Область техники
Настоящее изобретение относится к катализатору, представляющему собой модифицированную ионообменную смолу, и способу получения бисфенолов с применением такого катализатора. Более конкретно, изобретение относится к катализатору, представляющему собой модифицированную ионообменную смолу, который обладает высокой селективностью, и к способу получения бисфенолов путем взаимодействия фенольного соединения с кетонами в присутствии такого катализатора.
Уровень техники
Бисфенол A [2,2-бис-(4-гидроксифенил)пропан] обычно получают взаимодействием фенола с ацетоном в присутствии гомогенной кислоты или твердого кислотного катализатора. Кроме бисфенола A реакционная смесь содержит непрореагировавший ацетон, непрореагировавший фенол, получающуюся при этом воду и другие побочные продукты. Основным компонентом побочных продуктов является 2-(2-гидроксифенил)-2-(4-гидроксифенил)пропан (далее именуемый как o,п′-BPA) и, кроме того, побочные продукты включают в себя трисфенол, полифенольное соединение, хромановое соединение, незначительное количество примесей, которые могут вызывать окрашивание и т.п.
Примеры гомогенной кислоты, применяемой в качестве катализатора, включают в себя соляную кислоту, серную кислоту и т.п. Поскольку в случае применения гомогенной кислоты возможно одновременное протекание реакции осаждения кристаллов аддукта фенола с бисфенолом A путем их взаимодействия при пониженных температурах, можно получать бисфенол A с высокой конверсией ацетона и высокой селективностью уменьшением количества побочного o,п′-BPA в виде его изомера. Однако в случае катализатора, представляющего собой гомогенную кислоту, такую как соляная кислота, необходимо удалять катализатор из реакционной смеси или нейтрализовать его и, следовательно, проведение процесса осложняется. Кроме того, гомогенное растворение кислоты в реакционном растворе вызывает коррозию аппаратуры или т.п. Следовательно, для реакционной аппаратуры нужно применять дорогостоящие и антикоррозионные материалы, что является нерентабельным.
В качестве твердого кислотного катализатора обычно применяют катионообменную смолу сульфокислотного типа. Реакция получения бисфенола A преимущественно протекает только с кислотным катализатором, однако если в качестве кислотного катализатора применяется твердый кислотный катализатор, осуществляется процесс, при котором ацетон диффундирует с поверхности катализатора к активному участку, что приводит к низкой скорости реакции. Поэтому существует общий способ, применяемый для улучшения каталитической активности и селективности, при котором в реакционной системе обеспечивается совместное присутствие соединения, содержащего меркаптогруппу. В частности, известен способ, включающий в себя введение в реактор с неподвижным слоем, заполненный катионообменной смолой сульфокислотного типа, такого соединения в свободном виде, содержащего меркаптогруппу, как алкилмеркаптан, в дополнение к фенолу и ацетону, которые являются исходными веществами (например, патентный документ 1: JP-B № 45-10337, патентный документ 2: патент США № 6414200), и способ, включающий в себя образование ковалентной связи между частью сульфокислотной группы катионообменной смолы сульфокислотного типа и соединением, содержащим меркаптогруппу, или образование ионной связи между частью сульфокислотной группы катионообменной смолы сульфокислотного типа и соединением, содержащим меркаптогруппу (например, патентный документ 3: JP-B № 46-19953). Способ введения в реактор с неподвижным слоем, заполненный катионообменной смолой сульфокислотного типа, такого соединения в свободном виде, содержащего меркаптогруппу, как алкилмеркаптан, в дополнение к фенолу и ацетону, которые являются исходными веществами, обеспечивает постоянное совместное присутствие в реакционной системе определенного количества соединения, содержащего меркаптогруппу, и тем самым дает преимущество снижая значительную деградацию катализатора. Однако имеется опасение, что соединение, содержащее меркаптогруппу, может вызывать окрашивание бисфенола A и, следовательно, требуется способ удаления и извлечения соединения, содержащего меркаптогруппу.
С другой стороны, способ образования связи между частью сульфокислотной группы катионообменной смолы сульфокислотного типа и соединением, содержащим меркаптогруппу, приводит к меньшей потери соединения, содержащего меркаптогруппу, по сравнению со способом, обеспечивающим совместное присутствие в реакционной системе соединения в свободном виде, содержащего меркаптогруппу, и поэтому является предпочтительным, так как не возникает необходимости извлекать соединение, содержащее меркаптогруппу. В частности, в публикациях JP-A № 57-35533 (патентный документ 4, применение в качестве соединения, содержащего меркаптогруппу, пиридилэтантиола), JP-A № 08-187436 (патентный документ 5, применение в качестве соединения, содержащего меркаптогруппу, N,N-дизамещенного меркаптоалкиламина), JP-A № 08-089819 (патентный документ 6, применение в качестве соединения, содержащего меркаптогруппу,N,N,N-триметилмеркаптопропиламмония), JP-A № 10-211433 (патентный документ 7, применение в качестве соединения, содержащего меркаптогруппу, 1,4-димеркаптоалкилпиперидина) и патенте США № 6414200 (патентный документ 2, применение в качестве соединения, содержащего меркаптогруппу, кремнийсодержащего алкилмеркаптосоединения) описано, что скорость взаимодействия ацетона увеличивается при улучшении структуры соединения, содержащего меркаптогруппу, которое должно связываться с сильнокислой ионообменной смолой.
Кроме того, имеется также сообщение, относящееся к катионообменной смоле сульфокислотного типа, которая является кислотным катализатором для улучшения своей активности, которая ниже, чем активность описанной выше гомогенной кислоты. Когда диаметр частиц применяемой катионообменной смолы сульфокислотного типа большой, материалы, вступающие в реакцию, недостаточно диффундируют в частицы и, следовательно, нельзя получить достаточную конверсию ацетона. Соответственно, в публикации JP-A № 62-178532 (патентный документ 8) предлагается применять катионообменную смолу сульфокислотного типа в виде тонкодисперсных частиц или тонкодисперсного порошка с эффективным диаметром 0,3 мм или менее. В публикации JP-A № 6-340563 (патентный документ 9) аналогичным образом обеспечивается диаметр частиц применяемой катионообменной смолы сульфокислотного типа и уровень распределения таких частиц по диаметру, и описан более предпочтительный диапазон диаметров таких частиц. Кроме того, способы образования катионообменной смолы сульфокислотного типа с требуемым диаметром частиц описаны в публикациях JP-A № 4-268316 (патентный документ 10) и JP-A № 2002-253971 (патентный документ 11). Как было установлено, диаметр частиц катионообменной смолы сульфокислотного типа является важным фактором для получения достаточной конверсии при проведении реакции.
Были выполнены различные улучшения структуры получаемой смолы, которая является основным материалом для катионообменной смолы сульфокислотного типа. Катионообменная смола сульфокислотного типа представляет собой смолу, получаемую сульфонированием сополимера стирола и дивинилбензола, который получают радикальной сополимеризацией стирола и дивинилбензола. Дивинилбензол при полимеризации не только предохраняет полистирольную цепь от растворения в органическом растворителе, но его присутствие также является важным фактором для регулирования размера пор (размера гелевых микропор) внутри катионообменной смолы сульфокислотного типа, образуемых поглощением полярного растворителя, или для регулирования механической прочности катионообменной смолы сульфокислотного типа. Другими словами, катионообменная смола сульфокислотного типа с низким содержанием дивинилбензола обладает высокой каталитической активностью, обусловленной большим размером гелевых микропор, но имеет низкую механическую прочность. С другой стороны, если его содержание высокое, механическая прочность увеличивается, но размер гелевых микропор уменьшается, что вызывает пониженную активность.
Для улучшения диффузии внутри частиц получают ионообменные смолы, в которых степень сшивания увеличивается по мере того, как увеличивается содержание дивинилбензола; такие смолы с большим размером пор внутри частиц, упоминаемые как «макропористые», образуются с помощью физической обработки и обладают диаметром частиц 20 нм или более. Однако в том случае, когда такая макропористая ионообменная смола адсорбирует такую молекулу с высокой полярностью, как вода, сшитая структура имеет тенденцию подавлять вспучивание частиц, вызванное набуханием, и такая структура в конечном счете разрушается, если не может долго противостоять набуханию. В публикациях JP-A № 5-97741 (патентный документ 12) и JP-A № 6-320009 (патентный документ 13) описан способ, при котором соответствующие недостатки взаимно компенсируются путем одновременного заполнения реактора катионообменной смолой сульфокислотного типа с низким содержанием дивинилбензола и катионообменной смолой сульфокислотного типа с высоким содержанием дивинилбензола. Кроме того, об увеличении конверсии при проведении реакции сообщается в заявке Nippon Steel Chemical Co., Ltd. WO 2000/00454 (патентный документ 14), в которой предлагается получение катионообменной смолы сульфокислотного типа с большими гелевыми микропорами путем применения вместо дивинилбензола больших молекул, таких как дивинилбифенил.
Были исследованы различные по существу способы, относящиеся к катализаторам, в частности к соединению, содержащему меркаптогруппу, и стало понятно, что за исключением легкодоступных соединений, таких как аминоэтантиол и пиридинэтантиол, способ получения таких соединений требует осуществления многих реакций и способов выделения, и многие из технологических операций для получения продукта с высокой степенью чистоты являются сложными. Во всех случаях имеется возможность для улучшения селективности. Требуется разработать высокоселективный катализатор. Если попытаться улучшить селективность, можно не только уменьшить нагрузку, обусловленную проведением процессов извлечения побочных продуктов в способе получения, но также уменьшить отношение материалов фенол/ацетон без ухудшения селективности увеличением температуры реакции и, следовательно, привести к снижению затрат, относящихся к процессу извлечения избыточного фенола. Если активность несколько уменьшается, ее можно компенсировать увеличением размера реактора, а вызванные тем самым затраты при получении бисфенола весьма незначительны. Следовательно, требуется разработать катализатор, который можно легко получать и который обладает высокой селективностью при равноценной конверсии.
Описание изобретения
Целью настоящего изобретения является разработка катализатора, представляющего собой модифицированную ионообменную смолу, которая обладает более высокой селективностью в отношении бисфенолов, чем обычные модифицированные ионообменные смолы, в способах получения бисфенолов взаимодействием фенольного соединения с кетонами, и разработка способа получения бисфенолов.
Для решения упомянутых выше проблем авторы настоящего изобретения провели обширные исследования и в результате установили, что с применением модифицированной кислотной ионообменной смолы, в которой, по меньшей мере, одно соединение, выбранное из группы соединений, представленных следующими формулами:
(Формула 1)
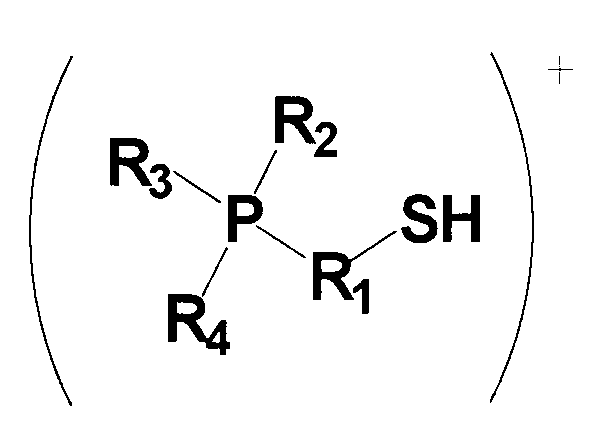
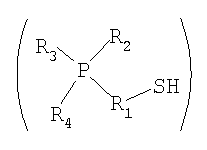
(в которой P представляет собой атом фосфора; S представляет собой атом серы; H представляет собой атом водорода; R1 представляет собой алкиленовую или алкениленовую группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу, и в которой один из атомов углерода может быть заменен атомом кремния, и один из фрагментов может содержать фениленовую группу; и каждый из R2, R3 и R4 независимо представляет собой (1) алкильную или алкенильную группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя гидроксильную группу или арильную группу, содержащую от 5 до 10 атомов углерода; (2) циклоалкильную группу, содержащую от 5 до 10 атомов углерода, или (3) арильную группу, содержащую от 5 до 10 атомов углерода, и любой из R2, R3 и R4 может представлять собой атом водорода); и
(Формула 2)
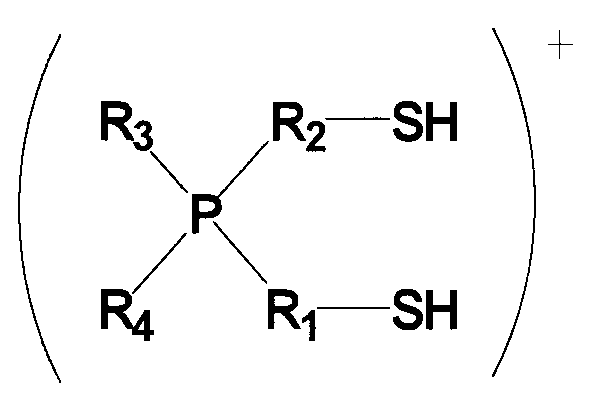
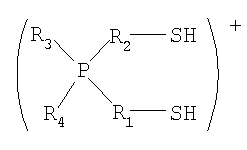
(в которой P представляет собой атом фосфора; S представляет собой атом серы; H представляет собой атом водорода; каждый из R1 и R2 представляет собой алкиленовую или алкениленовую группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу, и в которой один из атомов углерода может быть заменен атомом кремния, и один из фрагментов может содержать фениленовую группу; и каждый из R3 и R4 независимо представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, или арильную группу, содержащую от 5 до 10 атомов углерода, и любой из них может представлять собой атом водорода), присоединяется с помощью ионной связи к функциональной кислотной группе кислотной ионообменной смолы, селективность по бисфенолам увеличивается, и в результате бисфенолы получаются с высоким выходом. Таким образом, изобретение было выполнено.
То есть, изобретение относится к кислотной ионообменной смоле, которая частично нейтрализована, по меньшей мере, одним соединением, выбранным из группы соединений, представленных упомянутыми выше формулой 1 и формулой 2; к катализатору для получения бисфенолов, образованному из такой ионообменной смолы; и к способу получения бисфенолов с применением такого катализатора.
Наилучший способ осуществления изобретения
Применяемая в изобретении ионообменная смола предпочтительно представляет собой кислотную ионообменную смолу, а ее примеры включают в себя распространенный тип так называемой сильнокислотной ионообменной смолы, которую получают введением сульфогруппы в сополимер стирола и дивинилбензола, и перфторалкилсульфокислотные смолы, такие как Нафион (Nafion).
Модифицированную ионообменную смолу по изобретению получают путем образования ионной связи между кислотной ионообменной смолой и, по меньшей мере, одним соединением, выбранным из соединений, представленных формулой 1 и формулой 2:
(Формула 1)
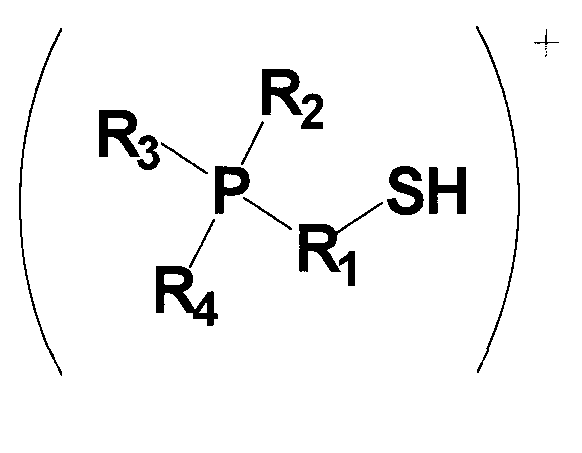
(в которой P представляет собой атом фосфора; S представляет собой атом серы; H представляет собой атом водорода; R1 представляет собой алкиленовую или алкениленовую группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу, и в которой один из атомов углерода может быть заменен атомом кремния, и один из фрагментов может содержать фениленовую группу; и каждый из R2, R3 и R4 независимо представляет собой (1) алкильную или алкенильную группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя гидроксильную группу или арильную группу, содержащую от 5 до 10 атомов углерода; (2) циклоалкильную группу, содержащую от 5 до 10 атомов углерода, или (3) арильную группу, содержащую от 5 до 10 атомов углерода, и любой из R2, R3 и R4 может представлять собой атом водорода);
(Формула 2)
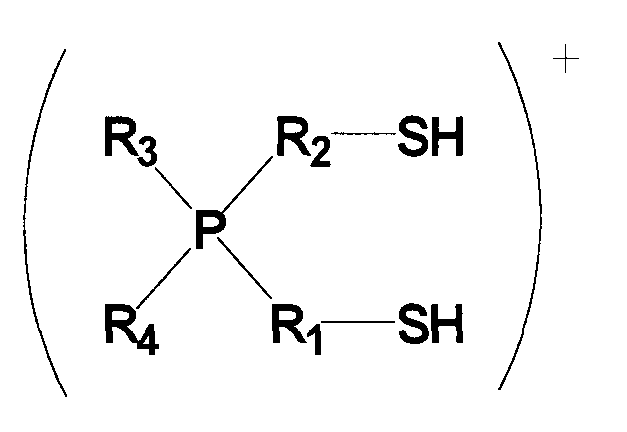
(в которой P представляет собой атом фосфора; S представляет собой атом серы; H представляет собой атом водорода; каждый из R1 и R2 представляет собой алкиленовую или алкениленовую группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу, и в которой один из атомов углерода может быть заменен атомом кремния, и один из фрагментов может содержать фениленовую группу; и каждый из R3 и R4 независимо представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, или арильную группу, содержащую от 5 до 10 атомов углерода, и любой из них может представлять собой атом водорода).
В модифицированной ионообменной смоле по изобретению соединение, выбранное из соединений, представленных формулой 1 и формулой 2, применяется либо в чистом виде, либо при сочетании нескольких типов таких соединений. Также смола может быть частично нейтрализована катионом, отличающимся от соединения, представленного формулой 1, формулой 2. Примеры катиона, отличающегося от соединений, представленных формулой 1, формулой 2, включают в себя катионы аминов и аммония, фосфония, фосфинов, катионы металлов и т.п.
Получение модифицированной кислотной ионообменной смолы по изобретению, в конечном счете, предпочтительно заключается в том, что непосредственно перед реакцией или во время реакции, по меньшей мере, одно катионное соединение, выбранное из соединений, представленных формулой 1 и формулой 2, присоединяется с помощью ионной связи к функциональной кислотной группе кислотной ионообменной смолы; и можно получать смолу, применяя катионное соединение и/или его предшественника, образующих такую структуру. Например, для катионизации можно приводить в контакт с кислотной ионообменной смолой в качестве предшественника нейтрально заряженное соединение, представленное формулой 3 и формулой 4.
(Формула 3)
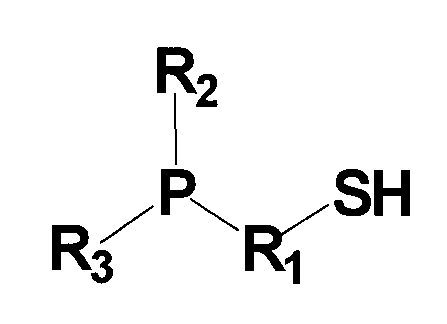
(в которой P представляет собой атом фосфора; S представляет собой атом серы; H представляет собой атом водорода; R1 представляет собой алкиленовую или алкениленовую группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу, и в которой один атом углерода может быть заменен атомом кремния, и один фрагмент которой может содержать фениленовую группу; и каждый из R2 и R3 независимо представляет собой (1) алкильную или алкенильную группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя гидроксильную группу или арильную группу, содержащую от 5 до 10 атомов углерода, (2) циклоалкильную группу, содержащую от 5 до 10 атомов углерода, или (3) арильную группу, содержащую от 5 до 10 атомов углерода).
(Формула 4)
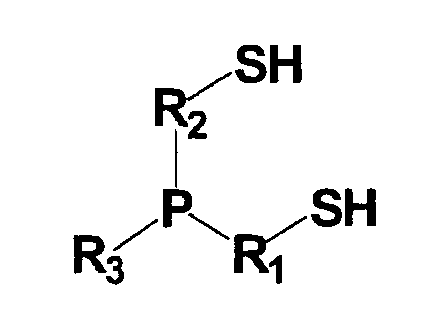
(в которой P представляет собой атом фосфора; S представляет собой атом серы; H представляет собой атом водорода; каждый из R1 и R2 представляет собой алкиленовую или алкениленовую группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу, и в которой один из атомов углерода может быть заменен атомом кремния, и один из фрагментов которой может содержать фениленовую группу; и R3 представляет собой (1) алкильную или алкенильную группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя гидроксильную группу или арильную группу, содержащую от 5 до 10 атомов углерода, (2) циклоалкильную группу, содержащую от 5 до 10 атомов углерода, или (3) арильную группу, содержащую от 5 до 10 атомов углерода).
Примеры предшественника меркаптогруппы включают в себя простые тиоэфиры, дисульфиды и тиоацетаты, и их также можно применять.
Для применяемого в изобретении соединения, которое представлено формулой 1, каждое из обозначений P, S и H в формуле 1 соответственно означает атом фосфора, атом серы и атом водорода. R1 представляет собой алкиленовую или алкениленовую группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу, и в которой один атом углерода может быть заменен атомом кремния, и один фрагмент которой может содержать фениленовую группу; и фениленовая группа может находиться на конце группы R1. Каждый из R2, R3 и R4 независимо представляет собой любую группу из (1) алкильной или алкенильной группы, содержащей от 1 до 6 атомов углерода, которая может содержать в качестве заместителя гидроксильную группу или арильную группу, содержащую от 5 до 10 атомов углерода; (2) циклоалкильной группы, содержащей от 5 до 10 атомов углерода; и (3) арильной группы, содержащей от 5 до 10 атомов углерода, и любой из R2, R3 и R4 может представлять собой атом водорода.
Среди соединений, представленных формулой 1, относительно легко синтезируемым соединением является соединение, у которого в формуле 1 R1 представляет собой алкиленовую группу, содержащую 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу, и один фрагмент которой может содержать фениленовую группу; и каждый из R2, R3 и R4 независимо представляет собой (1) алкильную группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя гидроксильную группу или арильную группу, содержащую от 5 до 10 атомов углерода; и (2) арильную группу, содержащую от 5 до 10 атомов углерода; и любой из R2, R3 и R4 может представлять собой атом водорода.
Среди соединений, представленных формулой 1, более предпочтительным соединением является соединение, у которого в формуле 1 R1 представляет собой алкиленовую группу, содержащую от 3 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу, и один фрагмент которой может содержать фениленовую группу; и каждый из R2, R3 и R4 независимо представляет собой любую группу из (1) алкильной группы, содержащей от 1 до 6 атомов углерода, которая может содержать в качестве заместителя гидроксильную группу или арильную группу, содержащую от 5 до 10 атомов углерода; и (2) арильной группы, содержащей от 5 до 10 атомов углерода; и любой из R2, R3 и R4 может представлять собой атом водорода.
Среди указанных соединений, представленных формулой 1, примеры группы R1, содержащей в одном из своих фрагментов фениленовую группу, включают в себя -CH2-C6H4-CH2-, -C6H4-CH(CH3)-, -C6H4-CH2-CH2- и т.п., но также могут быть включены и другие группы.
Для применяемого в изобретении соединения, которое представлено формулой 2, каждое из обозначений P, S и H в формуле 2 соответственно означает атом фосфора, атом серы и атом водорода. Каждый из R1 и R2 представляет собой алкиленовую или алкениленовую группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу; и в которой один атом углерода может быть заменен атомом кремния; и один фрагмент которой может содержать фениленовую группу. Каждый из R3 и R4 независимо представляет собой любую группу из алкильной группы, содержащей от 1 до 6 атомов углерода, циклоалкильной группы, содержащей от 5 до 10 атомов углерода, и арильной группы, содержащей от 5 до 10 атомов углерода, и любой из них может представлять собой атом водорода.
Среди соединений, представленных формулой 2, относительно легко синтезируемым соединением является соединение, у которого в формуле 2 каждый из Rl и R2 представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, циклоалкильную группу, содержащую от 5 до 10 атомов углерода, арильную группу, содержащую от 5 до 10 атомов углерода, или гидроксильную группу; и один фрагмент которой может содержать фениленовую группу; и каждый из R3 и R4 независимо представляет собой любую группу из (1) алкильной группы, содержащей от 1 до 6 атомов углерода, которая может содержать в качестве заместителя гидроксильную группу или арильную группу, содержащую от 5 до 10 атомов углерода; и (2) арильной группы, содержащей от 5 до 10 атомов углерода, и любой из R3 и R4 может представлять собой атом водорода.
Среди указанных соединений, представленных формулой 2, примеры групп R1 и R2, содержащих в одном из своих фрагментов фениленовую группу, включают в себя -CH2-C6H4-CH2-, -C6H4-CH(CH3)-, -C6H4-CH2-CH2- и т.п., но также могут быть включены и другие группы.
Способ модификации специально не ограничивается. Примеры легкодоступного способа включают в себя контактирование с ионообменной смолой в жидкой фазе при растворении в растворе, таком как вода или органический растворитель, и контактирование с ионообменной смолой в газовой фазе, когда применяется летучее вещество.
Примеры общеизвестных способов включают в себя способ, описанный в патенте JP-B № 46-19953 и т.п. Кроме того, соединение, представленное формулой 1, можно получить взаимодействием исходного материала, способного к образованию соединения, представленного формулой 1, в ионообменной смоле. Кроме того, можно применять способ, при котором, в конечном счете, образуется модифицированная кислотная ионообменная смола, такой как способ нейтрализации ионообменной смолы с применением эквивалентного или избыточного количества катионного соединения или его предшественника, и затем частичного возвращения к кислотному типу контактированием ионообменной смолы с кислым раствором.
Приведенное количество катализатора, представляющего собой модифицированную кислотную ионообменную смолу, в изобретении предпочтительно составляет от 0,1 до 50% от общего количества сульфокислотных групп. Таким образом, эффект модификации может проявиться в максимально возможной степени, не вызывая уменьшения активности, обусловленного уменьшением количества кислоты.
Способ измерения количества кислоты в ионообменной смоле специально не ограничивается, хотя количество кислоты может быть определено с помощью обычного способа измерения обменной емкости кислотной ионообменной смолы. В изобретении количество определяют из кривой титрования, получаемой перемешиванием 0,25 г сухой смолы в 200 мл 10%-ного водного раствора NaCl в течение одного часа и титрованием общего количества фильтрата 0,05 N водным раствором NaOH.
В изобретении в качестве фенола, применяемого в качестве исходного вещества для получения бисфенола A, можно применять обычно доступный промышленный фенол. Промышленный фенол включает в себя фенол, получаемый кумольным способом, окислением толуола или т.п., любой из которых можно применять. Обычно в продаже имеется фенол со степенью чистоты 98% или более. Такой промышленный фенол можно применять в качестве фенола в реакции синтеза бисфенола A, однако предпочтительно применяют фенол, который перед проведением реакции предварительно обрабатывают катионообменной смолой сильнокислотного типа в непрерывном или периодическом режиме при температуре обработки от 50 до 120°C при продолжительности контакта от 5 минут до 10 часов и подвергают полимеризации с карбонильным соединением, производным ацетона. Еще более предпочтительно применяют фенол, полученный с помощью способа, при котором промышленный фенол приводят в соприкосновение с катионообменной смолой сильнокислотного типа, как описано выше, и затем подвергают дистилляции в условиях от нормального давления до пониженного давления 10 мм Hg при температуре от 70 до 200°C.
Применяемый в изобретении ацетон специально не ограничивается, однако он может представлять собой имеющийся в продаже промышленный ацетон. Обычно доступен ацетон со степенью чистоты 99% или более.
Применяемые количества (количественные соотношения) фенола и ацетона, применяемых в качестве исходных материалов, специально не ограничиваются, однако предпочтительно рекомендуется молярное отношение фенол/ацетон в диапазоне от 0,1 до 100, и более предпочтительно в диапазоне от 0,5 до 50. Если количество фенола слишком мало, трудно достичь высокой конверсии ацетона в качестве исходного вещества; если количество фенола слишком большое, реактор становится неоправданно большим, потому что фенол применяется в большем количестве, чем требуется, и, кроме того, также требуется масштабная циркуляция фенола, даже если можно достичь высокой конверсии ацетона. Следовательно, нельзя достичь высокой производительности.
Как описано в патенте EP № 583712, смесь указанных исходных веществ предварительно может содержать 1% воды или менее.
В изобретении температура реакции специально не ограничивается, однако предпочтительно она находится в диапазоне от 0° до 300°C, и более предпочтительно в диапазоне от 30° до 200°C. Если температура реакции слишком низкая, скорость реакции уменьшается и, следовательно, выход продукта реакции также уменьшается. С другой стороны, если температура реакции слишком высокая, происходит нежелательная побочная реакция или т.п., тем самым, приводящая к увеличению количества побочных продуктов. Слишком высокая температура реакции неблагоприятна для стабильности фенола и ацетона как исходных веществ и, кроме того, для бисфенола A как продукта, уменьшает селективность реакции и экономически невыгодна. Следовательно, слишком высокая температура нерентабельна.
Реакцию можно проводить при любом давлении, - пониженном, повышенном и нормальном давлении. С точки зрения эффективности реакции (эффективность реакции на единицу объема), предпочтительно не проводить реакцию при слишком низком давлении. Обычно давление для проведения реакции предпочтительно находится в диапазоне от 0,01 до 20 МПа, и более предпочтительно в диапазоне от 0,05 до 10 МПа. Само собой разумеется, что изобретение не ограничивается такими диапазонами давления.
Кроме того, при осуществлении изобретения количество применяемого катализатора специально не ограничивается, однако, например, при проведении реакции в периодическом режиме рекомендуется осуществлять изобретение предпочтительно в диапазоне от 0,001 до 200 мас.% и более предпочтительно, в диапазоне от 0,1 до 50 мас.% относительно фенола как исходного материала.
При осуществлении изобретения можно проводить реакцию в разбавленном состоянии добавлением растворителя или газа, которые являются инертными по отношению к катализатору и участвующему в реакции веществу в реакционной системе. В частности, в качестве разбавителей можно применять такие алифатические углеводороды, как метан, этан, пропан, бутан, гексан и циклогексан, и такой инертный газ, как азот, аргон и гелий, и в случае необходимости можно применять водород.
При осуществлении изобретения способ можно осуществлять в любой периодической, полупериодической или непрерывной проточной системе. Способ можно осуществлять в любой жидкой фазе, газовой фазе и смешанной газожидкостной фазе. С точки зрения эффективности реакции, предпочтительно рекомендуется проводить реакцию в жидкой фазе. По способу эксплуатации катализатора можно использовать способы различного типа, например, с использованием неподвижного слоя, кипящего слоя, взвешенного слоя и неподвижного слоя на тарелке, любой из которых можно применять.
Продолжительность реакции (время пребывания или время соприкосновения с катализатором в проточной системе) специально не ограничивается, хотя обычно она составляет от 0,1 секунды до 30 часов, и предпочтительно от 0,5 секунды до 15 часов. После проведения реакции продукт реакции можно выделять и извлекать из катализаторов или т.п. с помощью такого способа выделения, как фильтрование, экстракция и отгонка. Бисфенол A в виде требуемого продукта можно выделять, очищать от извлеченного соединения путем проведения последующей обработки экстракцией растворителем, дистилляцией, обработкой щелочью, обработкой кислотой и т.п. или с помощью обычного способа выделения и очистки, в котором они сочетаются соответствующим образом, и таким образом получать бисфенол А. Кроме того, непрореагировавшие исходные вещества можно извлекать и возвращать в реакционную систему для повторного использования.
В случае проведения реакции в периодическом режиме катализатор, который выделяют и извлекают из продукта реакции после проведения реакции, можно применять, как он есть, или частично или полностью восстанавливать для многократного использования в реакции. В случае проведения реакции в проточной реакционной системе с неподвижным слоем или кипящим слоем, если катализатор использовали для проведения реакции и при этом часть катализатора или весь катализатор инактивировался или его активность снизилась, реакцию приостанавливают, и затем катализатор можно восстанавливать и снова применять для проведения реакции. Альтернативно, часть катализатора можно непрерывно или периодически извлекать и восстанавливать и затем возвращать в реактор для повторного использования. Кроме того, в реактор можно периодически подавать свежий катализатор. Когда реакция проводится в проточной реакционной системе с движущимся слоем, катализатор можно отделять, извлекать и, если необходимо, восстанавливать, как в случае проведения реакции в периодическом режиме.
Катализатор можно восстанавливать, применяя любой способ, при условии, что восстанавливается его каталитическая активность, например, катализатор можно промывать водой или органическим растворителем или можно подвергать повторной модификации после промывания кислотным растворителем. Кроме того, катализатор можно модифицировать после нескольких промываний поочередно кислотным растворителем и основным растворителем и в заключение кислотным растворителем.
ПРИМЕРЫ
Далее настоящее изобретение будет описано более подробно со ссылкой на примеры. Однако изобретение не должно ограничиваться примерами.
При этом несмотря на то, что применяется ионообменная смола одной и той же торговой марки, так как может встретиться случай, когда ее каталитическая способность в реакции синтеза BPA будет отличаться, поскольку отличается партия вещества, поэтому далее во всех примерах и примерах для сравнения применяется одна и та же торговая марка Амберлист 31 (Amberlyst) и одна и та же партия вещества.
Пример 1
(1) Синтез (3-меркаптопропил)трифенилфосфонийбромида
14,0 г (3-меркаптопропил)трифенилфосфонийбромида и 2,35 г тиомочевины растворяли в 335 мл этанола и кипятили смесь с обратным холодильником приблизительно 16 часов. Полученный раствор охлаждали льдом, фильтровали и полученное при этом твердое вещество в меру сушили, получая при этом кристаллическое вещество белого цвета 1. 5,3 г кристаллического вещества белого цвета 1 растворяли в 100 мл тщательно деаэрированной воды, прошедшей ионообменную обработку, и в течение 30 минут в атмосфере азота к полученному раствору по каплям добавляли водный раствор, полученный растворением 0,5 г гидроксида натрия в 25 мл воды, прошедшей ионообменную обработку. Затем смесь перемешивали при 60°C в течение 2 часов и охлаждали в атмосфере азота. Полученное при фильтровании твердое вещество промывали водой, прошедшей ионообменную обработку, дополнительно растворяли в хлороформе, многократно промывали водой, прошедшей ионообменную обработку, и разделяли на фазы. Фазу с хлороформом подвергали десольватации и полученное при этом твердое вещество перекристаллизовывали из хлороформа и сушили, получая при этом белое кристаллическое вещество 2. После анализа белого кристаллического вещества 2 методами 1H-ЯМР и FD-MS доказали, что оно имеет структуру, соответствующую формуле 5.
(Формула 5)
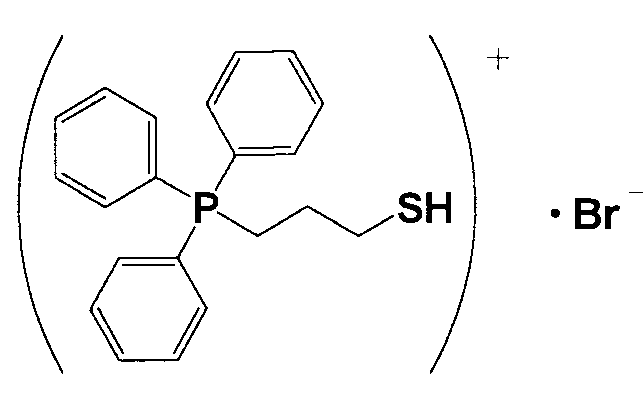
(2) Получение катализатора, представляющего собой ионообменную смолу, модифицированную (3-меркаптопропил)трифенилфосфонием
3 г тщательно промытой и высушенной смолы Амберлист 31 энергично перемешивали в 60 мл 50%-ного водного раствора ацетонитрила. К полученному продукту медленно по каплям добавляли 30 мл 0,077 моль/л раствора (3-меркаптопропил)трифенилфосфонийбромида в 50% водном растворе ацетонитрила, полученного с применением кристаллического вещества белого цвета 2 по п.(1). После добавления по каплям смесь далее перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, прошедшей ионообменную обработку. Затем полученный продукт сушили в вакууме при 80°C, по меньшей мере, в течение 10 часов, получая при этом катализатор 1. Количество кислоты в катализаторе в сухом состоянии составляло 3,47 миллиэквивалентов/г.
(3) Реакция синтеза бисфенола A
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 1, полученного по п.(2), 6,63 г фенола и 0,37 г ацетона и создавали в реакторе избыточное давление газообразного азота 0,5 МПа по манометру, затем проводили реакцию при нагревании и перемешивании при 75°C в течение 2 часов. Затем полученный продукт охлаждали до комнатной температуры. После сброса давления реакционный раствор извлекали и количественно анализировали с помощью способа жидкостной хроматографии. В результате конверсия ацетона составляла 88,1% и селективность в отношении пп′-бисфенола A составляла 94,1%.
Пример 2
(1) Синтез (4-меркаптобутил)трифенилфосфонийбромида
11,0 г (4-бромбутил)трифенилфосфонийбромида и 1,75 г тиомочевины растворяли в 250 мл этанола и кипятили смесь с обратным холодильником в течение приблизительно 16 часов. Из полученного продукта отгоняли около 170 мл этанола, реакционный раствор дополнительно охлаждали не более чем до 0°C, и затем оставляли для фильтрования. Полученное при этом твердое вещество в меру промывали хлороформом и сушили, получая при этом кристаллическое вещество белого цвета 3. 6,0 г кристаллического вещества белого цвета 3 растворяли в 140 мл тщательно деаэрированной воды, прошедшей ионообменную обработку, и к полученному продукту в атмосфере азота добавляли по каплям 30 мл 1,6%-ного водного раствора гидроксида натрия. Затем смесь перемешивали в атмосфере азота при 60°C в течение 2 часов и охлаждали. К полученному продукту добавляли 12 мл 5,6%-ного водного раствора HBr. К полученному продукту добавляли 70 мл хлороформа, хорошо перемешивали и затем разделяли на водную фазу и фазу с хлороформом. Фазу с хлороформом подвергали десольватации, получая при этом белое кристаллическое вещество 4. После анализа белого кристаллического вещества 4 методами 1H-ЯМР и LC-MS доказали, что оно имеет структуру, соответствующую формуле 6.
(Формула 6)
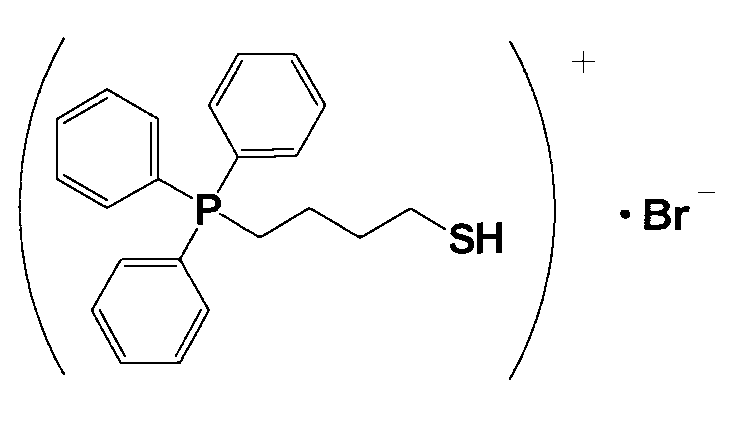
(2) Получение катализатора, представляющего собой ионообменную смолу, модифицированную (4-меркаптобутил)трифенилфосфонием
3 г тщательно промытой и высушенной смолы Амберлист 31 энергично перемешивали в 60 мл воды, прошедшей ионообменную обработку. К полученному продукту медленно по каплям добавляли 196 мл 0,0116 моль/л водного раствора (4-меркаптобутил)трифенилфосфонийбромида, полученного с применением кристаллического вещества белого цвета 4 по п.(1). После добавления по каплям смесь далее перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, прошедшей ионообменную обработку. Затем полученный продукт сушили в вакууме при 80°C в течение, по меньшей мере, 10 часов, получая при этом катализатор 2. Количество кислоты в катализаторе в сухом состоянии составляло 3,49 миллиэквивалентов/г.
(3) Реакция синтеза бисфенола A
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 2, полученного по п.(2), 6,63 г фенола и 0,37 г ацетона и создавали в реакторе избыточное давление газообразного азота 0,5 МПа по манометру, затем проводили реакцию при нагревании и перемешивании при 75°C в течение 2 часов. Затем полученный продукт охлаждали до комнатной температуры. После сброса давления реакционный раствор извлекали и количественно анализировали с помощью способа жидкостной хроматографии. В результате конверсия ацетона составляла 94,9%, и селективность в отношении пп′-бисфенола A составляла 93,9%.
Пример 3
(1) Синтез (5-меркаптопентил)трифенилфосфонийбромида
20,7 г 1,5-дибромпентана и 23,6 г трифенилфосфина растворяли в 55 мл толуола и перемешивали смесь приблизительно при 60°C в течение 18 часов. Затем толуол с поверхности насадки удаляли, остаток растворяли в воде, прошедшей ионообменную обработку, и полученный водный раствор многократно экстрагировали толуолом. Затем водный раствор перемешивали с добавлением хлороформа, разделяли на фазы и полученную фазу с хлороформом многократно экстрагировали (extruded) водой, прошедшей ионообменную обработку, фазу с хлороформом подвергали десольватации, получая при этом кристаллическое вещество белого цвета 5. 7,0 г кристаллического вещества белого цвета 5 и 1,1 г тиомочевины растворяли в 150 мл этанола и кипятили смесь с обратным холодильником в течение приблизительно 16 часов. Из раствора отгоняли этанол и затем сушили, получая при этом кристаллическое вещество белого цвета 6. 3,0 г кристаллического вещества белого цвета 6 растворяли в 75 мл тщательно деаэрированной воды, прошедшей ионообменную обработку, и к полученному продукту в атмосфере азота добавляли по каплям 17 мл 1,6%-ного водного раствора гидроксида натрия. Затем смесь перемешивали в атмосфере азота при 60°C в течение 2 часов и охлаждали. К полученному продукту добавляли 8 мл 5,6%-ного водного раствора HBr. К полученному продукту добавляли 30 мл хлороформа, хорошо перемешивали и затем разделяли на водную фазу и фазу с хлороформом. Фазу с хлороформом подвергали десольватации, получая при этом кристаллическое вещество белого цвета 7. После анализа белого кристаллического вещества 7 методами 1H-ЯМР и LC-MS доказали, что оно имеет структуру, соответствующую формуле 7.
(Формула 7).
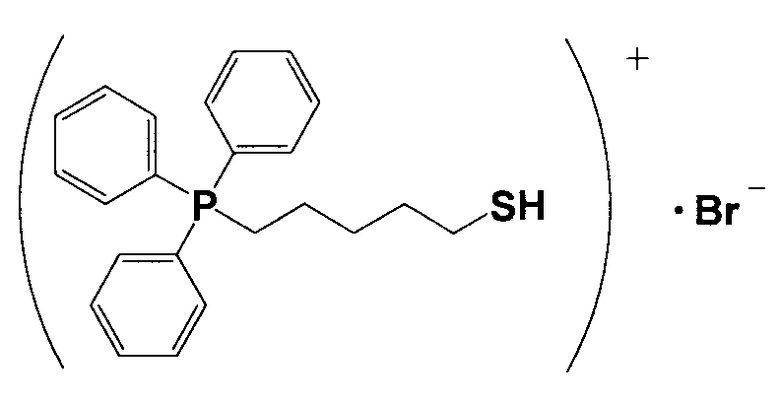
(2) Получение катализатора, представляющего собой ионообменную смолу, модифицированную (5-меркаптопентил)трифенилфосфонием
3 г тщательно промытой и высушенной смолы Амберлист 31 энергично перемешивали в 60 мл воды, прошедшей ионообменную обработку. К полученному продукту медленно по каплям добавляли 55 мл 0,042 моль/л водного раствора (5-меркаптопентил)трифенилфосфонийбромида, полученного с применением белого кристаллического вещества 7 по п.(1). После добавления по каплям смесь далее перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, прошедшей ионообменную обработку. Затем полученный продукт сушили в вакууме при 80°C, по меньшей мере, в течение 10 часов, получая при этом катализатор 3. Количество кислоты в катализаторе в сухом состоянии составляло 3,43 миллиэквивалентов/г.
(3) Реакция синтеза бисфенола A
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 3, полученного по п.(2), 6,63 г фенола и 0,37 г ацетона и создавали в реакторе избыточное давление газообразного азота 0,5 МПа по манометру, затем проводили реакцию при нагревании и перемешивании при 75°C в течение 2 часов. Затем полученный продукт охлаждали до комнатной температуры. После сброса давления реакционный раствор извлекали и количественно анализировали с помощью жидкостной хроматографии. В результате конверсия ацетона составляла 94,9%, и селективность в отношении пп′-бисфенола A составляла 94,0%.
Пример 4
(1) Синтез (6-меркаптогексил)трифенилфосфонийбромида
23,4 г 1,6-дибромгексана и 25,0 г трифенилфосфина растворяли в 35 мл толуола и перемешивали смесь приблизительно при 60°C в течение 18 часов. Затем надосадочный толуол удаляли, остаток растворяли в воде, прошедшей ионообменную обработку, и полученный водный раствор многократно экстрагировали толуолом. Затем водный раствор перемешивали с добавлением хлороформа, разделяли на фазы и полученную фазу с хлороформом многократно экстрагировали водой, прошедшей ионообменную обработку, и фазу с хлороформом подвергали десольватации, получая при этом кристаллическое вещество белого цвета 8. 7,2 г кристаллического вещества белого цвета 8 и 1,1 г тиомочевины растворяли в 150 мл этанола и кипятили смесь с обратным холодильником приблизительно в течение 16 часов. Из раствора отгоняли этанол и затем сушили, получая при этом белое кристаллическое вещество 9. 3,1 г белого кристаллического вещества 9 растворяли в 75 мл тщательно деаэрированной воды, прошедшей ионообменную обработку, и к полученному продукту в атмосфере азота добавляли по каплям 17 мл 1,6%-ного водного раствора гидроксида натрия. Затем смесь перемешивали в атмосфере азота при 60°C в течение 2 часов и охлаждали. К полученному продукту добавляли 8 мл 5,6%-ного водного раствора HBr. К полученному продукту добавляли 30 мл хлороформа, хорошо перемешивали и затем разделяли на водную фазу и фазу с хлороформом. Фазу с хлороформом подвергали десольватации, получая при этом белое кристаллическое вещество 10. После анализа кристаллическое вещество белого цвета 10 методами 1H-ЯМР и LC-MS доказали, что оно имеет структуру, соответствующую формуле 8.
(Формула 8)
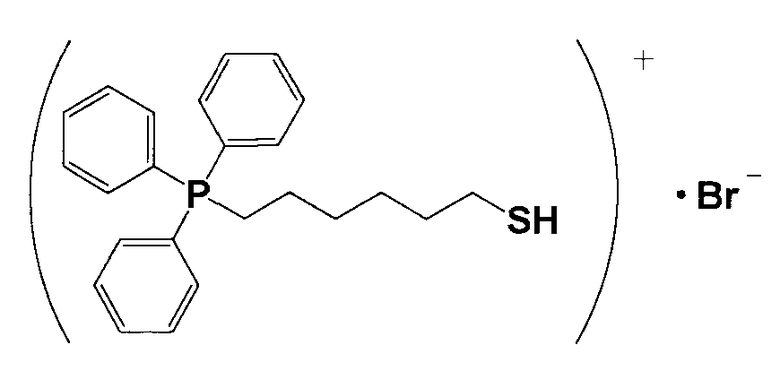
(2) Получение катализатора, представляющего собой ионообменную смолу, модифицированную (6-меркаптогексил)трифенилфосфонием
3 г тщательно промытой и высушенной смолы Амберлист 31 энергично перемешивали в 60 мл воды, прошедшей ионообменную обработку. К полученному продукту медленно по каплям добавляли 55 мл 0,042 моль/л водного раствора (6-меркаптогексил)трифенилфосфонийбромида, полученного с применением кристаллического вещества белого цвета 10 по п.(1). После добавления по каплям смесь далее перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, прошедшей ионообменную обработку. Затем полученный продукт сушили в вакууме при 80°C, по меньшей мере, в течение 10 часов, получая при этом катализатор 4. Количество кислоты в катализаторе в сухом состоянии составляло 3,41 миллиэквивалентов/г.
(3) Реакция синтеза бисфенола A
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 4, полученного по п.(2), 6,63 г фенола и 0,37 г ацетона и создавали в реакторе избыточное давление газообразного азота 0,5 МПа по манометру, затем проводили реакцию при нагревании и перемешивании при 75°C в течение 2 часов. Затем полученный продукт охлаждали до комнатной температуры. После сброса давления реакционный раствор извлекали и количественно анализировали с помощью способа жидкостной хроматографии. В результате конверсия ацетона составляла 95,1%, и селективность в отношении пп′-бисфенола A составляла 94,1%.
Пример 5
(1) Синтез (4-меркаптобутил)дифенил(п-толил)фосфонийбромида
15,3 г 1,4-дибромбутана и 19,2 г дифенил(п-толил)фосфина растворяли в 40 мл толуола и перемешивали смесь в атмосфере азота приблизительно при 70°C в течение 16 часов. Полученный раствор охлаждали, фильтровали и полученное при этом твердое вещество промывали толуолом и сушили, получая при этом белое кристаллическое вещество 11. 10 г кристаллического вещества белого цвета 11 и 1,55 г тиомочевины растворяли в 250 мл этанола и кипятили смесь с обратным холодильником в течение приблизительно 16 часов. Из раствора отгоняли этанол и полученное при этом твердое вещество промывали этанолом и сушили, получая при этом кристаллическое вещество белого цвета 12. 6,0 г кристаллического вещества белого цвета 12 растворяли в 140 мл тщательно деаэрированной воды, прошедшей ионообменную обработку, и к полученному продукту в атмосфере азота добавляли по каплям 31 мл 1,6%-ного водного раствора гидроксида натрия. Затем смесь перемешивали в атмосфере азота при 60°C в течение 2 часов и охлаждали. К полученному продукту добавляли 13 мл 5,6%-ного водного раствора HBr. К полученному продукту добавляли 70 мл хлороформа, хорошо перемешивали и затем разделяли на водную фазу и фазу с хлороформом. Фазу с хлороформом подвергали десольватации, получая при этом кристаллическое вещество белого цвета 13. После анализа белого кристаллического вещества 13 методами 1H-ЯМР и LC-MS доказали, что оно имеет структуру, соответствующую формуле 9.
(Формула 9)
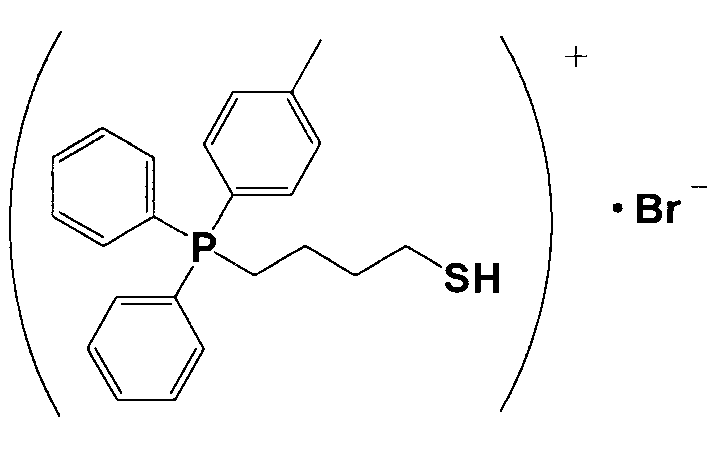
(2) Получение катализатора, представляющего собой
ионообменную смолу, модифицированную (4-меркаптобутил)дифенил(п-толил)фосфонием
3 г тщательно промытой и высушенной смолы Амберлист 31 энергично перемешивали в 60 мл воды, прошедшей ионообменную обработку. К полученному продукту медленно по каплям добавляли 196 мл 0,0116 моль/л водного раствора (4-меркаптобутил)дифенил(п-толил)фосфонийбромида, полученного с применением кристаллического вещества белого цвета 13 по п.(1). После добавления по каплям смесь далее перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, прошедшей ионообменную обработку. Затем полученный продукт сушили в вакууме при 80°C, по меньшей мере, в течение 10 часов, получая при этом катализатор 5. Количество кислоты в катализаторе в сухом состоянии составляло 3,43 миллиэквивалентов/г.
(3) Реакция синтеза бисфенола A
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 5, полученного по п.(2), 6,63 г фенола и 0,37 г ацетона и создавали в реакторе избыточное давление газообразного азота 0,5 МПа по манометру, затем проводили реакцию при нагревании и перемешивании при 75°C в течение 2 часов. Затем полученный продукт охлаждали до комнатной температуры. После сброса давления реакционный раствор извлекали и количественно анализировали с помощью способа жидкостной хроматографии. В результате конверсия ацетона составляла 93,3%, и селективность в отношении пп′-бисфенола A составляла 93,8%.
Пример 6
(1) Синтез (4-меркаптометилбензил)трифенилфосфонийбромида
6,3 г 1,4-бис-(бромметил)бензола и 6,5 г трифенилфосфина растворяли в 60 мл толуола и перемешивали смесь в атмосфере азота приблизительно при 60°C в течение 3 часов. Полученный раствор охлаждали, фильтровали и полученное при этом твердое вещество промывали толуолом и сушили, получая при этом кристаллическое вещество белого цвета 14. 10 г кристаллического вещества белого цвета 14 и 1,5 г тиомочевины растворяли в 100 мл этанола и кипятили смесь с обратным холодильником в течение приблизительно 2 часов. Из раствора отгоняли этанол и полученное при этом твердое вещество сушили, получая при этом кристаллическое вещество белого цвета 15. 11,0 г кристаллического вещества белого цвета 15 растворяли в 300 мл тщательно деаэрированной воды, прошедшей ионообменную обработку, и к полученному продукту в атмосфере азота добавляли по каплям 15 мл 5,4%-ного водного раствора гидроксида натрия. Затем смесь перемешивали в атмосфере азота при 60°C в течение 3 часов и охлаждали. К полученному продукту добавляли 7,8 мл 5,3%-ного водного раствора HBr. После перемешивания в течение нескольких минут смесь оставляли оседать, водную фазу извлекали и охлаждали, и оставляли оседать, как она есть, в течение еще 16 часов. Полученный продукт фильтровали и полученное при этом твердое вещество промывали холодной водой и сушили, получая при этом кристаллическое вещество белого цвета 16. После анализа кристаллического вещества белого цвета 16 методами 1H-ЯМР и LC-MS доказали, что оно имеет структуру, соответствующую формуле 10.
(Формула 10)
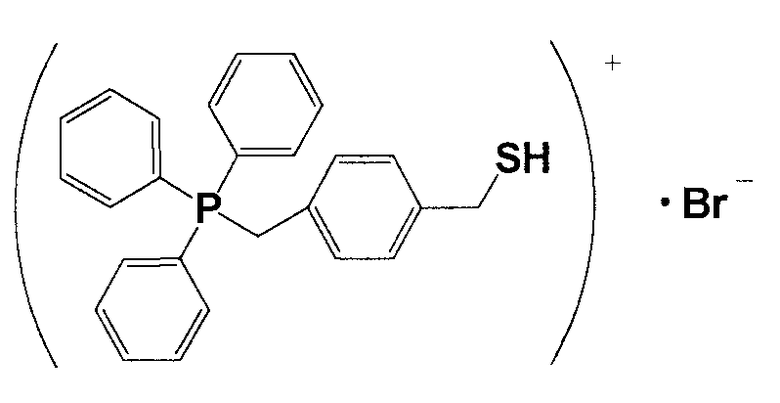
(2) Получение катализатора, представляющего собой ионообменную смолу, модифицированную (4-меркаптометилбензил)трифенилфосфонием
3 г тщательно промытой и высушенной смолы Амберлист 31 энергично перемешивали в 60 мл 25%-ного водного раствора ацетонитрила. К полученному продукту медленно по каплям добавляли 120 мл 0,0188 моль/л раствора (4-меркаптометилбензил)трифенилфосфонийбромида в 25%-ном водном растворе ацетонитрила, полученного с применением кристаллического вещества белого цвета 16 по п.(1). После добавления по каплям смесь далее перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, прошедшей ионообменную обработку. Затем полученный продукт сушили в вакууме при 80°C, по меньшей мере, в течение 10 часов, получая при этом катализатор 6. Количество кислоты в катализаторе в сухом состоянии составляло 3,40 миллиэквивалентов/г.
(3) Реакция синтеза бисфенола A
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 6, полученного по п.(2), 6,63 г фенола и 0,37 г ацетона и создавали в реакторе избыточное давление газообразного азота 0,5 МПа по манометру, затем проводили реакцию при нагревании и перемешивании при 75°C в течение 2 часов. Затем полученный продукт охлаждали до комнатной температуры. После сброса давления реакционный раствор извлекали и количественно анализировали с помощью способа жидкостной хроматографии. В результате конверсия ацетона составляла 93,9%, и селективность в отношении пп′-бисфенола A составляла 94,0%.
Пример 7
(1) Синтез (4-меркаптометилбензил)дифенилпропилфосфонийбромида
6,3 г 1,4-бис-(бромметил)бензола и 5,4 г дифенилпропилфосфина растворяли в 60 мл толуола и перемешивали смесь в атмосфере азота приблизительно при 60°C в течение 5 часов. Полученный раствор охлаждали, фильтровали и полученное при этом твердое вещество промывали толуолом и сушили, получая при этом кристаллическое вещество белого цвета 17. 10 г кристаллического вещества белого цвета 17 и 1,53 г тиомочевины растворяли в 100 мл этанола и кипятили смесь с обратным холодильником приблизительно в течение 3 часов. Из раствора отгоняли этанол и полученное при этом твердое вещество сушили, получая при этом кристаллическое вещество белого цвета 18. 11,0 г кристаллического вещества белого цвета 18 растворяли в 300 мл тщательно деаэрированной воды, прошедшей ионообменную обработку, и к полученному продукту в атмосфере азота добавляли по каплям 15 мл 5,4%-ного водного раствора гидроксида натрия. Затем, смесь перемешивали в атмосфере азота при 60°C в течение 3 часов и охлаждали. К полученному продукту добавляли 7,8 мл 5,3%-ного водного раствора HBr. После перемешивания в течение нескольких минут смесь оставляли оседать, водную фазу извлекали и охлаждали, и оставляли оседать, как она есть, в течение еще 16 часов. Полученный продукт фильтровали и полученное при этом твердое вещество промывали холодной водой и сушили, получая при этом кристаллическое вещество белого цвета 19. После анализа кристаллического вещества белого цвета 19 методами 1H-ЯМР и LC-MS доказали, что оно имеет структуру, соответствующую формуле 11.
(Формула 11)
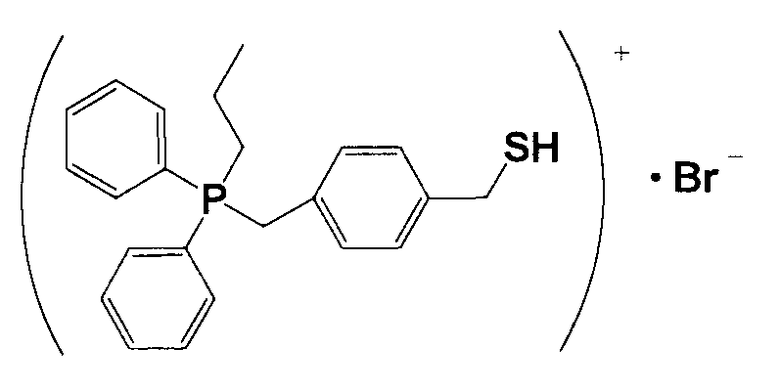
Получение катализатора, представляющего собой ионообменную смолу, модифицированную (4-меркаптометилбензил)дифенилпропилфосфонием
3 г тщательно промытой и высушенной смолы Амберлист 31 энергично перемешивали в 60 мл 25%-ного водного раствора ацетонитрила. К полученному продукту медленно по каплям добавляли 120 мл 0,0188 моль/л раствора (4-меркаптометилбензил)трифенилфосфонийбромида в 25%-ном водном растворе ацетонитрила, полученного с применением кристаллического вещества белого цвета 19 по п.(1). После добавления по каплям смесь далее перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, прошедшей ионообменную обработку. Затем полученный продукт сушили в вакууме при 80°C, по меньшей мере, в течение 10 часов, получая при этом катализатор 7. Количество кислоты в катализаторе в сухом состоянии составляло 3,43 миллиэквивалентов/г.
(3) Реакция синтеза бисфенола A
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 7, полученного по п.(2), 6,63 г фенола и 0,37 г ацетона и создавали в реакторе избыточное давление газообразного азота 0,5 МПа по манометру, затем проводили реакцию при нагревании и перемешивании при 75°C в течение 2 часов. Затем полученный продукт охлаждали до комнатной температуры. После сброса давления реакционный раствор извлекали и количественно анализировали с помощью способа жидкостной хроматографии. В результате конверсия ацетона составляла 94,8%, и селективность в отношении пп′-бисфенола A составляла 94,1%.
Пример для сравнения 1
Во время перемешивания 3 г тщательно промытой и высушенной смолы Амберлист 31 в 60 мл воды, прошедшей ионообменную обработку, к ней медленно по каплям добавляли 30 мл 0,077 моль/л водного раствора гидрохлорида аминоэтантиола. После добавления по каплям смесь далее перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, прошедшей ионообменную обработку. Затем полученный продукт сушили в вакууме при 80°C, по меньшей мере, в течение 10 часов, получая при этом катализатор 2. Количество кислоты в катализаторе в сухом состоянии составляло 4,14 миллиэквивалентов/г.
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 2, 6,63 г фенола и 0,37 г ацетона и создавали в реакторе избыточное давление газообразного азота 0,5 МПа по манометру, затем проводили реакцию при нагревании и перемешивании при 75°C в течение 2 часов. Затем полученный продукт охлаждали до комнатной температуры. После сброса давления реакционный раствор извлекали и количественно анализировали с помощью способа жидкостной хроматографии. В результате конверсия ацетона составляла 84,9%, и селективность в отношении пп′-бисфенола A составляла 91,5%.
Пример для сравнения 2
Во время перемешивания 3 г тщательно промытой и высушенной смолы Амберлист 31 в 60 мл воды, прошедшей ионообменную обработку, к ней медленно по каплям добавляли 30 мл 0,077 моль/л водного раствора гидрохлорида 4-пиридинэтантиола. После добавления по каплям смесь далее перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, прошедшей ионообменную обработку. Затем полученный продукт сушили в вакууме при 80°C, по меньшей мере, в течение 10 часов, получая при этом катализатор 3. Количество кислоты в катализаторе в сухом состоянии составляло 3,94 миллиэквивалентов/г.
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 3, 6,63 г фенола и 0,37 г ацетона и создавали в реакторе избыточное давление газообразного азота 0,5 МПа по манометру, и затем проводили реакцию при нагревании и перемешивании при 75°C в течение 2 часов. Затем полученный продукт охлаждали до комнатной температуры. После сброса давления реакционный раствор извлекали и количественно анализировали с помощью способа жидкостной хроматографии. В результате конверсия ацетона составляла 94,0%, и селективность в отношении пп′-бисфенола A составляла 91,6%.
Промышленная применимость
Согласно настоящему изобретению, можно получать бисфенолы с хорошим выходом и селективностью, и получать бисфенолы с исключительно хорошей безопасностью технологического процесса и низкими затратами.
Дополнительные примеры
Дополнительный пример 1
(1) Синтез S-4-(дифенилфосфино)фенетилтиоацетата
40 г п-стирилдифенилфосфина и 22 г тиоуксусной кислоты подвергали взаимодействию в течение 2 часов при 80°С в 160 мл толуола в качестве растворителя, в присутствии 0,12 г азобисизобутиронитрила. Полученный продукт охлаждали и очищали с помощью колоночной хроматографии, получая при этом 21,2 г S-4-(дифенилфосфино)фенетилтиоацетата
(2) Синтез бромида (4-меркаптобутил)[4-(2-меркаптоэтил)фенил]дифенилфосфония
6,6 г S-4-(дифенилфосфино)фенетилтиоацетата, полученного по примеру 1, и 7,4 г 1,4-дибромбутана подвергали взаимодействию в 33 мл толуола в течение 16 часов при 80°С. При фильтровании полученного продукта получали твердое вещество. Твердое вещество промывали толуолом и сушили, получая при этом кристаллическое белое вешество. 6,4 г кристаллического белого вещества и 1 г тиомочевины кипятили с обратным холодильником в 36 мл этанола в течение 6 часов, после чего этанол отгоняли, получая при этом твердое вещество. Твердое вещество растворяли в 370 мл воды, обработанной методом ионообмена. К раствору добавляли 20 г 20%-ного водного раствора гидроксида натрия. I Полученный продукт перемешивали при комнатной температуре в течение 3 часов и по каплям добавляли 57 г 15,7%-ного водного раствора HBr для нейтрализации. Растворимый компонент экстрагировали из такого водного раствора 200 мл хлороформа. Из растворимого компонента отгоняли растворитель. Затем растворимый компонент сушили при пониженном давлении, получая при этом 6,6 г требуемого компонента, представленного следующей формулой (I).
Ниже приведены значения, полученные, при измерении 1H ЯМР-спектра.
1H NMR (270 MHz, CDCL3)d/ppm 1.46 (t, J=8.1 Hz, SH(CH2CH2CH2CH2SH или С6Н4СН2СН2SH),1H), 1.49 (t,J=8.1 Hz, SH(CH2CH2CH2CH2SH или С6Н4СН2СН2SH),1H), 1.7-1.9 (m, СН2(СН2СН2СН2СН2SH),2H), 2.0 (кв. J=6.8 Hz, CH2(CH2CH2CH2CH2SH),2H), 2.64 (кв. J=7.2 Hz, CH2(CH2CH2CH2CH2SH),2H), 2.85 (кв. J=7.2 Hz, CH2(С6Н4СН2СН2SH),2H), 3.05 (t, J=6.8 Hz, СН2(С6Н4СН2СН2SH),2H), 3.7-3.9 (m CH2(CH2CH2CH2CH2SH),2H), 7.5-7.9 (m, CH(Ph2 and С6Н4),14Н)
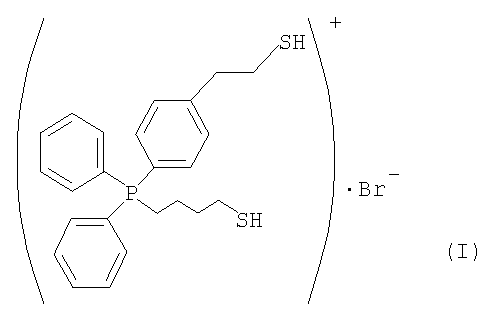
(3) Получение катализатора на основе ионообменной смолы, модифицированной [4-(2-меркаптоэтил)фенил]дифенилфосфонием
3 г тщательно промытого и высушенного Amberlist 31 энергично перемешивали в 60 мл воды, обработанной методом ионообмена. К полученному продукту медленно по каплям добавляли 25 мл 0,077 моль/л раствора бромида (4-меркаптобутил)[4-(2-меркаптоэтил)фенил]дифенилфосфония в 50%-ном водном ацетонитриле, который получали, применяя кристаллическое вещество, полученное по пункту (2). После добавления по каплям смесь последовательно перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, обработанной методом ионообмена. После чего полученный продукт сушили в вакууме при 80°С, по меньшей мере, в течение 10 часов, получая при этом катализатор 1. Количество кислоты в катализаторе в сухом состоянии составляло 3,6 миллиэквивалентов/г.
(4) Реакция синтеза бисфенола А
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 1, полученного по пункту (3), 6,63 г фенола и 0,37 г ацетона; в реактор нагнетали газообразный азот под манометрическим давлением 0,5 МПа и затем осуществляли реакцию при перемешивании и нагревании при 75°С в течение 2 часов. После чего полученный продукт охлаждали до комнатной температуры. После спуска давления реакционный раствор выгружали и методом жидкостной хроматографии проводили количественный анализ. В результате степень превращения ацетона составляла 97,6% и селективность в отношении пп'-бисфенола А составляла 92,9%.
Дополнительный пример 2
(1) Синтез бромида (3-меркаптопропил)[4-(2-меркаптоэтил)фенил]дифенилфосфония
3,0 г 5-4-(дифенилфосфино)фенетилтиоацетата, полученного в дополнительном примере 1, и 6,8 г 1,3-дибромпропана подвергали взаимодействию в 16 мл толуола в течение 8 часов при 110°С. После завершения реакции удаляли надосадочную жидкость. Затем остаток несколько раз промывали толуолом. Остаток растворяли в 80 мл хлороформа, после чего хлороформовую фазу три раза промывали 20 мл воды. При удалении растворителя получали твердое вещество. 3,2 г твердого вещества и 0,48 г тиомочевины кипятили с обратным холодильником в 28 мл этанола в течение 3 часов, после чего этанол отгоняли, получая при этом твердое вещество. Твердое вещество растворяли в 88 мл воды, обработанной методом ионообмена. К раствору добавляли 9,6 г 20%-ного водного раствора гидроксида натрия. Полученный продукт перемешивали при комнатной температуре в течение 3 часов и добавляли по каплям 8,4 г 48%-ного водного раствора HBr для нейтрализации. Растворимый компонент экстрагировали из такого водного раствора 48 мл хлороформа. Из растворимого компонента отгоняли растворитель. Затем растворимый компонент сушили при пониженном давлении, получая при этом 1,88 г требуемого компонента, представленного следующей формулой (II).
Ниже приведены значения, полученные при измерении 1Н ЯМР-спектра.
1H NMR (270 MHz, CDCL3)d/ppm 1.46 (t, J-7.8 Hz, SH(С6Н4СН2СН2SH),1H), 1.85-2.0 (m, СН2(СН2СН2СН2SH),2Н), 2.17 (t, J=8.5 Hz, SH(CH2CH2CH2SH),1H), 2.85 (кв., J=7.3 Hz, СН2(С6Н,СН2СН2SH),2Н), 2.9-3.0 (m, СН2(СН2СН2СН2SH),2Н), 3.04 (t, J=7.2 Hz, CH2(С6Н4СН2СН2SH),2H), 4.0-4.12 (m, СН2(СН2СН2СН2SH),2Н), 7.5-8.0 (m, СН(Ph2 and C6H4), 14H)
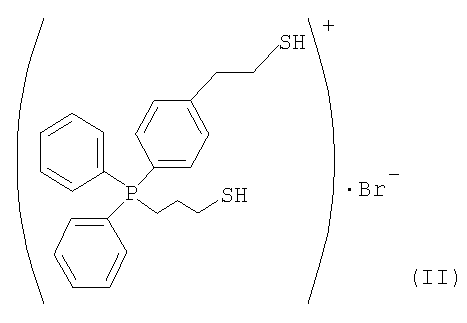
(2) Получение катализатора на основе ионообменной смолы, модифицированной (3-меркаптопропил)[4-(2-меркаптоэтил)фенил]дифенилфосфонием.
3 г тщательно промытого и высушенного Amberlist 31 энергично перемешивали в 60 мл воды, обработанной методом ионообмена. К полученному продукту медленно по каплям добавляли 25 мл 0,077 моль/л раствора бромида (3-меркаптопропил)[4-(2-меркаптоэтил)фенил]дифенилфосфония в 50%-ном водном ацетонитриле, который получали, применяя кристаллическое вещество, полученное по пункту (1). После добавления по каплям смесь последовательно перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, обработанной методом ионообмена. После чего полученный продукт сушили в вакууме при 80°С, по меньшей мере, в течение 10 часов, получая при этом катализатор 2. Количество кислоты в катализаторе в сухом состоянии составляло 3,6 миллиэквивалентов/г.
(3) Реакция синтеза бисфенола А
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 2, полученного по пункту (2), 6,63 г фенола и 0,37 г ацетона, в реактор нагнетали газообразный азот под манометрическим давлением 0,5 МПа и затем осуществляли реакцию при перемешивании и нагревании при 75°С в течение 2 часов. После чего полученный продукт охлаждали до комнатной температуры. После спуска давления реакционный раствор выгружали и методом жидкостной хроматографии проводили количественный анализ. В результате степень превращения ацетона составляла 95,5% и селективность в отношении пп'-бисфенола А составляла 93,2%.
Дополнительный пример 3
(1) Синтез бромида [4-(меркаптометил)бензил][4-(2-меркаптоэтил)фенил]дифенилфосфония 5 г 5-4-(дифенилфосфино)фенетилтиоацетата, полученного в дополнительном примере 1, и 3,43 г α,α'-дибромпараксилола подвергали взаимодействию в 122 мл толуола в течение 3 часов при 80°С. Затем при фильтровании полученного продукта получали твердое вещество. Твердое вещество промывали этилацетатом и сушили, получая при этом белое кристаллическое вещество. 6,3 г белого кристаллического вещества и 0,84 г тиомочевины кипятили с обратным холодильником в 55 мл этанола в течение 1 часа, после чего этанол отгоняли, получая при этом твердое вещество. Твердое вещество растворяли в 350 мл воды, обработанной методом ионообмена. К раствору добавляли 18,6 г 20%-ного водного раствора гидроксида натрия. Полученный продукт перемешивали при комнатной температуре в течение 6 часов и добавляли по каплям 17 г 48%-ного водного раствора HBr. Растворимый компонент экстрагировали из такого водного раствора 200 мл хлороформа. Из растворимого компонента отгоняли растворитель. Затем растворимый компонент сушили при пониженном давлении, получая при этом 4,5 г требуемого компонента, представленного следующей формулой (III).
Ниже приведены значения, полученные при измерении 1H ЯМР-спектра.
1H NMR (270 MHz, CDCL3)d/ppm 1.48 (t, J=7.8 Hz, SH(С6Н4СН2СН2SH),1H), 1.76 (t, J=7.4 Hz, SH(СН2С6Н4СН2SH),1Н), 2.83 (кв., J=7.5 Hz, СН2(С6Н4СН2СН2SH),2H), 3.03 (t, J=7.2 Hz, CH2(С6Н4СН2СН2SH),2H), 3.64 (dd, J1=7.4 Hz, J2=1.5 Hz, СН2(СН2С6Н4СН2SH),2H), 5.36 (d, J=14,3 Hz, СН2(СН2С6Н4СН2SH),2Н), 7.09 (S, CH(СН2С6Н4СН2SH),4H), 7.4-7.9 (m, CH(Ph2 и С6Н4),14Н)
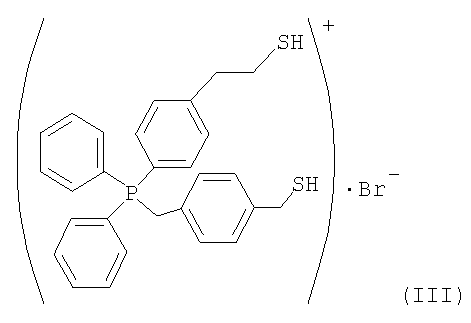
(2) Получение катализатора на основе ионообменной смолы, модифицированной [4-(меркаптометил)бензил][4-(2-меркаптоэтил)фенил]дифенилфосфонием]
3 г тщательно промытого и высушенного Amberlist 31 энергично перемешивали в 60 мл воды, обработанной методом ионообмена. К полученному продукту медленно по каплям добавляли 25 мл 0,077 моль/л раствора бромида [4-(меркаптометил)бензил] [4-(2-меркаптоэтил)фенил]дифенилфосфония] в 50%-ном водном ацетонитриле, который получали, применяя кристаллическое вещество, полученное по пункту (1). После добавления по каплям смесь последовательно перемешивали в течение 5 часов и затем многократно фильтровали и промывали водой, обработанной методом ионообмена. После чего полученный продукт сушили в вакууме при 80°С, по меньшей мере, в течение 10 часов, получая при этом катализатор 3. Количество кислоты в катализаторе в сухом состоянии составляло 3,6 миллиэквивалентов/г.
(3) Реакция синтеза бисфенола А
В 70 мл реактор, стойкий к давлению, загружали 0,35 г катализатора 3, полученного по пункту (2), 6,63 г фенола и 0,37 г ацетона, в реактор нагнетали газообразный азот под манометрическим давлением 0,5 МПа и затем осуществляли реакцию при перемешивании и нагревании при 75°С в течение 2 часов. После чего полученный продукт охлаждали до комнатной температуры. После спуска давления реакционный раствор выгружали и методом жидкостной хроматографии проводили количественный анализ. В результате степень превращения ацетона составляла 97,2% и селективность в отношении пп'-бисфенола А составляла 92,9%.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР НА ОСНОВЕ ИОНООБМЕННОЙ СМОЛЫ ДЛЯ СИНТЕЗА БИСФЕНОЛОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2128550C1 |
| КОМПОЗИЦИИ ДЛЯ КОНТЕЙНЕРОВ И ДРУГИХ ИЗДЕЛИЙ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2602157C2 |
| ОБРАБОТКА ОСТАТОЧНЫХ ПОТОКОВ ОТ ПРОИЗВОДСТВА БИСФЕНОЛОВ | 2018 |
|
RU2782457C2 |
| Композиции для контейнеров и других изделий и способы их применения | 2013 |
|
RU2618704C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА А | 2007 |
|
RU2424223C2 |
| КОМПОЗИЦИИ ПОКРЫТИЙ ДЛЯ КОНТЕЙНЕРОВ И ДРУГИХ ИЗДЕЛИЙ И СПОСОБЫ НАНЕСЕНИЯ ПОКРЫТИЙ | 2012 |
|
RU2561969C2 |
| СМОЛА С ПРОМОТОРОМ И АНТИОКСИДАНТОМ | 2016 |
|
RU2750165C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА А ВЫСОКОЙ ЧИСТОТЫ И ПРОИЗВОДСТВЕННАЯ УСТАНОВКА | 2007 |
|
RU2422429C2 |
| СОСТАВЫ ДЛЯ ПОКРЫТИЙ ДЛЯ УПАКОВОЧНЫХ ИЗДЕЛИЙ И СПОСОБЫ НАНЕСЕНИЯ ПОКРЫТИЙ | 2011 |
|
RU2544676C2 |
| НОВЫЙ ФОСФАЗЕНОВЫЙ НАНЕСЕННЫЙ НА НОСИТЕЛЬ КАТАЛИЗАТОР, НОВОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2340396C2 |
Настоящее изобретение относится к катализатору, представляющему собой модифицированную ионообменную смолу, способу получения бисфенолов. Описана модифицированная ионообменная смола, в которой, по меньшей мере, одно соединение, выбранное из указанных ниже соединений (А) и (В), присоединяется с помощью ионной связи к функциональной кислотной группе кислотной ионообменной смолы и соединение присоединяется с помощью ионной связи к 0,1-50 мол.% от общего числа функциональных кислотных групп, присутствующих в кислотной ионообменной смоле:
соединение (А) представлено формулой 1, в которой Р представляет собой атом фосфора; S представляет собой атом серы; Н представляет собой атом водорода; R1 представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода и в которой один из фрагментов может содержать фениленовую группу; и каждый R2, R3 и R4 независимо представляет собой (1) алкильную группу, содержащую от 1 до 6 атомов углерода, или (3) арильную группу, содержащую от 5 до 10 атомов углерода; и соединение (В) представлено формулой 2, в которой Р представляет собой атом фосфора; S представляет собой атом серы; Н представляет собой атом водорода; каждый из R1 и R2 представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода и в которой один из фрагментов которой может содержать фениленовую группу; и каждый из R3 и R4 независимо представляет собой арильную группу, содержащую от 5 до 10 атомов углерода. Также описан катализатор для получения бисфенолов, образуемый указанной выше смолой, а также способ получения бисфенолов. Описаны способы получения модифицированной ионообменной смолы (варианты). Технический результат - катализатор с высокой селективностью в отношении бисфенолов, получение бисфенола А с высокой конверсией ацетона. 5 н. и 4 з.п. ф-лы.
соединение (А) представлено формулой 1:
в которой Р представляет собой атом фосфора; S представляет собой атом серы; Н представляет собой атом водорода; R1 представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода и в которой один из фрагментов может содержать фениленовую группу; и каждый R2, R3 и R4 независимо представляет собой (1) алкильную группу, содержащую от 1 до 6 атомов углерода, или (3) арильную группу, содержащую от 5 до 10 атомов углерода; и
соединение (В) представлено формулой 2
в которой Р представляет собой атом фосфора; S представляет собой атом серы; Н представляет собой атом водорода; каждый из R1 и R2 представляет собой алкиленовую, содержащую от 1 до 6 атомов углерода и в которой один из фрагментов которой может содержать фениленовую группу; и каждый из R3 и R4 независимо представляет собой арильную группу, содержащую от 5 до 10 атомов углерода.

в которой Р представляет собой атом фосфора; S представляет собой атом серы; Н представляет собой атом водорода; и n равно целому числу от 1 до 4.
в которой Р представляет собой атом фосфора; S представляет собой атом серы; Н представляет собой атом водорода; R1 представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода, и в которой один из фрагментов которой может содержать фениленовую группу; и каждый из R2, R3 и R4 независимо представляет собой (1) алкильную группу, содержащую от 1 до 6 атомов углерода, или (3) арильную группу, содержащую от 5 до 10 атомов углерода, и
в которой Р представляет собой атом фосфора; S представляет собой атом серы; Н представляет собой атом водорода; каждый из R1 и R2 представляет собой алкиленовую группу, содержащую от 1 до 6 атомов углерода, и в которой один из фрагментов которой может содержать фениленовую группу; и каждый из R3 и R4 независимо представляет собой арильную группу, содержащую от 5 до 10 атомов углерода.
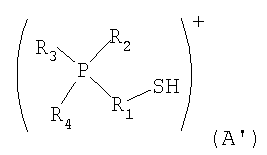
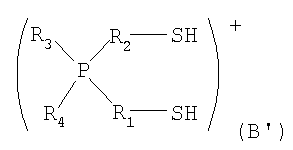
стадия [I] включает взаимодействия галогеналкил фосфония и тиомочевины с получением изотиурониевой соли и затем получение соединения содержащего катион представленный формулой (А') или катион представленный формулой (В') из изотиурониевой соли;
в которой Р представляет собой атом фосфора; S представляет собой атом серы; Н представляет собой атом водорода; R1 представляет собой алкиленовую группу, содержащую в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, и в которой один из фрагментов которой может содержать фениленовую группу; и каждый из R2, R3 и R4 независимо представляет собой (1) алкильную, содержащую от 1 до 6 атомов углерода, или (3) арильную группу, содержащую от 5 до 10 атомов углерода; и
в которой Р представляет собой атом фосфора; S представляет собой атом серы; Н представляет собой атом водорода; каждый из R1 и R2 представляет собой алкиленовую, содержащую от 1 до 6 атомов углерода, которая может содержать в качестве заместителя алкильную группу, содержащую от 1 до 6 атомов углерода, и в которой один из фрагментов которой может содержать фениленовую группу; и каждый из R3 и R4 независимо представляет собой арильную группу, содержащую от 5 до 10 атомов углерода, и любой из них может представлять собой атом водорода.
| JP 2003190805 А, 08.07.2003 | |||
| US 6414200 B1, 07.02.2002 | |||
| КАТАЛИЗАТОР НА ОСНОВЕ ИОНООБМЕННОЙ СМОЛЫ ДЛЯ СИНТЕЗА БИСФЕНОЛОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2128550C1 |
| Способ получения ионита для производства бисфенола "А | 1988 |
|
SU1600619A3 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |

