Существует обширная литература, относящаяся к пиридиновым соединениям, однако имеется очень мало данных, касающихся производных 3-амино-2-арилалкинил-, 3-амино-2-гетероарилалкинилпиридина по изобретению. Настоящее изобретение относится к замещенным 3-аминоарилалкинилпиридинам и 3-аминогетероарилалкинилпиридинам.
В патенте США № 6384235В2 и в публикации Rodriguez et al. Angewandte Chemie, International Edition 2000, 39, 14, 2488-2490 описан 3-амино-(2-фенилэтинил)пиридин в качестве синтетического промежуточного соединения. В WO 99/40091 раскрываются определенные 3-амино-(2-фенилэтинил)пиридины в качестве синтетических промежуточных соединений, в которых впоследствии проводят замыкание цикла. В WO 99/02497 описаны определенные 2-гетероалкинилпиридины как соединения, модулирующие mGluR, и приведено очень широкое описание ряда соединений. Однако ни одно из конкретно раскрытых соединений не относится к 3-амино-2-арил- или гетероарилэтинилпиридинам. В WO 02/46166 описаны различные соединения, имеющие структуру фенил-А-В, в качестве антагонистов mGluR, но в ней не раскрываются фенилэтинилпиридины. Уже известная структура в области лигандов mGluR5, подобная 2-метил-6-(фенилэтинил)пиридину (МРЕР), характеризуется низкой устойчивостью и селективностью (R. Kuhn et al. Amino Acids, 2002, 23, 207-211; N.D.P. Cosford et al. J. Med. Chem. 2003, 46, 204-206).
В патенте США № 6187777 описано соединение 3-амино-4-хлор-6-метил-2-(2-фенилэтинил)пиридин в качестве синтетического промежуточного соединения для получения соединений, регулирующих поведение, связанное с потреблением пищи.
В настоящее время неожиданно было обнаружено, что аминопиридиновые соединения по изобретению проявляют высокую активность и селективность в отношении рецептора mGluR5 и проявляют свойства, имеющие преимущества перед соединениями предшествующего уровня техники. Наблюдалось улучшение одной или нескольких из следующих характеристик соединений по изобретению: селективность в отношении мишени, растворимость, биологическая доступность, проникновение в мозг, активность на поведенческих моделях психиатрических и неврологических расстройств. Эти соединения можно применять при лечении или предотвращении расстройств, опосредованных mGluR5.
L-глутамат представляет собой основной возбуждающий нейромедиатор в мозге млекопитающих, и он действует через 2 гетерогенных семейства рецепторов: ионотропные и метаботропные рецепторы глутамата (mGluR) (Nakanishi S et al. 1998, Brain Res Rev., 26:230-235). К настоящему времени было клонировано 8 подтипов mGluR и классифицировано на 3 группы на основании аналогий последовательностей и фармакологических свойств. mGluR1 и mGluR5 относятся к группе I и инициируют клеточные реакции посредством механизма, опосредованного G-белком; они связаны с фосфолипазой С и стимулируют гидролиз фосфоинозитида (Schoepp DD et al. 1999, Neuropharmacology, 38:1431-1476).
Белок рецептора mGluR5 был локализован на периферии в структурах, участвующих в передаче болевого восприятия, и последние данные свидетельствуют о том, что антагонисты mGluR5 можно применять для лечения воспалительной и нейропатической боли, хронической и острой боли (B.A.Chizh in Amino Acids 2002, 23, 169-176).
Рецепторы mGluR5 также в большом количестве имеются в ЦНС, распределяясь по коре, гиппокампу, хвостатому ядру-скорлупе и ядру n.accumbens. Поскольку считается, что эти области мозга связаны с эмоциональными и мотивационными процессами, рецептор mGluR5 считался потенциальной лекарственной мишенью для лечения психиатрических и неврологических расстройств. Излечимые заболевания представляют собой психоз, эпилепсию, шизофрению, болезнь Альцгеймера, когнитивные расстройства, дефициты памяти, болезнь Паркинсона, гипоксию, ишемию, деменцию, вызванную СПИДом, мигрень, депрессию, расстройства настроения и тревожные расстройства. Другие излечимые показания представляют собой злоупотребление никотином, кокаином, амфетамином, бензодиазепином, опиатом или алкоголем и устойчивостью к веществам или зависимостью от них, невротическую булимию, невротическую анорексию, тягу к азартным играм, курение, сексуальную зависимость или навязчиво-компульсивными расстройствами (Brauner-Osborne H et al., 2000, J Med Chem. 43:2609-45; Bordi F and Ugolini A. 1999, Prog Neurobiol. 59:55-79; Spooren W et al. 2003, Behav Pharmacol: 14:257-77).
Настоящее изобретение относится к способу лечения или предотвращения состояния у млекопитающего, включая человека, лечение или предотвращение которого находится под воздействием или облегчается нейромодулирующим эффектом антагонистов mGluR5.
Настоящее изобретение относится к новым аминопиридиновым производным и к способам их применения; эти соединения соответствуют общей формуле
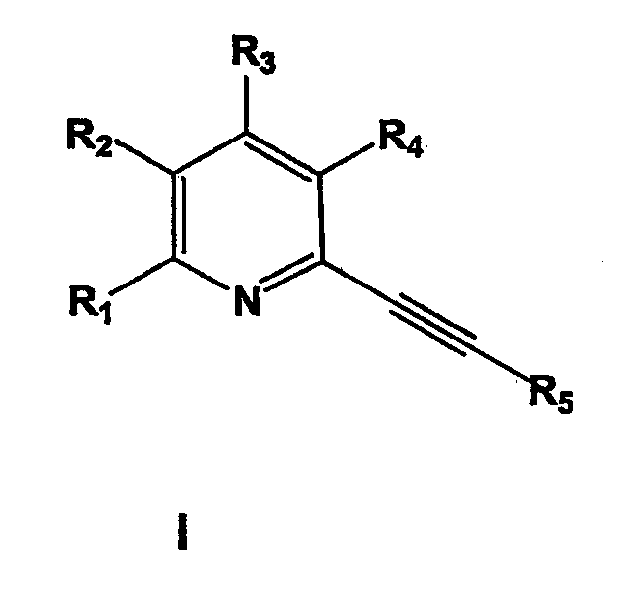
где
R1 представляет собой метил,
R2 и R3 независимо выбраны из водорода, галогена, нитро, С1-С6-алкила;
R4 представляет собой 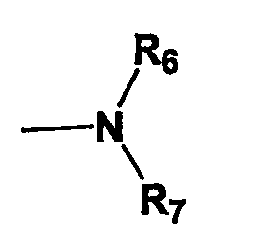 ,
,
R6 и R7, которые могут быть одинаковыми или различными, представляют собой водород или группу формулы: -X-R8, где Х представляет собой =CH-N(R8)2 и R8 представляет собой водород, С1-С6-алкил, галоген-С1-С6-алкил, арил, арил-С1-С6-алкил, гетероарил или гетероарил-С1-С6-алкил;
R5 представляет группу формулы
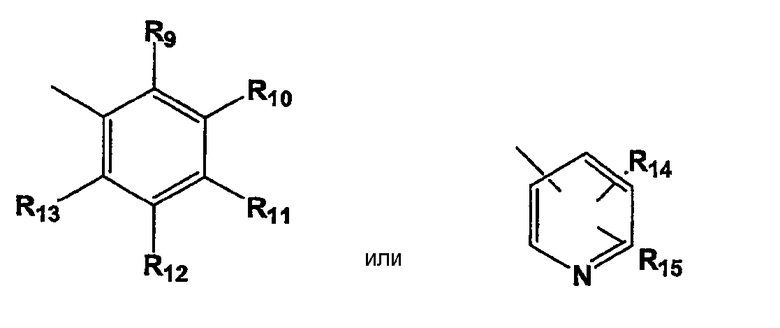 ,
,
где R9, R10, R11, R12 и R13 независимо представляют собой водород, галоген, циано, нитро, С1-С6-алкил, галоген-С1-С6-алкил, С1-С6-алкокси, карбокси-С1-С6-алкил или карбоксиарил;
R14 и R15 независимо имеют значения, определенные выше для R9-R13;
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений для предотвращения или лечения расстройств центральной нервной системы (ЦНС), а также других расстройств, модулируемых рецепторами mGluR5.
Соединение 3-амино-4-хлор-6-метил-2-(2-фенилэтинил)пиридин как таковое исключается из изобретения.
В указанном выше определении термин «С1-С6-алкил» включает такие группы, как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, трет-пентил, гексил или им подобные. «Гидрокси-С1-С6-алкил» включает такие группы, как гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил, 4-гидроксибутил, 5-гидроксипентил, 6-гидроксигексил и им подобные. «С1-С6-алкокси» включает такие группы, как метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, пентилокси, гексилокси или им подобные.
«Галоген» включает такие атомы, как фтор, хлор и йод. «Галоген-С1-С6-алкил» включает такие группы, как хлорметил, дихлорметил, трихлорметил, фторметил, дифторметил, трифторметил, бромметил, 1-хлорэтил, 1,1-дихлорэтил, 2-хлорэтил, 2,2,2-трихлорэтил, 1-фторэтил, 1,1-дифторэтил, 2-фторэтил, 2,2,2-трифторэтил, 2-бромэтил, 3-хлорпропил, 3-фторпропил, 3-бромпропил и им подобные. «Галоген-С1-С6-алкокси» включает такие группы, как хлорметокси, фторметокси, дифторметокси, трифторметокси, 2-фторэтокси, 2,2,2-трифторэтокси и им подобные.
«Карбокси-С1-С6-алкил» включает такие группы, как карбоксиметил, 2-карбоксиэтил, 3-карбоксипропил, 4-карбоксибутил, 5-карбоксипентил, 6-карбоксигексил или им подобные.
«Арил» включает С6-С10арильные группы, такие как фенил, 1-нафтил, 2-нафтил и им подобные.
«Гетероарил» включает 5-10-членные гетероциклические группы, содержащие от 1 до 4 гетероатомов, выбранных из кислорода, азота или серы, для образования такого кольца как фурил (фурановое кольцо), бензофуранил (бензофуран), тиенил (тиофен), бензотиофенил (бензотиофен), пирролил (пиррольное кольцо), имидазолил (имидазольное кольцо), пиразолил (пиразольное кольцо), тиазолил (тиазольное кольцо), изотиазолил (изотиазольное кольцо), триазолил (триазольное кольцо), тетразолил (тетразольное кольцо), пиридил (пиридиновое кольцо), пиразинил (пиразиновое кольцо), пиримидинил (пиримидиновое кольцо), пиридазинил (пиридазиновое кольцо), индолил (индольное кольцо), изоиндолил (изоиндольное кольцо), бензоимидазолил (бензимидазольное кольцо), пуринильная группа (пуриновое кольцо), хинолил (хинолиновое кольцо), фталазинил (фталазиновое кольцо), нафтиридинил (нафтиридиновое кольцо), хиноксалинил (хиноксалиновое кольцо), циннолил (циннолиновое кольцо), птеридинил (птеридиновое кольцо), оксазолил (оксазольное кольцо), изоксазолил (изоксазольное кольцо), бензоксазолил (бензоксазольное кольцо), фуразанил (фуразановое кольцо) и им подобные.
«Гетероарил-С1-С6-алкил» включает группы, где примерами гетероарила являются такие же, как проиллюстрированные в указанном выше определении, например, 2-фурилметил, 3-фурилметил, 2-тиенилметил, 3-тиенилметил, 2-имидазолилметил, 2-тиазолилметил, 2-пиридилметил, 3-пиридилметил, 4-пиридилметил, 2-хинолилметил и им подобные.
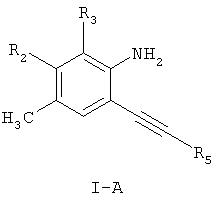
Предпочтительными соединениями настоящего изобретения являются соединения формулы I-A

где
R2 и R3 независимо выбраны из водорода, С1-С6-алкила;
R5 представляет группу формулы
где
R9, R10, R11, R12 и R13 независимо представляют собой водород, галоген, циано, нитро, С1-С6-алкил, галоген-С1-С6-алкил, С1-С6-алкокси, карбокси-С1-С6-алкил или карбоксиарил;
R14 и R15 независимо имеют значения, определенные выше для R9-R13;
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений.
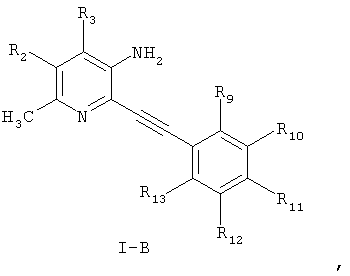
Более предпочтительными соединениями настоящего изобретения являются соединения формулы I-В
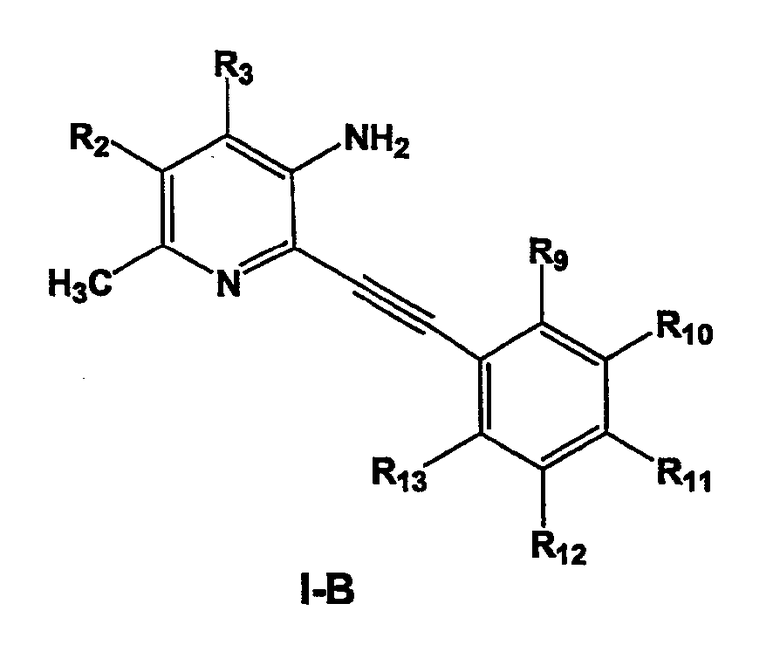
где
R2 и R3 независимо выбраны из водорода, С1-С6-алкила;
R9, R10, R11, R12 и R13 независимо представляют собой водород, галоген, циано, нитро, С1-С6-алкил, галоген-С1-С6-алкил, С1-С6-алкокси, карбокси-С1-С6-алкил или карбоксиарил;
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений.
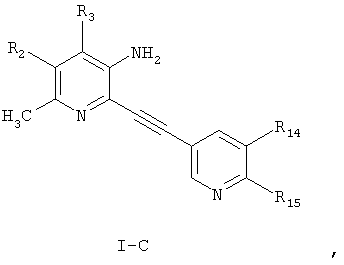
Особенно предпочтительными соединениями настоящего изобретения являются соединения формулы I-С
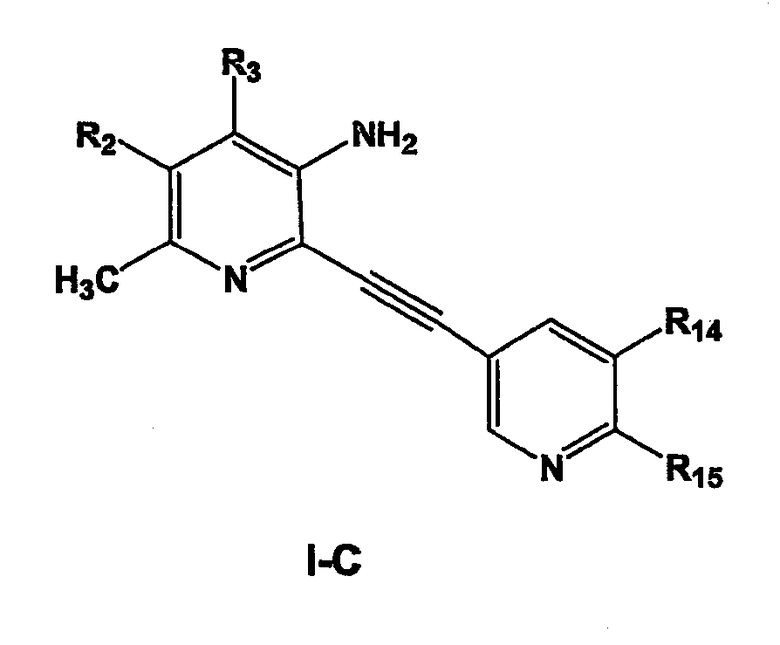
где
R2 и R3 независимо выбраны из водорода, С1-С6-алкила;
R14 и R15 независимо представляют собой водород, галоген, циано, нитро, С1-С6-алкил, галоген-С1-С6-алкил, С1-С6-алкокси, карбокси-С1-С6-алкил или карбоксиарил;
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений.
Конкретно предпочтительными соединениями являются
(6-метил-2-фенилэтинилпиридин-3-ил)амин
N,N-диметил-N'-(6-метил-2-фенилэтинилпиридин-3-ил)формамидин
(2-(3-фторфенилэтинил)-6-метилпиридин-3-ил)амин
(2-(3-метоксифенилэтинил)-6-метилпиридин-3-ил)амин
(6-метил-2-пиридин-3-илэтинилпиридин-3-ил)амин
(2-(4-фторфенилэтинил)-6-метилпиридин-3-ил)амин
(2-(3,5-дифторфенилэтинил)-6-метилпиридин-3-ил)амин
(2-(5-фторпиридин-3-илэтинил)-6-метилпиридин-3-ил)амин
3-(3-амино-6-метилпиридин-2-илэтинил)бензонитрил
(2-(5-хлорпиридин-3-илэтинил)-6-метилпиридин-3-ил)амин
(2-(3-хлорфенилэтинил)-6-метилпиридин-3-ил)амин
(2-(3-фторфенилэтинил)-4,6-диметилпиридин-3-ил)амин
(2-(3-хлорфенилэтинил)-4,6-диметилпиридин-3-ил)амин
Настоящее изобретение относится к фармацевтически приемлемым кислотно-аддитивным солям соединений формулы (I) или к композициям, включающим соединения формулы (I), вместе с фармацевтически приемлемыми носителями или эксципиентами.
Настоящее изобретение относится к способу лечения или предотвращения состояния у млекопитающего, включая человека, лечение или предотвращение которого находится под воздействием или облегчается нейромодулирующим эффектом антагонистов mGluR5 рецепторов.
Настоящее изобретение относится к способу, который можно использовать для лечения или предотвращения расстройств периферической и центральной нервной системы, выбранных из устойчивости к веществам или зависимости от них, тревожных расстройств, депрессии, расстройств настроения, психиатрических заболеваний, таких как психоз, воспалительной или нейропатической боли, дефицитов памяти, болезни Альцгеймера, болезни Паркинсона, мигрени, ишемии, злоупотребления наркотиками и наркомании.
Настоящее изобретение относится к фармацевтическим композициям, которые обеспечивают примерно от 0,01 до 1000 мг активного ингредиента на одну стандартную дозу. Композиции можно вводить любым подходящим путем. Например, перорально в форме капсул и т.д., парентерально в форме растворов для инъекций, местно в форме мазей или лосьонов, в глаза в форме глазных капель, ректально в форме суппозиторий, интраназально или через кожу в форме системы доставки, подобной накладке.
Фармацевтические композиции по изобретению можно получить обычными способами, принятыми в данной области; природа используемой фармацевтической композиции будет зависеть от желательного пути введения. Общая суточная доза обычно находится в диапазоне примерно от 0,05 до 2000 мг.
Изобретение также предоставляет применение соединений или композиций, как определено выше, при изготовлении лекарственных средств для лечения или предотвращения указанных расстройств.
Соединения формулы I можно получить обычными путями синтеза, как раскрыто в следующих способах.
Схема 1 иллюстрирует получение соединений формулы I взаимодействием алкинового производного, например этинилбензола, с замещенным аминопиридином (или его предшественником), например 2-бром-6-метилпиридин-3-иламином. Так, в схеме 1 R1, R2, R3, R5, R6 и R7 представляют собой определенные выше радикалы, а Q включает такие галогениды, как Cl, Br, I или трифторметансульфонил и паратолуолсульфонил. Общий путь синтеза был описан в публикации M.Н.Norman et al. J. Med. Chem. 2000, 43, 4288-4312.
Схема 1
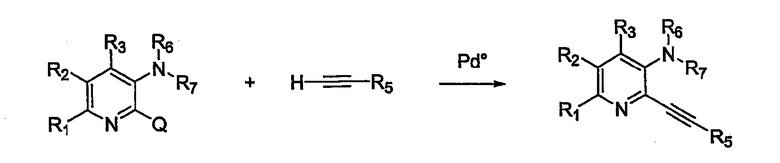
Эта катализируемая палладием реакция требует применения такого катализатора, как PdCl2(PPh3)2, Pd(PPh3)2, Pd(OAc)2 или Pd на угле, в подходящем растворителе, подобном DMF (диметилформамиду), ацетонитрилу или бензолу. Обычно в реакционной смеси также должны присутствовать дополнительный катализатор, такой как йодид меди (I), и основание (например, триэтиламин, диизопропиламин, KOAc...). Реакция соединения обычно происходит в условиях медленного повышения температуры взаимодействия от примерно 0°С до окружающей температуры или при повышении температуры нагреванием примерно от 30°С до 150°С. Затем реакционную смесь поддерживают при подходящей температуре в течение периода времени в диапазоне примерно от 1 до 24 ч, причем обычно достаточно примерно 12 ч. Продукт реакции можно выделить и очистить, используя стандартные методики, такие как экстракция растворителя, хроматография, кристаллизация, дистилляция, сублимация и им подобные.
В другом варианте осуществления настоящего изобретения, изображенном на схеме 2, замещенный алкинилом аминопиридин (или его предшественник) взаимодействует с соединением, несущим реактивную функциональную группу Q, как определено выше.
Схема 2
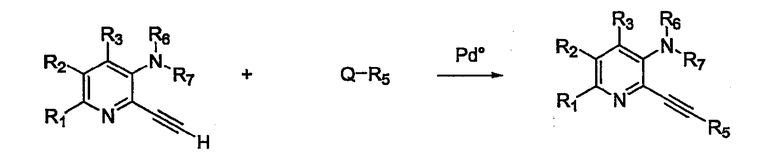
Так, в схеме 2 R1, R2, R3, R5, R6, R7, Q, катализаторы и условия взаимодействия такие же, как описано для схемы 1.
Другой вариант осуществления настоящего изобретения показан на схеме 3. Замещенный аминопиридин (или его предшественник) взаимодействует с алкеновым производным, как описано в публикации C. Niu et al. Tetrahedron, 1998, 54, 6311-6318, таким же образом, как в процедуре, представленной на схеме 1.
Схема 3
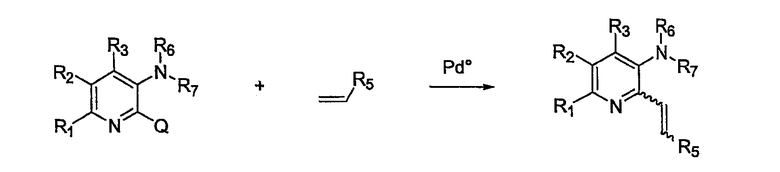
Продукт в виде алкенового производного из схемы 3 может быть превращен в алкиновое производное с использованием подхода, показанного на схеме 4.
Схема 4
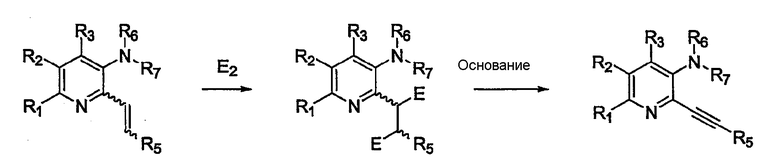
Этот путь синтеза относится к способам, описанным G.R. Newkome et al. J. Org. Chem. 1980, 45, 4380-4385 F. Gasparini et al. Bioorg. Med. Chem. Lett. 2002, 12, 407-410. Алкеновые производные можно обработать галогенирующим агентом, таким как хлор или бром в CHCl3 или CCl4. Затем галогенированные производные обрабатывают подходящим основанием, таким как NaOH, KOH или KOtBu, которые способствуют двойной реакции элиминирования для получения алкина. Реакция проводится в растворителе, подобном этанолу, трет-бутанолу, ТГФ и т.д., при соответствующей температуре, обычно от 0°С до 150°С.
В другом варианте осуществления настоящего изобретения замещенный аминопиридин (или его предшественник) взаимодействует с альдегидом для обеспечения замещенного алкена в соответствии с процедурой, разработанной в публикации D.Guay et al. Bioorg. Med. Chem. Lett. 1998, 8, 453-458 (Схема 5).
Схема 5
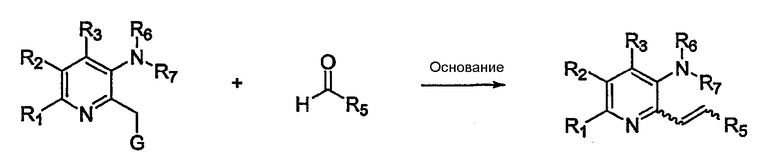
Таким образом, на схеме 5 G представляет собой PR3 или P(O)(OR)2. Реакцию проводят с подходящим катализатором, включая основания, такие как KH, NaH, н-бутиллитий и т.д., в ТГФ, ацетонитриле, бензоле и т.д., при соответствующей температуре, обычно от 0°С до 150°С.
В еще одном варианте осуществления настоящего изобретения замещенный гетероциклический альдегид взаимодействует с соединением, содержащим активированный метилен, с получением замещенного алкена, следуя процедуре, разработанной в публикации M. Cushman et al. J. Med. Chem. 1991, 34, 2579-2588 (Схема 6).
Схема 6
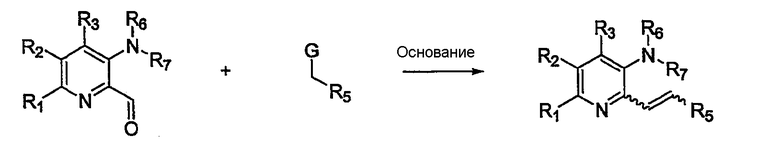
Таким образом, на схеме 6 G, катализатор и условия реакции такие, как описано для схемы 5.
Алкеновые продукты реакций, показанных на схеме 5 и схеме 6, могут быть превращены в алкиновое производное с использованием таких реагентов и условий, как описано для схемы 4.
Замещенные аминопиридины формулы I-A можно получить из соответствующих нитропиридиновых соединений избирательным восстановлением нитро составляющей части способом, описанным в публикации S. Glase et al. J. Med. Chem. 1996, 39, 3179-3187, с использованием смеси Fe и водной HCl в качестве восстанавливающего агента.
Реакции, описанные в схемах 1-6, могут привести к получению свободных амино соединений, когда R6 и R7 представляют собой водород. Последующую функционализацию в амидиновые соединения изобретения можно выполнить в соответствии со стандартными способами, знакомыми специалистам в области превращения свободных амино производных.
Соединения формулы I, которые являются основными по природе, могут образовывать широкое разнообразие различных фармацевтически приемлемых солей с различными неорганическими и органическим кислотами. Эти соли легко получаются обработкой основных соединений по существу эквивалентным количеством выбранной неорганической или органической кислоты в подходящем органическом растворителе, таком как метанол, этанол или изопропанол.
Фармакология
Некоторые из соединений формулы I были испытаны в соответствии со следующими способами.
Анализ связывания mGluR5
Аффинность соединений изобретения исследовали, следуя методике связывания радиоактивного лиганда, с использованием целого мозга крыс и насыщенного тритием 2-метил-6-(фенилэтинил)пиридина ([3H]-MPEP) в качестве лиганда, следуя способам, аналогичным способам, описанным в публикациях F. Gasparini et al. Bioorg. Med. Chem. Lett. 2002, 12, 407-409 и J.F. Anderson et al. J. Pharmacol. Exp. Ther. 2002, 303, 3, 1044-1051.
Получение мембран:
Кору отделяли от мозга крыс линии Sprague-Dawley массой 200-300 г (Charles River Laboratories, L'Arbresle, France). Ткани гомогенизировали в 10 объемах (об./мас.) 50 мМ ледяного Hepes-NaOH (рН 7,4) с использованием устройства для размельчения Polytron (Kinematica AG, Luzern, Switzerland) и центрифугировали в течение 30 мин при 40000 g (4°С). Надосадочную жидкость выливали и осадок после центрифугирования промывали дважды ресуспендированием в 10 объемах 50 мМ Hepes-NaOH. Затем мембраны собирали центрифугированием и промывали перед окончательным ресуспендированием в 10 объемах 20 мМ Hepes-NaOH, рН 7,4. Концентрацию белка определяли способом Bradford (Bio-Rad protein assay, Reinach, Switzerland) с альбумином бычьей сыворотки в качестве стандарта.
Эксперименты связывания [3H]-MPEP:
Мембраны оттаивали и ресуспендировали в связывающем буфере, содержащем 20 мМ Hepes-NaOH, 3 мМ MgCl2, 100 мМ NaCl, рН 7,4. Конкурентные исследования проводили инкубацией в течение 1 ч при 4°С: 3 нМ [3H]-MPEP (46,85 Ки/ммоль, Tocris, Cookson Ltd, Bristol, U.K.), 50 мкг мембран и диапазоне концентраций соединений 0,03 нМ - 30 мкМ, для общего объема взаимодействия 300 мкл. Неспецифическое связывание определяли с использованием 30 мкМ МРЕР. Реакцию прекращали быстрой фильтрацией через стекловолоконные фильтровальные пластины (96-луночные фильтровальные планшеты Unifilter GF/B, Perkin-Elmer, Schwerzenbach, Switzerland) с использованием 4×400 мкл ледяного буфера с использованием сборщика клеток (Filtermate, Perkin-Elmer, Downers Grove, USA). Радиоактивность определяли жидкостной сцинтилляционной спектрометрией с использованием считывающего устройства 96-луночных планшет (TopCount, Perkin-Elmer, Downers Grove, USA).
Анализ данных:
Строили кривые ингибирования с использованием программы Prism GraphPad (Graph Pad Software Inc, San Diego, USA). Определение IC50 проводили по данным, полученным по 8-точечным кривым реакции концентрации с использованием нелинейного регрессионного анализа.
Соединения настоящей заявки, по данным измерения в анализе, описанном выше, имеют величины IC50 в диапазоне менее чем 10 мкМ. Предпочтительные соединения включают примеры n° 3, 7, 10, 12 и 13, которые имеют величины IC50 менее чем 30 нМ.
Профиль селективности in vitro
Соединения изобретения проявляют улучшенную селективность в отношении рецептора mGluR5. Это указывает на большую специфичность и лучший профиль безопасности.
In vivo
Соединения изобретения эффективны на моделях, демонстрирующих полезность соединений для лечения нейропатической воспалительной боли ((B.A. Chizh, Amino Acids 2002, 23, 169-176), тревоги (W.P.J.M. Spooren et al. J. Pharmacol. Exp. Ther. 2000, 295, 3, 1267-1275; W.P.J.M. Spooren et al. Eur. J. Pharmacol. 2002, 435, 161-170), болезни Паркинсона (N. Breysse et al. J. Neurisci. 2003, 10, 23, 23, 8302-8309), мигрени (P. De Vries et al. 1999, 375, 61-74), депрессии (I.A. Paul and P. Skolnick, Ann. N Y Acad. Sci. 2003, 1003, 250-72) и расстройств зависимости (N.E. Paterson et al. Psychopharmacology 2003, 167, 257-264; C. Chiamulera et al. Nature Neurosci. 2001, 4, 9, 873-874).
Соединения настоящего изобретения проявляют высокую селективность и аффинность к рецептору mGluR5. В качестве функциональных антагонистов их можно использовать для производства лекарственных средств, в частности для лечения или предотвращения расстройств центральной нервной системы, а также других расстройств, опосредуемых этим рецептором.
Целесообразные изменения не следует рассматривать как отход от объема притязаний изобретения. Должно быть очевидно, что изобретение, описанное представленным выше описанием, может разнообразно изменяться специалистами в данной области.
Следующие неограничивающие примеры предназначены для иллюстрации изобретения. Физические данные, приведенные для проиллюстрированных соединений, согласуются с представленной структурой этих соединений.
ПРИМЕРЫ
При отсутствии других указаний все исходные материалы были получены от торговых поставщиков и использованы без дальнейшей очистки.
В частности, следующие аббревиатуры могут использоваться в примерах и по всему описанию.
Диметилсульфоксид)
хроматография
масс спектроскопия)
резонанс)
бис(трифенилфосфин)палладия(II)
При отсутствии других замечаний все реакции проводятся в инертной атмосфере при комнатной температуре.
Спектры 1H ЯМР регистрировали на Bruker ARX400 или на Bruker 500MHz. Химические сдвиги выражены в миллионных долях (м.д. δ единиц). Константы взаимодействия представлены в единицах герц (Гц). Схемы расщепления описывают видимые мультиплеты и обозначены как с (синглет), д (дублет), т (триплет), кв (квартет) и м (мультиплет).
Спектры ЖХ-МС регистрировали на устройстве Waters Micromass ZQ 2996 следующими условиями: Колонка 3,0*50 мм из нержавеющей стали с 5 мкм XTerra RP C-18; скорость потока 0,8 мл/мин; подвижная фаза: фаза А = 0,07% муравьиная кислота в воде, фаза В = 0,07% муравьиная кислота в ацетонитриле, 0-0,5 мин (А: 95%, В: 5%), 0,5-6,0 мин (А: 0%, В: 100%), 6,0-6,5 мин (А: 95%, В: 5%), 6,5-7 мин (А: 95%, В: 5%); УФ-детекция Серия диодов: 200-400 нм; Объем инжекции: 5 мкл. Все масс спектры определяли в условиях способов электрораспылительной ионизации (ESI).
Определение точки плавления проводили на приборе Buchi B-540.
Реакции контролировали тонкослойной хроматографией на силикагелевых пластинках толщиной 0,20 мм (60F254, Merck или G/UV254 Macherey Nagel) и визуализировали УФ-светом. Колоночную флэш-хроматографию выполняли на силикагеле (220-440 меш, Fluka).
Пример 1
Гидрохлорид (6-метил-2-фенилэтинилпиридин-3-ил)амина
К раствору CuI (10 мг, 50 мкмоль) в триэтиламине (5 мл) добавляют (2-бром-6-метилпиридин-3-ил)амин (200 мг, 1,07 ммоль) и (PPh3)2PdCl2 (36 мг, 50 мкмоль). Реакционную смесь охлаждают до 0°С и добавляют фенилацетилен (176 мкл, 1,60 ммоль). Реакционной смеси дают возможность нагреться до комнатной температуры и затем нагревают с обратным холодильником в течение 14 ч. Растворитель выпаривают и неочищенный остаток очищают флэш-хроматографией (гексан/этилацетат 4:1) с получением 105 мг (0,50 ммоль, 47%) (6-метил-2-фенилэтинилпиридин-3-ил)амина в виде желтого твердого вещества.
Rf: 0,09 (Гексан/этилацетат 4:1). Т.пл.: 154-155°С. (1H ЯМР (CDCl3, 400 МГц) δ: 2,47 (с, 3H), 4,13-4,17 (шир.с, 2H), 6,93-7,01 (2H), 7,34-7,39 (3H), 7,57-7,63 (2H).
(6-метил-2-фенилэтинилпиридин-3-ил)амин (105 мг, 0,50 ммоль) растворяют в CHCl3 (2 мл) и обрабатывают 1,56 мл (1,25 ммоль) 0,8 М растворе хлористоводородной кислоты в диэтиловом эфире. После выпаривания растворителя и растирания остатка с этилацетатом получают 107 мг (0,44 ммоль, 87%) указанного в заголовке гидрохлорида в виде желтоватого твердого вещества.
Rf: 0,50 (Гексан/этилацетат 1:1). Т.пл.: 183°С. 1H ЯМР (ДМСО[D6], 400 МГц) δ: 2,57 (с, 3H), 3,20-4,00 (шир.с, 3H), 7,51-7,57 (4H), 7,71 (д, J=8,8 Гц, 1H), 7,80-7,84 (2H), ЖХ-МС (RT): 2,14 мин; МС (ES+) дала m/z: 209,1.
Пример 2
N,N-диметил-N'-(6-метил-2-фенилэтинилпиридин-3-ил)формамидин
Раствор N,N-диметилформамиддиметилацеталя (80 мкл, 0,60 ммоль) и (6-метил-2-фенилэтинилпиридин-3-ил)амина (102 мг, 0,49 ммоль) из примера 1 в толуоле (1 мл) нагревают в течение 20 ч при 80°С. Растворитель выпаривают и остаток очищают флэш-хроматографией (гексан/этилацетат 4:1) для получения 24 мг (0,09 ммоль, 15%) N,N-диметил-N'-(6-метил-2-фенилэтинилпиридин-3-ил)формамидина в виде желтого масла.
Rf: 0,17 (Гексан/этилацетат 4:1). 1H ЯМР (ДМСО[D6], 400 МГц) δ: 2,42 (с, 3H), 3,05 (с, 3H), 3,07 (с, 3H), 7,14 (д, J=8,8 Гц, 1H), 7,32 (д, J=8,8 Гц, 1H), 7,44-7,58 (4H), 7,87 (с, 1H), 7,93 (д, J=7,2 Гц, 1H), ЖХ-МС (RT): 0,61 мин; МС (ES+) дала m/z: 264,1.
Пример 3
Гидрохлорид (2-(3-фторфенилэтинил)-6-метилпиридин-3-ил)амина
Следуя процедуре, описанной в примере 1, обеспечивают взаимодействие (2-бром-6-метилпиридин-3-ил)амина (200 мг, 1,07 ммоль) с (PPh3)2PdCl2 (36 мг, 0,05 ммоль), CuI (10 мг, 0,05 ммоль) и 1-этинил-3-фторбензолом (148 мкл, 1,28 ммоль) в триэтиламине (5 мл). Неочищенный остаток очищают флэш-хроматографией (гексан/этилацетат 4:1) для получения 145 мг (0,64 ммоль, 60%) (2-(3-фторфенилэтинил)-6-метилпиридин-3-ил)амина в виде бледно-желтого твердого вещества. Гидрохлорид (2-(3-фторфенилэтинил)-6-метилпиридин-3-ил)амина получают, как описано в примере 1, для выхода 148 мг (0,56 ммоль, 88%) указанного в заголовке гидрохлорида в виде желтого твердого вещества.
Rf: 0,52 (Гексан/этилацетат 1:1). Т.пл.: 199-200°С. 1H ЯМР (ДМСО[D6], 400 МГц) δ: 2,52 (с, 3H), 6,20-7,24 (шир.с, 2H), 7,35-7,43 (м, 1H), 7,48 (д, J=8,0 Гц, 1H), 7,52-7,61 (2H), 7,62-7,67 (м, 1H), 7,68-7,72 (м, 1H), ЖХ-МС (RT): 2,26 мин; МС (ES+) дала m/z: 227,1, Анализ., Рассчитано для C14H12ClFN2: C, 64,01%; H, 4,60%; Cl, 13,50%; F, 7,23%; N, 10,66%, Найдено: C, 63,30%; H, 4,62%; Cl, 13,56%; F, 6,98%; N, 10,57%.
Пример 4
(2-(3-метоксифенилэтинил)-6-метилпиридин-3-ил)амин
Следуя процедуре, описанной в примере 1, обеспечивают взаимодействие (2-бром-6-метилпиридин-3-ил)амина (200 мг, 1,07 ммоль) с (PPh3)2PdCl2 (37 мг, 0,05 ммоль), CuI (10 мг, 0,05 ммоль) и 1-этинил-3-метоксибензолом (204 мкл, 1,60 ммоль) в триэтиламине (5 мл) в течение 1,5 ч. Неочищенный остаток очищают флэш-хроматографией (гексан/этилацетат 4:1) для получения 118 мг (0,50 ммоль, 46%) (2-(3-метоксифенилэтинил)-6-метилпиридин-3-ил)амина в виде желто-коричневого твердого вещества.
Rf: 0,30 (Гексан/этилацетат 1:1). Т.пл.: 165-166°С. 1H ЯМР (ДМСО[D6], 400 МГц) δ: 2,60 (с, 3H), 3,85 (с, 3H), 7,11-7,16 (м, 1H), 7,37-7,49 (3H), 7,56 (д, J=8,4 Гц, 1H), 7,77 (д, J=8,8 Гц, 1H), ЖХ-МС (RT): 2,31 мин; МС (ES+) дала m/z: 239,1.
Пример 5
Гидрохлорид (6-метил-2-пиридин-3-илэтинилпиридин-3-ил)амина
Следуя процедуре, описанной в примере 1, обеспечивают взаимодействие (2-бром-6-метилпиридин-3-ил)амина (200 мг, 1,07 ммоль) с (PPh3)2PdCl2 (37 мг, 0,05 ммоль), CuI (10 мг, 0,05 ммоль) и 3-этинилпиридином (110 мг, 1,07 ммоль) в триэтиламине (1,6 мл). Неочищенный остаток очищают флэш-хроматографией (DCM-DCM/MeOH 97:3) для получения 100 мг (0,48 ммоль, 49%) (6-метил-2-пиридин-3-илэтинилпиридин-3-ил)амина в виде желтого порошка. Гидрохлорид (6-метил-2-пиридин-3-илэтинилпиридин-3-ил)амина получают, как описано в примере 1, для выхода после растирания с пентаном 145 мг (100%) указанного в заголовке гидрохлорида в виде желтого твердого вещества.
Т.пл.: 156,4-158°С. 1H ЯМР (ДМСО[D6], 500 МГц) δ: 2,54 (с, 3H), 7,52 (д, J=9 Гц, 1H), 7,61-7,68 (м, 1H), 7,69 (д, J=9,0, 1H), 8,25 (д, J=7,5, 1H), 8,68-8,86 (шир.с, 1H), 8,93-9,15 (шир.с, 1H), ЖХ-МС (RT): 0,65 мин; МС (ES+) дала m/z: 210,1.
Пример 6
Гидрохлорид (2-(4-фторфенилэтинил)-6-метилпиридин-3-ил)амина
Следуя процедуре, описанной в примере 1, обеспечивают взаимодействие (2-бром-6-метилпиридин-3-ил)амина (200 мг, 1,07 ммоль) с (PPh3)2PdCl2 (36 мг, 0,05 ммоль), CuI (10 мг, 0,05 ммоль) и 1-этинил-4-фторбензолом (184 мкл, 1,07 ммоль) в триэтиламине (5 мл). Неочищенный остаток очищают флэш-хроматографией (гексан/этилацетат 7:3) для получения 164 мг (0,72 ммоль, 68%) (2-(4-фторфенилэтинил)-6-метилпиридин-3-ил)амина в виде желтого твердого вещества. Гидрохлорид (2-(4-фторфенилэтинил)-6-метилпиридин-3-ил)амина получают, как описано в примере 1, для выхода после растирания с этилацетатом 154 мг (0,51 ммоль, 71%) указанного в заголовке гидрохлорида в виде желтого твердого вещества.
Rf: 0,43 (Гексан/этилацетат 1:1). Т.пл.: 122°С (разл.). 1H ЯМР (ДМСО[D6], 400 МГц) δ: 2,53 (с, 3H), 4,45-5,54 (шир.с, 2H), 7,36-7,43 (2H), 7,52 (д, J=8,8 Гц, 1H), 7,67 (д, J=8,8 Гц, 1H), 7,82-7,89 (2H), ЖХ-МС (при комнатной температуре): 2,24 мин; МС (ES+) дала m/z: 227,1.
Пример 7
Гидрохлорид (2-(3,5-дифторфенилэтинил)-6-метилпиридин-3-ил)амина
Следуя процедуре, описанной в примере 1, обеспечивают взаимодействие (2-бром-6-метилпиридин-3-ил)амина (800 мг, 4,28 ммоль) с (PPh3)2PdCl2 (150 мг, 0,21 ммоль), CuI (41 мг, 0,21 ммоль) и этинилтриметилсиланом (840 мг, 8,55 ммоль) в триэтиламине (30 мл). Неочищенный остаток очищают флэш-хроматографией (гексан/этилацетат 8:2) для получения 330 мг (1,61 ммоль, 38%) (6-метил-2-триметилсиланилэтинилпиридин-3-ил)амина в виде бежевого твердого вещества.
(6-метил-2-триметилсиланилэтинилпиридин-3-ил)амин (330 мг, 1,61 ммоль) растворяют в MeOH (3 мл) и охлаждают до 0°С, к полученному раствору добавляют 1 М раствор NaOH (1,6 мл). Ледяную баню удаляют и реакционную смесь перемешивают при комнатной температуре в течение 4 ч. Добавляют 90 мкл уксусной кислоты. Реакционную смесь частично концентрируют и остаток экстрагируют этилацетатом. Органические слои промывают водой, рассолом, сушат над Na2SO4, фильтруют и концентрируют для получения 160 мг (1,21 ммоль, 75%) (2-этинил-6-метилпиридин-3-ил)амина в виде коричневого твердого вещества, которое используют на следующей стадии без дальнейшей очистки.
К раствору CuI (4,3 мг, 23 мкмоль) в триэтиламине (5 мл) добавляют (2-этинил-6-метилпиридин-3-ил)амин (60 мг, 0,45 ммоль), (PPh3)2PdCl2 (16 мг, 23 мколь) и 1,3-дифтор-5-йодбензол (109 мг, 0,45 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 20 ч. Растворитель выпаривают для получения коричневого масла, которое собирают в DCM (дихлорметан), и раствор промывают водой. Водную фазу экстрагируют дважды DCM. Органические фазы сушат над Na2SO4, фильтруют и концентрируют. Неочищенный остаток очищают флэш-хроматографией (гексан/этилацетат 7:3) для получения 64 мг (0,26 ммоль, 58%) (2-(3,5-дифторфенилэтинил)-6-метилпиридин-3-ил)амина в виде желтого твердого вещества.
Гидрохлорид (2-(3,5-дифторфенилэтинил)-6-метилпиридин-3-ил)амина получают, как описано в примере 1, для выхода после растирания с диэтиловым эфиром 64 мг (0,20 ммоль, 78%) указанного в заголовке гидрохлорида в виде желтого порошка.
Rf: 0,61 (Гексан/этилацетат 1:1). Т.пл.: 212°С (разл.). 1H ЯМР (ДМСО[D6], 400 МГц) δ: 2,49 (с, 3H), 6,12-7,08 (шир.с, 2H), 7,44-7,55 (2H), 7,56-7,68 (3H), ЖХ-МС (RT): 2,39 мин; МС (ES+) дала m/z: 245,0.
Пример 8
Гидрохлорид (2-(5-фторпиридин-3-илэтинил)-6-метилпиридин-3-ил)амина
К раствору CuI (11 мг, 0,06 ммоль) в триэтиламине (5 мл) добавляют (2-этинил-6-метилпиридин-3-ил)амин (40 мг, 0,30 ммоль, как описано в примере 7), (PPh3)2PdCl2 (21 мг, 0,06 ммоль) и 3-фтор-5-йодпиридин (111 мг, 0,45 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 14 ч. Растворитель выпаривают. Неочищенный остаток очищают флэш-хроматографией (циклогексан/этилацетат 7:3) для получения 15 мг (66 мкмоль, 22%) (2-(5-фторпиридин-3-илэтинил)-6-метилпиридин-3-ил)амина в виде желтого твердого вещества.
Гидрохлорид (2-(5-фторпиридин-3-илэтинил)-6-метилпиридин-3-ил)амина получают, как описано в примере 1, для выхода после растирания с этилацетатом 6 мг (20 мкмоль, 30%) указанного в заголовке гидрохлорида в виде коричневого полутвердого вещества.
Rf: 0,31 (циклогексан/этилацетат 7:3). ЖХ-МС (при комнатной температуре): 1,71 мин; МС (ES+) дала m/z: 228,0.
Пример 9
Гидрохлорид (3-(3-амино-6-метилпиридин-2-илэтинил)бензонитрила
К раствору CuI (2,4 мг, 12 мкмоль) в триэтиламине (4 мл) добавляют (2-этинил-6-метилпиридин-3-ил)амин (33 мг, 0,30 ммоль, как описано в примере 7), (PPh3)2PdCl2 (8,8 мг, 12 мкмоль) и 3-йодбензонитрил (57 мг, 0,25 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 48 ч. Растворитель выпаривают и неочищенный остаток очищают флэш-хроматографией (циклогексан/этилацетат 1:1) для получения 16 мг (69 мкмоль, 21%) 3-(3-амино-6-метилпиридин-2-илэтинил)бензонитрила в виде бледно-желтого твердого вещества.
Гидрохлорид 3-(3-амино-6-метилпиридин-2-илэтинил)бензонитрила получают, как описано в примере 1, для выхода после растирания с диэтиловым эфиром 10 мг (33 мкмоль, 47%) указанного в заголовке гидрохлорида в виде желтого порошка.
Rf: 0,36 (гексан/этилацетат 1:1). Т.пл.: 132,4-134°С. ЖХ-МС (при комнатной температуре): 2,13 мин; МС (ES+) дала m/z: 234,1.
Пример 10
Гидрохлорид (2-(5-хлорпиридин-3-илэтинил)-6-метилпиридин-3-ил)амина
Следуя той же процедуре, как описано в примере 1, обеспечивают взаимодействие (2-бром-6-метилпиридин-3-ил)амина (150 мг, 0,80 ммоль) с (PPh3)2PdCl2 (28 мг, 40 мкмоль), CuI (8 мг, 40 мкмоль) и 3-хлор-5-этинилпиридином (165 мг, 1,20 ммоль) в триэтиламине (5 мл). Неочищенный остаток очищают флэш-хроматографией (гексан/этилацетат 7:3) для получения 141 мг (0,58 ммоль, 72%) (2-(5-хлорпиридин-3-илэтинил)-6-метилпиридин-3-ил)амина в виде желтого твердого вещества.
Гидрохлорид (2-(5-хлорпиридин-3-илэтинил)-6-метилпиридин-3-ил)амина получают, как описано в примере 1, для выхода 152 мг (0,48 ммоль, 83%) указанного в заголовке гидрохлорида в виде желтого твердого вещества.
Rf: 0,12 (гексан/этилацетат 1:1). Т.пл.: 193°С (разл.). 1H ЯМР (ДМСО[D6], 400 МГц) δ: 2,59 (с, 3H), 5,81-7,41 (шир.с, 2H), 7,58 (д, J=8,8 Гц, 1H), 7,73 (д, J=8,8 Гц, 1H), 8,43-8,44 (м, 1H), 8,79 (д, J=1,6 Гц, 1H), 8,89 (д, J=2,0 Гц, 1H), ЖХ-МС (RT): 2,03 мин; МС (ES+) дала m/z: 244,0.
Пример 11
Гидрохлорид (2-(3-хлорфенилэтинил)-6-метилпиридин-3-ил)амина
К раствору CuI (5,0 мг, 28 мкмоль) в триэтиламине (5 мл) добавляют (2-этинил-6-метилпиридин-3-ил)амин (75 мг, 0,57 ммоль, как описано в примере 7), (PPh3)2PdCl2 (20 мг, 28 мкмоль) и 1-хлор-3-йодбензол (135 мг, 0,57 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 4 ч. Растворитель выпаривают для получения коричневого масла, которое собирают в DCM, и раствор промывают водой. Водную фазу экстрагируют дважды DCM. Органические фазы сушат над Na2SO4, фильтруют и концентрируют. Неочищенный остаток очищают флэш-хроматографией (гексан/этилацетат 7:3) для получения 54 мг (0,22 ммоль, 39%) (2-(3-хлорфенилэтинил)-6-метилпиридин-3-ил)амина в виде желтого твердого вещества.
Гидрохлорид (2-(3-хлорфенилэтинил)-6-метилпиридин-3-ил)амина получают, как описано в примере 1, для выхода после растирания с диэтиловым эфиром 51 мг (0,16 ммоль, 73%) указанного в заголовке гидрохлорида в виде желтого порошка.
Rf: 0,57 (гексан/этилацетат 1:1). Т.пл.: 195°С (разл.). 1H ЯМР (ДМСО[D6], 400 МГц) δ: 2,51 (с, 3H), 6,43-7,11 (шир.с, 2H), 7,50 (д, J=8,4 Гц, 1H), 7,52-7,58 (м, 1H), 7,59-7,69 (2H), 7,72 (д, J=8,0 Гц, 1H), 7,97 (с, 1H), ЖХ-МС (RT): 2,54 мин; МС (ES+) дала m/z: 243,0, Анализ. Рассчитано для C14H12Cl2N2+ 0,5H2O: C, 58,35%; H, 4,55%; Cl, 24,61%; N, 9,72%, Найдено: C, 58,15%; H, 4,49%; Cl, 24,60%; N, 9,48%.
Пример 12
Гидрохлорид (2-(3-фторфенилэтинил)-4,6-диметилпиридин-3-ил)амина
К раствору 1,80 г (11,0 ммоль) 2-хлор-4,6-диметилпиридин-3-иламина (полученного, как описано в публикации J.M. Klunder et al. J. Med. Chem., 35, 1992, 1887-1897) в толуоле (10 мл) добавляют PBr3 (18 мл). Реакционную смесь перемешивают в течение 48 ч при кипячении с обратным холодильником. После охлаждения реакционной смеси ее выливают в лед, подщелачивают 2 М раствором NaOH (400 мл) и водную фазу экстрагируют дважды этилацетатом. Объединенные органические фазы промывают рассолом, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный остаток очищают флэш-хроматографией (циклогексан/этилацетат 7:3) для получения 2,31 г (38%) 2-бром-4,6-диметилпиридин-3-иламина, содержащего небольшое количество 2-хлор-4,6-диметилпиридин-3-иламина в виде желтого масла.
К раствору CuI (41 мг, 0,2 ммоль) в триэтиламине (12 мл) добавляют 2-бром-4,6-диметилпиридин-3-иламин (870 мг, 4,33 ммоль, (PPh3)2PdCl2 (152 мг, 0,22 ммоль) и 1-этинил-3-фторбензол (500 мкл, 4,33 ммоль). Реакционную смесь перемешивают в течение 30 мин при комнатной температуре и в течение 3 ч при кипячении с обратным холодильником. Растворитель выпаривают и неочищенный остаток очищают флэш-хроматографией (циклогексан/этилацетат 4:1) для получения 745 мг (3,10 ммоль, 72%) (2-(3-фторфенилэтинил)-4,6-диметилпиридин-3-ил)амина в виде желтого твердого вещества.
Гидрохлорид (2-(3-фторфенилэтинил)-4,6-диметилпиридин-3-ил)амина получают, как описано в примере 1, для выхода после растирания с пентаном 680 мг (2,46 ммоль, 79%) указанного в заголовке гидрохлорида в виде желтого порошка.
Rf: 0,37 (циклогексан/этилацетат 7:3). Т.пл.: 210°С. 1H ЯМР (ДМСО[D6], 500 МГц) δ: 2,33 (с, 3H), 2,51 (с, 3H), 6,31-6,72 (шир.с, 3H), 7,36-7,42 (м, 1H), 7,47 (с, 1H), 7,53-7,59 (м, 1H), 7,60-7,63 (м, 1H), 7,70-7,74 (м, 1H), ЖХ-МС (при комнатной температуре): 2,29 мин; МС (ES+) дала m/z: 241,1, Анализ. Рассчитано для C15H14ClFN2: C, 65,10%; H, 5,10%; Cl, 12,81%; F, 6,87%; N, 10,12%, Найдено: C, 64,73%; H, 4,97%; Cl, 12,78%; F, 6,71%; N 9,87%.
Пример 13
Гидрохлорид (2-(3-хлорфенилэтинил)-4,6-диметилпиридин-3-ил)амина
К раствору CuI (5,7 мг, 30 мкмоль) в триэтиламине (1,6 мл) добавляют 2-бром-4,6-диметилпиридин-3-иламина (120 мг, 0,60 ммоль, описанные в примере 12), (PPh3)2PdCl2 (21 мг, 30 мкмоль) и 1-этинил-3-хлорбензол (98 мг, 0,72 ммоль). Реакционную смесь перемешивают в течение 30 мин при комнатной температуре и в течение 3 ч при кипячении с обратным холодильником. Растворитель выпаривают и неочищенный остаток очищают флэш-хроматографией (циклогексан/этилацетат 4:1) для получения 39 мг (0,15 ммоль, 25%) (2-(3-хлорфенилэтинил)-4,6-диметилпиридин-3-ил)амина в виде коричневого масла.
Гидрохлорид (2-(3-хлорфенилэтинил)-4,6-диметилпиридин-3-ил)амина получают, как описано в примере 1, для выхода после растирания с пентаном 25 мг (85 мкмоль, 57%) указанного в заголовке гидрохлорида в виде желтого порошка.
Rf: 0,37 (циклогексан/этилацетат 7:3). Т.пл.: 204°С. ЖХ-МС (при комнатной температуре): 2,56 мин; МС (ES+) дала m/z: 257,0.
Типичными примерами рецептур для композиции изобретения являются следующие:
1) Таблетки
В этом примере соединение из примера 3 можно заменить таким же количеством соединения из любого из описанных примеров 1-13.
2) Суспензия
Водную суспензию готовят для перорального введения так, что каждый 1 мл содержит от 1 до 5 мг одного из описанных примеров, 50 мг карбоксиметилцеллюлозы натрия, 1 мг бензоата натрия, 500 мг сорбита и воды до 1 мл.
3) Раствор для инъекций
Парентеральную композицию получают перемешиванием 1,5 мас.% активного ингредиента изобретения в 10 об.% пропиленгликоля и воды.
4) Мазь
В этом примере соединение из примера 3 можно заменить таким же количеством соединения из любого из описанных примеров 1-13.
Целесообразные изменения не следует рассматривать как отход от объема притязаний изобретения. Должно быть очевидно, что специалисты в данной области могут различным образом изменить описанное выше изобретение.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ АНТАГОНИСТОВ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2327697C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ, ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2004 |
|
RU2345074C2 |
| ФЕНИЛЭТЕНИЛ- ИЛИ ФЕНИЛЭТИНИЛПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ГЛУТАМАТНОГО РЕЦЕПТОРА | 2001 |
|
RU2284323C9 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2016 |
|
RU2740019C1 |
| МОДУЛЯТОРЫ МЕТАБОТРОПНОГО ГЛУТАМАТНОГО РЕЦЕПТОРА ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2008 |
|
RU2508107C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА III | 2004 |
|
RU2360911C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2629118C2 |
| АЦЕТИЛЕНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ (MGLUR5) | 2002 |
|
RU2341515C2 |
| 5-АРОМАТИЧЕСКОЕ АЛКИНИЛЗАМЕЩЕННОЕ БЕНЗАМИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2695371C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
Изобретение относится к новым аминопиридиновым производным формулы (I-A)
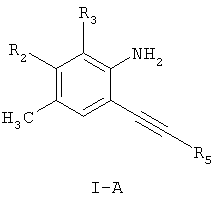
где R2 и R3 независимо выбраны из водорода, C1-С6-алкила; R5 представляет группу формулы 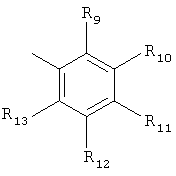 или
или 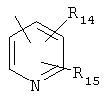 где R9, R10, R11, R12 и R13 независимо представляют собой водород, галоген, циано, С1-С6-алкил, галоген-С1-С6-алкил или С1-С6-алкокси; R14 и R15 независимо имеют значения, определенные выше для R9-R13, к фармацевтической композиции на основе данных соединений, обладающей антагонистической активностью в отношении mGluR5 рецепторов. Также изобретение относится к способу лечения расстройств центральной нервной системы, а также других расстройств, модулируемых рецепторами mGluR5, и к применению соединений формулы I для изготовления лекарственного средства. Технический результат: получение новых соединений и фармацевтической композиции на их основе, в целях лечения расстройств центральной нервной системы. 4 н. и 9 з.п. ф-лы.
где R9, R10, R11, R12 и R13 независимо представляют собой водород, галоген, циано, С1-С6-алкил, галоген-С1-С6-алкил или С1-С6-алкокси; R14 и R15 независимо имеют значения, определенные выше для R9-R13, к фармацевтической композиции на основе данных соединений, обладающей антагонистической активностью в отношении mGluR5 рецепторов. Также изобретение относится к способу лечения расстройств центральной нервной системы, а также других расстройств, модулируемых рецепторами mGluR5, и к применению соединений формулы I для изготовления лекарственного средства. Технический результат: получение новых соединений и фармацевтической композиции на их основе, в целях лечения расстройств центральной нервной системы. 4 н. и 9 з.п. ф-лы.
 ,
,
где R2 и R3 независимо выбраны из водорода, С1-С6-алкила;
R5 представляет группу формулы
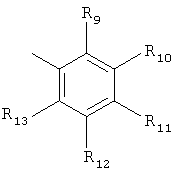 или
или 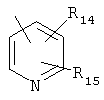 ,
,
где R9, R10, R11, R12 и R13 независимо представляют собой водород, галоген, циано, С1-С6-алкил, галоген-С1-С6-алкил или C1-С6-алкокси; R14 и R15 независимо имеют значения, определенные выше для R9-R13,
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений.
где R2 и R3 независимо выбраны из водорода, С1-С6-алкила;
R9, R10, R11, R12 и R13 независимо представляют собой водород, галоген, циано, С1-С6-алкил, галоген-С1-С6-алкил или С1-С6-алкокси,
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений.
где R2 и R3 независимо выбраны из водорода, С1-С6-алкила;
R14 и Rl5 независимо представляют собой водород, галоген, циано, C1-C6-алкил, галоген-С1-С6-алкил или С1-С6-алкокси,
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений.
(6-метил-2-фенилэтинилпиридин-3-ил)амина;
(2-(3-фторфенилэтинил)-6-метилпиридин-3-ил)амина;
(2-(3-метоксифенилэтинил)-6-метилпиридин-3-ил)амина;
(6-метил-2-пиридин-3-илэтинилпиридин-3-ил)амина;
(2-(4-фторфенилэтинил)-6-метилпиридин-3-ил)амина;
(2-(3,5-дифторфенилэтинил)-6-метилпиридин-3-ил)амина;
(2-(5-фторпиридин-3-илэтинил)-6-метилпиридин-3-ил)амина;
3-(3-амино-6-метилпиридин-2-илэтинил)бензонитрила;
(2-(5-хлорпиридин-3-илэтинил)-6-метилпиридин-3-ил)амина;
(2-(3-хлорфенилэтинил)-6-метилпиридин-3-ил)амина;
(2-(3-фторфенилэтинил)-4,6-диметилпиридин-3-ил)амина;
(2-(3-хлорфенилэтинил)-4,6-диметилпиридин-3-ил)амина
и их фармацевтически приемлемых солей.
| US 6187777 В1, 13.02.2001 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Способ получения производных пиридина | 1985 |
|
SU1424731A3 |

