Изобретение относится к новому палладиевому катализатору, представляющему собой комплекс три-[3,5-бис(фторметил)-фенил]-фосфина и палладия(0).
Изобретение дополнительно относится к способу получения нового катализатора. Изобретение также относится к применению нового катализатора в реакциях, требующих такие катализаторы, в частности в реакциях с образованием С-С связи (реакции кросс-сочетания, такие как реакция Сузуки, Хека, Стилле и др. сочетания), в реакциях с образованием связи С-гетероатом (C-N, С-О, C-S, С-Р, главным образом C-N) (например, реакция Бухвальда), а также при реакциях гидрогенизации.
Так как в настоящее время реакции кросс-сочетания, которые катализируются комплексами переходных металлов (наиболее часто комплексами Pd и Ni), играют исключительную роль в формировании С-С связи и такие реакции привели к радикальным изменениям в путях синтеза, изобретение будет обсуждаться ниже в первую очередь в связи с реакциями кросс-сочетания, однако без ограничения его объема данным способом применения.
Общий процесс реакций кросс-сочетания можно описать как
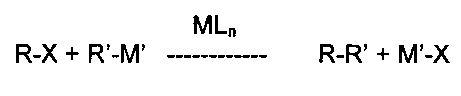
где
R и R′ представляют собой органические группы, подлежащие связыванию С-С связью,
М представляет сбой металлический компонент каталитического комплекса,
L представляет собой лиганды, присутствующие в каталитическом комплексе,
n представляет собой число присутствующих лигандов,
X представляет собой замещаемый атом или группу (например, Cl, Br, I, трифлат, мезилат, тозилат), и
М′ представляет собой металл или группу, содержащую металл, соответствующий типу рассматриваемой реакции кросс-сочетания (например, данным металлическим компонентом является бор для сочетания Сузуки-Мияуры, медь для сочетания Соногаширы, магний для сочетания по Харашу, кремний для сочетания Хияма, олово для сочетания Стилле, цинк для сочетания Негиши и т.д.).
Общий механизм реакций кросс-сочетания изображен на Фиг. 1.
Однако в аспектах практического применения данные способы имеют некоторые недостатки, которые особенно выражены в области фармацевтической промышленности. Одним из них является то, что требуются довольно большие количества катализатора (1-5 мол. % относительно субстрата), помимо этого, металлические примеси, источником которых является катализатор, как правило, можно удалить из конечного продукта в основном только утомительными и дорогими способами. Последнее является особенно справедливым для палладиевых катализаторов, которые, кроме того, в значительной степени подвержены разложению. В качестве примера, когда тетракис(трифенилфосфин)палладий (0) формулы (II)
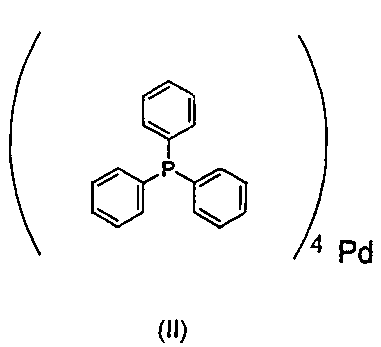
который до сих пор является катализатором, часто применяемым в промышленном масштабе, хранят на воздухе при комнатной температуре, значительное количество палладиевой черни отделяется в течение короткого времени, таким образом, его желательно хранить в холодильнике в атмосфере аргона. Несмотря на то что реакции кросс-сочетания с применением катализатора формулы (II) проводят в инертной атмосфере, отделение палладиевой черни все еще широко распространено, что приводит не только к значительной потере катализатора, но и к тому, что следует вводить утомительные трудоемкие и дорогостоящие стадии очистки.
Целью данного изобретения было получение нового катализатора на основе комплекса палладия (0), который является значительно более стабильным, чем катализаторы на основе комплекса палладия (0), которые применяли раньше для реакций кросс-сочетания, а также делает возможным значительно уменьшить количество катализатора, требуемое на 1 моль субстрата. В рамках данной области нашей основной целью являлось исключить образование палладиевой черни, так как палладиевая чернь, образующаяся из комплексов Pd(0), является конечным состоянием, причем разложение катализатора заметно снижает общую каталитическую активность. Кроме того, неконтролируемое разложение катализатора часто приводит к тому, что неконтролируемые количества P выщелачиваются в продукт.
На данный момент обнаружили, что катализатор на основе комплекса палладия (0) согласно изобретению полностью удовлетворяет указанным выше требованиям, а также имеет дополнительные преимущества.
Таким образом, в одном аспекте настоящее изобретение относится к комплексу палладия (0).
Данное соединение представляет собой твердое вещество яркого лимонно-желтого цвета с превосходной стабильностью: образование палладиевой черни не наблюдалось даже в образцах, которые хранили на воздухе при комнатной температуре в течение более 20 месяцев.
Данное соединение хранили на воздухе при различных значениях T температуры и влажности. Образцы периодически отбирали из хранящегося продукта и разложение продукта анализировали на основе 31P, 19F, 13C и 1H спектров ЯМР (ядерно-магнитный резонанс). Результаты приведены в следующей таблице.
При испытании соединения согласно изобретению ДСК (дифференциальная сканирующая калориметрия) разложение наблюдали при 169,5°C на воздухе при атмосферном давлении. При проведении испытания в инертной атмосфере, обнаружили, что точка плавления соединения составляет 220°C. Просто для сравнения, нефторированный катализатор формулы (II) начинал разлагаться при 98°C.
Проводили испытания на стабильность на катализаторах на основе комплекса[три-[замещенный фенил]-фосфина и Pd(0), где две из трех 3,5-(трифторметил)-фенильных групп, присоединенных к атому фосфора в лиганде, сохранены, а третья группа замещена моно-, ди- или триметоксифенильной, три-изопропилфенильной или 2-пиридильной группой. Ни одно из таких соединений не могло даже приблизиться к соединению согласно изобретению по стабильности при хранении. Таким образом, превосходная стабильность данного соединения при хранении является очень удивительной характеристикой, которая не проявляется даже у очень близких структурных аналогов.
При исследовании стабильности катализатора в условиях протекания реакции кросс-сочетания обнаружили, что катализатор не чувствителен к увеличению температуры; он сохраняет свою стабильность при любой температуре ниже своей точки плавления. Аналогичным образом, увеличение давления не оказывало влияния на стабильность катализатора.
При исследовании стабильности катализатора согласно изобретению было обнаружено следующее.
Катализатор не растворим в воде при промышленных температурах; в то же самое время он остается неограниченно стабильным при хранении в воде.
Растворимость катализатора в спиртах при комнатной температуре увеличивается с увеличением числа атомов углерода в спирте; однако при исследованных температурных интервалах каталитических реакций (110-130°C) его стабильность в спиртах снижается параллельно с увеличением числа атомов углерода спирта. Однако стабильность катализатора можно повысить или даже полностью восстановить при добавлении воды к реакционной смеси. В водных спиртах растворение катализатора начинается при температуре около 90°C и, в зависимости от рассматриваемого спирта, заканчивается при 110-130°C, где каталитическая активность достигает своего максимума. Однако даже при температурах, приводящих к полному растворению, отделения палладиевой черни не наблюдалось. Иногда происходило незначительное допустимое разложение, на которое указывало несильное потемнение цвета реакционной смеси (от лимонно-желтого до желтовато-коричневого). Особенно примечательным является то, что даже в таких условиях можно было достигнуть полного (100%) химического превращения. Для сравнения: когда соединение формулы (II) применяли в качестве катализатора при значительно более мягких условиях, чем те, которые обсуждались выше (атмосферное давление; точка кипения реакционной смеси), было невозможно избежать образования палладиевой черни, что ясно указывает на значительное разложение катализатора.
Для того чтобы избежать применения давлений выше атмосферного, что нежелательно в промышленных аспектах, стабильность катализатора согласно изобретению также испытывали в важных в промышленном отношении полярных апротонных и неполярных апротонных органических растворителях (например, диметилсульфоксид, диметилформамид, этилметилкетон, метилизобутилкетон, N-метилпирролидин и тетрагидрофуран), в которых катализатор полностью растворяется при более низких температурах. В данных растворителях также не наблюдалось образования палладиевой черни, хотя иногда цвет реакционной смеси становился в некоторой степени темнее во время каталитической реакции (наблюдали изменения цвета от лимонно-желтого до розового, оранжевого, красного или коричневатого). Как и в случае спиртов, обсуждаемых выше, в некоторых из данных растворителей небольшое снижение стабильности катализатора может быть значительно остановлено путем добавления воды к реакционной смеси.
При исследовании каталитической активности данного соединения в реакциях кросс-сочетания обнаружили, что при одном и том же субстрате и при прочих равных условиях реакции требуемое количество нового катализатора может быть снижено до части количества похожих известных катализаторов (с 1-5 мол.% относительно субстрата до 0,1-0,3 мол.% относительно субстрата) без какого-либо значимого уменьшения выхода и химического превращения, достигнутых за то же время реакции. Хотя выход и химическое превращение, которые достигаются при тех же условиях реакции, за данное время реакции снижаются, когда количество катализатора дополнительно уменьшают ниже данного уровня, это может быть достаточно скомпенсировано повышением температуры и/или времени реакции. В качестве примера: в реакции сочетания Сузуки 2-бромпиридина и 2-(4-этокси-3-метилфенил)-1,3,2-диоксаборолана, проводимой в 10:1 об./об. смеси метанола и воды, в присутствии K2CO3 при 110°C под давлением, 100% химического превращения достигали в течение 1 часа при применении 0,25 мол.% катализатора согласно изобретению. При уменьшении количества катализатора до 0,05 мол.% (что составляет 20% от прежней величины) достигнутое за один час химическое превращение еще оставалось довольно высоким (81%), а при применении всего 0,005 мол.% катализатора (что составляет 2% от прежней величины и 1-5 тысячных обычных промышленных значений) химическое превращение на 50% даже могло быть достигнуто за 1 час.
В большинстве случаев удаление палладия из продукта не требуется по той причине, что благодаря малому количеству и высокой стабильности нового катализатора, в продукте не остается палладия либо количество остаточного палладия ниже допустимого уровня. Если остаточный палладий все-таки следует удалить, дорогостоящие способы очистки [определенные операции для связывания Pd(0)], обычно применяющиеся для данной цели, могут быть полностью опущены. Остаточный, еще находящийся в комплексах палладий можно удалить путем простых операций (хроматография; фильтрация через недорогой угольный фильтр и т.д.), обычно применяющихся в промышленности, и, как правило, не требуется более одной стадии очистки.
Изобретение дополнительно относится к способу получения данного катализатора.
Катализатор согласно изобретению можно легко получить путем взаимодействия соли палладия (II) с три-[3,5-бис(трифторметил)-фенил]-фосфином, предпочтительно, с по меньшей мере четырехкратным молярным избытком указанного соединения, и восстановления палладия (II) до палладия (0) в полученной в результате комплексной соли в одно- или многостадийной реакции, предпочтительно проводящейся в одном реакционном сосуде без выделения промежуточных соединений. В качестве соли палладия (II) можно предпочтительно применять дихлорид палладия; предпочтительным восстанавливающим агентом является гидразин-гидрат.
Три-[3,5-бис(трифторметил)-фенил]-фосфин, используемый в качестве комплексообразующего агента, является известным веществом [см., например, Н.G. Alt, R. Baumgaertner, Н.A. Brune: Chemische Berichte 119(5), 1694-1703 (1986)].
Изобретение также относится к применению соединения согласно изобретению в качестве катализатора в реакциях сочетания C-C и C-гетероатом, а также при гидрогенизации.
Обнаружили, что соединение согласно изобретению можно применять в любом типе данных реакций. Условия таких реакций могут быть такими же, как применялись при использовании других катализаторов на основе комплексов Pd(0), с той разницей, что при применении соединения согласно изобретению в качестве катализатора обычно меньших, иногда гораздо меньших количеств катализатора все еще достаточно для проведения реакции. На основе своих общих знаний и на основе информации, представленной в данном описании, специалист в данной области может легко определить оптимальные параметры для реакций с применением катализатора, применяя стандартные способы или иногда простые испытания и принимая во внимание характеристики растворения катализатора. Здесь следует отметить, что идея применения катализатора согласно изобретению, приготовленного in situ (например, Pd2(dba)3 с PPh3(CF3)6), не является целесообразной, так как неконтролируемое образование комплекса и практически мгновенное появление Pd-черни приводит к низким выходам.
Следующие Примеры служат для иллюстрации дополнительных подробностей изобретения
Пример 1
Получение катализатора согласно изобретению
Аргон барботировали через 30 мл диметилсульфоксида при комнатной температуре, и затем добавляли 6,7 г (0,01 моль) три-[3,5-бис(трифторметил)-фенил]-фосфина и 0,355 г (0,002 моль) хлорида палладия (II). После этого смесь нагревали до 110-130°C. После того как достигали абсолютной прозрачности раствора, что свидетельствовало о том, что комплекс образовался, к смеси добавляли 0,5 г (0,01 моль) гидразин-гидрата. Затем колбу погружали в ледяную воду. Отделившийся продукт фильтровали через пористый стеклянный фильтр и три раза промывали хлороформом. Получали твердое кристаллическое вещество яркого лимонно-желтого цвета с выходом 90%.
Характеристические данные спектра ЯМР: 1Н-ЯМР (300 МГц, ТГФ-d8 (тетрагидрофуран), δ=3,58 млн-1) 8.17 (s, 12H), 7.84 (s, 24H); 13C-ЯМР (75 МГц, ТГФ-d8, δ=67,3 млн-1) 138.1 (C), 133.7 (q, J=38,7 Гц, C), 133.4 (CH), 126.3 (CH), 123.4 (q, J=271,57 Гц, CF3); 31P-ЯМР (300 МГц, ТГФ-d8) 28.77; 19F-ЯМР (300 МГц, ТГФ-d8) -62.94.
Пример 2
Получение 2-(4-этокси-3-метилфенил)пиридина с помощью реакции сочетания Сузуки с применением 10/1 об./об. смеси метанола и воды в качестве растворителя и катализатора согласно изобретению
Общий протокол
Количество катализатора, которое будет приведено ниже, 618 мг (3 ммоля) 2-(4-этокси-3-метилфенил)-1,3,2-диоксаборолана и 553 мг (4 ммоля) карбоната калия взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. В конце вводили 316 мг (190 мкл, 2 ммоля) 2-бромпиридина (субстрат) с использованием автоматической пипетки. Колбу закрывали и реакционную смесь перемешивали при температуре и в течение времени, приведенных ниже, необязательно при давлении выше атмосферного.
В целях обработки, охлажденную реакционную смесь экстрагировали четыре раза 5 мл хлороформа каждый раз; таким образом, практически все количество катализатора удаляли из продукта. Так как хлороформный экстракт все еще содержал примесь диоксоборолана, отделенное таким образом вещество дополнительно очищали колоночной хроматографией с силикагелем, применяя 3/1 об./об. смесь гексана и этилацетата в качестве элюирующего агента.
Серия испытаний (А)
В данной серии испытаний реакции проводили при температуре 110°C и при давлении выше атмосферного в течение 1 часа. Количество соединения согласно изобретению было различным, и проверяли, как это его изменение влияет на достигнутое химическое превращение.
Во всех случаях, представленных в данном описании, величины химического превращения определяли на основе спектров 1H ЯМР или с помощью газовой хроматографии. Результаты представлены в Таблице 1. Несмотря на эти довольно маломасштабные испытываемые реакции, обработка смеси влияет на практический выход, эти данные также приведены для справки.
Отделения палладиевой черни не наблюдалось ни в одном случае; цвет реакционной смеси сохранялся лимонно-желтым во всех реакциях. Особенно примечательным является то, что 50% химическое превращение еще могло быть достигнуто в течение 1 часа, когда количество катализатора согласно изобретению составляло только 0,005 мол.%. Согласно опыту, приобретенному в других испытаниях, данное снижение эффективности химического превращения может быть скомпенсировано увеличением времени реакции и/или температуры реакции.
В испытаниях, проводимых в целях проверки, вышеприведенную реакцию повторяли таким образом, что катализатор не добавляли к реакционной смеси. Таким образом, намеревались удостовериться в том, что образование продукта действительно может объясняться тем, что катализатор вводится в очень малом количестве, а не действием каких-либо металлических примесей, которые могли бы присутствовать в растворителях или в колбах. В данных условиях эффективность химического превращения была равна нулю, таким образом, можно утверждать с полной уверенностью, что катализатор согласно изобретению является активным даже в количестве 0,005 мол.%.
Серия испытаний (Б)
В данной серии испытаний применяли 0,25 мол.% катализатора согласно изобретению для 1 моля 2-бромпиридина в качестве субстрата и реакции проводили в течение 1 часа при температурах, приведенных в Таблице 2, при давлении выше атмосферного, при необходимости. Проверяли, каким образом изменения температуры влияют на достигнутое химическое превращение. Результаты приведены в Таблице 2; практические выходы также приведены для справки.
Наблюдаемые результаты показывают, что при применении 10/1 об./об. смеси метанола и воды в качестве реакционной среды, рекомендуется проводить реакцию сочетания при температуре выше 90°C и при давлении выше атмосферного, что позволяет реакционной смеси оставаться в жидком состоянии. Это можно объяснить тем, что замечательное растворение катализатора происходит при таких температурах. Образования палладиевой черни или любого другого признака разложения катализатора не могли наблюдать ни в одной из реакций. Для сравнения; когда в реакции, которую проводили при температуре 110°C, катализатор согласно изобретению заменяли тем же количеством катализатора формулы (II), реакционная смесь чернела в течение нескольких минут. После завершения реакции очень трудно удалить металлические примеси. Продукт, полученный в данной последней реакции, оставался оранжево-желтым/темно-оранжево-желтым даже после полного удаления палладиевой черни, в то время как при применении катализатора согласно изобретению, получали белоснежный продукт.
Физические константы всех образцов продукта, полученные в Примере 2, в пределах точности измерений хорошо согласовывались друг с другом и с соответствующими параметрами образца аутентичного продукта. Для справки ниже приведены физические константы, измеренные для образца 2-(4-этокси-3-метоксифенил)пиридина, приготовленного в 10/1 об./об. смеси метанола и воды при 110°C в течение 1 часа с применением 0,25 мол.% катализатора согласно изобретению:
1H ЯМР (300 МГц, CDCl3, δTMS=0 млн-1): 8.65 (d, J=4,8 Гц, 1H), 7.75 (m, 4H), 7.16 (m, 1H), 6.90 (d, J=8,4 Гц, 1H), 4.10 (q, J=6,9 Гц, 2H), 2.31 (s, 3H), 1.45 (t, J=7,2 Гц).
13C-ЯМР (75 МГц, CDCl3, δCDCl3=77,00 млн-1): 158.2 (C), 157.3 (C), 149.3 (CH), 136.7 (CH), 131.1 (C), 129 (CH), 127.1 (C), 125.5 (CH), 121.2 (CH), 119.9 (CH), 111.0 (CH), 63.6 (CH2), 16.4 (CH3), 14.9 (CH3).
ИК (KBr, ν см-1): 1604, 1587, 1561, 1467, 1433, 1394, 1309, 1281, 1247, 1181, 1151, 1131, 1109, 1042, 926, 884, 777, 742, 618.
Пример 3
Получение 2-(4-этокси-3-метилфенил)пиридина с помощью реакции сочетания Сузуки в реакционной среде, отличающейся от 10/1 об./об. смеси метанола и воды, и с применением соединения согласно изобретению в качестве катализатора
Реакцию сочетания Сузуки, описанную в Примере 2, повторяли с применением 316 мг (190 мкл, 2 ммоля) 2-бромпиридина в качестве субстрата и общим количеством реакционной среды, составляющим 11 мл, однако условия реакции (состав реакционной смеси; количество катализатора; количество реагента диоксоборолана; время реакции; температура) различались, как показано в Таблице 3. Химическое превращение оценивали, как описано в Примере 2. Результаты приведены в Таблице 3.
Когда применяли водный этанол, водный изопропанол и водный трет-бутанол, при времени реакции 1 час, цвет реакционной смеси постепенно становился насыщеннее и становился коричневым; порядок насыщения цвета был следующим: этанол-изопропанол-трет-бутанол. Однако палладиевая чернь не отделялась ни в одном из примеров и степень химического превращения оставалась 100%, отражая то, что катализатор сохранял свою активность. При проведении реакции в смесях гексан/вода, диметоксиэтан/вода и тетрагидрофуран/вода, обнаружили, что качество органического растворителя как компонента реакционной смеси в высокой степени влияет на степень химического превращения, достигаемую за заданный период времени. Это обычное явление для реакций кросс-сочетания. И в этом случае с данными растворителями образования палладиевой черни не могли наблюдать, хотя иногда цвет реакционной смеси становился насыщеннее в течение реакции. Результаты испытаний, которые проводили в смеси тетрагидрофуран/вода, являются особенно примечательными. Испытание также проводили с чрезвычайно малым количеством катализатора (0,002 мол.%, около одной тысячной количества, необходимого для известных катализаторов). Как и в Примере 2, данное чрезвычайно малое количество катализатора вводили в смесь в виде стокового раствора в тетрагидрофуране. Данные ясно показывают, что снижение степени химического превращения может быть достаточно скомпенсировано увеличением времени реакции и/или температуры реакции: при повышении температуры до 130°C и времени реакции до 19 часов можно было достигнуть 100% химического превращения даже с этим чрезвычайно малым количеством катализатора. После проведения контрольного испытания, описанного в Примере 2 (реакция без катализатора), снова убедились в том, что образование продукта может объясняться исключительно наличием катализатора, а не действием каких-либо возможных примесей металлов, которые могли бы присутствовать в растворителях или в колбах. Превосходную стабильность катализатора согласно изобретению хорошо иллюстрирует тот факт, что никакого признака разложения катализатора не могли наблюдать даже после того, как реакцию проводили при температуре 130°C в течение 19 часов, что является очень жестким условием.
Пример 4
Получение производных пиридина с помощью реакции сочетания Сузуки с применением катализатора согласно изобретению
Общий протокол
14 мг (0,25 мол.% относительно субстрата 2-бромпиридин) катализатора, 3 ммоля реагента диоксаборолан и 553 мг (4 ммоля) карбоната калия взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. В конце вводили 316 мг (190 мкл, 2 ммоля) 2-бромпиридина (субстрат) автоматической пипеткой. Затем колбу закрывали и реакционную смесь перемешивали в течение 1 часа при температуре 110°C при давлении, необходимом для поддержания жидкого состояния реакционной смеси. Затем реакционную смесь обрабатывали, как описано в Примере 2.
Применявшиеся реагенты, полученные продукты и их физические константы, а также практические выходы (%) приведены в Таблице 4.
Физические константы всех полученных продуктов, в пределах точности измерений, хорошо согласовывались с соответствующими параметрами образцов аутентичного продукта. Реакционные смеси всегда сохраняли лимонно-желтый цвет, даже после времени реакции, составляющего 16 часов. Никакого признака, относящегося к дополнительному разложению катализатора, не обнаруживали.
Пример 5
Получение производных индола с помощью реакции сочетания Сузуки с применением катализатора согласно изобретению
Общий протокол
14 мг (0,25 мол. % относительно субстрата 5-броминдол) катализатора согласно изобретению, 3 ммоля реагента диоксаборолана, 553 мг (4 ммоля) карбоната калия и 390 мг (2 ммоля) 5-броминдола взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. Затем колбу закрывали и реакционную смесь перемешивали в течение 1 ч при 110°C под давлением, необходимым для поддержания жидкого состояния реакционной смеси.
Из полученных конечных продуктов только 5-(п-толил)-1Н-индол растворим в воде. При получении данного соединения реакционную смесь обрабатывали, как описано в Примере 2.
Реакционные смеси, содержащие другие (нерастворимые в воде) индольные соединения, обрабатывали следующим образом.
К реакционной смеси добавляли 9 мл воды и отделенную твердую фазу, которая содержит катализатор и продукт, отфильтровывали через пористый стеклянный фильтр. Для того чтобы удалить катализатор, полученную в результате твердую фазу растворяли в хлороформе, нерастворимый в хлороформе катализатор отфильтровывали, фильтрат сушили над сульфатом натрия и затем упаривали в вакууме.
Используемые реагенты, полученные продукты и их физические константы, а также практические выходы (%) приведены в Таблице 5.
Физические константы всех полученных продуктов, в пределах точности измерений, хорошо согласовывались с соответствующими параметрами образцов аутентичного продукта. В реакционных смесях не наблюдали образования палладиевой черни; отделенный катализатор во всех случаях сохранял лимонно-желтый цвет.
Пример 6
Получение производных изохинолина с помощью реакции сочетания Сузуки с применением катализатора согласно изобретению
Общий протокол
14 мг (0,25 мол. % относительно субстрата 5-бром-изохинолин) катализатора согласно изобретению, 3 ммоля реагента диоксаборолана, 553 мг (4 ммоля) карбоната калия и 416 мг (2 ммоля) 5-бром-изохинолина взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. Затем колбу закрывали и реакционную смесь перемешивали в течение 1 ч при 110°C под давлением, необходимым для поддержания жидкого состояния реакционной смеси. Полученные в результате реакционные смеси обрабатывали, как описано в Примере 2.
Используемые реагенты, полученные продукты и их физические константы, а также практические выходы (%) приведены в Таблице 6.
Физические константы всех полученных продуктов, в пределах точности измерений, хорошо согласовывались с соответствующими параметрами образцов аутентичного продукта. Реакционные смеси во всех случаях сохраняли лимонно-желтый цвет, и никакого признака, относящегося к дополнительному разложению катализатора, не могли обнаружить.
Пример 7
Получение производных бифенила с помощью реакции сочетания Сузуки с применением катализатора согласно изобретению
Общий протокол
14 мг (0,25 мол.% относительно субстрата п-бромтолуол) катализатора согласно изобретению, 3 ммоля реагента диоксаборолана, 553 мг (4 ммоля) карбоната калия и 342 мг (2 ммоля) п-бромтолуола взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. Затем колбу закрывали и реакционную смесь перемешивали в течение 1 ч при 110°C под давлением, необходимым для поддержания жидкого состояния реакционной смеси. Полученные в результате реакционные смеси обрабатывали, как описано в Примере 5.
Используемые реагенты, полученные продукты и их физические константы, а также практические выходы (%) приведены в Таблице 7.
Физические константы всех полученных продуктов, в пределах точности измерений, хорошо согласовывались с соответствующими параметрами образцов аутентичного продукта. Реакционные смеси во всех случаях сохраняли лимонно-желтый цвет, и никакого признака, относящегося к дополнительному разложению катализатора, не могли обнаружить.
Пример 8
Получение производных стильбена с помощью реакции сочетания Хека с применением катализатора согласно изобретению
Производные стильбена получали с помощью реакции стирола с различными арилбромидами, как показано на следующей схеме реакции:
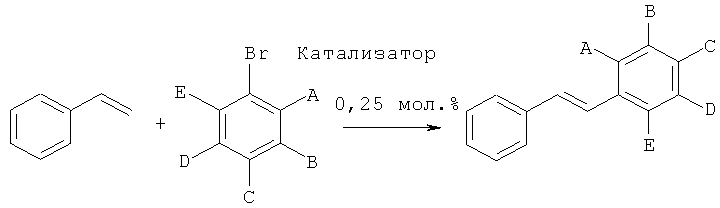
Общий протокол
552 мг (4 ммоля, 2 экв.) K2CO3, 14 мг (0,25 мол.%, рассчитанных на субстрат арилбромид) катализатора согласно изобретению, 312 мг (0,343 мл, 3 ммоля, 1,5 экв.) стирола, 2 ммоля (1 экв.) субстрата арилбромид и 10 мл 10:1 смеси метанола и воды загружали в высушенный в сушильной печи сосуд Шленка. Реакцию проводили при 110°C в течение 3 часов или 20 часов, как показано в Таблице 8. Степень химического превращения определяли, подвергая реакционные смеси ГХ (газовая хроматография), и затем продукт выделяли. Для испытаний №1, 2, 3 и 5 продукты осаждались из смеси при охлаждении, таким образом, они могли быть выделены путем простой фильтрации; тогда как для испытаний №4, 6 и 7 продукты выделяли с помощью флеш-хроматографии.
Результаты представлены в Таблице 8.
Данные ЯМР для полученных в результате производных стильбена являются следующими:
(E)-3-Фторстильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.53 (d, J=7,5 Гц, 2H), 7.39 (t, J=7,5 Гц, 2H), 7.41-7.22 (m, 4H), 7.11 (s, 1H), 7.10 (s, 1H), 6.99-6.94 (m, 1H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 163.5 (C, d, J=244 Гц), 139.9 (C, d, J=7,65 Гц), 137.1 (C), 130.3 (CH, d, J=8,18 Гц), 129.0 (CH), 128.2 (CH), 127.7 (CH, d, J=2.70 Гц), 126.9 (CH), 122.7 (CH, d, J=2,78 Гц), 114.62 (CH, d, J=21,5 Гц), 113.0 (CH, d, J=21,5 Гц).
(E)-4-Нитростильбен: 1H ЯМР (300 МГц, CDCl3) δ 8.23-8.21 (m, 2H), 7.63 (d, J=8,7 Гц, 2H), 7.55 (d, J=7,5 Гц, 2H), 7.43-7.25 (m, 4H), 7.14 (d, J=16,5 Гц, 1H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 147.0 (C), 136.4 (C), 133.6 (CH), 129.1 (CH), 127.3 (CH), 127.1 (CH), 126.5 (CH), 124.4 (CH).
(E)-4-Метилстильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.54 (d, J=7,8 Гц, 2H), 7.46 (d, J=7,8 Гц, 2H), 7.41-7.36 (m, 2H), 7.31-7.26 (m, 1H), 7.21 (d, J=7,8 Гц, 2H), 7.12 (s, 2H), 2.40 (s, 3H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 137.8 (C), 137.8 (C), 134.8 (C), 129.7 (CH), 128,9 (CH), 128.0 (CH), 127.7 (CH), 126.7 (CH), 21.5 (CH3).
(E)-2,4-Диметоксистильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.53 (d, J=8,4 Гц, 3H), 7.42 (d, J=16,5 Гц, 1H), 7.35 (t, J=7,5 Гц, 2H), 7.24 (dd, J=4,9 Гц, 12,1 Гц, 1H), 7.02 (d, J=16,5 Гц, 1H), 6.53 (dd, J=2,2 Гц, 9,9 Гц, 1H), 6.49 (d, J=2,4 Гц, 1H), 3.88 (s, 1H), 3.84 (s, 1H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 160.5 (C), 138.3 (C), 128.5 (CH), 127.2 (CH), 127.0 (CH), 126.9 (CH), 126.3 (CH), 123.3 (CH), 119.5 (C), 105.0 (CH), 98.5 (CH), 55.5 (CH3), 55.4 (CH3).
(E)-3,5-Диметилстильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.55-7.53 (m, 2H), 7.41-7.36 (m, 2H), 7.31-7.26 (m, 1H), 7.18 (s, 2H), 7.11 (d, J=2,4 Гц, 2H), 6.95 (s, 1H), 2.38 (s, 6H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 138.3 (C), 137.5 (C), 129.7 (CH), 129.1 (CH), 128.9 (CH), 128.5 (CH), 127.7 (CH), 126.7 (CH), 124.7 (CH), 21.5 (CH3).
(E)-2,6-Диметилстильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.54-7.52 (m, 2H), 7.42-7.37 (m, 2H), 7.32-7.26 (m, 1H), 7.13 (d, J=16,8 Гц, 1H), 7.1 (m, 3H), (m, 3H), 6.63 (d, J=16,8 Гц, 1H), 2.39 (s, 6H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 137.6 (C), 137.0 (C), 136.2 (C), 134.0 (CH), 128.7 (CH), 127.9 (CH), 127.6 (CH), 126.9 (CH), 126.7 (CH), 126.3 (CH), 21.0 (CH3).
(E)-2,4,6-Триизопропилстильбен: 1H ЯМР (300 М Гц, CDCl3) δ 7.54-7.52 (m, 2H), 7.43-7.38 (m, 2H), 7.33-7.26 (m, 1H), 7.22 (d, J=16,5 Гц, 1H), 7.07 (s, 2H), 6.52 (d, J=16,8 Гц, 1H), 3.31 (h, J=6,9 Гц, 2H), 2.94 (h, J=6,9 Гц, 1H), 1.33-1.23 (m, 18H); 13C ЯМР (АТФ) (75 М Гц, CDCl3) δ 142.4 (C), 141.4 (C), 132.3 (C), 128.6 (CH), 127.7 (C), 123.4 (CH), 122.1 (CH), 121.7 (CH), 121.0 (CH), 115.3 (CH), 29.0 (CH), 24.9 (CH), 18.7 (CH3), 18.5 (CH3).
Пример 9
Получение производных фенилаиетилена с помощью реакции сочетания Соногаширы с применением катализатора согласно изобретению
Производные фенилацетилена получали с помощью реакции фенилацетилена с различными арилбромидами, как показано на следующей схеме реакции:
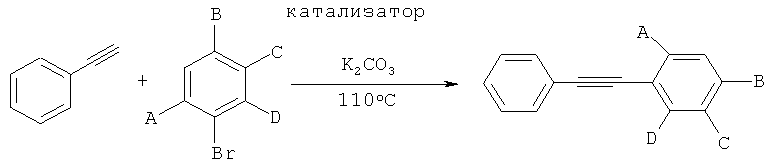
Общий протокол:
276 мг (2 ммоля, 1 экв.) K2CO3, 7 мг (0,25 мол.%, рассчитанных на субстрат арилбромид) катализатора согласно изобретению, 0,165 мл (1,5 ммоля, 1,5 экв.) фенилацетилена, 1 ммоль (1 экв.) субстрата арилбромида и 5 мл растворителя [растворитель (а): 5:1 смесь метанола и воды; растворитель (б): н-бутанол; растворитель (в): глицерол формаль] загружали в высушенный в сушильной печи сосуд Шленка. Реакцию проводили при 110°C в течение 3 часов или 24 часов, как показано в Таблице 9. Количества продуктов определяли, подвергая реакционную смесь ГХ.
Результаты представлены в Таблице 9.
Данные ЯМР для полученных в результате производных фенилацетилена выглядят следующим образом:
1-Метил-4-(фенилэтинил)-бензол: 1H ЯМР (300 МГц, CDCl3) δ 7.43 (d, J=8,0 Гц, 2H), 7.26-7.22 (m, 5H), 6.70 (d, J=8,3 Гц, 1H), 3.75 (s, 3H), 2.13 (s, 3H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 158.2 (C), 133.9 (CH), 131.5 (CH), 130.6 (CH), 128.5 (CH), 128.3 (CH), 127.9 (CH), 123.8 (C), 109.9 (CH), 89.9 (C), 55.4 (CH), 16.1 (CH).
1-Нитро-4-(фенилэтинил)-бензол: 1H ЯМР (300 МГц, CDCl3) δ 8.17 (d, J=9,0 Гц, 2H), 7.62 (d, J=9,0 Гц, 2H), 7.58-7.55 (m, 2H), 7.41-7.38 (m, 3H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 147.0 (C), 132.3 (CH), 131.9 (CH), 130.3 (C), 129.4 (CH), 129.0 (CH), 123.7 (CH), 122.2 (C), 94.8 (C), 87.7 (C).
1-Метил-4-(фенилэтинил)-бензол: 1H ЯМР (300 МГц, CDCl3) δ 7.60-7.57 (m, 2H), 7.49 (d, J=8,1 Гц, 2H), 7.40-7.35 (m, 3H), 7.20 (d, J=7,9 Гц, 2H), 2.41 (s, 3H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 138.6 (C), 131.8 (CH), 131.7 (CH), 129.3 (CH), 128.5 (CH), 128.3 (CH), 123.7 (C), 120.4 (C), 89.8 (C), 89.0 (CH), 21.7 (CH3).
Фенил-(2,4,6-триизопропил-фенил)-ацетилен: 1H ЯМР (300 МГц, CDCl3) δ 7.58 (dd, J=8,0 Гц, 1,4 Гц, 2H), 7.43-7.35 (m, 3H), 7.07 (s, 2H), 3.65 (sept, J=6,9 Гц, 2H), 2.96 (sept, J=6,9 Гц, 1H), 1.36 (d, J=6,9 Гц, 12H), 1.32 (d, J=6,9 Гц, 6H);
13C ЯМР (АТФ) (75 МГц, CDCl3) δ 150.9 (C), 149.5 (C), 131.5 (CH), 128.6 (CH), 128.1 (CH), 124.6 (CH), 120.7 (CH), 118.7 (C), 97.0 (C), 87.3 (C), 4.9 (CH3), 32.2 (CH3), 24.2 (CH3), 23.6 (CH3).
Описанную выше реакцию повторяли с применением 2-бром-3-метил-бут-2-ена в качестве субстрата. Полученные результаты приведены в Таблице 10.
Данные ЯМР для полученного в результате продукта выглядят следующим образом: 1H ЯМР (300 МГц, CDCl3) δ 7.45-7.39 (m, 2H), 7.34-7.22 (m, 3H), 2.02 (s, 3H), 1.89 (s, 3H), 1.79 (s, 3H).
Пример 10
Получение N-фенилпиперидина с помощью реакции Бухвальда с применением соединения согласно изобретению в качестве катализатора
224 мг (2 ммоля) трет-бутоксида калия, 70 мг (0,25 мол.%, рассчитанных на субстрат бромбензол) катализатора согласно изобретению, 105 мкл (1 ммоль) бромбензола, 198 мкл (2 ммоля) пиперидина и 5 мл растворителя загружали в высушенный в сушильной печи сосуд Шленка. Реакционную смесь нагревали на масляной бане при температуре 110°C в течение 24 часов, затем позволяли ей охладиться до комнатной температуры, и выпаривали при пониженном давлении. Для получения желаемого продукта остаток очищали колоночной хроматографией на силикагеле (растворитель для элюирования: гексан/этилацетат); 1H ЯМР (300 МГц, CDCl3) δ 7.31-7.26 (m, 2H), 7.00-6.97 (m, 2H), 6.89-6.84 (m, 1H), 3.19 (t, J=5,6 Гц, 4H), 1.79-1.72 (m, 4H), 1.64-1.59 (m, 2H).
Результаты испытаний, проведенных в различных растворителях, приведены в Таблице 11.
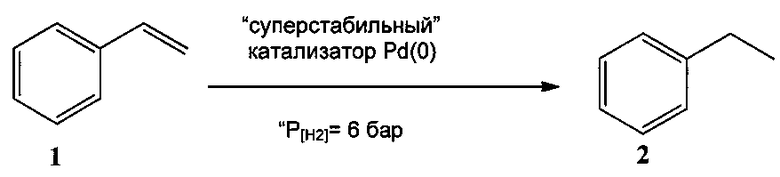
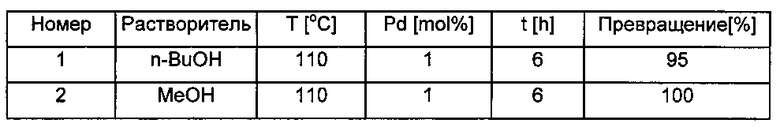
Пример 11
В тестовых реакциях в качестве растворителей использовали метанол и н-бутанол. Тестировали восстановление стирола, применяя во всех случаях давление водорода 6 бар. В реактор помещали 21 мг катализатора формулы (I) (1 моль % по отношению к субстрату, представляющему собой стирол), 1 ммоль стирола и 10 мл растворителя. Сначала колбу промывали газообразным аргоном, потом заполняли ее газообразным Н2 (6 бар). Реакционную смесь перемешивали 6 часов при 110°C при температуре 110°C. Реакционное превращение мониторили с помощью газовой хроматоргафии
Скрининг суперстабильного катализатора Pd(0) в отношении гидрогенгизации стрирола
| название | год | авторы | номер документа |
|---|---|---|---|
| N-ГИДРОКСИЛМОЧЕВИНЫ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ СРЕДСТВ | 1995 |
|
RU2152935C2 |
| 6, 6-БИЦИКЛИЧЕСКИЕ КОЛЬЦЕВЫЕ ЗАМЕЩЕННЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2005 |
|
RU2379308C2 |
| 4-АМИНО-6-(ГЕТЕРОЦИКЛИЧЕСКИЕ)ПИКОЛИНАТЫ И 6-АМИНО-2-(ГЕТЕРОЦИКЛИЧЕСКИЕ)ПИРИМИДИН-4-КАРБОКСИЛАТЫ И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2014 |
|
RU2672587C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2545074C1 |
| СПОСОБ МИНИМИЗАЦИИ ГИСТЕРОТРОФНОГО ЭФФЕКТА ТАМОКСИФЕНА И АНАЛОГОВ ТАМОКСИФЕНА | 1995 |
|
RU2158589C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ИНДОЛОПИРРОЛОКАРБАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ САХАРОВ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ РОСТА ОПУХОЛЕЙ | 1997 |
|
RU2167880C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ АРИЛКОНДЕНСИРОВАННЫХ АЗАПОЛИЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2282619C9 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2006 |
|
RU2382781C2 |
| ИНГИБИТОРЫ ПРОЛИЛГИДРОКСИЛАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2429226C9 |

Изобретение относится к способу получения комплекса три-[3,5-бис(трифторметил)-фенил]-фосфина и палладия(0). Способ осуществляют взаимодействием соли палладия(II) с три-[3,5-бис(трифторметил)-фенил]-фосфином и палладий(II) восстанавливают до палладия(0) в полученной в результате комплексной соли. Также предложены катализатор и его применение. Изобретение позволяет получить комплекс три-[3,5-бис(трифторметил)-фенил]-фосфина и палладия(0), обладающий превосходной стабильностью и который можно применять в качестве катализатора в реакциях сочетания С-С и С-гетероатом, а также для гидрогенизации. 4 н. и 14 з.п. ф-лы, 1 ил., 12 табл., 11 пр.
1. Способ получения комплекса три-[3,5-бис(трифторметил)-фенил]-фосфина и палладия(0), характеризующийся тем, что осуществляют реакцию соли палладия(II) с три-[3,5-бис(трифторметил)-фенил]-фосфином и палладий(II) восстанавливают до палладия(0) в полученной в результате комплексной соли.
2. Способ по п.1, характеризующийся тем, что реакцию соли палладия(II) осуществляют по меньшей мере с четырехкратным молярным избытком три-[3,5-бис(трифторметил)-фенил]-фосфина.
3. Способ по п.1, характеризующийся тем, что реакцию соли палладия(II) с три-[3,5-бис(трифторметил)-фенил]-фосфином и восстановление палладия(II) до палладия(0) осуществляют в одном реакционном сосуде без выделения промежуточных соединений.
4. Способ по п.1, отличающийся тем, что в качестве соли палладия(II) применяют дихлорид палладия.
5. Способ по п.1, отличающийся тем, что восстановление проводят гидразин-гидратом.
6. Катализатор для реакций сочетания С-С и С-гетероатом и для реакций гидрогенизации, представляющий собой комплекс три-[3,5-бис(трифторметил)-фенил]-фосфина и палладия(0), полученный способом по п.1.
7. Катализатор по п.6, характеризующийся тем, что его точка плавления при проведении испытания ДСК (дифференциальная сканирующая калориметрия) в инертной атмосфере составляет 220°C.
8. Катализатор по п.6, характеризующийся тем, что его температура разложения при проведении испытания ДСК на воздухе при атмосферном давлении составляет 169,5°C.
9. Катализатор по п.6, характеризующийся следующими данными ЯМР спектра: 1Н-ЯМР (300 МГц, ТГФ-d8 (тетрагидрофуран), δ=3,58 млн-1) 8,17 (s, 12 Н), 7,84 (s, 24 Н); 13С-ЯМР (75 МГц, ТГФ-d8, δ=67,3 млн-1) 138,1 (С), 133,7 (q, J=38,7 Гц, С), 133,4 (СН), 126,3 (СН), 123,4 (q, J=271,57 Гц, CF3); 31Р-ЯМР (300 МГц, ТГФ-d8) 28,77; 19F-ЯМР (300 МГц, ТГФ-d8) -62,94.
10. Катализатор для реакций сочетания С-С и С-гетероатом и для реакций гидрогенизации, представляющий собой комплекс три-[3,5-бис(трифторметил)-фенил]-фосфина и палладия(0), характеризующийся следующими данными ЯМР спектра: 1Н-ЯМР (300 МГц, ТГФ-d8 (тетрагидрофуран), δ=3,58 млн-1) 8,17 (s, 12 Н), 7,84 (s, 24 Н); 13С-ЯМР (75 МГц, ТГФ-d8, δ=67,3 млн-1) 138,1 (С), 133,7 (q, J=38,7 Гц, С), 133,4 (СН), 126,3 (СН), 123,4 (q, J=271,57 Гц, CF3); 31Р-ЯМР (300 МГц, ТГФ-d8) 28,77; 19F-ЯМР (300 МГц, ТГФ-d8) -62,94.
11. Катализатор по п.10, характеризующийся тем, что его точка плавления при проведении испытания ДСК в инертной атмосфере составляет 220°C.
12. Катализатор по п.10 или 11, характеризующийся тем, что его температура разложения при проведении испытания ДСК на воздухе при атмосферном давлении составляет 169,5°C.
13. Применение катализатора по любому из пп.6-12 в реакциях сочетания С-С и С-гетероатом, а также для гидрогенизации.
14. Применение по п.13, где реакция является реакцией С-С кросс-сочетания.
15. Применение по п.14, где реакция кросс-сочетания является реакцией сочетания Сузуки, сочетания Хека или сочетания Соногаширы.
16. Применение по п.13, где количество катализатора, применяющееся в реакции на один моль субстрата, составляет 0,25 мол. % или менее.
17. Применение по п.13, где реакция является реакцией сочетания C-N.
18. Применение по п.17, где реакция является сочетанием Бухвальда.
| SOBERATS B | |||
| et al, Conventional Tetrakis(triphenylphosphine)palladium-Copper(I) Iodide-Catalyzed Sonogashira Coupling of Free and BOC-Protected Propargylic Amines "On Water", Advanced Synthesis & Catalysis, 2009, v | |||
| Деревобетонный каток | 1916 |
|
SU351A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| Телефонная трансляция с катодным реле | 1920 |
|
SU1727A1 |
| CLARKE M.L | |||
| et al, The electron-poor phosphines P{ C6H3(CF3)2-3,5} 3 and P(C6F5)3 do not mimic | |||

