Область изобретения
Изобретение относится в общем к способам введения лекарств. Более конкретно изобретение относится к введению противоопухолевых агентов, включая ингибиторы рецептора еrbВ2. Изобретение также относится к способам улучшенного введения ингибиторов рецепторных протеинтирозинкиназ, которые полезны в лечении аномального клеточного роста, такого как рак, у млекопитающих. Изобретение также относится к наборам, полезным для введения таких ингибиторов при лечении аномального клеточного роста у млекопитающих, в особенности у людей.
Предшествующий уровень техники
Известно, что клетка может становиться злокачественной путем трансформации части ее ДНК в онкоген, представляющий собой ген, который при активации приводит к образованию злокачественных опухолевых клеток. Многие онкогены кодируют белки, которые представляют собой аберрантные тирозинкиназы, способные вызывать клеточную трансформацию. Альтернативно сверхэкспрессия нормальной протоонкогенной тирозинкиназы также может иметь результатом пролиферативные нарушения, иногда дающие в результате злокачественный фенотип.
Рецепторные тирозинкиназы представляют собой ферменты, которые пронизывают клеточную мембрану и имеют внеклеточный домен для связывания факторов роста, таких как эпидермальный фактор роста, трансмембранный домен и внутриклеточный участок, который действует в качестве киназы, фосфорилирующей специфические остатки тирозина в белках и тем самым влияющей на клеточную пролиферациию. Кроме того, некоторые рецепторные тирозинкиназы представляют собой субстраты для тех же самых или других протеинкиназ в процессе, который может регулировать киназную функцию. Рецепторные тирозинкиназы классифицируются на семейства, одно из которых представляет собой семейство erb, включающее erbB1 и erbB2. Известно, что киназы, такие как erbB2, часто аберрантно экспрессируются при таких распространенных видах рака у людей, как рак молочной железы, рак желудочно-кишечного тракта, такой как рак толстой кишки, прямой кишки или желудка, лейкемия и рак яичников, бронхиальный рак и рак поджелудочной железы. Было показано, что рецептор эпидермального фактора роста (erbB1), который обладает тирозинкиназной активностью, мутирован и/или сверхэкспрессируется при многих типах рака у людей, таких как опухоли головного мозга, легких, плоскоклеточная опухоль, опухоль мочевого пузыря, желудка, молочной железы, головы и шеи, опухоль пищевода, гинекологические опухоли и опухоли щитовидной железы. Соответственно было признано, что ингибиторы рецепторных тирозинкиназ полезны в качестве избирательных ингибиторов роста раковых клеток у млекопитающих. Аномальный клеточный рост может быть ассоциирован с клеточной экспрессией рецепторов erb.
Тем не менее, в достаточной степени не было принято во внимание, что способ введения ингибитора может влиять на эффективность этого ингибитора.
Краткое изложение сущности изобретения
Изобретение относится в общем к способам и наборам для ингибирования аномального клеточного роста. Более конкретно изобретение относится к улучшенным схемам приема противоопухолевых агентов.
Настоящее изобретение относится к способу лечения сверхэкспрессии рецептора erbB2 у нуждающегося в таком лечении млекопитающего, включающему:
(а) введение указанному млекопитающему терапевтически эффективного количества первого ингибитора рецептора erbB2; и
(б) последующее введение указанному млекопитающему после промежутка времени меньше 24 часов от одного до шести терапевтически эффективных количеств второго ингибитора рецептора erbB2.
В первом предпочтительном воплощении настоящего изобретения на стадии (б) указанного способа могут быть введены от одного до четырех терапевтически эффективных количеств указанного второго ингибитора рецептора erbB2. В более предпочтительном воплощении на стадии (б) указанного способа вводят от одного до двух терапевтически эффективных количеств указанного второго ингибитора, рецептора erbB2. В еще одном воплощении на стадии (б) указанного способа вводят одно терапевтически эффективное количество указанного второго ингибитора рецептора erbB2.
В еще одном воплощении настоящего изобретения промежуток времени на стадии (б) указанного способа составляет менее 12 часов. В предпочтительном воплощении промежуток времени на стадии (б) указанного способа составляет менее 6 часов. В более предпочтительном воплощении промежуток времени на стадии (б) указанного способа составляет менее 3 часов. В наиболее предпочтительном воплощении промежуток времени на стадии (б) указанного способа составляет менее 1 часа.
Введение ингибитора на стадиях (а) и (б) может включать пероральное, трансбуккальное, сублингвальное, интраназальное, внутрижелудочное, интрадуоденальное, местное, внутриглазное, ректальное или вагинальное введение.
В одном воплощении изобретения первый ингибитор на стадии (а) является таким же, как второй ингибитор на стадии (б). В одном воплощении способа в соответствии с настоящим изобретением первое количество может отличаться от последующих одного-шести количеств. В еще одном воплощении настоящего изобретения ингибитор на стадии (а) может отличаться от ингибитора на стадии (б). В другом конкретном воплощении ингибитор на стадии (а) является таким же, как ингибитор на стадии (б), возможно, тем же самым стереоизомером или той же самой солевой формой. В еще одном воплощении лечения первый ингибитор на стадии (а) является синергистом второго ингибитора на стадии (б). Первый ингибитор на стадии (а), второй ингибитор на стадии (б) или оба могут являться антагонистами рецептора erbB2.
В одном воплощении настоящего изобретения терапевтически эффективное количество указанного первого ингибитора рецептора erbB2 отличается от одного-шести терапевтически эффективных количеств указанного второго ингибитора рецептора erbB2. В одном предпочтительном воплощении настоящего изобретения первый ингибитор на стадии (а) отличен от второго ингибитора на стадии (б). В еще одном предпочтительном воплощении первый ингибитор на стадии (а) является синергистом второго ингибитора на стадии (б). В еще одном предпочтительном воплощении настоящего изобретения первый ингибитор на стадии (а), второй ингибитор на стадии (б) или оба являются антагонистами рецептора erbB2.
В одном предпочтительном воплощении настоящего изобретения первый ингибитор на стадии (а) и второй ингибитор на стадии (б) независимо выбраны из небольших молекул и моноклональных антител. В одном предпочтительном воплощении как первый ингибитор на стадии (а), так и второй ингибитор на стадии (б) представляют собой небольшие молекулы или моноклональные антитела. В еще одном предпочтительном воплощении настоящего изобретения первый ингибитор на стадии (а), второй ингибитор на стадии (б) или оба являются избирательными в отношении рецепторов erbB2.
Способ лечения в соответствии с изобретением может дополнительно включать условие, что ингибитор на стадии (а), ингибитор на стадии (б) или оба они обладают периодом полувыведения in vivo от получаса до восьми часов.
Способ в соответствии с изобретением может включать введение ингибитора, где ингибитор на стадии (а), ингибитор на стадии (б) или оба они не являются существенно цитотоксичными.
Способ может включать введение ингибитора, где ингибитор на стадии (а), ингибитор на стадии (б) или оба они не являются по существу ингибиторами митоза.
В одном аспекте изобретения введение представляет собой контролируемое высвобождение. Препарат с контролируемым высвобождением может быть введен перорально, трансбуккально, сублингвально, интраназально, внутрижелудочно, интрадуоденально, местно, внутриглазным путем, ректально или вагинально.
В одном воплощении способа в соответствии с изобретением ингибитор на стадии (а) и ингибитор на стадии (б) независимо выбраны из небольших молекул и моноклональных антител. В одном предпочтительном воплощении как ингибитор на стадии (а), так и ингибитор на стадии (б) представляют собой небольшие молекулы или моноклональные антитела. Небольшие молекулы могут быть менее 4000 дальтон.
Первый ингибитор на стадии (а), второй ингибитор на стадии (б) или оба они могут быть избирательными в отношении рецепторов erbB2.
В еще одном воплощении изобретения первый ингибитор на стадии (а), второй ингибитор на стадии (б) или оба они включают соединение формулы 1:
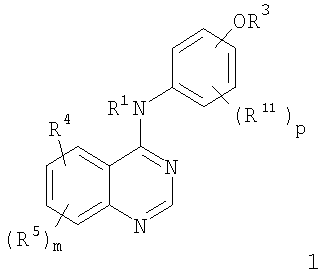
или его фармацевтически приемлемую соль, сольват или пролекарство.
В формуле 1 m представляет собой целое число от 0 до 3;
р представляет собой целое число от 0 до 4;
каждый из R1 и R2 независимо выбран из Н и С1-С6алкила;
R3 представляет собой -(CR1R2)t(4-10-членный гетероцикл), где t представляет собой целое число от 0 до 5, причем указанная гетероциклическая труппа, возможно, конденсирована с бензольным кольцом или С5-С8циклоалкильной группой, группировка -(CR1R2)t вышеупомянутой группы R3, возможно, включает углерод-углеродную двойную или тройную связь, где t представляет собой целое число от 2 до 5, а вышеупомянутые группы R3, включающие любые вышеупомянутые возможные конденсированные кольца, возможно, замещены 1-5 группами R8;
R4 представляет собой -(CR16R17)m-C(C-(CR16R17)tR9, -(CR16R17)m-C=C-(CR16R17)tR9, -(CR16R17)m-C(C-(CR16R17)kR13, -(CR16R17)m-C=C-(CR16R17)kR13 или -(CR16R17)tR9, где точкой присоединения к R9 является углерод группы R9, каждый k представляет собой целое число от 1 до 3, каждый t представляет собой целое число от 0 до 5 и каждый m представляет собой целое число от 0 до 3;
каждый R5 независимо выбран из галогено, гидрокси, -NR1R2, C1-С6алкила, трифторметила, C1-С6алкокси, трифторметокси, -NR6C(O)R1, -C(O)NR6R7, -SO2NR6R7, -NR6C(O)NR7R1 и -NR6C(O)OR7;
каждый из R6, R6a и R7 независимо выбран из Н, C1-С6алкила, -(CR1R2)t(С6-С10арил) и -(CR1R2)t(4-10-членный гетероцикл), где t представляет собой целое число от 0 до 5, 1 или 2 кольцевых атома углерода гетероциклической группы, возможно, замещены группировкой оксо (=O), алкильные, арильные и гетероциклические группировки вышеупомянутых групп R6 и R7, возможно, замещены 1-3 заместителями, независимо выбранными из галогено, циано, нитро, -NR1R2, трифторметила, трифторметокси, C1-С6алкила, С2-С6алкенила, С2-С6алкинила, гидрокси и С1-С6алкокси;
или R6 и R7 или R6a и R7, когда они присоединены к одному и тому же атому азота, могут быть взяты вместе с образованием 4-10-членного гетероциклического кольца, которое может включать 1-3 дополнительные гетерогруппировки в дополнение к атому азота, к которому присоединены указанные R6, R6a и R7, выбранные из N, N(R1), О и S, при условии, что два атома О, два атома S или атомы О и S не соединены непосредственно друг с другом;
каждый R8 независимо выбран из оксо (=O), галогено, циано, нитро, трифторметокси, трифторметила, азидо, гидрокси, C1-С6алкокси, C1-C10алкила, С2-С6алкенила, С2-С6алкинила, -C(O)R6, -C(O)OR6, -OC(O)R6, -NR6C(O)R7, -NR6SO2NR7R1, -NR6C(O)NR1R7, -NR6C(O)OR7, -C(O)NR6R7, -NR6R7, -NR6OR7, -SO2NR6R7, -S(O)j)(С1-С6алкил), где j представляет собой целое число от 0 до 2, -(CR1R2)t(С6-С10арил), -(CR1R2)t(4-10-членный гетероцикл), -(CR1R2)qС(O)(CR1R2)t(С6-С10арил), -(CR1R2)qC(O)(CR1R2)t(4-10-членный гетероцикл), -(CR1R2)tO(CR1R2)q(С6-С10арил), -(CR1R2)tO(CR1R2)q(4-10-членный гетероцикл), -(CR1R2)qS(O)j(CR1R2)t(C6-C10арил) и -(CR1R2)qS(O)j(CR1R2)t(4-10-членный гетероцикл), где j представляет собой 0, 1 или 2, q и t каждый независимо представляет собой целое число от 0 до 5, 1 или 2 кольцевых атома углерода гетероциклических группировок вышеупомянутых групп R8, возможно, замещены группировкой оксо (=O), а алкильные, алкенильные, алкинильные, арильные и гетероциклические группировки вышеупомянутых групп R8, возможно, замещены 1-3 заместителями, независимо выбранными из галогено, циано, нитро, трифторметила, трифторметокси, азидо, -OR6, -C(O)R6, -C(O)OR6, -OC(O)R6, -NR6C(O)R7, -C(O)NR6R7, -NR6R7, -NR6OR7, C1-С6алкила, С2-С6алкенила, С2-С6алкинила, -(CR1R2)t(C6-C10арил) и -(CR1R2)t(4-10-членный гетероцикл), где t представляет собой целое число от 0 до 5;
R9 представляет собой неароматическое моноциклическое кольцо, конденсированное или мостиковое бициклическое кольцо или спироциклическое кольцо, где указанное кольцо содержит от 3 до 12 атомов углерода, причем от 0 до 3 атомов углерода, возможно, заменены гетерогруппировкой, независимо выбранной из N, О, S(O)j, где j представляет собой целое число от 0 до 2, и -NR1-, при условии, что два атома О, две группировки S(O)j, атом О и группировка S(O)j, атом N и атом S или атом N и атом О не соединены непосредственно друг с другом в пределах указанного кольца, и где атомы углерода указанного кольца, возможно, замещены 1 или 2 группами R8;
каждый R11 независимо выбран из заместителей, приведенных в определении R8, за исключением того, что R11 не представляет собой оксо (=O);
R12 представляет собой R6, -OR6, -OC(O)R6, -OC(O)NR6R7, -OCO2R6, -S(O)jR6, -S(O)jNR6R7, -NR6R7, -NR6C(O)R7, -NR6SO2R7, -NR6C(O)NR6aR7, -NR6SO2NR6aR7, -NR6CO2R7, CN, -C(O)R6 или галогено, где j представляет собой целое число от 0 до 2;
R13 представляет собой -N11R14 или -OR14;
R14 представляет собой Н, R15, -C(O)R15, -SO2R15, -C(O)NR15R7, -SO2NR15R7 или -CO2R15;
R15 представляет собой R18, -(CR1R2)t(C6-C10арил), -(CR1R2)t(4-10-членный гетероцикл), где t представляет собой целое число от 0 до 5, 1 или 2 кольцевых атома углерода гетероциклической группы, возможно, замещены группировкой оксо (=O), а арильные и гетероциклические группировки вышеупомянутых групп R15, возможно, замещены 1-3 заместителями R8;
каждый R16 и R17 независимо выбран из Н, С1-С6алкила и -CH2OH, или R16 и R17, взятые вместе, представляют собой -СН2СН2- или -СН2CH2СН2-;
R18 представляет собой С1-С6алкил, причем каждый атом углерода, не связанный с атомом N или О или с S(O)j, где j представляет собой целое число от 0 до 2, возможно, замещен группой R12;
и где любой из вышеупомянутых заместителей, включающих группу СН3 (метил), СН2 (метилен) или СН (метин), которая не связана с группой галогено, SO или SO2 или с атомом N, О или S, возможно, замещен группой, выбранной из гидрокси, галогено, С1-С4алкила, С1-С4алкокси и -NR1R2.
Используемый здесь термин "галогено", если не указано иначе, включает фторо, хлоро, бромо или йодо. Предпочтительные группы галогено представляют собой фторо и хлоро.
Используемый здесь термин "алкил", если не указано иначе, включает насыщенные одновалентные углеводородные радикалы, имеющие прямые, циклические (включая моно- или мультициклические группировки) или разветвленные группировки. Понятно, что для того, чтобы указанная алкильная группа включала циклические группировки, она должна содержать по меньшей мере три атома углерода.
Используемый здесь термин "циклоалкил", если не указано иначе, включает насыщенные одновалентные углеводородные радикалы, имеющие циклические (включая моно- или мультициклические) группировки.
Используемый здесь термин "алкенил", если не указано иначе, включает алкильные группы, как определено выше, имеющие по меньшей мере одну углерод-углеродную двойную связь.
Используемый здесь термин "алкинил", если не указано иначе, включает алкильные группы, как определено выше, имеющие по меньшей мере одну углерод-углеродную тройную связь.
Используемый здесь термин "арил", если не указано иначе, включает органический радикал, полученный из ароматического углеводорода путем удаления одного атома водорода, такой как фенил или нафтил.
Используемый здесь термин "алкокси", если не указано иначе, включает группы -O-алкил, где алкил является таким, как определено выше.
Используемый здесь термин "4-10-членный гетероцикл", если не указано иначе, включает ароматические и неароматические гетероциклические группы, содержащие один или более гетероатомов, каждый из которых выбран из О, S и N, где каждая гетероциклическая группа содержит от 4 до 10 атомов в своей кольцевой системе. Неароматические гетероциклические группы включают группы, имеющие только 4 атома в своей кольцевой системе, а ароматические гетероциклические группы должны иметь по меньшей мере 5 атомов в своей кольцевой системе. Гетероциклические группы включают бензоконденсированные кольцевые системы и кольцевые системы, замещенные одной или более чем одной группировкой оксо. Примером 4-членной гетероциклической группы является азетидинил (полученный из азетидина). Примером 5-членной гетероциклической группы является тиазолил, а примером 10-членной гетероциклической группы является хинолинил. Примерами неароматических гетероциклических групп являются пирролидинил, тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, тетрагидротиопиранил, пиперидине, морфолино, тиоморфолино, тиоксанил, пиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 1,2,3,6-тетрагидропиридинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, 3Н-индолил и хинолизинил. Примерами ароматических гетероциклических групп являются пиридинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил. Вышеупомянутые группы, получаемые из перечисленных выше соединений, могут быть присоединены по атому С или N, когда такое присоединение возможно. Например, группа, полученная из пиррола, может представлять собой пиррол-1-ил (присоединение по атому N) или пиррол-3-ил (присоединение по атому С).
Термин "Me" означает метил, "Et" означает этил, и "Ас" означает ацетил. Используемое здесь выражение "фармацевтически приемлемая(ые) соль(и)", если не указано иначе, включает соли кислых или основных групп, которые могут присутствовать в соединениях по настоящему изобретению. Соединения по настоящему изобретению, которые являются основными по природе, способны образовывать широкое разнообразие солей с различными неорганическими и органическими кислотами. Кислоты, которые могут быть использованы для получения фармацевтически приемлемых солей присоединения кислот таких основных соединений, представляют собой кислоты, которые образуют нетоксичные соли присоединения кислот, то есть соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, кислый цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат (gentisinate), фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, пара-толуолсульфонат и памоат [то есть 1,1'-метиленбис-(2-гидрокси-3-нафтоат)]. Соединения по настоящему изобретению, которые включают основную группировку, такую как аминогруппа, могут образовывать фармацевтически приемлемые соли с различными аминокислотами в дополнение к вышеупомянутым кислотам.
Способ лечения в соответствии с изобретением может включать введение ингибитора рецептора erbB2, где ингибитор на стадии (а), ингибитор на стадии (б) или оба включают соединение, выбранное из группы, состоящей из гефитиниба (IRESSA, ZD1839), трастузумаба, цетуксимаба, эрлотиниба, IDM-1, ABX-EGF, гидрохлорида канертиниба, вакцины EGF-P64k, EKB-569, EMD-72000, GW-572016, MDX-210, ME-103, YMB-1001, антитела 2С4, АРС-8024, СР-724714, Е75, вакцины Her-2/neu, Herzyme, ТАК-165, ADL-681, B-17, D-69491, Dab-720, EGFrvIII, EHT-102, FD-137, вакцины HER-1, HuMax-DGFr, ME-104, MR1-1, SC-100, трастузумаба-DM1, YMB-1005, AEE-788 (Novartis), ингибиторов мишени рипамицина млекопитающих (mTOR), включая рапамицин (Rapamune, Siolimus, Wyeth), CCI-779 (Wyeth), AP23573 (ARIAD) и RAD001 (Novartis).
В одном воплощении настоящего изобретения сверхэкспрессию рецептора erbB2 определяют с использованием цитогенетического теста, измерения флуоресцентной гибридизации in-situ, иммуногистохимического теста, теста с использованием проточной цитометрии, теста, основанного на полимеразной цепной реакции с обратной транскриптазой, или любой их комбинации.
В одном воплощении настоящего изобретения млекопитающее представляет собой человека, а аномальный клеточный рост представляет собой злокачественное новообразование. Млекопитающее также может представлять собой экспериментальное животное, домашнее животное, скот или любое другое млекопитающее.
Способ лечения в соответствии с изобретением может также включать достижение уровней в плазме первого ингибитора на стадии (а), второго ингибитора на стадии (б) или обоих, составляющих приблизительно от 10 нг/мл до 4000 нг/мл.
В одном воплощении изобретения первый ингибитор на стадии (а) и второй ингибитор на стадии (б) каждый независимо выбран из группы, состоящей из:
(±)-(3-Метил-4-(пиридин-3-илокси)-фенил)-(6-пиперидин-3-илэтинил-хиназолин-4-ил)амина;
(+)-(3-Метил-4-(пиридин-3-илокси)-фенил)-(6-пиперидин-3-илэтинил-хиназолин-4-ил)амина;
(-)-(3-Метил-4-(пиридин-3-илокси)-фенил)-(6-пиперидин-3-илэтинил-хиназолин-4-ил)амина;
2-Метокси-N-(3-{4-(3-метил-4-(пиридин-3-илокси)-фениламино)-хиназолин-6-ил}проп-2-инил)-ацетамида;
(±)-(3-Метил-4-(6-метил-пиридин-3-илокси)-фенил)-(6-пиперидин-3-илэтинил-хиназолин-4-ил)-амина;
(+)-(3-Метил-4-(6-метил-пиридин-3-илокси)-фенил)-(6-пиперидин-3-илэтинил-хиназолин-4-ил)-амина;
(-)-(3-Метил-4-(6-метил-пиридин-3-илокси)-фенил)-(6-пиперидин-3-илэтинил-хиназолин-4-ил)-амина;
2-Метокси-N-(3-{4-(3-метил-4-(2-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-проп-2-инил)-ацетамида;
(3-Метил-4-(2-метил-пиридин-3-илокси)-фенил)-(6-пиперидин-4-илэтинил-хиназолин-4-ил)-амина;
(3-Метил-4-(6-метил-пиридин-3-илокси)-фенил)-(6-пиперидин-4-ил-этинил-хиназолин-4-ил)-амина;
2-Метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-проп-2-инил)-ацетамида;
2-Фтор-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}проп-2-инил)-ацетамида;
Е-2-Метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида;
(3-Метил-4-(пиридин-3-илокси)-фенил)-(6-пиперидин-4-илэтинил-хиназолин-4-ил)-амина;
2-Метокси-N-(1-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-илэтинил}-циклопропил)-ацетамида;
Е-N-(3-{4-(3-Хлор-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-2-метокси-ацетамида;
N-(3-{4-(3-Хлор-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}проп-2-инил)-ацетамида;
N-(3-{4-(3-Метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}проп-2-инил)-ацетамида;
E-N-(3-{4-(3-Хлор-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида;
Е-2-Этокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида;
1-Этил-3-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}проп-2-инил)-мочевины;
(3-{4-(3-Метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-проп-2-инил)амида пиперазин-1-карбоновой кислоты;
(3-{4-(3-Метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-проп-2-инил)амида (±)-2-гидроксиметил-пирролидин-1-карбоновой кислоты;
(3-{4-(3-Метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-проп-2-инил)-амида (+)-2-гидроксиметил-пирролидин-1-карбоновой кислоты;
(3-{4-(3-Метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-проп-2-инил)-амида (-)-2-гидроксиметил-пирролидин-1-карбоновой кислоты;
2-Диметиламино-N-(3-{4-(3-метил-4-(пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-проп-2-инил)-ацетамида;
Е-N-(3-{4-(3-Метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)метансульфонамида;
(3-{4-(3-Метил-4-(6-метил-пиридин-3-илокси)фениламино)-хиназолин-6-ил}проп-2-инил)-амида изоксазол-5-карбоновой кислоты;
1-(1,1-Диметил-3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-проп-2-инил)-3-этилмочевины.
Способ лечения включает применение одного агента, который ингибирует рецептор erbB2, а также применение двух различных агентов. Этот один агент и по меньшей мере один из двух агентов предпочтительно представляет собой агент формулы 1. Таким образом, в одном воплощении ингибитор выбран из группы, состоящей из (±)-(3-метил-4-(6-метил-пиридин-3-илокси)-фенил)-(6-пиперидин-3-илэтинил-хиназолин-4-ил)амина и его фармацевтически приемлемых солей, пролекарств и сольватов. В еще одном воплощении ингибитор выбран из группы, состоящей из (3-метил-4-(6-метил-пиридин-3-илокси)-фенил)-(6-пиперидин-4-илэтинил-хиназолин-4-ил)амина и его фармацевтически приемлемых солей, пролекарств и сольватов. В еще одном воплощении ингибитор выбран из группы, состоящей из E-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида и его фармацевтически приемлемых солей, пролекарств и сольватов. В еще одном воплощении ингибитор выбран из группы, состоящей из Е-N-(3-{4-(3-хлор-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-2-метокси-ацетамида и его фармацевтически приемлемых солей, пролекарств и сольватов. В еще одном воплощении ингибитор выбран из группы, состоящей из Е-N-(3-{4-(3-хлор-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида и его фармацевтически приемлемых солей, пролекарств и сольватов. В конкретном воплощении изобретения ингибитор выбран из группы, состоящей из (3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}проп-2-инил)амида пиперазин-1-карбоновой кислоты и его фармацевтически приемлемых солей, пролекарств и сольватов. В еще одном конкретном воплощении изобретения ингибитор выбран из группы, состоящей из E-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-метансульфонамида и его фармацевтически приемлемых солей, пролекарств и сольватов. В еще одном аспекте изобретения первый ингибитор со стадии (а), второй ингибитор со стадии (б) или оба находятся в фармацевтически приемлемом носителе.
В первом воплощении настоящего изобретения сверхэкспрессия рецептора erbB2 приводит в результате к аномальному клеточному росту. Аномальный клеточный рост, который лечат первым и вторым ингибитором рецептора erbB2, может представлять собой злокачественное новообразование. Злокачественное новообразование может быть выбрано из группы, состоящей из периферической лентигинозной меланомы, лучевого кератоза, аденокарциномы, аденокистозной карциномы, аденомы, аденосаркомы, железисто-плоскоклеточной карциномы, астроцитарной опухоли, карциномы бартолиновой железы, базально-клеточной карциномы, карциномы бронхиальных желез, капиллярной карциномы, карциноида, карциномы, карциносаркомы, кавернозной карциномы, холангиокарциномы, хондросаркомы; папилломы сосудистого сплетения, карциномы сосудистого сплетения, гипернефроидной опухоли почки, цистаденомы, эндодермальной полостной опухоли, эндометриальной гиперплазии, эндометриальной стромальной саркомы, эндометриоидной аденокарциномы, эпендимной карциномы, эпителиоидной саркомы, саркомы Юинга, фиброламеллярной, фокальной нодулярной гиперплазии, гастриномы, опухоли половых клеток, глиобластомы, глюкагономы, гемангиобластомы, гемангиоэндотелиомы, гемангиомы, печеночной аденомы, печеночного аденоматоза, печеночно-клеточного рака, инсулиномы, внутриэпителиальной неоплазии, межэпителиальной плоско клеточной неоплазии, инвазивного плоскоклеточного рака, крупноклеточного рака, лейомиосаркомы, злокачественного лентиго, злокачественной меланомы, злокачественной мезотелиальной опухоли, медуллобластомы, медулло-эпителиомы, меланомы, менингеальной мезотелиальной метастатической карциномы, слизеобразующего плоскоклеточного рака, нейробластомы, нейроэпителиальной аденокарциномы, узелковой меланомы, овсяно-клеточного рака, олигодендроглиальной остеосаркомы, панкреатического полипептида, серозной папиллярной аденокарциномы, пинеалоцита, опухоли гипофиза, плазмацитомы, псевдосаркомы, легочной бластомы, почечно-клеточной карциномы, ретинобластомы, рабдомиосаркомы, саркомы, серозной карциномы, мелкоклеточной карциномы, рака мягких тканей, соматостатин-секретирующей опухоли, плоскокого рака, сквамозной карциномы, сквамозноклеточной карциномы, субмезотелиальнои поверхностно-распространяющейся меланомы, недифференцированной карциномы, юношеской меланомы, сосочкового рака, випомы, высокодифференцированной карциномы, бронхиолоальвеолярного рака, опухоли Вильма.
В одном воплощении аномальный клеточный рост представляет собой злокачественное новообразование, выбранное из группы, состоящей из опухоли легкого, молочной железы, кожи, желудка, кишечника, пищевода, поджелудочной железы, печени, мочевого пузыря, головы, шеи, головного мозга, цервикальной опухоли и опухоли яичников. В одном предпочтительном воплощении аномальный клеточный рост представляет собой опухоль, выбранную из группы, состоящей из опухоли молочной железы, желудка, поджелудочной железы и яичников. В более предпочтительном воплощении аномальный клеточный рост представляет собой рак молочной железы.
В еще одном воплощении изобретения ингибитор рецептора erbB2 может быть избирательным в отношении рецептора erbB2. Способ по изобретению может дополнительно включать: (в) расчет соотношения аффинности связывания ингибитора для рецептора erbB2 и второй аффиности связывания ингибитора для рецептора erbB1, и (г) использование соотношения для оценки избирательности. В одном воплощении ингибитор по меньшей мере в два раза более избирателен в отношении рецептора erbB2. В еще одном воплощении ингибитор по меньшей мере в десять раз более избирателен в отношении рецептора erbB2.
Еще одно воплощение настоящего изобретения относится к способу лечения субъекта, страдающего от аномального клеточного роста, включающему пероральное, трансбуккальное, сублингвальное, интраназальное, внутриглазное, внутрижелудочное, интрадуоденальное, местное, ректальное или вагинальное введение указанному субъекту, нуждающемуся в лечении аномального клеточного роста, в пределах двадцати четырех часов первого количества ингибитора рецептора erbB2, терапевтически синергически эффективного второго количества ингибитора и возможно третьего или четвертого количества ингибитора. Ингибитор может представлять собой избирательный ингибитор рецептора erbB2.
В еще одном воплощении изобретение включает набор для лечения аномального клеточного роста, включающий по меньшей мере две дозы ингибитора рецептора erbB2, пригодные для перорального, трансбуккального, сублингвального, интраназального, внутриглазного, внутрижелудочного, интрадуоденального, местного, ректального или вагинального введения субъекту, и письменные инструкции по введению доз по меньшей мере дважды в сутки субъекту, страдающему от аномального клеточного роста. Целесообразно, если письменные инструкции находятся на этикетке или вложены в упаковку. В одном воплощении набора аномальный клеточный рост представляет собой опухоль, выбранную из группы, состоящей из опухоли легкого, молочной железы, кожи, желудка, кишечника, пищевода, мочевого пузыря, головы, шеи, головного мозга, цервикальной опухоли и опухоли яичников.
В еще одном воплощении изобретение включает способ лечения содержащей рецептор erbB2 опухоли у нуждающегося в этом субъекта, включающий введение указанному субъекту терапевтически эффективного количества ингибитора рецептора erbB2 путем инфузии указанному субъекту в течение 1-8 часов таким образом, что инфузия оказывается более эффективным, чем болюсная инъекция. Инфузия может быть внутривенной, внутримышечной, интраперитонеальной или подкожной. В одном воплощении ингибитор может представлять собой соединение формулы 1.
В еще одном воплощении изобретение включает способ усиления эффективности ингибитора рецептора erbB2 у нуждающегося в этом субъекта, включающий: (а) определение базовой дозы ингибитора рецептора erbB2 и (б) деление дозы для повышения эффективности. Повышенная эффективность представляет собой форму синергизма, являющегося результатом деления дозы. В одном воплощении дозу делят на 2-6 суточных доз.
В еще одном воплощении базовая доза обладает побочным действием, а разделенная доза обладает уменьшенным побочным действием. Ингибитор может быть по меньшей мере в два раза более избирательным в отношении рецептора erbB2 по сравнению с рецептором erbB1. В еще одном воплощении ингибитор по меньшей мере в десять раз более избирателен в отношении рецептора erbB2 по сравнению с рецептором erbB1.
Способ повышения эффективности может дополнительно включать стадии: (в) расчета соотношения аффинности связывания ингибитора для рецептора erbB2 и второй аффиности связывания ингибитора для рецептора erbB1 и (г) использования соотношения для оценки избирательности.
В еще одном воплощении изобретение включает способ повышения эффективности ингибитора рецептора erbB2, включающий введение нуждающемуся в этом пациенту суточной дозы терапевтически эффективного количества ингибитора, где суточная доза разделена для обеспечения уровня ингибитора в плазме указанного пациента, который меньше, чем терапевтически эффективное количество разовой суточной дозы, а эффективность повышена.
В еще одном воплощении изобретение включает способ повышения безопасности введения ингибитора рецептора erbB2 нуждающемуся в этом субъекту, включающий ежедневное введение указанному субъекту от двух до шести терапевтически эффективных количеств ингибитора.
В еще одном воплощении изобретение включает способ повышения безопасности введения ингибитора рецептора erbB2 нуждающемуся в этом субъекту, включающий определение базовой суточной дозы ингибитора, имеющей профиль безопасности, и деление этой дозы для улучшения профиля безопасности.
В еще одном воплощении изобретение включает набор для лечения аномального клеточного роста у субъекта, включающий дозу ингибитора рецептора erbB2, которая подходит для внутривенной, внутримышечной, интраперитонеальной или подкожной инфузии, и письменные инструкции по инфузии этой дозы указанному субъекту в течение 1-8 часов. В одном воплощении набора аномальный клеточный рост может включать опухоль, выбранную из группы, состоящей из опухоли легкого, молочной железы, кожи, желудка, кишечника, пищевода, мочевого пузыря, поджелудочной железы, печени, головы, шеи, головного мозга, цервикальной опухоли и опухоли яичников.
В еще одном воплощении изобретение включает профилактическое лечение субъекта, имеющего риск развития опухоли, включающее введение указанному субъекту эффективного количества избирательного ингибитора рецептора erbB2 по меньшей мере дважды в сутки. В одном воплощении профилактического лечения ингибитор может быть отличным от антитела или его фрагмента.
В еще одном воплощении изобретение включает способ повышения эффективности ингибитора рецептора erbB2, включающий введение суточной дозы терапевтически эффективного количества ингибитора нуждающемуся в этом пациенту, где суточная доза разделена для обеспечения уровня ингибитора в плазме указанного пациента, который меньше, чем терапевтически эффективное количество разовой суточной дозы, а эффективность повышена. В одном воплощении уровень в плазме выражается как Сср. В еще одном воплощении уровень в плазме выражается Сmax. Ингибитор может представлять собой избирательный ингибитор рецептора erbB2. В одном воплощении ингибитор отличен от антитела или его фрагмента.
В еще одном воплощении настоящее изобретение относится к способу лечения содержащей рецептор erbB2 опухоли у нуждающегося в этом субъекта, включающему введение указанному субъекту терапевтически эффективного количества ингибитора рецептора erbB2 путем инфузии указанному субъекту в течение 1-8 часов таким образом, что инфузия более эффективна, чем болюсная инъекция. Под болюсной инъекцией понимают относительно быструю терапевтическую инфузию, совместимую со свойствами области инъекции. Инфузия может быть внутривенной, внутримышечной, интраперитонеальной или подкожной. Объектом способа может быть человек, но подходит любое млекопитающее. В одном воплощении опухоль представляет собой злокачественное новообразование. В способе по изобретению инфузия может характеризоваться неравномерной скоростью. Например, скорость введения в ходе инфузии может увеличиваться или уменьшаться. Ингибитор может быть избирательным в отношении рецептора erbB2. Кроме того, способ может дополнительно включать: расчет соотношения аффинности связывания - ингибитора для рецептора erbB2 и второй аффинности связывания ингибитора для рецептора erbB1, и использование соотношения для оценки избирательности. Другие способы, известные в области техники, также подходят для оценки избирательности. В одном воплощении ингибитор по меньшей мере в два раза более избирателен в отношении рецептора erbB2. В еще одном воплощении ингибитор по меньшей мере в десять раз более избирателен в отношении рецептора erbB2. Объектом способа лечения в соответствии с изобретением может быть человек. Ингибитор может представлять собой антагонист. В одном воплощении ингибитор отличен от антитела или его фрагмента. В частности, ингибитор может представлять собой небольшую молекулу. Способ в соответствии с изобретением может дополнительно включать условие, чтобы ингибитор обладал периодом полувыведения in vivo от получаса до восьми часов.
Одно воплощение настоящего изобретения относится к способу лечения сверхэкспрессии рецептора erbB2 у нуждающегося в таком лечении млекопитающего, включающему:
(а) определение сверхэкспрессии рецептора erbB2 с использованием цитогенетического теста, флуоресцентной гибридизации in-situ, иммуногистохимического теста, теста с использованием проточной цитометрии, полимеразной цепной реакции с обратной транскриптазой или любой их комбинации;
(б) введение указанному млекопитающему терапевтически эффективного количества первого ингибитора рецептора erbB2 на основании сверхэкспрессии рецептора erbB2 в соответствии со стадией (а); и
(в) последовательное введение указанному млекопитающему после промежутка времени, составляющего меньше 24 часов, от одного до шести терапевтически эффективных количеств второго ингибитора рецептора erbB2 на основании сверхэкспрессии рецептора erbB2 в соответствии со стадией (а).
Способ может включать инфузию ингибитора, где ингибитор не является существенно цитотоксичным. Способ также может включать инфузию ингибитора, где ингибитор не является по существу ингибитором митоза.
Способ лечения путем инфузии ингибитора также может включать условие, чтобы инфузия была по меньшей мере на 20% более эффективной, чем болюсная инъекция.
Способ лечения путем инфузии также может включать инфузию дважды или трижды в сутки.
Способ лечения путем инфузии может дополнительно включать достижение уровня ингибитора в плазме от 10 нг/мл до 4000 нг/мл.
Используемый здесь термин "лечение", если не указано иначе, означает реверсию, облегчение, ингибирование прогрессирования или предотвращение расстройства или состояния, в отношении" которого применяется такой термин, или одного или более чем одного симптома такого расстройства или состояния. Используемый здесь термин "лечение", если не указано иного, относится к акту лечения в том смысле, в котором "лечение" определено выше.
Используемый здесь термин "Сmax", если не указано иного, означает максимальную концентрацию агента в крови, сыворотке или плазме после введения агента. Агент обычно представляет собой ингибитор рецептора erbB2 формулы 1.
Используемый здесь термин "AUC", если не указано иного, означает площадь под кривой и представляет собой меру концентрации агента, интегрированной по времени.
Используемый здесь термин "Сср." или "Сср.", если не указано иного, представляет собой меру средней концентрации агента в течение определенного периода времени.
Используемый здесь термин "ФК", если не указано иного, означает фармакокинетику или распределение агента стечением времени.
Используемые здесь термины "QD" и "BID", если не указано иного, оозначают соответственно ежедневное введение и введение дважды в сутки.
Используемые здесь термины "п.о. и "в.в.", если не указано иного, означают соответственно пероральный и внутривенный пути введения.
Используемый здесь термин "ФД", если не указано иного, означает фармакодинамику, анализ функциональных последствий применения агента.
Используемый здесь термин "избирательность", если не указано иного, означает эффективность относительно другого агента и обычно представляется как соотношение констант ингибирования (значений ингибирующей концентрации IC, такие как, например, IC50). Альтернативно избирательность может быть измерена как аффинность ингибитора в отношении рецептора erbB2 относительно аффинности в отношении другого рецептора, например erbB1. Избирательность может быть измерена любым подходящим образом, известным в данной области техники, включая абсолютную активность, активность относительно другого агента, эффективность относительно другого агента и наличие или степень эффектов не в отношении рецептора erbB2, но не ограничивающимся ими.
Используемый здесь термин "ингибирование рецептора erbB2", если не указано иного, обозначает конкурентное или неконкурентное блокирование связывания активатора, то есть агониста, замещение связавшегося активатора, уменьшение константы аффинности активатора, увеличение скорости диссоциации активатора, диссоциацию мультимерного рецептора, агрегацию мономерного рецептора или уменьшение внутриклеточных метаболических последствий активации рецептора.
Используемый здесь термин "синергизм" или "синергичный", если не указано иного, означает, что совместное действие двух ингибиторов больше, чем сумма действий каждого ингибитора в отдельности.
Используемый здесь термин "агонист", если не указано иного, означает лекарства, которые связываются с физиологическим рецептором и имитируют действие эндогенных регуляторных соединений. Используемый здесь термин "антагонист", если не указано иного, означает лекарства, которые связываются с рецептором и не имитируют связывание эндогенного агониста, а воздействуют на него. Такие лекарства или соединения, которые сами по себе лишены собственной регуляторной активности, но которые оказывают действие путем ингибирования действия агониста, именуются "антагонистами".
Используемый здесь термин "побочное действие", если не указано иного, означает действие или эффект лекарства, отличные от желаемого эффекта.
Используемый здесь термин "уменьшенное побочное действие", если не указано иного, означает уменьшенное действие или эффект лекарства, отличные от желаемого эффекта.
Используемый здесь термин "ингибитор", если не указано иного, означает химическое вещество, которое останавливает активность фермента или рецептора.
Те соединения формулы 1, которые являются кислыми по природе, способны образовывать соли оснований с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли щелочных или щелочно-земельных металлов и, в частности, кальциевые, магниевые, натриевые и калиевые соли соединений в соответствии с настоящим изобретением.
Некоторые функциональные группы, содержащиеся в соединениях по настоящему изобретению, могут быть заменены биоизостерными группами, то есть группами, которые обладают сходными пространственными или электронными требованиями с родительской группой, но демонстрируют отличающиеся или улучшенные физико-химические или другие свойства. Подходящие примеры хорошо известны специалистам в данной области техники и включают группировки, описанные в Patini et al., Chem. Rev, 1996, 96, 3147-3176 и цитируемых ссылках, но не ограничиваются ими.
Соединения формулы 1 могут иметь асимметрические центры и поэтому существовать в различных энантиомерных и диастереомерных формах. Изобретение относится к применению всех оптических изомеров и стереоизомеров соединений в соответствии с настоящим изобретением и их смесей и ко всем фармацевтическим композициям и способам лечения, которые могут быть использованы или содержать их. Соединения формулы 1 также могут существовать в виде таутомеров. Изобретение относится к применению всех таких таутомеров и их смесей.
Рассматриваемое изобретение также включает применение меченных изотопами соединений и их фармацевтически приемлемых солей, сольватов и пролекарств, которые идентичны соединениям и их фармацевтически приемлемым солям, сольватам и пролекарствам, приведенным в формуле 1, за исключением того, что один или более чем один атом замещен атомом, имеющим атомную массу или массовое число, отличающееся от атомной массы или массового числа, обычно обнаруживаемого в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13С, 14С, 15N, 18О, 17O, 35S, 18F и 36Cl соответственно. Соединения в соответствии с настоящим изобретением, их пролекарства и фармацевтически приемлемые соли указанных соединений или указанных пролекарств, которые содержат вышеупомянутые изотопы и/или другие изотопы других атомов, входят в объем изобретения. Некоторые меченные изотопами соединения в соответствии с настоящим изобретением, например соединения, в которые включены радиоактивные изотопы, такие как 3H и 14С, полезны в анализах тканевого распределения лекарства и/или субстрата. Тритированные изотопы, то есть 3H и углерод-14, то есть 14С, особенно предпочтительны ввиду легкости их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, то есть 2H, может обеспечить определенные терапевтические преимущества, являющиеся результатом большей метаболической стабильности, например увеличенного периода полувыведения in vivo или уменьшенной потребности в дозе и, следовательно, в некоторых случаях может быть предпочтительным. Меченные изотопами соединения формулы 1 в соответствии с изобретением и их пролекарства, как правило, могут быть получены путем осуществления методик, раскрытых на представленных ниже Схемах и/или в Примерах и Подготовительных примерах, путем замены не меченного изотопом реагента легкодоступным меченным изотопом реагентом.
Соединения формулы 1, обладающие свободными амино, амидо, гидроксильными или карбоксильными группами, могут быть превращены в пролекарства. Пролекарства включают соединения, где аминокислотный остаток или полипептидная цепь из двух или более (например двух, трех или четырех) аминокислотных остатков ковалентно связана посредством амидной или сложноэфирной связи со свободной амино, гидроксильной или карбоксильной группой соединений формулы 1. Аминокислотные остатки включают 20 аминокислот природного происхождения, обычно обозначаемых трехбуквенными символами, и также включают 4-гидроксипролин, гидроксилизин, демозин, изодемозин, 3-метилгистидин, норвалин, бета-аланин, замма-аминомасляную кислоту, цитрулин, гомоцистеин, гомосерин, орнитин и метионинсульфон, но не ограничиваются ими. Также охвачены дополнительные типы пролекарств. Например, могут быть получены производные свободных карбоксильных групп в виде амидов или алкильных эфиров. Могут быть получены производные свободных гидроксильных групп с использованием групп, включающих гемисукцинаты, фосфатные сложные эфиры, диметиламиноацетаты и фосфорилоксиметилоксикарбонилы в соответствии с тем, как представлено в Advanced Drug Delivery Reviews, 1996, 19, 115, но не ограничивающихся ими. Также включены карбаматные пролекарства гидроксильных и аминогрупп, а также карбонатные пролекарства, сульфонатные сложные эфиры и сульфатные сложные эфиры гидроксильных групп. Также охвачено получение производных гидроксильных групп в виде (ацилокси)метильных и (ацилокси)этильных простых эфиров, где ацильная группа может представлять собой алкильный сложный эфир, возможно, замещенный группами, включающими функциональные группы простого эфира, амина и карбоновой кислоты, но не ограничивающимися ими, или где ацильная группа представляет собой сложный эфир аминокислоты в соответствии с вышеописанным. Пролекарства этого типа описаны в. J. Med. Chem. 1996, 39, 10. Также могут быть получены производные свободных аминокислот в виде амидов, сульфонамидов или фосфонамидов. Все такие группировки пролекарств могут включать группы, включающие функциональные группировки простого эфира, амина и карбоновой кислоты, но не ограничивающиеся ими.
Краткое описание графических материалов
На Фиг.1 представлена противоопухолевая эффективность ингибитора Е-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)ацетамида, вводимого п.о. QD мышам, имеющим опухоли FRE/erbB2. По ординате отложено измерение опухолевого роста на 7-е сутки относительно контроля (носитель).
На Фиг.2 представлена противоопухолевая эффективность ингибитора E-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида, вводимого в.в. QD мышам, имеющим опухоли FRE/erbB2. По ординате отложено измерение опухолевого роста на 7-е сутки относительно контроля (носитель).
На Фиг.3 представлена противоопухолевая эффективность с течением времени ингибитора Е-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида, вводимого п.о. и QD мышам nu/nu, несущим опухоль SK-OV-3. На Фиг.3 символы имеют следующие значения: темный кружок, носитель, BID; светлый кружок, ингибитор в концентрации 50 мг/кг, QD; треугольник, ингибитор в концентрации 100 мг/кг, QD; и квадрат, ингибитор в концентрации 200 мг/кг, QD.
На Фиг.4 представлена противоопухолевая эффективность с течением времени ингибитора Е-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида, вводимого п.о. и QD мышам nu/nu, несущим опухоль SK-OV-3. На Фиг.4 символы имеют следующие значения: закрашенный кружок, носитель, BID; незакрашенный кружок, ингибитор в концентраций 25 мг/кг BID; ромб, ингибитор в концентрации 50 мг/кг BID; и треугольник, ингибитор в концентрации 100 мг/кг BID.
На Фиг.5А представлена противоопухолевая эффективность ингибитора Е-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)ацетамида, вводимого мышам, несущим опухоли ВТ-474, иллюстрирующая влияние множественных доз.
На Фиг.5Б представлена противоопухолевая эффективность ингибитора Е-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)ацетамида, вводимого мышам, несущим опухоли ВТ-474, иллюстрирующая влияние частоты дозирования.
На Фиг.6А представлена противоопухолевая эффективность ингибитора Е-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида, вводимого QD мышам, несущим опухоли MDA-MB-453.
На Фиг.6Б представлена противоопухолевая эффективность ингибитора E-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида, вводимого BID мышам, несущим опухоли MDA-MB-453.
Подробное описание изобретения
Способ по изобретению может включать введение ингибитора, где ингибитор на стадии (а), ингибитор на стадии (б) или оба не являются существенно цитотоксичными. Цитотоксичность может быть определена любыми средствами, традиционными в данной области техники, включая измерение апоптоза и метаболических функций, таких как дыхание и утилизация, но не ограничивающихся ими. Под существенно цитотоксичным понимают, что специалист в данной области техники признал бы, что цитотоксичность в общем обнаружена при введении агента тестируемому животному или при использовании анализа in vitro в условиях и концентрациях, соответствующих применению агента в изобретении.
Способ может включать введение ингибитора, где ингибитор на стадии (а), ингибитор на стадии (б) или оба не являются по существу ингибиторами митоза. Митоз может быть определен любыми средствами, традиционными в данной области техники, включая измерения митотического индекса, содержания ДНК и количества клеток, но не ограничивающихся ими. Под по существу ингибитором митоза понимают, что специалист в данной области техники признал бы, что уменьшенный митоз в общем обнаружено при введении агента тестируемому животному или при использовании анализа in vitro в условиях и концентрациях, соответствующих применению агента в изобретении.
Активность соединений in vitro для применения в способах в соответствии с настоящим изобретением может быть определена по величине ингибирования фосфорилирования тестируемым соединением относительно контроля. Внутриклеточные домены рекомбинантного erbB2 (аминокислотные остатки 675-1255) и рецептора эпидермального фактора роста (EGFR) (аминокислотные остатки 668-1211) экспрессировали в клетках Sf9, инифицированных бакуловирусом, в виде слитых белков с глутатион-S-трансферазой (GST) и очищали путем аффинной хроматографии на глутатионсефарозных шариках, Фосфорилирование поли(Clu, Tyr) измеряли, как описано в J.D.Moyer, E.G.Barbacci, K.K.Iwata, L Arnold, В.Boman, A.Cunningham, et al., Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase, Cancer Res. 57 (1997) 4838-4848, за исключением того, что киназную реакцию осуществляли в 50 мкл 50 мМ HEPES, рН 7,4, содержащего 125 мМ хлорида натрия, 10 мМ хлорида магния, 0,1 мМ ортованадата натрия и 1 мМ АТФ (аденозинтрифосфат).
Фосфорилирование тирозина в интактных клетках может быть измерено с использованием следующего анализа. Клетки NIH3T3, трансфицированные EGFR человека (B.D.Cohen, D.R.Lowy, J.T.Schiller, Transformation-specific interaction of the bovine papillomavirus E5 oncoprotein with the platelet-derived growth factor receptor transmembrane domain and the epidermal growth factor receptor cytoplasmic domain, J. Virol., 67 (1993) 5303-5311) или химерным рецептором с внеклеточным доменом EGFR и внутриклеточным доменом еrbВ2, высевали в 96-луночные тканевые культуральные планшеты в среде Игла, модифицированной Дульбекко (DMEM) (F.Fazioli, U.H.Kirn, S.G.Rhee, C.J.Molloy, O.Segatto, P.P.DiFlore, The erbB-2 mitogenic signaling pathway: tyrosine phosphorylation of phospholipase C-gamma and GTPase-activating protein does not correlate with erbB-2 mitogenic potency, Mol. Cell. Biol 11 (1991) 2040-2048).
Ингибиторы в диметилсульфоксиде (ДМСО) (или ДМСО в качестве носителя для контролей) добавляли через 24 ч после высевания и инкубировали с клетками в течение 2 ч при 37°С. Клетки стимулировали рекомбинантным эпидермальным фактором роста (EGF) человека (конечная концентрация 50 нг/мл) в течение 15 мин при комнатной температуре. Среду отсасывали и клетки фиксировали в течение 30 мин с использованием 100 мкл охлажденной смеси 1:1, этанол:ацетон, содержащей 200 мкМ Na3VO4. Планшеты промывали промывающим буфером (0,5% Tween-20 в забуференном фосфатом физиологическом растворе (ЗФР)) и добавляли 100 мкл блокирующего буфера (3% бычий сывороточный альбумин (БСА) в ЗФР + 200 мкМ свежеприготовленного ортованадата натрия). Планшеты дополнительно инкубировали в течение 1 ч при комнатной температуре и дважды промывали промывающим буфером. В лунки добавляли антитело против фосфотирозина (PY54), меченное пероксидазой хрена, и инкубировали в течение 1 ч при комнатной температуре. Антитело удаляли путем отсасывания и планшеты промывали 4 раза промывающим буфером. Колориметрический сигнал проявляли путем добавления 3,3'5,5'-тетра-метилбензидинового субстрата для пероксидазы (ТМВ Microwell Peroxidase Substrate) (Kirkegaard and Perry, Gaithersburg, MD) по 50 мкл в лунку и останавливали путем добавления 0,09 М серной кислоты по 50 мкл в лунку. Фосфотирозин оценивали путем измерения поглощения при длине волны 450 нм. Сигнал, полученный в контрольных лунках, не содержащих соединения, простимулированных EGF после вычитания фонового сигнала в лунках без EGF, определяли как 100% контроля. Проверка экстрактов этих стимулированных EGF клеток при помощи вестерн-блоттинга с антителами против фосфотирозина указывала на то, что большая часть белка фосфотирозина демонстрировала соответственно аутофосфорилированные EGFR или химеры EGFR/erbB2 соответственно, но другие белковые субстраты также демонстрировали увеличение фосфорилирования тирозина. EGF, как правило, увеличивал общие уровни фосфотирозина приблизительно в 4 раза в каждой трансфицированной клетке. Значения IC50 представляют собой концентрацию соединения, требующуюся для уменьшения сигнала до 50% от контроля, и они были определены графически на основе титрования на протяжении 100-кратного диапазона концентраций. За анализом фосфорилирования erbB путем иммунопреципитации следовал вестерн-блоттинг. Клетки SКВr3 обрабатывали соединением или активирующим лигандом в соответствии с тем, как указано. Среды отсасывали и добавляли 1 мл/75 см2 колбы охлажденного на льду лизирующего буфера для иммунопреципитации (1,0% ТХ100; 10 мМ Трис; 5 мМ этилендиаминтетрауксусная кислота (ЭДТА); 50 мМ NaCl; 30 мМ ортованадат натрия со свежедобавленными 100 мкМ фенилметилсульфонилфторида (ФМСФ) и 1 таблеткой ингибитора протеаз Complete™ (Roche Diagnostics, Indianapolis, IN на 50 мл буфера). Иммунопреципитацию проводили в 100 мкл лизата: EGFR подвергали иммунопреципитации с использованием Santa Cruz SC-120, 2 мкг/100 мкл лизата; erbB2 - с использованием Oncogene OP15, 1 мкг/100 мкл лизата; и erbB3 - с использованием Santa Cruz SC-285, 2 мкг/100 мкл лизата. Все процедуры иммунопреципитации проводили при 4°С в течение ночи на качалке в присутсвии 30 мкл шариков белка А. Шарики с иммобилизованным белком отделяли путем центрифугирования при 14000 об/мин, 4°С в течение 10 секунд. Супернатанты отсасывали и осадки трижды промывали ЗФР с 0,1% Tween 20. Затем образцы ресуспендировали в 40 мкл буфера Лэмли с дитиотреитолом (ДТТ) и кипятили в течение 4 минут. Затем образцы наносили на 4-12%-ный полиакриламидный гель для электрофореза (PAGE). Их подвергали электрофорезу в течение 1 часа при 150 В с использованием MES буфера. Гели переносили на поливинилиденфторид (ПВДФ) в присутствии 10% метанола. Мембрану блокировали с использованием блокирующего буфера (Roche Diagnostics, Indianapolis, IN) и фосфотирозин обнаруживали с использованием анти-РУ54 антитела, конъюгированного с пероксидазой хрена, и визуализировали путем усиленной хемилюминесценции в соответствии с инструкциями производителя (ECL™; Amersham, Pharmacia Biotech, Piscataway, NJ; LumiGLO™; Cell Signaling). Сигнал количественно оценивали при помощи Lumi-imager™ (Boehringer Mannheim, Indianapolis, IN).
Следующий анализ также может быть использован для с-еrbВ2 киназы для определения эффективности и избирательности соединений для их применения в качестве ингибиторов с-erbB2. Следующий анализ похож на анализ, описанный ранее в Schrang et al. Anal. Biochem. 211, 1993, р233-239. 96-луночные планшеты Nunc MaxiSorp покрывают путем инкубации в течение ночи при 37°С со 100 мл на лунку 0,25 мг/мл поли(Clu, Tyr) 4:1 (PGT) (Sigma Chemical Co., St. Louis, МО) в ЗФР (забуференный фосфатом физиологический раствор). Избыток PGT удаляют путем отсасывания и планшет трижды промывают промывающим буфером (0,1% Tween.20 в ЗФР). Киназную реакцию осуществляют в 50 мл 50 мМ HEPES (рН 7,5), содержащего 125 мМ хлорида натрия, 10 мМ хлорида магния, 0,1 мМ ортованадата натрия, 1 мМ АТФ, 0,48 мг/мл (24 нг/лунку) внутриклеточного домена с-erbB2. Внутриклеточный домен тирозинкиназы erbB2 (аминокислоты 674-1255) экспрессируют в виде GST слитого белка в бакуловирусе и очищают путем связывания с шариками, покрытыми глутатионом, с последующей элюцией. Добавляют соединение в ДМСО с получением конечной концентрации ДМСО 2,5%. Фосфорилирование инициируют путем добавления АТФ (аденозинтрифосфат) и осуществляют в течение 6 минут при комнатной температуре при постоянном встряхивании. Киназную реакцию останавливают путем отсасывания реакционной смеси и последующего промывания промывающим буфером (смотри выше). Фосфорилированный PGT измеряют путем инкубации в течение 25 минут с 50 мл на лунку конъюгированного с пероксидазой хрена (ПХ) PY54 (Опсодепе Science Inc. Uniondale, NY) антифософотирозинового антитела, разведенного до 0,2 мг/мл в блокирующем буфере (3% бычий сывороточный альбумин (БСА) и 0,05% Tween 20 в ЗФР). Антитело удаляют путем отсасывания и планшет 4 раза промывают промывающим буфером. Колориметрический сигнал проявляют путем добавления ТМВ Microwell Peroxidase Substrate (Kirkegaard and Perry, Gaithersburg, MD) no 50 мкл в лунку и останавливают путем добавления 0,09М серной кислоты по 50 мкл в лунку. Фосфотирозин оценивают путем измерения поглощения при длине волны 450 нм. Сигнал для контролей, как правило, составляет 0,6-1,2 единицы поглощения по существу при отсутствии фона в лунках без субстрата PGT, и он пропорционален времени инкубации в течение 10 минут. Ингибиторы идентифицируют по уменьшению сигнала по сравнению с лунками без ингибитора и определяют значения IC50, соответствующие концентрации соединения, требующейся для 50% ингибирования. Соединения, приведенные здесь для примера, которые соответствуют формуле 1, имеют значения IC50 меньше 10 мМ в отношении erbB2 киназы. Значения IC50 могут быть использованы для определения избирательности при помощи любого из способов, известных в области техники. Например, может быть использовано соотношение значений IC50 для рецепторов erbB1 и рецепторов erbB2 (IC50 erbB1 (IC50 erbB2). Благоприятно, когда это соотношение превышает два.
Противоопухолевая активность соединений in vivo для применения в способах в соответствии с настоящим изобретением может быть определена по величине ингибирования опухолевого роста тестируемым соединением по сравнению с контролем. Эффекты разных соединений по ингибированию опухолевого роста могут быть измерены в соответствии со способом Corbett Т.Н., et al., "Tumor Induction Relationships in Development of Transplantable Cancers of the Colon in Mice for Chemotherapy Assays, with a Note on Carcinogen Structure", Cancer Res., 35, 2434-2439 (1975) и Corbett Т.Н., et al., "A Mouse Colon-tumor Model for Experimental Therapy", Cancer Chemother. Rep. (Part 2)", 5, 169-186 (1975), с небольшими модификациями. Опухоли могут быть индуцированы в левом боку мышей путем подкожной (п.к.) инъекции 1-5 миллионов находящихся в логарифмической фазе роста опухолевых клеток, суспендированных в 0,1 мл среды RPMI 1640. После протекания периода времени, достаточного для того, чтобы опухоль стала пальпируемой (размер приблизительно 100-150 мм3/5-6 мм в диаметре), тестируемых животных (самки бестимусных мышей) лечат тестируемым соединением (приготовленным в концентрации 10-15 мг/мл в 5 Gelucire или 0,5%-ной метил целлюлозе) путем внутривенного (в.в.) или перорального (п.о.) введения один или два раза в сутки в течение 7-29 последовательных суток. Для определения противоопухолевого действия опухоль измеряют в миллиметрах с использованием штангенциркуля с нониусом по двум диаметрам и размер опухоли (мм3) рассчитывают с использованием формулы: размер опухоли (мм3)=(Ш×Ш)/2×Д (Д=длина и Ш=ширина) в соответствии со способами Geran, R.I., et al. "Protocols for Screening Chemical Agents and Natural Products Against Animal Tumors and Other Biological Systems", Third Edition, Cancer Chemother. Rep., 3, 1-104 (1972). Результаты выражают в виде процента ингибирования в соответствии с формулой: ингибирование роста (%)=[100-{(% роста подвергнутых лечению/% роста контролей)×100}]. Боковая область имплантации опухоли обеспечивает воспроизводимые эффекты доза/ответ для различных химиотерапевтических агентов, и способ измерения (диаметр опухоли) представляет собой надежный способ оценки скоростей роста опухоли.
Введение ингибиторов erbB2 может быть осуществлено с использованием любого способа, который делает возможной доставку соединений к месту действия. Эти способы включают пероральные пути, интрадуоденальные пути, парентеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутрисосудистую инъекцию или инфузию), местное и ректальное введение.
Вводимое количество активного соединения зависит от субъекта, которого лечат, тяжести расстройства или состояния, скорости введения, нахождения соединения и решения лечащего врача. Тем не менее, эффективная дозировка находится в диапазоне от 0,001 до 200 мг на кг массы тела в сутки, предпочтительно от 1 до 35 мг/кг/сутки. Для человека массой 70 кг это количество составляет от 0,05 до 7 г/сутки, предпочтительно от 0,2 до 2,5 г/сутки. В некоторых случаях уровени дозировки, находящиеся ниже нижней границы вышеуказанного диапазона, могут быть более чем достаточными, тогда как в других случаях могут быть использованы еще большие дозы, не вызывая какого-либо вредного побочного действия.
Ингибиторы erbB2 в соответствии с настоящим изобретением могут быть применены в виде монотерапии или могут включать одно или более других противоопухолевых веществ, в частности выбранных из, например, ингибиторов митоза, например винбластина; алкилирующих агентов, например цис-платина, карбоплатина и циклофосфамида; антиметаболитов, например 5-фторурацила, арабинозида цитозина и гидроксимочевины, или, например, одного из предпочтительных антиметаболитов, раскрытых в европейской заявке на патент №239362, такого как N-(5-[N-(3,4-дигидро-2-метил-4-оксохиназолин-6-илметил)-N-метиламино]-2-теноил)-1-глутаминовая кислота; ингибиторов факторов роста; ингибиторов клеточного цикла; интеркалирующих антибиотиков, например адриамицина и блеомицина; ферментов, например, интерферона; и антигормонов, например анти-эстрогенов, таких как Nolvadex™ (тамоксифен) или, например анти-андрогенов, таких как Casodex™ (4'-циано-3-(4-фторфенилсульфонил)-2-гидрокси-2-метил-3'-(трифторметил)пропион-анилид). Такое комбинированное лечение может быть осуществлено путем одновременного, последовательного или раздельного дозирования индивидуальных компонентов лечения.
Фармацевтическая композиция может, например, находиться в форме, подходящей для перорального введения в виде таблетки, капсулы, пилюли, порошка, препаратов с длительным высвобождением, раствора, суспензии, для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема или для ректального введения в виде суппозитория. Фармацевтическая композиция может быть представлена в стандартных лекарственных формах, подходящих для однократного введения точных дозировок. Фармацевтическая композиция включает обычный фармацевтический носитель или эксципиент и соединение в соответствии с изобретением в качестве активного ингредиента. Дополнительно она может включать другие медицинские или фармацевтические агенты, носители, адъюванты и так далее.
Примеры форм для парентерального введения включают растворы или суспензии активных соединений в стерильных водных растворах, например водных растворах пропиленгликоля или декстрозы. Такие лекарственные формы могут, если желательно, быть подходящим образом забуферены.
Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители. Фармацевтические композиции могут, если желательно, содержать дополнительные ингредиенты, такие как корригенты, связывающие вещества, эксципиенты и тому подобное. Таким образом, для перорального введения таблетки, содержащие различные эксципиенты, такие как лимонная кислота, могут быть использованы вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и некоторые комплексные силикаты, и со связывающими агентами, такими как сахароза, желатин и аравийская камедь. Для целей таблетирования часто, кроме того, оказываются полезными смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции сходного типа также могут быть использованы в твердых и мягких желатиновых капсулах с наполнением. Предпочтительные материалы для этого включают лактозу или молочный сахар и высокомолекулярные полиэтиленгликоли. Когда для перорального введения желательны водные суспензии или эликсиры, активное соединение может быть комбинировано с различными подсластителями или корригентами, красящими веществами или красителями и, если желательно, эмульгаторами или суспендирующими агентами вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации.
Способы приготовления различных фармацевтических композиций с конкретным количеством активного соединения известны или очевидны для специалиста в данной области техники. Например, смотри Remington's Pharmaceutical Sciences, Mack Publishing Company, Easter, Pa., 15th Edition (1975).
Предложенные ниже примеры и подготовительные примеры иллюстрируют и поясняют способы в соответствии с настоящим изобретением, причем понятно, что объем настоящего изобретения не ограничивается каким-либо образом объемом следующих примеров и подготовительных примеров.
"Тестируемое соединение", используемое в следующих Примерах, если не указано иного, представляет собой избирательный ингибитор erbB2, E-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)фениламино)-хиназолин-6-ил}-аллил)-ацетамид.
Пример 1
Модель FRE: Влияние длительности воздействия на противоопухолевую эффективность тестируемого соединения
Задача предклинических исследований заключалась в том, чтобы определить, являются ли Сmax или площадь под кривой (AUC) для тестируемого соединения критичными для противоопухолевой эффективности. Дополнительная задача заключалась в том, чтобы установить фармакокинетические/фармакодинамические (ФК/ФД) соотношения в модели опухоли FRE/erbB2. FRE/erbB2 представляет собой сконструированную модель опухоли мыши, которая сверхэкспрессирует erbB2 человека с трансмембранной мутацией.
Определяли роль продолжительности воздействия тестируемым соединением на рост опухоли FRE/erbB2 у бестимусных мышей. Тестируемое соединение вводили путем инфузии в хвостовую вену или перорально. При использовании инфузии в хвостовую вену в течение суток поддерживали рассчитанную фиксированную концентрацию Сmax (1200 нг/мл), тогда как длительность воздействия, а следовательно, и AUC варьировали. Варианты лечения и концентрации в плазме крови у животных, которых подвергали лечению, представлены в Таблице 1.
Раствор тестируемого соединения в концентрации 1,15 м г/мл вводили в.в. инфузией со скоростью 550 мкл/час посредством 2-минутных «нарастающих» инфузий с последующими ежедневными инфузиями 50 мкл/час в течение 15 мин или 4 часов (план основан на константе ингибирования (КИ) тестируемого соединения). Самок бестимусных мышей, несущих опухоли FRE/erbB2 (приблизительно 100 мм3 в размере), обрабатывали носителем, тестируемым соединением перорально или тестируемым соединением внутривенно. Изменения массы тела и размеры опухоли фиксировали с регулярными интервалами (сутки 1, 3, 5, и 7). Исследование осуществляли в течение 7 суток. Образцы плазмы и опухолей отбирали для анализа ФК и ФД в конце исследования. Результаты по противоопухолевой эффективности, объему опухоли, изменениям массы тела, концентрации тестируемого соединения в плазме, а также ингибированию р-erbB2 (фосфорилированной формы рецептора erbB2) у контрольных животных и животных, которых лечили тестируемым соединением, представлены в Таблице 1.
Исследование п.о., QD N=6; исследование в.в., QD N=4
%ИР =% ингибирование роста
Приблизительно 54% ингибирования роста опухоли удалось достичь у животных, которых лечили путем ежедневного перорального введения тестируемого соединения. Концентрация в плазме - через 0,5 часа после введения дозы на 7-е сутки составляла 1460 нг/мл. Лечение с использованием тестируемого соединения было безопасным и не вызывало какой-либо потери массы тела или смертности.
Ежедневная 15-минутная инфузия тестируемого соединения приводила в результате к ингибированию роста приблизительно на 34%. При этом эквивалентная инфузия 4 ч/сутки приводила в результате к существенно большему ингибированию роста опухоли (76%). Это означает, что длительность действия выше пороговой концентрации в плазме имеет важное значение в общей противоопухолевой эффективности тестируемого соединения в этой модели. Основываясь на этих результатах, также можно заключить, что действие (AUC) для 4 ч/сутки при приблизительной концентрации в плазме, составляющей 500 нг/мл, достаточно для того, чтобы вызвать значительное ингибирование роста опухоли FRE/erbB2. Длительность воздействия или AUC (действие) значительно влияют на эффективность: одна лишь ежедневная Сmax не может объяснить эффективность в этой модели.
Длительность действия (приблизительно 4 ч/сутки) при концентрации в плазме приблизительно 500 нг/мл обладает преимуществом по сравнению с меньшей длительностью действия (приблизительно 15 мин/сутки) в модели опухоли FRE/erbB2.
Противоопухолевая эффективность 25 мг/кг тестируемого соединения, вводимого перорально один раз в сутки, проявляемая в замедлении роста объема опухолей FRE у мышей nu/nu, представлена в виде столбиковых диаграмм на Фиг.1. На фигуре показано, что на седьмые сутки лечения объем опухоли FRE у мышей, которых подвергали лечению, составлял приблизительно половину от объема опухоли в контрольной группе.
На Фиг.2 в виде столбиковой диаграммы показано, что противоопухолевое действие 10 мг/кг тестируемого соединения, вводимого в.в. в течение 4-часового периода каждые сутки в течение семи суток, высокоэффективно как в абсолютном значении, так и при сравнении с ежесуточной инфузией приблизительно 1,4 мг/кг ингибитора в течение приблизительно 15 мин/сутки или инфузией носителя. Тестируемое соединение при приблизительно 10 мг/кг замедляло увеличение объема опухоли до уровня, составляющего менее 24% относительно контрольной группы, которой вводили носитель. При этом быстрая инфузия приблизительно 1,4 мг/кг замедляла увеличение объема опухоли до менее чем 66% относительно контрольной группы, которой вводили носитель.
Пример 2
Модель SK-OV-3: Влияние длительности воздействия на противоопухолевую эффективность тестируемого соединения
Предклинические исследования были проведены для определения того, является ли длительность действия тестируемого соединения критичной для противоопухолевой эффективности. Еще одна задача заключалась в том, чтобы установить минимальные эффективные (Сmax и Сср0-4 ч) концентрации в модели опухоли аденокарциномы яичников человека SK-OV-3.
В качестве предпосылок в Примере 1 было показано, что тестируемое соединение (п.о., QD) эффективно в отношении опухолей FRE erbB2. Аналогично в.в. введение тестируемого соединения было эффективным в отношении опухолей FRE erbB2. Эти результаты демонстрируют, что поддержание концентраций тестируемого соединения в крови приблизительно 500 нг/мл в течение 4 ч/сутки имеет преимущество перед меньшей длительностью воздействия (приблизительно 15 мин/сутки) при сравнимом уменьшении р-erbB2 (48-53%) в модели опухоли FRE erbB2. Фармакокинетические, фармакодинамические данные и данные по эффективности представлены в Таблице 1.
Основываясь на воздействии, измеренном на более ранних стадиях, Сmax, составляющая приблизительно 1200 нг/мл или AUC0-2 ч, составляющая приблизительно 985 нг·ч/мл для тестируемого соединения при действии приблизительно в течение 2 часов, были критичны для ингибирования роста опухоли FRE erbB2 приблизительно на 50%.
Это исследование было расширено на модель ксенотрансплантата человека, представляющую собой модель аденокарциномы яичников человека SK-OV-3, которая сверхэкспрессирует erbB2.
Клетки SK-OV-3, полученные из Американской коллекции типовых культур (АТСС) (Rockville, MD) выращивали в среде МсСоу, содержащей 10% фетальной телячьей сыворотки и пен./стрепт (пенициллин/стрептомицин). Клетки, находящиеся в стадии экспоненциального роста, собирали и подкожно (ПК) инокулировали (5 миллионов клеток/животное) самкам бестимусных мышей. Бестимусных мышей, несущих опухоли SK-OV-3 (размером приблизительно 100 мм3), случайным образом распределяли на 7 групп, как представлено в Таблице 2. Измерения опухоли и изменения массы тела были получены на 1, 3, 6, 10, 13 и 18-е сутки. Объем опухоли рассчитывали в соответствии со следующей формулой: Объем опухоли (мм3)=(Ш×Ш/2×Д (Д=длина и Ш=ширина). Образцы крови (приблизительно 50 мкл) отбирали через 0,5, 1, 2, 4 и 8 часов после введения дозы на 18 сутки для анализа ФК. Опухоли выделяли через 0,5 ч после введения дозы на 18 сутки для анализа ФД при помощи твердофазного иммуноферментного анализа (ИФА). Снижение р-erbB2, изменения объема опухоли и массы организма в контрольной группе и у животных, которых лечили тестируемым соединением, представлены в Таблице 2 ниже.
Фармакокинетика тестируемого соединения у мышей, несущих опухоль SK-OV-3
*Не обнаружено значимого различия между AUC0-время последн. и AUC0-4 ч.
Противоопухолевую эффективность тестируемого соединения при пероральном введении (QD и BID) определяли на модели аденокарциномы яичников человека SK-OV-3, которая сверхэкспрессирует erbB2. Кроме того, введение тестируемого соединения (QD или BID) было эффективным и вызывало дозозависимое ингибирование ксенотрансплантатов SK-OV-3 (Фиг.3 и 4). Тестируемое соединение хорошо переносилось, и не было потери массы тела или смертности среди животных.
Введение дозы тестируемого соединения QD 50 мг/кг в течение 18 суток не было эффективным. Было достигнуто ингибирование роста опухоли приблизительно на 29%, когда общую суточную дозу 50 мг/кг/сутки вводили в соответствии со схемой BID (25 мг/кг, BID). Снижение аутофосфорилирования рецептора erbB2 через 0,5 ч после введения дозы на 18-е сутки было сравнимым в обоих группах, которых лечили QD и BID (14-20%), тем не менее, Сmax для тестируемого соединения в группе 50 мг/кг QD была приблизительно в два раза выше по сравнению с животными, которым вводили дозу 25 мг/кг BID (Cmax, 3640 нг/мл по сравнению с 1780 нг/мл). Аналогично AUC0-4 ч (3410 нгч/мл по сравнению с 1560 нгч/мл) и Сср0-4 ч (853 нг/мл по сравнению с 390 нг/мл) в группе QD были приблизительно в два раза выше по сравнению с группой, которой вводили дозы BID. Эти результаты демонстрируют, что ни высокая Cmax, ни AUC0-4 ч не являются критичными для противоопухолевой эффективности тестируемого соединения. Среднее действие 390 нг/мл тестируемого соединения (Сср0-4 ч) дважды в сутки (BID) обладает преимуществом перед средним воздействием 853 нг/мл (Сср0-4 ч) один раз в сутки (QD), хотя оба подхода (QD и BID) обеспечивают сравнимое уменьшение аутофосфорилирования erbB2.
Преимущество введения доз BID по сравнению с QD также наблюдали при более высоких дозах тестируемого соединения в модели SK-OV-3. При сравнении с введением дозы тестируемого соединения 50 мг/кг BID (100 мг/кг/сутки) введение дозы 100 мг/кг/сутки QD приводило в результате к большему снижению аутофосфорилирования erbB2 (75% по сравнению с 24%) и ассоциировалось с более высокими Cmax (12100 нг/мл по сравнению с 3880 нг/мл), AUC0-4 ч (16300 нг·ч/мл по сравнению с 4180 нгч/мл) и Сср0-4 ч (4080 нг/мл по сравнению с 1050 нг/мл). Тем не менее, схема приема QD была менее эффективной, чем схема приема BID (23% по сравнению с 45% ингибирования роста опухоли). Эти результаты поддерживают заключение о том, что более высокая Cmax или AUC0-4 ч тестируемого соединения не обеспечивает какого-либо значимого преимущества в этой модели опухоли, тогда как частота воздействия (Сср0-4, BID по сравнению с QD) выше порогового уровня представляет собой детерминирующий фактор для противоопухолевой эффективности. Кроме того, снижение р-erbB2 опухоли SK-OV-3 приблизительно на 24% может быть достаточным для ингибирования роста приблизительно на 50%, если поддерживается большая средняя длительность действия при введении доз BID.
Абсорбция тестируемого соединения при пероральном введении нелинейна при введении дозы 200 мг/кг QD. Значения Сmax и Сср0-4 ч для тестируемого соединения были сравнимы у животных, которым вводили дозу 200 мг/кг QD, и у животных, которым вводили дозу 100 мг/кг BID. Несмотря на меньшее снижение аутофосфорилирования опухолевого erbB2 у животных, которым вводили дозу 100 мг/кг BID (62% по сравнению с 90%), ингибирование роста опухоли в этой группе было в два раза больше, чем у животных, которым вводили дозу 200 мг/кг, QD (71% по сравнению с 36%). Эти наблюдения дополнительно поддерживают заключение о том, что меньшее снижение аутофосфорилирования erbB2 (62% по сравнению с 90%) при более длительном/более частом суточном воздействии (схема приема BID) при сравнимой Сmax обладает значительным преимуществом.
Настоящие результаты находятся в соответствии с результатами, полученными на бестимусных мышах, несущих опухоли FRE erbB2 (Пример 1). В этом исследовании по сравнению с 15 мин/сутки поддержание концентраций тестируемого соединения в крови приблизительно 500 нг/мл при введении в течение 4 ч/сутки при сравнимом снижении аутофосфорилирования erbB2 имеет преимущество.
Таким образом, в этом примере результаты, полученные на модели опухоли SK-OV-3, свидетельствуют о том, что общее ежедневное воздействие, то есть частота суточного введения дозы, является критичной для противоопухолевой эффективности тестируемого соединения. То есть схема приема BID обладает преимуществом по сравнению со схемой приема QD. Большее снижение аутофосфорилирования erbB2 при меньшей длительности имеет ограниченную значимость.
Пример 3
Влияние длительности действия на противоопухолевую эффективность тестируемого соединения
Предклинические исследования были проведены для определения того, является ли длительность действия тестируемого соединения критичной для противоопухолевой эффективности. Еще одна задача заключалась в том, чтобы установить минимальные эффективные (Сmax и Сср0-4 ч) концентрации в модели опухоли аденокарциномы молочной железы человека ВТ-474.
В качестве предпосылок в Примере 1 было показано, что тестируемое соединение (п.о., QD) эффективно в отношении опухолей FRE erbB2. Аналогично в.в. введение тестируемого соединения было эффективным в отношении опухолей FRE erbB2. Эти результаты демонстрируют, что поддержание концентраций тестируемого соединения в крови приблизительно 500 нг/мл в течение 4 ч/сутки обладает преимуществом по сравнению с меньшей длительностью действия (приблизительно 15 мин/сутки) при сравнимом снижении р-erbB2 (48-53%) в модели опухоли FRE erbB2. Фармакокинетические, фармакодинамические данные и данные по эффективности представлены в Таблице 1.
Основываясь на воздействии, измеренном в проведенном раньше исследовании в модели FRE erbB2, исследование было расширено в Примере 2 на модель ксенотрансплантата аденокарциномы яичников человека SK-OV-3, которая сверхэкспрессирует erbB2. Тестируемое соединение было эффективным, и результаты, полученные на модели SK-OV-3 свидетельствуют о том, что общее суточное воздействие, т.е. частота суточного введения дозы, является критичной для противоопухолевой эффективности тестируемого соединения. Схема приема BID является более полезной по сравнению со схемой приема QD. Большее снижение аутофосфорилирования erbB2 при меньшей длительности имеет ограниченную значимость.
Настоящий пример распространяет оценку значимости частоты суточного введения дозы для противоопухолевой эффективности тестируемого соединения на модель аденокарциномы молочной железы человека ВТ-474, которая сверхэкспрессирует рецепторы erbB2.
Клетки ВТ-474 (RPMI 1640 с 10 мМ HEPES, 10% ФБС, и pen/strep [Gibco]), находящиеся в экспоненциальной фазе роста, собирали и инокулировали п.к. (5 миллионов клеток/животное) самкам бестимусных мышей. Кусочки опухолей ВТ-474, полученные при помощи троакара, затем имплантировали животным в правый бок. Мышей, несущих опухоль ВТ-474 (размер 50-320 мм3, N=40), случайным образом распределяли на 7 групп, каждая из которых состояла из 5-6 животных. Животных обрабатывали растворителем (п.о. BID) или тестируемым соединением (п.о. QD или BID) в соответствии с описанным в Таблице 4. Измерения опухоли и изменения массы тела проводили на 1, 6, 11, 15 и 22-е сутки после введения. Объем опухоли рассчитывали в соответствии со следующей формулой: объем (мм3)=(Ш×Ш)/2×Д (Д=длина и Ш=ширина). Образцы крови (приблизительно 50 мкл) отбирали через 0,5, 1, 2, 4 и 8 ч после введения дозы на 22-е сутки для анализа ФК. Опухоли выделяли через полчаса после введения дозы на 22-е сутки для анализа ФД с использованием твердофазного иммуноферментного анализа (ИФА).
Статистический анализ: ANOVA проводили на основе данных процентного роста и планируемые сравнения осуществляли между одинаковыми дозами. Данные были логарифмированы для анализа вследствие распределения значений. Способ Даннета-Тамхейна использовали для анализа путем множественного сравнения. Снижение р-erbB2, изменения объема опухоли и массы тела у контрольных животных и животных, которых лечили тестируемым соединением, представлены в Таблице 4.
Фармакокинетика тестируемого соединения у мышей, несущих опухоль ВТ-474, представлена в Таблице 5.
Значения представляют собой средние значения.
*3начения оценивали, основываясь на экстраполированной концентрации для 4 ч, исходя из воздействий 2 ч и 8 ч.
Таким образом, противоопухолевую эффективность тестируемого соединения при пероральном введении (QD и BID) определяли на модели аденокарциномы молочной железы человека ВТ-474, которая сверхэкспрессирует erbB2. Введение тестируемого соединения (QD или BID) было эффективным и вызывало ингибирование роста ксенотрансплантатов ВТ-474 (Фиг.5а и 5б). Тестируемое соединение хорошо переносилось, и не было потери массы тела или смертности среди животных. Вследствие широкой вариации исходного объема опухоли рассчитывали % роста каждой индивидуальной опухоли и среднее значение по каждой группе использовали для определения относительной противоопухолевой эффективности.
Лечение тестируемым соединением в дозе 15 мг/кг QD (15 мг/кг/сутки) и BID (30 мг/кг/сутки) в течение 22 суток было эффективным и вызывало ингибирование роста опухоли соответственно на 22% и 54% (р=0,007). Снижение аутофосфорилирования рецептора erbB2 через 0,5 ч после приема дозы на 22-е сутки было ниже предела обнаружения в обеих группах, подвергнутых лечению QD и BID, а определение Сср0-4 ч у животных, которым вводили дозу QD, было невозможно вследствие того, что экстраполируемый фрагмент AUC больше или равен 30% от общей AUC. Эффективные Сmax, AUC0-4 ч и Сср0-4 ч (ингибирование роста на 54%) для тестируемого соединения у животных, которым вводили дозу 15 мг/кг BID, составляли соответственно 616 нг/мл, 480 нг·ч /мл и 120 нг/мл.
ФК, ФД и противоопухолевую эффективность тестируемого соединения также определяли после обработок 30 мг/кг QD (30 мг/кг/сутки) и BID (60 мг/кг/сутки). Значения ФК были сравнимы для тестируемого соединения после введения дозы QD или BID при определении на 22-е сутки, т.е. Сmax (1800 нг/мл по сравнению с 1570 нг/мл), AUC0-4 ч (1280 нг·ч/мл по сравнению с 1440 нг·ч/мл) и Сср0-4 ч (320 нг/мл по сравнению с 360 нг/мл, Таблица 5). Снижение аутофосфорилирования erbB2 опухоли ВТ-474 у животных, которым вводили дозы QD, было больше по сравнению с животными, которым вводили дозы BID (57% по сравнению с 26%, р=0,06). Схема приема тестируемого соединения 30 мг/кг BID была более эффективной по сравнению с введением дозы QD (ингибирование роста на 68% по сравнению с 33%, р=0,053).
По сравнению с введением дозы тестируемого соединения 30 мг/кг QD или BID (30 мг/кг/сутки или 60 мг/кг/сутки), введение дозы 50 мг/кг/сутки QD или BID (50 мг/кг/сутки или 100 мг/кг/сутки) привело в результате к большему снижению аутофосфорилирования опухолевого erbB2 (снижение приблизительно на 75%). Параметры ФК для тестируемого соединения в группах, которым вводили 50 мг/кг QD или BID на 22-е сутки, также сравнимы, т.е. Сmax (5890 нг/мл по сравнению с 6170 нг/мл), AUC0-4 ч (4220 нг·ч/мл по сравнению с 5280 нг·ч/мл) и Сср0-4 ч (1060 нг/мл по сравнению с 1320 нг/мл). Схема приема QD представлялась менее эффективной по сравнению со схемой BID (35% ингибирование роста опухоли по сравнению с 68%, р=0,066).
Был проведен сводный тест, сравнивающий одинаковые дозы для QD и BID. Этот тест продемонстрировал, что в целом введение доз BID было более эффективным по сравнению с введением доз QD (р=0,0346). Это открытие свидетельствует о том, что кратность введения дозы тестируемого соединения оказывает положительное действие на общий результат лечения.
Сравнение ФК, ФД и противоопухолевой эффективности тестируемого соединения, обнаруживаемые в группах 50 мг/кг QD (50 мг/кг/сутки) по сравнению с 30 мг/кг BID (60 мг/кг/сутки) (две ближайшие группы по общей ежедневной дозе), также оценивали для определения значимости частоты введения доз. Снижение р-erbВ2 в группе, которой вводили дозу 50 мг/кг QD (50 мг/кг/сутки), было гораздо большим по сравнению с группой, которой вводили дозу 30 мг/кг BID (60 мг/кг/сутки) (снижение р-erbВ2 на 75% по сравнению с 26%, Таблица 4). Аналогично более высокие Сmax (5890 нг/мл по сравнению с 1570 нг/мл), AUC0-4 ч (4220 нг·ч/мл по сравнению с 1440 нг·ч/мл) и Сср0-4 ч (1060 нг/мл по сравнению с 360 нг/мл) для тестируемого соединения были обнаружены в группе, которой вводили 50 мг/кг QD по сравнению с группой, которой вводили 30 мг/кг BID (Таблица 5). Несмотря на меньшее снижение р-erbB2 и значения ФК для тестируемого соединения (т.е. Cmax, AUC0-4 ч и Сср0-4 ч) введение дозы 30 мг/кг BID (60 мг/кг/сутки) было более эффективным по сравнению с введением дозы 50 мг/кг QD (50 мг/кг/сутки). В целом наблюдали ингибирование роста опухоли в группах 30 мг/кг BID и 50 мг/кг QD соответственно приблизительно на 68% и 35% (р=0,0636). Хотя общая ежедневная доза тестируемого соединения в этих двух группах немного различалась, может быть сделано заключение, что частота введения суточной дозы, т.е. введение дозы BID обладает преимуществом перед введением дозы QD.
Эти результаты схожи с результатами, обнаруженными в исследовании модели опухоли SK-OV-3, Пример 2 выше, заключающимися в том, что частота введения суточной дозы, т.е. Сср0-4 при воздействии дважды в сутки с введением дозы BID обладает преимуществом по сравнению с Сср0-4 при воздействии один раз в сутки с введением дозы QD. Кроме того, снижение аутофосфорилирования в опухоли ВТ-474 приблизительно на 26% при введении дозы дважды в сутки BID может быть достаточным для ингибирования роста приблизительно на 50%, если средняя длительность воздействия (приблизительно 360 нг/мл) поддерживается в течение более длительного периода времени при введении дозы BID. Настоящие результаты соответствуют результатам в.в. введения тестируемого соединения посредством инфузии бестимусным мышам, несущим опухоли FRE erbB2. Это исследование продемонстрировало, что поддержание концентраций тестируемого соединения в крови приблизительно 500 нг/мл в течение 4 ч/сутки обладает преимуществом по сравнению с болюсным введением.
Таким образом, результаты, полученные на модели опухоли ВТ-474, свидетельствуют о том, что кратность введения дозы и частота введения суточной дозы являются критичными для противоопухолевой эффективности тестируемого соединения. Кратность введения дозы относится к введению дозы (X мг/кг) по меньшей мере от двух раз в сутки до шести или, возможно, семи раз в сутки по сравнению с ведением той же самой дозы (X мг/кг) один раз в сутки. Частота введения ежедневной дозы относится к делению суточной дозы, например по половине Х мг/кг дважды в сутки по сравнению с Х мг/кг один раз в сутки.
Большее снижение аутофосфорилирования erbB2 при меньшей длительности имеет ограниченную значимость.
Пример 4
Влияние длительности воздействия на противоопухолевую активность тестируемого соединения
Предклинические исследования были проведены для определения того, является ли длительность воздействия тестируемого соединения критичной для противоопухолевой активности. Еще одна задача заключалась в том, чтобы установить минимальные эффективные (Сmax и Сср0-4 ч) концентрации в модели опухоли аденокарциномы молочной железы человека MDA-MB-453.
В качестве предпосылок в Примере 1 было показано, что тестируемое соединение (п.о., QD) эффективно в отношении опухолей FRE erbB2. Аналогично в.в. введение тестируемого соединения было эффективным в отношении опухолей FRE erbB2. Эти результаты демонстрируют, что поддержание концентраций тестируемого соединения в крови приблизительно 500 нг/мл в течение 4 ч/сутки имеет преимущество перед меньшей длительностью воздействия (приблизительно 15 мин/сутки) при сравнимом снижении р-erbВ2 (48-53%) в модели опухоли FRE erbВ2. Фармакокинетические, фармакодинамические данные и данные по эффективности представлены в Таблице 1.
Исследование было расширено на модель ксеноторнсплантата аденокарциномы яичников человека SK-OV-3, которая сверхэкспрессирует erbB2. Тестируемое соединение было эффективным, и результаты, полученные на модели опухоли SK-OV-3, свидетельствуют о том, что общее ежедневное суточное воздействие, т.е. частота введения ежедневной дозы, является критичным для противоопухолевой эффективности тестируемого соединения (схема приема BID имеет преимущество по сравнению со схемой приема QD). Противоопухолевое действие схемы перорального введения тестируемого соединения QD по сравнению с BID также было исследовано в отношении модели аденокарциномы молочной железы человека ВТ-474, которая сверхэкспрессирует erbВ2. Эти результаты также свидетельствуют о том, что как кратность, так и частота введения дозы являются критичными для противоопухолевой эффективности тестируемого соединения. В общем, результаты, полученные на моделях SK-OV-3 и ВТ-474, свидетельствуют о том, что большее снижение аутофосфорилирования erbВ2 при меньшей длительности имеет ограниченную значимость.
Настоящее ииледование было поведено для определения противоопухолевой эффективности тестируемого соединения при пероральном введении в отношении дополнительной модели карциномы молочной железы человека MDA-MB-453, которая сверхэкспрессирует егЬВ2. Вторая задача авторов данного изобретения в этом исследовании заключалась в определении того, обладает ли кратность и частота введения дозы тестируемого соединения каким-либо преимуществом в отношении этой модели.
План исследования: Клетки MDA-MB-453 (DMEM/F12 с 10% ЗФР и pen/strep [Gibco]), находящиеся на стадии экспоненциального роста, собирали и п.к. инокулировали (5 миллионов клеток/животное) самкам бестимусных мышей. Мышей, несущих опухоль MDA-MB-453 (размер приблизительно 100 мм3, N=64), случайным образом распределяли по 8 группам, каждая из которых состояла из 8 животных. Животных обрабатывали растворителем (п.о. QD или BID) или тестируемым соединением (п.о. QD или BID) в соответствии с тем, как описано в Таблице 6. Размеры опухоли и изменения массы тела фиксировали на 1, 3, 7, 10, 14, 17, 21, 24, и 29-е сутки. Объем опухоли рассчитывали в соответствии со следующей формулой: объем опухоли (мм3)=(Ш×Ш)/2×Д (Д=длина и Ш=ширина). Образцы крови (приблизительно 50 мкл) отбирали через 0,5, 1, 2, 4 и 8 часов после введения дозы на 29-е сутки для анализа ФК. Опухоли выделяли через 0,5 ч после введения дозы на 29-е сутки для анализа ФД с использованием ИФА.
Статистический анализ: ANOVA проводили на основе данных процентного роста и планируемые сравнения осуществляли между одинаковыми дозами. Данные были логарифмированы для анализа ввиду распределения значений. Способ Даннета-Тамхейна использовали для анализа путем множественного сравнения.
Снижение р-erbВ2, изменения объема опухоли и массы тела у контрольных животных и животных, которых лечили тестируемым соединением, представлены в Таблице 6.
Фармакокинетика тестируемого соединения у мышей, несущих опухоль MDA-MB-453, представлена в Таблице 7.
Таким образом, противоопухолевую эффективность тестируемого соединения при пероральном введении (QD и BID) определяли в отношении модели аденокарциномы молочной железы человека MDA-MB-453, которая сверхэкспрессирует erbB2. Введение тестируемого соединения (QD или BID) было эффективным и вызывало ингибирование роста ксенотрансплантатов MDA-MB-453 (Фиг.6а и 6б). Тестируемое соединение хорошо переносилось, и не было потери массы тела или смертности среди животных.
Лечение тестируемым соединением 50, 100 и 200 мг/кг QD (50, 100 и 200 мг/кг/сутки) в течение 29 суток было эффективным и вызывало ингибирование роста опухоли соответственно на 38%, 63% и 100%. Снижение аутофосфорилирования рецептора erbВ2 через 0,5 ч после введения дозы на 29-е сутки в группах 50, 100 и 200 мг/кг составляло соответственно 78%, 88% и 92%. Ведение дозы тестируемого соединения 25, 50 и 100 мг/кг BID в течение 29 суток было эффективным в отношении опухолей МВА-МВ-453 и вызывало ингибирование роста соответственно на 19%, 66% и 83%. Снижение p-erbB2 в этих группах составляло соответственно 69%, 75% и 79%.
ANOVA использовали для статистического анализа общей эффективности для различных доз тестируемого соединения. Способ Даннета-Тамхейна использовали для множественных сравнений с корректировкой на носитель. Результаты демонстрируют, что отсутствовало значимое различие между схемами приема тестируемого соединения 25 мг/кг BID и 50 мг/кг QD (р=0,295), 50 мг/кг BID и 100 мг/кг QD (р=0,703) и 100 мг/кг BID и 200 мг/кг QD (р=0,117). Аналогично, отсутствовало значимое различие между одинаковыми дозами, т.е. 50 мг/кг BID по сравнению с 50 мг/кг QD (р=0,13) и 100 мг/кг BID по сравнению с 100 мг/кг QD (р=0,17). Сравнительная статистическая оценка с использованием только дозы/схемы приема и противоопухолевой эффективности, обнаруживаемой в различных группах, недостаточна для получения какого-либо окончательного заключения для ответа на вопрос; обладает ли схема приема тестируемого соединения BID каким-либо преимуществом по сравнению с введением дозы тестируемого соединения QD.
Снижение p-erbB2 после введения доз QD (50-200 мг/кг) или BID (25-100 мг/кг) составляло 69-92%, и его сложно использовать в качестве параметра для какого-либо дальнейшего статистического анализа данных. Поэтому анализ данных был расширен с использованием фармакокинетических параметров тестируемого соединения, т.е. Сmax и Сср0-4 ч.
Сср0-4 ч, составляющая 591 нг/мл и 3120 нг/мл, полученная после введения дозы 50 мг/кг (50 мг/кг/сутки) и 100 мг/кг (100 мг/кг/сутки) QD, вызывала ингибирование роста опухоли соответственно на 38% и 63%. Сср0-4 ч, составляющая 509 нг/мл, полученная при схеме приема 50 мг/кг дважды в сутки BID, приводила в результате к 66% эффективности. Сср0-4 ч, составляющая 509 нг/мл, поддерживаемая в течение 8 ч/сутки при введении дозы BID, статистически не отличалась от поддержания Сср0-4 ч при 591 нг/мл (введение дозы 50 мг/кг QD) или 3120 нг/мл (введение дозы 100 мг/кг QD) в течение 4 ч/сутки (соответственно р=0,13 и р=0,58). Это также может быть интерпретировано таким образом, что поддержание средней концентрации в плазме 509 нг/мл в течение 8 ч/сутки обладало равной или большей пользой по сравнению с поддержанием средних концентраций в плазме 591-3120 нг/мл в течение 4 ч/сутки. Сmax для тестируемого соединения в группах 50 мг/кг QD и 50 мг/кг BID была сравнимой (2760 нг/мл по сравнению с 2390 нг/мл), тогда как Сmax в группе 100 мг/кг QD была приблизительно в4 раза выше (9770 нг/мл). Эти результаты свидетельствуют о том, что сами по себе более высокие Сmax или Сср0-4 ч обладают ограниченной значимостью при сравнимом снижении р-erbВ2.
Также было проведено сравнение Сmax и Сср0-4 ч с противоопухолевой эффективности тестируемого соединения, наблюдаемой в группах 100 мг/кг BID и 200 мг/кг QD. Сmax для тестируемого соединения в группе 200 мг/кг QD была в 2,4 раза выше по сравнению с Сmax в группе 100 мг/кг BID (16700 нг/мл по сравнению с 6870 нг/мл). Аналогично, Сср0-4 ч была в 3,8 раза выше в группе 200 мг/кг QD по сравнению с группой 100 мг/кг BID (6510 нг/мл по сравнению с 1710 нг/мл). Несмотря на более высокие Сmax и Сср0-4 ч общая эффективность тестируемого соединения, наблюдаемая при дозе 200 мг/кг QD была сравнима с противоопухолевой эффективностью, наблюдаемой при введении дозы 100 мг/кг BID (100% по сравнению с 83%). Эти данные дополнительно свидетельствуют от том, что поддержание средней концентрации тестируемого соединения в плазме 1710 нг/мл (Сmax, 6870 нг/мл) в течение 8 ч/сутки путем введения дозы 100 мг/кг BID столь же полезно, как поддержание средней концентрации в плазме 6510 нг/мл (Сmax, 16700 нг/мл) после введения дозы 200 мг/кг QD.
Таким образом, результаты свидетельствуют о том, что в модели опухоли MDA-MB-453 поддержание концентрации тестируемого соединения в плазме приблизительно 509 нг/мл (50 мг/кг, введение дозы BID) в течение 8 ч/сутки столь же эффективно для ингибирования роста опухоли как поддержание средних концентраций в плазме 591-3120 нг/мл (50-100 мг/кг, введение дозы QD) в течение 4 ч/сутки. Таким образом, меньшая доза тестируемого соединения, вводимая по схеме приема BID, обладает пользой, равной пользе введения более высоких доз в схеме приема QD.
Настоящее изобретение не ограничено объемом описанных здесь специфических воплощений. Действительно, различные модификации в соответствии с изобретением в дополнение к описанным здесь понятны специалистам в данной области техники из предшествующего описания и сопутствующих графических материалов. Предполагается, что такие модификации находятся в объеме приложенной формулы изобретения.
Все патенты, заявки на патенты, публикации, способы тестирования, литературные и другие материалы, цитируемые здесь, включены путем ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕКАРСТВЕННЫЕ ФОРМЫ, СОДЕРЖАЩИЕ AG013736 | 2004 |
|
RU2341263C2 |
| ЛЕЧЕНИЕ РАКА ГОЛОВНОГО МОЗГА | 2013 |
|
RU2672575C2 |
| КОМБИНАЦИЯ, СОДЕРЖАЩАЯ ПАЛБОЦИКЛИБ И 6-(2,4-ДИХЛОРФЕНИЛ)-5-[4-[(3S)-1-(3-ФТОРПРОПИЛ)ПИРРРОЛИДИН-3-ИЛ]-8,9-ДИГИДРО-7H-БЕНЗО[7]АННУЛЕН-2-КАРБОНОВУЮ КИСЛОТУ, И ЕЕ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2764116C2 |
| КОМБИНИРОВАННАЯ ПРОТИВОРАКОВАЯ ТЕРАПИЯ | 2011 |
|
RU2607596C2 |
| ПРОТИВООПУХОЛЕВЫЕ КОМБИНАЦИИ ИЗ 4-АНИЛИНО-3-ЦИАНОХИНОЛИНОВ И КАПЕЦИТАБИНА | 2009 |
|
RU2498804C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ИНГИБИРОВАНИЯ ТИРОЗИНКИНАЗНОЙ АКТИВНОСТИ | 2011 |
|
RU2598852C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ, СОДЕРЖАЩАЯ TNO155 И РИБОЦИКЛИБ | 2020 |
|
RU2813111C2 |
| ПРИМЕНЕНИЕ ОБРАТНЫХ АГОНИСТОВ ИЛИ АНТАГОНИСТОВ РЕЦЕПТОРА ГРЕЛИНА ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ СНА | 2013 |
|
RU2600895C2 |
| ИСПОЛЬЗОВАНИЕ ПРОИЗВОДНЫХ 2-КАРБОКСАМИД-ЦИКЛОАМИНО МОЧЕВИНЫ В ЛЕЧЕНИИ EGFR-ЗАВИСИМЫХ ЗАБОЛЕВАНИЙ ИЛИ ЗАБОЛЕВАНИЙ С ПРИОБРЕТЕННОЙ РЕЗИСТЕНТНОСТЬЮ К АГЕНТАМ, НАЦЕЛЕННЫМ НА ЧЛЕНЫ EGFR-СЕМЕЙСТВА | 2011 |
|
RU2589695C2 |
| СПОСОБ ЛЕЧЕНИЯ АНОМАЛЬНОГО РОСТА КЛЕТОК | 2006 |
|
RU2384331C2 |
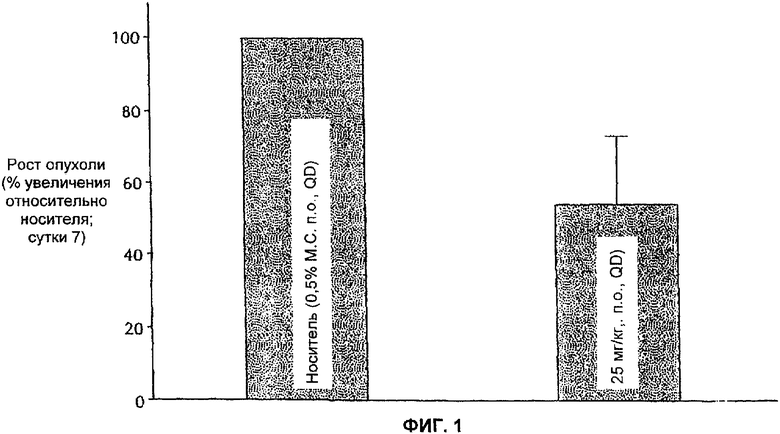
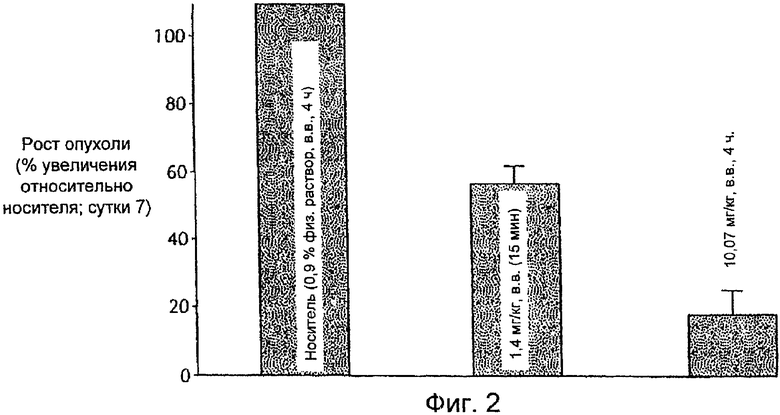
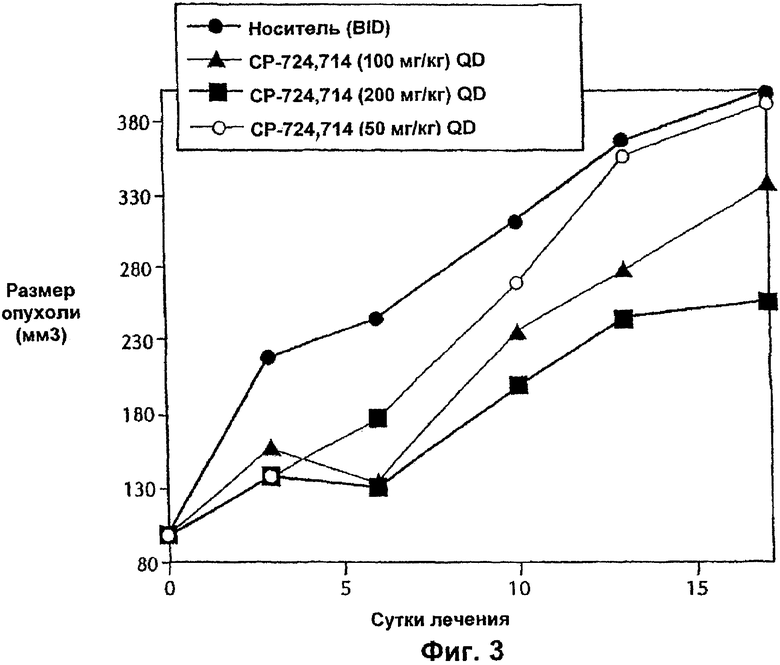
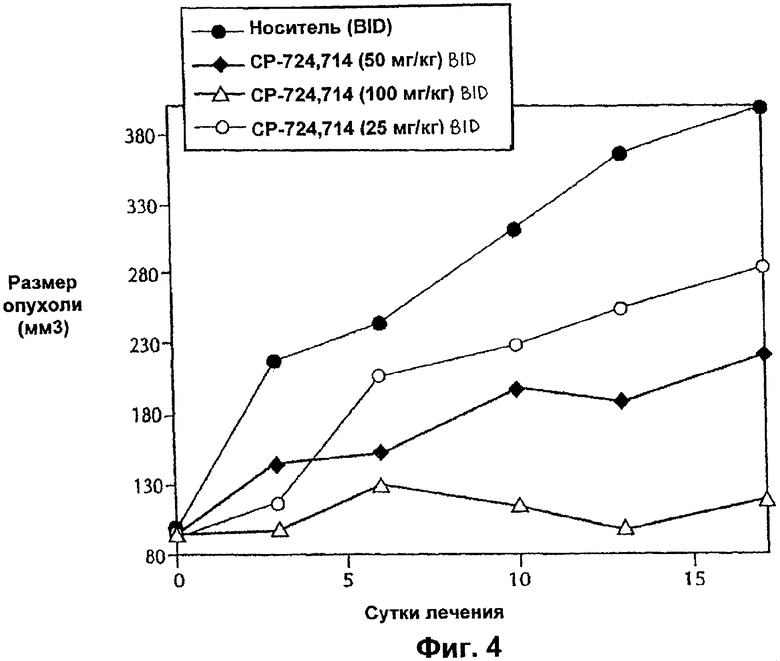
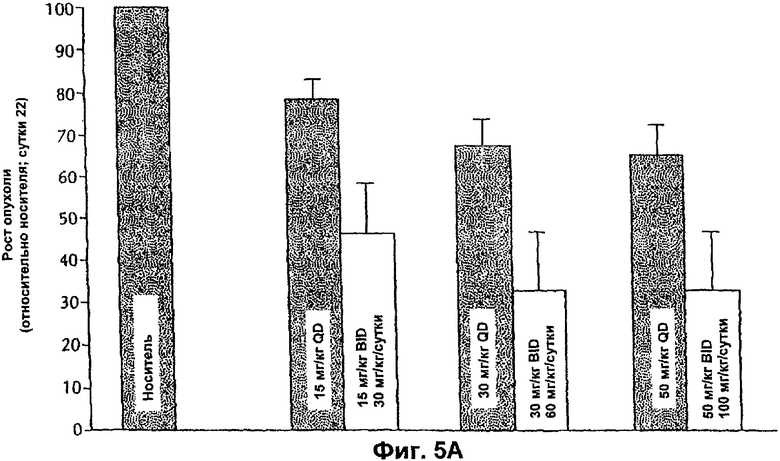
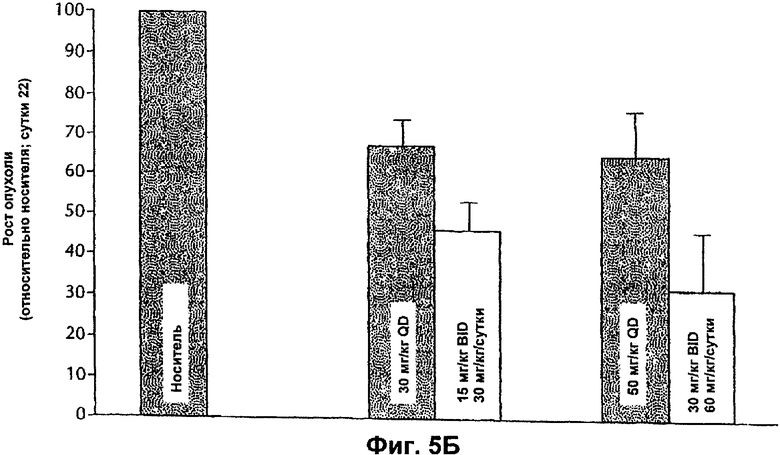
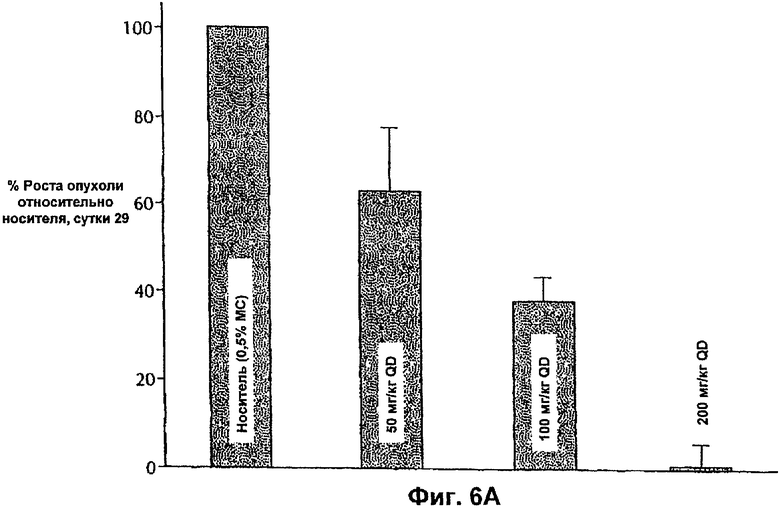
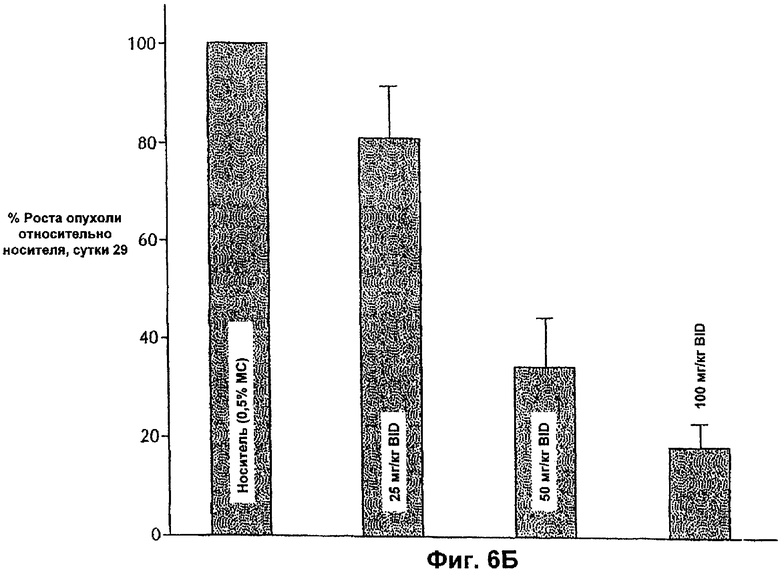
Группа изобретений относится к медицине, лечению аденокарциномы яичников у млекопитающего. Способы включают пероральное, трансбуккальное, сублинвальное, интраназальное, внутриглазное, внутрижелудочное, интрадуоденальное, местное, ректальное или вагинальное введение субъекту в пределах двадцати четырех часов первого терапевтически эффективного количества Е-2-Метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида (соединение Е), составляющего 25-100 мг/кг, и второго терапевтически эффективного количества соединения Е, составляющего 25-100 мг/кг. Изобретение обеспечивает улучшенную схему лечения аденокарциномы яичников за счет выбора конкретных малых доз и режима введения соединения Е, позволяющих увеличить процент ингибирования клеток аденокарциномы по сравнению с иными дозами и режимами его введения. 2 н.п. ф-лы, 6 ил., 7 табл.
(а) введение указанному млекопитающему терапевтически эффективного количества Е-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида, составляющего 25-100 мг/кг; и
(б) последующее введение указанному млекопитающему после промежутка времени меньше 24 ч еще одного терапевтически эффективного количества Е-2-метокси-N-(3-{4-(3-метил-4-(6-метил-пиридин-3-илокси)-фениламино)-хиназолин-6-ил}-аллил)-ацетамида, составляющего 25-100 мг/кг.

