Изобретение относится к новым промежуточным продуктам для полусинтеза таксанов и способам их получения.
Таксаны, являющиеся природными веществами, дитерпеновый скелет которых обычно этерифицирован до сложного эфира боковой β- аминокислотной цепью, происходящей от N-алкил- или N-ароилфенилизосерина, известны в качестве противораковых средств. Насчитывают несколько десятков таксанов, выделенных из тиссовых рода Taxus, как, например, паклитаксел (R1 = ацетил; R2 = фенил; R3 = R4 = H), цефаломанин, их дезацетилированные в положении 10 производные, или баккатины (производные без боковой цепи), отвечающие формулам 1 и 2: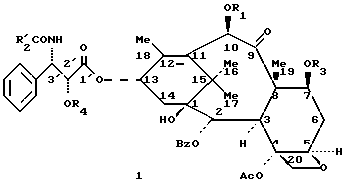

Для того чтобы быстро не исчерпать источник происхождения, Taxus brevifolia, французские исследователи стремились выделить паклитаксел из возобновляемых частей (листья) T.baccata, тиса европейского. Таким образом, они выявили возможный биогенетический предшественник таксанов, 10-дезацетилбаккатин III, исходное вещество для полусинтеза, выбранное вследствие его относительно большого количества в экстрактах из листьев.
Полусинтез таксанов, таких как паклитаксел или доцетаксел (R1 = ацетил; R2 = трет-бутилоксигруппа; R3 = R4 = H), следовательно, состоит в этерификации гидроксильной функции, находящейся в положении 13, защищенного производного баккатина или 10-дезацетилбаккатина III с помощью производного β- аминокислоты.
В уровне техники описываются различные способы полусинтеза паклитаксела или доцетаксела (европейские патенты NN 0 253 738, 0 336 840, 0 336 841, 0 495 718; международные заявки NN 92/09589, 94/07877, 94/07878, 94/07879, 94/10169, 94/12482; европейские патенты N 0 400 971 и N 0 428 376; международная заявка 94/14787). Две недавние работы [I. Georg, T.T. Chen, I. Ojima и D.M. Vyas "Taxane Anticancer Agents, Basic Science and Current Status", ACS Symposium Series 583, Washington (1995)] и в особенности [Matthew Suffhess, "TAXOL® Science and Applications" CRC press (1995) и 1500 цитированных ссылок] включают исчерпывающий сбор методик полусинтезов таксанов.
Боковые β- аминокислотные цепи, происходящие от N-алкил- или N-ароилфенилизосерина, пакситаксела или доцетаксела имеют конфигурацию (2R, 3S), и одна из трудностей полусинтеза таксанов состоит в получении энантиомерно чистого продукта. Первая проблема заключается в получении чистого энантиомера производных фенилизосерина, используемых в полусинтезе таксанов. Вторая проблема заключается в сохранении этой энантиомерной чистоты в процессе этерификации производного баккатина и при последующих обработках полученных продуктов (удаление защиты с гидроксильных групп и т.д.).
Многочисленные работы по асимметрическому синтезу, где используют β- аминокислотные производные, сосредотачиваются на химии изосерина и его β- аминокислотных производных, циклической дегидратированной формой которых является β-капролактам (европейский патент 0 525 589). Большинство различных синтезов производных фенилизосерина, пригодных в качестве предшественников боковых цепей таксанов, совпадают в части одного общего промежуточного продукта, (2R,3R)-цис- β- фенилглицидной кислоты, которую затем превращают в β- фенилизосерин путем взаимодействия с аммиаком (европейский патент 0 495 718) или с нуклеофилом (Gou и др., J. Org. Chem., 58, 1287-1289 (1983)). Эти различные способы требуют значительного числа стадий для получения β- фенилизосерина конфигурации (2R,3S), с обязательной стадией расщепления рацемата обычными способами селективной кристаллизации, либо цис -β- фенилглицидной кислоты, либо β- фенилизосерина, или в дальнейшем, после превращения. Кроме того, для сохранения энантиомерной чистоты предшественников боковых цепей таксанов в процессе этерификации производного баккатина предлагаются различные способы, в частности, основанные на использовании циклических промежуточных продуктов блокированной конфигурации, которые исключают опасность изомеризации во время реакций этерификации в жестких реакционных условиях. Речь идет, в частности, о производных β- лактамов (европейский патент 0 400 971), оксазолидинов (международные заявки NN 92/09589, 94/07877, 94/07878, 94/07879, 94/10169, 94/12482), оксазинонов (европейский патент 0 428 376) или же оксазолинов (международная заявка 94/14787). Эти циклические предшественники получают из соответствующего производного β- фенилизосерина. Как и в случае этого последнего, предлагаемые способы включают большое число стадий и обязательное расщепление рацемата для получения целевого предшественника боковой цепи таксанов. Следовательно, необходима разработка нового улучшенного пути синтеза промежуточных продуктов, предшественников боковой цепи таксанов, в особенности энантиомеров цис -β- фенилглицидной кислоты, β- фенилизосерина и их циклических производных.
Наконец, для полусинтеза таксанов и, в особенности, паклитаксела, единственным соответствующим производным баккатина, используемым до настоящего времени, является производное, в котором гидроксильный радикал в положении 7 защищен триалкилсилильной группой (европейский патент 0 336 840, международная заявка 94/14787), удаление защитной группы которого осуществляют исключительно в кислой среде. Следовательно, также необходимо использование новых защитных для гидроксильной функции групп, позволяющих осуществлять в частности селективную защиту гидроксильного радикала в положении 7, допуская, кроме того, более широкий выбор рабочих условий для стадии удаления защитной группы.
Настоящее изобретение прежде всего относится к улучшенному способу получения предшественников боковой цепи таксанов.
Способ, согласно изобретению, состоит в том, что превращают цис -β- производное общей формулы (I):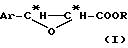
в которой Ar означает арил, в частности, фенил, и
R означает углеводородный радикал, предпочтительно линейный или разветвленный алкильный радикал или циклоалкильный радикал, незамещенный или замещенный одной или несколькими алкильными группами;
так, чтобы регио- и стереоспецифически получить β- N-алкиламид и α- гидроксил, или их циклические предшественники, в одну стадию по реакции Риттера. В зависимости от реакционной среды, четко различают таким образом два типа реакций Риттера: одна с раскрытием оксетанового цикла, приводящая к линейной форме непосредственно и полностью функционализированной цепи; другая - приводящая к прямому образованию оксазолина. Символ "*" указывает на наличие асимметрического атома углерода конфигурации R или S. В обоих случаях реакция Риттера является стереоспецифической, с сохранением конфигурации углерода в положении 2 и обращением конфигурации углерода в положении 3. Способ согласно изобретению предпочтительно осуществляют при использовании одного из энантиомеров цис -β- арилглицидатного производного общей формулы (I), чтобы получить соответствующий энантиомер линейной цепи или полученного оксазолина без необходимости проведения впоследствии расщепления рацемата. Согласно варианту получения цис -β- арилглицидатного производного общей формулы (I), описываемому в дальнейшем, R означает оптически чистый энантиомер сильно пространственно затрудненного хирального углеводородного радикала, предпочтительно циклоалкил, замещенный одной или несколькими алкильными группами, в частности, циклогексил. В таком случае R предпочтительно означает один из энантиомеров ментильного радикала, в частности, (+) -ментил.
1. Прямой синтез линейной цепи
Прямой синтез линейной цепи путем реакции Риттера состоит во введении во взаимодействие цис -β- арилглицидатного производного общей формулы (I), приведенной выше, с нитрилом формулы R2-CN, в которой
R2 означает арильный радикал, предпочтительно фенил, в присутствии протонной кислоты, такой, как серная, хлорная, тетрафторборная кислоты и т.д., и воды.
Тогда получают производное β- арилизосерина общей формулы (IIa):
в которой Ar, R и R2 имеют вышеуказанное значение.
Реакция протекает с обращением конфигурации углерода в положении 3 цис -β- фенилглицидатного производного. Таким образом, исходя из (2R,3R)-цис -β- фенилглицидатного производного получают соответствующее производное β- арилизосерина конфигурации (2R,3S).
Реакцию Риттера проводят в соответствующем растворителе, при температуре от -75oC до +25oC.
Соответствующим растворителем может быть сам нитрил, когда он является жидким при реакционной температуре, или же сама кислота (серная, хлорная или тетрафторборная кислота) или растворитель, такой, как, например, дихлорметан или диэтиловый эфир. Классически используемые протонные кислоты могут содержать необходимую для гидролиза воду.
Когда используют бензонитрил (R2 = фенил) для воздействия на цис -β- арилглицидат общей формулы (I) конфигурации (2R,3R), в котором Ar означает фенил, то напрямую получают соответствующее производное β- арилизосерина общей формулы (IIa), в которой Ar и R2 означают фенил конфигурации (2R,3S), которое является ничем иным, как предшественником боковой цепи паклитаксела.
2. Прямой синтез циклической цепи
Для реализации этой второй возможности, также проводят реакцию Риттера при использовании нитрила формулы R'2-CN, в которой R'2 означает вышеуказанный R2 или низший алкильный радикал или низший пергалогенированный алкильный радикал, такой, как трихлорметил, в присутствии кислоты Льюиса, в частности, комплекса трифторида бора с уксусной кислотой, эфирата трифторида бора, пентахлорида сурьмы, тетрахлорида олова, тетрахлорида титана и т.д., или в присутствии протонной кислоты, такой, как, например, тетрафторборная кислота, причем реакцию осуществляют в безводной среде.
Как и для синтеза линейной цепи, растворителем может быть сам нитрил, когда он является жидким при реакционной температуре, или же соответствующий растворитель, такой, как, например, дихлорметан или диэтиловый эфир. Реакционная температура также составляет от -75oC до +25oC.
В отсутствие воды осуществляют внутримолекулярную реакцию Риттера и получают оксазолин общей формулы (IIb):
в которой Ar, R и R'2 имеют вышеуказанное значение.
Как в случае реакции Риттера в присутствии воды, реакция протекает с обращением конфигурации углерода в положении 3 цис -β- фенилглицидатного производного. Таким образом, исходя из цис -β- фенилглицидата конфигурации (2R,3R) получают соответствующий оксазолин конфигурации (2R,3S).
Для обеих реакций Риттера, чтобы избежать образования свободного карбокатиона, вызывающего многочисленные потенциальные вторичные реакции, предпочтительно осуществляют добавление реагентов в нижеуказанном порядке: i) сначала получают комплекс нитрила с кислотой, затем ii) к образованной оксираном и нитрилом смеси добавляют кислотный катализатор.
Полученные на этой первой стадии продукты, производные β- арилизосерина общей формулы (IIa) или оксазолина общей формулы (IIb), могут быть снова превращены на второй, необязательной, нижеописанной стадии или затем переведены в кислоту путем осторожно осуществляемого омыления перед проведением реакции сочетания с защищенным производным баккатина для полусинтеза таксанов, в частности, паклитаксела и его дезацетилированных в положении 10 производных или доцетаксела. В случае производных β- арилизосерина общей формулы (IIa), омылению может предшествовать обычная стадия защиты гидроксила с помощью соответствующей защитной группы. Тогда получают производное общей формулы (II'a):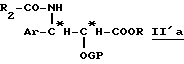
в которой Ar, R и R2 имеют вышеуказанное значение и GP означает защитную для гидроксильной функции группу, соответствующую синтезу таксанов, в частности, выбираемую среди алкоксиалкильных, арилалкилоксиалкильных, арилоксиалкильных или галогеналкоксикарбонильных радикалов, таких, как, например, метоксиметильная, 1-этоксиэтильная, бензилоксиметильная группа, (β-триметилсилилэтокси) метильная группа; тетрагидропиранильный радикал, β- алкоксикарбонильный радикал (TrOC), простые β- галогенированные, алкилсилилированные эфиры, или алкоксиацетильный, арилоксиацетильный, галогенацетильный или формильный радикалы.
3. Возможное превращение производных формулы (IIa) или (IIb)
Вышеполученные производные общей формулы (IIa) или (IIb) при необходимости могут быть превращены в новые промежуточные продукты, предшественники боковой цепи в полусинтезе таксанов. Эти превращения протекают с сохранением конфигурации атомов углерода в положениях 2 и 3. Полученные новые промежуточные продукты, следовательно, имеют такую же стереохимию, что и производные формулы (IIa) или (IIb), от которых они происходят. Полученные на этой второй стадии продукты затем переводят в кислоту путем осторожно осуществляемого омыления перед осуществлением их сочетания с защищенным производным баккатина для полусинтеза таксана, в особенности паклитаксела или доцетаксела.
3.1. Циклизация производных общей формулы (IIa)
Производные общей формулы (IIa) затем могут быть превращены в оксазолин формулы (IIb) обычными способами уровня техники (международная заявка 94/14787).
Производные β- арилизосерина общей формулы (IIa) также могут быть превращены в новые циклические промежуточные продукты: оксазолидинон общей формулы (III'a):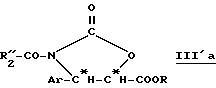
в которой Ar и R имеют вышеуказанное значение и R''2 означает вышеуказанный R'2, алкоксильный радикал, предпочтительно трет-бутоксигруппу, или линейный или разветвленный алкильный радикал по крайней мере с одной ненасыщенной связью, например, 1-метил-1-пропиленовый радикал; и соответствующие диалкилацетали.
Оксазолидиноны общей формулы (III'a) получают сначала путем введения во взаимодействие производного β- арилизосерина общей формулы (IIa) со сложным галогеналкоксикарбонильным эфиром, в особенности с 2,2,2-трихлорэтоксикарбонилом (TrOC), затем путем циклизации в присутствии сильного органического основания, такого, как диазабициклоундецен (ДБУ). Тогда получают производное оксазолидинона общей формулы (IIIa):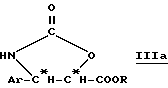
в которой Ar и R имеют вышеуказанное значение.
Производные общей формулы (IIIa) также могут быть получены путем прямого синтеза, вводя во взаимодействие β- арилглицидатные производные формулы (II'a) с мочевиной.
Ацилированные производные общей формулы (III'a) получают путем введения радикала R''2-CO- обычными способами ацилирования, в присутствии соответствующего ацилирующего средства, например, ацилгалогенида формулы R''2-CO-X, в которой R''2 имеет вышеуказанное значение и X означает галоген, или ангидрида соответствующей кислоты.
Диалкилацетали получают обычными способами получения ацеталей.
3.2. Раскрытие цикла оксазолина общей формулы (IIb)
Путем гидролиза оксазолина общей формулы (IIb) в кислой среде получают производное β- арилизосерина общей формулы (IIIb):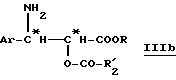
в которой Ar, R и R'2 имеют вышеуказанное значение.
Предпочтительно, когда R'2 означает низший пергалогеноалкил, такой, как трихлорметил, радикал R'2-CO- представляет собой защитную группу гидроксильной функции.
Затем этот предшественник боковой цепи таксанов можно превращать в амид (III'b):
в которой Ar, R, R'2 и R''2 имеют вышеуказанное значение.
Следовательно, можно получать, независимо, предшественник боковой цепи паклитаксела (R''2 = фенил) или доцетаксела (R''2 = трет-бутоксигруппа).
4. Получение производного цис -β- арилглицидной кислоты формулы (I)
Производное цис -β- арилглицидной кислоты формулы (I) может быть получено обычными способами уровня техники или путем простой этерификации цис -β- арилглицидной кислоты с помощью соответствующего спирта R-OH. Для улучшения общего выхода синтеза предшественников цепи таксанов в способе согласно изобретению получают цис -β- арилглицидатное производное общей формулы (I):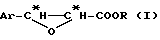
в которой Ar имеет вышеуказанное значение и
R означает чистый оптический энантиомер пространственно сильно затрудненного хирального углеводородного радикала,
путем введения во взаимодействие альдегида формулы:
Ar - CHO с галогенацетатом формулы X-CH2-COOR, где Ar, R имеют вышеуказанное значение и X означает галоген, в частности, хлор или бром.
Оптически чистый энантиомер пространственно сильно затрудненного хирального углеводородного радикала предпочтительно представляет собой циклоалкил, замещенный одной или несколькими алкильными группами, в частности, циклогексил.
Речь идет о реакции Дарценса, согласно которой получают смесь двух диастереоизомеров эфиров цис -β- арилглицидных кислот, в конфигурациях (2R,3R) и (2S,3S), и оптически чистого энантиомера хирального спирта R-OH, поскольку реакция Дарценса, осуществляемая с пространственно сильно затрудненным галогенацетатом, приводит главным образом к цис -β- арилглицидатной форме. Пространственно сильно затрудненный хиральный углеводородный радикал предпочтительно выбирают таким образом, чтобы он позволял осуществить физическое разделение двух диастереоизомеров в реакционной среде, например, путем селективной кристаллизации, без необходимости осуществлять стереоспецифическое отделение целевого энантиомера по окончании реакции с помощью обычных способов кристаллизации или хроматографии на колонке с хиральной фазой.
R-OH предпочтительно означает ментол, один из редко встречающихся пространственно сильно затрудненных хиральных спиртов, экономичный и имеющийся в продаже, в его обеих энантиомерных формах.
В способе синтеза предшественника боковой цепи таксанов стремятся получить цис -β- фенилглицидат конфигурации (2R,3R). В этом случае пространственно сильно затрудненный хиральный углеводородный радикал R выбирают таким образом, чтобы сначала из реакционной среды выкристаллизовывался диастереоизомер цис -β- фенилглицидата конфигурации (2R,3R). Когда R-OH является ментолом, предпочтительно используют (+)-ментол.
Асимметрическую реакцию Дарценса осуществляют в присутствии основания, в частности, алкоголята щелочного металла, такого, как трет-бутилат калия, или амида, такого, как бис-триметилсилиламид лития, в соответствующем растворителе, в частности, в простом эфире, таком, как диэтиловый эфир, при температуре от -78oC до 25oC. Реакция приводит к диастереоизомерной смеси, образованной почти исключительно цис-глицидатами, причем можно достигать выхода выше 95%, близкого к 97%. Обработка выделенного продукта в соответствующем растворителе, в частности, в смеси метанола с водой, позволяет легко достигать надлежащего физического разделения диастереоизомеров.
Путем фракционной кристаллизации (2 стадии) достигают быстрого обогащения целевым диастереоизомером с диастереоизомерной чистотой выше 99%.
Этот последний момент является особенно важным, так как он обусловливает изомерную чистоту целевого таксана, поскольку нежелательные диастереоизомеры обладают своей собственной биологической активностью, отличной от таковой целевого таксана.
Примечательно констатировать, что селективное использование двух энантиомеров сложного ментилового эфира с помощью одного и того же способа позволяет придти к двум диастереоизомерным предшественникам двух энантиомеров глицидной кислоты.
Кроме довольно высокого выхода чистого выделенного диастереоизомера (вплоть до 45%), диастереоизомерная чистота преобладающего продукта реакции, легкость осуществления реакции, простота и быстрота очистки, незначительная стоимость реагентов и катализаторов делают достаточно легким и экономичным промышленный синтез этого главного в асимметрическом синтезе β- аминокислот промежуточного продукта.
Когда в способе согласно изобретению используют производное общей формулы (I), получаемое путем асимметрической реакции Дарценса, то получают производные вышеприведенных общих формул (IIa), (II'a), (IIb), (IIIa), (IIIb) и (III'b), в которых R означает оптически чистый энантиомер пространственно сильно затрудненного хирального углеводородного радикала, такого, как циклоалкил, замещенного одной или несколькими алкильными группами, в частности, циклогексил, предпочтительно ментил, особенно предпочтительно (+)- ментил.
Настоящее изобретение относится также к этим производным, пригодным в качестве промежуточных продуктов в синтезе боковых цепей таксанов.
Следует отметить, что настоящий способ очень быстро приводит к замещенным хиральным оксазолинам, уже описанным в литературе (международная заявка 94/14787), в три стадии, исходя из доступных, имеющихся в продаже продуктов, вместо 6-8 стадий.
5. Мягкое омыление
Осуществляют мягкое омыление производных общих формул (IIa), (II'a), (IIb), (IIIa), (IIIb) и (III'b) в мягких условиях, чтобы высвободить кислотную функцию при сохранении структуры вышеуказанных производных, например, в присутствии карбоната щелочного металла в смеси метанола с водой.
После осуществляемого мягкого омыления получают производные вышеприведенных формул (IIa), (II'a), (IIb), (IIIa), (IIIb) и (III'b), в которых R означает атом водорода, которые могут быть прямо использованы в полусинтезе таксанов путем сочетания с соответствующим производным баккатина III.
6. Полусинтез таксанов
6.1. Этерификация с образованием сложного эфира
Настоящее изобретение, следовательно, также относится к способу полусинтеза таксанов общей формулы (IV):
C-B, (IV)
в которой C означает боковую цепь, выбираемую среди радикалов следующих формул: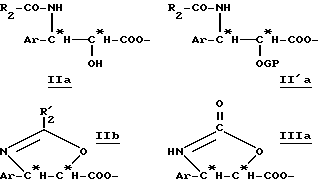

в которых Ar, R2, R'2, R''2, R3 и GP имеют вышеуказанное значение; и
B означает происходящий от баккатина III радикал общей формулы (V):
в которой Ac означает ацетил;
Bz означает бензоил;
Me означает метил;
R4 означает ацетил или защитную для гидроксильной функции группу GP1; и
R5 означает защитную для гидроксильной функции группу GP2;
путем этерификации с образованием сложного эфира соответствующего производного баккатина III общей формулы (V), содержащего гидроксильную функцию в положении C-13, с помощью одного из производных вышеуказанных общих формул (IIa), (II'a), (IIb), (IIIa), (III'a), (IIIb) и (III'b), в которых R означает атом водорода, в обычных условиях получения таксанов, таких, как указанные в уровне техники (особенно: европейские патенты N 0 253 738, 0 336 840, 0 336 841, 0 495 718; международные заявки 92/09589, 94/07877, 94/07878, 94/07879, 94/10169, 94/12482; европейские патенты 0 400 971 и 0 428 376; международная заявка 94/14787).
Защитные группы GP1 и GP2, независимо друг от друга, представляют собой обычные группы, используемые в полусинтезе таксанов, такие, как триалкилсилильные группы (европейский патент 0 336 840) или TrOC (европейский патент 0 336 841).
GP1 и GP2, независимо друг от друга, также означают линейные или разветвленные, пространственно затрудненные галогеналкоксикарбонильные радикалы, включающие по крайней мере один атом галогена. Предпочтительно речь идет о радикалах, алкильный остаток которых содержит 1-4 атома углерода и 3 или 4 атома галогена, предпочтительно выбираемых среди 2,2,2-трибромэтоксикарбонильного, 2,2,2,1-тетрахлорэтоксикарбонильного, 2,2,2-трихлор-трет-бутоксикарбонильного и трихлорметоксикарбонильного радикалов, причем все эти радикалы более пространственно затрудненные, чем галогеналкоксикарбонил (TrOC), используемый до настоящего времени для защиты таксанов в положении 7.
GP1 и GP2 также, независимо друг от друга, означают ацильные радикалы, атом углерода которых в α- положении карбонильной функции содержит по крайней мере один атом кислорода.
Эти ацильные радикалы описываются, например, в заявке на европейский патент 0 445 021. Речь идет преимущественно об алкокси- или арилоксиацетильных радикалах формулы:
R6-O-CH2-CO-,
в которой R6 означает пространственно затрудненный алкильный радикал, циклоалкильный радикал или арильный радикал, или об арилидендиоксиацетильных радикалах формулы (a):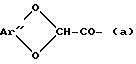
в которой Ar'' означает арилиденовый радикал.
Под пространственно затрудненным алкильным радикалом предпочтительно понимают линейный или разветвленный алкильный радикал с 1-6 атомами углерода, замещенный одним или несколькими пространственно затрудненными заместителями, выбираемыми среди галогенов, линейных или разветвленных алкильных радикалов с 1-6 атомами углерода, линейных или разветвленных алкоксильных радикалов с 1-6 атомами углерода или циклоалкильных радикалов с 3-6 атомами углерода или арильных радикалов. Речь идет, например, о трет-бутильном радикале или трифенилметильном радикале.
Под циклоалкилом предпочтительно понимают циклоалкильный радикал с 3-6 атомами углерода, незамещенный или замещенный одним или несколькими пространственно затрудненными заместителями, выбираемыми среди галогенов, линейных или разветвленных алкильных радикалов с 1-6 атомами углерода, линейных или разветвленных алкоксильных радикалов с 1-6 атомами углерода или арильных радикалов. Предпочтительно речь идет о циклогексильном радикале, замещенном одним или несколькими линейными или разветвленными алкильными радикалами с 1-6 атомами углерода, как, например, ментил, его рацемат или его энантиомеры и их смеси в любых соотношениях.
Под арилом предпочтительно понимают фенильный, нафтильный, антрильный или фенантрильный радикал, незамещенный или замещенный одним или несколькими пространственно затрудненными заместителями, выбираемыми среди галогенов, линейных или разветвленных алкильных радикалов с 1-6 атомами углерода, линейных или разветвленных алкоксильных радикалов с 1-6 атомами углерода или арильных радикалов, в частности, фенил.
Предпочтительно речь идет о фенильном радикале, незамещенном или замещенном одним или двумя вышеуказанными пространственно затрудненными заместителями в орто- или орто'-положении простой эфирной связи.
Наконец, под арилиденом предпочтительно понимают фениленовый, нафтиленовый, антриленовый или фенантриленовый радикал, незамещенный или замещенный одним или несколькими пространственно затрудненными заместителями, выбираемыми среди галогенов, линейных или разветвленных алкильных радикалов с 1-6 атомами углерода, линейных или разветвленных алкоксильных радикалов с 1-6 атомами углерода или арильных радикалов, в частности, фенил.
GP1 и GP2 также, независимо друг от друга, означают триалкилгерманильный радикал или вместе образуют двухвалентный радикал формулы -SiR7-O-SiR8-, в которой R7 и R8, независимо друг от друга, означают пространственно затрудненный алкильный радикал, такой, как указанный выше, в частности, каждый из R7 и R8 означает изопропильный радикал.
6.2. Возможное раскрытие цикла
Когда C означает радикал формулы (IIb) или (IIIa), осуществляют раскрытие оксазолинового цикла для получения производного таксанов формулы (VI):
в которой Ac, Bz, Me, Ar, R2, R4 и R5 имеют вышеуказанное значение.
Раскрытие циклов радикалов (IIb), (IIIa) и (III'a) обычно осуществляют путем гидролиза в кислой или основной среде. Для радикала формулы (IIb) это раскрытие можно осуществлять по способам, описанным в уровне техники (в особенности в международной заявке 94/14787), путем гидролиза в кислой среде, с последующей обработкой в щелочной среде для получения производного общей формулы (VI).
6.3. Удаление защитных групп
Удаляют защитные группы с гидроксильных функций производных общей формулы (V) или (VI), заменяя защитные для гидроксильной функции группы GP (когда C означает радикал формулы (II'a)), GP1 (когда R4 отличен от ацетила) и GP2 атомом водорода при использовании обычных способов.
В случае производных общей формулы (V), в которых C означает радикал формулы (IIb) или (IIIa) и GP1 и/или GP2, независимо друг от друга, означают обычные группы, используемые в полусинтезе таксанов, такие, как триалкилсилильные группы, удаление защитных групп осуществляют одновременно с вышеописанным раскрытием цикла.
Когда GP1 или GP2 представляют собой пространственно затрудненные галогеналкоксикарбонильные радикалы, удаление защитных групп осуществляют обычными способами, описанными для TrOC, путем воздействия цинка или цинка, активированного с помощью тяжелых металлов, таких, как медь, в органическом растворителе, в частности, в уксусной кислоте, тетрагидрофуране или этиловом спирте, с водой или без нее.
Когда GP1 и/или GP2 означают ацильные радикалы, углерод которых в α- положении карбонильной функции содержит по крайней мере один атом кислорода, удаление защитных групп осуществляют в основной среде путем омыления в метаноле при низкой температуре, предпочтительно с помощью раствора гидроксида аммония в метаноле при температуре ниже 10oC, предпочтительно, около 0oC.
В случае, где C означает радикал формулы (IIb), раскрытие оксазолинового цикла осуществляют одновременно с удалением защитных групп в основной среде с получением в одну стадию соответствующего производного таксана общей формулы (VI), в которой R4 означает ацетильный радикал или атом водорода и R5 означает атом водорода, в противоположность раскрытию цикла в кислой среде, описанному в уровне техники, которое требует второй стадии в основной среде.
Известные защитные группы удаляют с помощью известных способов и оксазолиновую цепь, когда она имеется, развертывают путем гидролиза, получая таксаны, полностью идентичные стандартным таксанам. В качестве примера и для демонстрирования пригодности изобретения, однако, не ограничивая его объема охраны, можно получать паклитаксел, 10-дезацетилтаксол, цефаломанин и доцетаксел из соответствующих защищенных производных.
Деблокирование ацильных групп, атом углерода которых в α- положении карбонильной функции содержит по крайней мере один атом кислорода, сначала осуществляли в условиях, классически рассматриваемых как наиболее мягкие, т.е. при использовании ацетата цинка в метанольной среде при кипячении с обратным холодильником. В этом случае, причем реакция полностью заканчивается за несколько часов (по сравнению с несколькими днями в случае ацетатов), постоянно получали, наряду с целевым продуктом, его эпимер в C-7, образующийся в результате классического равновесия ретроальдолизации. Предполагая, что точно также в нейтральных, даже в слегка кислых условиях, существенными факторами процессов являются метанол и, главным образом, температура, заявители прибегли к стандартным условиям деблокирования ацильных групп, описанных предшествующими авторами, путем омыления в основной среде в этаноле при низкой температуре. В этих условиях не обнаружено никакой заметной эпимеризации. В качестве примера, авторы настоящей заявки получили паклитаксел, 10-дезацетилтаксол, цефаломанин и доцетаксел, которые полностью идентичны стандартным таксанам, из соответствующих алкокси- или арилоксиацетильных производных.
Наконец, следует заметить, что все ранее описанные способы, целью которых, однако, является улучшение общего выхода полусинтеза, состоят в предварительном синтезе фенилизосериновой цепи для ее превращения в одну из вышеуказанных циклических структур (β-лактамы, оксазолидины или оксазолины). Следовательно, парадоксально, наилучшие очевидные результаты сочетания этих циклических структур только компенсируют понижение общего выхода, вызываемое добавлением стадий создания цикла с синтетической последовательностью линейной цепи (или в целом 9 стадий). В случае общего способа синтеза таксанов согласно изобретению получают продукт, такой, как паклитаксел, только за 5 стадий: (1S, 2R,5S)-(+)-ментил-(2R,3R)-3-фенилглицидат; (1S,2R,5S)-(+)-ментил-(4S, 5R)-2,4-дифенил-4,5-дигидрооксазол- 5-карбоксилат; омыление; полусинтез (этерификация с образованием сложного эфира); раскрытие цикла и удаление защитных групп.
Настоящее изобретение, наконец, относится к промежуточным продуктам синтеза вышеприведенных общих формул (IV), (V) и (VI), пригодным для общего синтеза таксанов, являющегося предметом настоящего изобретения.
Вообще, под углеводородным радикалом понимают предпочтительно, согласно изобретению, насыщенный или ненасыщенный углеводородный радикал, который может включать одну или несколько ненасыщенных связей, такой, как линейный или разветвленный алкил, который может быть ненасыщенным, циклоалкил, который может быть ненасыщенным, аралкил или арил, каждый из которых при необходимости может быть незамещен или замещен одним или несколькими заместителями, в частности, алкилом.
Под линейным или разветвленным алкилом предпочтительно, согласно изобретению, понимают алкил с 1-6 атомами углерода, в частности, выбираемый среди метильного, этильного, пропильного, изопропильного радикалов; бутильного радикала и его различных разветвленных изомеров, как, например, трет-бутил; пентильного и гексильного радикалов и их различных разветвленных изомеров. Это определение относится также к алкильным остатками алкоксильных или аралкоксильных
радикалов.
Под циклоалкилом понимают предпочтительно, согласно изобретению, циклоалкил с 3-6 атомами углерода, в частности, выбираемый среди циклопропильного, циклобутильного, циклопентильного или циклогексильного радикалов.
Под арилом понимают предпочтительно, согласно изобретению, ароматический или гетероароматический радикал, в частности, выбираемый среди фенильного, нафтильного, антрильного, фенантрильного, пиридильного, пиримидильного радикалов и т.д.
Наконец, под галогеном предпочтительно понимают хлор, бром или йод. В случае галогеналкоксикарбонильных радикалов предпочтительно речь идет о радикалах, алкильный остаток которых содержит 1-4 атома углерода и 3 или 4 атома галогена.
Общий способ синтеза таксанов согласно изобретению представлен на схеме 1 (схемы 1 и 2 см. в конце описания), где R означает (+)-ментил и R2 или R'2 означает фенил.
Последняя стадия полусинтеза таксанов по способу согласно изобретению представлена на нижеприводимых схемах 2 и 3. На схеме 2 представлен синтез паклитаксела из вышеуказанных производных формулы (IV), в которых C означает радикал формул (IIb) или (III'a). На схеме 3 представлен синтез 10-дезацетилтаксола из производного формулы (IV), в котором C означает радикал формулы (IIb).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
1. Предшественники боковой цепи таксанов
Пример 1: (1S,2R,5S)-(+)-Ментилхлорацетат
К перемешиваемому при комнатной температуре раствору 100 г (0,640 моль) (1S, 2R,5S)-(+)-ментола в 1 л безводного дихлорметана добавляют 57 мл (0,704 моль) безводного пиридина. После перемешивания в течение нескольких минут добавляют 56 мл (0,704 моль) хлорацетилхлорида и оставляют реакцию продолжаться в течение 30 минут. После контроля с помощью тонкослойной хроматографии (ТСХ) добавляют 50 г измельченного льда и реакционную среду интенсивно перемешивают в течение 1 часа. После разбавления с помощью 100 мл дихлорметана, органическую фазу промывают несколько раз водным насыщенным раствором хлорида натрия (200 мл), сушат над сульфатом магния, затем концентрируют при пониженном давлении. После очистки таким образом полученного сырого продукта путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 20:1) получают 146 г (1S,2R,5S)-(+)-ментилхлорацетата в виде сиропа.
Полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц, дейтерохлороформ), δ (м.д. = миллионные доли): 4,77 (1Н, дт); 4,06 и 4,02 (2Н, 2д, J = 13,6 Гц); 2,02 (1Н, м, J = 11,8 Гц); 1,87 (1Н, м, J = 7 и 2,6 Гц); 1,69 (2Н, м); 1,50 (1Н, м); 1,43 (1Н, м, J = 11,7 и 3 Гц); 1,07 (1Н, м); 1,02 (1Н, кв, J = 11,8 Гц); 0,92 и 0,90 (6Н, 2д, J = 6,4 Гц); 0,89 (1Н, м); 0,77 (3Н, д, J = 7 Гц).
Пример 2: (1S,2R,5S)-(+)-ментил-(2R,3R)-3-фенилглицидат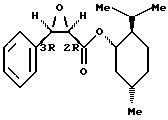
К перемешиваемому при комнатной температуре раствору 152 г (0,653 моль) (1S, 2R, 5S)-(+)-ментилхлорацетата в 600 мл безводного диэтилового эфира добавляют 69 мл (0,686 моль) бензальдегида. После перемешивания в течение нескольких минут раствор охлаждают до -78oC в инертной атмосфере, затем в течение двух часов добавляют суспензию 85 г (0,718 моль) трет-бутилата калия в 400 мл безводного диэтилового эфира и реакционную среду оставляют для повышения ее температуры до комнатной. После контроля с помощью ТСХ, органическую часть разбавляют с помощью 299 мл дихлорметана, промывают несколько раз с помощью насыщенного раствора хлорида натрия, сушат над сульфатом магния и концентрируют при пониженном давлении. Таким образом получают 200 г сырого продукта в виде сиропа, содержащего четыре диастереоизомера (из которых два цис-изомера и два транс-изомера), который подвергают фракционной кристаллизации.
Сначала раствор сырого продукта в 2 л метанола, доведенный до температуры 60oC, к которому постепенно добавляют 700 мл осмотизированной воды, выдерживают в течение 16 часов при комнатной температуре и в диапазоне отклонений. Нижнюю твердую фазу желтого цвета, обогащенную транс-изомерами, удаляют, и кристаллы белого цвета верхней фазы, обогащенной цис-изомерами, отделяют путем отфильтровывания. Таким образом полученные кристаллы снова растворяют в 2 л доведенного до температуры 60oC метанола с добавлением 500 мл осмотизированной воды вплоть до достижения устойчивой мутности и выдерживают в течение 16 часов при комнатной температуре. Три дополнительные кристаллизации, осуществляемые тем же способом, но с меньшими объемами метанола (1 л) и воды (200 мл), необходимы для получения 23 г (1S,2R,5S)-(+)-ментил-(2R, 3R)-3-фенилглицидата в виде кристаллического вещества с чистотой согласно ВЭЖХ (высокоэффективная жидкостная хроматография) выше 99% (выход = 12%).
Полученное соединение имеет следующие характеристики: Т. пл.= 104oC;
1H-ЯМР-спектр (400 МГц, дейтерохлороформ), δ (м.д.): 7,40 (2Н, дд, J = 7,8 и 1,7 Гц); 7,32 (3Н, м); 4,58 (1Н, дт, J = 10,9 и 4,2 Гц); 4,26 (1Н, д, J = 4,6 Гц); 3,83 (1Н, д, J = 4,8 Гц); 1,6-0,85 (9Н, м); 0,78 (3Н, д, J = 7 Гц); 0,75 (3Н, д, J = 6,4 Гц); 0,62 (3Н, д, J = 6,9 Гц).
Дифракция рентгеновских лучей монокристалла (1S,2R,5S)-(+)-ментил-2R, 3R)-3-фенилглицидата с целью непрямого определения абсолютной конфигурации:
Монокристалл получают из суспензии кристаллов, образующейся в результате добавления при нагревании нерастворителя (вода) к полунасыщенному раствору глицидата в метаноле. Путем медленного охлаждения из раствора осаждаются тонкие иглы с чистотой 99,95% (ВЭЖХ), которые сохраняют увлажненными до конечного отбора.
Отобранный образец (тонкая игла с размерами 0,12•0,12•0,40 мм) исследуют на автоматическом дифрактометре CAD4 ENRAF-NONIUS (излучение молибдена с графитовым монохроматором). Параметры (элементарной) ячейки получают путем "аффинажа" совокупности 25 отражений с большим углом θ. Накопление данных ( 2θmax = 50o; развертка ω/2θ = I; tmax = 60 с; область HKL: H=0,6; К=0,14; L= 0,28; контроли интенсивности без значительного отклонения (0,1%)) дает 1888 отражений, 1037 из которых с I > 1,5 σ (I).
C19H26O3 : Mr = 302,42; орторомбический; P 2I 2I 2I;
a = 5,709 (1 I), b = 12,908 (4), с = 24,433 (8)
Ангстрем; V = 1801 (5) Ангстрем-3: Z = 4; Dx = 1,116
Mg•M-3; λ(MoKα) = 0,70926 Ангстрем; μ = 0,69 см-1;
F(000) = 656; T = 294 K; R конечное = 0,0702 для 1037 наблюдений.
После коррекций Лоренца и коррекций поляризации, структуру устанавливают с помощью прямых методов, которые позволяют локализовать большинство неводородных атомов молекулы, причем оставшиеся атомы локализуют с помощью разностей Фурье и последовательных масштабирований. После изотропного (R=0,125), затем анизотропного (R=0,095) аффинажа большинство атомов водорода локализуют с помощью разности Фурье (между 0,39 и 0,14 еАнгстрем-3), причем другие атомы водорода занимают расчетное положение. Полную структуру аффинируют с помощью полной матрицы (x, y, z, βij для C и O; x, y, z для H; 200 переменных и 1037 наблюдений; w = 1/ σ (Fo)2 = [ σ2 (I)+(0,04 Fo 2)2]-1/2)), что приводит к R=0,080; Rw=0,072; Sw=1,521 (остаток Δρ ≤ 0,21 еАнгстрем-3).
Факторы диффузии берут из Международных Таблиц по кристаллографии [International Tables for X ray Cristallography (1974), том IV, Birmingam: Kynoch Press (Present distributer D. Reidel, Dordrecht)]. Расчеты для определения структуры проводят на приборе фирмы Хьюлетт-Паккард 9000-710 [SHELDRICK G. M. (1985). Кристаллографический расчет 3: сбор данных, определение структуры, протеины и базы данных. Под редакцией G.M. Sheidrick, C. Kruger и R.Goddard; Oxford: Clarendron Press] и на цифровой вычислительной машине Micro VAX 3100 для других расчетов с использованием программ MOLEN [FAIR C. K. (1990) MOLEN. Диалоговая интеллектуальная система для анализа структуры кристаллов. Enraf-Nonius, Delft, Нидерланды].
ДИАГРАММУ ORTEP (см. в конце описания).
Образец (1S, 2R,5S)-(+)-ментил-(2R,3R)-3-фенилглицидата путем обработки метилатом натрия в метаноле позволяет получить соответствующий метилфенилглицидат, характеристики которого следующие:
[α]
1H-ЯМР-спектр (400 МГц, дейтерохлороформ), δ (м.д.): 7,40 (2Н, д, J = 8 Гц); 7,32 (3Н, м); 4,26 (1Н, д, J = 4,6 Гц); 3,84 (1Н, д, J = 4,6 Гц); 3,55 (3Н, с).
Пример 3: (1S,2R,5S)-(+)-Ментил-(4S,5R)-2,4-дифенил-4,5-дигидрооксазол- 5-карбоксилат
К перемешиваемому в инертной атмосфере при температуре -65oC раствору 30 г (0,0993 моль) (1S, 2R,5S)-(+)-ментил-(2R,3R)-3- фенилглицидата и 305 мл (2,98 моль) бензонитрила в 5 л безводного дихлорметана в течение 10 минут добавляют 15 мл (0,109 моль) 54%-ного раствора тетрафторборной кислоты в диэтиловом эфире. Оставляют реакцию продолжаться в течение 1 часа при температуре -65oC и, после контроля с помощью ТСХ, добавляют 300 мл водного насыщенного раствора гидрокарбоната натрия и реакционную смесь оставляют стоять при перемешивании до повышения температуры до комнатной. После экстракции водной фазы дихлорметаном (2 раза по 200 мл), объединенные органические фазы промывают насыщенным раствором хлорида натрия (200 мл), водой (50 мл) и сушат над сульфатом магния. После концентрирования при пониженном давлении и удаления остаточного бензонитрила в вакууме при доведении температуры до 50oC, полученный сырой продукт очищают путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 20:1).
Таким образом выделяют 32 г (1S,2R,5R)-(+)-ментил-(4S,5R)-2,4- дифенил-4,5-дигидрооксазол-5-карбоксилата в виде бесцветного сиропа (выход = 80%), который имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,10 (2Н, д, J = 7,1 Гц); 7,54 (1Н, т, J = 7,4 Гц); 7,46 (2Н, т, J = 7,4 Гц); 7,34 (5Н, м); 5,40 (1Н, д, J = 6,4 Гц); 4,88 (1Н, д, J = 6,4 Гц); 4,85 (1Н, дт, J = 10,9 и 4,4 Гц); 2,09 (1Н, м); 1,84 (1Н, м, J = 7 и 2,7 Гц); 1,71 (1Н, м); 1,69 (1Н, м); 0,94 (3Н, д, J = 6,5 Гц); 0,9 (1Н, м); 0,85 (3Н, д, J = 7 Гц); 0,77 (3Н, д, J = 7 Гц).
Пример 4: (4S,5R)-2,4-Дифенил-4,5-дигидрооксазол- 5-карбоновая кислота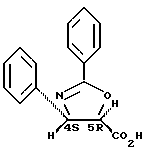
К перемешиваемому при комнатной температуре раствору 3,5 г (8,64 ммоль) (1S,2R,5S)-(+)-ментил-(4S,5R)-2,4-дифенил-4,5- дигидрооксазол-5-карбоксилата в 70 мл метанола добавляют 25 мл раствора 6 г (43,2 ммоль) карбоната калия в осмотизированной воде, и оставляют реакцию продолжаться в течение 16 часов при комнатной температуре. После контроля с помощью ТСХ, реакционную среду концентрируют при пониженном давлении. Таким образом полученную водную фазу промывают дихлорметаном (3 раза по 100 мл), подкисляют до pH 2 путем медленного добавления 20 мл 1 М водного раствора HCl и экстрагируют этилацетатом (3 раза по 100 мл). Объединенные органические фазы после экстракции сушат над сульфатом магния и концентрируют при пониженном давлении.
Таким образом получают 2,26 г (4S,5R)-2,4-дифенил-4,5-дигидрооксазол-5-карбоновой кислоты в виде порошка белого цвета (выход = 98%), которая имеет следующие характеристики:
[α]
Т.пл. = 201-292oC;
1H-ЯМР-спектр (400 МГц; гексадейтеродиметилсульфоксид), δ (м.д.): 7,99 (2Н, д, J = 7,3 Гц); 7,64 (1Н, т, J = 7,4 Гц); 7,55 (2Н, т, J = 7,7 Гц); 7,36 (5Н, м); 5,40 (1Н, д, J = 6,3 Гц); 4,99 (1Н, д, J = 6,4 Гц).
Пример 5: (1S,2R,5S)-(+)-Ментил-(2R,3S)-N-бензоил-3- фенилизосеринат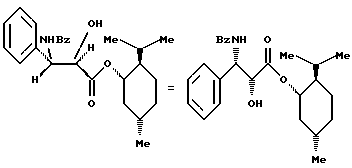
К перемешиваемому при комнатной температуре раствору 1 г (2,47 ммоль) (1S,2R,5S)-(+)-ментил-(4S,5R)-2,4-дифенил-4,5- дигидрооксазол-5-карбоксилата в смеси 15 мл метанола с 15 мл тетрагидрофурана добавляют 15 мл 1 М водного раствора HCl. Реакционную среду кипятят с обратным холодильником в течение 1 часа и после контроля с помощью ТСХ и возврата температуры к комнатной постепенно добавляют 45 мл водного насыщенного раствора гидрокарбоната натрия вплоть до достижения щелочного значения pH. После перемешивания в течение 48 часов при комнатной температуре, полученную после концентрирования при пониженном давлении водную фазу экстрагируют дихлорметаном (100 мл). Органическую фазу промывают насыщенным раствором хлорида натрия (2 раза по 50 мл), сушат над сульфатом магния, концентрируют при пониженном давлении и полученный остаток хроматографируют на силикагеле (15-40 мкм) (элюирующее средство: смесь дихлорметана с метанолом в соотношении 95:5).
Таким образом выделяют 0,835 г (1S,2R,5S)-(+)-ментил- (2R,3S)-N-бензоил-3-фенилизосерината в виде твердого вещества белого цвета (выход = 80%) со следующими характеристиками:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 7,77 (2Н, д, J = 7,2 Гц); 7,51 (1Н, т, J = 7,3 Гц); 7,45 (4Н, м); 7,36 (2Н, т, J = 7,2 Гц); 7,29 (1H, т, J = 7,2 Гц); 7,04 (1H, д, J = 9,2 Гц); 5,78 (1H, дд, J = 9,2 и 2,1 Гц); 4,79 (1H, дт, J = 10,9 и 4,4 Гц); 4,63 (1H, уш. с.); 3,35 (1H, уш. с); 1,81 (2Н, м); 1,67 (3Н, м); 1,5-1,36 (2Н, м); 1,09-0,91 (2Н, м); 0,89 (3Н, д, J = 6,9 Гц); 0,77 (3Н, д, J = 6,5 Гц); 0,74 (3Н, д, J = 6,9 Гц).
Пример 6: (1S, 2R,5S)-(+)-Ментил-(2R,3S)-N-бензоил-O- триэтилсилил-3-фенилизосеринат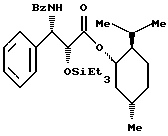
К раствору 0,8 г (1,89 ммоль) (1S,2R,5S)-(+)-ментил- (2R,3S)-N-бензоил-3-фенилизосерината в 10 мл безводного дихлорметана добавляют 0,255 г (2,08 ммоль) 4-диметиламинопиридина. После перемешивания в течение нескольких минут при комнатной температуре, в течение пяти минут добавляют 477 мкл (2,84 ммоль) триэтилсилилхлорида. После перемешивания в течение 1 часа при комнатной температуре и контроля с помощью ТСХ, реакционную среду разбавляют с помощью 100 мл дихлорметана. Органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия (2 раза по 20 мл), насыщенным раствором хлорида натрия (50 мл), сушат над сульфатом магния и концентрируют при пониженном давлении. После очистки полученного остатка путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 10:1), получают 0,74 г (1S,2R,5S)-(+)-ментил-(2R, 3S)-N-бензоил-О-триэтилсилил- 3-фенилизосерината в виде бесцветного сиропа (выход = 75%).
Полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 7,82 (2Н, д, J = 7 Гц); 7,52 (1Н, т, J = 7,4 Гц); 7,45 (2Н, т, J = 7 Гц); 7,37 (2Н, д, J = 7,2 Гц); 7,32 (2Н, т, J = 7,2 Гц); 7,26 (2Н, м); 5,60 (1Н, дд); 4,73 (1Н, дт, J = 11 и 4,3 Гц); 1,88-1,67 (м); 1,44 (2Н, м); 1,06-0,87 (м); 0,80 (м); 0,67 (3Н, д, J = 7 Гц); 0,62-0,34 (м).
Пример 7: (2R,3S)-N-Бензоил-O-триэтилсилил-3-фенилизосерин
К перемешиваемому при комнатной температуре раствору 0,5 г (0,931 ммоль) (1S,2R,5S)-(+)-ментил-(2R,3S)-N-бензоил-O- триэтилсилил-3-фенилизосерината в 15 мл метанола добавляют раствор 0,644 г (4,655 ммоль) карбоната натрия в 10 мл осмотизированной воды. После перемешивания в течение 16 часов при комнатной температуре и контроля с помощью ТСХ, реакционную среду концентрируют при пониженном давлении и остаточную водную фазу промывают дихлорметаном (3 раза по 50 мл), затем подкисляют до pH 2 путем добавления 1 М водного раствора HCl (10 мл). Водную фазу экстрагируют этилацетатом (3 раза по 50 мл), и объединенные органические фазы сушат над сульфатом магния и концентрируют при пониженном давлении.
Получают 0,320 г (2R,3S)-N-бензоил-O- триэтилсилил-3-фенилизосерина в виде порошка белого цвета (выход = 90%), который имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; гексадейтеродиметилсульфоксид), δ (м.д.): 8,46 (1Н, д, J = 9,3 Гц); 7,82 (2Н, д, J = 7,1 Гц); 7,54 (1Н, т, J = 7,2 Гц); 7,47 (4Н, м); 7,32 (2Н, т); 7,36 (1Н, т); 5,44 (1H, дд, J = 9,2 и 5,5 Гц); 4,64 (1Н, д, J = 5,6 Гц); 0,77 (9Н, м), 0,45 (6Н, м).
Пример 8: (1S, 2R,5S)-(+)-Ментил-(2R,3S)-N-бензоил-O- (2,2,2-трихлорэтокси)карбонил-3-фенилизосеринат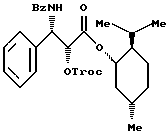
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 1,38 г (3,3 ммоль) (1S,2R,5S)-(+)-ментил- (2R,3S)-N-бензоил-3-фенилизосерината в 30 мл безводного дихлорметана добавляют 480 мг (3,96 ммоль) 4-диметиламинопиридина. После перемешивания в течение 10 минут добавляют в течение 5 минут 540 мкл (3,96 ммоль) 2,2,2-трихлорэтоксикарбонилхлорида. После перемешивания в течение двух часов при комнатной температуре и контроля с помощью ТСХ, органическую фазу промывают насыщенным раствором гидрокарбоната натрия (2 раза по 10 мл), насыщенным раствором хлорида натрия (10 мл), сушат над сульфатом магния и концентрируют при пониженном давлении. После очистки полученного остатка путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 5: 1) получают 1,60 г (1S,2R,5S)-(+)-ментил-(2R,3S)-N-бензоил-О- (2,2,2-трихлорэтокси-О-карбонил-3-фенилизосерината в виде бесцветного сиропа (выход = 82%).
Полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 7,82 (2Н, д, J = 7,4 Гц); 7,53 (1Н, т, J = 7,4 Гц); 7,44 (4Н, м); 7,35 (2Н, т, J = 7 Гц); 7,29 (1Н, т, J = 7 Гц); 7,09 (1Н, д, J = 9,3 Гц); 6,0 (1Н, дд, J= 9,3 и 2,5 Гц); 5,45 (1Н, д, J = 2,6 Гц); 4,78 и 4,72 (2Н, 2д, J = 11,9 Гц); 4,77 (1Н, м); 1,85 (1H, м); 1,79 (1Н, м); 1,65 (2Н, м); 1,43 (1Н, м); 1,02 (1Н, м); 0,96 (1Н, м); 0,86 (1H, м); 0,83 (3Н, д, J= 7 Гц); 0,78 (3Н, д, J= 6,5 Гц); 0,68 (3Н, д, J=6,9 Гц).
Пример 9: (1S,2R,5S)-(+)-Ментил-(4S,5R)-4-фенилоксазолидин- 2-он-5-карбоксилат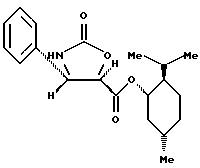
К перемешиваемому в инертной атмосфере и при комнатной температуре раствору 3,96 г (6,62 ммоль) (1S,2R,5S)-(+)- ментил-(2R,3S)-N-бензоил-O-(2,2,2-трихлорэтокси)карбонил-3- фенилизосерината в 30 мл безводного дихлорметана добавляют 1 мл (7,28 ммоль) 1,8-диазабицикло[5,4,0]ундец-7-ена. После перемешивания в течение 30 минут при комнатной температуре, органическую фазу промывают с помощью 10 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния и концентрируют при пониженном давлении. После очистки остатка путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 7:3) получают 2,18 г целевого соединения в виде желтого сиропа (выход = 95%).
Полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 7,40 (5Н, м); 6,09 (1Н, с); 4,93 (1Н, д, J = 5,3 Гц); 4,86 (1Н, дт, J = 11 и 4,4 Гц); 4,73 (1Н, д, J = 5,4 Гц); 2,05 (1Н, м); 1,81 (1H, м); 1,71 (2H, м); 1,54-1,41 (3Н, м); 1,07 (2Н, м); 0,94 (3Н, д, J= 6,5 Гц); 0,88 (3Н, д, J = 7 Гц); 0,77 (3Н, д, J = 7 Гц).
Пример 10: (1S,2R,5S)-(+)-Ментил-(4S,5R)-N- трет-бутоксикарбонил-4-фенилоксазолидин-2-он-5-карбоксилат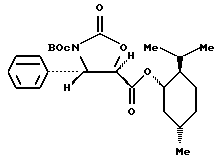
К перемешиваемому при температуре -40oC и в инертной атмосфере раствору 1,91 г (5,52 ммоль) (1S,2R,5S)-(+)-ментил- (4S,5R)-4-фенилоксазолидин-3-он-5-карбоксилата в 20 мл безводного тетрагидрофурана добавляют 3,8 мл (6,07 ммоль) 1,6 М раствора н-бутиллития в гексане. После перемешивания в течение 10 минут при температуре -40oC, добавляют раствор 1,81 г (8,28 ммоль) трет-бутоксикарбонового ангидрида в 5 мл тетрагидрофурана и реакционную среду оставляют стоять в течение 15 минут для повышения ее температуры до комнатной. После разбавления с помощью 50 мл дихлорметана и промывки с помощью 2%-ного водного раствора HCl вплоть до достижения pH 5, органическую фазу сушат над сульфатом магния и концентрируют при пониженном давлении. После очистки сырого продукта путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 5:1) получают 2,12 г целевого соединения в виде бесцветного сиропа (выход = 86%). Таким образом полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 7,45-7,26 (5Н, м); 5,19 (1Н, д, J = 3,7 Гц); 4,86 (1Н, дт, J = 10,9 и 4,5 Гц); 4,66 (1Н, д, J = 3,7 Гц); 2,05 (1Н, м); 1,79 (1Н, м); 1,73 (2Н, м); 1,62-1,24 (3Н, м); 1,33 (9Н, с); 1,11 (2Н, м); 0,94 (3Н, д, J = 6,5 Гц) и (1Н, м); 0,89 (3Н, д, J = 7 Гц); 0,77 (3Н, д, J = 7 Гц).
Пример 11: (1S,2R,5S)-(+)-Ментил-(4S,5R)-3-N-бензоил-4- фенилоксазолидин-3-он-5-карбоксилат
К перемешиваемому в инертной атмосфере и при комнатной температуре раствору 500 мг (1,45 ммоль) (1S,2R,5S)-(+)- ментил-(4S,5R)-4-фенилоксазолидин-3-он-5-карбоксилата и 176 мг (1,16 ммоль) 4-пирролидинопиридина в 7 мл безводного дихлорметана добавляют 0,25 мл (2,17 ммоль) бензоилхлорида. После перемешивания в течение трех часов при температуре 50oC, реакционную среду доводят до комнатной температуры и разбавляют с помощью 20 мл дихлорметана. Органическую фазу промывают с помощью 10 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния и концентрируют при пониженном давлении. После очистки сырого продукта путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 5:1) получают 300 мг целевого соединения в виде бесцветного сиропа (выход = 46%).
Таким образом полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,16 (2Н, д, J = 7,1 Гц), 7,68 (1Н, т); 7,53 (4Н, м); 7,43 (3Н, м); 5,57 (1Н, д, J = 4,4 Гц); 4,90 (1Н, дт, J = 10,9 и 4,4 Гц); 4,85 (1Н, д, J = 4,3 Гц); 2,07 (1Н, м); 1,80 (1H, м); 1,72 (2Н, м); 1,47 (3Н, м); 1,09 (2Н, м); 0,95 (3Н, д, J = 6,5 Гц); 0,88 (3Н, д, J = 7 Гц); 0,78 (3Н, д, J = 7 Гц).
Пример 12: (4S, 5R)-3-N-Бензоил-4-фенилоксазолидин-3-он-5- карбоновая кислота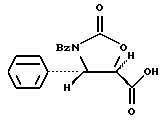
К перемешиваемой при комнатной температуре смеси 120 мг (0,266 ммоль) (1S, 2R, 5S)-(+)-ментил-(4R, 5R)-3-N-бензоил-4- фенилоксазолидин-3-он-5-карбоксилата в 2 мл метанола добавляют раствор 75 мг (0,543 ммоль) карбоната калия в 1 мл воды. После перемешивания в течение 30 минут реакционную среду разбавляют с помощью 10 мл воды и водную фазу промывают с помощью 5 мл дихлорметана. После подкисления до pH 4 с помощью 1 М раствора HCl, остаточную водную фазу экстрагируют этилацетатом (3 раза по 10 мл). Объединенные органические фазы промывают с помощью 5 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния и концентрируют при пониженном давлении.
Получают 40 мг (4S,5R)-3- N-бензоил-4-фенилоксазолидин-3-он-5-карбоновой кислоты в виде порошка белого цвета (выход = 52%), которая имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; гексадейтеродиметилсульфоксид), δ (м.д.): 12,98 (1Н, уш.с); 7,95 (2Н, д, J = 7,1 Гц); 7,63 (1Н, т, J = 7,4 Гц); 7,50 (2Н, т, J = 7,5 Гц); 7,42 (2Н, м); 7,37 (3Н, м); 4,90 (1Н, д, J = 5 Гц); 4,77 (1Н, д, J = 5 Гц).
Пример 13: (4S,5R)-4-Фенилоксазолидин-3-он-5-карбоновая кислота.

К перемешиваемому при температуре 0oC и в инертной атмосфере раствору 300 мг (0,867 ммоль) (1S,2R,5S)-(+)-ментил-(4S,5R)- 4-фенилоксазолидин-3-он-5-карбоксилата, 3 мл метанола, затем 0,5 мл воды в 6,5 мл пиридина быстро добавляют 10 мл гомогенного раствора 360 мг (8,67 ммоль) гидроксида натрия, 3 мл метанола и 0,5 мл воды в пиридине. После перемешивания в течение 20 минут при температуре 0oC, реакционную среду разбавляют с помощью 30 мл воды и промывают с помощью 30 мл дихлорметана. После подкисления до pH 1, остаточную водную фазу экстрагируют этилацетатом (3 раза по 20 мл) и объединенные органические фазы сушат над сульфатом магния и концентрируют при пониженном давлении.
Таким образом получают 86 мг (4S,5R)-4-фенил-оксазолидин-3-он-5-карбоновой кислоты в виде желтого сиропа (выход = 53%), которая имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; гексадейтеродиметилсульфоксид), δ (м.д.): 13,33 (1Н, уш.с); 8,46 (1H, с); 7,38 (5Н, м); 4,89 (1H, д, J = 5 Гц); 4,75 (1H, д, J = 5 Гц).
II. Производные баккатина III
Пример 14: 7-O-Триэтилсилил-10-дезацетилбаккатин III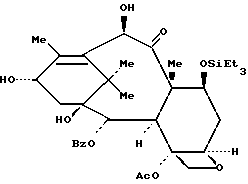
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 10 г (18,3 ммоль) 10-дезацетилбаккатина III и 8,17 г (54,9 ммоль) 4-пирролидинопиридина в 500 мл безводного дихлорметана в течение 10 минут добавляют 6,2 мл (36,6 ммоль) триэтилсилилхлорида. После протекания реакции в течение трех часов при комнатной температуре, добавляют 10 г измельченного льда и смесь выдерживают в течение 10 минут при перемешивании. Остаточную органическую фазу промывают водой (200 мл), сушат над сульфатом магния и концентрируют при пониженном давлении.
После обработки полученного сырого продукта с помощью минимального количества этилацетата получают 11,2 г 7-О-триэтилсилил-10-дезацетилбаккатина III в виде кристаллического вещества (выход = 92,3%). Таким образом полученный продукт имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,10 (2Н, д, J = 7,4 Гц); 7,60 (1Н, т, J = 7,5 Гц); 7,47 (2Н, т, J = 7,6 Гц); 5,60 (1Н, д, J =7 Гц); 5,17 (1Н, д, J = 1,9 Гц); 4,96 (1Н, д, J = 8 Гц); 4,86 (1Н, м); 4,41 (1Н, дд, J = 10,6 и 6,6 Гц); 4,31 и 4,16 (2Н, 2д, J = 8,4 Гц); 4,26 (1Н, д, J = 1,9 Гц); 3,95 (1Н, д, J = 6,9 Гц); 2,48 (1Н, ддд, J = 14,5; 9,7 и 6,7 Гц); 2,29 (3Н, с); 2,27 (2Н, м); 2,08 (3Н, с); 1,90 (1Н, м); 1,73 (3Н, с); 1,62 (1Н, с); 1,08 (6Н, с); 0,94 (9Н, т, J = 8 Гц); 0,56 (6Н, м).
Пример 15: 7-O-Триэтилгерманил-10-дезацетилбаккатин III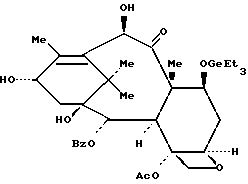
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 100 мг (0,183 ммоль) 10-дезацетилбаккатина III и 41 мг (0,275 ммоль) 4-пирролидинопиридина в 4 мл безводного дихлорметана в течение 10 минут добавляют 80 мкл (0,476 ммоль) триэтилгерманилхлорида, и смесь перемешивают в течение 13 часов при температуре 50oC. После охлаждения реакционной среды и разбавления с помощью 15 мл дихлорметана добавляют 1 г измельченного льда и смесь интенсивно перемешивают в течение 10 минут. Остаточную органическую фазу промывают насыщенным раствором гидрокарбоната натрия (5 мл), насыщенным раствором хлорида натрия (5 мл), сушат над сульфатом магния и концентрируют при пониженном давлении. После хроматографирования сырого продукта на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 25:75) получают 67 мг 7-O-триэтилгерманил-10-дезацетилбаккатина III в виде бесцветного сиропа. Таким образом полученный продукт имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,09 (2Н, д, J = 7,1 Гц); 7,60 (1Н, т, J = 7,4 Гц); 7,48 (2Н, т, J = 7,6 Гц); 5,63 (1Н, д, J = 7,1 Гц); 5,24 (1Н, с); 4,99 (1Н, д, J = 8 Гц); 4,78 (1Н, т); 4,32 (1Н, д, J = 8,3 Гц); 4,28 (1Н, м); 4,17 (2Н, м); 3,97 (1H, д, J = 7 Гц); 2,59 (1Н, м); 2,30 (3Н, с); 2,24 (1Н, м); 2,10 (1Н, м); 2,03 (3Н, с); 1,82 (1Н, м); 1,73 (3Н, с); 1,11 (9Н, м); 1,0 (6Н, т, J = 7,7 Гц).
Пример 16: 7-O-(2,2,2-Трихлор-трет-бутоксикарбонил)-10- дезацетилбаккатин III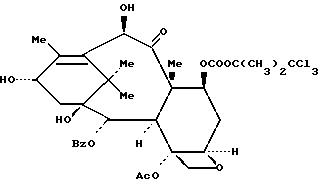
К перемешиваемому при температуре 40oC и в инертной атмосфере раствору 5 г (9,19 ммоль) 10-дезацетилбаккатина III и 1,1 мл безводного пиридина в 250 мл безводного дихлорметана в течение двух часов добавляют 3,3 г (13,8 ммоль) 2,2,2-трихлор-трет-бутоксикарбонилхлорида. После протекания реакции в течение дополнительных 30 минут и возврата к комнатной температуре, органический раствор промывают с помощью 2%-ного водного раствора HCl (30 мл), промывают осмотизированной водой (2 раза по 100 мл), сушат над сульфатом магния и концентрируют при пониженном давлении (выход = 55%). После хроматографии сырого продукта на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 60: 40) получают 7-О-(2,2,2-трихлор-трет-бутоксикарбонил)-10-дезацетилбаккатин III в виде порошка белого цвета.
Полученный продукт имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,10 (2Н, д, J = 7 Гц); 7,62 (1Н, т, J = 7,4 Гц); 7,49 (2Н, т, J = 7,6 Гц); 5,65 (1Н, д, J = 6,9 Гц); 5,44 (1Н, дд, J = 10,8 и 7,3 Гц); 5,39 (1Н, д); 4,98 (1Н, д, J = 7,5 Гц); 4,89 (1Н, м); 4,35 и 4,20 (2Н, 2д, J = 8,4 Гц); 4,10 (1Н, д, J =7 Гц); 4,01 (1Н, д, J = 1,8 Гц); 2,64 (1Н, м); 2,31 (3Н, с); 2,29 (1Н, м); 2,11 (3Н, д); 2,05 (2Н, м); 1,89 (3Н, с); 1,09 (3Н, с); 1,07 (3Н, с).
Пример 17: а) 7-O-Триэтилсилилбаккатин III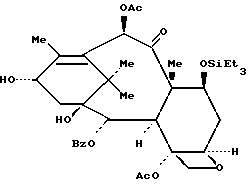
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 1 г (1,5 ммоль) 7-O-триэтилсилил-10- дезацетилбаккатина III и 1,25 мл (15 ммоль) пиридина в 15 мл безводного дихлорметана в течение 10 минут добавляют 0,54 мл (7,5 ммоль) ацетилхлорида. После протекания реакции в течение двух часов при комнатной температуре и контроля с помощью ТСХ, добавляют 1 г измельченного льда и смесь интенсивно перемешивают в течение 10 минут. Остаточную органическую фазу промывают водой (2 раза по 10 мл), сушат над сульфатом магния и концентрируют при пониженном давлении. После хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 60:40) получают 0,756 г 7-O-триэтилсилилбаккатина III в виде порошка белого цвета (выход = 70%).
Полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,11 (2Н, д, J = 7,1 Гц); 7,6 (1Н, т, J = 7,4 Гц); 7,48 (2Н, т, J = 7,7 Гц); 6,46 (1Н, с); 5,63 (1Н, д, J = 7 Гц); 4,96 (1Н, д, J = 8,1 Гц); 4,83 (1Н, м); 4,49 (1Н, дд, J = 10,4 и 6,7 Гц); 4,31 и 4,15 (2Н, 2д, J = 8,3 Гц); 3,88 (1Н, д, J = 7 Гц); 2,53 (1Н, м); 2,29 (3Н, с); 2,27 (2Н, м); 2,19 (3Н, д, J=0,8 Гц); 2,18 (3Н, с); 2,12 (1Н, д); 1,88 (1Н, м); 1,68 (3Н, с); 1,65 (1Н, с); 1,2 (3Н, с); 1,04 (3Н, с); 0,92 (9Н, т); 0,59 (6Н, м).
Пример 18: 7-O-(2,2,2-Трихлор-трет-бутоксикарбонил)-баккатин III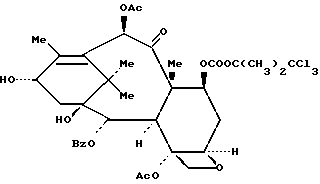
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 260 мг 7-O-(2,2,2-трихлор-трет-бутоксикарбонил)-10- дезацетилбаккатина III и 127,5 мг (1,04 ммоль) 4-диметиламинопиридина в 2,5 мл безводного дихлорметана добавляют 50 мкл (0,695 ммоль) ацетилхлорида. После протекания реакции в течение 1 часа при комнатной температуре, органическую фазу промывают 2%-ным водным раствором HCl вплоть до достижения pH 6, сушат над сульфатом магния и концентрируют при пониженном давлении. После хроматографии полученного остатка на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 6:4) получают 0,23 г 7-O-(2,2,2-трихлор-трет-бутоксикарбонил)баккатина III в виде твердого вещества (выход = 83%).
Полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,11 (2Н, д, J = 7,1 Гц); 7,62 (1Н, т, J = 7,4 Гц); 7,49 (2Н, т, J = 7,6 Гц); 6,39 (1Н, с); 5,64 (1H, д, J = 6,9 Гц); 5,61 (1Н, дд, J = 10,7 и 7,2 Гц); 4,99 (1H, д, J = 8,2 Гц); 4,87 (1Н, м); 4,33 и 4,16 (2Н, 2д, J = 8,4 Гц); 4,02 (1H, д, J = 6,9 Гц); 2,64 (1H, ддд, J = 14,4; 9,5 и 7,2 Гц); 2,30 (3H, с) и (2Н, м); 2,17 (3Н, с); 2,13 (3Н, д, J = 0,8 Гц); 2,04 (1H, м); 1,83 (3Н, с); 1,63 (1H, с); 1,14 (3Н, с); 1,09 (3Н, с).
Пример 19: 7-O-Феноксиацетил-10-дезацетилбаккатин III
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 1,03 г (1,88 ммоль) 10-дезацетилбаккатина III и 0,6 мл (7,5 ммоль) безводного пиридина в 100 мл безводного дихлорметана в течение 10 минут добавляют 1,05 мл (7,5 ммоль) феноксиацетилхлорида. После протекания реакции в течение 30 минут при комнатной температуре и контроля с помощью ТСХ, органический раствор промывают 2%-ным водным раствором HCl вплоть до достижения pH 2, промывают осмотизированной водой (2 раза по 50 мл), сушат над сульфатом магния и концентрируют при пониженном давлении (выход = 70,5%). После хроматографии сырого продукта на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 60:40) получают 7-О-феноксиацетил-10- дезацетилбаккатин III в виде порошка белого цвета.
Полученный продукт имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,09 (2Н, д, J = 7,3 Гц); 7,61 (1Н, т, J = 7,4 Гц); 7,48 (2Н, т, J = 7,6 Гц); 7,31 (2Н, т, J = 7,7 Гц); 6,99 (3Н, м); 6,42 (1Н, с); 5,61 (1Н, д, J =7 Гц); 4,97 (1Н, д, J = 7,8 Гц); 4,86 (3Н, м); 4,44 (1Н, дд, J = 10,6 и 6,8 Гц); 4,30 и 4,15 (2Н, 2д, J = 8,4 Гц); 3,86 (1Н, д, J = 7 Гц); 2,56 (1Н, м); 2,27 (3Н, с); 2,27 (2Н, м); 2,05 (3Н, с); 1,86 (1Н, м); 1,68 (3Н, с); 1,01 (3Н, с); 0,98 (3Н, с).
Пример 20: 7,10-O-Ди-(феноксиацетил)-10-дезацетилбаккатин III
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 500 мг (0,92 ммоль) 10-дезацетилбаккатина III и 0,6 мл (7,36 ммоль) безводного пиридина в 50 мл безводного дихлорметана в течение 10 минут добавляют 0,5 мл (3,68 ммоль) феноксиацетилхлорида. После протекания реакции в течение 6 часов при комнатной температуре и контроля с помощью ТСХ, раствор промывают с помощью 2%-ного водного раствора HCl вплоть до pH 2, промывают осмотизированной водой (2 раза по 20 мл), сушат над сульфатом магния и концентрируют при пониженном давлении. После хроматографии сырого продукта на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 6:4) получают 0,55 г 7,10-O-бис(феноксиацетил)-10- дезацетилбаккатина III в виде порошка белого цвета (выход = 74%).
Полученный продукт имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,09 (2Н, д, J = 7,1 Гц), 7,61 (1Н, т, J = 7,4 Гц); 7,48 (2Н, т, J = 7,6 Гц); 7,29 (2Н, т, J = 6,8 Гц); 7,22 (2Н, т, J = 7,5 Гц); 6,96 (4Н, м); 6,84 (2Н, д, J = 7,9 Гц); 6,42 (1Н, с); 5,69 (1Н, дд, J = 10,5 и 7,1 Гц); 5,60 (1Н, д, J = 6,9 Гц); 4,96 (1Н, д, J = 8,2 Гц); 4,84 (1Н, т, J = 7,4 Гц); 4,8 (2Н, с); 4,65 и 4,41 (2Н, 2д, J = 15,8 Гц); 4,32 и 4,14 (2Н, 2д, J = 8,4 Гц); 3,98 (1Н, д, J = 6,8 Гц); 2,65 (1Н, м); 2,28 (3Н, с); 2,26 (2Н, м); 2,09 (3Н, с); 1,80 (3Н, с) и (1Н, м); 0,98 (6Н, с).
Пример 21: 7-О-Феноксиацетилбаккатин III
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 1,11 г (1,64 ммоль) 7-O-феноксиацетил-10- дезацетилбаккатина III в 40 мл безводного пиридина в течение 10 минут добавляют 0,233 мл (3,27 моль) ацетилхлорида. После протекания реакции в течение 16 часов при комнатной температуре и контроля с помощью ТСХ, реакционную среду разбавляют с помощью 50 мл осмотизированной воды и водную фазу экстрагируют этилацетатом (3 раза по 30 мл). Объединенные органические фазы промывают водой (2 раза по 20 мл), сушат над сульфатом магния и концентрируют при пониженном давлении (выход = 84,5%). После хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 60:40) получают 7-O-феноксиацетилбаккатин III в виде кристаллического вещества.
Полученный продукт имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,10 (2Н, д, J = 7,1 Гц); 7,61 (1Н, т, J = 7,4 Гц); 7,48 (2Н, т, J = 7,7 Гц); 7,27 (2Н, т, J = 8 Гц); 6,95 (3Н, м); 6,26 (1Н, с); 5,71 (1Н, дд, J = 10,4 и 7,2 Гц); 5,62 (1Н, д, J = 6,9 Гц); 4,96 (1Н, д, J = 8,3 Гц); 4,80 (1Н, м); 4,81 и 4,53 (2Н, 2д, J = 16 Гц); 4,32 и 4,14 (2Н, 2д, J = 8,5 Гц); 4,0 (1Н, д, J = 6,9 Гц); 2,64 (1Н, м); 2,29 (2Н, м); 2,28 (3Н, с); 2,24 (1Н, д, J = 5 Гц); 2,16 (3Н, с); 2,09 (3Н, д, J = 0,7 Гц); 1,81 (1Н, м); 1,78 (3Н, с); 1,13 (3Н, с); 1,08 (3Н, с).
Пример 22: 7,10-O-(1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-10-дезацетилбаккатин III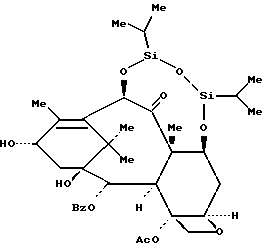
К перемешиваемому при температуре -40oC и в инертной атмосфере раствору 500 мг (0,93 ммоль) 10-дезацетилбаккатина III в 20 мл безводного тетрагидрофурана в течение 10 минут добавляют 1,28 мл (2,05 ммоль) н-бутиллития в виде 1,6 М раствора в гексане. После перемешивания в течение 5 минут добавляют 350 мкл (1,12 ммоль) 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксана и реакционную среду выдерживают в течение 20 минут для повышения ее температуры до комнатной. После перемешивания в течение 1 часа при комнатной температуре, добавляют 225 мг (2,05 ммоль) 4-диметиламинопиридина и реакционную среду перемешивают дополнительно в течение 1 часа. После добавления 20 мл водного насыщенного раствора хлорида натрия среду экстрагируют дихлорметаном (3 раза по 30 мл). Объединенные органические фазы промывают с помощью водного насыщенного раствора хлорида натрия (20 мл), сушат над сульфатом магния и концентрируют при пониженном давлении. После очистки путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 60:40) получают 480 мг 7,10-O-(1,1,3,3-тетраизопропил-1,3- дисилоксандиил)-10-дезацетилбаккатина III в виде аморфного вещества (выход = 65%).
Полученный продукт имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,10 (2Н, д, J = 7,2 Гц); 7,60 (1Н, т, J = 7,4 Гц); 7,47 (2Н, т, J = 7,6 Гц); 5,60 (1Н, с); 5,59 (1Н, д); 4,97 (1Н, д, J = 7,9 Гц); 4,87 (1Н, м); 4,68 (1Н, дд, J = 10,4 и 6,9 Гц); 4,30 и 4,17 (2Н, 2д, J = 8,5 Гц); 3,92 (1Н, д, J = 7,1 Гц); 2,49 (1Н, м); 2,28 (3Н, с); 2,27 (1Н, м); 2,04 (1Н, м); 1,91 (1Н, м); 1,67 (3Н, с); 1,55 (1Н, с); 1,32-0,85 (34Н, м).
Пример 23: 13-O-{[(4S,5R)-2,4-Дифенил-4,5-дигидрооксазол-5-ил]карбонил}- 7-O-триэтилсилилбаккатин III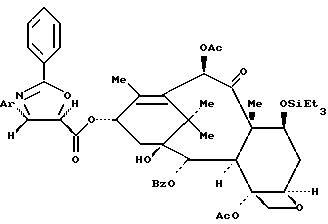
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 2,67 г (10 ммоль) (4S,5R)-2,4-дифенил-4,5- дигидрооксазол-5-карбоновой кислоты в 55 мл безводного толуола добавляют 2,06 г (10 ммоль) дициклогексилкарбодиимида. После перемешивания в течение 5 минут добавляют 3,5 г (5 ммоль) 7-О- триэтилсилилбаккатина III и 0,61 г (5 ммоль) 4-диметиламинопиридина, и реакционную смесь нагревают в течение 1 часа при температуре 70oC. После возврата температуры к комнатной и удаления нерастворимых составных частей путем отфильтровывания, органическую фазу концентрируют при пониженном давлении. После очистки сырого продукта путем хроматографии на силикагеле (15-25 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 90: 10) получают 4,62 г 13-O-{[(4S,5R)- 2,4-дифенил-4,5-дигидрооксазол-5-ил] карбонил} -7-O- триэтилсилилбаккатина III в виде кристаллического вещества (выход = 97%).
Таким образом полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,23 (2Н, д, J = 7,2 Гц); 8,07 (2Н, д, J = 7,3 Гц); 7,63 (1Н, т, J = 7,4 Гц); 7,58 (1Н, т, J = 7,4 Гц); 7,49 (4Н, м); 7,38 (5Н, м); 6,42 (1Н, с); 6,18 (1Н, т, J = 8,2 Гц); 5,68 (1Н, д, J = 7,1 Гц); 5,60 (1Н, д, J = 6,5 Гц); 4,95 (2Н, д); 4,50 (1Н, дд, J = 10,5 и 6,7 Гц); 4,29 (1Н, д, J = 8,4 Гц); 4,14 (1H, д, J = 8,4 Гц); 3,83 (1H, д, J = 7,1 Гц); 2,55 (1H, м); 2,37 (1H, дд, J = 15,3 и 9,3 Гц); 2,26 (1H, дд, J = 15,3 и 8,6 Гц); 2,16 (3Н, с); 2,07 (3Н, с); 1,99 (3Н, с); 1,89 (1H, м); 1,72 (1H, с); 1,69 (3Н, с); 1,23 (3Н, с); 1,19 (3Н, с); 0,92 (9Н, т, J = 8 Гц); 0,57 (6Н, м).
Пример 24: 13-O-{[(4S,5R)-2,4-Дифенил-4,5-дигидрооксазол-5-ил] карбонил} -7-O-феноксиацетилбаккатин III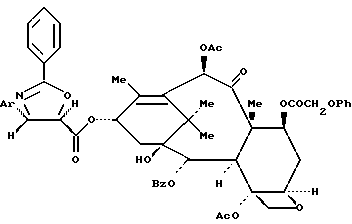
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 490 мг (1,83 ммоль) (4S,5R)-2,4-Дифенил-4,5- дигидрооксазол-5-карбоновой кислоты в 10 мл безводного толуола добавляют 380 мг (1,84 ммоль) дициклогексилкарбодиимида. После перемешивания в течение 5 минут добавляют 660 мг (0,92 ммоль) 7-O-феноксиацетилбаккатина III и 112 мг (0,92 ммоль) 4-диметиламинопиридина, и реакционную смесь в течение двух часов выдерживают при температуре 70oC.
После возврата к комнатной температуре и удаления нерастворимых составных частей путем отфильтровывания, органическую фазу концентрируют при пониженном давлении. После очистки сырого продукта путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 99: 1) получают 800 мг 13-O-{[(4S,5R)-2,4-дифенил-4,5-дигидрооксазол-5-ил] карбонил} -7- O-феноксиацетилбаккатина III в виде кристаллического вещества (выход = 90%).
Таким образом полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,18 (2Н, д, J = 7 Гц); 8,07 (2Н, д, J = 7,3 Гц); 7,63 (1Н, т, J = 7,4 Гц); 7,59-7,32 (10Н, м); 7,28 (2Н, т, J = 7,5 Гц); 6,94 (3Н, м); 6,23 (1Н, с) и (1Н, м); 5,70 (1Н, дд, J = 10,4 и 7,1 Гц); 5,67 (1Н, д, J = 7,3 Гц); 5,58 (1Н, д, J = 7 Гц); 4,93 (2Н, д); 4,79 и 4,53 (2Н, 2д, J = 15,9 Гц); 4,30 и 4,13 (2Н, 2д, J = 8,5 Гц); 3,97 (1Н, д, J = 6,9 Гц); 2,67 (1Н, м); 2,38 (1Н, дд, J = 15,2 и 9,3 Гц); 2,26 (1Н, дд, J = 15,2 и 8,4 Гц); 2,15 (3Н, с); 2,02 (3Н, с); 1,95 (3Н, с) и (1Н, м); 1,80 (3Н, с); 1,74 (1Н, с); 1,25 (3Н, с); 1,17 (3Н, с).
Пример 25: 13-O-{[(4S,5R)-2,4-Дифенил-4,5-дигидрооксазол-5-ил] карбонил} -7-О-(2,2,2-трихлор-трет-бутоксикарбонил)баккатин III
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 35 мг (4S,5R)-2,4-дифенил-4,5-дигидрооксазол-5- карбоновой кислоты в 3 мл безводного толуола добавляют 27 мг (0,13 ммоль) дициклогексилкарбодиимида. После перемешивания в течение 5 минут, добавляют 51 мг (0,065 ммоль) 7-O-(2,2,2-трихлор-трет-бутоксикарбонил)баккатина III и 8 мг (0,065 ммоль) 4-диметиламинопиридина, и смесь выдерживают в течение 1 часа при температуре 70oC. После возврата к комнатной температуре и удаления нерастворимых составных частей путем отфильтровывания, органическую фазу концентрируют при пониженном давлении и полученный остаток очищают путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 9:1).
Таким образом получают 0,99 г целевого соединения в виде твердого вещества белого цвета (выход = 67%), которое имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,18 (2Н, д, J = 7,2 Гц); 8,07 (2Н, д, J = 7,3 Гц); 7,65 (1Н, т, J = 7,4 Гц); 7,59 (1Н, т, J = 7,3 Гц); 7,52 (4Н, м); 7,39 (5Н, м); 6,35 (1Н, с); 6,24 (1Н, т, J = 8,4 Гц); 5,68 (1Н, д, J = 7,1 Гц); 5,59 (1Н, д, J = 7 Гц) и (1Н, дд); 4,95 (1Н, д); 4,94 (1Н, д, J = 7 Гц); 4,31 и 4,15 (2Н, 2д, J = 8,4 Гц); 3,97 (1Н, д, J = 6,9 Гц); 2,64 (1Н, м); 2,37 (1Н, дд, J= 15,1 и 6 Гц); 2,27 (1Н, дд, J = 15,2 и 8,5 Гц); 2,16 (3Н, с); 2,01 (3Н, с); 1,98 (3Н, с); 1,83 (3Н, с); 1,72 (1Н, с); 1,25 (3Н, с); 1,18 (3Н, с).
Пример 26: 13-O-{[(4S,5R)-2,4-Дифенил-4,5-дигидрооксазол-5-ил] карбонил} -7,10-O-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-10- дезацетилбаккатин III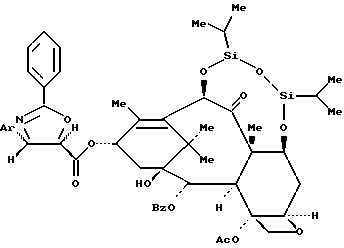
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 4 мг (0,015 ммоль) (4S,5R)-2,4-дифенил-4,5- дигидрооксазол-5-карбоновой кислоты в 0,5 мл безводного толуола добавляют 7 мг (0,06 ммоль) дициклогексилкарбодиимида. После перемешивания в течение 5 минут, добавляют раствор 5 мг (0,0065 ммоль) 7,10-O-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-10- дезацетилбаккатина III и 1 мг (0,0078 ммоль) 4-диметиламинопиридина в 1 мл безводного толуола. После перемешивания в течение 20 минут при комнатной температуре, реакционную смесь выдерживают в течение дополнительных 20 минут при 50oC. После возврата к комнатной температуре, органическую фазу разбавляют с помощью 5 мл дихлорметана, промывают с помощью 2 мл водного насыщенного раствора хлорида натрия, сушат над сульфатом магния и концентрируют при пониженном давлении. После очистки сырого продукта путем хроматографии на силикагеле (15-25 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 7:3) получают 6 мг целевого соединения (выход = 90%) в виде аморфного вещества.
Полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,21 (2Н, д, J = 7,2 Гц); 8,07 (2Н, д, J = 7,6 Гц); 7,63 (1Н, т, J = 7,5 Гц), 7,59 (1Н, т, J = 7,4 Гц); 7,50 (2Н, т, J = 7,4 Гц); 7,39 (5Н, м); 6,26 (1H, т); 5,64 (1H, д, J = 7 Гц), 5,59 (1H, д, J = 6,9 Гц); 5,54 (1H, с); 4,93 (1H, д, J = 6,8 Гц) и (1H, м); 4,68 (1H, дд); 4,28 и 4,16 (2Н, 2д, J = 8 Гц); 3,84 (1H, д, J = 7,3 Гц); 2,48 (1H, м); 2,35 и 2,25 (2Н, 2дд); 2,02 (3Н, с); 1,88 (3Н, с) и (1H, м); 1,67 (3Н, с); 1,63 (1H, с); 1,30-0,90 (34Н, м).
Пример 27: 13-O-{[(4S,5R)-3-N-Бензоил-4-фенилоксазолидин-3-он- 5-ил]карбонил}-7-O-триэтилсилилбаккатин III
К перемешиваемому в инертной атмосфере и при комнатной температуре раствору 40 мг (0,137 ммоль) (4S,5R)-3-N-бензоил-4- фенилоксазолидин-3-он-5-карбоновой кислоты в 2 мл безводного толуола добавляют 28 мг (0,136 ммоль) дициклогексилкарбодиимида. После перемешивания в течение 5 минут добавляют 30 мг (0,043 ммоль) 7-О-триэтилсилилбаккатина III и 8 мг (0,066 ммоль) 4-диметиламинопиридина, и реакционную смесь выдерживают при 60oC в течение 13 часов. После возврата к комнатной температуре, реакционную среду разбавляют с помощью 10 мл дихлорметана, и органическую фазу промывают с помощью 5 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния и концентрируют при пониженном давлении. После очистки путем хроматографии на силикагеле (15-14 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 2:1) получают 13 мг целевого производного в виде аморфного вещества (выход = 31%).
Полученное соединение имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,06 (2Н, д, J = 7,3 Гц); 7,72 (2Н, д, J = 7 Гц); 7,63 (1Н, т, J = 7,4 Гц); 7,58 (1Н, т, J = 7,4 Гц); 7,54-7,44 (8Н, м); 7,40 (1Н, т); 6,44 (1Н, с); 6,33 (1Н, т); 5,73 (1Н, д, J = 5,7 Гц); 5,67 (1Н, д, J = 5,7 Гц); 4,96 (1Н, д, J = 5,8 Гц); 4,88 (1Н, д, J = 8,3 Гц); 4,45 (1H, дд, J = 10,4 и 6,6 Гц); 4,27 и 4,12 (2Н, 2д, J = 8,3 Гц); 3,80 (1Н, д, J =7 Гц); 2,50 (1H, м); 2,26 (2Н, м); 2,19 (3Н, с); 2,07 (3Н, с); 1,98 (3Н, с); 1,85 (1H, м); 1,76 (1H, с); 1,67 (3Н, с); 1,24 (3Н, с); 1,23 (3Н, с); 0,91 (9Н, т, J= 7,9 Гц); 0,56 (6Н, м).
Пример 28: 13-O-{ [(4S, 5R)-4-Фенилоксазолидин-3-он-5-ил] карбонил}-7,10-O-ди-(феноксиацетил)-10-дезацетилбаккатин III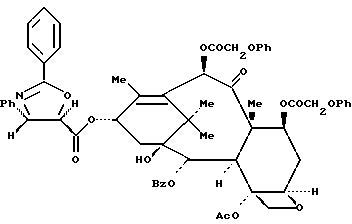
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 78 мг (0,293 ммоль) (4S,5R)-2,4-дифенил-4,5- дигидрооксазол-5-карбоновой кислоты в 3 мл безводного толуола добавляют 65 мг (0,315 ммоль) дициклогексилкарбодиимида. После перемешивания в течение 5 минут добавляют раствор 237 мг (0,293 ммоль) 7,10-O-бис(феноксиацетил)-10-дезацетилбаккатина III и 36 мг (0,295 ммоль) 4-диметиламинопиридина в 3 мл толуола, и реакционную смесь выдерживают при 60oC в течение 1 часа. После возврата к комнатной температуре и удаления нерастворимых составных частей путем отфильтровывания, органическую фазу концентрируют при пониженном давлении, и полученный сырой продукт очищают путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 1:1).
Таким образом получают 280 мг целевого соединения в виде аморфного вещества (выход = 90%), которое имеет следующие характеристики:
1H-ЯМР-спектр (400 МГц; дейтерохлороформ), δ (м.д.): 8,18 (2Н, д, J = 7 Гц); 8,06 (2Н, д, J = 7,1 Гц); 7,64 (1Н, т, J = 7,4 Гц); 7,58 (1Н, т, J = 7,3 Гц); 7,51 (4Н, м); 7,39 (5Н, м); 7,25 (4Н, м); 6,96 (4Н, м); 6,85 (2Н, д, J = 8 Гц); 6,33 (1Н, с); 6,19 (1Н, т, J = 9 Гц); 5,68 (1Н, дд, J = 10,5 и 7,1 Гц); 5,65 (1Н, д, J=6,9 Гц); 5,59 (1Н, д, J =7 Гц); 4,93 (2Н, д, J = 7,1 Гц); 4,79 (2Н, с); 4,63 и 4,40 (2Н, 2д, J = 15,9 Гц); 4,30 и 4,13 (2Н, 2д, J = 8,4 Гц); 3,94 (1Н, д, J = 6,9 Гц); 2,68 (1Н, m); 2,37 (1Н, дд, J = 15,3 и 9,3 Гц); 2,24 (1Н, дд, J = 15,3 и 8,7 Гц); 2,02 (3Н, с); 1,95 (3Н, с); 1,80 (3Н, с) и (1Н, m); 1,69 (1Н, с); 1,12 (3Н, с); 1,01 (3Н, с).
III. Полусинтез
Пример 29: Получение паклитаксела:
а) из 13-O-{[4S,5R)-2,4-дифенил-4,5-дигидрооксазол-5-ил]карбонил}-7-O- триэтилсилилбаккатина III
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 90 мг (0,095 ммоль) 13-O-{[(4S,5R)-2,4-дифенил-4,5-дигидрооксазол-5-ил] карбонил} -7-O- триэтилсилилбаккатина III в смеси тетрагидрофурана (1,2 мл) с метанолом (1,2 мл) добавляют 0,6 мл (0,6 ммоль) 1 М водного раствора HCl, и реакционную смесь перемешивают при комнатной температуре в течение 4,5 часов. После добавления 3,5 мл водного насыщенного раствора гидрокарбоната натрия, раствор поддерживают гомогенным за счет добавления 6 мл тетрагидрофурана и 6 мл воды, и реакционную среду перемешивают дополнительно в течение полутора часов. После добавления 15 мл этилацетата и 15 мл осмотизированной воды, остаточную водную фазу экстрагируют этилацетатом (15 мл). Органическую фазу сушат над сульфатом магния, концентрируют при пониженном давлении и таким образом полученный сырой продукт очищают путем хроматографии на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 1:1).
Таким образом выделяют 75 мг таксола в виде кристаллического вещества (выход = 95%), характеристики которого полностью согласуются с литературными данными;
б) из 13-O-{[(4S,5R)-2,4-дифенил-4,5-дигидрооксазол-5-ил] карбонил}-7-O-(2,2,2-трихлор-трет-бутоксикарбонил)баккатина III
К перемешиваемому при комнатной температуре и в инертной атмосфере раствору 15 мг (0,0148 ммоль) 13-О-{[(4S,5R)-2,4-дифенил-4,5-дигидрооксазол-5-ил] карбонил}-7-O-(2,2,2- трихлор-трет-бутоксикарбонил)баккатина III в смеси тетрагидрофурана (0,18 мл) с метанолом (0,18 мл) добавляют 90 мкл (0,09 ммоль) 1 М водного раствора HCl, и реакционную смесь перемешивают при комнатной температуре в течение 8 часов. После добавления 0,6 мл водного насыщенного раствора гидрокарбоната натрия, раствор поддерживают гомогенным за счет добавления 1 мл тетрагидрофурана и 1 мл воды, и реакционную среду перемешивают дополнительно в течение полутора часов. После добавления 2,5 мл этилацетата и 2,5 мл осмотизированной воды, остаточную водную фазу экстрагируют этилацетатом (2,5 мл). Объединенные органические фазы сушат над сульфатом магния и концентрируют при пониженном давлении.
Таким образом получают 14 мг 7-O-(2,2,2-трихлор-трет-бутоксикарбонил) таксола в виде сырого продукта и (выход = 93%), который используют без всякой другой очистки в следующей стадии.
К перемешиваемому при комнатной температуре раствору 13 мг (0,0128 ммоль) 7-O-(2,2,2-трихлор-трет- бутоксикарбонил)таксола в 2 мл этилацетата добавляют 30 мкл (0,525 ммоль) уксусной кислоты и 22,5 мг (0,344 ммоль) цинкового порошка.
После перемешивания в течение 2,5 часов при комнатной температуре и контроля с помощью ТСХ и после разбавления реакционной среды с помощью 3 мл этилацетата, органическую фазу промывают осмотизированной водой (1 мл), водным насыщенным раствором гидрокарбоната натрия (1 мл), снова водой, сушат над сульфатом магния и концентрируют при пониженном давлении.
После хроматографии сырого продукта на силикагеле (15-40 мкм) (элюирующее средство: смесь циклогексана с этилацетатом в соотношении 6:4) таким образом выделяют 9,5 мг таксола в виде кристаллического вещества (выход = 89%).
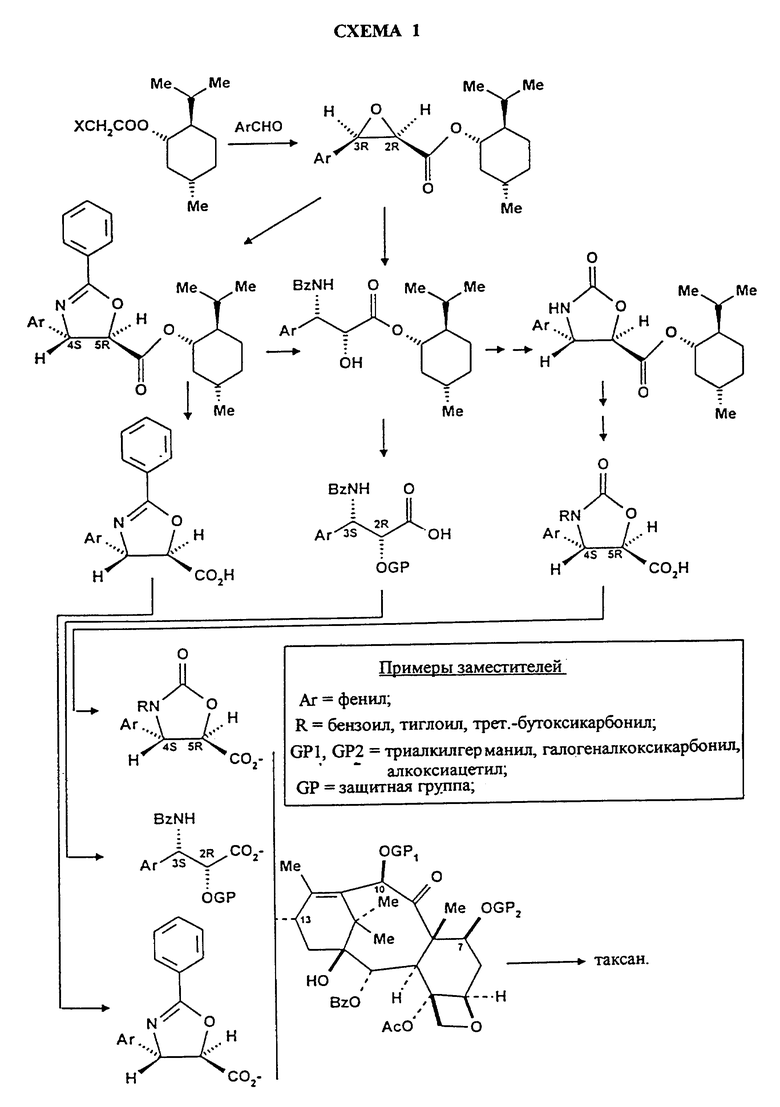
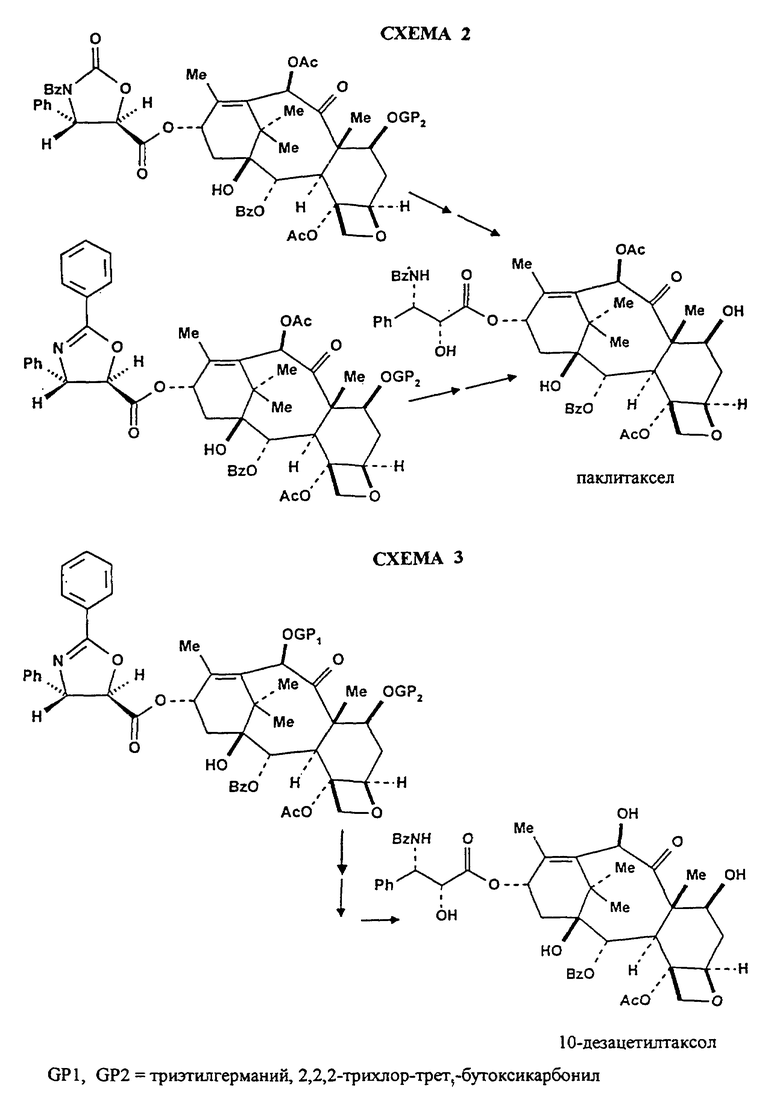
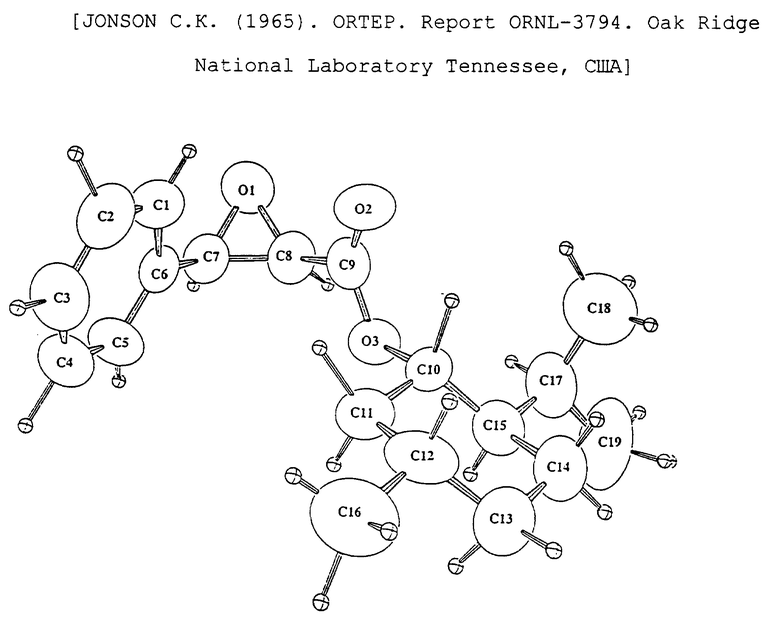
Описывается способ получения предшественников боковой цепи таксанов, в котором превращают цис-β-арилглицидатное производное общей формулы (I) конфигурации (2R, 3S), в которой Ar - арил и R - углеводородный радикал, предпочтительно линейный или разветвленный алкильный радикал или циклоалкильный радикал, незамещенный или замещенный одной или несколькими алкильными группами, так, чтобы регио- и стереоспецифически получить β-N-алкиламид и α-гидроксил или их циклические предшественники в одну стадию по реакции Риттера, заключающийся в том, что осуществляют прямой синтез линейной цепи путем введения во взаимодействие цис-β-арилглицидатного производного общей формулы (I) с нитрилом формулы R2-CN, в котором R2 - арильный радикал, в присутствии протонной кислоты и воды для получения производного β- арилизосерина общей формулы (IIa). Настоящее изобретение относится также к новым промежуточным продуктам для полусинтеза таксанов, производных оксазолидинов или оксазолидинонов, а также новых производных баккатина III. Технический результат - упрощение процесса. 6 с. и 22 з.п.ф-лы.
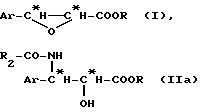
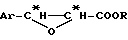
конфигурации (2R, 3S),
в которой Ar - арил;
R - углеводородный радикал, предпочтительно линейный или разветвленный алкильный радикал или циклоалкильный радикал, незамещенный или замещенный одной или несколькими алкильными группами;
так, чтобы регио- и стереоспецифически получить β-N-алкиламид и α-гидроксил или их циклические предшественники в одну стадию по реакции Риттера, заключающийся в том, что осуществляют прямой синтез линейной цепи путем введения во взаимодействие цис-β-арилглицидатного производного общей формулы (I) с нитрилом формулы R2 - CN, в которой R2 - арильный радикал, в присутствии протонной кислоты и воды для получения производного β-арилизосерина общей формулы (IIa)
в которой Ar, R и R2 имеют вышеуказанные значения;
и конфигурации (2R, 3S).
в которой Ar, R и R2 имеют указанное в п.1 значение,
GP означает защитную для гидроксильной функции группу, пригодную для синтеза таксанов, в частности, выбираемую среди алкоксиэфирных, арилалкоксиэфирных, арилоксиэфирных, или галогеналкоксикарбонильных радикалов, таких, как, например, метоксиметильная, 1-этоксиэтильная, бензилоксиметильная группа, (β-триметилсилилэтокси)метильная группа, тетрагидропиранильный радикал, β-алкоксикарбонильный радикал (TrOC), простые (β-галогенированные, алкилсилилированные эфиры или алкоксиацетильный, арилоксиацетильный, галогенацетильный или формильный радикалы.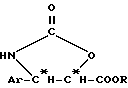
в которой Ar и R имеют указанные в п.1 значения,
путем введения во взаимодействие производного β-арилизосерина общей формулы (IIa) по любому из пп.1 - 5 со сложным галогеналкоксикарбонильным эфиром, в частности с 2,2,2-трихлорэтоксикарбонилом (TrOC), затем путем циклизации в присутствии сильного органического основания, такого, как диазабициклоундецен (ДБУ), превращаемый затем при необходимости в соответствующий амид общей формулы (III'a)
в которой Ar и R имеют указанные в п.1 значения;
R''2 означает R'2, который означает указанный R2 или низший алкильный радикал, или низший пергалогенированный алкильный радикал, такой, как трихлорметил, или алкоксильный радикал, или линейный или разветвленный алкильный радикал по крайней мере с одной ненасыщенной связью.
в которой Ar имеет указанное в п.1 значение;
R - оптически чистый энантиомер пространственно сильнозатрудненного хирального углеводородного радикала,
получают путем введения во взаимодействие альдегида формулы Ar - CHO с галогенацетатом формулы X - CH2 -COOR, причем Ar, R имеют указанные в п.1 значения, X - галоген, в частности хлор или бром, и тем, что путем мягко осуществляемого омыления получают производные формул (IIa), (II'a), (IIb), (IIIa), (III'a), которых R - атом водорода.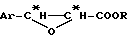
конфигурации (2R, 3S),
в которой Ar - арил;
R - углеводородный радикал, предпочтительно линейный или разветвленный алкильный радикал, или циклоалкильный радикал, незамещенный или замещенный одной или несколькими алкильными группами;
так, чтобы регио- и стереоспецифически получить β-N-алкиламид и α-гидроксил, или их циклические предшественники в одну стадию по реакции Риттера, заключающийся в том, что осуществляют прямой синтез циклической цепи путем введения во взаимодействие цис-β-арилглицидатного производного общей формулы (I) с нитрилом формулы R'2 - CN, в которой R'2 - вышеуказанный R2, или низший алкильный радикал, или низший пергалогенированный алкильный радикал, такой, как трихлорметил, в присутствии кислоты Льюиса или протонной кислоты в безводной среде для получения оксазолина общей формулы (IIb)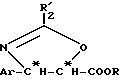
в которой Ar, R и R'2 имеют указанные значения,
конфигурация 2R, 3S.
в которой Ar, R и R'2 имеют указанные в п.1 значения,
превращаемого затем при необходимости в соответствующий амид общей формулы (III'b)
в которой Ar, R, R'2 и R''2 имеют указанные значения.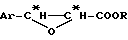
в которой Ar имеет указанное в п.1 значение;
R - оптически чистый энантиомер пространственно сильнозатрудненного хирального углеводородного радикала,
получают путем введения во взаимодействие альдегида формулы Ar - CHO с галогенацетатом формулы X - CH2 - COOR, причем Ar, R имеют указанные в п.1 значения, X - галоген, в частности хлор или бром, и тем, что путем мягко осуществляемого омыления получают производные формул (IIb), (IIIb) и (III'b), в которых R - атом водорода.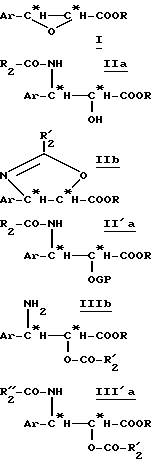
в которых Ar, R2, R'2, R''2 и GP имеют указанные в любом из пп.1 - 3 и 5 значения;
R - оптически чистый энантиомер пространственно сильнрозатрудненного хирального углеводородного радикала, такого, как циклоалкил, замещенный одной или несколькими алкильными группами.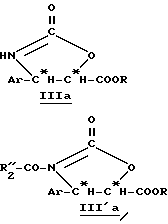
в которых Ar, R и R''2 имеют указанные в пп.1 и 7 значения или R - атом водорода,
и они имеют конфигурацию (2R, 3S).
C - B, (IV)
в которой B - радикал общей формулы (V)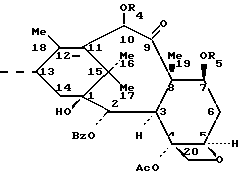
в которой Ac - ацетил;
Bz - бензоил;
Me - метил;
R4 - ацетил или защитная для гидроксильной функции группа GP1;
R5 - защитная для гидроксильной функции группа GP2;
C - боковая цепь, выбираемая среди радикалов формул (IIa), (II'a), (IIb), (IIIa), (III'a), (IIIb) и (III'b)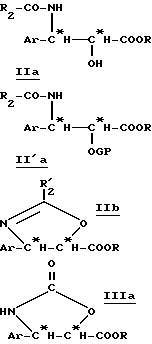
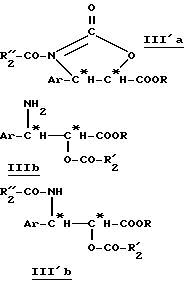
в которых Ar, R2, R'2, R''2, R3 и GP имеют указанные в пп.1, 7 и 8 значения,
путем этерификации соответствующего производного баккатина III общей формулы (V), содержащего гидроксильную функцию в положении C-13, с помощью одного из производных общих формул (IIa), (II'a), (IIb), (IIIa), (III'a), (IIIb) и (III'b), в которых R - атом водорода, получаемых по способу согласно п.14.
в которой Ar'' - арилиденовый радикал.
в которой Ac, Bz, Me и R'2 имеют указанные выше значения;
R4 - атом водорода или ацетильный радикал;
R5 - атом водорода.
C - B,
в которой C обозначает боковую цепь, выбираемую из радикалов формул (IIIa) и (III'a), указанных в п.19;
B имеет значение, указанное в п.19;
GP1 и GP2 независимо друг от друга - пространственно затрудненные ацильные радикалы, атом углерода которых в альфа-положении карбонильной функции несет по меньшей мере один атом кислорода, или GP1 и GP2 вместе образуют двухвалентный радикал формулы -SiR7 - O - SiR8-, в которой R7 и R8 независимо друг от друга - пространственно затрудненный алкильный радикал.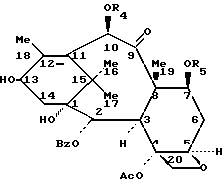
в которой Ac - ацетил;
Bz - бензоил;
Me - метил;
R4 - ацетил или защитная для гидроксильной функции группа GP1;
R5 - защитная для гидроксильной функции группа GP2;
GP1 и GP2 независимо друг от друга - пространственно затрудненные ацильные радикалы, атом углерода которых в α-положении карбонильной функции содержит по крайней мере один атом кислорода, или GP1 и GP2 вместе образуют двухвалентный радикал формулы -SiR7 - O - SiR8-, в которой R7 и R8 независимо друг от друга - пространственно затрудненный алкильный радикал.
| Захват манипулятора | 1974 |
|
SU522958A1 |
| 1972 |
|
SU414610A1 | |
| WO 9310076 A, 27.05.1993 | |||
| WO 9113066 A, 05.09.1991 | |||
| WO 9316059 A1, 19.08.1993 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСОЛА | 1990 |
|
RU2017724C1 |

