Настоящее изобретение относится к усовершенствованному способу очистки сырого пропеноксида удалением метанольной и ацетальдегидной примесей до уровней ниже 100 ч./млн за одну стадию дистилляции и к способу получения пропеноксида осуществлением такой стадии очистки.
Предпосылки создания изобретения
Из ЕР-А 100119 известно, что пероксидом водорода пропен можно превращать в пропеноксид, если в качестве катализатора использовать силикалит титана. Метанол является предпочтительным растворителем, обуславливающим высокую активность катализатора. Сырой пропеноксид, получаемый по этому способу, вследствие побочных реакций обычно содержит больше 1 мас.% метанола и больше 200 ч./млн ацетальдегида. В зависимости от реакционных условий сырой пропеноксид может также включать метилформиат в количествах, превышающих 200 ч./млн.
Большую часть пропеноксида используют в качестве исходного материала для получения простых полиэфиров с концевыми гидроксильными группами, которые, в свою очередь, используют для получения полиуретановых пенопластов. Пропеноксид для применения с этой целью должен удовлетворять жестким требованиям чистоты, и содержание метанола, ацетальдегида и метилформиата должно составлять ниже 100 ч./млн для каждого компонента. Следовательно, пропеноксид, получаемый с использованием силикалита титана как катализатора, необходимо очищать дополнительно и должны быть удалены примеси метанола, ацетальдегида и (в случае наличия) метилформиата.
Метанол, ацетальдегид и метилформиат трудно удалить из пропеноксида простой дистилляцией. В смесях, содержащих больше 98 мольных % пропеноксида, эти соединения демонстрируют фактически такую же относительную летучесть, как и пропеноксид. Следовательно, очистка дистилляцией до низких уровней метанола, ацетальдегида и метилформиата требует наличия колонок с большими числами разделительных ступеней, работающих при высоких коэффициентах орошения. Это приводит к экономически невыгодным капиталовложениям и энергетическим затратам.
Было предложено множество способов удаления метанола, ацетальдегида и метилформиата из пропеноксида. Получившим распространение методом очистки пропеноксида и удаления кислородсодержащих примесей является экстрактивная дистилляция.
В ЕР-А 1009746 описана очистка пропеноксида, содержащего метанол и ацетальдегид, экстрактивной дистилляцией с полярным экстракционным растворителем, обладающим гидроксильной функциональной группой. Этот способ позволяет также удалять часть ацетальдегида, содержащегося в сыром пропеноксиде. Однако, как можно видеть из примеров, очищенный пропеноксид все еще содержит ацетальдегид и метанол в количествах, превышающих 100 ч./млн. Следовательно, для достижения целевой чистоты пропеноксида необходимы дополнительные стадии очистки.
В ЕР-В 004019 описана альтернатива экстрактивной дистилляции для удаления карбонильных соединений из пропеноксида обработкой сырого пропеноксида простой дистилляцией и подачей в дистилляционную колонну на уровне выше точки введения сырого пропеноксида соединения, содержащего незамещенную группу NH2. В качестве таковых подают жидкие соединения с группой NH2, такие как гидразин и гидразингидрат. Твердые соединения с группой NH2 подают растворенными в инертном растворителе. В этом документе также говорится, что на той же стадии дистилляции из пропеноксида можно выделять растворитель, содержащийся в сыром пропеноксиде. Однако в этом документе не содержится никакой информации или указания на то, что по описанному способу из пропеноксида можно выделять метанол.
Все растворители, описанные в стр. с 8 по 12 колонки 7 ЕР-В 004019, относятся к тому типу растворителей, которые могут быть легко выделены из пропеноксида простой дистилляцией благодаря достаточным различиям летучести. Вследствие того факта, что существует лишь очень небольшая разница между летучестями метанола и пропеноксида при высоких концентрациях пропеноксида специалист в данной области техники может прийти к выводу о том, что осуществление способа, описанного в ЕР-В 004019, включающего простую дистилляцию, не дает возможности удалять метанол до целевого уровня ниже 100 ч./млн, когда его осуществляют при экономически обоснованных параметрах для данных числа разделительных ступеней и коэффициента орошения.
В ЕР-В 004019 речь идет также о том, что водным растворам гидразина, когда их используют для удаления ацетальдегида, свойственны некоторые недостатки: реакция альдегида протекает медленно и реакция с ацетальдегидом приводит к образованию нерастворимых продуктов. В способе очистки пропеноксида, описанном в ЕР-В 004019, медленная реакция альдегидов обычно ведет к неполному удалению ацетальдегида, а образование нерастворимых продуктов обычно приводит к образованию нежелательных отложений в дистилляционной колонне и на испарителе кубовых остатков.
В US 2622060 описан способ очистки сырого пропеноксида экстрактивной дистилляцией с использованием в качестве экстракционного растворителя водного раствора щелочного соединения, такого как гидроксид натрия. Несмотря на эффективность удаления метилформиата омылением, этот способ обладает серьезными недостатками. Когда его осуществляют в виде периодического процесса дистилляции, это приводит к избыточной потере пропеноксида из-за омыления пропеноксида. Когда его осуществляют в виде непрерывной дистилляции, очистка оказывается неэффективной, поскольку очищенный пропеноксид содержит больше 1000 ч./млн каждого из ацетальдегида и метанола, а степень чистоты не превышает 97%. Следовательно, для очистки пропеноксида необходимы дополнительные стадии дистилляции.
Всем известным методам очистки сырого пропеноксида свойственен недостаток, состоящий в том, что для очистки сырого пропеноксида, содержащего больше 1 мас.% метанола и больше 200 ч./млн ацетальдегида, до целевой чистоты меньше 100 ч./млн каждого из метанола и ацетальдегида требуется больше одной стадии дистилляции.
Следовательно, целью настоящего изобретения является разработка способа очистки сырого пропеноксида, содержащего больше 1 мас.% метанола и больше 200 ч./млн ацетальдегида, с получением очищенного пропеноксида, содержащего меньше 100 ч./млн метанола и меньше 100 ч./млн ацетальдегида, с использованием только одной стадии дистилляции.
Сущность изобретения
Этой цели достигают благодаря способу очистки сырого пропеноксида, содержащего метанол и ацетальдегид, проведением непрерывной экстрактивной дистилляции, в котором
(I) в дистилляционную колонну на уровне выше точки подачи сырого пропеноксида вводят экстракционный растворитель в количестве, эффективном для понижения летучести метанола относительно летучести пропеноксида,
(II) в дистилляционную колонну на уровне выше точки подачи сырого пропеноксида подают соединение, содержащее незамещенную группу NH2 и способное взаимодействовать с ацетальдегидом в условиях дистилляции с образованием соединений с температурой кипения выше точки кипения пропеноксида, или его смешивают с сырым пропеноксидом, направляемым в дистилляционную колонну, и
(III) на уровне выше точек подачи экстракционного растворителя и соединения, содержащего незамещенную группу NH2, из дистилляционной колонны отводят очищенный пропеноксид.
В предпочтительном варианте объектом настоящего изобретения является способ, как он представлен выше, в котором сырой пропеноксид смешивают с водным щелочным раствором и в смеси перед ее подачей на экстрактивную дистилляцию проводят реакцию в течение от 1 до 200 мин при температуре от 20 до 100°С.
Кроме того, этой цели достигают благодаря способу каталитического эпоксидирования пропена, в котором
а) на реакционной стадии проводят реакцию пропена с водным пероксидом водорода в метаноле в присутствии силикалита титана как катализатора,
б) поток продуктов с этой реакционной стадии необязательно направляют на стадию сброса давления и
в) затем в предварительной испарительной колонне, обладающей меньше, чем 20 теоретическими разделительными ступенями, поток продуктов разделяют на головной продукт, содержащий пропен, пропеноксид и метанол, и остаточный продукт, содержащий метанол и воду, причем от 20 до 60% общего количества метанола, вводимого с потоком продуктов, удаляют с головным продуктом, а остальное остается в остаточном продукте,
г) головной продукт со стадии в) по меньшей мере частично конденсируют, а пропен и весь имеющийся пропан необязательно отпаривают с получением конденсата, содержащего пропеноксид, больше 1 мас.% метанола и больше 200 мас.ч./млн ацетальдегида,
д) конденсат со стадии г) обрабатывают на стадии экстрактивной дистилляции так, как указано выше, благодаря чему получают остаточный продукт, содержащий метанол и экстракционный растворитель, и
е) весь или часть остаточного продукта со стадии в) после необязательного частичного удаления воды возвращают на реакционную стадию а).
Подробное описание изобретения
Способ очистки в соответствии с изобретением особенно приемлем для очистки сырого пропеноксида, содержащего больше 1 мас.% метанола и больше 200 мас.ч./млн ацетальдегида (мас.ч./млн означает массовых частей на миллион). Сырой пропеноксид в предпочтительном варианте получают эпоксидированием пропена пероксидом водорода с использованием титансодержащего силикалитного катализатора и метанольного растворителя. Очищенный пропеноксид обычно содержит меньше 100 мас.ч./млн метанола и меньше 100 мас.ч./млн ацетальдегида, предпочтительно меньше 50 мас.ч./млн метанола и меньше 50 мас.ч./млн ацетальдегида.
В процессе очистки сырой пропеноксид подвергают непрерывной экстрактивной дистилляции. В процессе экстрактивной дистилляции в дистилляционную колонну на уровне выше точки введения сырого пропеноксида направляют экстракционный растворитель. Для использования в качестве экстракционного растворителя приемлемы соединения или смеси соединений, которые в смесях, включающих пропеноксид, метанол и экстракционный растворитель, понижают летучесть метанола относительно летучести пропеноксида. Для того чтобы получить очищенный пропеноксид с низким содержанием экстракционного растворителя, предпочтителен экстракционный растворитель, обладающий температурой кипения выше 50°С. В предпочтительном варианте экстракционный растворитель включает полярное соединение, содержащее функциональную гидроксильную группу.
В предпочтительном варианте выполнения изобретения экстракционный растворитель включает один или смесь нескольких компонентов, которые встречаются в процессе получения сырого пропеноксида либо как компоненты потоков исходных материалов, либо как побочные продукты, образующиеся в этом процессе. В предпочтительном варианте экстракционным растворителем служит вода, пропиленгликоль, 1-метокси-2-пропанол, 2-метокси-1-пропанол или смесь двух или большего числа этих соединений. Для использования в качестве экстракционного растворителя особенно предпочтительна вода. Этот вариант выполнения изобретения обладает тем преимуществом, что остаточный продукт экстрактивной дистилляции можно объединять с одним или несколькими технологическими потоками, образующимися в процессе получения сырого пропеноксида, для извлечения метанола и необязательного извлечения экстракционного растворителя. Следовательно, для извлечения метанола и необязательно экстракционного растворителя из остаточного продукта экстрактивной дистилляции не требуется никакой дополнительной дистилляционной колонны.
В процессе очистки по изобретению либо направляют в дистилляционную колонну на уровне выше точки введения сырого пропеноксида, либо смешивают с сырым пропеноксидом, направляемым в дистилляционную колонну, дополнительное соединение, содержащее незамещенную группу NH2.
Приемлемы все соединения, содержащие незамещенную группу NH2, которые способны взаимодействовать с ацетальдегидом в условиях дистилляции с образованием соединения с температурой кипения выше точки кипения пропеноксида. Для осуществления предлагаемого способа приемлемы также образуемые кислотами соли этих соединений, у которых группа NH2 протонирована до группы NH3 +. Предпочтительны соединения, у которых незамещенная группа NH2 связана непосредственно с атомом азота или кислорода. Примерами этих предпочтительных соединений являются гидразин, гидразинмоногидрат, метилгидразин, N,N-диметилгидразин и гидроксиламин, а также их соли, такие как гидразинсульфат, гидразингидрохлорид и гидроксиламиносульфат. Особенно предпочтителен гидразин.
В предпочтительном варианте выполнения изобретения в дистилляционную колонну направляют смесь экстракционного растворителя и соединения, содержащего незамещенную группу NH2. В предпочтительном варианте эта смесь представляет собой водный раствор гидразина. Особенно предпочтительны водные растворы гидразина, содержащие от 0,5 до 5 мас.% гидразина. В противовес утверждению в ЕР-В 904019 было установлено, что водные растворы гидразина оказались неожиданно эффективными. В дистилляционной колонне протекает быстрая реакция с ацетальдегидом, ведущая к почти полному превращению ацетальдегида, и никакие отложения нерастворимых соединений в дистилляционной колонне или на испарителе остаточного продукта не образуются.
Количество экстракционного растворителя, подаваемого в дистилляционную колонну, в предпочтительном варианте выбирают таким образом, чтобы значение массового отношения вводимого экстракционного растворителя к количеству метанола, содержащегося в подаваемом сыром пропеноксиде, находилось в интервале от 0,1 до 10. Количество соединения, содержащего незамещенную группу NH2, подаваемого в дистилляционную колонну, в предпочтительном варианте выбирают таким образом, чтобы значение молярного отношения этого соединения, содержащего незамещенную группу NH2, к ацетальдегиду, содержащемуся в подаваемом сыром пропеноксиде, находилось в интервале от 0,5 до 2. Если в дистилляционную колонну направляют смесь экстракционного растворителя и соединения, содержащего незамещенную группу NH2, то состав смеси и вводимые количества в предпочтительном варианте подбирают таким образом, чтобы они удовлетворяли параметрам как предпочтительного массового отношения экстракционного растворителя к метанолу, так и предпочтительного молярного отношения этого соединения, содержащего незамещенную группу NH2, к ацетальдегиду.
В процессе очистки по изобретению очищенный пропеноксид отводят из дистилляционной колонны на уровне выше точек введения экстракционного растворителя и соединения, содержащего незамещенную группу NH2. В предпочтительном варианте очищенный пропеноксид отводят из верхней части этой колонны. В этом случае значение отношения коэффициента орошения конденсатом, возвращаемым в колонну, к конденсату, отводимому в виде очищенного пропеноксида, в предпочтительном варианте выбирают в интервале от 1 до 5.
В предпочтительном варианте экстрактивную дистилляцию проводят под абсолютным давлением от 1 до 5 бар, более предпочтительно под абсолютным давлением от 1,5 до 2,5 бара.
Дистилляционная колонна, применяемая для экстрактивной дистилляции, включает отпарную секцию между кубовыми остатками колонны и точкой введения сырого пропеноксида, экстракционную секцию между точкой введения сырого пропеноксида и точкой введения экстракционного растворителя и ректификационную секцию между точкой введения экстракционного растворителя и точкой, где отводят очищенный пропеноксид. В предпочтительном варианте применяют дистилляционную колонну, которая обладает эффективностью разделения от 10 до 30 теоретических ступеней как в отпарной секции, так и в экстракционной секции и эффективностью разделения от 20 до 60 теоретических ступеней в ректификационной секции. Дистилляционной колонной может служить тарельчатая колонна, включающая отдельные тарелки, такие как ситчатые тарелки или колпачковые тарелки. Дистилляционной колонной может служить также насадочная колонна и можно использовать как неупорядоченные насадки, так и структурированные насадки, такие как насадки из металлической сетки. Дистилляционная колонна может также сочетать секции с отдельными тарелками и секции с насадками. В предпочтительном варианте экстракционная секция сконструирована с расчетом на отдельные тарелки.
В еще одном варианте выполнения изобретения сырой пропеноксид смешивают с водным щелочным раствором и перед подачей на экстрактивную дистилляцию проводят реакцию. Продолжительность реакции между смешением сырого пропеноксида с водным щелочным раствором и подачей смеси на экстрактивную дистилляцию как правило находится в интервале от 1 до 200 мин, предпочтительно от 1 до 30 мин. Реакционная температура как правило составляет от 20 до 100°С. В предпочтительном варианте водный щелочной раствор представляет собой водный раствор гидроксида натрия, гидроксида калия или карбоната натрия. Наиболее предпочтительны водные растворы гидроксида натрия, содержащие от 0,1 до 2 мас.% гидроксида натрия. Количество водного щелочного раствора в предпочтительном варианте выбирают таким образом, чтобы значение молярного отношения гидроксидных ионов, вводимых с водным щелочным раствором, к количеству метилформиата, содержащегося в сыром пропеноксиде, находилось в интервале от 1,1 до 4. Реакцию в смеси сырого пропеноксида с водным щелочным раствором перед ее подачей на дистилляцию в предпочтительном варианте проводят в трубчатом реакторе. В результате взаимодействия сырого пропеноксида с водным щелочным раствором метилформиат, содержащийся в сыром пропеноксиде, благодаря его гидролизу превращается в метанол и формиат. Очищенный пропеноксид, получаемый в этом варианте выполнения изобретения, обладает пониженным содержанием метилформиата. В предпочтительном варианте количество водного щелочного раствора выбирают с целью получения очищенного пропеноксида, обладающего содержанием метанола меньше 50 мас.ч./млн, содержанием ацетальдегида меньше 50 мас.ч./млн и содержанием метилформиата меньше 100 мас.ч./млн.
Объектом изобретения является также усовершенствованный способ каталитического эпоксидирования пропена водным пероксидом водорода с использованием силикалита титана как катализатора. Этот усовершенствованный способ составляет одно целое с экстрактивной дистилляцией по изобретению при обработке реакционной смеси с получением пропеноксида высокой степени чистоты с минимумом разделительных ступеней и низкой энергетической потребностью.
Эпоксидирование пропена пероксидом водорода проводят в присутствии силикалита титана как катализатора и метанольного растворителя.
В качестве катализаторов для осуществления способа эпоксидирования в соответствии с изобретением приемлемы кристаллические силикалиты титана, предпочтительно состава (TiO2)x(SiO2)1-x, где х обозначает число от 0,001 до 0,05, обладающие кристаллической структурой MFI или MEL, известные как силикалит титана-1 и силикалит титана-2. Такие катализаторы могут быть получены, например, в соответствии со способом, описанным в US 4410501.
Метанольный растворитель, используемый в процессе эпоксидирования, благодаря возврату веществ в процесс может включать от 0 до 20 мас.% воды.
В предпочтительном варианте пероксид водорода используют в виде водного раствора, содержащего от 10 до 90 мас.% пероксида водорода. В предпочтительном варианте в качестве пероксида водорода используют сырой продукт, получаемый на стадии экстракции в антрахиноновом методе, содержащий от 30 до 45 мас.% пероксида водорода. По другому варианту можно использовать растворы пероксида водорода в спиртах, предпочтительно в метаноле. Эти спиртовые растворы могут быть получены реакцией водорода и кислорода в присутствии катализатора на основе благородного металла и спирта.
Пропен можно использовать смешанным с пропаном с содержанием пропана в пределах 0 и 50 об.%. В предпочтительном варианте пропен содержит пропан в пределах 5 и 20 об.%.
В одном варианте выполнения изобретения во время реакции титансиликалитный катализатор суспендируют в реакционной смеси. В этом случае такой катализатор применяют в форме порошка или в форме способного суспендироваться гранулированного материала, который приготовлен известным путем, например распылительной сушкой или гранулированием в псевдоожиженном слое. Когда используют суспендированный катализатор, для проведения реакции можно применять реакторы с перемешиваемым потоком, например резервуарные реакторы с мешалками или реакторы с рециркуляцией, а также реакторы с неперемешиваемым потоком, например трубчатые проточные реакторы. В предпочтительном варианте применяют каскад, включающий от одного до трех реакторов с перемешиваемым потоком и соединенный с ними и установленный после них реактор с неперемешиваемым потоком.
В другом варианте выполнения изобретения титансиликалитный катализатор используют в виде неподвижного слоя, над которым пропускают смесь исходных материалов. В таком случае этот катализатор применяют в виде формованных тел, которые готовят по известному методу, например экструзией с добавлением связующих материалов. Для возможности процесса формования катализатор может включать от 1 до 99% связующего вещества или материала носителя, причем приемлемы все эти связующие вещества и материалы носителей, которые в реакционных условиях, создаваемых для эпоксидирования, не взаимодействуют с пероксидом водорода или эпоксидом. В предпочтительном варианте в качестве катализаторов неподвижного слоя используют экструдаты диаметром от 1 до 5 мм.
Когда применяют катализатор в неподвижном слое, можно применять реакторы с характеристиками пузырьковой колонны, т.е. реактор содержит непрерывную жидкую фазу, а дисперсная газообразная фаза движется по реактору в режиме восходящего потока. По другому варианту можно применять реакторы с характеристиками стекания в слое тонкой струйкой, т.е. реактор содержит газовую фазу, а жидкая фаза движется по реактору в режиме нисходящего потока.
В особенно предпочтительном варианте выполнения настоящего изобретения процесс проводят в реакторе с неподвижным слоем, и для того чтобы поддержать в каталитическом слое режим стекания тонкой струйкой, в слое выбирают следующие условия истечения:
G/λ<2000 м/ч и
LΨ<50 м/ч,
где
G обозначает расход на единицу сечения газового потока, определяемый в проточном реакторе непрерывного действия как скорость газового потока в м3/ч, деленную на поперечное сечение каталитического слоя в м2;
L обозначает расход на единицу сечения потока жидкости, определяемый в проточном реакторе непрерывного действия как скорость потока жидкости в м3/ч, деленную на поперечное сечение каталитического слоя в м2;
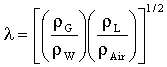
и
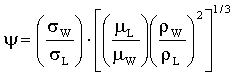
ρG обозначает плотность газовой фазы в г/см3,
ρL обозначает плотность жидкой фазы в г/см3,
ρW обозначает плотность воды в г/см3,
ρAir обозначает плотность воздуха в г/см3,
σW обозначает поверхностное натяжение воды в дин/см,
σL обозначает поверхностное натяжение жидкой фазы в дин/см,
μL обозначает вязкость жидкой фазы в сантипуазах,
μW обозначает вязкость воды в сантипуазах.
Реакцию эпоксидирования в предпочтительном варианте проводят при температурах в пределах 0 и 80°С, более предпочтительно в пределах 40 и 65°С. В соответствии с наиболее предпочтительным вариантом выполнения настоящего изобретения реакцию эпоксидирования проводят в реакторе с неподвижным слоем, оборудованном охлаждающим средством, и внутри реактора поддерживают такой температурный профиль, при котором температура охлаждающей среды охлаждающего средства составляет по меньшей мере 30°С, а максимальная температура внутри каталитического слоя составляет 60°С, предпочтительно самое большее 55°С.
В предпочтительном варианте реакцию эпоксидирования проводят под повышенными давлениями от 10 до 40 бар, более предпочтительно от 15 до 30 бар. Пропен используют в избытке, а продолжительность пребывания в реакторе выбирают таким образом, чтобы добиться превращения пероксида водорода больше 90%, предпочтительно больше 95%. В предпочтительном варианте количество используемого растворителя выбирают таким образом, чтобы добиться соотношения от 1 до 10 мас.ч. растворителя на одну массовую часть водного раствора пероксида водорода.
В предпочтительном варианте выполнения изобретения условия для реакции эпоксидирования, т.е. температуру, давление и количества пропена, пероксида водорода и растворителя, выбирают с тем, чтобы получить мультифазную реакционную смесь, включающую богатую пероксидом водорода жидкую водную фазу, содержащую метанол, и богатую олефином жидкую органическую фазу. Для гарантии образования второй богатой олефином жидкой органической фазы количество пропена необходимо выбирать в избытке относительно количества, растворимого в водной фазе, включающей при выбранных температуре и давлении воду, пероксид водорода и метанол. Сохранение двух несмешивающихся жидких фаз во время реакции эпоксидирования приводит к повышенной селективности в отношении пропеноксида.
В предпочтительном варианте перед стадией обработки давление реакционной смеси процесса эпоксидирования сбрасывают на стадии сброса давления до давления, используемого в ходе обработки пропеноксида. Часть пропена, растворенного в реакционной смеси, и возможно пропан удаляют дегазацией. Получаемый газ повторно сжимают посредством компрессора до давления, превалирующего в реакторе, и возвращают в реакцию.
Далее в предварительной испарительной колонне реакционную смесь разделяют на головной продукт, содержащий пропен, возможно пропан, пропеноксид и метанол, и остаточный продукт, содержащий метанол, воду, высококипящие побочные продукты, такие как, например, пропиленгликоль, непрореагировавший пероксид водорода и возможно суспендированный титансиликалитный катализатор. Предварительная испарительная колонна в соответствии с изобретением обладает меньше чем 20, предпочтительно самое большее 10 теоретическими разделительными ступенями и в предпочтительном варианте сконструирована таким образом, что ректификационная секция соответствует одной дистилляционной ступени, а остального эффекта разделения добиваются в отпарной секции. Предварительная испарительная колонна работает при коэффициенте орошения самое большее 1,5 и при необходимости может также работать вообще без орошения. Для того чтобы избежать разложения пероксида водорода, давление в предварительной испарительной колонне в предпочтительном варианте выбирают в интервале от 1,5 до меньше 3 бар. Предварительная испарительная колонна работает таким образом, чтобы с головным продуктом удалять в пределах 20 и 60% от количества растворителя, подаваемого с реакционной смесью, а остальное остается в остаточном продукте. В предпочтительном варианте больше 95%, более предпочтительно больше 98%, а наиболее предпочтительно больше 99% вводимого пропеноксида содержатся в головном продукте и в предпочтительном варианте больше 90%, более предпочтительно больше 97% вводимой воды содержатся в остаточном продукте.
Поток продуктов, направляемый в предварительную испарительную колонну, в предпочтительном варианте содержит от 0,5 до 20 мас.% пропена, от 0 до 4 мас.% пропана, от 5 до 35 мас.% пропеноксида, от 35 до 80 мас.% метанола, от 5 до 40 мас.% воды, от 0,1 до 8 мас.% высококипящих побочных продуктов, от 0,1 до 5 мас.% пероксида водорода, от 0 до 5 мас.% силикалита титана как катализатора и больше 200 мас.ч./млн ацетальдегида. В предпочтительном варианте в предварительной испарительной колонне этот поток продуктов разделяют на головной продукт, содержащий от 1 до 40 мас.% пропена, от 0 до 10 мас.% продукт, от 15 до 75 мас.% пропеноксида, от 20 до 85 мас.% метанола, от 0 до 5 мас.% воды и больше 200 мас.ч./млн ацетальдегида, и остаточный продукт, содержащий от 0 до 2 мас.% пропеноксида, от 30 до 80 мас.% метанола, от 15 до 65 мас.% воды, от 0,1 до 10 мас.% высококипящих побочных продуктов, от 0,1 до 5 мас.% пероксида водорода и от 0 до 10 мас.% силикалита титана как катализатора.
Головной продукт из предварительной испарительной колонны по меньшей мере частично конденсируют с получением конденсата, содержащего пропеноксид, больше 1 мас.% метанола и больше 200 мас.ч./млн ацетальдегида. В предпочтительном варианте этот головной продукт только частично конденсируют, а не сконденсировавшийся пропен, возможно смешанный с пропаном, повторно сжимают с помощью компрессора до давления, превалирующего в реакционной части, и возвращают в реакцию. Пропен, все еще растворенный в конденсате, и возможно пропан в предпочтительном варианте отпаривают из конденсата в секции для отпарки С3продуктов. В предпочтительном варианте выделенный отпаркой газ возвращают в конденсатор для частичной конденсации.
В наиболее предпочтительном варианте головной продукт из предварительной испарительной колонны частично конденсируют в первом конденсаторе, а газообразный отходящий поток из первого конденсатора конденсируют во втором конденсаторе, в котором поддерживают температуру ниже температуры первого конденсатора. В предпочтительном варианте температуру внутри первого конденсатора поддерживают на уровне от 40 до 70°С, а температуру внутри второго конденсатора поддерживают на уровне от 20 до 35°С. С использованием двухступенчатой конденсации значительно уменьшают количество ценного пропеноксида, который может быть не выделен, и сокращают потребление энергии на охлаждение в сравнении с ее затратами при одноступенчатой конденсации. Когда применяют двухступенчатую конденсацию, конденсаты обоих конденсаторов направляют в секцию для отпарки С3продуктов для удаления компонентов, обладающих температурой кипения, которая ниже температуры кипения пропеноксида, благодаря чему газообразный отходящий поток из отпарной секции частично конденсируют во втором конденсаторе и конденсат возвращают в секцию для отпарки С3продуктов.
Затем конденсат, получаемый по меньшей мере частичной конденсацией головного продукта из предварительной испарительной колонны и необязательной его обработкой отпаркой в секции для отпарки С3продуктов, подвергают экстрактивной дистилляции в соответствии с изобретением, как это изложено выше, с получением очищенного пропеноксида, содержащего меньше 100 мас.ч./млн метанола и меньше 100 мас.ч./млн ацетальдегида. В основании экстрактивной дистилляционной колонны получают остаточный продукт, содержащий метанол и экстракционный растворитель.
В предпочтительном варианте экстракционный растворитель выбирают из воды, пропиленгликоля, 1-метокси-2-пропанола, 2-метокси-1-пропанола и их смеси. Особенно предпочтительна для использования в качестве экстракционного растворителя вода. В предпочтительном варианте соединение, содержащее незамещенную группу NH2, выбирают из гидразина, гидразинмоногидрата, метилгидразина, N,N-диметилгидразина и гидроксиламина, а также их солей, таких как гидразинсульфат, гидразингидрохлорид и гидроксиламинсульфат. Особенно предпочтителен гидразин.
В наиболее предпочтительном варианте конденсат отпаривают в секции для отпарки С3продуктов, а остаточный продукт из секции для отпарки С3продуктов, содержащий от 15 до 75 мас.% пропеноксида, от 25 до 85 мас.% метанола, от 0 до 8 мас.% воды, больше 200 мас.ч./млн ацетальдегида и по существу не содержащий пропена или пропана, подвергают экстрактивной дистилляции в соответствии с изобретением с использованием водного раствора гидразина, включающего в качестве экстракционного растворителя от 0,5 до 5 мас.% гидразина. Очищенный пропеноксид, содержащий меньше 100 мас.ч./млн метанола и меньше 100 мас.ч./млн ацетальдегида, отводят из верхней части экстрактивной дистилляционной колонны.
По меньшей мере часть, а предпочтительно весь остаточный продукт предварительной испарительной колонны, включающий метанол и воду, возвращают в реакцию эпоксидирования. В предпочтительном варианте по меньшей мере часть воды, содержащейся в остаточном продукте предварительной испарительной колонны, перед направлением рециклового потока на стадию эпоксидирования удаляют.
В предпочтительном варианте остаточный продукт предварительной испарительной колонны перегонкой разделяют на головной продукт, включающий метанол, и остаточный продукт, включающий воду, высококипящие побочные продукты и непрореагировавщий пероксид водорода. По меньшей мере часть, предпочтительно весь головной продукт, получаемый на этой стадии дистилляции, возвращают назад на реакционную стадию окисления пропена. В предпочтительном варианте такую стадию дистилляции осуществляют под давлением и давление выбирают таким образом, чтобы температура головного продукта, включающего метанол, превышала соответственно температуру в основании предварительной испарительной колонны и секции для отпарки С3продуктов. В этом варианте выполнения изобретения теплоту конденсации головного продукта на упомянутой стадии дистилляции можно использовать для нагрева предварительной испарительной колонны и отпарной секции, благодаря чему значительно уменьшают энергетическую потребность процесса обработки.
В предпочтительном варианте процесса эпоксидирования остаточный продукт из предварительной испарительной колонны совмещают с остаточным продуктом из экстрактивной дистилляции и объединенные потоки направляют на стадию каталитической гидрогенизации. По меньшей мере часть, а предпочтительно весь полученный продукт возвращают на стадию эпоксидирования. В предпочтительном варианте по меньшей мере часть воды, содержащейся в гидрированном продукте, перед направлением рециклового потока на стадию эпоксидирования удаляют.
В наиболее предпочтительном варианте в качестве экстракционного растворителя используют водный раствор гидразина, содержащий от 0,5 до 5 мас.% гидразина, остаточный продукт из предварительной испарительной колонны совмещают с остаточным продуктом из экстрактивной дистилляции и объединенные потоки направляют на стадию каталитической гидрогенизации. Затем гидрированный поток подвергают разгонке на остаточный продукт, включающий воду и высококипящие побочные продукты, и головной продукт, включающий метанол. Головной продукт возвращают назад на реакционную стадию эпоксидирования пропена.
В предпочтительном варианте каталитическую гидрогенизацию проводят как гетерогенную каталитическую гидрогенизацию при парциальном давлении водорода от 0,5 до 30 МПа. В особенно предпочтительном варианте стадию гидрогенизации осуществляют при температуре в интервале 80 и 150°С, предпочтительно от 100 до 180°С, и при парциальном давлении водорода от 1 до 25 МПа.
Приемлемые катализаторы гидрогенизации выбирают из нанесенных на носитель катализаторов, включающих один или несколько металлов, выбранных из ряда, включающего Ru, Rh, Pd, Pt, Ag, Ir, Fe, Cu, Ni и Со. По другому варианту можно использовать никель Ренея и кобальт Ренея, причем их обоих необязательно легируют одним или несколькими из упомянутых выше металлов. В предпочтительном варианте каталитический носитель выбирают из активированного угля и оксидов металлов, выбранных из SiO2, TiO2, ZrO2 и Al2O3, смешанных оксидов, включающих по меньшей мере два элемента из Si, Al, Ti и Zr, и их смесей.
Гидрогенизацию можно проводить в виде непрерывного процесса или периодического процесса, например по суспензионному методу или методу с неподвижным слоем. Особенно предпочтительно применение реактора со стеканием в слое тонкой струйкой. Предпочтительные предусмотренные для использования в неподвижном слое катализаторы представляют собой окатыши диаметром от 0,5 до 5 мм, преимущественно от 1 до 3 мм, и длиной от 1 до 10 мм. Содержание благородного металла находится в обычном интервале, предпочтительно от 0,5 до 5 мас.%.
В еще одном варианте способа эпоксидирования остаточный продукт из секции для отпарки С3продуктов смешивают с водным щелочным раствором и перед направлением в экстрактивную дистилляционную колонну в этой смеси проводят реакцию в течение от 1 до 200 мин при температуре от 20 до 100°С. В предпочтительном варианте водный щелочной раствор представляет собой водный раствор гидроксида натрия, содержащий 0,1-2 мас.% гидроксида натрия. В предпочтительном варианте количество гидроксида натрия выбирают с целью добиться значения молярного соотношения между гидроксидом натрия и количеством метилформиата, содержащегося в остаточном продукте секции для отпарки С3продуктов, в интервале от 1,1 до 4.
Преимущества настоящего изобретения вполне очевидны, принимая во внимание следующие примеры.
Примеры
Пример 1
Сырой пропеноксид, содержавший 52,7 мас.% пропеноксида, 44,3 мас.% метанола, 2,2 мас.% воды, 1500 мас.ч./млн ацетальдегида и 430 мас.ч./млн метилформиата, подвергали непрерывной экстрактивной дистилляции. Экстрактивную дистилляцию осуществляли в колонне со структурированной насадкой, обладавшей эффективностью разделения 80 теоретических ступеней, работавшей под абсолютным давлением 1,8 бара и с коэффициентом орошения 2. На 20-ю ступень (считая от основания) подавали 1241 г/ч сырого пропеноксида. Одновременно на 40-ю ступень (считая от основания) подавали 207 г/ч раствора гидразина в воде концентрацией 1,5 мас.%. Из верхней части этой колонны отводили 658 г/ч очищенного пропеноксида, содержавшего 99,92 мас.% пропеноксида, 36 мас.ч./млн метанола, 70 мас.ч./млн воды, меньше 20 мас.ч./млн ацетальдегида и 390 мас.ч./млн метилформиата.
Пример 2
Эксперимент примера 1 повторяли со следующими изменениями: 1301 г/ч сырого пропеноксида, содержавшего 51,3 мас.% пропеноксида, 46,1 мас.% метанола, 2,0 мас.% воды, 1200 мас.ч./млн ацетальдегида и 280 мас.ч./млн метилформиата, смешивали с 80 г/ч водного гидроксида натрия концентрацией 0,5 мас.% и перед направлением в экстракционную колонну в течение 30 мин проводили реакцию в трубчатом реакторе при температуре 60°С. Водный раствор гидразина подавали с пониженной скоростью, 100 г/ч. Из верхней части этой колонны отводили 666 г/ч очищенного пропеноксида, содержавшего 99,98 мас.% пропеноксида, 33 мас.ч./млн метанола, 50 мас.ч./млн воды, 8 мас.ч./млн ацетальдегида и 52 мас.ч./млн метилформиата.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНОВ | 2003 |
|
RU2327694C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ОЛЕФИНОВ | 2003 |
|
RU2322442C2 |
| ОБЪЕДИНЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОПЕНА И ПРОПЕНОКСИДА ИЗ ПРОПАНА | 2017 |
|
RU2717556C1 |
| СПОСОБ И УСТАНОВКА, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ИЗВЛЕЧЕНИЯ МЕТОКСИПРОПАНОЛОВ ИЗ ВОДНОГО ПОТОКА | 2021 |
|
RU2826984C1 |
| ОЧИСТКА ОКИСИ ПРОПИЛЕНА | 2011 |
|
RU2569848C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ПРОПЕНА | 2002 |
|
RU2314300C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ПРОПЕНА | 2006 |
|
RU2388755C2 |
| ВЫДЕЛЕНИЕ ПРОПИЛЕНОКСИДА ИЗ СМЕСИ, СОДЕРЖАЩЕЙ ПРОПИЛЕНОКСИД И МЕТАНОЛ | 2005 |
|
RU2357963C2 |
| Способ очистки оксида пропилена от примесей карбонильных и карбоксильных соединений | 2019 |
|
RU2722835C1 |
| СПОСОБ ОЧИСТКИ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2740188C1 |
Изобретение относится к способу очистки сырого пропеноксида, содержащего метанол и ацетальдегид, проведением непрерывной экстрактивной дистилляции с использованием экстракционного растворителя, понижающего летучесть метанола, и подачей соединения, содержащего незамещенную группу NH2, способного взаимодействовать с ацетальдегидом, в дистилляционную колонну на уровне выше точки введения сырого пропеноксида с получением очищенного пропеноксида, содержащего меньше 100 част./млн метанола и меньше 100 част./млн ацетальдегида. Описан также способ каталитического эпоксидирования пропена, который включает эту стадию очистки. Технический результат - получение очищенного пропиленоксида, содержащего указанное выше количество примесей метанола и ацетальдегида, с использованием только одной стадии дистилляции. 2 н. и 16 з.п. ф-лы.
(I) в дистилляционную колонну на уровне выше точки подачи сырого пропеноксида вводят экстракционный растворитель в количестве, эффективном для понижения летучести метанола относительно летучести пропеноксида,
(II) в дистилляционную колонну на уровне выше точки подачи сырого пропеноксида подают соединение, содержащее незамещенную группу NH2 и способное взаимодействовать с ацетальдегидом в условиях дистилляции с образованием соединений с более высокой температурой кипения, чем у пропеноксида, или его смешивают с сырым пропеноксидом, направляемым в дистилляционную колонну, и
(III) на уровне выше точек подачи экстракционного растворителя и соединения, содержащего незамещенную группу NH2, из дистилляционной колонны отводят очищенный пропеноксид.
сырой пропеноксид содержит больше 1 мас.% метанола и больше 200 мас.ч./млн ацетальдегида.
очищенный пропеноксид содержит меньше 50 мас.ч./млн метанола и меньше 50 мас.ч./млн ацетальдегида.
очищенный пропеноксид отводят из верхней части колонны.
в котором в дистилляционную колонну направляют смесь экстракционного растворителя и соединения, содержащего незамещенную группу NH2.
экстракционный растворитель выбирают из воды, пропиленгликоля, 1-метокси-2-пропанола, 2-метокси-1-пропанола и их смесей, а предпочтительна вода.
соединение, содержащее незамещенную группу NH2, выбирают из гидразина, гидразинмоногидрата и гидразиниевых солей.
смесь представляет собой водный раствор гидразина, содержащий от 0,5 до 5 мас.% гидразина.
значение молярного отношения между соединением, содержащим незамещенную группу NH2, и ацетальдегидом находится в интервале от 0,5 до 2.
значение массового отношения между подаваемым экстракционным растворителем и количеством метанола, содержащегося в подаваемом сыром пропеноксиде, находится в интервале от 0,1 до 10.
сырой пропеноксид смешивают с водным щелочным раствором и в смеси перед ее подачей на экстрактивную дистилляцию проводят реакцию в течение от 1 до 200 мин, предпочтительно в течение от 1 до 30 мин при температуре от 20 до 100°С.
водный щелочной раствор представляет собой водный гидроксид натрия концентрацией от 0,1 до 2 мас.%.
значение молярного отношения между гидроксидными ионами, вводимыми с водным щелочным раствором, и количеством метилформиата, содержащегося в подаваемом сыром пропеноксиде, находится в интервале от 1,1 до 4.
реакцию в смеси проводят в трубчатом реакторе.
очищенный пропеноксид содержит меньше 50 мас.ч./млн метанола, меньше 50 мас.ч./млн ацетальдегида и меньше 100 мас.ч./млн метилформиата.
а) на реакционной стадии проводят реакцию пропена с водным пероксидом водорода в метаноле в присутствии силикалита титана как катализатора,
б) поток продуктов с этой реакционной стадии необязательно направляют на стадию сброса давления и
в) затем в предварительной испарительной колонне, обладающей меньше чем 20 теоретическими разделительными ступенями, поток продуктов разделяют на головной продукт, содержащий пропен, пропеноксид и метанол, и остаточный продукт, содержащий метанол и воду, причем от 20 до 60% от общего количества метанола, вводимого с потоком продуктов, удаляют с головным продуктом, а остальное остается в остаточном продукте,
г) головной продукт со стадии в) по меньшей мере частично конденсируют, а пропен и весь имеющийся пропан необязательно отпаривают с получением конденсата, содержащего пропеноксид, больше 1 мас.% метанола и больше 200 мас.ч./млн ацетальдегида,
д) конденсат со стадии г) подвергают экстрактивной дистилляции в соответствии со способом по одному из предыдущих пунктов, благодаря чему получают остаточный продукт, содержащий метанол и экстракционный растворитель, и
е) весь или часть остаточного продукта со стадии в) после необязательного частичного удаления воды возвращают на реакционную стадию а).
остаточный продукт со стадии в) и остаточный продукт со стадии д) объединяют, объединенные продукты подвергают каталитической гидрогенизации и весь или часть полученного продукта после необязательного частичного удаления воды возвращают на реакционную стадию а).
| RU 2000105329, 10.03.2002 | |||
| ТУПАЯ КРЕСТОВИНА | 1925 |
|
SU4019A1 |
| US 5958192 А, 28.09.1999 | |||
| US 5116466 А, 26.05.1992 | |||
| Композиция для производства пористого заполнителя | 2016 |
|
RU2622060C1 |
| US 3149131 А, 15.09.1964 | |||
| DE 681866 А, 14.09.1939 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

