Область техники
Настоящее изобретение относится к новым оксазолидиноновым производным с циклической амидоксимной или циклической амидразоновой группой, представленных формулой I, их фармацевтически приемлемым солям, способам их получения и содержащим их фармацевтическим композициям.
[Формула I]
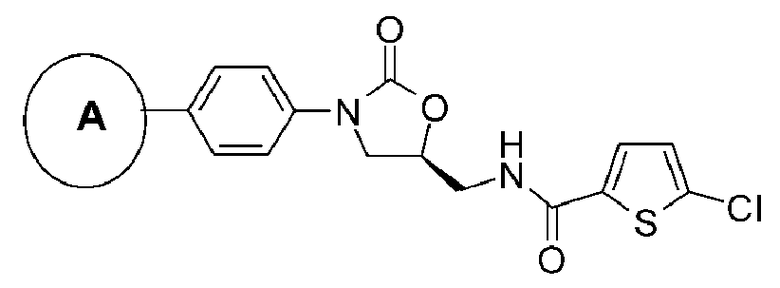
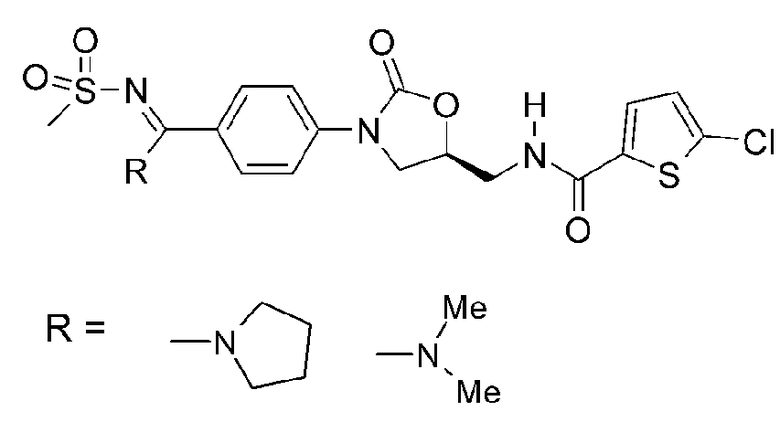
где цикл А представляет собой остаток, выбранный из группы, включающей следующие структуры:
Антитромботический и антикоагулянтный эффект новых оксазолидиноновых производных по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленных формулой I, приписывают ингибированию активной коагулирующей протеазы, известной как фактор Xa, или других активных сериновых протеаз, таких как фактор VIIa, фактор IXa или тромбин.
Уровень техники
Факторы свертывания крови распределены в плазме, притом что различные типы факторов, от 1-го фактора свертывания до 13-го фактора свертывания, работают по каскадному типу, обеспечивая в результате свертывание крови. Механизм, по которому индивидуальные факторы свертывания крови участвуют в свертывании крови, показан на фиг. 1.
Как показано на фигуре 1, свертывание крови происходит в результате ряда сложных реакций. В целом, инактивированные предшественники активируются специфическими активными факторами свертывания крови (указаны индексом "a", прибавленным после номера фактора коагуляции). Затем активируются следующие факторы свертывания крови. Большинство из этих активированных факторов свертывания крови представляют собой ферменты семейства сериновых протеаз. Они прилипают к поверхности активированного тромбоцита на месте раны, постепенно активируют факторы свертывания крови и, в конце концов, продуцируют фибриновый сгусток, приводя к остановке кровотечения.
Тромбин является многофункциональным фактором коагуляции, который вовлечен в конечные стадии коагуляционного каскада. Протромбин, предшественник тромбина, активируется комплексом протромбиназы, состоящим из фактора Va, фактора Xa, Ca++ и фосфолипидов (PL), получая тромбин, который преобразует фибриноген в фибрин. Генерированные фибрины покрывают агрегированный тромбоцит, индуцируя свертывание крови. В заключение фибрины сшиваются фактором XIIIa, продуцируя стабильный фибриновый сгусток.
Для образования комплекса протромбиназы фактор X активируется в фактор Xa, что опосредовано, главным образом, комплексом Хазы. Фактор VIIIa, фактор IXa, Ca++ и фосфолипиды (PL), генерируемые по внутреннему пути, или фактор VIIa, фактор ткани (TF) и Ca++, генерируемые по внешнему пути, работают как комплекс Хазы.
Тромбин также активирует фактор V и фактор VIII. Если происходит перепроизводство тромбина, сам кровеносный сосуд может забиться. Чтобы избежать закупоривания, тромбин запускает действие по ингибированию свертывания крови. То есть, тромбин связывается с тромбомодулином, активируя протеин C. Активированный протеин C (APC) образует комплекс с протеином S, инактивирует фактор Va и фактор VIIIa.
Фактически фактор Xa сам по себе является сериновой протеазой и включен в сложный процесс свертывания крови. Фактор Xa, как существенный член комплекса протромбиназы, действует как катализатор для преобразования протромбина в тромбин. Тромбин преобразует фибриноген в фибриновые мономеры, и генерированные таким образом фибриновые мономеры включаются в образование и стабилизацию тромба. Таким образом, в результате чрезмерного или несоответствующего продуцирования тромбина может происходить тромбоэмболия. Следовательно, ингибирование самого тромбина или генерации тромбина может приводить к снижению образования фибрина, вовлеченного в образование тромба, и предупреждению тромбоэмболии.
Вкратце, ингибирование фактора Xa приводит к ингибированию образования тромбина, посредством чего можно предупредить или ослабить тромбоэмболию. Соединение, представленное в настоящем изобретении формулой I, и его фармацевтически приемлемая соль могут ингибировать фактор Xa, что в конечном счете согласно изложенной выше логике ведет к предупреждению тромбоэмболических заболеваний (MI, удара, PE и др.).
Среди соединений, известных в качестве ингибиторов фактора Xa, типичными примерами ингибиторов белка являются антистазин (ATS) и клещевой антикоагулянтный пептид (TAP). ATS (состоящий из 119 аминокислот) представляет собой природный пептид, выделяемый из пиявок, имеющий значение Ki 0,05 нМ относительно фактора Xa. TAP также представляет собой пептид, выделяемый из клещей, который составлен из 60 аминокислот и имеет значение Ki 0,5 нМ относительно фактора Xa. Однако эти ингибиторы имеют ограниченное клиническое применение; только гепарин или его сульфатированные полисахаридные аналоги имеют клиническое применение с некоторым ограничением.
В качестве ингибитора свертывания крови, в частности ингибитора фактора Xa, разработано низкомолекулярное соединение, которое описано в W095291S9. Между тем, в WO9933800 описан ингибитор фактора Xa, имеющий индольные фрагменты. Кроме того, различные ингибиторы фактора Xa раскрыты и в процессе разработки. Например, гетероциклическое соединение, содержащее атом азота (WO2004058743), имидазольные производные (WO2004050636), пиразольные производные (WO2004056815), индол-2-карбоксамидные производные (WO2003044014), оксибензамидные производные (WO200205I831), гуанидин/амидиновые производные (WO2002046159), амино-бициклические пиразинон/пиридиноновые производные (WO2004002405) и др.
Чтобы быть клинически пригодными в качестве ингибиторов FXa, эти молекулы должны обладать высоким антитромботическим эффектом, высокой стабильностью в плазме и печени, подходящей селективностью в отношении других родственных сериновых протеаз (тромбин, трипсин, катепсин G и др.), низкой токсичностью и удовлетворительной биодоступностью.
Наиболее прогрессивным соединением, содержащим оксазолидинон, аналогично соединению по настоящему изобретению, является ривароксабан (формула A), который в настоящее время находится в фазе III клинической оценки. Некоторые оксазолидиноновые производные, представленные формулой 2, описаны в WO 01/47917. Однако сообщается, что некоторые из этих соединений имеют ограниченную растворимость; конкретный пример данной проблемы представляет ривароксабан. Растворимость ривароксабана составляет только 8 мг/л. В результате слабой растворимости может возникать множество практических ограничений, включая вариабельность и медленное разложение. Эти проблемы можно обойти, вводя высокорастворимый фрагмент.
[Формула A]
[Формула B]
В WO 2004/83174 описано применение пиразольных производных, включая апиксабан. Некоторые из этих ингибиторов являются циклическими амидиновыми и сульфониламидиновыми производными, представленными формулой C.
Однако не имеется прецедентов, аналогичных настоящему изобретению, которые описывают специфическое введение циклического амидоксима или циклического амидразона в оксазолидиноновый каркас с целью получения ингибиторов фактора Xa. Фактически, мало что известно в отношении обоснования присутствия циклического амидоксима или циклического амидразона в конструкции лекарства.
[Формула C]
Главной тенденцией последних исследований FXa и ингибитора тромбина является реализиция амидиновых функций. Предполагается, что амидиновая функция, так называемая группа P1, связывает Asp189, расположенный на дне кармана S1. И FXa, и тромбин распознают аргининовый остаток в природном субстрате как сайт P1. Амидиновая группа (в том числе гуанидиновые производные), замещающая гуанидин в аргинине, является высокогидрофильной. Таким образом, ингибиторы с амидиновой функцией обычно не очень хорошо абсорбируются, и даже если они абсорбируются, то очень быстро выводятся вследствие присущей им высокой полярности амидина (Drugs of the Future, 1999, 24(7), 771).
Сам амидин характеризуется сильной основностью (PKa: примерно 12,5). Из-за формально положительного заряда в физиологических условиях амидиновый ингибитор обычно демонстрирует слабую абсорбцию. Следовательно, необходимо произвести изменения в направлении менее основных альтернатив. Типичными примерами таких веществ являются пиридиновое производное, амидразон, циклический амин, алкиламинное производное, аминобензизоксазол и др. (US 6958356). Имеются также фундаментально отличные подходы к преодолению этих проблем, включающие использование амидоксимов. Амидоксим легко синтезируют, добавляя гидроксильную группу к амидиновой структуре, которая представляет собой пролекарство, основываясь на том, что слабая связь N-O легко восстанавливается in vivo до амидина. Этот подход выгодно использует тот факт, что PKa амидоксима заметно ниже (8-9), чем соответствующее значение для амидина. Другим примером пролекарства такого же типа является ксимелагатран. Эта тенденция видна не только при исследовании FXa-ингибитора, но также при исследовании ингибитора тромбина. Однако большинство этих попыток оказываются не такими хорошими, как ожидалось. В качестве попытки третьего типа вводят нейтральные группы P1. В отличие от лекарств другого класса FXa и ингибиторы тромбина имеют тенденцию демонстрировать хорошую эффективность, если они присутствуют в крови при высоких концентрациях. Кроме того, очень важна концентрация в крови свободного лекарственного средства, не связанного с белками сыворотки. В случае ингибитора с нейтральной группой P1 существует тенденция высокого связывания белка, в результате чего эффективность является меньшей, чем ожидалось.
Для преодоления этих проблем авторы настоящего изобретения вводят относительно полярную группу в положения, отличные от сайта P1, при фиксированном положении нейтральной группы на сайте P1. Параллельно с этой логикой включаются некоторые другие важные факторы для улучшения фармацевтического эффекта, которые приведены далее: 1) существенное повышение растворимости в воде; 2) низкое связывание белка плазмы; если концентрация свободного лекарственного средства является высокой, эффективность в PT-исследовании также повышается, даже если FXa-связывающая афинность отчасти снижается.
Сайт, выбранный для введения полярной группы, в данном изобретении является подсайтом P4 ингибитора, и логика выбора является следующей. Сайт S4 от FXa имеет U-образный сайт связывания, окруженный с трех сторон Tyr99, Phe174 и Trp215. Сайт связывания составлен только из боковых цепей ароматических аминокислот, в отличие от тромбина, окруженного Leu99, Ile174 и Trp215. Различие используют в конструкции лекарства.
S4 карман от FXa является подходящим при взаимодействии с катионным остатком, которое обычно называют "π-катионным взаимодействием". Фактически, некоторые ингибиторы конструируют и синтезируют таким образом, чтобы они имели положительный заряд на сайтах P4. В данном изобретении циклический амидоксим или амидразон вводят в сайт P4 для улучшения растворимости в воде и повышения эффекта лекарственного средства по снижению связывания белка, как упоминается выше. Причина выбора циклической формы заключается в том, что авторы полагают, что абсорбцию можно улучшить, снижая количество NH связей, которые обычно оказывают отрицательное влияние на абсорбцию. Согласно последним исследованиям для лекарства более предпочтительно иметь меньше доноров водородных связей (HBD), чем акцепторов водородной связи (HBA). Согласно правилу Липински можно допустить наличие до 10 HBA, а количество HBD ограничено только 5 (Adv. Drug Deliveiy Rev., 2001, 46, 3-26), и, в частности, в случае нового лекарства среднее количество HBD составляет примерно 2, предполагая, что число HBD ограничено более строго (J. Med. Chem. 2004, 47, 6338-48). Амидоксимная или амидразоновая группа сама по себе имеет основный характер, что делает возможным более легкое разделение-очистку-хранение в виде соли и, как результат, ожидается повышенная растворимость в воде.
Вкратце, авторы вводят амидоксим или амидразон в сайт P4. Для снижения числа HBD авторы используют циклическую форму функции, получая ингибиторы, более похожие на лекарство.
Фактически, установлено, что соединения формулы I по настоящему изобретению имеют указанные выше преимущества. Представлена растворимость в воде и уровень связывания белка вместе со значениями 2× PT и Ki.
Раскрытие изобретения
Техническая задача
Авторы настоящего изобретения синтезировали новые оксазолидиноновые производные с циклической амидоксимной или циклической амидразоновой группой, имеющей подходящие свойства, которые можно применять для приготовления фармацевтических препаратов. В частности, оксазолидиноновые производные с циклической амидоксимной или циклической амидразоновой группой демонстрируют FXa ингибирующее действие, так что их можно применять для лечения или профилактики тромбоза, инфаркта миокарда, артериосклероза, воспаления, апоплексии, стенокардии, рецидивирующей стриктуры после ангиопластики и тромбоэмболии, например перемежающейся хромоты. Кроме того, оксазолидиноновые производные с циклическим амидоксимом или циклическим амидразоном по настоящему изобретению могут служить в качестве ингибитора в отношении фактора VIIa, фактора IXa и тромбина, которые представляют собой факторы коагуляции в каскаде свертывания крови.
Целью настоящего изобретения является обеспечение новых оксазолидиноновых производных с циклической амидоксимной или циклической амидразоновой группой, демонстрирующих свойство ингибирования фактора Xa, или их фармацевтически приемлемых солей.
Другой целью настоящего изобретения является обеспечение фармацевтической композиции для противодействия коагуляции, содержащей в качестве активного ингредиента оксазолидиноновые производные с циклической амидоксимной или циклической амидразоновой группой или их фармацевтически приемлемые соли.
Также целью настоящего изобретения является обеспечение фармацевтической композиции, содержащей в качестве активного ингредиента оксазолидиноновые производные с циклической амидоксимной или циклической амидразоновой группой или их фармацевтически приемлемые соли, для лечения или профилактики тромбоза, инфаркта миокарда, артериосклероза, воспаления, удара, стенокардии, рестеноза, перемежающейся хромоты, флеботромбоза, легочной эмболии, артериального тромбоза, ишемии миокарда или тромбоэмболии.
Кроме того, целью настоящего изобретения является обеспечение фармацевтической композиции для лечения или профилактического лечения заболевания коронарной артерии, заболевания церебральных артерий и заболевания периферических артерий, характеристически подвергаемых совместному лечению оксазолидиноновыми производными с циклической амидоксимной или циклической амидразоновой группой или их фармацевтически приемлемыми солями и тромболитическим агентом.
Также целью настоящего изобретения является обеспечение применения оксазолидиноновых производных с циклической амидоксимной или циклической амидразоновой группой или их фармацевтически приемлемых солей в качестве антикоагулянта для in vitro консервации крови, плазмы или продуктов крови.
Техническое решение
Настоящее изобретение относится к новым оксазолидиноновым производным с циклической амидоксимной или циклической амидразоновой группой, представленным формулой I, или их фармацевтически приемлемым солям, способам их получения и содержащим их фармацевтическим композициям.
[Формула I]
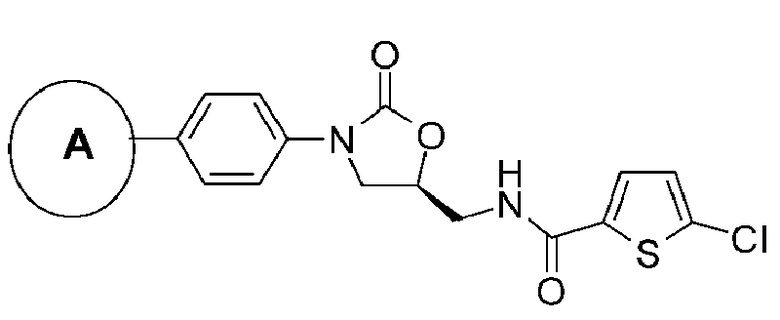
где цикл А представляет собой остаток, выбранный из группы, включающей следующие структуры;
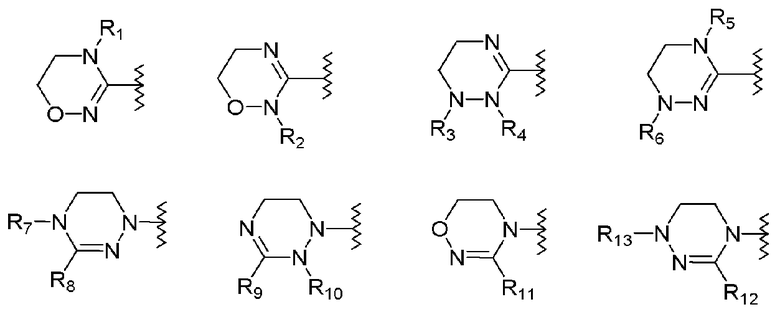
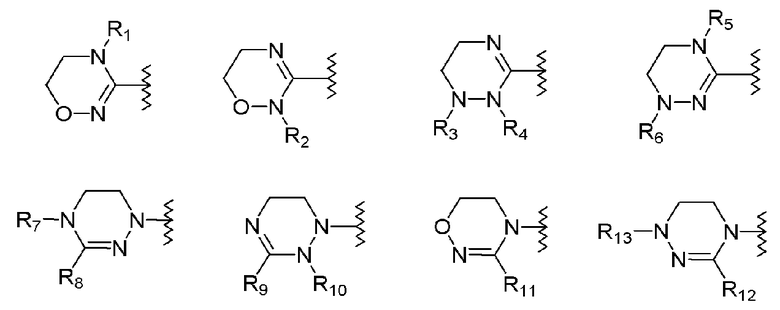
R1-R12 независимо представляют собой H, (C1-C7)алкил, (C3-C7)циклоалкил, (C6-C12)арил или (C4-C12)гетероарил, содержащий от одного до четырех гетероатомов, выбранных из группы, состоящей из O, S и N, R3 и R4 образуют цикл посредством соединения с (C3-C5)алкиленом, атом углерода алкилена может быть замещен карбонилом, и алкил, циклоалкил, арил или гетероарил от R1-R12 может быть замещен одним заместителем, выбранным из группы, состоящей из (C1-C7)алкила, галоген(C1-C7)алкила, (C1-C7)алкокси и галогена;
R13 означает H, (C1-C7)алкил, (C1-C7)циклоалкил, формил, (C1-C7)алкилкарбонил, (C1-C7)алкоксикарбонил или (C6-C12)арил.
Термин "арил" в этом изобретении означает органический радикал, получаемый из ароматического углеводорода, отщеплением одного H, в котором каждый цикл моноциклической системы или конденсированной циклической системы содержит 6-12, предпочтительно 6-10, циклических атомов. Конкретно, термин включает фенил, нафтил, бифенил и инденил, но не всегда ограничен ими.
Термин "гетероарил" в этом изобретении означает арильную группу, которая содержит 1-4 гетероатома, выбранных из группы, состоящей из N, O и S, в качестве атомов, составляющих структуру ароматического цикла, и остальные атомы, составляющие структуру ароматического цикла, являются атомами C, которая проиллюстрирована (5-6)-членным моноциклическим гетероарилом и полициклическим гетероарилом, конденсированным с одним или большим количеством бензольных колец, которые могут быть частично насыщенными.
Оксазолидиноновые производные по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой выбраны из соединений следующих далее формул II-XI.
[Формула II]
[Формула III]
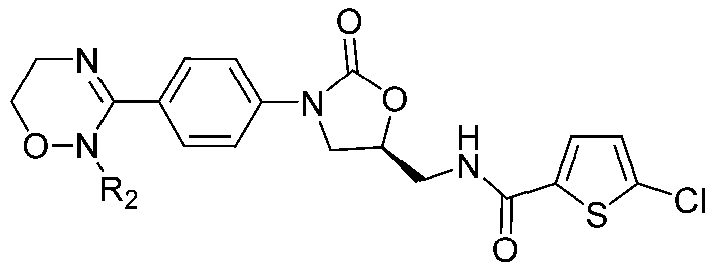
[Формула IV]
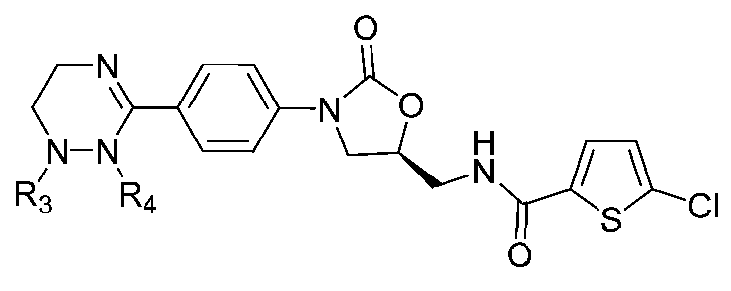
[Формула V]
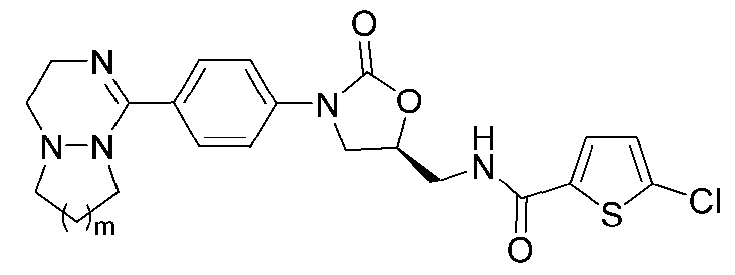
[Формула VI]
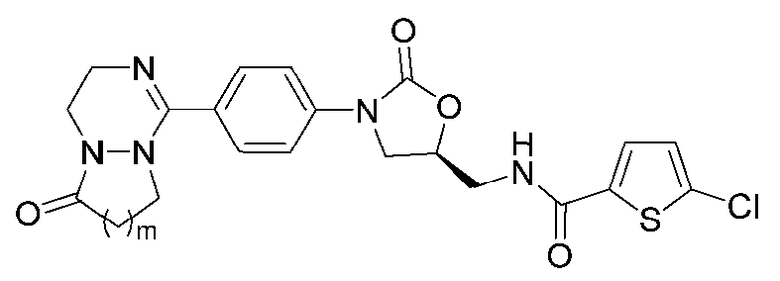
[Формула VII]
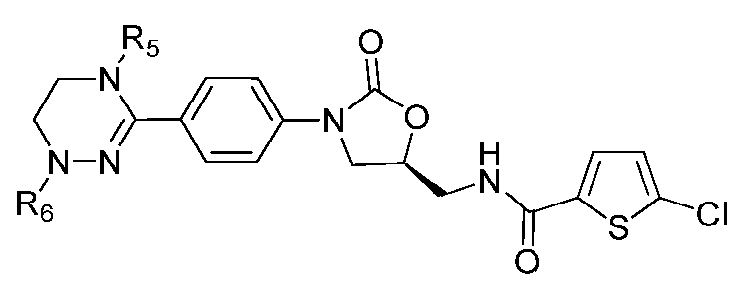
[Формула VIII]
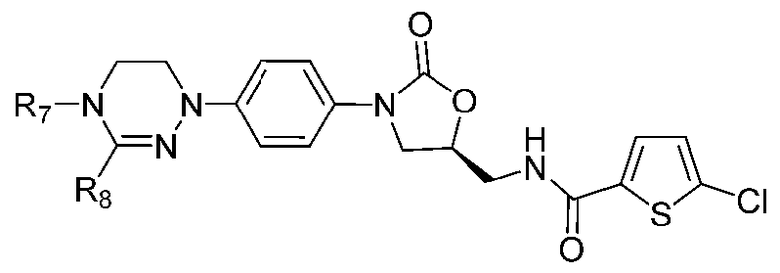
[Формула IX]
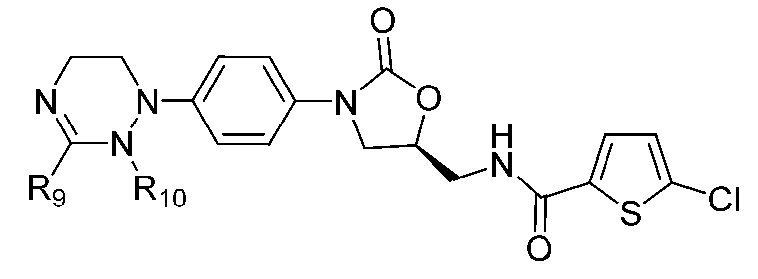
[Формула X]
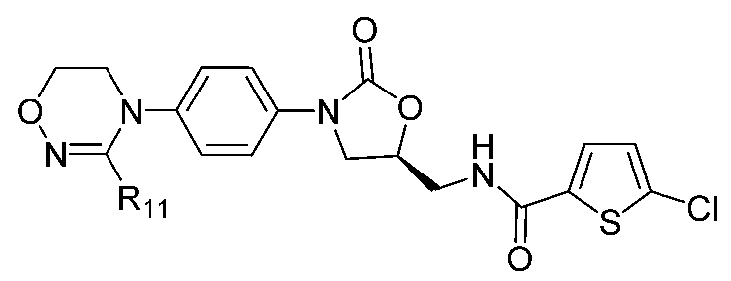
[Формула XI]
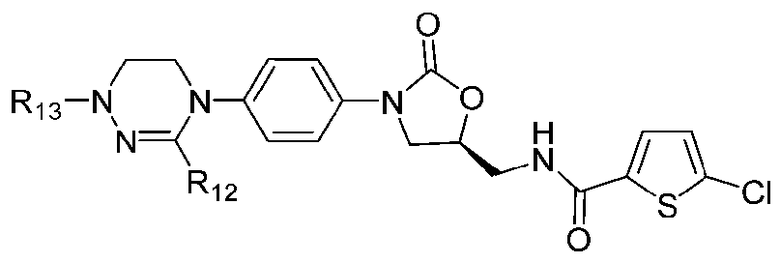
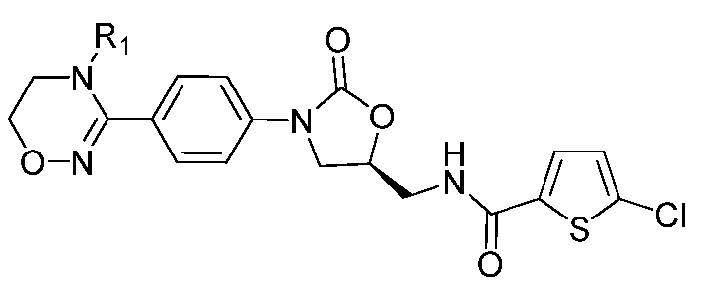
где R1-R12 независимо представляют собой H, (C1-C7)алкил или (C3-C7)циклоалкил; R13 означает H, (C1-C7)алкил, (C3-C7)циклоалкил, формил или (C1-C7)алкилкарбонил; и m равно целому числу от 1 до 3.
В качестве примера оксазолидиноновых производных по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой в приведенных выше формулах II-XI R1-R12 независимо представляют собой H, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, циклопропил, циклобутил, циклопентил или циклогексил; R13 означает H, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, циклопропил, циклобутил, циклопентил, циклогексил, формил или ацетил; и m равно целому числу 1.
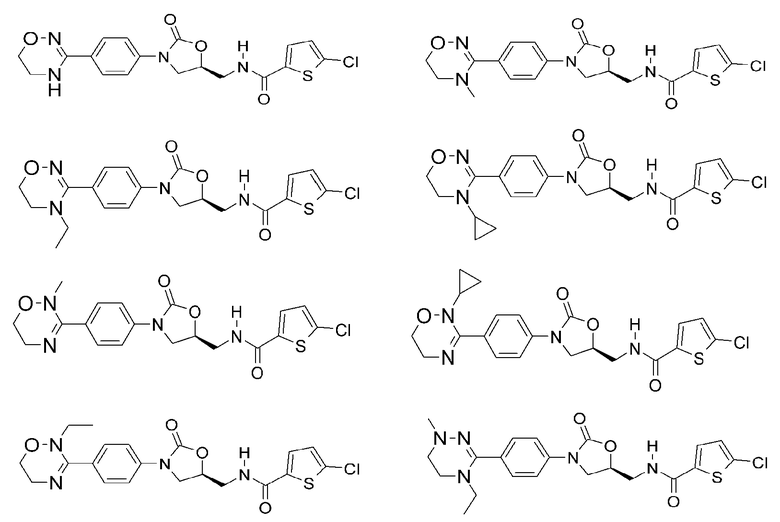
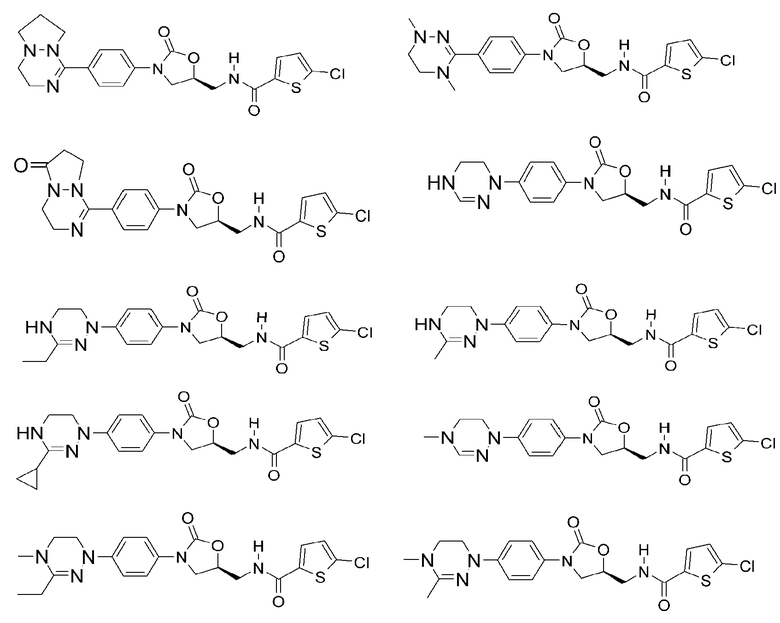
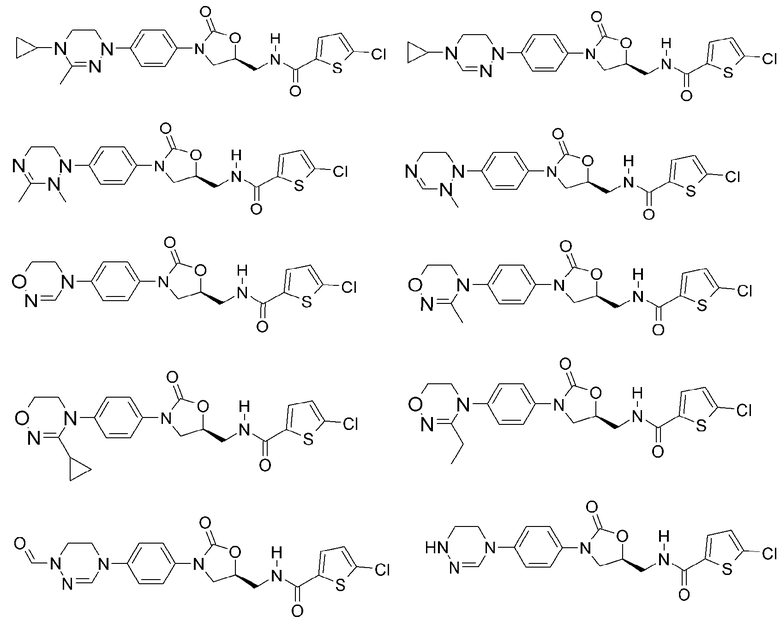
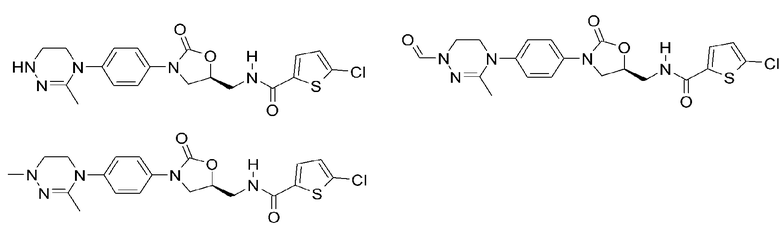
Оксазолидиноновые производные по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой можно проиллюстрировать следующими соединениями, но не всегда ограничиваясь ими:
В этом изобретении способы получения оксазолидиноновых производных с циклической амидоксимной или циклической амидразоновой группой, представленных формулой I, проиллюстрированы при помощи реакционных схем с 1 по 5. Однако приведенный далее способ не может ограничивать способы получения оксазолидиноновых производных по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленных формулой I, и специалистам в данной области хорошо известно, что модификация приведенного ниже способа дозволена и также включена в признаки настоящего изобретения. Если указано иного, заместители на реакционных схемах такие, как определено для формулы I.
Как показано на реакционных схемах 1-5, способ получения оксазолидиноновых производных с циклической амидоксимной или циклической амидразоновой группой, представленных формулой I, модифицируют в соответствии со структурой циклического соединения A в формуле I. Сначала синтезируют циклические амидоксимные соединения, такие как соединения B1 и B2 на реакционной схеме 1 и соединение E на реакционной схеме 4. Также синтезируют циклические амидразоновые соединения, такие как соединения C1 и C2 на реакционной схеме 1, соединение D1 на реакционной схеме 2 и соединение F на реакционной схеме 5.
[Реакционная схема 1]
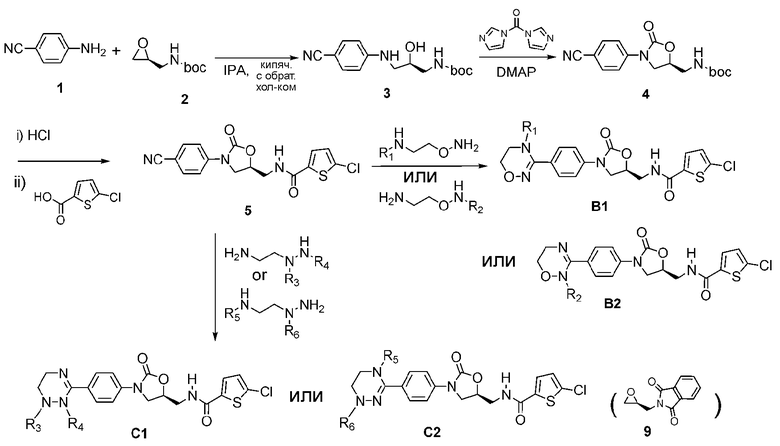
Как показано на реакционной схеме 1, синтез циклических амидоксимных соединений B1 и B2 формулы I и циклических амидразоновых соединений C1 и C2 осуществляют, проводя взаимодействие 4-цианоанилина (1) и 2-(((S)-оксиран-2-нил)метил)трет-бутилоксикарбонила (2) с получением соединения 3. Затем синтезируют оксазолидиноновое циклическое соединение 4, используя 1,1-карбонилдиимидазол и DMAP, и реакционную смесь обрабатывают HCl для удаления защитной группы boc. Проводят конденсацию с 5-хлортиофен-2-карбоновой кислотой с последующей обработкой HCl. В заключение проводят взаимодействие реагента с диаминным соединением, получая циклические амидоксимные соединения B1 и B2 формулы I и циклические амидразоновые соединения C1 и C2.
В синтезе соединения 3 можно получить аминное соединение, защищенное фталимидом, используя 2-(((S)-оксиран-2-ил)метил)изоиндолин-1,3-дион (9) из реакционной схемы 2 вместо 2-(((S)-оксиран-2-нил)метил)трет-бутилоксикарбонила (2), аналогично способу по реакционной схеме 2. На реакционной схеме 1 описан способ с использованием 2-(((S)-оксиран-2-нил)метил)трет-бутилоксикарбонила (2), и на реакционной схеме 2 описан способ с использованием 2-(((S)-оксиран-2-ил)метил)изоиндолин-1,3-диона (9).
Циклические амидразоновые соединения D1 и D2 формулы I можно различить по расположению двойной связи, и их синтезируют по реакционной схеме 2 и реакционной схеме 3.
[Реакционная схема 2]
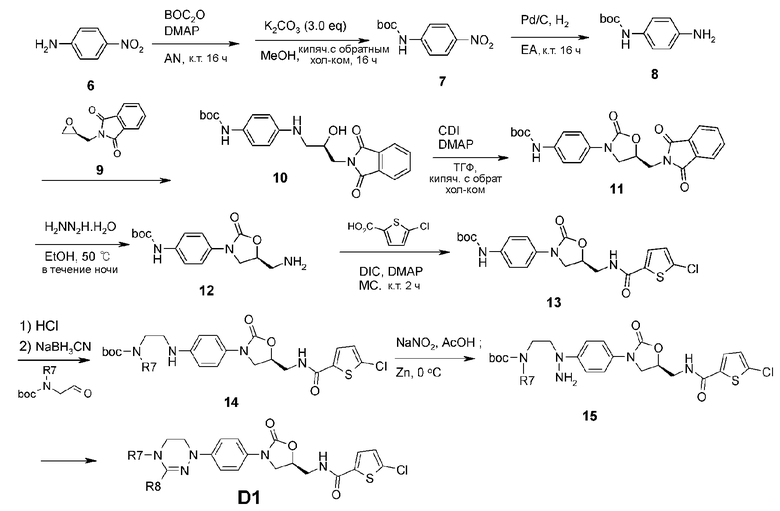
Сначала 4-нитроанилин (6) защищают при помощи группы boc, а затем гидрируют, используя палладиевый катализатор. Затем синтезируют аминоспиртовое соединение 10, используя 2-(((S)-оксиран-2-ил)метил)изоиндолин-1,3-дион (9). Оксазолидиноновый цикл получают, используя карбонилдиимидазол, с получением соединения 11. Используя гидразин, удаляют фталимидную защитную группу, а затем проводят конденсацию с 5-хлортиофен-2-карбоновой кислотой, получая соединение 13. Соединение 13 обрабатывают HCl для удаления защитной группы boc, далее проводят взаимодействие с boc-защищенным аминалом, получая соединение 14. Вводят нитрозогруппу, используя NaNO2, а за тем проводят восстановление посредством Zn, получая гидразиновое соединение 15. Проводят взаимодействие соединения 15 с ортоформиатом, получая циклическое амидразоновое соединение D1.
Другое циклическое амидразоновое соединение D2 синтезируют по реакционной схеме 3.
[Реакционная схема 3]
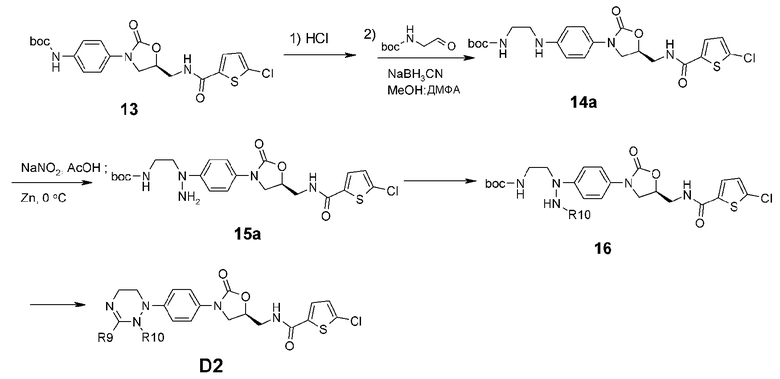
Из соединения 14, показанного на реакционной схеме 2, синтезируют соединение 14a, где R7 означает H, в которое вводят аминогруппу (15a) и затем постепенно вводят алкильную группу (16). Индуцируют циклизацию, используя ортоформиат и получая циклическое амидразоновое соединение D2.
Циклическое амидоксимное соединение E формулы I синтезируют по реакционной схеме 4.
[Реакционная схема 4]
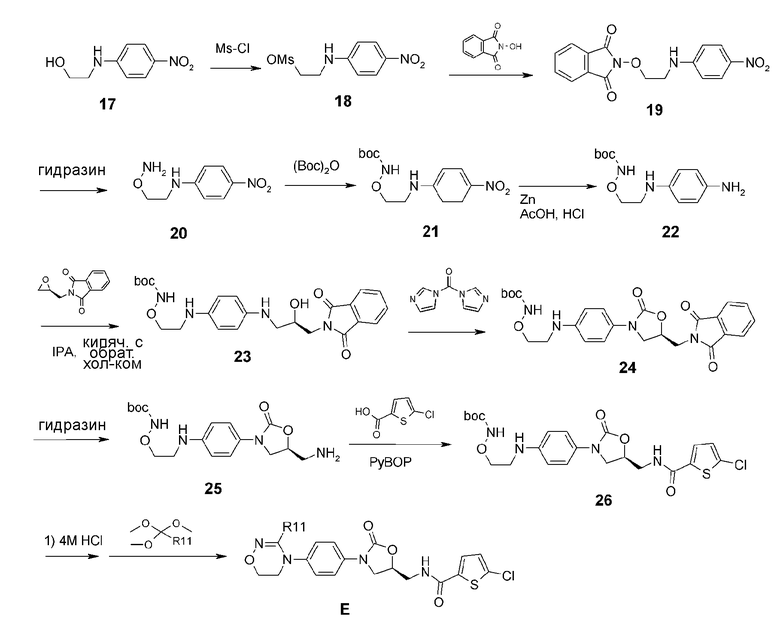
Проводят взаимодействие соединения 17, полученного из 4-фторнитробензола, с метансульфонил хлоридом, получая соединение 18, затем проводят взаимодействие с гидроксифталимидом, получая соединение 19. Фталимидную защитную группу удаляют при помощи гидразина, а вместо нее используют для защиты группу boc, получая в результате соединение 21. Нитрогруппу соединения 21 восстанавливают, используя Zn и получая соединение 22, затем проводят взаимодействие с 2-(((S)-оксиран-2-ил)метил)изоиндолин-1,3-дионом (9) способом, показанным на реакционной схеме 2, с получением соединения 26. Соединение 26 обрабатывают HCl, затем проводят взаимодействие с ортоэфиром, получая соединение E.
Циклическое амидразоновое соединение F формулы I синтезируют по реакционной схеме 5.
[Реакционная схема 5]
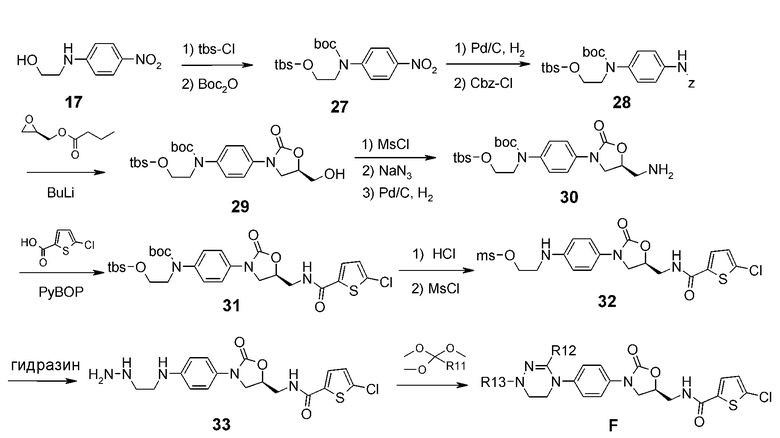
Вводят защиту соединения 17, полученного из 4-фторнитробензола, посредством группы tbs (трет-бутилдиметилсилил) и затем вводят защиту аминной области посредством boc, получая в результате соединение 27. Используя палладиевый катализатор, получают амин, который подвергают взаимодействию с Cbz-Cl, получая соединение 28. Проводят взаимодействие соединения 28 с глицидилбутилатом, получая соединение 29. Спиртовую группу заменяют амином с получением соединения 30. Проводят взаимодействие этого соединения с хлортиофенкарбоновой кислотой, получая соединение 31, затем проводят взаимодействие с метансульфонилхлоридом. Реагент обрабатывают гидразином с получением соединения 33. Проводят взаимодействие соединения 33 с ортоэфиром, получая соединение F.
Оксазолидиноновые производные по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленные формулой I, можно применять в медицине в качестве фармацевтически активного ингредиента, в частности, для профилактики и лечения тромбоза, инфаркта миокарда, артериосклероза, воспаления, апоплексии, стенокардии, рецидивирующей стриктуры после ангиопластики, перемежающейся хромоты, флеботромбоза, легочной эмболии, артериального тромбоза, ишемии миокарда, нестабильной стенокардии на базе тромбоза и тромбоэмболии, например криза.
Оксазолидиноновые производные по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленные формулой I, или их фармацевтически приемлемые соли можно использовать для профилактики или лечения атеросклеротического заболевания, в том числе заболевания коронарной артерии, заболевания церебральных артерий или заболевания периферических артерий. Для лечения инфаркта миокарда оксазолидиноновые производные с циклической амидоксимной или циклической амидразоновой группой можно применять совместно с тромболитическим агентом (например, альтеплазой, тенектеплазой и др.). Указанные соединения также можно использовать для профилактики реокклюзии после тромболизиса, чрескожной транслюминальной коронарной ангиопластики (PTCA) и обходного шунтирования коронарной артерии.
Оксазолидиноновые производные по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленные формулой I, или их фармацевтически приемлемые соли можно использовать для профилактики постоперативного повторного образования тромбов. Их также можно использовать в качестве антикоагулянта в отношении искусственного органа или гемодиализа. Указанное соединение можно использовать для промывки катетера и медицинского вспомогательного устройства, применяемого in vivo. Кроме того, их также можно использовать в качестве антикоагулянтной композиции для ex vivo хранения крови, плазмы и продуктов крови других типов. Указанные соединения по настоящему изобретению также эффективны при лечении заболевания, связанного со свертыванием крови, или заболевания, вызванного вторичным поражением, например рака (в том числе, метастатического рака), воспалительного заболевания, в том числе артрита и диабета.
Оксазолидиноновые производные по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленные формулой I, можно использовать в виде фармацевтически приемлемых солей. Что касается фармацевтически приемлемых солей, предпочтительны аддитивные соли кислот, получаемые с использованием фармацевтически приемлемой свободной кислоты. Свободную кислоту (неорганическая или органическая кислота) можно использовать, если она является фармацевтически приемлемой. Примеры неорганической свободной кислоты включают соляную кислоту, бромистоводородную кислоту, серную кислоту и фосфорную кислоту. Примерами доступных органических свободных кислот являются лимонная кислота, уксусная кислота, молочная кислота, малеиновая кислота, фумаровая кислота, глюконовая кислота, метансульфоновая кислота, гликолевая кислота, янтарная кислота, 4-толуолсульфоновая кислота, трифторуксусная кислота, галактуроновая кислота, эмбоновая кислота, глютаминовая кислота и аспарагиновая кислота. Оксазолидиноновые производные по настоящему изобретению могут включать гидрат соли, в частности, если указанная соль является гигроскопичной, ее предпочтительно использовать в виде кристаллогидрата.
Оксазолидиноновые производные по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленные формулой I, можно получить как пролекарство, предназначенное для повышения in vivo абсорбционной способности или растворимости, и можно применять в виде гидрата или сольвата. Например, как объясняется ниже, присоединяют группу, которую можно легко отделить после in vivo абсорбции, или получают соединение в виде соли, а именно в виде одного или большего количества гидратов или сольватов. Пролекарство и гидрат или сольват соли также включены в признаки настоящего изобретения.
Для получения эффекта от лечения можно определить эффективную дозу оксазолидиноновых производных, представленных формулой I, их гидратов, их сольватов или их фармацевтически приемлемых солей, принимая во внимание конкретные используемые соединения, способ введения, целевой субъект, целевое заболевание и др., но предпочтительной является доза 0,1-20 мг/кг (массы тела) в день оксазолидинонового производного соединения, представленного формулой 1. Дневную дозу можно вводить один раз в день (одновременно) или несколько раз в день, поделив дневную эффективную дозу должным образом. В соответствии с препаратом можно допустить пероральное введение, парентеральное введение (инъекцию) или локальное введение. Фармацевтическую композицию по настоящему изобретению для перорального введения можно приготовить, например, в виде таблеток, порошков, сухих сиропов, жевательных таблеток, гранул, капсул, мягких капсул, пилюль, напитков, подъязычных препаратов и др. Композицию по изобретению, приготовленную в виде таблеток, можно вводить субъекту любым способом или путем, который доставляет эффективную дозу таблетки с биодоступностью, которую может обеспечить пероральный путь. Также можно определить способ или путь введения в соответствии с характеристиками, стадиями целевого заболевания и другими условиями. Если композицию по изобретению формуют в виде таблеток, то она может дополнительно включать фармацевтически приемлемые наполнители. Содержание и характеристики наполнителя можно определить по растворимости и химическим свойствам выбранной таблетки, пути введения и исходя из стандартной фармацевтической практики.
Краткое описание фигур
Указанные выше и другие цели, отличительные признаки и преимущества настоящего изобретения станут ясны из следующего описания предпочтительных вариантов, данных в сопряжении с фиг. 1, где приведена диаграмма, демонстрирующая механизм свертывания крови.
Способ по изобретению
Фактические и предпочтительные в настоящее время варианты настоящего изобретения являются иллюстративными, как показано в следующих примерах.
Однако понятно, что специалисты в данной области при обсуждении данного раскрытия могут производить модификации и исправления в рамках духа и области настоящего изобретения.
Пример получения 1: получение соединения 5
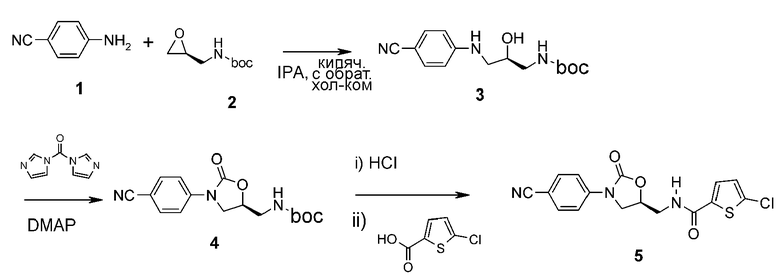
<1-1> Получение соединения 3
4-Аминобензонитрил(1) (5 г, 42,30 ммоль) и 2-(((S)-оксиран-2-нил)метил)трет-бутилоксикарбонил (2) (8,79 г, 50,78 ммоль) добавляют к 2-изопропиловому спирту (20 мл) с последующим кипячением с обратным холодильником при перемешивании в течение 12 часов. Реакционную смесь концентрируют при пониженном давлении и затем отправляют на колонку, получая указанное в заголовке соединение 3 в виде белого твердого вещества (7,30 г, 25,1 ммоль, 59%).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,41 (д, J=8,4 Гц, 4H), 6,59 (д, J=8,4 Гц, 1H), 4,95 (широкий с, 1H), 4,80 (широкий с, 1H), 3,97-3,93 (м, 1H), 3,31-3,15 (м, 5H), 1,46 (c,9H).
<1-2> Получение соединения 4
Полученное выше соединение 3 (7,30 г, 25,05 ммоль), 1,1-карбонилдиимидазол (4,87 г, 30,06 ммоль) и диметиламинопиридин (1,53 г, 12,52 ммоль) постепенно добавляют к тетрагидрофурану (70 мл) с последующим кипячением с обратным холодильником при перемешивании в течение 12 часов. Реакционную смесь концентрируют при пониженном давлении, затем растворяют в этилацетате (300 мл). После последовательного промывания 1н. раствором HCl (50 мл) и раствором бикарбоната натрия (50 мл) реакционную смесь сушат над сульфатом натрия, а затем концентрируют при пониженном давлении. Реакционную смесь промывают диэтиловым эфиром (100 мл) с получением соединения 4 в виде белого твердого вещества (6,60 г, 20,8 ммоль, 83%).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,67 (c, 4H), 4,95 (широкий с, 1H), 4,82-4,79 (м, 1H), 4,07 (дд, J=8,8, 8,8 Гц, 1H), 3,94 (дд, J=8,8, 6,8 Гц, 1H), 3,56-3,54 (м, 2H), 1,38 (c, 9H).
<1-3> Получение соединения 5
Полученное выше соединение 4 (6 г, 18,90 ммоль) добавляют к этилацетату (10 мл), реакционную смесь добавляют к 4н. HCl, растворенному в 1,4-диоксане (60 мл), с последующим перемешиванием при комнатной температуре в течение одного часа. Полученное твердое вещество отфильтровывают при пониженном давлении и затем последовательно промывают этилацетатом (20 мл) и диэтиловым эфиром (30 мл). В результате получают гидрохлорид аминного соединения с удаленными boc-группами в виде белого твердого вещества (4,60 г, 18,1 ммоль, 95,9%).
1H ЯМР (400 МГц, хлороформ-d1) δ 8,36 (широкий с, 3H), 7,90 (д, J=8,8 Гц, 2H), 7,73 (д, J=8,8 Гц, 2H), 5,03-4,96 (м, H), 4,25 (дд, J=9,2, 9,2 Гц, 1H), 3,92 (дд, J=9,2, 6,4 Гц, 1H), 3,28-3,25 (м, 2H).
Аминное соединение (4,60 г, 18,13 ммоль), HOBt (2,75 г, 19,94 ммоль), EDC (4,17 г, 21,75 ммоль), 5-хлортиофен-2-карбоновую кислоту (3,20 г, 19,04 ммоль) и триэтиламин (5,70 мл, 39,88 ммоль) постепенно добавляют к N,N'-диметилформамиду (50 мл) с последующим перемешиванием при комнатной температуре в течение 12 часов. Реакционную смесь медленно добавляют к дистиллированной воде (800 мл) и полученное твердое вещество отфильтровывают при пониженном давлении. Реакционную смесь промывают диэтиловым эфиром (100 мл) с получением соединения 5 в виде белого твердого вещества (5,70 г, 15,8 ммоль, 87%).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,93 (т, J=5,2 Гц, 1H), 7,81 (д, J=9,2 Гц, 2H), 7,69 (д, J=9,2 Гц, 2H), 7,63 (д, J=4,0 Гц, 1H), 7,15 (д, J=4,0 Гц, 1H), 4,86-4,8l (м, 1H), 4,17 (дд, J=9,2, 9,2 Гц, 1H), 3,83 (дд, J=9,2, 5,2 Гц, 1H), 3,57 (дд, J=5,2, 5,2 Гц, 2H); ЖХМС: 362(M+H+) для C16H12ClN3O3S.
Пример получения 2: получение соединения 13
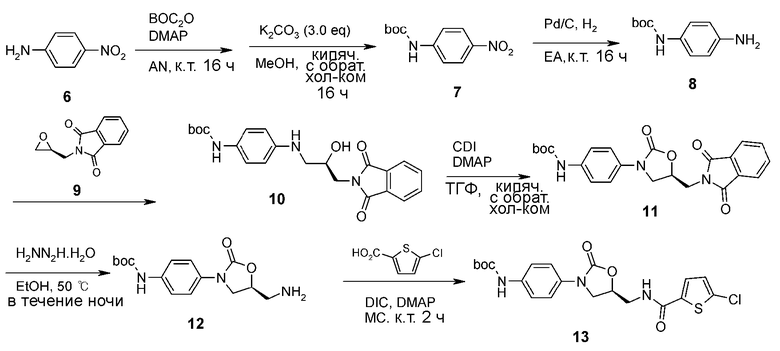
<2-1> Получение соединения 7
4-Нитроанилин (20 г, 145 ммоль) растворяют в ацетонитриле (200 мл), к смеси добавляют ди-трет-бутилдикарбонат (63,2 г, 290 ммоль) и 4-диметиламинопиридин (3,54 г, 29 ммоль) с последующим кипячением с обратным холодильником при перемешивании в течение 16 часов. Реакционный раствор охлаждают до комнатной температуры с последующим концентрированием при пониженном давлении, получая коричневое твердое соединение, имеющее две группы boc (49 г, 145 ммоль, 100%).
1H ЯМР (600 МГц, CDCl3) δ 8,25 (д, J=9 Гц, 2H), 7,36 (д, J=9 Гц, 2H), 1,45 (c, 18H).
Полученное соединение (49 г, 145 ммоль) растворяют в метаноле (200 мл), добавляют к раствору карбонат калия (60 г, 434 ммоль), затем кипятят с обратным холодильником при перемешивании в течение 16 часов. Реакционный раствор охлаждают при комнатной температуре, с последующим концентрированием при пониженном давлении. Проводят колоночную хроматографию (н-гексан/этилацетат, 6/1) реакционного раствора, получая указанное в заголовке соединение 7 в виде светло-желтого твердого вещества (17,6 г, 73,9 ммоль, 51%).
1H ЯМР (600 МГц, CDCl3) δ 8,18 (д, J=9 Гц, 2H), 7,53 (д, J=9 Гц, 2H), 6,93 (широкий с, 1H), 1,54 (c, 9H).
<2-2> Получение соединения 8
Соединение 7 (17,6 г, 73,9 ммоль) растворяют в этилацетате (200 мл), добавляют к раствору палладий на угле (10 мас.%, 3,9 г), затем перемешивают в атмосфере водорода из баллона. Через 16 часов реакционный раствор отфильтровывают через селлаит с последующим концентрированием при пониженном давлении, получая указанное в заголовке соединение 8 в виде светло-розового твердого вещества (15,4 г, 73,9 ммоль, 100%).
1H ЯМР (600 МГц, CDCl3) δ 7,12 (широкий с, 2H), 6,62 (д, J=9 Гц, 2H), 6,31 (широкий с, 1H), 3,53 (широкий с, 2H), 1,50 (c, 9H).
<2-3> Получение соединения 10
Соединение 8 (13,5 г, 65,1 ммоль) растворяют в 2-пропаноле (170 мл), добавляют к раствору (S)-глицидилфталимид (9) (14,6 г, 71,9 ммоль) с последующим взаимодействием в течение 12 часов. Затем дополнительно добавляют (S)-глицидилфталимид (9) (2,65 г, 13,0 ммоль) с последующим кипячением с обратным холодильником при перемешивании в течение 4 часов. Реакционный раствор охлаждают при комнатной температуре и затем концентрируют при пониженном давлении. Выполняют перекристаллизацию из н-гексана (500 мл), получая указанное в заголовке соединение 10 в виде желтого твердого вещества (26,8 г, 65,1 ммоль, 100%).
1H ЯМР (600 МГц, CDCl3) δ 7,89-7,84 (м, 2H), 7,78-7,73 (м, 2H), 7,15 (широкий, 2H), 6,63 (д, J=8 Гц, 2H), 6,26 (широкий, 1H), 4,18-4,12 (м, 1H), 4,05 (широкий, 1H), 3,94-3,86 (м, 2H), 3,25 (дд, J=13, 4,5 Гц, 1H), 3,15 (дд, J=13, 6,6 Гц, 1H), 2,84 (д, J=4,8 Гц, 1H), 1,50 (c, 9H).
<2-4> Получение соединения 11
Соединение 10 (26,8 г, 65,1 ммоль) растворяют в тетрагидрофуране (200 мл), добавляют к раствору 1,1-карбонилдиимидазол (15,9 г, 98,1 ммоль) и 4-диметиламинопиридин (1,59 г, 13,0 ммоль) с последующим кипячением с обратным холодильником при перемешивании в течение 16 часов. Реакционный раствор охлаждают и концентрируют при пониженном давлении. Добавляют туда насыщенный раствор хлорида аммония (200 мл) с последующей экстракцией этилацетатом (250 мл ×2). Собранный органический слой сушат над безводным сульфатом натрия, фильтруют, концентрируют при пониженном давлении и затем отправляют на колоночную хроматографию (н-гексан/этилацетат/дихлорметан, 1/1/1), получая указанное в заголовке соединение 11 в виде светло-желтого твердого вещества (20,0 г, 45,7 ммоль, 70%).
1H ЯМР (600 МГц, CDCl3) δ 7,91-7,87 (м, 2H), 7,79-7,74 (м, 2H), 7,43 (д, J=9 Гц, 2H), 7,36 (д, J=9 Гц, 2H), 6,48 (широкий с, 1H), 5,00-4,95 (м, 1H), 4,15 (дд, J=14, 7 Гц, 1H), 4,11 (т, J=9 Гц, 1H), 3,97 (дд, J=14, 6 Гц, 1H), 3,89 (дд, J=9,6 Гц, 1H), 1,52 (c, 9H).
<2-5> Получение соединения 12
Соединение 11 (15,3 г, 35,0 ммоль) растворяют в этаноле (200 мл), добавляют к раствору гидразингидрат (3,40 мл, 70,0 ммоль) с последующим кипячением с обратным холодильником при перемешивании в течение 3 часов. Реакционный раствор охлаждают при комнатной температуре. Полученное белое твердое вещество отфильтровывают и фильтрат концентрируют при пониженном давлении. Добавляют дихлорметан (100 мл) и затем полученное твердое вещество удаляют фильтрованием. Фильтрат концентрируют при пониженном давлении. Этот способ повторяют еще два раза и затем реагент сушат, получая указанное в заголовке соединение 12 в виде белого твердого вещества (10,0 г, 32,5 ммоль, 93%).
1H ЯМР (600 МГц, CDCl3) δ 7,45 (д, J=9 Гц, 2H), 7,37 (д, J=9 Гц, 2H), 6,65 (широкий с, 1H), 4,67-4,63 (м, 1H),4,03 (т, J=9 Гц, 1H), 3,82 (дд, J=9,7 Гц, 1H), 3,09 (дд, J=14, 4 Гц, 1H), 2,98 (дд, J=14, 6 Гц, 1H), 1,52 (c, 9H).
<2-6> Получение соединения 13
Соединение 12 (3,63 г, 11,8 ммоль) растворяют в хлороформе (50 мл), добавляют к раствору 5-хлортиофенкарбоновую кислоту (2,30 г, 14,1 ммоль) и добавляют 4-диметиламинопиридин (0,30 г, 13,0 ммоль). Температуру понижают до 0°C. Добавляют N,N'-диизопропилкарбодиимид (2,20 мл, 14,1 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционный раствор концентрируют при пониженном давлении с последующей перекристаллизацией из раствора н-гексан/диэтиловый эфир (1/1, 200 мл), получая указанное в заголовке соединение 13 в виде белого твердого вещества (5,0 г, 11,1 ммоль, 94%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,30 (c, 1H), 8,95 (т, J=6 Гц, 1H), 7,67 (д, J=4 Гц, 1H), 7,45-7,36 (м, 4H), 7,17 (д, J=4 Гц, 1H), 4,82-4,74 (м, 1H), 4,11 (т, J=9 Гц, 1H), 3,77 (дд, J=9, 6 Гц, 1H), 3,57 (т, J=5,6 Гц, 2H), 1,45 (c, 9H).
Пример получения 3: получение соединения 16a
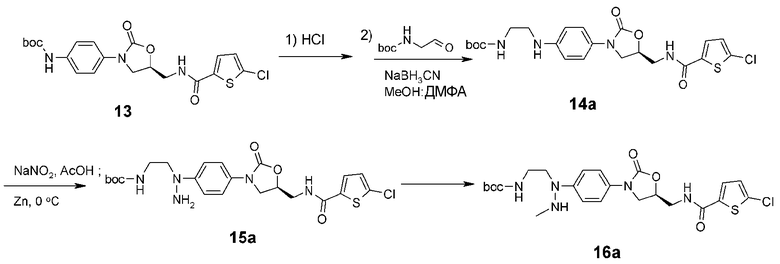
<3-1> Получение соединения 14a
Соединение 13 (16,5 г, 36,5 ммоль) растворяют в дихлорметане (150 мл), добавляют к раствору HCl (150 мл, 4M раствор 1,4-диоксана) с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционный раствор концентрируют при пониженном давлении и сушат, получая белое твердое соединение (14,1 г, 36,3 ммоль, 99%).
1H ЯМР (600 МГц, ДМСО-d6) δ 9,07 (т, J=6 Гц, 1H), 7,73 (д, J=3,6 Гц, 1H), 7,63 (д, J=9 Гц, 2H), 7,37 (д, J=9 Гц, 2H), 7,20 (д, J=3,6 Гц, 1H), 4,88-4,83 (м, 1H), 4,18 (т, J=9 Гц, 1H), 3,87 (дд, J=9,6 Гц, 1H), 3,61 (т, J=5,4 Гц, 2H).
Метанол (40 мл) и N,N-диметилформамид (15 мл) добавляют к полученному выше соединению (3,0 г, 7,73 ммоль), добавляют к смеси N-Boc-2-аминоацетальдегид (1,48 г, 9,30 ммоль) и цианборгидрид натрия (486 мг, 7,73 ммоль) с последующим перемешиванием при комнатной температуре в течение 16 часов. Добавляют насыщенный раствор хлорида аммония (20 мл) и затем раствор концентрируют при пониженном давлении. Еще добавляют туда насыщенный раствор хлорида аммония (50 мл) с последующей экстракцией этилацетатом (250 мл ×2). Собранный органический слой сушат над безводным сульфатом натрия, фильтруют, концентрируют при пониженном давлении и затем отправляют на колоночную хроматографию (н-гексан/этилацетат, 1/2→1/4), получая указанное в заголовке соединение 14a в виде белого твердого вещества (2,76 г, 5,58 ммоль, 72%).
1H ЯМР (400 МГц, CDCl3) δ 7,30 (д, J=4 Гц, 1H), 7,18 (д, J=9 Гц, 2H), 7,00 (т, J=6 Гц, 1H), 6,80 (д, J=4 Гц, 1H), 6,53 (д, J=9 Гц, 2H), 4,84-4,74 (м, 2H), 4,04 (широкий, 1H), 3,98 (т, J=9 Гц, 1H), 3,81 (ддд, J=14,4, 6, 3 Гц, 1H), 3,74 (дд, J=9,6 Гц, 1H), 3,66 (дт, J=14,8, 9 Гц, 1H), 3,36-3,26 (м, 2H), 3,18 (т, J=6 Гц, 2H), 1,41 (c, 9H).
<3-2> Получение соединения 15a
Соединение 14a (1,0 г, 2,0 ммоль) растворяют в уксусной кислоте (10 мл), медленно загружают в раствор нитрит натрия (NaNO2) (170 мг, 2,46 ммоль), растворенный в дистиллированной воде (2 мл), при 0°C с последующим перемешиванием в течение 30 минут. Затем загружают туда цинковую амальгаму (650 мг цинка промывают 0,5% раствором ацетата ртути(II) и затем промывают дистиллированной водой, применяют должным образом) с последующим перемешиванием при 0°C в течение 5 часов. Медленно добавляют насыщенный раствор карбоната натрия (50 мл) с последующей экстракцией этилацетатом (250 мл ×2). Собранный органический слой сушат над безводным сульфатом натрия, фильтруют, концентрируют при пониженном давлении и затем отправляют на колоночную хроматографию (н-гексан/этилацетат, 1/2→1/4), получая указанное в заголовке соединение 15a в виде светло-желтого твердого вещества (450 мг, 0,88 ммоль, 45%).
1H ЯМР (600 МГц, CDCl3) δ 7,34 (д, J=3,6 Гц, 1H), 7,32 (д, J=8,4 Гц, 2H), 6,96 (д, J=8,4 Гц, 2H), 6,90 (широкий, 1H), 6,87 (д, J=3,6 Гц, 1H), 4,94 (широкий с, 1H), 4,87-4,81 (м, 1H), 4,05 (т, J=9 Гц, 1H), 3,90-3,83 (м, 1H), 3,80 (дд, J=9, 6 Гц, 1H), 3,75-3,67 (м, 1H), 3,68 (широкий с, 2H), 3,44 (широкий, 4H), 1,41 (c, 9H).
<3-3> Получение соединения 16a
Соединение 15a (150 мг, 0,29 ммоль) растворяют в метаноле (3 мл), добавляют к раствору формалин (0,10 мл, 37 мас.% водный раствор) с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционный раствор концентрируют при пониженном давлении и разбавляют дистиллированной водой (15 мл) с последующей экстракцией дихлорметаном (15 мл ×2). Собранный органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая 110 мг светло-желтого твердого вещества. Это твердое вещество растворяют в метаноле (3 мл) и тетрагидрофуране (1 мл) и затем понижают температуру до 0°C. Добавляют боргидрид натрия (160 мг, 4,22 ммоль). Регулируют pH при 5, используя уксусную кислоту, и медленно повышают реакционную температуру до 50°C. Через 10 часов реакционный раствор концентрируют при пониженном давлении и разбавляют 2н. раствором HCl (30 мл) с последующей экстракцией дихлорметаном (25 мл ×2). Собранный органический слой сушат над безводным сульфатом натрия, фильтруют, концентрируют при пониженном давлении и затем отправляют на колоночную хроматографию (н-гексан/этилацетат, 1/1→1/3), получая указанное в заголовке соединение 16a в виде белого твердого вещества (15 мг, 0,029 ммоль).
Пример получения 4: получение соединения 15b
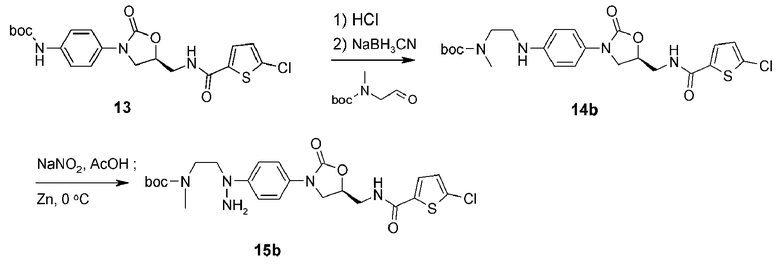
<4-1> Получение соединения 14b
Как объясняется в синтезе соединения 14a в примере получения 3, соединение 13 растворяют в дихлорметане, добавляют к раствору HCl (4M 1,4-диоксановый раствор) с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционный раствор концентрируют при пониженном давлении и сушат, получая белое твердое соединение (500 мг, 1,29 ммоль). Добавляют метанол (10 мл) и N,N-диметилформамид (2 мл), добавляют N-Boc-N-метил-2-аминоацетальдегид (268 мг, 1,55 ммоль) и цианборгидрид натрия (81 мг, 1,29 ммоль) с последующим перемешиванием при комнатной температуре в течение 16 часов. Добавляют насыщенный раствор хлорида аммония (3 мл) и затем раствор концентрируют при пониженном давлении. Еще добавляют насыщенный раствор хлорида аммония (30 мл) с последующей экстракцией этилацетатом (30 мл ×2). Собранный органический слой сушат над безводным сульфатом натрия, фильтруют, концентрируют при пониженном давлении и затем отправляют на колоночную хроматографию (н-гексан/этилацетат, 1/2→1/4), получая указанное в заголовке соединение 14b в виде белого твердого вещества (544 мг, 1,07 ммоль, 83%).
1H ЯМР (600 МГц, CDCl3) δ 7,31 (д, J=4 Гц, 1H), 7,24 (д, J=9 Гц, 2H), 6,89 (д, J=4 Гц, 2H), 6,65 (широкий, 1H), 6,59 (д, J=9 Гц, 1H), 4,85-4,79 (м, 1H), 4,04 (т, J=9 Гц, 1H), 3,90 (ддд, J=16,6, 7, 3 Гц, 1H), 3,79 (дд, J=9, 6 Гц, 1H), 3,74-3,67 (м, 1H), 3,54-3,39 (м, 2H), 3,26 (т, J=6 Гц, 2H), 2,88 (c, 3H), 1,46 (c, 9H).
<4-2> Получение соединения 15b
Соединение 14b (405 мг, 0,80 ммоль) растворяют в уксусной кислоте (3 мл), медленно загружают в раствор нитрит натрия (NaNO2) (66 мг, 0,96 ммоль), растворенный в дистиллированной воде (0,5 мл), при 0°C с последующим перемешиванием в течение 30 мин. Затем загружают туда цинк (130 мг, 1,99 ммоль) с последующим перемешиванием при 0°C в течение 3 часов. Медленно добавляют насыщенный раствор карбоната натрия (30 мл) с последующей экстракцией этилацетатом (30 мл ×2). Собранный органический слой сушат над безводным сульфатом натрия, фильтруют, концентрируют при пониженном давлении и затем отправляют на колоночную хроматографию (н-гексан/этилацетат, 1/2→1/4), получая указанное в заголовке соединение 15b в виде светло-коричневого твердого вещества (81 мг, 0,15 ммоль, 19%).
Пример получения 5: получение соединения 26
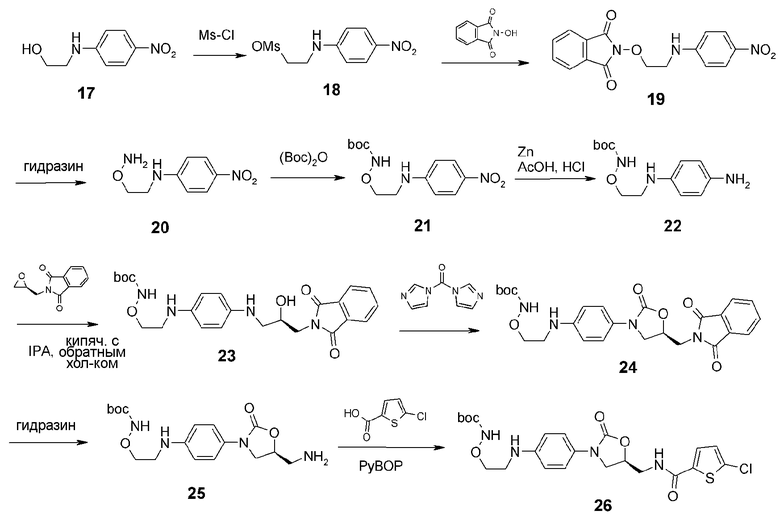
<5-1> Получение соединения 18
4-Фторнитробензол (5,2 г, 37 ммоль) растворяют в ацетонитриле (40 мл), добавляют к раствору 2-аминоэтанол (5,2 г, 85 ммоль) с последующим кипячением с обратным холодильником при перемешивании в течение ночи. Реакционный раствор охлаждают при комнатной температуре, концентрируют при пониженном давлении, растворяют в этилацетате, промывают последовательно водой, 1н. HCl и соленой водой. Реакционную смесь сушат над безводным сульфатом натрия, фильтруют и перегоняют при пониженном давлении с получением соединения 17. Соединение 17 (15 г, 82,33 ммоль) и диизопропилэтиламин (27 мл, 164 ммоль) растворяют в дихлорметане (150 мл), медленно загружают в раствор метансульфонилхлорид (9,5 мл) при 0°C с последующим перемешиванием при комнатной температуре в течение 2 часов. По завершении взаимодействия добавляют к реакционному раствору дихлорметан (800 мл). Реакционную смесь промывают раствором бикарбоната натрия (500 мл) и концентрируют при пониженном давлении, получая желтое твердое соединение 18 (22 г, 82,00 ммоль, 99%).
1H ЯМР (400 МГц, хлороформ-d1) δ 8,12 (д, J=9,2 Гц, 2H), 6,60 (д, J=9,2 Гц, 2H), 4,45 (т, J=5,6 Гц, 2H), 3,63 (т, J=5,6 Гц, 2H), 3,06 (c, 3H); ЖХМС: 183 (M+H+) для C8H10N2O3S.
<5-2> Получение соединения 19
Соединение 18 (22 г, 82,00 ммоль), гидроксифталимид (17,4 г, 107,04 ммоль) и триэтиламин (17,3 мл, 123,5 ммоль) добавляют к ацетонитрилу (300 мл) с последующим кипячением с обратным холодильником при перемешивании в течение 6 часов. По завершении взаимодействия добавляют к реакционному раствору дихлорметан (1000 мл). Реакционную смесь промывают 0,5н. раствором HCl (500 мл) и насыщенным раствором бикарбоната натрия (500 мл) и концентрируют при пониженном давлении, получая желтое твердое соединение 19 (26 г, 79,44 ммоль, 97%).
1H ЯМР (400 МГц, хлороформ-d1) δ 8,12 (д, J=9,2 Гц, 2H), 7,88 (м, 2H), 7,87 (м, 2H), 6,65 (д, J=9,2 Гц, 2H), 5,83 (широкий с, 1H), 4,45 (т, J=4,8 Гц, 2H), 3,56 (м, 2H); ЖХМС: 328 (M+H+) для C16H13N3O5.
<5-3> Получение соединения 20
Соединение 19 (79,44 ммоль) и гидразин (20 мл) добавляют к этанолу с последующим кипячением с обратным холодильником при перемешивании в течение 2 часов. Реакционную смесь охлаждают при комнатной температуре, фильтруют и концентрируют при пониженном давлении с получением соединения 20 (19 г, сырое). Это соединение не очищают и используют для следующего взаимодействия.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,94 (д, J=9,6 Гц, 2H), 7,34 (т, J=5,6 Гц, 1H), 6,69 (д, J=9,5 Гц, 2H), 3,68 (т, J=5,2 Гц, 2H), 3,37 (м, 2H); ЖХМС: 198 (M+H+) для C8H11N3O3.
<5-4> Получение соединения 21
Соединение 20 (19 г) и карбонат натрия (21 г, 198 ммоль) растворяют в диоксане (200 мл) и дистиллированной воде (200 мл), медленно добавляют к раствору ди-трет-бутоксикарбонил (26 г, 119 ммоль) с последующим перемешиванием при комнатной температуре в течение 4 часов. По завершении взаимодействия реакционную смесь отфильтровывают при пониженном давлении, промывают дистиллированной водой (1000 мл) и сушат, получая желтое твердое соединение 21 (26 г, сырое).
1H ЯМР (400 МГц, хлороформ-d1) δ 8,10 (д, J=9,2 Гц, 2H), 6,54 (д, J=9,2 Гц, 2H), 5,50 (c, 1H), 5,04 (c, 1H), 3,9l (т, J=5,2 Гц, 2H), 3,45 (м, 2H), 1,43 (c, 9H); ЖХМС: 298 (M+H+) для C13H19N3O5.
<5-5> Получение соединения 22
Соединение 21 (13 г, 40 ммоль), уксусную кислоту (132 мл) и концентрированную HCl (10 мл, 300 ммоль) растворяют в тетрагидрофуране (200 мл), добавляют к раствору цинк (26 г, 400 ммоль) при 0°C с последующим перемешиванием в течение 2 часов. Медленно добавляют 20% раствор аммиака (200 мл) при 0°C и затем добавляют диметиленхлорид (500 мл). Органический слой отделяют, затем концентрируют при пониженном давлении, получая белое твердое соединение 22 (6,5 г, 24,31 ммоль, 61%).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,17 (c, 1H), 6,60 (д, J=8,4 Гц, 2H), 6,60 (д, J=8,4 Гц, 2H), 4,03 (т, J=4,8 Гц, 2H), 3,30 (т, J=4,8 Гц, 2H), 1,47 (c, 9H); ЖХМС: 268 (M+H+) для C13H21N3O3.
<5-6> Получение соединения 23
Соединение 22 (6,5 г, 24,31 ммоль) и (S)-глицидилфталимид (3,95 г, 19,45 ммоль) добавляют к изопропиловому спирту (100 мл), затем кипятят с обратным холодильником при перемешивании в течение 6 часов. По завершении взаимодействия реакционную смесь концентрируют при пониженном давлении с последующей колоночной хроматографией, получая белое твердое соединение 23 (9 г, неочищенное).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,85 (м, 2H), 7,79 (м, 2H), 6,33 (д, J=9,2 Гц, 2H), 6,2l (д, J=9,2 Гц, 2H), 4,07 (м, 3H), 3,58-3,31 (м, 6H), 1,46 (c, 9H); ЖХМС: 471(M+H+) для C24H30N4O6.
<5-7> Получение соединения 24
Соединение 23 (9 г, неочищенное) и карбодиимидазол (3,5 г, 29,16 ммоль) добавляют к тетрагидрофурану (150 мл) с последующим перемешиванием в течение 12 часов. По завершении взаимодействия реакционную смесь концентрируют при пониженном давлении, проводят колоночную хроматографию, получая белое твердое соединение 24 (1,6 г, 3,22 ммоль).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,88 (м, 2H), 7,75 (м, 2H), 7,28 (д, J=8,8 Гц, 2H), 6,55 (д, J=8,8 Гц, 2H), 4,95 (м, 1H), 4,17-3,82 (м, 6H), 3,37(т, J=5,2 Гц, 2H), 1,46 (c, 9H); ЖХМС: 497 (M+H+) для C25H28N4O7.
<5-8> Получение соединения 25
Соединение 24 (1,6 г, 3,22 ммоль) и гидразин (1,6 мл, 32,20 ммоль) добавляют к этиловому спирту (30 мл) с последующим кипячением с обратным холодильником при перемешивании в течение 2 часов. Реакционную смесь охлаждают при комнатной температуре, фильтруют и концентрируют при пониженном давлении с получением соединения 25 (1,3 г, неочищенное). Это соединение не очищают и используют для следующего взаимодействия.
1H ЯМР (400 МГц, хлороформ-d1) δ 7,33 (д, J=9,2 Гц, 2H), 6,66 (д, J=9,2 Гц, 2H), 4,63 (м, 1H), 4,03 (м, 3H), 3,77 (дд, J=6,4 2,0 Гц, 1H), 3,34 (т, J=5,2 Гц, 2H), 3,03 (м, 2H), 1,49 (c, 9H); ЖХМС: 367 (M+H+) для C17H26N4O5.
<5-9> Получение соединения 26
Соединение 25 (1,3 г, 3,22 ммоль), 5-хлортиофен-2-карбоновую кислоту (0,68 г, 4,19 ммоль) и PyBOP [(бензотриазол-1-илокси)трипирролидинофосфоний гексафторфосфат] (2,5 г, 4,83 ммоль) добавляют к N,N-диметилформамиду (20 мл), медленно добавляют к раствору диизопропилэтиламин (1,06 мл, 6,44 ммоль) при 0°C с последующим перемешиванием в течение 1 часа. По завершении взаимодействия добавляют к реакционному раствору этилацетат (300 мл). Реакционную смесь дважды промывают дистиллированной водой (200 мл) и концентрируют при пониженном давлении, получая белое твердое соединение 26 (1,6 г, 3,13 ммоль).
1H ЯМР (400 МГц, ДМСО-d6) δ 10,05 (c, 1H), 8,97 (т, J=5,6 Гц, 1H), 7,70 (д, J=4,0 Гц, 1H), 7,20 (м, 3H), 6,60 (д, J=8,4 Гц, 2H), 4,76 (м, 1H), 4,07 (т, J=8,8 Гц, 1H), 3,83 (т, J=5,6 Гц, 2H), 3,74 (м, 1H), 3,57 (т, J=5,2 Гц, 2H), 3,21 (т, J=5,6 Гц, 2H), 1,40 (c, 9H); ЖХМС; 511 (M+H+) для C22H27ClN4O6.
Пример 1: получение соединения 100
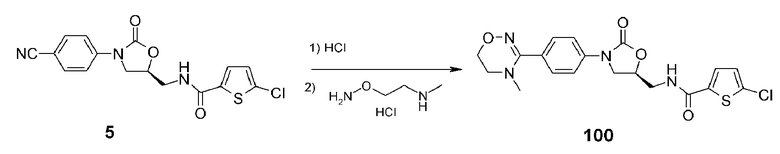
Соединение 5 (5,0 г, 13,8 ммоль), полученное в примере получения 1, добавляют к безводному метанолу (200 мл), затем барботируют газообразный HCl при 0°C в течение 30 мин. Добавляют метанол (100 мл), затем снова барботируют газообразный HCl в течение более 30 мин с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении для удаления остающегося HCl. Добавляют туда уксусную кислоту (120 мл) и 2-(N-метиламино)этилгидроксиламин гидрохлорид (3,5 г, 27,6 ммоль) с последующим кипячением с обратным холодильником в течение ночи. Реакционный раствор концентрируют при пониженном давлении, растворяют в этилацетате и промывают насыщенным раствором NaHCO3, затем разделяют смесь, используя колоночную хроматографию и получая указанное в заголовке соединение 100 в виде белого твердого вещества (1,85 г, 4,3 ммоль, 31%).
1H ЯМР (400 МГц, CDCl3) δ 7,54 (д, J=9,0 Гц, 2H), 7,44 (д, J=9,0 Гц, 2H), 7,34 (д, J=4,2 Гц, 1H), 6,89 (широкий т, 1H), 4,80 (м, 1H), 4,12 (т, J=4,8 Гц, 2H), 4,04 (т, J=9,0 Гц, 1H), 3,85-3,80 (м, 2H), 3,69-3,64 (м, 1H), 3,45 (т, J=4,8 Гц, 2H), 2,76 (c, 3H); ЖХМС; 435 (M+H+) для C19H19ClN4O4S.
Пример 2: получение соединения 101
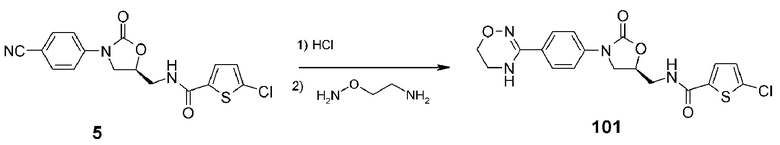
Соединение 101 получают в виде белого твердого вещества (17 мг, 0,04 ммоль, 14%) аналогично тому, как описано в примере 1, используя соединение 5 (0,1 г, 0,27 ммоль), полученное в примере получения 1, и (аминоэтил)гидроксиламин (62 мг, 0,8 ммоль).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,98 (т, J=6,0 Гц, 1H), 7,68 (д, J=4,0 Гц, 1H), 7,63 (д, J=8,8 Гц, 2H), 7,57 (д, J=8,8 Гц, 2H), 7,19 (д, J=4,0 Гц, 1H), 7,07 (c, 1H), 4,85 (м, 1H), 4,19 (т, J=8,8 Гц, 1H), 3,86-3,81 (м, 3H), 3,61 (т, J=5,2 Гц, 2H), 3,38 (м, 2H); ЖХМС: 421 (M+H+) для C18H17ClN4O4S.
Пример 3: получение соединения 102
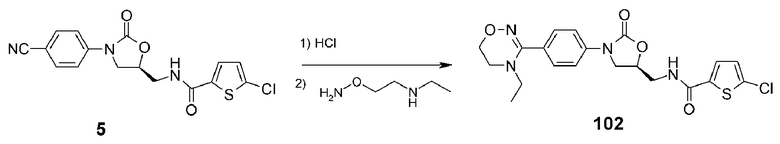
Соединение 102 получают в виде белого твердого вещества (30 мг, 48%) аналогично тому, как описано в примере 1, используя соединение 5 (100 мг, 0,27 ммоль), полученное в примере получения 1, и O-[2-(2-этиламино)-этил]гидроксиламин (15 мг, 0,139 ммоль).
1H ЯМР (400 МГц, CDCl3) δ 7,52 (д, J=8,4 Гц, 2H), 7,42 (д, J=8,4 Гц, 2H), 7,37 (д, J=3,6 Гц, 1H), 7,28-7,22 (м, 1H), 6,85 (д, J=3,6 Гц, 1H), 4,82-4,7l (м, 1H), 4,11 (т, J=4,4 Гц, 2H), 3,98 (т, J=8,8 Гц, 1H), 3,83-3,72 (м, 2H), 3,66-3,56 (м, 1H), 3,43 (т, J=4,4 Гц, 2H), 3,01 (кв, J=7,2 Гц, 2H), 1,03 (т, J=4,4 Гц, 3H); ЖХМС: 449 (M+H+) для C20H21ClN4O4S.
Пример 4: получение соединения 103
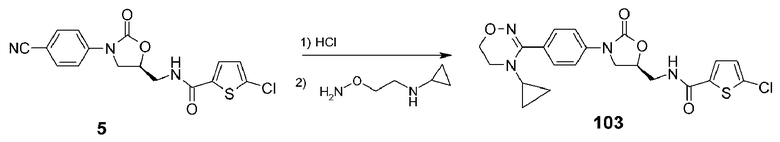
Соединение 103 получают в виде белого твердого вещества (31 мг, 29%) аналогично тому, как описано в примере 1, используя соединение 5 (100 мг, 0,27 ммоль), полученное в примере получения 1, и O-[2-(2-циклопропиламино)этил]гидроксиламин (27 мг, 0,23 ммоль).
1H ЯМР (400 МГц, CDCl3) δ 7,52 (д, J=7,6 Гц, 2H), 7,46 (д, J=7,6 Гц, 2H), 7,38 (д, J=3,6 Гц, 1H), 7,23-7,18 (м, 1H), 6,87 (д, J=3,6 Гц, 1H), 4,85-4,72 (м, 1H), 4,06 (т, J=4,4 Гц, 2H), 4,02 (т, J=8,4 Гц, 1H), 3,85-3,74 (м, 2H), 3,68-3,55 (м, 1H), 3,53 (т, J=4,4 Гц, 2H), 2,62-2,5l (м, 1H), 0,51-0,32 (м, 4H); ЖХМС: 461 (M+H+) для C21H21ClN4O4S.
Пример 5: получение соединения 104
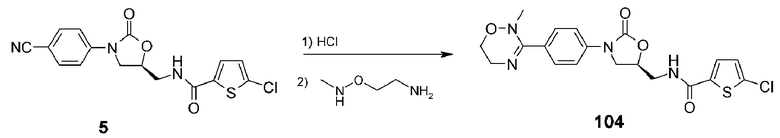
Соединение 104 получают в виде белого твердого вещества (63,7 мг, 0,146 ммоль, 58%) аналогично тому, как описано в примере 1, используя соединение 5 (100 мг, 0,27 ммоль), полученное в примере получения 1, и N-метил-O-(2-аминоэтил)гидроксиламиндигидрохлорид (180,0 мг, 1,104 ммоль).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,07 (т, J=6,4 Гц, 1H), 7,70 (д, J=4,4 Гц, 1H), 7,54 (м, 4H), 7,14 (д, J=4,4 Гц, 1H), 4,80 (м, 1H), 4,15 (т, J=8,8 Гц, 1H), 3,86 (дд, J=8,8 Гц, 8,8 Гц, 1H), 3,81 (т, J=4,8 Гц, 2H), 3,55-3,48 (м, 4H), 2,82 (c, 3H); ЖХМС: 435 (M+H+) для C19H19ClN4O4S.
Пример 6: получение соединения 105
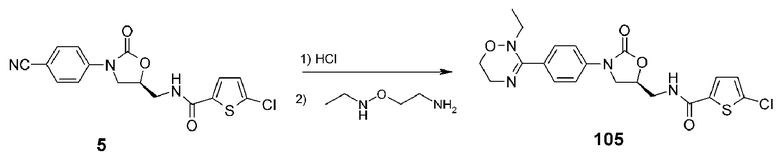
Соединение 105 получают в виде светло-желтого твердого вещества (9,1 мг, 0,020 ммоль, 9%) аналогично тому, как описано в примере 1, используя соединение 5 (80,0 г, 0,221 ммоль), полученное в примере получения 1, и N-этил-O-(2-аминоэтил)гидроксиламиндигидрохлорид (300,0 мг, 1,69 ммоль).
1H ЯМР (600 МГц, ДМСО-d6) δ 9,37 (широкий, 1H), 7,88 (м, 1H), 7,79 (д, J=6,0 Гц, 2H), 7,73 (д, J=5,6 Гц, 2H), 7,20 (д, J=4,2 Гц, 1H), 4,91 (м, 1H), 4,24 (т, J=8,7 Гц, 1H), 4,05 (дд, J=8,7 Гц, 8,7 Гц, 1H), 3,66 (т, J=4,8 Гц, 2H), 3,63-3,54 (м, 4H), 2,81 (кв, J=7,2 Гц, 2H), 1,24 (т, J=7,2 Гц, 3H); ЖХМС: 449 (M+H+) для C20H21ClN4O4S.
Пример 7: получение соединения 106
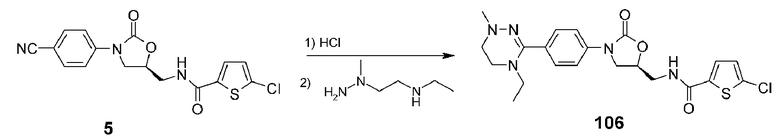
Соединение 106 получают в виде белого твердого вещества (20 мг, 0,04 ммоль, 16%) аналогично тому, как описано в примере 1, используя соединение 5 (100 мг, 0,27 ммоль), полученное в примере получения 1, и 2-(1-метилгидразинил)-N-этилэтанамин (93 мг, 0,8 ммоль).
1H ЯМР (600 МГц, ДМСО-d6) δ 8,99 (т, J=5,4 Гц, 1H), 7,69 (д, J=3,6 Гц, 1H), 7,53 (д, J=8,4 Гц, 2H), 7,36 (т, J=8,4 Гц, 2H), 7,19 (д, J=3,6 Гц, 1H), 4,84 (м, 1H), 4,19 (т, J=9,6 Гц, 1H), 3,86 (дд, J=8,4 6,6 Гц, 1H), 3,61 (т, J=5,4 Гц, 2H), 3,3l (м, 2H), 2,92 (м, 2H), 2,80 (широкий с, 2H), 2,59 (c, 3H), 0,94 (т, J=7,2 Гц, 1H) (2 эквивалента уксусной кислоты по данным ЯМР); ЖХМС: 462 (M+H+) для C21H24ClN5O3S.
Пример 8: получение соединения 107
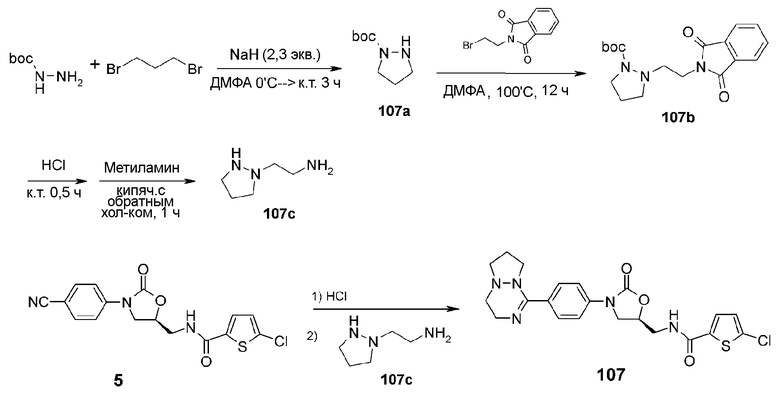
NaH (3,3 г, 87,01 ммоль) добавляют к N,N-диметилформамиду (30 мл) с последующим перемешиванием в течение 15 мин. Трет-бутилшабазит (5 г, 37,83 ммоль) растворяют в N,N-диметилформамиде (10 мл) и медленно добавляют к реакционной смеси при 0°C. Также добавляют туда дибромпропан (7,6 г, 37,83 ммоль) при 0°C с последующим перемешиванием при комнатной температуре в течение 3 часов. Реакционную смесь охлаждают при 0°C и промывают дистиллированной водой (50 мл). По завершении взаимодействия реакционную смесь растворяют в этилацетате (500 мл), затем три раза промывают раствором бикарбоната натрия (50 мл). Проводят колоночную хроматографию, получая соединение 107a в виде масла (2,4 г, 13,93 ммоль, 36,7%).
1H ЯМР (400 МГц, хлороформ-d1) δ 3,85 (c, 1H), 3,45 (т, J=7,2 Гц, 2H), 3,04 (т, J=6,4 Гц, 2H), 2,03 (м, 2H), 1,49 (c, 9H); ЖХМС: 173 (M+H+) для C8H16N2O2.
Соединение 107a (1,5 г, 8,71 ммоль), N-2-бромфталимид (2,33 г, 8,71 ммоль) и карбонат калия (1,32 г, 9,58 ммоль) добавляют к N,N-диметилформамиду (10 мл) с последующим перемешиванием при 100°C в течение 12 часов. Реакционную смесь растворяют в этилацетате (200 мл), затем промывают три раза раствором бикарбоната натрия (30 мл). Проводят колоночную хроматографию, получая соединение 107b в виде масла (1,7 г, 4,92 ммоль, 56,2%).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,89 (м, 2H), 7,74 (м, 2H), 3,94(т, J=4,4 Гц, 2H), 3,90 (т, J=5,2 Гц, 2H), 3,11 (т, J=6,0 Гц, 2H), 3,06 (т, J=4,4 Гц, 2H), 2,01 (м, 2H), 1,48 (c, 9H); ЖХМС: 346 (M+H+) для C18H23N3O4.
Соединение 107b (0,5 г, 1,45 ммоль) добавляют к 4M HCl (в диоксане) (5 мл) с последующим перемешиванием в течение 0,5 часа. Реакционную смесь концентрируют при пониженном давлении, добавляют к раствору метиловый спирт (5 мл) и метиламин (4 мл) с последующим кипячением с обратным холодильником при перемешивании в течение 1 часа. Реакционную смесь концентрируют при пониженном давлении с получением соединения 107c, которое больше не очищают и используют для следующего взаимодействия. ЖХМС: 116 (M+H+) для C5H13N3.
Соединение 5 (100 мг, 0,27 ммоль), полученное в примере получения 1, добавляют к безводному метиловому спирту (10 мл), затем барботируют газообразным HCl при 0°C в течение 30 минут и перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении для удаления оставшейся HCl. Добавляют туда последовательно безводный метиловый спирт (10 мл) и соединение 107c (300 мг, 0,8 ммоль) с последующим перемешиванием при комнатной температуре в течение 12 часов и концентрированием при пониженном давлении. Затем проводят препаративную ТСХ, получая указанное в заголовке соединение 107 в виде белого твердого вещества (60 мг, 0,13 ммоль, 48%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,12 (т, J=5,6 Гц, 1H), 7,77-7,64 (м, 5H), 7,14 (д, J=4,4 Гц, 1H), 4,84 (м, 1H), 4,18 (т, J=8,8 Гц, 1H), 3,90(дд, J=8,8, 6,0 Гц, 1H), 3,70 (т, J=6,8 Гц, 1H), 3,65 (т, J=6,8 Гц, 1H), 3,59-3,52 (м, 3H), 3,25 (м, 1H), 3,11 (м, 1H), 2,95 (м, 1H), 2,65 (м, 1H), 2,55 (м, 1H), 2,08 (м, 2H); ЖХМС: 460(M+H+) для C21H22ClN5O3S.
Пример 9: получение соединения 108
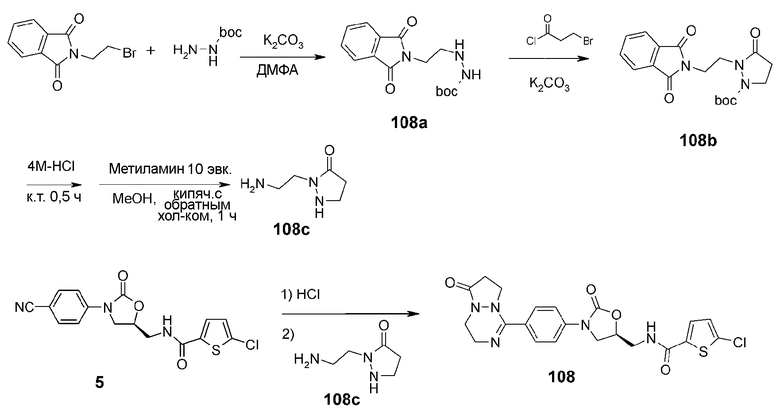
Трет-бутилшабазит (трет-бутилкарбазат) (2,5 г, 18,91 ммоль), N-(бромэтил)фталимид (5,25 г, 20,80 ммоль) и карбонат калия (3,14 г, 22,70 ммоль) добавляют к N,N-диметилформамиду (30 мл) с последующим перемешиванием при 90°C в течение 12 часов. По завершении взаимодействия реакционную смесь растворяют в этилацетате (250 мл), затем три раза промывают раствором бикарбоната натрия (150 мл). После концентрирования при пониженном давлении проводят колоночную хроматографию, получая белое твердое соединение 108a (1 г, 3,2 ммоль, 19%).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,85 (м, 2H), 7,73 (м, 2H), 6,49 (c, 1H), 4,12 (c, 1H), 3,83 (т, J=3,6 Гц, 2H), 3,05 (м, 2H), 1,46 (c, 9H); ЖХМС: 306 (M+H+) для C15H19N3O4.
Соединение 108a (0,9 г, 2,95 ммоль) и карбонат калия (1,03 г, 7,4 ммоль) растворяют в N,N-диметилформамиде (10 мл), добавляют к раствору 3-бромпропаноилхлорид (0,7 г, 3,6 ммоль), затем перемешивают при 90°C в течение 5 часов. Добавляют этилацетат (100 мл), затем промывают раствором бикарбоната натрия (30 мл). После концентрирования при пониженном давлении проводят колоночную хроматографию, получая соединение 108b (в виде масла 230 мг, 0,64 ммоль, 22%).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,85 (м, 2H), 7,73 (м, 2H), 6,49 (c, 1H), 4,12 (c, 1H), 3,83 (т, J=3,6 Гц, 2H), 3,05 (м, 2H), 1,46 (c, 9H); ЖХМС: 360 (M+H+) для C18H21N3O5.
Соединение 108b (230 мг, 0,64 ммоль) добавляют к 4M HCl (в диоксане) (2 мл) с последующим перемешиванием в течение 1 часа. Реакционную смесь концентрируют при пониженном давлении, добавляют к раствору метиловый спирт (5 мл) и метиламин (2 мл), затем перемешивают в течение 1 часа. Реакционную смесь концентрируют при пониженном давлении с получением соединения 108c (103 мг, 0,64 ммоль), которое больше не очищают и используют для следующего взаимодействия.
ЖХМС: 130 (M+H+) для C5H11N3O.
Соединение 108 получают в виде белого твердого вещества (26 мг, 0,05 ммоль, 19%) аналогично тому, как описано в примере 1, используя соединение 5 (0,1 г, 0,27 ммоль), полученное в примере получения 1, и соединение 108c (103 мг, 0,64 ммоль).
1H ЯМР (600 МГц, ДМСО-d6) δ 9,09 (т, J=4,8 Гц, 1H), 7,79 (д, J=8,4 Гц, 2H), 7,72-7,71 (м, 3H), 7,19 (д, J=4,2 Гц, 1H), 4,89 (м, 1H), 4,24 (т, J=9,0 Гц, 1H), 4,12 (т, J=7,8 Гц, 2H), 3,94-3,89 (м, 3H), 3,62 (м, 2H), 3,53 (т, J=4,8 Гц, 2H), 2,80 (т, J=8,4 Гц, 2H), 3,59-3,52 (м, 3H), 3,25 (м, 1H), 3,11 (м, 1H), 2,95 (м, 1H), 2,65 (м, 1H), 2,55 (м, 1H), 2,08 (м, 2H); ЖХМС: 474 (M+H+) для C21H20ClN5O4S.
Пример 10: получение соединения 109
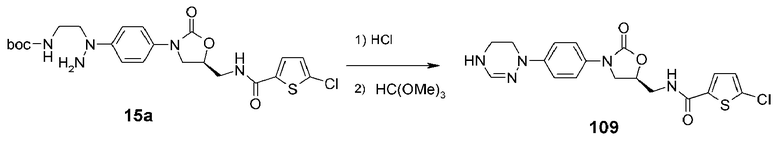
Соединение 15a (450 мг, 0,88 ммоль), полученное в примере получения 3, растворяют в дихлорметане (10 мл), добавляют HCl (4M 1,4-диоксановый раствор) (10 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционную смесь концентрируют при пониженном давлении и сушат, получая светло-желтое твердое соединение (425 мг, 0,88 ммоль, 100%). Это соединение (392 мг, 0,81 ммоль) растворяют в уксусной кислоте (4 мл), добавляют триметилортоформиат (2 мл) с последующим кипячением с обратным холодильником при перемешивании. Через 10 часов после полного выпаривания растворителя проводят колоночную хроматографию (дихлорметан/метанол (об./об.) 20/1→12/1), получая указанное в заголовке соединение 109 в виде светло-желтого твердого вещества (215 мг, 5,12 ммоль, 63%).
1H ЯМР (400 МГц, CDCl3) δ 7,35 (д, J=9,2 Гц, 2H), 7,33 (д, J=4,4 Гц, 1H), 7,14 (д, J=9,2 Гц, 2H), 7,01 (т, J=6,4 Гц, 1H), 6,88 (c, 1H), 6,85 (д, J=4,4 Гц, 1H), 4,87-4,79 (м, 1H), 4,06 (т, J=9 Гц, 1H), 3,86 (ддд, J=14,4, 6, 3 Гц, 1H), 3,81 (дд, J=9, 6,4 Гц, 1H), 3,69 (дт, J=14,4, 6 Гц, 1H), 3,62-3,58 (м, 2H), 3,55-3,51 (м, 2H); ЖХМС: 420 (M+H+) для C18H18ClN5O3S.
Пример 11: получение соединения 110
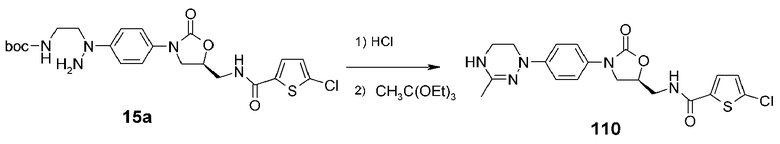
Используют соединение 15a, полученное в примере получения 3, для получения указанного в заголовке соединения 110 способом, аналогичным способу, описанному в примере 10, используя триэтилереортоацетат вместо триметилортоформиата из примера 10.
1H ЯМР (600 МГц, CDCl3) δ 7,36 (д, J=9 Гц, 2H), 7,30 (д, J=4 Гц, 1H), 7,17 (д, J=9 Гц, 2H), 6,90 (д, J=4,2 Гц, 1H), 6,49 (широкий т, 1H), 4,85-4,80 (м, 1H), 4,28 (широкий, 1H), 4,08 (т, J=9 Гц, 1H), 3,95-3,90 (м, 1H), 3,79 (т, J=7,8 Гц, 1H), 3,72-3,67 (м, 1H), 3,62-3,57 (м, 2H), 3,49-3,44 (м, 2H), 1,98 (c, 3H); ЖХМС: 434 (M+H+) для C19H20ClN5O3S.
Пример 12: получение соединения 111
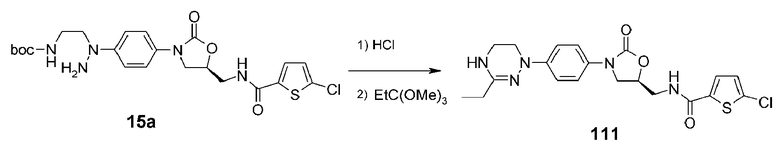
Используют соединение 15a, полученное в примере получения 3, получая указанное в заголовке соединение 111 способом, аналогичным способу, описанному в примере 10, используя триэтилортопропионат вместо триметилортоформиата из примера 10.
1H ЯМР (600 МГц, CDCl3) δ 7,35 (д, J=9 Гц, 2H), 7,30 (д, J=4,2 Гц, 1H), 7,18 (д, J=9 Гц, 2H), 6,90 (д, J=4,2 Гц, 1H), 6,53 (широкий т, 1H), 4,85-4,80 (м, 1H), 4,27 (широкий, 1H), 4,08 (т, J=9 Гц, 1H), 3,92 (ддд, J=14,4, 6,6, 3 Гц, 1H), 3,79 (т, J=7,8 Гц, 1H), 3,72-3,67 (м, 1H), 3,62-3,57 (м, 2H), 3,50-3,44 (м, 2H), 2,26 (кв, J=7,8 Гц, 2H), 1,20 (т, J=7,8 Гц, 3H); ЖХМС: 448 (M+H+) для C20H22ClN5O3S.
Пример 13: получение соединения 112
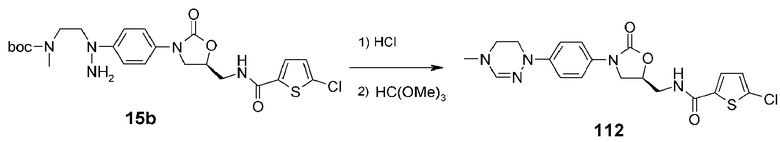
Используют соединение 15b, полученное в примере получения 4, получая указанное в заголовке соединение 112 способом, аналогичным способу, описанному в примере 10.
1H ЯМР (600 МГц, CDCl3) δ 7,35 (д, J=9 Гц, 2H), 7,33 (д, J=4,5 Гц, 1H), 7,13 (д, J=9 Гц, 2H), 6,93 (т, J=6 Гц, 1H), 6,85 (д, J=4,5 Гц, 1H), 6,64 (c, 1H), 4,86-4,80 (м, 1H), 4,06 (т, J=9 Гц, 1H), 3,91-3,85 (м, 1H), 3,80 (дд, J=9, 7 Гц, 1H), 3,71-3,65 (м, 1H), 3,54 (т, J=4,8 Гц, 2H), 3,43 (т, J=4,8 Гц, 2H), 2,90 (c, 3H); ЖХМС: 434 (M+H+) для C19H20ClN5O3S.
Пример 14: получение соединения 113
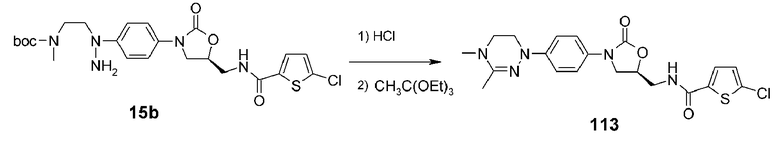
Используют соединение 15b, полученное в примере получения 4, получая указанное в заголовке соединение 113 способом, аналогичным способу, описанному в примере 10, используя триэтилортоацетат вместо триметилортоформиата из примера 10.
1H ЯМР (600 МГц, CDCl3) δ 7,34 (д, J=9 Гц, 2H),7,31 (д, J=4 Гц, 1H),7,15 (д, J=9 Гц, 2H), 6,88 (д, J=4 Гц, 1H), 6,69 (т, J=6 Гц, 1H), 4,84-4,78 (м, 1H), 4,06 (т, J=9 Гц, 1H), 3,90 (ддд, J=11, 7, 3 Гц, 1H), 3,79 (дд, J=9, 6,6 Гц, 1H), 3,68 (дт, J=14,4, 6,6 Гц, 1H), 3,52 (т, J=5 Гц, 2H), 3,44 (т, J=5 Гц, 2H), 2,93 (c, 3H), 2,05 (c, 3H); ЖХМС: 448 (M+H+) для C20H22ClN5O3S.
Пример 15: получение соединения 114
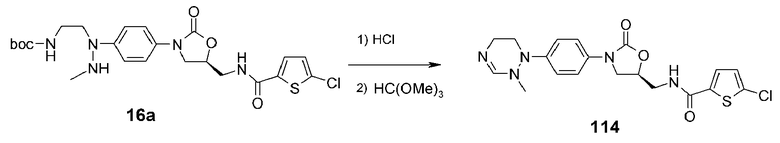
Используют соединение 16a, полученное в примере получения 3, получая указанное в заголовке соединение 114 в виде белого твердого вещества (7,4 мг, 0,014 ммоль, 47%) способом, аналогичным способу, описанному в примере 10.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,03 (т, J=5,4 Гц, 1H), 8,65 (c, 1H), 7,68 (д, J=3,6 Гц, 1H), 7,55 (д, J=8,4 Гц, 2H), 7,19 (д, J=3,6 Гц, 1H), 7,14 (д, J=8,4 Гц, 2H), 4,88-4,81 (м, 1H), 4,17 (т, J=9 Гц, 1H), 3,85-3,81 (м, 1H), 3,70-3,50 (м, 4H), 3,29 (c, 3H), 3,13-3,05 (м, 2H); ЖХМС: 434 (M+H+) для C19H20ClN5O3S.
Пример 16: получение соединения 115
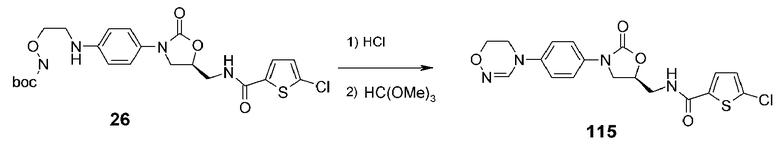
Соединение 26, полученное в примере получения 5, добавляют к 4M HCl (2 мл) с последующим перемешиванием в течение одного часа. Реакционную смесь концентрируют при пониженном давлении, добавляют триметилортоформиат (2 мл) и уксусную кислоту (4 мл) с последующим кипячением с обратным холодильником при перемешивании в течение 12 часов. Проводят колоночную хроматографию реакционной смеси, получая указанное в заголовке соединение 115 в виде белого твердого вещества (11 мг, 0,03 ммоль, 8%).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,55 (c, 1H), 7,50 (д, J=9,2 Гц, 2H), 7,42 (широкий с, 1H), 7,39 (д, J=4,0 Гц, 1H), 7,03 (д, J=9,2 Гц, 2H), 6,89 (д, J=4,0 Гц, 1H), 4,86 (м, 1H), 4,20 (т, J=4,8 Гц, 2H), 4,12 (м, 1H), 3,87 (м, 1H), 3,83-3,74 (м, 4H); ЖХМС: 421 (M+H+) для C18H17ClN4O4S.
Пример 17: получение соединения 116
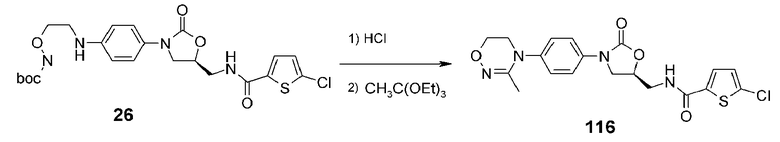
Соединение 116 получают в виде белого твердого вещества способом, аналогичным способу, описанному в примере 14, используя соединение 26, полученное в примере получения 5.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,93 (т, J=5,6 Гц, 1H), 7,63 (д, J=4,2 Гц, 1H), 7,50 (д, J=8,8 Гц, 2H), 7,25 (д, J=8,8 Гц, 2H), 7,14 (д, J=4,2 Гц, 1H), 4,79 (м, 1H), 4,13 (т, J=8,8 Гц, 1H), 3,94 (т, J=4,6 Гц, 2H), 3,79, 3,56-3,51 (м, 4H), 1,56 (c, 3H); ЖХМС: 435 (M+H+) для C19H19ClN4O4S.
Пример 18: получение соединения 117
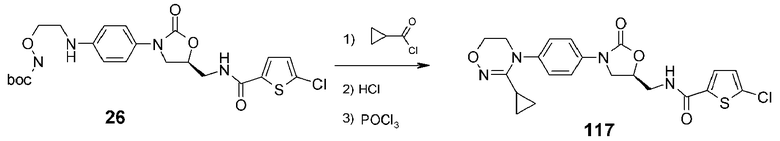
Соединение 26 (0,20 мг, 0,39 ммоль), полученное в примере получения 5, циклопропанкарбонилхлорид (50 мг, 0,47 ммоль), пиридин (61 мг, 0,78 ммоль) и 4-диметиламинопиридин (5 мг) добавляют к метиленхлориду (5 мл) с последующим перемешиванием в течение 2 часов. По завершении взаимодействия добавляют туда метиленхлорид (50 мл), затем дважды промывают дистиллированной водой (10 мл). После концентрирования при пониженном давлении проводят колоночную хроматографию, получая амидное соединение в виде белого твердого вещества (150 мг, 0,29 ммоль, 74%).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,90 (c, 1H), 8,98 (т, J=6,0 Гц, 1H), 7,69 (д, J=4,0 Гц, 1H), 7,62 (д, J=8,8 Гц, 2H), 7,42 (д, J=8,8 Гц, 2H), 7,19 (д, J=4,0 Гц, 1H), 4,84 (м, 1H), 4,21 (т, J=8,8 Гц), 3,87 (дд, J=9,2, 6,4 Гц, 1H), 3,78-3,74 (м, 4H), 3,61 (т, J=5,6 Гц, 2H), 1,22 (м, 1H), 0,78 (м, 2H), 0,59 (м, 2H).
Полученное выше соединение (0,15 г, 0,29 ммоль) добавляют к 4н. HCl (в диоксане) (2 мл) с последующим перемешиванием при комнатной температуре в течение одного часа. Реакционную смесь концентрируют при пониженном давлении. Добавляют толуол (5 мл) и оксихлорид фосфора (45 мг, 0,29 ммоль), затем кипятят с обратным холодильником при перемешивании в течение 12 часов. Проводят колоночную хроматографию, получая указанное в заголовке соединение 117 в виде белого твердого вещества (11 мг, 0,03 ммоль, 10%).
1H ЯМР (400 МГц, хлороформ-d1) δ 7,54 (д, J=8,8 Гц, 2H), 7,31 (д, J=4,4 Гц, 1H), 7,25 (д, J=8,8 Гц, 2H), 6,90 (д, J=4,4 Гц, 1H), 6,55 (т, J=4,8 Гц, 1H), 4,88 (м, 1H), 4,10 (т, J=4,8 Гц, 2H), 3,89 (м, 2H), 3,79 (м, 1H), 3,67 (т, J=4,8 Гц, 2H), 1,06 (м, 1H), 0,93 (м, 2H), 0,58 (м, 2H); ЖХМС: 461 (M+H+) для C21H21ClN4O4S.
Пример 19: получение соединения 118
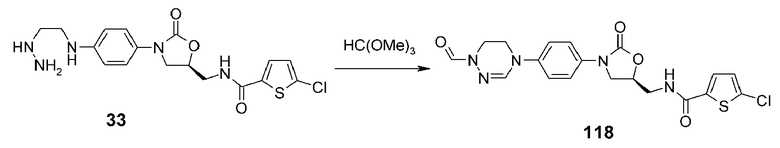
Соединение 118 получают в виде белого твердого вещества способом, аналогичным способу, описанному в примере 10, используя соединение 33, синтезированное способом по реакционной схеме 5.
1H ЯМР (400 МГц, хлороформ-d1) δ 8,55 (c, 1H), 7,52 (д, J=8,8 Гц, 2H), 7,33 (д, J=4,4 Гц, 1H), 7,10 (c, 1H),7,05 (д, J=8,8 Гц, 2H), 6,89 (д, J=4,4 Гц, 1H), 6,76 (т, J=4,8 Гц, 1H), 4,88 (м, 1H), 4,11 (т, J=8,8 Гц, 1H), 4,00 (т, J=4,8 Гц, 2H), 3,91-3,77 (м, 3H), 3,74 (т, J=4,8 Гц, 2H); ЖХМС: 448 (M+H+) для C19H18ClN5O4S.
Пример 20: получение соединения 119
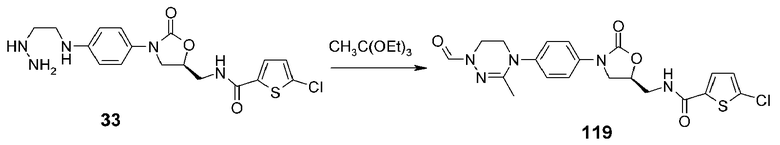
Соединение 119 получают в виде белого твердого вещества способом, аналогичным способу, описанному в примере 14, используя соединение 33, синтезированное способом по реакционной схеме 5.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,94 (широкий т, 1H), 8,35 (c, 1H), 7,64 (д, J=4,4 Гц, 1H), 7,52 (д, J=8,8 Гц, 2H), 7,26 (д, J=8,8 Гц, 2H), 7,14 (д, J=4,4 Гц, 1H), 4,80 (м, 1H), 4,15 (т, J=8,4 Гц, 1H), 3,81-3,75 (м, 3H), 3,58-3,51 (м, 4H), 1,75 (c, 3H); ЖХМС: 462 (M+H+) для C20H20ClN5O4S.
Пример 21: получение соединения 120
Соединение 119, синтезированное в примере 20, растворяют в метаноле, затем деформилируют посредством HCl, получая указанное в заголовке соединение 120 в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,98 (широкий т, 1H), 7,69 (д, J=4,4 Гц, 1H), 7,52 (д, J=8,8 Гц, 2H), 7,23-7,19 (м, 3H), 4,83 (м, 1H), 4,18 (т, J=8,4 Гц, 1H), 3,84 (м, 1H), 3,61-3,55 (м, 4H), 3,06 (т, J=4,8 Гц, 2H), 1,64 (c, 3H); ЖХМС: 434 (M+H+) для C19H20ClN5O3S.
Пример 22: получение соединения 121
Проводят взаимодействие соединения 120, синтезированного в примере 21, с йодметаном, получая указанное в заголовке соединение 121 в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,98 (широкий т, 1H), 7,69 (д, J=4,4 Гц, 1H), 7,55 (д, J=8,8 Гц, 2H), 7,28 (д, J=8,8 Гц, 2H), 7,19 (д, J=4,4 Гц, 1H), 4,84 (м, 1H), 4,18 (т, J=8,4 Гц, 1H), 3,84 (м, 1H), 3,65-2,92 (м, 6H), 2,60 (c, 3H), 1,67 (c, 3H); ЖХМС: 448(M+H+) для C20H22ClN5O3S.
Экспериментальный пример 1: ингибирующая активность ингибитора фактора Xa (FXa)
1) Реагент и материал
S-2765 (N-Z-D-Arg-Gly-Arg-pNA.2HCl), хромогенный субстрат, необходимый для определения активности фактора Xa, приобретают от Chromogenix. Человеческий FXa приобретают от Enzyme Research Laboratories. 96-луночный микропланшет приобретают от Corning Life Sciences.
2) Ингибирующая активность ингибитора FXa
Ингибирующую активность оксазолидиноновых производных по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленных формулой I, относительно FXa определяют следующим образом.
Активность соединений относительно очищенного человеческого FXa определяют, используя хромогенный субстрат S-2765 (Z-D-Arg-Gly-Arg-pNA.2HCl), в 96-луночных микропланшетах при 37°C. Ферментативную активность исследуют в 100 мМ Tris-HCl буфере (pH 7,8), содержащем человеческий FXa (2,6 нМ), NaCl (150 мМ), PEG 8000 (0,1%), разведения исследуемого соединения (1% ДМСО) и S-2765 (300 мкМ). Реакцию инициируют, добавляя субстрат, и непрерывно в течение 5 мин наблюдают поглощение при 405 нм, применяя SpectraMax 190 (Molecular Devices, USA). Рассчитывают константу ингибирования (Ki) относительно человеческого FXa по уравнению Ченга-Пруссофа (Ki=IC50/1+[S]/Km), где [S] обозначает концентрацию субстрата и Km обозначает константу Михаэлиса-Ментен. Km определяют из графика Лайнуивера-Бэрка. IC50 обозначает количество ингибитора, необходимое для снижения начальной скорости контроля на 50%. Значения IC50 рассчитывают, используя программное обеспечение GraFit, версия 5.0.12 (Erithacus Software Ltd., UK).
Используемое для расчета значение Km составляет 125 нМ, его получают, изменяя концентрацию субстрата при постоянной концентрации фермента.
Экспериментальный пример 2: влияние на свертывание крови
Эффект оксазолидиноновых производных по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленных формулой I, на свертывание крови исследуют, определяя протромбиновое время (PT).
1) Определение PT
a) Способ с применением коагулометра: протромбиновое время (PT) определяют при помощи Thrombotimer 4 - канального коагулометра (Behnk Elektronik, Germany). В исследованиях используют цитратную плазму человека и крысы. Для определения PT 100 мкл свежеразмороженной плазмы смешивают с 3 мкл серийного разведения исследуемого соединения или ДМСО. После 5-минутной инкубации при 37°C добавляют 200 мкл STA-Neoplastine (Diagnostica Stago, France) для начала образования сгустков. Антикоагулянтную активность соединений определяют как концентрацию, необходимую для увеличения в два раза времени свертывания плазмы [2 x PT (мкМ)]. Человеческую плазму получают от Daejeon Red Cross Blood Center. Крысиную кровь отбирают из сонной артерии или верхней полой вены под анестезией. Кровь собирают в пластиковые пробирки, содержащие 1/10 объема 3,8% цитрата натрия. Плазму получают немедленным центрифугированием при 2500g в течение 10 мин при 4°C и хранят при 70°C.
b) Способ применения spectramax: серийные разведения раствора соединения по настоящему изобретению (5 мкл) смешивают с цитратной плазмой (45 мкл), затем через 5 мин добавляют STA-Neoplastine (Diagnostica Stago, France) при 37°C. Непрерывно наблюдают поглощение при 340 нм и определяют PT как время (в секундах), когда поглощение при 340 нм достигает 0,1. Антикоагулянтную активность соединений определяют как концентрацию, необходимую для увеличения в два раза времени свертывания плазмы [2×PT (мкМ)]. Антикоагулянтную активность соединений определяют как концентрацию, необходимую для увеличения в два раза времени свертывания плазмы [2×PT (мкМ)]. Человеческую плазму получают из Daejeon Red Cross Blood Center. Крысиную кровь отбирают из сонной артерии или верхней полой вены под анестезией. Кровь собирают в пластиковые пробирки, содержащие 1/10 объема 3,8% цитрата натрия. Плазму получают немедленным центрифугированием при 2500g в течение 10 мин при 4°C и хранят при -70°C.
2) Определение антитромботического эффекта с использованием модели артериовенозного шунта (AV-шунт) у крыс.
Антитромботический эффект оксазолидиноновых производных по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленных формулой I, оценивают, применяя артериовенозный (AV) шунт у крыс. Голодным самцам крыс Sprague-Dawley массой 200-240 г и примерно 7-недельного возраста проводят анестезию посредством внутрибрюшинной инъекции уретана (1,25 г/кг) или хлоралгидрата. Находящимся под анестезией крысам проводят артериовенозное (AV) шунтирование, как описано ранее [Journal of Thrombosis and Haemostasis (2004) 3, 514] с незначительной модификацией. В левую общую сонную артерию и правую яремную вену вводят канюлю с двумя трубками длиной 200 мм, заполненными физиологическим раствором (PE-50, Becton Dickinson, USA). Полиэтиленовые трубки соединяют посредством силиконовой трубки длиной 8 мм (L/S®13, MasterFlex, USA) с силиконовой трубкой длиной 50 мм (L/S®16, MasterFlex, USA), содержащей хлопковую нить длиной 75 мм. Соединение или носитель дают перорально за 60 мин до открытия шунта на 15 мин. Затем хлопковую нить удаляют и взвешивают. Значения ED50 рассчитывают, применяя линейный регрессионный анализ и используя Excel 2003 (Microsoft®).
3) Определение времени кровотечения (BT), используя модель кровотечения из хвоста крыс.
Голодным самцам крыс Sprague-Dawley массой 200-240 г и примерно 7-недельного возраста проводят анестезию посредством внутрибрюшинной инъекции пентобарбитала натрия (60 мг/кг). Ингибиторы FXa или носитель дают перорально за 60 минут до отсечения 2 мм кончиков хвостов анестезированных крыс и вертикального погружения их в физиологический раствор при 37°C. Определяют, что время до прекращения непрерывного течения крови составляет >30 сек при максимальном времени наблюдения 30 мин (большее определяемое время кровотечения составляет 30 мин).
Константа ингибирования относительно человеческого FXa, антикоагулянтный эффект (выраженный как 2×PT) и антитромботический эффект в AV-шунте крыс (выраженный как % ингибирования образования тромбов), определяемые в описанных выше экспериментах, показаны в таблице 1. Эффекты соединений на время кровотечения при отсечении крысиных хвостов суммированы в таблице 2. Ривароксабан, представленный формулой A, применяют в качестве сравнительного лекарственного средства.
[Формула A]
Константа ингибирования относительно человеческого FXa, антикоагулянтный эффект (выраженный как 2×PT) и антитромботический эффект (выраженный как % ингибирования образования тромбов) в AV-шунте крыс для соединений формулы I
Как показано в таблице 1, оксазолидиноновые производные по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленные формулой I, имеют значения Ki и PT, аналогичные значениям сравнительного лекарственного препарата ривароксабана. На модели AV-шунтированных крыс антитромботический эффект сравним с эффектом ривароксабана, даже если обнаруживают небольшие вариации в антитромботическом эффекте в зависимости от применяемого анестетика. Наиболее существенным побочным эффектом ривароксабана является кровотечение. Для исследования влияния на кровотечение оценивают время хвостового кровотечения при многократных дозах типичных соединений, представленных формулой I. Эффекты соединений 100, 109 и сравнительного лекарственного препарата ривароксабана на время кровотечения при отсечении крысиного хвоста суммированы в таблице 2.
Эффект соединений 100 и 109 на время кровотечения из крысиного хвоста (n=13)
В исследовании на модели кровотечения из хвоста крыс соединение 100 не удлиняет время кровотечения даже при 10 мг/кг по сравнению с контрольным носителем. С другой стороны, ривароксабан вызывает пролонгацию времени кровотечения в три-четыре раза даже при 1,25 мг/кг. Следовательно, установлено, что соединения формулы I значительно снижают побочный эффект (кровотечение). Кроме того, соединения формулы I можно получить в виде солей, используя такую кислоту, как метансульфоновая кислота или HCl, так чтобы можно было повысить растворимость в воде. Растворимость в воде определяют в следующем эксперименте.
Экспериментальный пример 3: определение растворимости в воде
Соединения 100 и 109, типичные примеры оксазолидинонового производного формулы I по настоящему изобретению с циклической амидоксимной группой (100) и циклической амидразоновой группой (109) в виде гидрохлорида или метансульфоната (MSA) тестируют на растворимость в воде, и результаты показаны в таблице 3. В качестве сравнительного лекарственного препарата используют ривароксабан, представленный формулой A.
Растворимость в воде соединений 100 и 109 в виде гидрохлорида и метансульфоната
Как показано в таблице 3, оксазолидиноновое производное формулы I по настоящему изобретению с циклической амидоксимной (100) или циклической амидразоновой (109) группой имеет большое преимущество в том, что может быть приготовлено в виде соли и, следовательно, может обладать превосходной растворимостью в воде, в 200 раз более высокой, чем растворимость ривароксабана - контрольного вещества. Этот результат показывает, что оксазолидиноновые производные формулы I по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой имеют высокую применимость в качестве композиции для перорального введения и инъекции.
Как объясняется в данном описании ранее, оксазолидиноновые производные формулы I по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой демонстрируют почти отсутствие кровотечения, одного из серьезных побочных эффектов общепринятого лекарственного препарата, такого как ривароксабан, но обладают такой же ингибирующей активностью, как ривароксабан, и превосходной растворимостью, так что они могут иметь превосходную применимость в качестве композиции для перорального введения и инъекции.
Промышленная применимость
Новые оксазолидиноновые производные по настоящему изобретению с циклической амидоксимной или циклической амидразоновой группой, представленные формулой I, могут быть очень безопасным лекарственным средством, которое не увеличивает кровотечение - серьезный побочный эффект ривароксабана, который является контрольным веществом в этом изобретении, и имеют более высокую растворимость в воде, чем ривароксабан, так что их можно легко разрабатывать в качестве композиции для перорального введения или инъекции.
Специалистам в данной области понятно, что концепции и специфические варианты, раскрытые в предшествующем описании, можно легко использовать в качестве базиса для модификации или конструирования других вариантов с целью выполнения вышеупомянутых целей настоящего изобретения. Специалистам в данной области понятно, что такие эквивалентные варианты не отклоняются от духа и области изобретения, которое изложено в приложенной формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПРОИЗВОДНОЕ ОКСАЗОЛИДИНОНА И ВКЛЮЧАЮЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2012 |
|
RU2617408C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА С ЦИКЛИЧЕСКИМ АМИДОКСИМОМ ИЛИ ЦИКЛИЧЕСКИМ АМИДРАЗОНОМ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2009 |
|
RU2468023C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| СОЕДИНЕНИЯ ИНДОЛА И ИНДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА НЕКРОЗА КЛЕТКИ | 2008 |
|
RU2437883C1 |
| СОЕДИНЕНИЯ ИНДОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ КЛЕТОЧНОГО НЕКРОЗА | 2008 |
|
RU2477282C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И ИНДАЗОЛА, ОБЛАДАЮЩИЕ КОНСЕРВИРУЮЩИМ ДЕЙСТВИЕМ ПО ОТНОШЕНИЮ К КЛЕТКАМ, ТКАНЯМ И ОРГАНАМ | 2009 |
|
RU2460525C2 |
| ТРИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ИМИДАЗО[4,5-с]ПИРИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА ГИСТАМИНА 4 (hH4R) | 2012 |
|
RU2628074C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| БИАРИЛ- ИЛИ ГЕТЕРОЦИКЛИЧЕСКИЕ БИАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСЕНА В КАЧЕСТВЕ ИНГИБИТОРОВ СЕТР | 2014 |
|
RU2627361C2 |
| СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ РАСЩЕПЛЯЕМЫЙ ЛИНКЕР, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2795168C2 |
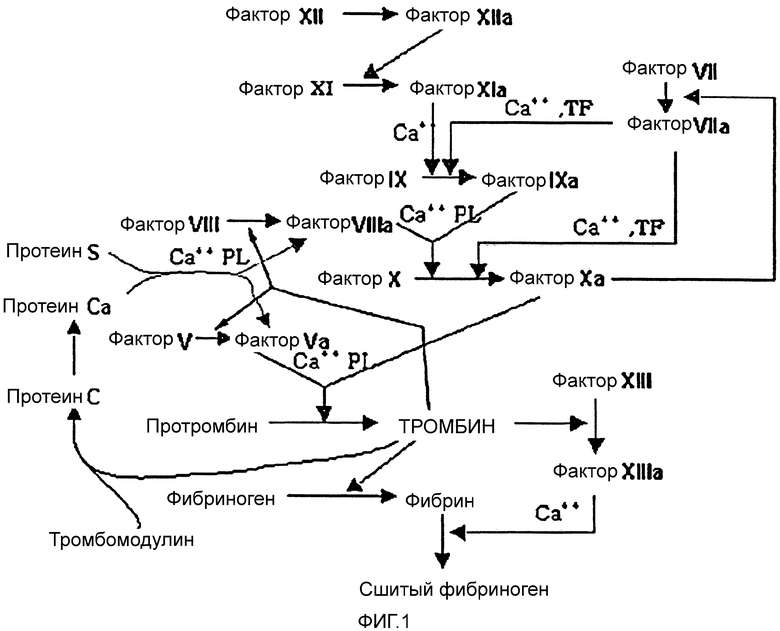
Изобретение относится к соединениям формулы 1, обладающим свойствами ингибитора фактора Ха, их фармацевтически приемлемым солям и фармацевтическим композициям на их основе. В формуле 1
[Формула 1]
цикл А обозначает остаток, выбранный из группы, включающей следующие структуры:

R1-R2 независимо представляют собой Н, (С1-С7) алкил или (С3-С7) циклоалкил, R3 и R4 образуют цикл посредством соединения с (С3-C5) алкиленом, атом углерода алкилена может быть замещен карбонилом; R13 означает Н, (С1-С7)алкил или формил. 5 н. и 3 з.п. ф-лы, 1 ил., 3 табл., 22 пр.
1. Оксазолидиноновое производное с циклической амидоксимной или циклической амидразоновой группой, представленное формулой 1, или его фармацевтически приемлемая соль:
[Формула 1]
где цикл А обозначает остаток, выбранный из группы, включающей следующие структуры:
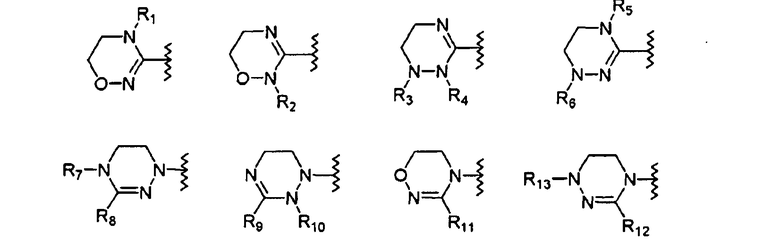
R1-R12 независимо представляют собой Н, (C1-C7)алкил или (С3-С7)циклоалкил, R3 и R4 образуют цикл посредством соединения с (С3-С5)алкиленом, атом углерода алкилена может быть замещен карбонилом;
R13 означает Н, (С1-С7)алкил или формил.
2. Оксазолидиноновое производное или его фармацевтически приемлемая соль по п.1, где оксазолидиноновое производное выбрано из следующих формул II-ХI
[Формула II]
[Формула III]
[Формула IV]
[Формула V]
[Формула VI]
[Формула VII]
[Формула VIII]
[Формула IX]
[Формула X]
[Формула XI]
где R1-R12 независимо друг от друга представляют атом водорода, (С1-С7)алкил или (С3-С7)циклоалкил; R13 означает Н, (С1-С7)алкил или формил; и m равно целому числу от 1 до 3.
3. Оксазолидиноновое производное или его фармацевтически приемлемая соль по п.2, где R1-R12 независимо друг от друга представляют атом водорода, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, циклопропил, циклобутил, циклопентил или циклогексил; R13 обозначает Н, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил или формил; и m равно 1.
4. Оксазолидиноновое производное или его фармацевтически приемлемая соль по п.3, где указанное оксазолидиноновое производное выбрано из следующих соединений:
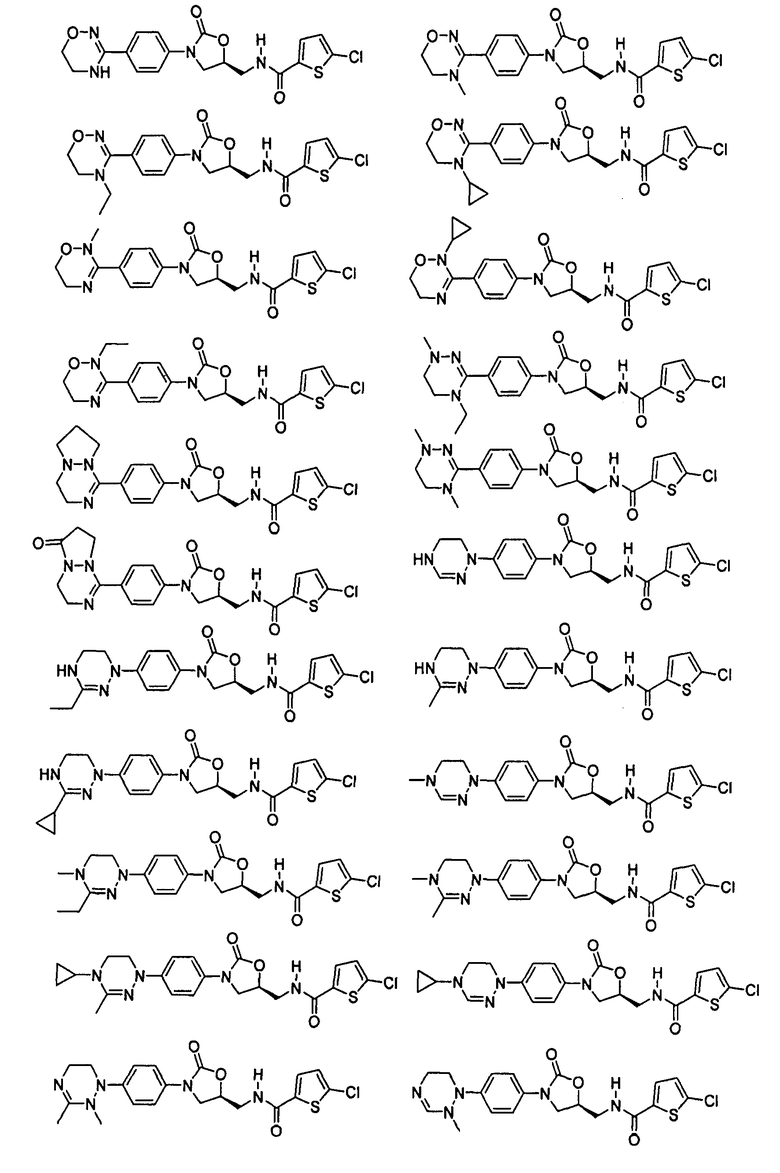
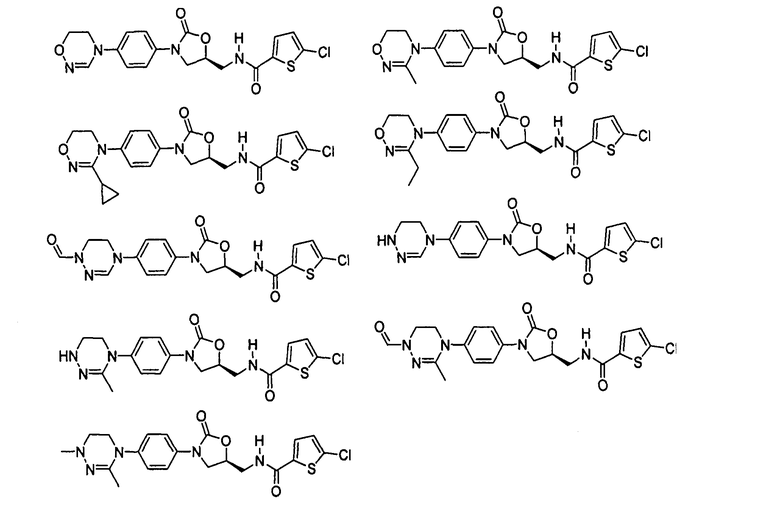
5. Фармацевтическая антикоагулянтная композиция, содержащая терапевтически эффективное количество оксазолидинонового производного или его фармацевтически приемлемой соли по любому из пп.1-4 и фармацевтически приемлемые наполнители.
6. Фармацевтическая антикоагулянтная композиция, содержащая терапевтически эффективное количество оксазолидинонового производного или его фармацевтически приемлемой соли по любому из пп.1-4 и фармацевтически приемлемые наполнители, для профилактики или лечения тромбоза, инфаркта миокарда, артериосклероза, воспаления, удара, стенокардии, рестеноза, перемежающейся хромоты, флеботромбоза, легочной эмболии, артериального тромбоза, ишемии миокарда или тромбоэмболии.
7. Фармацевтическая антикоагулянтная композиция, содержащая терапевтически эффективное количество оксазолидинонового производного или его фармацевтически приемлемой соли по любому из пп.1-4 и фармацевтически приемлемые наполнители в комбинации с тромболитическим лекарственным средством, для профилактики или лечения заболевания коронарной артерии, заболевания церебральных артерий или заболевания периферических артерий.
8. Фармацевтическая антикоагулянтная композиция, содержащая эффективное количество оксазолидинонового производного или его фармацевтически приемлемой соли по любому из пп.1-4 и фармацевтически приемлемые наполнители, для консервирования крови, плазмы или продуктов крови in vitro.
| WO 2001047919 A1, 05.07.2001 | |||
| US 20040242660 A1, 02.12.2004 | |||
| ЗАМЕЩЕННЫЕ ОКСАЗОЛИДИНОНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ ЗАМЕЩЕННЫХ ОКСАЗОЛИДИНОНОВ | 2000 |
|
RU2297415C2 |

