Настоящее изобретение относится к усовершенствованному способу эпоксидирования олефинов с использованием титансодержащего цеолитного катализатора, благодаря чему при рециркуляции растворителя значительно уменьшается дезактивация катализатора.
Предпосылки создания изобретения
Из ЕР-А 100118 известно, что когда в качестве катализатора применяют титансодержащий цеолит, то с использованием пероксида водорода олефины могут быть превращены в эпоксиды. Реакцию в предпочтительном варианте проводят в присутствии полярного растворителя, который в реакционной смеси растворяет как олефин, так и пероксид водорода. В качестве растворителя предпочтительно применение метанола.
В процесс часто вводят азотсодержащие соединения в качестве компонентов исходных материалов, в качестве добавок в реакцию эпоксидирования или в качестве добавок или вспомогательных веществ во время обработки реакционной смеси.
Азотсодержащие соединения могут быть введены с используемым пероксидом водорода. Технические растворы пероксида водорода часто включают азотсодержащие соединения в качестве стабилизаторов пероксида или в качестве замедлителей коррозии. Одним примером служит аминотриметиленфосфоновая кислота, используемая в качестве стабилизатора.
Азотсодержащие соединения можно добавлять в реакцию эпоксидирования для повышения селективности в отношении пропиленоксида. Описан большой ряд используемых для этой цели азотсодержащих соединений: аммиак, аммониевые соли и нитраты - в ЕР-А 230949 и ЕР-А 712852, органические азотистые основания, такие как амины, - в ЕР-А 1072599 и WO 00/17178, аминооксиды - в WO 00/17178, амиды карбоновых кислот - в ЕР-А 940393 и WO 01/92242 и нитрилы - в WO 01/68623. Аналогичным образом в смесь процесса эпоксидирования с целью подавить разложение пероксида водорода добавляют азотсодержащие хелатообразователи, как это изложено в ЕР-А 757045.
Пример введения азотсодержащего соединения во время обработки смеси процесса эпоксидирования описан в заявке WO 99/14208, согласно которой в качестве растворителя для экстракции оксида олефина из реакционной смеси описан нитробензол.
Когда процесс эпоксидирования проводят в промышленном масштабе, полярный растворитель, используемый в реакции эпоксидирования, при обработке реакционной смеси по экономическим соображениям необходимо выделять и возвращать в реакцию эпоксидирования. Способы извлечения растворителя описаны в WO 99/07690, WO 99/08985, WO 99/11639, WO 02/02544, WO 02/02545, WO 02/14298, WO 02/00635, WO 99/14208, WO 99/23052, WO 02/00634, US 5599955, WO 01/57009 и WO 01/57010. Когда растворитель возвращают в процесс, примеси, содержащиеся в рецикловом растворителе, при рециркуляции могут накапливаться до нежелательных уровней. Эта проблема обусловлена загрязненностью ацетальдегидом, как сказано в WO 99/07690 и WO 99/08985, и метилформиатом - в WO 02/02544 и WO 02/02545.
Рецикловый растворитель может также включать примеси, отравляющие катализатор эпоксидирования, что приводит к снижению каталитической активности и более быстрой дезактивации катализатора эпоксидирования, когда в реакции эпоксидирования используют рецикловый растворитель. Такое отравление катализатора наблюдали, когда на определенной стадии процесса окисления вводили одно или несколько азотсодержащих соединений. Известные методы выделения и возврата в процесс растворителя не имеют отношения к проблеме отравления катализаторов, и их осуществление не дает решения этой проблемы.
Следовательно, объектом настоящего изобретения является усовершенствование способа каталитического эпоксидирования олефинов в присутствии титансодержащего цеолитного катализатора, согласно которому, когда растворитель выделяют и возвращают на стадию эпоксидирования, для достижения повышенной активности катализатора эпоксидирования на определенной стадии способа вводят одно или несколько азотсодержащих соединений.
Сущность изобретения
Этой цели достигают благодаря способу каталитического эпоксидирования олефинов, в котором на определенной стадии вводят одно или несколько азотсодержащих соединений, включающему следующие стадии:
I) взаимодействие олефина с пероксидом водорода в присутствии титансодержащего цеолитного катализатора и полярного растворителя,
II) выделение из продукта взаимодействия стадии I) потока, включающего полярный растворитель,
III) обработка потока, выделяемого на стадии II), до содержания азота в форме органических соединений азота меньше 50 мас. част./млн и
IV) возврат по меньшей мере части обработанного потока со стадии III) на стадию эпоксидирования I).
В предпочтительном варианте способа обработка на стадии III) представляет собой кислотную обработку.
Кроме того, этой цели достигают благодаря способу каталитического эпоксидирования пропена, в котором на определенной стадии вводят одно или несколько азотсодержащих соединений и
а) на реакционной стадии проводят реакцию пропена с пероксидом водорода в присутствии силикалита титана как катализатора и растворителя, включающего метанол,
б) поток продуктов с этой реакционной стадии необязательно направляют на стадию сброса давления,
в) затем поток продуктов в предварительной испарительной колонне, обладающей меньше чем 10 теоретическими ступенями разделения, разделяют на головной продукт, содержащий пропен, пропеноксид и метанол, и остаточный продукт, содержащий метанол и воду, причем от 20 до 60% общего количества метанола, вводимого с потоком продуктов, удаляют с головным продуктом, а остальное остается в остаточном продукте,
г) по меньшей мере часть остаточного продукта со стадии в) обрабатывают до содержания азота в форме органических соединений азота меньше 50 мас. част./млн и необязательно после частичного удаления воды,
д) по меньшей мере часть обработанного продукта со стадии г) возвращают на реакционную стадию а).
Подробное описание изобретения
В способе каталитического эпоксидирования олефинов в соответствии с изобретением на определенной стадии этого способа вводят одно или несколько азотсодержащих соединений. Если вводят несколько азотсодержащих соединений, то они могут быть введены либо на одной и той же стадии, либо на разных стадиях способа. Азотсодержащие соединения могут быть введены как умышленно, так и непреднамеренно.
Непреднамеренное введение одного или нескольких азотсодержащих соединений происходит, если по меньшей мере один из таких исходных материалов, как олефин и пероксид водорода, или полярный растворитель, используемый в реакции эпоксидирования, включает азотсодержащее соединение. Одним примером служит применение технического пероксида водорода, включающего азотсодержащее соединение в качестве добавки. Такими добавками являются, например, аминофосфоновые кислоты, используемые в качестве стабилизаторов пероксида, или нитратные соли, используемые в качестве замедлителей коррозии.
Азотсодержащие соединения могут быть также введены в процесс с определенной целью. Добавки, такие как аммиак, амины, аминооксиды, карбоксамиды и нитрилы, могут быть введены в реакцию эпоксидирования для повышения селективности в отношении эпоксида. На той же стадии с целью уменьшить разложение пероксида могут быть введены азотсодержащие хелатообразующие соединения. Азотсодержащие соединения могут быть также введены во время обработки реакционной смеси как растворители, например на стадиях экстракции или экстрактивных дистилляций. Во время обработки для превращения карбонильных соединений в менее летучие продукты можно использовать азотсодержащие соединения, включающие незамещенную группу NH2, предпочтительно гидразин.
Азотсодержащие соединения, вводимые в процесс, можно превращать в другие азотсодержащие соединения химическими реакциями в рамках процесса. Такой реакцией может быть окисление пероксидом водорода, как некатализируемое, так и катализируемое титансодержащим цеолитом. Аммиак может быть окислен до гидроксиламина, нитрита или нитрата. Первичные амины могут быть окислены до замещенных гидроксиламинов, оксимов или нитроалканов. Вторичные амины могут быть окислены до замещенных гидроксиламинов. Третичные амины могут быть окислены до аминооксидов. Аммиак или первичные амины могут также взаимодействовать с эпоксидами с образованием аминоспиртов, которые, в свою очередь, могут быть подвергнуты дополнительному окислению. Гидроксиламин может взаимодействовать с карбонильными соединениями, образующимися в побочных реакциях из эпоксида, с образованием оксимов. Аналогичным путем гидразин обычно взаимодействует с карбонильными соединениями с образованием гидразонов и азинов. Если обработка реакционной смеси включает стадию гидрогенизации, оксимы, гидразоны и азины обычно превращаются в первичные амины.
Из вышеизложенного совершенно очевидно, что поток, включающий полярный растворитель, выделяемый из реакционной смеси процесса эпоксидирования, может включать большой ряд органических соединений азота, даже если в процесс вводят только неорганические азотсодержащие соединения. Вследствие большого числа реакций и сложной сети реакций невозможно предсказать по существу, какие конкретные органические соединения азота содержатся и в каком количестве.
При создании настоящего изобретения было отмечено, что когда на определенной стадии процесса эпоксидирования с титансодержащим цеолитным катализатором вводят одно или несколько азотсодержащих соединений, возврат выделенного полярного растворителя на стадию эпоксидирования неожиданно приводит к пониженной активности катализатора эпоксидирования и более быстрой дезактивации катализатора.
Было установлено, что активность катализатора эпоксидирования может быть повышена, а дезактивация катализатора может быть уменьшена обработкой выделенного потока, включающего полярный растворитель, перед его возвратом на стадию эпоксидирования до содержания азота в форме органических соединений азота меньше 50 мас. част./млн (мас. част./млн означает массовых частей на миллион, т.е. мг/кг). В предпочтительном варианте выделенный поток обрабатывают до содержания азота в форме органических соединений азота меньше 30 мас. част./млн, более предпочтительно меньше 15 мас. част./млн, а наиболее предпочтительно меньше 10 мас. част./млн.
Под органическими соединениями азота понимают соединения, которые между углеродным и азотным атомами содержат ковалентную связь. Количество азота в форме органических соединений азота в предпочтительном варианте определяют как разницу между общим количеством азота и количеством азота в форме неорганических соединений азота. Общее количество азота в предпочтительном варианте определяют по методу Кьельдаля так, как изложено в описании стандарта DIN 53625. Количество азота в форме неорганических соединений азота в предпочтительном варианте определяют ионной хроматографией, позволяющей определить отдельные неорганические соединения азота. Когда выделенный поток растворителя обрабатывают на стадии дистилляции перед его возвратом на стадию эпоксидирования, он обычно не содержит неорганических соединений, отличных от аммиака. Следовательно, для практических целей достаточно определить количество азота в форме неорганических соединений азота определением аммония либо как такового, либо в форме аммониевых солей.
Принимая во внимание цель изобретения, определение азота в форме органических соединений азота можно осуществлять как непрерывно, так и время от времени. Когда определение осуществляют время от времени, промежутки времени могут быть как одинаковыми, так и неодинаковыми. Обычно достаточно определить количество азота в форме органических соединений азота только когда параметры процесса изменяются.
Обработка выделенного потока до содержания азота в форме органических соединений азота меньше 50 мас. част./млн может включать стадии разделения или химические реакции. Обработка может также сочетать несколько стадий разделения или может сочетать стадии разделения с одной или несколькими химическими реакциями. Приемлемыми стадиями разделения для обработки являются, например, дистилляция, кристаллизация, экстракция, абсорбция или мембранные разделения. Приемлемыми химическими реакциями для обработки являются реакции кислот с основаниями, селективное окисление органических соединений азота или селективное восстановление органических соединений азота. В предпочтительном варианте обработка включает кислотную обработку выделенного потока растворителя.
В предпочтительном варианте способа обработка выделенного потока является кислотной обработкой, включающей добавление кислоты в выделенный поток и обработку полученной смеси на стадии дистилляции. На стадии дистилляции отводят дистиллят, включающий полярный растворитель и содержащий меньше 50 мас. част./млн азота в форме органических соединений азота.
В другом предпочтительном варианте способа обработка выделенного потока представляет собой кислотную обработку, включающую обработку выделенного потока проведением непрерывного процесса дистилляции и подачей потока, включающего кислоту, на уровне выше точки введения выделенного потока в дистилляционную колонну. Поток продуктов, включающий полярный растворитель и содержащий меньше 50 мас. част./млн азота в форме органических соединений азота, отводят из дистилляционной колонны на уровне выше точки введения включающего кислоту потока, предпочтительно из верхней части этой колонны.
Кислотную обработку выделенного потока можно осуществлять любой приемлемой кислотой, такой как минеральная кислота, карбоновая кислота и кислотный ионообменник в его протонированной форме. Приемлемыми минеральными кислотами являются азотная кислота, серная кислота, соляная кислота, фосфорная кислота и перхлорная кислота. Предпочтительными минеральными кислотами являются серная кислота и фосфорная кислота. Карбоновые кислоты в предпочтительном варианте выбирают из моно- и дикарбоновых кислот с C1 по С12, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, масляная кислота, пентановая кислота, гексановая кислота, гептановая кислота, октановая кислота, нонановая кислота, декановая кислота, ундекановая кислота и додекановая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, глутаровая кислота, адипиновая кислота, малеиновая кислота и фумаровая кислота. Наиболее предпочтительной карбоновой кислотой является уксусная кислота.
Для кислотной обработки кислым ионообменником в его протонированной форме можно использовать как сильнокислотные ионообменники, так и слабокислотные ионообменники. Предпочтительны сильнокислотные ионообменники, содержащие группы SO3Н, и слабокислотные ионообменники, содержащие группы СООН. Кислотный ионообменник в предпочтительном варианте основан на органическом полимере, таком как сшитый полистирол, или органо-неорганическом гибридном полимере, таком как полисилоксан. Кислотный ионообменник может представлять собой либо жидкость, либо твердое вещество гелевого типа, либо макропористое твердое вещество. В предпочтительном варианте обработку осуществляют пропусканием выделенного потока растворителя через неподвижный слой, включающий кислотный ионообменник.
С применением способа в соответствии с изобретением может быть эпоксидирован любой олефин, в частности олефины, содержащие от 2 до 6 углеродных атомов. Способ в соответствии с изобретением больше всего приемлем для эпоксидирования пропена до пропеноксида. По экономическим соображениям для проведения процесса в промышленном масштабе было бы предпочтительным использовать пропен не в чистом виде, а в виде технической смеси с пропаном, которая, как правило, содержит от 1 до 15 об.% пропана. Олефин можно направлять в реакционную систему в виде жидкости, а также в газообразной форме.
В предпочтительном варианте пероксид водорода используют в виде водного раствора, содержащего от 1 до 90 мас.%, предпочтительно от 10 до 70 мас.% пероксида водорода. В наиболее предпочтительном варианте в качестве пероксида водорода используют сырой продукт, получаемый на стадии экстракции в антрахиноновом методе, содержащий от 30 до 45 мас.% пероксида водорода. По другому варианту можно использовать растворы пероксида водорода в спиртах, предпочтительно в метаноле. Эти спиртовые растворы могут быть получены реакцией водорода и кислорода в присутствии катализатора на основе благородного металла и спирта.
Для осуществления способа эпоксидирования в соответствии с изобретением в качестве катализаторов пригодны для использования кристаллические титансодержащие цеолиты, предпочтительно цеолиты состава (TiO2)x(SiO2)1-x, где х обозначает число от 0,001 до 0,05, характеризующиеся кристаллической структурой MFI или MEL, известные как титансиликалит-1 и титансиликалит-2. Такие катализаторы могут быть приготовлены, например, в соответствии со способом, описанным в US 4410501. Титансиликалитный катализатор можно применять в виде формованного катализатора в форме гранул, экструдатов или других формованных тел. Для возможности процесса формования катализатор может включать от 1 до 99% связующего вещества или материала носителя, причем приемлемы все связующие вещества и материалы носителей, которые в реакционных условиях, создаваемых для эпоксидирования, не взаимодействуют с пероксидом водорода или эпоксидом. В предпочтительном варианте в качестве катализаторов неподвижного слоя используют экструдаты диаметром от 1 до 5 мм.
Для того чтобы повысить растворимость олефина, предпочтительно пропена, в жидкой фазе, реакцию проводят в присутствии растворителя. Для применения в качестве растворителя подходят все полярные растворители, которые в выбранных реакционных условиях пероксидом водорода не окисляются или окисляются лишь в слабой степени и которые растворяются в воде в количестве больше 10 мас.%. Предпочтительны растворители, которые полностью смешиваются с водой. Приемлемые растворители включают спирты, такие как метанол, этанол и трет-бутанол; гликоли, такие как, например, этиленгликоль, 1,2-пропандиол или 1,3-пропандиол; циклические простые эфиры, такие как, например, тетрагидрофуран, диоксан или пропиленоксид; гликолевые простые эфиры, такие как, например, этиленгликольмонометиловый эфир, этиленгликольмоноэтиловый эфир, этиленгликольмонобутиловый эфир или пропиленгликольмонометиловый эфир; и кетоны, такие как, например, ацетон или 2-бутанон. В качестве растворителей предпочтительны спирты, особенно предпочтителен метанол.
Олефин, раствор пероксида водорода и полярный растворитель могут быть введены в реактор в виде независимых исходных материалов или перед введением в реактор один или несколько этих исходных материалов могут быть смешаны. В предпочтительном варианте с целью обеспечить значительное потребление пероксида водорода олефин используют в избытке относительно пероксида водорода, причем предпочтительное значение молярного соотношения между олефином, предпочтительнее пропеном, и пероксидом водорода выбирают в интервале от 1,1 до 30. В предпочтительном варианте растворитель добавляют при значении массового отношения к количеству используемого раствора пероксида водорода от 0,5 до 20. Используемое количество катализатора можно варьировать в широком интервале, причем в предпочтительном варианте его выбирают таким образом, чтобы в создаваемых реакционных условиях в течение от 1 мин до 5 ч потребление пероксида водорода превышало 90%, предпочтительнее превышало 95%.
В одном варианте выполнения изобретения во время реакции титансиликалитный катализатор суспендируют в реакционной смеси. В этом случае такой катализатор применяют в форме порошка или в форме способного суспендироваться гранулированного материала, который приготовлен известным путем, например распылительной сушкой или гранулированием в псевдоожиженном слое. Когда используют суспендированный катализатор, для проведения реакции можно применять реакторы с перемешиваемым потоком, например резервуарные реакторы с мешалками или реакторы с рециркуляцией, а также реакторы с неперемешиваемым потоком, например трубчатые проточные реакторы. В предпочтительном варианте применяют каскад, включающий от одного до трех реакторов с перемешиваемым потоком и соединенный с ними и установленный после них реактор с неперемешиваемым потоком.
В другом варианте выполнения изобретения титансиликалитный катализатор используют в виде неподвижного слоя, над которым пропускают смесь исходных материалов. В таком случае этот катализатор применяют в виде формованных тел, которые готовят по известному методу, например экструзией с добавлением связующих материалов.
Когда катализатор используют в неподвижном слое, можно применять реакторы с характеристиками пузырьковой колонны, т.е. реактор содержит непрерывную жидкую фазу, а дисперсная газообразная фаза движется по реактору в режиме восходящего потока. По другому варианту можно применять реакторы с характеристиками отекания в слое тонкой струйкой, т.е. реактор содержит газовую фазу, а жидкая фаза движется по реактору в режиме нисходящего потока.
В особенно предпочтительном варианте выполнения настоящего изобретения процесс проводят в реакторе с неподвижным слоем и, для того чтобы поддержать в каталитическом слое режим отекания тонкой струйкой в слое, выбирают следующие условия истечения:
G/λ<2000 м/ч и
LΨ<50 м/ч,
где G обозначает расход на единицу сечения газового потока, определяемый в проточном реакторе непрерывного действия как скорость газового потока в м3/ч, деленную на поперечное сечение каталитического слоя в м2;
L обозначает расход на единицу сечения потока жидкости, определяемый в проточном реакторе непрерывного действия как скорость потока жидкости в м3/ч, деленную на поперечное сечение каталитического слоя в м2;

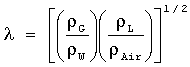
и

ρG обозначает плотность газовой фазы в г/см3,
ρL обозначает плотность жидкой фазы в г/см3,
ρW обозначает плотность воды в г/см3,
ρAir обозначает плотность воздуха в г/см3,
σw обозначает поверхностное натяжение воды в дин/см,
σL обозначает поверхностное натяжение жидкой фазы в дин/см,
μL обозначает вязкость жидкой фазы в сантипуазах,
μw обозначает вязкость воды в сантипуазах.
Реакцию эпоксидирования в предпочтительном варианте проводят при температурах в пределах 0 и 80°С, более предпочтительно в пределах 40 и 65°С. В соответствии с наиболее предпочтительным вариантом выполнения настоящего изобретения реакцию эпоксидирования проводят в реакторе с неподвижным слоем, оборудованном охлаждающим средством, и внутри реактора поддерживают такой температурный профиль, при котором температура охлаждающей среды охлаждающего средства составляет по меньшей мере 30°С, а максимальная температура внутри каталитического слоя составляет 60°С, предпочтительно самое большее 55°С.
В зависимости от олефина, который должен быть эпоксидирован, реакцию эпоксидирования можно проводить в нормальных условиях или под повышенным давлением. Если температура кипения олефина ниже температуры, выбранной для реакции эпоксидирования, давление обычно поддерживают на уровне от 5 до 50 бар, предпочтительно от 15 до 30 бар.
В предпочтительном варианте выполнения изобретения условия для реакции эпоксидирования, т.е. температуру, давление и количества олефина, пероксида водорода и растворителя, выбирают с тем, чтобы получить мультифазную реакционную смесь, включающую богатую пероксидом водорода жидкую водную фазу, содержащую полярный растворитель, и богатую олефином жидкую органическую фазу. Для гарантии образования второй богатой олефином жидкой органической фазы количество олефина необходимо выбирать в избытке относительно количества, растворимого в водной фазе, включающей при выбранных температуре и давлении воду, пероксид водорода и полярный растворитель. Сохранение двух несмешивающихся жидких фаз во время реакции эпоксидирования приводит к повышенной селективности в отношении эпоксида.
В предпочтительном варианте выполнения изобретения для повышения селективности в отношении эпоксида на стадии эпоксидирования добавляют аммиак в количестве от 100 до 3000 част./млн, предпочтительно от 300 до 2000 част./млн в пересчете на массу пероксида водорода.
Выделение полярного растворителя из реакционной смеси процесса эпоксидирования можно осуществлять по любому приемлемому методу разделения или сочетанием методов разделения, таких как дистилляция, кристаллизация, экстракция, адсорбция и т.п. В предпочтительном варианте для выделения используют сочетание стадий дистилляции. В предпочтительном варианте непрореагировавший олефин и эпоксидный продукт из потока растворителя выделяют во время процесса выделения. Некоторое количество воды, содержащейся в потоке растворителя, можно выделять также во время выделения растворителя. Выделенный поток растворителя, который обрабатывают до содержания азота в форме органических соединений меньше 50 мас. част./млн, может включать дополнительные компоненты, такие как вода, непрореагировавший пероксид водорода и побочные продукты, процесса эпоксидирования.
Предпочтительным вариантом выполнения изобретения является усовершенствованный способ каталитического эпоксидирования пропена пероксидом водорода в присутствии силикалита титана как катализатора и растворителя, включающего метанол. Этот усовершенствованный способ составляет одно целое с предлагаемыми обработкой и возвратом в процесс растворителя при обработке реакционной смеси с получением пропеноксида с минимумом стадий разделения и низкой энергетической потребностью при одновременных сохранении высокой каталитической активности и длительном сроке службы катализатора.
Эпоксидирование пропена проводят так, как изложено выше, с использованием растворителя, включающего метанол.
В предпочтительном варианте перед стадией обработки давление реакционной смеси процесса эпоксидирования сбрасывают на стадии сброса давления до давления, используемого в ходе обработки пропеноксида. Часть пропена, растворенного в реакционной смеси, и возможно пропан удаляют дегазацией. Получаемый газ повторно сжимают посредством компрессора до давления, превалирующего в реакторе, и возвращают в реакцию.
Далее в предварительной испарительной колонне реакционную смесь разделяют на головной продукт, содержащий пропен, возможно пропан, пропеноксид и метанол, и остаточный продукт, содержащий метанол, воду, высококипящие побочные продукты, такие как, например, пропиленгликоль, непрореагировавший пероксид водорода и возможно суспендированный титансиликалитный катализатор. Предварительная испарительная колонна в соответствии с изобретением обладает меньше чем 20, предпочтительно самое большее 10 теоретическими разделительными ступенями и в предпочтительном варианте сконструирована таким образом, что ректификационная секция соответствует одной стадии дистилляции, а остального эффекта разделения добиваются в отпарной секции. Предварительная испарительная колонна работает при коэффициенте орошения самое большее 1,5 и при необходимости может также работать вообще без орошения. Для того чтобы избежать разложения пероксида водорода, давление в предварительной испарительной колонне в предпочтительном варианте выбирают в интервале от 1,5 до меньше 3 бар. Предварительная испарительная колонна работает таким образом, чтобы с головным продуктом удалять в пределах 20 и 60% от количества растворителя, подаваемого с реакционной смесью, а остальное остается в остаточном продукте. В предпочтительном варианте больше 95%, более предпочтительно больше 98%, а наиболее предпочтительно больше 99% вводимого пропеноксида содержатся в головном продукте и в предпочтительном варианте больше 90%, более предпочтительно больше 97% вводимой воды содержатся в остаточном продукте.
Поток продуктов, направляемый в предварительную испарительную колонну, в предпочтительном варианте содержит от 0,5 до 20 мас.% пропена, от 0 до 4 мас.% пропана, от 5 до 35 мас.% пропеноксида, от 35 до 80 мас.% метанола, от 5 до 40 мас.% воды, от 0,1 до 8 мас.% высококипящих побочных продуктов, от 0,1 до 5 мас.% пероксида водорода и от 0 до 5 мас.% силикалита титана как катализатора. В предпочтительном варианте в предварительной испарительной колонне этот поток продуктов разделяют на головной продукт, содержащий от 1 до 40 мас.% пропена, от 0 до 10 мас.% пропана, от 15 до 75 мас.% пропеноксида, от 20 до 85 мас.% метанола и от 0 до 5 мас.% воды, и остаточный продукт, содержащий от 0 до 2 мас.% пропеноксида, от 30 до 80 мас.% метанола, от 15 до 65 мас.% воды, от 0,1 до 10 мас.% высококипящих побочных продуктов, от 0,1 до 5 мас.% пероксида водорода, от 0 до 10 мас.% силикалита титана как катализатора и больше 50 мас. част./млн азота в форме органических соединений азота.
По меньшей мере часть, а предпочтительно весь остаточный продукт предварительной испарительной колонны, включающий метанол и воду, обрабатывают до содержания азота в форме органических соединений азота меньше 50 мас. част./млн. Далее по меньшей мере часть, а предпочтительно весь полученный обработанный продукт возвращают в реакцию эпоксидирования. В предпочтительном варианте по меньшей мере часть воды, содержащейся в остаточном продукте предварительной испарительной колонны, перед направлением рециклового потока на стадию эпоксидирования удаляют. Удаление воды можно осуществлять перед обработкой, после обработки или в сочетании с обработкой на одной технологической стадии.
Обработку выделенного метанола до содержания азота в форме органических соединений азота меньше 50 мас. част./млн можно осуществлять по всем методам, описанным выше, как подходящим для обработки выделяемого потока полярного растворителя. В предпочтительном варианте обработка включает кислотную обработку выделенного потока метанола. Наиболее предпочтительно использовать такие же варианты кислотной обработки, как те, которые представлены выше в качестве предпочтительных вариантов кислотной обработки потока растворителя.
В предпочтительном варианте способа эпоксидирования пропена головной продукт из предварительной испарительной колонны по меньшей мере частично конденсируют с получением конденсата, содержащего от 0 до 12 мас.% пропена, от 0 до 5 мас.% пропана, от 15 до 75 мас.% пропеноксида, от 20 до 85 мас.% метанола и от 0 до 8 мас.% воды. В предпочтительном варианте этот головной продукт только частично конденсируют, а несконденсировавшийся пропен, возможно смешанный с пропаном, повторно сжимают с помощью компрессора до давления, превалирующего в реакционной части, и возвращают в реакцию. Пропен, все еще растворенный в конденсате, и возможно пропан в предпочтительном варианте отпаривают из конденсата в секции для отпарки С3продуктов. В предпочтительном варианте выделенный отпаркой газ возвращают в конденсатор для частичной конденсации. В предпочтительном варианте частичную конденсацию проводят в две стадии. Головной продукт из предварительной испарительной колонны частично конденсируют в первом конденсаторе, а газообразный отходящий поток из первого конденсатора конденсируют во втором конденсаторе, в котором поддерживают температуру ниже температуры первого конденсатора. В предпочтительном варианте температуру внутри первого конденсатора поддерживают на уровне от 40 до 70°С, а температуру внутри второго конденсатора поддерживают на уровне от 20 до 35°С. С использованием двухступенчатой конденсации значительно уменьшают количество ценного пропеноксида, который может быть не выделен, и сокращают потребление энергии на охлаждение в сравнении с ее затратами при одноступенчатой конденсации. Когда применяют двухступенчатую конденсацию, конденсаты обоих конденсаторов направляют в секцию для отпарки С3продуктов для удаления компонентов, обладающих температурой кипения, которая ниже температуры кипения пропеноксида, благодаря чему газообразный отходящий поток из отпарной секции частично конденсируют во втором конденсаторе и конденсат возвращают в секцию для отпарки С3продуктов.
Затем конденсат, получаемый по меньшей мере частичной конденсацией головного продукта из предварительной испарительной колонны и необязательной его обработкой отпаркой в секции для отпарки С3продуктов, подвергают экстрактивной дистилляции с водным экстракционным растворителем. Этот конденсат направляют в среднюю секцию непрерывно работающей дистилляционной колонны. Водный экстракционный растворитель добавляют в экстрактивную дистилляционную колонну на уровне выше точки, в которой в колонну поступает конденсат. Из верхней части этой колонны отводят очищенный пропеноксид. В основании экстрактивной дистилляционной колонны получают остаточный продукт, содержащий метанол и воду. Этот остаточный продукт совмещают с остаточным продуктом из предварительной испарительной колонны и объединенный продукт подвергают обработке для удаления органических соединений азота.
В предпочтительном варианте в качестве экстракционного растворителя используют водный раствор гидразина, содержащий от 0,5 до 5 мас.% гидразина. Когда в качестве экстракционного растворителя используют водный раствор гидразина, при экстрактивной дистилляции в одну стадию очистки из пропеноксида удаляют не только метанол, но также ацетальдегид и другие карбонильные соединения и обеспечивают получение очищенного пропеноксида, содержащего меньше 50 част./млн метанола и меньше 50 част./млн ацетальдегида.
Количество экстракционного растворителя, подаваемого в дистилляционную колонну, в предпочтительном варианте выбирают таким образом, чтобы значение массового отношения вводимого экстракционного растворителя к количеству метанола, содержащегося в подаваемом сыром пропеноксиде, находилось в интервале от 0,1 до 10. В предпочтительном варианте экстрактивную дистилляцию проводят под абсолютным давлением от 1 до 5 бар, более предпочтительно под абсолютным давлением от 1,5 до 2,5 бара. В предпочтительном варианте коэффициент орошения выбирают в интервале от 1 до 5.
Дистилляционная колонна, применяемая для экстрактивной дистилляции, включает отпарную секцию между кубовыми остатками колонны и точкой введения конденсата предварительной испарительной колонны, экстракционную секцию между точкой введения конденсата предварительной испарительной колонны и точкой введения экстракционного растворителя и ректификационную секцию между точкой введения экстракционного растворителя и точкой, где отводят очищенный пропеноксид. В предпочтительном варианте применяют дистилляционную колонну, которая обладает эффективностью разделения от 10 до 30 теоретических ступеней как в отпарной секции, так и в экстракционной секции и эффективностью разделения от 20 до 60 теоретических ступеней в ректификационной секции. Дистилляционной колонной может служить тарельчатая колонна, включающая отдельные тарелки, такие как ситчатые тарелки или колпачковые тарелки. Дистилляционной колонной может служить также насадочная колонна, и можно использовать как неупорядоченные насадки, так и структурированные насадки, такие как насадки из металлической сетки. Дистилляционная колонна может также сочетать секции с отдельными тарелками и секции с насадками. В предпочтительном варианте экстракционная секция сконструирована с расчетом на отдельные тарелки.
В предпочтительном варианте процесса эпоксидирования остаточный продукт из предварительной испарительной колонны или объединенные остаточные продукты из предварительной испарительной колонны и экстрактивной дистилляции перед тем, как их подвергают обработке для удаления органических соединений азота, направляют на стадию каталитической гидрогенизации. Осуществление дополнительной стадии гидрогенизации уменьшает накопление побочных продуктов, таких как метилформиат и диметоксиметан, в рецикловом растворителе и повышает чистоту пропеноксида, выделяемого по такому способу.
В предпочтительном варианте каталитическую гидрогенизацию проводят как гетерогенную каталитическую гидрогенизацию при парциальном давлении водорода от 0,5 до 30 МПа. В особенно предпочтительном варианте стадию гидрогенизации осуществляют при температуре в интервале от 80 и 150°С, предпочтительно от 100 до 180°С, и при парциальном давлении водорода от 1 до 25 МПа.
Приемлемые катализаторы гидрогенизации выбирают из нанесенных на носитель катализаторов, включающих один или несколько металлов, выбранных из ряда, включающего Ru, Rh, Pd, Pt, Ag, Ir, Fe, Cu, Ni и Со. По другому варианту можно использовать никель Ренея и кобальт Ренея, причем их обоих необязательно легируют одним или несколькими из упомянутых выше металлов. В предпочтительном варианте каталитический носитель выбирают из активированного угля и оксидов металлов, выбранных из SiO2, TiO2, ZrO2 и Al2O3, смешанных оксидов, включающих по меньшей мере два элемента из Si, Al, Ti и Zr, и их смесей.
Гидрогенизацию можно проводить в виде непрерывного процесса или периодического процесса, например по суспензионному методу или методу с неподвижным слоем. Особенно предпочтительно применение реактора со стеканием в слое тонкой струйкой. Предпочтительные предусмотренные для использования в неподвижном слое катализаторы представляют собой окатыши диаметром от 0,5 до 5 мм, преимущественно от 1 до 3 мм, и длиной от 1 до 10 мм. Содержание благородного металла находится в обычном интервале, предпочтительно от 0,5 до 5 мас.%.
Преимущества настоящего изобретения вполне очевидны, принимая во внимание следующие примеры.
Примеры
Пример 1 (сравнительный пример)
Эпоксидирование осуществляли проведением непрерывного процесса в трубчатом реакторе емкостью 300 мл, заполненном связанными диоксидом кремния титансиликалитными экструдатами диаметром 3 мм. Более того, оборудование включало три контейнера для жидкостей и соответствующие насосы, а также сосуд для отделения жидкости. Эти три контейнера для жидкостей содержали метанол, водный Н2О2 концентрацией 60 мас.% и пропен. Значение рН водного Н2О2 концентрацией 60 мас.% аммиаком доводили до 4,5. Реакционную температуру регулировали водной охлаждающей жидкостью, которая циркулировала в охлаждающей рубашке, благодаря чему с помощью термостата регулировали температуру охлаждающей жидкости. Абсолютное давление в реакторе составляло 25 бар. Массовый расход питающих насосов регулировали с тем, чтобы в результате концентрация исходного пропена составляла 21,5 мас.%, концентрация исходного метанола составляла 57 мас.%, а концентрация исходного Н2О2 была равной 9,4 мас.%. Реактор работал в переточном режиме. Температура охлаждающей рубашки составляла 41°С, общий массовый расход был равным 0,35 кг/ч, а максимальная температура составляла 59°С. Выход продукта определяли газовой хроматографией, а превращение Н2О2 - окислительно-восстановительным титрованием. Селективность катализатора рассчитывали, основываясь на данных газового хроматографического анализа продуктов окисления пропена в виде соотношения между количеством образовавшегося пропеноксида и количеством всех образовавшихся продуктов окисления пропена. Начальное превращение Н2О2 составляло 96% при селективности катализатора 96%.
Реакционную смесь, образовавшуюся в результате реакции, после сброса давления разделяли на стадии предварительного испарения на головной продукт, содержавший пропен, пропан, пропеноксид и метанол, и остаточный продукт, содержавший метанол, пропиленгликольмонометиловые эфиры, пропиленгликоль, воду, высококипящие соединения и не подвергшийся превращению пероксид водорода. Из головного продукта в парообразном состоянии получали жидкий конденсат, который содержал пропеноксид и метанол, а также растворенные в нем пропен и пропан. Несконденсировавшийся поток, который состоял по существу из пропена и пропана, возвращали в реакцию эпоксидирования. Пропен и пропан, растворенные в конденсате, отпаривали из этого последнего в секции для отпарки С3продуктов и возвращали в процесс в парообразном состоянии совместно с потоком головного продукта предварительной испарительной колонны на стадию частичной конденсации. Поток после отпарки конденсата, который состоял по существу из пропеноксида и метанола и был освобожден от пропена и пропана, в процессе экстрактивной дистилляции, в который в качестве экстрагента непосредственно ниже головной части колонны вводили водный раствор гидразина концентрацией 1,5 мас.%, разделяли на пропеноксидный очищенный продукт и остаточный продукт, который по существу состоял из метанола и воды. Этот остаточный продукт объединяли с остаточным продуктом, получаемым в предварительной испарительной колонне, и подвергали гидрогенизации в реакторе для непрерывной гидрогенизации с режимом стекания в слое тонкой струйкой. Гидрогенизационный реактор обладал внутренним объемом 150 мл и был заполнен катализатором гидрогенизации в форме экструдатов диаметром 2,3 мм, включавших 2% Ru на активированном угле (этот катализатор готовили в соответствии с методом по начальной влажности с использованием RuCl3, "Preparation of Catalyst", Demon, В. и др., Elsevier, Amsterdam, 1976, с.13). Гидрогенизацию проводили при 140°С и под абсолютным давлением 40 бар при скорости потока водорода 10 мл/ч.
Гидрированный продукт подавали со скоростью 3,5 кг/ч в среднюю секцию колонны для отгонки метанола с 35 теоретическими ступенями и непрерывно дистиллировали под абсолютным давлением 2 бара при коэффициенте орошения 2. Из верхней части этой колонны получали 2,5 кг/ч потока метанола, который содержал больше 99 мас.% метанола, 0,13 мас.% воды, 55 мас. част./млн совокупного азота и меньше 5 мас. част./млн азота аммиака. Общее содержание азота определяли по методу Кьельдаля согласно стандарту DIN 53625, азота аммиака - ионной хроматографией. По разнице общего содержания азота и азота аммиака содержание азота органических соединений азота находилось, согласно расчетам, в пределах 50 и 55 мас. част./млн.
Выделенный метанол повторно использовали для эпоксидирования пропена в лабораторном аппарате, включавшем два последовательных трубчатых реактора диаметром 18 мм и длиной 300 мм каждый, причем каждый содержал 8 г такого же катализатора, как использованный в первом эпоксидировании. Этот катализатор предварительно кондиционировали его использованием для эпоксидирования пропена в течение больше 3000 ч. Реакторы работали в режиме восходящего потока под абсолютным давлением 25 бар и при температуре 36°С в первом реакторе и 44°С во втором реакторе. В реактор одновременно вводили три потока исходных материалов. Исходный материал 1 представлял собой смесь, включавшую 715 г водного пероксида водорода концентрацией 60 мас.% и 1420 г свежего метанола, причем этот метанол не содержал поддававшихся определению количеств соединений азота, его вводили со скоростью 22,5 г/ч. Исходный материал 2 представлял собой смесь 1284 г выделенного метанола и 1,335 г водного аммиака концентрацией 32 мас.%, вводимую со скоростью 14,8 г/ч. Исходный материал 3 был пропеном, вводимым со скоростью 23 г/ч.
Отходившие из реакторов потоки анализировали газовой хроматографией на пропиленоксид и окислительно-восстановительным титрованием на пероксид водорода. Выход пропиленоксида рассчитывали по количеству введенного пероксида водорода. Результаты представлены в таблице 1.
Пример 2
Эксперимент примера 1 повторяли с изменением, состоявшим в том, что раствор уксусной кислоты в метаноле концентрацией 10 мас.% вводили в поток на орошение колонны для отгонки метанола со скоростью 81 г/ч. Из верхней части колонны отводили поток метанола, содержавший больше 98 мас.% метанола, 1,6 мас.% воды, 33 мас. част./млн совокупного азота и 7 мас. част./млн азота аммиака. Согласно расчетам по разнице общего содержания азота и азота аммиака содержание азота органических соединений азота составляло 26 мас. част./млн. Вследствие повторного использования выделенного метанола при эпоксидировании пропена, как в примере 1, получали результаты, представленные в таблице 1.
Пример 3
Эксперимент примера 1 повторяли с изменением, состоявшим в том, что водный раствор уксусной кислоты концентрацией 10 мас.% вводили в поток на орошение колонны для отгонки метанола со скоростью 98 г/ч. Из верхней части колонны отводили поток метанола, содержавший больше 97 мас.% метанола, 2 мас.% воды, 14 мас. част./млн совокупного азота и меньше 4 мас. част./млн азота аммиака. По разнице общего содержания азота и азота аммиака содержание азота органических соединений азота находилось, согласно расчетам, в пределах 10 и 14 мас. част./млн. Вследствие повторного использования выделенного метанола при эпоксидировании пропена, как в примере 1, получали результаты, представленные в таблице 1.
Пример 4
Эксперимент примера 1 повторяли и из верхней части колонны для отгонки метанола отводили поток метанола, содержавший больше 97 мас.% метанола, 2,5 мас.% воды и 110 мас. част./млн совокупного азота. 1000 мл этого выделенного метанола обрабатывали 10 мл сильнокислотного ионообменника Dowex 50Х8 в Н+-форме при комнатной температуре в течение 3 дней с периодическим встряхиванием. Обработанный метанол содержал 1,9 мас. част./млн совокупного азота. Этот обработанный выделенный метанол повторно использовали при эпоксидировании пропена так, как изложено в примере 1, с отличием, состоявшим в том, что вместо свежего метанола в исходном материале 1 использовали выделенный метанол. Результаты представлены в таблице 1.
В сравнительном примере 1 отравление катализатора возвращаемым в процесс метанолом обуславливает низкую степень превращения пероксида водорода и низкий выход пропеноксида. Применение выделенного метанола с низким содержанием азота в форме органических соединений в примерах со 2 по 4 приводит к повышенной активности катализатора, как это можно видеть по повышенным степени превращения пероксида водорода и выходу пропеноксида.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНОВ | 2003 |
|
RU2322442C2 |
| СПОСОБ ОЧИСТКИ СЫРОГО ПРОПЕНОКСИДА | 2003 |
|
RU2330032C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНОВ | 2003 |
|
RU2320650C2 |
| НОВЫЕ ВОДНЫЕ РАСТВОРЫ ПЕРОКСИДА ВОДОРОДА | 2003 |
|
RU2336225C2 |
| ОБЪЕДИНЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОПЕНА И ПРОПЕНОКСИДА ИЗ ПРОПАНА | 2017 |
|
RU2717556C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ОЛЕФИНА ИЗ ГАЗОВОГО ПОТОКА | 2005 |
|
RU2355671C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОКСИДА | 2008 |
|
RU2480459C2 |
| ИЗВЛЕЧЕНИЕ ПРОПЕНА ПОСРЕДСТВОМ ОЧИСТКИ В СКРУББЕРЕ СО СМЕСЬЮ РАСТВОРИТЕЛЬ/ВОДА | 2018 |
|
RU2766954C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ПРОПЕНА | 2004 |
|
RU2340608C2 |
| ВОДНЫЙ РАСТВОР ПЕРОКСИДА ВОДОРОДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ИСПОЛЬЗОВАНИЕ | 2008 |
|
RU2468990C2 |
Изобретение относится к способу каталитического эпоксидирования олефинов в присутствии титансодержащего цеолитного катализатора и полярного растворителя. В этом способе на определенной стадии вводят одно или несколько азотсодержащих соединений, а поток растворителя выделяют, обрабатывают кислотой до содержания азота в форме органических соединений азота меньше 50 мас. част./млн и по меньшей мере часть потока возвращают на стадию эпоксидирования. Описан также способ каталитического эпоксидирования пропена, который составляет одно целое с предлагаемыми кислотной обработкой и возвратом растворителя в процесс обработки реакционной смеси. Технический результат - значительное уменьшение дезактивации катализатора при рециркуляции растворителя. 2 н. и 17 з.п. ф-лы, 1 табл.
I) взаимодействие олефина с пероксидом водорода в присутствии титансодержащего цеолитного катализатора и полярного растворителя,
II) выделение из продукта взаимодействия стадии I) потока, включающего полярный растворитель,
III) кислотную обработку потока, выделяемого на стадии II), до содержания азота в форме органических соединений азота меньше 50 мас. ч./млн и
IV) возврат по меньшей мере части обработанного потока со стадии III) на стадию эпоксидирования I).
а) добавление кислоты в поток, выделяемый на стадии II);
б) обработку смеси со стадии а) дистилляцией и
в) отвод дистиллята, включающего полярный растворитель и содержащего меньше 50 мас. ч./млн азота в форме органических соединений азота.
а) обработку потока, выделяемого на стадии II), непрерывной дистилляцией;
б) подачу потока, включающего кислоту, на уровне выше точки введения потока, выделяемого на стадии II), и
в) отвод потока продуктов, включающего полярный растворитель и содержащего меньше 50 мас. ч./млн азота в форме органических соединений азота, на уровне выше точки введения включающего кислоту потока.
а) на реакционной стадии проводят реакцию пропена с пероксидом водорода в присутствии силикалита титана как катализатора и растворителя, включающего метанол,
б) поток продуктов с этой реакционной стадии необязательно направляют на стадию сброса давления,
в) затем поток продуктов в предварительной испарительной колонне, обладающей меньше чем 20 теоретическими ступенями разделения, разделяют на головной продукт, содержащий пропен, пропеноксид и метанол, и остаточный продукт, содержащий метанол и воду, причем от 20 до 60% общего количества метанола, вводимого с потоком продуктов, удаляют с головным продуктом, а остальное остается в остаточном продукте,
г) по меньшей мере часть остаточного продукта со стадии в) обрабатывают до содержания азота в форме органических соединений азота меньше 50 мас. ч./млн и, необязательно после частичного удаления воды, где обработка представляет собой кислотную обработку,
д) по меньшей мере часть обработанного продукта со стадии г) возвращают на реакционную стадию а).
е) головной продукт со стадии в) по меньшей мере частично конденсируют, причем конденсат, необязательно после отпарки пропена и всего имеющегося пропана, содержит
0-12 мас.% пропена,
0-5 мас.% пропана,
15-75 мас.% пропеноксида,
20-85 мас.% метанола и
0-8 мас.% воды,
ж) конденсат со стадии е) подвергают экстрактивной дистилляции, в которой
ж1) конденсат вводят в среднюю секцию экстрактивной дистилляционной колонны,
ж2) водный экстракционный растворитель добавляют в экстрактивную дистилляционную колонну на уровне выше точки, в которой поступает конденсат,
ж3) пропеноксид отбирают в головной части колонны,
ж4) остаточный продукт, содержащий метанол и воду, удаляют и
з) остаточный продукт со стадии в) перед его подачей на стадию г) совмещают с остаточным продуктом со стадии ж4).
экстракционный растворитель на стадии ж2) представляет собой водный раствор гидразина, содержащий от 0,5 до 5 мас.% гидразина.
| Устройство обнаружения цветоразностных сигналов | 1982 |
|
SU1085017A1 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНА | 1995 |
|
RU2154641C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНА | 1996 |
|
RU2140415C1 |
| WO 00/17178 А1, 30.03.2000 | |||
| УСТРОЙСТВО ДЛЯ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ УПАРИВАЕМОГО РАСТВОРА | 1982 |
|
SU1072599A1 |
| Устройство для разделения суспензий | 1980 |
|
SU940393A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ЕР 0757043 А1, 05.02.1997. | |||

