Настоящее изобретение касается производных фталазина, содержащих их фармацевтических композиций, а также применения этих производных в качестве ингибиторов фосфородиэстеразы-4.
Фосфородиэстеразы представляют собой семейство изоферментов, составляющих фундамент основного механизма гидролитического инактивирования цАМФ (циклического аденозин-3′,5′-монофосфата). Было показано, что цАМФ представляет собой вторичный мессенджер, опосредующий биологическую ответную реакцию на многие гормоны, трасмиттеры и лекарственные средства [Krebs Endocrinology Proceedings of the 4th International Congress Excerpta Medica, 17-29, 1973]. При связывании соответствующего агониста с поверхностью клетки аденилатциклаза активируется и превращает Mg2+-АТФ в цАМФ. цАМФ модулирует активность большинства (если не всех) клеток, которые вносят вклад в патофизиологию различных респираторных заболеваний, имеющих как аллергическую, так и не аллергическую природу. Из этого следует, что рост концентрации цАМФ оказывает благоприятное воздействие, например, типа релаксации гладких мышц дыхательных путей, подавления высвобождения медиатора мастоцитов (гранулезных клеток базофилов), супрессии дегрануляции нейтрофилов и базофилов, подавления активации моноцитов и макрофагов. Таким образом, соединения, способные активировать аденилатциклазу или ингибировать фосфородиэстеразы, могут подавить нежелательную активность гладких мышц дыхательных путей, а также большого количества воспалительных клеток.
В семейство фосфодиэстераз входит отдельная группа изоферментов, фосфодиэстеразы-4 (называемые далее PDE 4), характерных для гидролиза цАМФ в гладких мышцах дыхательных путей и воспалительных клетках (Torphy, "Phosphodiesterase Isoenzymes: Potential Targets for Novel Anti-asthmatic Agents" in New Drugs for Asthma, Barnes, ed. IBC Technical Services Ltd, 1989). Исследования, проведенные на этом ферменте, показали, что его ингибирование обеспечивает не только релаксацию гладких мышц дыхательных путей, наравне с подавлением дегрануляции мастоцитов, базофилов и нейтрофилов, также как и подавление активации моноцитов и нейтрофилов. Поэтому ингибиторы PDE-4 эффективны для терапии астмы.
Селективное подавление PDE-4 ослабляет функциональные возможности клеток воспалительного очага, например таких как нейтрофилы, альвеолярные макрофаги, а также Т-клетки, которые, как известно, играют ключевую роль в хронических обструктивных заболеваниях легких (COPD); и такая активность предполагает, каким образом этот класс соединений может обеспечить эффективную терапию такого рода патологий (Duglas WP Hay, Curr. Opin.Chem. Biol., 2000, vol.4, pages 412-419).
Такие соединения предлагают уникальное приложение к терапии различных респираторных заболеваний как аллергического, так и неаллергического происхождения, и они обладают существенными преимуществами над используемой в настоящее время терапией.
Избыточное или не отвечающее нормам продуцирование фактора некроза опухоли (называемого далее TNFa), цитокина с противовоспалительной активностью, вырабатываемого клетками различного рода, действует на перенос многих патологий, таких как например, респираторный дистресс-синдром взрослых и хронические воспалительные заболевания легких, или на их обострение. Следовательно, соединения, способные регулировать негативное действие TNFa, т.е. ингибиторы этого цитокина, следует рассматривать пригодными против многих патологий.
В описании патента ЕП 722936 (Eisai) между прочим заявляются соединения формулы
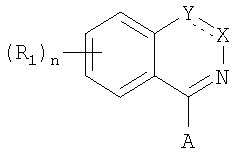
в которой n=0-4; R1 представляет собой необязательно замещенную низшую алкоксигруппу, необязательно замещенный циклоалкил, или -OR9 группу, где R9 представляет собой необязательно замещенную арилалкильную группу; Х представляет собой -N= или -NR6-, где R6 представляет собой водород, низшую алкильную группу, или факультативно замещенную арилалкильную или гетероарилалкильную группы; Y представляет собой -СО или -СВ=, где В представляет собой -NR7R8, в которой каждый из R7 и R8 может независимо представлять собой Н, необязательно замещенную гетероарилалкильную группу, или же В представляет собой водород или факультативно замещенную арильную, гетероарильную, арилалкильную или гетероарилалкильную группу; А представляет собой атом водорода или галогена, или факультативно моно- или бизамещенную аминогруппу, замещенную (необязательно) арильную, гетероарильную или гетероарилалкильную группу.
Среди групп, необязательно замещающих указаны упомянутые выше остатки, атомы галогенов, а также защищенная (необязательно) карбоксильная группа.
Из приведенных в качестве примера соединений с очень широкой общей формулой, особый интерес представляют случаи, в которых: значениями А являются фталазиновый цикл, замещенный на (3-хлоро-4-метокси)-бензиламиногруппу или 3,4-метилендиокси-бензиламино-группу; значениями R1 являются галоген, нитро-группы, или циано-группы при n=1; значениями В является -NR7R8, где каждый из R7 и R8 независимо представляет собой водород, необязательно замещенную группу низшего алкила, необязательно замещенную гетероарилалкильную группу, или R7 и R8 вместе с атомом азота, с которым они связаны химической связью могут образовывать цикл, который может быть замещен.
Таким образом ни один из аспектов описания, а также ни один из примеров не приводят к соединениям, в которых R1 представляет собой метокси или дифторметокси при n=1, А является фенилом или гетероциклом, замещенным на карбоксильную группу, а также (необязательно) на другую функциональную группу, а В представляет собой (3,5-дихлор)-пиридин-4-ил-метильную группу.
Кроме того, эти соединения известны своей активностью как ингибиторы цГМФ-фосфордиэстеразы, т.е. PDE 5, где именно фосфордиэстераза действует по цГМФ-зависимому механизму и областью применения этих соединений в основном является сердечно-сосудистая система (Schudt С.et al., Phosphodiesterase Inhibitors, Academic Press).
Международная патентная заявка WO 00/05218 (Zambon Group S.p.A) в числе прочих заявляет свои права на соединения формулы
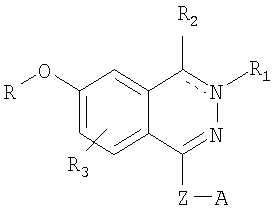
в которой химическая связь между атомом углерода, с которым связан заместитель R1, и соседним атомом азота является одинарной или двойной; R представляет собой (С1-С6)алкильную или полифтор(С1-С6)алкильную группу; R1 отсутствует в случае, если химическая связь между атомом углерода, с которым связан заместитель R2, и соседним атомом азота является двойной, или если эта же связь является простой, то R1 представляет собой атом водорода, необязательно замещенный (С1-С6)алкильной группой или (C1-С4)алкилсульфонильной группой; если химическая связь между атомом углерода, с которым связан заместитель R2, и соседним атомом азота является двойной, то R2 представляет собой атом водорода, циано-группу, амидо-группу, (С1-С8)алкильную, (С2-C8)алкенильную или (С2-С8)алкинильную группу, алкокси-группу, или необязательно замещенный арил или гетероцикл; R3 представляет собой водород или (С1-С8)алкильную, (С2-С8)алкенильную группу, или (С2-C8)алкинильную необязательно замещенную группу; Z представляет собой NH, метилен или (С2-C6)алкиленовую цепь, необязательно разветвленную и/или ненасыщенную, и/или прерываемую (С3-С7)циклоалкильным остатком; А представляет собой фенил или гетероцикл, необязательно замещенный одним или большим количеством заместителей.
Указанные соединения очень активны в роли ингибиторов PDE, а также ингибиторов высвобождения TNFa, кроме того, они не обладают никакой активностью по отношению к ферментам PDE 3 и 5.
Очевидно, что такая специфичность и избирательность действия делает эти соединения пригодными в качестве терапевтических средств для лечения патологий, включающих PDE 4 и TNFa.
Тем не менее соединения из приведенного выше патентного описания обладают некоторыми физико-химическими характеристиками (например, такими как растворимость в воде), пригодными для получения ограниченного количества составов.
Действительно, ограниченная растворимость активного элемента представляет собой основу трудностей, возникающих при приготовлении составов, которые способных гарантировать достаточно высокую биодоступность.
Более того, обычно для устранения слабой растворимости в доклинических моделях in vitro и in vivo используют специальные составы, позволяют применять нефизиологичные носители, а следовательно, создающие трудности в определении как активности соединений, так и их биологической доступности.
Было бы желательно, чтобы для применения указанных соединений по их предназначениям мы располагали широким диапазоном рецептур, для большинства классических случаев использования которых необходимо, чтобы активные ингредиенты были в значительной степени растворимы в воде.
Пероральный способ, например, представляет собой наиболее удобный и широко используемый путь введения лекарственных средств, и поэтому особенно важна способность улучшить растворимость потенциально активных ингредиентов и возможность разработать оральные составы, не прибегая к специальным навыкам создания рецептур.
В настоящее время нами было неожиданно обнаружено, что путем внедрения карбоксильной группы в фенильный или гетероциклический заместитель, входящий в значения R4, (смотри международную заявку WO 00/05218), получаемые соединения приобретают мощную ингибирующую активность по высвобождению PDE 4 и TNFa, инертность по отношению к PDE 1, 2, 3 и 5, а следовательно, они селективны, и наделены оптимальными свойствами в отношении растворимости в воде.
Такие соединения составляют подкласс в границах общей формулы из приведенного выше описания патентной заявки от Zambon Group, который не приводится в качестве примера, а следовательно, указанные соединения являются новыми.
Дополнительная полярность, введенная в молекулы с помощью карбоксильной группы, дает возможность получения соединений, наделенных такими физико-химическими свойствами, которые легче осуществимы с точки зрения составления рецептур, а также оценки биологической доступности соединений формулы I в носителях, пригодных для фармацевтических составов; доказано, что биологическая доступность сравнима или намного выше биологической доступности, выявленной для соединений из упомянутых выше патентных заявок, которую определяли, растворяя эти соединения в нефизиологичных носителях.
Внедрение на позицию 1 фенила или гетероцикла карбоксильной группы, имеющей специфические характеристики полярности, позволяет соединениям формулы I приобрести необходимый водно-липофильный молекулярный баланс, который необходим для растворения в жидкостях, а также для проницаемости сквозь биологические мембраны в процессах абсорбции и распределения, при этом высокая активность подавления в отношении фермента PDE 4 сохраняется.
Таким образом, были получены соединения, которые, за счет введения карбоксильного заместителя, приобретают свойства, оптимальные с точки зрения растворимости, их легко включить в приготавливаемую рецептуру, соединения получают высокую ингибирующую активность, селективность и биологическую доступность, которые сравнимы с аналогичными характеристиками других соединений.
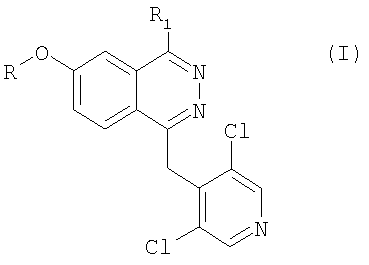
Итак, предметом настоящего изобретения являются соединения формулы
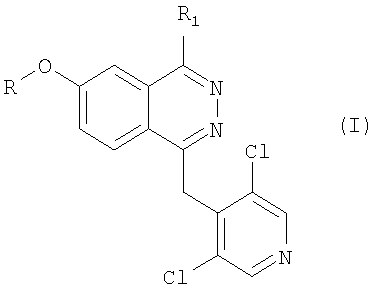
в которой R представляет собой метильную или дифторметильную группу; R1 представляет собой фенил или 5- или 6-членный гетероцикл, содержащий от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы, и связанный химической связью с фталазиновым циклом посредством углерод-углеродной связи, при этом указанный фенил и гетероцикл замещены на карбоксильную группу, а также (необязательно) на вторую функциональную группу, выбранную из метокси-, нитро-, N-ацетиламино-, N-метансульфонилгруппы;
N-оксидные производные соединений формулы I, а также их фармацевтически приемлемые соли.
Соединения формулы I активны в качестве ингибиторов PDE 4 и TNFa, и поэтому они находят применение в роли терапевтических средств при аллергических и воспалительных патологиях, таких, например как респираторный дистресс-синдром у взрослых, хроническое обструктивное заболевание легких, астма и аллергический ринит.
Под 5- или 6-членным ароматическим гетероциклом, содержащим от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы, понимают такие ароматические гетероциклы как пиррол, тиофен, фуран, имидазол, пиразол, тиазол, изотиазол, изоксазол, оксазол, пиридин, пиразин, пиримидин, пиридазин, пиперазин, триазол и тиадиазол.
N-оксидная форма (если она присутствует) может включать в себя атомы азота, находящиеся как на фталазиновом цикле, так и атомы азота, находящиеся на пиридильном цикле.
Фармацевтически приемлемые соли соединения I представляют собой соли щелочных или щелочноземельных металлов, соли цинка, а также соли с органическими основаниями, приемлемыми с фармацевтической точки зрения, например, такими как трометамол(2-амино-2-гидроксиметилпропан-1,3-диол), N-метилглюкамин.
Предпочтительными соединениями формулы I являются такие соединения, в которых R1 представляет собой фенил, замещенный на карбоксильную группу, а конкретно, такие соединения, карбоксильная группа которых находится в мета-положении относительно фталазинового цикла.
Характерными примерами соединениями, являющимися предметом настоящего изобретения, являются:
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойная кислота;
4-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойная кислота;
2-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойная кислота;
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-5-нитробензойная кислота;
5-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-2-метоксибензойная кислота;
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-4-метоксибензойная кислота;
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-5-метансульфониламинобензойная кислота;
3-ацетиламино-5-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метокси-фталазин-1-ил]-бензойная кислота;
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-5-метоксибензойная кислота;
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-дифторометоксифталазин-1-ил]-бензойная кислота;
2-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-оксазол-4-карбоновая кислота;
2-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-тиофен-3-карбоновая кислота;
(2-амино-2-гидроксиметилпропан-1,3-диол) соль 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойной кислоты;
N-метилглюкаминовая соль 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойной кислоты.
Соединения формулы I, являющиеся предметом настоящего изобретения, получают реакцией ароматического нуклеофильного замещения или реакцией связывания в присутствии катализатора (например, такого, как палладий) между соединением формулы II
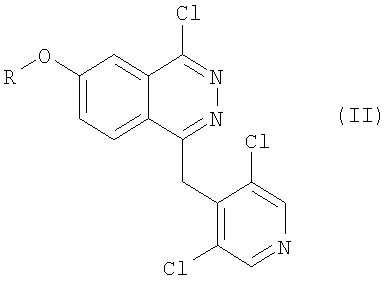
в которой R имеет значения, упомянутые для соединения формулы I,
и реагентом типа производного оловянной или бороновой кислоты, который пригоден для замещения атома галогена, химически связанного непосредственно с фталазиновым циклом фенилом или гетероциклом, замещенным на карбоксильную группу и замещенную (необязательно) на вторую функциональную группу, значения которой были определены для R1 формулы I.
Производные фталазина формулы II получают по схеме синтеза, описанной в заявке на международный патент WO 00/05218 (Zambon Group S.p.A), пример 45, стр.35 и пример 99, стр.57.
Для производных олова используют реакцию связывания, было бы лучше, чтобы указанные производные содержали предшественников свободной карбоксильной группы, например, такие как циано-группа или сложноэфирная группа. Предпочтительно, чтобы реакция связывания по Suzuki между соединениями формулы II и нужной из бороновых кислот протекала в присутствии палладия, трифенилфосфина, а также водного раствора карбоната калия.
Используемыми кислотами бора, например, являются необязательно замещенные карбокси-(фенил или гетероцикл)бороновые кислоты, или необязательно замещенные формил-(фенил или гетероцикл)бороновые кислоты, которые потом подвергают оксидированию для получения соответствующих соединений формулы I.
В качестве альтернативы для производных, у которых R1 представляет собой замещенный гетероцикл, гетероароматический цикл получают многостадийной реакцией по стандартных схемам синтеза, используя в качестве субстрата пригодный для этого цикл фталазина.
Получение N-оксидированных соединений формулы I проводят обработкой перкислотами, такими, например, как м-хлорпербензойная кислота.
Если реакция оксидирования направлена на атом азота, присутствующий на пиридиновом цикле, то для гарантии селективности процесса его проводят на цикле изобензофуранола, являющемся предшественником фталазинового цикла, способ осуществляется по схеме, описанной в заявке на международный патент WO 00/05218.
Получение солей соединения формулы I проводят известными способами.
Соединения формулы I являются ингибиторами PDE 4, как это было выявлено тестами по подавлению ферментативной активности (пример 18), они способны также подавлять высвобождение TNFa (пример 19).
Помимо этого, соединения по настоящему изобретению не демонстрируют никакой ингибирующей активности по отношению к ферментам PDE 1, 2, 3 и 5, как это было показано проведенными тестами по подавлению ферментативной активности.
Очевидно, что такая селективность и специфические особенности в сочетании с недостаточной активностью по отношению к сердечно-сосудистой системе, делают указанные соединения формулы I особенно пригодными для лечения различных патологий, в которые вовлечены PDE 4 и TNFa, такие, как астма, хроническое обструктивное заболевание легких, респираторный дистресс-синдром взрослых, аллергический риноконъюнктивит, псориаз, атопический дерматит, ревматоидный артрит, септический шок, язвенный колит, хотя в настоящем контексте интерес сфокусирован практически на респираторный патологиях. В частности, соединения по настоящему изобретению особенно пригодны для лечения аллергических и воспалительных состояний, а более всего - для терапии хронических обструктивных заболеваний легких, астмы и аллергического ринита.
Терапевтические дозы обычно составляют от 0,1 до 1,000 мг в сутки, а при однократном введении оральным способом - от 1 до 200 мг.
Терапевтически эффективные количества будут зависеть от возраста и общего физиологического состояния пациента, от способа введения и конкретно используемого фармацевтического состава.
Другим предметом настоящего изобретения являются фармацевтические составы, содержащие терапевтически эффективные количества соединения формулы I или их фармацевтически приемлемые соли в смеси с подходящим носителем.
Фармацевтические составы, являющиеся предметом настоящего изобретения, могут быть жидкими, пригодными для перорального и/или парентерального введения (например, такие, как капли, сиропы, готовые к употреблению или получаемые разведением лиофилизованных препаратов растворы для инъекций), твердыми или псевдотвердыми (такими как таблетки, капсулы, гранулы, порошки, вагинальные свечи, суппозитории, крема, мази, гели, притирания), или нешипучими растворами, суспензиями, эмульсиями, а также другими формами, пригодными для трансдермального или ингаляционного введения.
В зависимости от типа состава, в целях фармацевтического применения он должен, помимо терапевтически эффективного количества одного соединения формулы I (или большего их количества), содержать некоторое количество твердых или жидких наполнителей или разбавителей, а также (необязательно) другие наполнители, используемые обычно при получении фармацевтических составов; к этим веществам относятся загустители, связующие, смазывающие вещества, ароматизаторы и красители.
Приготовление фармацевтических составов, являющихся предметом настоящего изобретения, может быть осуществлено известными способами.
Для лучшего понимания настоящего изобретения ниже приведены примеры.
ПРИМЕР 1
Синтез 4-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойной кислоты (соединение 1)
К смеси 4-хлор-1-(3,5-дихлорпиридин-4-ил-метил)-6-метокси-фталазина (промежуточное соединение 1) (300 мг; 0,85 ммол; 1 экв), приготовленного согласно заявке на международный патент WO 00/05218 (пример 45, стр.35) 4-карбоксифенилбороновую кислоту (155 мг; 0,93 ммол; 1,1 экв), палладий тетракис(трифенилфосфин) (50 мг; 5 мол.%), добавили 2N водный раствор карбоната калия 91,26 мл), DME (10 мл) и этанол (0,5 мл), предварительно промытые сильной струей азота. Процесс проводили при комнатной температуре в инертной атмосфере аргона. Реакционную смесь перемешивали при 90°С в течение 4-16 ч. Реакцию "гасили" 5% водным раствором лимонной кислоты до тех пор, пока значение рН не составило 6. Продукт, полученный в результате реакции связывания, экстрагировали этилацетатом (2×15 мл), высушили (Na2SO4), и упаривали при пониженном давлении. В результате было получено соединение I в виде светло-желтого твердого вещества (216 мг; 0,49 ммол; выход 58%).
MS: 440 [М+Н]+
ЯМР ДМСО_d6: 8.70 (s, 2Н, Ру); 8.58 (d, 1Н, JHH=9.1 Hz, *СН=СН-С-ОМе); 8.14 (d-уширенный, 2Н, JHH=8.3 Hz, C-(*CH=CH)2-C), 7.87 (d-уширенный, 2Н, JHH=8.3 Hz, C-(CH=*CH)2-C); 7.79 (dd, 1Н, CH=*CH-C-OMe); 7.29 (d, 1Н, JHH=2.5 Hz, C=*CH-C); 5.04 (s, 2Н, *CH2-Ру); 3.89 (s, 3H, OMe).
ПРИМЕР 2
Синтез 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойной кислоты (соединение 2)
Соединение 2 синтезировали действуя так же, как это было описано для соединения 1; при этом в качестве субстрата использовали промежуточное соединение 1 (5 г; 14 ммол) и 3-карбоксифенилбороновую кислоту (2,5 мг; 15 ммол). Продукт, полученный в результате реакции связывания, экстрагировали этилацетатом (2×15 мл), высушили (NB2SO4), а затем отфильтровали через целит, промывая этилацетатом (2×30 мл). Образовавшийся фильтрат подкисляли концентрированной соляной кислотой до рН=4, а затем перемешивали в течение 1 ч. Осадок собрали путем фильтрования и высушивали его при 50°С; в результате было получено соединение 2 в виде светло-желтого твердого вещества (3,4 г; 8 ммол; выход 57%).
MS: 440 [М+Н]+
ЯМР ДМСО_d6: 13.20 (s-уширенный, 1Н, ОН); 8.71 (s, 2Н, Ру); 8.58 (d, 1Н, JHH=8.7 Hz, *CH=CH-C-OMe); 8.25 (s-уширенный, 1Н, C=*CH-C-CO2H); 8.14 (d-уширенный, 1Н, JHH=8.1 Hz, *CH), 8.02 (d-уширенный, 1Н, JHH=8.1 Hz, *CH); 7.80 (dd, 1Н, СН=*СН-С-OMe); 7.77-7.69 (m, 1Н, CH=*CH-CH); 7.29 (d, 1Н, JHH=2.3 Hz, O=*CH-C); 5.04 (s, 2Н, *CH2-Ру); 3.88 (s, 3H, OMe).
ПРИМЕР 3
Синтез 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-5-нитробензойной кислоты (соединение 3)
Соединение 3 синтезировали действуя так же, как это было описано для соединения 1; при этом в качестве субстрата использовали промежуточное соединение 1 (300 мг; 0,85 ммол) и 3-карбокси-5-нитрофенилбороновую кислоту (196 г; 0,93 ммол). Продукт, полученный в результате реакции связывания, экстрагировали этилацетатом (2×15 мл), высушили (Na2SO4). Затем его упаривали при пониженном давлении и отфильтровали через целит, промывая этилацетатом (10 мл). Образовавшийся фильтрат упаривали при пониженном давлении; в результате было получено соединение 3 в виде светло-желтого твердого вещества (235 мг; 0.48 ммол; выход 57%).
MS: 485 [М+Н]+
ЯМР ДМСО_d6: 8.78-8.73 (m, 2H, *СН,*СН); 8.71 (s, 2H, Ру); 8.64-8.60 (m, 2H, *СН=СН-С-ОМе, *СН); 8.84 (dd, 1Н, JHH=9.2 Hz, 2.5 Hz, CH=*CH-C-OMe); 7.32 (d, 1H, JHH=2.5 Hz, MeO-C=*CH-C); 5:07 (s, 2H, *CH2-Ру); 3.89 (s, 3H, OMe).
ПРИМЕР 4
Синтез 5-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-2-метоксибензальдегида (промежуточное соединение 2)
Промежуточное соединение 2 синтезировали действуя так же, как это было описано для соединения 1; при этом в качестве субстрата использовали промежуточное соединение 1 (450 мг; 1,26 ммол) и 2-формил-4-метоксифенилбороновую кислоту (248 г; 1,38 ммол). Реакционную смесь вылили в Varian ChemElut CE100S и промывали дихлорметаном (3×10 мл). Фильтрат упаривали при пониженном давлении, а продукт очистили на Varian Mega Bond ChemElut (SiO2), промывая смесью СН2Cl2/метанол (в соотношении от 100:0 до 97:3 объем/объем). Получившийся в результате продукт перекристаллизовали из метанола; в результате было получено промежуточное соединение 2 в виде светло-желтого твердого вещества (260 мг; 0, 6 ммол; выход 48%).
MS: 455 [М+Н]+
ЯМР ДМСО_d6: 10.40 (s, 1H, CHO); 8.70 (s, 2H, Ру); 8.57 (d, 1H, JHH=9.1 Hz, *CH=CH-C-OMe); 8.10 (dd, 1H, JHH=2.3 Hz, 8.5 Hz, C-*CH=CH-C (OMe)-C-CHO); 8.04 (d, 1H, JHH=2.3 Hz, *CH-C-CHO); 7.79 (dd, 1H, JHH=2.5 Hz, 9.1 Hz, CH-*CH=C-OMe); 7.46 (d, 1H, JHH=8.7 Hz, *CH=C(OMe)-C-CHO); 7.32 (d, 1H, JHH=2.6 Hz, C=*CH-C); 5.02 (s, 2H, *CH2-Ру); 4.04 (s, 3H, OMe); 3.89 (s, 3H, OMe).
ПРИМЕР 5
Синтез 5-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-2-метоксибензойная кислота (соединение 4)
К быстро перемешиваемому раствору промежуточного соединения 2 (70 мг; 0,15 ммол; 1 экв) в трет-бутиловом спирте (3 мл) и 2 N раствора 2-метил-2-бутена в тетрагидрофуране (0,77 мл; 1,5 ммол; 10 экв) в атмосфере инертного азота по каплям добавляли раствор хлорида натрия (16 мг; 0,18 ммол; 1, 2 экв) в 1 N водном кислом фосфатном буфере (рН=3,5; 1 мл; 1,12 ммол; 7,5 экв). Процесс проводили при комнатной температуре в инертной атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 5 ч и упаривали при пониженном давлении. Получившийся продукт разбавили водой (5 мл), экстрагировали дихлорметаном (2×7 мл) и упаривали при пониженном давлении. Потом продукт очистили на Varian Mega Bond ChemElut (SiO2), промывая смесью СН2Cl2/метанол (в соотношении от 100:0 до 97:3 объем/объем). После очистки осуществляли промывку 100% метанолом; в результате было получено промежуточное соединение 4 в виде желтого твердого вещества (47 мг; 0, 1 ммол; выход 67%).
MS: 471 [М+Н]+
ЯМР ДМСО_d6: 8.69 (s, 2H, Ру); 8.53 (d, 1H, JHH=9.3 Hz, *CH=CH-C-OMe); 7.80-7.70 (m, 3H, C-C-CH=*CH-C, С=*СН-С-СООН, CH=*CH-C-OMe); 7.36 (d, 1H, JHH=2.5 Hz, C=*CH-C); 7.20 (d, 1H, JHH=8.7 Hz, C-C-*CH=CH-C); 5.00 (s, 2H, *CH2-Ру); 3.88 (s, 3H, OMe); 3.85 (s, 3H, OMe).
ПРИМЕР 6
Синтез 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-4-метоксибензальдегида (промежуточное соединение 3)
Промежуточное соединение 3 синтезировали действуя так же, как это было описано для соединения 1; при этом в качестве субстрата использовали промежуточное соединение 1 (450 мг, 1,26 ммол) и 5-формил-2-метоксифенилбороновую кислоту (248 г, 1,38 ммол). Реакционную смесь вылили в Varian ChemElut CE100S и промывали дихлорметаном (3×10 мл). Фильтрат упаривали при пониженном давлении, а продукт очистили на Varian Mega Bond ChemElut (SiO2), промывая смесью СН2Cl2/метанол (в соотношении от 100:0 до 97:3 объем/объем). Получившийся в результате продукт перекристаллизовали из метанола; в результате было получено промежуточное соединение 3 в виде светло-желтого твердого вещества (214 мг; 0,5 ммол; выход 40%).
MS: 455 [М+Н]+
ЯМР ДМСО_d6: 9.96 (S, 1H, CHO); 8.71 (s, 2H, Ру); 8.54 (d, 1H, JHH=9.2 Hz, C=C-*CH=CH-C-OMe); 8.15 (dd, 1H, JHH=2.3 Hz, 8.5 Hz, CHO-C-*CH=CH); 7.94 (d, 1H, JHH=2.3 Hz, C-*CH=C-CHO); 7.75 (dd, 1H, JHH=2.6 Hz, 9.2 Hz, C=C-CH=*CH-C-OMe); 7.47 (d, 1H, JHH=8.5 Hz, C-*CH=CH-C-CHO); 6.83 (d, 1H, JHH=2.3 Hz, C=*CH-C-OMe); 5.05 (s, 2H, *CH2-Ру); 3.83 (s, 3H, OMe); 3.82 (s, 3H, OMe).
ПРИМЕР 7
Синтез 3-[4-[3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-4-метоксибензойной кислоты (соединение 5)
Соединение 5 синтезировали действуя так же, как это было описано для соединения 4; при этом в качестве субстрата использовали промежуточное соединение 3 (150 мг; 0,33 ммол). Подученный продукт очистили на Varian Mega Bond ChemElut (SiCO2), промывая смесью СН2Cl2/метанол/уксусная кислота (в соотношении 99:1:0,05 объем/объем/объем). В результате было получено соединение 5 в виде желтого твердого вещества (78 мг; 0,16 ммол; выход 50%).
MS: 470 [М+Н]+
ЯМР ДМСО_d6: 8.70 (s, 2H, Ру); 8.53 (d, 1H, JHH=9.1 Hz, *CH=CH-C-OMe); 8.16 (dd, 1H, JHH=2.2 Hz, 8.7 Hz, CH-*CH=C-COOH); 7.90 (d, 1H, JHH=2.2 Hz, C-*CH-C-CO2H); 7.74 (dd, 1H, JHH=2.5 Hz, 9.1 Hz, CH=*CH-C-OMe); 7.36 (d, 1H, JHH=8.7 Hz, *CH-CH=C-COOH); 6.82 (d, 1H, JHH=2.5 Hz, C=*CH-C-OMe); 5.04 (s, 2H, *CH2-Ру); 3.81 (s, 3H, OMe); 3.79 (s, 3H, OMe).
ПРИМЕР 8
Синтез 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-5-метансульфониламинобензойной кислоты (соединение 6)
К суспензии палладия на древесном угле (12 мг) в метаноле (15 мл) добавили 3-карбокси-5-нитрофенилбороновую кислоту (250 мг; 1,1 ммол) и проводили восстановление в атмосфере водорода (40 фунтов на квадратный дюйм). Спустя 15 ч реакционную смесь отфильтровали через целит, а полученный фильтрат упаривали при пониженном давлении. После этого сырой продукт (200 мг; приблизительно 1,1 мол) растворили в воде (2 мл) и диоксане (1 мл) и добавили 10 N водный раствор гидроксида натрия (0,33 мл; 3 экв). К получившейся смеси по каплям при 0°С добавляли раствор метансульфонилхлорида (0,1 мл; 1,2 ммол; 1,1 экв) в дихлорметане (1 мл). После этого реакционную смесь перемешивали при комнатной температуре в течение 2 ч и подкислили 5% водным раствором лимонной кислоты. Полученный в результате продукт экстрагировали этилацетатом (2×10 мл). Объединенные органические слои высушили (Na2SO4) и упаривали при пониженном давлении. Затем провели реакцию связывания сырого продукта (130 мг; 0,5 ммол) с промежуточным соединением 1 (178 мг; 0,5 ммол) в условиях, описанных Suzuki для соединения 1. Полученный продукт экстрагировали этилацетатом (2×10 мл), высушили (Na2SO4) и упаривали при пониженном давлении. Продукт очистили на Varian Mega Bond ChemElut (SiO2), промывая смесью СН2Cl2/метанол/уксусная кислота (в соотношении 99:1:0,05 объем/объем/объем). В результате было получено соединение 6 в виде светло-желтого твердого вещества (13 мг; 0, 02 ммол, выход 5%).
MS:534[M+H]+
Высокоэффективная жидкостная хроматография/масс-спектрометрия: установка Gilson, снабженная колонкой C18 Zorbax SBC18 (3,5 мкм, 2,1×50 мм), связанной с диодным УФ-детектором (220 нм), а также с масс-спектрометром Finnigan Aqa (электронное распыление, положительная ионизация). Использовались следующие параметры: скорость потока 1 мл/мин; температура колонки 40°С; градиент элюирования А/В (элюент А: 0,5% муравьиная кислота в воде; элюент В: 0,5% муравьиная кислота в ацетонитриле); t=0 мин, А/В=95:5; t=8 мин, А/В=5:95; Rt=4,36.
ПРИМЕР 9
Синтез метилового эфира 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-5-метоксибензойной кислоты (промежуточное соединение 4)
К суспензии этилового эфира 3,5-динитробензойной кислоты (5 г; 0,02 ммол) в метаноле (50 мл) добавили метиллитий (1,66 г; 0,04 мол; 2,1 экв); процесс проводили в инертной атмосфере азота. Реакционную смесь нагревали до температуры перегонки в течение 16 ч, а потом ее вылили в охлаждаемый льдом водный раствор соляной кислоты (25 мл). Полученный продукт экстрагировали простым эфиром (3×25 мл), промыли водой, высушили (Na2SO4) и упаривали при пониженном давлении. Продукт очистили на Varian Mega Bond ChemElut (SiO2), промывая смесью циклогексан/этилацетат (в соотношении 9:1 объем/объем).
Полученный в результате метиловый эфир 3-метокси-5-нитробензойной кислоты (1 г; 5 ммол) растворили в метаноле (30 мл) и этилацетате (20 мл). Добавили концентрированный водный раствор соляной кислоты (0,4 мл) и палладий на древесном угле (50 мг). Реакционную смесь оставили при комнатной температуре под давлением водорода, составляющем 40 фунтов на квадратный дюйм. Спустя 1 ч палладиевый катализатор отфильтровали через целит, а фильтрат упарили при пониженном давлении. Получившийся в результате метиловый эфир 3-метокси-5-нитробензойной кислоты использовали без дальнейшей очистки. К суспензии неочищенного метилового эфира 3-метокси-5-нитробензойной кислоты (960 мг; 4,4 ммол; 1 экв) в смеси воды и концентрированной соляной кислоты (5 мл) в соотношении 1:1 (объем/объем) по каплям при 0°С добавляли раствор Na2NO2; (365 мг; 5,3 ммол; 1,2 экв) в воде (5 мл). Смесь перемешивали 10 мин при 0°С. Затем медленно добавили раствор иодида калия (1,47 г; 8,8 ммол; 2 экв) в воде (3 мл). Одновременно в реакционную смесь вылили толуол (8 мл). Баню со льдом удалили, а реакционную смесь перемешивали в течение 3 ч при комнатной температуре, и затем при температуре перегонки еще в течение 1 ч. Добавили воду (10 мл) и продукт экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промыли соляным раствором (5 мл), высушили (Na2SO4) и упаривали при пониженном давлении. Хроматографическая очистка (SiO2) при элюировании смесью циклогексан/этилацетат (в соотношении 9:1 объем/объем) дала в результате метиловый эфир 3-метокси-5-нитробензойной кислоты (730 мг; 2,5 ммол; выход за две стадии 50%).
В инертной атмосфере аргона метиловый эфир 3-иодо-5-метоксибензойной кислоты (500 мг; 1,7 ммол; 1 экв) добавили в раствор тетракис(трифенилфосфин) палладия (20 мг; 4 мол.%) в толуоле (25 мл). В эту реакционную смесь добавили гексаметилдистаннат (0,41 мл; 2 ммол; 1,2 экв), и получившуюся смесь нагревали вплоть до температуры перегонки. Спустя 3 ч добавили этилацетат (10 мл). Реакционную смесь промыли водным буфером NaOH/KH2PO4, имеющим рН=7, высушили (Na2SO4) и упарили при пониженном давлении. Получившийся в результате продукт разбавили толуолом (30 мл) и добавили в смесь промежуточного соединения 1 (610 мг; 1,7 ммол; 1 экв) и тетракис(трифенилфосфин) палладия (90 мг; 5 мол.%); процесс проводили в инертной атмосфере аргона. Смесь нагревали в течение 18 ч при 110°С. Получившийся продукт экстрагировали этилацетатом (2×15 мл), высушили (Na2SO4), упарили при пониженном давлении, а потом его хроматографическая очистка (SiO2) при элюировании смесью циклогексан/этилацетат/метанол (в соотношении 50:50:1 объем/объем/объем) дала в результате промежуточное соединение 4 в виде светло-желтого твердого вещества (483 мг; 1 ммол; выход 60%).
MS: 484 [М+Н]+
ЯМР ДМСО_d6: 8.71 (s, 2Н, Ру); 8.59 (а, 1Н, JHH=9.2 Hz, *CH=CH-C-OMe); 7.84-7.81 [m, 1Н, *СН); 7.78 (d, 1Н, JHH=2.6 Hz, *CH); 7.64-7.59 (m, 2Н, *CH, *CH); 7.31 (d, 1Н, JHH=2.5 Hz, C-C=*CH-C); 5.04 (s, 2Н, *CH2-Ру); 3.90 (s, 3H, OMe); 3.89 (s, 3H, OMe); 3.88 (s, 3H, OMe).
ПРИМЕР 10
Синтез 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-5-метоксибензойной кислоты (соединение 7)
К раствору промежуточного соединения 4 (430 мг; 0,9 ммол; 1 экв) в смеси тетрагидрофуран/вода в соотношении 3:1 (объем/объем) добавили гидроксид лития (65 мг; 2,7 ммол; 3 экв). Реакционную смесь перемешивали в течение 4 ч, а затем вылили на охлаждаемую льдом водяную баню (10 мл). Смесь подкислили 5% водным раствором лимонной кислоты, а полученный в результате продукт экстрагировали этилацетатом (2×10 мл). Объединенные органические слои высушили (Na2SO4) и упаривали при пониженном давлении. В результате без последующей очистки было получено соединение 7 в виде светло-желтого твердого вещества (420 мг; 0,9 ммол; выход 100%).
MS: 471 [М+Н]+
ЯМР DMS0_d6: 13.21 (s-уширенный, 1H, ОН); 8.70 (s, 2H, Ру); 8.58 (d, 1H, JHH=9.1 Hz, *CH=CH-C-OMe); 7.82-7.80 (m, 1H, *CH); 7.77 (d, 1H, JHH=2.5 Hz, *CH); 7.63-7.61 (m, 1H, *CH); 7.56-7.54 (m, 1H, *CH); 7.31 (d, 1H, JHH=2.3 Hz, С-С=*СН-С); 5.03 (s, 2H, *CH2-Ру); 3.88 (s/ 6H, OMe, OMe).
ПРИМЕР 11
Синтез 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-дифторметоксифталазин-1-ил]-бензойной кислоты (соединение 8)
К смеси 4-хлор-1-(3,5-дихлорпиридин-4-ил-метил)-6-дифторметоксифталазина (5 г; 12.8 ммол; 1 экв), полученному методом, который был описан (заявка на международный патент WO 00/05218, пример 99, стр.45), 3-карбоксифенилбороновой кислоты (2,34 г; 14 ммол; 1.1 экв) и тетракис(трифенилфосфин)палладия (450 мг; 5 мол.%) в инертной атмосфере азота добавили: раствор карбоната калия (5,3 г; 39 ммол; 3 экв) в воде (30 мл), DME (100 мл) и этанол (10 мл), предварительно промытые азотом. Процесс проводили при комнатной температуре, а получившуюся реакционную смесь перемешивали 16 ч при 80°С. Затем смесь упаривали при пониженном давлении и добавили в нее воду (250 мл). Избыток реактивов экстрагировали этилацетатом (3×50 мл), а водный слой подкислили концентрированной соляной кислотой до рН=3. Смесь перемешивали в течение 1 ч, а осадок собрали путем фильтрования и высушили его при 50°С. В результате было получено соединение 8 в виде светло-желтого твердого вещества. После последующей перекристаллизации из хлороформа было получено более чистое соединение 8 (3,9 г; 8,7 ммол; выход 68%).
MS: 477 [М+Н]+
ЯМР ДМСО_d6: 8.77 (d, 1H, JHH=9.0 Hz, *СН=СН-С-O-CHF2); 8.72 (s, 2H, Ру); 8.24 (m, 1H, *CH=C-COH); 8.19-7.97 (m, 3H, *CH=CH-CH=C-COOH, СН=*СН-С-СООН, СН=*СН-С-OCHF2); 7.74 (m, 1H, СН-*СН=СН); 7.61 (d, 1H, JHH=2.4 Hz, CHF2-O-C-*CH=C); 7.51 (t, 1H, JHF=73.4 Hz, *CHF2); 5.10 (s, 2H, *CH2-Ру).
ПРИМЕР 12
Синтез метилового эфира 1-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-карбоновой кислоты (промежуточное соединение 5)
В суспензию промежуточного соединения 1 (9,7 г; 27 ммол) в диметилсульфоксиде (80 мл) и метаноле (40 мл) добавили карбонат калия (7,4 г; 54 ммол), ацетат палладия (0,31 г; 1,4 ммол) и 1,3-бисдифенилфосфинпропан (0,75 г; 1,8 ммол). В течение нескольких часов реакционную смесь выдерживали в атмосфере моноксида углерода (8,5 бар) и нагревали до 50°С, а затем вылили в воду (1 л). Продукт экстрагировали этилацетатом (4×200 мл), промыли насыщенным соляным раствором (300 мл), высушили и упарили в вакууме. Хроматографическая очистка (SiO2) при элюировании смесью петролейный эфир/этилацетат (в соотношении 1:1 объем/объем) дала в результате промежуточное соединение 5 в виде белого твердого вещества (5,0 г; 13 ммол; выход 49%).
ЯМР CDCl3: 8.42 е 7.97 (2s, 2H, Ру); 7.90 (d, 1H, JHH=8.6 Hz, *CH=CH-C-OMe); 7.31-7.23 (m, 2H, Ar); 7.19 (dd, 1H, СН-*СН=С-ОМе); 7.11-7.09 (m, 2H, Ar); 7.02 (d, 1H, JHH=2.3 Hz, C=*CH-C=CH); 5.62 (s, 1H, CH); 5.08 (s, 1H, CH); 3.95 (s, 4H, OCH2CH2O); 3.91 (s, 3H, OCH3); 3.42 (bs, 1H, OH).
ПРИМЕР 13
Синтез 1-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил-4-карбоновой кислоты (промежуточное соединение 6)
В суспензию промежуточного соединения 5 (750 мг; 1,98 ммол) в метаноле (27 мл) и воде (3 мл) добавили 85% гидроксид калия (210 мг; 3 мол). Спустя 1 ч реакционную смесь упарили, добавили воду (10 мл) и промыли дихлорметаном (2×5 мл). После этого подкислили концентрированным водным раствором соляной кислоты. Полученный при этом осадок отфильтровали и промыли водой (2×5 мл). В результате было получено промежуточное соединение 6 в виде белого твердого вещества (0,66 г; 1,8 ммол; выход 92%).
ЯМР ДМСО_d6: 10.13 (s, 1H, СНО); 8.70 (s, 2H, Ру); 8.57 (d, 1H, JHH=9.0 Hz, *CH=CH-C-OMe); 8.29-7.80 (m, 4H, Ar-CHO; 7.82-7.76 (dd, 1H, CH-*CH=C-Ome); 7.28 d, 1H, JHH=2.65 Hz, MeO-C-*CH=C); 4.96 (s, 2H, *CH2-Ру); 3.88 (s, 3H, OMe).
ПРИМЕР 14
Синтез 2-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-оксазол-4-карбоновой кислоты (соединение 9)
В раствор гидрохлорида этилового эфира серина (233 мг; 1,4 ммол; 1 экв) в диметилформамиде (3 мл) добавили раствор промежуточного соединения 6 (500 мг; 1,4 ммол; 1 экв) в дихлорметане (15 мл), а также триэтиламин (0,22 мл; 1,5 ммол; 1,1 экв) и 1-гидроксибензотриазол (200 мг; 1,5 ммол; 1,1 экв). Реакционную смесь охладили до 0°С, и в течение 10 мин добавляли 1,3-дициклогексилкабодиимид (310 мг; 1,5 ммол; 1,1 экв). Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 36 ч, а затем подкислили 5% водным раствором лимонной кислоты. Образовавшиеся слои разделили, и органический слой промывали 5% водным раствором бикарбоната натрия (5 мл) и водой (2 мл). Органический слой высушили (Na2SO4), упарили при пониженном давлении, а потом его хроматографическая очистка (SiO2) при элюировании смесью циклогексан/этилацетат/метанол (в соотношении 60:40:1 объем/объем/объем) в результате дала промежуточное соединение (350 мг; 0,75 ммол; выход 55%). Указанное соединение (300 мг; 0,6 ммол; 1 экв) растворили в тетрагидрофуране (10 мл) в инертной атмосфере азота. Добавили реагент Бургесса (160 мг; 0,7 ммол; 1,1 экв) и реакционную смесь нагревали до температуры перегонки в течение 3 ч. Добавили воду (5 мл). Продукт экстрагировали этилацетатом (2×15 мл), высушили (Na2SO4), упарили при пониженном давлении. Хроматографическая очистка (SiO2) при элюировании смесью циклогексан/этилацетат/метанол (в соотношении 50:50:1 объем/объем/объем) дала в результате этиловый эфир 2-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-4,5-дигидрооксазол-4-карбоновой кислоты. Этот промежуточный эфир (100 мг; 0,22 ммол; 1 экв) растворили в дихлорметане (3 мл), а получившийся раствор охладили до 0°С. Затем по каплям добавляли 1,8-диазобицикло[5,6]ундец-7-ен (0,036 мл; 0,24 ммол; 1,1 экв) и бромтрихлорметан (0,024 мл; 0,24 ммол; 1,1 экв). Реакционную смесь перемешивали в течение 6 ч при 0°С. Добавили насыщенный водный раствор хлорида аммония (10 мл), и получившиеся слои разделили. Объединенный органический слой промыли соляным раствором (5 мл), высушили (Na2SO4) и упаривали при пониженном давлении. После этого полученный сложный эфир (85 мг; 0,18 ммол; 1 экв) растворили в смеси тетрагидрофуран/вода (6 мл) в соотношении 3:1 (объем/объем) и добавили гидроксид лития (14 мг; 0,5 ммол; 3 экв). Спустя 2 ч ввели водный раствор 5% лимонной кислоты (5 мл). Полученный продукт экстрагировали этилацетатом (2×15 мл), высушили (Na2SO4) и упарили при пониженном давлении. Затем последующая перекристаллизация из метанола дала соединение 9 в виде твердого вещества белого цвета (65 мг; 0,14 ммол; выход 80%).
MS: 431 [М+Н]+
ЯМР DMSO_d6: 9.08 (s, 1H, O-*CH=C); 8.86 (d, 1H, JHH=2.5 Hz, C=*CH-C); 8.71 (s, 2H, Ру); 8.63 (d, 1H, JHH=9.3 Hz, *CH=CH-C-OMe); 7.87 (dd, 1H, CH=*CH-C-OMe); 5.08 (s, 2H, *CH2-Ру); 4.04 (s, 3H, OMe).
ПРИМЕР 15
Синтез 2-[4-(3,5-дихлорпиридин-4-ил-метил-7-метоксифталазин-1-ил]-тиофен-3-карбоновой кислоты (соединение 10)
Соединение 10 получили, действуя так, как это было описано для соединения 1, при этом в качестве субстрата использовали промежуточное соединение 1 (500 мг; 1,4 ммол), а также 3-формил-2-тиофенбороновую кислоту (220 г; 1,4 ммол). Продукт реакции связывания экстрагировали этилацетатом (2×15 мл), высушили (Na2SO4) и упарили при пониженном давлении. Затем была проведена хроматографическая очистка (SiO2), которая при элюировании смесью циклогексан/этилацетат/метанол в соотношении 60:40:1 (объем/объем/объем) дала промежуточное соединение 2-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-тиофен-3-карбоксиальдегид (150 мг; 0,35 ммол; выход 25%). Затем указанный промежуточный альдегид (80 мг; 0,18 ммол) окислили в соответствующую кислоту, при этом действовали так, как это было описано для соединения 5. Затем была проведена хроматографическая очистка (SiO2), которая при элюировании смесью дихлорметан/метанол/уксусная кислота в соотношении 95:5:95 (объем/объем/объем) и последующей перекристаллизации из смеси дихлометана/метанол в соотношении 99:1 (объем/объем) дала соединение 10 в виде твердого вещества белого цвета (40 мг; 0,09 ммол; выход 50%).
MS: 446 [М+Н]+
ЯМР ДМСО_d6: 8.70 (s, 2H, Ру); 8.58 (d, 1H, JHH=9.1 Hz, *CH=CH-C-OMe); 7.97 (d, 1H, JHH=4.1 Hz, CH-S); 7.85-7.80 (m, 2H, S-CH=*CH, CH=*CH-C-OMe); 7.75 (d, 1H, JHH=2.5 Hz, C-*CH=C); 5.02 (s, 2H, *CH2-Ру); 4.03 (s, 3H, OMe).
ПРИМЕР 16
Синтез N-метил-Р-глюкаминовой соли соединения 2 (соединение 11)
К суспензии соединения 2 (500 мг; 1,136 ммол) в метаноле (5 мл), нагретой до температуры перегонки в инертной атмосфере азота добавили раствор N-метил-D-глюкамина (233 мг; 1,19 ммол) в метаноле (1,2 мл) и воду (1,5 мл). После полного растворения указанной смеси ее упаривали при пониженном давлении при комнатной температуре. Добавили этанол (10 мл) и полученный раствор выдерживали в течение 4 дней при 4°С. Образовавшийся осадок отфильтровали и промыли этанолом (5 мл) и диэтиловым эфиром (5 мл). Продукт высушивали 15 ч в вакууме при 50°С, получив при этом соединение 11 в виде твердого вещества светло-желтого цвета (400 мг; 0,61 ммол; выход 54%).
ЯМР ДМСО_d6: 8.68 (s, 2H, Ру); 8.54 (d, 1H, JHH=8.7 Hz, *CH=CH-C-OMe); 8.17 (s-уширенный, 1H, C=*CH-C-CO2H); 8.06 (d-уширенный, 1H, JHH=8.1 Hz, *CH), 7.78 (d-уширенный, 1H, JHH=8.1 Hz, *CH); 7.72 (dd, 1H, CH=*CH-C-OMe); 7.58-7.50 (m, 1H, CH=*CH-CH); 7.29 (d, 1H, JHH=2.3 Hz, C=*CH-C); 5.04 (s, 2H, *CH2-Ру); 3.91-3.82 (m, 4H, OMe, *CHH-OH); 3.58-3.33 (m, 5H, HO-*CHH-*CH(OH)-*CH(OH)-*CH(OH)-*CH(OH)), 3.06-2.84 (m, 2H, CH2-NH), 2.09 (s, 3H, NHMe).
ПРИМЕР 17
Синтез 2-амино-2-гидрокисметилпропан-1.3-диоловой соли соединения 2 (соединение 12)
К суспензии соединения 2 (500 мг; 1,136 ммол) в этаноле (5 мл), нагретой до температуры перегонки в атмосфере инертного азота добавили 2-амино-2-гидроксиметилпропан-1,3-диол (138 мг; 1,13 ммол). После полного растворения температура снизилась до комнатной. Спустя 2 ч образовавшийся осадок отфильтровали и промыли этанолом (5 мл) и диэтиловым эфиром (5 мл). Продукт высушивали в вакууме в течение 15 ч при 50°С, получив при этом соединение 12 в виде твердого вещества светло-желтого цвета (400 мг; 0,71 ммол; выход 63%).
ЯМР ДМСО_d6: 8.68 (s, 2H, Ру); 8.54 (d, 1H, JHH=8.7 Hz, *CH=CH-C-OMe); 8.17 (s-уширенный, 1H, C=*CH-C-CO2H); 8.05 (d-уширенный, 1H, JHH=8.1 Hz, *CH), 7.79 (d-уширенный, 1H, JHH=8.1 Hz, *CH); 7.77-7.70 (m, 1H, CH=*CH-C-OMe); 7.60-7.51 (m, 1H, CH=*CH-CH); 7.29 (d, 1H, JHH=2.3 Hz, C=*CH-C); 5.01 (s, 2H, *CH2-Ру); 3.86 (s, 3H, OMe), 3.46 (s, 6H, C(CH2)3).
Пример 18
Ингибирование фермента PDE 4
а) Очистка фермента PDE 4
Фермент PDE 4 выделяли из клеточной линии U937. используя способ Nielson et al. (J. Allergy Clin. Immunol, 1990, vol.86, pages 801-807), частично модифицированный для жидкостной экспресс-хроматографии белков (FPLC).
Клеточная линия U-937 (Istituto Zooprofilattico Sperimentale. Brescia. Italy) выдерживалась в RPMI 1640 вместе с 10% плодной бычьей сывороткой и 2 мкМ глутамина, плотность составляла (1·106-8×106) клеток в мл, процесс проводили в инкубаторе при 37°С, при 5% содержании СО2.
Суспензию клеток U937 гомогенизировали в буфере с рН 7,8, содержащем 10 мМ Трис (три-(гидроксиметил)-аминометан), 5 мкМ MgCl2, 4 мкМ EGTA (этиленгликоль-бис-(β-аминоэтиловый эфир)-N,N,N′N′-тетрауксусной кислоты), 5 мкМ β-меркаптоэтанола, 1 мкМ лейпептина, 1 мкМ пепстатина А, 1% Тритон х-100, а также 100 мкМ фенилметилсульфонилфторида (PMSF).
Гомогенизат центрифугировали, а супернатант использовали для очистки фермента PDE 4; ферментом засеяли колонку, присоединенную к хроматографической системе для биологических объектов (FPLC, BIO RAD).
Фермент PDE 4 элюировали ацетатом натрия при его линейном градиенте от 50 мМ до 1 М, элиюрованные фракции собрали для последующего определения ферментативной активности PDE 4
Объединили фракции, содержащие активный PDE 4, и после проведенного в течение ночи диализа (в целях отделения ацетата натрия), упарили до 30% объема; для последней процедуры использовали систему фильтрования Amicon с мембранным фильтром YM10.
Добавили этиленгликоль (30% объем/объем), и полученный образец хранили при 20°С в виде единичной аликвоты вплоть до использования.
б) Анализ активности PDE 4
Активность фермента анализировалась в устройстве Scintillation Proximity Assay (SPA) (Amersham), которое включало [3H]цАМФ в качестве метки, буфер для анализа PDE (10× раствор: 500 мМ Трис/HCl с рН 7,5; 83 мкМ MgCl2 и 17 мкМ EGTA), а также гранулы PDE Yttrium SPA (содержащие 18 мкМ сульфата цинка).
Радиоактивность указанных гранул определяли с помощью сцинтилляционного счетчика (Packard model MINAXI β TRI-CARB 4000 SERIES).
Значения IC50 подсчитывались по кривым зависимости ингибирования от концентрации, при этом использовали нелинейный регрессионный анализ по программе ORIGIN 3.5 (MICROCAL SOFTWARE INC.). Каждый параметр представлял собой собственно значение ± среднее арифметическое от величины, полученной в 3 экспериментах с применением различных препаратов PDE 4.
Соединения формулы I настоящего изобретения продемонстрировали селективное подавление фермента PDE 4.
Полученные результаты приведены ниже в таблице 1.
Пример 19
Высвобождение TNFa в цельную кровь человека
Образцы крови, полученные от здоровых добровольцев, собрали в гепаринизированные пробирки и разбавили RPMI 1640 в соотношении 1:5, при этом никаких добавок сыворотки не производили.
Анализ проводили на пластинах с 96 ячейками. Образцы, содержащие 150 мкл разбавленной крови и 150 мкл RPMI 1640 с контрольным носителем или с различными концентрациями соединений, по настоящему изобретению инкубировали при 37°С в течение 30 мин во влажной атмосфере с 5% содержанием СО2.
Цельная кровь в образцах для анализа была разбавлена в соотношении 1:10 (объем/объем).
Образцы были обработаны LPS (липополисахарид из Е.Coli: serotype В 0:55, Sigma) в количестве 0,25 мкг/мл, а затем проводили инкубирование в течение 24 ч.
После центрифугирования супернатанты были собраны для определения в них концентрации TNFa,; для анализа использовали коммерчески доступное устройство ELISA (Biosource).
Тест-объекты растворили в диметилсульфоксиде (ДМСО) при концентрации 10-2 М, а затем растворили в RPMI 1640.
Конечная концентрация ДМСО не превышала 0,1%, и она не оказывала влияния на высвобождение TNFa.
Для каждой из 9 концентраций соединений анализ проводился дважды. Полученные в результате данные затем подтверждались использованием образцов крови, полученных от различных доноров.
Подсчитали ингибирование выработки TNFa для каждой концентрации, а величины IC50 определяли с помощью 4-парметрового уравнения логистики (Origin calculation program, Microcal Software Inc.).
Оказалось, что соединения формулы I по настоящему изобретению ингибируют высвобождение TNFa.
В приведенной ниже таблице 2 приведены результаты, выраженные как IC50 при двух различных концентрациях.
ПРИМЕР 20
Ингибирование ферментов PDE 3 и РРЕ 5
а) Очистка ферментов PDE 3 и PDE 5
Ферменты PDE 3 и PDE 5 очищали от плазмы с большим содержанием тромбоцитов (ПБТ), полученной от здоровых волонтеров.
Очистку ферментов PDE 3 и PDE 5 осуществляли по методике Simpson A.W.M. et al. (Biochem. Pharmacol, 1988, 37, 2315-2320), частично модифицированной для жидкостной экспресс-хроматографии белков (FPLC).
ПБТ разбавили в соотношении 1:1 и центрифугировали при комнатной температуре в течение 15 мин при скорости вращения 2000 об/мин.
Тромбоциты суспендировали в 10 мл лизисного раствора (155 мМ NH4Cl, 10 мМ КНСО3 и 0,1 мМ натриевой соли этилендиаминтетрауксусной кислоты (ЭТДК) рН 7,4). Для удаления примеси эритроцитов провели инкубирование в течение 10 мин на ледяной бане.
После центрифугирования тромбоциты суспендировали в 10 мл раствора 20 мМ бис-Трис (2 мл ЭДТК, 5 мМ β-меркаптоэтанола, 50 мМ ацетата натрия, 2 мМ бензамидина), а потом гомогенизировали на ледяной бане в гомогенизаторе Polytron.
Гомогенат центрифугировали, а полученный супернатант перенесли в колонку UNO Q12, присоединенную к системе для хроматографии биологических объектов (FPLC, Bio-Rad).
Ферменты PDE 3 и PDE 5 элюировали ацетатом натрия с линейным градиентом 0,05-1M, а элюированные фракции собрали для последующего анализа активности ферментов PDE.
Объединили фракции, обладающие активностью фермента PDE, в течение ночи провели их диализ от дистиллированной воды и, используя систему Amicon filtration system (с мембранным фильтром YM10), провели 10-кратное сгущение.
Добавили этиленгликоль до получения 30% концентрации (объем/объем) и получившийся раствор сохраняли при -20°С.
При указанных условиях ферментативная активность сохранялась стабильной.
б) Анализ активности PDE 3 и PDE 5
Активность ферментов анализировалась в устройстве Scintillation Proximity Assay (SPA) (Amersham), которое включало 3H-цАМФ для PDE 3 или 3H-цГМФ для PDE 5, буфер для анализа PDE (10× раствор: 500 мМ Трис/HCl с рН 7,5; 83 мкМ MgCl2 и 17 мкМ EGTA), а также гранулы PDE Yttrium SPA (содержащие 18 мкМ сульфата цинка).
Радиоактивность указанных гранул определяли с помощью сцинтилляционного счетчика (Packard model MINAXI β TRI-CARB 4000 SERIES).
В приведенной ниже таблице 3 указаны результаты, выраженные как величины IC50 некоторых соединений, являющихся представительными всего класса соединений.
ПРИМЕР 21
Ингибирование фермента PDE 2
а) Очистка фермента PDE 2
PDE 2 очищали от клеток мыши РС12 по методике, описанной в работе Whalin et al. (Molecular Pharmacol. 1991, 39, 711-717).
Клетки РС12, полученные от Istituto Zooprofilattico Brescia (Italy), выдерживали в сосудах емкостью 150 см3 при 37°С и 5% CO2 в среде, содержащей 5% плодной бычьей сыворотки, 15% лошадиной сыворотки, 1% пенициллин, 1% стрептомицин и 1% глутамин.
Клетки увеличивались в объеме, при этом обращалось внимание на то, чтобы их расщепление происходило только по достижению слияния.
После промывания клеток их отделили, используя для этого смесь 0,05% трипсин/0,02% ЭДТК, а потом собрали в чистую пробирку.
После центрифугирования клетки суспендировали в буфере с рН 6,5, содержащем бис-Трис (20 мкМ), ЭДТК (2 мкМ), 2-меркаптоэтанол (5 мкМ), лейпептин (1 мкМ), пепстатин А (1 мкМ), фенилметилсульфонилфторид (100 мкМ), а затем гомогенизировали с помощью гомогенизатора Polytron.
Полученный в результате центрифугирования супернатант перенесли в колонку UNO Q12, присоединенную к системе для хроматографии биологических объектов (FPLC, Bio-Rad).
Фермент PDE 2 элюировали ацетатом натрия с линейным градиентом 0,05-1M, a элюированные фракции собрали для последующего анализа активности фермента PDE 2.
Объединили фракции, обладающие активностью фермента PDE 2, в течение ночи провели их диализ от дистиллированной воды и, используя систему Amicon filtration system (с мембранным фильтром YM10), провели 10-кратное сгущение.
Добавили этиленгликоль (30%, объем/объем) и получившийся раствор сохраняли при -20°С.
При указанных условиях ферментативная активность сохранялась стабильной в течение нескольких недель.
б) Анализ активности PDE 2
Активность фермента PDE 2 анализировалась в устройстве Scintillation Proximity Assay (SPA) (Amersham), которое включало метку 3H-цАМФ, буфер для анализа PDE (10× раствор: 500 мМ Трис/HCl с рН 7,5; 83 мкМ MgCl2 и 17 мкМ EGTA), а также гранулы PDE Yttrium SPA (содержащие 18 мкМ сульфата цинка).
Радиоактивность указанных гранул определяли с помощью сцинтилляционного счетчика (Packard model MINAXI β TRI-CARB 4000 SERIES).
В приведенной ниже таблице 4 содержатся результаты, выраженные как величины IC50 некоторых соединений, являющихся представительными всего класса соединений.
ПРИМЕР 22
Ингибирование фермента PDE 1
а) Очистка фермента PDE 1
Лиофилизированный и частично очищенный фермент PDE 1 был приобретен у фирмы Sigma (P0520).
Указанный фермент содержал фракцию Р1, о которой сообщалось в работе Но et al. (Biochim Biophys Acta, 1976, vol.429 (2), 461-473).
Лиофилизованный фермент был восстановлен 30% объем/объем этиленгликолем с концентрацией 500 г/мл, и его сохраняли при t -20°С.
б) Анализ активности PDE 1
Активность фермента PDE 1 анализировалась в устройстве Scintillation Proximity Assay (SPA) (Amersham), которое включало метку 3H-цАМФ, буфер для анализа PDE (10× раствор: 500 мМ Трис/HCl с рН 7,5; 83 мкМ MgCl2 и 17 мкМ EGTA), а также гранулы PDE Yttrium SPA (содержащие 18 мкМ сульфата цинка).
Радиоактивность указанных гранул определяли с помощью сцинтилляционного счетчика (Packard model MINAXI β TRI-CARB 4000 SERIES).
В приведенной ниже таблице 5 содержатся результаты, выраженные как величины IC50 некоторых соединений, являющихся представительными всего класса соединений.
ПРИМЕР 23
Приводятся данные о растворимости соединений формулы I настоящего изобретения в сравнении с аналогичными данными, указанными в заявке на международный патент WO 00/05218.
Для испытания растворимости использовали метод жидкостной хроматографии с высоким разрешением (ВРЖХ).
Тест на растворимость относительно эталона проводился с помощью анализа методом ВРЖХ (колонка 4,6×15 мм; градиент: вода/0,1%ТРА-СН3CN/0,06% TFA).
Был приготовлен 10 М раствор анализируемого вещества, растворенного в диметилсульфоксиде (ДМСО).
Для приготовления образца к 10 мкл описанного выше раствора добавили 190 мкл фосфатно-солевого буфера (ФСБ; рН 7,4). Для приготовления эталона к другим 10 мкл описанного выше раствора добавили 190 мкл ДМСО.
Результаты, полученные для соединений формулы I, указаны в приведенной ниже таблице 6.
Результаты, полученные для соединений из заявки на международный патент WO 00/05218

в которых R представляет собой группу -СН3 (если это не оговорено особо); приведены ниже в таблице 7.
Соединение, обозначенное в заявке на международный патент WO 00/05218 под номером 75, обладает ингибирующей активностью в отношении PDE 4, которая ниже величины, установленной для соединений по настоящему изобретению.
ПРИМЕР 24
Фармакинетические исследования на примере крыс
Крысам вводили смесь, содержащую 3-5 соединений из работы Ward K.W. et al. (Drug Metab. Dispos. 2001, vol.29, 82-88), доза составляла 10 мг/кг и ее вводили орально (1 мг/мл в 77% N-метилглюкозамине 25 мкМ и 20% PG, содержащего 3% Твин 80), или внутривенно в дозе 3 мг/кг (3 мг/мл в 15% ДМСО/85% ПЭГ).
В процессе исследования животных обрабатывали однократно.
Крысы с долгосрочно имплантированным катетером (SilasticR Dow Corning) получали исследуемую смесь в виде внутривенного бонуса (через имплантированный катетер) или орально через зонд.
Образцы крови отбирали через указанный катетер в различные моменты времени вплоть до 6 ч после введения смеси.
Анализ образцов крови проводили после осаждения белков при помощи двух объемов СН3CN; концентрации плазмы определяли методами жидкостной хроматографии/масс-спектрометрии.
Результаты, полученные для нескольких соединений формулы I, приведены в таблице 8.
Увеличение растворимости соединений по настоящему изобретению позволило провести изучение биологической доступности с помощью физиологических носителей, обычно используемых в стандартных фармацевтических составах.
Изучение соединений с общей формулой из заявки на международный патент WO 00/05218, которое было проведено с использованием физиологических носителей, показало, что величина биологической доступности равна 0.
Поскольку соединения с общей формулой из заявки на международный патент WO 00/05218 обладают слабой растворимостью, то фармакинетические исследования было необходимо проводить с использованием раствора 15% ДМСО/85% ПЭГ для соединений, вводимых и внутривенно и орально (т.е. использовали абсолютно нефизиологичный носитель).
В таблице 9 приведены полученные в указанных условиях результаты по биологической доступности.
Таким образом, соединения формулы I, являющиеся предметом настоящего изобретения, при их введении в физиологическом носителе демонстрируют хорошую биологическую доступность, величины которой сравнимы, а во многих случаях и выше, чем у соединений из сравниваемой заявки. При этом подчеркивается, что биологическая доступность последних соединений выше лишь в случае, если ее измеряют в нефизиологичном растворе ДМСО/ПЭГ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АЗАИНДАЗОЛА ИЛИ ДИАЗАИНДАЗОЛА В КАЧЕСТВЕ МЕДИКАМЕНТА | 2012 |
|
RU2600976C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2609018C2 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2597364C2 |
| ЗАМЕЩЕННЫЕ МЕТИЛФЕНИЛКЕТОНЫ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2007 |
|
RU2493149C2 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2589878C2 |
| НОВЫЕ ГЕМ-ДИФТОРИРОВАННЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2369612C2 |
| НОВОЕ ПРОИЗВОДНОЕ ГЕТЕРОЦИКЛИЧЕСКОГО АМИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2760184C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИСПИРОИНДОЛИНОНОВ | 2018 |
|
RU2682678C1 |
| Спиросоединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2800042C1 |
| НОВЫЙ ПАЛЛАДИЕВЫЙ КАТАЛИЗАТОР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2011 |
|
RU2575249C2 |
Изобретение относится к производным фталазина формулы (I), в которой R представяет собой метальную или дифторметильную группу; R1 представляет собой фенил или оксазолил или тиофенил, связанный химической связью с фталазиновым циклом посредством углерод-углеродной связи, причем как фенил, так и указанный гетероцикл замещены на карбоксильную группу, а также необязательно на вторую функциональную группу, выбранную из метокси-, нитро-, N-ацетиламино-, N-метансульфониламино-группы; а также их фармацевтически приемлемым солям. Указанные соединения формулы I являются ингибиторами фосфодиэстеразы. Объектами изобретения также являются способ получения соединений формулы (I) и фармацевтический состав для лечения аллергических и противовоспалительных заболеваний на основе указанных соединений. 3 н. и 6 з.п. ф-лы, 9 табл.
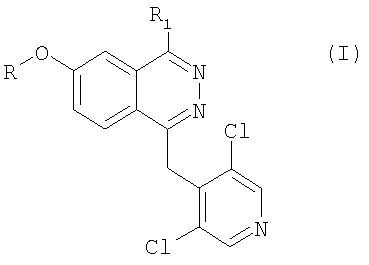
в которой R представяет собой метиальную или дифторметильную группу; R1 представляет собой фенил, или оксазолил, или тиофенил, связанный химической связью с фталазиновым циклом посредством углерод-углеродной связи, причем как фенил, так и указанный гетероцикл замещены на карбоксильную группу, а также необязательно на вторую функциональную группу, выбранную из метокси-, нитро-, N-ацетиламино-, N-метансульфониламиногруппы,
а также их фармацевтически приемлемые соли.
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойной кислоты;
4-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойной кислоты;
2-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойной кислоты;
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-5-нитробензойной кислоты;
5-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-2-метоксибензойной кислоты;
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-4-метоксибензойной кислоты;
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-5-метансульфониламинобензойной кислоты;
3-ацетиламино-5-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метокси-фталазин-1-ил]-бензойной кислоты;
3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-дифторметоксифталазин-1-ил]-бензойной кислоты;
2-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-оксазол-4-карбоновой кислоты;
2-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-тиофен-3-карбоновой кислоты;
(2-амино-2-гидроксиметилпропан-1,3-диольной соли 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойной кислоты;
N-метилглюкаминовой соли 3-[4-(3,5-дихлорпиридин-4-ил-метил)-7-метоксифталазин-1-ил]-бензойной кислоты;
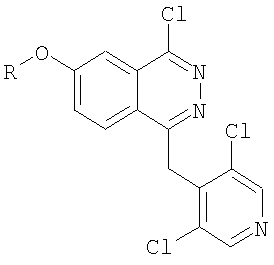
в которой R имеет значения, указанные для соединений формулы I,
и реагентом типа производного оловянной или бороновой кислоты, пригодного для замещения атома галогена, непосредственно связанного с фталазиновым циклом, на фенил, или оксазолил, или тиофенил, замещенный на карбоксигруппу, а также необязательно на вторую функциональную группу, указанную для значений R1 в формуле I.
| WO 00/052218 A1, 03.02.2000, реферат, формула изобретения | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| RU 99119224 А, реферат, формула изобретения. | |||

