Настоящее изобретение относится к соединениям, обладающим повышенной эффективностью для снижения хронической боли, особенно невропатической боли. При описании хронической боли обычно для упрощения ссылаются на невропатическую боль.
Известно, что невропатическая боль представляет собой форму хронической боли, возникающей при повреждении или заболевании центральной или периферической нервной системы. Невропатическая боль включает ряд болезненных симптоматологий, таких как боль при диабетической невропатии, болезненный постинфарктный синдром, боль, возникающая при химиотерапевтическом лечении, или боль, возникающая при инфекции вирусами, например герпес, опоясывающий лишай и т.д.
Обычно от невропатической боли пациенты страдают многие годы, и это представляет социальную проблему, так как хронический характер симптомов вызывает у пациента опасный эмоциональный стресс.
За последние двадцать лет в исследовании патогенеза хронической боли был достигнут значительный успех. Исследования, проведенные на экспериментальных моделях невропатической боли у людей и животных, показали, что центральная нервная система реагирует на болезненный раздражитель серией биохимических и физиопатологических ответных реакций. Эта способность центральной нервной системы к функциональной и морфологической адаптации к болезненным раздражителям известна как пластика нерва и играет существенную роль при стимулировании начала или поддержании болезненной симпоматологии.
Применение болеутоляющих средств, используемых для лечения хронической боли, на самом деле частично или полностью не эффективно.
Широко используемый в клинических исследованиях карбамазепин активен при лечении невралгии тройничного нерва, боли при диабетической невропатии и постгерпетической невралгии. Использование этого лекарства имеет такой недостаток, как наличие побочных эффектов, например сонливость, головокружение, расстройство координации движения, тошнота и рвота, что ограничивает его использование.
За последние годы был исследован ряд медикаментов для лечения невропатической боли. Среди них в особенности упоминается габапентин, который очень активен как болеутоляющее средство при лечении невропатической боли, главным образом боли при диабетической невропатии и постгерпетической боли. Однако в этих случаях также имеют место опасные побочные эффекты, например сонливость, усталость, ожирение и т.д. (Martindale XXXth Ed, стр.374).
Задачей настоящего изобретения является разработка лекарственных средств, обладающих улучшенным фармакотерапевтическим профилем и/или незначительными побочными действиями, для лечения хронической боли, в особенности невропатической боли.
Заявителем неожиданно было установлено, что эта проблема может быть решена с помощью группы описываемых далее лекарственных средств.
Настоящее изобретение относится к нитрооксипроизводным или их солям общей формулы (I):
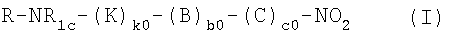 ,
,
где
с0 представляет 0 или 1, предпочтительно 1;
b0 представляет собой 0 или 1, при условии, что с0 и b0 не могут одновременно быть 0;
k0 представляет собой 0 или 1;
R представляет собой радикал болеутоляющего средства против хронической боли, например, невропатической боли;
R1c представляет собой Н или прямой или разветвленный алкил с 1-5 атомами углерода;
K представляет собой (СО) или двухвалентный радикал (1C), имеющий следующую формулу:
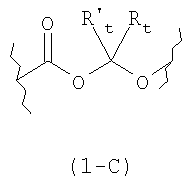
где карбонильная группа связана с T1; Rt и R′t представляют собой одинаковые или разные Н, C1-С10-алкил, фенил или бензил, -COORy, где Ry равно Н, C1-С10-алкил, фенил бензил;
В=-ТВ-Х2-ТBI-, где
ТВ=(СО) или X, где Х=О, S, NH;
при условии, что:
когда b0=1 и k0=0, тогда ТB=(СО);
когда b0=1 и k0=1, K=(СО), тогда ТB=Х, как указано выше;
TBI-(СО) или (X), где Х как указано выше;
когда с0=0, тогда TBI=-О-;
Х2 представляет двухвалентную мостиковую группу, которая соответствует предшественнику В, имеющему формулу Z-TB-X2-TBI-Z′, в которой Z, Z′ независимо представляют Н или ОН, выбранному из следующих соединений:
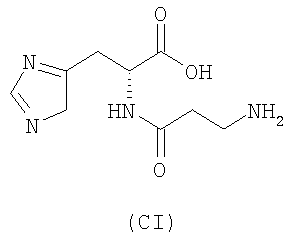
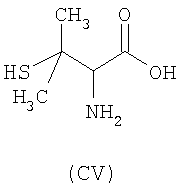
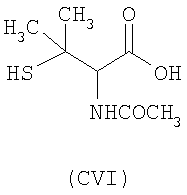
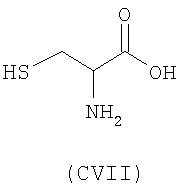
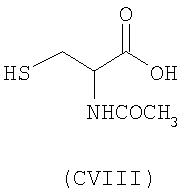
- аминокислоты: L-карнозин (CI), пеницилламин (CV), N-ацетилпеницилламин (CVI), цистеин (CVII), N-ацетилцистеин (CVIII):
 ,
,  ,
,
 ,
,  ,
, 
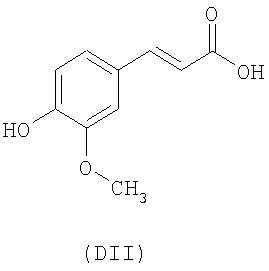
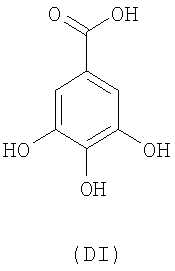
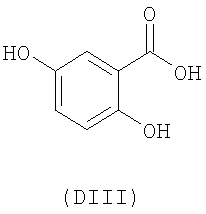
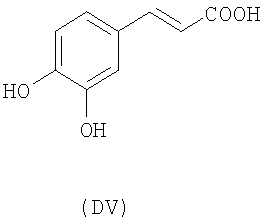
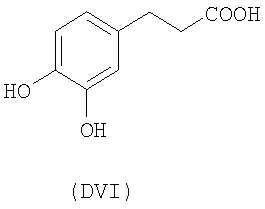
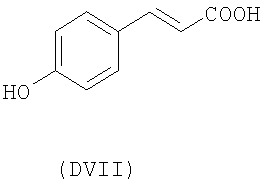
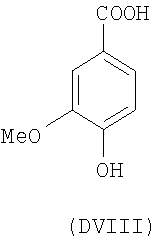
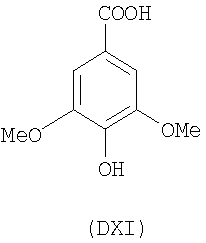
- оксикислоты: галловая кислота (DI), феруловая кислота (DII), гентизиновая кислота (DIII), кофейная кислота (DV), гидрокофейная кислота (DVI), п-кумаровая кислота (DVII), ванилиновая кислота (DVIII), сиреневая кислота (DXI):
 ,
,  ,
,  ,
,
 ,
,  ,
,
 ,
,  ,
,  ;
;
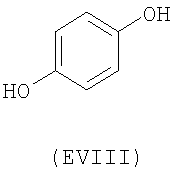
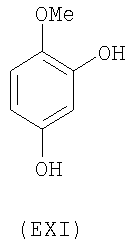
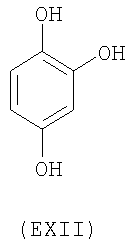
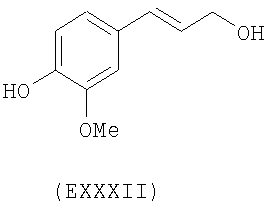
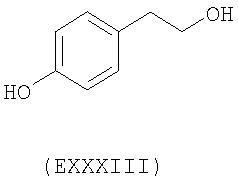
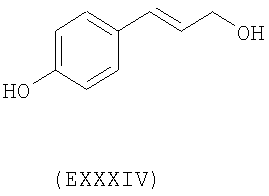
- ароматические многоатомные спирты: гидрохинон (EVIII), метоксигидрохинон (EXI), гидроксигидрохинон (EXII), конифериловый спирт (EXXXII), 4-гидроксифенетиловый спирт (EXXXIII), п-кумариновый спирт (EXXXIV):
 ,
,  ,
,  ,
,
 ,
,  ,
, 
С представляет собой двухвалентный радикал, имеющий формулу -Тc -Y-
где Тc=(СО) или X, как указано выше;
при условии, что когда b0=0 и k0=1:
-Тc=(СО), когда K=(1C),
-Тc=Х, как указано выше, когда K=(СО);
Y имеет одно из следующих значений:
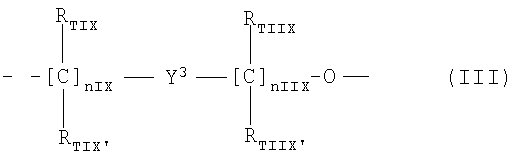 ,
,
где:
nIX представляет собой целое число от 0 до 5, преимущественно от 1;
nIIX представляет собой целое число от 1 до 5, преимущественно от 1;
RTIX, RTIX', RTIIX, RTIIX' одинаковые или разные, представляют собой Н или прямой или разветвленный С1-С4-алкил; предпочтительно RTIX, RTIX', RTIIX, RTIIX' представляют собой Н;
Y3 представляет собой насыщенное, ненасыщенное или ароматическое кольцо с 5 или 6 атомами, содержащее от одного до трех гетероатомов, преимущественно один или два, указанные гетероатомы являются одинаковыми или разными и выбраны из азота, кислорода или серы;
или Y может быть:
группой алкиленокси -R′О-, в которой R′ представляет собой прямой или разветвленный C1-C20, преимущественно с от 2 до 6 атомами углерода, или циклоалкиленом с от 5 до 7 атомами углерода, где в циклоалкиленовом кольце один или более атомов углерода могут быть замещены гетероатомами и кольцо может иметь боковые цепи R′ типа, где R′, как указано выше;
или одной из следующих групп:
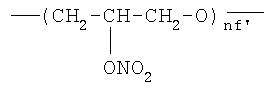 ;
; 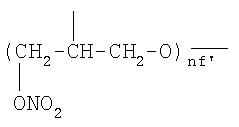 ,
,
где nf′ представляет целое число от 1 до 6, преимущественно от 1 до 4;
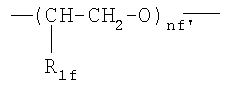 ;
; 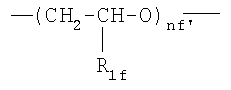 ,
,
где R1f=H, СН3 и nf′ представляет целое число от 1 до 6; преимущественно от 1 до 4;
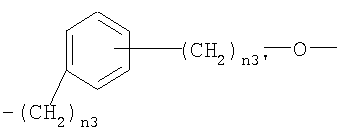
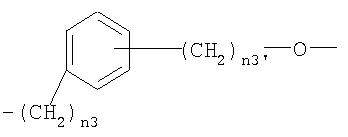 ,
,
где n3 представляет целое число от 0 до 5 и n3′ представляет целое число от 1 до 3;
или
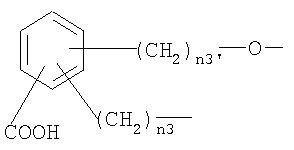 ,
,
где n3 и n3′ имеют указанные выше значения.
Радикал R в формуле (I) преимущественно представляет собой радикал болеутоляющего средства против хронической боли, в особенности против невропатической боли, которое может быть выбрано из обычных имеющихся в продаже средств, пригодных для указанного использования. Можно назвать трициклические антидепрессанты и антиэпилептические средства.
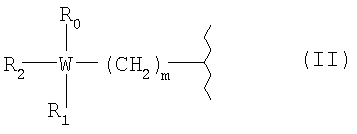
R представляет собой радикал болеутоляющего средства, имеющий формулу (II)
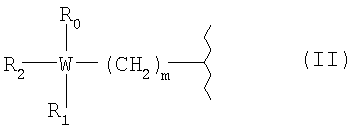 ,
,
где:
W представляет атом углерода или азота;
m представляет целое число от 0 до 2;
R0=Н, -(CH2)n-COORy, Ry, как указано выше;
n представляет целое число от 0 до 2;
R1=Н; когда W=N, R1 представляет собой дуплет электронов на атоме азота (свободная валентность):
R2 выбирают из следующих групп:
- фенил, необязательно замещенный атомом галогена или группой, выбранной из -ОСН3, -CF3, нитро;
- моно или дигидрокси-замещенный бензил, предпочтительно 3,4-дигидроксибензил;
- группа амидино: H2N(C=NH)-;
- радикал формулы (IIA), где этиленовая ненасыщенность между углеродными атомами может находиться в положении 1 и 2, или 3 и 4, или 4 и 5:
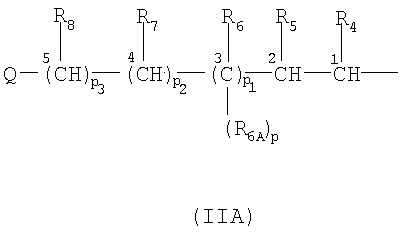 ,
,
где:
р, p1, p2 представляют одинаковые или разные целые числа 0 или 1;
р3 представляет целое число от 0 до 10;
R4 представляет водород, прямой или разветвленный C1-С4-алкил, свободную валентность;
R5 может иметь следующие значения:
- водород,
- прямой или разветвленный C1-С6-алкил,
- С3-С6-циклоалкил,
- ORA, RA, имеющий следующие значения:
- прямой или разветвленный C1-С6-алкил, необязательно замещенный одним или более атомами галогена, предпочтительно F,
- фенил, необязательно замещенный атомом галогена или одной из следующих групп: -ОСН3, -CF3, нитро;
R6, R6A, R7, R8 представляют одинаковые или разные Н, метил или свободную валентность,
при условии, что когда этиленовая ненасыщенность находится между C1 и С2 в радикале формулы (IIA), R4 и R5 представляют собой свободные валентности, способные образовывать двойную связь между C1 и C2; если ненасыщенность находится между С3 и C4, R6 и R7 представляют собой свободную валентность, способную образовывать двойную связь между С3 и С4; ненасыщенность находится между С4 и C5, R7 и R8 представляет собой свободную валентность, способную образовывать двойную связь между C4 и С5;
- Q представляет собой Н, ОН, ORВ, RB представляет собой бензил, прямой или разветвленный С1-С6-алкил, необязательно замещенный одним или более атомами галоида, предпочтительно F, фенил, необязательно замещенный атомом галоида или одной из следующих групп: -ОСН3, -CF3, нитро;
или Q может иметь одно из следующих значений:
- прямой или разветвленный C1-С6-алкил,
- С3-С6-циклоалкил,
- гуанидино (H2NC(=N)NH-),
- тиогуанидино (H2NC(=S)NH-);
в формуле (II) R2 с R1 и с W=C вместе образуют насыщенное или ненасыщенное кольцо С4-С10, предпочтительно насыщенное кольцо С6.
Когда в формуле (II) W=C, m=1 и R0=-(CH2)n-COORу, где n=1 и Ry=Н; R2 и R1 с W, имеющим указанное выше значение, образуют циклогексановое кольцо; лекарственный предшественник R, имеющий формулу R-NH2, известен как габапентин;
когда в формуле (II) W=C, m=0 и если R0 имеет значение, как указано для габапентина с n=0; R1=Н; R2 представляет собой радикал формулы (IIA), в котором р=p1=1, р2=р3=0, R4=R5=R6=R6А=Н, Q=Н;
лекарственный предшественник R, имеющий формулу R-NH2, известен как норвалин;
когда в формуле (II) W=C, m=0 и если R0 имеет значение, как указано для габапентина с n=0; R1=H; R2 представляет собой радикал формулы (IIA), в котором р=p1=1, p2=р3=0, R4=R5=R6=R6А=H, Q представляет собой группу гуанидино; лекарственный предшественник R, имеющий формулу R-NH2, известен как аргинин;
когда в формуле (II) W=C, m=0 и если R0 имеет значение, как указано для габапентина с n=0; R1=H; R2 представляет собой радикал формулы (IIA), в котором p=p1=1, p2=p3=0, R4=R5=R6=R6А=H, Q представляет собой группу тиогуанидино; лекарственный предшественник R, имеющий формулу R-NH2, известен как тиоцитрулин;
когда в формуле (II) W=C, m=1 и если R0 имеет значение, как указано для габапентина с n=1; R1=H; R2 представляет собой радикал формулы (IIA), в котором р=p1=р2=р3=0, R4=H, R5=Q=СН3; лекарственный предшественник R, имеющий формулу R-NH2, известен как прегабалин;
когда в формуле (II) W=C и имеет (S) конфигурацию, m=1 и если R0 имеет значение, как указано для габапентина с n=1; R1=H; R2 представляет собой радикал формулы (IIA), в котором р=p1=р2=р3=0, R4=H, R5=Q=СН3; лекарственный предшественник R, имеющий формулу R-NH2, известен как (S)3-изобутилGABA;
когда в формуле (II) W=C и имеет (S) конфигурацию, m=0; R0=R1=H; R2 представляет собой радикал формулы (IIA), в котором р=p1=1, р2=р3=0, R4=R5=R6=R6А=H, Q представляет собой группу гуанидино; лекарственный предшественник R, имеющий формулу R-NH2, известен как агматин;
когда в формуле (II) W=C, m=0; если R0 имеет значение, как указано для габапентина с n=2; R1=H; R2 представляет собой радикал формулы (IIA), в котором р=p1=р2=р3=0, R4 и R5 представляют свободные валентности и между C1 и С2 имеется этиленовая ненасыщенность, Q=H; лекарственный предшественник R, имеющий формулу R-NH2, известен как вигабатрин;
когда в формуле (II) W=C, m=0; если R0 имеет значение, как указано для габапентина с n=0; R1=Н; R2 представляет собой 3,4-дигидроксибензильный радикал; лекарственный предшественник R, имеющий формулу R-NH2, известен как 2-амино-3-(3,4-дигидроксифенилпропановая кислота (допа).
Дальнейшими соединениями, используемыми против хронической боли и которые могут использоваться в качестве предшественников R в формуле (I), являются ламотригин, топирамат, зонисамид, карбамазепин, фелбамат, аминептин, амоксапин, демексиптиллин, дезиптрамин, нортриптилин, тианептин.
Лекарственные предшественники R синтезируют главным образом в соответствии со способами, описанными в «The Merck Index, 12th Ed.» (1996). Когда лекарственные предшественники R присутствуют в молекуле радикала формулы (IIA), они могут быть получены, как описано в WO 00/79658.
Предшественники В, входящие в указанные выше группы, можно синтезировать в соответствии с хорошо известными из литературы методами, описанными, например, в «The Merck Index, 12th Ed.», которые включены в настоящее описание в качестве ссылки.
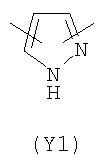
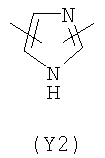
В формуле (III) Y3 выбирают из следующих двухвалентных радикалов:
 ;
;  ;
; 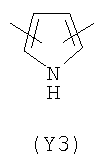 ;
; 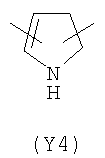 ;
; 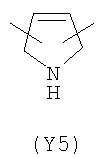 ;
; 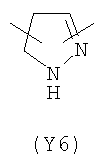 ;
;
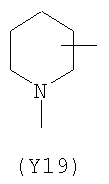 ;
;
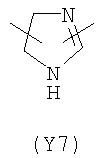 ;
; 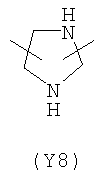 ;
; 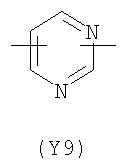 ;
; 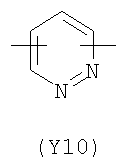 ;
; 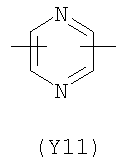 ;
; 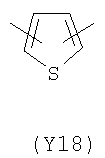 ;
;
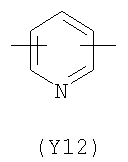 ;
;  ;
;  ;
;  ;
;  ;
; 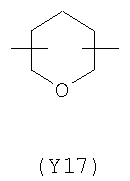 .
.
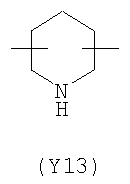
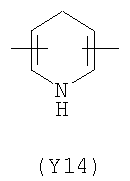
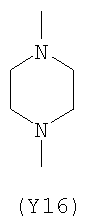
Преимущественными значениями Y3 являются следующие: (Y12), имеющий обе свободные валентности в орто положении относительно атома азота; (Y16), в котором обе валентности связаны с гетероатомами, (Y1) (пиразол) 3,5 замещенный.
Y предшественники формулы (III), в которых свободная валентность кислорода занята Н и свободная валентность концевого углеродного атома занята карбоксильной или гидроксильной группой, являются имеющимися в продаже доступными продуктами и могут быть получены известными методами.
В формуле (I) предпочтительными В предшественниками для синтеза используемых в настоящем изобретении нитрооксипрозводных являются следующие: феруловая кислота и N-ацетилцистеин, предпочтительными лекарственными предшественниками являются габапентин, норвалин, аргинин, прегабалин, (S)-3-изобутилGABA, агматин и вигабатрин.
Предпочтительными соединениями формулы (I) настоящего изобретения являются следующие:
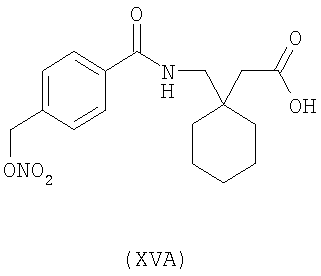
1-[4-(нитрооксиметил)бензоиламинометил]-циклогексануксусная кислота (XVA)
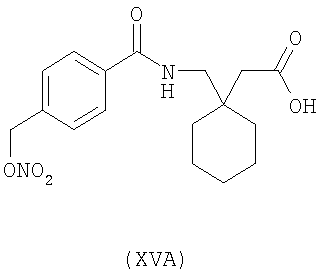 ,
,
1-[3-(нитрооксиметил)бензоиламинометил]-циклогексануксусная кислота (XVIA)
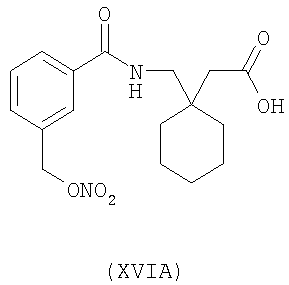 ,
,
1-[2-(нитрооксиметил)бензоиламинометил]-циклогексануксусная кислота (XVIIA)
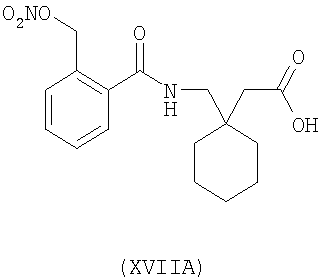 ,
,
1-(4-нитрооксибутаноиламинометил)-циклогексануксусная кислота (XVIIIA)
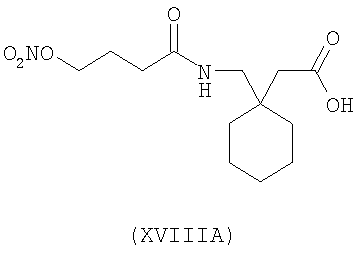 ,
,
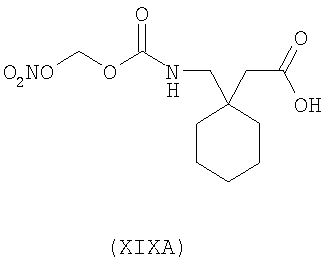
1-(нитрооксиметоксикарбониламинометил)-циклогексануксусная кислота (XIXA)
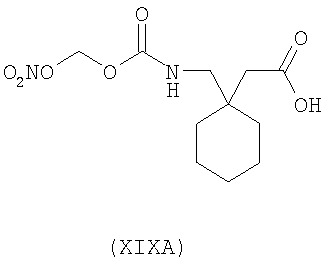 ,
,
1-{[4-(нитрооксиметил)бензоилокси]метоксикарбониламинометил}-циклогексануксусная кислота (ХХА)
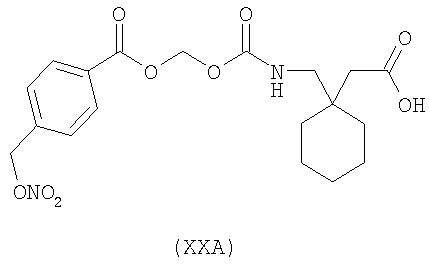 ,
,
1-{[3-(нитрооксиметил)бензоилокси]метоксикарбониламинометил}-циклогексануксусная кислота (XXIA)
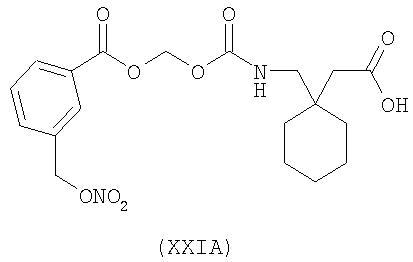 ,
,
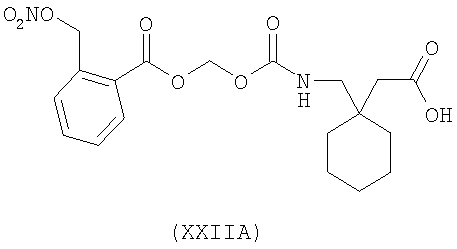
1-{[2-(нитрооксиметил)бензоилокси]метоксикарбониламинометил}-циклогексануксусная кислота (XXIIA)
 ,
,
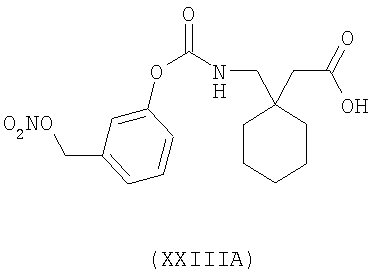
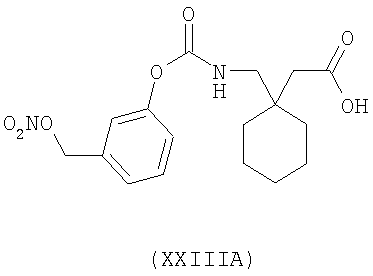
1-[3-(нитрооксиметил)феноксикарбониламинометил]-циклогексануксусная кислота (XXIIIA)
 ,
,
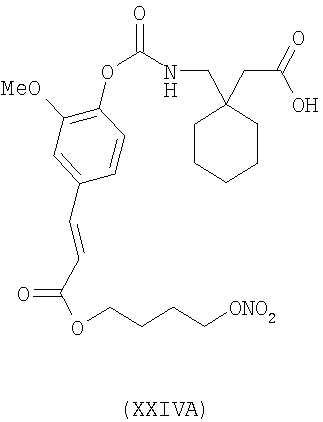
{2-метокси-4-[(1Е)-3-[4-(нитрооксибутокси)-3-окса-1-пропенилфенокси]-карбониламинометил}-циклогексануксусная кислота (XXIVA)
 ,
,
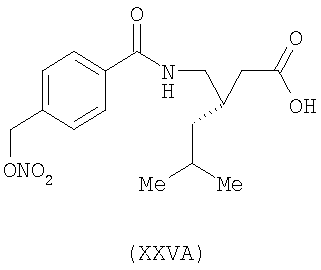
3-(S)-[4-(нитрооксиметил)бензоиламинометил]-5-метил-гексановая кислота (XXVA)
 ,
,
3-(S)-[3-(нитрооксиметил)бензоиламинометил]-5-метил-гексановая кислота (XXVIA)
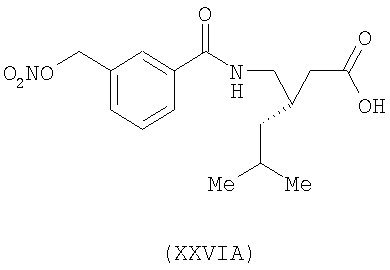 ,
,
3-(S)-[2-(нитрооксиметил)бензоиламинометил]-5-метил-гексановая кислота (XXVIIA)
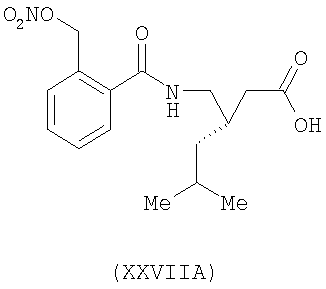 ,
,
3-(S)-[4-(нитрооксибутаноил)аминометил]-5-метил-гексановая кислота (XXVIIIA)
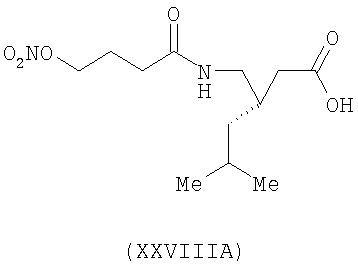 ,
,
3-(S)-[4-(нитрооксиметоксикарбонил)аминометил]-5-метил-гексановая кислота (XXIXA)
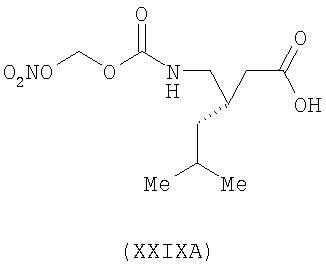 ,
,
3-(S)-{[2-(нитрооксиметил)бензоилокси]метоксикарбониламинометил}-5-метил-циклогексануксусная кислота (ХХХА)
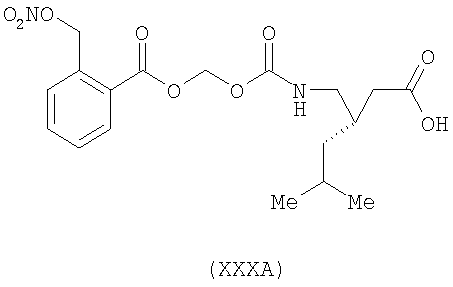 ,
,
3-(S)-{[3-(нитрооксиметил)бензоилокси]метоксикарбониламинометил}-5-метил-гексановая кислота (XXXIA)
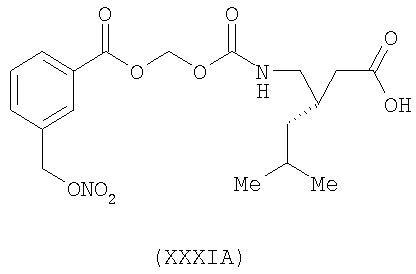 ,
,
3-(S)-{[4-(нитрооксиметил)бензоилокси]метоксикарбониламинометил}-5-метил-циклогексануксусная кислота (XXXIIA)
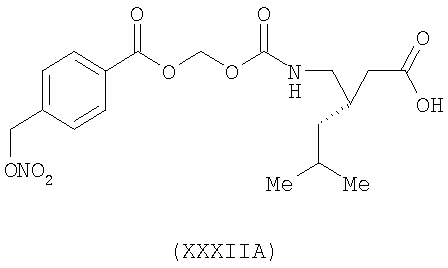 ,
,
3-(S)-[(3-нитрооксиметил)феноксикарбониламинометил]-5-метил-гексановая кислота (XXXIIIA)
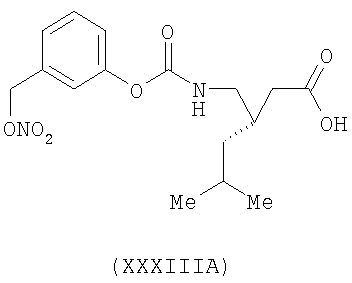 ,
,
3-(S)-{2-метокси-4-[(1Е)-3-[4-(нитрооксибутокси)-3-окса-1-пропенилфенокси]карбониламинометил}-5-метил-гексановая кислота (XXXIVA)
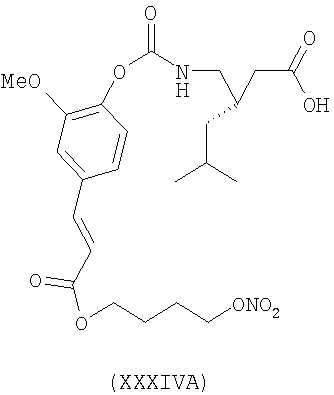 ,
,
1-[4-(нитрооксибутилоксикарбонил)аминометил]-циклогексануксусная кислота (XXXVA)
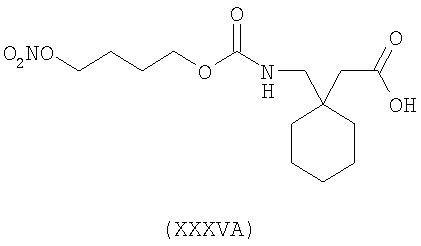
Соединения настоящего изобретения могут также использоваться в форме соответствующих солей с фармацевтически приемлемыми катионами, таких как соли щелочных металлов.
Соединения настоящего изобретения, имеющие в составе молекулы солеобразующий атом азота, например, когда в формуле (I) с0=1 и Y = часть формулы (III), могут быть превращены в соответствующие соли взаимодействием с эквимолярным количеством соответствующей органической или неорганической кислоты в органическом растворителе, таком как ацетонитрил, тетрагидрофуран. Примерами органических кислот являются щавелевая, винная, малеиновая, янтарная и лимонная кислота. Примерами неорганических кислот являются азотная, хлористоводородная, серная и фосфорная кислота. Предпочтительными являются нитратные соли.
Соединения настоящего изобретения обладают повышенной активностью для лечения хронической боли, в особенности невропатической боли, а также боли, связанной с центральной и периферической нервной системой. Более того, неожиданно было найдено, что соединения настоящего изобретения обладают повышенной эффективностью не только для уменьшения невропатической боли, но также обладают непредвиденной способностью задерживать развитие патологических состояний, вызывающих невропатическую боль. Например, при назначении лекарственных средств настоящего изобретения больным диабетом для лечения боли при диабетической невропатии было обнаружено, что указанные соединения способны не только ослаблять невропатию, но также уменьшают осложнения, вызываемые диабетом, например осложнения на кровеносные сосуды и/или почечную систему.
Соединения настоящего изобретения эффективны в основном при лечении невропатической боли, например боли при диабетической невропатии и постинфарктной боли.
Соединения настоящего изобретения также можно использовать в комбинации или в смеси с хорошо известными донорами NO. Такие соединения содержат в составе молекулы, например, одну или более ONO2 или ONO групп.
Доноры NO, которые могут использоваться вместе с соединениями настоящего изобретения, будут указаны в приводимых ниже тестах in vitro. Эти тесты относятся к генерированию окиси азота донорами NO в присутствии эндотелиальных клеток (метод а) или тромбоцитов (метод в), например, нитроглицерином, никорандилом, нитропруссидным соединением и т.д.
а) Эндотелиальные клетки
Клетки пупочной вены человека, высеенные на планшет плотностью 103 клеток/на ячейку, инкубируют со скалярными концентрациями донора NO (1-100 мкг/мл) 5 минут. Затем для определения способности генерировать NO анализируют инкубационную среду (физиологический раствор, например Tyrode) следующими методами:
1) определение оксида азота с помощью хемилюминесценции,
2) определение цГМФ (циклический гуанизин монофосфат №2715 из указанного выше Merck Index).
Для осуществления хемилюминесцентного анализа в реакционную камеру хемилюминесцентного анализатора, содержащую ледяную уксусную кислоту и иодид калия, впрыскивают аликвоту в 100 мкл. Присутствующие в инкубационной среде нитриты/нитраты в этих условиях превращаются в NO, который обнаруживается благодаря его реакции с озоном с последующей световой генерацией. Обычно в устройствах для измерения хемилюминесценции люминесценция прямо пропорциональна генерированному уровню NO и измеряется соответствующим элементом фотоэлектронного умножителя хемилюминесцентного анализатора. Фотоэлектронный умножитель превращает падающий свет в электрическое напряжение, которое затем количественно регистрируется. По калибровочной кривой, построенной со скалярными концентрациями нитрита, можно количественно определить концентрацию генерированного NO.
Для определения цГМФ часть инкубационной среды (100 мкл) 20 секунд центрифугируют при 100 об/мин. Отделяют супернатант и остаток обрабатывают ледяным фосфатным буфером (рН 7,4). Полученные уровни цГМФ определяют иммуноферментным анализом с помощью специфических реагентов. Из этих экспериментов следует, что в указанных условиях эксперимента инкубация одного из тестируемых доноров NO вызывает значительное увеличение цГМФ по сравнению с величинами, полученными в отсутствие NO-донора. Например, после инкубации со 100 мкл нитропруссида натрия регистрируют примерно 20-кратное увеличение по сравнению с соответствующей величиной, полученной при инкубации одного носителя без донора NO.
b) Тромбоциты
Использовали промытые тромбоциты человека, полученные, как описано Radomski и др. (Br.J.Pharmacol., 92, 639-1987). Аликвоты по 0,4 мл инкубировали 5 мин со скалярными концентрациями донора NO (1-100 мкг/мл). Затем для определения способности генерировать NO анализируют инкубационную среду (например, Tyrode) путем определения оксида азота хемилюминесцентным методом и определения цГМФ с использованием описанной выше процедуры, проводимой для исследования на эндотелиальных клетках. Что касается хемилюминесцентного анализа, то в этом случае на основании калибровочной кривой, построенной со скалярными концентрациями нитрита, можно количественно определить концентрацию генерируемого NO. Например, после инкубации 100 мкл никорандила генерируется 35 мкл NO.
Проведенное в тех же условиях определение цГМФ показало, что инкубация с одним из различных тестируемых доноров NO приводит к значительному увеличению цГМФ по сравнением с величинами, полученными в отсутствие донора NO. Например, после инкубации со 100 мкл нитропруссида натрия регистрируют примерно 30-кратное увеличение по сравнению с соответствующей величиной, полученной инкубацией одного носителя без донора NO.
Предпочтительными донорами NO являются те, которые содержат внутри молекулы радикалы следующих групп: аспирин, салициловая кислота, ибупрофен, парацетамол, напроксен, диклофенак, флурбипрофен. Эти предпочтительные соединения могут быть синтезированы, как описано в международных патентных заявках WO 95/20641, WO 97/16405, WO 95/09831, WO 01/12584.
Соединения настоящего изобретения могут быть получены в соответствии с описываемыми ниже синтетическими методами.
В случае, если молекула лекарственного средства содержит несколько реакционно-способных групп, таких как СООН и/или НХ, где Х представляет собой О, S или NH, они могут быть предварительно перед реакцией защищены известными из литературы методами, например, описанными Th.W.Green в «Protective Group In Organic Synthesis», Harvard University Press, 1980. Однако для получения соединений настоящего изобретения защита этих групп не является строго необходимой.
Для приготовления соединений настоящего изобретения, когда k0=0, аминная функция болеутоляющего средства реагирует с реакционно-способным соединением линкерного С предшественника, когда b0=0, или линкерного В предшественника, когда b0=1.
Когда в формуле (I) b0=0, болеутоляющее средство подвергают взаимодействию в основном с одним из следующих соединений:
1. Если k0=0 и связующей с болеутоляющим средством функцией является амидная функция, реагирующее с лекарственным средством соединение получают следующим образом.
Исходными соединениями являются ацилгалогениды формулы Hal-Y1-CO-Hal, где Y1 представляет собой Y, как указано выше, но без кислородного атома связывающего NO2 и Hal=CI, Br, I. Эти соединения, когда они не имеются в продаже, могут быть получены в соответствии с хорошо известными в данной области способами, например, из соответствующих кислот и тионил или оксалил хлорида, РIII или РIV галогенидов в инертных в условиях реакции растворителях, таких как толуол, хлороформ, ДФА и т.д.
Имеющий указанную выше формулу ацилгалогенид взаимодействует с карбоксильной группой конденсирующего агента, такого как N-гидроксисукцинимид (SIMD-N-OH), в соответствии с известными в данной области техники методами, например, в галогенированных растворителях в присутствии основания при комнатной температуре с получением эфира N-гидроксисукцинимида, как это указано в следующей схеме реакции:
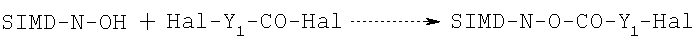
1a. Эфир гидроксисукцинимида взаимодействует с аминной функцией болеутоляющего средства при комнатной температуре в спиртовых и/или хлорированных растворителях в присутствии органического или неорганического основания в соответствии со следующей схемой:

1-1. Или же вместо указанного выше ацилгалогенида можно использовать оксикислоты формулы OH-Y1-COOH, где Y1, как указано выше, которые реагируют с N-гидроксисукцинимидом в присутствии агента, активирующего карбоксильную группу, такого как DCC, в галогенированных растворителях при комнатной температуре в соответствии со следующей схемой:

1-1.а Полученное в 1-1 соединение реагирует с аминной функцией болеутоляющего средства в указанных в 1а условиях в соответствии со следующей схемой:

1b. Когда в формуле (I) k0=1 с K=СО, связывающей с болеутоляющим средством функцией является карбаминовая функция. Лекарственное средство RNR1cH реагирует с галоидформиатом формулы Hal-Y1-OCO-Hal, где Y1, как указано выше.
Используемые галоидформиаты в основном имеются в продаже или же могут быть получены хорошо известными в данной области техники методами. Реакцию галоидформиата с лекарственным средством проводят в смеси растворителей при комнатной температуре и в присутствии основания, например, в воде и диоксане или метиленхлориде и диметилформамиде (ДФА). Схема реакции следующая:

1с. Приготовление нитрооксипроизводных из полученных указанными выше способами амидов и карбаматов (b0=0).
Когда полученные по описанной выше реакции соединения имеют формулу 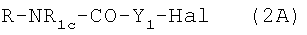 или
или 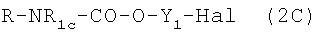 , соответствующие нитрооксипроизводные получают реакцией (2A) или (2С) с AgNO3 в органическом растворителе, таком как ацетонитрил, тетрагидрофуран, при температуре от 20 до 100°С в соответствии со следующей схемой реакции:
, соответствующие нитрооксипроизводные получают реакцией (2A) или (2С) с AgNO3 в органическом растворителе, таком как ацетонитрил, тетрагидрофуран, при температуре от 20 до 100°С в соответствии со следующей схемой реакции:
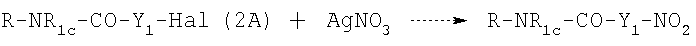
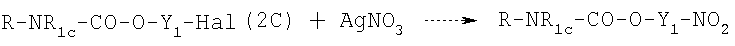
Когда полученные по описанным выше реакциям соединения имеют формулу 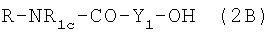 , гидроксильную группу галоидируют, например PBr3, PCl5, SOCl2, PPh3+I2 при комнатной температуре, затем подвергают взаимодействию с AgNO3 в органическом растворителе, таком как ацетонитрил, тетрагидрофуран, в указанных выше условиях. Получают нитрооксипроизводные, имеющие формулу R-NR1c-CO-Y1-NO2.
, гидроксильную группу галоидируют, например PBr3, PCl5, SOCl2, PPh3+I2 при комнатной температуре, затем подвергают взаимодействию с AgNO3 в органическом растворителе, таком как ацетонитрил, тетрагидрофуран, в указанных выше условиях. Получают нитрооксипроизводные, имеющие формулу R-NR1c-CO-Y1-NO2.
1d. Когда в формуле (I) b0=0, k0=1 и, например, K=СО, осуществляют следующие стадии. Аминную функцию лекарственного средства подвергают взаимодействию с коммерчески доступным хлорметил хлорформиатом ClC(O)ОСН2Cl. Полученное таким образом соединение R-NR1c-(CO)-OCH2Cl подвергают взаимодействию с HO-Y1-COOH в основной среде, как указано в 1a, c получением соединения формулы R-NR1c-K-(CO)-Y1-OH, которое затем обрабатывают, как указано выше в разделе 1с, получая при этом соответствующее нитрооксипроизводное.
2. Когда в формуле (I) b0=с0=1, синтез соответствующих нитрооксипроизводных включает три стадии. На первой стадии получают амиды (в формуле (I) k0=0), имеющие заместители, содержащие Hal группы (Hal=Cl, Br, I), или карбаматы (в формуле (I) k0=1), имеющие заместители, содержащие Hal группы, как указано ниже.
2а. Для получения галоидзамещенных амидов аминная функция лекарственного средства реагирует с эфиром N-гидроксисукцинимида, полученным из ацилгалогенида формулы P-X2-COHal, где:
- Х2 и Hal, как указано выше,
- Р=НХ, где Х, как указано выше, или представляет собой карбоксильную группу, защищенную, например, соответствующим трет-бутиловым эфиром,
и N-гидроксисукцинимида (SIMD-N-OH) в соответствии с известными в данной области техники методами, например, при комнатной температуре в галогенированных растворителях, в присутствии основания, с получением соединения формулы R-NR1c-CO-X2-P, которое, когда Р=НХ, подвергают взаимодействию с соединением формулы Hal-Y1-CO-Hal, где Hal и Y1, как указано выше. Схема реакции приведена ниже:
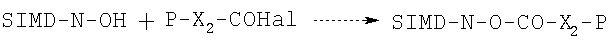
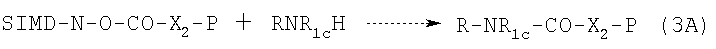

Когда в формуле (3А) Р = указанной выше эфирной группе, карбоксильная группа может быть восстановлена известными методами, например реакцией с безводным HCl в этилацетате или диоксане, если исходный эфир представляет собой трет-бутиловый эфир. Полученная таким образом кислота реагирует с галоидзамещенным спиртом формулы Hal-Y1-OH. Галоидзамещенный спирт имеется в продаже.
2а.1 Или же лекарственное средство RNR1cH реагирует с эфиром N-гидроксисукцинимида, полученным из кислоты формулы P-X2-COOH, где Р и X2, как указано выше, и N-гидроксисукцинимида (SIMD-N-OH), в присутствии дициклогексилкарбодиимида или другого конденсирующего агента в соответствии с хорошо известными в данной области методами, например при комнатной температуре в галоидзамещенных растворителях с получением соединения формулы 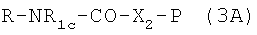 в соответствии со следующей схемой:
в соответствии со следующей схемой:
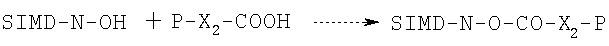
Затем соединение формулы (3А) обрабатывают, как указано в разделе 2а, получая при этом (3А′).
2b. Получение галогензамещенных карбаматов.
Из соединения 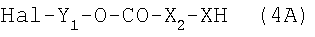 и трифосгена в присутствии органического основания получают галоидформиат формулы Hal-Y1-O-CO-X2-XCO-Hal в соответствии со схемой, указанной в 1b. Соединение (4A) получают реакцией спирта формулы Hal-Y1-OH с HX-X2-COOH. Полученный таким образом галоидформиат подвергают взаимодействию с аминной функцией лекарственного средства, следуя хорошо известным методам, например в ДФА и/или метиленхлориде в присутствии основания при комнатной температуре, как указано в следующей схеме:
и трифосгена в присутствии органического основания получают галоидформиат формулы Hal-Y1-O-CO-X2-XCO-Hal в соответствии со схемой, указанной в 1b. Соединение (4A) получают реакцией спирта формулы Hal-Y1-OH с HX-X2-COOH. Полученный таким образом галоидформиат подвергают взаимодействию с аминной функцией лекарственного средства, следуя хорошо известным методам, например в ДФА и/или метиленхлориде в присутствии основания при комнатной температуре, как указано в следующей схеме:

2с. Получение нитрооксипроизводных из амидов и карбаматов, полученных в разделе 2а или 2b.
Соединения (3А′) или (3В) посредством стоящего на конце молекулы галоидного атома взаимодействуют в органическом растворителе, таком как ацетонитрил, тетрагидрофуран с AgNO3 с образованием соответствующего нитрооксипроизводного.
Заявитель неожиданно обнаружил, что соединения настоящего изобретения проявляют более высокую активность при лечении хронической боли, чем соответствующие предшественники.
Когда соединения настоящего изобретения содержат один или более хиральных центров, они могут использоваться в форме рацемата, в виде смеси диастереомеров или энантиомеров, чистых энантиомеров или диастереомеров. Если соединения обладают геометрической асимметрией, они могут использоваться в цис или транс форме.
Соединения настоящего изобретения можно формулировать в различные фармацевтические композиции для орального, парентерального и местного назначения в соответствии с хорошо известными в данной области методами с обычными эксципиентами: например, как это описано в "Remington′s Pharmaceutical Sciences 15″ Ed".
Количество активного ингредиента в этих композициях равно или меньше, чем максимально предполагаемое количество для предшественника. Благодаря улучшенной толерантности можно использовать повышенные дозы. Назначаемые дневные дозы равны или ниже доз предшественника. Указанные дневные дозы можно найти, например, в "Physician′s Desk Reference".
Следующие примеры иллюстрируют изобретение и не ограничивают его объема.
Примеры
Пример 1
Синтез 1-[4-(нитрооксиметил)бензоиламинометил]-циклогексануксусной кислоты (формула XVA)
A) Синтез N-гидроксисукцинимидил-4-(хлорметил)бензоата
К раствору N-гидроксисукцинимида (1,375 г, 11,94 ммол) в метиленхлориде (30 мл) добавляют триэтиламин (1,66 мл, 11,94 ммол). К полученному таким образом раствору, охлаждаемому на водно-ледяной бане, медленно добавляют раствор 4-(хлорметил)бензоилхлорида (2,26 г, 11,94 ммол) в метиленхлориде (20 мл). После окончания добавления смесь оставляют стоять на ночь при комнатной температуре. Затем смесь сушат в вакууме, получая 4,84 г белого твердого вещества (смесь желаемого соединения и триэтиламмоний хлорида с количественным выходом), которое используют в следующей реакции без дополнительной очистки.
B) Синтез 1-[4-(хлорметил)бензоиламинометил]циклогексануксусной кислоты
К суспензии 1-(аминометил)циклогексануксусной кислоты (габапентин, 2,25 г, 13,13 ммол) в абсолютном этаноле (100 мл) добавляют триэтиламин (3,66 мл, 26,27 ммол) с получением прозрачного раствора. К полученному таким образом и охлажденному на водно-ледяной бане раствору прикапывают раствор эквимолярной смеси триэтиламмоний хлорида и N-гидроксисукцинимидил-4-(хлорметил)бензоата (4,84 г, 11,94 ммол) в метилен хлориде (100 мл), полученной на стадии А. После 4 часов перемешивания при комнатной температуре к смеси добавляют этилацетат (100 мл) и экстрагируют раствор 4% водным раствором хлористоводородной кислоты. Органическую фазу сушат под вакуумом, получая 3.85 г требуемого продукта в виде твердого белого вещества.
С) Синтез 1-[4-(нитрооксиметил)бензоиламинометил]-циклогексануксусной кислоты
К суспензии 1-[4-(хлорметил)бензоиламинометил]циклогексануксусной кислоты (4,01 г, 12,39 ммол) в ацетонитриле (250 мл) добавляют нитрат серебра (2,11 г, 12,39 ммол). Смесь перемешивают под вакуумом на свету при 60°С, добавляя пять аликвот нитрата серебра в течение 20 часов. Смесь нагревают 24 часа, добавляя другие пять эквивалентов нитрата серебра дополнительно к тем, что уже добавлены. Полученную таким образом соль отфильтровывают, к смеси добавляют этилацетат (200 мл) и 2% раствор хлористоводородной кислоты. Выпавшие в осадок нерастворимые соли отфильтровывают и органическую фазу сушат под вакуумом. Полученный таким образом сырой материал очищают с помощью хроматографии на силикагеле, используя н-гексан/этилацетат 6/4 (об./об.) в качестве элюента. Полученный продукт кристаллизуют из этилацетата/н-гексана, получая белое твердое вещество с Т.пл. 127-128°С.
1H-NMR (CDCl3) ppm: 7.86 (2H, d); 7.50 (2H, d); 7.06 (1H, t); 5.49 (2H, s); 3.54 (2H, d); 2.43 (2H, s); 1.53 (10Н, m).
Пример 2
Синтез 1-(нитрооксиметоксикарбониламинометил)-циклогексануксусной кислоты (формула XIXA)
А) Синтез 1-(хлорметоксикарбониламинометил)-циклогексануксусной кислоты
К раствору 1-(аминометил)циклогексануксусной кислоты (габапентин, 2,00 г, 11,68 ммол) в смеси воды (30 мл) и диоксана (20 мл) добавляют диизопропилэтиламин (4,06 мл, 23,36 ммол). К полученному таким образом и охлажденному на водно-ледяной бане раствору медленно прикапывают растворенный в диоксане (100 мл) дихлорметилхлорформиат (2,25 мл, 14,02 ммол). По окончании добавления смесь оставляют стоять 3 часа при комнатной температуре. Затем смесь вливают в 4% раствор хлористоводородной кислоты, понижая в конце величину рН до примерно 2. Добавляют этилацетат и органическую фазу сушат в вакууме, получая 2,87 г прозрачного, желтого масла, которое используют в следующей реакции без дополнительной очистки.
В) Синтез 1-(нитрооксиметоксикарбониламинометил)-циклогексануксусной кислоты
К раствору 1-(хлорметоксикарбониламинометил)-циклогексануксусной кислоты (2,87 г, 10,92 ммол) в ацетонитриле (25 мл) добавляют нитрат серебра (3,71 г, 21,84 ммол). Смесь перемешивают под вакуумом на свету при 40°С 3 часа. Отфильтровывают выпавшую в осадок соль и добавляют к смеси этилацетат (30 мл) и 2% раствор хлористоводородной кислоты. Образовавшиеся соли отделяют фильтрованием, а органическую фазу сушат под вакуумом. Полученный таким образом маслянистый продукт очищают с помощью хроматографии на силикагеле, используя н-гексан/этилацетат 6/4 (об./об.) в качестве элюента и получают 2,68 г бесцветного масла.
1H-NMR (CDCl3) ppm: 6.03 (2H, s); 5.51 (1H, t); 3.30 (2H d), 2.36 (2H, s); 1.47 (10Н, m).
Пример 3
Синтез 1-[3-(нитрооксиметил)феноксикарбониламинометил]-циклогексануксусной кислоты (XXIIIA)
А) Синтез 1-[3-(бромометил)феноксикарбониламинометил]-циклогексануксусной кислоты
К суспензии 3-бромметилфенола (0,50 г, 2,67 ммол) в метиленхлориде (8 мл) добавляют охлажденный бис(трихлорметил)карбонат (трифосген, 0,368 г, 1,24 ммол), растворенный в метилхлориде (2 мл) и диизопропиламине (0,466 мл, 2,67 ммол). Полученный раствор перемешивают при комнатной температуре в течение ночи и затем греют с обратным холодильником 2 часа. Затем этот охлажденный раствор добавляют по каплям к суспензии 1-(аминометил)циклогексануксусной кислоты (габапентин, 0,911 г, 5,35 ммол) и диизопропилэтиламина (0,932 мл, 5,35 ммол) в безводном диметилформамиде (4 мл). После 3 часов перемешивания к смеси добавляют этилацетат и промывают 4% раствором хлористоводородной кислоты. Органическую фазу сушат под вакуумом и полученный таким образом сырой продукт очищают с помощью хроматографии на силикагеле, используя н-гексан/этилацетат 1/1 (об./об.) в качестве элюента. Получают желаемый продукт в виде масла (0,100 г), который используют далее без дополнительной очистки.
В) Синтез 1-[3-(нитрооксиметил)феноксикарбониламинометил]-циклогексануксусной кислоты
К раствору 1-[3-(бромометил)феноксикарбониламинометил]-циклогексануксусной кислоты (0,100 г, 0,26 ммол) в ацетонитриле (2 мл) добавляют нитрат серебра (0,100 г, 0,59 ммол). Смесь перемешивают в течение ночи при комнатной температуре в атмосфере азота на свету. Отфильтровывают образовавшуюся соль и добавляют к смеси этилацетат (5 мл) и 2% раствор хлористоводородной кислоты. Нерастворимые соли отфильтровывают, органическую фазу очищают с помощью хроматографии на силикагеле, используя метиленхлорид/метанол 97/3 (об./об.) в качестве элюента, и получают 0,080 г продукта в виде масла.
1H-NMR (CDCl3) ppm: 7.38 (1Н, t); 7.22 (3Н, m); 5.68 (1Н, t); 5.43 (2H, s); 3.34 (2H, d); 2.41 (2H, s); 1.49 (10Н, m).
Пример 4
Синтез 1-[4-(нитрооксибутилоксикарбонил)аминометил]-циклогексануксусной кислоты (XXXVA)
A) Синтез 1-[4-(хлорбутилоксикарбонил)аминометил]циклогексануксусной кислоты
К раствору 1-(аминометил)циклогексануксусной кислоты (1,95 г, 11,4 ммол) в смеси диоксан/вода (1:1, 40 мл) добавляют N,N-диизопропилэтиламин (4,00 мл, 23,0 ммол) и раствор охлаждают при 0°С. Затем медленно добавляют 1-хлорбутилхлорформиат (1,30 мл, 9,50 ммол), дают реакционной массе достичь комнатной температуры и оставляют при перемешивании на 5 часов. Смесь разбавляют метиленхлоридом и промывают 4% водной хлористоводородной кислотой, обезвоживают и сушат под вакуумом, получая 2,87 г бесцветного масла, которое используют в следующей реакции без дополнительной очистки.
B) 1-[4-(иодбутилоксикарбонил)аминометил]циклогексануксусной кислоты
К раствору 1-[4-хлорбутилоксикарбонил)аминометил]циклогексануксусной кислоты (1,68 г, 5,70 ммол) в ацетонитриле (20 мл) добавляют иодид натрия (8,48 г, 57,0 ммол) и реакционную смесь нагревают с обратным холодильником 5 часов при перемешивании. Затем под вакуумом удаляют растворитель и остаток обрабатывают метиленхлоридом. Органическую фазу промывают водой, обезвоживают и сушат под вакуумом, получая 2,12 г маслянистого продукта, который используют на следующей стадии без очистки.
C) Синтез 1-[4-(нитрооксибутилоксикарбонил)аминометил]-циклогексануксусной кислоты
К раствору 1-[4-(иодбутилоксикарбонил)аминометил]-циклогексануксусной кислоты (2,12 г, 5,30 ммол) в ацетонитриле (25 мл) добавляют нитрат серебра (2,42 г, 14,2 ммол). Смесь перемешивают 5 часов при 40°С в атмосфере азота на свету, затем фильтруют на целите и концентрируют. Остаток обрабатывают метиленхлоридом и экстрагируют 4% раствором хлористоводородной кислоты. Отфильтровывают образовавшиеся соли и водную фазу экстрагируют метиленхлоридом. Органические фазы промывают насыщенным раствором хлористого натрия, обезвоживают и сушат под вакуумом. Маслянистый остаток растворяют в этиловом эфире, фильтруют на целите и сушат под вакуумом, получая 1,64 г продукта в виде масла.
1H-NMR (CDCl3) ppm: 5.65 (1Н, m); 4.49 (2Н, t); 4.12 (2H, t); 3.23 (2H, d); 2.34 (2H, s); 1.9-1.7 (4H, m); 1.6-1.3 (10H, m).
Пример Fl
Определение анальгетической активности соединений настоящего изобретения в тесте корчей, вызванных введением уксусной кислоты (Vinegar et asl., 1979).
Девять групп самцов мышей Swiss (20-25 г, Charles River), по 10 животных в каждой группе, по 10 животных в группе, получали путем орального введения через желудочный зонд (принудительное питание) габапентин в количестве от 1 до 10 мг/кг или соединение настоящего изобретения (XVA, пример 1), в дальнейшем NO-габапентин, в количестве от 1 до 10 мг/кг, растворенные в физиологическом растворе. Через час после введения растворов соединений животным путем внутрибрюшинной инъекции вводили раствор ледяной уксусной кислоты (0,5 мл, 0,6%). В течение последующих после введения уксусной кислоты 15 минут у каждого животного подсчитывают число абдоминальных сокращений. Исследование проводили вслепую.
Результаты, приведенные в таблице 1, представляют собой число общих сокращений в течение времени наблюдения (15 мин). Результаты показывают, что NO-габапентин более активен, чем лекарственный предшественник, при ингибировании числа абдоминальных сокращений.
Пример F2
Определение анальгетической активности соединений настоящего изобретения в тесте вылизывания лапы.
Три группы самцов мышей Swiss (20-25 г, Charles River), по 10 животных в каждой, получали путем орального введения, как указано в примере F1, габапентин в количестве 3 мг/кг (17,5 мкм/кг) или соединение формулы (XVA, пример 1), в дальнейшем NO-габапентин, в количестве 3 мг/кг (8,5 мкм/кг), растворенные в физиологическом растворе. Контрольная группа получала равное количество физиологического раствора. Через час после введения растворов соединений животным путем инъекции в лапу вводили формалин (10 мкл).
Инъекция формалина индуцирует двухфазовую реакцию. Во время первой фазы (фаза I, 0-15 минут) наблюдается острое воспаление; во время второй фазы (фаза II, 15-30 минут) происходит выделение химических медиаторов, проявляющееся в виде невропатической боли. В течение последующих после инъекции формалина 30 минут для каждого животного регистрировали время в секундах, в течение которого животное вылизывает свою лапу. Исследование проводили вслепую.
Результаты, приведенные в таблице 2, представляют полное время вылизывания животными лапы в секундах в течение первой и второй указанных выше фаз.
Результаты показывают, что NO-габапентин более активен, чем исходный лекарственный препарат при ингибировании вылизывания лапы на первой фазе, даже несмотря на то, что введенная молярная доза соответствовала 50% габапентина. По этой причине на второй фазе NO-габапентин менее эффективен.
Пример F3
Определение анальгетической активности соединений настоящего изобретения на моделях невропатической боли у животных.
Противоболевое действие соединения формулы (XVA, пример 1), в дальнейшем NO-габапентин, на модели невропатической боли, вызванной длительным констрикционном повреждением седалищного нерва у крысы. В качестве лекарственного препарата сравнения использовался исходный габапентин.
Одностороннюю периферическую мононевропатию вызывали в соответствии с методом, описанным Bennet GJ и Xie YK, Pain (33) 1988: 87-107. Для обработки использовали образцы популяций в диапазоне от 8 до 12 крыс (крысы SD, весом 250-300 г). Противоболевое действие лекарственных средств определяли измерением порогового значения голосового сигнала (VTPP), вызываемого сдавливанием лапы как с поврежденной, так и с противоположной стороны. Тестирование проводили на 14 день после поражения. Все соединения были протестированы на острый противоболевой эффект. Острый противоболевой эффект определяли в течение 60 минут, следующих после одноразовой предварительной внутрибрюшинной (i.p.) инъекции лекарственных средств перед тестированием.
Каждая группа крыс получала габапентин в дозе 30 мг/кг (175 мкм/кг), или эквимолярную дозу NO-габапентина (175 мкм/кг), или то же количество наполнителя (контрольная группа). Лекарственные средства растворяли в наполнителе, содержащем физиологический раствор: диметилсульфоксид (ДМСО): касторовое масло (68:8:24).
Результаты, представленные в таблице 3, показывают, что NO-габапентин более эффективен, чем габапентин.
Настоящее изобретение относится к нитрооксипроизводным или их солям общей формулы (I):
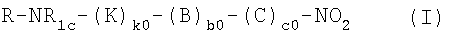
где с0 представляет 1; b0 представляет собой 0; k0 представляет собой 0 или 1; R представляет собой радикал болеутоляющего средства формулы (II)
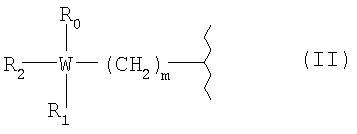
где W представляет атом углерода; m представляет целое число от 0 до 1; R0=H или -(СН2)n-СООН, где n представляет целое число от 0 до 2; R2 с R1 и с W вместе образуют насыщенное кольцо С6; R1c представляет собой Н; К представляет собой (СО); С представляет собой двухвалентный радикал, имеющий формулу -Tc-Y-,
где Тс=(СО) или О; или Y может быть группой алкиленокси -R′O-, в которой R′ представляет собой прямой или разветвленный С1-С20алкил, или группу формулы
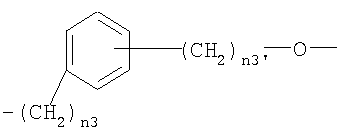
где n3 представляет целое число от 0 до 5 и n3′ представляет целое число от 1 до 3. Также описываются фармацевтическая композиция на основе нитроксипроизводных и их применение в качестве лекарственных средств для лечения хронической боли. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 4 н. и 4 з.п. ф-лы, 3 табл.
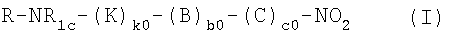
где с0 представляет 1;
b0 представляет собой 0;
k0 представляет собой 0 или 1;
R представляет собой радикал болеутоляющего средства формулы (II)
где W представляет атом углерода;
m представляет целое число от 0 до 1;
R0 - H или -(CH2)n-СООН,
где n представляет целое число от 0 до 2;
R2 с R1 и с W вместе образуют насыщенное кольцо С6;
R1c представляет собой Н;
K представляет собой (СО);
С представляет собой двухвалентный радикал, имеющий формулу -Тc-Y-,
где Тс - (СО) или О;
или Y может быть:
группой алкиленокси -R′O-, в которой R′ представляет собой прямой или разветвленный С1-С20алкил, или группу формулы
где n3 представляет целое число от 0 до 5 и n3′ представляет целое число от 1 до 3.
где когда в формуле (II) W=C, m=1 и R0=-(CH2)n-COORy, где n=1 и Ry=H; R2 и R1 с W, имеющим указанное выше значение, образуют циклогексановое кольцо; лекарственный предшественник R, имеющий формулу R-NH2, известен, как габапентин;
когда в формуле (II) W=C, m=0; если R0 имеет значение, как указано для габапентина с n=0; R1=H; R2 представляет собой 3,4-дигидроксибензильный радикал; лекарственный предшественник R, имеющий формулу R-NH2, известен, как 2-амино-3-(3,4-дигидроксифенилпропановая кислота (допа).
1-(4-(нитрооксиметил)бензоиламинометил]-циклогексануксусная кислота (XVA)
1-(нитрооксиметоксикарбониламинометил)-циклогексануксусная кислота (XIXA)
1-[3-(нитрооксиметил)феноксикарбониламинометил]циклогексануксусная кислота (XXIIIA)
| RU 215921 C2, 27.04.2001 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO 00/54773 A, 21.09.2000 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

