Изобретение относится к области органической химии и фармацевтики, а именно к гетеробициклическим соединениям и фармацевтическим композициям на их основе, а также способам получения этих соединений.
Известно использование флавоксата, представляющего собой 8-(2-пиперридиноэтоксикарбонил)-3-метил-4-оксо-2-фенил-4-1-бензпиран, в качестве фармацевтического средства при лечении мочевых путей, поскольку он обладает релаксационной активностью гладких мышц из-за активности антагониста кальция. Эта активность оказывает влияние на гладкие мышцы свода мочевого пузыря, а также центра мочеиспускания центральной нервной системы (ЦНС).
Соединения, характеризуемые настоящим изобретением, содержат более сложные аминовые фрагменты вместо пиперидиновой группы. Кроме того, они содержат этоксикарбоновую группу, расположенную в пространстве между аминовым фрагментом бензопиринового кольца и другими группами, стоящими в 2-, 3- и 7-замещениями в бензопириновом кольце, замещение гетероатома кольца на атом серы или сульфинил, сульфонил или иминогруппу, и/или 2,3-дигидрирования бензопиринового кольца. Эти структурные изменения придают новым соединениям способность взаимодействовать с различными биологическими системами.
Соединения, характеризующие настоящее изобретение, имеют общую формулу I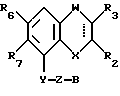
в которой  представляет собой ординарную или двойную связь,
представляет собой ординарную или двойную связь,
X - атом серы или кислорода;
W - валентная связь или карбонил-, метиленовая или гидроксиметиленовая группа;
R2 - атом водорода, C1 - C6-алкил, незамещенный или замещенный фенилом C1 - C6-алкенил, C3 - C6-карбоцикл, пиридил, фурил, тиенил, бензоил, фенил, незамещенный или замещенный феноксигруппой, фенилом, трифторметилом, C1 - C6-алкокси-, гидрокси-, амино-, C1 - C6-ациламино- или нитрогруппой;
R3 - атом водорода или галогена, C1 - C6-алкил или гидрокси-C1 - C6-алкил;
R6 - атом водорода или галогена, нитро-, амино-, C1 - C6-алкиламино-, ди(C1 - C6)алкиламино-, C1 - C6-ациламино, C1 - C6-алкилсульфониламино-, гидрокси-, C1 - C6-алкоксигруппа или C1 - C6-алкил,
R7 - атом водорода или C1 - C6-алкоксигруппа,
Y - имеет значение:
Y1 - -CO-,
Y2 - -COO-,
Y3 - -CONH-,
Y4 - -CON(CH3)-,
Y5 - -COH(OH)-,
Y6 - -CH(OH)-,
Y7 - -CH(OAlkyl)-,
Y8 - -CH=CH-,
Y10 - CH=CH-CONH-,
Y11 - CN=NO-,
Y12 - CH2-,
Y13 - CH2COO-,
Y14 - CH2CONH-,
Y15 - CH2NH-,
Y16 - CH2N(CH3)-,
Y17 - CH2N(COCH3)-,
Y18 - CH2N(CONH2)-,
Y19 - CH2NHCO-,
Y20 - CH2N(CH3)CO-,
Y21 - CH2NH-CONH-,
Y22 - CH2NHSO2-,
Y23 - CH2O-,
Y24 - CH2S-,
Y25 - CH2SO-,
Y26 - CH2SO2-,
Y27 - CH2SO2NH-,
Y28 - CH2SO2N(CH3)-,
Y29 - NH-,
Y30 - N(CH3)-,
Y32 - N(CONH2)-,
Y33 - NHCO-,
Y34 - N(CH3)CO-,
Y35 - NH-CO-NH-,
Y37 - -O-,
Y42 - -SO2N(CH3)-,
Y45 - -CSNH-,
Y47 -
Z - линейная или разветвленная C1 - C6-алкиленовая группа;
B представляет собой одну из следующих групп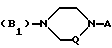
в которой Q представляет собой метиленовую группу, а A представляет собой одну из следующих групп
A1 - фенил, замещенный одним или двумя атомами галогена и/или одной или двумя C1 - C6-алкил-, C1 - C6-алкокси- или гидроксигруппой,
A2 2-пиримидинильная группа
(B2)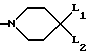
в которой каждый из L1 и L2 представляет независимо друг от друга атом водорода, фенил-, 4-фторбензол- или 2-оксо-1-бензимидазолинильную группу при условии, что L1 и L2 одновременно не являются атомами водорода,
(B3)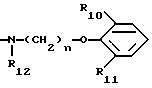
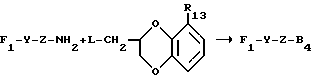
в котором каждый из R10 и R11 независимо представляет собой атом водорода или C1 - C6-алкокси-;
R12 представляет собой атом водорода или C1 - C6-алкильную группу;
n = 2 или 3,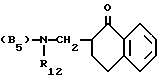
в которой R12 определено выше.
В случае, когда 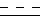 представляет собой двойную связь, W представляет собой карбонильную группу, а X представляет собой иминогруппу, кольцо может претерпевать таутомеризацию, давая в результате структуру 4-гидрокси-хинолина.
представляет собой двойную связь, W представляет собой карбонильную группу, а X представляет собой иминогруппу, кольцо может претерпевать таутомеризацию, давая в результате структуру 4-гидрокси-хинолина.
Изобретение относится к энантомерам, диастереомерам, N - оксидам, фармацевтически приемлемым солям, а также соединениям формулы I, в которых амино- и/или имино-, и/или гидроксигруппы защищены. Соединения формулы I, в которых амино- и/или имино-, и/или гидроксигруппы защищены, могут быть использованы в качестве лекарственных препаратов пролонгированного действия.
Группа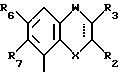
в дальнейшем будет обозначена как F1. Предпочтительны для этой группы следующие значения заместителей двойная связь, X - атом кислорода, W - карбонильная группа, R2 - фенильная группа, R3 - метильная группа, R6 - атом водорода, R7 - атом водорода.
двойная связь, X - атом кислорода, W - карбонильная группа, R2 - фенильная группа, R3 - метильная группа, R6 - атом водорода, R7 - атом водорода.
Группа, содержащая все эти предпочтительные заместители, является 3-метил-4-оксо-2-фенил-4H-1-бензопиран-8-ил-группой.
Z предпочтительно представляет собой триметиленовую или тетраметиленовую группу. Y предпочтительно представляет собой одну из групп B1 или B3 предпочтительно, 1-(2-метоксифенил)-пиперазинилгруппу. В тексте использована общепринятая аббревиатура: Me - метил, Et - этил, Ac - ацетил, Alk - алкил, THF - тетрагидрофуран, DMF - диметилформамид и DMSO - диметилсульфоксид.
Соединения, охарактеризованные настоящим изобретением, могут быть в основном синтезированы (за исключением случаев, когда R6 и заместители при R2 представляют собой OH или аминоалкил, Y15 и Y29) следующим образом.
а) конденсацией исходных соединений с получением соединения F1-Y-Z-B6 с отщеплением тозилоксигруппы. Предпочтительно проводить реакцию при 20 - 140oC в полярном растворителе (диметилформамид, метанол) в присутствии карбоната кальция.
б) альтернативным методом получения соединений формулы I является конденсация (при тех же условиях) соединения F1-Y-H с соединением L-Z-B, где L представляет собой атом галогена или отщепляемую группу,
в) соединения формулы I, имеющие группы NH2 в R6 или в R2, могут быть получены восстановлением соответствующих соединений формулы I, в которых указанные радикалы в качестве заместителей содержат нитрогруппы. Такое восстановление можно провести следующими способами:
используя в качестве катализатора никель Ренея в протонном растворителе (метанол, этанол, изопропанол, вода или их смесь),
используя водный раствор хлорида двухвалентного олова при необязательном присутствии соляной кислоты с использованием протонного растворителя (метанол, этанол, изопропанол или их смеси с водой) или апротонного растворителя (этилацетат),
используя железо и водный раствор соляной кислоты в протонном растворителе (метанол, этанол, изопропанол или их смеси с водой).
Температуру вышеприведенных реакций следует выбирать в диапазоне 20 - 100oC.
г) соединения формулы I, имеющие NHAlk-группу в качестве заместителя при R6, можно получить при моноалкилировании соединений, схожих с соединениями формулы I, но имеющими в качестве заместителя аминогруппу. Например, это можно осуществить при реакции аминосоединения I с избытком трифторуксусного ангидрида с последующим взаимодействием трифторуксусного производного с L-алкильным реагентом с последующим снятием защиты с полученного таким образом трифторуксусного алкильного производного при обработке его карбонатом кальция в метаноле или борогидридом натрия в метаноле или диметилсульфоксиде,
д) соединения формулы I, содержащие группы NHAlk или N(Alk)2 в качестве заместителей при R6 или в качестве заместителя в фенильной группе при R2, могут быть получены алкилированием соответствующих соединений, в которых содержатся аминные заместители, соответствующими алканами в присутствии восстановителя, в частности цианоборгидрида натрия,
е) соединения, содержащие OH-группу в качестве R6 или в качестве заместителя в R2, могут быть получены при использовании в качестве исходных веществ соответствующих родственных соединений, содержащих алкоксизамещение в соответствующих положениях. В этом случае предпочтительно обрабатывать их трехбромистым бором в дихлорметане при 0 - 40oC,
ж) соединения общей формулы I, с одинарной связью можно получить или при селективном гидрировании соответствующих соединений с двойной связью, или превращением подходящих исходных, у которых связь 2,3- уже насыщена. Второй вариант предпочтителен особенно если указанное соединение содержит нитрогруппу, поскольку гидрирование способно превратить ее в аминогруппу. Селективное гидрирование может быть проведено или с использованием водорода в присутствии металлического катализатора или катализатора на основе оксида металла (палладий на угле или диоксид палладия) в протонном растворителе при 20 - 120oC, или диизобутилалюминийгидридом в апротонном растворителе (тетрагидрофуране и/или дихлорметане) при температуре от -70 до 0oC,
з) соединения, в котором W представляет собой гидроксиметиленовую группу и связь 2,3- насыщена, могут быть получены при восстановлении с использованием борогидрида натрия соответствующих соединений, в которых W представляет собой карбонильную группу и связь 2,3- насыщена.
В некоторых случаях соединения общей формулы I могут быть получены путем превращения родственных соединений. Такие превращения включают:
a) F1 - CO - Z - B в F1 - CH(OH) - Z - B при восстановлении,
b) F1 - CH(OH) - Z - B в F1 - CH(OAlkyl) - Z - B при этерификации,
c) F1 - (CH2)n-NH - Z - B в F1 - (CH2)n-N(CH3) - Z - B, где n = 0 или 1, при N - метилировании,
d) F1 - (CH2)n- NH - Z - B в F1 - (CH2)n - N(COCH3) - Z - B, где n = 0 или 1, при N - ацетилировании,
e) F1 - (CH2)n- NH - Z - B в F1 - (CH2)n - N(CONH2) - Z - B, где n = 0 или 1, при реакции с изоцианатом кальция,
f) F1 - CH(OHO) - Z - B в F1 - CO - Z - B при окислении,
g) F1 - Y - Z - B в F1 - Y - Z - B(N-оксид) при окислении,
h) H2N - F1 - Y - Z - B в CH3 - CONH - F1 - Y - Z - B (где H2N - F1 представляет собой группу F1, в которой R6 - аминогруппа или R2 включает в себя аминогруппу) с использованием метода N-ацилирования.
Предпочтительная методика синтеза зависит от синтезируемого вещества.
Исходные материалы, используемые для синтеза, коммерчески доступны или легко могут быть синтезированы из доступных материалов.
Адренергическая антагонистическая активность вышеуказанных соединений делает их полезными в качестве средств, воздействующих на ткани организма, такие как кровеносные сосуды, простата и уретата. Следовательно, антиадренергические соединения могут быть использованы в качестве терапевтических средств для лечения гипертонии и нарушения мочеиспускания, связанных с нарушением проходимости нижнего участка мочевых путей, включая те из них, которые вызваны доброкачественной гипертрофией простаты, но не ограничивается ей.
Серотоническая активность данных соединений делает их полезными в качестве средства воздействия на ткани центральной нервной системы. Эти соединения обладают биологической активностью в блокировании связей между рецепторами и их разнообразными специфическими лигандами. Они могут быть полезными терапевтическими средствами для лечения состояния депрессии и тревоги.
Удивительно, что соединения настоящего изобретения (особенно те из них, которые имеют сродство как к альфа1-адренергическому, так и к 5HТ1A-серотонинергическому рецептору) демонстрируют высокую селективность в нижней части мочевых путей млекопитающих, т.е. они значительно более активны при противодействии уретральных сокращений, чем при снижении кровяного давления. Например, известные альфа1-антагонисты, такие как празосин, который представляет собой 1-(4-амино-6,7-диметокси-2-хиназолинил)-4-(2-фуроил)-пиперазин (GB 1156973), не обладают такой селективностью (и в действительности вызывают гипотензию в качестве основного побочного действия), в то время как производные флавона, структурно аналогичные флавоксату, такие как тетрафлавоксат, который представляет собой 8-(1,1-диметил-2-пиперидино-этоксикарбонил)-3-метил-4-оксо-2-фенил- 4H-1-бензопиран гидрохлорид (EP 0072620), не имеют никакого влияния на уретральные сокращения. Естественно, те соединения настоящего изобретения, которые не являются селективными для нижней части мочевых путей, предпочтительны в качестве антигипертензивных средств, однако именно избирательные соединения благодаря своей низкой токсичности могут быть часто использованы в качестве антигипертензивных средств.
Соединения настоящего изобретения демонстрируют также хорошее антагонистическое влияние против сокращений мочевого пузыря крыс, которые были вызваны хлоридом кальция. Это влияние может быть приписано активности в качестве антагониста кальция и делает эти новые соединения полезными в качестве спазмолитиков нижнего участка мочеполового тракта (т.е. полезными для лечения недержания мочи, синдрома позыва к мочеиспусканию и других похожих нарушений).
Главная черта соединений настоящего изобретения состоит в их низкой токсичности. Поэтому их можно использовать в больших количествах; это преимущество часто более чем компенсирует относительно низкий уровень активности, который присущ некоторым из этих соединений. Естественно, предпочтительны те соединения, которые имеют как высокую активность, так и низкую токсичность.
Изобретение обеспечивает также фармацевтический состав, содержащий соединение согласно настоящему изобретению, или пролекарство, энантиомер, диастереоизомер, N-оксид или фармацевтически приемлемую соль такого соединения в смеси с фармацевтически приемлемым разбавителем или носителем.
Синтез соединений настоящего изобретения.
Соединения, согласно настоящему изобретению, могут быть в основном получены (за исключением тех случаев, когда группы R6 и заместители при R2 представляют собой OH или аминоалкил и Y - Y15 или Y29) следующим образом.
Путь а:
Конденсацией соединений F1-Y-Z-B, в которых L представляет собой атом галогена или отщепляемую группу, такую как тозилоксигруппа, с соединением H-B. Предпочтительно, но не обязательно проводить конденсацию при 20 - 140oC в полярном растворителе, таком как диметилформамид или метанол, обычно в присутствии основания типа карбоната кальция. Конденсации такого типа приведены ниже в примерах 1 - 3, 7 - 9, 11, 13 - 16, 21, 23 - 31, 38 - 42, 46 - 49, 54 - 59, 69, 73, 77, 78 и 84. (См. также Patai, The Chemistry of the Amino Group, p. 45 et seq., Wiley Interscience, New York, 1968).
Альтернативным методом получения соединений настоящего изобретения является конденсация (при тех же условиях, которые описаны в предыдущем параграфе) соединения F1-Y-H с соединением L-Z-B, в котором L было определено выше. Эта конденсация проиллюстрирована ниже примерами 5, 6, 66, 79 и 81. Согласно этому пути могут быть также получены соединения, имеющие Y - Y15 или Y29 (см. предыдущую библиографическую ссылку).
Соединения формулы I, имеющие группы NH2 в R6 или в качестве заместителя в R2, могут быть получены восстановлением соответствующих соединений I, в которых R6 или заместители в R2 представляют собой NH2-группы. Такое восстановление можно провести:
- используя катализатор никель Ренея в протонном растворителе, выбранном из метанола, этанола, изопропанола, воды или их смеси; или
- используя SnCl2, H2O при необязательном присутствии соляной кислоты, или в протонном растворителе, таком как метанол, этанол, изопропанол, вода, уксусная кислота и их смесь, или в апротонном растворителе, таком как этилацетат; или
- используя Fe и водную соляную кислоту в протонном растворителе, таком как метанол, этанол, изопропанол, вода и их смеси.
Температуры приведенных выше реакций следует выбирать в диапазоне 20 - 100oC (J. March, Advanced Organic Chemistry, III Ed., p. 1103, Wiley Interscience, 1985). Примеры этих реакций приведены в примерах изобретения 94 - 124.
Соединения формулы I, имеющие NHAlk-группу в качестве заместителя R6, можно получить при моноалкилировании, начиная с соответствующих родственных соединений I, в которых R6=NH2. Например, это можно осуществить при реакции аминосоединения I сначала с избытком трифторуксусного ангидрида, затем полученное таким образом трифторуксусное производное взаимодействует с L-алкил реагентом и, наконец, снятием защиты с полученного таким образом трифторуксусноалкилированного производного при обработке его карбонатом кальция в метаноле или при обработке борогидридом натрия в метаноле или диметилсульфоксиде. Эти реакции описаны в примерах 32 и 33, в которых они были проведены на Y-группах.
Или же соединения формулы I, содержащие группы NHAlk или N(Alk)2 в качестве заместителей R6 или как заместители фенильной группы в R2, могут быть получены алкилированием соответствующих родственных соединений I, в которых R6=NH2, подходящими алканами в присутствии восстановительного агента, такого как цианоборогидрид натрия. Описания этих реакций приведены ниже в примерах 96 и 97.
Соединения, имеющие OH-группу как R6 или как заместитель в R2, могут быть получены при использовании в качестве исходных веществ соответствующих родственных соединений I, алкоксизамещенных в указанных положениях. Это может быть завершено обработкой родственных соединений, например BBr3 в дихлорметане при 0 - 40oC (T.W. Greene, Protective Groups in Organic Synthesis, p. 87, Wiley Interscience, 1981) или согласно другим методам, описанным в этой же работе.
Соединения общей формулы I, в которых  представляет собой одинарную связь, могут быть получены или при селективном гидрировании соответствующих соединений, в которых
представляет собой одинарную связь, могут быть получены или при селективном гидрировании соответствующих соединений, в которых  представляет собой двойную связь, или превращением подходящих исходных, в которых связь 2,3- уже насыщена; такие исходные материалы могут быть применены в соответствии со схемами реакции 4, 6 - 9, 11, 12 и 14, приведенными ниже. Этот последний путь проиллюстрирован ниже в примере 87, он предпочтителен, особенно если указанное соединение содержит нитрогруппу, так как гидрирование способно превратить нитрогруппу и аминогруппу. Селективное гидрирование может быть проведено на выбор с использованием:
представляет собой двойную связь, или превращением подходящих исходных, в которых связь 2,3- уже насыщена; такие исходные материалы могут быть применены в соответствии со схемами реакции 4, 6 - 9, 11, 12 и 14, приведенными ниже. Этот последний путь проиллюстрирован ниже в примере 87, он предпочтителен, особенно если указанное соединение содержит нитрогруппу, так как гидрирование способно превратить нитрогруппу и аминогруппу. Селективное гидрирование может быть проведено на выбор с использованием:
- водорода в присутствии металлического катализатора или катализатора на основе оксида металла (например, палладий на угле или диоксид платины) в протонном растворителе при 20 - 120oC (E.H. Rodd, Chemistry of Carbob Campounds, vol. IVB, p. 903, Elsevier,. 1959); или
- ди(изобутил)алюминий гидрида в апротонном растворителе (например, тетрагидрофуране и/или дихлорметане) при температуре от -70 до 0oC (H. Sarges et al, J. Med. Chem., 33, 1859, 1990).
Соединения, в которых W представляет собой гидроксиметиленовую группу и связь 2,3- насыщена, могут быть получены при восстановлении с помощью борогидрида натрия соответствующих соединений, в которых W представляет собой карбонильную группу и связь 2,3- насыщена; об этом сообщается ниже в примере 123.
В некоторых случаях соединения общей формулы I могут быть получены путем превращения других (родственных) соединений настоящего изобретения. Такие превращения включают:
Путь b: F1-CO-Z-B ---> F1-CH(OH)-Z-B
при восстановлении, как это проиллюстрировано ниже в примерах 17-20.
Путь c: F1-CH(OH)-Z-B ---> F1-CH(OAlkyl)-Z-B
при этерификации, как это проиллюстрировано ниже в примере 22.
Путь d: F1-(CH2)n-NH-Z-B ---> F1(CH2)n-N(CH3)-Z-B,
где n = 0 или 1, при N-метилировании, как это проиллюстрировано ниже в примере 35.
Путь e: F1-(CH2)n-NH-Z-B ---> F1(CH2)n-N(COCH3)-Z-B,
где n = 0 или 1, при N-ацетилировании, как это проиллюстрировано ниже в примере 36.
Путь f: F1-(CH2)n-NH-Z-B ---> F1(CH2)n-N(CONH2)-Z-B,
где n = 0 или 1, при реакции с изоцианатом кальция, как это проиллюстрировано в примере 50 ниже.
Путь g: F1-CH(OH)-Z-B ---> F1-CO-Z-B
при окислении, как это проиллюстрировано ниже в примере 51.
Путь h: F1-Y-Z-B ---> F1-Y-Z-B (N-оксид)
при окислении, как это проиллюстрировано ниже в примерах 43 и 122.
Путь i: H2N-F1-Y-Z-B ---> CH3-CONH-F1-Y-Z-B
(где H2N-F1 представляют собой группу F1, в которой R6 - аминогруппа или R2 включает в себя аминогруппу) с использованием метода N-ацилирования, описанного ниже в примерах 36 и 95.
Путь j: F1 (R6=NH2)-Y-Z-B ---> F1 (R6=CH3SO2NH)-Y-Z-B
при амидировании с использованием метода, описанного ниже в примере 112.
Путь k: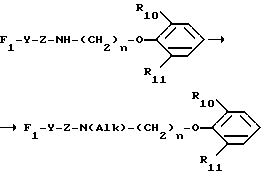
или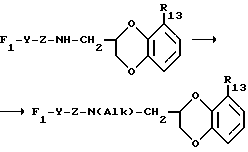
или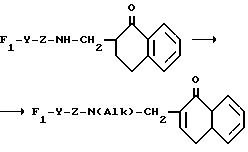
при N-алкилировании с использованием метода, описанного ниже в примерах 35 и 62.
Некоторые соединения могут быть получены по реакциям присоединения. Например, те соединения, в которых Z представляет собой гидроксизаместитель, могут быть получены при перекрестном присоединении эпоксигруппы.
Путь l: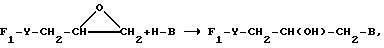
как это проиллюстрировано в примере 45.
Возможно также перекрестное присоединение двойной связи, например:
Путь m: F1-Y-CH=CH2 + H-B ---> F1-Y-CH2-CH2-B,
как это проиллюстрировано ниже в примерах 37, 63 и 82.
Другие схемы синтеза включают образование Y, Z или B в процессе реакции, например:
Путь n: F1-(X)-(Q)-Cl + A-HN-Z-B ---> F1(X)-(Q)-N(A)-Z-B,
где X - связь, CH2 или CH=CH, Q=CO или SO2, A=H, alkyl или OPr, где Pr - это защитная группа, как это проиллюстрировано в примере 12 (что особенно предпочтительно) и в примерах 60, 61, 64, 67, 68, 72, 87, 88, 93, 98, 116, 129, 130.
Эти же самые соединения могут быть получены другими путями, включая:
F1-(X)-COOH + A-NH-Z-B в присутствии отщепляющего агента (например, дициклогексилкарбодиимида, N, N'-карбонил-диимидазола или диэтилцианофосфоната) при необязательном присутствии промотирующего агента (например, 4-диметиламинопиридина или N-гидробензотриазола) в апротонном растворителе (например, в диметилформамиде, хлороформе) при -10/140oC (Albertson, Org. React., 12, 205-218, 1962; Doherty et al., J. Med. Chem., 35, 9, 1992; Staab et al., Newer Methods Prep. Org. Cytm., 5, 61, 1968; Ishihara, Chem. Pharm. Bull., 39, 3236, 1991), как это проиллюстрировано в примерах 80, 86, 90, 92, 99 - 111, 113 - 115, 117 - 119 и 128.
F1-(X)-COOH + A-NH-Z-B без использования растворителя при 150 - 220oC (Mitchell et al. , J. Am. Chem. Soc., 53, 1879, 1931) или в высококипящих эфирных растворителях (например, диглиме);
F1-(X)-COO-Alk + A-NH-Z-B при необязательном присутствии отщепляющего агента (например, триметилалюминия) в апротонном и/или хлорированном растворителе (например, гексане, дихлорметане) при -10/80oC, или без растворителей при 80 - 180oC (S.M. Weinreb et al., Tetrahedron, 4171, 1977; M.F. Lipton et al., Org. Synth., 59, 49, 1979);
F1-(X)-COOH + алкилхлороформиат в присутствии третичного амина (например, триэтиламина) с последующим добавлением A-NH-Z-B при 0 - 80oC; при необязательном добавлении промотирующего агента (например, 1-гидроксипиперидина) перед добавкой амина (Albertson, Org. React., 12, 157, 1962).
Путь o: F1-COCl + HS-Z-B ---> F1-Y49-Z-B.
Путь p: F1-COCl + HO-Z-B ---> F1-Y2-Z-B,
как это проиллюстрировано в примере 10.
Путь g: F1CHO + H2NO-Z-B ---> F1-Y11-Z-B,
как это проиллюстрировано ниже в примере 70.
Путь r: F1-CHO + A-HN-Z-B ---> F1-CS-N(A)-Z-B,
(где A=H или CH3) в присутствии серы в апротонном растворителе, например в DMF или в пиридине при 60-120oC (M. Сfrmack et al., Org. Reaction., 83, 1947; R. Benassi et al., Org. Magn. Res., 15,25, 1981), как это проиллюстрировано ниже в примере 83.
Путь s: F1-NH2 + HCO-Z-B ---> F1-Y29-Z-B,
как это проиллюстрировано ниже в примере 34.
Путь t: F1-Y-CH3 + HO-CH2-B ---> F1-Y-CH2-CH2-B,
как это проиллюстрировано ниже в примере 4.
Путь u: F1-CH=CH-CONH2 + HOCH2-B ---> F1-Y10-CH2-B.
Путь v: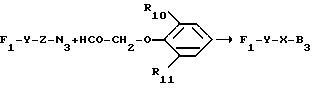
в условиях восстановления, как это проиллюстрировано ниже в примере 44.
Путь w: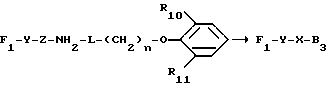 ,
,
как проиллюстрировано ниже в примерах 74 - 76.
 ,
,
как проиллюстрировано ниже в примере 52.
Путь x: ,
,
как проиллюстрировано ниже в примере 65.
Путь y: F1-Y-Z-CHO + HB ---> F1-Y-Z-B,
как проиллюстрировано ниже в примере 53.
Люди, квалифицированные в данной области, должны понять, что все названные выше пути синтеза от b) до y) должны быть упрощены с тем, чтобы вступающие в реакцию промежуточные соединения не имели бы в дальнейшем групп, чувствительных к тем же самым реагентам (например, CO-, NH2-, NHAlk- или OH-группы). Соединения формулы I, имеющие такие реактивные группы, могут быть получены путями синтеза от b) до y) при условии, что реактивные группы, присутствующие в исходных соединениях, предварительно были защищены, а затем, после реакции, эта защита была снята, как это проиллюстрировано в примере 71. Некоторые примеры защиты и ее снятия для разных реактивных групп можно найти в работе T.W. Green, Protective Groups in Organic Synthesis, Wiley Interscience, 1981.
Или же нереактивные группы (например, NO2) в процессе первой реакции могут не испытывать превращений, а затем превращены в реактивные группы (например, NH2) на заключительной стадии синтеза (См., например, путь a).
Предпочтительная методика синтеза будет зависеть от того, какое соединение требуется получить, но путь n) является в основном предпочтительным для тех соединений, которые можно получить с его помощью. Дополнительные методы синтеза являются очевидными для людей, квалифицированных в данной области.
Исходные материалы.
Исходные материалы (F1-Y-Z-L, F1-Y-H и другие), используемые при методах получения, описанных выше, могут быть сами получены из простых соединений, таких как F1-COOH, F1-CHO, F1 -COCl, F1-NH2 и F1OH-путем превращений, известных людям, квалифицированным в данной области. Многие из этих простых соединений (F1-COOH, F1-CHO, F1-COCl, F1-NH2 и F1-OH) доступны в коммерческом отношении или их синтез известен в литературе. Те соединения, которые не являются доступными, можно синтезировать согласно одной или более схемам реакций 1 - 16, приведенных ниже.
Схема 1 реакции (см. в конце описания) дает соединения, в которых W представляет собой карбонильную группу, а X представляет собой атом кислорода.
Стадия 1a. Метод без выделения промежуточного фенилового эфира:
- R3CH2COCl или (R3CH2CO)2O и кислота Льюиса (например, AlCl3 или ZnCl2), без растворителя или в апротонном растворителе (например, нитробензол или хлорированный растворитель) при 20 - 180oC.
Метод с выделением промежуточного фенилового эфира:
R3CH2COCl или (R3CH2CO)2O, нагретые с исходными материалами или другие методы этерификации, такие как метод Шоттена-Баумана. Выделенный эфир затем нагревают в нитробензоле или другом апротонном растворителе (например, хлорированном растворителе) или без всякого растворителя при 20 - 180oC в присутствии кислоты Льюиса типа AlCl3 или ZnCl2 (A.M. Blatt, Org. React., 1, 342, 1942).
Стадия 1b.
R2COCl или (R2CO)2O и R2COONa по отдельности или в высококипящем непротонном растворителе (таком как о-дихлорбензол) при 150 - 220oC; эта реакция также допускает прямое превращение соединений (2) в соединения (6), если соединения (2) имеют A=COOH;
R2C(OAlk)3 в присутствии HClO4 при 20 - 40oC или в пиридине в присутствии пиперидина при 60 - 80oC;
R2COCl или (R2CO)2O в хлорированном растворителе при температуре от -10 до 120oC в присутствии основания типа 1,8-диазобициклоундецен (DBU).
Стадия 1c.
R2COCl в пиридине при 20 - 100oC или в непротонном растворителе при 0 - 80oC, при необязательном присутствии основания типа NEt3 или 4-диметиламинопиридин.
Стадия 1d.
K2CO3 в ацетоне или метил-этил кетоне при 20 - 80oC;
NaH и DMSO или THF при 0 - 40oC;
KOH или t-бутоксид кальция в пиридине при 20 - 100oC.
Стадия 1e.
HCl или H2SO4 в AcOH при дефлегмации или в спирте (MeOH, EtOH, изопропанол) при температуре от 20oC до температуры дефлегмации.
Стадия 1f.
R2COCl и K2CO3 или KOH в воде и фазовый катализатор в бензоле или толуоле при дефлегмации;
R2COOAlk и бис(триметилсилил)амид лития или диизопропиламид лития THF при температуре от -78 до 0oC.
Стадия 1g.
Если A представляет собой COOCH3- или COOC2H5-группу:
NaOH в водной EtOH при 0 - 75oC;
LiOH в водной DMF, MeOH или THF или их смеси при 10 - 100oC;
HCl в апротонном растворителе типа диоксана при 60 - 120oC.
Если A представляет собой NO2:
восстановление с использованием катализатора никель Ренея в протонном растворителе (например, изопропанол) или в смеси протонных растворителей при 20 - 100oC;
восстановление с помощью водорода и катализатора (например, никель Ренея или Pd/C) в протонном растворителе (например, MeOH, EtOH, изопропанол или их смесь) при 20 - 100oC;
восстановление с помощью SnCl2 в присутствии водной HCl в протонном растворителе (например, AcOH) при 20 - 100oC;
восстановление в присутствии Fe и водной HCl в протонном растворителе при 20 - 100oC;
Если A представляет собой CH=CHCH3-группу:
окисление с помощью Na2Cr2O7 или других окисляющих агентов, таких как KMnO4 в ацетоне/H2SO4 при 0 - 100oC.
Схема 2 реакции (см. в конце описания) приводит к соединениям, в которых X представляет собой атом серы или сульфинил- или сульфонилгруппу, а W представляет собой карбонильную группу. Исходные о-меркаптобензоаты (1) доступны в коммерческом отношении или их можно получить известными способами, например превращением соответствующих о-алкилкарбонил-бензолдиазониевых солей при их обработке этилксантатом калия (M.S. Cohen et al., J. Org. Chem., 18, 1394, 1953).
Стадия 2a.
R2COCH(R3)CN или R2COCH(R3)COOAlk в полифосфорной кислоте при 50 - 120oC;
R2C ≡ C-COOAlk и Al2O3 в апротонном растворителе (например, EtOH) при 0 - 40oC;
R2C≡COOAlk и основание в апротонном растворителе (например, THF или DMF) при 20 - 140oC.
Последние оба варианта предусматривают последующую обработку полифосфорной кислотой при 50 - 120oC.
Стадия 2b.
NaOH в водной EtOH при 40 - 75oC;
LiOH в водной DMF при 40 - 100oC.
Стадия 2c.
Стехиометрическое количество 30% H2O2 в AcOH при 25 -60oC;
Стадия 2d.
30% H2O2 в AcOH при 50 - 80oC.
Схема 3 реакций приводит к соединениям (2) в которых R7 представляет собой метоксигруппу, W представляет собой карбонильную группу, а X - атом серы или кислорода. Соединения (1) могут быть получены согласно схемам 1 и 2 реакций, используя в качестве исходных соответствующие фенолы или тиофенолы (не замещенные в положении 2 или 6 на COOAlk или NO2).
Схема 3.

Стадия 3a.
HCHO и газообразный HCl в AcOH, содержащей водную HCl (d = 1.18) при 50 - 100oC (P. Da Re et al., Ann. Chim., 46, 904, 1956). Этот метод можно использовать, если R3 отличен от H или CH2OH.
Простые промежуточные 2,3-дигидросоединения ( представляет собой одинарную связь) могут быть получены согласно схеме 4 (см в конце описания) реакций при условии, что другие реактивные группы, возможно присутствующие (например, NH2, OH), были предварительно защищены так, как это было указано выше. Полученные таким образом соединения (4) могут быть превращены в соответствующие производные, где A=COOH или NH2 согласно методу, приведенному в стадии 1g.
представляет собой одинарную связь) могут быть получены согласно схеме 4 (см в конце описания) реакций при условии, что другие реактивные группы, возможно присутствующие (например, NH2, OH), были предварительно защищены так, как это было указано выше. Полученные таким образом соединения (4) могут быть превращены в соответствующие производные, где A=COOH или NH2 согласно методу, приведенному в стадии 1g.
Стадия 4a.
R2-CHO, водная NaOH в EtOH или другом протонном растворителе;
R2-CHO, NaH или t-бутоксид калия в THF (или другом биполярном апротонном растворителе) при 0 - 150oC.
Стадия 4b.
Минеральная кислота (например, HCl или H2SO4) в воде или другом протонном растворителе (например, EtOH, AсOH) при 0 - 100oC.
Стадия 4c.
R2CHO, 0,1 н.- 1 н. водный NaOH или другое подходящее основание в протонном растворителе;
R2CHO, пирролидин в протонном (например, MeOH) или полярном апротонном растворителе при 0 - 100oC (H.J. Kabbe, Synthesis, 1978, p. 886).
Стадия 4d.
Диизопропиламид лития в THF при 0 - 20oC; затем триметилсилилхлорид и органическое основание (например, NEt3) (S.E. Kelly et al., J. Org. Chem., 56, 1325, 1991).
Стадия 4e.
R2-CHO в хлорированном растворителе (например, в дихлорметане) при -78oC, затем TiCl4 или другая кислота Льюиса (S.E. Kelly et al., J. Org. Chem., 56, 1325, 1991).
Стадия 4f.
Диизопропиламид лития в THF при -78oC, затем R2-CHO (A. Banerij et al., Tetrahedron Letter, 1979, 3685).
Стадия 4g.
R2-CH= CR3COCl, кислота Льюиса (например, AlCl3) в подходящем растворителе (например, нитробензол) или без растворителя при 20 - 180oC.
Стадия 4h.
R2-CH= CR3COOAlk, гидроксид триэтилбензиламмония в апротонном растворителе (например, бензоле или без растворителя) при 50 - 150oC; затем водный NaOH в MeOH при 20 - 50oC или LiOH в водном DMF. (В этом случае соединения, в которых A - COOCH3 или COOC2H5, также гидролизуются до соединений, имеющих A = COOH).
Стадия 4i.
Концентрированная H2SO4 или P2O5, или полифосфорная кислота, или кислота Льюиса в нитробензоле или толуоле, или без растворителя при 0 - 180oC. (В этом случае происходит также гидролиз A = COOAlk до A = COOH).
Простые исходные материалы, содержащие R3 - OH или OR8, где R8 представляет собой алкил или аралкил, могут быть получены согласно схеме 5 реакции (см. в конце описания), в которой A имеет то же самое значение, что и в схеме реакций 1. Соединения (1) и (2) (которые являются теми же самыми, что (2) и (4) в схеме реакций 4, но в которых R3 - H) можно получить согласно схеме реакций 4, используя в качестве исходных подходящие фенолы или тиофенолы, имеющие R3 - H. Соединения (4), используемые в схеме реакций 5, могут быть получены известными методами из соответствующих салицилатов или тиосалицилатов (см. J. March, Advanced Organic Chemistry, 486, John Wiley and Sons, New York, 1985, L. Rene et al., Eur. J. Med. Chem.-Chim. Ter., 4, 385, 1977 и ссылки, указанные в них). Заместитель A в соединениях (3) и (6) схемы реакций 5 может быть превращен в заместитель B с помощью процессов стадии 1g схемы реакций 1.
Стадия 5a.
Водный NaOH в спиртовом растворителе (например, MeOH или EtOH), затем обработка 30% H2O2 при температуре от -10 до -78oC. (N.D. Meyer et al., J. Med. Chem. , 34, 736, 1991 и ссылки, приведенные в этой работе). (В случае если A не является CH=CH3; если A = COOR, он превращается одновременно в COOH).
Стадия 5b.
Если  представляет собой одинарную связь:
представляет собой одинарную связь:
амилнитрит или другой алкилнитрит без растворителя или в подходящем растворителе (например, EtOH или бензоле) в присутствии катализатора, такого как 37% HCl (Org. React., 7, 327, 1953 и ссылки, приведенные в этой работе); затем водная H2SO4 в протонном растворителе (например, AcOH) при 10 - 100oC (Acheson R. M. An Introduction to the Chemistry of Heterocyclic Compounds, 347, John Wiley and Sons, New York, 1976).
Если  представляет собой двойную связь:
представляет собой двойную связь:
диизопропиламид лития в сухом THF при -78oC; затем AcOH и 30% H2O2 (B.D. M. Cunningham et al., Anti-Cancer Drug Design, 7, 365, 1992).
Стадия 5c.
R2 - CH=CH-NO2 (1 - 5 эквивалентов) в подходящем растворителе (например, диизобутиловом эфире, DMSO или DMF) в присутствии основания (например, KOH или NaOH) в каталитических или в стехиометрических количествах при 20 - 150oC (L. Rene and T. Sakakibara et al., Bull. Chem. Soc. Jpn., 51, 3095, 1978).
Стадия 5d.
15% H2O2, NaOH или другое основание (например, NEt3) в протонном растворителе, таком как MeOH, при 20 - 100oC (S.R. Deshpande et al., Synthesis, 835, 1983) или фотолиз и щелочной гидролиз (Rao T.S. et al., Heterocycles, 22, 1377, 1984) или KO2 в бензоле, содержащем 18-краун-6 эфир при 20 - 100oC (Rao T.S., Heterocycles, 26, 2117, 1987). (Если A не представляет собой CH= CH-CH3; если A = COOR, то он одновременно превращается в COOH).
Стадия 5e.
R8L, где L представляет собой отщепляемую группу (например, алкилсульфат, галоген, тозил) и основание (например, K2CO3, NaH, KOH, NaOH или LiOH) в подходящем растворителе (например, THF, DMSO, бензоле) в присутствии фазового катализатора (например, бромида бензилтриэтиламмония) при 0 - 180oC.
Стадия 5f.
Согласно методам стадии 1b.
Схема 6 реакций (см. в конце описания), в которой A имеет то же значение, что и в схеме реакций 1, приводит к соединениям, в которых W представляет собой тиокарбонильную группу. Соединения (1) и (2) схемы реакций 6 могут быть получены согласно схемам реакций 1, 2, 4 и 5. Заместитель A соединения (4) из схемы реакций 6 может быть превращен согласно процессам стадии 1g в заместитель B, как это было определено в схеме реакций 1.
Стадия 6a.
P2S5 в пиридине при 50 - 100oC (Stavaux et al., Bull, Soc, Chim. Fr., 2082, 1967).
Стадия 6b.
P2S5 или B2S3, или SiS2, или реагент Лавессона в хлорированном растворителе (например, хлороформе) или в другом ароматическом растворителе (например, бензоле, толуоле, ксилоле) при дефлегмации (Dean et al., J. Chem. Soc. C, 2192, 1963, R.K. Razdan et al., J. Med. Chem., 21, 643, 1978, K. Clausen et al., Tetrahedron, 37, 3635, 1991).
Стадия 6c.
COCl2 без растворителя или в инертном растворителе (например, бензоле) при 40 - 90oC (A. Schonberg et al., Chem. Ber., 101, 701, 1968).
Стадия 6d.
Тиоуксусная или тиобензойная кислота или тиэтилксантогенат кальция в подходящем растворителе (например, бензоле) при дефлегмации (см. предыдущую ссылку).
Схема реакции 7 (см. в конце описания), в которой A имеет то же самое значение, что и в схеме реакции 1, приводит к соединениям, в которых W представляет собой метиленовую или гидроксиметиленовую группу. Соединения (1), (2) и (4) из схемы реакций 7 могут быть получены согласно реакционным схемам 1, 2, 5 и 6. Заместитель A в соединениях (7) из схемы реакций 7 может быть превращен с помощью процессов стадии 1g в заместитель B согласно определениям схемы реакций 1.
Стадия 7a.
1,2-Этандитиол или 1,3-пропандитиол в апротонном растворителе (например, дихлорметане или бензоле, или толуоле) при 0 - 110oC в присутствии катализатора (например, п-толуолсульфоновой кислоты).
Стадия 7b
R2COCH2R3 в подходящей смеси растворителей (например, EtOAc или дихлорметан вместе с EtOH или MeOH), насыщенной газообразным HCl или 0 - 40oC; затем водная HClO4 в ацетоне при 20 - 100oC (L. Jurd, Tetrahedron, 28, 493, 1972).
Стадия 7c.
LiAlH4 в THF при дефлегмации (если A отличен от COOR и NO2);
ZnI2 и цианоборогидрид натрия (6 эквивалентов) в хлорированном растворителе (например, 1,2-дихлорэтане) при температуре от комнатной до температуры дефлегмации (C.K. Lau et al., J. Org. Chem., 51, 3083, 1986).
Стадия 7d.
Никель Ренея в спиртовом растворителе (например, изопропаноле) при температуре от комнатной до температуры дефлегмации (Hilton et al., J. Am. Chem. Soc., 90, 6887, 1968).
Стадия 7e.
NaHB4 в подходящем растворителе (например, MeOH или EtOH или DMSO) при температуре от -10 до 50oC (см. ссылку в пункте 7b).
LiAlH4 в THF (или любом другом подходящем растворителе) при 0 - 50oC (если A отличен от COOR или NO2) (Degani et al., Ann. Chim., 61, 793, 1971, Kurosawa, Bull. Chem. Soc, Jpn., 51, 1175, 1978).
Стадия 7f.
Тритилперхлорат в ацетонитриле при комнатной температуре (см. предыдущую ссылку).
Стадия 7g.
Плавление с P2O5 при 80 - 180oC (Hortmann et al., J. Am. Chem. Soc., 96, 6118, 1974).
Стадия 7h.
NaBH4 в EtOH или другом подходящем растворителе при температуре от 0oC до температуры дефлегмации (K. Anaya, Bull. Chem. Soc. Jpn., 40, 1884, 1967);
водород (1 - 10 атм) в EtOH (или другом подходящем растворителе) в присутствии катализатора, такого как 5% или 10% Pd/C или никель Ренея, или PtO2 при температуре от комнатной до 80oC (см. предыдущую ссылку) в случае, если A отличен от CH=CH-CH3. Если A = NO2, то он одновременно восстанавливается до NH2);
триизопропоксид алюминия в изопропаноле при температуре от комнатной до 92oC.
В схеме 8 реакций (см. в конце описания) указано получение простых исходных материалов, таких как (4), (5), (6) и (9), в которых A имеет то же самое значение, что и в схеме реакций 1. Соединения (1), (2), (3), (7), (8) могут быть получены по реакционным схемам 1, 2, 4, 5, 7, 9, 11. Заместитель A в соединениях (4) - (6) и (9) схемы реакций 8 может быть превращен согласно стадии 1g в заместитель, указанный в схеме реакций 1.
Стадия 8a.
Pb(OAc)4 в подходящем растворителе (например, бензоле, толуоле) при дефлегмации (G.A. Russel et al., J. Am. Chem. Soc., 1906, 1975).
Стадия 8b.
NaNH4 в спиртах (см. схему реакций 7, стадия 7a), затем щелочной гидролиз (если A - COOR, то он может одновременно быть превращен в COOH);
изопропоксид алюминия, как это было описано в схеме реакций 7, стадия 7f;
диборан в THF при температуре от -10oC до комнатной; затем водная H2O2 в присутствии NaOH (если A отличен от CH=CH-CH3; если A = COOR, то он одновременно превращается в COOH). (Kirkiacharian et al., C.R. Hebd. Seances Acad. Ser. C, 289, 227, 1979);
LiAlH4 и AlCl3 в подходящем растворителе (например, THF) при 0oC при дефлегмации (в случае, если A отличен от COOR и NO2) (Bokadia et al., J. Chem. Soc., 4663, 1961).
Стадия 8c.
Водород (100 атм), хромит меди в EtOH при 140oC (M.A. Vickars, Tetrahedron, 20, 2873, 1964). Если A= NO2, то он одновременно превращается в NH2-группу.
Стадия 8d.
KMnO4 в t-бутаноле (или другом подходящем растворителе) в присутствии водной NaOH при температуре от -10 до 0oC (K. Hanaya, Bull. Chem. Soc. Jpn., 40, 1884, 1967). (В случае, если A отличен от CH=CH-CH3). (См. также A.H. Haines, Methods for Oxidation of Organic Compounds, Academic Press Inc. (London), 1985, chapter 3.2.2).
тетроксид осмия (см. предыдущую ссылку, глава 3.2.1) в подходящем растворителе (например, Et2O) при комнатной температуре (Baranton et al., Bull. Soc. Chim. Fr., 4203, 1968) (в случае, если A отличен от CH=CH-CH3);
водная H2O2 в муравьиной или уксусной кислоте при температуре от -20 до -50oC; затем NaOH, H2O, 45oC (см. Baranton et al. выше и A.H. Haines выше, глава 3.2.7) (в случае, если A отличен от CH=CH-CH3; если A = COOR, то он одновременно превращается в COOH);
ацетат серебра и иод во влажном AcOH при 0 - 20oC (см. K. Hanaya выше и A. H. Haines главы 3.2.3, 3.2.4, 3.2.9) в случае, если A отличен от CH=CH-CH3.
Стадия 8e.
30% H2O2 в присутствии NaHCO3 в бензонитриле при 0 - 110oC, затем LiAlH4 в THF при 0 - 40oC (в случае, если A отличен от COOR и CH=CH-CH3 (Clark et al., Austr. Journ. of Chem., 27, 865, 1974).
Стадия 8f.
Водород (1 - 50 атм) в подходящем растворителе (например, EtOH) в присутствии металлического катализатора (например, PdCl2) при температуре от комнатной до 78oC (если A = NO2, то он одновременно превращается в NH2). (Bolger et al., Tetrahedron, 23, 341, 1967).
Стадия 8g.
См. стадию 8b (Clakr et al, см. выше).
Стадия 8h.
0.4 М гептагидрат трихлорида церия в MeOH в подходящем растворителе (например, MeOH); затем NaHB4 при температуре 0 - 78oC (WO 89/06650);
NaBH4 в диглиме при температуре от 0oC до температуры дефлегмации (G.P. Thakar, Indian J. Chem., 3, 74, 1965) (если A = NO2, то он превращается в NH2);
NaBH4 и AlCl3 в подходящем растворителе (например, THF или бензоле) при температуре от 0oC до температуры дефлегмации (в случае, если A отличен от COOR) (см. предыдущую ссылку);
диборан в THF при комнатной температуре (в случае, если A отличен от CH= CH-CH3) (см. предыдущую ссылку).
Простые исходные материалы, в которых W - CH2 и одинарная связь находится в положении 2, 3 могут быть получены согласно схеме реакций 9, в которой A имеет такие же самые значения, как и в схеме реакций 1. Соединения (1) из схемы 9 реакций (см. в конце описания) могут быть получены согласно схеме реакций 6. Эти же самые соединения могут быть также получены исходы из соединений (2) превращением последних в эфир 4-толуолсульфоновой кислоты или в эфир метансульфоновой кислоты, или в галогенопроизводное, которое может быть превращено в производное тиоэфира (1) при нуклеофильном замещении тиолом. Эти простые превращения могут быть осуществлены методами, известными людям, квалифицированным в этой области. Соединения (2) из схемы реакций 9 могут быть получены согласно схеме реакций 7. Соединения (3) из схемы реакций 9, в которых P= OC(S)-арил или OC(S)-гетероарил, или OC(S)O-алкил, или OC(S)O-алкил, или OC(S)O-арил или OC(S)S-алкил, могут быть получены при реакции соединений (2) с подходящим хлортиоформиатом или хлоротиокарбонатом или 1-1'-тиокарбонил-диимидазолом, как это описано в J. Org. Chem., 55, 924, 1990 и в Synthesis, 362, 1991, а также в ссылках, приведенных в этих работах. Соединения (4) могут быть получены из соединений (1) или (3) простыми реакциями элиминирования с основаниями. Заместитель A в соединениях (6) их схемы реакций 9 может быть превращен согласно процессам, указанным в стадии 1g, в заместитель B, как это определено в реакционной схеме 1.
Стадия 9a.
Никель Ренея в подходящем растворителе (например, изопропаноле) при температуре от комнатной до 100oC. Если A представляет собой NO2, то он одновременно превращается в NH2;
гидрид триэтилолова в бензоле или другом ароматическом растворителе при 30 - 150oC. Описание других методов, таких как хлорид никеля и NaBH4 в MeOH или боранпиридиновый комплекс в трифторуксусной кислоте или в дихлорметане в присутствии AlCl3, см. в работе J. March, Advanced Organic Chemistry, p. 728, J. Wiley and Sons, New York, 1992 и ссылках, приведенных в ней. (В случае, когда A отличен от CH=CH-CH3).
Стадия 9d.
Водород с катализатором согласно схеме реакций 8, стадия 8f. Если A представляет собой NO2, то он превращается в NH2 одновременно.
Стадия 9c.
Гидрид трибутилолова или трис(триметилсилил)силан в присутствии азаизобутиронитрила в подходящем растворителе (например, толуоле) при 80 - 150oC (M. Drescher, Synthesis, 362, 1992, M. Sekine J. Org. Chem. 55, 924, 1990);
силан (например, триэтилсилан или дифенилсилан) в подходящем растворителе (например, дихлорметане) при температуре от -20oC до температуры дефлегмации в присутствии CF3COOH или BF3 (F.M. Mauser, J. Org. Chem., 55, 555, 1990);
триэтилхлоросилан, иодид натрия в ацетонитриле, затем цинковый порошок в AcOH и ацетонитриле при температуре от комнатной до 80oC (T. Morita et al., Synthesis, 32, 1981).
Если P является галогеном или O-S-производным:
восстановительный агент (например, цианоборогидрид натрия в гексаметилфосфотриамиде или NaBH4 в DMSO), который выбирают из тех, что приведены в J. March, Advanc. Org. Chem., J. Wiley, New York, 1992, главы 0-76, 0-77.
Стадия 9d.
Водород (1 - 5 атм) в подходящем растворителе (например, EtOH) в присутствии катализатора (например, 10% Pd/C при 50 - 78oC) (Sarcevic, Helv. Chim. Acta, 56, 1457, 1973). (Если A = NO2, то одновременно он превращается в NH2);
Zn и газообразный HCl в Et2O или Ac2O в толуоле при 0 - 80oC (Todah, Bull Chem. Soc. Jpn., 45, 264, 1972) (В случае, если A отличен от NO2).
Стадия 9e.
Zn и водная HCl в подходящем растворителе (например, EtOH) при 0 - 78oC;
согласно стадии 9d, приведенной выше (если A = NO2, то он одновременно превращается в NH2);
гидразин, NaOH в этан-1,2-диоле при 200oC (Chemical Abstracts, 74, (1971): 22699) (в случае, если A отличен от COOR, NO2) или другие методы, приведенные выше в работе J. March (в случае, если A отличен от COOR, NO2);
согласно стадии 7c (если A отличен от NO2).
Схема 10 реакций (см. в конце описания) указывает пути получения соединений, в которых W представляет собой валентную связь, а X - атом кислорода или серы
Стадия 10a.
Согласно стадии 1a, но используя R3COCl или (R3CO)2O вместо R3CH2COCl или (R3CH2CO)2O с выделением или без выделения промежуточного фенилового эфира;
гексаметилентетрамин в CF3COOH при дефлегмации с последующим добавлением водной HCl. Если A = COOAlk, то в такой сильнокислотной среде он может быть гидролизован до COOH, и тогда необходима повторная ректификация с подходящим спиртом (например, используя тионилхлорид при температуре дефлегмации) перед стадией 10c;
Стадия 10b.
R3COCH(R2)Hal в ацетоне или метилэтил кетоне или дихлорметане или хлороформе в присутствии подходящего основания, такого как K2CO3, NEt3 или NaH при 20 - 80oC.
Стадия 10c.
R2CH(Hal)COOAlk в апротонном растворителе, например в DMF, в присутствии основания, например K2CO3 при 70 - 100oC с последующим гидролизом неочищенных промежуточных соединений сильным основанием (например, KOH) в протонном растворителе, таком как EtOH, при дефлегмации, в заключение создавая условия для декарбоксилирования-дегидратации, используя непротонный растворитель (например, ксилол) и кислотный катализ (например, п-толуолсульфокислота) при дефлегмации или простом нагреве при 240oC в хинолине;
R2CH2Hal и KOH при дефлегмации EtOH с последующей циклизацией выделенного промежуточного фенил(тио)эфира метоксидом натрия в кипящей смеси DMF/MeOH. Если A представляет собой COOAlk, то могут быть получены промежуточные соединения (4), содержащие A = COOH;
Стадия 10d.
Энергичное перемешивание в подогретой предварительно полифосфорной кислоте при 90 - 140oC;
кислота Льюиса (например, AlCl3) в хлорбензоле при 70 - 90oC. Циклизации соединений (3), содержащих R3 - Cl, с использованием кислоты Льюиса (например, AlCl3) в о-дихлорбензоле при 45oC или с помощью BF3 в Et2O при 20 - 25oC приводят к соединениям (4), в которых R3 - OH, как сообщено K. Davies, J. Chem. Soc. P.T., 1, 2624, 1957, если X=S и R2=H.
Стадия 10e.
Алкоголят натрия (1 эквивалент) в том же самом спирте при 0 - 90oC; если A = COOAlk, то соответствующий AlkOH можно использовать в качестве реакционного растворителя;
если R2 = COOAlk и X - S, то соединения (4) могут быть гидролизованы смесью серной и уксусной кислот до соответствующего R2=COOH (если имеется A = COOAlk, то он то же может давать A = COOH) и могут быть избирательно декарбоксилированы с помощью меди в безводном хинолине при 210 - 220oC, давая соединения (4), в которых R2=H согласно работе J. Cooper et al., J. Chem. Soc. (C), 1971, 3405.
Стадия 10f.
R2CH2XH и один эквивалент натрия в EtOH при дефлегмации или с помощью NaHCO3 в смеси Et : вода при 60 - 90oC.
Стадия 10g.
Если A = COOAlk или NO2, то могут быть использованы методы, изложенные в стадии 1g. Следует отметить, что восстановление группы NO2 в NH2-группу при каталитическом гидрировании может вызвать одновременно гидрирование двойной связи в положении 2, 3; об этом сообщается в работах S.L. Meisel et al., Heterocyclic Compounds, Ed. Interscience Publ. : "Compounds with Condensed Thiophene Rings", p. 34(1954) и M. Ahmed, Ed. Wiley Interscience: "Benzofurans", p. 56 (1974).
Если A = NO2 и R2 = COAr, то восстановление, проводимое с помощью водорода в присутствии катализатора Pd/C, дает соединения (5), в которых B -NH2 и R2= CH2Ar; об этом сообщается в WO 86/07056;
Если A = CH3 и R2, R3, R6 отличны от CH3 или R2 не имеет CH3-группы, то соединения могут быть превращены соответственно в:
A = CH2Br взаимодействием с N-бромсукцинимидом в CCl4 при использовании 2,2'-азобисизобутиронитрила или перекиси бензоила как катализаторов при дефлегмации;
A = CHO реакцией перечисленных выше соединений с гексаметилентетрамином при дефлегмации в хлороформе с последующим кислотным гидролизом соли в кипящем AcOH или взаимодействием соединений, содержащих A = CH3, с бихроматом тетрабутиламмония при дефлегмации в хлороформе согласно работе Valeri et al. , Arzneim. Forsch., 40, 122 (1990);
A = COOH при окислении указанных выше соединений (A = CHO) оксидом серебра в смеси протонного водного растворителя (например, EtOH-DMF) при 0 - 70oC согласно работе H.R.Rodrigues et al., Tetrahedron, 24, 6587, 1968) или с помощью KMnO4 в t-бутаноле в присутствии водного раствора NaH2PO4 при 70 - 75oC согласно работе S. Maruzama et al., Tetrahedron Letters, 27, 4537 (1986).
Соединения (4) приведенной выше схемы реакций 10, содержащие R3 = C6H5 или t-бутил, R2= H и X = O, могут быть превращены в соответствующие промежуточные соединения, содержащие R2 = C6H5 или t-бутил и R3 = H взаимодействием с полифосфорной кислотой при 132oC согласно работе Davies et al., J. Chem. Soc., 1958, 822.
Если X представляет собой имино- или алкилиминогруппу, а W такое же значение, как было приведено выше, за исключением валентной связи, то простые исходные материалы могут быть получены согласно следующей схеме реакций 11 (см. в конце описания):
Стадия 11a.
EtOC(R2)= C(COOEt)2 при 80 - 140oC без растворителя и в полярном растворителе (например, изопропаноле).
Стадия 11b.
R2COC(R3)COOAlk и п-толуолсульфоновая кислота или метансульфоновая кислота в хлорированном растворителе (например, хлороформе или дихлорметане) или в апротонном растворителе (например, бензоле) при дефлегмации в азеотропных условиях.
Стадия 11c.
Нагревание в Ph2O в присутствии п-толуолсульфоновой кислоты, или фосфорной кислоты, или ZnO в качестве катализаторов при 245 - 255oC согласно Hung, Teljies 6251 (Chemical Abstracts, 79, 92026v, 1973):
нагревание в высококипящем растворителе (например, Ph2O) с последующим гидролизом невыделенных соединений (4) (R3 = COOEt) сильной кислотой (например, HCl) в протонном растворителе (например, уксусной кислоте) при дефлегмации, чтобы получить соединения (4), в которых R3=COOH. Выделенные ранее кислоты могут быть декарбоксилированы при нагревании в высококипящем растворителе (например, Ph2O), давая соединения (4), в которых R3 = H согласно работе R. Albrecht et al., Ber., 105, 3118 (1972).
Стадия 11d.
Нагревание в высококипящем растворителе (например, Ph2O) при 255oC;
если R = Alk, то соединения (4) получают непосредственно из соединений (1) без выделения соединений (3) при конденсации с R2COCH(R3)COOAlk в полифосфорной кислоте при 90 - 150oC согласно работе F. Piozzi et al., Gazz. Chim. It., 100, 678, 1970.
Стадия 11e.
Al/Hg амальгама в водном растворе EtOH при дефлегмации с последующим подкислением сильной кислотой (например, HCl) и обработкой FeCl3 при дефлегмации согласно работе W.A. Denny et al., J. Med. Chem., 32, 396, 1989;
если A = COOAlk, то соединения (4) необходимо гидролизовать до соответствующих A-COOH перед осуществлением стадии 11e,
если A = NO2, то получают промежуточные соединения, имеющие A = NH2;
Стадия 11f.
R2CH= CHCHO и мышьяковая кислота в сильнокислой среде (например, концентрированной H2SO4) и вода при 105 - 115oC согласно EP 0206802.
Если A = COOAlk, то соединения (1) необходимо гидролизовать до соответствующих A = COOH перед осуществлением стадии f. Все соединения (1) имеют R = H, а все полученные соединения (5) имеют R3 = H.
Стадия 11g.
R2CH(Hal)-CH(R3)COOH в протонном растворителе (например, воде) в присутствии сильного основания типа NaOH при 100 - 125oC с последующей циклизацией выделенных соединений бета-анилинопропионовых кислот с помощью предварительно подогретой полифосфорной кислоты при 120 - 125oC или с помощью пентоксида фосфора в высококипящем апротонном растворителе (например, ксилоле) при 120 - 140oC. В некоторых случаях полезно исходить из соединений (1), в которых R представляет собой тозил или другие подходящие защитные группы; полученные соединения (6), в которых R - тозил, могут быть легко превращены в соединения (6), в которых R - H, гидролизом сильной кислотой (например, HCl) в протонном растворителе (например, AcOH) при дефлегмации.
Если A = COOAlk, то получают соединения (6), в которых A = COOH.
Стадия 11g.
R2CHO и этилен в AcOH и HCl при 25 - 30oC согласно работе K.D. Hess, Liebig., Ann. Chem., 741, 117 (1970). Если в соединениях (1) R = H, то получают исходные материалы (7), в которых R=R3= H;
эпихлоргидрин с последующей циклизацией выделенных производных анилинопропанола при дефлегмации N, N-диэтиланилина или о-дихлорбензола в присутствии акцептора протонов (например, NEt3) согласно работе S.D. Boyd et al., J. Org. Chem., 30, 2801(1965). В этом случае получают соединения (7), в которых R=R2=H и R3= OH.
Стадия 11i.
Гидрирование в присутствии катализатора (например, оксид платины) в протонном растворителе (например, EtOH) при 20 - 30oC и 2 - 4 атм согласно работе G.M. Coppola,. J. Heter. Chem., 15, 645, 1978.
Если A = NO2, то получают соединения (7), в которых A = NH2.
Полученные таким образом соединения (4), (6), (7) могут быть превращены в соответствующие производные, в которых A = COOH или NH2 согласно методам из схемы реакций 1, стадия 1g.
Синтез простых соединений (7) из схемы 11 реакций, в которых R = H и A = COOH может быть осуществлен также следуя методу, указанному в схеме 12 реакции,
Схема 12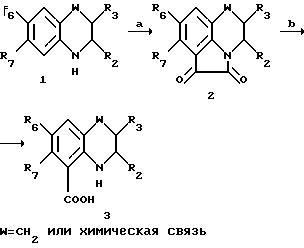
Стадия 12a.
Оксалил хлорид в полярном растворителе (например, THF) при дефлегмации с последующим внутренним ацилированием по Фриделю - Крафту неочищенного хлороксалиламида кислотой Льюиса (например, AlCl3) в неполярном растворителе (например, CS2) при дефлегмации согласно EP 0402859.
Стадия 12b
30 - 35% водная H2O2 и сильное основание (например, NaOH) в полярном растворителе (например, воде) при 20 - 30oC с последующим добавлением сильной кислоты (например, HCl), как это изложено в EP 0402859.
Схемы реакций 13 и 14 (см. в конце описания) приводят к простым исходным материалам, в которых X представляет собой иминогруппу, а W представляет собой валентную связь. В обеих этих схемах A имеет такое же самое значение, как и в схеме реакций 1.
Стадия 13a.
ClCH2(Cl)= CH2 в присутствии K2CO3 при 40 - 80oC согласно работе L. Purdie, J. Chem. Soc., (C) 1970, 1126.
Стадия 13b.
R2COHal в пиридине или в хлорированном растворителе (например, дихлорметане) в присутствии акцептора протонов (например, NEt3) при 20 - 100oC или в полярном растворителе (например, ацетоне) в присутствии K2CO3 при 20 - 80oC.
Стадия 13c.
BF3 в MeOH при 130 - 155oC;
нагревание от 100 до 110oC.
Соединения (5), полученные по стадии 13c, всегда содержат R2 = CH3.
Стадия 13d.
R2COCH(OAlk)2 в неполярном растворителе (например, толуоле) в присутствии иода в качестве катализатора при дефлегмации в азеотропных условиях с последующим восстановлением выделенного (или невыделенного) иминосоединения с помощью NaBH4 в полярном растворителе (например, MeOH) в присутствии в качестве катализатора NaOH при дефлегмации. Если A = COOAlk, то он будет гидролизован до COOH.
Стадия 13e.
Амид натрия в высококипящем растворителе (например, N,N-диэтиланилине) при 220 - 250oC согласно работе F. Piozzi et al., Gazz. Chim. It., 93, 1382, 1963;
t-бутоксид кальция в полярном растворителе (например, DMF) при 20 - 100oC согласно EP 0042298.
Стадия 13f.
BF3 в аполярном растворителе (например, бензоле) при 5 - 10oC.
Стадия 13g.
Zn или Fe пыль в кислой среде (например, AcOH) и воде при 70 - 100oC. Если A = NO2, то он будет восстановлен до NH2.
Стадия 13h.
Тионилхлорид при дефлегмации. Получающиеся в результате ацилхлориды выделяют, и они взаимодействуют с азидом натрия в кислой среде (например, AcOH) при 10 - 20oC, при последующем нагревании при 50 - 70oC.
Стадия 13i.
Диазотирование с помощью NaNO2 в концентрированной H2SO4 с последующим добавлением водного ZnCl2 при 5 - 10oC и взаимодействием выделенных солей диазония с CH2=C(R2)COOH в полярном растворителе (например, ацетоне) в присутствии соли меди (например, CuCl2) при 25 - 30oC. Примеры стадий 13g, 13h, 13i даны в работе A. Allais et al., Eur. J. Med. Chem., 10, 187, 1975.
Стадия 13j.
R2CH2NO2 в полярном растворителе (например, EtOH) в присутствии основания (например, n-бутиламина) и каталитических количеств кислоты (например, AcOH) при дефлегмации.
Полученные таким образом соединения (5) могут быть превращены в соответствующие производные, содержащие A=COOH или NH2, согласно методам, изложенным в схеме реакций 1, стадия 1g.
Что касается схемы реакций 14, то она предназначается для соединений (4), имеющих R3= H, которые соответствуют соединениям (5) из схемы реакций 13.
Стадия 14a.
NaNO2 в водной кислой среде (например, в HCl) при температуре от -5 до 5oC;
изоамилнитрит в полярном растворителе (например, EtOH) при 5 - 10oC.
Стадия 14b.
Водный раствор SO2 при 0 - 10oC согласно работе Pfannstiel et al., Ber. 75, 1096, 1942;
трифенилфосфин и нагревание выделенной фосфониевой соли в водно-спиртовом растворе HCl при дефлегмации согласно Horner et al., Ber., 85, 1073, 1953.
Стадия 14c.
R2COCO(R3)Hal в высококипящем растворителе (например, N,N-диэтиланилине) при 160 -0 180oC или просто нагреванием без растворителей при 180oC;
R3COCH(R2)Hal в полярном растворителе (например, ацетоне) в присутствии подходящего акцептора протонов (например, K2CO3) при дефлегмации с последующей циклизацией выделенных промежуточных бета-анилинокетона с помощью свежерасплавленного ZnCl2 в протонном растворителе (например, EtOH) при дефлегмации;
R2CH(Hal)CN в присутствии BCl3 и кислоты Льюиса (например, TiCl4) в аполярном растворителе (например, бензоле) при дефлегмации с последующей циклизацией выделенных 2-амино-альфа-галоацетофенонов с помощью подходящего восстановительного агента (например, NaBH4) в полярной среде (например, диоксан-воде) при дефлегмации согласно работе T. Sagusawa et al., J. Org. Chem., 44, 578, 1979. Описанным выше методом получают соединения (4), в которых R3= H;
R2COCH(R3)Hal (0.5 эквивалента) в полярном растворителе (например, MeOH) при дефлегмации с последующей циклизацией выделенных оснований Шиффа сильной кислотой (например, CF3COOH) при 20 - 30oC.
Стадия 14d.
R2COCH2R3 при нагревании при 100oC без растворителей или при дефлегмации в полярном растворителе (например, MeOH) с последующей циклизацией выделенных гидразонов с помощью полифосфорной кислоты при 100 - 130oC или с последующим нагреванием в этиленгликоле и водной муравьиной кислоте или в этанольной муравьиной кислоте;
циклизацию можно также осуществить при нагревании в этанольной HCl при дефлегмации или в смеси AcOH/HCl при дефлегмации, или в ортофосфорной кислоте при 95 - 105oC, или при простом нагревании с безводным ZnCl2 при 100 - 220oC. Если A = COOAlk, то могут быть получены соединения (4), в которых A = COOH.
Стадия 14e.
Боранпиридиновый комплекс при 0 - 30oC с последующим добавлением агента протонирования (например, HCl);
олово или цинк и водная HCl при 50 - 100oC;
NaBH4 в присутствии кислоты Льюиса (например, AlCl3) в пиридине при 0 - 30oC или в присутствии соли типа хлорида кобальта или хлорида цинка;
бороцианогидрид натрия в AcOH при 20 - 80oC;
водород в присутствии катализатора (например, Pt) в полярном растворителе (например, EtOH) при 20 - 80oC.
Другие основные методы изложены в работе Houlihan, Heterocyclic Compounds, part 1, Ed. Wiley Interscience: "Indols", p. 462 (1972). Если A = NO2, то соединения (4) могут быть восстановлены до соответствующих соединений (5), в которых A = NH2.
Стадия 14f.
NaH и RHal в безводном полярном растворителе (например, DMF) при 20 - 80oC;
амид натрия и RHal в полярном безводном растворителе (например, THF) при низкой температуре (-70oC).
Соединения (4), имеющие другие реактивные группы типа NH2 или OH, необходимо защищать, используя подходящие защитные группы, которые могут быть селективно разрушены с использованием методов снятия защиты.
Стадия 14g.
RHal в присутствии карбонатов щелочных металлов (например, карбоната калия) согласно работе Houlihan, Heterocyclic Compounds, part 2, Ed. Wiley Interscience: "Indoles", p. 90 (1972) и ссылкам, указанным в ней;
соединения (5), имеющие другие реактивные группы типа NH2 или OH, необходимо защищать так, как это указано выше.
Стадия 14h.
Тетрахлоро-(1,4)-бензохинон в полярном растворителе (например, монометиловый эфир этиленгликоля) при дефлегмации;
хлорид меди (II) в пиридине при дефлегмации согласно работе Kikugawa et al., J. Heter. Chem., 16, 1325, 1979.
Соединения (6), в которых R2 и R3 отличны от H, могут быть восстановлены до соответствующих исходных материалов (7) с помощью литий-алюминий гидрида согласно работе H.C.Printy et al., J. Am. Chem. Soc., 71, 3206, 1949.
Соединения (4) из схемы реакций 14, имеющие R2=H и R3=OH, могут быть получены, исходя из соединений (7) схемы реакций 11, в которых R=R2=H и R3=OH, при сжатии кольца с использованием оксиданта (например, периодата натрия) и основания (например, NaOH) в водном EtOH при дефлегмации согласно работе S. D. Boyd et al., J. Org. Chem., 30, 2801, 1965.
Исходные материалы (4), (5), (6) и (7) можно превратить в соответствующие A = COOH или NH2 согласно методу их схемы реакций 1, стадия 1g, а из них - в альтернативные конечные продукты. Если присутствует NH и возможно его влияние на последующие реакции, то его можно защитить так, как это описано в работе T.W. Green, Protective Groups in Organic Synthesis, Wiley Interscience, 1981. Или же нереактивные группы (например, NO2) могут быть превращены в реактивные группы (например, NH2) на заключительной стадии синтеза.
Исходные материалы, в которых W представляет собой валентную связь, X представляет собой иминогруппу, а заместитель в положении 7 представляет собой карбоксиметильную группу, могут быть получены согласно схеме реакций 15 (см. в конце описания).
Стадия 15a.
Водород в присутствии 10% Pd/C в качестве катализатора и 45 фунтов воды, содержащей один эквивалент NaOH, с последующим диазотированием нитритом натрия в HCl при 0 - 5oC и хлорид олова (II). Циклизация осуществляется при подкислении соли олова с помощью H2S и завершается при дефлегмации в ксилоле, см. H.E. Baumgarten et al., J. Am. Chem. Soc., 82, 3977, 1960.
Стадия 15b.
R3CH2COR2 в присутствии кислоты (например, уксусной кислоты) в полярном растворителе (например, EtOH) при дефлегмации согласно работе W.J. Welstead et al., J. Med. Chem., 22, 1074 (1979) при R2= CH2 и R3=C6H5, в которой изложены также стадии 15c и 15d.
Стадия 15c.
Нижний алканол (например, MeOH, EtOH) при дефлегмации в присутствии тока хлористого водорода.
Стадия 15d.
Сильное основание (например, KOH) в полярном растворителе (например, воде) при дефлегмации.
Получение простых исходных материалов, в которых R3 представляет собой гидроксиалкил, и/или соответствующих эфиров можно осуществить или путем взаимодействия соединений (3) из схемы реакций 1, соединений (2), (4) или (5) из схемы реакций 2, соединений (4) из схемы реакций 6, 10, 11 и 15, соединений (5) из схемы реакций 13 и соединений (4) и (6) из схемы реакций 14, в которых R3= H, CH3, согласно схеме реакций 16 (см. в конце описания), в которой A и B имеют такие же самые значения, как и в схеме реакций 1, R4 представляет собой алкил- или аралкилгруппу, а R5 представляет собой H или алкильную группу.
Стадия 16a.
R3 = H, W = CO, CS (и нет никаких активированных фенильных колец):
формальдегид и HCl в воде, EtOH или AcOH при 50 - 100oC;
хлорометил-метиловый эфир и дымящая серная кислота при 50 - 70oC (H. Nakarumo et al., Bull. Chem. Soc. Jap., 57, 2323, 1984);
R3 = CH3, W = CO, CS, или представляет собой химическую связь; в молекуле нет больше никаких метильных групп:
N-бромсукцинимид в присутствии перекиси бензоила или 2,2'-азобисизобутиронитрила в CCl4 при 50 - 80oC.
Стадия 16b.
R3 = H, W представляет собой химическую связь X =O, S, NH или N = Alk и в других кольцах молекулы нет никаких электронодонорных групп:
оксихлорид фосфора и DMF при 50 - 140oC или другие реагенты (см. Jutz, Adv. Org. Chem., 9, 225, 1976);
R3 = CH3, W представляет собой химическую связь, X = O, S, NH или N = Alk и нет больше никаких групп CH3;
облучение с помощью ртутной лампы высокого давления в протонном растворителе (например, AcOH) при 20 - 100oC согласно работе Frasca et al., Tetrahedron, 23, 603, 1973.
Стадия 16c.
Ацетат натрия или калия в апротонном растворителе (например, ацетоне, DMF) при 40 - 120oC.
Стадия 16d.
R5 в соединениях (5) представляет собой H:
восстанавливающий гидрид (например, NaBH4) в полярном растворителе (например, MeOH или EtOH, или диоксан) при 0 - 80oC;
R5 в соединениях (5) представляет собой алкил:
магний алкилбромид в апротонных растворителях (например, Et2O, THF) при 0 - 60oC.
Стадия 16e.
NaOH или LiOH в протонных растворителях (например, спирты, вода) или в их смеси при 25 - 50oC. (В этом случае, если A = COOAlk, то он может быть одновременно гидролизован до COOH.)
Стадия 16f.
Те же самые методы, которые приведены в стадии 1g схемы реакций 1, но для соединений (5) происходит окисление CH=CHCH3 до COOH.
Стадия 16g.
Сильное основание (например, NaH) в реагент R4-L (в котором L представляет собой атом галогена или тозилоксигруппу) в безводных апротонных растворителях (например, DMF или THF) при 20 - 140oC.
Стадия 16h.
R4OH и основание (например, Na, NaH) в избытке R4OH или в апротонном растворителе (например, DMF или THF) при 20 - 140oC.
Простые соединения (6), содержащие гидроксиалкильную группу в положении 3 и полученные таким образом, могут взаимодействовать так же, или могут давать производные по гидроксиметильной группе с известными реагентами и методами, так что указанная группа не служит препятствием для дальнейших стадий реакции, которые необходимы для получения соединений формулы I, которые содержат защищенную гидроксиальную группу типа R3. Защищенные конечные соединения в итоге превращаются с помощью методов снятия защиты в соединения формулы I, в которых R3 представляет собой гидроксиалкильную группу.
Пролекарства согласно данному ранее определению могут быть получены из соответствующих гидроксисоединений с помощью метода 1, изложенного ниже, или из соответствующих амидных соединений с помощью приведенного ниже метода 2.
Метод 1.
Взаимодействием с хлороформиатом, изоцианатом или изотиоцианатом, хлоридом или бромидом карбонила или другим производным активированной кислоты (например, ангидридом) в подходящем растворителе (например, хлорированный растворитель, DMF, THF, диоксан, ацетонитрил, пиридин) в присутствии основания (например, NEt3, пиридин, 4-диметиламинопиридин, NaOH, карбонат калия или 1,10-диазобициклоундецен или других не оговоренных особо) или в отсутствие основания при -20/100oC;
взаимодействием с карбоновой кислотой в растворителях, указанных выше, в присутствии конденсирующего агента типа N,N'-карбонилдиимидазола, карбодиимидов или других агентов, известных людям, квалифицированным в этой области;
взаимодействием с диалкил- или диарилхлорофосфатом или диалкилцианофосфатом в тех же самых условиях, которые были описаны выше (см. ниже пример 114 и S.O. Thorberg et al., J. Med. Chem., 30, 2008, 1987).
Метод 2.
Производные пролекарств "кислой" NH-группы, как это обсуждалось выше, могут быть синтезированы из соединений формулы I получением N-гидрокси(замещенного)метилового производного и последующей реакцией его таким же образом, как это было описано выше для кислородных производных.
Промежуточное N-гидрокси(замещенное)метиловое производное может быть выделено или может реагировать напрямую, давая целевое соединение.
N-гидрокси(замещенное)метиловое производное типа Ny-CH(R1)OH, где R1 = H или CCl3, может быть получено взаимодействием подходящих соединений формулы 1 с формальдегидом или CCl3CHO, как это описано в работе H.E. Zaugg, Organic Reactions, 14, CHapter 2, 52; J. Wiley and Sons, New York, 1965 или J.P. Chup, J. Org. Chem., 28, 2592, 1965.
В случае, если R1 представляет собой фенил, указанные соединения могут быть синтезированы взаимодействием бензальдегида и циклического амина (например, морфолина) в MeOH или в смеси дихлорметан:MeOH 1:1 при температуре от 0oC до температуры дефлегмации и гидролизом промежуточного соединения 0,1 н. HCl при pH 4 (O. Jacobseen, Annalen, 157, 243, 1884; Y. Bundgaard et al., Int. J. Pharm., 22, 45, 1984).
Все описанные выше пути реакций и их стадии следует рассматривать в качестве примеров, они не ограничивают возможности настоящего изобретения. Люди, квалифицированные в этой области, знают, что указанные химические превращения осуществляются в полифункциональных субстратах и что реагенты в действительности могут оказывать препятствие, взаимодействуя также и с другими группами, присутствующими в молекуле, например каталитическое гидрирование может превращать нитрогруппы в аминогруппы, как это и требуется, но однако и изолированные двойные связи могут гидрироваться и могут выделяться атомы галогенов; литий-алюминий гидрид может восстанавливать сопряженные кетоны до алканов, как это и требуется (например, стадия 7c в схеме реакций 7), но он может и восстанавливать COOAlk-группы до CH2OH или NO2-группы до -N=N- и т.д. Нежелательные побочные реакции можно избежать или минимизировать путем выбора подходящих условий или использованием альтернативных реагентов или различных путей синтеза. Если этот "альтернативный" путь дает негативные результаты, то получаемые нежелательные промежуточные соединения необходимо превратить в нужные соединения, используя те методы, которые известны квалифицированным в этой области людям.
Получение промежуточных соединений.
8-(3-Бромопропоксикарбонил)-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение I).
30 г 1,3-дибромпропана добавляют по каплям к суспензии из 30 г 3-метил-4-оксо-2-фенил-4H-1-бензопиран-8-карбоксилата натрия в 150 мл диметилформамида и 35 мл воды при комнатной температуре. Реакционная смесь встряхивается при комнатной температуре в течение 5 дней. Добавляют 100 мл воды и продолжают встряхивание еще в течение 15 мин. Осадок отфильтровывают с отсасыванием, промывают водой и очищают с использованием тонкослойной хроматографии на силикагеле, элюируя смесью хлороформ:этилацетат в соотношении 95: 5. Собранные фракции упаривают в вакууме досуха, а остаток рекристаллизуют из этанола; это дает 27.7 г целевого соединения с температурой плавления 114 - 115oC.
Бензопиран-карбоксилатная соль, используемая в предыдущем синтезе, была получена растворением 104 г соответствующей кислоты в 560 мл горячего метанола и добавлением 280 мл водного раствора, содержащего 31 г кислого карбоната натрия. К этому раствору было добавлено 850 мл ацетона для осаждения целевой соли, которую собирали при фильтровании с отсасыванием (62 г, температура плавления более 280oC). Соответствующая кислота была получена согласно работе Da Re, P et al., J. Med. Pharm. Chem., 2, 263, 1960.
8-Гидроксиметил-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение II).
467 мл 1,48 н. раствора борогидрида натрия в безводном диметилформамиде было добавлено спустя 30 мин при встряхивании при комнатной температуре к раствору из 100 г 3-метил-4-оксо-2-фенил-4H-бензопиран-8-карбонил хлорида в 1 л безводного диметилформамида. Реакционную смесь встряхивали в течение 2.5 ч при комнатной температуре; 88 мл 2 н. HCl было добавлено при поддержании температуры в пределах 0 - 5oC. Затем было добавлено 102 мл 12.7 н. раствора гидроксида натрия. Смесь выливали в 6 л воды, встряхивали в течение 3 ч и фильтровали на воронке Бюхнера. Фильтрат промывали 4 н. раствором гидроксида натрия, а затем водой. Полученный белый сухой остаток кристаллизовали из метанола, при этом получили 50 г соединения, указанного в заголовке, с температурой плавления 145 - 147oC.
E-8-(2-Карбоксивинил)- 3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение III).
Смесь, содержащая 7.92 г 8-формил-3-метил-4-оксо-2-фенил-4H-1-бензопирана (приготовленного согласно Uneyama K. et al., Bull. Chem. Soc. Jap., 58, 2361, 1985), 3.75 г малоновой кислоты и 0.46 мл пиперидина в 15 мл безводного пиридина встряхивали при 100oC в течение 3 ч. После охлаждения до 20 - 25oC реакционную смесь выливали в смесь из 90 г измельченного льда и 33 мл HCl (d = 1.18). Полученный в результате осадок был собран при фильтровании с отсасыванием, промыт водой и дважды кристаллизован из 95% этанола; в результате получили 5.5 г соединения, указанного в заголовке, с температурой плавления 226 - 229oC.
E-8-(2-Хлорокарбонилвинил)-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение IV).
Раствор из 9.2 г промежуточного соединения III и 7.8 г тионилхлорида в 75 мл толуола подвергали дефлегмации в течение 3 ч. После охлаждения до 20 - 25oC полученные в результате этого кристаллы собрали при фильтровании с отсасыванием, промыли ацетоном и высушили в вакууме; в результате получили 6.8 г соединения, указанного в заголовке, с температурой плавления после рекристаллизации из толуола 196 - 198oC.
8-Ацетил-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение V).
1.17 г магниевой стружки, 7.4 мл безводного этанола и 0.2 мл безводного четыреххлористого углерода поместили в круглодонную колбу под ток азота. Когда температура начала повышаться, было добавлено 7.5 мл безводного хлорбензола с последующим добавлением медленным прикапыванием (25 мин) раствора из 5.28 мл безводного диэтилмалоната и 375 мл безводного хлорбензола в 16 мл безводного этанола. Реакционную колбу нагрели до 75oC в течение 2 ч, затем охладили до 25oC и медленно добавили раствор 8.8 г 3-метил-4-оксо-2-фенил-4H-бензопиран-8-карбонил хлорида в 88 мл безводного хлорбензола, температура при этом не превышала 35oC. Далее реакционную смесь встряхивали в течение 2 ч при 35oC, а затем охладили до 0oC. Добавляли 13 мл воды и 1.9 мл серной кислоты (d = 1.84). Полученный раствор декантировали от нерастворимых неорганических веществ и встряхивали в вакууме.
Полученный сырой ацилмалонат подвергали дефлегмации в течение 6 ч с 10.4 мл уксусной кислоты, 7 мл воды и 1.3 мл серной кислоты (d = 1.84). После охлаждения раствор выливали в ледяную воду и собирали осадок фильтрованием с отсасыванием, промывали водным карбонатом натрия. При кристаллизации из 90% этанола было получено 6.5 г соединения, указанного в заглавии, с температурой плавления 159 - 161oC.
8-Бромацетил-3-метил-4-оксо-2-фенил-4H-бензопиран (промежуточное соединение VI).
Спустя 2 ч при 20 - 25oC к раствору 19.5 мг промежуточного соединения V в 700 мл хлороформа добавляют раствор из 11.2 г бромина в 250 мл хлороформа. После встряхивания в течение 1 ч при 20 - 25oC раствор промывают 400 мл 2 н. водного раствора гидроксида натрия и затем вновь водой, высушивают безводным сульфатом натрия и упаривают в вакууме. Сырой продукт обрабатывают диэтиловым эфиром, собирают фильтрованием с отсасыванием и кристаллизуют из ацетона, получая в результате 16 г соединения, указанного в заглавии и имеющего температуру плавления 134 - 135oC.
8-(2-Гидроксиэтилкарбамоил)-3-метил-4-оксо-2-фенил-4H-бензопиран (промежуточное соединение VII).
Указанное соединение получают так же, как и промежуточное соединение XXXVI, используя 2-аминоэтанол вместо 3-аминоэтанола. Температура плавления 206 - 208oC.
3-Метил-4-оксо-2-фенил-4H-1-бензопиран-8-сульфонил хлорид (промежуточное соединение VIII).
Раствор 4.55 г нитрита натрия в 12 мл воды был прибавлен каплями к встряхиваемой смеси 15.1 г 8-амино-3-метил-4-оксо-2-фенил-4H-бензопирана (приготовленного, как описано в работе Da Re, p et al., 11. Farmaco (Ed. Sci), Il, 670, 1956) в 150 мл HCl (d = 4.18) при - 5oC. Встряхивание продолжали при 0oC в течение 30 мин. Спустя 10 мин при температуре от -5 до 0oC раствор выливали в 120 мл 30% раствора диоксида серы в уксусной кислоте, содержащий 1.53 г дигидрата хлорида меди и 13 мл воды. К этой смеси добавляли 300 мл ледяной воды спустя 1 ч при 0oC и спустя еще 1 ч при 20 - 25oC. Образовавшийся осадок собирали фильтрованием с отсасыванием, промывали водой и высушивали в эксикаторе над гидроксидом натрия до постоянного веса. Получено 18 г сырого продукта, указанного в заглавии, с точкой плавления 165 - 170oC и готовый к применению без дальнейшей очистки.
8-(3-Хлоропропокси)-3-метил-4-оксо-2-фенил-H-1-бензопиран (промежуточное соединение IX).
Это соединение получают так же, как и промежуточное соединение XI, но используя 1-бром-3-хлорпропан вместо 1-бром-2-хлорэтана; после промывания смесью петролейный эфир: диэтиловый эфир в соотношении 7:3 температура плавления составила 98 - 102oC.
8-Акриламидо-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение X).
Раствор 1.75 мл хлорида акрилоила в 15 мл безводного тетрагидрофурана прибавляли каплями к встяхиваемой смеси из 5 г 8-амино-3-метил-4-оксо-2-фенил-4H-1-бензопирана и 3 мл триэтиламина в 60 мл безводного тетрагидрофурана при -10oC. После встряхивания при 0oC в течение 1 ч и при комнатной температуре в течение того же времени реакционную смесь выливали в воду и фильтровали при осасывании. Фильтрат промывали водой. Высушивание дало 5.5 г соединения, указанного в заглавии, с температурой плавления 229 - 230oC.
8-(2-Хлорэтокси)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XI).
Смесь из 7.52 г 8-гидрокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана (приготовленного, как это описано Da Re, P et al., Ann.Chim., 1962, p. 506), 6.22 г безводного карбоната калия и 25.5 мл 1-бром-2-хлорэтана в 70 мл диметилформамида встряхивали в течение 25 ч при 60oC. Смесь охлаждали до 20 - 25oC и выливали в 600 мл воды. Органический раствор, полученный при экстракции дихлорметаном, промывали водным раствором хлорида натрия и высушивали на безводном сульфате натрия. Растворители и избыток 1-бром-2-хлорэтана выпаривали в вакууме, для того чтобы получить 8.8 г соединения, указанного в заглавии, имеющего после кристаллизации из хлороформа: гексана температуру плавления 141 - 142oC.
8-(2-Азидоэтокси)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XII).
Смесь 15.2 г промежуточного соединения XI и 6.24 г азида натрия в 150 мл безводного диметилформамида встряхивали в течение 12 ч при 70 - 75oC. После охлаждения до 20 - 25oC реакционную смесь выливали в 1.5 л воды и экстрагировали дихлорметаном. Органический раствор промывали водным раствором хлорида натрия и высушивали на безводном сульфате натрия. Растворители выпаривались в вакууме. Остаток перенесли в воду, собрали фильтрованием с отсасыванием и высушили; это дало 14 г соединения, указанного в заглавии, с температурой плавления 119 - 120oC.
8-[N-(2-Гидроксиэтил)-N-метил-карбамоил] -3-метил-4-оксо-фенил-4H- 1-бензопиран (промежуточное соединение XIII).
Раствор из 1.6 мл 2-метиламино-этанола в 10 мл воды был добавлен по каплям спустя 5 мин к суспензии из 6 г 8-хлоркарбонил-3-метил-4-оксо-2-фенил-4H-бензопирана и 1.52 г карбоната натрия в 60 мл ацетона. После встряхивания в течение 2.5 ч при 20 - 25oC растворитель удаляли в вакууме, а осадок переносили в 150 мл ацетона. Смесь подвергалась дефлегмации в течение 15 мин, а затем фильтровалась. Растворитель выпаривали из фильтрата, а осадок растворяли в 20 мл диметилформамида, обрабатывали 14 мл 1,4% раствора карбоната натрия, встряхивали в течение 30 мин при 20 - 25oC и разбавляли, добавляя 150 мл воды. Смесь экстрагировали хлороформом, органический слой промывали 0,5 н. HCl, а затем водой. Раствор высушивали над безводным сульфатом натрия, при этом выпаривался хлороформ. Оставшееся масло переносили в 200 мл диэтилового эфира и встряхивали в течение 2 ч при 20 - 25oC. Сухие остатки собирали фильтрованием и кристаллизовали из этилацетата, чтобы получить 4.97 г соединения, указанного в заглавии, с температурой плавления 128 - 130oC.
8-(2-Хлорэтилкарбамоил)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XIV).
Указанное соединение было получено тем же самым способом, что и промежуточное соединение XXXVII, только промежуточное соединение VII использовали вместо соединения XXXVI и реакция проводилась при комнатной температуре. Температура плавления 181 - 182oC (этилацетат).
8-(N-Метил-2-хлор-этилкарбамоил)-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XV).
Раствор 1.1 мл тионилхлорида в 2 мл дихлорметана был добавлен к раствору из 3.37 г промежуточного соединения XIII в 20 мл дихлорметана и смесь встряхивали в течение 4 ч при комнатной температуре. При удалении растворителя получили масло, которое перенесли в диэтиловый эфир. Соединение, указанное в заглавии, было осаждено в виде белого сухого остатка, собранного для использования фильтрованием без дальнейшей очистки. Температура плавления (118) 126 - 128oC (диэтиловый эфир).
8-(4-Бромбутокси)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XVI).
Смесь 5 г 8-гидрокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана, 4.2 г безводного карбоната калия и 43.6 г диметилформамида встряхивали при 75oC в течение 2 ч. Смесь охлаждали до 20 - 25oC, выливали в 100 мл воды и экстрагировали хлороформом. Органический слой промывали водным раствором хлорида натрия и высушивали на безводном сульфате натрия. Растворители и избыток 1,4-дибромбутана выпаривали в вакууме. Остаток промывали 55 мл смеси петролейный эфир : диэтиловый эфир 7:4 и собирали при фильтровании с отсасыванием, чтобы получить 5.6 г соединения, указанного в заглавии, с температурой плавления 91 - 92oC.
8-(5-Бромпентилокси)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XVII).
Это соединение было получено с помощью метода, описанного для получения промежуточного соединения XVI, но вместо 1,4-дибромбутана использовали 1,5-дибромпентан и сырой продукт очищали хроматографией на колонках на силикагеле (элюируя смесью дихлорметан : этилацетат 99:1). Температура плавления после промывания смесью петролейный эфир : диэтиловый эфир 30:4 составила 75 - 76oC.
8-(2-Хлорэтоксиметил)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XVIII).
6 мл тионилхлорида в 18 мл хлороформа добавили при 0oC к встряхиваемому раствору 23 г промежуточного соединения XXII и 11 мл триэтиламина в 185 мл хлороформа. Реакционную смесь подогревали до 70oC и встряхивали в течение 2 ч; после охлаждения до комнатной температуры ее вылили в воду. Органический слой отделили, промыли раствором хлорида натрия, высушили на безводном сульфате натрия и упарили досуха в вакууме. Выход: 24 г соединения, указанного в заглавии. Образец, кристаллизованный из этанола, имел температуру плавления 102 - 103oC.
8-Хлорметил-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XIX).
53.4 г промежуточного соединения II и 38.8 мл безводного триэтиламина растворили в 440 мл хлороформа. В этот раствор при температуре, поддерживаемой от -10 до -2oC, по каплям добавили раствор 19.8 мл тионилхлорида в 80 мл безводного хлороформа. Реакционную смесь встряхивали при комнатной температуре в течение 4 ч, а затем разбавили 400 мл воды. Водную фазу экстрагировали хлороформом, а экстракты добавили к хлороформовой фазе. Раствор хлороформа промывали солевым раствором, высушивали на безводном сульфате натрия и выпаривали в вакууме досуха. Выход: 56 г соединения, указанного в заглавии, при рекристаллизации из этанола его температура плавления составила 112 - 113oC.
8-Метиламинометил-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XX).
Раствор 15.1 г безводного хлорида цинка и 14,5 г цианоборогидрида натрия в 400 мл безводного этанола добавляли по каплям во встряхиваемую смесь из 58.8 г 8-формил-3-метил-4-оксо-2- фенил-4H-1-бензопирана, 60 г гидрохлорида метиламина и 125 мл триэтиламина в 600 мл безводного этанола; прикапывание проводили при 0oC. После встряхивания в течение 5 ч при 20 - 25oC растворитель выпаривали в вакууме, а остаток переносили в 200 мл воды и собирали фильтрованием с отсасыванием. Сырой продукт растворяли в водной уксусной кислоте, промывали этилацетатом и осаждали добавлением холодного раствора 6 н. гидроксида натрия. Было получено 49 г соединения, указанного в заглавии; его температура плавления после кристаллизации из 75% этанола составила 97 - 99oC
8-(2-Хлорэтилтиометил)-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XXI).
Раствор из 37 г промежуточного соединения XIX и 10.5 г тиомочевины в этаноле подвергался дефлегмации в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и 42 г 8-амидинотиометил-3-метил-4-оксо-2-фенил-4H-1-бензопирана закристаллизовалось самопроизвольно. Образец, рекристаллизованный из этанола, имел температуру плавления 233 - 235oC. 48 мл 35% водного раствора гидроксида натрия добавляли к энергично встряхиваемой суспензии из 35 г полученного ранее соединения и 1.05 г бензилтриэтиламмония хлорида в 440 мл 1,2-дихлорэтана. Смесь встряхивали в течение 2.5 ч, а затем выливали в 300 мл воды. Водный слой экстрагировали 1,2-дихлорэтаном, экстракты добавляли к органическому слою, который промывали раствором хлорида натрия, высушивали на безводном сульфате натрия и выпаривали досуха в вакууме. Остаток кристаллизовали из метанола; при этом получили 22 г соединения, указанного в заглавии, с температурой плавления 82 - 83oC.
8-(2-Гидроксиэтоксиметил)-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XXII).
Был приготовлен раствор из 2.5 г промежуточного соединения XIX в 25 мл ксилола и 3 мл диоксана. 0.15 г натрия было растворено в 3.10 мл безводного этиленгликоля и этот раствор по каплям добавляли к раствору промежуточного соединения XIX при комнатной температуре. После дефлегмации в течение 5.5 ч реакционную смесь охлаждали до комнатной температуры и выливали в 150 мл воды. Проводили экстракцию дихлорметаном, а экстракт промывали раствором хлорида натрия, высушивали на безводном сульфате натрия и выпаривали досуха в вакууме. Твердый остаток кристаллизовали из этанола, получая при этом 2.1 г соединения, указанного в заглавии, имеющего температуру плавления 132 - 133oC.
8-Трифторацетамидо-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XXIII).
Раствор из 9.5 мл трифторуксусного ангидрида в 20 мл безводного дихлорметана добавляли по каплям при температуре от -5oC до 0oC к раствору, содержащему 5 г 8-амино-3-метил-4-оксо-2- фенил-1H-1-бензопирана в 50 мл безводного дихлорметана. Реакционную смесь встряхивали в течение 2 ч при 20 - 25oC, а затем выливали на измельченный лед. Органический раствор, полученный при экстракции дихлорметаном, промывали холодным 5% раствором бикарбоната натрия, а затем высушивали на безводном сульфате натрия. Растворитель удаляли в вакууме, а остаток кристаллизовали из этанола для получения 5.2 г соединения, указанного в заглавии и имеющего температуру плавления 175 - 176oC.
8-Аминометил-3-метил-4-2-фенил-4H-1-бензопиран (промежуточное соединение XXIV).
Смесь из 21 г промежуточного соединения XXIX и 19 г трифенилфосфина в 160 мл тетрагидрофурана встряхивали при комнатной температуре в течение 8 ч. Тонкослойная хроматография указала на исчезновение промежуточного соединения XXIX. Было добавлено 3 мл воды и встряхивание продолжали еще в течение 24 ч. Растворители удаляли с помощью роторного испарителя, а осадок растворяли в воде. Водный раствор промывали этилацетатом, делая этот раствор основным с помощью 37% раствора гидроксида натрия, и затем фильтровали на воронке Бюхнера. Фильтрат промывали водой и высушивали для получения 18 г соединения, указанного в заглавии. Гидрохлорид, рекристаллизованный из этанола, имел температуру плавления 256 - 258oC.
8-(2-Хлорэтилсульфонилметил)-3-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XXV).
41.6 мл водной 30% перекиси водорода было добавлено по каплям в течение 20 мин к раствору из 26.2 г промежуточного соединения XXI в 300 мл ледяной уксусной кислоты при 40oC. Смесь нагрели до 60oC, встряхивая при этой температуре в течение 4.5 ч, охладили до комнатной температуры и вылили в 60 мл воды. При фильтровании на воронке Бюхнера был получен фильтрат, который промывали водой и высушивали. В результате было получено 29.4 г соединения, указанного в заглавии. Образец кристаллизовали из этанола, его температура плавления составила 159 - 161oC.
8-(2-Хлорэтилсульфинилметил)-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XXVI).
36 мл водной 30% перекиси водорода было быстро добавлено по каплям к раствору из 12 г промежуточного соединения XXI в 84 мл ледяной уксусной кислоты при 10oC. Реакционную смесь встряхивали в течение 4 ч при комнатной температуре, а затем вылили в 220 мл воды. Соединение, указанное в заглавии, было собрано при фильтровании с отсасыванием, промыто водой и высушено. Выход 12.4 г, температура плавления 142 - 145oC (метанол).
8-[N-Метил-N-(2-хлорэтил)-аминометил] -3-метил-4-оксо-2-фенил- 4H-1-бензопиран (промежуточное соединение XXVII).
Смесь из 22 г промежуточного соединения XX, 66 мл 1-бром-2-хлорэтана и 11 г безводного карбоната калия в 88 мл диметилформамида встряхивали в течение 12 ч при 20 - 25oC. Затем реакционную смесь выливали в 600 мл воды и экстрагировали дихлорметаном. Органический слой промывали водой, высушивали на безводном сульфате натрия и подкисляли этанольным хлористым водородом. Растворитель и избыток 1-бром-2-хлорэтана перегоняли в вакууме при 70 - 80oC. Остаток помещали в холодный 1 н. водный раствор гидроксида натрия и экстрагировали дихлорметаном. Органический слой промывали водой, высушивали на безводном сульфате натрия и выпаривали досуха в вакууме при 25 - 30oC. Сырой продукт, указанный в заглавии, очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 7 : 3. Было получено 18 г соединения, указанного в заглавии, с температурой плавления после кристаллизации из этанола 118 - 120oC.
1-(2-Гидрокси-2-метилпропил)-4-(2-метоксифенил)-пиперазин (промежуточное соединение XXVIII).
Смесь из 7 г 1-(2-метоксифенил)пиперазина, 7.33 г безводного карбоната калия, 1.75 г иодида калия и 5.6 мл 1-хлор-2-метил-2-пропанола встряхивали в течение 90 мин при 70oC и далее в течение 6 ч - при 90oC. Реакционную смесь выливали в смесь воды со льдом и экстрагировали этилацетатом. Органический слой промывали водным раствором хлорида натрия, высушивали на безводном сульфате натрия и выпаривали в вакууме досуха. Продукт, указанный в заглавии, был получен в виде масла и идентифицирован как его дигидрохлорид, кристаллизующийся из этанола и плавящийся при 225 - 227oC.
8-Азидометил-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XXIX).
Смесь из 22.8 г промежуточного соединения XIX и 6.8 г азида натрия в 110 мл диметилформамида встряхивали в течение 3 ч при 100oC. После охлаждения до комнатной температуры к реакционной смеси было добавлено 130 мл воды и 88 мл этанола. Спустя 1 ч кристаллы были собраны при вакуумном фильтровании, промыты водой и высушены. Выход: 22 г продукта, указанного в заглавии. Образец, рекристаллизованный из этанола, имел температуру плавления 132 - 134oC.
8-[N-(2-Гидроксиэтил)-аминометил] -3-метил-4-оксо-2-фенил- 4H-1-бензопиран (промежуточное соединение XXX).
Раствор из 2.38 г безводного хлорида цинка и 2.30 г цианборогидрида натрия в 71 мл безводного этанола был добавлен по каплям при встряхивании к смеси из 9.24 г 8-формил-3-метил-4-оксо-2-фенил-4H-1-бензопирана и 9.12 г этаноламина в 90 мл безводного метанола. Встряхивание продолжали в течение 5 ч при 20 - 25oC, затем растворитель был удален в вакууме. К остатку было добавлено 250 мл воды, а нерастворенное вещество было собрано при фильтровании с отсасыванием и промыто водой. Сырой продукт растворяли в 1 н. уксусной кислоте, а раствор промывали этилацетатом. Этот водный раствор подщелачивали добавлением 2 н. раствора гидроксида натрия, а осадок собирали при фильтровании с отсасыванием. После промывания водой было получено 8.5 г соединения, указанного в заглавии, имеющего после высушивания при 60oC температуру плавления 117 - 121oC.
8-(N-Метил-N-хлорацетил-аминометил)-3-метил-4-оксо-2-фенил- 4H-1-бензопиран (промежуточное соединение XXXI).
Раствор из 6 мл хлорацетил хлорида в 60 мл 1,2-дихлорэтана был по каплям добавлен к раствору из 20 г промежуточного соединения XX и 10 мл триэтиламина в 200 мл 1,2-дихлорэтана при температуре от -5 до 0oC. После встряхивания в течение 2 ч при 20 - 25oC к реакционной смеси было добавлено 150 мл воды и произведено разделение фаз. Органическую фазу промывали водой и высушивали на безводном сульфате натрия. Растворитель удаляли в вакууме, а остаток кристаллизовали из этанола; было получено 22.5 г соединения, указанного в заглавии, с температурой плавления 146 - 148oC.
8-Хлорацетамидометил-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XXXII).
Раствор из 3.2 мл хлорацетилхлорида в 32 мл 1,2-дихлорэтана прибавляли по каплям при встряхивании к смеси из 10 г промежуточного соединения XXIV и 5.5 мл триэтиламина в 80 мл 1,2-дихлорэтана при -5oC. Реакционную смесь встряхивали в течение 1 ч при комнатной температуре и затем добавили 150 мл воды. Проводилось разделение фаз, водную фазу экстрагировали 1,2-дихлорэтаном, экстракты были добавлены к органической фазе, которую затем промывали холодным насыщенным раствором бикарбоната натрия. Затем промывали водой, высушивали на безводном сульфате натрия и упаривали в вакууме досуха. Остаток кристаллизовали из этанола, получив 10.7 г соединения, указанного в заглавии, с температурой плавления 152 - 155oC.
8-[N-Ацетил-N-(2-хлорэтил)-аминометил] -3-метил-4-оксо-2-фенил- 4H-1-бензопиран (промежуточное соединение XXXIII).
8.65 г промежуточного соединения XXX и 4.15 мл триэтиламина были растворены в 70 мл тетрагидрофурана. К этому раствору при -10oC был добавлен по каплям в течение 40 мин раствор из 2,35 мл ацетилхлорида в 23 мл тетрагидрофурана. После встряхивания в течение 3 ч при 0 - 10oC и в течение 2 ч при 20 - 25oC растворитель выпаривали в вакууме. К остатку было добавлено 100 мл воды и проведена экстракция дихлорметаном. Органические экстракты соединяли, а затем выпаривали в вакууме растворитель. Остаток растворяли в 50 мл метанола, к нему было добавлено 3 г карбоната калия и 10 мл воды. После встряхивания в течение 20 мин при 50oC, которое проводилось для гидролиза образовавшегося N,O-диацетилпроизводного, растворитель удаляли в вакууме, а остаток обрабатывали водой и дихлорметаном так, как это было описано выше. Дихлорметановый раствор вновь упаривали досуха; в результате получено 5.9 г 8-[N-ацетил-N-(2-гидроксиэтил)-аминометил] -3-метил-4- оксо-2-фенил-4H-1-бензопирана с температурой плавления 171 - 172oC. 3.6 мл тионилхлорида в 30 мл дихлорметана было добавлено по каплям при 0oC к раствору из 6.1 г полученного выше соединения в 70 мл дихлорметана. После встряхивания в течение 90 мин при 20 - 25oC реакционную смесь промывали водой и высушивали. Растворитель удаляли в вакууме, чтобы полученный сырой продукт, указанный в заглавии, был готов к использованию без дальнейшей очистки.
8-(3-Хлорпропилтио)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XXXIV).
Раствор из 20.1 г дигидрата хлорида олова (II) в 18 мл HCl (d = 1.18) добавляли в течение 5 мин при 65oC к раствору из 6 г промежуточного соединения VIII в 70 мл уксусной кислоты. Спустя 10 мин реакционную смесь охладили до 30 - 35oC, а растворитель удалили в вакууме. Остаток перенесли в воду, а нерастворенное вещество собрали при фильтровании с отсасыванием, промыли водой и высушили. Выход 8-меркапто-3-метил-4-оксо-2-фенил-4H-1-бензопирана составил 3.2 г, температура плавления после кристаллизации из этанола составила 115 - 118oC.
Смесь, содержащая 8 г приготовленного таким образом соединения, 27 мл 1-бром-3-хлорпропана, 0.2 г бромида тетрабутиламмония и 6.2 мл 35% гидроксида натрия в 80 мл бензола встряхивали энергично в течение 4 ч при 20 - 25oC. Было добавлено 100 мл воды и 40 мл дихлорметана. Органический слой отделяли, промывали водой и высушивали на безводном сульфате натрия. Растворители и избыток 1-бром-3-хлорпропана удаляли в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат в соотношении 9:1; было получено 5.7 г соединения, указанного в заглавии. После кристаллизации из метанола его температура плавления составила 84 - 86oC.
8-(3-Хлорпропилсульфонил)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XXXV).
7 мл 30% перекиси водорода было добавлено к раствору 3.45 г промежуточного соединения XXXIV в 35 мл уксусной кислоты при 20 - 25oC. После встряхивания в течение 4 ч при 60oC реакционную смесь охладили до 20 - 25oC и добавили 30 мл воды. Образовавшийся осадок был собран при фильтровании с отсасыванием, промыт водой и высушен. В результате было получено 3.4 г соединения, указанного в заглавии, которое после кристаллизации из ацетона имело температуру плавления 160 - 163oC.
8-(3-Гидроксипропилкарбамоил)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XXXVI).
Раствор 7.6 мл 3-аминопропанола в 50 мл воды добавляли по каплям в течение 30 мин к суспензии из 30 г 3-метил-4-оксо-2-фенил-4H-1-бензопиран-8-карбонил хлорида и 15.2 г карбоната калия в 400 мл ацетона. Эту вязкую суспензию встряхивали в течение 3 ч при 20 - 25oC. Растворители удаляли в вакууме, а остаток переносили в 300 мл воды. После встряхивания в течение 1 ч осадок собирали фильтрованием с отсасыванием и промывали водой. Сырой продукт очищали кристаллизацией из 95% этанола; было получено 23.8 г соединения, указанного в заглавии и имеющего температуру плавления 191 - 193oC. При концентрировании в вакууме фильтрата было получено дополнительно 4.7 г этого же соединения.
8-(3-Хлорпропилкарбамоил)-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XXXVII).
Раствор 1.1 мл тионилхлорида в 2 мл хлороформа добавили к кипящему раствору 3.37 г промежуточного соединения XXXVI в 20 мл хлороформа. После встряхивания при дефлегмации в течение 90 мин растворитель удаляли в вакууме, а осадок кристаллизовали их ацетонитрила. Было получено 3 г чистого соединения, указанного в заглавии и имеющего температуру плавления 193 - 194oC.
8-[1-Гидрокси-4-(4-метилфенилсульфонилокси)-бутил] -3-метил-4- оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XXXVIII).
1.12 г цианида натрия в 3 мл воды было добавлено к встряхиваемой смеси 3.96 г 8-формил-3-метил-4-оксо-2-фенил-4H-1-бензопирана, 2.61 г морфолина и 4.48 г n-толуолсульнофоновой кислоты в 20 мл тетрагидрофурана и 30 мл 1,2-дихлорэтана при 20 - 25oC. Реакционная смесь подвергалась дефлегмации в течение 4 ч, после этого было добавлено 10 мл холодной воды. Тетрагидрофуран отогнали при нормальном давлении и добавили 10 мл 1,2-дихлорэтана и 10 мл хлороформа. Органическую фазу отделяли, промывали водным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали в вакууме досуха. Осадок суспендировали в диэтиловом эфире, отфильтровывали и кристаллизовали из смеси хлороформ : этилацетат. Выход: 3.55 г 8-(морфолино-цианометил)-3-метил-4-оксо-2-фенил-4H-1- бензопирана; температура плавления 236 - 238oC.
К суспензии из 22.8 г приготовленного таким образом соединения в 520 мл безводного тетрагидрофурана было добавлено при комнатной температуре и встряхивании 3.5 мл 30% раствора гидроксида калия в безводном этаноле. В эту суспензию было добавлено по каплям 6.3 мл акрилонитрила в 20 мл тетрагидрофурана, и реакционную смесь встряхивали в течение 1 ч при комнатной температуре. Растворители упаривали в вакууме. Кристаллизация остатка из метанола дала 23.22 г 8-(1,3-дициано-1-морфолино-пропил)-3-метил-4-оксо-2-фенил-4H-1- бензопирана.
23.2 г полученного таким образом соединения было растворено в 250 мл диоксана. Было добавлено 250 мл 6 M HCl и полученная смесь подвергалась дефлегмации в течение 2.5 ч. После охлаждения до комнатной температуры смесь выливали в 700 мл водного раствора хлорида натрия и экстрагировали этилацетатом. Экстракты промывали водным раствором хлорида натрия и обрабатывали 700 мл 1 M раствора гидроксида натрия. Водный слой промывали этилацетатом и подкисляли 37% HCl. Осадок собирали при фильтровании с отсасыванием и кристаллизовали из этанола, что дало 10.2 г 8-(3-карбокси-1-окси-пропил)-3-метил-4-оксо-2-фенил-4H-1-бензопирана с температурой плавления 191 - 192oC.
Диборан, полученный при прикапывании в 19 мл 0.66 М раствора борогидрида натрия в диглиме раствора из 2.1 мл свежеперегнанного диэтилового эфира трифторида бора в 10 мл безводного диглима, барботировали в суспензию 2.28 г полученного выше соединения в 23 мл безводного тетрагидрофурана и встряхивали под током азота при 0oC. Встряхивание продолжали в течение 20 мин при 0oC и далее в течение 20 мин при комнатной температуре. В смесь при 0oC осторожно прикапывали метанол, для того чтобы погасить реакцию. Растворители удаляли выпариванием в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 3:7. Собранные фракции упаривали в вакууме, в результате осталось 2 г 8-(1,4-дигидроксибутил)-3-метил- 4-оксо-2-фенил-4H-1-бензопирана с температурой плавления 133 - 134oC.
2,8 г n-толуолсульфонилхлорида было добавлено при 0oC к встряхиваемому раствору 3.17 г полученного выше соединения в 32 мл безводного пиридина. Смесь встряхивали в течение 6 ч при 0oC и оставляли на ночь без встряхивания при температуре -4oC. Затем ее выливали в 200 мл водного раствора хлорида натрия, подкисляли 10 мл 12 н. HCl и фильтровали при отсасывании. Фильтрат растворяли в хлороформе, а раствор промывали водным раствором хлорида натрия и высушивали на безводном сульфате натрия. Растворитель отгоняли на роторном испарителе. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 1:1. Собранные фракции упаривали досуха в вакууме; в результате было получено 3.04 г продукта, указанного в заглавии, с температурой плавления 123 - 124oC.
4-[4-(2-Метоксифенил)-1-пиперазинил]-бутиральдегид (промежуточное соединение XXXIX).
Раствор 5.4 г 2-(3-хлоропропил)-диоксолана и 15.9 г 1-(2-метоксифенил)-пиперазина в 60 мл диметилформамида встряхивали в течение 4 ч при 80oC. После охлаждения до 20 - 25oC реакционную смесь выливали в 500 мл охлажденного льдом 0.5 н. раствора гидроксида натрия и экстрагировали дихлорметаном. Органическую фазу промывали водой и высушивали на безводном сульфате натрия. Растворитель удаляли в вакууме, а остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью дихлорметан : этанол в соотношении 95: 5. Было получено 9.8 г 2-{3-[4-(2-метоксифенил)-1-пиперазинил]-пропил}-диоксолана в виде масла.
ЯМР CDCl3 (δ)
1.5 - 2.0 (4H, m, CH2CH2CH)
2.2 - 3.2 (10H, m, 5•CH2N)
3.7 - 4.0 (7H, m, OCH3 и 2•OCH2)
4.8 (1H, t, OCHO)
6.7 - 6.9 (4H, m, ароматические протоны).
Раствор, содержащий 12.8 г полученного таким образом соединения в 200 мл тетрагидрофурана и 420 мл 1 н. HCl, выдерживали в течение 24 ч при 20 - 25oC. Затем его подщелачивали 5 н. раствором гидроксида натрия и немедленно экстрагировали дихлорметаном. Органический слой промывали водой и высушивали на безводном сульфате натрия. Растворитель выпаривали в вакууме, а остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью дихлорметан : метанол в соотношении 97 : 3. Было получено 6.4 г соединения, указанного в заголовке, в виде масла.
ЯМР CDCl3 (δ)
1.5 - 2.0 (2H, m, CH2CH2CH2)
2.2 - 2.8 (8H, m, 3•CH2N и CH2CHO)
2.9 - 3.2 (4H, m, 2•CH2NAr)
3.8 (3H, s, OCH3)
6.8 (H, s, ароматические протоны)
9.3 (1H, s, CHO).
8-(2,3-Эпоксипропокси)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XL).
7 мл 2,3-эпоксипропилхлорида было добавлено по каплям при 20 - 25oC к встряхиваемой смеси 5 г 8-гидрокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана и 9.7 мл 2 н. гидроксида натрия в 10 мл этанола. После выдержки в течение 6 ч при температуре 20 - 25oC реакционную смесь выливали в 100 мл воды и полученный осадок собирали фильтрованием с отсасыванием. После высушивания и очистки тонкослойной хроматографией на силикагеле (элюат - петролейный эфир : этилацетат 65:35) было получено 4.45 г соединения, указанного в заглавии, с температурой плавления 128 - 129oC.
8-[N-Метил-2-(4-метилфенилсульфонилокси)-этилсульфамоил] -3- метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XLI).
Раствор 5 г промежуточного соединения VIII в 60 мл дихлорметана и 20 мл тетрагидрофурана было добавлено по каплям к смеси 275 мл 2-метиламиноэтанола и 2.1 мл триэтиламина в 20 мл дихлорметана при 0oC. После встряхивания в течение 2 ч при 20 - 25 oC к реакционной смеси было добавлено 100 мл воды и 100 мл дихлорметана. Фазы разделяли, а органический слой высушивали на безводном сульфате натрия. Растворитель удаляли в вакууме, а остаток очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир: этилацетат 3: 7. Было получено таким образом 4,5 г 8-(N-метил-2-гидроксиэтилсульфамоил)-3-метил-4-оксо-2-фенил-4H-1- бензопирана с температурой плавления после кристаллизации из этанола 146 - 147oC.
Полученное соединение было превращено в соединение, указанное в заглавии, при п-толуолсульфонилировании согласно второй стадии процесса, описанного ниже, получения промежуточного соединения XLII. Соединение, указанное в заглавии, было использовано без дальнейшей очистки.
8-[2-(4-Метилфенилсульфонилокси)-этилсульфамоил] -3-метил-4- оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XLII).
Раствор 5 г промежуточного соединения VIII в 37 мл тетрагидрофурана при 0oC прибавляли по каплям к смеси 2,5 мл этаноламина и 275 мл триэтиламина в 25 мл тетрагидрофурана. После встряхивания при 20 - 25oC реакционную смесь выливали в 400 мл воды. Образовавшийся осадок собирали при фильтровании с отсасыванием, промывали водой и высушивали воздухом. В результате было получено 4,6 г 8-(2-гидроксиэтилсульфамоил)-3-метил-4-оксо-2-фенил-4H-1-бензопирана, плавящегося при 186 - 187oC после кристаллизации из этилацетата.
2.1 г п-толуолсульфонил хлорида было добавлено порциями при 0oC к раствору 3,6 г полученного выше соединения в 25 мл пиридина. После выдержки в течение 6 ч при 20 - 25oC смесь медленно выливали на измельченный лед, содержащий небольшой избыток соляной кислоты. Образовывался осадок, который собирали фильтрованием с отсасыванием и промывали водой. Было получено 4,9 г соединения, указанного в заглавии, которое плавится при 166 - 169oC после кристаллизации из этилацетата.
8-(3-Аминопропилкарбамоил)-3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид (промежуточное соединения XLIII).
Раствор 21,6 г 3-метил-4-оксо-2-фенил-4H-1-бензопиран-8-карбонил хлорида в 250 мл безводного тетрагидрофурана прикапывали при 0oC к встряхиваемому раствору 17 г 3-(2-метил-2-пропоксикарбомоил)пропиламина (приготовленного согласно работе Saari, W. S. et al., J.Med.Chem., 33, 97. 1990) в 13 мл триэтиламина. После встряхивания в течение 2 ч при комнатной температуре реакционную смесь выливали в воду и фильтровали для выделения 12.3 г 3-(2-метил-2-пропоксикарбамоил)-пропил-3-метил-4-оксо-2-фенил-4H-1- бензопиран-8-карбоксамида, который перекристаллизовывали из этанола. Температура плавления 178 - 180oC.
Раствор 4.3 мл трифторуксусной кислоты в 15 мл безводного дихлорметана добавляли по каплям при -5oC при встряхивании к раствору, содержащему 3.3 г соединения, приготовленного выше, и 35 мл безводного дихлорметана. После нагрева до комнатной температуры смесь встряхивали в течение 8 ч. Дихлорметан и избыток трифторуксусной кислоты выпаривали при 20 - 25oC, используя роторный испаритель. Маслянистый остаток растворили в дихлорметане и добавили 1 н. раствор гидроксида натрия. Органический слой промывали водой, высушивали на безводном сульфате натрия и фильтровали. К фильтрату добавили избыточное количество этанольной HCl, а растворитель удаляли в вакууме. Остаток кристаллизовали из этанола; было получено 1,5 г соединения, указанного в заглавии, с температурой плавления 253 - 255oC.
8-(2-Хлорэтилуреидо)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XLIV).
4 мл 2-хлорэтилизоцианата было добавлено при встряхивании при комнатной температуре к раствору 3.9 г 8-амино-3-метил-4-оксо-2-фенил-4H-1-бензопирана в 52 мл безводного диметилформамида. Встряхивание продолжали в течение 5 ч при 70oC. К реакционной смеси добавили воду, а затем проводили экстракцию этилацетатом. Органическую фазу выпаривали досуха в вакууме. Остаток суспендировали при встряхивании в диэтиловом эфире. Продукт, указанный в заглавии, отфильтровывали и рекристаллизовали из метанола. Выход 3.74 г, температура плавления 213 - 214oC.
(Z, E-8-{ 4-[2-(1,3-Диоксанил)] -1-бутенил}-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XLV).
1.6 мл 2.5 н. бутиллития в гексане был добавлен по каплям при -20oC к раствору 1,53 г 2-[2-(1,3-диоксанил)]-этил трифенилфосфония бромида в 10 мл безводного тетрагидрофурана. Смесь встряхивали в течение 20 мин при -20oC. В эту смесь был прикапан раствор 0.8 г 8-формил-3-метил-4-оксо-2-фенил-4H-1-бензопирана в 11 мл безводного тетрагидрофурана. Затем в течение 90 мин проводили нагрев до 0oC, а потом в течение 30 мин - до комнатной температуры. Растворители упаривали в вакууме, а остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат: петролейный эфир 3:7. Было получено соединение, указанное в заглавии, как смесь E и Z-диастереоизомеров, с температурой плавления 98 - 100oC. Соотношение этих двух изомеров было определено ЯМР-спектроскопией и составило E:Z = 65:35.
ЯМР, CDCl3 (δ):
8.1 - 8.2 (m, 1H, CH в положении 5 в бензопирановом кольце);
7.2 - 7.8 (m, 7H, другие ароматические CH - группы бензопиранового и фенильного колец);
6.9 (dt, 1H, Fl-CH E-изомера);
6.8 (dt, 1H, Fl-CH Z-изомера);
6.4 (dt, 1H, Fl-CH=CH E-изомера);
5.9 (dt, 1H, Fl-CH=CH Z-изомера);
4.6 - 4.7 (m, 1H, OCHO);
3.6 - 4.2 (m, 4H, OCH2O из диоксанового кольца);
2.4 - 2.7 (m, 2H, CHCH2CH);
1.9 - 2.3 (m, 5H, CH3 и CH2 в положении 5 в диоксановом кольце).
8-{ 4-[2-(1,3-Диоксанил)]-бутил)-3-метил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XLVI).
Смесь 0.2 г 10% катализатора Pd/C и 1 г промежуточного соединения XLV в 24 мл метанола гидрировали в аппарате Парра при комнатной температуре при давлении водорода 1.5 атм. После использования всего теоретического количества водорода катализатор отфильтровывали, а растворитель удаляли выпариванием в вакууме. Остаток кристаллизовали из циклогексана для получения соединения, указанного в заглавии; его температура плавления составила 118 - 119oC.
8-Карбоксиметил-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XLVII).
4.5 г перманганата калия порциями в течение 1.5 ч при встряхивании при 0 - 10oC было добавлено в смесь 2.76 г 8-аллил-3-метил-4-оксо-2-фенил-4H-1-бензопирана (P. Da Re, US 3350411), 0.17 г Aliquat 336, 1,12 мл уксусной кислоты, 56 мл дихлорметана, 3.2 мл серной кислоты (d = 1.84) и 60 мл воды. Встряхивание продолжали в течение 5 ч при комнатной температуре. При 0 - 5oC в течение 15 мин порциями было добавлено 3.4 г метабисульфита натрия. Органический слой отделяли, промывали водой и экстрагировали 60 мл 1 н. водного раствора гидроксида натрия. Водную фазу подкисляли добавлением разбавленной соляной кислоты и экстрагировали этилацетатом. Органическую фазу промывали водой, высушивали на безводном сульфате натрия и после фильтрования упаривали в вакууме досуха. Остаток обрабатывали четыреххлористым углеродом, а твердое вещество собирали фильтрованием с отсасыванием. Был получен 1 г соединения, указанного в заглавии, с температурой плавления 191 - 192oC (ацетонитрил).
8-(4-Хлорбутирамидо)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XLVIII)
Соединение, указанное в заглавии, было получено таким же способом, что и промежуточное соединение X, но только вместо хлорида акрилоила использовали хлорид 4-хлорбутирила. Полученный твердый осадок, отфильтрованный от воды и высушенный, промывали горячим диэтиловым эфиром и собирали при отсасывании. Образец, кристаллизованный из 50% водного этанола и промытый диэтиловым эфиром, плавился при 162 - 164oC.
8-Метиламино-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XLIX).
Раствор 0.5 г промежуточного соединения XXIII в 1.5 мл безводного диметилформамида был добавлен по каплям при встряхивании при -5oC к суспензии 0,045 г гидрида натрия (80% в минеральном масле). После встряхивания в течение часа при комнатной температуре было добавлено по каплям 0,092 мл метилиодида в 0.6 мл безводного диметилформамида. Затем реакционную смесь встряхивали в течение 1 ч при 50oC, охлаждали до 20oC, выливали в воду, фильтровали с отсасыванием и высушивали в течение 3 ч при 60oC. Было получено 0.6 г 8-(N-метилтрифторацетамидо)-3-метил-4-оксо-2-фенил-4H-1-бензопирана.
ЯМР (CDCl3, δ ):
8.15 (dd, 1H, бензопирановый CH в положении 5);
7.10 - 7.60 (m, 7H, другие бензопирановые и фенильные CHs);
3.30 (s, 3H, CH3-N);
2.10 (s, 3H, бензопирановый CH3 в положении 3).
Смесь, содержащая 0.44 г названного выше соединения и 0.05 г борогидрида натрия в 4 мл этанола и 1 мл диметилсульфоксида встряхивали в течение 1 ч при комнатной температуре, а затем обрабатывали избытком 4 н. соляной кислоты. После удаления этанола выпариванием в вакууме остаток промывали водой, а затем - 3 н. гидроксидом натрия и экстрагировали этилацетатом. Органический слой промывали водой, высушивали на безводном сульфате натрия и упаривали в вакууме досуха. Твердый остаток кристаллизовали из этанола; при этом получено 0.22 г соединения, указанного в заглавии, плавящегося при температуре 143 - 146oC.
8-(N-Метилакриламидо)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение L).
Это соединение было получено тем же способом, что и промежуточное соединение X, но вместо 8-амино-3-метил-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение XLIX. Вместо разбавления водой THF удаляли выпариванием в вакууме, а сырой остаток растворяли в этилацетате и промывали водой. Органический слой высушивали на безводном сульфате натрия и упаривали досуха в вакууме, чтобы получить соединение, указанное в заглавии. Образец, очищенный колоночной хроматографией на силикагеле (элюировали смесью этилацетат:петролейный эфир 4:6) и кристаллизованный из циклогексана, плавился при 136 - 137oC.
1-[2-(1,3-Дигидро-1,3-диоксо-2H-изоиндол-2-илокси)-этил]-4-(2- метоксифенил)-пиперазин (промежуточное соединение LI).
Смесь 6.73 г N-гидроксифталимида, 3.73 г ацетата натрия и 10 г 1-(2-хлорэтил)-4-(2-метоксифенил)-пиперазина в 100 мл безводного диметилсульфоксида встряхивали в течение 4 ч при 100oC. Затем реакционную смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали этилацетатом. Собранные органические слои промывали 1 н. гидроксидом натрия, высушивали на безводном сульфате натрия и упаривали досуха в вакууме; было получено 7.58 г соединения, указанного в заглавии. Образец, кристаллизованный из циклогексана, плавился при 80 - 83oC.
1-(2-Аминооксиэтил)-4-(2-метоксифенил)-пиперазин гидрохлорид (промежуточное соединение LII).
Раствор 6.59 г промежуточного соединения LI и 1.10 мл 85% гидразин гидрата в 130 мл 95% этанола подвергали дефлегмации в течение 4 ч. Этанол удаляли выпариванием в вакууме. Остаток промывали водой, а затем избытком 37% HCl и фильтровали. Кислый водный раствор делали щелочным с помощью 5% гидроксида натрия, а потом экстрагировали хлороформом. Органический слой высушивали на безводном сульфате натрия и упаривали в вакууме досуха, чтобы получить 4,3 г соединения, указанного в заглавии, в виде масла. Образец превращали в гидрохлорид путем образования соли с этанольным хлористым водородом в дихлорметане. Растворители удаляли выпариванием в вакууме, а сырой остаток кристаллизовали из этанола; было получено соединение, указанное в заглавии, с температурой плавления 208 - 209oC.
8-(4-Хлорбутилтио)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение LIII).
Соединение, указанное в заглавии, было получено тем же способом, что и промежуточное соединение XXXIV, но вместо 1-бром-3-хлорпропана использовали 1-бром-4-хлорбутан. Температура плавления 81 - 84oC (этанол).
8-(4-Хлорбутилсульфинил)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение LIV).
Соединение, указанное в заглавии, было получено тем же способом, что и промежуточное соединение XXVI, только промежуточное соединение LIII было использовано вместо соединения XXI. Образец, кристаллизованный из смеси циклогексан : бензол 0.5:1, плавился при 124 - 125oC.
8-Карбокси-4-оксо-3-фенил-4H-1-бензопиран (промежуточное соединение LV).
Раствор 38.22 г нитрата серебра в 75 мл воды добавляли каплями при встряхивании при 20 - 25oC к раствору 22.5 г 8-формил-4-оксо-3-фенил-4H-1-бензопирана (приготовленного согласно работе G.Atassi et al., Eur. J. Med. Chem. - Chim. Ter., 20, 393, 1985) в 150 мл 85% этанола и 450 мл диметилформамида. Затем по каплям при встряхивании при 15 - 20oC был добавлен раствор 32.67 г 85% гидроксида калия в 195 мл воды. После дополнительного встряхивания при комнатной температуре реакционную смесь фильтровали с отсасыванием; маточную жидкость подкисляли 37% соляной кислотой и разбавляли 1.2 л воды. После фильтрования с отсасыванием и промывания водой до нейтральной реакции было получено соединение, указанное в заглавии, как сырой продукт. Его суспендировали в 150 мл этилацетата и встряхивали с 444 мл 0.3 М кислого карбоната натрия до получения прозрачных слоев. Водный слой промывали 75 мл этилацетата, затем подкисляли 37% соляной кислотой, фильтровали и высушивали при 60 - 65oC. Было получено 19.12 г соединения, указанного в заглавии, плавящегося при 218oC. Образец, кристаллизованный из этанола, имел такую же температуру плавления.
8-Хлоркарбонил-4-оксо-3-фенил-4H-1-бензопиран (промежуточное соединение LVI).
Смесь 15.97 г промежуточного соединения LV и 15.6 г тионилхлорида в 75 мл безводного толуола встряхивали при 80 - 85oC в течение 4 ч. После удаления растворителя в вакууме остаток дважды промывали 20 мл толуола и упаривали досуха в вакууме. После высушивания было получено 16 г соединения, указанного в заглавии и плавящегося при 138 - 140oC, который использовали без дальнейшей очистки. Температура плавления 138 - 140oC (толуол).
8-(N-Ацетилкарбамоил)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение LVII).
Смесь 3.5 г 8-карбамоил-4-оксо-2-фенил-4H-1-бензопирана (описанного в JP 61-238783), 4.8 мл уксусного ангидрида и 0.25 мл серной кислоты (d = 1.098) встряхивали в течение 3 мин при 140oC. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и фильтровали с отсасыванием, что дало, после промывания водой и осушки, 3.88 г соединения, указанного в заглавии.
ЯМР (CDCl3, δ ):
10.50 (bs, 1H, имидный NH);
8.35 - 8.70 (m, 2H, CH в положении 5 и 7 бензопиранового кольца);
7.45 - 8.00 (m, 6H, другие ароматические CHs);
2.60 (s, 3H, CH3CO);
2.20 (s, 3H, CH3 в положении 3 бензопиранового кольца).
2-(2-Метилтиофенокси)-ацетальдегид диэтилацеталь (промежуточное соединение LVIII).
Смесь 15.2 мл 97% 2-бромацетальдегид диэтилацеталя, 14 г 2-(метилтио)-фенола, 13.7 г безводного карбоната калия и 3.13 трикаприлметиламмоний хлорида в 140 мл безводного диметилформамида встряхивали в течение 38 ч при 95oC. В конце этого времени реакционную смесь охлаждали до комнатной температуры, выливали в 1 л воды и экстрагировали диэтиловым эфиром. Органический слой промывали водой, высушивали на безводном сульфате натрия и упаривали в вакууме досуха. Маслянистый остаток очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 99:1. Выпаривание в вакууме собранных фракций дало 12.9 г чистого соединения, указанного в заглавии. Образец, кристаллизованный из н-гексана, плавился при 50 - 52oC.
2-(2-Метилтиофенокси)-ацетальдегид (промежуточное соединение LIX).
Смесь 10.5 г промежуточного соединения LVIII и 140 мл 2 н. соляной кислоты в 85 мл безводного тетрагидрофурана встряхивали в течение 2 ч при 50oC. Затем органический растворитель удаляли выпариванием в вакууме, а водный остаток экстрагировали этилацетатом. Органический слой промывали водой, высушивали на безводном сульфате натрия и упаривали досуха в вакууме. В результате было получено 9.5 г соединения, указанного в заглавии, как сухое вещество, которое использовали без дальнейшей очистки. Образец кристаллизовали из циклогексана, получив чистое соединение, указанное в заглавии, с температурой плавления 102 - 104oC.
8-(4-Хлорбутилсульфонил)-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение LX).
Соединение, указанное в заглавии, было получено таким же методом, как промежуточное соединение XXV, только вместо промежуточного соединения XXI использовали промежуточное соединение LIII. Оно кристаллизовалось из диизопропилового эфира и плавилось при 112 - 115oC.
8-Этоксикарбонил-4-оксо-4H-1-бензопиран (промежуточное соединение LX).
4.35 г металлического натрия было добавлено кусочками к раствору 9.85 г этил-3-ацетил-2-гидрокси бензоата (синтезированного из 3-ацетил-2-гидроксибензойной кислоты, полученной, как описано R.E.Ford, J. Med. Chem., 29, 538, 1986) при комнатной температуре. В течение 1.5 ч проводили дефлегмацию в 6 н. этанольном хлористом водороде, упаривали досуха в вакууме, а сырой продукт очищали колоночной хроматографией на силикагеле (элюат представлял смесь этилацетат : петролейный эфир 8:2) в 98 мл этилформиата; температура плавления 47oC (гексан).
После самопроизвольной дефлегмации реакционной смеси в течение 20 мин, ее встряхивали при комнатной температуре в течение 4 ч, и этилформиат удаляли упариванием в вакууме досуха. Полученный сырой продукт промывали 120 мл этанола и 67 мл 5.6 М этанольного хлористого водорода. Смесь встряхивали при дефлегмации в течение 30 мин, затем охлаждали до комнатной температуры и упаривали досуха в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 3 : 7 - 6 : 4. Было получено 8.31 г соединения, указанного в заглавии; образец, кристаллизованный из циклогексана, плавился при 88 - 89oC.
8-Карбокси-4-оксо-4H-1-бензопиран (промежуточное соединение LXII).
30 мл 6 н. соляной кислоты добавили к раствору 4.0 г промежуточного соединения LXI в 30 мл диоксана, полученную смесь встряхивали при дефлегмации в течение 5 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в 200 мл воды. После выдерживания в течение 12 ч при 0 - 5oC было получено соединение, указанное в заглавии; его фильтровали при отсасывании. После промывки водой и диэтиловым эфиром и осушения было получено 2.8 г соединения, указанного в заглавии, которое использовали без дальнейшей очистки. Образец промывали кипящей смесью ацетонитрил : этанол 25 : 1, фильтровали и кристаллизовали из уксусной кислоты; образец плавился при 253 - 254oC.
8-Карбокси-6-гидрокси-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение LXIII).
Смесь 175 г 8-карбокси-6-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана (полученного, как это описано в JP 61-15880) и 28 мл 57% иодистоводородной кислоты в 47 мл уксусной кислоты встряхивали при дефлегмации в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в воду; для доведения pH до 4 - 5 был добавлен 1 н. гидроксид натрия. Было добавлено 5.2 г тиосульфата натрия и встряхивание продолжалось в течение 15 мин. Спустя это время сырой продукт, указанный в заглавии, был отфильтрован с отсасыванием и растворен в 0.5 М гидроксиде натрия. Раствор промывали этилацетатом и подкисляли до pH 1 добавлением 37% соляной кислоты. Соединение, указанное в заглавии, собирали при отсасывании и осушали. Выход: 1.12 г соединения, указанного в заглавии, которое использовали без дальнейшей очистки; плавилось при 279 - 281oC после кристаллизации из 50% этанола.
2-Гидрокси-5-нитро-3-пропионил-бензойная кислота (промежуточное соединение LXIV).
97.1 г 2-гидрокси-3-пропионил-бензойной кислоты, приготовленной, как это описано в Brit 1,343, 119 (1974), было добавлено в течение 5 мин к 500 мл серной кислоты (d = 1.84) с последующим встряхиванием при - 25oC. К реакционной смеси в течение 40 мин была добавлена смесь 40 мл 65% азотной кислоты и 100 мл серной кислоты (d = 1.84); при этом температура реакционной смеси поддерживали от -20 до -13oC. Смесь встряхивали дополнительно в течение 30 мин при -18oC. После этого ее быстро выливали в смесь 2.0 кг измельченного льда и 500 мл воды, встряхивали в течение 10 мин и фильтровали для получения соединения, указанного в заглавии, после промывания водой и высушивания при 50oC в течение 6 ч. Кристаллизация этого сухого вещества из 50% этанола дала 91.5 г соединения, указанного в заглавии и плавящегося при 186 - 189oC. Соединение использовали без дальнейшей очистки; образец, рекристаллизованный из 50% этанола, плавился при 189 - 191oC.
Этил 2-гидрокси-5-нитро-3-пропионил-бензоат (промежуточное соединение LXV).
Раствор 93.3 г промежуточного соединения LXIV и 25 мл серной кислоты (d = 1.84) в 490 мл этанола подвергали дефлегмации в течение 17 ч. После охлаждения до комнатной температуры было добавлено порциями 47.7 г карбоната калия, а этанол выпаривали в вакууме. Остаток промывали 1.2 л воды, подщелачивали добавлением 37% гидроксида натрия и встряхивали 15 мин. К этой суспензии была добавлена 37% соляная кислота, чтобы довести pH до 6. При фильтровании было получено 85.4 г соединения, указанного в заглавии, которое использовали без дальнейшей очистки (температура плавления 75 - 77oC). Образец, кристаллизованный дважды из этанола, плавился при 76 - 77oC.
8-Этоксикарбонил-3-метил-6-нитро-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение LXVI).
Смесь 48.1 г промежуточного соединения LXV, 63 мл бензоилхлорида и 85.6 г бензоата натрия встряхивали при 180oC (температура бани) в течение 8 ч. Пастообразную смесь охлаждали до 60 - 70oC, было добавлено 700 мл 50% этанола и полученную смесь встряхивали снова при 50oC в течение 30 мин. 60 мл 35% гидроксида натрия было добавлено при 5oC, следя за тем, чтобы температура не поднялась выше 15oC. Фильтрование с отсасыванием при последующем промывании 50% этанолом и водой дало в результате сырой продукт, который очищали двойным прохождением через хроматографическую колонку с силикагелем. Первое элюирование проводили смесью дихлорметан : петролейный эфир в соотношении 8 : 2 - 9 : 1, затем элюировали дихлорметаном и в заключение - смесью дихлорметан : этилацетат в соотношении 95 : 5. При выпаривании в вакууме собранных фракций получили соединение, указанное в заглавии, которое промывали 140 мл этанола. Выход 43 г, температура плавления 132 - 133oC (этанол).
8-Карбокси-3-метил-6-нитро-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение LXVII).
Смесь 15.9 г промежуточного соединения LXVI и 48 мл 1 н. гидроксида натрия в 320 мл этанола встряхивали при дефлегмации в течение 30 мин. Органический растворитель удаляли выпариванием в вакууме, а полученную в результате суспензию разбавляли 200 мл воды и подкисляли 37% соляной кислотой. Фильтрование и промывание диэтиловым эфиром дали в результате 11.1 г соединения, указанного в заглавии и плавящегося при 286 - 292oC, которое использовали без дальнейшей очистки. После кристаллизации из смеси диметилформамид : вода 6 : 4 это соединение показало ту же температуру плавления.
8-Хлоркарбонил-3-метил-6-нитро-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение LXVIII).
Смесь 6.2 г промежуточного соединения LXVII, 5.2 мл тионилхлорида и 0.1 мл безводного диметилформамида в 60 мл толуола встряхивали в течение 2 ч при 90oC. Упаривание досуха в вакууме и сушка дали в результате 6.5 г соединения, указанного в заглавии, с температурой плавления 161 - 162oC, которое использовали без дальнейшей очистки. Образец кристаллизовали из толуола, и он имел ту же температуру плавления.
8-Карбокси-7-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение LXIX).
216 мл 0.3 М раствора перманганата калия в воде добавляли по каплям в течение 40 мин к смеси 7.94 г 8-формил-7-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана (полученного так, как это описано Da Re et al., J. Org. Chem. , 25, 1097, 1960) и 54 мл 5% дигидрофосфата натрия в 162 мл t-бутанола и встряхивали при 75oC. Спустя 2.5 ч встряхивания при этой же температуре реакционную смесь охладили до комнатной температуры и в нее медленно по каплям добавили 81 мл 1 М дитионита натрия. Смесь экстрагировали этилацетатом, органический слой промывали 4 раза 160 мл 0.5 н. гидроксида натрия; собранные водные слои промывали диэтиловым эфиром и подкисляли 37% соляной кислотой. Соединение, указанное в заглавии, выпадало в осадок. Его отфильтровывали и промывали водой, после сушки было получено 3.3 г соединения, которое использовали для последующих реакций без дальнейшей очистки. После кристаллизации из 95% этанола соединение плавилось при 180 - 181oC.
Этил 3-пропионил-2-(4-трифторметилбензоилокси)-бензоат (промежуточное соединение LXX).
Раствор 6.7 г 4-трифторметилбензоилхлорида (полученного из соответствующей бензойной кислоты и тионилхлорида в бензоле при дефлегмации и используемого без очистки) в 50 мл хлороформа было добавлено по каплям к раствору 7.13 г этил 2-гидрокси-3-пропионилбензоата и 4.9 мл триэтиламина в 50 мл хлороформа. Смесь встряхивали в течение 2 ч при комнатной температуре, растворитель выпаривали в вакууме, а остаток очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 85 : 15. При упаривании досуха в вакууме собранных фракций было получено 7.4 г соединения, указанного в заглавии, в виде масла.
ЯМР-спектры при 60 МГц (CDCl3, δ ):
7.6 - 8.5 (m, 6H, ароматические CHs);
7.5 (t, 1H, фенольное кольцо, CH в положении 5);
4.2 (q, 2H, COOCH2);
2.9 (q, 2H, COCH2);
1 - 1.3 (2t, 6H, 2 • CH3).
8-Этоксикарбонил-3-метил-4-оксо-2-(4-трифторметилфенил)-4H-1-бензопиран (промежуточное соединение LXXI).
Смесь 6.96 г промежуточного соединения LXX и 2.58 г t-бутоксида калия в 35 мл пиридина встряхивали в течение 2 ч при 100oC. Спустя это время реакционную смесь охлаждали до комнатной температуры, выливали в раствор 50 мл уксусной кислоты в 600 мл воды и экстрагировали этилацетатом. Органический слой промывали 10% соляной кислотой и водой, высушивали на безводном сульфате натрия и упаривали в вакууме досуха. Было получено 6.9 г 1-(2-гидрокси-3-этоксикарбонил)-2-метил-3-(4-трифторметилфенил)- 1,3-пропандиона. Раствор этого соединения и 2.2 мл 37% соляной кислоты в 35 мл ледяной уксусной кислоты встряхивали в течение 1.5 ч при 100oC. После охлаждения до комнатной температуры смесь выливали в 630 мл 1 н. гидроксида натрия и экстрагировали этилацетатом. Органический слой промывали водой, высушивали на безводном сульфате натрия и упаривали досуха в вакууме. Сырой продукт очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 85 : 15. При выпаривании в вакууме досуха было получено 2.95 г соединения, указанного в заглавии, плавящегося после кристаллизации из циклогексана при 111 - 113oC.
8-Карбокси-3-метил-4-оксо-2-(4-трифторметилфенил)-4H-1-бензопиран (промежуточное соединение LXXII).
Смесь 2.95 г промежуточного соединения LXXI и 0.43 г моногидрата гидроксида лития в 12.5 мл метанола и 12.5 мл тетрагидрофурана, содержащего 8 мл воды, встряхивали в течение 1.5 ч при комнатной температуре. Смесь выливали в раствор 30 мл 1 н. соляной кислоты в 300 мл воды и фильтровали с отсасыванием. Было получено 2.47 г соединения, указанного в заглавии, которое использовали без дальнейшей очистки. Образец, кристаллизованный из 60% этанола, плавился при 253 - 254oC.
8-Этоксикарбонил-2-(4-бензилфенил)-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение LXXIII).
Указанное соединение было синтезировано согласно методам получения промежуточного соединения LXX и LXXI, только в качестве исходного использовали 4-бензоилбензоил хлорид вместо 4-трифторметилбензоил хлорида и проводили реакцию в присутствии 4-диметиламинопиридина вместо триэтиламина. После обычной процедуры остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан : этилацетат 9 : 1. После упаривания досуха в вакууме собранных фракций было получено соединение, указанное в заглавии; его использовали без дальнейшей очистки. Образец, кристаллизованный из циклогексана, плавился при 125 - 136oC.
8-Карбокси-2-(4-бензоилфенил)-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение LXXIV).
Указанное соединение было получено так же, как и промежуточное соединение LXXII, только в качестве исходного использовали промежуточное соединение LXXIII вместо соединения LXXI. Соединение, указанное в заглавии, очищали растворением сырого продукта в 0.5 М гидроксиде натрия, промывкой водного слоя этилацетатом и осаждением чистого соединения при добавлении 37% соляной кислоты. Образец, кристаллизованный из уксусной кислоты, плавился при 260 - 262oC.
Этил 2-(4-феноксибензоилокси)-3-пропионил-бензоат (промежуточное соединение LXXV).
Соединение, указанное в заглавии, было получено методом, описанным для промежуточного соединения LXX, но вместо 4-трифторметилбензоилхлорида в качестве исходного использовали 4-феноксибензоилхлорид. Упаривая растворитель, получили чистый продукт, указанный в заглавии.
ЯМР-спектры при 200 МГц (CDCl3, δ ):
8.17 (dd, 3H, фенильный CHs в ортоположении относительно карбоксилатных групп);
7.92 (dd, 1H, фенильный CH в ортоположении относительно CO-группы);
7.38 - 7.48 (m, 3H, фенильный CHs в метаположении относительно карбоксилатных групп);
7.25 (d, 1H, CH в положении 4 феноксильного кольца);
7.05; 7.10 (2d, 4H, другие CHs феноксильного кольца);
4.25 (q, 2H, CH2O);
2.90 (q, 2H, CH2CO);
1.05 - 1.20 (m, 6H, 2 • CH3).
8-Этоксикарбонил-3-метил-4-оксо-2-(4-феноксифенил)-4H-1-бензопиран (промежуточное соединение LXXVI).
Соединение, указанное в заглавии, было получено так же, как соединение LXXI, только в качестве исходного вместо промежуточного соединения LXXV использовали соединение LXX. Очистку проводили колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 6 : 4. Выпаривание в вакууме дало в результате чистый продукт, указанный в заглавии, с температурой плавления 98 - 100oC.
8-Карбокси-3-метил-4-оксо-2-(4-феноксифенил)-4H-1-бензопиран (промежуточное соединение LXXVII).
Это соединение было получено таким же способом, что и промежуточное соединение LXXII, но в качестве исходного вместо промежуточного соединения LXXVI использовали соединение LXXI. Температура плавления 216 - 218oC.
8-Карбокси-2-(t-бутил)-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение LXXVIII).
6 мл пивалоилхлорида было добавлено по каплям в встряхиваемый раствор 8.9 г этил 2-гидрокси-3-пропионил-бензоата в 20 мл безводного пиридина. Реакционную смесь встряхивали в течение 6 ч при 80oC, охлаждали до комнатной температуры и выливали в смесь 200 г измельченного льда и 30 мл 10 н. соляной кислоты. После экстракции диэтиловым эфиром органическую фазу промывали соляным раствором, высушивали на безводном сульфате натрия и упаривали досуха в вакууме. В результате получили 11.4 г сырого этил 2-пивалоилокси-3-пропионил-бензоата.
2.4 г этого соединения растворяли в 4 мл безводного пиридина и добавили 1 г безводного t-бутоксида калия. Полученную смесь нагревали в течение 15 мин при 100oC, охлаждали до комнатной температуры и выливали в 50 г смеси воды со льдом, содержащей 8 мл 10 н. соляной кислоты. После экстракции диэтиловым эфиром органическую фазу промывали соляным раствором, высушивали на безводном сульфате натрия и упаривали досуха в вакууме. В результате было получено 2.1 г сырого этил 2-гидрокси-3-(2-пивалоилпропионил)-бензоата, который использовали на следующей стадии без дальнейшей очистки. 2 г этого соединения нагревали при 100oC в течение 15 мин после растворения в смеси, содержащей 15 мл уксусной кислоты и 1.5 мл 37% соляной кислоты. После охлаждения до комнатной температуры смесь выливали в 100 мл воды и экстрагировали диэтиловым эфиром. Органическую фазу промывали 5% водным кислым карбонатом натрия, а затем - водой, высушивали на безводном сульфате натрия и упаривали в вакууме. В результате получили 1.6 г сырого 8-этоксикарбонил-2-(t-бутил)-3-метил-4-оксо-4H-бензопирана.
1.5 г указанного выше эфира растворили в 20 мл этанола. Затем медленно, поддерживая температуру 25 - 35oC, добавили 3 мл 10 н. гидроксида натрия. После выдержки при комнатной температуре в течение 1.5 ч реакционную смесь разбавили 100 мл воды и экстрагировали этилацетатом. Водный слой подкисляли 3 н. соляной кислотой. Осадок собрали при отсасывании, промыли водой и кристаллизовали из этанола. Было получено 0.8 г соединения, указанного в заглавии, плавящегося при 225 - 228oC.
8-Карбокси-2-циклогексил-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение LXXIX).
Это соединение было получено согласно последовательности реакций и методам, описанным для промежуточного соединения LXXVIII, только в качестве исходного соединения использовали хлорид циклогексилкарбоновой кислоты, а не пивалоилхлорид; были также иные незначительные различия. Этил 2-циклогексилкарбонилокси-3-пропионил бензоат был получен после 8 ч встряхивания при комнатной температуре и перегруппирован в этил 2-гидрокси-3-(2-циклогексилкарбонилпропионил)бензоат нагреванием с t-бутоксидом калия в течение 2.5 ч при 100oC. При нагревании в смеси уксусной и соляной кислот при 100oC в течение 1.5 ч была проведена циклизация в 8-этоксикарбонил-2-циклогексил-3-метил-4-оксо-4H-1-бензопиран. Гидролиз соединения, указанного в заглавии, осуществляли при комнатной температуре в течение 20 мин. После кристаллизации из 40% этанола соединение, указанное в заглавии, плавилось при 224oC.
8-Этоксикарбонил-2-(2-фурил)-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение LXXX).
Смесь 3.2 г промежуточного соединения XC и 1.3 г безводного t-бутоксида калия в 8 мл безводного пиридина встряхивали в течение 15 мин при 60oC, охлаждали до комнатной температуры и выливали в 60 мл смеси воды со льдом, содержащей 15 мл 10 н. соляной кислоты. После экстракции этилацетатом органическую фазу промывали 5% водным бикарбонатом натрия и водой и высушивали на безводном сульфате натрия. При выпаривании в вакууме было получено 2.5 г сырого этил 3-(2-фуроил-пропионил)-2-гидрокси бензоата.
2.5 г полученного таким образом соединения встряхивали в течение 30 мин при 100oC с 10 мл уксусной кислоты и 0.7 мл 37% соляной кислоты. После охлаждения до комнатной температуры смесь выливали в 180 мл воды. Соединение, указанное в заглавии, осаждали, собирали при фильтровании с отсасыванием и промывали водой; кристаллизацию проводили из изопропанола. Выход 1.5 г, температура плавления 137 - 139oC.
8-Карбокси-2-(2-фурил)-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение LXXXI).
Смесь 3.5 г промежуточного соединения LXXX и 6 мл 10 н. гидроксида натрия в 40 мл метанола встряхивали при комнатной температуре в течение 1 ч и выливали в 500 мл воды. После экстракции этилацетатом водный слой подкисляли 3 н. соляной кислотой. Соединение, указанное в заглавии, осаждали, собирали при фильтровании с отсасыванием и промывали водой; кристаллизацию проводили из смеси метанол : хлороформ 7: 3. Выход 2.55 г, температура плавления 272 - 277oC.
8-Этоксикарбонил-3-метил-4-оксо-2-(2-тиенил)-4H-1-бензопиран (промежуточное соединения LXXXII).
Это соединение было получено в две стадии, согласно методам, изложенным для промежуточного соединения LXXX, но вместо промежуточного соединения XC использовали соединение XCI. Соединение, указанное в заглавии, после кристаллизации из изопропанола плавилось при 116 - 118oC.
8-Карбокси-3-метил-4-оксо-3-(2-тиенил)-4H-1-бензопиран (промежуточное соединение LXXXIII).
Это соединение было получено согласно методу, описанному для промежуточного соединения LXXXI, только промежуточное соединение LXXXII было использовано вместо соединения LXXX. После кристаллизации из смеси метанола и хлороформа 7 : 3 температура плавления этого соединения была 287 - 294oC.
8-Карбокси-4-оксо-2-фенил-4H-1-бензотиопиран (промежуточное соединение LXXXIV).
Смесь 1 г 8-метоксикарбонил-4-оксо-2-фенил-4H-1-бензотиопирана (промежуточное соединение XCII), 2.2 мл 12.5 н. гидроксида натрия, 15 мл метанола и 5 мл диоксана встряхивали в течение 2.5 ч при комнатной температуре. После выпаривания в вакууме добавили до полного растворения воды и этот раствор экстрагировали хлороформом. Выделенную водную фазу подкисляли разбавленной соляной кислотой до полного осаждения сырого продукта, который отфильтровывали и очищали кристаллизацией из уксусной кислоты. Выход 0.62 г, температура плавления 302oC.
(E)-8-Этоксикарбонил-3-метил-4-оксо-2-(2-стирил)-4H-1- бензопиран (промежуточное соединение LXXXV).
Это соединение было получено в три стадии согласно методам, описанным для промежуточного соединения XC (первая стадия) и промежуточного соединения LXXX (вторая и третья стадии). На первой стадии использовали (E)-циннамоилхлорид вместо 2-фуроилхлорида; полученный (E)-этил 2-гидрокси-3-(2-стирил-пропионил)-бензоат был использован для второй стадии без очистки. Соединение, указанное в заглавии, после кристаллизации из изопропанола плавилось при 129 - 130oC.
(E)-8-Карбокси-3-метил-4-оксо-2-стирил-4H-1-бензопиран (промежуточное соединение LXXXVI).
Это соединение получали согласно методу, описанному для промежуточного соединения LXXXI, только в качестве исходного использовали промежуточное соединение LXXXV, а не соединение LXXX, и реакция проходила при комнатной температуре в течение 10 ч. Соединение, указанное в заглавии, после кристаллизации из этанола плавилось при 284 - 286oC.
8-Карбокси-3-метил-2-(4-метилфенил)-4-оксо-4H-1-бензопиран (промежуточное соединение LXXXVII).
Смесь 1.9 г 2-гидрокси-3-пропионил бензойной кислоты (полученной согласно Brit, 1,343, 119, 1974), 5.2 г безводного 4-метилбензоата натрия и 3.9 мл 4-метилбензоилхлорида встряхивали в течение 8.5 ч при 185 - 195oC. После охлаждения до комнатной температуры твердеющую массу оставляли на ночь в 100 мл хлороформа. Затем смесь встряхивали с 5% водным раствором карбоната натрия до тех пор, пока pH водной фазы не стала 8.9. Органическую фазу вновь экстрагировали диэтиловым эфиром, а водные фазы сливали вместе, повторно экстрагировали диэтиловым эфиром и подкисляли 10 н. соляной кислотой. Осадок очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 100 : 2 - 100 : 20. Соединение, указанное в заглавии, было получено при выпаривании в вакууме объединенных фракций, которые содержат его. Температура плавления этого соединения после кристаллизации из этанола была 249 - 251oC.
8-Этоксикарбонил-2-(4-фторфенил)-3-метил-4-оксо-4Н-1-бензопиран (промежуточное соединение LXXXVIII)
Это соединение было получено в три стадии согласно методам, описанным для промежуточного соединения XC (первая стадия) и соединения LXXX (вторая и третья стадии). На первой стадии 4-фторбензоилхлорид использовали вместо 2-фуроилхлорида, а реакция продолжалась 20 ч при комнатной температуре, давая в результате этил 2-(4-фторбензоилокси)-3-пропионил бензоат. Это соединение без дальнейшей очистки было использовано для второй стадии. После промывания диэтиловым эфиром и кристаллизации из этанола соединение, указанное в заглавии, плавилось при 128 - 130oC.
8-Карбокси-2-(4-фторфенил)-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение LXXXIX).
Раствор 3.3 г промежуточного соединения LXXXVIII и 0.6 г гидрата гидроксида лития в 50 мл тетрагидрофурана, 10 мл метанола и 10 мл воды выдерживали при комнатной температуре в течение 5 ч, а затем выливали в 300 мл 1 н. соляной кислоты. Образовавшийся осадок собирали при отсасывании, промывали водой и высушивали. В результате было получено 2.3 г соединения, указанного в заглавии, которое после перекристаллизации из 95% этанола плавилось при 249 - 250oC.
Этил 2-(2-фуроилокси)-3-пропионил-бензоат (промежуточное соединение XC)
4.35 мл 2-фуроилхлорида было добавлено по каплям при 10 - 15oC к встряхиваемой смеси 8.9 г этил-гидрокси-3-пропионил-бензоата и 5.4 г 4-диметиламинопиридина в 25 мл дихорметана. После выдержки в течение 2 ч при комнатной температуре реакцию гасили 200 мл воды. Органический слой промывали 5% бикарбонатом натрия, высушивали на безводном сульфате натрия и выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 4 : 1. В результате было получено 9.4 г соединения, указанного в заглавии, в виде твердого низкоплавящегося вещества, которое без дальнейшей очистки использовали на следующей стадии.
ЯМР-спектры при 60 МГц (CDCl3, δ ):
8.2 (1H, dd, CH в положении 4 бензольного кольца);
8.0 (1H, dd, CH в положении 6 бензольного кольца);
7.7 - 7.8 (1H, dd, CH в положении 5 фуранового кольца);
7.43 (1H, t, CH в положении 5 бензольного кольца);
7.45 (1H, s, CH в положении 3 фуранового кольца);
6.6 - 6.8 (1H, m, CH в положении 4 фуранового кольца);
4.3 (2H, q, COOCH2CH3);
2.9 (2H, q, COCH2CH3);
0.95 - 1.35 (6H, m, 2 • CH3).
Этил 3-пропионил-2-(2-тиенилкарбонилокси)-бензоат (промежуточное соединение XCI).
Это соединение было получено согласно методу, описанному для промежуточного соединения XC, но с использованием хлорида 2-тиенилкарбоновой кислоты вместо 2-фуроилхлорида.
ЯМР-спектры при 60 МГц (CDCl3, δ ):
7.1 - 8.35 (6H, m, ароматический CHs).
4.25 (2H, q, COOCH2CH3);
2.9 (2H, q, COCH2CH3);
0.95 - 1.3 (6H, m, 2 • CH3).
8-Метоксикарбонил-4-оксо-2-фенил-4H-1-бензотиопиран (промежуточное соединение XCII).
Смесь 16.8 мл метилтиосалицилата, 25.6 мл этилбензоилацетата и 360 г полифосфорной кислоты встряхивали в течение 3 ч при 90oC. После охлаждения до комнатной температуры смесь выливали в измельченный лед, собирали сырой продукт при фильтровании, промывали его водой и очищали кристаллизацией из этанола. Температура плавления 170 - 171oC.
Элементный анализ для C17H12O3S:
Вычислено (%): C 68.90; H 4.08; S 10.82.
Найдено (%): C 68.59; H 4.13; S 10.69.
ЯМР-спектры при 200 МГц (CDCl3, δ ):
8.83 - 8.95 (dd, 1H, CH бензотиопирана в положении 5);
8.45 - 8.53 (dd, 1H, CH бензотиопирана в положении 7);
7.68 - 7.80 (m, 2H, 2-фенил CHs в положении 2 и 6);
7.55 - 7.65 (t, 1H, CH бензотиопирана в положении 6);
7.45 - 7.55 (m, 3, 2-фенил CHs в положении 3, 4 и 5);
7.24 (s, 1H, CH бензотиопирана в положении 3);
4.00 (s, 3H, COOH3).
8-Этоксикарбонил-3-бромметил-4-оксо-2-фенил-4Н-1-бензопиран (промежуточное соединение XCIII).
Смесь 9.2 г 8-этоксикарбонил-3-метил-4-оксо-2-фенил-4H-1- бензопирана (полученного согласно Da Re, P et al., J. Med. Pharm. Chem., 2, 263, 1960), 6,4 г N-бромсукцинимида и 0.04 г перекиси бензоила в 80 мл безводного четыреххлористого углерода встряхивали при дефлегмации в течение 1.5 ч. После охлаждения до комнатной температуры образовавшийся сукцинимид собирали при отсасывании и промывали холодным четыреххлористым углеродом. Маточные жидкости упаривали в вакууме досуха, а остаток промывали диэтиловым эфиром и собирали при фильтровании с отсасыванием. В результате было получено 9.2 г соединения, указанного в заглавии, которое после кристаллизации из смеси ацетон : н-гексан, плавилось при 133 - 134oC.
8-Этоксикарбонил-3-ацетоксиметил-4-оксо-2-фенил-4H-1- бензопиран (промежуточное соединение XCIV).
Раствор 10.2 г тригидрата ацетата натрия в 30 мл воды при комнатной температуре был прибавлен по каплям к раствору 29 г промежуточного соединения XCIII в 300 мл диметилформамида. После встряхивания в течение 1.5 ч при 50oC реакционную смесь выливали в 2 л воды. Соединение, указанное в заглавии, выпадало в осадок, его собирали при фильтровании с отсасыванием; после кристаллизации из ацетона было получено 20 г соединения с температурой плавления 151 - 152oC.
8-Карбокси-3-гидроксиметил-4-оксо-2-фенил-4H-1-бензопиран (промежуточное соединение XCV).
116 мл 1 н. гидроксида натрия было добавлено в течение 10 мин к встряхиваемой суспензии 14.8 г промежуточного соединения XCIV в 300 мл 95% этанола. Затем реакционную смесь нагревали в течение 15 мин при 60 - 65oC, полученный при этом прозрачный раствор выдерживали при комнатной температуре еще в течение 1 ч. После выпаривания в вакууме остаток растворяли в 200 мл воды, а раствор подкисляли при медленном добавлении 10 мл соляной кислоты (d = 1.18). После встряхивания в течение 1 ч при комнатной температуре соединение, указанное в заглавии, было собрано при фильтровании с отсасыванием, промыто водой и кристаллизовано из изопропанола. В результате было получено 9.3 г соединения с температурой плавления 237 - 240oC.
Этил 2-(4-Нитробензоилокси)-3-пропионил-бензоат (промежуточное соединение XCVI).
Соединение, указанное в заглавии, было получено согласно методике, описанной для промежуточного соединения XC, только вместо 2-фуроилхлорида использовали 4-нитробензоилхлорид. Был получен продукт в виде твердого низкоплавящегося вещества (температура плавления 78 - 80oC)
ЯМР-спектры при 60 МГц (CDCl3, δ ):
7.85 - 8.50 (m, 6H, ароматический CHs);
7.50 (t, 1H, CHs в положении 5 фенольного кольца);
4.25 (g, 2H, CH2O);
3.95 (g, 2H, CH2);
0.95-1.30 (m, 6H, CH3).
8-Этоксикарбонил-3-метил-2-(4-нитрофенил)-4-оксо-4H-1-бензопиран (промежуточное соединение XCVII).
Смесь 29,7 г промежуточного соединения XCVI и 10.18 г безводного t-бутоксида калия в 89 мл безводного пиридина встряхивали в течение 13 ч при 100oC. Реакционную смесь охлаждали до комнатной температуры, выливали в 400 мл 4 ню соляной кислоты и экстрагировали дихлорметаном. Органический слой повторно промывали водой, а затем 2.5% кислым карбонатом натрия, высушивали на безводном сульфате натрия и упаривали досуха в вакууме. Сырой продукт очищали колоночной хроматографией на силикагеле, элюируя смесью гексан : этилацетат 7:3. Выпаривание в вакууме собранных фракций дало 7 г соединения, указанного в заглавии, которое плавилось при 145 - 148oC.
8-Карбокси-3-метил-2-(4-нитрофенил)-4-оксо-4H-1-бензопиран (промежуточное соединение XCVIII).
Суспензия 0.38 г промежуточного соединения XCVII в 4.75 мл диоксана и 4.75 мл метанола встряхивали при 50oC. Было добавлено 1.29 мл 1 н. гидроксида натрия и при той же температуре в течение 3 ч продолжали встряхивание. Реакционную смесь охлаждали до комнатной температуры и добавляли 3 н. соляную кислоту до тех пор, пока pH не стал равным 1. При фильтровании полученной суспензии с отсасыванием было получено 0.13 г соединения, указанного в заглавии; после кристаллизации из диоксана оно плавилось при 320 - 321oC.
8-Этоксикарбонил-3-метил-4-оксо-2-трифторметил-4H-1-бензопиран (промежуточное соединение XCXIX).
3.16 мл 1,8-диазобицикло [5.4.0]ундец-7-ена было добавлено по каплям с помощью шприца при 0oC к встряхиваемой смеси 3 г этил 2-гидрокси-3-пропионил-бензоата и 5.53 мл трифторуксусного ангидрида. Реакционную смесь встряхивали при 60oC в течение 4 ч, после этого ее охладили до комнатной температуры и разбавили этилацетатом и водой. Органический слой промывали 1 н. гидроксидом натрия и водой, высушивали на безводном сульфате натрия и упаривали досуха в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 95:5; в результате было получено 0.8 г соединения, указанного в заглавии.
ЯМР-спектры при 200 МГц (CDCl3, δ ):
8.41; 8.37 (2dd; 2H, CHs в положении 5 и 7 бензопиранового кольца);
7.51 (t, 1H, CH в положении 6 бензопиранового кольца);
4.46 (q, 2H, COOCH2);
2.22 - 2.27 (m, 3H, JH-F = 2.16 Гц, CH3 в положении 3 бензопиранового кольца);
1.39 (t, 3H, CH2CH3).
8-Карбокси-3-метил-4-оксо-2-трифторметил-4H-1-бензопиран (промежуточное соединение C).
Соединение, указанное в заглавии, было получено таким же способом, что и промежуточное соединение LXII, только вместо соединения LXI использовали промежуточное соединение XCIX и после разбавления водой вместо фильтрации проводили экстракцию этилацетатом. После высушивания на безводном сульфате натрия и упаривания досуха в вакууме органического слоя было получено соединение, указанное в заглавии, в виде твердого остатка, который плавился при 175 - 178oC.
3-[4-(2-Метоксифенил)-1-пиперазинил] -N-метил-пропиламин (промежуточное соединение CI).
42 мл 35% водного метиламина было добавлено к раствору 8.2 г 3-[4-(2-метоксифенил)-1-пиперазинил] -пропил хлорида в 48 мл диметилформамида. Смесь нагревали в закрытом сосуде при 60oC в течение 5 ч, а затем охлаждали до 30oC. Растворитель выпаривали в вакууме, а остаток встряхивали в течение 30 мин со 100 мл диэтилового эфира. Твердые остатки, собранные при фильтрации с отсасыванием, растворили в 200 мл смеси хлороформ : 5 н. метанольный аммиак 100 : 5 - 100 : 15. Фракции, содержащие соединение, указанное в заглавии, были собраны вместе и выпарены в вакууме. В результате было получено 3 г промежуточного соединения CI в виде плотного масла.
ЯМР-спектры при 60 МГц (DMCO-d6, δ ):
6.80 (s, 4H, ароматический CHs);
3.75 (s, 3H, OCH3);
3.20 - 2.75 (m, 4H, CH3s пиперазина в положении 3, 5);
2.75 - 2.10 (m, 8H, CH2s пиперазина в положении 2, 6 и CH2CH2CH2);
2.40 (s, 1H, NH);
2.30 (s, 3H, NCH3);
1.80 - 1.40 (m, 2H, CH2CH2CH2).
Этил 2-бензоил-3-этилбензо[b] фуран-7-карбоксилат (промежуточное соединение CII).
Смесь 11.1 г этил 2-гидрокси-3-пропионил-бензоата, 9.9 г фенацилбромида, 6.9 г безводного карбоната калия и 200 мл ацетона встряхивали при температуре дефлегмации в течение 7 ч. После охлаждения до комнатной температуры неорганические соли отделяли фильтрованием, а раствор выпаривали в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя толуолом. Соединение, указанное в заглавии, полученное при выпаривании в вакууме собранных вместе фракций, содержащих его, было затем кристаллизовано из 90% этанола. Выход 9.8 г, температура плавления 64 - 66oC.
7-Карбокси-2-бензоил-3-этил-бензо[b]фуран (промежуточное соединение CIII).
Смесь 7 г промежуточного соединения CII, 275 мл 0.95 н. гидроксида натрия и 400 мл диоксана встряхивали в течение 4 ч при температуре дефлегмации. После охлаждения до комнатной температуры диоксин выпаривали в вакууме и замещали таким же объемом воды. После фильтрования с использованием угля раствор подкисляли разбавленной соляной кислотой, осадок отфильтровывали и очищали кристаллизацией из ацетона. Выход 4.94 г, температура плавления 193 - 195oC.
8-Метоксикарбонил-3-метил-2-(4-метилфенил)-4-оксо-4H-1-бензопиран (промежуточное соединение CIV).
Это соединение было получено в три стадии согласно методам, описанным для промежуточного соединения XC (первая стадия) и промежуточного соединения LXXX (вторая и третья стадии). На первой стадии 4-метилбензоилхлорид был использован вместо 2-фуроилхлорида, а метил 2-гидрокси-3-пропионил-бензоат использовали вместо этил 2- гидрокси-3-пропионил-бензоата. Реакция протекала при комнатной температуре в течение 4 ч. В результате был получен 2-(4-метилбензоилокси)-3-пропионил-бензоат. Это соединение использовали для второй стадии, которая длилась 175 ч при 100oC, без дальнейшей очистки. На третьей стадии вместо 37% соляной кислоты использовали 96% серную кислоту. Соединение, указанное в заглавии, плавилось при 174 - 175oC после кристаллизации из этанола.
8-Этокси-карбонил-2-(4-бифенилил)-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение CV).
Это соединение было получено в три стадии согласно методам, описанным для промежуточного соединения XC (первая стадия) и промежуточного соединения CIV (вторая и третья стадии). На первом стадии 4-фенилбензоилхлорид использовали вместо 2-фуроилхлорида, а реакция продолжалась 20 ч при комнатной температуре и 13 ч при дефлегмации. Очистку проводили колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 100 : 5 - 100 : 10, в результате был получен 2-(4-бифенилил)-3-пропионил-бензоат. Соединение, указанное в заглавии, плавилось при 165 - 167oC после промывания 95% этанолом.
8-Карбокси-2-(4-бифенилил)-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение CVI).
Смесь 4.3 г промежуточного соединения CV и 35 мл 35% соляной кислоты в 50 мл 1,4-диоксана и 15 мл воды встряхивали при дефлегмации в течение 16 ч. После охлаждения смесь выливали в 200 мл воды и экстрагировали этилацетатом. Органический слой отделяли и экстрагировали 20% водным карбонатом натрия. Осадок, образовавшийся после подкисления водного слоя разбавленной соляной кислотой, собрали при отсасывании, промыли водой и высушили. В результате было получено 2.5 г соединения, указанного в заглавии, которое плавилось при 242.5 - 244oC.
8-Карбокси-2-(4-гидроксифенил)-3-метил-4-оксо-4H-1-бензопиран (промежуточное соединение CVII).
Смесь 3 г 8-этоксикарбонил-2-(4-метоксифенил)-3-метил-4-оксо-4H-1-бензопирана (полученного согласно JP 58-225083) и 60 мл 48% бромистоводородной кислоты и 80 мл уксусной кислоты встряхивали при дефлегмации в течение 8 ч. После охлаждения смесь выливали в 500 мл воды, а осадок собирали при отсасывании и промывали водой. Сырой продукт очищали тонкослойной хроматографией, элюируя вначале смесью хлороформ : изопропанол 9 : 1 - 7 : 3, а затем метанолом. В результате был получен 1 г соединения, указанного в заглавии, которое плавилось при 300oC.
4-[4-(2-Метоксифенил)-1-пиперазинил] -N-метил-бутиламин (промежуточное соединение CVIII).
Раствор 3.8 мл трифторуксусного ангидрида в 25 мл безводного дихлорметана добавляли по каплям при встряхивании при 0oC к раствору 2.53 г 4-[4-(2-метоксифенил)-1-пиперазинил] -бутиламина в 25 мл безводного дихлорметана. После встряхивания в течение 2 ч при комнатной температуре реакционную смесь разбавили дихлорметаном и промыли водой. Органический слой высушивали на безводном сульфате натрия и упаривали досуха в вакууме; в результате было получено 3,3 г чистого 4-[4-(2-метоксифенил)-1-пиперазинил]-N-трифторацетил-бутиламина.
ЯМР-спектры при 60 МГц (CDCl3, δ ):
7.70 - 8.00 (bs, 1H, NH);
6.80 - 7.20 (m, 4H, ароматический CHs);
3.85 (s, 3H, CH3O);
2.90 - 3.80 (m, 12H, CH2s пиперазина, CH2N и CH2NHCO);
1.50 -2.05 (m, 4H, C-CH2CH2-C).
К раствору, содержащему 3.3 г указанного выше промежуточного соединения в 46 мл безводного диметилформамила при встряхивании при 0oC порциями добавляли 0,88 г 50% гидрида натрия. После встряхивания в течение 1 ч при указанной температуре было добавлено 0,57 мл метилиодида. Реакционную смесь встряхивали еще 1.5 ч при 0oC, а затем вылили в воду и экстрагировали этилацетатом. Органический слой промывали водой, высушивали на безводном сульфате натрия и упаривали досуха в вакууме. В результате было получено 1.13 г сырого 4-[4-(2-метоксифенил)-1-пиперазинил] -N-трифторацетил-N-метил-бутиламина, который использовали в следующих стадиях без дальнейшей очистки. 0.18 г борогидрида натрия добавили к раствору 1.13 г указанного выше промежуточного соединения в 30 мл этанола, и полученную смесь встряхивали при 60oC в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь выливали в воду и экстрагировали дихлорметаном. Органический слой промывали водой, высушивали на безводном сульфате натрия и упаривали в вакууме досуха. В результате было получено 0.82 г чистого соединения, указанного в заглавии.
ЯМР-спектры при 60 МГц (CDCl3, δ ):
6.80 - 7.20 (m, 4H, ароматический CHs);
3.85 (s, 3H, CH3O);
2.90 - 3.20 (m, 4H, CH2s пиперазина положения 3 и 5);
2.30 - 2.80 (m, 8H, CH2s пиперазина положения 2 и 6; 2 CH2N);
2.50 (s, 3H, CH3N);
1.80 (s, 1H, NH);
1.40 - 1.80 (m, 4H, C-CH2-CH2-C).
1-(3-Амино-2,2-диметилпропил)-4-(2-метоксифенил)-пиперазин.
Соединение, указанное в заглавии, может быть получено при обработке 1-(2-метоксифенил)-пиперазина изобутиральдегидом, 37% формальдегидом в воде и уксусной кислотой при 90 - 150oC или теми же реагентами в этанольном хлористом водороде (реакция Манниха). Полученное соединение - 1-(2-формил-2-метилпропил)-4-(2-метоксифенил)-пиперазин аминируют обработкой избытком аммиака в условиях восстановления. Последние может давать водород или катализатор (например, PD/C, никель Ренея или диоксид платины) в растворителе (например, этаноле, метаноле, изопропаноле, дихлорметане, хлороформе или диметилформамиде) при температуре от комнатной до 80oC или же гидрид металла (например, борогидрид натрия, цианоборогидрид натрия или калия или триацетоксиборогидрид натрия) в растворителе (например, метаноле, этаноле, хлороформе, бензоле или 1,2-дихлорэтане) в присутствии кислоты (например, газообразного хлористого водорода или уксусной).
Это соединение может реагировать с 8-хлоркарбонил-3-метил-4-оксо-2-фенил-4H-1-бензопираном так, как это описано ниже в примере 12, давая 8-(2,2-диметил-3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил)- 3-метил-4-оксо-2-фенил-4H-1-бензопиран.
8-Трифторацетамидометил-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Это соединение было получено согласно методике, описанной для промежуточного соединения XXIII, но только при этом использовали промежуточное соединение XXIV вместо 8-амино-3-метил-4-оксо-2-фенил-4H-1-бензопирана. Его можно использовать в качестве исходного вместо промежуточного соединения XXIII в реакции, описанной в примере 32, для получения 8-(2-[4-(2-метоксифенил)-1-пиперазинил]-этиламинометил)-3-метил-4- оксо-2-фенил-4H-1-бензопирана.
8-(2-Хлорэтилуреидометил)-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Это промежуточное соединение может быть получено с помощью метода, описанного для получения промежуточного соединения XLIV, только при использовании промежуточного соединения XXIV вместо 8-амино-3-метил-4-оксо-2-фенил-4H-1-бензопирана. Оно может взаимодействовать с соединением, имеющим формулу H-B, согласно пути реакции (a), для того чтобы получить конечные продукты.
8-Этинилсульфониламинометил-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Это соединение можно получить при взаимодействии промежуточного соединения XXIV с 2-хлорэтилсульфонилхлоридом в галогенированном растворителе типа дихлорметана в присутствии триэтиламина при 0 - 40oC, согласно работе A. A.Goldberg, J.Chem.Soc., 464, 1945. Это соединение может взаимодействовать с подходящим соединением H-B, согласно пути реакции (m), для того чтобы получить соответствующие конечные соединения.
Поступая так, как это было описано выше, но используя в качестве исходного 8-амино-3-метил-4-оксо-2-фенил-4H-1-бензопиран, можно получить конечные соединения F1'-Y36-(CH2)2-B.
8-Хлорсульфонилметил-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Это промежуточное соединение может быть получено взаимодействием 8-амидинотиометил-3-метил-4-2-фенил-4H-1-бензопирана (синтез которого описан в получении промежуточного соединения XXI) с хлором в воде при температуре от -10 до +10oC согласно работе. T.B. Johnson et al., J. Chem.Soc., 61, 2548, 1939. Взаимодействием этого промежуточного соединения с соответствующими соединениями формулы A-NH-Z-B, следуя пути реакции n, могут быть получены конечные целевые продукты.
Пример 1. 8-{ 2-[4-(2-Метоксифенил)-1-пиперазинил] -1-оксоэтил}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Раствор 11.5 г 1-(2-метоксифенил)-пиперазина в 30 мл метанола было добавлено по каплям при 20 - 25oC к встряхиваемой смеси, содержащей 21.4 г промежуточного соединения VI и 4.1 г карбоната калия в 120 мл метанола. После встряхивания при указанной температуре в течение 4 ч реакционную смесь упаривали в вакууме. Остаток экстрагировали хлороформом, а органический раствор промывали водой, высушивали на безводном сульфате натрия/калия, фильтровали и упаривали в вакууме. Полученный сырой продукт растворили в ацетоне и добавили небольшой избыток этанольного хлористого водорода. После фильтрования с отсасыванием и рекристаллизации из 95% этанола было получено 16.3 г соединения, указанного в заглавии, с температурой плавления 195 - 199oC.
Пример 2. 8-{2-[4-(2-Метилфенил)-1-пиперазинил]-1-оксоэтил}-3- метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Это соединение было получено согласно примеру 1, только используя 1-(2-метилфенил)-пиперазин вместо 1-(2-метоксифенил)-пиперазина, и реакцию проводили в течение 1 ч в диметилформамиде, а не в течение 4 ч в метаноле. Температура плавления 203 - 206oC (изопропанол)
Пример 3. 8-{2-[4-(2-Этоксифенил)-1-пиперазинил]-1-оксоэтил}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Это соединение было получено согласно примеру 1, но вместо 1-(2-метоксифенил)-пиперазина использовали 1-(2-этоксифенил)- пиперазин и реакцию проводили в течение 2 ч в диметилформамиде, а не в течение 4 ч в метаноле. Температура плавления 208 - 210oC (изопропанол).
Пример 4. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-1-оксопропил}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Раствор 10 мл 37% формальдегида в 15 мл метанола был добавлен по каплям при 0oC в течение 3 мин к раствору 5.75 г 1-(2-метоксифенил)-пиперазина в 10 мл метанола. После выдержки в течение 12 ч при 0oC смесь упаривали в вакууме и повторно растворяли в 15 мл метанола. 20 мл 3.6 н. хлористого водорода и диэтиловом эфире добавили при 0oC. После встряхивания в вакууме остаток суспендировали в 15 мл 1,4-диоксана. В условиях встряхивания при 20 - 25oC был добавлен раствор 8.3 г промежуточного соединения V в 100 мл 1,4-диоксана. После встряхивания в течение 8 ч при дефлегмации реакционную смесь охладили до 30 - 40oC. Затем было добавлено в смесь 50 мл метанола и смесь подвергали дефлегмации еще 2 ч. После охлаждения до 20 - 25oC полученный раствор был разбавлен 300 мл диэтилового эфира. При указанной температуре встряхивание продолжали еще 3 ч. Соединение, указанное в заглавии, было получено после фильтрования с отсасыванием и рекристаллизации из этанола. Выход 4 г, температура плавления 209 - 210oC.
Пример 5. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропоксикарбонил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид.
Смесь 4.24 г 8-карбокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана и 6.3 г безводного карбоната калия в 60 мл диметилформамида встряхивали в течение 30 мин при 80oC. Потом было добавлено 5.23 г 1-(3-хлорпропил)-4-(2-метоксифенил)-пиперазина и встряхивание продолжали при 80oC в течение 3.5 ч. Реакционную смесь охлаждали до комнатной температуры, выливали в смесь воды со льдом и экстрагировали этилацетатом. Органические экстракты промывали водным раствором хлорида натрия, высушивали на безводном сульфате натрия и упаривали в вакууме досуха. Остаток переносили в этанол и к раствору добавили избыток этанольного хлористого водорода. Выход соединения, указанного в заглавии, составил 8.16 г, температура плавления 198 - 203oC.
Пример 6. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]-этоксикарбонил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид.
Соединение, указанное в заглавии, было получено способом, описанным в примере 5, только вместо 1-(3-хлорпропил)-4-(2- метоксифенил)-пиперазина использовали 1-(2-хлорэтил)-4-(2- метоксифенил)-пиперазин. Температура плавления составила 200 - 203oC (этанол).
Пример 7. 8-{3-[4-(2-Хлорфенил-1-пиперазинил]-пропоксикарбонил}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид.
Смесь 2.8 г 1-(2-хлорфенил)-пиперазин гидрохлорида и 4.2 г безводного карбоната калия в 25 мл диметилформамида встряхивали при комнатной температуре в течение 15 мин. Было добавлено 4.81 г промежуточного соединения 1 и встряхивание продолжили в течение 2 дней. Затем реакционную смесь выливали в 200 мл холодной воды и экстрагировали диэтиловым эфиром и этилацетатом. Органические экстракты промывали по очереди водным раствором хлорида натрия, 0.1 н. уксусной кислотой, водным раствором хлорида натрия, водным 4% раствором карбоната натрия и водой, а затем сушили на безводном сульфате натрия. После упаривания досуха в вакууме остаток растворили в 160 мл ацетонитрила и добавили избыток хлористого водорода в диэтиловом эфире. Нерастворимое соединение, указанное в заглавии, было рекристаллизовано из ацетонитрила. Выход 3.6 г, температура плавления 138 - 143oC
Пример 8. 8-[3-(4-Фенил-1-пиперазинил)-пропоксикарбонил] -3- метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид.
Соединение, указанное в заглавии, было получено методом, описанным в примере 7, но вместо 1-(2-хлорфенил)-пиперазин гидрохлорида использовали 1-фенил-пиперазин. Рекристаллизацию проводили из метанола, температура плавления 229 - 231oC.
Пример 9. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропоксикарбонил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид.
Соединение, указанное в заглавии, было получено согласно методу, описанному в примере 7, только вместо 1-(2-хлорфенил)-пиперазин гидрохлорида использовали 1-(2-метоксифенил)-пиперазин гидрохлорид. Этот путь является альтернативным для продукта из примера 5.
Пример 10. 8-{ 3-[4-(2-Метоксифенил)-1-пиперазинил]-2-метил-2- пропоксикарбонил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид.
5.29 г промежуточного соединения XXVIII в 25 мл 1,2-дихлорэтана при 60oC по каплям добавили к раствору 6 г 8-хлоркарбонил-3-метил-4-оксо-2-фенил-4H-1-бензопирана в 22 мл 1,2-дихлорэтана. Реакционная смесь подвергалась дефлегмации в течение 16 ч, затем ее охлаждали до комнатной температуры и выливали в холодный 0.5 н. раствор гидроксида натрия. Затем добавляли воду и дихлорметан. Органическую фазу отделяли, промывали водным раствором хлорида натрия и высушивали на безводном сульфате натрия. Растворители выпаривали, а маслянистый остаток очистили тонкослойной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 85 : 15. Собранные фракции упаривали в вакууме досуха, остаток растворили в этаноле. Для получения соединения, указанного в заглавии, был добавлен избыток этанольного хлористого водорода. В результате получили 6.71 г соединения, указанного в заглавии, с температурой плавления 203 - 204oC.
Пример 11. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил-4-оксо -2-фенил-4H-1-бензопиран дигидрохлорид.
Смесь 6.28 г 1-(2-метоксифенил)-пиперазина и 5.34 г промежуточного соединения XXXVII нагревали при 180oC в течение 5 ч. После охлаждения массу темного цвета очищали тонкослойной хроматографией на силикагеле, элюируя смесью дихлорметан : метанол 100 : 3. Фракции, содержащие соединение, указанное в заглавии, были собраны вместе, растворители выпарили в вакууме, а остаток растворили в кипящем этаноле. Раствор отфильтровали, подкислили этанольным хлористым водородом и оставили на ночь при 20 - 25oC. Сырой продукт был собран при фильтровании и кристаллизован из этанола; было получено в результате 5 г соединения, указанного в заглавии, имеющего температуру плавления 182 - 186oC.
Пример 12. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил-4- оксо-2-фенил-4H-1-бензопиран дигидрохлорид полугидрат.
Раствор 4.48 г 8-хлоркарбонил-3-метил-4-оксо-2-фенил-4H-1- бензопирана в 40 мл хлороформа добавили каплями в течение 10 мин при комнатной температуре к раствору 3.74 г 3-[4-(2-метоксифенил)-1-пиперазинил]-пропиламина, полученного согласно GB 2161807, и 1.97 г триэтиламина в 50 мл хлороформа. После встряхивания в течение 2 ч раствор промывали сначала 0.5 н. соляной кислотой, затем насыщенным водным раствором бикарбоната натрия и в заключение - водой. Хлороформовый раствор высушивали на безводном сульфате натрия, а растворитель выпаривали в вакууме. Остаток обрабатывали так, как это описано в примере 11; в результате было получено 6.67 г соединения, указанного в заглавии, с температурой плавления 182 - 186oC. Этот путь является альтернативным путем получения продукта из примера 11.
Были получены также следующие соли: гидрат монохлорида, температура плавления 151 - 154oC, монометансульфонат, температура плавления 162 - 164oC и (±)-гидрат полумалата, температура плавления 110 - 112oC.
Этот пример описывает конденсацию амина, 3-[4-(2-метоксифенил)-1-пиперазинил] -пропиламина с карбонилхлоридом и с 3-метил-4-оксо-2-фенил-4H-1-бензопиран-8-карбонилхлоридом. Необходимо отметить, что амин можно конденсировать с соответствующей свободной кислотой или с соответствующим этиловым эфиром при нагревании их эквимолярных количеств с растворителем или без него. При использовании растворителя подходит гидрофильный или гидрофобный растворитель с высокой температурой кипения. Амин можно синтезировать также при комнатной температуре с эквимолярным количеством соответствующей свободной кислоты в присутствии N,N'-дициклогексилкарбодиимида и 4-диметиламинопиридина в растворителе типа дихлорметана, хлороформа, тетрагидрофурана или диметилформамида.
Пример 13. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]-этилкарбамоил}-3-метил-4- оксо-2-фенил-4H-1-бензопиран моногидрохлорид полугидрат.
Соединение, указанное в заглавии, было получено методом, описанным в примере 16, только использовали промежуточное соединение XIV вместо промежуточного соединения XV и проводили нагревание при 55 - 60oC в течение 32 ч. Кроме этого, были следующие изменения. После сбора основания фильтрованием осуществляли его очистку тонкослойной хроматографией на силикагеле, сначала элюируя смесью хлороформ:метанол 100:0,5, а затем 100:1. Фракции, содержащие соединение, указанное в заглавии, были собраны вместе, а растворители удалены в вакууме. Остаток кристаллизовали их этанола. После фильтрования твердый остаток переносили в кипящую воду и, чтобы растворить его, добавляли достаточное количество разбавленной соляной кислоты. Кристаллическую соль отделяли при охлаждении и собирали при фильтровании с отсасыванием. Температура плавления 119 - 123oC.
Пример 14. 8-{3-[2-(2-Метоксифенокси)-этиламино]-пропилкарбамоил}-3-метил-4- оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Соединение, указанное в заглавии, было получено так, как это описано в примере 1, но вместо 1-(2-метоксифенил)-пиперазина использовали 2-(2-метоксифенокси)-этиламин (полученный согласно работе Augstein, J. et. al., J. Med. Chem. , 8,356, 1965), а вместо нагревания в течение 5 ч - нагревание в течение 2 ч. В качестве элюата использовали дихлорметан : метанол 100 : 5. Температура плавления 200 - 202oC (этанол).
Пример 15. 8-[3-(4-Фенил-1-пиперазинил)-пропилкарбамоил]-3-метил-4-оксо-2-фенил-4H-1-бензопиран монохлорид полугидрат.
Способом, описанным в примере 11, было получено соединение, указанное в заглавии; только вместо 1-(2-метоксифенил)-пиперазина использовали 1-фенилпиперазин; а вместо нагревания в течение 5 ч - нагревание в течение 2 ч. В качестве элюата применяли смесь дихлорметан : метанол 100 : 4. Температура плавления (с разложением) 255 - 258oC (87% этанол).
Пример 16. 8-{N-Метил-2-[4-(2-метоксифенил)-1-пиперазинил]-этилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран моногидрохлорид.
Смесь 3.56 г промежуточного соединения XV, 2,35 г 1-(2-метоксифенил)-пиперазина, 2.76 г безводного карбоната калия и 1.66 г иодида калия в 25 мл диметилформамида встряхивали в течение 6 ч при 100oC. После охлаждения растворитель удаляли в вакууме, а остаток переносили в 50 мл воды, встряхивали при комнатной температуре в течение 1 ч и собирали при фильтровании. Затем промывали водой и кристаллизовали из 95% этанола в присутствии малых количеств активированного угля (для обесцвечивания). Основание растворяли в 105 мл кипящей 0.086 н. соляной кислоты. После охлаждения закристаллизованную соль собирали при фильтровании; в результате было получено 4.3 г соединения, указанного в заглавии (температура плавления 201 - 203oC).
Пример 17. 8-{1-Гидрокси-2-[4-(2-метоксифенил)-1-пиперазинил]-этил}-3-метил-4- оксо-2-фенил-4H-1-бензопиран гидрохлорид.
1.36 г борогидрида натрия добавляли порциями к раствору 15.5 г соединения, полученного в примере 1, в 1500 мл метанола при 0 - 5oC. После встряхивания при температуре 0 - 5oC в течение 90 мин для незначительного подкисления реакционной смеси добавили 3 н.соляную кислоту; затем смесь выпаривали в вакууме. Остаток встряхивали с 2 н. водным раствором гидроксида натрия и экстрагировали хлороформом. Органический слой высушивали на безводном сульфате натрия/хлориде кальция, фильтровали, подкисляли этанольным хлористым водородом и выпаривали в вакууме. После промывания диэтиловым эфиром сырой продукт кристаллизовали из этанола; было получено 9.5 г соединения, указанного в заглавии, с температурой плавления 248 - 249 oC.
Пример 18. 8-{1-Гидрокси-2-[4-(2-метилфенил)-1-пиперазинил]-этил}-3-метил-4-оксо-2- фенил-4H-1-бензопиран гидрохлорид.
Это соединение было получено согласно примеру 17, только в качестве исходного лучше использовать соединение, полученное в примере 2, чем соединение, полученное в примере 1. Температура плавления 257 - 258oC (этанол).
Пример 19. 8-{1-Гидрокси-2-[4-(2-этоксифенил)-1-пиперазинил]-этил}-3-метил-4-оксо-2- фенил-4H-1-бензопиран гидрохлорид.
Это соединение было получено согласно примеру 17, только лучше в качестве исходного использовать соединение, полученное в примере 3, чем соединение, полученное в примере 1. Температура плавления 241 - 242oC (метанол).
Пример 20. 8-{1-Гидрокси-3-[4-(2-метоксифенил)-1-пиперазинил]-пропил}-3-метил-4- оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Это соединение было получено согласно примеру 17, только лучше в качестве исходного использовать соединение, полученное в примере 4, чем соединение, полученное в примере 1. Сырое основание очищали тонкослойной хроматографией (силикагель, элюат - смесь этилацетат : хлороформ 4 : 1). Фракции, содержащие чистое основание, были собраны вместе, подкислены этанольным хлористым водородом и выпарены в вакууме. Остаток кристаллизовали из этанола. Температура плавления 156 - 160oC.
Пример 21. 8-{1-Гидрокси-4-[4-(2-метоксифенил)-1-пиперазинил]-бутил} -3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид моногидрат.
Раствор 3.04 г промежуточного соединения XXXVIII и 2.45 г 1-(2-метоксифенил)-пиперазина в 21 мл безводного диметилформамида встряхивали при комнатной температуре в течение 5 ч. Далее добавили 1.22 г 1-(2-метоксифенил)-пиперазина и смесь встряхивали еще 4 ч, выливали в 300 мл воды и экстрагировали этилацетатом. Слитые вместе органические экстракты промывали водным раствором бикарбоната натрия, затем водным раствором хлорида натрия и упаривали досуха в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 95 : 5. Собранные фракции встряхивали в роторном испарителе, а остаток растворили в 0.81 М этанольном хлористом водороде и вновь встряхивали в вакууме. Твердый остаток кристаллизовали из смеси вода : этанол 9 : 1, с тем чтобы получить 2.43 г соединения, указанного в заглавии, с температурой плавления 144 - 146oC.
Пример 22. 8-{1-Этокси-2-[4-(2-метоксифенил)-1-пиперазинил]-этил}-3-метил-4-оксо-2- фенил-4H-1-бензопиран гидрохлорид. \
6 мл безводного диметилсульфоксида добавили к 6.55 г гидрида натрия (50% в минеральном масле, неоднократно промываемого гексаном) под азотом. Раствор 3 г соединения, полученного в примере 17, в 50 мл безводного диметилсульфоксида при 20-25oC был добавлен к этой смеси. После встряхивания при 20oC в течение 1 ч было добавлено 0,66 г этилбромида. Реакционную смесь встряхивали при указанной выше температуре еще 20 мин, а потом вылили в смесь воды со льдом. Сырой продукт, полученный после фильтрования с отсасыванием, очищали тонкослойной хроматографией (силикагель, элюат - смесь хлороформ : этилацетат 8 : 2). Фракции, содержащие чистый продукт, указанный в заглавии, собрали вместе, подкисляли этанольным хлористым водородом и встряхивали в вакууме. Остаток кристаллизовали из смеси хлороформ : диэтиловый эфир и высушивали в вакууме. В результате было получено 1.6 г соединения, указанного в заглавии, с температурой плавления 209oC.
Пример 23. 8-{N-Метил-2-[4-(2-метоксифенил)-1-пиперазинил]-этиламинометил}-3- метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид полугидрат.
Смесь 5.2 г промежуточного соединения XXVII, 3.1 г 1-(2-метоксифенил)-пиперазина и 2.2 г безводного карбоната калия в 50 мл диметилформамида встряхивали в течение 7 ч при 70oC. После охлаждения до 20 - 25oC реакционную смесь вылили в 500 мл воды и экстрагировали дихлорметаном. Органическую фазу промывали водой и высушивали на безводном сульфате натрия. Растворитель удаляли в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат: петролейный эфир 98:2. Соединение, указанное в заглавии, было получено солеобразованием при помощи этанольного хлористого водорода. Его температура плавления 217-219oC.
Пример 24. 8-{N-Ацетил-2-[4-(2-метоксифенил)-1-пиперазинил]-этиламинометил}-3 -метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Смесь промежуточного соединения XXXIII и 5.3 г 1-(2-метоксифенил)-пиперазина в 75 мл диметилформамида встряхивали в течение 2 ч при 95oC. После охлаждения до 20 - 25oC реакционную смесь выливали в воду и высушивали на безводном сульфате натрия. Растворитель удаляли в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью дихлорметан : метанол 100 : 0.2. После образования соли с помощью этанольного хлористого водорода и рекристаллизации из метанола было получено 4.4 г соединения, указанного в заглавии. Это соединение плавилось при 227-228oC и содержало 1 эквивалент метанола.
Пример 25. 8-[4-2-Метоксифенил)-1-пиперазинилацетамидометил]-3-метил-4-оксо-2 -фенил-4H-1-бензопиран гидрохлорид.
Смесь 3.42 г промежуточного соединения XXXII, 2.74 г 1-(2-метоксифенил)-пиперазина и 0.71 г безводного карбоната калия в 34 мл безводного диметилформамида встряхивали в течение 2 ч при 0oC. Реакционную смесь выливали в воду и отфильтровывали с отсасыванием. Полученный твердый остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесь этилацетат : петролейный эфир 6 : 4. Собранные фракции упаривали в вакууме досуха, а остаток кристаллизовали из метилэтилкетона. Полученное основание обрабатывали в этанольном растворе молярным эквивалентом водной 2.25 н. соляной кислоты для получения соединения, указанного в заглавии; температура плавления 168-170oC.
Пример 26. 8-{N-Метил-N-[4-(2-метоксифенил)-1-пиперазинил]- ацетамидометил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид гидрат.
Смесь 5 г промежуточного соединения XXXI, 2.9 г 1-(2-метоксифенил)-пиперазина и 2 г безводного карбоната калия в 50 мл диметилформамида встряхивали в течение 3 ч при 20 - 25oC. Затем реакционную смесь вылили в 500 мл воды и экстрагировали дихлорметаном. Органическую фазу промыли водой и высушили на безводном сульфате натрия. Растворитель удалили в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 6 : 4, и кристаллизовали из ацетона. Было получено 3.6 г основания соединения, указанного в заглавии, которое плавилось при 144 - 145oC. Основание растворили в этаноле, добавив 8 н. соляную кислоту и воду, в результате после высушивания в вакууме при 100oC было получено соединение, указанное в заглавии, с температурой плавления 218 - 220oC.
Пример 27. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]-этоксиметил}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид.
Смесь 4 г промежуточного соединения XVIII, 2.4 г 1-(2-метоксифенил)-пиперазина, 1.96 г иодида калия и 1.65 г безводного карбоната калия в 40 мл безводного диметилформамида встряхивали 7 ч при 90oC. После охлаждения до комнатной температуры смесь вылили в воду и экстрагировали дихлорметаном. Объединенные экстракты промыли водным раствором хлорида натрия, высушили на безводном сульфате натрия и упаривали в вакууме досуха. Остаток кристаллизовали из этилацетата, а собранные кристаллы растворили в этаноле и обработали избытком этанольного хлористого водорода. В результате было получено 5.21 г соединения, указанного в заглавии, с температурой плавления 199 - 201oC.
Пример 28. 8-{2-[2-(2-Этоксифенокси)-этиламино]-этоксиметил}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Соединение, указанное в заглавии, было получено согласно методике, описанной в примере 27, только вместо 1-(2-метоксифенил)-пиперазина использовали 2-(2-этоксифенокси)-этиламин и провели стадию очистки методом тонкослойной хроматографии на силикагеле, элюируя смесью этилацетат : метанол 97 : 3. В результате было получено 4.25 г соединения, указанного в заглавии. Температура плавления 191 - 193oC.
Пример 29. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]-этилтиометил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
2.5 г карбоната калия, 2.13 г иодида калия и 3.15 г 1-(2-метоксифенил)-пиперазина добавили к раствору 5 г промежуточного соединения XXI в 50 мл диметилформамида, полученную смесь встряхивали при 90oC в течение 4.5 ч. После охлаждения до комнатной температуры реакционную смесь вылили в 450 мл воды и экстрагировали этилацетатом. Органически экстракты промыли водой, высушили на безводном сульфате натрия и упаривали досуха в вакууме. Остаток растворили в ацетоне, а полученный раствор обработали молярным эквивалентом 3.8 н. хлористого водорода в диэтиловом эфире. Затем отфильтровали и рекристаллизовали из этанола; в результате было получено 6.15 г соединения, указанного в заглавии, с температурой плавления 223 - 224oC.
Пример 30. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]- этилсульфинилметил} -3-метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид полугидрат.
Соединение, указанное в заглавии, было получено методом, описанным в примере 29, только вместо промежуточного соединения XXI использовали промежуточное соединение XXVI, а встряхивание проводили не 4.5 ч, а 2.5 ч. После обычной процедуры остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат:метанол 97:3. Собранные фракции подкислили избытком этанольного хлористого водорода и упаривали досуха в вакууме. Остаток кристаллизовали из этанола, в результате было получено 5.2 г соединения, указанного в заглавии, с температурой плавления 170 - 172oC. Указанное соединение содержит 1 эквивалент этанола.
Пример 31. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]- этилсульфонилметил} -3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Смесь 4.5 г промежуточного соединения XXV, 2.36 г 1-(2-метоксифенил)-пиперазина и 0.84 г карбоната калия в 45 мл безводного диметилформамида встряхивали в течение 2.5 ч при комнатной температуре. Реакционную смесь вылили в 300 мл воды и отфильтровали с отсасыванием, промывая водой. Твердое основание кристаллизовали из этанола, его температура плавления 143 - 146oC. Полученное вещество растворили в 1,2-дихлорэтане и подкислили этанольным хлористым водородом. После рекристаллизации из смеси метанол:вода 1:3.5 было получено 4.4 г соединения, указанного в заглавии, температура плавления 229 - 233oC.
Пример 32. 8-{ 2-[4-(2-Метоксифенил)-1-пиперазинил] -этиламино}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид.
Раствор 3.7 г промежуточного соединения XXIII в 10 мл диметилформамида добавили каплями при 0oC к суспензии 0.9 г гидрида натрия (50% в минеральном масле) в 9 мл диметилформамида. Охлаждающую баню убрали, и спустя 30 мин при 20 - 25oC добавили раствор 4.1 г 1-(2-хлорэтил)-4-(2-метоксифенил)-пиперазина в 10 мл диметилформамида. Смесь встряхивали в течение 5 ч при 90oC, а затем охладили до 20 - 25oC. Добавили 0.25 г гидрида натрия (50% в минеральном масле), а потом 1.36 г 1-(2-хлорэтил)-4-(2-метоксифенил)-пиперазина в 5 мл формамида. Смесь встряхивали 8 ч при 90oC, а затем вновь охладили до 20 - 25oC. Осторожно добавили 200 мл воды, а потом провели экстракцию этилацетатом. Органический слой промыли водой и высушили на безводном сульфате натрия. Растворитель выпарили в вакууме, а остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью н-гексан : этилацетат 3 : 2. В результате была получена смесь основания соединения, указанного в заглавии, с соответствующим N-трифторуксусным соединением.
3.8 г этой смеси растворили в 35 мл этанола и 35 мл диметилсульфоксида. К этому раствору порциями, при 20 - 25oC, было добавлено 0.55 г борогидрида натрия. При указанной температуре смесь встряхивали в течение 3 ч, затем вылили в 200 мл воды и экстрагировали этилацетатом. Органические слои промывали водой, высушивали на безводном сульфате натрия и выпаривали досуха в вакууме. Остаток был растворен в дихлорметане. Для получения соединения, указанного в заглавии, было добавлено 2 эквивалента этанольного хлористого водорода. Полученное соединение рекристаллизовали из этанола; выход 3.8 г, температура плавления 231 - 234oC.
Пример 33. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропиламино}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид 2.75-гидрат.
Используя 1-(3-хлорпропил)-4-(2-метоксифенил)-пиперазин вместо 1-(2-хлорэтил)-4-(2-метоксифенил)-пиперазина, а в остальном делая так, как это описано в примере 32, было получено соединение, указанное в заглавии. Температура плавления 206 - 208oC (10% этанол).
Пример 34. 8-{4-[4-(2-Метоксифенил)-1-пиперазинил]-бутиламино}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид полугидрат.
Смесь 4.5 г промежуточного соединения XXXIX, 3.9 г 8-амино-3-метил-4-оксо-2-фенил-4H-1-бензопирана, 8.3 г триацетоксиборогидрида натрия и 3.4 мл уксусной кислоты в 40 мл 1,2-дихлорэтана встряхивали в течение 6 ч при 20 - 25oC. Затем добавили 15 мл 5% водного раствора бикарбоната натрия и встряхивали смесь еще 10 мин. Затем смесь подщелачивали добавлением 0.5 н. раствора гидроксида натрия и экстрагировали хлороформом. Органические экстракты промыли водой и высушили на безводном сульфате натрия. Растворитель выпаривали в вакууме, а остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 9 : 1. Полученное основание растворили в дихлорметане и добавили 1 эквивалент этанольного хлористого водорода. После удаления в вакууме растворителя остаток кристаллизовали из 50% этанола; в результате было получено 1.6 г соединения, указанного в заглавии, с температурой плавления 151 - 153oC.
Пример 35. 8-[N-Метил-3-[4-(2-метоксифенил)-1-пиперазинил]-пропиламино} -3- метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид полугидрат.
Смесь 4 г соединения, полученного в примере 33, в виде основания, 4.35 мл 37% водного формальдегида и 1.15 г цианоборогидрида натрия в 25 мл ацетонитрила встряхивали при 20-25oC, поддерживая при этом pH 5-6 путем добавления в течение реакции уксусной кислоты. Через 4 ч растворитель выпаривали в вакууме. К остатку добавили 80 мл этилацетата и 200 мл 1 н. раствора гидроксида натрия, охлажденного льдом, органическую фазу промыли водой, высушили на безводном сульфате натрия и упарили в вакууме досуха. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 3 : 1. Полученное таким образом чистое основание растворили в диэтиловом эфире. Затем добавили 1 эквивалент этанольного хлористого водорода, а растворитель удалили в вакууме. Кристаллизация остатка из воды дала 2 г соединения, указанного в заглавии, с температурой плавления 186 - 187oC.
Пример 36. 8-{N-Ацетил-3-[4-(2-метоксифенил)-1-пиперазинил]-пропиламино)-3- метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид гидрат.
Смесь 4.8 г соединения, полученного в примере 33, в виде основания, 2.8 мл уксусного ангидрида и 33 мл пиридина встряхивали в течение 3 ч при 80oC. Реакционную смесь после охлаждения до 20 -25oC вылили в 200 мл смеси воды со льдом, подкислили 10 н. соляной кислотой и экстрагировали дихлорметаном. Органические экстракты промыли водой, высушили на безводном сульфате натрия и упаривали досуха в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 95 : 5. Полученное чистое основание растворили в дихлорметане. Затем добавили 1 эквивалент этанольного хлористого водорода, а растворитель удалили в вакууме. Остаток кристаллизовали из ацетонитрила. Было получено 3 г соединения, указанного в заглавии, которое содержало 0.33 эквивалента ацетонитрила. Температура плавления 208.5 - 210.5oC.
Пример 37. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропионамидо}-3-метил-4-оксо-2- фенил-4H-1-бензопиран гидрохлорид.
Смесь 3.97 г промежуточного соединения X и 3.07 г 1-(2-метоксифенил)-пиперазина в 40 мл диметилформамида встряхивали в течение 6 ч при 60oC. Затем реакционную смесь охладили до комнатной температуры и вылили в воду. Органическую фазу, полученную при последующей экстракции дихлорметаном, промыли водой и высушили на безводном сульфате натрия. Растворитель удалили в вакууме. Сырой остаток кристаллизовали из этанола, в результате было получено основание соединения, указанного в заглавии, которое затем растворили в горячем этаноле. К раствору добавили 1 молярный эквивалент 0.81 М этанольного хлористого водорода, при этом было получено 4 г соединения, указанного в заглавии, имеющего температуру плавления 255 - 257oC.
Пример 38. 8-(2-[4-(2-Метоксифенил)-1-пиперазинил] -этилуреидо)-3-метил-4-оксо-2- фенил-4H-1-бензопиран метансульфонат.
Смесь 3.34 г промежуточного соединения XLIV и 7.22 г 1-(2-метоксифенил)-пиперазина встряхивали в течение 5 ч при 100oC. Затем добавили еще 1.8 г 1-(2-метоксифенил)-пиперазина и продолжали встряхивание еще в течение 2 ч при той же температуре. Реакционную смесь после охлаждения до комнатной температуры вылили в воду и экстрагировали этилацетатом. Органическую фазу промыли водным раствором гидроксида натрия, высушили на безводном сульфате натрия и упаривали досуха в вакууме. Сырой продукт очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 98 : 2. Собранные фракции упаривали досуха в вакууме и кристаллизовали из смеси вода : этанол 4 : 6. Кристаллы повторно растворяли в дихлорметане и обрабатывали 1 молярным эквивалентом метансульфоновой кислоты. Сырой метансульфонат, полученный при выпаривании в вакууме, кристаллизовали из смеси этилацетат : этанол 1 : 1. Было получено 2.35 г продукта, указанного в заглавии, плавящегося при 191 - 193oC.
Пример 39. 8-(2-[4-(2-Метоксифенил)-1-пиперазинил]-этокси)-3-метил-4-оксо-2- фенил-4H-1-бензопиран гидрохлорид гидрат.
Смесь 6.61 г промежуточного соединения XI, 8.34 г 1-(2-метоксифенил)-пиперазина и 1.26 г иодида натрия в 70 мл диметилформамида встряхивали в течение 17 ч при 80oC. После охлаждения до 20 - 25oC реакционную смесь вылили в 600 мл воды, подщелочили 5% водным раствором бикарбоната натрия и экстрагировали дихлорметаном. Органические экстракты промыли водным раствором хлорида натрия, высушили на безводном сульфате натрия и упаривали в вакууме досуха. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью дихлорметан : метанол 99 : 1, затем смесью дихлорметан : метанол 98 : 2. Фракции, содержащие основания соединения, указанного в заглавии, были собраны вместе, а растворитель удалили в вакууме. Остаток растворили в этаноле и добавили этанольный хлористый водород. Соединение, указанное в заглавии, было кристаллизовано и затем собрано при фильтровании с отсасыванием. Рекристаллизацию проводили из 95% этанола. Выход 6.5 г, температура плавления 224 - 225oC.
Элементный анализ:
Найдено, %: C 66.38, H 6,34, N 5.35, Cl 6.76, H2O 3.35,
Вычислено, %: C 66.34, H 6,14, N 5,33, Cl 6,75, H2O 3,43,
ЯМР-спектры при 60 МГц (CDCl3-CD3OD):
7.8 - 7.1 (m, 8H, ароматические протоны бензопиранового кольца);
7.1 - 6.6 (m, 4H, ароматические протоны 2-метоксифенильного кольца);
4.8 - 4.4 (m, 2H, OCH2);
3.9 - 3.0 (m, 10H, 5 • CH2N);
3.8 (s, 3H, OCH3);
2.1 (s, 3H, CH3).
Пример 40. 8-(3-[4-(2-Метоксифенил)-1-пиперазинил]-пропокси)-3-метил-4-оксо-2-фенил-4H-1- бензопиран дигидрохлорид.
Это соединение было получено методом, описанным в примере 39, но вместо промежуточного соединения XI использовали промежуточное соединение IX. В этом случае очистка тонкослойной хроматографией была признана ненужной. Температура плавления 226 - 227oC.
Пример 41. 8-(4-[4-(2-Метоксифенил)-1-пиперазинил]-бутокси)-3-метил-4-оксо-2-фенил-4H- 1-бензопиран дигидрохлорид.
Смесь 7.75 г промежуточного соединения XVI, 4.7 г 1-(2-метоксифенил)-пиперазина, 3.3 г иодида калия и 2.8 г безводного карбоната калия в 78 мл диметилформамида встряхивали в течение 2 ч при 75oC. После охлаждения до 20 - 25oC реакционную смесь вылили в 600 мл воды и экстрагировали дихлорметаном. Органические экстракты промыли водой и высушили на безводном сульфате натрия, а затем растворитель выпарили в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя этилацетатом. Полученное таким образом соединение, указанное в заглавии, в виде основания было затем превращено в дигидрохлорид обработкой его этанольным хлористым водородом. После кристаллизации из этанола было получено 6.5 г соединения, указанного в заглавии. Температура плавления 217 - 219oC.
Пример 42. 8-(5-[4-(2-Метоксифенил)-1-пиперазинил] -пентилокси)-3-метил-4-оксо-2-фенил- 4H-1-бензопиран гидрохлорид.
Это соединение было получено методом, описанным в примере 41, только вместо промежуточного соединения XVI использовали промежуточное соединение XVII. Температура плавления 173oC (этанол). Соответствующее основание плавится при 117 - 118oC (этанол).
Пример 43. 8-(3-[4-(2-метоксифенил)-1-оксо-1-пиперазинил]-пропокси)-3-метил-4-оксо-2- фенил-4H-1-бензопиран 1.75 - гидрат.
2.93 г монопероксифталата магния в 10 мл воды добавляли по каплям при -15oC к раствору 4.34 г соединения, полученного в примере 40, и 0.1 г бензил(триэтил)хлорида аммония в 20 мл дихлорметана и 20 мл метанола. Смесь встряхивали в течение 2 ч при 0oC, а затем нагрели до комнатной температуры. Смесь вылили в воду и довели ее до щелочной реакции добавлением водного раствора гидроксида натрия. После экстракции дихлометаном был получен твердый остаток, который очищали тонкослойной хроматографией, элюируя смесью дихлорметан : метанол 9 : 1. Собранные фракции, содержащие чистое вещество, упаривали в вакууме досуха, а остаток кристаллизовали из ацетонитрила. В результате было получено 0.5 г соединения, указанного в заглавии, с температурой плавления 89 - 92oC.
Пример 44. 8-2-(2-[2-(2.6-Диметоксифенокси)-этиламино] -этокси)-3-метил-4-оксо-2-фенил- 4H-1-бензопиран гидрохлорид.
Смесь 4.5 г промежуточного соединения XII, 3.7 г трифенилфосфина и 2.85 г 2.6-диметоксифеноксиацетальдегида (полученного согласно Nelson, W.L., et al. , J.Med. Chem, 22, 1125, 1979) в 45 мл бензола встряхивали при 20 - 25oC в течение 18 ч и при дефлегмации в течение 5 ч. Растворитель упаривали в вакууме, а остаток растворили в 80 мл безводного метанола. Добавили 3  молекулярное сито. Затем при 0oC добавили 0.61 г борогидрида натрия. Смесь выдерживали в течение 1 ч при 0oC и в течение 1 ч при 20 - 25oC, затем вылили в смесь воды со льдом и экстрагировали дихлорметаном. Органические экстракты промыли водой и высушили на безводном сульфате натрия. Растворитель удаляли в вакууме, а остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан : метанол 49 : 1. Полученное основание обработали этанольным хлористым водородом. Соединение, указанное в заглавии, было получено после кристаллизации из этанола. Выход 40%, температура плавления 200 - 203oC.
молекулярное сито. Затем при 0oC добавили 0.61 г борогидрида натрия. Смесь выдерживали в течение 1 ч при 0oC и в течение 1 ч при 20 - 25oC, затем вылили в смесь воды со льдом и экстрагировали дихлорметаном. Органические экстракты промыли водой и высушили на безводном сульфате натрия. Растворитель удаляли в вакууме, а остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан : метанол 49 : 1. Полученное основание обработали этанольным хлористым водородом. Соединение, указанное в заглавии, было получено после кристаллизации из этанола. Выход 40%, температура плавления 200 - 203oC.
Пример 45. 8-(2-Гидрокси-3-[4-(2-метоксифенил)-1-пиперазинил]-пропокси)-3-метил-4- оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Раствор 3.7 г промежуточного соединения XL и 4.64 г 1-(2-метоксифенил)-пиперазина в 40 мл диметилформамида встряхивали в течение 3 ч при 80oC. После охлаждения до 20 - 25oC реакционную смесь вылили в 400 мл воды и экстрагировали дихлорметаном. Водную фазу подщелачивали 1 н. раствором гидроксида натрия и экстрагировали этилацетатом. Объединенные органические экстракты промывали водой и высушивали на безводном сульфате натрия. Растворитель выпаривали в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя этилацетатом. Фракции, содержащие продукт, указанный в заглавии, в виде его основания, были собраны вместе и выпарены в вакууме. Остаток растворяли в дихлорметане и добавили 1 эквивалент этанольного хлористого водорода. Растворители удаляли в вакууме, а остаток кристаллизовали из этанола. Было получено 5 г соединения, указанного в заглавии, которое содержало 1 эквивалент этанола. Температура плавления с разложением составили 126 - 128oC.
Пример 46. 8-{ 3-[4-(2-Метоксифенил)-1-пиперазинил] -пропилтио}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Смесь 4.4 г промежуточного соединения XXXIV, 2.5 г 1-(2-метоксифенил)-пиперазина, 1 г иодида калия и 1.8 г безводного карбоната калия в 40 мл диметилформамида встряхивали в течение 3 ч при 100oC. После охлаждения до 20 - 25oC реакционную смесь выливали в 350 мл воды и экстрагировали дихлорметаном. Органические экстракты промыли водой и высушили на безводном сульфате натрия; затем растворители выпаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 3 : 2. При кристаллизации из этанола было получено 3.9 г соединения, указанного в заглавии. Температура плавления 96 - 99oC.
Пример 47. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилсульфонил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Раствор 3.8 г промежуточного соединения XXXV и 4 г 1-(2-метоксифенил)-пиперазина в 40 мл диметилформамида нагревали при 60oC в течение 7 ч. После охлаждения до 20 - 25oC реакционную смесь выливали в 500 мл воды и экстрагировали дихлорметаном. Органические экстракты промыли водой и высушили на безводном сульфате натрия, а затем в вакууме выпарили растворитель. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 1 : 1. Было получено основание соединения, указанного в заглавии. Его растворили в этаноле и добавили 1 эквивалент этанольного хлористого водорода, в результате было получено 4.5 г соединения, указанного в заглавии и имеющего температуру плавления 226 - 228oC.
Пример 48. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]- этилсульфамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Раствор 4.5 г промежуточного соединения XLII и 3.8 г 1-(2-метоксифенил)-пиперазина в 40 мл диметилформамида нагревали в течение 7 ч при 70oC. После охлаждения до 20 - 25oC реакционную смесь вылили в 150 мл воды и экстрагировали дихлорметаном. Органический раствор промыли водой и высушили на безводном сульфате натрия, затем растворитель выпарили в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 3 : 7. Соединение, указанное в заглавии, было получено при образовании соли с помощью этанольного хлористого водорода. Выход 2.9 г, температура плавления 236 - 238oC.
Пример 49. 8-{N-Метил-2-[4-(2-метоксифенил)-1-пиперазинил]- этилсульфамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Соединение, указанное в заглавии, было получено методом, описанным в примере 48, но вместо промежуточного соединения XLII использовали промежуточное соединение XLI. Температура плавления 194 - 198oC (этанол).
Пример 50 8-{N-Карбамоил-3-[4-(2-метоксифенил)-1-пиперазинил]- пропиламино}-3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат полугидрат.
Смесь 4.06 г соединения из примера 33 и 1.5 г цианата калия в 42 мл ледяной уксусной кислоты встряхивали в течение 4 ч при 50oC. Реакционную смесь вылили в смесь воды со льдом и подщелочили. Осадок собрали при фильтровании с отсасыванием, высушили и очистили тонкослойной хроматографией, используя колонку с силикагелем и элюируя смесью этилацетат : метанол 98 : 2. Фракции, содержащие продукт, указанный в заглавии, в виде основания, упаривали в вакууме досуха. После растворения в 30 мл дихлорметана к остатку добавили 1 эквивалент метансульфоновой кислоты. Растворитель выпарили в вакууме, а остаток кристаллизовали из этанола, было получено 3.1 г соединения, указанного в заглавии (температура плавления 157 - 160oC без разложения). Полученное соединение содержало 1 молярный эквивалент этанола.
Пример 51. 8-{4-[4-(2-Метоксифенил)-1-пиперазинил]-1-оксобутил}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Раствор 1.33 мл безводного диметилсульфоксида в 9 мл дихлорметана добавили при -70oC к раствору 0.74 мл оксалил хлорида в 6 мл дихлорметана. После встряхивания в течение 15 мин при -70oC добавили раствор 2.8 г соединения из примера 21 (в виде основания) в 14 мл дихлорметана. При указанной температуре спустя 15 мин добавили 4.7 мл безводного триэтиламина, и в течение 30 мин температура поднялась до -30oC. Встряхивание продолжали при -30oC еще 30 мин. После того как температура поднялась до 0oC смесь разбавили 120 мл воды и экстрагировали дихлорметаном. Органическую фазу промыли водой, высушили на безводном сульфате натрия и упарили в вакууме досуха. Остаток очищали тонкослойной хроматографией в колонке с силикагелем, элюируя смесью этилацетат : дихлорметан 9 : 1. Фракции, содержащие продукт, указанный в заглавии, в виде основания, упаривали в вакууме досуха, а к остатку после растворения в 30 мл дихлорметана добавили 1 эквивалент метансульфоновой кислоты. Растворитель упаривали в вакууме, остаток кристаллизовали из этанола. Было получено 2.9 г соединения, указанного в заглавии, с температурой плавления 194 - 195oC.
Пример 52. 8-{3-[2-(1,4-Бензодиоксанил)-метиламино]- пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Смесь 5.56 г промежуточного соединения XLIII в виде основания, 4.58 г 2-(п-толуолсульфонилоксиметил)-1,4-бензодиоксана и 1.9 г безводного карбоната калия в 80 мл безводного диметилформамида встряхивали в течение 5 ч при 110oC. Реакционную смесь охладили до комнатной температуры, вылили в воду и экстрагировали дихлорметаном. Органическую фазу промыли водой, высушили на безводном сульфате натрия, отфильтровали и упаривали в вакууме досуха. Остаток очищали тонкослойной хроматографией, используя колонку с силикагелем при элюировании смесью этилацетат : метанол 95 : 5. Фракции, содержащие соединение, указанное в заглавии, в виде основания, упаривали досуха в вакууме. Остаток растворили в этаноле и добавили 1 эквивалент метансульфоновой кислоты, растворенной в этилацетате. Закристаллизовавшийся продукт отфильтровали, а затем рекристаллизовали из этанола. В результате было получено 2.4 г соединения, указанного в заглавии, с температурой плавления 172 - 174oC.
Пример 53. 8-{4-[4-(2-Метоксифенил)-1-пиперазинил]-бутил}-3- метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Раствор 2.8 г промежуточного соединения XLVI и 0.13 г п-толуолсульфоновой кислоты в 150 мл метанола подвергали дефлегмации в течение 5 ч. После охлаждения до 20 - 25oC добавили 0.8 г безводного карбоната калия и еще в течение 3 ч продолжали встряхивание. После фильтрования реакционную смесь упаривали досуха в вакууме, в результате было получено 2.5 г 8-(4,4-диметоксибутил)-3- метил-4-оксо-2-фенил-4H-1-бензопирана.
ЯМР (CDCl3, δ ):
1.6 - 1.9 (4H, m, CHCH2CH2CH);
2.2 (3H, s, CH3 флавона);
2.9 (2H, t, F1-CH2);
3.3 (6H, s, 2 • OCH3);
4.4 (1H, t, CH(OCH3)2);
7.3 (1H, dd, CH флавона в положении 6);
7.5 - 7.8 (6H, m, CH флавона в положении 7 и 5 • фенил CH);
8.1 (1H, dd, CH флавона в положении 5).
Раствор 2.5 г соединения, полученного таким образом, в 10 мл воды и 30 мл уксусной кислоты нагревали в течение 2.5 ч при 50oC. Затем реакционную смесь охладили до комнатной температуры, разбавили смесью воды со льдом, подщелачивали водным карбонатом натрия и экстрагировали хлороформом. Органическую фазу высушили на безводном сульфате натрия, отфильтровали и упаривали досуха в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : этилацетат 3 : 1. Было получено 2.1 г 8-(4-оксобутил)-3-метил-4-оксо-2-фенил-4H-1-бензопирана (выход более 75%), который использовался на следующей стадии без дальнейшей очистки.
ЯМР (CDCl3, δ) :
1.9 - 2.1 (2H, dd, CH2CH2CH2CHO);
2.2 (3H, s, CH3 флавона);
2.5 (2H, t, CH2CHO);
2.9 (2H, t, F1-CH2);
7.3 (1H, dd, CH флавона в положении 6);
7.5 - 7.7 (6H, m, CH флавона в положении 7 и 5•фенил CH);
8.1 (1H, dd, CH флавона в положении 5);
9.7 (1H, s, CHO).
2.3 мл 6 н. соляной кислоты в этаноле, раствор 2.1 г полученного ранее соединения в 40 мл метанола и 0.45 г цианоборогидрида натрия были последовательно добавлены к раствору 8 г 1-(2-метоксифенил)-пиперазина в 30 мл метанола. После встряхивания реакционной смеси при комнатной температуре в течение 24 ч ее вылили в 500 мл смеси воды со льдом и экстрагировали дихлорметаном. Органическую фазу промыли водой, высушили на безводном сульфате натрия и упаривали досуха в вакууме. Остаток очищали тонкослойной хроматографией, используя колонку с силикагелем, элюировали смесью этилацетат : петролейный эфир 9 : 1. Фракции, содержащие продукт, указанный в заглавии (в виде основания), упаривали досуха в вакууме. Остаток растворили в 30 мл дихлорметана и добавили 1 эквивалент метансульфоновой кислоты. Растворитель выпаривали в вакууме, а остаток кристаллизовали из ацетона. Было получено 2.35 г соединения, указанного в заглавии (температура плавления 141 - 143oC).
Пример 54. 8-[3-(4-Фенил-1-пиперидинил)-пропилкарбамоил] -3- метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Это соединение было получено методом, описанным в примере 11, используя вместо 1-(2-метоксифенил)-пиперазина 4-фенилпиперидин и проводя реакции в течение 1 ч вместо 5 ч. Очистку проводили тонкослойной хроматографией, используя колонку с силикагелем и элюируя смесью дихлорметан : метанол 100 : 5. Температура плавления 157 - 159oC (этилацетат). Соответствующее основание плавилось при 147 - 149oC (этанол).
Пример 55. 8-[3-(4.4-Дифенил-1-пиперидинил)-пропилкарбамоил] -3-метил- 4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Это соединение было получено методом, описанным в примере 11, используя 4,4-дифенилпиперидин вместо 1-(2-метоксифенил)-пиперазина и проводя реакцию в течение 2 ч вместо 5 ч. Температура плавления 221 - 223oC (этилацетат).
Пример 56. 8-{3-[4-(4-Фторбензоил)-1-пиперидинил]- пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Это соединение было получено методом, описанным в примере 11, используя 4-(4-фторбензоил)-пиперидин вместо 1-(2- метоксифенил)-пиперазина и проводя реакцию в течение 30 мин вместо 5 ч. Очистку проводили тонкослойной хроматографией, используя колонку с силикагелем при элюировании смесью дихлорметан : 5 н. метанольный аммиак в соотношении 100 : 1 - 100 : 20. Температура плавления 181 - 183oC (этанол).
Пример 57. 8-{3-[4-(2-Оксо-1-бензимидазолинил)-1-пиперидинил] -пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Это соединение было получено методом, описанным в примере 11, используя 4-(2-оксо-1-бензимидазолинил)-пиперидин вместо 1-(2-метоксифенил)-пиперазина. Очистку проводили тонкослойной хроматографией, используя колонку с силикагелем, элюировали смесью хромоформ : 5 н. метанольный аммиак 100 : 3. Температура плавления 238 - 241oC (этанол).
Пример 58. 8-{3-[4-(2-Пиримидинил)-1-пиперазинил]- пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Соединение, указанное в заглавии, было получено методом, описанным в примере 11, используя 1-(2-пиримидинил)-пиперазин вместо 1-(2-метоксифенил)-пиперазина и проводя реакцию в течение 2 ч. Продукт очищали тонкослойной хроматографией, используя колонку с силикагелем, элюировали смесью хлороформ : метанол 100 : 3. Целевые фракции растворяли в дихлорметане и к раствору добавляли 1 эквивалент метансульфоновой кислоты. После выпаривания в вакууме растворителя осадок кипятили в течение 1 ч с этилацетатом, а затем собирали фильтрованием. Температура плавления 209 - 210oC. Полученный продукт содержал 0.2 эквивалента этилацетата и 0.1 эквивалент воды. Соответствующее основание плавилось при 178 - 180oC (этанол).
Пример 59. 8-{3-[4-(2-Гидроксифенил)-1-пиперазинил-пропилкарбамоил}-3-метил -4-оксо-2-фенил-4H-1-безопиран.
Соединение, указанное в заглавии, было получено следуя описанию в примере 11, только вместо 1-(2-метоксифенил)-пиперазина использовали 1-(2-гидроксифенил)-пиперазин, а нагревание вместо 5 ч проводили в течение 1.5 ч. В качестве элюата при колоночной хроматографии использовали смесь дихлорметан : метанол 100 : 3 - 100 : 10. Температура плавления 118 - 120oC (95% этанол).
Пример 60. 8-{4-[4-(2-Метоксифенил)-1-пиперазинил]- бутилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Это соединение было получено методом, используемых в примере 12, только вместо 3-[4-(2-метоксифенил)-1-пиперазинил]-пропиламина применяли 4-[4-(2-метоксифенил)-1-пиперазинил] -бутиламин. Реакционную смесь встряхивали при комнатной температуре в течение 22 ч, промывали водой и фильтровали с отсасыванием, промывая нерастворившиеся твердые вещества водой. Сырой остаток высушивали и очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетата : метанол 9 : 1. Фракции, содержащие чистый продукт в виде основания, собирали вместе, упаривали в вакууме досуха и растворяли в дихлорметане. К раствору добавили метансульфоновую кислоту, соль осадили добавлением 2 объемов этилацетата и отфильтровали. После рекристаллизации из этанола было получено соединение, указанное в заглавии, с температурой плавления 230 - 232oC. Указанный продукт содержал 0.3 молярных эквивалента этанола.
Пример 61. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилсульфамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Это соединение было получено следуя описанию в примере 12, только вместо 8-хлоркарбонил-3-метил-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение VIII и встряхивание проводили в течение 24 ч вместо 2.5 ч. Сырой продукт очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 98.5 : 1,5. Собранные фракции, содержащие чистый продукт в виде основания, упаривали досуха в вакууме, а затем растворяли в дихлорметане. К раствору добавили метансульфоновую кислоту, а растворитель удаляли выпариванием в вакууме. Для получения продукта, указанного в заглавии, сырую соль кристаллизовали из этанола. Температура плавления 198 - 200oC.
Пример 62. 8-{3-[N-Метил-2-(2-метоксифенокси)-этиламино]- пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Раствор 10.5 мл 40% формальдегида в воде добавили к суспензии 6.66 г соединения, полученного в примере 14, в 55 мл ацетонитрила и 20 мл воды. После встряхивания в течение 15 мин при комнатной температуре к раствору добавили 2.70 г 95% цианоборогидрида натрия, а спустя еще 15 мин в тех же условиях добавили 1.38 мл уксусной кислоты. После встряхивания в течение 3 ч растворители удалили в вакууме, а остаток промыли 250 мл воды и 250 мл хлороформа. После добавления 3 н. гидроксида натрия органическую фазу отделили, а водную фазу дважды экстрагировали хлороформ. Растворитель удаляли из собранных органических фаз при выпаривании их в вакууме, а остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : 5.2 н. метанольный аммиак 100 : 0.5 - 100 : 2. Собранные фракции, содержащие чистый продукт, указанный в заглавии (в виде основания), упаривали досуха в вакууме, а остаток растворяли в горячем этаноле. Раствор подкисляли этанольным хлористым водородом, а остаток, после выпаривания в вакууме растворителя, промывали диэтиловым эфиром и встряхивали при комнатной температуре. Сырой продукт собрали при фильтровании и кристаллизовали из ацетонитрила. Было получено 3.1 г соединения, указанного в заглавии, с температурой плавления 146 - 148oC.
Пример 63. 8-{ N-метил-3-[4-(2-метоксифенил)-1-пиперазинил]- пропионамидо}-3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Соединение, указанное в заглавии, было получено в виде сырого основания, следуя описанию в примере 37, только вместо промежуточного соединения X использовали промежуточное соединение L и встряхивание проводили не в течение 6 ч при 60oC, а 4 ч при 90oC. Сырой метансульфонат был получен после очистки колоночной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 95 : 5. Соединение, указанное в заглавии, было получено после кристаллизации из ацетона. Температура плавления 200 - 202oC.
Пример 64. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил]-3-фенил-4-оксо-4H-1-бензопиран диметансульфонат.
Соединение, указанное в заглавии, было получено согласно тому, что описано в примере 12, только вместо 8-хлоркарбонил-3-метил-4- оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LVI и встряхивание вместо 2.5 ч проводили в течение 24 ч. Сырой продукт очищали колоночной хроматографией, элюируя смесью этилацетат : метанол 98 : 2; чистое основание, полученное при выпаривании собранных фракций в вакууме, растворяли в дихлорметане. Добавили 2 эквивалента метансульфоновой кислоты; сырой диметансульфонат, полученный после выпаривания растворителя, рекристаллизовали из ацетона. Температура плавления 153 - 156oC.
Пример 65. 8-{3-[(3,4-Дигидро-1-оксо-2H-нафтил)-метиламино]-пропилкарбамоил}-3-метил -4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Смесь 6 г промежуточного соединения XLIII, 2.4 г 2-метилен-альфа-тетралона (полученного, как описано в Org.Synth., 60, 88, 1981) и 3.14 мл триэтиламина в 48 мл безводного диметилформамида встряхивали в течение 6 ч при комнатной температуре, а затем в течение 1 ч при 50oC. Реакционную смесь разбавляли водой и экстрагировали дихлорметаном. Органические слои промывали водой, высушили на безводном сульфате натрия и упаривали в вакууме досуха. Сырой остаток дважды очищали колоночной хроматографией, элюируя первый раз смесью дихлорметан : метанол 95 : 5, а затем дихлорметан: метанол: 5.8 н. метанольный аммиак 98:2:0.2. Было получено 1.74 г соединения, указанного в заглавии, в виде основания. Это основание было превращено в метансульфонат способом, описанным в примеры 61. Для получения соединения, указанного в заглавии, соль рекристаллизовали сначала из ацетона, а затем из ацетонитрила. Температура плавления 157 - 159oC.
Пример 66. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]-этоксикарбонил-метил} -3-метил- 4-оксо-2-фенил-4H-1-бензопиран дигидрохлорид.
Соединение, указанное в заглавии, было получено методом, описанным в примере 5, но вместо 8-карбокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение XLVII, а вместо 1-(3-хлорпропил)-4-(2-метоксифенил)-пиперазина использовали 1-(2-хлорэтил)-4-(2-метоксифенил)-пиперазин. Температура плавления из смеси этанол : диэтиловый эфир 193 - 196oC.
Пример 67. 8-{4-[4-(2-Метоксифенил)-1-пиперазинил]-бутилсульфамоил}-3-метил-4-оксо-2 -фенил-4H-1-бензопиран диметансульфонат.
Соединение, указанное в заглавии, было получено согласно описанию в примере 61, но вместо 3-[4-(2-метоксифенил)-1-пиперазинил]-пропиламина использовали 4-[4-(2-метоксифенил)-1-пиперазинил]-бутиламин. Сырой диметансульфонат кристаллизовали сначала из ацетонитрила, а затем из этанола. Температура плавления 172 - 174oC.
Пример 68. 8-{N-(2-тетрагидропиранилокси)-3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат полугидрат.
Раствор 3.6 г 1-(3-хлорпропил)-4-(2-метоксифенил)-пиперазина в 30 мл безводного диметилформамида добавляли по каплям при встряхивании при 0oC к смеси 3.92 г O-(2-тетрагидропиранил)-гидроксиламина (получено так, как это описано R. N. Watrener et al. , Angewandte Chem. Int. Ed., 5, 511, 1966). Встряхивание продолжали в течение 2 ч при 0oC, а затем в течение 12 ч при 110oC. Реакционную смесь охладили до комнатной температуры, а диметилформамид удалили перегонкой в вакууме. Остаток промыли водой и экстрагировали этилацетатом. Собранные вместе органические слои промыли водой и высушили на безводном сульфате натрия. После выпаривания растворителя и вакууме было получено 4.39 г 1-[3-(2-тетрагидропиранилоксиамино)-пропил]-4-(2-метоксифенил)-пиперазина.
1H-ЯМР (CDCl3, δ ):
6.50 - 6.75 (m, 4H, ароматические протоны);
5.20 (bs, 1H, NH);
4.60 (m, 1H, O-CH-O);
3.30 - 4.00 (m, 5H, OCH3 и CH2O тетрагидрофурана);
2.80 - 3.20 (m, 6H, пиперазин 2 • CH2, алкильная цепь CH2N);
2.20 - 2.80 (m, 6H, пиперазин 2 • CH2, алкильная цепь CH2N);
1.30 - 2.00 (m, 6H, тетрагидрофуран 3 • CH2, алкильная цепь C-CH2-C).
Раствор 2.79 г 8-хлоркарбонил-3-метил-4-оксо-2-фенил-4H-1-бензопирана в 47 мл хлороформа добавляли по каплям при комнатной температуре к смеси 3.26 г полученного ранее соединения и 1.42 г карбоната калия в 47 мл хлороформа. Реакционную смесь встряхивали в течение 3 ч, а затем разбавляли 75 мл хлороформа и трижды промывали 1 M гидроксидом натрия. Органический слой промыли водой, высушили на безводном сульфате натрия и упаривали в вакууме досуха. Сырой остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 98 : 2. Для получения 2.99 г чистого соединения, указанного в заглавии, собранные фракции в виде основания упаривали досуха в вакууме. Основание растворили в дихлорметане и к раствору добавили метансульфоновую кислоту. Растворитель удалили выпариванием в вакууме, а для получения соединения, указанного в заглавии, сырую соль кристаллизовали из этилацетата. Температура плавления 159 - 160oC.
Пример 69. 8-{ 4-[4-(2-Метоксифенил)-1-пиперазинил]-бутирамидо}-3-метил-4-оксо-2- фенил-4Н-1-бензопиран метансульфонат полугидрат.
Соединение, указанное в заглавии, было получено методом, описанным в примере 38, но вместо промежуточного соединения XLIV использовали промежуточное соединение XLVIII и вместо встряхивания в течение 7 ч при 100oC встряхивание проводили 1 ч при 70oC и 2 ч при 130oC. После обычной обработки сырой остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 95 : 5. Фракции, содержащие чистый продукт, указанный в заглавии (в виде основания), собрали и упаривали в вакууме досуха. Остаток растворили в дихлорметане и к раствору добавили 1 эквивалент метансульфоновой кислоты. После упаривания растворителя в вакууме досуха сырую соль кристаллизовали из ацетона, температура плавления 175-176oC.
Пример 70. E-8-{2-[4-(2-Метоксифенил)-1-пиперазинил-этоксииминометил}-3-метил-4- оксо-2-фенил-4H-1-бензопиран.
Раствор 5.4 г 8-формил-3-метил-4-оксо-2-фенил-4H-1-бензопирана и 5.13 г промежуточного соединения LII в 10 мл хлороформа, содержащего 3  молекулярное сито, встряхивали при дефлегмации в течение 6 ч. Молекулярное сито удаляли фильтрованием, а раствор упаривали досуха в вакууме. Сырой продукт очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 7 : 3. Были собраны две группы фракций, которые упарили в вакууме досуха. Первая группа фракций (менее полярная) почти полностью содержала чистый продукт, указанный в заглавии, а вторая группа (более полярная) представляла собой смесь 1:1 E- и Z-диастереоизомеров, согласно определениям ЯМР.
молекулярное сито, встряхивали при дефлегмации в течение 6 ч. Молекулярное сито удаляли фильтрованием, а раствор упаривали досуха в вакууме. Сырой продукт очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 7 : 3. Были собраны две группы фракций, которые упарили в вакууме досуха. Первая группа фракций (менее полярная) почти полностью содержала чистый продукт, указанный в заглавии, а вторая группа (более полярная) представляла собой смесь 1:1 E- и Z-диастереоизомеров, согласно определениям ЯМР.
1H-ЯМР (CDCl3, δ ):
8.75 (dd, 0.5H, CH бензопирана в 7, Z);
8.65 (s, 0.5H, CH имина, E);
8.30 (dd, 1H, CH бензопирана в 5, E+Z);
8.15 (dd, 0.5H, CH бензопирана в 7, E);
8.00 (s, 0.5 H, CH имина, Z);
7.60 - 7.75 (m, 2H, CH фенила в 2' и 6', E+Z);
7.50 - 7.60 (m, 3H, CH фенила в 3', 4' и 5', E+Z);
7.45 (dd, 0.5H, CH бензопирана в 6, Z);
7.41 (dd, 0.5H CH бензопиран в 6, E);
6.70-7.10 (m, 4H, протоны фенила, E+Z);
4,41 (t, 2H, CH2O, E+Z);
3.86 (s, 3H, CH3O, E+Z);
3.05 - 3.20 (m, 4H, 2 • CH2 пиперазина, E+Z);
2.70 - 2.90 (m, 6H, 2 • CH2 пиперазина и CH2N, E+Z);
2.20 (s, 1.5H, CH3 бензопирана в 3, Z);
2.18 (s, 1.5H, CH3 бензопирана в 3, E).
Для получения 2.5 г чистого продукта, указанного в заглавии, E-диастереоизомер кристаллизовали из смеси этанол : вода 2 : 1, температура плавления 107 - 109oC.
Пример 71. 8-{N-Гидрокси-3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат 0.25 H2O.
Раствор 2.04 г соединения из примера 68 в виде основания в 104 мл 1.6 н. этанольного хлористого водорода встряхивали при комнатной температуре в течение 12 ч. Этанол удаляли выпариванием в вакууме, а остаток промыли 1 н. гидроксидом натрия и дихлорметаном. Органический слой собрали, промыли водой, высушили на безводном сульфате натрия и упарили досуха в вакууме. К раствору этого остатка в дихлорметане добавили 1 молярный эквивалент метансульфоновой кислоты. Растворитель удаляли, а сырой метансульфонат кристаллизовали из ацетона. Было получено 1.02 г соединения, указанного в заглавии и имеющего температуру плавления 211 - 213oC. Полученный продукт содержал 0.25 моль воды.
Пример 72. E-8-<2-{2-[4-(2-Метоксифенил)-1-пиперазинил]-этилкарбамоил} -этинил>-3-метил- 4-оксо-2-фенил-4H-1-бензопиран метансульфонат 1.2 H2O.
Соединение, указанное в заглавии, было получено методом, описанным в примере 61, но вместо промежуточного соединения VIII использовали соединение IV и 2-[4-(2-метоксифенил)-1-пиперазинил]-эталамин применяли вместо соответствующего пропиламина; 1,1,2,2-тетрахлорэтилен использовали в качестве растворителя. В конце реакционную смесь разбавили водой и хлороформом и промыли сначала 1 н. водным гидроксидом натрия, а затем водой. После высушивания на безводном сульфате натрия к органическому слою добавили метансульфоновую кислоту, а растворители выпарили в вакууме. Сырой продукт дважды кристаллизовали из изопропанола, было получено соединение, указанное в заглавии, содержащее 1.2 молярных эквивалента воды. Температура плавления 124 - 127oC.
Пример 73. 8-(4-[4-(2-Метоксифенил)-1-пиперазинил]-бутилсульфинил)-3-метил-4- оксо-2-фенил-4H-1-бензопиран метансульфонат.
Соединение, указанное в заглавии, было получено согласно примеру 38, только вместо промежуточного соединения XLIV использовали соединение LIV; встряхивание проводили при 70oC в течение 3 ч, а затем после добавления каталитических количеств (0.01 эквивалента) иодида калия - еще в течение 3 ч при 90oC. После очистки колоночной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 9 : 1, соединение, указанное в заглавии, было получено в виде основания. К сырому основанию, растворенному в дихлорметане, добавили 1 молярный эквивалент метансульфоновой кислоты. После удаления в вакууме растворителя полученную соль кристаллизовали из ацетона; было получено соединение, указанное в заглавии, с температурой плавления 183 - 184oC.
Пример 74. 8-(3-[3-(2-Метоксифенокси)-пропиламино]-пропилкарбамоил)-3-метил-4-оксо- 2-фенил-4H-1-бензопиран метансульфонат полугидрат.
Соединение, указанное в заглавии, было получено согласно методу, описанному в примере 76, только вместо 2-(2,6-диметоксифенокси)-этилбромида использовали 3-(2-метоксифенокси)-пропилхлорид (полученный согласно B. Willhalm, Tetrahedron, 20, 1185, 1964). Остаток после экстракции дихлорметаном очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан : метанол : 5 н. метанольный аммиак 9 : 1 : 0.3. Полученное чистое основание превратили в метансульфонат, который дважды кристаллизовали из смеси этилацетат : ацетонитрил 9 : 1, получено соединение, указанное в заглавии и плавящееся при 87 - 90oC.
Пример 75. 8-(3-[2-(2-Метилтиофенокси)-этиламино]-пропилкарбамоил)-3-метил-4-оксо- 2-фенил-4H-1-бензопиран метансульфонат.
1.85 г 95% борогидрида натрия добавляли к раствору 7 г промежуточного соединения LIX в 70 мл метанола, встряхиваемому при 0oC. После встряхивания при указанной температуре в течение 1 ч растворитель удаляли выпариванием в вакууме. Остаток разбавили водой и 2 н. соляной кислотой и экстрагировали этилацетатом. Органический слой промыли водой, высушили на безводном сульфате натрия и выпарили досуха в вакууме, получив при этом 6.6 г чистого 2-(2-метилтиофенокси)-этанола в виде масла. К раствору полученного таким образом соединения в 35 мл пиридина добавили порциями 8.57 г п-толуолсульфонилхлорида, встряхивая при 0oC. После встряхивания при комнатной температуре в течение 14 ч реакционную смесь вылили в холодную 2 н. соляную кислоту и экстрагировали дихлорметаном. Органический слой дважды промыли водой, высушили на безводном сульфате натрия и упаривали досуха в вакууме. Было получено 7.8 г смеси 2-(2-метилтиофенокси)-этил-п-толуолсульфоната и 2-(2-метилтиофенокси)-этилхлорида в соотношении 3: 1 (согласно ЯМР) в виде имеющего низкую температуру плавления твердого остатка, который использовали без дальнейшей очистки. Однородную смесь 3.3 г полученной выше смеси и 8 г промежуточного соединения XLIII выдерживали в течение 20 мин при 140oC на масляной бане. Затем расплавленная масса охлаждалась до комнатной температуры и затвердевала. Твердый остаток промыли дихлорметаном и 4 н. гидроксидом натрия. Органический слой промыли водой, высушили на безводном сульфате натрия и упарили досуха в вакууме.
Сырой продукт очищали колоночной хроматографией на силикагеле, элюируя смесью дихорметан : метанол 9 : 1; было получено 2.07 г соединения, указанного в заглавии, в виде основания. Его превращали обычным методом в метансульфонат, который кристаллизовали сначала из ацетона, а затем из ацетонитрила. Температура плавления 143 - 146oC.
Пример 76. 8-{3-[2-(2,6-Диметоксифенокси)-этиламино]-пропилкарбамоил}-3-метил-4- оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Однородную смесь 3.3 г 2-(2,6-диметоксифенокси)-этилбромида (полученного согласно J. Augstein et al., J. Med. Chem., 8, 356, 1965) и 8.4 г промежуточного соединения XLIII нагревали в течение 10 мин при 150oC на масляной бане. Расплавленная масса охлаждалась до комнатной температуры и затвердевала. Твердый остаток промыли этилацетатом и 2 н. гидроксидом натрия. Органический слой промыли водой, высушили на безводном сульфате натрия и упарили досуха в вакууме. Маслянистый остаток дважды очищали колоночной хроматографией на силикагеле, элюируя сначала смесью этилацетат : метанол : 5 н. метанольный аммиак 97 : 3 : 0.3, а затем смесью дихлорметан : метанол : триэтиламин 90 : 10 : 0.3. Было получено 3.3 г соединения, указанного в заглавии, в виде основания. Сырой гидрохлорид, полученный обычным способом, кристаллизовали сначала из ацетона, а затем из ацетонитрила. Температура плавления 179 - 181oC.
Пример 77. 8-{ 3-[4-(5-Хлор-2-метоксифенил)-1-пиперазинил]- пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Это соединение было получено методом, описанным в примере 11, но вместо 1-(2-метоксифенил)-пиперазина использовали 1-(5-хлор-2-метоксифенил)-пиперазин, а реакцию вместо 5 ч проводили в течение 6 ч. Очистку проводили тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : 5 н. метанольный аммиак 100 : 1. Соединение, указанное в заглавии, после кристаллизации из 95% этанола плавилось при 163 - 166oC.
Пример 78. (E)-8-{4-[4-(2-Метоксифенил)-1-пиперазинил]-1- бутенил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
33.4 мл 1 М раствора литий бис(триметилсилил)амида в безводном тетрагидрофуране добавляли по каплям в течение 15 мин к суспензии 6.4 г бромида 3-гидроксипропилтрифенилфосфония в 60 мл безводного тетрагидрофурана, охлажденного до -15oC. Затем по каплям добавляли раствор 4 г 8-формил-3-метил-4-оксо-2-фенил-4H-1-бензопирана в 40 мл тетрагидрофурана. Реакционную смесь встряхивали в течение 30 мин при 0oC, а затем в течение 1.5 ч при комнатной температуре. Обработка метанолом с последующим упариванием досуха в вакууме дало остаток, который очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 6 : 4. Собранные фракции выпаривали в вакууме; было получено 4.17 г 8-(4-гидрокси-1-бутенил)-3-метил-оксо-2-фенил-4H-1-бензопирана как смесь E-Z диастереоизомеров в соотношении 3.5 : 1 согласно определениям ЯМР.
1H-ЯМР, 200 МГц (CDCl3, δ ):
8.10 - 8.20 (d. 1H, CH бензопирана в 5, E + Z);
7.30 - 7.80 (m, 7H, другие ароматики, E + Z);
6.80; 7.00 (2d, 1H, арил-CH=, E + Z);
6.41 (dt, 0.78H, CH-CH2, E);
5.90 (dt, 0.22H, CH-CH2, Z);
3.60 - 3.80 (m. 2H, CH2O, E + Z);
2.45 - 2.60 (m. 2H, CH-CH2, E + Z);
2.18 (s, 3H, CH3 бензопирана в 3, E + Z);
1.60 - 1.90 (sa, 1H, OH, E + Z).
1.65 г п-толуолсульфонилхлорида добавили к раствору 2.2 г указанной выше смеси в 24 мл безводного пиридина и встряхивали при 0oC. Встряхивание при этой же температуре продолжали в течение 48 ч, а затем реакционную смесь вылили в холодную 1 н. соляную кислоту и отфильтровали с отсасыванием. Клейкий твердый остаток промыли водой и дихлорметаном. Раствор высушили на безводном сульфате натрия и упарили досуха в вакууме. Было получено 2.30 г (E, Z)-4-{ 8-[3-метил-4-оксо-2-фенил-4H-1-бензопиранил}-3-бутенил п-толуолсульфоната, имеющего такой же диастереоизомерный состав, как и указанное выше промежуточное соединение.
Раствор 2.85 г эфира п-толуолсульфоновой кислоты и 2.98 г 1-(2-метоксифенил)-пиперазина в безводном диметилформамиде встряхивали в течение 48 ч при комнатной температуре. Затем смесь вылили в 250 мл воды и экстрагировали этилацетатом. Органический слой промыли водой, высушили на безводном сульфате натрия и упарили досуха в вакууме, получив остаток, который затем очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 6 : 4. Собранные фракции упаривали в вакууме досуха, а сырой продукт кристаллизовали из 70% этанола. Было получено 1.48 г соединения, указанного в заглавии и плавящегося при 119 - 121oC.
1H-ЯМР, 200 МГц (CDCl3, δ ):
8.14 (dd, 1H, CH бензопирана в 5);
7.85 (dd, 1H, CH бензопирана в 7);
7.41 - 7.70 (m, 5H, CHs фенила);
7.34 (dd, 1H, CH бензопирана в 6);
6.70 - 7.10 (m, 5H, арил-CH= и CHs метоксифенила);
6.30 - 6.50 (dt, 1H, Jtrans = 16.5, CH-CH2);
3.86 (s, 3H, CH3O);
3.00 - 3.15 (m, 4H, CH2 пиперазина);
2.50 - 2.80 (m, 8H, CH2, пиперазина, CHCH2CH2N);
2.18 (s, 3H, CH3 бензопирана в 3).
Пример 79. (E)-8-[2-{ 2-{ 2-[4-(2-Метоксифенил)-1-пиперазинил]-этоксикарбонил}- этинил]-3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Соединение, указанное в заглавии, было получено согласно примеру 6, но вместо 8-карбокси-3-метил-4-оксо-2-фенил-4H-бензопирана использовали промежуточное соединение III. После обычной обработки остаток дважды кристаллизовали из этанола. Полученное твердое вещество очищали колоночной хроматографией на силикагеле, элюируя смесью хлороформ : этилацетат 8 : 2. Было получено чистое основание, которое затем растворяли в смеси хлороформ : этанол 1 : 1. К раствору добавили метансульфоновую кислоту, а растворители выпаривали в вакууме. Сырую соль кристаллизовали из изопропанола, получая при этом соединение, указанное в заглавии, которое плавилось при 193 - 195oC. Это соединение содержало 0.33 эквивалента изопропанола и 0.25 эквивалента воды.
Пример 80. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]-этилкарбамоилметил} -4- оксо-2-фенил-4H-1-бензопиран метансульфонат гидрат.
Смесь 2.8 г промежуточного соединения XLVII и 1.28 г 1-гидроксибензотриазола в 20 мл безводного диметилформамида встряхивали при 0 - 5oC в течение 15 мин. К этой смеси в течение приблизительно 40 мин добавляли по каплям раствор 1.96 г дициклогексилкарбодимида в 20 мл безводного диметилформамида. После встряхивания в течение 8 ч при комнатной температуре добавили раствор 2.24 г 1-(2-аминоэтил)-4-(2-метоксифенил)-пиперазина в 15 мл безводного диметилформамида. После встряхивания в течение 5 ч и выдержки при той же температуре в течение ночи нерастворившееся вещество отфильтровали, а фильтрат вылили в 300 мл воды и подщелочили добавлением 1 н. гидроксида натрия. Смесь экстрагировали дихлорметаном, органические слои отделили, высушили на безводном сульфате натрия и выпарили в вакууме. Сырой продукт очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 95 : 5. К раствору полученного сырого основания в этаноле добавили 1 молярный эквивалент метансульфоновой кислоты. Добавляли диэтиловый эфир до момента кристаллизации соли. Полученную соль затем отфильтровывали и рекристаллизовали из смеси этанол : диэтиловый эфир 1 : 2. Было получено 1.15 г соединения, указанного в заглавии, с температурой плавления 160 - 162oC.
Пример 81. 8-{N-Ацетил-3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3- метил-4-оксо-2-фенил-4H-1-бензопиран.
Раствор 2.86 г промежуточного соединения LVII, 5.04 г 1-(3-хлорпропил)-4-(2-метоксифенил)-пиперазина и 2.58 г безводного карбоната калия в 50 мл диметилформамида встряхивали в течение 7 ч при 90oC. После охлаждения до комнатной температуры реакционную смесь вылили в 500 мл воды и экстрагировали дихлорметаном. Органический слой промыли водой, высушили на безводном сульфате натрия и упаривали досуха в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 7 : 3. Было получено 1.89 г соединения, указанного в заглавии, плавящегося при 62 - 63oC.
Пример 82. 8-{2-[4-(2-Метоксифенил)-1-пиперазинил]-этилсульфониламино} -3- метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
1.05 мл 2-хлорэтансульфонил хлорида добавляли по каплям к раствору 5 г 8-амино-3-метил-4-оксо-2-фенил-4H-1-бензопирана в 1.4 мл триэтиламина, встряхивая при 0oC. В течение двух дней реакционную смесь встряхивали при комнатной температуре. После отфильтровывания твердого осадка раствор упаривали в вакууме досуха; был получен сырой остаток, содержащий 8-(этинилсульфониламино)-3-метил-4- оксо-2-фенил-4H-1-бензопиран, который использовали без дальнейшей очистки. Смесь 7.54 г этого остатка и 5.8 г 1-(2-метоксифенил)-пиперазина и 4.15 г карбоната калия в 100 мл диметилформамида встряхивали в течение 4 ч при комнатной температуре, вылили в 600 мл воды и экстрагировали этилацетатом. Органический слой упаривали в вакууме досуха, а остаток очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир : ацетон 8 : 2. Собранные фракции досуха упаривали в вакууме и кристаллизовали из 70% этанола; было получено 0.75 г соединения, указанного в заглавии, в виде основания. Его растворили в дихлорметане и добавили к полученному раствору 1 эквивалент метансульфоновой кислоты. Сырой метансульфонат, полученный при упаривании в вакууме, кристаллизовали из ацетона. Было получено 0.6 г соединения, указанного в заглавии, плавящегося при 202 - 203oC.
Пример 83. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилтиокарбамоил} -3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Встряхиваемую смесь 0.8 г 8-формил-3-метил-4-оксо-2-фенил-4H-1-бензопирана, 0.75 г 1-(3-аминопропил)-4-(2-метоксифенил)-пиперазина и 0.14 г серы в 5 мл пиридина подвергали дефлегмации в течение 6 ч. После упаривания в вакууме растворителя остаток очищали тонкослойной хроматографией на силикагеле, элюируя хлороформом. Полученное соединение, указанное в заглавии (в виде основания), растворили в дихлорметане и добавили 1 эквивалент метансульфоновой кислоты. Соединение, указанное в заглавии, было получено при упаривании в вакууме и кристаллизации остатка из ацетонитрила. Выход 0.7 г, температура плавления 189 - 190oC.
Пример 84. 8-{4-[4-(2-Метоксифенил)-1-пиперазинил]- бутилсульфонил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат. 0.25 H2O.
Соединение, указанное в заглавии, было получено согласно примеру 73, только вместо промежуточного соединения LIV использовали соединение LX. Очистку этого соединения проводили колоночной хроматографией на силикагеле, элюируя смесью этилацетат : петролейный эфир 7 : 3. К раствору сырого основания в дихлорметане добавили 1 эквивалент метансульфоновой кислоты. После удаления растворителя выпариванием в вакууме полученную в результате соль кристаллизовали из ацетона, для того чтобы получить соединение, указанное в заглавии. Температура плавления 212 - 214oC.
Пример 85. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-3-гидроксиметил-4-оксо-2-фенил-4H-1-бензопиран, 0.4 этанол
Смесь 3.6 г промежуточного соединения XCV и 1.65 г 1-гидроксибензотриазола в 35 мл безводного диметилформамида в течение 15 мин при 0 - 5oC. Раствор 2.5 г дициклогексилкарбодиимида в 35 мл безводного диметилформамида добавляли по каплям. После встряхивания в течение 1 ч при указанной температуре был добавлен раствор 1-(3-аминопропил)-4-(2-метоксифенил)-пиперазина. После встряхивания в течение 2 ч при той же температуре и выдержки в течение ночи при комнатной температуре реакционную смесь упаривали в вакууме досуха. Остаток очищали тонкослойной хроматографией, элюируя смесью дихлорметан : метанол 100 : 3 с последующей кристаллизацией из этанола. Было получено 2.5 г соединения, указанного в заглавии, с температурой плавления 152 - 154oC.
Пример 86. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-4-оксо-2-фенил-4H-1-бензопиран метансульфонат. 0.25 H2O.
3.6 мл диэтилцианофосфоната добавляли по каплям при 0 - 5oC во встряхиваемый раствор 4 г 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана (полученного согласно описанию Da Re et al., Ber., 99, 1962, 1966) и 3.75 г 1-(3-аминопропил)-4-(2-метоксифенил)-пиперазина в 35 мл безводного диметилформамида. Сразу же после этого при той же температуре добавили по каплям 2.5 мл триэтиламина. После встряхивания в течение 30 мин при 0 - 5oC и в течение 1 ч при комнатной температуре реакционную смесь вылили в 350 мл 2.5% водного карбоната натрия. Образовавшийся осадок встряхивали в течение 1 ч при комнатной температуре, отфильтровывали с отсасыванием и кристаллизовали из этанола. Полученное таким образом основание соединения, указанного в заглавии, растворили в дихлорметане и добавили 1 эквивалент метансульфоновой кислоты. После выпаривания в вакууме был получен твердый стекловидный остаток, который измельчили и подвергали дефлегмации в течение 1 ч в ацетоне. Было получено 5 г соединения, указанного в заглавии и плавящегося при 191 - 194oC.
Пример 87. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2,3-дигидро-4-оксо-4H-1-бензопиран метансульфонат.
Раствор 0.84 мл тионилхлорида в 17 мл безводного дихлорметана добавляли по каплям к раствору 2.0 г 8-карбокси-2,3-дигидро-4-оксо-4H-1-бензопирана (полученного согласно Lichtenberger et al., Bull. Chem. Soc. Fr., 275, 1963) и 1.75 мл триэтиламина в 17 мл дихлорметана, встряхиваемого при комнатной температуре. Встряхивание продолжали при этой же температуре еще 1.5 ч, затем раствор упаривали досуха в вакууме, чтобы получить 8-хлоркарбонил-2,3-дигидро-4-оксо-4H-1-бензопиран. Последний использовали согласно методу из примеру 10 вместо 8-хлоркарбонил-3-метил-4-оксо-2-фенил-4H-1-бензопирана для получения основания соединения, указанного в заглавии. Основание очищали колоночной хроматографией, элюируя смесью этилацетат : метанол 85 : 15. Выход чистого основания составил 1.91 г. После растворения в дихлорметане, подкисления метансульфоновой кислотой и упаривания в вакууме досуха полученную сырую соль кристаллизовали из ацетонитрила. Было получено 1.57 г соединения, указанного в заглавии и плавящегося при 175 - 177oC.
Пример 88. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-4-оксо-4H-1-бензопиран метансульфонат. 1.25 H2O.
Соединение, указанное в заглавии, было получено методом, описанным в примере 87, только вместо 8-карбокси-2,3-дигидро-4-оксо-4H-1-бензопирана использовали промежуточное соединение LXII. Сырой метансульфонат промывали диэтиловым эфиром, фильтровали и кристаллизовали из ацетонитрила. Температура плавления 155 - 157oC.
Пример 89. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-6-бром-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Это соединение было получено согласно примеру 86, только вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали 8-карбокси-6-бром-3-метил-4-оксо-2-фенил-4H-1-бензопиран (полученный согласно EP 107804). Его очищали в виде основания тонкослойной хроматографией на силикагеле, элюируя смесью дихлорметан : метанол 100 : 3, а кристаллизовали из 95% этанола. Температура плавления 154 - 159oC.
Пример 90. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-6-метокси- 3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
1.01 мл диэтилцианофосфоната и 0.85 мл триэтиламина добавили к раствору 1.7 г 8-карбокси-6-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана (полученного согласно JP 61-15880) и 1.51 г 1-(2-метоксифенил)-4-(3-аминопропил)-пиперазина в 20 мл безводного диметилформамида, встряхиваемому при 0oC. После встряхивания в течение 1 ч при температуре от 0oC до комнатной реакционную смесь вылили в смесь 100 мл воды и 10 мл 1 н. гидроксида натрия. Основание соединения, указанного в заглавии, выпало в осадок, его отфильтровали и промыли водой. После осушки его обычным путем превратили в метансульфонат, который кристаллизовали из ацетонитрила. Выход 1.7 г, температура плавления 185 - 186oC.
Пример 91. 8-(3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил)-6-гидрокси -3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
0.8 г соединения, полученного в примере 114, и 5.8 мл 1 н. гидроксида натрия в 10 мл этанола встряхивали в течение 4 ч при комнатной температуре. После выдержки в течение ночи добавили 15 мл 1 н. гидроксида натрия и 15 мл этанола и смесь встряхивали в течение 1 ч при комнатной температуре. Метанол выпарили в вакууме, а к остатку добавили воду. Суспензию отфильтровали с отсасыванием и получили 0.48 г основания соединения, указанного в заглавии. С помощью обычной методики это соединение превратили в метансульфоновую соль соединения, указанного в заглавии, и рекристаллизовали из ацетонитрила. Температура плавления 200 - 202oC.
Пример 92. 8-(3-[4-(2-Метоксифенил)-1-пиперазинил] -пропилкарбамоил)-3,6-диметил -4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Соединение, указанное в заглавии, было получено согласно примеру 90, но вместо 8-карбокси-6-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана использовали 8-карбокси-3,6-диметил-4-оксо-2-фенил-4H-1-бензопиран (полученный так, как это описано Da RE et al., Arch. Pharm., 296, 714, 1963). Сырой метансульфонат кристаллизовали из ацетонитрила, он плавился при 196 - 197oC.
Пример 93. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3- метил-6-нитро-4-оксо-2-фенил-4H-1-бензопиран.
Этот продукт был получен согласно процедуре, описанной в примере 12, но вместо 8-хлоркарбонил-3-метил-4-оксо-2-фенил-4H-1- бензопирана использовали промежуточное соединение LXVIII, а вместо хлороформа - 1,1,2-трихлорэтан. После обычной обработки сырой продукт очищали колоночной хроматографией, элюируя смесью дихлорметан : метанол 98 : 2. Собранные фракции упаривали досуха в вакууме и кристаллизовали из этанола; было получено соединение, указанное в заглавии, с температурой плавления 159.5 - 161oC.
Пример 94. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-6- амино-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Смесь 33 г соединения, полученного в примере 93, 109 мл 1 н. соляной кислоты, 105 мл воды и 8.78 г никеля Ренея в 950 мл этанола гидрировали в аппарате Парра при давлении водорода 2 атм и 40oC, встряхивая смесь в течение 12 ч. Затем катализатор отфильтровывали и промывали 80% этанолом. Маточные жидкости упаривали в вакууме до объема 80 мл и отфильтровывали. Сырой продукт промыли водой и суспендировали в воде, затем добавляли 37% соляную кислоту до pH 1. Нерастворившееся вещество отфильтровали с отсасыванием, а фильтрат подщелочили, добавляя 35% гидроксид натрия. Соединение, указанное в заглавии, выпало в осадок, его отфильтровали и промыли водой. Осушка дала 26 г продукта, плавящегося при 215 - 217oC, который использовали в примере 95 без дальнейшей очистки. 4.7 г этого продукта кристаллизовали сначала из этанола, а затем из 85% этанола, при этом было получено 3 г соединения, указанного в заглавии, с температурой плавления 218-219oC.
Пример 95. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-6- ацетамидо-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Соединение, указанное в заглавии, было получено согласно процедуре, описанной в примере 36, но вместо соединения, полученного в примере 33, использовали соединение, полученное в примере 94. Реакционную смесь разбавили водой и фильтровали с отсасыванием, промывая осадок водой. После осушки при 80oC этот твердый осадок очищали колоночной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 95 : 5. Выпаривание в вакууме собранных фракций и кристаллизация остатка из 95% этанола привели к продукту, указанному в заглавии. Температура плавления 218 - 220oC.
Пример 96. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-6- этиламино-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Смесь 8.42 г соединения, полученного в примере 94, 0.45 мл ацетальдегида, 0.59 г 85% цианоборогидрида натрия и 3.3 мл 4.85 н. этанольного хлористого водорода в 73 мл метанола встряхивали в течение 5 дней при комнатной температуре. Затем реакционную смесь вылили в холодный 1.5 н. гидроксид натрия, полученную суспензию разбавили водой и отфильтровали с отсасыванием. После осушки осадок очищали колоночной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 100 : 3. После упаривания в вакууме собранных фракций было получено 6 г соединения из примера 94 и 2.67 г соединения, указанного в заглавии. Последнее плавилось при 198 - 201oC после рекристаллизации из этанола.
Пример 97. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-6- диметиламино-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
Соединение, указанное в заглавии, было получено согласно примеру 35, только вместо соединения, полученного в примере 33, использовали соединение, полученное в примере 94, и использовали 10 молярных эквивалентов 40% формальдегида вместо 7 молярных эквивалентов, 3 моль цианборогидрида натрия вместо 2 моль и встряхивание проводили при комнатной температуре в течение 18 ч вместо 4.5 ч. После обычной обработки и очистки колоночной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 97 : 3, упаривания в вакууме собранных фракций и кристаллизации остатка из этанола получен продукт, указанный в заглавии и плавящийся при 183 - 186oC.
Пример 98. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]пропилкарбамоил}-7-метокси-3- метил-4-оксо-2-фенил-4H-1-бензопиран.
Соединение, указанное в заглавии, было получено согласно процедуре, описанной в примере 87, только вместо 8-карбокси-2,3-дигидро-4-оксо-4H-1-бензопирана использовали промежуточное соединение LXIX. После обычной обработки твердый остаток очищали колоночной хроматографией, элюируя смесью этилацетат : метанол 8 : 2. Собранные фракции упаривали в вакууме досуха, а остаток кристаллизовали из ацетонитрила. Было получено соединение, указанное в заглавии, которое плавилось при 151 - 152oC.
Пример 99. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил- 4-оксо-2-(4-трифторметилфенил)-4H-1-бензопиран метансульфонат полуторагидрат.
Соединение, указанное в заглавии, было получено в соответствии с методикой, описанной в примере 90, но вместо 8-карбокси-6-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана в качестве исходного использовали промежуточное соединение LXXII. После кристаллизации из ацетонитрила полученный метансульфонат плавился при 90 - 120oC.
Пример 100. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-2-(4- бензоилфенил)-3-метил-4-оксо-4H-1-бензопиран метансульфонат полугидрат.
Соединение, указанное в заглавии, было получено согласно методике, описанной в примере 90, только в качестве исходного соединения вместо 8-карбокси-6-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LXXIV. Полученный сырой метансульфонат кристаллизовали из ацетонитрила, температура плавления 208 - 210oC.
Пример 101. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3- метил-4-оксо-2-(4-феноксифенил)-4H-1-бензопиран метансульфонат. 0.25 H2O.
Соединение, указанное в заглавии, было получено согласно методике, описанной в примере 90, но в качестве исходного соединения вместо 8-карбокси-6-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LXXVII. Полученный сырой метансульфонат кристаллизовали из ацетонитрила. Температура плавления 200 - 202oC.
Пример 102. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2,3-диметил-4-оксо-4H-1-бензопиран.
Это соединение было получено согласно примеру 86, но вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали 8-карбокси-2,3-диметил-4-оксо-4H-1-бензопиран (полученный согласно Da Re, Farmaco Ed. Sci., 11, 678, 1956) и реакцию проводили в течение 5 ч при комнатной температуре. Соединение очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 98 : 2, а затем кристаллизовали из ацетона. Температура плавления 155 - 158.5oC.
Пример 103. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2-(t-бутил)-3-метил-4-оксо-4H-1-бензопиран дигидрохлорид дигидрат.
Соединение было получено согласно примеру 86, но вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LXXVIII. Полученное основание очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 49 : 1,; основание было превращено в дигидрохлорид в смеси метанол : диэтиловый эфир. После рекристаллизации из смеси метанол : диэтиловый эфир 1 : 1 целевой продукт плавился при 226 - 229oC.
Пример 104. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2-циклогексил-3-метил-4-оксо-4H-1-бензопиран.
Это соединение было получено согласно примеру 86, но вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LXXIX и проводили реакцию в течение 5 ч при комнатной температуре. Очистку проводили тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 49 : 1; а затем кристаллизовали из ацетонитрила. Температура плавления 155 - 157oC.
Пример 105. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2-(2-фурил)-3-метил-4-оксо-4H-1-бензопиран.
Это соединение было получено согласно примеру 86, но вместо 8-карбокси-4-оксо-2-фенил-4H-1-бинзопирана использовали промежуточное соединение LXXXI и реакцию проводили при комнатной температуре в течение 5 ч. После ее завершения продукт, указанный в заглавии, выделили экстракцией хлороформом и очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 49 : 1. Затем проводили кристаллизацию из ацетонитрила. Температура плавления 151 - 153oC.
Пример 106. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-3-метил-4-оксо-2-тиенил-4H-1-бензопиран.
Это соединение было получено согласно примеру 86, только вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LXXXIII. Его очищали встряхиванием в воде (для удаления диметилформамида) с последующей очисткой колоночной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 49 : 1, и кристаллизовали из ацетонитрила. Температура плавления 174 - 175oC.
Пример 107. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-4-оксо-2-фенил-4H-1-бензотиопиран.
Смесь 2.8 г промежуточного соединения LXXXIV и 3.4 г 1,1'-карбонилдиимидазола в 60 мл безводного диметилформамида встряхивали под азотом в течение 1.5 ч при комнатной температуре. Затем добавили 2.7 г 1-(3-аминопропил)-4-(2-метоксифенил)-пиперазина. После 2 ч встряхивания при комнатной температуре реакционную смесь вылили в 300 мл воды и экстрагировали хлороформом. Органический слой высушили на безводном сульфате натрия и выпаривали в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 49 : 1. Затем промыли водой и кристаллизовали из ацетонитрила. Было получено 2 г соединения, указанного в заглавии и плавящегося при 144 - 146oC.
Пример 108. (E)-8-{ 3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-3-метил-4-оксо-2-(2-стирил)-4H-1-бензопиран.
Это соединение было получено согласно примеру 86, только вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LXXXVI. Соединение очищали кристаллизацией из ацетонитрила. Температура плавления 191 - 194oC.
Пример 109. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-3-метил-2-(4-метилфенил)-4-оксо-4H-1-бензопиран.
Это соединение было получено согласно примеру 86, только вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LXXXVII и реакцию проводили в течение 4 ч при комнатной температуре. После ее окончания соединение, указанное в заглавии, выделяли экстракцией этилацетатом, высушивали на безводном сульфате натрия и выпаривали в вакууме. Затем промывали диэтиловым эфиром и кристаллизовали из ацетонитрила. Температура плавления 161 - 163oC.
Пример 110. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2-(4-метоксифенил)-3-метил-4-оксо-4H-1-бензопиран.
Это соединение было получено согласно примеру 86, только вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали 8-карбокси-2-(4-метоксифенил)-3-метил-4-оксо-4H-1-бензопиран (полученный так, как это описано в EP 108986) и реакцию проводили в течение 3.5 ч при комнатной температуре. Соединение очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 49 : 1, а затем кристаллизовали из ацетонитрила. Температура плавления 158 - 161oC.
Пример 111. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2-(4-фторфенил)-3-метил-4-оксо-4H-1-бензопиран.
Это соединение было получено согласно примеру 86, только вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LXXXIX. Соединение очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 100 : 2 - 100 : 6, а затем кристаллизовали из 95% этанола. Температура плавления 166 - 168oC.
Пример 112. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-6-метансульфониламино-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
0.032 мл метансульфонилхлорида в 1 мл диметилформамида добавляли по каплям в течение 10 мин к раствору 0.21 г соединения из примера 94 и 0.062 мл триэтиламина в 4 мл диметилформамида, встряхивая при -20oC. Встряхивание продолжали при той же температуре в течение 3.5 ч. Затем реакционную смесь вылили в воду, полученную суспензию фильтровали с отсасыванием. После рекристаллизации из 80% этанола было получено 0.1 г соединения, указанного в заглавии. Температура плавления 272 - 275oC.
Пример 113. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-3-метил-2-(4-нитрофенил)-4-оксо-4H-1-бензопиран.
Соединение, указанное в заглавии, было получено следуя методике, описанной в примере 90, но в качестве исходного соединения вместо 8-карбокси-6-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение XCVIII. После встряхивания в течение 1 ч при комнатной температуре реакционную смесь вылили в холодный 2% раствор карбоната натрия, выпавший твердый осадок собирали при фильтровании с отсасыванием. После осушки и кристаллизации из этанола было получено указанное в заглавии соединение, плавящееся при 185 - 187oC.
Пример 114. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-6-диэтоксифосфонилокси-3-метил-4-оксо-2-фенил-4H- 1-бензопиран.
Соединение, указанное в заглавии, было получено согласно методике, описанной в примере 90, но в качестве исходного соединения вместо 8-карбокси-6-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение LXIII и вместо 1.1 эквивалента диэтилцианофосфата использовали 2 эквивалента этого вещества. После фильтрования из воды было получено соединение, указанное в заглавии, которое плавилось при 48 - 50oC. Соединение, указанное в заглавии, можно рассматривать в качестве пролекарства 8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-6-гидрокси- 3-метил-4-оксо-2-фенил-4H-1-бензопирана.
Пример 115. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-3-метил-4-оксо-2-трифторметил-4H-1-бензопиран метансульфонат.
Сырое основание соединения, указанного в заглавии, было получено согласно методике, приведенной в примере 90, но в качестве исходного соединения вместо 8-карбокси-6-метокси-3-метил-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение C. После обычной процедуры экстракции этилацетатом остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 9 : 1. Полученное соединение с по помощью обычной методики было превращено в метансульфонат и рекристаллизовано из этилацетата. Температура плавления 145 - 148oC.
Пример 116. 8-{ N-метил-3-[4-(2-метоксифенил)-1-пиперазинил] - пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран гидрохлорид.
Это соединение было получено согласно примеру 12, но вместо 3-[4-(2-метоксифенил)-1-пиперазинил] -пропиламина использовали промежуточное соединение CI. Сырое основание очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ: 5 н. метанольный аммиак 100 : 1, а затем обычным способом превратили в гидрохлорид. После кристаллизации из ацетона температура плавления 195 - 198oC.
Пример 117. 7-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2-бензоил-3-этил-бензо[b]фуран.
Это соединение было получено согласно примеру 86, но вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопиран-8-карбоновой кислоты использовали промежуточное соединение CIII и реакцию проводили в течение 4 ч при комнатной температуре. Соединение очищали кристаллизацией из этанола. Температура плавления 165 - 166oC.
Пример 118. 8-{3-[4-(2-метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2-(4-бифенилил)-3-метил-4-оксо-4H-1-бензопиран.
Соединение, указанное в заглавии, было получено согласно примеру 86, но вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение CVI. Реакция продолжалась 20 ч при комнатной температуре. Основание очищали кристаллизацией из этанола (температура плавления 164 - 166oC).
Пример 119. 8-{3-[4-(2-метоксифенил)-1-пиперазинил]- пропилкарбамоил}-3-метил-4-оксо-2-(3-пиридил)-4H-1-бензопиран.
Смесь 6.2 г метил 3-пропионил-силицилата и 5.8 г гидрохлорида никотиноилхлорида в 18 мл безводного пиридина встряхивали и нагревали в течение 2 ч при 100oC под азотом. Затем добавили 16 мл триэтиламина и продолжили нагревание при той же температуре в течение 1 ч. Реакционную смесь охладили до комнатной температуры и вылили в 600 мл воды. Остаток собрали при отсасывании и промыли водой, в результате получили 5.4 г метил 2-гидрокси-3-(2- никотиноилпропионил)-бензоата, который использовали на следующей стадии без дальнейшей очистки. 3.4 г полученного таким образом соединения нагревали в течение 1.5 ч при 100oC после растворения в смеси, содержащей 15 мл уксусной кислоты и 1 мл 37% соляной кислоты. После охлаждения до комнатной температуры смесь вылили в 150 мл воды и экстрагировали этилацетатом. Органическую фазу промыли 5% водным кислым карбонатом натрия, а затем водой, высушили на безводном сульфате натрия и выпарили в вакууме, получив в результате 1.3 г сырого 8-метоксикарбонил-3- метил-4-оксо-2-(3-пиридил)-4H-1-бензопирана.
1 г этого эфира растворили в 9 мл метанола и 15 мл 1,4-диоксана и медленно добавили 1.7 мл 10 н. гидроксида натрия, поддерживая при этом температуру 20 - 25oC. После выдержки в течение 1 ч при 50oC реакционную смесь вылили в 100 мл воды и экстрагировали этилацетатом. Водный слой подкислили 1 н. соляной кислотой. Осадок собрали при отсасывании; было получено 0.6 г 8-карбокси-3-метил-4- оксо-2-(3-пиридил)-4H-1-бензопирана, который использовали на следующей стадии без дальнейшей очистки.
Соединение, указанное в заглавии, было получено согласно примеру 86, только была использована полученная таким образом кислота, а не 8-карбокси-4-оксо-2-фенил-4H-1-бензопиран, и реакцию проводили в течение 2 ч при комнатной температуре. Основание очищали тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : метанол 98 : 2, а затем кристаллизовали из ацетона. Выход 0.15 г, температура плавления 134.5 - 137oC.
Пример 120. 8-{3-[4-(2-Ацетоксифенил)-1-пиперазинил]- пропилкарбамоил} -3-метил-4-оксо-2-фенил-4H-1-бензопиран.
1 г соединения из примера 59 и 0.32 г 4-диметиламинопиридина растворили в 10 мл дихлорметана. Затем медленно добавили 0.15 мл ацетилхлорида, поддерживая при этом температуру 8 - 10oC. После выдержки в течение 2 ч при комнатной температуре реакционную смесь вылили в 70 мл воды и экстрагировали дихлорметаном. Органический слой промыли 5% водным кислым карбонатом натрия, а затем водой и упаривали в вакууме досуха. Сырое основание очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат : метанол 9 : 1 с последующей кристаллизацией из этанола. Выход 0.74 г соединения, указанного в заглавии, температура плавления 120 - 123oC.
Пример 121. 8-{3-[4-(2-Метиламинокарбонилоксифенил)-1- пиперазинил]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
3 г соединения из примера 59 и 1.8 мл метилизоцианата растворили в 30 мл сухого диметилформамида и встряхивали при комнатной температуре в течение 24 ч. Смесь разбавили водой и встряхивали в течение 2 ч, затем отфильтровали с отсасыванием. Сырое основание очистили тонкослойной хроматографией на силикагеле, элюируя смесью хлороформ : 5 н. метанольный аммиак 100 : 3. После кристаллизации из этанола соединение, указанное в заглавии, плавилось при 132 - 135oC.
Пример 122. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]- пропилкарбамоил}-6-ацетокси-3-метил-4-оксо-2-фенил-4H-1-бензопиран.
0.17 мл ацетилхлорида добавляли по каплям в течение 5 мин при встряхивании при 0oC к раствору 1 г соединения из примера 91 и 0.32 мл триэтиламина в 36 мл хлороформа. После встряхивания в течение 2 ч при указанной температуре реакционную смесь разбавили дихлорметаном и водой. Органический слой отделили, промыли водой, высушили на безводном сульфате натрия и упаривали в вакууме досуха. После кристаллизации остатка из ацетонитрила было получено 0.8 г соединения, указанного в заглавии, плавящегося при 148 - 149oC.
Пример 123. (R, S)-8-{3-[4-(2-метоксифенил)-1-пиперазинил]- пропилкарбамоил}-2,3-дигидро-4-гидрокси-4H-1-бензопиран метансульфонат.
Соединение, указанное в заглавии, было получено методом, описанным в примере 17, но в качестве исходного соединения вместо соединения из примера 1 использовали соединение из примера 87. Реакционную смесь разбавили водой и встряхивали в течение 15 мин, а затем экстрагировали этилацетатом. Обычной обработкой был получен сырой продукт, который очищали тонкослойной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол 95:5. После выпаривания в вакууме собранных фракций было получено чистое основание, которое перевели в метансульфонат и кристаллизовали из ацетонитрила. Температура плавления 172 - 175oC.
Пример 124. 8-{3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-2- (4-аминофенил)-3-метил-4-оксо-4H-1-бензопиран.
2,22 г соединения из примера 113 и 0.56 г никеля Ренея в 96 мл этанола и 4.8 мл уксусной кислоты гидрировали в аппарате Парра (давления водорода 1 атм) при комнатной температуре. После встряхивания в течение 6 ч катализатор отфильтровали. Фильтрат подщелочили 3 н. гидроксидом натрия и разбавили водой. После выдержки в течение 2 дней осадок соединения, указанного в заглавии, собрали при фильтровании с отсасыванием, промыли водой и осушили. Продукт рекристаллизовали сначала из этилацетата, а затем из этанола. Выход 1.5 г, температура плавления 192 - 194oC.
Пример 125. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-2-(4- ацетиламинофенил)-3-метил-4H-1-бензопиран.
Соединение, указанное в заглавии, было получено методом, описанным в примере 36, но вместо соединения, полученного в примере 33, использовали соединение, полученное в примере 134. Целевой продукт очищали кристаллизацией из 95% этанола, температура плавления 207 - 209oC.
Пример 126. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-2-(4-гидроксифенил)-3-метил-4H-1- бензопиран дигидрохлорид моногидрат.
Соединение, указанное в заглавии, было получено методом, описанным в примере 86, но вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали промежуточное соединение CVIII, при этом реакцию проводили при комнатной температуре в течение 14 ч, а гексаметилфосфорамид использовали в качестве сорастворителя. Выделенный диэтилфосфониловый эфир соединения, указанного в заглавии, гидролизовали щелочной обработкой с последующей нейтрализацией разбавленной соляной кислотой. Полученное сырое основание экстрагировали хлороформом. Органический слой промывали водой и выпаривали в вакууме. Соль, полученную при добавлении этанольного хлористого водорода к ацетонному раствору основания, упаривали досуха и промывали ацетоном. Температура плавления 193 - 205oC.
Пример 127. 8-{3-[4-(2-Метоксифенил)-1-пиперазинил]-пропилкарбамоил}-2-фенил- 4,N'N'-триоксо-4H-1-бензотиопиран моногидрат,
К 0,8 г соединения из примера 107 в 15 мл уксусной кислоты добавили 0.32 мл 30% перекиси водорода. Смесь встряхивали в течение 3 ч при 50oC. К этой смеси было добавлено тремя равными порциями с интервалом в 2 ч 0.48 мл 30% перекиси водорода. После охлаждения смесь вылили в 240 мл воды, нейтрализовали (pH 7) 5% водным кислым карбонатом калия и экстрагировали хлороформом. Органический слой промыли водой, высушили на безводном сульфате натрия и выпарили в вакууме. В результате было получено 0.18 г соединения, указанного в заглавии и плавящегося после кристаллизации из ацетонитрила при 172 - 175oC.
Пример 128. 7-{3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-2-фенил-бензо [b]фуран.
Соединение, указанное в заглавии, было получено методом, описанным в примере 86, но вместо 8-карбокси-4-оксо-2-фенил-4H-1-бензопирана использовали 7-карбокси-2-фенил-бензо[b]фуран (полученный согласно EP 0306226) и реакцию проводили в течение 1.5 ч при комнатной температуре. Очистку проводили кристаллизацией из четыреххлористого углерода. Температура плавления 132 - 136oC.
Пример 129. 8-{N-Метил-3-[4-(2-метоксифенил)-1-пиперазинил]-пропилсульфамоил}-3- метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
2.29 г промежуточного соединения VIII добавляли порциями при встряхивании при 0oC к раствору промежуточного соединения C1 и 0.95 г триэтиламина в 30 мл хлороформа. После встряхивания в течение 2 ч при комнатной температуре реакционную смесь разбавили дихлорметаном, водой и 0.5 н. гидроксидом натрия. Органический слой промыли водой, высушили на безводном сульфате натрия и упаривали досуха в вакууме. Остаток очищали тонкослойной хроматографией на силикагеле, элюируя смесью этилацетат:метанол 96:4. При выпаривании в вакууме собранных фракций было получено соединение, указанное в заглавии, в виде основания. Обычным способом его перевели в соль метансульфонат, которую кристаллизовали из этилацетата. Было получено 2.75 г соединения, плавящегося при 135 - 141oC.
Пример 130. 8-{N-Метил-4-[4-(2-метоксифенил)-1-пиперазинил]-бутилсульфамоил}- 3-метил-4-оксо-2-фенил-4H-1-бензопиран метансульфонат.
Соединение, указанное в заглавии, было получено методом, описанным в примере 129, но вместо промежуточного соединения C1 использовали соединение CVIII. Целевой продукт кристаллизовали из ацетонитрила. Температура плавления 173 - 175oC.
Фармакологические данные.
Методология.
Самцы крыс весом 200 - 300 г, самки мышей-альбиносов весом 20 - 30 г и самцы гончих собак весом 10 - 12 кг были получены от Charles River, Italy и Nossan (Correzzana, Milan, Italy) соответственно. Животные содержались при свободном доступе к пище и воде и принудительном цикле свет-темнота при 22 - 24oC до дня эксперимента включительно.
Кратковременная токсичность.
Кратковременная токсичность синтезированных соединений оценивалась после интраперитонеального и орального введения этих соединений самкам мышей-альбиносов. Четыре (по логарифмической шкале) дозы соединения растворили или суспендировали в 0,5% Methocel и вводили в объеме 10 мл/кг группам из четырех мышей. Смертность была зарегистрирована спустя 7 дней после введения препарата. Данные анализа: значения ЛД50 и доверительные пределы этих величин были рассчитаны согласно методу Weil (Biometrics, 8, 249, 1952).
Изучение рецепторной связи.
Связи [3H] празосина (рецепторы альфа1). Кору головного мозга крыс гомогенизировали в 50 mM охлажденном льдом буфере Трис-HCl с pH 7.4 в объеме, в 50 раз превышающем начальный вес этого буфера. Гомогенаты центрифугировали в течение 10 мин при 48000 g, а осадки, полученные в пробирке, вновь суспендировали в том же самом объеме охлажденного льдом буфера, и центрифугировали; повторное суспендирование повторяли еще 2 раза. Затем полученные осадки вновь суспендировали в том же объеме буфера и проводили инкубацию согласно условиям, приведенным ниже в табл. А.
Упомянутые выше излучения рецепторной связи, так же как и экспериментальные данные, полученные на собаках и изложенные ниже, дают возможность установить, что соединения настоящего изобретения являются альфа1-блокаторами, т. е. что они входят в класс веществ, которые широко используются в качестве антигипертензивных и анти-ВРН агентов (см. например, Frishman W.H. et al., Medical Clinics of N. America, 72, 427, 1988 и ссылки, в ней помещенные).
Связь [3H]8-OH-DPAT (5HT1A-рецепторы). Гиппокамп крыс был гомогенизирован в 50 mM охлажденном льдом Трис-HCl буфере с pH 7.4 в объеме, в 50 раз превышающем начальный вес буфера. Гомогенаты центрифугировали в течение 10 мин при 48000 g, полученные в пробирке осадки вновь суспендировали в том же самом объеме охлажденного льдом буфера, инкубировали в течение 10 мин при 37oC, центрифугировали и вновь суспендировали еще 2 раза. Затем полученные осадки снова суспендировали в том же объеме буфера и инкубировали согласно условиям, приведенным ниже в табл. А.
Проведенные изучения рецепторной связи помогают установить, что соединения настоящего изобретения являются лигандами 5НТ1А-рецептора. Ранее сообщалось, что соединения, которые являются 5HT1A-лигандами, оказывают анксиологическое и антидепрессантное воздействие на животных и человека (Hamon M et al., Ann. N.Y. Acad. Sci., 600, 114,1990: Traber J. et al., T.I.P.S., 8, 437, 1987),
Изучение рецепторной связи
Инкубирование завершили спустя соответствующее время (см. табл. А) быстрым фильтрованием через фильтры Whatman GF/B с использованием ячейки Brandel. Затем фильтры дважды промыли 15 мл охлажденного льдом буфера (см. табл. А). Остаточная радиоактивность фильтров определилась с помощью жидкостных сцинтилляционных счетчиков. Неспецифическую связь (которая обычно составляет 10 - 30%) оценивали при добавлении специфических высококонцентрированных заместителей (см. табл. А). Вначале все соединения испытывали при концентрации 1 • 10-6 М, а полная сравнительная кривая (ниже концентрации 10-11М) была построена в условиях значительной активности заместителей. Все эти образцы были повторены трижды.
Сравнительные кривые всегда анализировали с помощью нелинейной кривой, построенной по логарифмическому уравнению согласно методу, изложенному De Lean et al (Am. J. Physiol., 235, E97, 1978) при использовании программы ALLFIT, написанной для IBM РС и известной из National Institutes of Health, Bethesda, Maryland, USA.
K'-индуцированные сокращения участков мочевого пузыря крыс.
Мочевой пузырь крысы был целиком удален и немедленно помещен в раствор Кребса, подогретый до 37oC. Полоски мышцы (20-30 мм длиной, 1 - 2 мм шириной) вырезали из верхней части пузыря. Каждую из полосок поместили в ванночку на 10 мл и присоединили при постоянной нагрузке в 1 г к изометрическому измерителю напряжения (DY-1 Basile, Comerio, Varest, Italy). Сокращения записывались с помощью полиграфа Basile 7070. После 60-минутного периода равновесия полоски обрабатывали 80 mM KCl (конечная концентрация). Это вызывало быстрое фазовое сокращение с последующим замедлением и продолжительную тоническую компоненту. Когда тоническое сокращение стало стабильным, полоски промыли, и спустя 30 мин было индуцировано новое сокращение. После того как были записаны две или более воспроизводимых реакции, в ванну добавили одно из исследуемых лекарственных веществ в определенной концентрации и спустя 30 мин было индуцировано новое сокращение. Экспериментальные группы содержали по меньшей мере 2 препарата, взятые от разных животных при каждой концентрации исследуемых лекарственных веществ. Величины IC50 ингибирования агонист-индуцированных сокращений оценивались с помощью линейного регрессионного анализа.
Влияние на уретральные сокращения и кровяное давление у собак.
Описанные эксперименты были осуществлены согласно методу Imagawa et al (J. Pharmacol. Methods, 22, 103-111, 1989), но со следующими существенными модификациями: взрослых самцов гончих собак, весящих 8 - 10 кг, анестезировали пентобарбиталом натрия, интубировали и осуществляли самопроизвольную вентиляцию воздухом из комнаты. Для осуществления контроля за кровяным давлением в дугу аорты через правую сонную артерию ввели полиэтиленовый катетер.
Боковая левая бедренная вена была канюлирована для вливания анестетика, а правая бедренная вена была канюлирована для введения лекарственных веществ. Для внутриартериального вливания норадреналина (НА) полиэтиленовый катетер был присоединен к нижнему участку желудочной аорты через правую внешнюю подвздошную артерию. За счет такой процедуры НА был селективным образом распределен в нижней части мочеиспускательных путей. Путем срединной лапаротомии были извлечены мочевой пузырь и проксимальная уретра. Для предотвращения заполнения мочевого пузыря оба мочеточника были канюлированы и моча выводилась наружу. Для записи давления в предстательной части мочеиспускательного канала катетер Mikro-tip(6F) ввели в мочевой пузырь через мочеиспускательное отверстие; его удалили, когда датчик давления укрепили в предстательной части канала. Была наложена лигатура между шейкой пузыря и уретрой, чтобы изолировать реакцию последней и чтобы исключить любое взаимодействие с мочевым пузырем. Еще одна лигатура была наложена вокруг Mekro-tip катетера и внешнего мочеиспускательного отверстия, чтобы обезопасить сам катетер. После стабилизационного периода и последующей хирургической процедуры (30 мин), во время которых осуществляли непрерывный мониторинг артериального давления и давления в предстательной части мочеиспускательного канала, с интервалом в 10 мин было произведено введение НА. Доза НА была выбрана такая, чтобы она создавала по крайней мере 100% увеличение давления в предстательной части. Исследуемые соединения вводили в кумулятивном порядке с интервалом в 15 - 20 мин между введениями, а инъекции НА повторяли примерно спустя 5 мин после каждого введения исследуемого соединения.
Кривые, отражающие реакцию на дозы, были построены с помощью вычислений процентного отношения ингибирования к увеличению уретрального давления (индуцированного НА) и процентного отношения падений кровяного давления, вызванного исследуемыми соединениями. Величины ЭД25 (дозы, индуцирующей 25% уменьшение) для диастолического кровяного давления и ИД50 (дозы, вызывающей 50% ингибирование увеличения уретрального давления, индуцированного НА) были вычислены с помощью линейного регрессионного анализа.
Соединения, полученные в примерах, были испытаны согласно методам, приведенным выше. Результаты этих испытаний вместе со сравнительными результатами для стандартов приводятся дальше в табл. I и II. Соединения, имеющие сродство к рецептору (величины IC50) менее ~ 500 nM, считаются обладающими хорошим сродством. Обычно предпочтение отдается соединениям, имеющим величину IC50 менее 100 nM.
Ниже представлены эффективные суточные дозы (выраженные в мг/кг веса тела) для орального, парентерального или внутривенного введения при:
а) обcтруктивных нарушениях нижней части мочевых путей:
Общая доза - 0.001 - 20
Предпочтительная доза - 0.05 - 1
Наиболее предпочтительная ** - 0.3
б) в качестве антигипертензивных агентов
Общая доза - 0.01 - 20
Предпочтительная доза - 0.1 - 5
Наиболее предпочтительная ** - 1
в) в качестве анксиолитиков-антидепрессантов:
Общая доза - 0.01 - 20
Предпочтительная доза - 0.05 - 5
Наиболее предпочтительная ** - 0.5
г) в качестве спазмолитиков мочевого пузыря
Общая доза - 0.01 - 20
Предпочтительная доза - 0.02 - 10
Наиболее предпочтительная ** - 2
Наиболее предпочтительные значения ** относятся к оральным дозировкам. Внутривенные дозировки должны быть в 10 - 100 раз ниже.
В число пациентов, нуждающихся в лечении этими соединениями и составами, входят также люди, имеющие один или более симптомов депрессии (согласно определению Harrisson's Principles of Internal Medicine, XII Ed., McGraw-Hill, Inc. p. 2124), или люди, испытывающие симптомы беспокойства.
Селективное использование дозировок, а именно тех дозировок, которые активны в нижней части мочевых путей и не оказывают при этом существенного эффекта на кровяное давление, зависит от самого применяемого соединения, но обычно может быть применена доза выбранного соединения, до 4 раз превышающая ЭД50 и не оказывающая при этом существенного влияния на кровяное давление. Дальнейшие усовершенствования и оптимизации дозировок возможны не более чем обычные эксперименты.
Активные соединения настоящего изобретения могут применяться орально, например с инертным растворителем или со съедобным носителем, или могут быть заключены в желатиновые капсулы, или же спрессованы в таблетки. Активные соединения настоящего изобретения вместе с наполнителями могут быть использованы для орального терапевтического введения в форме таблеток, пастилок, капсул, эликсиров, суспензии, сиропов, жевательной резины и т.п. Эти препараты должны содержать по крайней мере 0.5% активных соединений, но количество активного ингредиента может варьироваться в зависимости от лекарственной формы и может составлять от 5 до 70% от веса препарата. Количество активного соединения в этих композициях такое, что достигается приемлемая дозировка, а требуемая дозировка может быть получена при приеме некоторого количества дозировок. Согласно настоящему изобретению предпочтительные композиции и препараты приготавливают таким образом, что единица формы оральной дозы содержит 1.00 - 300 мг активного соединения.
Таблетки, капсулы, пастилки и т.п. могут также содержать следующие ингредиенты: связующее типа микрокристаллической целлюлозы, трагакантовой камеди или желатина; наполнитель типа крахмала или лактозы; расщепляющее вещество типа альгиновой кислоты, зернового крахмала и т.п.; смазку типа стеарата; подслащивающий агент типа сахарина или сахарозы или ароматизатор типа перечной мяты, метилсалицилата или апельсиновый ароматизатор. Если единичной дозировкой является капсула, то она может содержать кроме названных выше материалов жидкий носитель типа жирного масла. Другие формы дозировок могут содержать иные разнообразные материалы, которые изменяют физическую форму единичной дозировки, такие как, например, покрытия. Таким образом, таблетки или драже могут быть покрыты сахаром, шеллаком или иным покрытием, улучшающим энтерические свойства. Помимо активных соединений в сиропе может содержаться сахароза в качестве подслащивающего агента и некоторые консерванты, красители и ароматизаторы. Материалы, используемые при получении этих разнообразных композиций, должны быть чистыми в фармацевтическом отношении и нетоксичными в применяемых количествах.
Активные соединения настоящего изобретения могут использоваться для парентерального терапевтического введения в виде раствора или суспензии. Эти препараты должны содержать по меньшей мере 0.1% активного соединения, но его количество может варьироваться от 0.5 до 30% от веса всего состава. Количество активного соединения в таких композициях должно быть таким, чтобы была получена подходящая дозировка. Предпочтительные композиции и препараты согласно настоящему изобретению готовят таким образом, чтобы парентеральная дозировка содержала от 0,2 до 100 мг активного соединения. Растворы или суспензии могут включать также и следующие компоненты: стерильный разбавитель типа воды для инъекций, солевой раствор, нелетучие масла, полиэтиленгликоль, глицерин, пропиленгликоль или иные синтетические растворители; антибактериальные агенты типа бензилового спирта или метилпарабенза; антиоксиданты типа аскорбиновой кислоты или бисульфита натрия; хелатообразователь типа этилендиаминтетрауксусной кислоты; буфер типа ацетатов; цитраты или фосфаты или средства для корректировки тонуса, такие как хлорид натрия или декстроза. Емкость для неединичной парентеральной дозы может быть сделана из стекла или пластических материалов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-(N-ФЕНИЛАМИНОАЛКИЛ)ПИПЕРАЗИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2199533C2 |
| Способ получения производных имидазола или их солей | 1979 |
|
SU865125A3 |
| Производные тиенопиранкарбоксамидов | 2000 |
|
RU2225409C2 |
| БЕНЗОПИРАНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2122000C1 |
| ПРИМЕНЕНИЕ СЕЛЕКТИВНЫХ АНТАГОНИСТОВ АЛЬФА1B-АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА ДЛЯ УЛУЧШЕНИЯ СОСТОЯНИЯ ПРИ СЕКСУАЛЬНОЙ ДИСФУНКЦИИ | 2000 |
|
RU2239633C2 |
| СПОСОБ ПОЛУЧЕНИЯ 9-(2-ГИДРОКСИ)-ЭТОКСИМЕТИЛГУАНИНА | 1994 |
|
RU2125570C1 |
| БЕНЗОТИОФЕНОВЫЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1996 |
|
RU2158737C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОПИРАНОНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ГИДРАТЫ | 1989 |
|
RU2118321C1 |
| ФЕНИЛОКСАЗОЛИДИНОНЫ, ЗАМЕЩЕННЫЕ В КОЛЬЦЕ ГЕТЕРОАРОМАТИЧЕСКИМИ КОЛЬЦАМИ, В КАЧЕСТВЕ АНТИМИКРОБНЫХ АГЕНТОВ | 1996 |
|
RU2154645C2 |
| ЗАМЕЩЕННЫЕ ФЕНИЛФЕНОЛЬНЫЕ СОЕДИНЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2095340C1 |
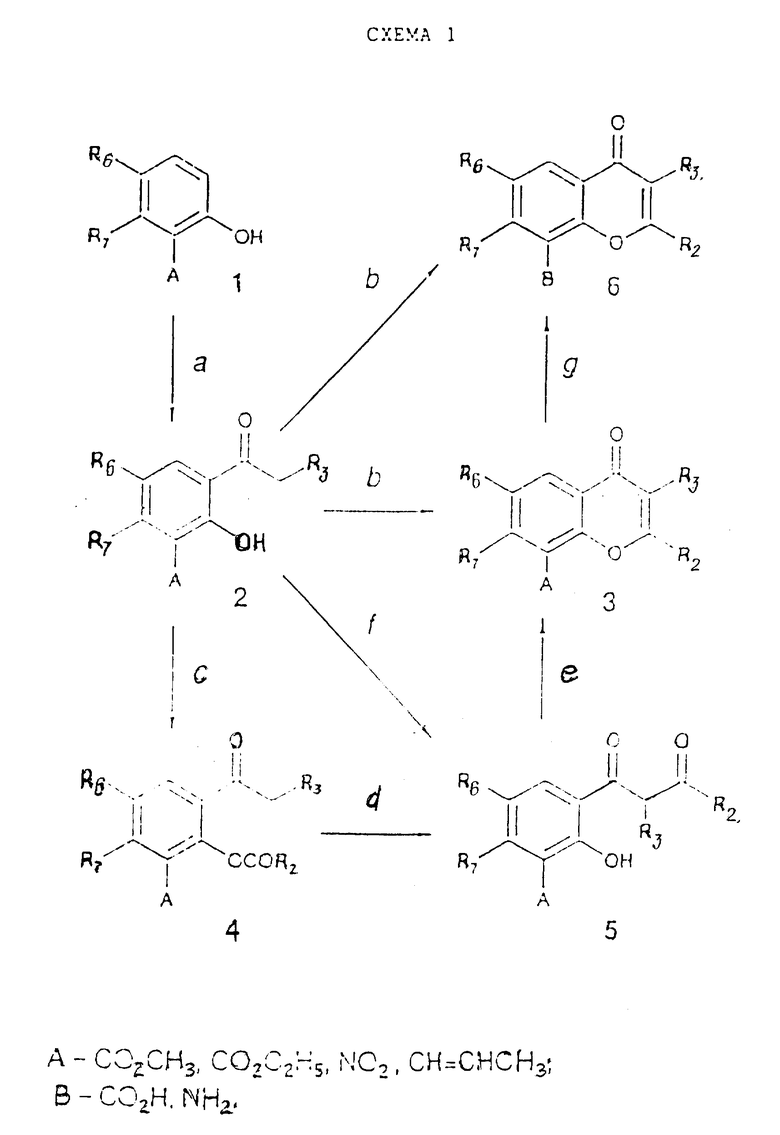
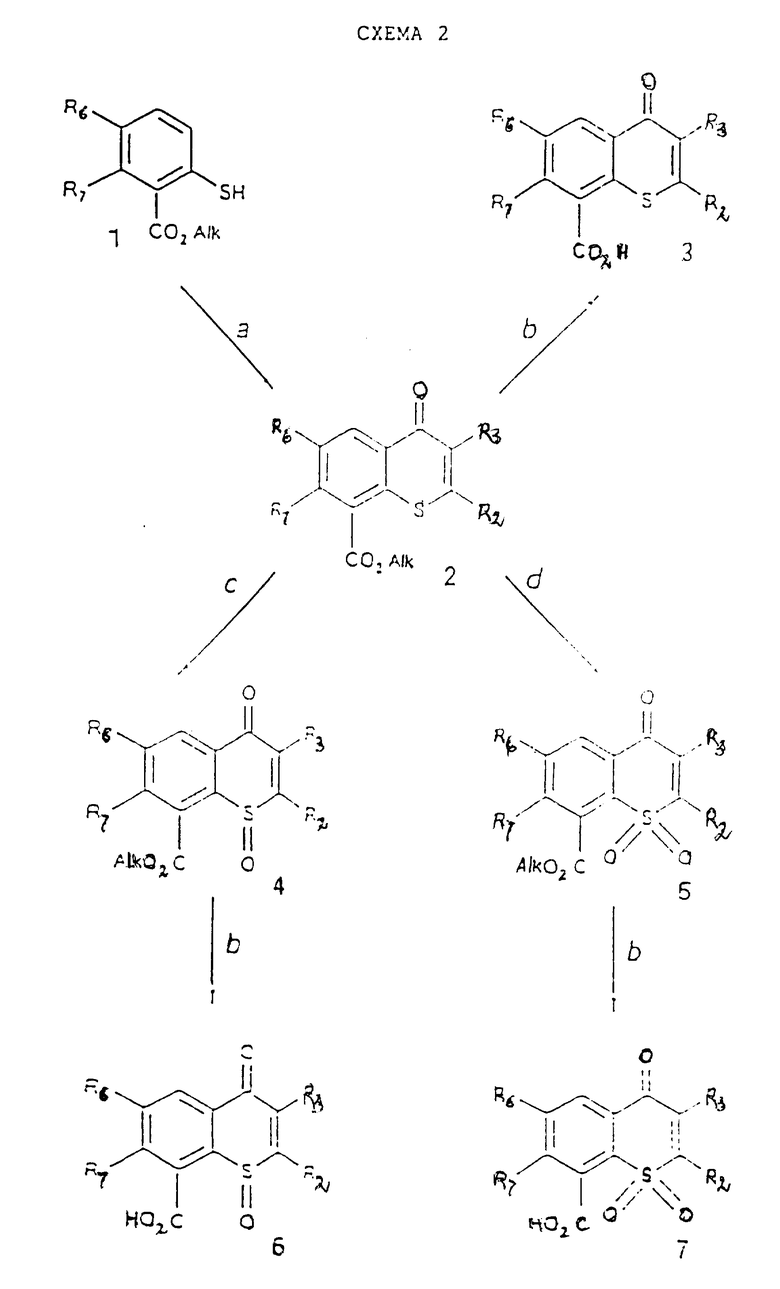
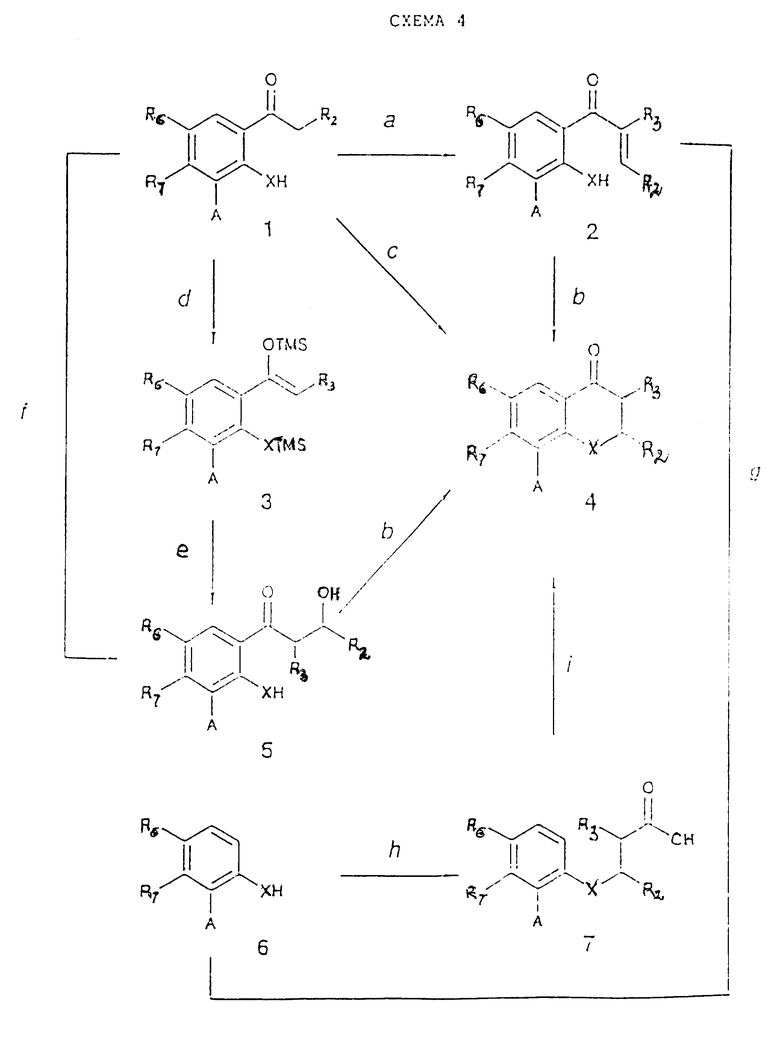
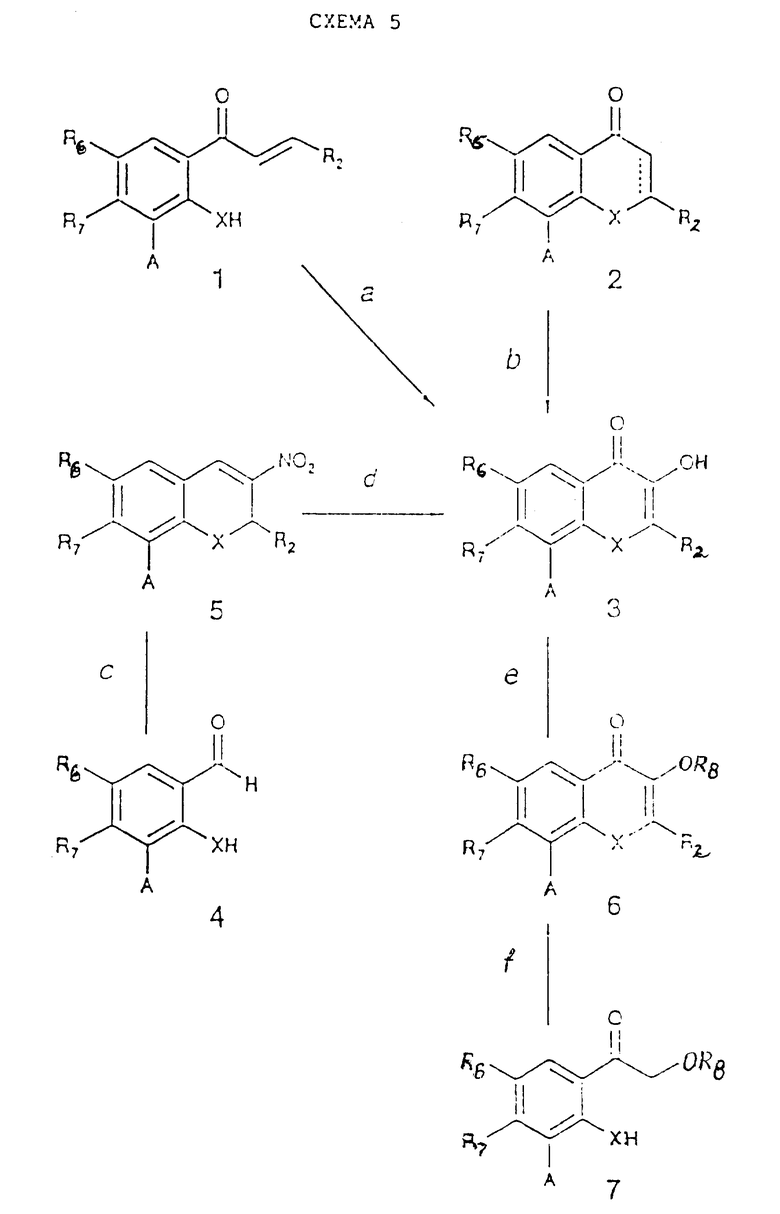

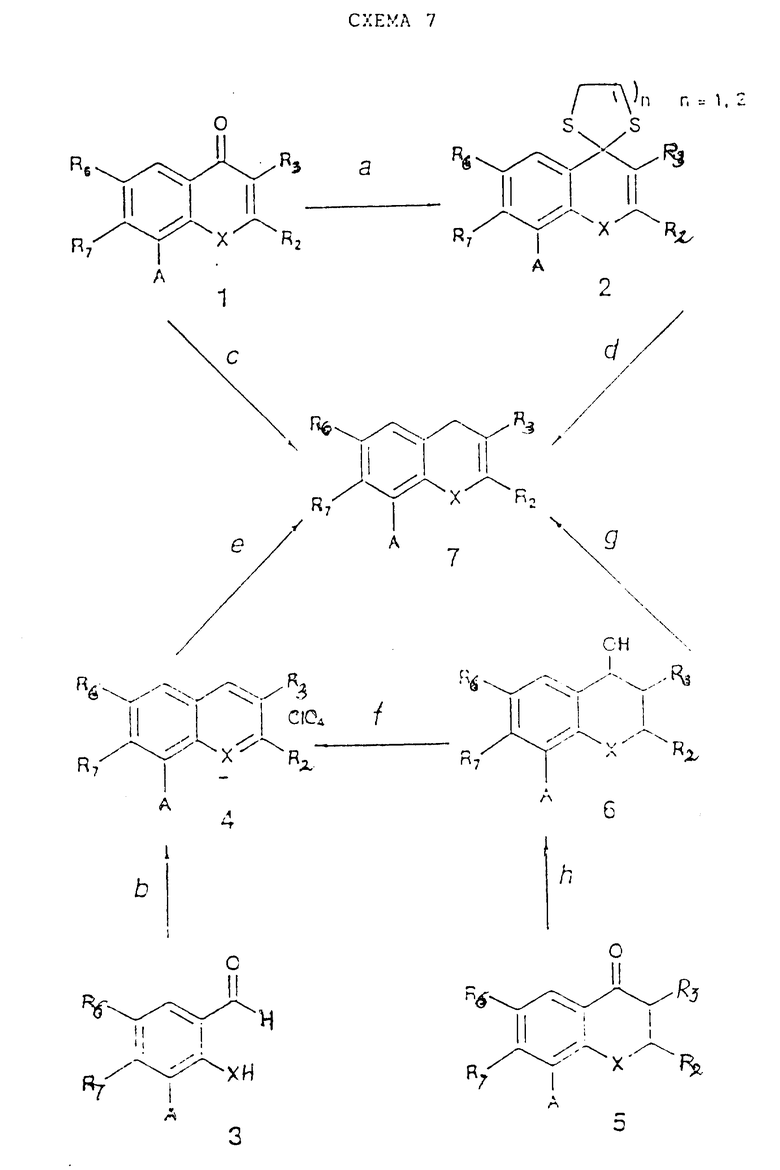
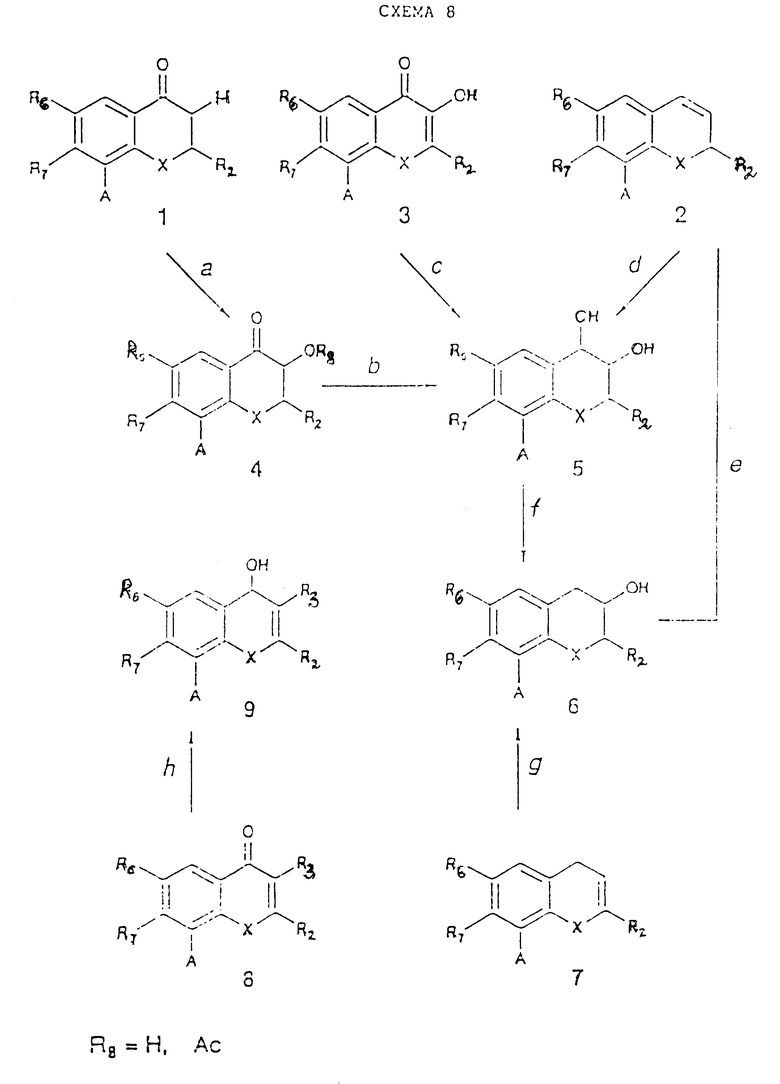
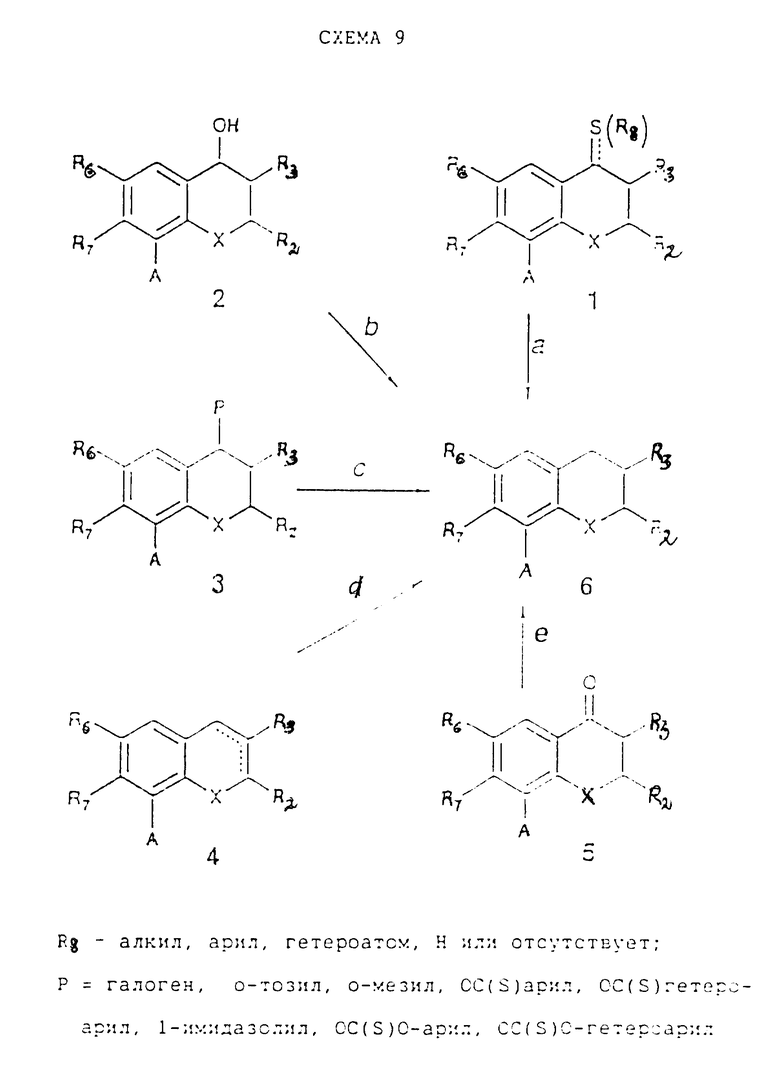
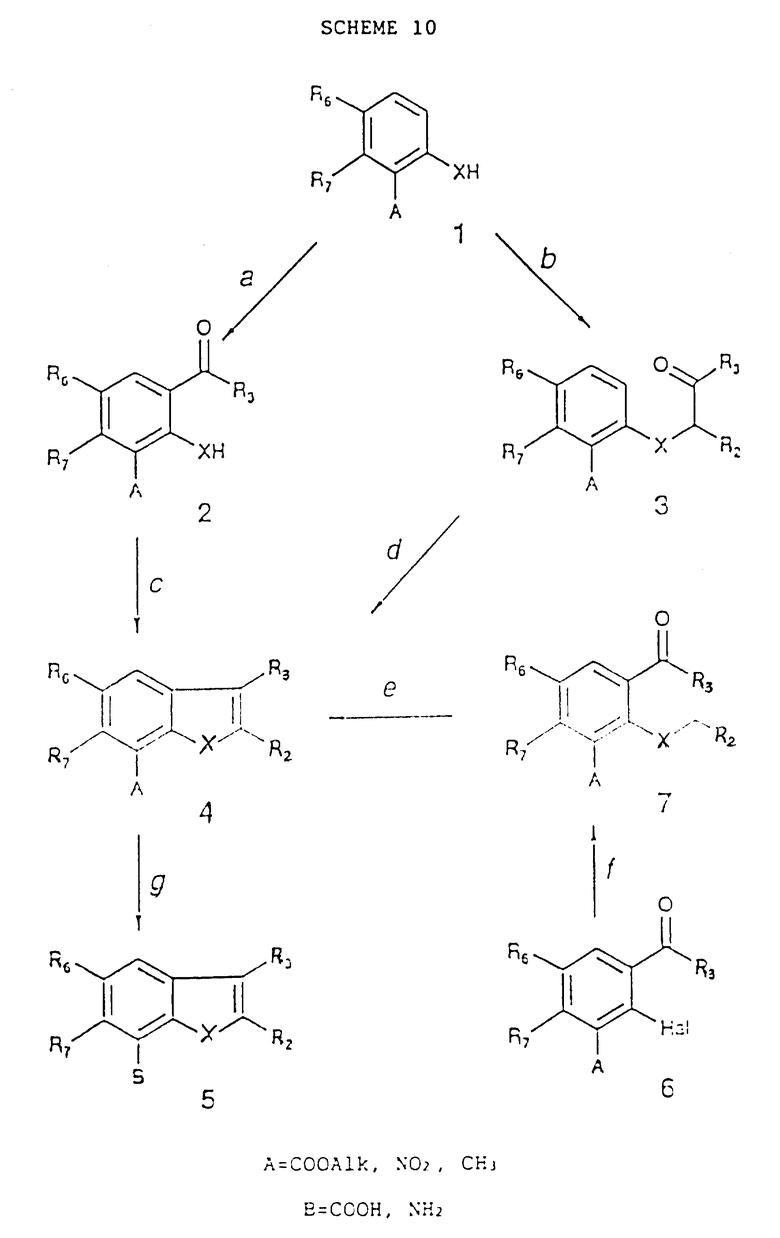
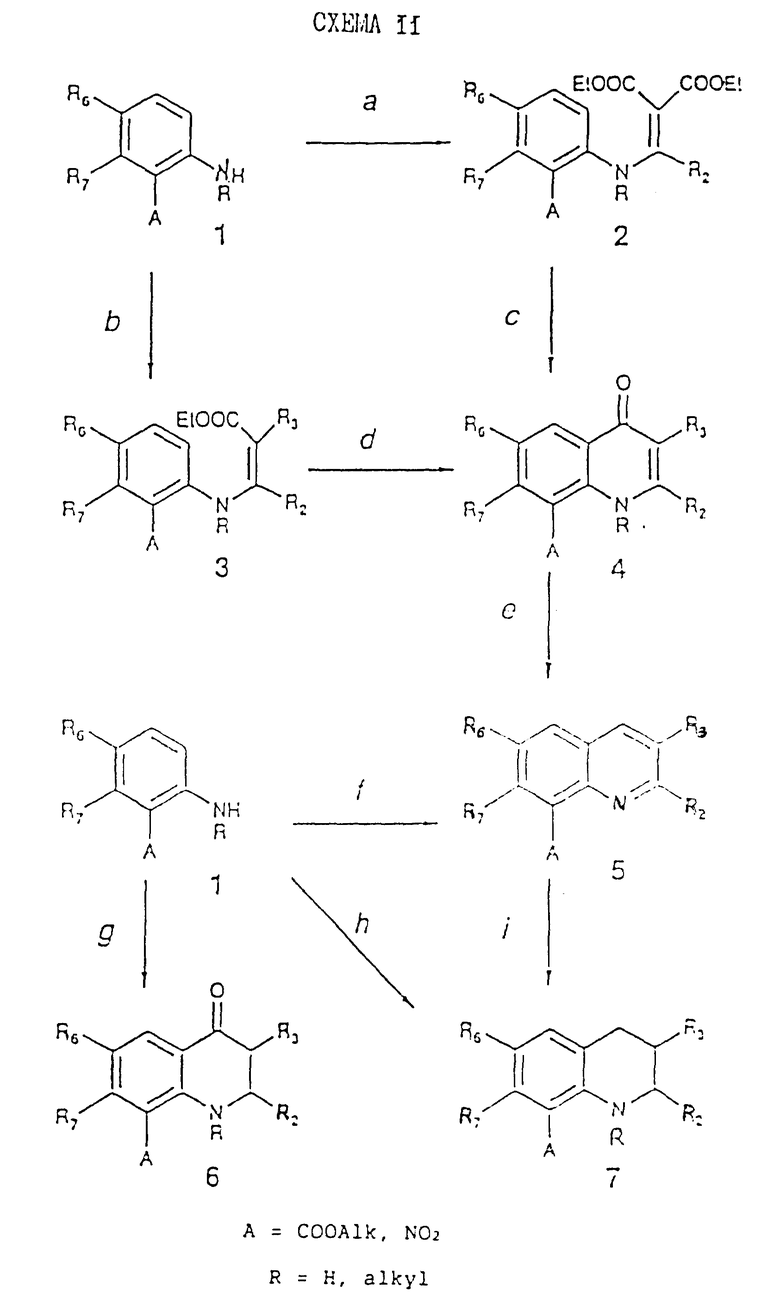
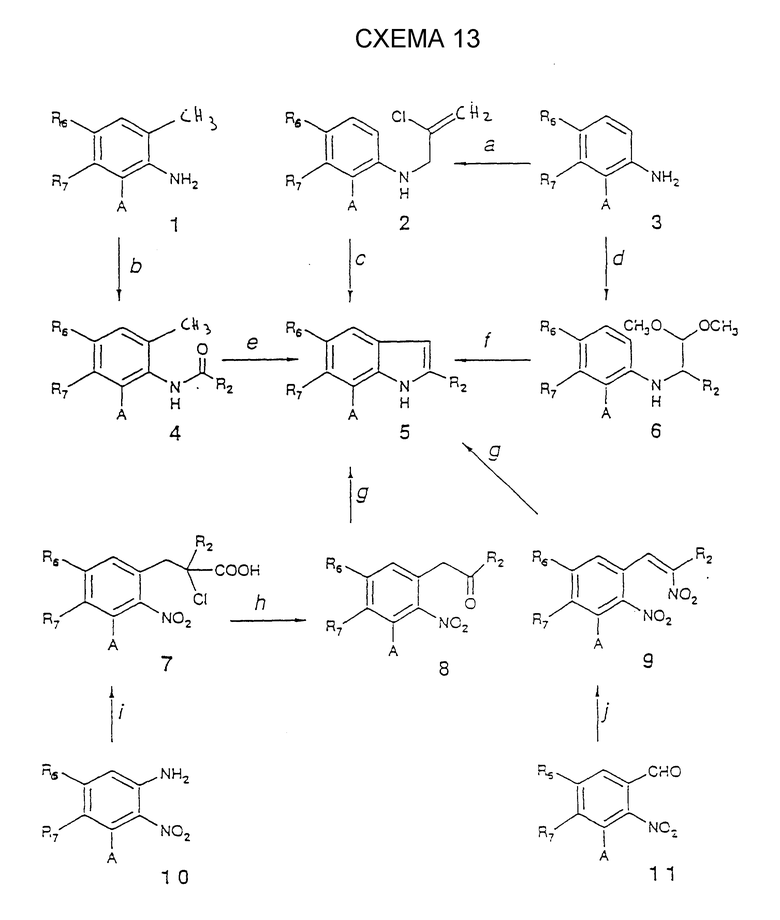
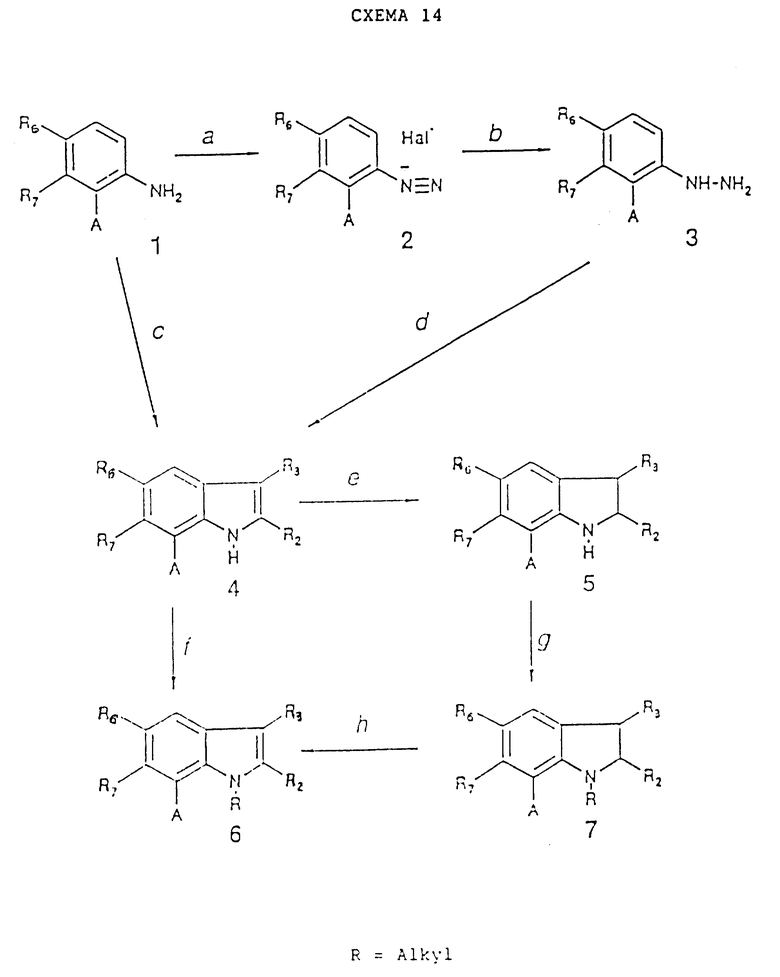
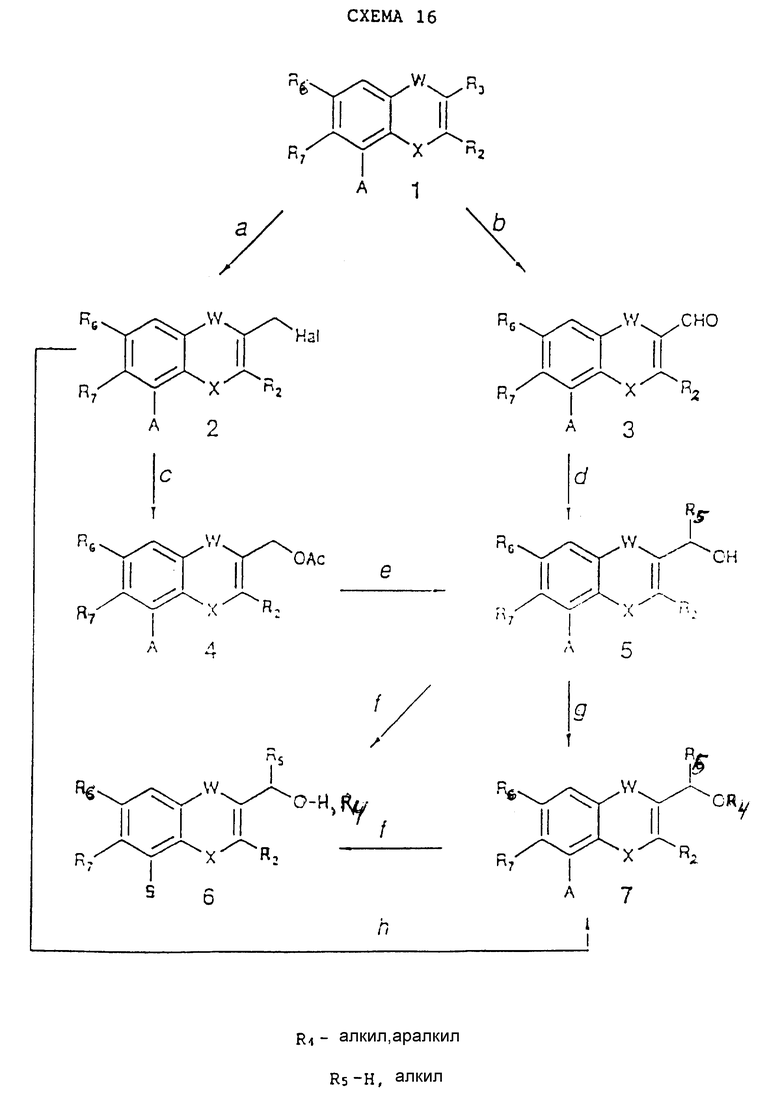
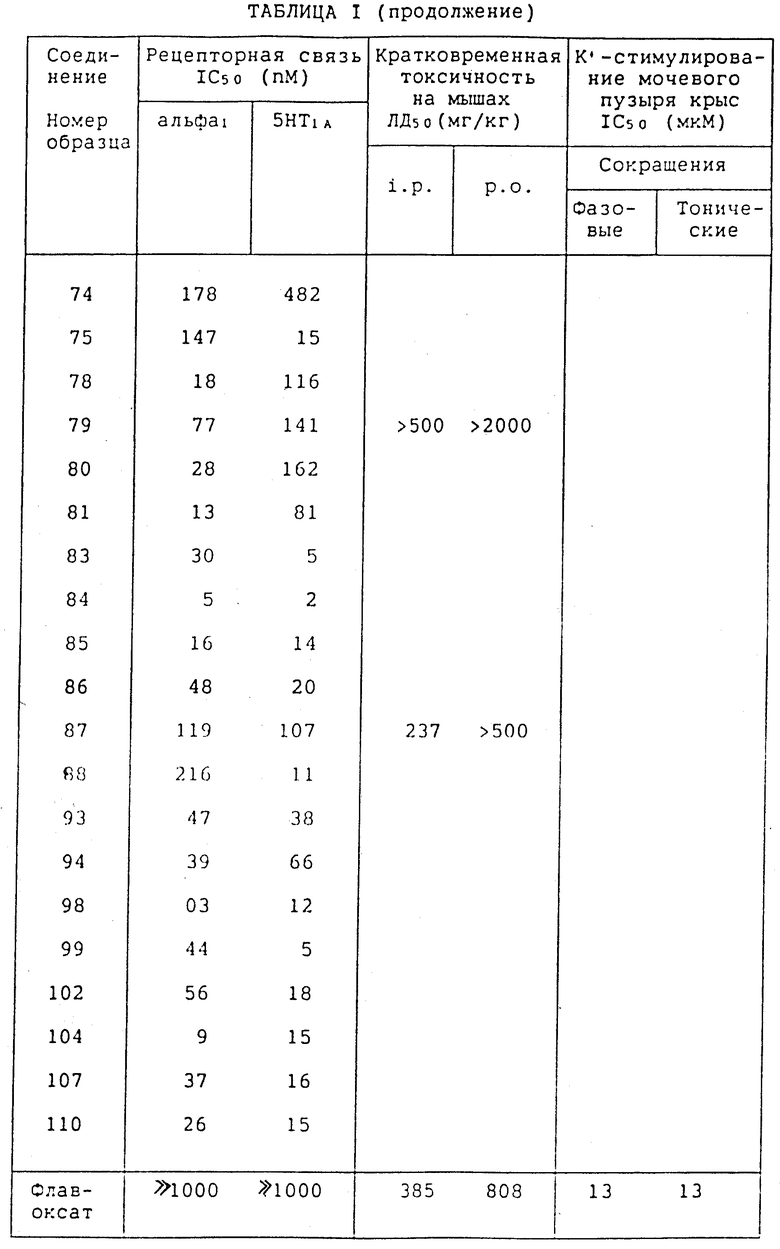
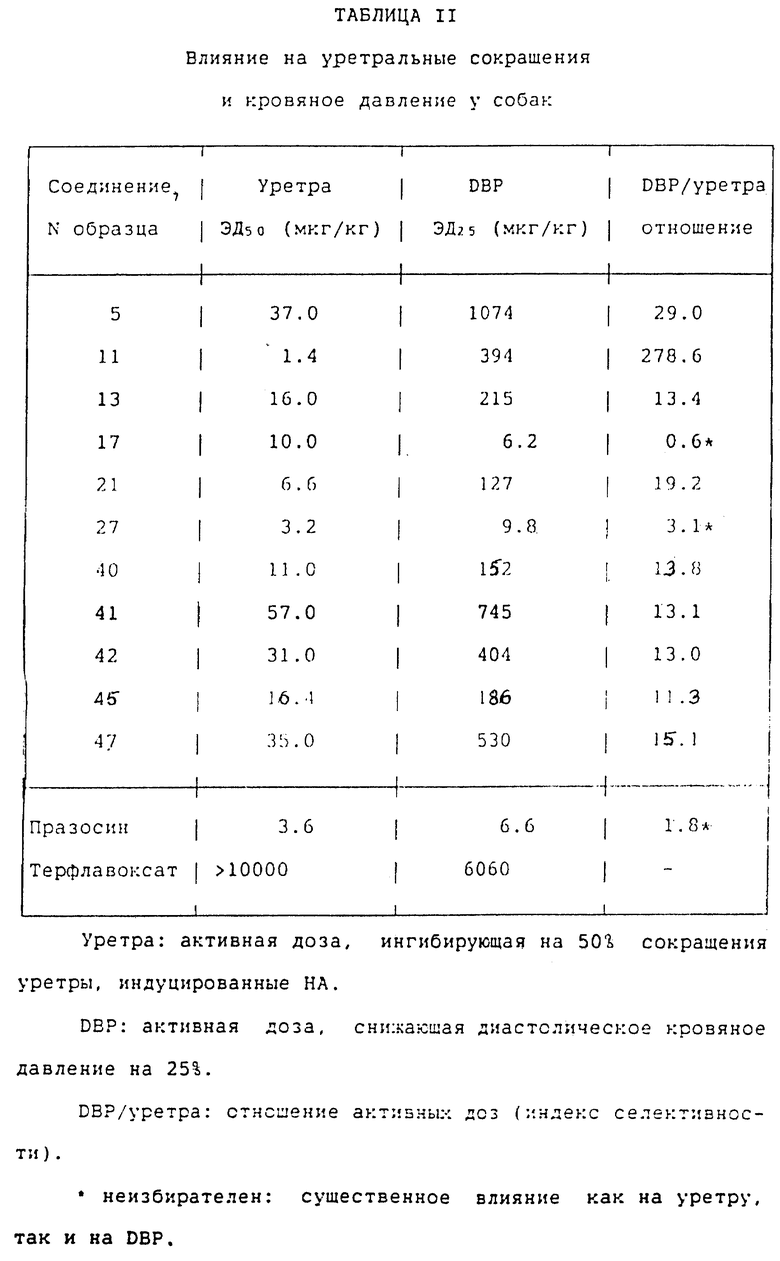

Описываются соединения общей формулы I. Гетероатом Х представляет собой кислород, но может быть и другим. Группа W представляет собой предпочтительно карбонильную группу, но может иметь и другие значения. Предпочтительным гетероциклическим кольцом является 4-оксо-4Н-1-бензопирановое кольцо. Оно может иметь широкий спектр заместителей R2, R3, R6 и R7. Y представляет собой связанную группу, выбранную из широкого спектра, но включающую -COO, -CONH, -O-, -SO2 и SO2NH-. Z представляет собой алкиленовую цепь, а В - сложный амин. Эти соединения и их пролекарства, энантомеры и диастереизомеры, N - оксиды и фармацевтически приемлемые соли полезны для лечения гипертензии и нарушений мочевых путей, связанных с доброкачественной гипертрофией простаты, и для лечения других заболеваний. Предложен способ получения соединений формулы I, заключающийся в том, что соединение общей формулы II F1-Y-Z-L, где F1 представляет собой формулу III, в которой  X, W, R2, R3, R6 и R7 имеют указанные выше значения, L - атом галогена или отщепляемая группа, подвергают конденсации с соединением формулы IV Н-В, где В имеет значения, указанные в п.1 формулы изобретения. 6 с. и 10 з.п.ф-лы, 16 ил., 2 табл.
X, W, R2, R3, R6 и R7 имеют указанные выше значения, L - атом галогена или отщепляемая группа, подвергают конденсации с соединением формулы IV Н-В, где В имеет значения, указанные в п.1 формулы изобретения. 6 с. и 10 з.п.ф-лы, 16 ил., 2 табл.
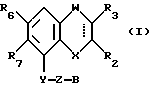
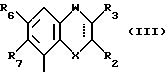
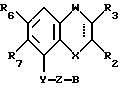
где  - ординарная или двойная связь;
- ординарная или двойная связь;
X - атом серы или кислорода;
W - валентная связь или карбонил, метиленовая или гидроксиметиленовая группа;
R2 - атом водорода, C1 - C6-алкил, незамещенный или замещенный фенилом C1 - C6-алкенил, C3 - C6-карбоциклил, пиридил, фурил, тиенил, бензоил, фенил, незамещенный или замещенный феноксигруппой, фенилом, трифторметилом, C1 - C6-алкокси-, гидрокси-, амино-, C1 - C6-ациламино- или нитрогруппой;
R3 - атом водорода или галогена, C1 - C6-алкил или гидрокси-C1 - C6-алкил;
R6 - атом водорода или галогена, нитро-, амино-, C1 - C6-алкиламино-, ди(C1 - C6-алкил)амино-, C1 - C6-ациламино-, C1 - C6-алкилсульфониламино-, гидрокси-, C1 - C6-алкоксигруппа или C1 - C6-алкил;
R7 - атом водорода или C1 - C6-алкоксигруппа;
Y представляет собой одну из следующих групп, в каждой из которых представленный слева конец присоединен к гетероциклическому кольцу, а каждый представленный справа конец присоединен к Z:
Y1 - -CO-;
Y2 - -COO-;
Y3 - -CONH-;
Y4 - -CON(CH3)-;
Y5 - -COH(OH)-;
Y6 - -CH(OH)-;
Y7 - -CH(OAlKyl)-;
Y8 - -CH = CH-;
Y10 - -CH = CH-CONH-;
Y12 - -CH2-;
Y13 - -CH2COO-;
Y14 - -CH2CONH-;
Y15 - -CH2NH-;
Y16 - -CH2N(CH3)-;
Y17 - -CH2N(COCH3)-;
Y18 - -CH2N(CONH2)-;
Y19 - -CH2NHCO-;
Y20 - -CH2N(CH3)CO-;
Y21 - -CH2NH-CONH-;
Y22 - -CH2NHSO2-;
Y23 - -CH2O-;
Y24 - -CH2S-;
Y25 - -CH2SO-;
Y26 - -CH2SO2-;
Y27 - -CH2SO2NH-;
Y28 - -CH2SO2N(CH3)-;
Y29 - -NH-CONH;
Y30 - -N(CH3)-;
Y32 - -N(CONH2)-;
Y33 - -NHCO-;
Y34 - -N(CH3)CO-;
Y35 - -NHSO2-;
Y37 - -O-;
Y38 - -S-;
Y39 - -SO-;
Y40 - -SO2-;
Y41 - -SO2NH-;
Y42 - -SO2N(CH3)-;
Y45 - -CSNH-;
Y47 - 
Z - линейная или разветвленная C1 - C6-алкиленовая группа;
B представляет одну из следующих групп: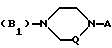
в которой Q - метиленовая группа;
A представляет одну из следующих групп:
A1 - фенил, замещенный одним или двумя атомами галогена и/или одной или двумя C1 - C6-алкил, C1 - C6-алкокси- или гидроксигруппой;
A2 - 2-пиримидинильная группа или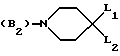
в которой каждый из L1 и L2 независимо друг от друга - атом водорода, фенил, 4-фторбензол или 2-оксо-1-бензимидазолинильная группа, при условии, что оба L1 и L2 не являются атомами водорода;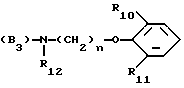
в которой каждый из R10 и R11 независимо - атом водорода или C1 - C6-алкоксигруппа;
R12 - атом водорода или C1 - C6-алкил;
n = 2 или 3,
или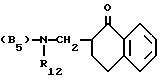
в которой R12 имеет указанные значения, при условии, что если R6 и R7 - атомы водорода, то Y не может быть атомом кислорода или серы,
или его энантиомер, диастереоизомер, N - оксид или фармацевтически приемлемая соль, или соединение формулы I, в котором амино-, имино- и гидроксигруппы могут быть замещены. представляет собой двойную связь, X - атом кислорода, W - карбонильная группа, R2 - фенильная группа, R3 - метильная группа, R6 - атом водорода, R7 - атом водорода.
представляет собой двойную связь, X - атом кислорода, W - карбонильная группа, R2 - фенильная группа, R3 - метильная группа, R6 - атом водорода, R7 - атом водорода.
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил] -1-оксоэтил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 2-[4-(2-метилфенил)-1-пиперазинил] -1-оксоэтил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 2-[4-(2-этоксифенил)-1-пиперазинил] -1-оксоэтил} -3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -1-оксопропил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{3-[4-(2-метоксифенил)-1-пиперазинил]-пропоксикарбонил)}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 2-[4-метоксифенил)-1-пиперазинил]-этоксикарбонил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 3-[4-(2-хлорфенил-1-пиперазинил)-пропоксикарбонил} -3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-[3-(4-фенил-1-пиперазинил)-пропоксикарбонил] -3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил]-пропоксикарбонил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -2- метил-2-пропоксикарбонил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил]-этилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ 3-[2-(2-метоксифенокси)-этиламино]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-[3-(4-фенил-1-пиперазинил)-пропилкарбамоил] -3-метил-4-оксо-2-фенил-4H-1-бензопиран,
8-{ N-метил-2-[4-(2-метоксифенил)-1-пиперазинил] -этилкарбамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 1-гидрокси-2-[4-(2-метоксифенил)-1-пиперазинил] -этил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 1-гидрокси-2-[4-(2-метилфенил)-1-пиперазинил]-этил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 1-гидрокси-2-[4-(2-этоксифенил)-1-пиперазинил] -этил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{1-гидрокси-3-[4-(2-метоксифенил)-1-пиперазинил]-пропил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 1-гидрокси-4-[4-(2-метоксифенил)-1-пиперазинил]-бутил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 1-этокси-2-[4-(2-метоксифенил-1-пиперазинил] -этил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-метил-2-[4-метоксифенил)-1-пиперазинил] -этиламинометил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-ацетил-2-[4-метоксифенил)-1-пиперазинил]-этиламинометил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-[4-2-метоксифенил)-1-пиперазинилацетамидометил] -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-метил-N-[4-(2-метоксифенил)-1-пиперазинил] -ацетамидометил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил]-этоксиметил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[2-(2-этоксифенокси)-этиламино] -этоксиметил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил] -этилтиометил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил]-этилсульфинилметил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил]-этилсульфонилметил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил] -этиламино} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил]-пропиламино}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 4-[4-(2-метоксифенил)-1-пиперазинил] -бутиламино}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-метил-3-[4-(2-метоксифенил)-1-пиперазинил] -пропиламино}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-ацетил-3-[4-(2-метоксифенил)-1-пиперазинил]-пропиламино}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил]-пропиламидо}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил] -этилуреидо}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил] -этокси} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропокси}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 4-[4-(2-метоксифенил)-1-пиперазинил] -бутокси} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 5-[4-(2-метоксифенил)-1-пиперазинил] -пентилокси}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-оксо-1-пиперазинил] -пропокси}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-2-{ 2-[2-(2,6-диметоксифенокси)-этиламино] -этокси}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-гидрокси-3-[4-(2-метоксифенил)-1-пиперазинил] -пропокси}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилтио} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилсульфонил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил] -этилсульфамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-метил-2-[4-(2-метоксифенил)-1-пиперазинил] -этилсульфамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-карбамоил-3-[4-(2-метоксифенил)-1-пиперазинил] -пропиламино}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 4-[4-(2-метоксифенил)-1-пиперазинил]-1-оксобутил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[2-(1,4-бензодиоксанил)-метиламино]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 4-[4-(2-метоксифенил)-1-пиперазинил]-бутил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-[3-(4-фенил-1-пиперидинил)-пропилкарбамоил] -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-[3-(4,4-дифенил-1-пиперидинил)-пропилкарбамоил] -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(4-фторбензоил)-1-пиперидинил] -пропилкарбамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-оксо-1-бензимидазолинил)-1-пиперидинил]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-пиримидинил)-1-пиперазинил] -пропилкарбамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-гидроксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 4-[4-(2-метоксифенил)-1-пиперазинил] -бутилкарбамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил]-пропилсульфамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[N-метил-2-(2-метоксифенокси)-этиламино] -пропилкарбамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-метил-3-[4-(2-метоксифенил)-1-пиперазинил]-пропионамидо}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-фенил-4-оксо-4Н-1-бензопиран,
8-{ 3-[(3,4-дигидро-1-оксо-2Н-нафтил)-метиламино]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил] -этоксикарбонилметил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 4-[4-(2-метоксифенил)-1-пиперазинил] -бутилсульфамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-(2-тетрагидропиранилокси)-3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 4-[4-(2-метоксифенил)-1-пиперазинил] -бутирамидо}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
Е-8-{2-[4-(2-метоксифенил)-1-пиперазинил-этоксииминометил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-гидрокси-3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
Е-8-{ 2-[4-(2-метоксифенил)-1-пиперизинил] -этилкарбамоил}-этинил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 4-[4-(2-метоксифенил)-1-пиперазинил] -бутилсульфинил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[3-(2-метоксифенокси)-пропиламино] -пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[2-(2-метилтиофенокси)-этиламино] -пропилкарбамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{3-[2-(2,6-диметоксифенокси)-этиламино]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(5-хлор-2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
(Е)-8-{ 4-[4-(2-метоксифенил)-1-пиперазинил]-1-бутенил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
(Е)-8-<2-{ 2-[4-(2-метоксифенил)-1-пиперазинил] -этоксикарбонил} -этинил>-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил] -этилкарбамоилметил}-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-ацетил-3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 2-[4-(2-метоксифенил)-1-пиперазинил]-этилсульфониламино}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил]-пропилтиокарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 4-[4-(2-метоксифенил)-1-пиперазинил] -бутилсульфонил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-гидроксиметил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2,3-дигидро-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -6-бром-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-6-метокси-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-6-гидрокси-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3,6-диметил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -3-метил-6-нитро-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-6-амино-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-6-ацетамидо-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-6-этиламино-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-6-диметиламино-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-7-метокси-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-метил-4-оксо-2-(4-трифторметилфенил)-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2-(4-бензоилфенил)-3-метил-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-метил-4-оксо-2-(4-феноксифенил)-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2,3-диметил-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2-(t-бутил)-3-метил-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -2-циклогексил-3-метил-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2-(2-фурил)-3-метил-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-метил-4-оксо-2-тиенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -4-оксо-2-фенил-4Н-1-бензотиопиран,
(Е)-8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-метил-4-оксо-2-(2-стирил)-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил-2-(4-метилфенил)-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2-(4-метоксифенил)-3-метил-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -2-(4-фторфенил)-3-метил-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -6-метансульфониламино-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил-2-(4-нитрофенил)-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -6-диэтоксифосфонилокси-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-метил-4-оксо-2-трифторметил-4Н-1-бензопиран,
8-{ N-метил-3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
7-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2-бензоил-3-этил-бензо[b]фуран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -2-(4-бифенил)-3-метил-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-3-метил-4-оксо-2-(3-пиридил)-4Н-1-бензопиран,
8-{ 3-[4-(2-ацетоксифенил)-1-пиперазинил]-пропилкарбамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метиламинокарбонилоксифенил)-1-пиперазинил]-пропилкарбамоил} -3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-6-ацетокси-3-метокси-4-оксо-2-фенил-4Н-1-бензопиран,
(R,S)-8-{3-[4-(2-метоксифенил)-1-пиперазинил]-пропилкарбамоил}-2,3-дигидро-4-гидрокси-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2-(4-аминофенил)-3-метил-4-оксо-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2-(4-ацетиламинофенил)-3-метил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -2-(4-гидроксифенил)-3-метил-4Н-1-бензопиран,
8-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил}-2-фенил-4,N1, N4-триоксо-4Н-1-бензотиопиран,
7-{ 3-[4-(2-метоксифенил)-1-пиперазинил] -пропилкарбамоил} -2-фенил-бензо[b]фуран,
8-{ N-метил-3-[4-(2-метоксифенил)-1-пиперазинил]-пропилсульфамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран,
8-{ N-метил-4-[4-(2-метоксифенил)-1-пиперазинил] -бутилсульфамоил}-3-метил-4-оксо-2-фенил-4Н-1-бензопиран;
или пролекарство, энантиомер, диастереоизомер, N-оксид или фармацевтически приемлемая соль такого соединения.
F1 - Y - Z - L,
где F1 представляет собой общую формулу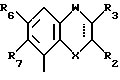
в которой  X, W, R2, R3, R6 и R7 имеют значения, указанные в п.1;
X, W, R2, R3, R6 и R7 имеют значения, указанные в п.1;
L - атом галогена или отщепляемая группа,
подвергают конденсации с соединением формулы III
H - B,
где B имеет значения, указанные в п.1.
F1 - Y - H,
где F1 - группа общей формулы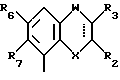
в которой  , W, X, R2, R3, R6 и R7 имеют значения, указанные в п.1,
, W, X, R2, R3, R6 и R7 имеют значения, указанные в п.1,
подвергают конденсации с соединением формулы V
L - Z - B,
где L - атом галогена или отщепляемая группа,
Z и B имеют значения, указанные в п.1. Z и B имеют значения, указанные в п.1, а Y представляет собой одну из групп Y3, Y4, Y5, Y10, Y14, Y27, Y28, Y41 или Y42, значения которых определены в п.1, заключающийся в том, что соединение общей формулы VI
Z и B имеют значения, указанные в п.1, а Y представляет собой одну из групп Y3, Y4, Y5, Y10, Y14, Y27, Y28, Y41 или Y42, значения которых определены в п.1, заключающийся в том, что соединение общей формулы VI
F1 - X1 - Q - Cl,
где F1 - группа общей формулы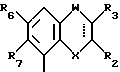
в которой  R2, R3, R6, R7, W и Х имеют значения, указанные в п.1;
R2, R3, R6, R7, W и Х имеют значения, указанные в п.1;
X1 - валентная связь, метиленовая или виниленовая группа;
Q - карбонильная или сульфонильная группа,
подвергают конденсации с соединением общей формулы VII
A - NH - Z - B,
где A - атом галогена, C1 - C6-алкил или группа OPr, где Pr - защитная группа.
F1 - X1 - COOH,
где F1 имеет значения, определенные в п.10;
X1 имеет значения, определенные в п.12,
подвергают конденсации с соединением общей формулы
L - Z - B или A1 - NH - Z - B,
где L имеет значения, указанные в п.10;
A1 имеет значения, указанные в п.12 для значения A,
или с соединениями HS - Z - B, HO - Z - B или H2NO - Z - B, где Z и B имеют значения, определенные в п.1.
| WO 9118597 A1, 1991 | |||
| WO 9119707 A1, 1991 | |||
| US 2921070 A, 1960 | |||
| US 3810896 A, 1974 | |||
| СОСУД ДЛЯ ОБРАБОТКИ ПРОВОЛОКИ, НИТЕЙ ИЛИ ЛЕНТ ЖИДКОСТЯМИ | 1930 |
|
SU19015A1 |
| 0 |
|
SU156331A1 | |
| 0 |
|
SU270342A1 | |
| ВХОДНОЙ ОГОЛОВОК ГЛУБИННОГО ВОДОСБРОСА12 | 0 |
|
SU306226A1 |
| WO 9006112 A1, 1990 | |||
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, т | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
