей являются толуол, хлористый метилен, хлороформ или их смеси.
Реакция взаимодействия соединения общей формулы IV с соединением общей формулы V происходит обычно в молярном отношении 0,5-2:1, в пересчете на молярные количества соединения общей формулы IV к соединению общей формулы V.
Время реакции составляет, в общем, от 0,1 до 20 часов, предпочтительно от 5 до 15 часов.
Ниже радикал R1b, который может присутствовать как в соединениях общей формулы I, так и в соединениях общей формулы IV (как радикал R5), будет описан более подробно.
Предпочтительно радикал R1b соединения общей формулы I или радикал R5 в соединении общей формулы IV является радикалом, который в состоянии реагировать с радикалом R3 при образовании С-С-связи. В частности радикал R1b соединения общей формулы I или радикал R5 в соединении общей формулы IV является радикалом, который в состоянии реагировать с радикалом R3 в реакциях Сузуки, Сталла или Гриньяра.
Понятия «реакция Сузуки», «реакция Сталла» или «реакция Гриньяра» известны любому специалисту и описаны, например, в Chem. Rev. 2002, 102(5), 1359-1470.
Особенно предпочтительны следующие значения радикала R1b в соединении общей формулы I или радикала R5 в соединении общей формулы IV:
- галоген, в частности бром,
- радикал общей формулы VI

где R8 и R9 обозначают водород, группу С1-С6-алкила или вместе обозначают группу С1-С6-алкила,
- радикал триалкил-олова, причем предпочтительно «алкил» обозначает C1-C12, в частности радикал С1-C6-алкила, или
- если в способе используют соединение общей формулы I с радикалом R1b, то радикал магний(II)галогенида,
и причем,
если R1b и R5 обозначают галоген, R3 обозначает радикал общей формулы VI, радикал триалкил-олова или если в способе используют соединение общей формулы I с радикалом R1b, то радикал магний(II)галогенида и наоборот.
В группе общей формулы VI радикал R8 и R9 вместе обозначают предпочтительно 2,3-диметилбутан-2,3-диил.
Кроме того, особенно предпочтительно выбрать радикал R3 общей формулы III из группы, включающей следующие радикалы:
- галоген, предпочтительно бром,
- радикал общей формулы VI,
- в радикале триалкил-олова, причем предпочтительно «алкил» обозначает С1-С6, в частности радикал С1-С6-алкила, или
- радикал магний(II)галогенида, предпочтительно радикал магний(II)бромида,
однако при следующем условии, что радикалы R1b и R5, с одной стороны, и радикал R3, с другой стороны, выбирают так, что радикал R1b и радикал R5, с одной стороны, и радикал R3, с другой стороны, представляют собой дополнительные радикалы, т.е. образуют дополнительную пару, которые могут желаемым образом вступать друг с другом в реакцию взаимодействия.
Таким образом, радикалы R1b и R3 или R5 и R3 выбирают так, что они образуют дополнительные пары. Предпочтительными дополнительными парами являются:
a) галоген и радикал общей формулы VI, так как они могут, например, вступать друг с другом в реакции Сузуки;
b) галоген и радикал триалкил-олова, причем предпочтительно «алкил» стоит вместо радикала C1-C12-алкила, в частности C1-C6-алкила, так как они могут, например, вступать друг с другом в реакции Сталла;
c) галоген или радикал магний(II)галогенида, так как они могут, например, вступать друг с другом в реакции Гриньяра.
Галоген обозначает предпочтительно бром и радикал магний(II)галогенида, предпочтительно радикал магний(II)бромида.
В реакциях Сузуки, Сталла или Гриньяра, которые можно применять в заявленных способах, используют выгодным образом один или несколько катализаторов. Катализаторами могут быть один или несколько переходных металлов, в частности марганец, хром, железо, кобальт, никель или палладий. Предпочтительно применяемыми соединениями являются те, которые выбраны из группы MnCl2, СrСl3, FeCl2, Fе(асас)3, FеСl3, Fe(salen)Cl, NiCl2(PPh3)2, CoCl2(dppe), CoCl2(dpph), Co(acac)2, CoCl2(dppb), PdCl2(PPh3)2 или Pd(PPh3)4.
Выгодным образом катализаторы применяют вместе с активаторами и/или стабилизаторами. Активатор переводит атомы металла катализаторов в нулевую стадию окисления, а стабилизатор стабилизирует атомы металла катализаторов в нулевой стадии окисления. Примерами таких активаторов являются цинк (предпочтительно в виде цинкового порошка), боргидрид натрия, гидрид лития-алюминия или органические соединения алюминия, магния или лития (предпочтительно бутиллитий или DIBAH). Примерами таких стабилизаторов являются льюисные основания, предпочтительно фосфаны, особенно предпочтительно триарилфосфаны и триалкилфосфаны, в частности трифенилфосфан.
В реакции Сузуки особенно предпочитают палладиевый катализатор, такой как Рd(РРh3)4. Она происходит предпочтительно в присутствии слабого основания Бренстеда, такого как карбонат щелочного металла. Выгодным образом реакцию проводят в двухфазной системе, при которой одна фаза образована из водного раствора, а другая - из раствора, который содержит органический растворитель, смешиваемый с водой не бесконечно, такой как бензол, толуол, ксилол, хлористый метилен или хлороформ.
В реакции Сталла особенно предпочитают палладиевый катализатор, например Рd(РРh3)4 или РdCl2(РРh3)2. Реакция происходит предпочтительно при повышенной температуре, предпочтительно при температурах от 50°С и до температуры кипения растворителя. Выгодным образом реакцию проводят в присутствии сокатализатора, такого как CuI (йодид меди) или СuО (оксид меди). Реакцию ведут предпочтительно в инертных растворителях, таких как, например, толуол, ксилол, диметоксиэтан, диметилформамид, тетрагидрофуран или диоксан.
В реакции Гриньяра с особым предпочтением применяют палладиевый катализатор, такой как Рd(РРh3)4, РdСl2(РРh3)2 или NiCl2(PPh3)2. Реакцию ведут предпочтительно в полярных, непротонных растворителях, таких как, например, тетрагидрофуран, простой диэтиловый эфир или диоксан.
Соединения общей формулы I получают, в общем, так, как это было описано выше, т.е. в результате реакции взаимодействия соединения общей формулы IV с соединениями общей формулы V. В зависимости от вида радикала R1 и R5 предпочитают различный порядок синтеза.
В рамках данного изобретения более подробно описываются три предпочтительных типа синтеза, обозначенные как тип синтеза А, тип синтеза В и тип синтеза С.
Тип синтеза А описывает случай, когда R5 в соединениях общей формулы IV обозначает радикал общей формулы II. (Это значит, что группа с R5 в формуле IV соответствует группе R1a в формуле I.)
В общем, получение соединения общей формулы IV, где R5 обозначает радикал общей формулы II, когда соединение общей формулы VII
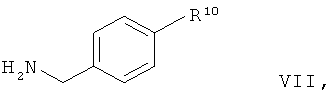
в которой R10 обозначает радикал формулы II,
вступает в реакцию обмена с соединением общей формулы VIII

где R11 обозначает радикал С1-С12-алкила и X- обозначает анион минеральной кислоты,
происходит в присутствии основания Бренстеда.
Реакцию проводят обычно в подходящем инертном растворителе, например алифатическом спирте, таком как этанол. В качестве основания Бренстеда подходит, например, третич. алифатический амин, например триэтиламин, для нейтрализации.
Реакция взаимодействия происходит обычно в молярном отношении 0,3-3:1, в пересчете на молярное количество соединения общей формулы VII в отношении к соединению общей формулы VIII. Продолжительность реакции составляет, в общем, от 0,1 до 20 часов, предпочтительно от 5 до 15 часов.
Особенно выгодным образом происходит получение соединения общей формулы VII, когда:
1) готовят соединение общей формулы IX

в которой R12 представляет собой радикал общей формулы II, и
2) из соединения общей формулы IX в условиях, обычных для реакции Габриеля, получают соединение общей формулы VII. Для этого соединение общей формулы IX вступает в реакцию взаимодействия, например, с гидразингидратом в спиртовом растворе.
Получение соединения общей формулы IX известно, например, из Bioorganic & Medicinal Chemistry Letters 1993, 3(4), 757-760 (обозначено там как соединение 20).
Получение соединения общей формулы VIII известно, например, из J. Org. Chem. 1999, 64(22), 8084-8089.
Способ синтеза В описывает случай, когда R5 в соединениях общей формулы IV обозначает атом галогена, в частности бром. (Это значит, что радикал R5 в формуле IV соответствует радикалу R1b в формуле I.)
Для получения соединения общей формулы IV, в которой R5 обозначает галоген, проводят реакцию взаимодействия предпочтительно производного бензиламина, замещенного в пара-положении атомом галогена (т.е. соединение, которое соответствует общей формуле VII), с соединением общей формулы VIII предпочтительно в присутствии основания Бренстеда. Для условий такого взаимодействия подходит то же, что и для получения соединения общей формулы IV, в которой R5 обозначает радикал общей формулы II.
Производное бензиламина, замещенного в пара-положении атомом галогена, очень просто получают в реакции Габриэля при условиях, обычных для реакции Габриэля с фталимидом из бензилгалогенида, замещенного в пара-положении атомом галогена, предпочтительно бромом, предпочтительно пара-бромбензилбромидом.
При типе синтеза В реакция взаимодействия соединения общей формулы IV с соединением общей формулы V приводит к получению соединения общей формулы I, причем в отношении радикала R1 речь идет о радикале R1b.
В одной из предпочтительных форм выполнения полученное соединение общей формулы I с радикалом R1b переводится в результате вышеописанной реакции сочетания С-С в соединение общей формулы I с радикалом R1a.
Получение соединения общей формулы I с радикалом R1a происходит предпочтительно так, что соединение общей формулы I, в котором R1b обозначает галоген, предпочтительно бромид, вступает в реакцию взаимодействия с соединением общей формулы III, в которой R3 обозначает радикал общей формулы VI, радикал триалкил-олова или радикал магний(II)галогенида, при условиях, которые являются обычными для реакций Сузуки, Сталла или Гриньяра.
Способ синтеза С описывает случай, когда R5 в соединениях общей формулы IV обозначает радикал общей формулы VI. (Это значит, что радикал R5 в формуле IV соответствует радикалу R1b в формуле I.)
Получение соединения общей формулы IV, в которой R5 обозначает радикал общей формулы VI, происходит предпочтительно так, что производное бензиламина, замещенное в пара-положении радикалом R5 общей формулы, вступает в реакцию взаимодействия с соединением общей формулы VIII предпочтительно в присутствии основания Бренстеда.
Производное бензиламина, замещенное в пара-положении радикалом R5 общей формулы VI, получают, в общем, в результате реакции Габриэля при обычных для нее условиях с фталимидом из бензилгалогенида, замещенного в пара-положении радикалом R5 общей формулы VI, предпочтительно бензилбромидом, замещенным радикалом R5 общей формулы VI.
При типе синтеза С реакция взаимодействия соединения общей формулы IV с соединением общей формулы V приводит к получению соединения общей формулы I, причем в смысле радикала R1 речь идет о радикале R1b.
В предпочтительном варианте выполнения полученное соединение общей формулы I с радикалом R1b переводят с помощью описанной выше реакции сочетания С-С в соединение общей формулы I с радикалом R1a.
Соединение общей формулы I, в которой R1 обозначает радикал общей формулы II, получают по особенно предпочтительному методу, проводя реакцию взаимодействия соединения общей формулы I, в которой R1b обозначает радикал общей формулы VI, с соединением общей формулы III, в которой R3 обозначает галоген, предпочтительно бром, при условиях, обычных для реакции Сузуки.
В том случае, если в общей формуле IV R5 обозначает триалкил-олово или радикал MgHal, то предпочитают такое течение реакции, которое описано в синтезе С.
Другим аспектом изобретения является получение производного имидазола, в котором, по меньшей мере, один атом углерода имидазольного кольца замещен хлором (производное имидазола А), когда проводят реакцию взаимодействия между имидазолом или производным имидазола, который, по меньшей мере, в одном атоме углерода имидазольного кольца имеет атом водорода (производное имидазола В), и СеСl3 и солью щелочного металла гипогалогеновой кислоты.
В качестве производного имидазола (А) предпочтительно получают соединение, которое в атоме углерода имидазольного кольца в 4-м или 5-м положении или в двух названных положениях замещено хлором, и причем в качестве производного имидазола (В) применяют соединение, в котором атом углерода имидазольного кольца в 4-м или 5-м положении или в обоих указанных положениях имеет еще атом водорода.
Особенно хорошо этот метод хлорирования подходит для получения производного лосартана, когда атом водорода тетразольной группы заменен терразольной защитной группой, причем в качестве производного имидазола (B) применяют соединение общей формулы XI.
CeCl3 и соль щелочного металла гипогалогеновой кислоты используют в стехиометрических количествах или в избытке. В качестве соли щелочного металла гипогалогеновой кислоты используют выгодным образом соль калия или натрия. В качестве соли щелочного металла гипогалогеновой кислоты используют выгодным образом соль щелочного металла гипохлорной кислоты.
Заявленная реакция хлорирования проводится обычно в 2-фазной системе, когда одна фаза образуется из водного раствора, а другая фаза образуется из раствора, который содержит органический растворитель, который смешивается с водой не бесконечно, например хлористый метилен, хлороформ или толуол.
Лосартан или его фармакологически эффективную соль можно очень просто получить из соединения общей формулы I с радикалом R1а,
а) получив на стадии (а) из соединения общей формулы I с радикалом R1а соединение общей формулы XI

в которой R15 обозначает радикал общей формулы II, восстанавливая группу формила, которой замещена имидазольная группа, в группу гидроксиметила,
b) заменяя на стадии b) еще оставшийся единственный атом водорода в имидазольной группе полученного на стадии а) соединения хлором, и
c) отщепляя на стадии с) в соединении, полученном на стадии b), защитную группу тетразола и, при необходимости,
d) получая из лосартана его фармакологически эффективные соли, например соль калия.
Восстановление группы формила на стадии (а) может происходить обычным образом. Предпочтительно восстановление группы формила на стадии (а) происходит с помощью боргидрида натрия или гидрида лития-алюминия.
Хлорирование на стадии (b) может происходить обычным образом. Предпочтительно стадию (b) выполняют с применением описанного выше способа хлорирования, т.е. с применением СеСl3.
Стадию (с) выполняют обычным образом, как это описано в WO 03/093262. Отщепление особенно предпочтительной защитной группы трифенилметила происходит так, что, например, раствор соединения, полученного на стадии (b), обрабатывают разбавленной минеральной кислотой, предпочтительно хлористоводородной кислотой.
Получение фармакологически эффективной соли лосартана, например лосартан-калия (стадия (d)), описано, например, в ЕР 324377 А, стр.191, пример 316, часть D и в WO 95/17396, стр.18, пример 4 и стр.24, пример 9, стадия С.
Далее способы синтеза А, В и С, описанные выше, раскрываются более подробно.
Соединения, используемые и/или получаемые в соответствующих способах синтеза, обозначены арабскими цифрами. При этом соединения соответствуют соединениям в следующей схеме формул:
- 4 - соединению общей формулы I с радикалом R1 общей формулы II (т.е. в смысле R1 речь идет о радикале R1a),
- 15 и 21 - соединениям общей формулы I с радикалом R1b,
- 13, 23 - соединениям общей формулы III,
- 3, 14 - соединениям общей формулы IV,
- 8 - соединению общей формулы V,
- 21 - соединению общей формулы IV с радикалом R5 общей формулы VI,
- 1 - соединению общей формулы VII,
- 2 - соединению общей формулы VIII, и
- 9, 10 - соединениям общей формулы IX.
Одновременно они представляют предпочтительные примеры выполнения группы соединений, определенные с помощью соответствующей общей формулы.
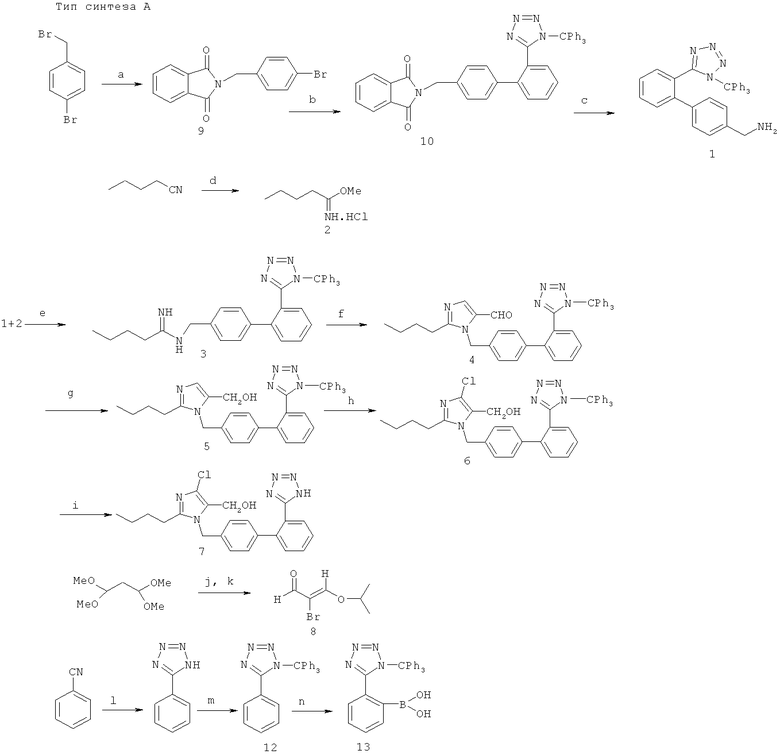
Реакции обмена а)-n) проводятся, в общем, в обычных для реакции условиях, предпочтительно в присутствии следующих реактивов:
a) Фталимид
b) 13
d) СН3ОН
e) Основание
f) 8
g) Средство гидрирования
h) Средство хлорирования
В особенно предпочтительном варианте выполнения можно применять следующие реактивы и условия для реакции:
a) фталимид, К2СО3/DMSO;
b) 13, Pd(PPh3)4, Na2CO3, Толуол-Н2О, 80°С;
c) гидразингидрат, СН3ОН/CH2Cl2;
d) СН3ОН/НСl;
e) NEt3/ЕtOН;
f) 8, К2СО3/СНСl3-Н2O;
g) NaBH4/СН3ОН;
h) СеСl3·7Н2O, NaClO/СН2Сl2-Н2O;
i) 2N НСl, СН3ОН-THF;
j) концентрированная НСl, Вr2;
k) PPTS, изопропанол;
l) NaN3, NH4Cl, LiCI, DMF, 100°С;
m) Рh3ССl, Et3N/CH2Cl2;
n) BuLi, от -20°С до -5°С, затем В(ОМе)3, от -20°С до комнатной t°.
В предпочтительном варианте выполнения соединения 1 и 2 вступают в реакцию взаимодействия без изоляции от соединения 3 к соединению 4.
Тип синтеза В
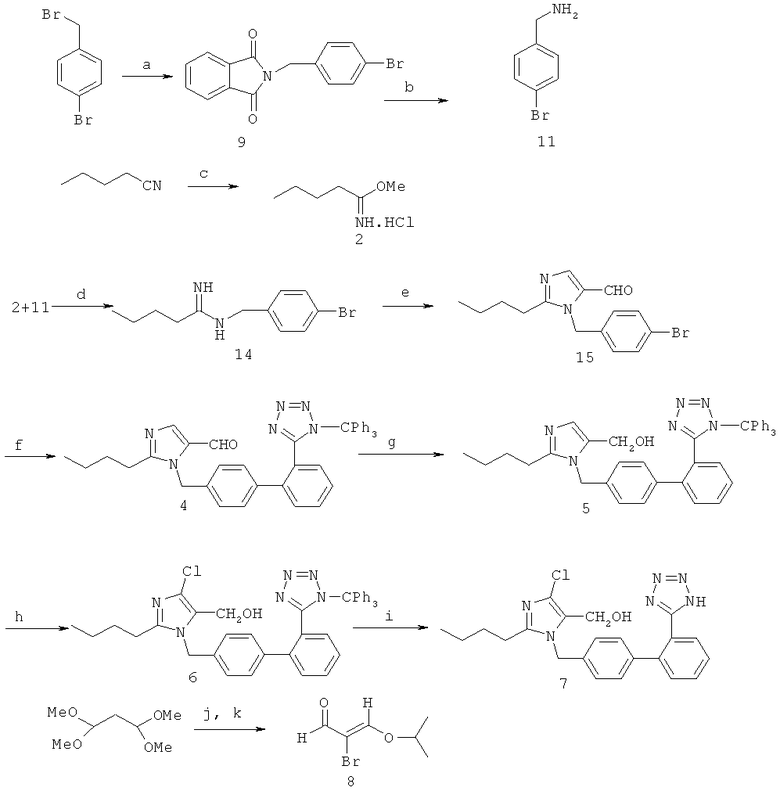
Реакции обмена а)-k) происходят, в общем, в условиях, обычных для реакции, предпочтительно в присутствии следующих реактивов:
а) фталимид
c) СН3ОН
d) основание
e) 8
f) 13
g) средство гидрирования
h) средство хлорирования
В особенно предпочтительном варианте выполнения можно применять следующие реактивы и условия для реакции:
a) фталимид, К2СО3/DMSO;
b) гидразингидрат, СН3СН2ОН/Н2O;
c) СН3ОН/HCl;
d) NEt3/ЕtOН;
e) 8, К2СО3/СНСl3-Н2O;
f) 13, Pd(PPh3)4, Na2CO3, толуол-Н2О, 80°С;
g) NaBH4/СН3ОН;
h) СеСl3·7Н2O, NaClO/CH2Cl2-Н2O;
i) 2N HCl, СН3ОН-THF;
j) концентрированная HCl, Br2;
k) PPTS, изопропанол;
 Реакции взаимодействия а)-о) происходят, в общем, при условиях, обычных для реакции, предпочтительно в присутствии следующих реактивов:
Реакции взаимодействия а)-о) происходят, в общем, при условиях, обычных для реакции, предпочтительно в присутствии следующих реактивов:
d) фталимид
e) СН3ОН
g) 2, основание, затем 8
h) 13
i) средство гидрирования
j) средство хлорирования
В одном из особенно предпочтительных вариантов выполнения можно применять следующие реактивы и условия для реакции:
a) Мg, I2, THF, кипячение с перемешиванием и обратным потоком в течение 1 часа, затем -78°С, В(ОМе)3;
b) пинакол, циклогексан, кипячение с перемешиванием и обратным потоком до удаления воды;
c) NBS, циклогексан, кипячение с перемешиванием и обратным потоком;
d) фталимид, К2СО3/ацетон, кипячение с перемешиванием и обратным потоком;
e) СН3ОН/HCl;
f) гидразингидрат, СН3СН2ОН, кипячение с перемешиванием и обратным потоком;
g) 2, NEt3/СН3ОН, затем К2СО3, 8;
n) 13, Pd(PPh3)4, К2СО3, толуол-Н2О, 80°С;
i) NaBH4/СН3ОН;
j) СеСl3·7Н2O, NaClO/СН2Сl2-Н2О;
k) 2N HCl, СН3ОН-THF;
l) концентрированная HCl, Br2;
m) PPTS, изопропанол;
n) NaN3, NH4Cl, LiCl, DMF, 100°C;
o) Ph3CCl, Et3N/CH2Cl2.
Экспериментальная часть
В экспериментальной части более подробно описывается получение соединений в типах синтеза А, В и С.
Описание опытов
Приборы и реактивы
Все сухие растворители (CН2Cl2, THF, Et2O, бензол, толуол, DMF, МеСN) были высушены стандартными методами, т.е. путем удаления воды и кислорода и перегонки перед использованием. Реакции проводили, если это было нужно, в атмосфере инертного газа (N2 или Аr) и наблюдали с помощью тонкослойной хроматографии. Растворителями для экстракций были, например, простой диэтиловый эфир, этилацетат или хлороформ. Экстракты, если ничего другого не дано, были высушены, например, с безводным MgSО4. Продукты реакции, если это было необходимо, очищали, например, при помощи колоночной хроматографии с применением, например, петролейного эфира (60-90°С)/этилацетата и хлороформа/метанола как элюэнта. Если использовали пластины типа GF254 для TLC (тонкослойной хроматографии), то в качестве средства обнаружения использовали йод или раствор этанола фосфор-молибденовой кислоты. Силикагель для хроматографии (зернистость 200-300) и тонкослойной хроматографии (GF254) был получен по Qingdao Sea Chemical Factory и Yantan Chemical Factory. Все растворители и реактивы были аналитически или химически чисты. Температуру плавления определяли с помощью ХТ4-100х micro-melting point tester. Спектрометры Nicolet AVATAR 360 FT-IR и Nicolet NEXUS 670 FT-IR использовались для приема инфракрасных спектров с прессованного КВr или РЕ-пленки. Спектрометры Mercury-300 (Varian) и АМ-400 (Bruker) использовали для измерения ядерно-магнитного резонанса с SiMe4 в качестве внутреннего стандарта, a CDCl3 использовали в качестве растворителя, если ничего другого не было дано. LRMS определяли с помощью масс-спектрометра HP-5988 с применением EI при 70eV, если не было дано ничего другого. HRMS измеряли с помощью масс-спектрометра Bruker Daltonics APEX II 47е FT-ICR.
Получение соединения 9
Фталимид (11 г, 75,6 ммоля) растворили в 80 мл DMSO в атмосфере защитного газа аргона. После добавления К2СО3 (20 г, 144 ммоля) реакционную смесь нагревали в течение 2-х часов до температуры 120°С. Затем реакционную смесь охлаждали до температуры примерно 50°С и добавляли п-бромбензилбромид (18 г, 72 ммоля). Еще через 10 часов помешивания добавляли 100 мл Н2О. Безцветный осадок отфильтровывали, промывали и высушивали, что дало соединение 9 (18.6 г). Выход составил 82%.
1H ЯМР (CDCl3, 300 МГц): δ 4,77 (s, 2H, NCH2), 7,29 (d, J=8,4 Гц, 2Н, ArH), 7,41 (d, J=8,4 Гц, 2H, ArH), 7,67-7,70 (m, 2H, ArH), 7,80-7,83 (m, 2H, ArH); 13С ЯМР (CDCl3, 75 МГц): δ 40,9, 121,8, 123,3 (2С), 130,3 (2С), 131,7 (2С), 131,9 (2С), 134,0 (2С), 135,2, 167,8 (2С); Масс-спектрометрия (FAB): M+=315, найдено: 316 (М++1), 318 (М++3); ИК (пленка, см-1) vмакс=3460, 3100, 3045, 2938, 1771, 1702, 1612, 1486, 1464, 1430, 1399, 1332, 1298, 1173, 1077, 1010, 957, 935, 845, 796, 731, 713, 528.
Получение соединения 10
Соединение 9 (1,45 г, 4,6 ммоля), соединение 13 (3,4 г, 1,2 эквивалента) и Na2CO3 (1,46 г, 3 эквивалента) растворяли в смеси 20 мл толуола и Н2O (7:3). Затем системы трижды промывали аргоном и добавляли Pd(PPh3)4 (266 мг, 0,05 эквивалента). Реакционную смесь нагревали в течение 13 часов до температуры 80°С, а затем экстрагировали с этилацетатом. Органические фазы сгущали, а твердое вещество очищали при помощи колоночной хроматографии, что дало соединение 10 в виде белого твердого вещества (2,43 г). Выход составлял 86%.
1Н ЯМР (CDCl3, 300 МГц): δ 4,73 (s, 2H), 6,87-6,90 (m, 6H), 7,06 (d, J=8,1 Гц, 2H), 7,18-7,33 (m, 12H), 7,42-7,46 (m, 2H), 7,67-7,70 (m, 2H), 7,82-7,84 (m, 2H), 7,92-7,95 (m, 1H); 13C ЯМР (CDCl3, 75 МГц): δ 41,2, 82,8, 123,3, 126,2, 127,6, 128,2, 129,5, 129,8, 130,2, 130,7, 132,1, 133,9, 134,8, 140,7, 141,2, 141,7, 163,9, 167,9; Масс-спектрометрия (FAB): M+=623, найдено: 662 (М++К); ИК (пленка, см-1) vmax=3467, 3061, 3032, 2249, 1770, 1714, 1603, 1492, 1469, 1446, 1429, 1393, 1349, 1187, 1159, 1087, 1031, 1004, 937, 909, 880, 733, 700, 633, 406.
Получение соединения 1
Соединение 10 (37,3 г, 60 ммолей) растворяли в смеси из 200 мл метанола и 300 мл СН2Сl2. Добавляли гидразингидрат (600 ммолей, 10 эквивалентов) и реакционную смесь перемешивали в течение 10 часов при комнатной температуре. Затем фильтровали, а фильтрат разбавляли с СНСl3 и промывали водой. Органическую фазу сушили с МgSO4 и сгущали, что дало соединение 1 в виде желтоватого твердого вещества (23,7 г). Выход составил 80%.
1H ЯМР (CDCl3, 200 МГц): δ 3,80 (s, 2H), 4,52 (s, br, 2H), 6,89-6,92 (m, 6H), 7,04-7,14 (m, 4H), 7,24-7,31 (m, 10Н), 7,43 (s, br, 2H), 7,90-7,93 (m, 1H); Macc-спектрометрия (FAB): M+=493, найдено: 494 (M++1), 516 (M++Na).
Получение соединения 2
HCl-газ вводили в раствор 8,3 г (100 ммолей) валеронитрила в 8 мл метанола при охлаждении в ледяной бане. При этом температуру удерживали постоянно ниже 10°С. Через 2 часа реакцию заканчивали и получали соединение 2 в виде белого твердого вещества.
1Н ЯМР (CDCl3, 300 МГц): δ 0,84 (t, J=7,2 Гц, 3Н, СН3), 1,26-1,34 (m, 2H, СН2), 1,57-1,67 (m, 2H, СН2), 2,68 (t, J=7,5Гц, 2H, СН2), 4,19 (s, 3Н, ОМе), 7,43 (s, 1H, NH), 11,12 (s, br, 1H, HCI), 13C ЯМР (CDCl3, 75MГц): δ 13,2, 21,7, 27,3, 32,5, 60,4, 180,6.
Получение соединения 8
Концентрированную соляную кислоту (3,6 мл) капали в смесь из 1,1,3,3-тет-раметоксипропана (6,5 г, 40 ммолей) и воды (64 мл). Реакционную смесь путем помешивания превращали в однородную, охлаждали до 0°С и по каплям добавляли бром (2,1 мл, 40 ммолей). Мешали еще 10 минут, а затем большую часть воды удаляли в вакууме при температуре ниже 70°С. После нового охлаждения до 0°С фильтровали. Полученное таким образом твердое вещество высушивали, а затем растворяли в смеси из 65 мл циклогексана и 10 мл изопропанола и добавляли каталитическое количество PPTS. Реакционную смесь кипятили 90 мин с перемешиванием и обратным потоком, а появляющуюся воду отделяли. Остальные растворители удаляли в вакууме. Осталось соединение 8 в виде желтого масла. Чистота была более 95%, что дает возможность использовать соединение 8 сразу, т.е. без дальнейшей очистки. Выход составил 65%.
1H ЯМР (CDCl3, 300 МГц): δ 1,38 (d, J=6 Гц, 6H, 2СН3), 4,45-4,50 (m, 1H, СН), 7,70 (s, 1H, CH=), 9,07 (s, 1H, СНО); 13C ЯМР (CDCl3, 75 МГц): δ 22,3 (2С), 80,6, 105,1, 166,2, 184,0; Масс-спектрометрия (EI) m/z (%):194 (M+, 5), 192 (М+, 5), 166 (2), 152 (95), 150 (100), 121 (13), 93 (16), 71 (30), 43 (59).
Получение соединения 3
Соединение 2 (6,4 г, 40 ммолей) растворяли в 75 мл абсолютного этанола и 20 мл NEt3. Затем добавляли соединение 1 (10 г, 20 ммолей) при 10°С и реакционную смесь перемешивали в течение 5 часов, в результате чего получали раствор, который мешали еще 8 часов при комнатной температуре. После этого разбавляли хлороформом и промывали водой. Органическую фазу отделяли, высушивали и сгущали при температуре ниже 45°С. Соединение 3 получили в виде масла. Выход составил 80%.
1H ЯМР (CDCl3, 300 МГц): δ 0,67-0,80 (m, 3H, СН3), 1,09-1,24 (m, 2H, CH2), 1,58 (s, br, 2H, CH2), 2,51 (s, br, 2H, CH2), 4, 53 (s, br, 2H, NCH2), 6,89-7,08 (m, 8H, ArH), 7,21-7,44 (m, 14H, ArH), 7,87 (d, J=3,9Гц, 1Н, АrН); 13C ЯМР (CDCl3, 75 МГц): δ 13,3, 21,6, 28,0, 32,3, 45,4, 82,9, 125,5, 126,0, 127,5, 128,2, 129,2, 129,8, 133,3, 140,3, 140,7, 141,0, 163,9, 167,7; Масс-спектрометрия (FAB): M+=576, найдено: 577 (M++1); ИК (пленка, см-1) vmax=3222, 3057, 3030, 2962, 2933, 2210, 1677, 1636, 1531, 1493, 1449, 1190, 1004, 910, 732, 700, 639.
Получение соединения 4
Метод А-1 (с изолированием соединения 3)
Соединение 3 (8 г, 13,9 ммоля) растворяли в 60 мл хлороформа и 7,5 мл воды. После добавления К2СО3 (2,69 г, 19,5 ммоля) и соединения 8 (3,73 г, 19,5 ммоля) реакционную смесь перемешивали 12 часов при комнатной температуре. Затем экстрагировали с хлороформом, а органическую фазу сгущали. Полученный таким образом продукт очищали с помощью колоночной хроматографии, что дало соединение 4 в виде белого твердого вещества (3.67 г). Выход составил 42%.
Метод А-2 (без изолирования соединения 3)
Соединение 2 (4,5 г, 1,5 эквивалента, 30 ммолей) растворяли в смеси из 50 мл абсолютного этанола и NEt3 (8,3 мл, 3 эквивалента) при 0°С и добавляли соединение 1 (10 г, 1 эквивалент, 20 ммолей). Реакционную смесь перемешивали в течение примерно 5 часов для получения прозрачного раствора и еще 16 часов при комнатной температуре. После этого добавляли К2СО3 (4,14 г, 30 ммолей, 1,5 эквивалента) и соединение 8 (4,6 г, 1,2 эквивалента, 24 ммолей) и перемешивали в течение последующих 12 часов при комнатной температуре. Реакционную смесь экстрагировали с СНСl3 и органическую фазу сгущали. Полученное твердое вещество очищали с помощью колоночной хроматографии и получали соединение 4 в виде белого твердого вещества.
Метод А-3 (без изолирования соединения 3)
Соединение 1 (563 г) растворяли в смеси из 3,6 л абсолютного этанола и 1,2 л триэтиламина. Реакционную смесь охлаждали до 0°С и медленно добавляли соединение 2 (350 г). Перемешивали в течение часа при 0°С, а затем реакционную смесь разбавляли хлороформом и водой. Органическую фазу отделяли, а водную фазу снова экстрагировали с хлороформом. В объединенную органическую фазу при комнатной температуре добавляли К2СО3 (180 г), 560 мл воды и 330 г соединения 8. Затем при комнатной температуре мешали в течение ночи, и реакционную смесь разбавляли хлороформом и водой. Органическую фазу отделяли, а водную фазу снова экстрагировали с хлороформом. Объединенную органическую фазу высушивали с МgSO4, фильтровали и сгущали.
Полученный таким образом остаток перекристаллизовывали из этилацетата, что дало соединение 4 (530 г). Выход составил 74%.
Метод В
Соединение 15 (1,47 г, 4,6 ммолей), соединение 13 (3,4 г, 1,2 эквивалента) и Nа2СО3 (1,46 г, 3 эквивалента) растворяли в 20 мл смеси из толуола и воды (7:3). После этого систему трижды опрыскивали аргоном и добавляли Рd(РРh3)4 (266 мг, 0,05 эквивалента). Реакционную смесь нагревали в течение 10 часов до температуры 80°С, после чего экстрагировали с этилацетатом. Органические фазы сгущали, а остаток очищали с помощью колоночной хроматографии, что дало соединение 4 в виде твердого вещества (2,1 г). Выход составил 74%.
Метод С
Соединение 21 (40 мг, 0,11 ммолей), соединение 23 (151 мг, 0,33 ммолей) и К2СО3 (45 мг, 0,33 ммолей) растворяли в 3 мл смеси из толуола и воды (7:3). После этого систему трижды опрыскивали аргоном и добавляли Рd(РРh3)4 (6 мг, 0,05 эквивалента). Реакционную смесь нагревали в течение 10 часов до температуры 80°С, а затем экстрагировали с этилацетатом. Органические фазы сгущали, а остаток очищали с помощью колоночной хроматографии, что дало соединение 4 в виде твердого вещества (48 мг). Выход составил 72%.
1H ЯМР (CDCl3, 400 МГц,): δ 0,86 (t, J=7,2 Гц, 3Н, СН3), 1,25-1,32 (m, 2H, СН2), 1,64-1,68 (m, 2H, СН2), 2,55 (t, J=7,6 Гц, 2H, СН2), 5,48 (s, 2H, NСН2), 6,82 (d, J=8,4 Гц, 2H, ArH), 6,91-6,93 (m, 6H, ArH), 7,09 (d, J=8,4 Гц, 2H, ArH), 7,23-7,27 (m, 6H, ArH), 7,31-7,35 (m, 4H, ArH), 7,41-7,49 (m, 2H, ArH), 7,78 (s, 1H, CH=), 7,91 (dd, J=1,2 Гц, 7,2 Гц, ArH), 9,64 (s, 1H, СНО); 13С ЯМР (CDCl3, 100 МГц,): δ 13,7, 22,3, 26,4, 29,2, 47,8, 82,8, 125,9, 126,2, 127,6, 127,8, 127,9, 128,2, 129,7, 129,9, 130,1, 130,2, 130,7, 131,3, 134,7, 140,7, 141,2, 141,4, 143,6, 156,7, 163,8, 178,5; Масс-спектрометрия (FAB): M+=628, найдено: 629 (M++1), 651 (M++Na); ИК (пленка, см-1) vmax=3060, 3031, 2959, 2932, 2868, 1670, 1619, 1597, 1532, 1466, 1446, 1187, 1160, 1030, 1003, 909, 880, 824, 762, 733, 701, 640.
Получение соединения 5
Соединение 4 (6,3 г, 10 ммолей) суспендировали в 30 мл метанола и добавляли 3 мл СНСl3 для полного растворения. Реакционную смесь охлаждали в ледяной бане и добавляли NaBH4 (760 мг, 20 ммолей). Перемешивали в течение часа и смесь экстрагировали с СНСl3. Органическую фазу сгущали, что дало соединение 5 (6 г) в виде масла.
Выход составил 95%.
1Н ЯМР (CDCl3, 300 МГц): δ 0,85 (t, J=7,5 Гц, 3Н, СН3), 1,27-1,32 (m, 2H, СН2), 1,61-1,66 (m, 2H, СН2), 2,50 (t, J=7,5Гц, 2H, СН2), 4,32 (s, 2H, СН2ОН), 5,12 (s, 2H, NCH2), 6,74 (d, J=8,1 Гц, 2H, ArH), 6,84 (s, 1H, CH=), 6,93 (d, J=7,2 Гц, 6Н, ArH), 7,08 (d, J=8,1 Гц, 2H, ArH), 7,23-7,36 (m, 10Н, ArH), 7,44-7,48 (m, 2H, ArH), 7,90-7,93 (m, 1H, ArH); 13C ЯМР (CDCl3, 75 МГц): δ 13,7, 22,3, 26,7, 29,7, 46,3, 54,4, 82,8, 125,2, 126,2, 126,6, 127,6, 128,2, 129,7, 130,2, 130,7, 131,0, 135,3, 140,5, 141,2, 141,4, 150,1, 163,9; Масс-спектрометрия (FAB): M+=630, найдено: 631 (М++1), 653 (M++Na);
ИК (пленка, см-1) vmax=3060, 2956, 2927, 2865, 1493, 1464, 1448, 1355, 1272, 1189, 1153, 1026, 906, 880, 822, 753, 699, 636.
Получение соединения 6
Соединение 5 (6,3 г, 10 ммолей) растворяли в смеси из растворителей: 40 мл CH2Cl2 и воды (1:1). После добавления СеСl3·7Н2O (7,44 г, 20 ммолей) и последующего 2-минутного размешивания по каплям добавляли 10%-ный водный раствор NaClO (37 мл). Затем перемешивали еще 10 мин и добавляли насыщенный водный раствор Na2SO3. Реакционную смесь экстрагировали с СНСl3, органические фазы сгущали, а полученное твердое вещество очищали с помощью колоночной хроматографии, что дало соединение 6 (4,65 г). Выход составил 70%.
1Н ЯМР (CDCl3, 300 МГц): δ 0,86 (t, J=7,2 Гц, 3Н, СН3), 1,23-1,33 (m, 2H, СН2), 1,58-1,69 (m, 2H, СН2), 2,49 (t, J=7,8Гц, 2H, СН2), 3,30 (s, br, 1H, ОН), 4,32 (s, 2H, СН2ОН), 5,14 (s, 2H, NCH2), 6,78 (d, J=7,8 Гц, 2H, ArH), 6,94 (d, J=7,5 Гц, 6Н, ArH), 7,12 (d, J=7,8 Гц, 2H, ArH), 7,23-7,37 (m, 10Н, ArH), 7,43-7,51 (m, 2H, ArH), 7,94-7,97 (m, 1H, ArH); 13С ЯМР (CDCl3, 75 МГц): δ 13,6, 22,3, 26,5, 29,5, 47,0, 52,7, 82,8, 124,9, 125,2, 126,1, 126,8, 127,5, 128,2, 129,7, 130,1, 130,6, 134,5, 140,7, 141,1, 141,2, 148,3, 163,8; Масс-спектрометрия (FAB): M+=664, найдено: 665 (М++1), 687 (М++Na);
ИК (пленка, см-1) vmax=3184, 3061, 2958, 2931, 2869, 2244, 1577, 1492, 1466, 1447, 1356, 1255, 1189, 1160, 1078, 1028, 1005, 909, 881, 756, 733, 701, 640.
Получение соединения 7
Соединение 6 (6,64 г, 10 ммолей) растворяли в 20 мл тетрагидрофурана и прибавляли 20 мл 2N HCl. Реакционную смесь перемешивали в течение 4 часов при комнатной температуре, затем разбавляли СНСl3, промывали водой и высушивали. Органические фазы сгущали, а полученное твердое вещество очищали с помощью колоночной хроматографии, что дало соединение 7 (3,8 г). Выход составил 90%.
1H ЯМР (d-DMSO, 300 МГц): δ 0,78 (t, J=7,2 Гц, 3Н, СН3), 1,18-1,25 (m, 2H, СН2), 1,41-1,46 (m, 2H, СН2), 2,44 (t, J=7,5 Гц, 2H, CH2), 4,32 (s, 2H, CH2OH), 5,23 (s, 2H, NCH2), 7,01 (d, J=8,1 Гц, 2H, ArH), 7,07 (d, J=8,1 Гц, 2H, ArH), 7,49-7,58 (m, 2H, ArH), 7,63-7,65 (m, 2H, ArH); 13C ЯМР (d-DMSO, 75 МГц): δ 13,5, 21,5, 25,7, 28,9, 46,4, 51,3, 123,5, 125,2, 125,6, 126,2 (2С), 127,7, 129,1 (2С), 130,5 (2С), 131,0, 136,1, 138,4, 141,0, 147,3, 155,0; Масс-спектрометрия (FAB): M+=422, найдено: 423 (М++1), 445 (M++Na);
ИК (пленка, см-1) vmax=3351, 2959, 2932, 2870, 1936, 1709, 1575, 1464, 1422, 1361, 1257, 1226, 1078, 1007, 824, 758.
Получение соединения 11
Соединение 9 (9,5 г, 30 ммолей) растворяли в смеси из 100 мл этанола и 30 мл воды. Затем добавляли гидразингидрат (9 мл) и кипятили с перемешиванием и обратным потоком. Примерно через час выпало в осадок белое твердое вещество, а через следующие 9 часов кипячения с перемешиванием и обратным потоком охлаждали до комнатной температуры. Добавляли раствор NaOH (4.88 М, 100 мл) и реакционную смесь экстрагировали с простым диэтиловым эфиром. Органическую фазу высушивали с MgSO4 и сгущали, что дало соединение 11 (5 г). Выход составил 90%.
1H ЯМР (CDCl3, 300 МГц): δ 1,36 (s, 2H, NH2), 3,75 (s, 2H, NCH2), 7,12 (d, J=8,1 Гц, 2H, ArH), 7,38 (d, J=8,1 Гц, 2H, АrН); 13С ЯМР (CDCl3, 75 МГц): δ 45,5, 120,2, 128,6 (2С), 131,2 (2С), 142,0; ИК (пленка, см-1) vmax=3380, 2924, 2854, 2645, 2210, 1653, 1562, 1529, 1481, 1441, 1410, 1380, 1332, 1072, 1007, 905, 812, 789, 645, 618.
Получение соединения 15
Соединение 2 (6,8 г, 1,5 эквивалента, 45 ммолей) растворяли в смеси из 60 мл абсолютного этанола и NEt3 (12,5 мл, 3 эквивалента) при 0°С и затем добавляли соединение 11 (5,6 г, 1 эквивалент, 30 ммолей). Реакционную смесь размешивали при комнатной температуре в течение 10 часов, а затем прибавляли К2СО3 (6,2 г, 45 ммолей, 1,5 эквивалента) и соединение 8 (6,9 г, 1,3 эквивалента, 36 ммолей) и мешали следующие 12 часов при комнатной температуре. После этого экстрагировали с СНСl3, органическую фазу сгущали, а остаток очищали с помощью колоночной хроматографии, что дало соединение 15 (3,74 г). Выход составил 39%.
1H ЯМР (CDCl3, 300 МГц): δ 0,88 (t, J=7,2 Гц, 3Н, СН3), 1,31-1,38 (m, 2H, СН2), 1,63-1,71 (m, 2H, СН2), 2,63 (t, J=8,1 Гц, 2H, СН2), 5,50 (s, 2H, NCH2), 6,88 (d, J=8,4 Гц, 2H, ArH), 7,42 (d, J=8,4 Гц, 2H, АrН), 7,77 (s, 1H, СН=), 9,64 (s, 1H, СНО); 13C ЯМР (CDCl3, 75 МГц): δ 13,6, 22,4, 26,4, 29,3, 47,5, 121,7, 128,0 (2С), 131,2, 131,9 (2С), 135,2, 143,7, 156,6, 178,7; Масс-спектрометрия (FAB): M+=320, найдено: 321 (М++1), 323 (М++3); ИК (пленка, см-1) vmах=2958, 2932, 2868, 1671, 1533, 1485, 1463, 1407, 1373, 1161, 1072, 1011, 813, 769, 648.
Получение соединения 16
Раствор п-бромтолуола (17 г, 100 ммолей) в 100 мл сухого THF по каплям добавляли к смеси из порошка магния (3,6 г, 150 ммолей), йода (200 мг) и 4-х капель 1,2-диброметана в течение часа. После этого кипятили с перемешиванием и обратным потоком, а затем охлаждали до -78°С и добавляли триметилборат (10,3 г, 10 ммолей). Реакционную смесь перемешивали в течение следующих 2-х часов, а затем, добавив воды, гасили. После экстракции с этилацетатом органическую фазу промывали, высушивали и сгущали. Остаток очищали с помощью колоночной хроматографии, что дало соединение 16 (10,2 г) в виде бесцветных кристаллов. Выход составил 75%.
1H ЯМР (CDCl3, 300 МГц): δ 2,41 (s, 3Н, СН3), 7,28 (d, J=7,5 Гц, 2Н, АrН), 8,09 (d, J=7,5 Гц, 2Н, АrН); 13С ЯМР (CDCl3, 75 МГц): δ 21,9, 128,8 (2С), 135,7 (3С), 142,9; Масс-спектрометрия (El): m/z (%): 354 (M+, 100), 262 (19), 193 (17), 145 (18), 119 (36), 91 (39), 43 (47); ИК (пленка, см-1) vmax=3045, 3022, 2918, 1920, 1613, 1517, 1406, 1367, 1347, 1307, 1179, 1109, 1081, 818, 736, 685, 528, 477.
Получение соединения 17
Тримеризованную борную кислоту 16 (5 г, 14,1 ммоля), пинаколгексагидрат (11,5 г, 50,8 ммоля) растворяли в 100 мл циклогексана и раствор кипятили с перемешиванием и обратным потоком в течение 10 часов для удаления воды. После этого циклогексан отгоняли при пониженном давлении, а остаток очищали с помощью колоночной хроматографии, что дало соединение 17 (7,8 г) в виде масла. Выход составил 84%.
1Н ЯМР (CDCl3, 300 МГц): δ 1,37 (s, 12H, 4СН3), 2,40 (s, 3H, СН3), 7,22 (d, J=7,5 Гц, 2H, Ar), 7,75 (d, J=7,5 Гц, 2Н, Ar); 13C ЯМР (CDCl3, 75 МГц): δ 21,7, 24,8 (4С), 83,5 (2С), 128,5 (2С), 134,8 (3С), 141,3; Масс-спектрометрия (El): m/z (%): 218 (M+, 23), 203 (33), 132 (52), 119 (100), 91 (22), 43 (58); ИК (пленка, см-1) vmах=3046, 2979, 2928, 1613, 1519, 1448, 1398, 1361, 1320, 1268, 1214, 1146, 1089, 1023, 962, 859, 816, 726, 656.
Получение соединения 18
Соединение 17 (5 г, 22,9 ммолей), NBS (5,3 г, 29,8 ммолей) и AlBN (200 мг) растворяли в 40 мл циклогексана и раствор кипятили с перемешиванием и обратным потоком в течение 5,5 часов. После этого фильтровали при пониженном давлении, а фильтрат сгущали. Остаток очищали с помощью колоночной хроматографии, что дало соединение 17 (5,86 г) в виде масла. Выход составил 86%.
1Н ЯМР (CDCl3, 300 МГц): δ 1,35 (s, 12H, 4СН3), 4,45 (s, 2H, СН2), 7,40 (d, J=7,2 Гц, 2H, ArH), 7,81 (d, J=7,2 Гц, 2H, ArH); 13C ЯМР (CDCl3, 75 М Гц): δ 24,8 (4С), 33,2, 83,8 (2С), 125,6, 128,2 (2С), 135,2 (2С), 140,6; Масс-спектрометрия (EI): m/z (%): 297 (М++1, 5), 295 (M+-1, 5), 281 (3), 283 (3), 217 (100), 197 (15), 131 (13), 117 (50), 91 (12), 43 (39); ИК (пленка, см-1) vmax=3044, 2974, 2919, 1937, 1609, 1512, 1396, 1356, 1320, 1269, 1217, 1143, 1085, 1017, 960, 845, 784, 655, 602.
Получение соединения 19
Соединение 18 (16 г, 53,9 ммолей), фталимид (10,3 г, 72 ммоля) и К2СО3 (9,7 г, 72 ммоля) кипятили в 60 мл сухого ацетона в течение 12 часов с перемешиванием и обратным потоком. После этого ацетон отгоняли при пониженном давлении и добавляли 100 мл воды к раствору неорганической соли. Реакционную смесь фильтровали при пониженном давлении, промывали водой и сушили, что дало соединение 19 (17,6 г) в виде белого твердого вещества. Выход составил 90%.
1H ЯМР (CDCl3, 300 МГц): δ 1,31 (s, 12H, 4СН3), 4,85 (s, 2H, СН2), 7,42 (d, J=6,6 Гц, 2H, ArH), 7,67-7,70 (m, 2H, ArH), 7,76 (d, J=6,6 Гц, 2H, ArH), 7,81-7,84 (m, 2H, ArH); 13C ЯМР (CDCl3, 75 М Гц): δ 24,8 (4С), 41,6, 83,7 (2С), 123,3 (2С), 127,8 (2С), 132,1, 133,9 (2С), 135,1 (3С), 139,3 (2С), 167,9 (2С); Масс-спектрометрия (El): m/z (%): 363 (M+, 100), 348 (17), 264 (37), 217 (34), 160 (31), 130 (29), 117 (92), 91 (16), 76 (36), 43 (78); ИК (пленка, см-1) vmax=2989, 2941, 1772, 1718, 1610, 1429, 1393, 1358, 1342, 1141, 1087, 1020, 962, 938, 856, 786, 718, 659.
Получение соединения 21
Соединение 19 (2,5 г, 6,9 ммолей) и гидразингидрат (0,47 мл, 80%, 7 ммолей) растворили при комнатной температуре в 30 мл метанола и смесь кипятили с перемешиванием и обратным потоком. Через 12 часов охлаждали до комнатной температуры и фильтровали при пониженном давлении. Получили белое твердое вещество, и фильтрат сгущали до сухого состояния. При этом воду удалили с помощью сухого бензола в атмосфере защитного газа. Затем добавили NEt3 (2,9 мл), сухой метанол (10 мл) и соединение 2 (2,1 г, 14 ммолей) и реакционную смесь перемешивали в течение 10 часов при комнатной температуре. Затем добавили К2СО3 (952 мг, 7 ммолей) и соединение 8 (1,6 г, 8,3 ммоля) и мешали еще 10 часов при комнатной температуре. После этого в реакционную смесь добавили немного воды и экстрагировали с СНСl3. Промывали водой, а органическую фазу отделяли, высушивали и сгущали. Остаток очищали с помощью колоночной хроматографии, что дало соединение 21 (85 мг) в виде масла. Выход составил 4%.
1H ЯМР (CDCl3, 300 М Гц): δ 0,88 (t, J=7,5 Гц, 3Н, СН3), 1,25-1,38 (m, 14H, СН2 и 4СН3), 1,63-1,73 (m, 2Н, CH2), 2,62 (t, J=7,5 Гц, 2Н, СН2), 5,59 (s, 2H, АrСН2), 6,99 (d, J=7,8 Гц, 2Н, ArH), 7,73 (d, J=7,8 Гц, 2Н, ArH), 7,78 (s, 1H, NCH=), 9,66 (s, 1H, CHO); 13C ЯМР (CDCl3, 75 М Гц): δ 13,7, 22,4, 24,8(4С), 26,5, 29,3, 48,2, 83,9(2С), 125,5(2С), 131,4, 135,3 (3С), 139,2, 143,6, 156,8, 178,7; Масс-спектрометрия (FAB): M+=368, найдено: 369 (М++1); ИК (пленка, см-1) vmax=3044, 2975, 2933, 2870, 1671, 1614, 1533, 1464, 1405, 1362, 1326, 1270, 1160, 1145, 1089, 1021, 963, 858, 821, 793, 721, 654.
Получение соединений 22 и 23
Суспензию из о-бромбензонитрила (9,1 г, 50 ммолей), NH4Cl (3,5 г, 65 ммолей), NaN3 (4,3 г, 65 ммолей) и LiCl в 80 мл DMF нагревали до 100°С и 12 часов перемешивали. Большую часть растворителя отгоняли при 120°С при пониженном давлении. Остаток делали щелочным с помощью 10%-ного водного раствора NaOH, пока водородный показатель рН не составил 12. Реакционную смесь экстрагировали с этилацетатом, а неорганическую фазу подкисляли концентрированной соляной кислотой до рН 2, причем выпало в осадок белое твердое вещество. Его отфильтровали при пониженном давлении с помощью воронки Бюхнера, промыли в воде и высушили, что дало соединение 22 (10 г, выход 90%).
Соединение 22 растворяли в 30 мл СН2Сl2, смесь охлаждали в бане с ледяной водой до 0°С и добавляли NEt3 (8 мл). После этого тремя порциями в течение 10 мин добавляли Рh3ССl (13,2 г, 47 ммолей), а реакционную смесь нагревали до комнатной температуры. После 3-часового перемешивания фильтровали при пониженном давлении в воронке Бюхнера, промывали в воде и сушили, что дало соединение 23 (18,9 г). Выход составил 90%.
1Н ЯМР (CDCl3, 300 М Гц): δ 7,18-7,36 (m, 17H, ArH), 7,66 (d, J=7,8 Гц, 1Н, АrН), 7,88 (d, J=7,8 Гц, 1Н, ArH); 13С ЯМР (CDCl3, 75 М Гц): 6 83,3, 122,2, 127,3, 127,7, 128,3, 128,7, 130,3, 131,1, 131,6, 133,9, 141,2, 162,9.
Получение соединения 12
Бензонитрил (10.3 г, 100 ммолей), NH4Cl (6.9 г, 1.3 экв.), NаN3 (8.5 г, 1.3 экв.) и LiCl (300 мг) растворяли в 100 мл DMF и реакционную смесь перемешивали в течение 12 часов при температуре 100°С. После этого большую часть растворителя удаляли при пониженном давлении. Остаток делали щелочным, добавив 10%-ный водный NaOH до получения водородного показателя рН 12. После экстракции с этилацетатом водную фазу отделяли и, добавив концентрированную соляную кислоту, подкисляли до получения водородного показателя рН 2. Осадок отфильтровывали в воронке Бюхнера, промывали водой и высушивали, что дало соединение 5-фенилтетразол (13.5 г, температура плавления 208-209°С). Выход составил 96%.
1H ЯМР (d-DMSO, 300 МГц) δ 7.55-7.57 (3Н, m), 8.01-8.03 (2H, m); 13C ЯМР (d-DMSO, 75 МГц) δ 129.5, 132.4, 134.8, 136.7, 160.7; Масс-спектрометрия (El) m/z (%): 146 (M+, 42), 118 (100), 103 (17), 91 (46), 77 (32), 63 (48); ИК (пленка, см-1) vmax=3055, 2982, 2837, 2607, 2545, 1607, 1562, 1485, 11463, 1409, 1163, 1056, 1013, 725, 703, 686.
5-Фенилтетразол (6.6 г, 45 ммолей) растворяли в 20 мл СН2Сl2 и прибавляли NEt3 (8 мл, 1.3 экв). Реакционную смесь охлаждали в бане с ледяной водой до 0°С и тремя порциями в течение 10 мин добавляли Рh3ССl (13.2 г, 1.05 экв.). После этого нагревали до комнатной температуры и три часа перемешивали. Реакционную смесь фильтровали, промывали водой и высушивали, получая соединение 12 (16.5 г, t° плавления 163-164°С). Выход составил 94%.
1H ЯМР (CDCl3, 300 МГц) δ 7.21-7.24 (6Н, m), 7.37-7.39 (9H, m), 7.47-7.49 (3Н, m), 8.19-8.20 (2H, m); 13C ЯМР (CDCl3, 75 МГц) δ 83.0, 127.0, 127.5, 127.7, 128.3, 128.7, 130.3, 141.3, 164.0; ИК (пленка, см-1) vmax=3058, 1490, 1465, 1445, 1186, 1028, 874, 763, 748, 697, 635.
Получение соединения 13
Раствор соединения 12 (10 г, 25.8 ммоля) в THF (30 мл) охлаждали в атмосфере аргона до -20°С. Затем добавляли BuLi (1 М, 27 мл, 1.05 экв.). Температуру повышали до -5°С и в течение часа перемешивали. За это время выпало в осадок большое количество твердого вещества. Снова охлаждали до -25°С и медленно, через шприц добавляли В(ОМе)3 (4.3 мл, 1.5 экв.). Затем реакционной смеси позволяли нагреться до 20°С и перемешивали ее в течение получаса. Количество растворителя уменьшили на треть от первоначального при пониженном давлении, причем образовалось белое твердое вещество. Твердое вещество отфильтровывали, промывали в 20 % THF в Н2О (40 мл) и воде (40 мл) и высушивали, что дало соединение 13 (10.4 г). Выход составил 94%. Соединение 13 можно использовать уже без дополнительной очистки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КАНДЕСАРТАНА | 2005 |
|
RU2407741C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-2-ФЕНИЛАЛКАНОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2009 |
|
RU2486174C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВИРУЮЩИМ ДЕЙСТВИЕМ НА ПОДТИПЫ РЕЦЕПТОРОВ, АКТИВИРУЕМЫХ ПРОЛИФЕРАТОРОМ ПЕРОКСИСОМ (PPARs), И СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ УКАЗАННЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2546117C2 |
| СПОСОБ МИНИМИЗАЦИИ ГИСТЕРОТРОФНОГО ЭФФЕКТА ТАМОКСИФЕНА И АНАЛОГОВ ТАМОКСИФЕНА | 1995 |
|
RU2158589C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ СИГМА РЕЦЕПТОРОВ | 2011 |
|
RU2582338C2 |
| ИНГИБИТОРЫ MMPL3, КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2795229C2 |
| 3,5-ДИЗАМЕЩЕННЫЕ 1,2,4-ТИАДИАЗОЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ СВЯЗЫВАНИЯ ТИОЛОВ | 1997 |
|
RU2173319C2 |
| ЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ 3-ГИДРОКСИ-4-ПИРИДИНОНОВ | 2004 |
|
RU2345992C2 |
| БИАРИЛ- ИЛИ ГЕТЕРОЦИКЛИЧЕСКИЕ БИАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСЕНА В КАЧЕСТВЕ ИНГИБИТОРОВ СЕТР | 2014 |
|
RU2627361C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ, ИХ СПОСОБ ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2749437C2 |
Описывается способ получения соединения общей формулы I
где R1 означает радикал общей формулы II:
где R2 - защитная группа тетразола, которое является новым промежуточным соединением при получении лосартана - 2-н-бутил-4-хлор-5-гидроксиметил-1-{[2'-(1 Н-тетразол-5-ил)бифенил-4]метил}-имидазола, и способ получения лосартана, исходя из соединения I путем восстановления формильной группы с последующим введением атома хлора в имидазольное кольцо и снятием защитной группы R2. Описываемый способ обеспечивает достижение высокого выхода целевого продукта из доступных исходных и промежуточных соединений в технических масштабах. 2 н. и 7 з.п. ф-лы.
1. Способ получения соединения общей формулы I

в которой R1 является радикалом общей формулы II
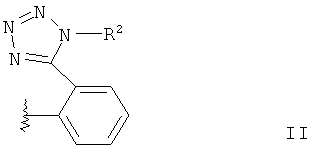
в которой R2 означает защитную группу тетразола,
причем соединение общей формулы IV

в которой R5 является радикалом общей формулы II,
подвергают взаимодействию с соединением общей формулы V
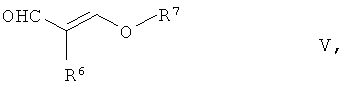
в которой R6 означает галоген из группы Cl, Вr, I, предпочтительно Вr, и R7 означает разветвленную или неразветвленную группу C1-С6-алкила, предпочтительно группу изопропила,
в присутствии основания Бренстеда, в двухфазной реакционной системе, в которой одна фаза образуется из водного раствора, а другая фаза образуется из раствора, включающего органический растворитель, ограниченно смешиваемый с водой, при молярном отношении 0,5-2:1, в пересчете на молярные количества соединения общей формулы IV к соединению общей формулы V, причем время реакции составляет от 0,1 до 20 ч.
2. Способ по п.1, в котором защитная группа тетразола R2 в формуле II обозначает трифенилметил или трет-бутил.
3. Способ по п.1, в котором реакцию проводят в присутствии слабого основания Бренстеда.
4. Способ по п.1, в котором в качестве основания Бренстеда используют щелочной карбонат или щелочной гидрогенкарбонат, предпочтительно, например, карбонат натрия, карбонат калия или гидрогенкарбонат натрия.
5. Способ по п.1, в котором в качестве основания Бренстеда используют карбонат калия.
6. Способ по п.1, в котором в качестве органического растворителя, ограниченно смешиваемого с водой, используют толуол, хлористый метилен, хлороформ или их смеси.
7. Способ по п.1, в котором время реакции составляет от 5 до 15 ч.
8. Способ получения лосартана формулы

или его фармакологически приемлемой соли, в котором
а) на стадии (а), исходя из соединения общей формулы I
в которой R1 является радикалом общей формулы II
в которой R2 означает защитную группу тетразола,
получают соединение общей формулы XI

в которой R15 означает радикал вышеприведенной общей формулы II, восстановлением группы формила, которой замещена группа имидазола, в группу гидроксиметила,
b) на стадии (b) заменяют хлором единственный еще оставшийся атом водорода в группе имидазола соединения, полученного на стадии (а), и
c) на стадии (с) отщепляют защитную группу тетразола и, при необходимости,
d) получают из лосартана одну из его фармакологически приемлемых солей.
9. Способ по п.8, в котором на стадии (b) заменяют хлором единственный еще оставшийся атом водорода в имидазольной группе полученного на стадии (а) соединения путем взаимодействия соединения, полученного на стадии (а), с СеСl3 и солью щелочного металла гипогалогеновой кислоты в качестве реагентов.

