Настоящее изобретение относится к агонистам рецептора ноцицептина ORL-1 - 8-(бис-(галогенфенил)-метил)-3-гетероарил-8-азабицикло-[3.2.1]-октан-3-олам и его производным, используемых при лечении кашля, боли, состояния тревоги, астмы, алкогольной зависимости или депрессии. Также раскрыты фармацевтические композиции, включающие эти соединения и комбинации заявленных соединений с другими препаратами, предназначенными для симптоматического лечения кашля, аллергии или астмы.
8-(Бис-(галогенфенил)-метил)-3-гетероарил-8-азабицикло-[3.2.1]-октан-3-олы в целом, но не специфически, раскрыты в патенте США US 6262066 B1 и в международной заявке WO 01/07050, как используемые при лечении кашля, боли, состояния тревоги, астмы, алкогольной зависимости или депрессии. Соединения, соответствующие настоящему изобретению, представляют собой селективное изобретение по сравнению с патентом США US 6262066 B1 и WO 01/07050.
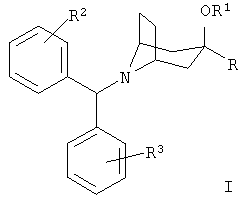
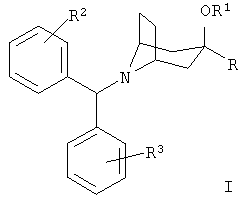
Соединения по настоящему изобретению представлены формулой I
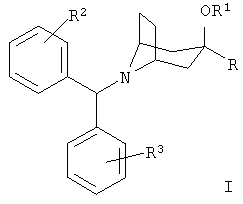
или ее фармацевтически приемлемыми солями, где:
R означает R4-гетероарил или 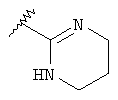 ;
;
R1 означает водород или алкил с 1-6 атомами углерода;
R2 и R3 независимо друг от друга выбирают из группы, включающей метил, метокси, фтор, хлор, бром и йод;
R4 означает от 1 до 4 заместителей, независимо выбранных из группы, включающей водород, галоген, алкил с 1-6 атомами углерода, циано, трифторметил, трифторметокси, -(CH2)n-OR5, -(CH2)n -NR5R6, -(CH2)n-NHSO2R5, -(CH2)n-NH(CH2)2NR5R6, -(CH2)n-NHC(O)NR5R7, -(CH2)n-NH(CH2)2OR5 и 1-пиперазинил;
n равно 0, 1, 2 или 3;
R5 и R6 независимо друг от друга выбирают из группы, включающей водород и алкил с 1-3 атомами углерода; и
R7 означает водород, алкил с 1-3 атомами углерода или аминоалкил с 1-3 атомами углерода.
В другом воплощении настоящее изобретение относится к фармацевтической композиции, включающей, по меньшей мере, одно соединение формулы I и фармацевтически приемлемый носитель.
Соединения, соответствующие настоящему изобретению, являются агонистами рецептора ORL-1 и поэтому в другом воплощении настоящее изобретение относится к способу лечения боли, состояния тревоги, кашля, астмы, алкогольной зависимости или депрессии, включающему назначение млекопитающему, нуждающемуся в таком лечении, эффективного количества, по меньшей мере, одного соединения формулы I.
В другом воплощении настоящее изобретение относится к способу лечения кашля, включающему назначение млекопитающему, нуждающемуся в таком лечении: (а) эффективного количества, по меньшей мере, одного соединения формулы I; и (b) эффективного количества одного или большего количества дополнительных препаратов, предназначенных для симптоматического лечения кашля, аллергии или астмы, выбранных из группы, включающей: антигистамины, ингибиторы 5-липоксигеназы, ингибиторы лейкотриена, ингибиторы Н3, агонисты β-адренергического рецептора, производные ксантина, агонисты α-адренергического рецептора, стабилизаторы тучных клеток, противокашлевые препараты, отхаркивающие препараты, антагонисты рецепторов тахикинина NK1, NK2 и NK3 и агонисты GABAB.
В еще одном воплощении настоящее изобретение относится к фармацевтической композиции, включающей, по меньшей мере, одно соединение формулы 1 и один или большее количество дополнительных препаратов, выбранных из группы, включающей: антигистамины, ингибиторы 5-липоксигеназы, ингибиторы лейкотриена, ингибиторы Н3, агонисты β-адренергического рецептора, производные ксантина, агонисты α-адренергического рецептора, стабилизаторы тучных клеток, противокашлевые препараты, отхаркивающие препараты, антагонисты рецепторов тахикинина NK1, NK2 и NK3 и агонисты GABAB.
Согласно приведенной выше формулы 1 предпочтительными соединениями по настоящему изобретению являются такие, в которых R2 и R3 находятся в 2-положении фенильных колец. Также предпочтительными являются соединения, в которых и для R2, и для R3 выбирают одинаковый атом галогена. Более предпочтительными являются соединения, в которых R2 означает хлор и R3 означает хлор, причем наиболее предпочтительными являются соединения, в которых R2 означает 2-хлор и R3 означает 2-хлор.
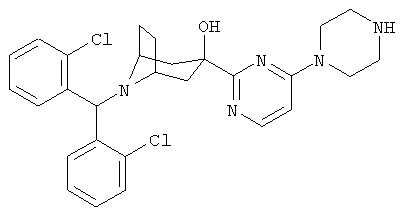
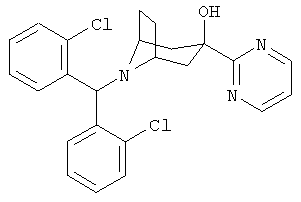
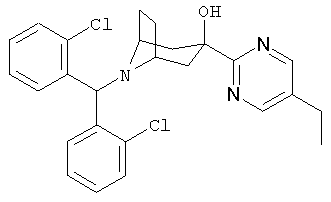
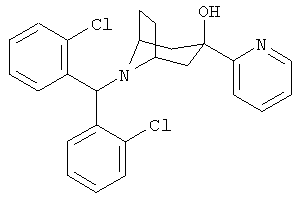
Также предпочтительными являются соединения, в которых R означает R4-гетероарил, где гетероарил означает пиридил, пиримидинил, пиразинил, пиридазинил, имидазолил, пиразолил или индолил, в частности, 2-пиридил или 2-пиримидинил. Предпочтительными значениями R4 является водород, алкил с 1-6 атомами углерода, -OR5 и 1-пиперазинил. Более предпочтительными значениями R являются 2-пиримидинил, 5-этил-2-пиримидинил, 4-(1-пиперазинил)-2-пиримидинил, 2-пиридил и 6-метокси-2-пиридил.
R1 предпочтительно означает водород или метил, а более предпочтительным является водород.
Особенно предпочтительными являются следующие конкретные соединения:
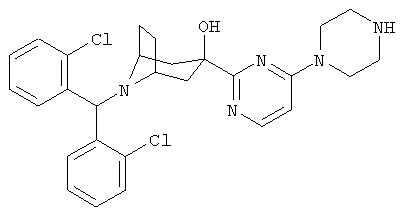 ,
, 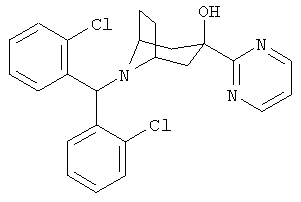 ,
, 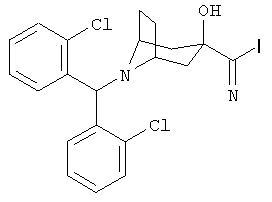 .
.
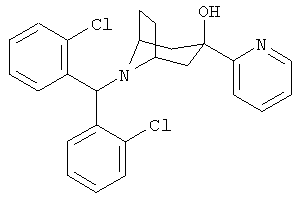 и
и 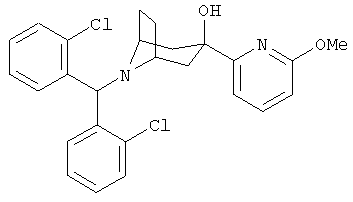
Предпочтительным для соединений формулы I является их использование для лечения кашля.
Если не указано иного, то при использовании в настоящем изобретении следующие термины имеют указанные ниже значения:
Галоген означает фтор, хлор, бром и йод.
Гетероарил означает циклические ароматические группы, содержащие от 5 до 6 атомов, или бициклические группы, содержащие от 9 до 10 атомов, включающие 1, 2 или 3 гетероатома, независимо выбранные из группы, включающей кислород, серу и азот, указанный гетероатом (указанные гетероатомы) входит в карбоциклическую кольцевую структуру и имеет количество делокализованных π-электронов, достаточным для придания ароматического характера, при условии, что циклы не включают соседних атомов кислорода и/или серы. Атомы азота могут образовать N-оксид. Включаются все региоизомеры, например, 2-пиридил, 3-пиридил и 4-пиридил. Типичными 6-членными гетероарильными группами являются пиридил, пиримидинил, пиразинил, пиридазинил и их N-оксиды. Типичными 5-членными гетероарильными циклами являются фурил, тиенил, пирролил, тиазолил, изотиазолил, имидазолил, пиразолил и изоксазолил. Типичными бициклическими группами являются сконденсированные с бензольным циклом системы, образованные из указанных выше гетероарильных групп, например, хинолил, фталазинил, хиназолинил, бензофуранил, бензотиенил и индолил. Гетероарильный цикл может в качестве заместителей содержать 1-4 группы R4, причем любые доступные и способные к замещению атомы углерода или азота в указанной гетероарильной группе могут быть необязательно и независимо замещены.
Некоторые соединения по настоящему изобретению могут существовать в различных стереоизомерных формах (например, энантиомерных, диастереоизомерных и атропомерных). Настоящее изобретение включает все такие стереоизомеры и в чистом виде и в смеси, включая рацемические смеси.
Некоторые соединения по природе являются кислыми (например, соединения, в которых содержится карбоксильная или фенольная гидроксильная группа). Эти соединения могут образовывать фармацевтически приемлемые соли. Примеры таких солей могут включать соли натрия, калия, кальция, алюминия, золота и серебра. Также включаются соли, образующиеся с фармацевтически приемлемыми аминами, такими как аммиак, алкиламины, гидроксиалкиламины, N-метилглюкамин и т.п.
Некоторые основные соединения также образуют фармацевтически приемлемые соли, например, кислотно-аддитивные соли. Например, пиридиновые атомы азота могут образовывать соли с сильными кислотами, тогда как соединения, содержащие основные заместители, такие как аминогруппы, также образуют соли с более слабыми кислотами. Примерами кислот, подходящих для образования солей, являются хлористоводородная, серная, фосфорная, уксусная, лимонная, щавелевая, малоновая, салициловая, яблочная, фумаровая, янтарная, аскорбиновая, малеиновая, метансульфоновая и другие минеральные и карбоновые кислоты, хорошо известные специалистам в данной области техники. Соли получают путем взаимодействия свободного основания с достаточным количеством необходимой кислоты с образованием соли. Свободное основание можно выделить путем обработки соли подходящим разбавленным водным раствором основания, таким как разбавленный водный раствор NaOH, карбоната калия, аммиака и бикарбоната натрия. Свободное основание отличается от соответствующей соли по некоторым физическим характеристикам, таким как растворимость в полярных растворителях, однако для задач настоящего изобретения по остальным характеристикам соль эквивалентна соответствующему свободному основанию.
В соответствии с объемом настоящего изобретения предполагается, что все такие кислые и основные соли являются фармацевтически приемлемыми и для задач настоящего изобретения все кислые и основные соли считаются эквивалентными свободным формам соответствующих соединений.
Соединения по настоящему изобретению могут быть получены по известным методикам из исходных веществ или известных в данной области техники, или получаемых по методикам, известным в данной области техники.
Типичная методика получения соединений формулы Ia, в которой R1 означает водород, включает взаимодействие 8-[бис-(галогенфенил)-метил]-8-азабицикло-[3.2.1]-октан-3-она формулы II с литиевым производным гетероарила:
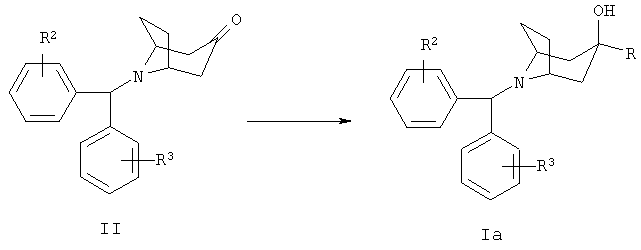
Исходное вещество формулы II может быть получено по следующей реакционной схеме:
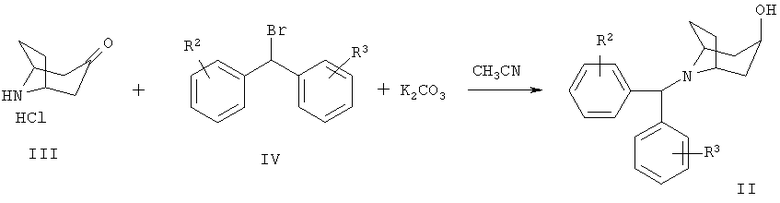
Соединение формулы II может быть получено алкилированием пиперидинового производного III производным дифенилбромметана IV в присутствии основания, такого как К2СО3, в растворителе, таком как СН3CN, при 80°С. Соединения формул III и IV известны в данной области техники или могут быть получены по известным методикам.
Соединения по настоящему изобретению и исходные вещества для их получения, приведенные ниже в качестве примеров, не следует рассматривать, как ограничивающие объем настоящего раскрытия.
Следующие растворители и реагенты в настоящем изобретении обозначены указанными аббревиатурами: тетрагидрофуран (THF); этанол (EtOH); метанол (МеОН); этилацетат (EtOAc); диизопропиламид лития (LDA); триэтиламин (Et3N) и N,N-диметилформамид (DMF).
Приготовление 1
8-Азабицикло-[3.2.1]-октан-3-он, гидрохлорид
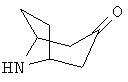
К раствору тропинона (10 г, 71,84 ммоль) в дихлорэтане (200 мл) по каплям при 0°С прибавляют α-хлорэтилхлорформиат (15,4 г, 108 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 2 часов. Растворитель выпаривают и получают коричневый остаток. Остаток растворяют в МеОН (200 мл) и кипятят с обратным холодильником в течение 2 часов. МеОН выпаривают и твердое вещество перемешивают в EtOAc, фильтруют, собирают твердое вещество и промывают эфиром и получают продукт (7 г). Неочищенный продукт используют без дополнительной очистки. 1H ЯМР (CDCl3) δ 4,45 (s, br, 2H), 3,35 (dd, 2H), 2,58 (d, 2H), 2,49 (dd, 2H), 2,0 (m, 2H).
Приготовление 2
Бис-(2-хлорфенил)-бромметан
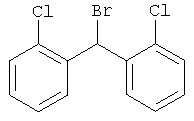
Стадия 1:
К раствору 2,2'-дихлорбензофенона (5 г, 19,9 ммоль) в МеОН (40 мл) при комнатной температуре прибавляют NaBH4 (1,5 г, 39,82 ммоль) и перемешивают в течение 2 часов. Реакцию останавливают с помощью H2O, нейтрализуют с помощью 1 N HCl и удаляют МеОН. Остаток экстрагируют с помощью EtOAc, промывают рассолом, сушат над MgSO4 и концентрируют и получают искомое соединение (5 г) в виде белого твердого вещества, которое используют на следующей стадии реакции без очистки. 1H ЯМР (CDCl3) δ 7,45 (m, 4H), 7,35 (m, 4H), 6,60 (d, 1H), 2,58 (d, 1H, ОН).
Стадия 2:
Продукт, полученный на стадии 1 (20,36 г, 80,47 ммоль), в CH2Cl2 обрабатывают с помощью SOBr2 (30,11 г, 144,85 ммоль) при 0°С и перемешивают при комнатной температуре в течение ночи. Реакцию останавливают с помощью льда и NaHCO3 (водный раствор), экстрагируют с помощью CH2Cl2, сушат и фильтруют. Удаляют растворитель и получают искомый бромид (23,6 г). 1H ЯМР (CDCl3) δ 7,6 (d, 2H), 7,4 (d, 2H), 7,13 (m, 4H), 7,0 (s, 1H).
Приготовление 3
8-[Бис-(2-хлорфенил)-метил]-8-азабицикло-[3.2.1]-октан-3-он
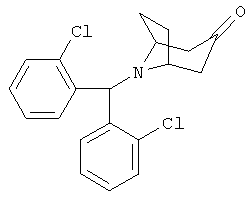
Смесь продуктов, полученных в Синтезе 1 (26 г, 161 ммоль) и в Приготовлении 2 (53 г, 168 ммоль), и K2СО3 (110 г, 796 ммоль) нагревают в безводном СН3CN (410 мл) при 80°С в течение 80 часов. Реакционную смесь охлаждают до комнатной температуры и фильтруют. Растворитель выпаривают и твердое вещество очищают с помощью флэшхроматографии на колонке (4%, 7% EtOAc/гексан) и получают искомый продукт.1H ЯМР (CDCl3) δ 7,9 (d, 2Н), 7,3 (m, 4H), 7,2 (m, 2H), 5,7 (s, 1H), 3,35 (s, br, 2H), 2,7 (dd, 2H), 2,3 (m, 2H), 2,2 (d, 2H), 1,65 (dd, 2H).
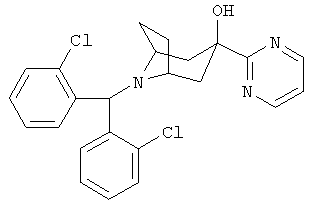
Пример 1
8-[Бис-(2-хлорфенил)-метил]-3-(2-пиримидинил)-8-азабицикло-[3.2.1]-октан-3-ол
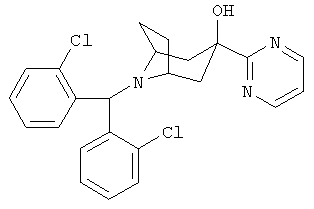
Стадия 1: 2-Трибутилстаннилпиримидин
Это соединение получают по методике, описанной в работе Sandosham et al., Tetrahedron (1994), 50, 275-284). Из диизопропиламина (25 мл, 178 ммоль) и n-BuLi (2,5 M, 70 мл, 175 ммоль) в THF (230 мл) готовят свежий LDA. Раствор LDA по каплям при 0°С обрабатывают раствором трибутиллитийгидрида (142 мл, 156 ммоль) в THF (30 мл) и после завершения прибавления перемешивают в течение еще 15 минут. Реакционную смесь охлаждают до -78°С, по каплям прибавляют раствор 2-хлорпиримидина (15 г, 131 ммоль) в THF (100 мл) и перемешивают реакционную смесь в течение 3 часов при -78°С, затем реакционной смеси дают нагреться до 0°С в течение 30 минут. Реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомое соединение в виде светло-желтого масла. 1H ЯМР (CDCl3) δ 8,65 (d, 2H), 7,1 (t, 1H), 1,6 (m, 6H), 1,3 (m, 6H), 1,1 (m, 6H), 0.85 (t, 9H).
Стадия 2:
n-BuLi (2,5 M в гексане, 16,5 мл, 41,2 ммоль) по каплям прибавляют к раствору продукта, полученного на стадии 1 (15 г, 40,6 ммоль), в THF (80 мл) при -78°С и реакционную смесь выдерживают при этой температуре в течение 45 минут. К этому раствору по каплям прибавляют раствор продукта, полученного в Приготовлении 3 (6 г, 16,7 ммоль), в THF (30 мл) и перемешивают реакционную смесь в течение еще 3 часов при -78°С. Реакционную смесь нагревают до комнатной температуры в течение 1,5 часов. Реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют.Очистка остатка с помощью хроматографии на колонке дает искомое соединение в виде легкого белого твердого вещества. 1Н ЯМР (CDCl3) δ 8,75 (d, 2H), 7,96 (d, 2H), 7,30 (m, 4H), 7,20 (t, 1H), 7,15 (m, 2H), 5,59 (s, 1H), 4,86 (s, 1H, ОН), 3,20 (m, br, 2H), 2,60 (dd, 2H), 2,40 (dd, 2H), 2,24 (m, 2H), 1,68 (d, 2H).
Пример 2
8-[Бис-(2-хлорфенил)-метил]-3-(5-этил-2-пиримидинил)-8-азабицикло-[3.2.1]-октан-3-ол
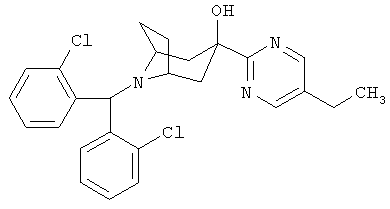
Стадия 1: 5-Этил-2-трибутилстаннилпиримидин
С использованием методики, описанной в Примере 1, стадия 1, с использованием LDA, трибутилгидрида олова (23,8 г, 81,78 ммоль) и 2-хлор-5-этилпиримидина (10 г, 70 ммоль)) получают искомое соединение (6 г). 1H ЯМР (CDCl3) δ 8,55 (s, 2H), 2,60 (q, 2H), 1,55 (m, 6H), 1,35 (m, 6H), 1,25 (t, 3H), 1,15 (t, 6H), 0,85 (t, 9H).
Стадия 2:
n-BuLi (2,5 M, 6,5 мл, 16,33 ммоль) по каплям прибавляют к раствору продукта, полученного на стадии 1 (5,9 г, 14,85 ммоль), в THF при -78°С температуру реакционной смеси поддерживают равной -78°С в течение 30 минут. К ней прибавляют продукт, полученный в Приготовлении 3 (5,34 г, 14,85 ммоль). Реакционную смесь медленно нагревают до комнатной температуры и перемешивают при комнатной температуре в течение ночи. Реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют. Очистка остатка с помощью хроматографии на колонке дает искомое соединение в виде белого твердого вещества. 1H ЯМР (CDCl3) δ 8,6 (s, 2Н), 8,0 (d, 2Н), 7,25 (m, 4Н), 7,15 (m, 2H), 5,6 (s, 1H), 4,85 (s, 1H, ОН), 3,2 (s, br, 2Н), 2,65 (q, 2Н), 2,60 (d, 2Н), 2,40 (m, 2Н), 2,25 (m, 2Н), 1,65 (d,2H), 1,30 (t, 3H).
Пример 3
8-[Бис-(2-хлорфенил)-метил]-3-[4-(1-пиперазинил)-2-пиримидинил]-8-аза-бицикло-[3.2.1]-октан-3-ол
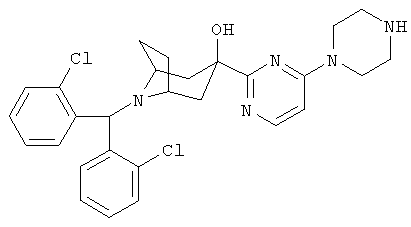
Стадия 1: 4-Хлор-2-трибутилстаннилпиримидин
С использованием методики, описанной в Примере 1, стадия 1, с использованием LDA, трибутилгидрида олова (10,8 г, 37,2 ммоль) и 2,4-дихлорпиримидина (5,2 г, 34,9 ммоль)) получают искомое соединение (6,3 г). 1H ЯМР (CDCl3) δ 8,52 (d, 1H), 7,18 (d, 1H), 1,58 (m, 6H), 1,30 (q, 6H), 1,18 (t, 6H), 0,86 (t, 9H).
Стадия 2: 8-[Бис-(2-хлорфенил)-метил]-3-(4-хлор-2-пиримидинил)-8-азабицикло-[3.2.1]-октан-3-ол
n-BuLi (2,5 M, 8,0 мл, 20,0 ммоль) по каплям прибавляют к раствору продукта, полученного на стадии 1 (6,3 г, 16,2 ммоль), в THF (30 мл) при -78°С и реакционную смесь выдерживают при этой температуре в течение 30 минут. К ней прибавляют продукт, полученный в Приготовлении 3 (4,0 г, 11,1 ммоль). Реакционную смесь медленно нагревают до комнатной температуры и перемешивают при комнатной температуре в течение ночи. Реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют. Очистка остатка с помощью хроматографии на колонке дает искомое соединение в виде светло-коричневого вспененного вещества. 1H ЯМР (CDCl3) δ 8,61(d, 1Н), 7,93 (d, 2Н), 7,25 (m, 5H), 7,12 (m, 2H), 5,65 (s, 1Н), 4,33 (s, 1Н, ОН), 3,18 (s, br, 2H), 2,58 (dd, 2H), 2,33 (m, 2H), 2,13 (m,2H), 1,65(d,br,2H).
Стадия 3:
К раствору продукта, полученного на стадии 2 (25 мг, 0,05 ммоль), в EtOH (4 мл) при комнатной температуре прибавляют пиперазин (20 мг, 0,23 ммоль). Реакционную смесь перемешивают при 80°С в течение ночи. Экстракция и очистка дает искомое соединение (20 мг). 1H ЯМР (CDCl3) δ 8,24 (d, 1Н), 7,93 (d, 2H), 7,26 (d, 2H), 7,22 (t, 2H), 7,10 (t, 2H), 6,33 (d, 1Н), 5,64 (s, 1Н), 3,67 (s, br, 4H), 3,15 (s, br, 2H), 2,95 (m, 4H), 2,59 (dd, 2H), 2,34 (m, 2H), 2,17 (m, 2H), 1,57 (d, br, 2H).
Пример 4
8-[Бис-(2-хлорфенил)-метил]-3-(2-пиридинил)-8-азабицикло-[3.2.1]-октан-3-ол
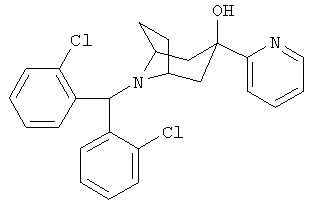
n-BuLi (2,5 М в гексане, 1,5 мл, 3,8 ммоль) по каплям прибавляют к раствору 2-бромпиридина (0,50 г, 3,10 ммоль) в THF (1 мл) при -78°С и перемешивают в течение 1 часа. К нему прибавляют раствор продукта, полученного в Приготовлении 3 (0,5 г, 1,4 ммоль) в THF (1,5 мл) по каплям и перемешивают реакционную смесь в течение еще 3,5 часов при -78°С. Реакционную смесь нагревают до 0°С в течение 1 часа, выливают реакционную смесь в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют. Очистка остатка с помощью хроматографии на колонке дает искомое соединение в виде бледно-желтого твердого вещества (400 мг). 1H ЯМР (CDCl3) δ 8,49 (d, 1H), 7,92 (d, 2H), 7,76 (t, 1H), 7,61 (d, 1H), 7,28 (m, 4H), 7,16 (m, 3H), 5,65 (s, 1H), 5,54 (s, 1H, ОН), 3,18 (s, br, 2H), 2,41 (m, 2H), 2,32 (dd, 2H), 2,21 (m, 2H), 1,72 (d, br, 2H).
Пример 5
8-[Бис-(2-хлорфенил)-метил]-3-(6-метокси-2-пиридинил)-8-азабицикло-[3.2.1]-октан-3-ол
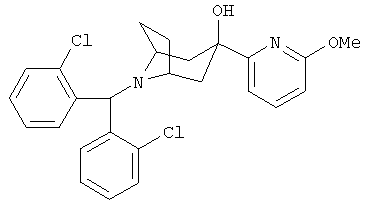
n-BuLi (2,5 М в гексане, 1,5 мл, 3,8 ммоль) по каплям прибавляют к раствору 2-бром-6-метоксипиридина (700 мг, 3,7 ммоль) в THF (2 мл) при -78°С и перемешивают в течение 0,5 часа. К нему по каплям прибавляют раствор продукта, полученного в Приготовлении 3 (600 мг, 1,7 ммоль), в THF (3 мл) и перемешивают реакционную смесь в течение еще 1 часа при -78°С. Реакционную смесь нагревают до 0°С в течение 2,5 часов. Реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют. Очистка остатка с помощью хроматографии на колонке дает искомое соединение (0,5 г).). 1H ЯМР (CDCl3) δ 7,90 (d, 2Н), 7,65 (t, 1H), 7,31 (d, 2Н), 7,26 (t, 2Н), 7,13 (m, 3H), 6,63 (d, 1H), 5,64 (s, 1H), 5,15 (s, 1H, ОН), 3,96 (s, 3H), 3,17 (s, br, 2Н), 2,33 (m, 4H), 2,21 (m, 2Н), 1,74 (d, br, 2Н).
Пример 6
8-[Бис-(2-хлорфенил)-метил]-3-метокси-3-(2-пиримидинил)-8-азабицикло-[3.2.1]-октан
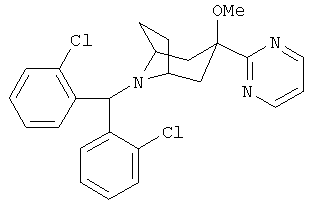
Продукт, полученный в Примере 1 (300 мг, 0,68 ммоль), в THF (3 мл) и DMF (1 мл) обрабатывают с помощью NaH (30 мг, 0,75 ммоль) при 0°С в течение 30 минут. Прибавляют СН3l и реакционную смесь нагревают до комнатной температуры. После перемешивания в течение ночи реакцию останавливают с помощью H2O, экстрагируют с помощью EtOAc, промывают рассолом, сушат и концентрируют. Полученный остаток очищают с помощью хроматографии на колонке и получают искомое соединение (0,25, г). 1H ЯМР (CDCl3) δ 8,77 (d, 2H), 7,83 (d, 2H), 7,27 (d, 2H), 7,18 (m, 3Н), 7,10 (t, 2H), 5,54 (s, 1H), 3,15 (s, br, 2H), 2,99 (s, 3Н), 2,38 (dd, 2H), 2,12 (m, 6H),
Пример 7
8-[Бис-(2-хлорсренил)-метил]-3-(1Н-пиразол-5-ил)-8-азабицикло-[3.2.1]-октан-3-ол
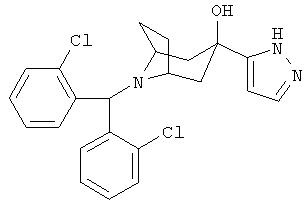
К пиразолу (0,68 г, 10 ммоль) в воде (4 мл) при комнатной температуре прибавляют формальдегид (37 мас.%, 1,5 мл, 50 ммоль), перемешивают при комнатной температуре в течение ночи. Экстрагируют с помощью CH2Cl2, сушат (Na2SO4) и концентрируют и получают 1-гидроксиметилпиразол. К раствору 1-гидроксиметилпиразота (129 мг, 1,31 ммоль) в THF (2 мл) при -78°С прибавляют свежеприготовленный LDA (2,63 ммоль) в THF, перемешивают при -20°С в течение 40 минут и охлаждают до -78°С. К нему по каплям прибавляют раствор продукта, полученного в Приготовлении 3 (236 мг, 0,65 ммоль), в THF (3 мл) и перемешивают реакционную смесь в течение еще 2 часов при -78°С. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи. Реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют эфиром. Органические слои объединяют, сушат, фильтруют и концентрируют. Очистка остатка с помощью препаративной тонкослойной хроматографии и ВЭЖХ дает искомое соединение (25 мг). 1H ЯМР (CDCl3) δ 8,2 (s, br, 2H), 8,05 (d, 2H), 7,25-7,40 (m, 6H), 7,20 (t, 2H), 6,2 (s, br, 1H), 5,9 (s, 1H), 3,2 (s, br, 2H), 2,55 (d, 2H), 2,41 (dd, 2H), 2,3 (m, 2H), 1,95 (d, 2H).
Пример 8
8-[Бис(2-хлорфенил)-метил]-3-(1-метил-пиразол-5-ил)-8-азабицикло-[3.2.1]-октан-3-ол
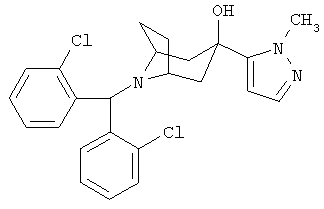
К раствору продукта, полученного в Примере 8 (70 мг, 0,164 ммоль), в THF при 0°С прибавляют NaH (9,84 мг, 0,246 ммоль) и перемешивают в течение 30 минут. Прибавляют СН3I (34,89 мг, 0,246 ммоль), нагревают до комнатной температуры и перемешивают в течение ночи. Реакцию останавливают с помощью насыщенного водного раствора NH4Cl, экстрагируют с помощью EtOAc, сушат (Na2SO4), фильтруют и концентрируют. Очистка остатка с помощью препаративной тонкослойной хроматографии дает искомое соединение (51 мг). 1H ЯМР (CDCl3) δ 7,85 (d, 2H), 7,3 (m, 6H), 7,15 (t, 2H), 6,21 (s, 1H), 5,6 (s, 1H), 3,85 (s, 3Н), 3,15 (s, br, 2H), 2,6 (s, 1H), 2,2-2,4 (m, 6H), 1,85(d,2H).
Пример 9
8-[Бис(2-хлорфенил)-метил]-3-(1-метил-1Н-индол-2-ил)-8-азабицикло-[3.2.1]-октан-3-ол
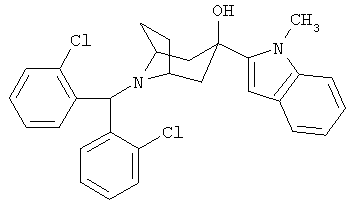
n-BuLi (1,6 М в гексане, 0,32 мл, 0,51 ммоль) по каплям прибавляют к раствору 1-метилиндола (67 мг, 0,51 ммоль) в THF (2 мл) при -20°С, нагревают до комнатной температуры, перемешивают в течение 3,5 часов и охлаждают до -78°С. К нему прибавляют раствор продукта, полученного в Синтезе 3 (92 мг, 0,26 ммоль), в THF (2 мл). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 1,5 часов. Реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют. Очистка остатка с помощью препаративной тонкослойной хроматографии дает искомое соединение (5 мг). 1H ЯМР (CDCl3) δ 7,80 (d, 2Н), 7,60 (d, 1H), 7,05-7,35 (m, 9H), 6,45 (s, 1H), 5,55 (s, 1H), 3,20 (s, br, 2H), 2,55 (dd, 2H), 2,15 (br, s, 4H), 2,1 (d, 2H).
Пример 10
8-[Бис(2-хлорфенил)-метил]-3-(1-метил-1Н-имидазол-2-ил)-8-азабицикло-[3.2.1]-октан-3-ол
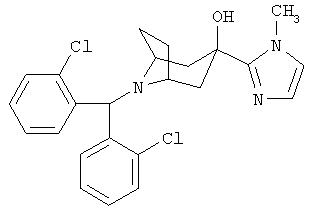
n-BuLi (2,5 М в гексане, 0,60 мл, 1,50 ммоль) по каплям прибавляют к раствору 1-метилимидазола (0,15 г, 1,88 ммоль) в THF (2 мл) при -78°С и перемешивают в течение 1,5 часов. К нему по каплям прибавляют раствор продукта, полученного в Приготовлении 3 (0,20 г, 0,55 ммоль), в THF (2 мл) и реакционную смесь перемешивают в течение еще 2 часов при -78°С. Реакционную смесь нагревают до комнатной температуры в течение в течение ночи, реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют. Очистка остатка с помощью хроматографии на колонке дает искомое соединение в виде бледно-желтого твердого вещества (80 мг). 1H ЯМР (CDCl3) δ 7,79 (d, 2Н), 7,27 (d, 2H), 7,18 (t, 2H), 7,10 (t, 2H), 6,63 (d, 2H), 5,48 (s, 1H), 3,74 (s, 3H), 3,08 (br s, 2H), 2,45 (d, 2H),2,14(m,4H), 1,81 (d, 2H).
Пример 11
8-[Бис(2-хлорфенил)-метил]-3-(3-пиридазинил)-8-азабицикло-[3.2.1]-октан-3-ол
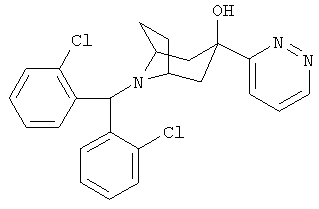
n-BuLi (2,5 М в гексане, 4,8 мл, 12,0 ммоль) по каплям прибавляют к раствору 2,2,6,6-тетраметилпиперидина (1,67 г, 11,9 ммоль) в THF (40 мл) при -78°С и перемешивают в течение 0,5 часа. Реакционную смесь нагревают до 0°С в течение 0,5 часа. Реакционную смесь охлаждают до -78°С, по каплям прибавляют раствор пиридазина (0,94 г, 11,7 ммоль) в THF (5 мл) и перемешивают реакционную смесь в течение 15 минут при -78°С. К ней по каплям прибавляют раствор продукта, полученного в Приготовлении 3 (1,0 г, 2,8 ммоль), в THF (5 мл) и перемешивают реакционную смесь в течение еще 1 часа при -78°С. Реакционную смесь нагревают до комнатной температуры в течение ночи. Реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют. Очистка остатка с помощью хроматографии на колонке дает искомое соединение (300 мг). 1H ЯМР (CDCl3) δ 9,10 (dd, 1Н), 7,87 (d, 2H), 7,81 (dd, 1H), 7,53 (dd, 1H), 7,29 (d, 2H), 7,26 (t, 2H), 7,14 (t, 2H), 5,62 (s, 1H), 4,71 (br s, 1H), 3,20 (br s, 2H), 2,38 (m, 4H), 2,23 (m, 2H), 1,80 (d, 2H).
Пример 12
8-[Бис(2-хлорфенил)-метил]-3-(2-пиразинил)-8-азабицикло[3.2.1]октан-3-ол
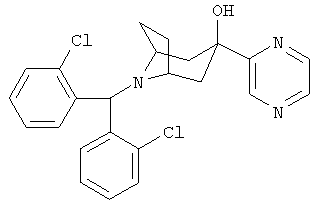
t-BuLi (1,7 M в пентане, 6,0 мл, 10,2 ммоль) по каплям прибавляют к раствору йодпиразина (1,0 г, 4,9 ммоль) в диэтиловом эфире (20 мл) при -50°С и перемешивают в течение 0,5 часа. К нему по каплям прибавляют раствор продукта, полученного в Приготовлении 3 (1,0 г, 2,8 ммоль), в THF (4 мл) и перемешивают реакционную смесь в течение еще 1,5 часов при -50°С. Реакционную смесь нагревают до комнатной температуры в течение в течение ночи. Реакционную смесь выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью EtOAc. Органические слои объединяют, сушат и концентрируют. Очистка остатка с помощью хроматографии на колонке дает искомое соединение (400 мг). 1H ЯМР (CDCl3) δ 8,96 (s, 1Н), 8,47 (m, 2H), 7,89 (d, 2H), 7,29 (d, 2H), 7,27 (t, 2H), 7,14 (t, 2H), 5,63 (s, 1Н), 4,34 (s, 1Н), 3,20 (br s, 2H), 2,37 (m, 4H), 2,22 (m, 2H), 1,76 (d, 2H).
Пример 13
8-[Бис(2-хлорфенил)-метил]-3-(4-пиримидинил)-8-азабицикло[3.2.1]октан-3-ол
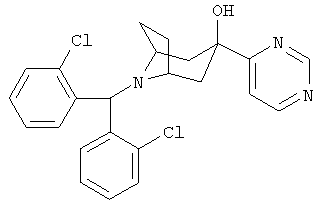
Стадия 1: 8-[Бис(2-хлорфенил)метил]-3-(5-бром-4-пиримидинил)-8-азабицикло-[3.2.1]октан-3-ол
К раствору 5-бромпиридина (450 мг, 2,77 ммоль) и продукта, полученного в Приготовлении 3 (1 г, 2,77 ммоль), в THF (5 мл) по каплям прибавляют предварительно охлажденный (с помощью твердой углекислоты), свежеприготовленный LDA (2,77 ммоль) в THF (5 мл) и перемешивают при комнатной температуре в течение ночи. Реакцию останавливают с помощью льда-Н2O, экстрагируют с помощью EtOAc, сушат, фильтруют и концентрируют. Очистка остатка с помощью хроматографии на колонке дает искомое соединение (187 мг).
Стадия 2:
Продукт, полученный на стадии 1 (22 мг), в CH3ОН-EtOAc (1:1,10 мл) и NH3/CH3ОН (7 N, 1 мл) гидрируют в присутствии катализатора Линдлара при давлении 1 атм в течение 2 часов, фильтруют и концентрируют и получают искомое соединение. 1H ЯМР (CDCl3) δ 9,15 (s, 1H), 8,70 (d, 1H), 8,00 (m, 2H), 7,80 (d, 1H), 7,25 (m, 4H), 7,19 (t, 2H), 5,61 (s, 1H), 3,15 (br s, 2H), 2,50 (dd, 2H), 2,25 (m, 4H), 1,65 (d, 2H).
Пример 14
8-[Бис(2-хлорсренил)-метил]-3-(5-бром-2-пиридинил)-8-азабицикло-[3.2.1]-октан-3-ол
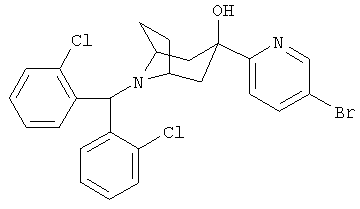
BuLi (1,6 M в гексане, 1,59 мл, 2,54 ммоль) прибавляют к 2,5-дибромпиридину (501 мг, 2,12 ммоль) в толуоле (13 мл) при -78°С и перемешивают в течение 2 часов. Прибавляют продукт, полученный в Приготовлении 3 (501 мг, 2,12 ммоль), в толуоле (2 мл) при -78°С и перемешивают в течение 3 часов. Нагревают до комнатной температуры, реакцию останавливают с помощью насыщенного водного раствора NH4Cl, экстрагируют с помощью CH3Cl2, сушат и концентрируют. Очистка остатка с помощью препаративной тонкослойной хроматографии и ВЭЖХ дает искомое соединение. 1H ЯМР (CDCl3) δ 8,59 (s, 1H), 7,85 (m, 3H), 7,50 (d, 1H), 7,25 (m, 4H), 7,19 (t, 2H), 5,61 (s, 1H), 4,85 (s, 1H), 3,20 (br s, 2H), 2,15-2,40 (m,4H), 1,75(d,2H).
Пример 15
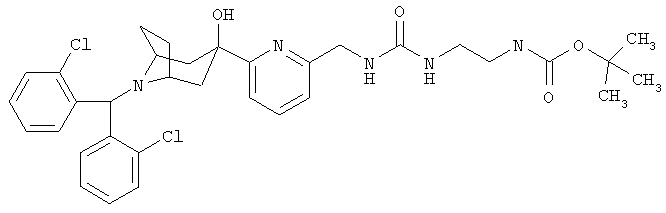
1,1-Диметилэтил-[2-[[[[[6-[8-[бис(2-хлорфенил)метил]-3-гидрокси-8-аза-би-цикло[3.2.1]-окт-3-ил]-2-пиридинил]метил]амино]карбонил]-амино]-этил]-карбамат
Стадия 1: 2-бром-6-гидроксиметилпиридин
NaBH4 (1,46 г, 38,58 ммоль) прибавляют к 6-бром-2-пиридин-карбоксилальдегиду (5,32 г, 28,58 ммоль) в CH3ОН при 0°С и перемешивают при 0°С в течение 1 часа, экстрагируют с помощью CH2Cl2, сушат над Na2SO4 и концентрируют и получают искомое соединение.
Стадия 2: 2-бром-6-(трет-бутилдиметилсилоксиметил)пиридин
К раствору продукта, полученного на стадии 1 (5,54 г, 29,46 ммоль), и трет-бутилдиметилсилилхлориду (4,97 г, 32,99 ммоль) в CH2Cl2 (60 мл) при комнатной температуре прибавляют имидазол (3,01 г, 44,19 ммоль) и перемешивают в течение ночи. Фильтруют реакционную смесь и фильтрат концентрируют. Остаток очищают с помощью хроматографии и получают искомое соединение.
Стадия 3: 8-[Бис(2-хлорфенил)-метил]-3-(6-(трет-бутилдиметилсилоксиметил)-2-пиридинил)-8-азабицикло-[3.2.1]-октан-3-ол
n-BuLi (1,6 М в гексане, 7,2 мл, 11,49 ммоль) прибавляют к продукту, полученному на стадии 2 (3,29 г, 10,88 ммоль), в THF (5 мл) при -78°С и перемешивают в течение 1 часа. Прибавляют продукт, полученный в Приготовлении 3 (1,84 г, 5,11 ммоль), в THF (14 мл) при -78°С медленно нагревают до 0°С (˜2 часа). Реакцию останавливают с помощью насыщенного водного раствора NH4Cl, экстрагируют с помощью EtOAc, сушат и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомое соединение.
Стадия 4: 8-[Бис(2-хлорфенил)-метил]-3-(6-гидроксиметил)-2-пиридинил)-8-азабицикло-[3.2.1]-октан-3-ол
К раствору продукта, полученного на стадии 3 (2,34 г, 4,01 ммоль), в THF (30 мл) при комнатной температуре прибавляют тетрабутиламмонийфторид (2,1 г, 8,04 ммоль) и перемешивают в течение ночи. Реакцию останавливают с помощью насыщенного водного раствора NaHCO3, экстрагируют с помощью EtOAc, сушат над Na2SO4 и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомое соединение.
Стадия 5: 3-[6-(Азидометил)-2-пиридинил]-8-[бис-(2-хлорфенил)-метил]-8-азабицикло-[3.2.1]-октан-3-ол
К продукту, полученному на стадии 4 (404 мг, 0,86 ммоль), при 0°С прибавляют дифенилфосфорилазид (272 мг, 0,99 ммоль) и 1,8-диазабицикло-[5,4,0]ундец-7-ен (150 мг, 0,99 ммоль), перемешивают в течение 20 минут, нагревают до комнатной температуры, затем перемешивают при 50°С в течение 1 часа. Охлаждают до комнатной температуры и перемешивают в течение ночи. Реакцию останавливают с помощью H2O и насыщенного водного раствора NH4Cl, экстрагируют с помощью CH2Cl2, сушат и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомое соединение.
Стадия 6: 3-[6-(Аминометил)-2-пиридинил]-8-[бис-(2-хлорфенил)-метил]-8-азабицикло-[3.2.1]-октан-3-ол
К суспензии продукта, полученного на стадии 5 (279 мг), в смеси EtOAc и CH3ОН в присутствии 7 N NH3 в CH3ОН (1 мл) прибавляют катализатор Линдлара (44 мг). Смесь гидрируют при 1 атм в течение 1,5 часов, фильтруют через целит, промывая с помощью NH3/CH3ОН (3,5 N,) и концентрируют и получают искомое соединение.
Стадия 7:
К раствору продукта, полученного на стадии 7 (157 мг, 0,335 ммоль), в толуоле (10 мл) при комнатной температуре в атмосфере аргона прибавляют трифосген (34,8 мг, 0,117 ммоль) и диизопропилэтиламин (222 мг, 1,675 ммоль). Нагревают до 120°С и перемешивают в течение 2,5 часов. Охлаждают до комнатной температуры, прибавляют N-Boc-этилендиамин (65 мг, 0,42 ммоль) и перемешивают в течение ночи. Реакцию останавливают с помощью насыщенного водного раствора NH4Cl, экстрагируют с помощью EtOAc, сушат над Nа2SO4 и концентрируют. Остаток очищают с помощью препаративной тонкослойной хроматографии и получают искомое соединение. 1H ЯМР (CDCl3) δ 7,9 (d, 2Н), 7,75 (t, 1H), 725 (d, 1H), 7,1-7,4 (m, 4H), 5,65 (s, 1H), 5,25 (b, s, 1H), 4,45 (d, 2Н), 3,25 (m, 2Н), 3,1 (m, 4H), 2,15-2,45 (m, 6H), 1,65 (d, 2Н).
Пример 16
N-(2-(Аминоэтил)-N'-[[6-[8-[бис-(2-хлорфенил)-метил]-3-гидрокси-8-азабицикло-[3.2.1]-окт-3-ил]-2-пиридинил]-метил]-мочевина
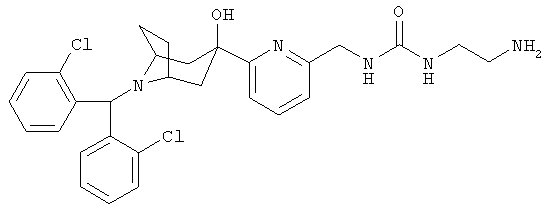
К раствору продукта, полученного в Примере 15 (53 мг), в CH2Cl2 и CH3ОН при комнатной температуре прибавляют HCl (1 N в эфире, 1,0 мл) и перемешивают, пока ЖХ-МС (жидкостная хроматография-масс-спектроскопия) не укажет на полное использование продукта, полученного в Примере 15, и получают искомое соединение в виде гидрохлорида. МС-ИЭР (масс-спектроскопия с ионизацией электрораспылением) 554,1 (100, M+).
Пример 17
3-[3-(Аминометил)-2-пиридинил]-8-[бис-(2-хлорфенил)-метил]-8-азабицик-ло-[3.2.1]-октан-3-ол
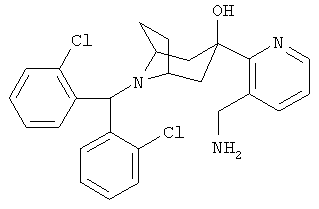
Стадия 1: 2-Бром-3-гидроксиметилпиридин
К раствору 2-бром-3-пиридинкарбоновой кислоты (5,63 г, 27,89 ммоль) и Et3N (2,96 г, 29,28 ммоль) в толуоле (150 мл) при комнатной температуре прибавляют этилхлорформиат (3,17 г, 29,28 ммоль) и перемешивают в течение 1 часа, фильтруют и концентрируют. Остаток растворяют в THF (93 мл), при -78°С по каплям прибавляют к суспензии LiAIH4 (1,11 г, 29,28 ммоль) в THF (37 ммоль) и перемешивают в течение 30 минут. Реакцию останавливают с помощью насыщенного водного раствора NH4Cl, перемешивают при комнатной температуре в течение 1 часа, фильтруют через целит, экстрагируют с помощью EtOAc, сушат над Na2SO4 и концентрируют. Очистка остатка с помощью хроматографии на колонке дает искомое соединение.
Стадия 2: 2-бром-3-(трет-бутилдиметилсилоксиметил)пиридин
По методике, описанной на стадии 2 Примера 15, с использованием 2-бром-3-гидроксиметилпиридина (3,66 г, 19,48 ммоль), трет-бутилдиметилсилилхлорида (5,87 г, 38,97 ммоль) и имидазола (3,31 г, 48,71 ммоль) получают искомое соединение (6,38 г).
Стадия 3: 8-[Бис(2-хлорфенил)-метил]-3-(3-(трет-бутилдиметилсилоксиметил)-2-пиридинил)-8-азабицикло-[3.2.1]-октан-3-ол
По методике, описанной на стадии 3 Примера 15, с использованием продукта, полученного на стадии 2 (6,38 г, 21,1 ммоль), n-BuLi (1,6 М в гексане, 14,5 мл, 21,1 ммоль) и продукта, полученного в Приготовлении 3 (7,60 г, 21,1 ммоль), получают искомый продукт.
Стадия 4: 8-[Бис-(2-хлорфенил)-метил]-3-(3-гидроксиметил)-2-пиридинил)-8-азабицикло-[3.2.1]-октан-3-ол
По методике, описанной на стадии 4 Примера 15, с использованием продукта, полученного на стадии 3 (12,3 г, 21,1 ммоль), и тетрабутиламмонийфторида (11 г, 42,2 ммоль) получают искомое соединение.
Стадия 5: 3-[3-(Азидометил)-2-пиридинил]-8-[бис(2-хлорфенил)-метил]-8-азабицикло-[3.2.1]-октан-3-ол
По методике, описанной на стадии 5 Примера 15, с использованием продукта, полученного на стадии 4 (95,2 мг, 0,213 ммоль), дифенилфосфорилазида (67,4 мг, 0,245 ммоль) и 1,8-диазабицикло-[5,4,0]-ундец-7-ена (52,96 мг, 0,32 ммоль) в качестве содержащегося в меньшем количестве продукта получают искомое соединение.
Стадия 6:
По методике, описанной на стадии 6 Примера 15, с использованием продукта, полученного на стадии 5 (69 мг) и катализатора Линдлара (7 мг) получают искомое соединение. 1H ЯМР (CDCl3) 8,40 (d, 1H), 7,95 (d, 2H), 775 (d, 1H), 7,05-7,15 (m, 7H), 5,60 (s, 1H), 5,25 (b, s, 1H), 4,40 (s, 2H), 3,20 (s, br, 2H), 2,50 (dd, 2H), 2,3 (m, 4H), 1,75 (d, 2H).
Пример 18
8-[Бис(2-хлорфенил)-метил]-3-[4-(метиламино)-2-пиридинил]-8-азабицикло-[3.2.1]-октан-3-ол
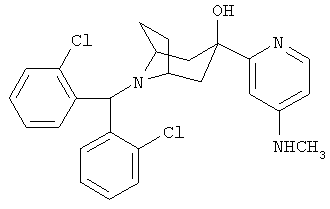
Стадия 1: 2-бром-4-(трет-бутоксикарбониламино)пиридин
Смесь 4-амино-2-бромпиридина (1,00 г, 5,79 ммоль), Et3N (1,75 г, 17,37 ммоль) и ди-трет-бутилдикарбоната (1,90 г, 8,69 ммоль) в CH2Cl2 (20 мл) перемешивают при комнатной температуре в течение ночи. Разбавляют с помощью CH2Cl2 (10 мл), промывают насыщенным водным раствором NaHCO3, сушат над MgSO4 и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомое соединение.
Стадия 2: - Диметилэтил-[2-[8-[бис(2-хлорфенил)-метил]-3-гидрокси-8-аза-бицикло-3.2.1]-окт-3-ил]-4-пиридинил]-карбамат
n-BuLi (1,6 М в гексане, 1,12 мл, 1,81 ммоль) прибавляют к продукту, полученному на стадии 1 (237 мг, 0,87 ммоль), в THF (2,7 мл) при -78°С и перемешивают в течение 2 часов. Прибавляют продукт, полученный в Синтезе 3 (337 мг, 0,94 ммоль), в THF (1 мл) при -78°С и перемешивают в течение 3 часов, нагревают до комнатной температуры и перемешивают в течение ночи. Реакцию останавливают с помощью насыщенного водного раствора NH4Cl, экстрагируют с помощью EtOAc, сушат и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомый продукт.
Стадия 3:
К раствору продукта, полученного на стадии 2 (48,4 мг, 0,087 ммоль), в диоксане (0,5 мл) при комнатной температуре прибавляют LiAlH4 (1 М в эфире, 0,26 мл, 0,26 ммоль) в диоксане (0,5 мл) и перемешивают при кипячении с обратным холодильником в течение ночи. Охлаждают до комнатной температуры, прибавляют LiAlH4 (1,0 М в эфире, 0,2 мл) и перемешивают при кипячении с обратным холодильником в течение 5 часов. Реакцию останавливают с помощью Н2О (0,05 мл), водного раствора NaOH (15%, 0,1 мл) и Н2О (0,05 мл). Разбавляют с помощью EtOAc, фильтруют и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомое соединение. 1H ЯМР (CDCl3) 8,10 (d, 1H), 7,95 (d, 2H), 7,05-7,15 (m, 6H), 6,75 (s, 1H), 6,39 (d, 2H), 5,70 (s, 1H), 3,20 (s, br, 2H), 2,95 (s, 3H), 2,35 (m, 4H), 2,2 (m, br, 2H), 1,65 (d, 2H).
Пример 19
3-[6-[(2-Аминоэтил)-амино]-2-пиридинил]-8-[бис-(2-хлорфенил)-метил]-8-азабицикло-[3.2.1]-октан-3-ол
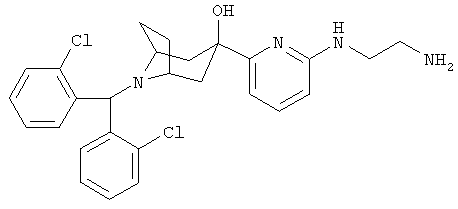
Стадия 1: 8-[Бис(2-хлорфенил)метил]-3-(6-бром-2-пиридинил)-8-азабицикло-[3.2.1]-октан-3-ол
n-BuLi (1,6 M в гексане, 26,8 мл, 42,92 ммоль) прибавляют к 2,6-дибромпиридину (12,2 г, 51,5 ммоль) в THF (150 мл) при -78°С и перемешивают в течение 2 часов. Прибавляют продукт, полученный в Синтезе 3 (9,28 г, 25,75 ммоль), в THF (50 мл) при -78°С и перемешивают в течение 3 часов, нагревают до комнатной температуры и перемешивают в течение ночи. Реакцию останавливают с помощью насыщенного водного раствора NH4Cl, экстрагируют с помощью EtOAc, сушат и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомый продукт.
Стадия 2: 1,1-Диметилэтил-[2-[6-[8-[бис(2-хлорфенил)-метил]-3-гидрокси-8-азабицикло-[3.2.1]-окт-3-ил]-2-пиридинил]-аминоэтил]-карбамат
Раствор продукта, полученного на стадии 1 (64,5 мг, 0,128 ммоль), N-Boc-этилендиамин (123 мг, 0,77 ммоль) и пиридин (12 мг, 0,154 ммоль) перемешивают при 110°С в запаянной трубке в течение 3,5 часов. Охлаждают до комнатной температуры, прибавляют N-Boc-этилендиамин (0,3 мл) и нагревают при 140°С в течение ночи. Охлаждают до комнатной температуры, реакцию останавливают с помощью H2O, экстрагируют с помощью EtOAc, сушат и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомый продукт.
Стадия 3:
К раствору продукта, полученного на стадии 2 (11 мг, 0,018 ммоль), в CH2Cl2 при комнатной температуре в течение 24 часов прибавляют HCl (1 N в эфире, 0,36 мл). Прибавляют HCl (1 N в эфире, 0,36 мл) и перемешивают при комнатной температуре в течение 24 часов. Прибавляют еще 0,36 мл HCl (1 N в эфире) и перемешивают при 30°С в течение 24 часов. Концентрируют, обрабатывают эфиром и фильтруют и получают искомое соединение в виде белого твердого вещества. МС-ИЭР 497,1 (100, М+).
Пример 20
8-[Бис-(2-хлорфенил)-метил]-3-(1,4,5,6-тетрагидро-2-пиримидинил)-8-азабицикло-[3.2.1]-октан-3-ол
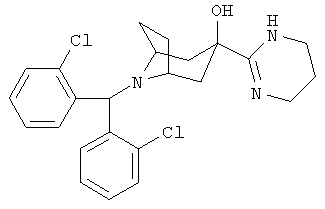
К раствору продукта, полученного в Примере 1 (160 мг), в этаноле (10 мл) при комнатной температуре прибавляют никель Ренея. Нагревают до 80°С и перемешивают в течение 20 часов, фильтруют и концентрируют. Остаток очищают с помощью хроматографии на колонке и получают искомое соединение. 1H ЯМР (CDCl3) 7,85 (d, 2Н), 7,25 (m, 4H), 7,15 (t, 2H), 5,55 (s, 1H), 3,40 (dd, 4H), 3,10 (s, br, 2H), 2,05-2,35 (m, 6H), 2,75 (q, 2H), 1,55 (d, 2H).
Соединения формулы I проявляют более чем 50-кратную селективность по отношению к классическим опиоидным рецепторам. Рецептор ORL-1 характеризуется высокой степенью гомологичности с классическими опиоидными рецепторами (т.е., μ, κ и δ), но рецептор ORL-1 не активируется эндогенными опиоидами и эндогенные опиоиды не активируют рецептор ORL-1. Известно, что кодеин и другие опиоиды, применяющиеся для подавления кашля, активируют опиоидный рецептор μ, приводя к побочным эффектам, таким как угнетение дыхания, запор, толерантность и соматическая зависимость. Агонисты рецептора ORL-1 не активируют опиоидный рецептор μ и поэтому предположительно приводят к превосходному профилю безопасности по сравнению с опиоидами.
Активность соединений формулы I, как агонистов рецептора ORL-1, и их влияние на кашель и дыхание можно количественно определить с помощью описанных ниже исследований.
Анализ связывания ноцицептина
Препарат мембран клеток яичника китайского хомячка, экспрессирующий рецептор ORL-1 (2 мг), инкубируют с [125I][Tyr14]ноцицептином в различных концентрациях (3-500 пМ) в буферном растворе, содержащем 50 mM HEPES (рН 7,4), 10 мМ NaCl, мМ MgCl2, 2,5 мМ CaCl2, 1 мг/мл бычьего сывороточного альбумина и 0,025% бацитрацина. В ряде исследований анализы проводят в буферном растворе, содержащем 50 мМ tris-HCl (рН 7,4), 1 мг/мл бычьего сывороточного альбумина и 0,025% бацитрацина. Образцы инкубируют в течение 1 часа при комнатной температуре (22°С). Меченые лиганды, связанные с мембраной, собирают на фильтрах GF/B, предварительно замоченных в 0,1% полиэтиленимине, с использованием прибора для сбора клеток Brandell и 5 раз промывают с помощью 5 мл холодной дистиллированной воды. Неспецифическое связывание определяют параллельно с помощью аналогичных анализов, проводимых в присутствии 1 мкМ ноцицептина. Анализы при каждых условиях выполняют дважды, определяя полное и неспецифическое связывание.
Расчет значений Ki проводят способами, хорошо известными в данной области техники.
Для соединений по настоящему изобретению полученные значения Ki находятся в диапазоне от 0,6 до 30 нМ, а предпочтительными являются соединения, обладающие значениями Ki, равными менее 10 нМ.
Значения Ki для некоторых типичных соединений, представлены в приведенной ниже Таблице:
С использованием методик, описанных в European Journal of Pharmacology, 336 (1997), p.233-242, определена активность соединений, соответствующих настоящему изобретению, как агонистов. Измеренная активность как агонистов (EC50) этих соединений находится в диапазоне 20-200 пМ.
Исследование кашля
Воздействие агониста ноцицептина оценено с помощью вызванного капсаицином кашля у морских свинок по методикам, предложенным Bolser et al. British Journal of Pharmacology (1995) 114, 735-738 (также см. McLeod et al., British Journal of Pharmacology (2001) 132,1175-1178). Эта модель является широко используемой методикой оценки активности возможных противокашлевых лекарственных препаратов. Голодавших в течение ночи самцов морских свинок Hartley (350-450 г, Charles River, Bloomington, MA, USA) помещают в прозрачные камеры размером 12×14 дюймов. Для создания кашлевого рефлекса животных подвергают воздействию аэрозоля капсаицина (300 мкМ, в течение 4 минут), полученного струйным распылителем (Puritan Bennett, Lenexa, KS, USA). На каждую морскую свинку воздействуют капсаицином только один раз. Количество кашлей регистрирует микрофоном, помещенный в камеру, и проверяет обученный наблюдатель. Сигнал от микрофона поступает на полиграф, который регистрирует количество кашлей. За 2 часа до воздействия аэрозолем капсаицина вводят или наполнитель (метилцеллюлоза, 1 мл/кг, перорально) или исследуемое соединений. В качестве положительного контроля также определяют противокашлевую активность баклофена (3 мг/кг, перорально).
Исследования дыхания
Исследования проводят на самцах морских свинок Hartley массой от 450 до 550 г. Животные голодают в течение ночи, но им в неограниченном количестве дают воду. Морских свинок головой наружу помещают в плетизмограф для регистрации изменений объема всего тела и на голову животного помещают резиновый воротник и получают герметичное уплотнение между морской свинкой и плетизмографом. Поток воздуха измеряют с помощью разности давлений на проволочной сетке, закрывающей 1-дюймовое отверстие в стенке плетизмографа. Сигнал потока воздуха интегрируется в сигнал, пропорциональный объему, с помощью предварительного усилителя и компьютера, регистрирующего функцию легких (Buxco Electronics, Sharon, СТ., model XA). К плетизмографу присоединяют головную камеру и в течение всего исследования через головную камеру циркулирует воздух, поступающий из источника сжатого воздуха (21% О2, остальное N2). Все исследования дыхания проводят с морскими свинками, вдыхающими этот циркулирующий воздух,
Сигнал об объеме, поступающий от каждого животного, направляют в систему накопления/анализа данных (Buxco Electronics, model XA), которая рассчитывает дыхательный объем и частоту дыхания для периодов между последовательными вдохами. Эти сигналы выводятся на экран монитора. Дыхательный объем и частота дыхания в виде средних значений регистрируются каждую минуту.
Морским свинкам дают успокоиться в плетизмографе в течение 30 минут. Измерения базовых значений проводят в конце этого 30-минутного периода. Затем морских свинок извлекают из плетизмографа и им перорально вводят исследуемое соединение (10 мг/кг, перорально), баклофен (3 мг/кг, перорально) или наполнитель метилцеллюлозу в качестве плацебо (2 мл/кг, перорально). Сразу же после введения морских свинок помещают в плетизмограф, повторно присоединяют головную камеру и циркулирующий воздух и через 30, 60, 90 и 120 минут после введения измеряют параметры дыхания (дыхательный объем (VT), частоту дыхания (f) и минутный объем (MV=VT×f)). Это исследование проводят по протоколу ACUC #960103.
В способах по настоящему изобретению можно вводить от одного до трех соединений формулы I, предпочтительно, одно соединение.
Соединения по настоящему изобретению обладают противокашлевой активностью, что делает их применимыми для подавления кашля у млекопитающих. Млекопитающим, которых лечат от кашля, по меньшей мере, один агонист ноцицептинового рецептора ORL-1 можно назначить совместно с одним или большим количеством дополнительных препаратов, предназначенных для симптоматического лечения кашля, аллергии или астмы, выбранных из группы, включающей антигистамины, ингибиторы 5-липоксигеназы, ингибиторы лейкотриена, ингибиторы Н3, агонисты β-адренергического рецептора, производные ксантина, агонисты α-адренергического рецептора, стабилизаторы тучных клеток, противокашлевые препараты, отхаркивающие препараты, антагонисты рецепторов тахикинина NK1, NK2 и NK3 и агонисты GABAB. Предпочтительно, чтобы комбинация по настоящему изобретению включала одно соединение формулы I и 1-3 дополнительных препарата, предпочтительно - 1-2 дополнительных препарата, а более предпочтительно - 1 дополнительный препарат.
Неограничивающие примеры антигистаминов включают: астемизол, азатадин, азеластин, акривастин, бромфенирамин, цертиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбэтоксиоратадин (также известный, как SCH-34117), доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокарбастин, мизоластин, эквитазин, миансерии, ноберастин, меклизин, норастемизол, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин.
Неограничивающие примеры антагонистов рецептора Н3 гистамина включают: тиоперамид, импромидин, буримамид, клобенпропит, импентамин, мифетидин, S-сопримидин, R-сопримидин, SKF-91486, GR-175737, GT-2016, UCL-1199 и клозапин. Другие соединения можно легко исследовать для определения активности по отношению к рецепторам Н3 с помощью известных способов, включая анализ мембран головного мозга морских свинок и анализ нервного сокращения подвздошной кишки морских свинок, которые описаны в патенте США US 5352707. В другом полезном способе анализа используются мембраны головного мозга крыс и он описан в работе West et al., "Identification of Two-H3-Histamine Receptor Subtypes," Molecular Pharmacology, Vol.38, pages 610-613 (1990).
Термин "ингибитор лейкотриена" включает любой препарат или соединение, которое ингибирует, ограничивает или другим образом влияет на действие или активность лейкотриенов. Неограничивающие примеры ингибиторов лейкотриена включают монтелукаст [R-(Е)]-1[[[1-[3-[2-(7-хлор-2-хинолинил)-этенил]фенил]-3[2-(1-гидрокси-1-метилэтил)фенил]пропил]-тио]-метил]-циклопропануксусную кислоту и ее натриевую соль, описанные в европейской заявке на патент ЕР 0480717; 1-(((R)-(3-(2-(6,7-дифтор-2-хинолинил)этенил)фенил)-3-(2-(2-гидрокси-2-пропил)-фенил)-тио)-метилциклопропануксусную кислоту и ее натриевую соль, описанные в международной заявке WO 97/28797 и патенте США US 5270324; 1-(((1(R)-3(3-(2-(2,3-дихлортиено-[3,2-b]-пиридин-5-ил)-(Е)-этенил)-фенил)-3-(2-(1-гидрокси-1-метилэтил)-фенил)пропил)-тио)-метил)-циклопропануксусную кислоту и ее натриевую соль, описанные в международной заявке WO 97/28797 и патенте США US 5472964; пранлукаст, N-[4-оксо-2-(1Н-тетразол-5-ил)-4Н-1-бензопиран-8-ил]-п-(4-фенилбутокси)-бензамид), описанный в международной заявке WO 97/28797 и европейской заявке на патент ЕР 173516; зафирлукаст, (циклопентил-3-[2-метокси-4-[(о-толилсульфонил)-карбамоил]-бензил]-1-метил-индол-5-карбамат), описанный в международной заявке WO 97/28797 и европейской заявке на патент ЕР 199543; и [2-[[2(4-трет-бутил-2-тиазолил)-5-бензофуранил]-окси-метил]-фенил]-уксусную кислоту, описанную в патенте США US 5296495 и японской патенте JP 08325265 А.
Термин "ингибитор 5-липоксигеназы" или "ингибитор 5-LO" включает любой препарат или соединение, которое ингибирует, замедляет или другим образом влияет на ферментативное действие 5-липоксигеназы. Неограничивающие примеры ингибиторов 5-липоксигеназы включают зилеутон, доцебенон, пирипост, ICl-D2318 и АВТ 761.
Неограничивающие примеры агонистов β-адренергического рецептора включают: албутерол, битолтерол, изоэтарин, матапротеренол, пербутерол, салметерол, тербуталин, изопротенерол, эфедрин и эпинефрин.
Неограничивающим примером производного ксантина является теофиллин.
Неограничивающие примеры агонистов α-адренергического рецептора включают арилалкиламины (например, фенилпропаноламин и псевдэфедрин), имидазолы (например, нафазолин, оксиметазолин, тетрагидрозолин и ксилометазолин) и циклоалкиламины (например, пропилгекседрин).
Неограничивающим примером стабилизатора тучных клеток является натриевая соль недокромила.
Неограничивающие примеры противокашлевых препаратов включают кодеин, декстрометорфан, бензонатат, клофедианол и носкарпин.
Неограничивающим примером отхаркивающего препарата является гвайфенезин.
Неограничивающие примеры антагонистов рецепторов тахикинина NK1, NK2 и NK3 включают СР-99,994 и SR 48968.
Неограничивающие примеры агонистов GABAB включают баклофен и 3-аминопропилфосфиновую кислоту.
При изготовлении фармацевтических композиций из соединений, описанных в настоящем изобретении, инертные, фармацевтически приемлемые носители могут быть твердыми или жидкими. К твердым формам препаратов относятся порошки, таблетки, диспергирующиеся гранулы, капсулы, облатки и суппозитории. Порошки и таблетки могут содержать от примерно 5 до примерно 70% активного компонента. Подходящие твердые носители известны в данной области техники и включают, например, карбонат магния, стеарат магния, тальк, сахар, лактозу. Таблетки, порошки, облатки и капсулы можно использовать в качестве твердых дозировочных форм, пригодных для перорального введения.
Для приготовления суппозиториев низкоплавкий воск, такой как смесь глицеридов жирных кислот или масло какао, сначала расплавляют и в ней равномерно диспергируют активный компонент, например, путем перемешивания. Затем расплавленную однородную смесь выливают в формы стандартного размера, дают ей охладиться и тем самым затвердеть.
Жидкие формы композиций включают растворы, суспензии и эмульсии. В качестве примера можно указать водные или водно-пропиленгликолевые растворы для парентеральных инъекций.
К жидким формам композиций также могут относиться растворы для внутриназального введения.
Аэрозольные композиции, пригодные для ингаляции, могут включать растворы и твердые вещества в порошкообразной форме, которые могут сочетаться с фармацевтически приемлемым носителем, таким как сжатый инертный газ.
В объем настоящего изобретения также включены твердые формы композиций, которые предназначены для превращения в жидкие формы композиций, предназначенных для перорального или парентерального введения, которое выполняется незадолго до использования. Такие жидкие формы включают растворы, суспензии и эмульсии.
Соединения по настоящему изобретению также можно вводить чрескожно. Чрескожные композиции могут представлять собой кремы, лосьоны, аэрозоли и/или эмульсии и они могут быть включены в матрицу пластыря чрескожного воздействия или пластыря резервуарного типа, что обычно используется в данной области техники для такой цели.
Соединение по настоящему изобретению предпочтительно вводить перорально.
Предпочтительно, чтобы фармацевтическая композиция содержалась в разовой дозировочной форме. В такой форме композиция разделяется на разовые дозы подходящей величины, содержащие соответствующие количества активного компонента, например, эффективного количества, достаточного для достижения необходимой цели.
Количество активного соединения of формулы I, содержащегося в разовой дозе препарата, в соответствии с конкретным случаем применения может меняться или регулироваться в диапазоне от примерно 0,1 до примерно 1000 мг, более предпочтительно - от примерно 1 до примерно 300 мг.
Используемая активная доза может меняться в зависимости от требований пациента и тяжести подвергающегося лечению патологического состояния. Определение надлежащего дозировочного режима для конкретного случая проводит специалист в данной области техники. Обычно лечение начинается с меньших доз, которые меньше оптимальной дозы соединения. Затем дозу увеличивают небольшими шагами, пока не будет достигнут оптимальный при данных обстоятельствах эффект. Для удобства полную суточную дозу, при необходимости, можно разделять и назначать порциями в течение дня.
Количество и частота введения соединений по настоящему изобретению и/или их фармацевтически приемлемых солей будет регулироваться в соответствии с решением лечащего врача, учитывающего такие факторы, как возраст, состояние и массу пациента, а также тяжесть симптомов, подвергающихся лечению. Типичный рекомендованный суточный дозировочный режим при пероральном введении может включать введение от 10 до 2000 мг/сутки, предпочтительно - от 10 до 1000 мг/сутки, назначаемых в виде двух-четырех разделенных доз, и обеспечивать устранение боли, состояния тревоги, депрессии, астмы или алкогольной зависимости. При введении в этом диапазоне доз соединения являются нетоксичными.
Когда агонист ноцицептинового рецептора ORL-1 формулы I назначается вместе с одним или большим количеством дополнительных препаратов, соединение формулы I и дополнительный препарат (дополнительные препараты) предпочтительно назначать в виде комбинированной дозировочной формы (например, в виде одной таблетки), хотя их можно назначать и по отдельности. Дополнительные препараты назначают в эффективных количествах и обеспечивают устранение симптомов кашля, аллергии или астмы, назначая предпочтительно от примерно 0,1 до 1000 мг, более предпочтительно - от примерно 1 до 300 мг в разовой дозе. Типичный рекомендованный дозировочный режим для дополнительного препарата составляет от 1 до 2000 мг/сутки, предпочтительно - от 1 до 1000 мг/сутки виде двух-четырех разделенных доз. Типичные значения доз других препаратов можно взять из литературных данных, например из справочника The Physicians's Desk Reference.
Ниже приведены примеры фармацевтических дозировочных форм, которые содержат соединение по настоящему изобретению. Специалисты в данной области техники должны понять, что такие дозировочные формы можно легко модифицировать с включением одного или большего количества активных компонентов. Объем настоящего изобретения в его воплощении в виде фармацевтической композиции не ограничивается приведенными примерами.
Примеры фармацевтических дозировочных форм
ПРИМЕР А - Таблетки
Способ изготовления
В подходящем смесителе в течение 10-15 минут перемешивают компоненты №№1 и 2. Смесь гранулируют с компонентом №3. При необходимости, влажные гранулы пропускают через крупнозернистое сито (например, 1/4 дюйма, 0,63 см). Влажные гранулы сушат. При необходимости, сухие гранулы просеивают и смешивают с компонентом №4 и перемешивают в течение 10-15 минут. Прибавляют компонент №5 и перемешивают в течение 1-3 минут. Смесь прессуют в таблетки соответствующего размера и массы на подходящей таблетирующей машине.
ПРИМЕР В - Капсулы
Способ изготовления
В подходящем смесителе в течение 10-15 минут перемешивают компоненты №1, 2 и 3. Прибавляют компонент №4 и перемешивают в течение 1-3 минут. Смесь расфасовывают в соответствующие двухкомпонентные капсулы из твердого желатина на подходящей капсулирующей машине.
Хотя настоящее изобретение описано в связи с конкретными вариантами осуществления, приведенными выше, для специалиста с общей подготовкой в данной области техники должны быть очевидны многие его альтернативы, модификации и изменения. Подразумевается, что все такие альтернативы, модификации и изменения входят в объем и сущность настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗАБИЦИКЛИЧЕСКИЕ И ДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2018 |
|
RU2795521C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АГОНИСТОВ STING И СПОСОБЫ С ИХ ИСПОЛЬЗОВАНИЕМ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2825271C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ МУСКАРИНОВЫЕ АГОНИСТЫ И КОМПОЗИЦИИ, ИХ ПРИМЕНЕНИЕ И СПОСОБЫ ЛЕЧЕНИЯ | 2002 |
|
RU2292346C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛИДИНОНА В КАЧЕСТВЕ АГЕНТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2288919C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| Производные изохинолинона, способ их получения и фармацевтическая композиция для профилактики или лечения заболеваний, связанных с поли(АДФ-рибоза)полимеразой-1, содержащая их в качестве активного ингредиента | 2020 |
|
RU2815480C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2767857C2 |
Изобретение относится к новым гетероарильным производным 8-азабицикло-[3.2.1]-октан-3-ола общей формулы I, где R-R4-гетероарил, причем гетероарил представляет собой циклическую ароматическую группу с С5-С6 или бициклическую группу с С9-С10, которые включают 1, 2 или 3 гетероатома, независимо О, S или N, или остаток 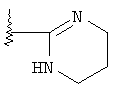 ,
,
R1-H, C1-С6-алкил, R2 и R3 независимо СН3, -ОСН3, F, Cl, Br, J, R4 - от 1 до 4 заместителей, -Н, галоген, C1-С6алкил, -CF3, -OCF3, -(СН2)n-OR5, -(CH2)n-NR5R6, -(CH2)n-NHSO2R5, -(CH2)n-NH(CH2)2NR5R6, -(CH2)n-NHC(O)NR5R7, -(CH2)n-NH(CH2)2OR5 или 1-пиперазинил; n - 0, 1, 2, 3; R5 и R6 - Н, C1-С3алкил, R7-H, C1-С3алкил, аминоалкил C1-С3, или к их фармацевтически приемлемым солям. Соединения являются агонистами рецептора ноцицептина ORL-1 и полезны при лечении кашля. 3 н. и 7 з.п. ф-лы, 3 табл.
или его фармацевтически приемлемые соли,
где R означает R4 гетероарил, причем гетероарил представляет собой циклическую ароматическую группу с 5 или 6 атомами углерода или бициклическую группу с 9 или 10 атомами углерода, включающие 1, 2 или 3 гетероатома, независимо выбранные из группы, включающей кислород, серу и азот;
или 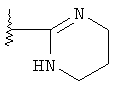 ;
;
R1 означает водород или алкил с 1-6 атомами углерода;
R2 и R3 независимо выбирают из группы, включающей метил, метокси, фтор, хлор, бром и йод;
R4 означает от 1 до 4 заместителей, независимо выбранных из группы, включающей водород, галоген, алкил с 1-6 атомами углерода, циано, трифторметил, трифторметокси, -(CH2)n-OR5, -(CH2)n-NR5R6, -(СН2)n-NHSO2R5, -(CH2)n-NH(CH2)2NR5R6, -(CH2)n-NHC(O)NR5R7, -(CH2)n-NH(CH2)2OR5 или 1-пиперазинил;
n равно 0, 1, 2 или 3;
R5 и R6 независимо выбирают из группы, включающей водород и алкил с 1-3 атомами углерода; и
R7 означает водород, алкил с 1-3 атомами углерода или аминоалкил с 1-3 атомами углерода.
 ,
,  ,
,  .
.
 и
и 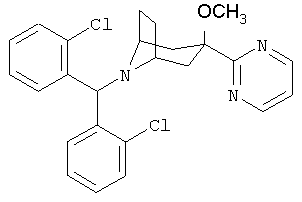
| УСТРОЙСТВО ДЛЯ ОПРЕДЕЛЕНИЯ, АВТОМАТИЧЕСКОГО КОНТРОЛЯ И РЕГУЛИРОВАНИЯ ПЛОТНОСТИ ТОКА В ГАЛЬВАНИЧЕСКОЙ ВАННЕ | 1955 |
|
SU107050A1 |
| US 6262066 В1, 17.07.2001. | |||

