Предпосылки изобретения
Изобретение относится к новому способу получения хирально чистых β-аминоспиртов, а также их промежуточных соединений, включая α-аминокислоты. Соединения по настоящему изобретению могут найти применение для различных целей, в том числе для использования в фармацевтических композициях.
Для получения предпочтительного энантиомера из α-аминокислот описано множество методов. Эти методы на определенной стадии синтеза целевого соединения требуют применения либо методов разделения, либо методов асимметрического синтеза. Для получения хирально чистых целевых соединений требуется использование более эффективных средств.
Краткое описание изобретения
В соответствии с одним из аспектов в настоящем изобретении предлагается способ получения хирально чистых α-аминокислот.
В соответствии с еще одним из аспектов настоящего изобретения предлагается способ получения хирально чистых S-энантиомеров β-аминоспиртов.
Наконец, в соответствии с еще одним из аспектов настоящего изобретения предлагается способ получения хирально чистых S-энантиомеров N-сульфонил-β-аминоспиртов.
Эти и другие аспекты настоящего изобретения станут понятны специалистам в данной области техники после прочтения приведенного далее подробного описания изобретения.
Подробное описание изобретения
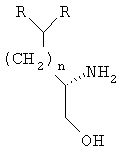
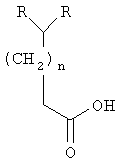
Настоящее изобретение касается способа получения хиральных α-аминокислот формулы (R)2CH(CH2)nCH(NH2)C(=O)OH, где n равно от 0 до приблизительно 10; хиральных β-аминоспиртов формулы (R)2CH(CH2)nCH(NH2)CH2OH и хиральных S-энантиомеров N-сульфонил-β-аминоспиртов формулы (R)2CH(CH2)nCH(CH2OH)NH-S(O)2-2-C4H2S-5-Cl, где R обозначает низший алкил, замещенный низший алкил, низший алкенил, замещенный низший алкенил, низший алкинил, замещенный низший алкинил, циклоалкил, замещенный циклоалкил, фенил, замещенный фенил, бензил, замещенный бензил, CH2циклоалкил, CH2-3-индол, CH(низший алкил)-2-фуран, CH(низший алкил)-4-метоксифенил, CH(низший алкил)фенил или CH(OH)-4-SCH3-фенил.
В соответствии с настоящим изобретением могут быть получены как натуральные, так и искусственные α-аминокислоты, как натуральные, так и искусственные β-аминоспирты и их промежуточные соединения. Эти α-аминокислоты и β-аминоспирты можно также обозначить как 2-аминокислоты или 2-аминоспирты.
Используемый по тексту настоящего описания термин "хирально чистый" обозначает соединения, которые представляют собой 100%-ную S-энантиомерную форму по данным высокоэффективной хиральной жидкостной хроматографии. Другие методы определения хиральной чистоты включают обычные аналитические методы, в том числе удельное вращение плоскости поляризации, и обычные химические методы. Методика, применяемая для определения хиральной чистоты, не является ограничением настоящего изобретения.
Как приводится в описании, после стадии перекристаллизации способ по настоящему изобретению позволяет получить хирально чистые α-аминокислоты или β-аминоспирты. В том случае, если хиральная чистота не является необходимой, способ по настоящему изобретению может быть использован также для получения хиральных α-аминокислот или β-аминоспиртов, которые содержат некоторый процент смеси энантиомерных форм, в частности, в соответствии со способом по настоящему изобретению без проведения перекристаллизации они могут состоять приблизительно от 90% до приблизительно 99% из S-энантиомеров.
В одном из вариантов осуществления настоящего изобретения предлагается способ получения хиральных S-энантиомеров α-аминокислот, который включает приготовление металорганического реагента из галоидного алкила формулы (R)2CH(CH2)nCH2X, где X обозначает Cl, Br или I, а n обозначает от 0 до приблизительно 10; добавление металлорганического реагента к диоксиду углерода с получением карбоновой кислоты; активирование карбоновой кислоты хлорангидридом кислоты, трихлоридом фосфора, ангидридом кислоты или тионилхлоридом в присутствии основания в форме третичного амина; взаимодействие продукта со стадии активирования с солью щелочного металла S-4-бензил-2-оксазолидинона; обработку продукта, полученного на стадии взаимодействия с щелочным металлом, сильным ненуклеофильным основанием с образованием енолят-аниона; взаимодействие енолят-аниона с 2,4,6-триизопропилбензолсульфонилазидом с образованием азида оксазолидинона; гидролиз азида оксазолидинона водным раствором основания с образованием α-азидокислоты; восстановление азидокислоты в α-аминокислоту и перекристаллизацию α-аминокислоты с получением хирально чистой α-аминокислоты.
Другой вариант осуществления настоящего изобретения относится к способу получения хиральных S-энантиомеров β-аминоспиртов, который включает описанное выше получение α-аминокислоты, восстановление α-аминокислоты в β-аминоспирт и перекристаллизацию β-аминоспирта с получением хирально чистого β-аминоспирта.
В еще одном варианте осуществления настоящего изобретения предлагается способ получения хиральных S-энантиомеров N-сульфонил-β-аминоспиртов общей формулы
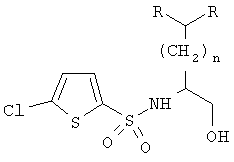
где R обозначает низший алкил, замещенный низший алкил, низший алкенил, замещенный низший алкенил, низший алкинил, замещенный низший алкинил, циклоалкил, замещенный циклоалкил, фенил, замещенный фенил, бензил, замещенный бензил, CH2циклоалкил, CH2-3-индол, CH(низший алкил)-2-фуран, CH(низший алкил)-4-метоксифенил, CH(низший алкил)фенил или CH(OH)-4-SCH3-фенил, а n равно от 0 до приблизительно 10. Этот способ включает восстановление α-аминокислоты в β-аминоспирт формулы (R)2CH(CH2)nCH(NH2)CH2OH; сульфонилирование β-аминоспирта 5-хлортиофен-2-сульфонилгалогенидом и перекристаллизацию продукта со стадии сульфонилирования с получением хирально чистых N-сульфонил-β-аминоспиртов.
В соответствии с одним из способов осуществления настоящего изобретения соединения по настоящему изобретению содержат один хиральный атом углерода, где R в приведенной выше формуле имеет те же значения. В некоторых специальных вариантах осуществления настоящего изобретения группы R обозначают метил, этил, н-пропил, наиболее предпочтительно группы R обозначают этил. Однако настоящее изобретение далее включает получение α-аминокислот и β-аминоспиртов общей формулы, при условии, что группы R отличаются друг от друга. В этих соединениях может присутствовать один или несколько дополнительных хиральных центров; тем не менее дополнительные хиральные центры должны быть оптически чистыми и не должны мешать получению хирально чистых α-аминокислот, β-аминоспиртов и S-энантиомеров N-сульфонил-β-аминоспиртов по настоящему изобретению.
В другом предпочтительном варианте осуществления настоящего изобретения хиральный углеродный центр имеет S-стереохимию, что приводит к энантиомерно чистым продуктам.
Таким образом, в способе по настоящему изобретению предлагается эффективный путь синтеза хирально чистых S-энантиомеров β-аминоспиртов и их промежуточных соединений, которые полезны для многих целей. Так, из предлагаемых в настоящем описании примеров соединений N-сульфонил-β-аминоспирты применимы для ингибирования продукции β-амилоида, который принимает участие, помимо прочих, в амилоидной ангиопатии, церебральной амилоидной ангиопатии, системном амилоидозе, болезни Альцгеймера, наследственном внутримозговом кровотечении с датским амилоидозом, миозите с необычными включениями-тельцами, болезни Дауна.
Используемый по тексту настоящего описания термин "фармацевтически применимый" относится к соединениям, обладающим требуемым биологическим действием либо как терапевтическое средство, иммунный стимулятор, либо как супрессор, вспомогательное соединение, вакцинное средство. Аналогично по способу настоящего изобретения среди прочих могут быть получены различные соединения, которые подходят для нефармацевтического использования, в частности для диагностики, в качестве маркеров. Этим способом могут быть получены другие фармацевтически применимые соединения.
Используемый по тексту настоящего описания термин "алкил" обозначает насыщенные алифатические углеводородные группы как с прямой, так и с разветвленной цепью, которые содержат от одного до десяти атомов углерода, предпочтительно от одного до восьми атомов углерода и наиболее предпочтительно от одного до шести атомов углерода; "алкенил" включает алкильные группы как с прямой, так и с разветвленной цепью, содержащие по крайней мере одну двойную углерод-углеродную связь и от двух до восьми атомов углерода, предпочтительно от двух до шести атомов углерода, а "алкинильная" группа включает алкильные группы как с прямой, так и с разветвленной цепью, содержащие по крайней мере одну тройную углерод-углеродную связь и от двух до восьми атомов углерода, предпочтительно от двух до шести атомов углерода. Используемый по тексту настоящего описания термин "низший" относится к любой из описанных ранее групп, содержащих от одного до шести атомов углерода.
Термины "замещенный алкил", "замещенный алкенил", "замещенный алкинил", "замещенный низший алкил", "замещенный низший алкенил" и "замещенный низший алкинил" относятся к только что рассмотренному алкилу, алкенилу, алкинилу, низшему алкилу, низшему алкенилу и низшему алкинилу, которые имеют одного до трех заместителей, независимо выбранных из галогена, нитрильной группы, гидроксильной группы, нитрогруппы, аминогруппы, арила, гетероциклической группы, замещенного арила, замещенной гетероциклической группы, алкоксигруппы, замещенной алкоксигруппы, арилоксигруппы, замещенной арилоксигруппы, алкилкарбонильной группы, алкилкарбоксильной группы, алкиламиногруппы и арилтиогруппы. Указанные заместители могут присоединяться к любому атому углерода в алкильной, алкенильной или алкинильной группе при условии, что в результате такого присоединения образуется устойчивое химическое соединение.
Термин "замещенный фенил" относится к фенильной группе, имеющей от одного до четырех заместителей, независимо выбранных из галогена, нитрильной группы, гидроксильной группы, нитрогруппы, аминогруппы, алкила, циклоалкила, алкенила, алкинила, алкоксигруппы, арилоксигруппы, замещенной алкилоксигруппы, алкилкарбонильной группы, алкилкарбоксильной группы, алкиламиногруппы и арилтиогруппы.
Термин "циклоалкил" относится к углеродному кольцу, в состав которого входят более 3 атомов углерода.
Термин "замещенный бензил" относится к бензильной группе, имеющей от одного до четырех заместителей, независимо выбранных из галогена, нитрильной группы, гидроксильной группы, нитрогруппы, аминогруппы, алкила, циклоалкила, алкенила, алкинила, алкоксигруппы, арилоксигруппы, замещенной алкилоксигруппы, алкилкарбонильной группы, алкилкарбоксильной группы, алкиламиногруппы и арилтиогруппы.
Термин "галоген" относится к хлору, брому, фтору или йоду.
Термин "сильное ненуклеофильное основание" относится к основному ненуклеофильному реагенту, который не действует как нуклеофильный агент или не связывается с реагентами, которые используются по настоящему изобретению. Из области техники известен ряд ненуклеофильных реагентов, и они включают гидрид натрия, гидрид калия, диизопропиламид лития и гексаметилдисилазид калия.
Термин "водное основание" относится к раствору, который как минимум содержит основание и воду. Из области техники известен ряд оснований, которые легко растворяются в воде, и они, среди прочих, включают гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия или гидроксид калия. Водный раствор основания может дополнительно содержать другие реагенты, которые не мешают протеканию реакций по настоящему изобретению, и, среди прочих, может включать органические растворители, такие как тетрагидрофуран, метанол или углеводородные растворители, а также соли, такие как хлорид натрия, и буферные добавки.
Термин "органический растворитель" может включать любой известный из области техники растворитель, который не вступает во взаимодействие с используемыми в реакции реагентами, и, среди прочих, включает насыщенные углеводородные растворители, ненасыщенные углеводородные растворители, в том числе ароматические растворители, спирты, простые эфиры и ацетаты.
Соединение по настоящему изобретению может быть использовано в форме соли, в частности соли, полученной из фармацевтически или физиологически приемлемых кислот или оснований. Эти соли включают, однако ими не ограничиваются, следующие соли с органическими и неорганическими кислотами, такими как уксусная, молочная, лимонная, винная, янтарная, фумаровая, малеиновая, малоновая, миндальная, яблочная, хлористоводородная, бромистоводородная, фосфорная, азотная, серная кислоты, метансульфокислота, толуолсульфокислота и аналогичные известные приемлемые соли и их смеси. Другие соли включают соли со щелочными металлами или щелочноземельными металлами, такими как натрий (в частности, с гидроксидом натрия), калий (в частности, с гидроксидом калия), литий, кальций или магний.
Указанные соли, также как и другие соединения по настоящему изобретению, могут быть в виде сложных эфиров, карбаматов и других обычных пролекарственных форм, которые, если они вводятся в указанной форме, превращаются в активное вещество в условиях in vivo. В данном предпочтительном варианте осуществления настоящего изобретения пролекарством являются сложные эфиры. См., в частности, B.Testa and J.Caldwell, "Prodrugs Revisited: The "Ad Hoc" Approach as a Complement to Ligand Design", Medicinal Research Reviews, 16(3): 233-241, ed., John Wiley & Sons (1996).
В одном из способов осуществления настоящего изобретения α-аминокислоты и β-аминоспирты по настоящему изобретению вступают во взаимодействие с различными реагентами с образованием комплексов, которые содержат по крайней мере один хиральный центр при атоме углерода. В одном из способов осуществления настоящего изобретения α-аминокислоты и спирты взаимодействуют с тиофенсульфонилгалогенидами, более предпочтительно с 5-галогентиофенсульфонилгалогенидами и наиболее предпочтительно 5-хлортиофенсульфонилгалогенидами, с образованием хирально чистых гетероциклических N-сульфонил-β-аминоспиртов формулы (8).
В другом способе осуществления настоящего изобретения α-аминокислоты или β-аминоспирты по настоящему изобретению вступают во взаимодействие с фурансульфонилгалогенидами с образованием хирально чистых гетероциклических N-сульфонил-β-аминоспиртов.
Следующая схема (Схема 1) должна помочь специалистам в данной области техники понять изобретение в целом, в то время как Схема 2 описывает предпочтительный вариант осуществления настоящего изобретения. Для специалиста должно быть понятно, как использовать настоящее изобретение для осуществления различных предпочтительных вариантов, которые охватываются настоящим изобретением.
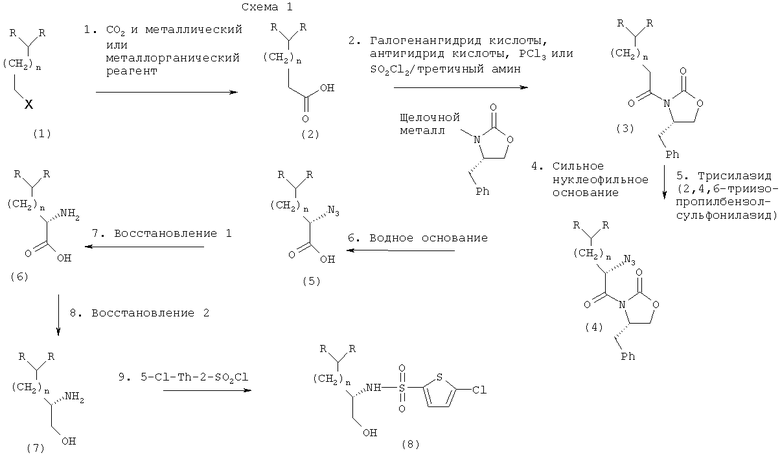
В соответствии со Схемой 1 превращение алкилгалогенида (1) в карбоновую кислоту (2) можно осуществить вначале превращением алкилгалогенида (1) в металлорганический реагент. Из области техники известны различные способы превращения алкилгалогенидов в металлорганические реагенты. См., в частности, Organometallic Syntheses, Volume 2, John J. Eisch, ed., Academic Press, New York, 1981. Алкилгалогенид предпочтительно является алкилбромидом, алкилхлоридом или алкилйодидом. Более предпочтительно алкилгалогенид является алкилбромидом. Известно большое количество металлов и металлорганических реагентов, которые облегчают превращение алкилгалогенидов в карбоновые кислоты, и среди них реактивы Гриньяра, литийорганические реагенты, металлический магний и литий. Как только металлорганический реагент приготовлен, его сразу же превращают в карбоновую кислоту преимущественно взаимодействием с диоксидом углерода. В качестве альтернативы превращение в карбоновую кислоту можно осуществить с помощью любого известного из области техники способа. Подобные способы включают взаимодействие металлорганического реагента с диэтилкарбонатом или этилхлорформиатом с образованием сложного этилового эфира, который гидролизуют в карбоновую кислоту водным основанием. Специалисты в данной области техники могут выбрать разные водные основания, которые, среди прочих, включают гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия или гидроксид калия.
Карбоновую кислоту (2) затем превращают в производное оксазолидинона (3). Карбоновую кислоту вначале превращают в активированное карбонильное производное взаимодействием карбоновой кислоты с реагентом, который включает, однако ими не ограничивается, галогенангидрид кислоты, трихлорид фосфора, ангидрид кислоты или тионилхлорид, с последующим взаимодействием с третичным амином. Может быть использовано большое количество галогенангидридов кислот, и они включают хлорангидриды, бромангидриды и йодангидриды кислот. Галогенангидридом кислоты предпочтительно является хлорангидрид кислоты. Из области техники известно большое количество хлорангидридов кислот, которые включают, среди прочих, пивалоилхлорид, изовалерилхлорид, этилхлоркарбонат и изобутилхлоркарбонат. Наиболее предпочтительным хлорангидридом является пивалоилхлорид. Из области техники известно большое количество ангидридов кислот, и они включают трифторуксусный ангидрид и трихлоруксусный ангидрид. Ангидридом кислоты преимущественно является трифторуксусный ангидрид. Из области техники известен ряд третичных аминов, и, среди прочих, они включают триэтиламин, триметиламин, N,N-диизопропилэтиламин и пиридин. Третичным амином предпочтительно является триэтиламин.
Активированные карбонильные соединения далее вступают во взаимодействие с солью щелочного металла вспомогательного хирального агента в подходящем органическом растворителе. Вспомогательным хиральным агентом преимущественно является S-4-бензил-2-оксазолидинон. Тем не менее специалистами в данной области техники могут быть использованы и легко выбраны и другие вспомогательные хиральные агенты. См., например, Principles and Applications of Asymmetric Synthesis, G.Lin, Y.Li, and A.Chan, Wiley-Interscience, New York, 2001 (например, стр.104, таблица 2-13). Соли щелочных металлов S-4-бензил-2-оксазолидинона, которые могут быть применены в данной реакции, включают соли лития, натрия и калия. Вспомогательным хиральным агентом преимущественно является литиевая соль S-4-бензил-2-оксазолидинона.
С целью максимально увеличить выход продукта превращение кислоты в производное оксазолидинона предпочтительно проводят в течение приблизительно 30 мин. Однако время реакции может зависеть от различных факторов, включая, в числе прочих, температуру реакции, чистоту реагентов, масштаб проведения реакции, внешние условия, точную структуру субстрата и концентрацию. Специалисты в области техники могут выбирать более длительные или более короткие времена реакции (например, от 10 до приблизительно 60 мин).
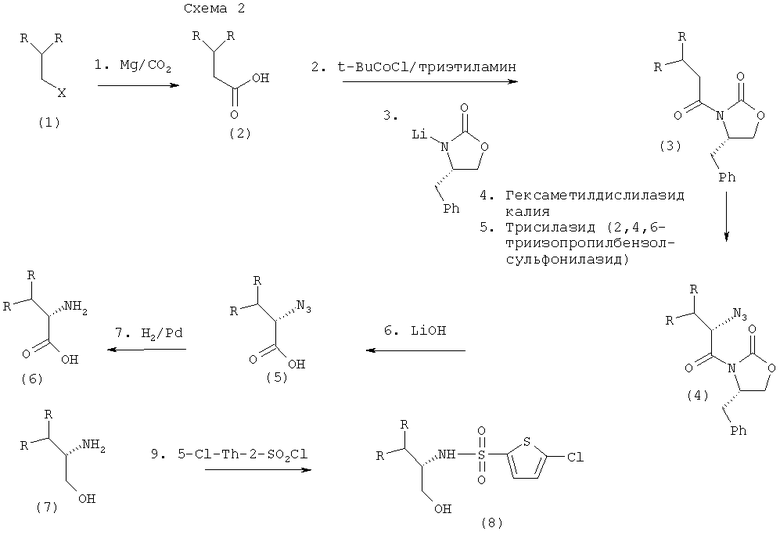
В предпочтительном варианте осуществления настоящего изобретения карбоновую кислоту (2) превращают в смешанный ангидрид по реакции с пивалоилхлоридом в присутствии триэтиламина, а затем вводят в реакцию с литиевой солью S-4-бензил-2-оксазолидинона (получают действием н-бутиллития на S-4-бензил-2-оксазолидинон) в тетрагидрофуране (ТГФ) с образованием производного оксазолидинона (3). См. Схему 2.
Производное оксазолидинона (3) затем превращают, как указывалось в данном описании, в его енолят-анион взаимодействием с сильным ненуклеофильным основанием. Сильным ненуклеофильным основанием предпочтительно является гексаметилдисилазид калия. Далее енолят-анион взаимодействует с трисилазидом с образованием азидооксазолидинонового промежуточного соединения (4).
Азидооксазолидиноновое промежуточное соединение (4) можно превратить в α-азид кислоты (5) любым подходящим способом, известным из области техники. В предпочтительном варианте осуществления настоящего изобретения азидооксазолидиноновое промежуточное соединение, как указывалось, превращают в α-азид кислоты гидролизом водного основания. Азидооксазолидиноновое промежуточное соединение превращают в α-азид кислоты гидролизом водного раствора гидроксида лития.
α-Азид кислоты (5) можно превратить в α-аминокислоту (6) с помощью любого подходящего способа, известного из области техники. Реакцию преимущественно проводят каталитическим восстановлением с использованием газообразного водорода в присутствии 10%-ного палладия на угле в качестве катализатора. В качестве альтернативы восстановление можно провести с использованием цинка/HCl, борогидридом натрия или водным трифенилфосфином. Для получения максимального выхода продукта реакции желательно проводить в течение 24 час. Однако время реакции может зависеть от различных факторов, включая, в числе прочих, температуру реакции, чистоту реагентов, масштаб проведения реакции, внешние условия, точную структуру субстрата и концентрацию. Специалисты в данной области техники могут выбирать более длительные или более короткие времена реакции (например, приблизительно от 12 час до приблизительно 96 час).
Хиральную α-аминокислоту можно затем выделить, используя методики, хорошо известные специалистам в данной области техники, которые включают, однако ими не ограничиваются, хроматографию и перекристаллизацию. Перекристаллизацию можно проводить с использованием различных органических и неорганических растворителей, известных из области техники, и получить хирально чистые α-аминокислоты.
В качестве альтернативы α-аминокислоты (6) могут быть восстановлены в β-аминоспирты (7) различными способами, известными из области техники. В предпочтительном варианте осуществления настоящего изобретения восстановление α-аминокислоты проводят каталитическим гидрированием с использованием диборана, родственных боранов, таких как катехинборан, борогидрида лития/триметилсилилхлорида (TMSCl), алюмогидридом лития, диизобутилалюмогидрида (DiBAL-H), бис(2-метоксиэтокси)алюмогидрида (Red-Al) и алана. Более предпочтительно восстановление осуществляют с помощью борогидрида лития/триметилсилилхлорида (TMSCl) в течение более 48 час.
Хиральный β-аминоспирт можно затем выделить, используя методики, хорошо известные специалистам в данной области техники, которые включают, однако ими не ограничиваются, хроматографию и перекристаллизацию. Перекристаллизацию можно проводить с использованием различных органических и неорганических растворителей, известных из области техники.
В качестве альтернативы β-аминоспирт (7) превращают в целевое хиральное соединение (8) путем взаимодействия с 5-хлортиофен-2-сульфонилхлоридом в присутствии сильного ненуклеофильного основания, такого как третичный амин или гидроксид щелочного металла. Перекристаллизацией с использованием подходящей системы растворителей, известной из области техники, получают хирально чистое целевое соединение.
Короткая цепочка проведения химических реакций, простота синтеза, масштабируемость и доступность потенциальных исходных соединений делает указанный процесс практическим способом получения хирально чистых S-энантиомеров N-сульфонил-β-аминоспиртов.
В тех случаях, когда на стадии проведения реакции по настоящему изобретению используют катализатор или растворитель, предполагается, что могут быть использованы и другие катализаторы или растворители, известные из области техники, но не упомянутые здесь; специалисты в данной области техники легко смогут выбрать подходящие катализаторы, растворители и условия реакции для каждой стадии проведения реакции, которая используется в настоящем изобретении.
Изобретение включает определенные типы реакций, такие как связывание енолята, реакции гидролиза и восстановления, которые в общем случае известны из области техники, однако ранее не были использованы в новом качестве, по которому они используются в настоящем изобретении. Вариации специфических методов, используемых при осуществлении индивидуальных стадий по настоящему изобретению, могут быть понятны специалистам. Хотя все указанные возможные вариации нельзя предусмотреть заранее, предполагается, что подобные вариации входят в объем притязаний по настоящему изобретению.
Следующие примеры приведены для иллюстрации получения и пояснения активности отдельных соединений по настоящему изобретению и для иллюстрации их свойств при проведении анализов. Для специалиста должно быть понятно, что, несмотря на то, что в следующих примерах приводятся специфические реагенты и условия, эти реагенты и условия не ограничивают настоящее изобретение.
ПРИМЕРЫ
Пример 1. 3-Этилпентановая кислота
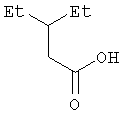
В трехгорлую колбу емкостью 2 л, снабженную обратным холодильником с присоединенной к нему трубкой для подачи азота, механической мешалкой и капельной воронкой емкостью 500 мл, помещают магниевые стружки (18,7 г, 0,769 моль), которые предварительно измельчают в ступке пестиком. Реакцию проводят в токе аргона. Добавляют ТГФ в количестве, достаточном для того, чтобы слегка покрыть магниевую стружку. При отключенной механической мешалке в капельную воронку добавляют 2-этил-1-бромбутан (155,5 г, 0,70 моль) и 2-3 мл и из воронки добавляют к магниевой стружке. Этот участок раствора в ТГФ затем нагревают с помощью струйного воздушного нагревателя до возникновения бурного кипения. Оставшийся 2-этил-1-бромбутан разбавляют с помощью ТГФ (200 мл) и по каплям при перемешивании добавляют к реакционной смеси с такой скоростью, чтобы поддерживалось равномерное кипение. Добавление заканчивается приблизительно через 3-4 час.
Через 24 час суспензию серого цвета разбавляют с помощью ТГФ (500 мл) и осторожно нагревают на водяной бане до образования гомогенного раствора. Теплый раствор выливают в 4-литровые стаканы фирмы Nalgene, каждый из которых заполнен крошкой твердого диоксида углерода (1 л). Полученный пластичный материал затем хорошо перемешивают с помощью покрытой тефлоном магнитной мешалки и оставляют при комнатной температуре. Через 18 час раствор разбавляют этилацетатом (500 мл) и промывают 2N раствором соляной кислоты (500 мл). Органический слой отделяют, водный слой насыщают хлоридом натрия и еще раз экстрагируют этилацетатом (500 мл). Этилацетатные вытяжки объединяют и сушат (над Na2SO4). Концентрируют при пониженном давлении и получают влажный продукт, который растворяют в метиленхлориде (600 мл), сушат (над Na2SO4) и концентрируют при 56°С при пониженном давлении, получая приведенный в названии продукт в виде масла (91 г, выход 99,8%). Спектр 1H-ЯМР (ДМСО-d6, 500 МГц): δ 11,95 (ушир. с, 1Н), 2,11 (д, J=6,9 Гц, 2Н), 1,60 (септ., J=6,5 Гц, 1Н), 1,28 (м, 4Н), 0,81 (т, J=7,5 Гц, 6Н); масс-спектр (-ESI):[M-H]- 129 (100%); элементный анализ: вычислено для C7H14O2: C 64,58, H 10,84; найдено: С 64,61, Н 11,11.
Пример 2. (3(2S),4S)-3-(3-Этил-1-оксопентил)-4-(фенилметил)-2-оксазолидинон
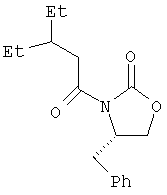
В трехгорлую колбу емкостью 3 л, снабженную трубкой для подачи азота, механической мешалкой и капельной воронкой с пробкой, помещают продукт, полученный по примеру 1 (91 г, 0,699 моль), и ТГФ (1 л). Пропускают ток азота и раствор охлаждают до минус 78°С. Добавляют триэтиламин (102,3 мл, 0,734 моль), а затем пивалоилхлорид (90,4 мл, 0,734 моль). Баню с температурой минус 78°С заменяют на баню со льдом. Полученную густую суспензию перемешивают при 0°С в течение часа.
Параллельно в трехгорлую колбу емкостью 5 л, снабженную пробкой, механической мешалкой и трубкой для подачи азота, помещают (S)-(-)-4-бензил-2-оксазолидинон (136 г, 0,767 моль) и ТГФ (1 л), пропускают ток азота и раствор охлаждают до минус 78°С. Добавляют раствор н-бутилития (480 мл 1,6 М раствора в гексане, 0,768 моль). Полученный раствор перемешивают при температуре минус 78°С в течение 40 мин. Полученную выше густую суспензию смешанного ангидрида в течение 10 мин вновь охлаждают до температуры минус 78°С и затем через воронку для добавления твердых веществ добавляют к раствору литиевого енолята (S)-(-)-4-бензил-2-оксазолидинона. Дают реакционной смеси в течение ночи медленно нагреться до комнатной температуры и делят на три порции по 1 л. Каждую порцию разбавляют этилацетатом (800 мл), промывают один раз насыщенным раствором одноосновного фосфата калия (300 мл), дважды смесью лед:1N раствор NaOH (1:1, 300 мл), один раз насыщенным раствором одноосновного фосфата калия (300 мл), один раз насыщенным раствором соли (300 мл) и сушат (над Na2SO4). После упаривания при пониженном давлении полученный сырой продукт пропускают через слой силикагеля (1 кг), используя гексан и этилацетат, и получают масло янтарного цвета (123,7 г, выход 61%). Оптическое вращение [α]D 25=+94,75° (1%-ный раствор в ДМСО); спектр 1Н-ЯМР: (ДМСО-d6, 500 МГц): δ 7,35-7,15 (м, 5Н), 4,65 (м, 1Н), 4,31 (т, J=8,6 Гц, 1Н), 4,17 (дд, J=2,8, 8,9 Гц, 1Н), 3,00 ((дд, J=3,2, 13,4 Гц, 1Н), 2,91 (дд, J=7,7, 13,5 Гц, 1Н), 2,81 (дд, J=6,7, 16,2 Гц, 1Н), 2,69 (дд, J=6,9, 16,2 Гц, 1Н), 1,80 (септ., J=6,4 Гц, 1Н), 1,32 (м, 4Н), 0,84 и 0,83 (два перекр. триплета, J=7,3 Гц, 6Н); спектр 13С ЯМР (ДМСО-d6, 100 МГц): δ 172,15, 153,27, 135,65, 129,39, 128,47, 126,81, 65,96, 54,26, 38,41, 36,70, 36,19, 25,07, 24,98, 10,62, 10,52; масс-спектр (+ESI): [M+H]+ 290 (100%); элементный анализ: вычислено для C17H23NO3: C 70,56, H 8,01, N 4,84; найдено: С 70,69, Н 8,31, N 4,83. Аналитическая ВЭЖХ (4,6 мм × 100 мм колонка Chromolith Monolith, элюент 70:30 ацетонитрил/вода, каждый из которых содержит 0,1% трифторуксусной кислоты, изократическое элюирование, объемный расход 4 мл/мин, УФ детектирование при длине волны 254 нм) подтверждает основной компонент (97,7%).
Пример 3. (3(2S),4S)-3-(2-Азидо-3-этил-1-оксопентил)-4-(фенилметил)-2-оксазолидинон
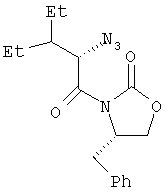
В трехгорлую колбу емкостью 12 л, снабженную трубкой для подачи азота, механической мешалкой и капельной воронкой с пробкой емкостью 500 мл, помещают продукт, полученный по примеру 2 (122,8 г, 0,424 моль), и ТГФ (943 мл). Пропускают ток азота и раствор охлаждают до минус 78°С. В течение 2,5 час по каплям добавляют раствор бис(триметилсилил)амида калия (1,018 л 0,5 М раствора в толуоле, 0,509 моль) в ТГФ (943 мл). Перемешивают при температуре минус 78°С в течение часа и через канюлю (две двойные иглы №18) в течение 14 мин добавляют предварительно охлажденный (минус 78°С, 50 мин) раствор 2,4,6-триизопропилбензолсульфонилазида (170,6 г, 0,55 моль) в ТГФ (573 мл). Перемешивают еще в течение 9 мин при температуре минус 78°С, а затем через капельную воронку в один прием добавляют ледяную уксусную кислоту (111,7 мл, 1,95 моль). Перемешивают при температуре минус 78°С еще в течение 2 мин и добавляют безводный ацетат калия (166,5 г, 1,70 моль). Баню с температурой минус 78°С отставляют и реакционной смеси в течение ночи при перемешивании дают нагреться до комнатной температуры. Реакционную смесь делят на четыре порции по 1 л. Каждую порцию разбавляют этилацетатом (400 мл), промывают дважды насыщенным раствором одноосновного фосфата калия (400 мл), один раз промывают насыщенным раствором соли (400 мл) и сушат (над Na2SO4). Концентрируют при пониженном давлении и полученный сырой продукт пропускают через слой силикагеля (1 кг), используя гексан и этилацетат, и получают масло (79 г, выход 56%). Оптическое вращение [α]D 25=+112,16° (1%-ный раствор в ДМСО); спектр 1Н-ЯМР: (ДМСО-d6, 500 МГц): δ 7,35-7,15 (м, 5Н), 5,11 (д, J=6,1 Гц, 1Н), 4,72 (м, 1Н), 4,41 (т, J=8,5 Гц, 1Н), 4,27 (дд, J=2,4, 8,9 Гц, 1Н), 3,07 (дд, J=3,1, 13,6 Гц, 1Н), 3,00 (дд, J=7,5, 13,6 Гц, 1Н), 1,80 (м, 1Н), 1,55-1,20 (м, 4Н), 0,87 и 0,84 (два перекр. триплета, J=7,6 Гц, 6Н); спектр 13С-ЯМР: (ДМСО-d6, 100 МГц): δ 170,58, 153,48, 136,00, 130,13, 129,26, 127,65, 67,38, 62,74, 55,55, 42,35, 36,86, 22,74, 21,58, 11,54, 11,19; элементный анализ: вычислено для C17H22N4O3: C 61,80, H 6,71; найдено: С 62,53, Н 6,88.
Пример 4. 2(S)-Азидо-3-этилпентановая кислота
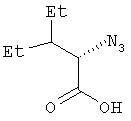
В трехгорлую колбу емкостью 3 л, снабженную термометром, механической мешалкой и трубкой для подачи азота, помещают продукт, полученный по примеру 3 (79 г, 0,239 моль), и ТГФ:H2O (3:1, 1,196 л). Колбу заполняют азотом и раствор охлаждают на бане со льдом и солью. После перемешивания в течение 50 мин температура реакционной смеси опускается до минус 0,5°С. Добавляют моногидрат гидроксида лития (20,8 г, 0,478 моль) с такой скоростью, чтобы температура реакционной смеси не поднималась выше 2,5°С. За ходом реакции следят с помощью тонкослойной хроматографии. Через 1 час добавляют твердый бикарбонат натрия (60 г). Реакционную смесь делят на две порции по 0,6 л. Каждую порцию разбавляют насыщенным раствором бикарбоната натрия (80 мл) и водой (160 мл) и экстрагируют этилацетатом (700 мл). Слой этилацетата еще раз промывают насыщенным раствором бикарбоната натрия (160 мл). Этот слой этилацетата содержит хиральное побочное соединение, и его отбрасывают. Объединенные бикарбонатные экстракты подкисляют 2N соляной кислотой (600 мл) до рН<2. Подкисленный водный слой экстрагируют этилацетатом (500 мл), затем насыщенным раствором хлорида натрия и еще раз экстрагируют этилацетатом (500 мл). Этилацетатные вытяжки объединяют, сушат (над Na2SO4), фильтруют и концентрируют при пониженном давлении, получая масло (31,7 г, выход 77%). Оптическое вращение [α]D 25=-82,5° (1%-ный раствор в ДМСО); спектр 1Н-ЯМР: (ДМСО-d6, 500 МГц): δ 13,12 (ушир.с, 1Н), 4,26 (д, J=4,3 Гц, 1Н), 1,67 (м, 1Н), 1,45-1,15 (м, 4Н), 0,88 (т, J=7,5 Гц, 3Н), 0,83 (т, J=7,5 Гц, 3Н); масс-спектр (-ESI): [M-H]- 170 (85%).
Пример 5. 2(S)-Амино-3-этилпентановая кислота
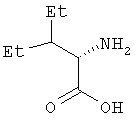
Смесь продукта, полученного по примеру 4 (31,7 г, 0,185 моль), 10%-ный палладий на угле (7,9 г), ледяную уксусную кислоту (370 мл) и воду (926 мл) помещают в аппарат Парра для гидрирования и встряхивают в качалке в атмосфере водорода (давление 40 фунтов на квадратный дюйм). Через 20 час реакционную смесь отфильтровывают через слой силикагеля (1/2"), который затем хорошо промывают водой (200 мл). Фильтрат упаривают при пониженном давлении на бане с температурой до 70°С и получают твердое вещество белого цвета. Его растирают с этилацетатом (500 мл), отфильтровывают, промывают еще раз этилацетатом (500 мл) и затем сушат на воздухе. Получают обозначенное в названии вещество в виде твердого вещества белого цвета (21,2 г, выход 79%). Оптическое вращение [α]D 25=+12,01° (1%-ный раствор в воде); спектр 1Н-ЯМР: (D2О, 500 МГц): δ 4,65 (с, 3Н), 3,69 (д, J=3,4 Гц, 1Н), 1,69 (м, 1Н), 1,44 (м, 1Н), 1,03-1,10 (м, 3Н), 0,83 и 0,81 (два перекр. триплета, J=7,5 Гц, 6Н); масс-спектр (-ESI): [M-H]- 144 (100%), элементный анализ: вычислено для C7H15NO2: C 57,90, H 10,41, N 9,65; найдено: С 57,75, Н 10,89, N 9,40. Хиральная ВЭЖХ (колонка Symmetry С18, элюент: растворитель А=50 мМ раствор триэтиламина, рН которого доведен до 3,0 с помощью фосфорной кислоты; растворитель В = ацетонитрил; градиент от 80%А/20%В до 50%А/50%В в течение 20 мин, объемный расход раствора 1,0 мл/мин, детектирование при длине волны 340 нм, S-изомер имеет время удерживания 12,26 мин, а R-изомер имеет время удерживания 15,46 мин) производного аминокислоты, полученного с помощью реагента Марфи (N-α-(2,4-динитро-5-фторфенил)-L-аланинамид) дает энантимерное отношение 99,5:0,5 (2S:2R).
Пример 6. 2(S)-Амино-3-этилпентанол
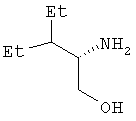
В трехгорлую колбу емкостью 3 л, снабженную трубкой для подачи азота, механической мешалкой и капельной воронкой с пробкой, помещают борогидрид лития (145 мл 2 М раствора в ТГФ, 0,29 моль). Пропускают ток азота и охлаждают раствор до 0°С. По каплям в течение 30 мин добавляют триметилхлорсилан (73,8 мл, 0,58 моль). Баню со льдом убирают и полученную суспензию перемешивают при комнатной температуре в течение 30 мин. Охлаждают реакционную смесь до 0°С и порциями в течение 15 мин в виде твердого вещества добавляют продукт, полученный по примеру 5 (21,1 г, 0,145 моль). Реакционной смеси дают медленно нагреться до комнатной температуры по мере таяния бани со льдом. Перемешивают при комнатной температуре 3 дня, реакционную смесь вновь охлаждают до 0°С и в течение 80 мин осторожно добавляют метанол (217 мл). Раствор дополнительно перемешивают при комнатной температуре в течение 40 мин, а затем упаривают при пониженном давлении на водяной бане с температурой 60°С. Полученную суспензию подщелачивают 20%-ным раствором гидроксида натрия (37,5 мл). Добавляют воду (37,5 мл), полученную общую водную фазу экстрагируют метиленхлоридом (300 мл) и экстракты сушат (над Na2SO4). Концентрируют при пониженном давлении и получают указанное в названии вещество в виде масла (17,3 г, выход 91%), которое сразу же используют или же на ночь оставляют в холодильнике. Оптическое вращение [α]D 25=-3,7° (1%-ный раствор в ДМСО); спектр 1Н-ЯМР: (ДМСО-d6, 500 МГц): δ 4,38 (ушир.с, 1Н), 3,35 (дд, перекр. с ушир. с. при δ 3,32, J=4,5; 10,3 Гц, 3Н), 3,14 (дд, J=7,9; 10,2 Гц, 1Н), 2,63 (м, 1Н), 1,45-1,05 (м, 5Н), 0,82 и 0,81 (два перекр. триплета, J=7,4 Гц, 6Н); масс-спектр (+ESI): [M+H]+ 132 (60%).
Пример 7. 5-Хлор-N-[(1S)-2-этил-1-(гидроксиметил)бутил]-2-тиофенсульфонамид
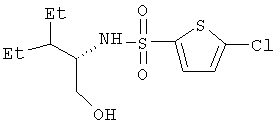
Смесь 2(S)-амино-3-этилпентанола (34,1 г, 0,26 моль) и метиленхлорида (700 мл) под аргоном охлаждают до 0°С. Добавляют триэтиламин (36,2 мл, 0,26 моль), а затем по каплям добавляют 5-хлортиофен-2-сульфонилхлорид (56,4 г, 0,26 моль) в метиленхлориде (400 мл). Дают реакционной смеси по мере таяния бани со льдом медленно нагреться до комнатной температуры. Перемешивают 3 дня при комнатной температуре и реакционную смесь делят на две порции по 0,6 л. Каждую порцию разбавляют этилацетатом (1 л) и трижды промывают насыщенным раствором одноосновного фосфата калия (200 мл), один раз насыщенным раствором соли (200 мл) и сушат (над Na2SO4). Концентрируют при пониженном давлении и получают твердое вещество белого цвета (74,5 г, выход 92%). Продукт, полученный из нескольких экспериментов (87,98 г), объединяют и перекристаллизовывают из горячей смеси гептан : этилацетат (4:1, 775 мл), получая указанное в названии вещество в виде кристаллов (74,9 г, выход 85%); т.пл. 115-117,6°С; оптическое вращение [α]D 25=+10,81° (1%-ный раствор в метаноле); спектр 1Н-ЯМР: (ДМСО-d6, 500 МГц): δ 7,71 (д, J=8,1 Гц, 1Н), 7,44 (д, J=4,1 Гц, 1Н), 7,22 (д, J=4,1 Гц, 1Н), 4,56 (т, J=5,2 Гц, ОН), 3,31-3,15 (м, 3Н), 1,40-1,15 (м, 4Н), 1,07 (м, 1Н), 0,79 и 0,76 (два перекр. триплета, J=7,3 Гц, 6Н); спектр 13С-ЯМР: (ДМСО-d6, 100 МГц): δ 141,75, 133,73, 130,95, 127,60, 60,41, 56,89, 41,57, 21,31, 20,80, 11,79, 11,51; масс-спектр (-ESI):[M+H]-, картина 1 изотопа хлора, 310 (100%), 312 (30%); элементный анализ: вычислено для C11H18ClNO3S2: C 42,37, H 5,82, N 4,49; найдено: С 42,34, Н 5,65, N 4,43. Хиральная ВЭЖХ (колонка Chiralpak AD, 25×0,46 см, элюент 8:2 гексан/изопропанол, содержащий 0,1% трифторуксусной кислоты, объемный расход 0,5 мл/мин, УФ-детектирование при длине волны 254 нм, время удерживания для S- и R-изомеров составляет 10,95 мин и 11,95 мин соответственно) показывает, что отношение S/R составляет 100,0:0,0.
В соответствии с настоящим изобретением получают следующие соединения:
Все публикации, на которые даются ссылки в данном описании, приведены здесь в качестве ссылок. Хотя изобретение было описано на примере предпочтительного варианта его осуществления, следует понимать, что могут быть осуществлены модификации, которые соответствуют сути настоящего изобретения. Подобные модификации входят в объем притязаний в соответствии с приведенной далее формулой изобретения.
Предлагается способ получения хирально чистых S-энантиомеров α-аминокислот, который включает следующие стадии: а) приготовление металлорганического реагента из галоидного алкила формулы (R)2CH(СН2)nСН2Х, b) добавление металлорганического реагента к диоксиду углерода с образованием карбоновой кислоты; с) активирование карбоновой кислоты хлорангидридом кислоты, трихлоридом фосфора, ангидридом кислоты или тионилхлоридом в присутствии третичного аминового основания; d) взаимодействие продукта со стадии с) с солью щелочного металла S-4-бензил-2-оксазолидинона; е) обработка продукта со стадии d) сильным ненуклеофильным основанием с образованием енолят-аниона; f) связывание енолят-аниона 2,4,6-триизопропилбензолсульфонилазидом с образованием азида оксазолидинона; g) гидролиз азида оксазолидинона водным раствором основания с образованием α-азидокислоты; h) восстановление α-азидокислоты в α-аминокислоту и i) перекристаллизация α-аминокислоты в хирально чистую α-аминокислоту. Предлагается также способ получения хирально чистых S-энантиомеров β-аминоспиртов, который далее включает стадии восстановления сырой α-аминокислоты в β-аминоспирт и перекристаллизацию β-аминоспирта в хирально чистый β-аминоспирт. Предлагается также способ получения хирально чистых S-энантиомеров N-сульфонил-β-аминоспиртов, который далее включает стадии сульфонилирования β-аминоспирта 5-хлортиофен-2-сульфонилхлоридом и перекристаллизации с получением хирально чистых N-сульфонил-β-аминоспиртов, 3 н. и 22 з.п. ф-лы.
где Х обозначает Cl, Br или I, a n обозначает число от 0 до 10;
R означает С1-С6алкил, замещенный C1-С6алкил, С2-С6алкенил, замещенный С2-С6алкенил, С2-С6алкинил, замещенный С2-С6алкинил, С3-С8циклоалкил, замещенный С3-С8циклоалкил, фенил, замещенный фенил, бензил, замещенный бензил, СН2-С3-С8циклоалкил, СН2-3-индол, CH(С1-С6алкил)-2-фуран, СН(С1-С6алкил)-4-метоксифенил, CH(С1-С6алкил)-фенил и СН(ОН)-4-SCH3-фенил, где
указанные «замещенный алкил», «замещенный алкенил», «замещенный алкинил» и «замещенный циклоалкил» относятся к алкилу, алкенилу, алкинилу и циклоалкилу, которые имеют от одного до трех заместителей, независимо выбранных из галогена, CN-группы, ОН-группы, NO2-группы, аминогруппы, арила, гетероциклической группы, алкоксигруппы, арилоксигруппы, алкилкарбонильной группы, алкилкарбоксильной группы, алкиламиногруппы и арилтиогруппы;
указанный «замещенный фенил» относится к фенильной группе, имеющей от одного до четырех заместителей, независимо выбранных из галогена, CN-группы, ОН-группы, нитрогруппы, NO2-группы, алкила, циклоалкила, алкенила, алкинила, алкоксигруппы, арилоксигруппы, замещенной алкилоксигруппы, алкилкарбонильной группы, алкилкарбоксильной группы, алкиламиногруппы и арилтиогруппы;
указанный «замещенный бензил» относится к бензильной группе, имеющей от одного до четырех заместителей, независимо выбранных из галогена, CN-группы, ОН-группы, NO2-группы, аминогруппы, алкила, циклоалкила, алкенила, алкинила, алкоксигруппы, арилоксигруппы, алкилкарбонильной группы, алкилкарбоксильной группы, алкиламиногруппы и арилтиогруппы;
b) добавление указанного металлорганического реагента к диоксиду углерода с образованием карбоновой кислоты;
c) активирование указанной карбоновой кислоты галогенангидридом кислоты, трихлоридом фосфора, ангидридом кислоты или тионилхлоридом в присутствии третичного аминового основания;
d) взаимодействие продукта со стадии с) с солью щелочного металла S-4-бензил-2-оксазолидинона;
e) обработка продукта со стадии d) сильным ненуклеофильным основанием с образованием енолят-аниона;
f) связывание указанного енолят-аниона 2,4,6-триизопропилбензолсульфонилазидом с образованием азида оксазолидинона;
g) гидролиз указанного азида оксазолидинона водным раствором основания с образованием α-азидокислоты;
h) восстановление указанной α-азидокислоты в α-аминокислоту и
i) перекристаллизация указанной α-аминокислоты с получением указанной хирально чистой α-аминокислоты.
 ;
;
где R определен в п.1.
 ;
;
где R определен в п.1.
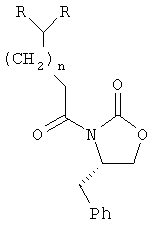 ;
;
где R определен в п.1.
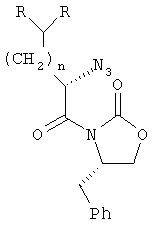 ;
;
где R определен в п.1.
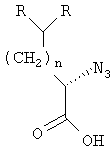 ;
;
где R определен в п.1.
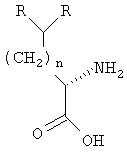 ;
;
где R определен в п.1.
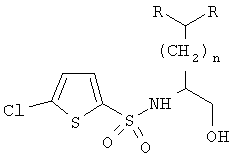
где R определен в п.1; при этом указанный способ включает следующие стадии:
1) восстановление указанной α-аминокислоты, полученной в соответствии с п.1, в β-аминоспирт:
m) перекристаллизация указанного β-аминоспирта с получением указанного хирально чистого β-аминоспирта;
n) сульфонилирование указанного β-аминоспирта 5-хлортиофен-2-сульфонилгалогенидом и
о) перекристаллизация продукта со стадии n) с получением хирально чистого N-сульфонил-β-аминоспирта формулы
где R определен в п.1.
| СПОСОБ ПОЛУЧЕНИЯ ТРАМАДОЛА | 2000 |
|
RU2165922C1 |
| US 5449778 A, 12.09.1995 | |||
| SOLOMONS, T.W.G | |||
| Organic Chemistry, 1992, 5th Edition, John Wiley & Sons, New York, p.771. | |||

