Настоящее изобретение относится к производным блокаторов рецепторов ангиотензина II (ARB). Более конкретно, настоящее изобретение относится к нитропроизводным ARB, содержащим их фармацевтическим композициям и их использованию для лечения сердечно-сосудистых заболеваний, заболеваний почек и хронических заболеваний печени, воспалительных процессов и метаболических синдромов.
Под блокаторами рецептора ангиотензина II подразумевают класс соединений, включающий, в качестве основных компонентов, лозартан, ЕХР3174, кандесартан, телмисартан, валсартан, эпросартан, ирбесартан и олмесартан.
ARB апробированы только для лечения гипертензии, при этом антигипертензивное действие обусловлено, главным образом, селективной блокадой AT1 рецепторов и последующим уменьшением прессорного эффекта ангиотензина II. Ангиотензин II стимулирует синтез и секрецию альдостерона и вызывает повышение кровяного давления из-за мощного направленного сосудосуживающего действия.
В настоящее время сообщалось, что блокаторы рецепторов ангиотензина II обладают такими побочными эффектами, как, например артериальная гипотензия, гиперкалиемия, миалгия, нарушение деятельности дыхательных путей, заболевания почек, боль в пояснице, заболевания желудочно-кишечного тракта, утомление и нейтропения (Martindale, 33 издание, стр.921).
Целью настоящего изобретения является обеспечение новых производных ARB, способных не только устранить или, по крайней мере, уменьшить побочные эффекты, характерные для их родоначального соединения, но также обладающих улучшенным фармакологическим действием. Неожиданно было обнаружено, что общий профиль нитропроизводных блокаторов рецепторов ангиотензина II значительно лучше, по сравнению с природными соединениями с точки зрения более широкого фармакологического действия и лучшей переносимости.
В частности, было обнаружено, что нитропроизводные блокаторов рецепторов ангиотензина II по настоящему изобретению демонстрируют сильное противовоспалительное, антитромботическое и антитромбоцитарное действие и, кроме того, могут быть использованы для лечения или профилактики сердечной недостаточности, инфаркта миокарда, ишемического инсульта, атеросклероза, внутриглазной и легочной гипертензии, артериальной гипертензии, диабетической нефропатии, заболеваний периферических сосудов, дисфункции и гипертрофии левого желудочка, фиброза печени, портальной гипертензии и метаболических синдромов.
Объектом настоящего изобретения являются нитропроизводные блокаторов рецепторов ангиотензина II общей формулы (I) и их фармацевтически приемлемые соли или стереоизомеры:
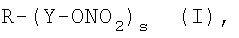
где
s равно 1 или 2;
R выбирают из следующих остатков блокаторов рецептора ангиотензина II формулы (II) или (III):
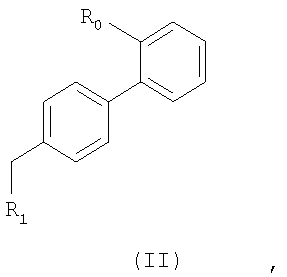
где
R0 представляет 
или -N0, где -N0 представляет собой способную к связыванию с Y группу, имеющую одно из следующих значений:
-СОО-, -О-, -CONH-, -ОСО-, -ОСОО- или
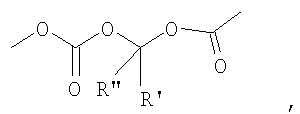
где R' и R'' одинаковые или разные и представляют собой Н или прямой или разветвленный C1-C4 алкил;
R1 выбирают из группы, состоящей из
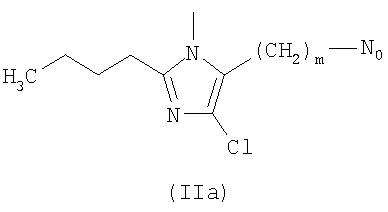
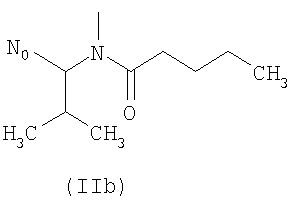
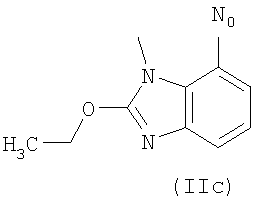
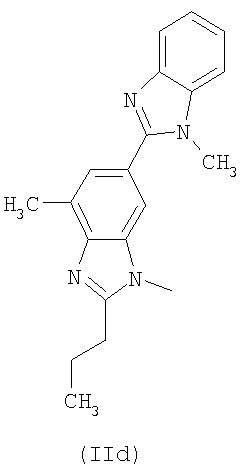
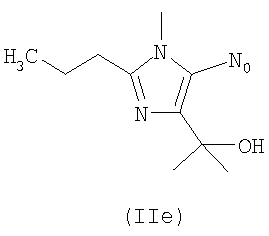
где m целое число, равное 0 или 1, и N0 как указано выше;
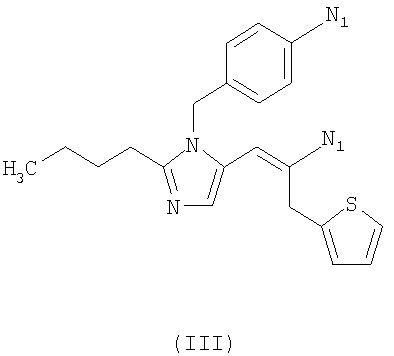
где N1 имеет значения, указанные для N0 или равно -СООН;
при условии, что по крайней мере одна из групп N1 равна -СОО- или -CONH-, то есть это группа, способная связываться с Y;
Y представляет собой двухвалентный радикал, имеющий следующие значения:
a)
- прямой или разветвленный C1-С20 алкилен, преимущественно С1-С10, необязательно замещенный одним или более заместителями, выбранными из группы, состоящей из атомов галогена, гидрокси, -ONO2 или Т0, где Т0 представляет -CO(O)(С1-С10алкил)-ONO2 или -O(С1-С10алкил)-ONO2;
- циклоалкилен с 5-7 атомами углерода в циклоалкиленовом кольце, необязательно замещенный боковой цепью Т, где Т представляет прямой или разветвленный алкил с 1-10 атомами углерода, преимущественно СН3;
b)
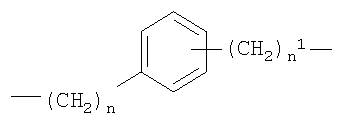
c)
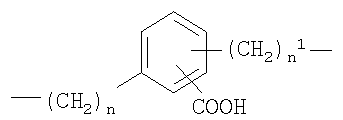
где n представляет целое число от 0 до 20, и n1 представляет целое число от 0 до 20;
d)

где
n1 как указано выше и n2 представляет целое число от 0 до 2;
X1=-ОСО- или -СОО- и R2 представляет собой Н или СН3;
е)

где
n1, n2, R2 и X1 как указано;
Y1 представляет собой -CH2-СН2- или -СН=СН-(СН2)n 2-;
f)
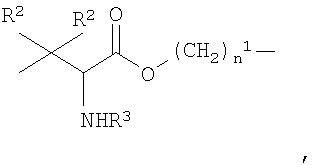
где
n1 и R2 как указано выше, R3 представляет собой Н или -СОСН3;
при условии, что когда Y выбирают их двухвалентных радикалов, указанных под b)-f), группа -ONO2 связана с группой -(СН2)n 1;
g)

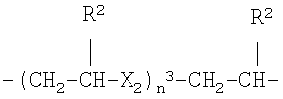
где Х2 представляет -О- или -S-, n3 представляет целое число от 1 до 6, преимущественно от 1 до 4, R2 как указано выше;
h)
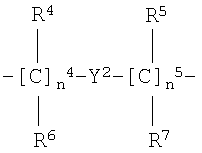
где
n4 целое число от 0 до 10;
n5 целое число от 0 до 10;
R4, R5, R6, R7 одинаковые или разные и представляют собой Н или прямой или разветвленный C1-C4 алкил, преимущественно R4, R5, R6, R7 представляют Н;
где группа -ONO- связана с
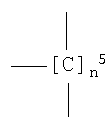
где n5 как указано выше;
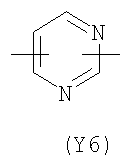
Y2 означает гетероциклическое насыщенное, ненасыщенное или ароматическое 5-6 членное кольцо, содержащее один или более гетероатомов, выбранных из азота, кислорода, серы, и выбрано из
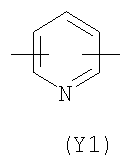 ,
, 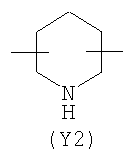 ,
,  ,
, 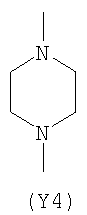 ,
, 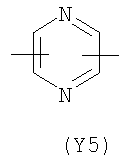 ,
,
 ,
, 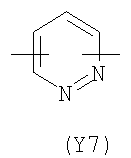 ,
, 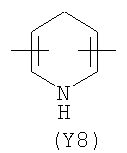 ,
, 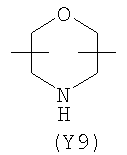 ,
, 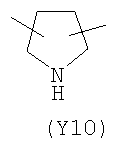 ,
,
 ,
, 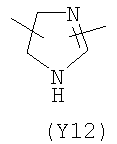 ,
, 
Используемый термин «С1-С20алкилен» означает прямую или разветвленную углеводородную цепь C1-C20, преимущественно содержащую от 1 до 10 атомов углерода, такую как метилен, этилен, пропилен, изопропилен, н-бутилен, пентилен, н-гексилен и тому подобное.
Используемый термин «C1-С10 алкил» означает прямые или разветвленные алкильные группы, содержащие от одного до десяти атомов углерода, включая метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил, гексил, октил и тому подобное.
Используемый термин «циклоалкилен» означает кольцо, имеющее от 5 до 7 атомов углерода, включая, но не ограничиваясь этим, циклопентилен, циклогексилен, необязательно замещенные боковыми цепями, такими как прямой или разветвленный (С1-С10)-алкил, преимущественно СН3.
Используемый термин «гетероциклил» означает насыщенное, ненасыщенное или ароматическое 5- или 6-членное кольцо, содержащее один или более гетероатомов, выбранных из азота, кислорода, серы, такое как например пиридин, пиразин, пиримидин, пирролидин, морфолин, имидазол и тому подобное.
Другим объектом настоящего изобретения является применение соединений формулы (I) в комбинации, по крайней мере, с соединением, используемым для лечения сердечно-сосудистых заболеваний, выбранным из группы, включающей ингибиторы АТФ, ингибиторы HMG-CoA редуктазы, бета-адренергические блокаторы, блокаторы кальциевых каналов, диуретики, антитромботические агенты, такой как аспирин, нитрозированные ингибиторы АТФ, нитрозированные ингибиторы HMG-CoA редуктазы, нитрозированные бета-адренергические блокаторы, нитрозированный аспирин и нитрозированные диуретики.
Подходящие ингибиторы АТФ, ингибиторы HMG-CoA редуктазы, бета-адренергические блокаторы, блокаторы кальциевых каналов, антитромботические агенты и диуретики описаны в литературе, например, The Merck Index (13th edition).
Подходящие нитрозированные соединения описаны в WO 98/21193, WO 97/16405 и WO 98/09948.
Указанные выше соединения могут назначаться одновременно или последовательно.
Настоящее изобретение также относиться к фармацевтическим наборам, включающим один или более контейнеров, заполненных одним или более соединениями и/или композициями настоящего изобретения и одним или более соединениями, используемыми для лечения указанных выше сердечно-сосудистых заболеваний.
Как указано выше, настоящее изобретение включает также фармацевтически приемлемые соли соединений формулы (I) и их стереомеры.
Примерами фармацевтически приемлемых солей являются любые соли с неорганическими основаниями, такими как гидроксиды натрия, калия, кальция и алюминия, или с органическими основаниями, такими как лизин, аргинин, триэтиламин, дибензиламин, пиперидин и другие приемлемые амины.
Соединения настоящего изобретения, содержащие в молекуле один солеобразующий атом азота, могут быть превращены в соответствующие соли реакцией с соответствующими органическими или неорганическими кислотами в органическом растворителе, таком как ацетонитрил, тетрагидрофуран.
Примерами органических кислот являются щавелевая, винная, малеиновая, янтарная, лимонная кислота. Примерами неорганических кислот являются азотная, хлористоводородная, серная, фосфорная кислота.
Предпочтительными солями являются соли с азотной кислотой.
Соединения настоящего изобретения, имеющие один или более ассиметричных атомов углерода, могут существовать как оптически чистые энантиомеры, чистые диастереоизомеры, смеси энантиомеров, смеси диастереоизомеров, энантиомерные рацемические смеси, рацематы или рацематные смеси. Настоящее изобретение также охватывает все возможные изомеры, стереоизомеры и их смеси соединений формулы (I).
Предпочтительными соединениями настоящего изобретения являются соединения формулы (I), где
s и R как указано выше;
Y представляет двухвалентный радикал, имеющий следующие значения:
a)
- прямой или разветвленный С1-С10 алкилен, необязательно замещенный Т0, где Т0 как указано выше;
b)

где n целое число, равное 0 или 1, и n1 целое число, равное 1; при условии, что группа -ONO2 связана с группой -(СН2)n 1;
c)
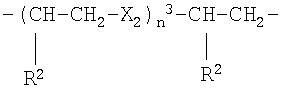
где Х2 представляет -О- или -S-, n3 целое число, равное 1, и R2 представляет Н;
В соответствии с настоящим изобретением предпочтительными соединениями являются следующие:

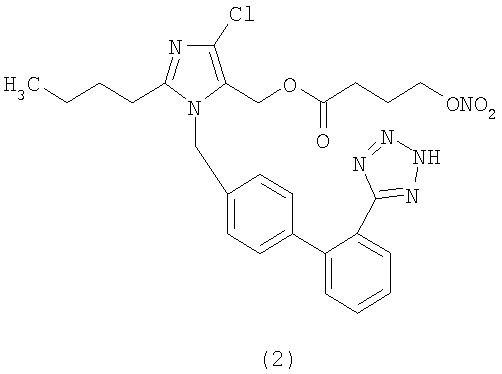
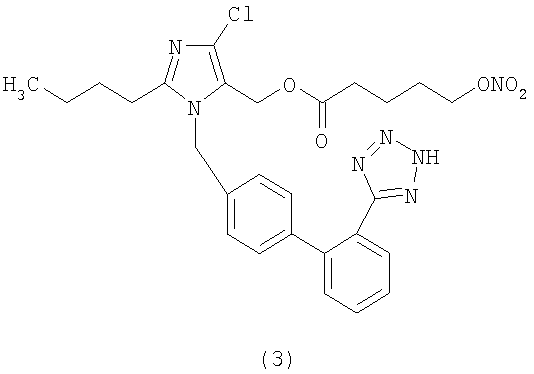
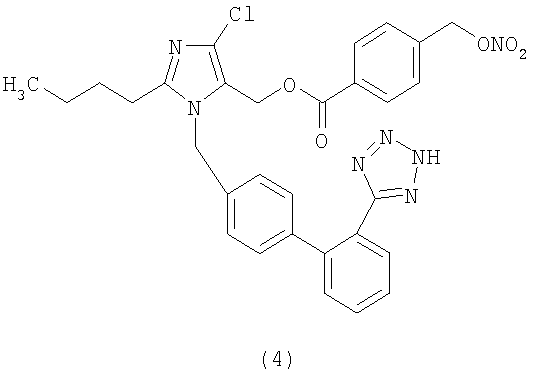
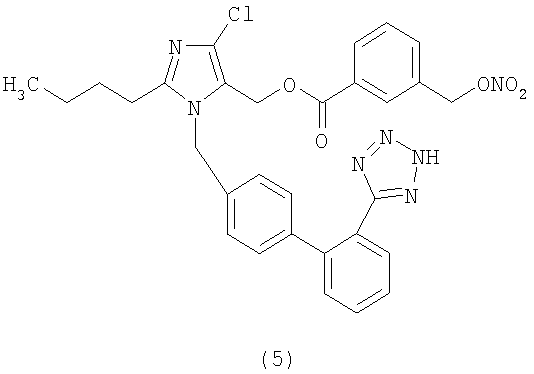
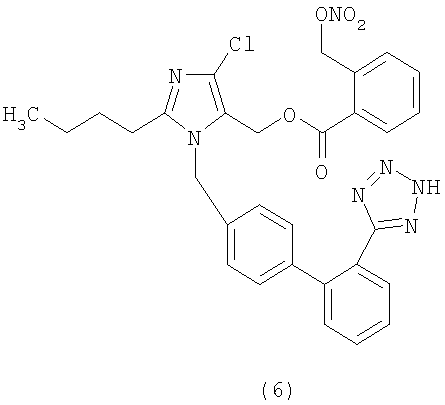
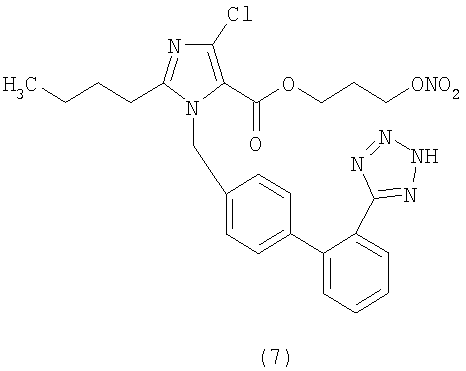
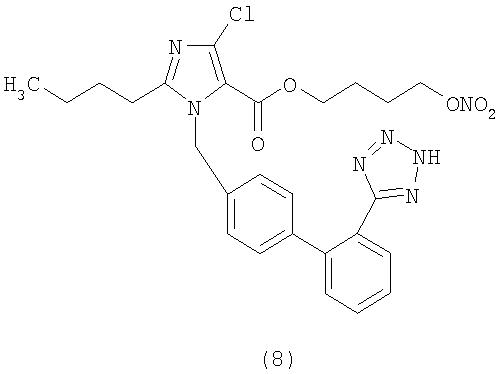

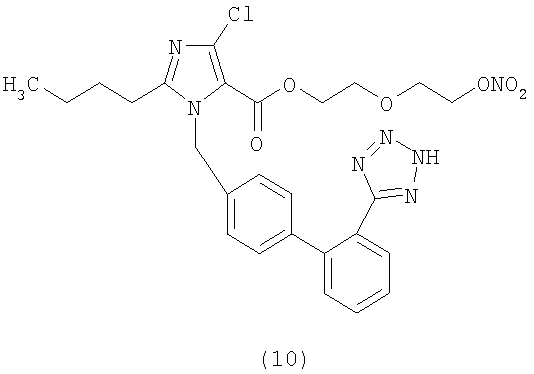
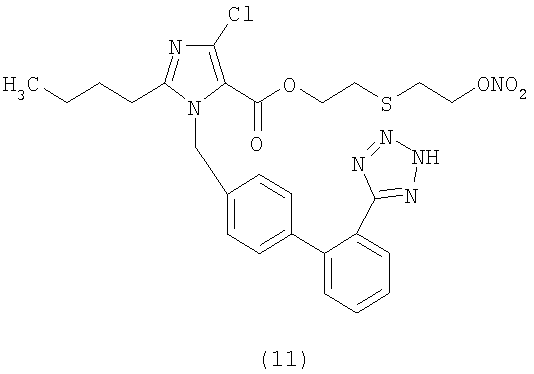


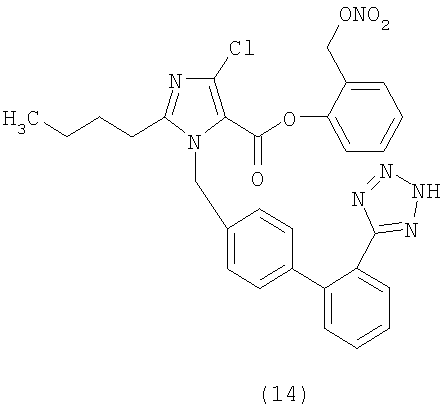
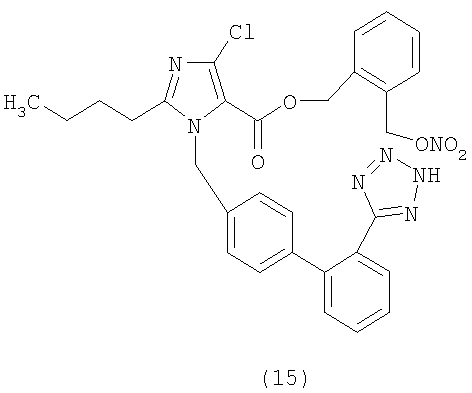
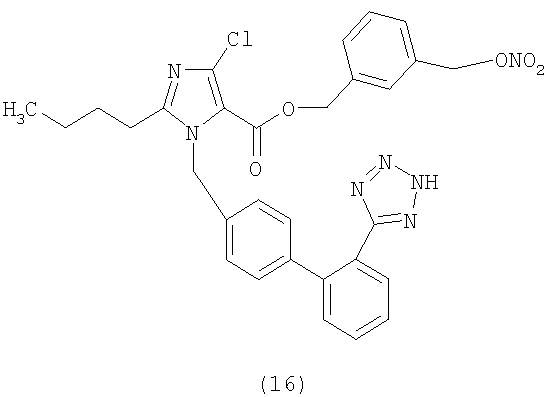
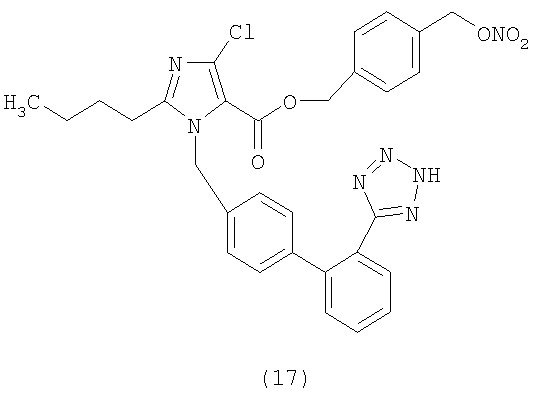
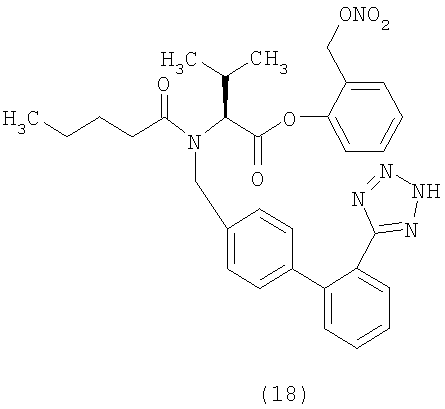
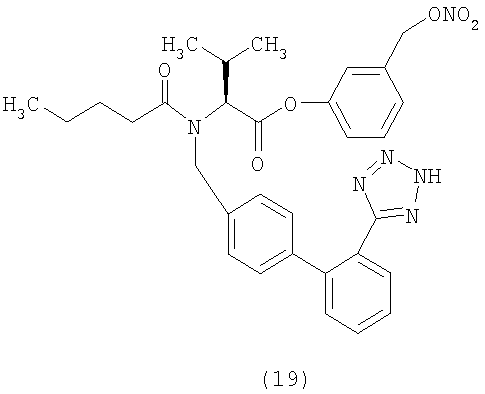
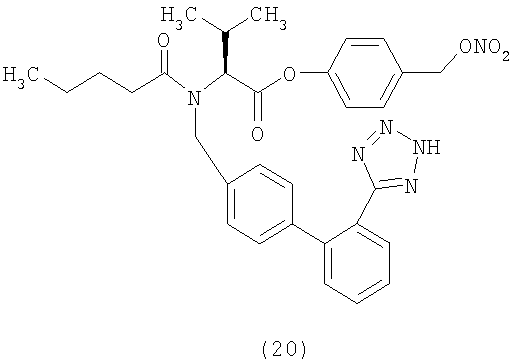

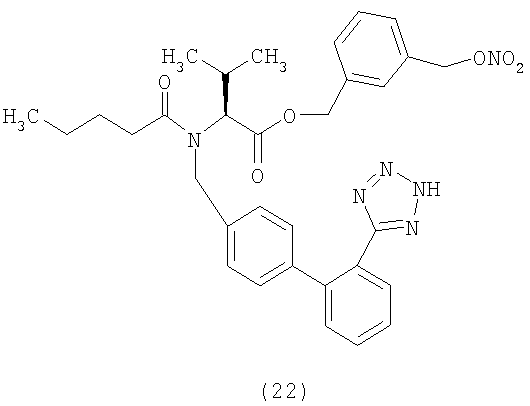
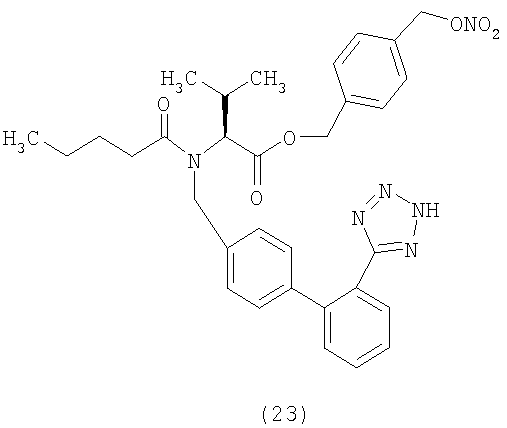
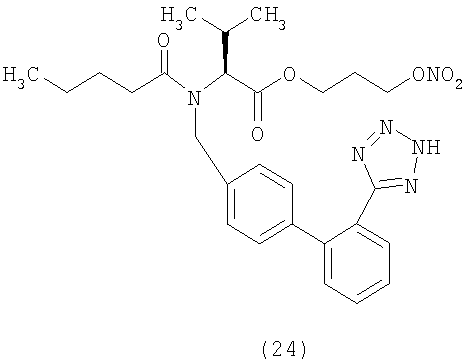
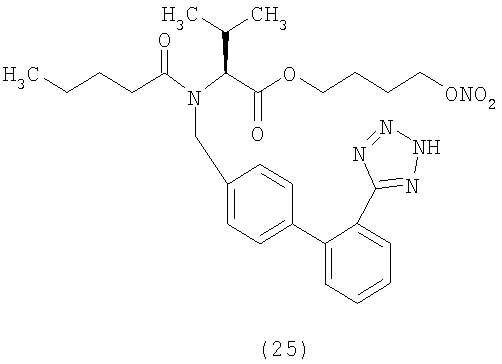

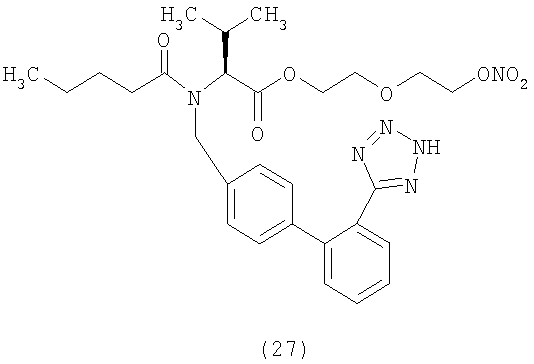
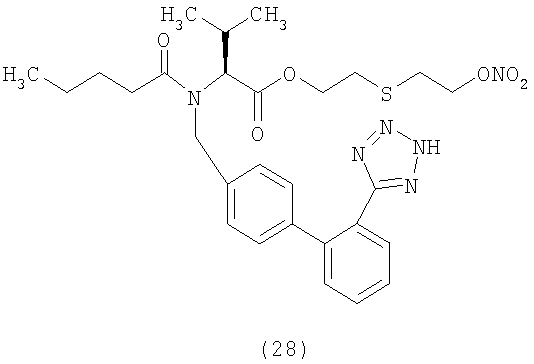
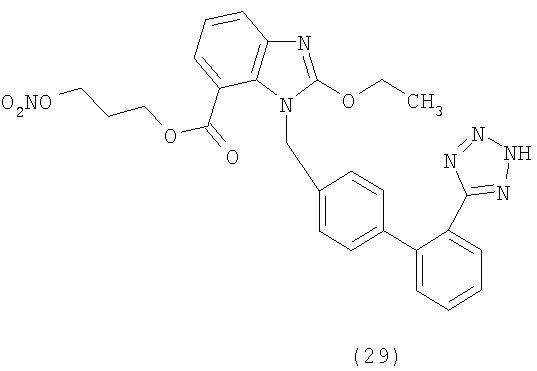
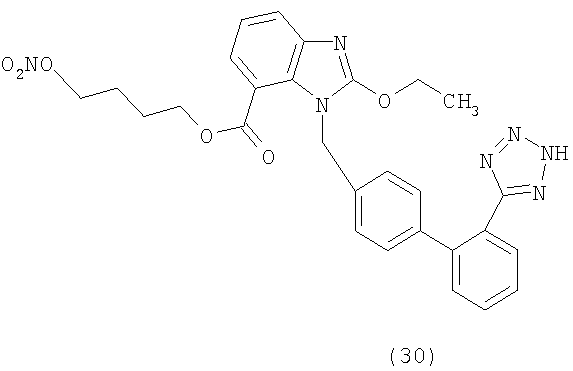
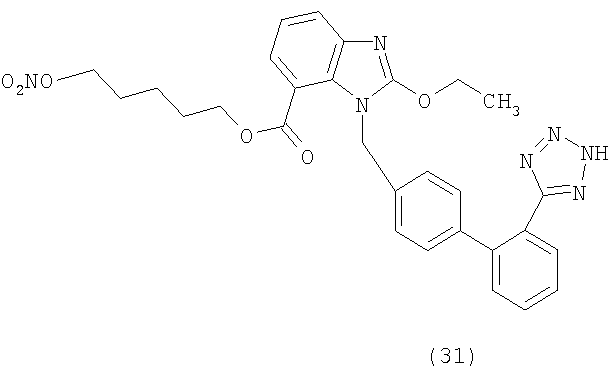
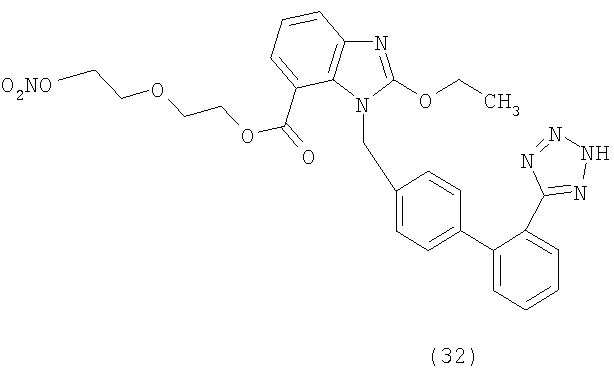
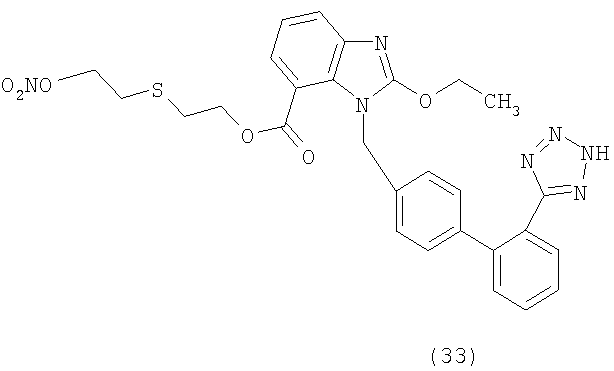
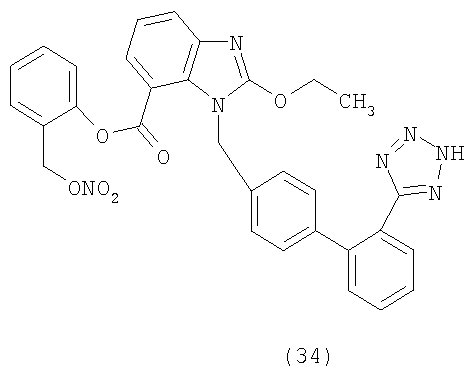
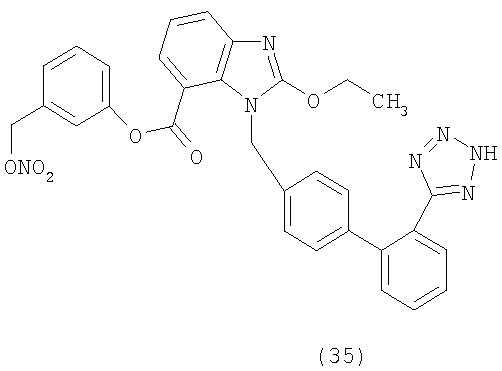
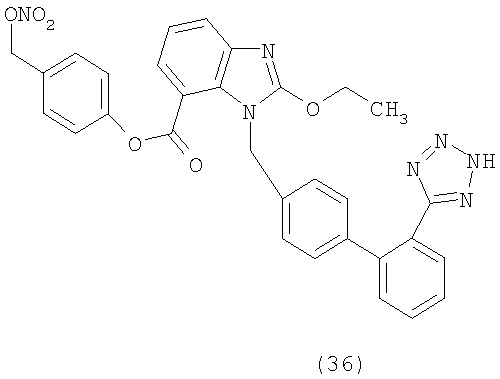


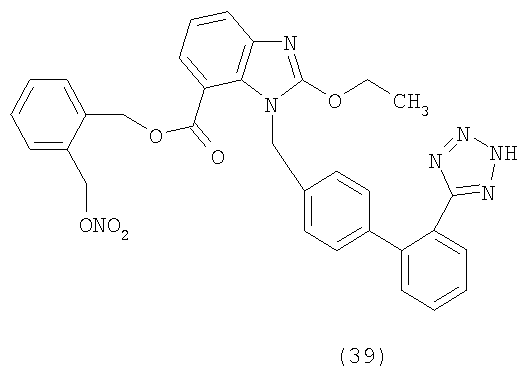
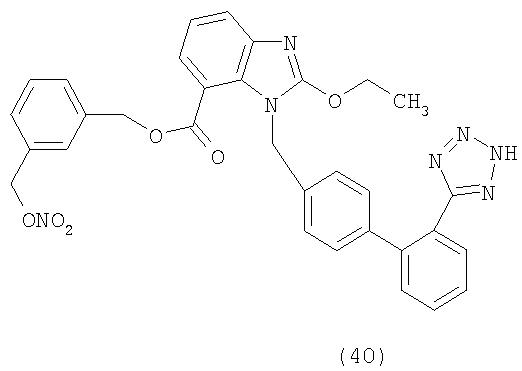
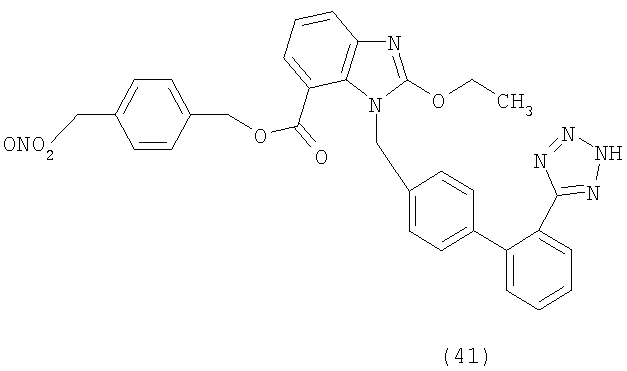
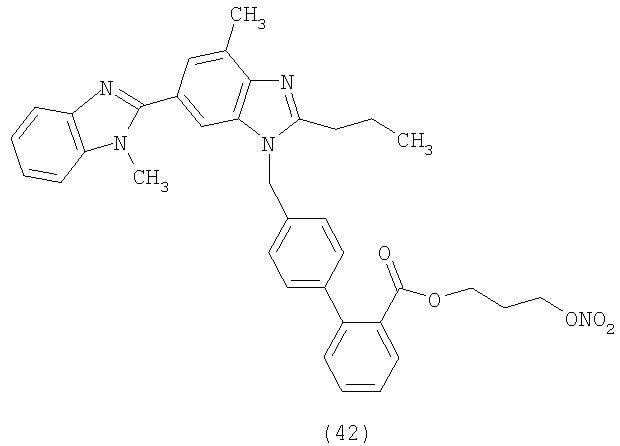
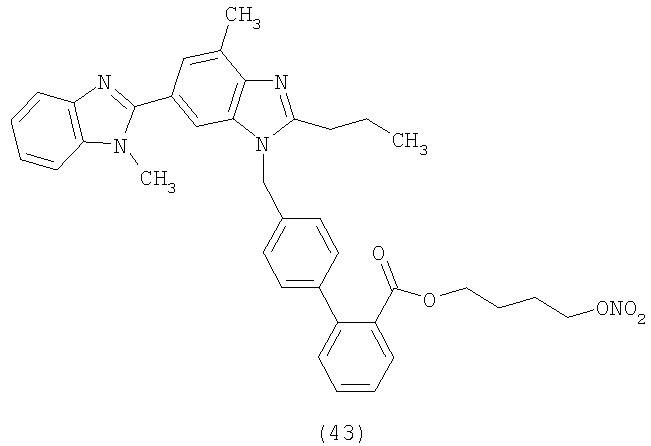

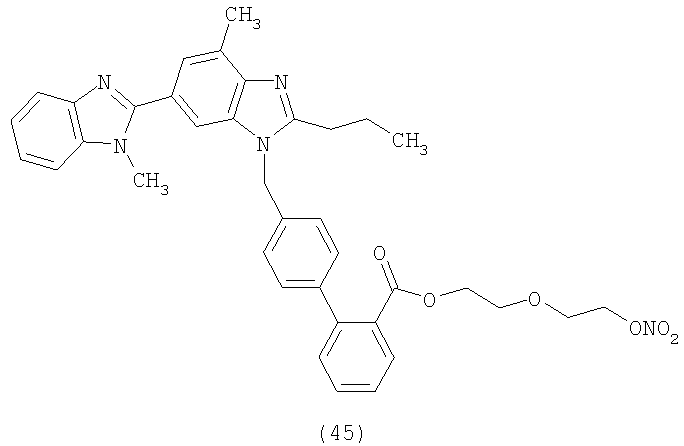
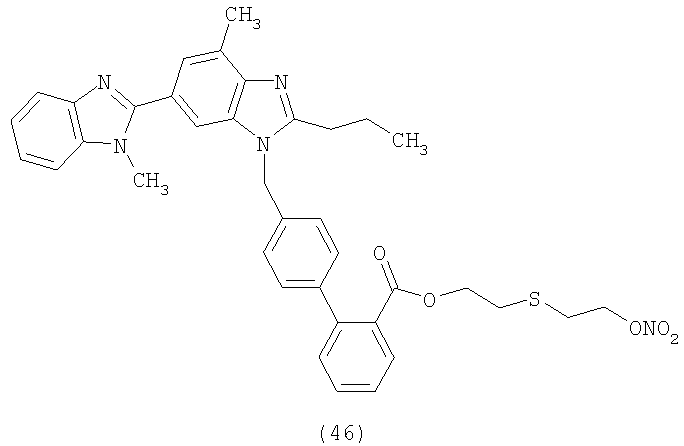
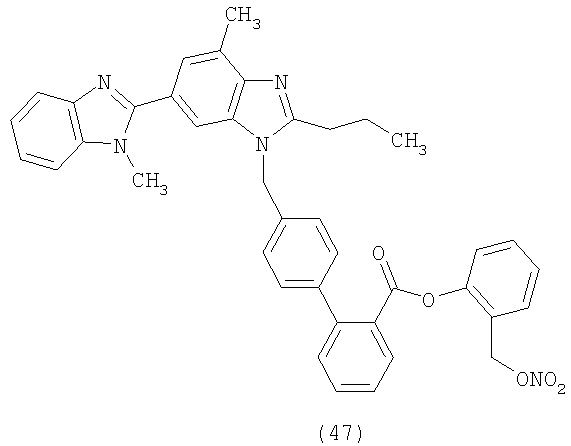





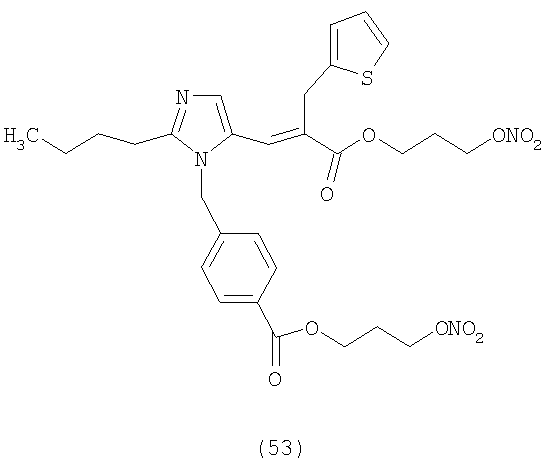
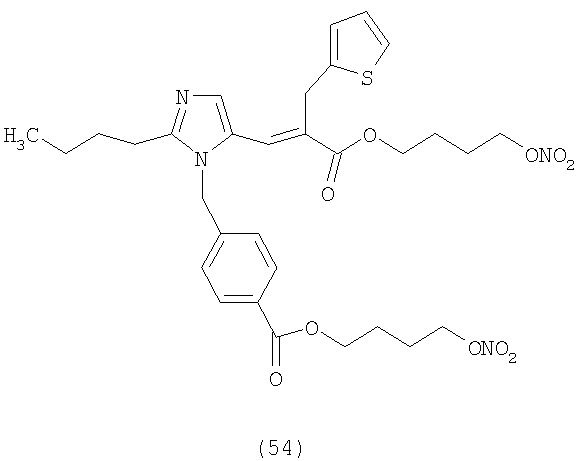
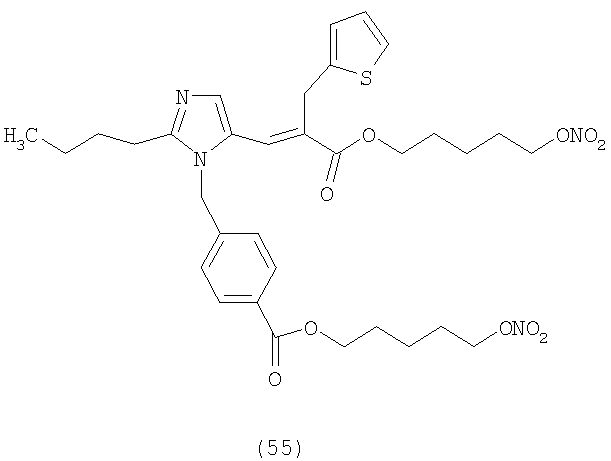
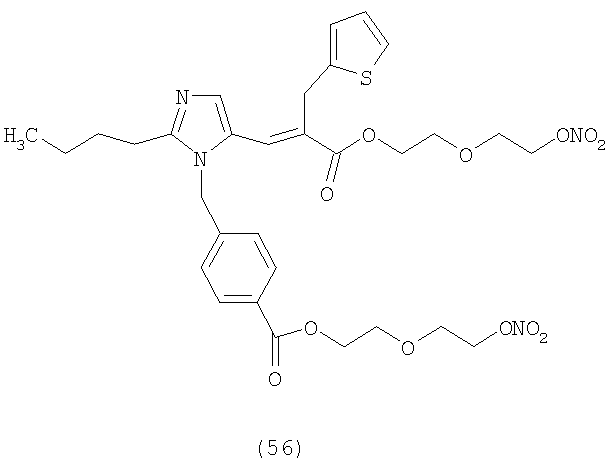
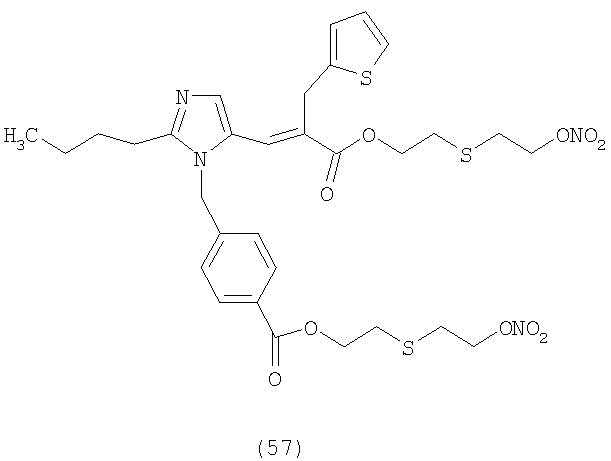
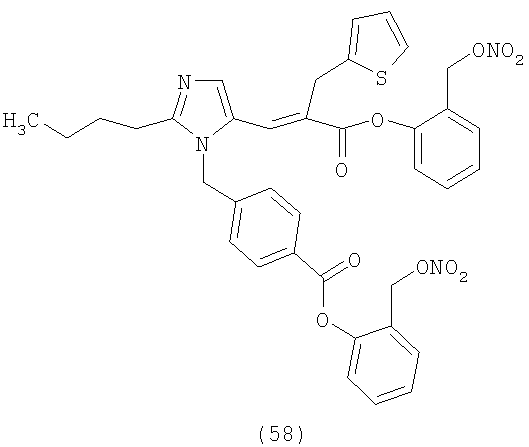

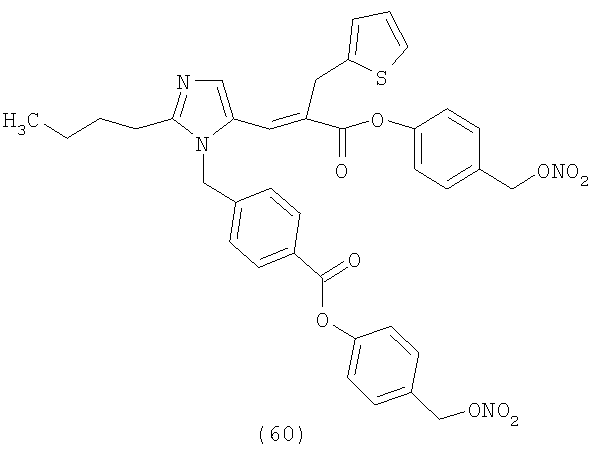
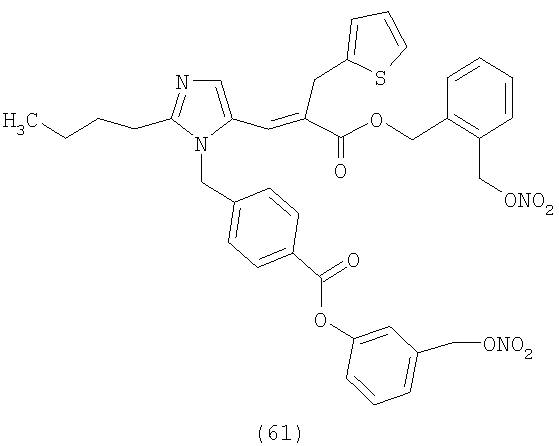
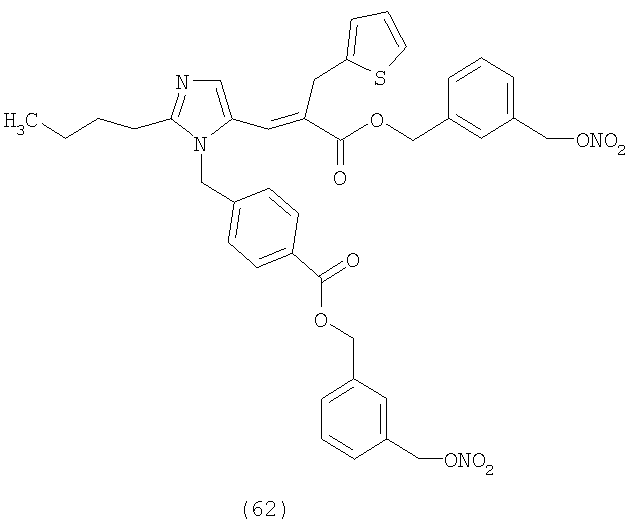
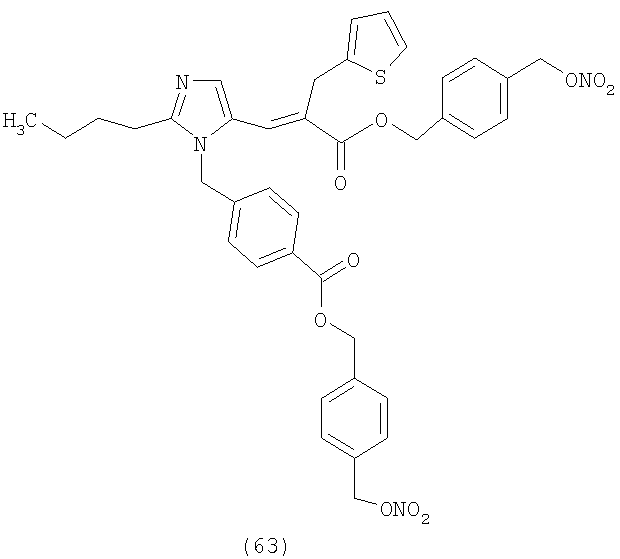

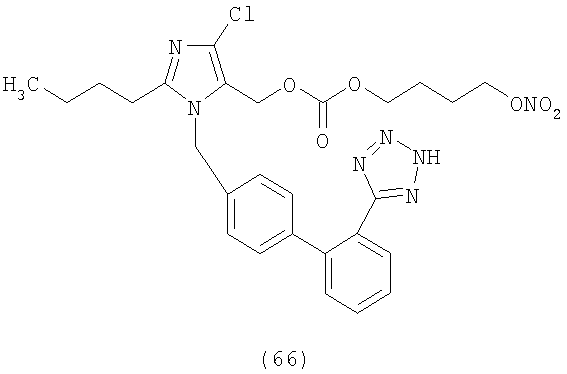

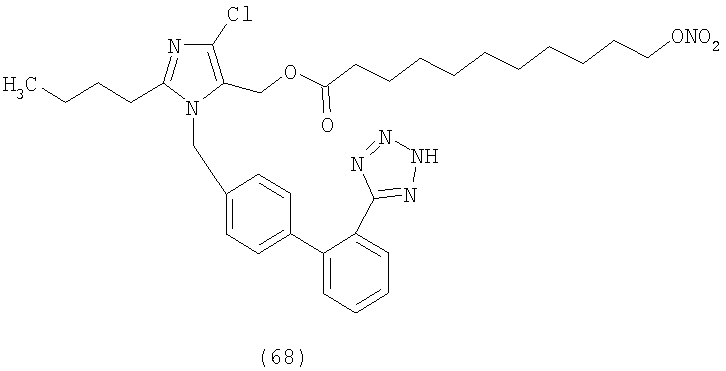
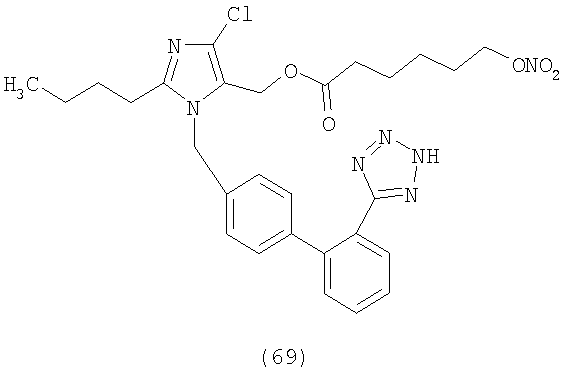


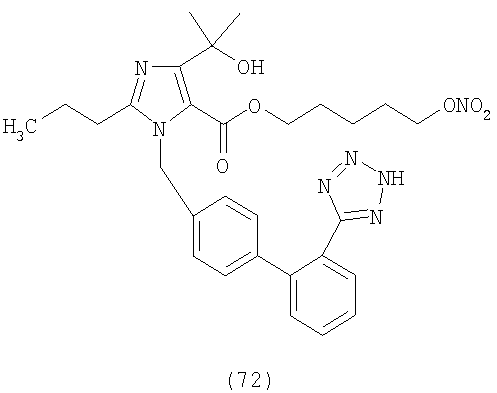
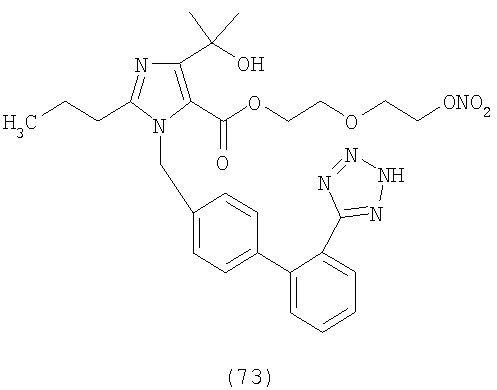
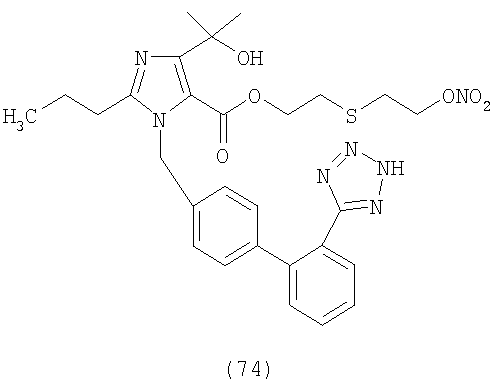

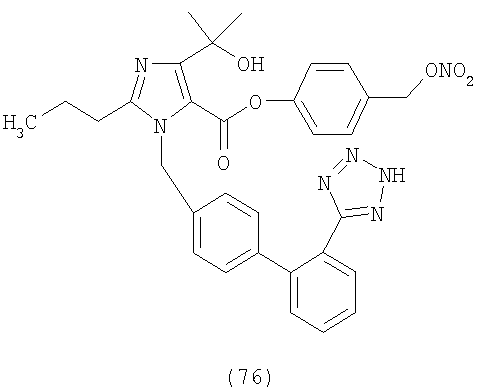

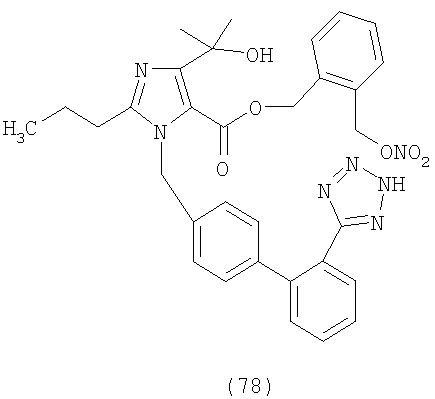

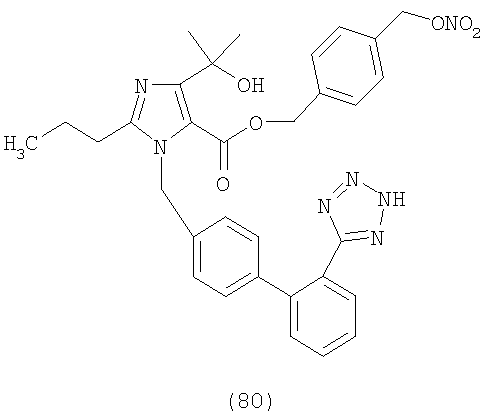
Как указано выше, объектом настоящего изобретения являются также фармацевтические композиции, содержащие по крайней мере соединение настоящего изобретения формулы (I) вместе с обычно используемыми в фармацевтике нетоксичными вспомогательными веществами и/или носителями.
Ежедневно назначаемая доза активного ингредиента может представлять собой единичную дозу или может представлять собой эффективное количество активного ингредиента, разделенное на несколько небольших доз, назначаемых в течение дня. Обычно общая дневная доза составляет преимущественно от 50 до 500 мг. Режим дозировки и частота назначения для лечения указанных выше заболеваний с помощью соединений и/или фармацевтических композиций настоящего изобретения выбирают в зависимости от различных факторов, включая, например, возраст, вес тела, пол и состояние пациента, а также серьезность заболевания, путь введения, фармакологические соображения и возможную сопутствующую терапию с помощью других лекарств. В некоторых случаях уровни доз могут быть ниже или выше указанного выше интервала или чаще адекватны ему и определяются врачом в зависимости от состояния болезни.
Соединения настоящего изобретения могут назначаться орально, парентерально, ректально или местно, с помощью ингаляции или аэрозоля в составах, в конечном счете, желательно, содержащих обычные нетоксичные фармацевтически приемлемые носители, вспомогательные вещества и наполнители. Местное назначение может включать трансдермальное применение, такое как трансдермальные повязки или устройства для электрофореза. Используемый здесь термин «парентерально» включает подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или технику вливания.
Инъецируемые препараты, например стерильные инъецируемые водные или масляные суспензии могут изготавливаться известными в данной области методами с использованием подходящих диспергирующих, смачивающих или суспендирующих агентов. Стерильный инъецируемый препарат может также представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе. Приемлемыми наполнителями являются вода, раствор Рингера и изотонический хлористый натрий. Кроме того, в качестве растворяющей или суспендирующей среды можно использовать традиционные стерильные нелетучие масла. Для этой цели можно использовать легкое нелетучее масло, включая синтетические моно или триглицериды, жирные кислоты, такие как олеиновая кислота, находящие применение при изготовлении препаратов для инъекций.
Суппозитории для ректального назначения лекарства можно получить смешиванием активного ингредиента с подходящим нераздражающим эксципиентом, таким как масло какао и полиэтиленгликоли.
Твердые лекарственные формы для орального назначения могут включать капсулы, таблетки, пилюли, порошки, гранулы и гели. В таких твердых лекарственных формах активное вещество может быть смешано по крайней мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут также включать дополнительные другие нежели инертные разбавители вещества, например лубриканты, такие как стеарат магния. В случае капсул, таблеток и пилюль лекарственные формы также могут включать буферные агенты. Таблетки и пилили могут быть приготовлены с энтеральным покрытием.
Жидкие лекарственные формы для орального назначения могут включать фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и элексиры, содержащие инертный, обычно используемый в данной области разбавитель, такой как вода. Такие композиции могут также включать адъюванты, такие как смачивающие агенты, эмульгирующие или суспендирующие агенты, подсластители, ароматизаторы и т.д.
Соединения настоящего изобретения можно синтезировать следующим образом.
А) Указанное выше соединение общей формулы (I) или его фармацевтически приемлемую соль:
где R остаток формулы (II), может быть получено способом, включающим:
i) реакцию соединения формулы (IV):
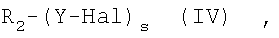
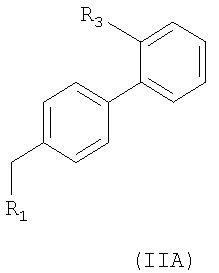
где s=1 и R2 остаток формулы (IIA):
 ,
,
где R3 группа формулы (VA):
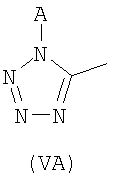
где А=Н или W, W представляет собой защитную для тетразола группу, такую как тритил, трет-бутоксикарбонил (ВОС) и этилоксикарбонил, или R3 представляет -СОО- группу, способную связываться с Y;
R1 выбирают из указанных выше групп (IIa)-(IIe), где N0 представляет группу, способную связываться с Y; Y как указано выше и Hal представляет атом галогена, преимущественно Cl, Br или I;
с AgNO3 в подходящем органическом растворителе, таком как ацетонитрил или тетрагидрофуран (ТГФ), в атмосфере азота в темноте при температурном интервале 20-80°С; или же реакцию с AgNO3 можно осуществлять под воздействием микроволнового излучения в растворителе, таком как ацетонитрил или ТГФ, при температурном интервале 100-180°C в течение короткого времени (1-60 мин), и
ii) необязательный кислотный гидролиз защитной W группы тетразола известными методами, как описано, например, в Т.W.Greene "Protective groups in organic synthesis", Harvard University Press, 1980, и
iii) в случае необходимости, превращение полученного соединения формулы (I) в его фармацевтически приемлемую соль.
Соединение формулы (IV) может быть получено реакцией соединения формулы (V):
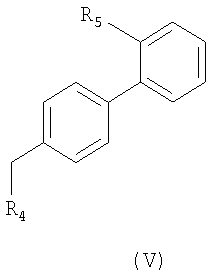
где R5 представляет указанную выше группу формулы (VA) или -СООН и R4 имеет те же значения, что и R1 с N0=-СООН или -ОН,
i.1) когда R5 представляет группу (VA), R4=R1 и R1 означает группу (IIa), где m=1 и
N0=-ОН, с соединением формулы (VI) или (VII):
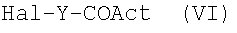
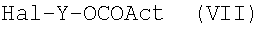
где Hal и Y как указано выше и Act представляет Hal или активирующую карбоновую кислоту группу, используемую в химии пептидов, как

Реакцию в основном проводят в присутствии неорганического или органического основания в апротонном полярном/неполярном растворителе, таком как ДФА, ТГФ или CH2Cl2, при температуре 0-65°C или в двухфазной системе H2O/Et2O при температуре 20-40°C.
Соединения формулы (VI), где Act представляет Hal, являются коммерчески доступными или могут быть получены из соответствующих кислот формулы (VIII):

известными способами, например реакцией с тионил или оксалил хлоридом, галоидами PIII или PV в инертном растворителе, таком как толуол, хлороформ, ДФА и т.д. Соответствующие кислоты являются коммерчески доступными соединениями.
Соединения формулы (VI), где Act не является Hal, могут быть получены из соответствующих соединений формулы (VI), где Act является Hal, с помощью известной из литературы реакции с N-гидроксисукцинимидом или с соответствующими замещенными фенолами в присутствии основания.
Соединения формулы (VII), где Act представляет Hal, являются коммерчески доступными соединениями или могут быть получены из соответствующих спиртов формулы (IX):
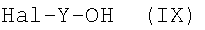
реакцией с трифосгеном в присутствии органического основания; соединения формулы (VII), где Act не является Hal, могут быть получены из соответствующих соединений формулы (VII), где Act является Hal, с помощью известной из литературы реакции с N-гидроксисукцинимидом или с соответствующими замещенными фенолами в присутствии основания.
С другой стороны соединения формулы (VI) могут быть получены реакцией указанного в i.1) соединения формулы (V) с указанным выше коммерчески доступным соединением формулы (VIII), в присутствии конденсирующего агента, такого как дициклогексилкарбодиимид (DCC), EDAC, в присутствии каталитического количества DMAP или активирующего агента, такого как N,N'-карбонилдиимидазол (CDI), в растворителе, таком как ДФА, ТГФ, хлороформ, при температуре от -5°С до 50°С;
i.2) когда R5 представляет группу (VA) или -СООН, R4=R1 и R1 выбирают из групп (IIa)-(IId), где m=0 и N0=-СООН, с указанным выше соединением формулы (IX) в присутствии конденсирующего агента, такого как дициклогексилкарбодиимид (DCC), EDAC, в присутствии каталитического количества DMAP или активирующего карбонильную группу агента, такого как N,N'-карбонилдиимидазол (CDI), в растворителе, таком как ДФА, ТГФ, хлороформ, при температуре от -5°С до 50°С.
Соединения формулы (IX) являются коммерчески доступными.
С другой стороны превращение группы -СООН в активированную ацилхлоридную или другую пригодную для этерификации группу осуществляют в соответствии с известными из литературы методами и этерификацию проводят в присутствии органического или неорганического основания в апротонном полярном/неполярном растворителе, таком как ДФА, ТГФ или CH2Cl2, при температуре 0°С - 65°С или в двухфазной системе H2O/Et2O при температуре 20°С - 40°С.
А1) Или же указанные выше соединения формулы (I), где R представляет остаток формулы (II), могут быть получены реакцией указанных выше соединений формулы (V):
i.1.1) когда R5 представляет группу (VA), R4=R1 и R1 представляет группу (IIa), где m=1 и N0=-ОН, с соединением формулы (X):
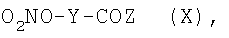
где Y как указано выше и Z представляет ОН или определенную выше группу Act, наиболее подходящим синтетическим путем, например в присутствии конденсирующего агента, такого как дициклогексилкарбодиимид (DCC), EDAC, или активирующего агента - N,N'-карбонилдиимидазола (CDI) в растворителе, таком как ДФА, ТГФ, хлороформ, при температуре от -5°С до 50°С и/или в присутствии органического или неорганического основания.
Соединения формулы (X) могут быть получены из соответствующих спиртов реакцией с азотной кислотой и уксусным ангидридом при температуре от -50°С до 0°С или реакцией соответствующих галоидпроизводных формулы (VI) или (VIII) с AgNO3, как уже описано.
i.2.1) Когда R5 представляет группу (VA) или -СООН, R4=R1 и R1 выбирают из групп (IIa)-(IId), где m=0 и N0=-СООН, с соединением формулы (XI):
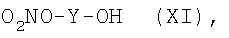
где Y как указано выше, в присутствии конденсирующего агента, такого как дициклогексилкарбодиимид (DCC) или EDAC, или активирующего агента, такого как N,N'-карбонилдиимидазол (CDI), в растворителе, таком как ДФА, ТГФ, хлороформ, при температуре от -5°С до 50°С.
Соединения формулы (XI) могут быть получены реакцией соединения формулы (IX) с AgNO3 в подходящем органическом растворителе, таком как ацетонитрил или ТГФ в среде азота при температуре 20°С - 80°С; или же реакция с AgNO3 может быть проведена под воздействием микроволнового излучения в растворителях, таких как ацетонитрил или ТГФ, при температуре 100-180°С в течение короткого промежутка времени (1-60 мин).
Или же, когда R5 представляет группу (VA) или -СООН, R4=R1 и R1 выбирают из групп (IIa)-(IIe), где m=0 и N0=-СООН, с соединением формулы (XI.1):
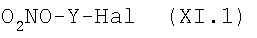
где Y и Hal как указано ранее, в присутствии неорганического или органического способного к солеобразованию с карбоксильной группой основания.
В) Соединения общей формулы (I), где R представляет собой остаток формулы (III), могут быть получены реакцией соединения формулы (XII):
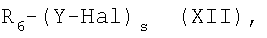
где s=2, R6 представляет остаток (III) и N1 представляет -COO-, Y и Hal как указано выше, с AgNO3, как уже описано.
Соединения формулы (XII) получают реакцией соединения формулы (XIII):
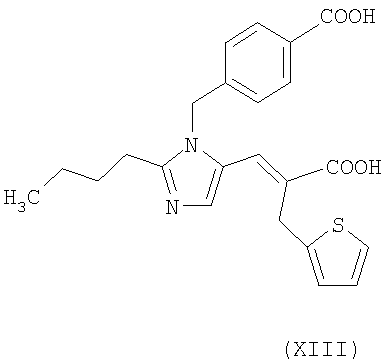
с указанными выше соединениями формулы (IX) в присутствии конденсирующего агента, такого как дициклогексилкарбодиимид (DCC) или EDAC, или активирующего агента, такого как N,N'-карбонилдиимидазол (CDI), в растворителе, таком как ДФА, ТГФ, хлороформ, при температуре от -5°С до 50°С.
С другой стороны превращение группы -СООН в активированную ацилхлоридную или другую пригодную для этерификации группу осуществляют в соответствии с известными из литературы методами и этерификацию ведут в присутствии органического или неорганического основания в апротонном полярном/неполярном растворителе, таком как ТГФ или CH2Cl2 при температуре 0°С-65°С или в двухфазной системе.
В1) Или же указанные выше соединения общей формулы (I), где R представляет собой остаток формулы (III), могут быть получены реакцией соединения формулы (XIII) с указанным выше соединением формулы (XI) в присутствии конденсирующего или активирующего агента, как уже описано.
С другой стороны, превращая группу -СООН в соль с неорганическим или органическим основанием в соответствии с известными из литературы методами и реакцией с
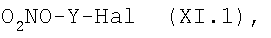
как описано в литературе.
С) Указанные выше соединения формулы (I), где s=1 и R представляет остаток формулы (II), где R0 представляет собой тетразольную группу и R1 представляет группу (IIa), где m=1 и N0 представляет
 ,
,
где R' и R'' как указано выше, могут быть получены реакцией соединения формулы (IVa):
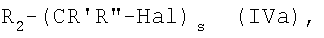
где s=1, R2 и Hal как указано выше, R3 представляет группу (VA), R1 представляет группу (IIa), где m=1 и N0 представляет -ОСОО-, с указанным выше соединением формулы (X) в присутствии органического или неорганического основания в полярном растворителе, таком как ДФА, ТГФ, ацетонитрил, при температуре от -5°С до 60°С или в двухфазной системе, как известно из литературы.
Соединения (IVa) могут быть получены взаимодействием указанного выше соединения формулы (V), где R3 представляет группу (VA), R4=R1 и R1 представляет группу (IIa), где m=1 и N0=-ОН, с соединением формулы (VIIa):
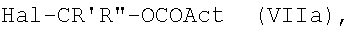
где Act имеет те же значения, которые указаны выше для (VII), тем же способом, который уже описан для соединений (IV); и необязательным кислотным гидролизом указанной выше защитной группы тетразола.
D) Указанные выше соединения формулы (I), где s=1 и R представляет остаток формулы (II), где R0 представляет собой тетразольную группу и R1 выбран из групп (IIa)-(IIc), где m=0 и N0 представляет
,
где R' и R'' как указано выше, могут быть получены реакцией соединения формулы (V), где R5 представляет группу (VA), R4=R1 и R1 представляет группу (IIc), где N0=-СООН, с соединением формулы (XIV):
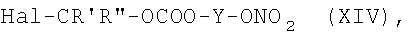
где Hal, Y, R' и R'' как указано выше, в присутствии органического или неорганического основания в полярном растворителе, таком как ДФА, ТГФ, ацетонитрил, при температуре от -5°С до 60°С или в двухфазной системе, как известно из литературы.
Соединения формулы (XIV) могут быть получены реакцией соединений формулы (XI) с указанными выше соединениями формулы (VIIa). Реакцию в основном ведут в присутствии основания в апротонном полярном/неполярном растворителе, таком как ТГФ или CH2Cl2, при температуре 0-65°С или в двухфазной системе H2O/Et2O при температуре 20-40°С и необязательным кислотным гидролизом указанной выше защитной группы тетразола.
Е) Указанные выше соединения формулы (I), где s=1 и R представляет остаток формулы (II), где R0 представляет собой тетразольную группу и R1 выбран из групп (IIa)-(IIc), также могут быть получены реакцией соединения формулы (XV) с коммерчески доступным соединением формулы (XVI):
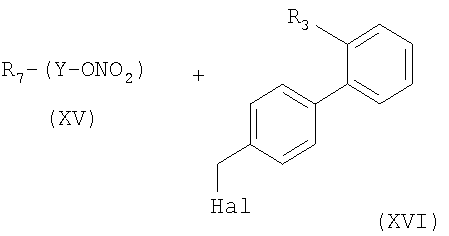
где R7 представляет собой остаток (IIa)-(IIc), R3 группу (Va) и Hal как уже указано выше. Реакцию в основном ведут в присутствии основания в апротонном полярном/неполярном растворителе, таком как ДФА, ТГФ или CH2Cl2 при температуре от -15 до +80°С или в двухфазной системе H2O/Et2O при температуре от 20-40°С и необязательным кислотным гидролизом указанной выше защитной группы тетразола. Соединения формулы (XV) могут быть получены реакцией соединений формулы (XVII):

где R8 представляет остаток формулы (IIa.1), (IIb.1) или (IIc.1):
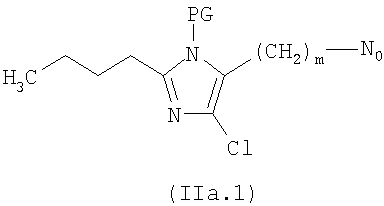


где PG означает N-защитную группу, такую как ВОС или тритил, с AgNO3, как уже описано, и необязательным гидролизом N - защитной группы. Соединения (XVII), где
R8 представляет (IIa.I), где m=1 и N0=-ОСО-, могут быть получены из соответствующих спиртов, как уже описано, реакцией с соединением формулы (VI) или (VII). Указанные выше спирты получают известными защитными и восстановительными реакциями из коммерчески доступных соединений формулы (IIa.2):

где m представляет 0 и N00 представляет -СНО.
Соединения формулы (XVII), где R8 представляет (IIa.1) с m=0 и N0=-COO- или R8 представляет (IIb.1) или (IIc.1) с N0=-COO-, могут быть получены из соответствующих кислот реакцией с соединениями формулы (IX). Указанные выше соответствующие кислоты (IIa.1) получают из соединений (IIa.2), где m представляет 0 и N00 представляет -СНО, известными защитными и окислительными реакциями. Указанные выше соответствующие кислоты (IIb.1) и (IIc.1) получают из коммерчески доступных (IIb.2) и (IIc.2):


где N0 представляет -СООН, известными методами.
Следующие примеры иллюстрируют изобретение без его ограничения.
Пример 1
Сложный эфир 4-(нитрооксиметил)бензоиной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (соответствует соединению (4))
К раствору натриевой соли Лазартана (7,0 г; 15,2 ммол) в ТГФ добавляют порциями трифенилметилхлорид (4,68 г; 16,8 ммол). Полученную смесь перемешивают при комнатной температуре 24 часа. Затем абсорбируют на силикагеле и очищают с помощью флешхроматографии (н-гексан/AcOEt 6:4), получая 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанол (6,7 г; 66%). Из этого соединения можно получить указанное в заголовке соединение двумя различными способами синтеза.
Синтетический способ А
К охлажденному до 0°С раствору 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,7 г; 2,6 ммол), 4-(нитрооксиметил)бензойной кислоты (0,66 г; 3,38 ммол) и N,N-диметиламинопиридина (0,049 г; 0,4 ммол) в CH2Cl2 (20 мл) и ТГФ (6 мл) медленно добавляют раствор дициклогексилкарбодиимида (0,722 г; 3,5 ммол) в CH2Cl2 (5 мл) и перемешивают при комнатной температуре 24 часа. Затем отфильтровывают образовавшуюся диклогексилмочевину и органическую фазу концентрируют. Сырой продукт очищают путем хроматографии на силикагеле (н-гексан/AcOEt 75:25), получая сложный эфир 4-(нитрооксиметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,2 г, 55%) в виде белого твердого вещества.
Сложный эфир 4-(нитрооксиметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,2 г, 1,42 ммол) растворяют в CH2Cl2 (10 мл) и через полученный раствор 20 минут барботируют HCl. Затем смесь концентрируют и очищают с помощью флешхроматографии (CH2Cl2/ацетон 8:2, затем ацетон), получая сырое соединение, которое растворяют в H2O/CH3CN и с помощью сушки замораживанием получают сложный эфир 4-(нитрооксиметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола в виде твердого белого вещества (0,304 г, 36%).
1H-NMR (DMSO-d6): 7.73-7.56 (7H, m); 7.24 (1H, d); 7.00 (4H, m); 5.60 (2H, s); 5.39 (2H, s); 5.28 (2H, s); 2.61 (2H, t); 1.53 (2H, m); 1.28 (2H, m); 0.82 (3H, t).
Синтетический способ В
К охлажденному до 0°C раствору 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,7 г; 2,6 ммол), 4-(хлорметил)бензойной кислоты (0,571 г; 3,35 ммол) и N,N-диметиламинопиридина (0,049 г; 0,4 ммол) в CH2Cl2 (20 мл) и ТГФ (6 мл) медленно добавляют дициклогексилкарбодиимид (0,644 г; 3,12 ммол) и перемешивают при комнатной температуре 24 часа. Затем отфильтровывают образовавшуюся диклогексилмочевину и органическую фазу концентрируют. Сырой продукт очищают путем флешхроматографии (н-гексан/AcOEt 75:25), получая сложный эфир 4-(хлорметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,56 г, выход 73%).
Сложный эфир 4-(хлорметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,807 г; 0,98 ммол) растворяют в CH3CN (15 мл) и добавляют в темноте в атмосфере азота AgNO3 (0,305 г, 1,8 ммол). Смесь перемешивают при 60°С 6 часов. Затем отфильтровывают осадок соли серебра и органическую фазу разбавляют AcOEt, промывают NaH2PO4 (5%, 2×10 мл) и рассолом (2×10 мл), сушат над Na2SO4 и концентрируют. Сырой продукт очищают путем флешхроматографии (н-гексан/AcOEt 75:25), получая сложный эфир 4-(нитрооксиметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,553 г, 66%).
Путем кислотного гидролиза, описанного в способе А, из сложного эфира 4-(нитрооксиметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола получают указанное в заголовке соединение - сложный эфир 4-(нитрооксиметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола.
Пример 2
Сложный эфир 4-нитрооксимасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (соответствует соединению (2))
Это соединение можно получить четырьмя различными способами синтеза.
Синтетический способ А
К охлажденному до 0°С раствору 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (полученного в примере 1) (1,7 г; 2,6 ммол), 4-нитрооксимасляной кислоты (0,536 г; 3,6 ммол) и N,N-диметиламинопиридина (0,05 г; 0,4 ммол) в CH2Cl2 (20 мл) и ТГФ (6 мл) медленно добавляют раствор дициклогексилкарбодиимида (DCC) (0,722 г; 3,5 ммол) в CH2Cl2 (5 мл) и перемешивают при комнатной температуре 24 часа. Затем отфильтровывают образовавшуюся диклогексилмочевину и органическую фазу концентрируют. Сырой продукт очищают путем хроматографии на силикагеле (н-гексан/AcOEt 7:3), получая сложный эфир 4-нитрооксимасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,45 г, 70%).
Сложный эфир 4-нитрооксимасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,0 г, 1,25 ммол) растворяют в CH2Cl2 (10 мл) и через полученный раствор 20 минут барботируют HCl. Затем смесь концентрируют и очищают в помощью флешхроматографии (CH2Cl2/ацетон 8:2, затем ацетон), получая сырое соединение в виде белой пены, которую растворяют в Н2О/CH3CN и с помощью сушки замораживанием получают сложный эфир 4-нитрооксимасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола в виде твердого белого вещества (0,507 г, выход 71%).
1H-NMR (DMSO-d6): 7.66 (2H, d); 7.57 (1H, d); 7.49 (1H, d); 7.09 (2H, d); 6.95 (2H, d); 5.25 (2H, s); 4.99 (2H, s); 4.49 (2H, t); 2.54 (2H, t); 2.01 (2H, t); 1.60 (2H, m); 1.49 (2H, m); 1.32 (4H, m); 0.84 (3H, t).
Синтетический способ В
К охлажденному до 0°C раствору 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (полученного в примере 1) (1,7 г; 2,6 ммол), 4-броммасляной кислоты (0,561 г; 3,36 ммол) и N,N-диметиламинопиридина (0,05 г; 0,4 ммол) в CH2Cl2 (20 мл) и ТГФ (6 мл) медленно добавляют раствор дициклогексилкарбодиимида (0,722 г; 3,5 ммол) в CH2Cl2 (5 мл) и перемешивают при комнатной температуре 24 часа. Затем отфильтровывают образовавшуюся диклогексилмочевину и концентрируют органическую фазу. Сырой продукт очищают путем хроматографии на силикагеле (н-гексан / EtOAc 75:25), получая сложный эфир 4-броммасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил] метил]-1H-имидазол-5-метанола (1,27 г, выход 60%).
Сложный эфир 4-броммасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,2 г; 1,47 ммол) растворяют в CH3CN (20 мл) и добавляют в темноте в атмосфере азота AgNO3 (0,475 г, 2,8 ммол). Смесь перемешивают при 60°С 8 часов. Затем распределяют между EtOAc и фосфатным буфером (рН=3,40 мл). Органическую фазу промывают фосфатным буфером (рН=3,2×25 мл), рассолом (3×25 мл), сушат над Na2SO4 и концентрируют. Сырой продукт очищают путем флешхроматографии (н-гексан/AcOEt 7:3), получая сложный эфир 4-нитрооксимасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,819 г, 70%) в виде пены.
Путем кислотного гидролиза, описанного в примере 2, способ А, из сложного эфира 4-нитрооксимасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола получают указанное в заголовке соединение - сложный эфир 4-нитрооксимасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,507 г, 71%).
Синтетический способ С
К охлажденному до 0°С раствору 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (3,6 г; 8,5 ммол), N,N-диметиламинопиридина (0,1 г; 0,85 ммол) и TEA (1,18 мл, 0,85 ммол) в ТГФ (60 мл) медленно добавляют в атмосфере азота раствор 4-бромбутаноилхлорида (0,98 г; 8,5 ммол) в ТГФ (1 мл) и перемешивают при комнатной температуре 1,5 часа. Затем распределяют между EtOAc и фосфатным буфером (рН=3,40 мл) и экстрагируют EtOAc (3×15 мл). Органическую фазу сушат над Na2SO4 и концентрируют. Сырой продукт очищают путем флешхроматографии (CH2Cl2/ацетон 8:2), получая сложный эфир 4-броммасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (2,5 г, 51%) в виде твердого белого вещества.
Сложный эфир 4-броммасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,56 г; 0,98 ммол) растворяют в CH3CN (15 мл) и добавляют в темноте в атмосфере азота AgNO3 (0,83 г, 4,9 ммол). Смесь перемешивают при 60°С 8 часов. Затем охлаждают и вливают в раствор фосфатного буфера (рН=3, 40 мл). Добавляют твердый NaCl и смесь экстрагируют EtOAc. Органическую фазу промывают фосфатным буфером (рН=3,2×25 мл), рассолом (3×25 мл), сушат над Na2SO4 и концентрируют. Сырой продукт очищают путем флешхроматографии (CH2Cl2/ацетон 8:2, затем ацетон), получая сырой продукт в виде белой пены. Пену растворяют в Н2О/CH3CN и с помощью сушки вымораживанием получают сложный эфир 4-нитрооксимасляной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,3 г, 55%) в виде белого твердого вещества.
Синтетический способ D
К охлажденному до 0°С раствору 4-броммасляной кислоты (0,91 г, 5,4 ммол), пентафторфенола (1,0 г, 5,4 ммол) и DMAP (0,13 г, 1,1 ммол) в CH2Cl2(10 мл) в атмосфере азота добавляют порциями N,N-дициклогексилкарбодиимид (1,70 г, 8,1 ммол). Через 1 час реакционную смесь медленно нагревают до комнатной температуры и перемешивают 5 часов. Отфильтровывают дициклогексилмочевину, маточник концентрируют и очищают с помощью флешхроматографии (н-гексан/EtOAc 98:2), получая пентафторфениловый эфир 4-броммасляной кислоты в виде бесцветного масла (1,40 г, 78%).
Смесь пентафторфенилового эфира 4-броммасляной кислоты (0,65 г, 1,9 ммол) и AgNO3 (0,83 г, 4,9 ммол) в CH3CN (8 мл) нагревают при 70°С 20 мин под воздействием микроволнового облучения. Образовавшиеся соли отфильтровывают, раствор концентрируют и остаток очищают с помощью флешхроматографии (н-гексан/EtOAc 95:5), получая петафторфениловый эфир 4-нитрооксимасляной кислоты в виде прозрачного масла (0,38 г, 62%).
К охлажденному до 0°С раствору 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,48 г; 1,1 ммол), TEA (0,16 г, 1,1 ммол) и DMAP (0,14 г, 1,1 ммол) в ДФА(3 мл) добавляют раствор петафторфенилового эфира 4-нитрооксимасляной кислоты (0,36 г, 1,1 ммол) в ДФА (3 мл). Реакционную смесь медленно нагревают до комнатной температуры и перемешивают 3 часа. Затем при пониженном давлении упаривают растворитель. Остаток растворяют в EtOAc (10 мл) и промывают буферным раствором (рН=3), затем рассолом. Органический слой сушат над Na2SO4, концентрируют и очищают с помощью флешхроматографии (CH2Cl2/MeOH 98:2), получая указанное в заголовке соединение (0,41 г, 66%).
Пример 3
Сложный эфир 11-нитрооксиундекановой кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (соответствуюет соединению (68))
Используя описанный в примере 2 способ А, но исходя из 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,7 г, 2,6 ммол) и 11-нитрооксиундекановой кислоты (0,78 г, 3,36 ммол), получают сложный эфир 11-нитрооксиундекановой кислоты и 2-бутил-4-хлор-1-[[2'-трифенилметилтетразол) [1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,65 г, 80%). Путем кислотного гидролиза этого соединения (1,6 г, 2,0 ммол) после перекристаллизации из Et2O/н-гексана получают сложный эфир 11-нитрооксиундекановой кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,91 г, 70%).
(DMSO): 7.66 (2H, d); 7.57 (1H, d); 7.59 (1H, d); 7.09 (2H, d); 6.95 (2H, d); 5.25 (2H, s); 4.99 (2H, s); 4.49 (2H, t); 2.54 (2H, t); 2.01 (2H, t); 1.62 (2H, m); 1.49 (2Н, m); 1.35-1.14 (16H, m); 0.84 (3H, t).
Пример 4
Сложный эфир 3-(нитрооксиметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (соответствующий соединению (5))
2-Бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанол (полученный в примере 1) (1,0 г; 1,5 ммол), триэтиламин (0,42 мл, 3,0 ммол) и N,N-диметиламинопиридин (36 мг; 0,30 ммол) растворяют в CH2Cl2 (10 мл). Затем добавляют 3-(хлорметил)бензоилхлорид (0,24 мл, 1,7 ммол) и реакционную смесь перемешивают при комнатной температуре 4 часа. Смесь разбавляют EtOAc (50 мл) и органическую фазу промывают NaH2PO4 (5%, 2×25 мл), NaHCO3 (5%, 2×25 мл), рассолом (2×25 мл), сушат над Na2SO4 и концентрируют. Сырой продукт очищают путем флешхроматографии (н-гексан/EtOAc 75:25), получая сложный эфир 3-(хлорметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (1,0 г, 81%) в виде масла.
Сложный эфир 3-(хлорметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,66 г, 0,20 ммол) суспендируют в CH3CN (10 мл) и добавляют NaI (0,24 г, 1,6 ммол). Реакционную смесь нагревают с обратным холодильником 1 час, затем разводят EtOAc (25 мл). Органическую фазу промывают водой (3×25 мл), сушат над Na2SO4 и концентрируют. Сырой продукт растворяют в
CH3CN (4 мл) и добавляют в атмосфере азота в темноте AgNO3 (0,34 г, 2 ммол). Реакционную смесь перемешивают при комнатной температуре 2 часа, затем разводят EtOAc (10 мл). Органическую фазу промывают NaH2PO4 (5%, 2×10 мл), рассолом (2×10 мл), сушат над Na2SO4 и концентрируют.Сырой продукт очищают путем флешхроматографии (н-гексан/EtOAc 75:25), получая сложный эфир 3-(нитрооксиметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (230 мг, 33%).
Сложный эфир 3-(нитрооксиметил)бензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,23 г, 0,27 ммол) растворяют в CH2Cl2 (5 мл) и через полученный раствор барботируют HCl. Через 10 минут реакционную смесь концентрируют и очищают с помощью флешхроматографии (CH2Cl2/ацетон 8:2 и затем ацетон). Полученную желтую пену обрабатывают обесцвечивающим углем, растворяют в H2O/CH3CN и после сушки вымораживанием получают сложный эфир м-нитробензилбензойной кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола в виде белого твердого вещества (0,11 г, 63%).
(CDCl3): 7.90 (2H, m); 7.78 (1H, d); 7.56 (3H, m); 7.40 (1H, m); 7.19 (1H, d); 7.06 (2H, d); 6.83 (2H, d); 5.40 (2H, s); 5.24 (2H, s); 5.14 (2H, s); 2.47 (2H, t); 1.61 (2H, m); 1.32 (2H, m); 0.87 (3H, m).
Пример 5
Сложный эфир 6-нитрооксикапроновой кислоты и 2-бутил-4-хлор-1-[[2'-(1H-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (соответствует соединению 69)
2-Бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанол (полученный в примере 1) (2,0 г, 3,9 ммол), 6-бромкапроновую кислоту (0,9 г, 4,6 ммол), N,N-диметиламинопиридин (38 мг, 0,3 ммол) и триэтаноламин (1,3 мл, 9,3 ммол) растворяют в CH2Cl2 (20 мл) и полученный раствор охлаждают до 0°С. Затем добавляют 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (EDAC) (0,94 г, 9,3 ммол) и реакционную смесь медленно нагревают до комнатной температуры и перемешивают в течение ночи. Органическую фазу промывают NaH2PO4 (5%, 20 мл), рассолом (20 мл), сушат над Na2SO4 и очищают путем флешхроматографии (н-гексан/EtOAc 7:3), получая сложный эфир 6-бромкапроновой кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1H-имидазол-5-метанола в виде масла (1,94 г, 76%).
Сложный эфир 6-бромкапроновой кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,77 г, 0,90 ммол) и NaI (0,3 г, 2,0 ммол) растворяют в CH3CN (10 мл) и смесь нагревают с обратным холодильником 1 час. Затем разводят EtOAc (50 мл) и органическую фазу промывают водой (2×25 мл), сушат над Na2SO4 и концентрируют. Сырой продукт суспендируют в CH3CN (7 мл) и добавляют AgNO3(0,60 г, 3,50 ммол). Реакционную смесь перемешивают при комнатной температуре в темноте в атмосфере азота 3 часа. Затем распределяют между EtOAc (30 мл) и фосфатным буфером (рН=3,25 мл). Органическую фазу промывают фосфатным буфером (рН=3,2×25 мл), рассолом (3×25 мл), сушат над
Na2SO4 и концентрируют. Сырой продукт очищают путем флешхроматографии (н-гексан/EtOAc 7:3), получая сложный эфир 6-нитрооксикапроновой кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)(1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола в виде пены (0,69 г, 64%).
Сложный эфир 6-нитрооксикапроновой кислоты и 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-метанола (0,88 г) растворяют в CH2Cl2 (20 мл) и через полученный раствор барботируют в течение 20 минут HCl. Реакционную смесь концентрируют и очищают с помощью флешхроматографии (CH2Cl2/ацетон 8:2 и затем ацетон), получая желтую пену, которую обрабатывают обесцвечивающим углем, растворяют в H2O/CH3CN и после сушки вымораживанием получают сложный эфир 6-нитрооксикапроновой кислоты и 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1H-имидазол-5-метанола в виде белого твердого вещества (0,41 г, 68%).
(CDCl3): 7.79 (1H, d); 7.63-7.49 (2H, m); 7.41 (1H, d); 7.08 (2H, d); 6.77 (2H, d); 5.14 (2H, s); 4.88 (2H, s); 4.38 (2H, t); 2.38 (2H, t); 2.06 (2H, m); 1.70-1.50 (6H, m); 1.37-1.30 (4H, m); 0.85 (3H, t).
Пример 6
(3-Нитроокси)пропиловый эфир 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-карбоновой кислоты (соответствует соединению (7)).
К раствору 2-бутил-4-хлор-5-формил имидазола (1,2 г, 6,4 ммол) в трет-ButOH (35 мл) и 5% водного раствора Na2HPO4 (25 мл) добавляют раствор KMnO4 (6,1 г, 38,6 ммол) в воде (40 мл). Через 6 минут при комнатной температуре смесь гасят добавлением 40% водного раствора NaHSO3. Полученную суспензию фильтруют, промывают водой и фильтрат сушат вымораживанием. Остаток поглощают водой (50 мл), подкисляют до рН 2,5 с помощью 3N HCl, экстрагируют EtOAC (3×70 мл). Объединенные органические экстракты сушат над Na2SO4 и упаривают до получения сухого остатка 2-бутил-4-хлор-имидазол-5-карбоновой кислоты (1,07 г, 83%) в виде белого твердого вещества.
К охлажденному до 0°С раствору 2-бутил-4-хлор-имидазол-5-карбоновой кислоты (0,61 г, 3 ммол), 3-бромпропанола (0,52 г, 3,74 ммол) и N,N-диметиламинопиридина (0,08 г, 0,65 ммол) в ТГФ (12 мл) медленно добавляют порциями дициклогексилкарбодиимид (0,91 г, 4,4 ммол) и реакционную смесь перемешивают при комнатной температуре 4 часа. Отфильтровывают образовавшуюся дициклогексилмочевину и органическую фазу концентрируют. Сырой продукт очищают с помощью хроматографии на силикагеле (н-гексан/AcOEt 8:2), получая 3-бромпропиловый эфир 2-бутил-4-хлор-имидазол-5-карбоновой кислоты (0,5 г, 50%) в виде белой пены.
3-Бромпропиловый эфир 2-бутил-4-хлор-имидазол-5-карбоновой кислоты (0,807 г, 2,47 ммол) растворяют в CH3CN (15 мл) и добавляют AgNO3 (0,63 г, 3,7 ммол). Реакционную смесь перемешивают при комнатной температуре 8 часов. Затем отфильтровывают выпавшие в осадок соли серебра, органическую фазу разбавляют AcOEt и промывают NaH2PO4 (5%, 2×10 мл) и рассолом (2×10 мл), сушат над Na2SO4 и концентрируют. Сырой продукт очищают путем флешхроматографии (н-гексан/AcOEt 70:30), получая 3-нитрооксипропиловый эфир 2-бутил-4-хлор-имидазол-5-карбоновой кислоты (0,377 г, 50%).
К охлажденному до 0°С раствору 3-нитрооксипропилового эфира 2-бутил-4-хлор-имидазол-5-карбоновой кислоты (0,76 г, 2,5 ммол) в диметилацетамиде (DMA) (13 мл) медленно порциями в атмосфере азота добавляют трет-бутилат калия (0,28 г, 2,5 ммол). После 10 минут перемешивания добавляют раствор N-(трифенилметил)-5-(4'-бромметилбифенил-2-ил)-тетразола (1,7 г, 3 ммол) в ДФА (10 мл) и смесь перемешивают при комнатной температуре 1 час. Затем смесь распределяют между водой и EtOAc. Органическую фазу отделяют, сушат над Na2SO4 и концентрируют. Сырой продукт очищают путем флешхроматографии (н-гексан/ EtOAc 7:3), получая 3-нитрооксипропиловый эфир 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-карбоновой кислоты (1,56 г, 80%).
Путем кислотного гидролиза, описанного для аналогичного соединения в примере 1, способ А, из 3-нитрооксипропилового эфира 2-бутил-4-хлор-1-[[2'-(1-трифенилметилтетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-карбоновой кислоты (1 г, 1,28 ммол) получают указанное в заголовке соединение (белое твердое вещество) (0,28 г, 40%).
1H-NMR (DMSO-d6: 7.60-7.20 (4H, m); 7.12 (2H, d); 6.92 (2H, d); 5.72 (2H, s); 4.58 (2H, t); 4.50 (2H, t); 2.54 (2H, t); 2.31 (2H, m); 1.49 (2H, m); 1.32 (2H, m); 0.84 (3H, t).
Изучение сосудистого тонуса.
Способность нитропроизводных ARB индуцировать вазорелаксацию по сравнению с природными ARB, исследовали in vitro на изолированных препаратах грудной аорты кролика (Wanstall J.C. et al., Br.J.Pharmacol, 134: 463-472, 2001). Самцов новозеландских кроликов анестезировали тиопенталом натрия (50 мг/кг, в/в), скарифицировали путем обескровливания, затем вскрывали грудную клетку и рассекали аорту. Кольцевые препараты грудной аорты (4 мм длиной) помещали в небольшие кюветы (5 мл) с физиологическим солевым раствором (ФСР) при температуре 37°С. Состав ФСР (мМ): NaCl 130, NaHCO3 14,9, KH2PO4 1,2, MgSO4 1,2, HEPES 10, CaCl2, аскорбиновая кислота 170 и глюкоза 1,1 (95% O2/5% CO2; рН 7,4). Каждое кольцо оставляли под пассивным натяжением 2 g. Изометрическое натяжение регистрировали с помощью Grass-преобразователя (Grass FT03), соединенного с системой BIOPAC MP150. Препараты оставляли для уравновешивания в течение 1 часа, затем вызывали их субмаксимальное сокращение с использованием норадреналина (NA, 1 мкМ) и, когда сокращение стало стабильным, добавляли ацетилхолин (ACh, 10 мкМ). Расслабляющая реакция на ацетилхолин указывает на наличие функционально активного эндотелия. Сосуды, не способные сокращаться под действием норадреналина или не расслабляющиеся под действием ацетилхолина, не учитывали. При достижении стабильного предварительного сокращения, получали кумулятивную кривую доза-ответ для каждого из вазорелаксантов в присутствии функционально активного эндотелия. На каждое артериальное кольцо воздействовали только одной комбинацией ингибитора и вазорелаксанта. Кроме того, действие растворимого ингибитора гуанилилциклазы ODQ (1-Н-(1,2,4)-оксадиазол(4,3-а)хиноксалин-1-он) на вазорелаксацию, вызванную данными соединениями, проверяли предварительным инкубированием колец аорты с ODQ (10 рМ) в течение 20 минут.
Реакцию на вазорелексанты выражали как процент остаточного сокращения и строили диаграмму в зависимости от концентрации исследуемого соединения. Значения IC50 (где IC50 представляет собой концентрацию, обеспечивающую 50% максимального расслабления или максимальной релаксации на исследуемое соединение) интерполировали от этих диаграмм.
В течение экспериментального периода, плато, полученное с NA, было стабильно без значимого спонтанного уменьшения сокращения в кольцах аорты. При этих экспериментальных условиях ARB лозартан не приводил к релаксации при любой исследованной концентрации, кривая не отличалась от кривой, полученной в присутствии одного наполнителя.
Как показано в таблице 1, нитропроизводные по данному изобретению обладают способностью вызывать релаксацию в зависимости от концентрации. В экспериментах, проводимых в присутствии ODQ (10 мкМ), наблюдается ингибирование вазорелаксирующих реакций на тестируемые лекарственные средства.
Действия нитропроизводного лозартана на каскады воспаления in vitro
Эксперименты проводили с использованием моноцит-макрофагальной клеточной линии RAW264.7. Клетки стимулировали в присутствии липополисахарида (LPS) (1 мкг/мл) в течение 16 часов. По окончании инкубации культуральную среду собирали и определяли наличие нитрита с использованием стандартной реакции Грисса (Griess).
Полученные результаты, представленные в таблице 2, выражены как % содержания нитрита для каждой обработки по сравнению с образцами, обработанными LPS.
Как показано в таблице 2, в отличие от родоначального соединения, нитропроизводное (соединение примера 4) обладает способностью ингибировать накопление нитритов, индуцированное LPS.
Изучение антитромбоцитарного действия нитропроизводных лозартана in vitro.
Способность нитропроизводных лозартана ингибировать агрегацию тромбоцитов оценивали in vitro на тромбоцитах человека. Агрегацию тромбоцитов измеряли в образцах богатой тромбоцитами плазмы (PRP) объемом 0,25 мл в соответствии со способом Борна (Gresele P, Arnout J, Deckmyn H, et al., J Clin Invest. 1987; 80: 1435-45). Используемый агент агрегации U46619 представлял собой аналог ТхА2, исходя из того, что этот агонист является чувствительным к действиям оксида азота. Перед добавлением агента агрегации соединения инкубировали 2 минуты при температуре 37°С. Агрегацию проводили в течение 5 минут и измеряли максимальную амплитуду (см). В качестве носителя использовали DMSO (конечная концентрация 0,05%). Соединения тестировали при концентрациях в диапазоне от 10 до 100 мкМ.
Как показано в таблице 3, нитропроизводные обладают способностью существенно ингибировать агрегацию тромбоцитов, индуцированную U46619. Лозартан оказывает слабое действие.
Изучение антигипертензивного действия нитропроизводных лозартана in vivo.
Способность нитропроизводного лозартана (соединение примера 2) снижать кровяное давление оценивали у крыс, испытывающих спонтанную гипертензию (SHR). Две группы крыс SHR (250-300 г) в течение 3 дней получали перорально ежедневную дозу или лозартана (10 мг/кг, перорально), или нитропроизводного лозартана (эквимолярная доза). Систолическое кровяное давление (SBP) и частоту сердечных сокращений контролировали телеметрически в различные временные точки после введения определенных доз лекарственного средства.
Как показано в таблице 4, в отличие от родоначального соединения нитропроизводное (соединение примера 2) обладает способностью индуцировать явное снижение уровней кровяного давления на всем протяжении периода лечения.
Новые химические примеры
Пример 7
3-(Нитрооксиметил)бензиловый эфир 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-карбоновой кислоты (соответствует соединению 16)
Указанное в заголовке соединение получено в соответствии с процедурой, описанной в примере 6, исходя из 2-бутил-4-хлор-имидазол 5-карбоновой кислоты и (3-(хлорметил)фенил)метанола.
1H-NMR (CDCl3): δ 8.11 (1Н, m); 7.58 (2H, m); 7.40 (5H, m); 7.18 (2H, m); 6.96 (2H, m); 5.53 (2H, s); 5,42 (2H, s); 5,28 (2H, s); 2,59 (2H, m); 1.66 (2H, m); 0.88 (3H, t, J=7.3 Hz).
Пример 8
2-[2-(Нитроокси)этокси]этил 2-этокси-3-[2'-(2Н-тетразол-5-ил)-бифенил]-4-ил]метил]-3Н-бензимидазол-4-карбоксилат (соответствует соединению 32)
К раствору коммерческой 2-этокси-3-[2'-(2-тритил-тетразол-5-ил)-бифенил-4-илметил]-3Н-бензимидазол-4-карбоновой кислоты (2 г, 2,93 ммол) и триэтиламина (TEA) (432 мкл, 4,39 ммол) в CH2Cl2 (30 мл) добавляют при перемешивании и 0°С HOBt (400 мг, 2,93 ммол) и HBTU (1,68 г, 4,39 ммол) и реакционную смесь перемешивают в течение 30 мин. Добавляют диэтиленгликоль (280 мкл, 2,93 ммол), реакционной смеси дают достичь комнатной температуры и перемешивают в течение ночи. Органическую фазу промывают
NaH2PO4 (5%, 2×50 мл), NaHCO3 (10%, 2×50 мл) и рассолом, сушат над Na2SO4 и концентрируют. Сырой материал очищают с помощью флешхроматографии и системы BIOTAGE SPI (элюируя EtOAc/гексаном от 5 до 40%), получая 2-(2-гидроксиэтокси)этил 2-этокси-
3-[2'-(2Н-тритил-тетразол-5-ил)-бифенил-4-илметил]-3Н-бензимидазол-4-карбоксилат в виде белой пены (1,3 г, 60%).
1H-NMR (CDCl3): δ 7.9-7.7 (2Н, m); 7.6-7.1 (12H, m); 7.08-6.89 (8H, m); 6.76 (2Н, m); 5.58 (2Н, s); 4.64 (2Н, m); 4,24 (2Н, m); 3,76-3.43 (6H, m); 1,66 (2Н, m); 1.42 (3H, m).
2-(2-Гидроксиэтокси)этил 2-этокси-3-[2'-(2Н-тритил-тетразол-5-ил)-бифенил-4-илметил]-3Н-бензимидазол-4-карбоксилат (3 г, 3,9 ммол), нитрат тетраэтиламмония (1,5 г, 7,8 ммол) и 2,6-ди-трет-бутил-4-метилпиридин (1,2 г, 5,85 ммол) растворяют в CH2Cl2 (90 мл) и раствор охлаждают до -78°С. Медленно добавляют раствор трифторметансульфонового ангидрида (832 мкл, 3,9 ммол) в СН2Сl2 (10 мл) и реакционную смесь перемешивают при -78°С 30 минут, затем медленно нагревают до комнатной температуры и перемешивают 3 часа. Затем гасят NaH2PO4, органическую фазу промывают рассолом, сушат над Na2SO4 и концентрируют. Сырой материал очищают с помощью флешхроматографии и системы BIOTAGE SPI (элюируя EtOAc/гексаном от 5 до 60%) получая 2-[2-(нитроокси)этокси]этил 2-этокси-3-[2'-(2-тритил-тетразол-5-ил)-бифенил]-4-ил]метил]-3Н-бензимидазол-4-карбоксилат в виде белой пены (2,2 г, 70%).
1H-NMR (CDCl3): δ 7.9-7.7 (2Н, m); 7.6-7.1 (12H, m); 7.08-6.89 (8H, m); 6.76 (2Н, m); 5.58 (2Н, s); 4.64 (2Н, m); 4,52 (2Н, m); 4,22 (2Н, m); 3,66 (2Н, m); 3.58 (3H, m); 1.66 (2Н, m); 1.43 (3H, m).
2-[2-(Нитроокси)этокси]этил 2-этокси-3-[2'-(2-тритил-тетразол-5-ил)-бифенил]-4-ил]метил]-3Н-бензимидазол-4-карбоксилат (2,2 г, 5,4 ммол) растворяют в МеОН (40 мл) и раствор подвергают действию микроволнового излучения при 90°С 20 минут, затем концентрируют. Сырой продукт очищают с помощью флешхроматографии и системы BIOTAGE SPI (элюируя CH2Cl2/MeOH 95/5), получая 2-[2-(нитроокси)этокси]этил 2-этокси-3-[2'-(2Н-тетразол-5-ил)-бифенил]-4-ил]метил]-3Н-бензимидазол-4-карбоксилат в виде твердого белого остатка (1,2 г, 80%).
1H-NMR (DMSO-d6): δ 7.71-7.42 (6H, m); 7.2 (1H, t); 7.01 (1H, d); 6.87 (1H, d); 6.87 (1H, d); 5.52 (2Н, s); 4,65 (2Н, m); 4,60 (2Н, q); 4,28 (2Н, m); 3.71-3.61 (4H, dm); 1.39 (3H, t).
Пример 9
4-(Нитроокси)бутил 1-((2'-(1Н-тетразол-5-ил)-бифенил-4-ил)метил)-2-этокси-1Н-бензо[d]имидазол-7-карбоксилат (соответствует соединению 30)
Указанное в заголовке соединение получают в виде твердого белого остатка, следуя процедуре, описанной в примере 8, исходя из 2-этокси-3-[2'-(2-тритил-тетразол-5-ил)-бифенил-4-ил)метил]-3Н-бензоимидазол-4-карбоновой кислоты и 1,4-бутандиола.
1H-NMR (DMSO-d6): δ 7.70-7.40 (6H, m); 7.20 (1H, t); 6.86 (4H, dd); 5.51 (2H, s); 4.56 (2H, q); 4.51 (2H, m); 4,17 (2H, m); 1,69 (4H, m); 1,39 (3Н, m).
Пример 10
4-(1-((2'-(1Н-Тетразол-5-ил)-бифенил-4-ил)метил)-2-этокси-1Н-бензо[d]имидазол-7-карбоксамидо)бутил нитрат
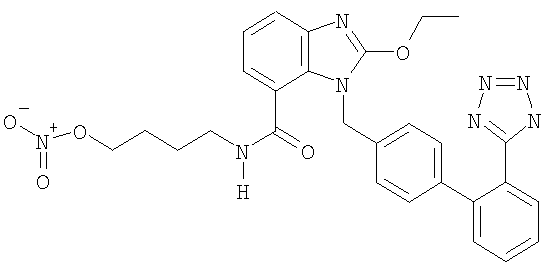
Указанное в заголовке соединение получают, следуя процедуре, описанной в примере 8, исходя из 2-этокси-3-[2'-(2-тритил-тетразол-5-ил)-бифенил-4-ил)метил]-3Н-бензоимидазол-4-карбоновой кислоты и 4-амино-1-бутанола.
1H-NMR (DMSO-d6): δ 8.4 (1H, m); 7.7-7.3 (6H, m); 7.13 (2H, m); 7.03-6.85 (4H, m); 5.42 (2H, s); 4.64-4.40 (4H, m); 3,15 (2H, m); 1,62 (2H, m); 1,52-1.32 (5H, m).
Пример 11
4-(3-(Нитроокси)пропил)фениловый эфир 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-карбоновой кислоты
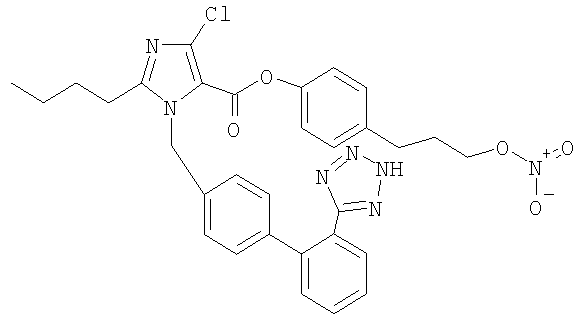
В 22-литровую 4-горлую круглодонную колбу помещают 10 литров воды. Воду охлаждают до 0°С. При 0°С добавляют гидроксид калия (855 г, 15,24 мол) с последующим добавлением калий лозартана (500 г, 1,09 мол), периодата натрия (554 г, 2,59 мол) и хлоргидрата рутения (III) (12 г, 0,05 мол) и реакционную смесь перемешивают при 0°С в течение ночи. Реакционную смесь отфильтровывают. При перемешивании добавляют к фильтрату IPA (90 мл). Раствор нагревают до 25°С и перемешивают 2,5 часов. Через 2,5 часа добавляют фосфорную кислоту (1200 мл), поддерживая температуру ниже +30°С. Смесь перемешивают 30 минут, продукт отфильтровывают и промывают водой. Осадок сушат под вакуумом при 55°С в течение ночи. Твердый остаток растворяют в метаноле (2 литра) и изопропилацетате (12 литров) и добавляют активированный уголь (100 г). Смесь перемешивают при комнатной температуре 3,5 часа, фильтруют и концентрируют. Продукт перерастворяют в CH2Cl2/MeOH и высаживают гептаном с получением 2-бутил-4-хлор-1-{[2'-(1Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновой кислоты (Е3174) в виде зелено/коричневой пены, которую используют на следующих стадиях без очистки.
К раствору Е3174 (234,58 г, 0,54 мол) в CH2Cl2 (4500 мл) добавляют триэтиламин (85 мл, 0,59 мол) с последующим добавлением раствора тритилхлорида (159 г, 0,56 мол) в CH2Cl2(800 мл) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь промывают водой, сушат (MgSO4), фильтруют и концентрируют в вакууме. Проводят хроматографию на силикагеле, используя в качестве элюента 20-80% ацетон/гептан, и получают 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновую кислоту в виде оранжевого твердого остатка.
Реакцию 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновой кислоты с 3-(4-гидроксифенил)-1-пропанолом осуществляют, следуя процедуре, описанной для аналогичного соединения в примере 8, получая 4-(3-(нитроокси)пропил)фениловый эфир 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-карбоновой кислоты в виде белого твердого остатка.
1H-NMR (CDCl3): δ 8.11 (1Н, m); 7.58 (2H, m); 7.42 (1H, m); 7.21 (4H, m); 7.05 (4H, s); 5.58 (2H, s); 4,44 (2H, t, J=6.4 Hz); 2,71 (4H, m); 2,03 (2H, m); 1.72 (2H, m); 1.38 (2H, m), 0.90 (3Н, t, J=7.3 Hz).
Пример 12
4-[2-(Нитроокси)этил]фениловый эфир 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-карбоновой кислоты

Указанное в заголовке соединение получают, следуя процедурам, описанным в примере 8 и примере 11, исходя из 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновой кислоты и 4-гидроксифенилэтанола.
1H-NMR (DMSO-d6): δ 7.71-7.51 (4H, m); 7.36 (2H, m); 7.10-6.99 (6H, m); 5.58 (2H, s); 4.75 (2H, t, J=6.6 Hz); 3.02 (2H, t, J=6.6 Hz); 2,70 (2H, m); 1,56 (2H, m); 1,29 (2H, m); 0.84 (3Н, t, J=7.3 Hz).
Пример 13
2-Метокси-4-((4-(нитроокси)бутокси)карбонил)фениловый эфир 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-карбоновой кислоты
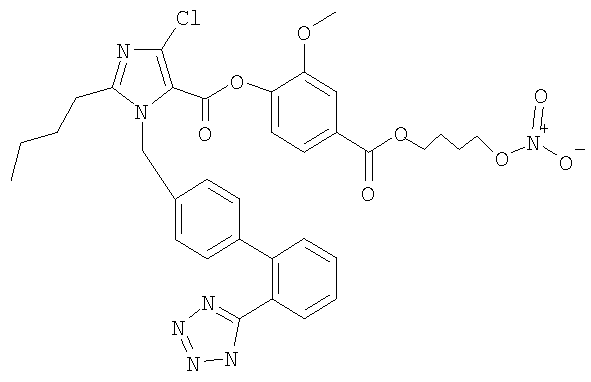
Указанное в заголовке соединение получают, следуя процедурам, описанным в примере 8 и примере 11, исходя из 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновой кислоты и промежуточного соединения 2.
1H-NMR (CDCl3): δ 8.09 (1Н, d); 7.75-7.50 (4Н, m); 7.42 (1Н, m); 7.24-7.00 (5Н, m); 5.58 (2Н, s); 4.62-4.33 (4Н, m); 3,81 (3Н, s); 2,69 (2Н, t); 1,80-1.65 (3Н, m); 1.48-1.20 (6Н, m); 0.91 (3Н, t).
Пример 14
2-Метокси-4-[2-(4-нитроокси-бутоксикарбонил)-винил]фениловый эфир 2-бутил-5-хлор-3-[2'-(2Н-тетразол-5-ил)-бифенил-4-илметил]-3Н-имидазол-4-карбоновой кислоты
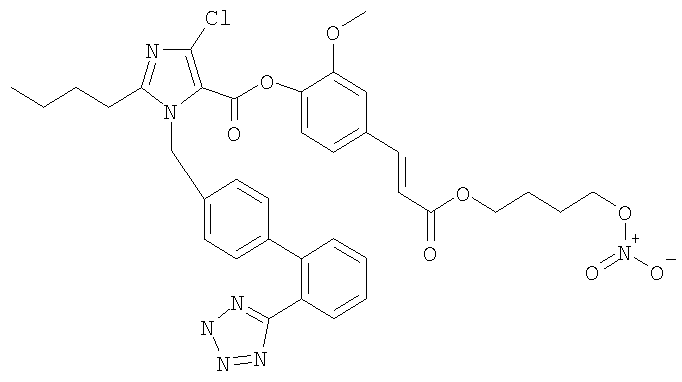
Указанное в заголовке соединение получают, следуя процедурам, описанным в примере 8 и примере 11, исходя из 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновой кислоты и промежуточного соединения 3.
1H-NMR (DMSO-d6): δ 7.74-7.5 (6H, m); 7.37-7.3 (1H, m); 7.2-7.0 (5H, m); 6.8-6.65 (1H, m); 5.60 (2H, s); 4.59 (2H, t); 4,20 (2H, t); 3,76 (3H, s); 2,68 (2H, t); 1.9-1.7 (4H, m); 1.62-1.5 (2H, m); 1.4-1,23 (2H, m); 0,84 (3H, t).
Пример 15
3-(2-(Нитроокси)этил)фениловый эфир 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1H-имидазол-5-карбоновой кислоты

Указанное в заголовке соединение получают, следуя процедурам, описанным в примере 8 и примере 11, исходя из 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновой кислоты и 4-гидроксифенетилового спирта.
1H-NMR (CDCl3): δ 8.13 (1H, m); 7.59 (2H, m); 7.39 (2H, m); 7.22 (2H, m); 7.09 (5H, m); 5.58 (2H, s); 4.65 (2H, t, J=6.8 Hz); 3.05 (2H, t, J=6.8 Hz); 2.69 (2H, m); 1.39 (2H, m); 0.91 (3H, t, J=7.3 Hz).
Пример 16
3-(3-(Нитроокси)пропил)фениловый эфир 2-бутил-4-хлор-1-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1Н-имидазол-5-карбоновой кислоты
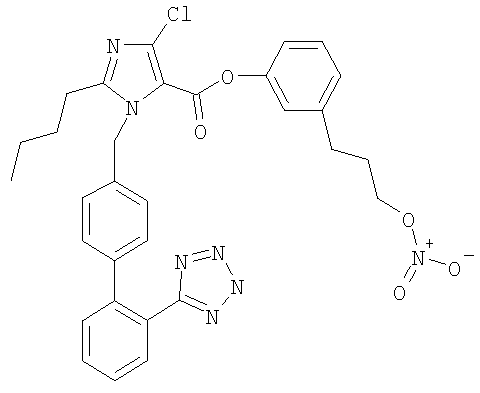
Указанное в заголовке соединение получают, следуя процедурам, описанным в примере 8 и примере 11, исходя из 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил)}-1Н-имидазол-5-карбоновой кислоты и промежуточного соединения 1.
1H-NMR (CDCl3): δ 8.12 (1H, m); 7.58(2H, m); 7.42 (1H, m); 7.28 (4H, m); 7.02 (5H, m); 5.58 (2H, s); 4.44 (2H, t, J=6A Hz); 2.72 (4H, m); 2.05 (2H, m); 1.72 (2H, m); 1.38 (2H, m); 0.90 (3H, t, J=7.3 Hz).
Пример 17
4-(1-((2'-(2Н-Тетразол-5-ил)-бифенил-4-ил)метил)-2-бутил-4-хлор-1Н-имидазол-5-карбоксамидо)бутил нитрат
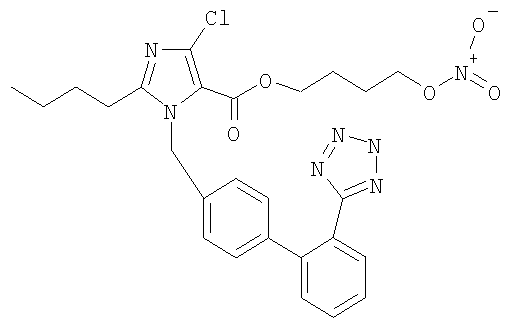
Указанное в заголовке соединение получают, следуя процедурам, описанным в примере 8 и примере 11, исходя из 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновой кислоты и 4-амино-1-бутанола.
1H-NMR (DMSO-d6): δ 8.18 (1H, m); 7.7-7.5 (4H, m); 7.05 (4H, m); 5.42 (2H, s); 4.51 (2H, m); 3.41-3.16 (6H, m); 2.54 (2H, m); 1.68-1.41 (6H, m); 1.23 (2H, m); 0.81 (3H, t, J=7.3 Hz).
Пример 18
2-(2-Аминоэтокси)этил нитрат 1-((2'-(2Н-тетразол-5-ил)-бифенил-4-ил)метил)-2-бутил-4-хлор-1Н-имидазол-5-карбоксамида
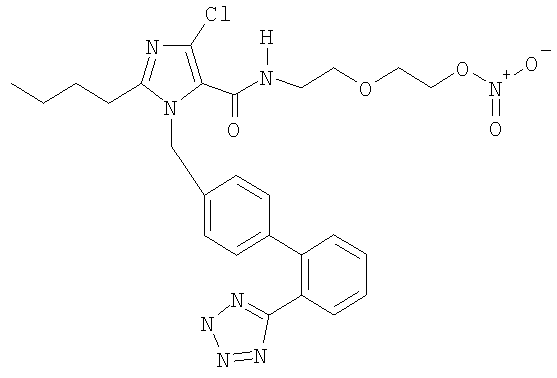
Указанное в заголовке соединение получают, следуя процедурам, описанным в примере 8 и примере 11, исходя из 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновой кислоты и 2-(2-аминоэтокси)этанола.
1H-NMR (DMSO-d6): δ 8.04 (1Н, m); 7.7-7.5 (4H, m); 7.05 (4H, m); 5.46 (2H, s); 4.63 (2H, m); 3.7 (2H, m); 3.49 (2H, m); 3.36 (2H, m); 2.54 (2H, m); 1.46 (2H, m); 1.24 (2H, m); 0.80 (3Н, t, J=7.3 Hz).
Пример 19
1-((2-(2-(Нитроокси)этокси)этокси)карбонилокси)этил 1-((2'-(2Н-тетразол-5-ил)-бифенил-4-ил)метил)-2-бутил-4-хлор-1Н-имидазол-5-карбоксилат
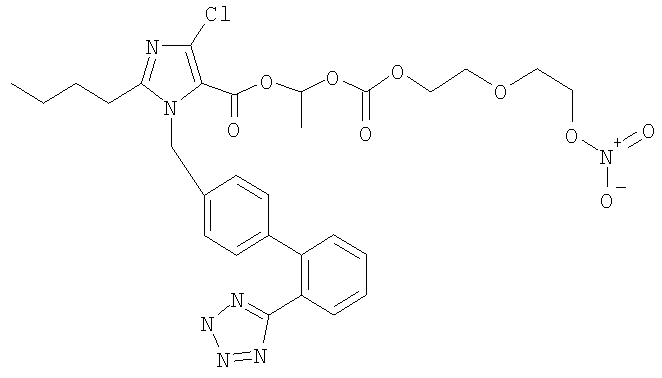
К перемешиваемому раствору 2-бутил-4-хлор-1-{[2'-(2-тритил-2Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-имидазол-5-карбоновой кислоты (полученной, как описано в примере 11) (6,3 г, 9,3 мл) и Cs2CO3 (3,02 г, 9,3 ммол) в ДФА (186 мл) добавляют 1-хлорэтил 2-(2-нитроокси)этокси)этил карбонат (промежуточное соединение 4 (5,98 г, 23,2 ммол)). Раствор перемешивают при 70°С 2 часа. Добавляют воду (50 мл) и раствор экстрагируют EtOAc (3×100 мл). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют под вакуумом. Остаток перерастворяют в МеОН (186 мл). После перемешивания при 70°С в течение 2 часов раствор хроматографируют с помощью системы BIOTAGE SPI (колонка 25+М, элюент CH2Cl2/МеОН 98:2), получая указанное в заголовке соединение (4,20 г, выход 50%).
1H-NMR (DMSO-d6): δ 7.7-7.6 (2H, m); 7.59-7.48 (2H, m); 7.08 (2H, d); 6.98 (2H, d); 6.75 (1H, g); 5.61-5.48 (2H, m); 4.65-4.59 (2H, m); 4.3-4.11 (2H, m); 3.75-3.67 (2H, m); 3.65-3.4 (2H, m); 2.62 (2H, t); 1.61-1.41 (5H, m); 1.32-1.2 (2H, m); 0.8 (3Н, t).
Пример 20
2-((2-(2-(Нитроокси)этокси)этокси)карбонилокси)пропан-2-ил 1-((2'-(2Н-тетразол-5-ил)-бифенил-4-ил)метил)-2-этокси-1Н-бензо[d]имидазол-7-карбоксилат
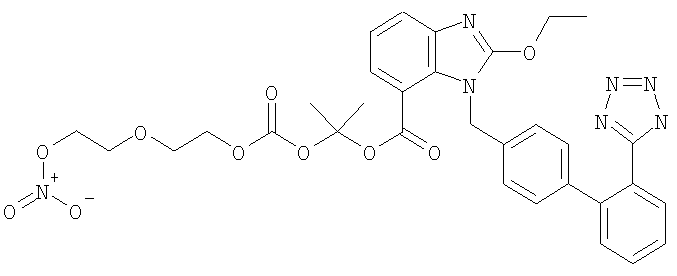
Оранжевую суспензию оксида ртути (1,17 г, 5,39 ммол) и 2-этокси-1-{[2'-(1-тритил-1Н-тетразол-5-ил)бифенил-4-ил]метил}-1Н-бензимидазол-7-карбоновой кислоты (7,36 г, 10,8 ммол) в сухом тетрагидрофуране (95 мл) перемешивают при комнатной температуре 24 часа. Затем добавляют 2-хлоризопропил p-нитрофенил карбоната (полученного, как описано в US 5684018) (1,40 г, 5,390 ммол) и реакционную смесь перемешивают при комнатной температуре в течение 7 дней и контролируют с помощью ТСХ (гексан/этилацетат 6/4). Смесь разбавляют дихлорметаном, промывают водой, органический слой сушат над Na2SO4 и концентрируют. Остаток очищают с помощью BIOTAGE (элюент EtOAc/гексан от 7 до 70%), получая 1-метил-1-{[4-нитрофенокси)карбонил]окси}этил 3-этокси-1-{[2'-(1-тритил-1Н-тетразол-5-ил)-бифенил-4-ил)метил)-1Н-бензимидазол-7-карбоксилат (2,20 г, выход 30%).
Получение 2-(2-гидроксиэтокси)этил нитрата
К раствору 2-(2-хлорэтокси)этанола (0,68 мл, 6,43 ммол) в ацетонитриле (20 мл) добавляют AgNO3 (2,73 г, 16,6 ммол). Смесь нагревают в микроволновом устройстве (320 минут, 110°С). Отфильтровывают серебряные соли и раствор концентрируют, получая указанное в заголовке соединение (0,97 г, выход 100%). К перемешиваемому раствору 1-метил-1-{[4-нитрофенокси)карбонил]окси}этил 2-этокси-1-{[2'-(1-тритил-1Н-тетразол-5-ил)-бифенил-4-ил)метил)-1Н-бензимидазол-7-карбоксилата (0,90 г, 0,99 ммол) в дихлорметане (10 мл) добавляют 2-(2-гидроксиэтокси)этил нитрат (0,22 г, 1,49 ммол) и N,N-диметиламинопиридин (0,12 г, 0,99 ммол). Раствор перемешивают при комнатной температуре 18 часов. Затем его промывают раствором NaH2PO4 (5%, 2×40 мл) и рассолом (40 мл). Органический слой сушат над
Na2SO4, концентрируют и очищают с помощью BIOTAGE (элюент EtOAc/гексан от 25 до 100%), получая 1-метил-1-[2-(2-нитрооксиэтокси)-этоксикарбонилокси]-этиловый эфир 2-этокси-3-[2'-(1-тритил-1Н-тетразол-5-ил)-бифенил-4-ил)метил]-3Н-бензимидазол-4-карбоновой кислоты (0,38 г, 38%) в виде белого твердого остатка. Раствор 1-метил-1-[2-(2-нитроокси-этокси)-этоксикарбонилокси]-этилового эфира 2-этокси-3-[2'-(1-тритил-1Н-тетразол-5-ил)-бифенил-4-ил)метил)-3Н-бензимидазол-4-карбоновой кислоты (Стадия С, 0,3 8 г, 0,41 ммол) в дихлорметане/метаноле (8 мл, 1:7) перемешивают при комнатной температуре 7 дней. Затем раствор концентрируют и остаток очищают с помощью BIOTAGE (элюент МеОН/CH2Cl2 2/98), получая 1-метил-1-[2-(2-нитроокси-этокси)-этоксикарбонилокси]-этиловый эфир 2-этокси-3-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил)метил]-3Н-бензимидазол-4-карбоновой кислоты (0,19 г, 68%).
1Н NMR (300 MHz, CDCl3): δ 8.03 (m, 1H), 7.70-7.47 (m, 3Н), 7.34 (m, 1H), 7.06-6.79 (m, 4H), 6.72 (d, 2H), 6.81 (d, 3Н), 5.63 (s, 2H), 4.56 (t, 2H), 4.46-4.28 (m, 2H), 4.20 (t, 2H), 3.87-3.59 (m, 4H), 1.70 (s, 6H), 1.46 (t, 6Н).
Промежуточное соединение 1
3-(3-Гидроксипропил)фенол
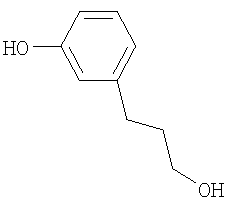
3-(3-Метоксифенил)-пропионовую кислоту (500 мг, 2,77 ммол) и NaJ (4,15 г, 2,77 ммол) суспендируют в CH3CN (10 мл). Добавляют триметилхлорсилан (3,5 мл, 2,74 ммол) и смесь нагревают с обратным холодильником 4 часа. Смесь распределяют между Na2S2O4 (10%) и EtOAc. Водную фазу экстрагируют EtOAc и объединенные органические фазы промывают рассолом, сушат над Na2SO4 и концентрируют, получая 3-(3-гидроксифенил)пропановую кислоту в виде прозрачного масла (410 мг, 89%).
1H-NMR (CDCl3): δ 7.19 (1H, m); 6.77 (3Н, m); 2.89 (2H, m); 2.68 (2H, m).
3-(3-Гидроксифенил)пропановую кислоту (460 мг, 2,77 ммол) растворяют в ТГФ, раствор охлаждают до 0°С, медленно добавляют ВН3·ТГФ (1М, 3 мл). Реакционную смесь перемешивают в течение ночи при комнатной температуре, затем гасят ледяной водой и твердым К2СО3. Водную фазу экстрагируют EtOAc и объединенные органические фазы промывают рассолом, сушат над Na2SO4 и концентрируют, получая 3-(3-гидроксипропил)фенол (340 мг, 80%) в виде прозрачного масла.
1H-NMR (CDCl3): δ 7.15 (1H, m); 6.77 (1H, m); 6.67 (1H, m); 3.68 (2H, t, J=6.4 Hz); 2.67 (2H, t, J=6.0 Hz); 1.89 (2H, m).
Промежуточное соединение 2
4-(Нитроокси)бутил 4-гидрокси-3-метоксибензоат
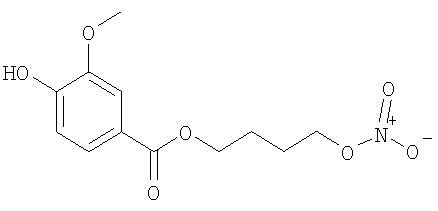
К раствору ванилиновой кислоты (1,5 г, 8,92 ммол) в тетрагидрофуране (100 мл) добавляют трифенилфосфин (4,7 г, 17,8 ммол) и тетрабромметан (5,92 г, 17,8 ммол). Смесь перемешивают при комнатной температуре 4 часа, затем фильтруют и растворитель упаривают под вакуумом. Сырой остаток очищают с помощью хроматографии на силикагеле (элюент н-гексан/этилацетат 8/2), получая 4-бромбутил-4-гидрокси-3-метоксибензоат в виде твердого продукта (2,44 г, 90,4%).
Раствор 4-бромбутил 4-гидрокси-3-метоксибензоата (0,285 г, 0,94 ммол) и нитрата серебра (0,4 г, 2,3 ммол) в ацетонитриле (15 мл) нагревают (115°С, 900 сек) в микроволновом агрегате Emrys™ Creator (Personal Chemistry). Отфильтровывают выпавший осадок (соли серебра) и растворитель упаривают под вакуумом. Остаток очищают с помощью флешхроматографии (элюент н-гексан/этилацетат 75/25), получая 4-(нитроокси)бутил 4-гидрокси-3-метоксибензоат в виде белого порошка (0,117 г, 43,7%).
1H-NMR (CDCl3): δ 7.63 (1H, dd); 7.55 (1H, d); 6.96 (1H, d); 6.05 (1H, s); 4.55 (2H, t); 4.36 (2H, t); 3.97 (3H, s); 1.92 (4H, m).
Промежуточное соединение 3
(Е)-4-(Нитроокси)бутил 3-(4-гидрокси-3-метоксифенил)акрилат
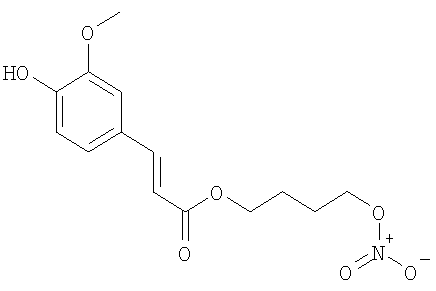
К раствору ванилиновой кислоты (1,5 г, 8,92 ммол) в тетрагидрофуране (100 мл) добавляют трифенилфосфин (4,7 г, 17,8 ммол) и тетрабромметан (5,92 г, 17,8 ммол). Смесь перемешивают при комнатной температуре 4 часа, затем фильтруют и растворитель упаривают под вакуумом. Сырой остаток очищают с помощью хроматографии на силикагеле (элюент н-гексан/этилацетат 8/2), получая (Е)-4-бромбутил 3-(4-гидрокси-3-метоксифенил)акрилат в виде твердого продукта. (2,44 г, 90,4%). Т.пл.83-88°С.
Раствор (Е)-4-бромбутил 3-(4-гидрокси-3-метоксифенил)акрилата (0,285 г, 0,94 ммол) и нитрата серебра (0,4 г, 2,3 ммол) в ацетонитриле (15 мл) нагревают в микроволновом агрегате Emrys™ Creator (Personal Chemistry) (115°C, 900 сек). Отфильтровывают выпавший осадок (соли серебра) и растворитель упаривают под вакуумом. Остаток очищают с помощью флешхроматографии (элюент н-гексан/этилацетат 75/25), получая (Е)-4-(нитроокси)бутил 3-(4-гидрокси-3-метоксифенил)акрилат в виде белого порошка (0,117 г, 43,7%).
1H-NMR (CDCl3): 7.64 (1H, d); 7.11 (1H, m); 7.06 (1H, s); 6.95 (1H, d); 6.30 (1H, d); 6.02 (1H, s); 4.54 (2H, t); 4.27 (2H, t); 3.95 (3H, s); 1.92-1.85 (4H, m).
Промежуточное соединение 4
1-Хлорэтил 2-(2-(нитроокси)этокси)этил карбонат

1-Хлорэтил хлорформиат (6,42 мл, 58,9 ммол) добавляют к перемешиваемому раствору 2-(2-гидроксиэтокси)этил нитрата (8,08 г, 53,5 ммол) и триэтиламина (8,95 мл, 64,2 ммол) в CH2Cl2 (268 мл) при 0°С. Через 2 часа раствор промывают водой, сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают колоночной хроматографией на силикагеле, элюируя 5-40% EtOAc/гексаном с получением указанного в заголовке соединения в виде бесцветного масла (5,51 г; выход 40%).
1H-NMR (CDCl3): δ 6.44 (1Н, q); 4.64 (2H, m); 4.37 (2H, m); 3.90-3.65 (4H, m); 1.86 (3H, d).
Пример 21
(S)-3-(Нитрооксиметил) бензил 2-(N-{(2'-(1H-тетразол-5-ил)бифенил-4-ил)метил)пентанамидо)-3-метилбутаноат (соответствует соединению 22)
К раствору 3-(хлорметил)бензил нитрата (0,115 г, 0,57 ммол) в CH2Cl2 (6 мл) и ДФА (0,5 мл) добавляют валсартан (250 мг, 0,57 ммол), дициклогексилкарбодиимид (135 мг, 0,66 ммол) и диметиламинопиридин (4 мг). Полученную суспензию перемешивают при комнатной температуре около 2 часов и затем упаривают растворитель, получая сырой остаток, который очищают с помощью флешхроматографии (элюент EtOAc 100%). Полученный продукт растирают в порошок с гексаном, получая указанное в заголовке соединение (129,7 г, выход 38%) в виде белого твердого остатка.
1Н NMR (CDCl3) δ 8.20-8.10 (m, 1Н); 7.62-7.52 (m, 2H); 7.41-7.32 (m, 6H); 7.21-7.02 (m, 3H); 5.37 (s, 2H); 4.97-4.32 (m, 5H); 2.60-2.21 (m, 3H); 1.64-1.57 (m, 2H); 1.39-1.14 (m, 2H); 1.10-0.86 (m, 9H).
Пример 22
(S)-4-(Нитрооксиметил)бензил 2-(N-((2'-(1Н-тетразол-5-ил)бифенил-4-ил)метил)пентанамидо)-3-метилбутаноат (соответствует соединению 23)
Указанное в заголовке соединение получают, исходя из валтарсана и 4-(хлорметил)бензил нитрата, следуя процедуре, описанной в примере 21.
1Н NMR (CDCl3) δ 8.25-8.15 (1Н, m); 7.63-7.49 (2H, m);7.4-7.0 (9H, m); 5.38 (2H, d); 5.05-5.6 (4H, m); 4.35 (0.5H, d); 4.12 (0.5H, d); 2.62-2.2 (3H, m); 1.8-1.55 (2, m); 1.45-1.25 (2H, m); 0.99 (3Н, d); 0.95-0.82 (6H, m).
Новые фармакологические данные.
Исследования на сосудистом тонусе.
Способность нитропроизводных ARB вызывать понижение тонуса сосудов в сравнении с нативными ARB была протестирована in vitro на отделенной препарированной грудной аорте кролика (Wanstall J.С. et al., Br. J. Pharmacol., 134: 463-472, 2001). Самцов кроликов Новой Зеландии анестезировали тиопенталом -Na (50 мг/кг, в/в), убивали путем кровопускания, затем вскрывали грудную клетку и извлекали аорту. Препарированное артериальное кольцо (4 мм в длину) было помещено в физиологический солевой раствор (ФСР) при 37°С в маленькие контейнеры для органов (5 мл). ФСР состоял из (мМ): NaCl 130, NaHCO3 14.9, КН2РО4 1.2, MgSO4 1.2, HEPES 10, CaCl2, аскорбиновая кислота 170 и глюкоза 1.1 (95% O2 / 5% CO2; рН 7.4). Каждое кольцо готовили для микроскопического исследования под инертным давлением 2g. Изометрическое давление регистрировали измерительным преобразователем Grass (Grass FT03), подключенным к BIOPAC MP150 System. Препараты стабилизировали в течение 1 часа, а затем законтрактовывали сублимацией с норадреналином (НА, 1 мкМ) и когда контрактирование стабилизировалось, добавляли ацетилхолин (АХ, 10 мкМ). Релаксационный ответ на АХ показывает наличие функционального эндотелия. Сосуды, которые не способны контрастировать с НА или не показывают релаксационный ответ на АХ, удаляют. Когда стабильность преконтрактирования была достигнута, строили суммарную кривую зависимости концентрация - ответ на любой сосудорасширяющий агент в присутствии функционального эндотелия. Каждое артериальное кольцо подвергалось только одной комбинации ингибитора и сосудорасширяющего вещества. Кроме того, эффект растворимого ингибитора гуанилил циклазы ODQ (1-Н-(1,2,4)-оксадиазол(4,3-а)хиноксалин-1-он)на вызванное соединениями сосуд орасширение был рассмотрен при инкубировании аортных колец с ODQ (10 мкМ) в течение 20 минут.
Ответы на релаксирующие агенты выражались как процентное содержание остаточного контактирования и наносились на кривую в зависимости от концентрации тестируемой композиции. Значения IC50 (где IC50 - концентрация, приводящая к 50% максимальной релаксации в ответ на исследуемое соединение) интерполировали из этих графиков. В течение экспериментального периода полученный плоский участок с НА был стабильным без особых потерь контактирования в артериальных кольцах. При этих экспериментальных условиях ARB лозартан не вызывал релаксацию при любой исследуемой концентрации, кривая не отличалась от кривой в присутствии носителя.
Как показано в таблице 1, нитропроизводные данного изобретения способны стимулировать релаксацию в концентрационно - зависимом методе. Кроме того, при исполнении эксперимента в присутствии ODQ (10 мкМ) происходило ингибирование ответного понижения тонуса сосудов на тестируемое вещество.
Исследование гипертензивной активности (in vitro).
Способность веществ данного изобретения уменьшать кровяное давление определяли на крысе, страдающей гипертонией (SHRs). SHRs (250-300 г) получали разовые пероральные дозы исследуемого вещества (1, 3 или 10 мг/кг). Систолическое кровяное давление (СКД) и сердце крысы исследовали дистанционным измерением через 24 часа после получения дозы исследуемого вещества. СКД определяли перед (исходный уровень) и в различные временные точки (то есть 2-6, 12, 21-24 ч) с последующей пероральной обработкой исследуемыми веществами. Данные обрабатывали, как абсолютную величину или как дельта между абсолютной величиной и исходной величиной.
Для исследовали систолического давления, диастолического давления, среднего артериального давления, скорости сердца крысы и двигательной активности была использована телеметрическая система Dataquest IV (Data Sciences International). Система наблюдения состоит из радиопередатчика (модель радиочастотного датчика ТА11РА), панели приемного датчика, объединяющей модели и персонального компьютера с сопровождающим программным обеспечением. Перед встраиванием аппарата его калибрововали с точностью до ±3 мм рт.ст. Крысы были анестезированы кетамин/ксилазин/ацепромазином и вводили гибкий катетер радиопередатчика хирургическим путем, закрепляли его на брюшной аорте непосредственно ниже почечной артерии. После введения накладывали подкожно шов. После операции крысам предоставили отдельные клетки. Каждая клетка была оснащена приемным датчиком, который соединялся с персональным компьютером для исследования данных. Крысы были не фиксированы и могли свободно перемещаться в своих клетках. Гемодинамические данные образцов измеряли каждые 2 минуты в течение 10 секунд.
По сравнению с Лозартаном 10 мг/кг (контрольное вещество) заявленные вещества обеспечивали низкий ВР и продолжительное действие с продленным пик - эффектом, принимая во внимание, что вещества не дозированы в эквимолярном отношении (см. данные таблицы 2).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТЕТРАЗОЛА | 1992 |
|
RU2091376C1 |
| АЦИЛАЛЫ ИМИДАЗОЛ-5-КАРБОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ СВОЙСТВА АНТАГОНИСТА АНГИОТЕНЗИНА II РЕЦЕПТОРА | 1992 |
|
RU2105764C1 |
| ЧЕТЫРЕХЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2174513C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2838028C2 |
| ПРОИЗВОДНЫЕ ПРОПИОНОВЫХ КИСЛОТ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АКТИВАТОРОВ hPPARs. | 2003 |
|
RU2316539C2 |
| АНТИГИПЕРТЕНЗИВНЫЕ СОЕДИНЕНИЯ ДВОЙНОГО ДЕЙСТВИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2476427C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 1992 |
|
RU2017733C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОНИЖАЮЩАЯ КРОВЯНОЕ ДАВЛЕНИЕ | 1992 |
|
RU2104272C1 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2728783C2 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2047604C1 |
Настоящее изобретение относится к новым нитропроизводным блокаторов рецепторов ангиотензина II формулы  (значения радикалов R, Y и s приведены в формуле изобретения), обладающим широким фармакологическим действием и высокой переносимостью. Новые соединения могут использоваться для лечения сердечно-сосудистых заболеваний, заболеваний почек, хронических заболеваний печени и воспалительных процессов. 1 н. и 5 з.п. ф-лы, 6 табл.
(значения радикалов R, Y и s приведены в формуле изобретения), обладающим широким фармакологическим действием и высокой переносимостью. Новые соединения могут использоваться для лечения сердечно-сосудистых заболеваний, заболеваний почек, хронических заболеваний печени и воспалительных процессов. 1 н. и 5 з.п. ф-лы, 6 табл.
1. (57) Соединения общей формулы (I) или их фармацевтически приемлемые соли или стереоизомеры

где s равно 1 или 2;
R выбирают из следующих остатков блокаторов рецептора ангиотензина II формулы (II)

где R0 представляет
R1 выбирают из группы, состоящей из
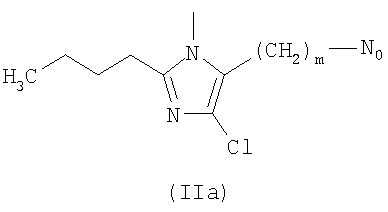
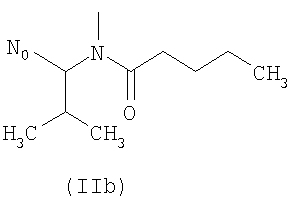
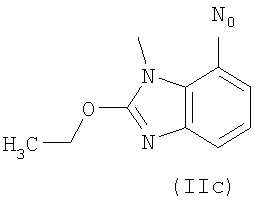
где m целое число, равное 0 или 1, и No представляет -СОО-, CONH-, -ОСО- или
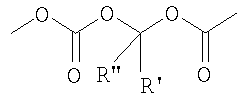
Y представляет собой двухвалентный радикал, имеющий следующие значения:
a)
прямой или разветвленный C1-C20 алкилен, преимущественно C1-C10,
b)
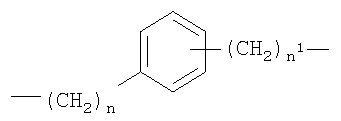
где n представляет целое число от 0 до 20, и n1 представляет целое число от 0 до 20;
d)
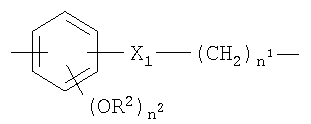
где n1 как указано выше и n2 представляет целое число от 0 до 2;
X1=-OCO- или -COO- и R2 представляет собой Н или СН3;
e) 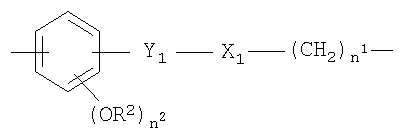
где n1, n2, R2 и X1 как указано выше;
Y1 представляет собой -СН2-СН2- или -СН=СН-(СН2)n 2 - при условии, что когда
Y выбирают из двухвалентных радикалов, указанных под b)-е), группа -ONO2 связана с группой - (CH2)n 1;
g)
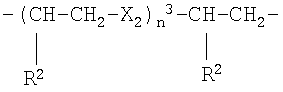
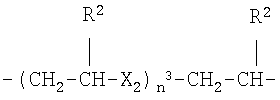
где Х2 представляет -О- или -S-, n3 представляет целое число от 1 до 6,
преимущественно от 1 до 4, R2 как указано выше;
2. Соединения общей формулы (I) или их фармацевтические приемлемые соли или стереоизомеры по п.1, где Y представляет двухвалентный радикал, имеющий следующие значения:
a)
прямой или разветвленный C1-C10 алкилен;
b)

где n целое число, равное 0 или 1, и n1 целое число, равное 1; при условии, что группа -ONO 2 связана с группой -(СН2)n1;
g)
где Х2 представляет -O-, n3 целое число, равное 1 и R2 представляет Н;
3. Соединение по п.1 или 2, выбранное из группы, состоящей из
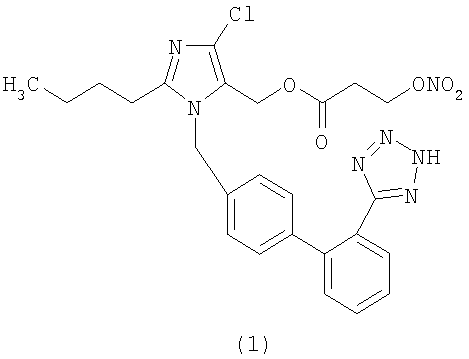
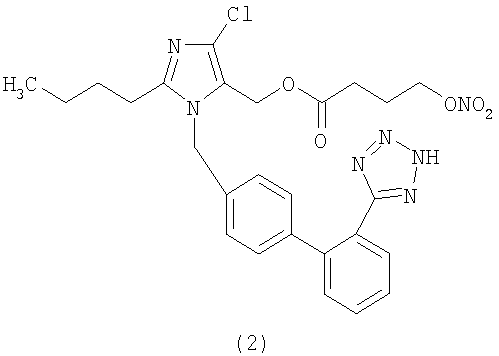
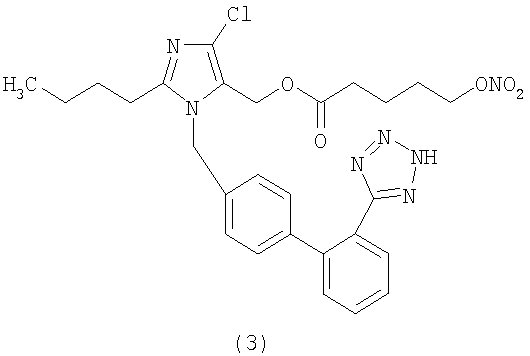
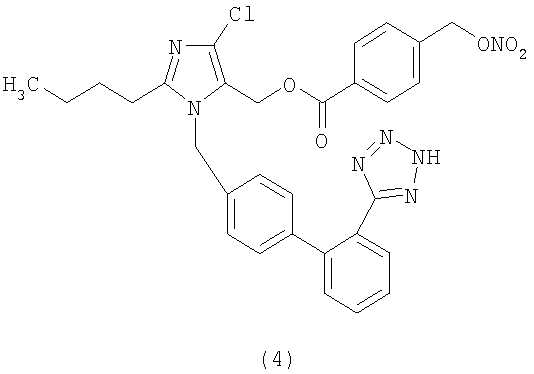

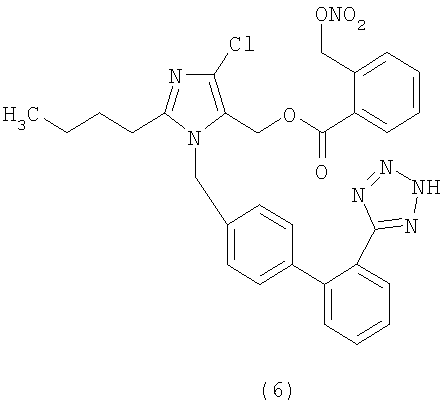
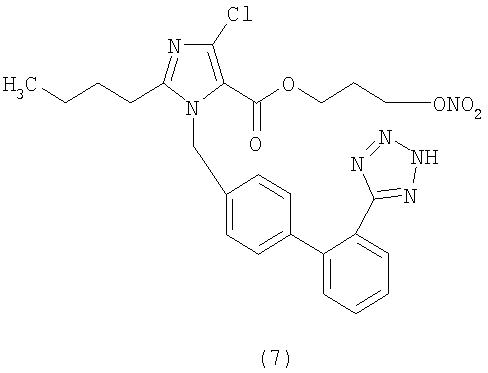


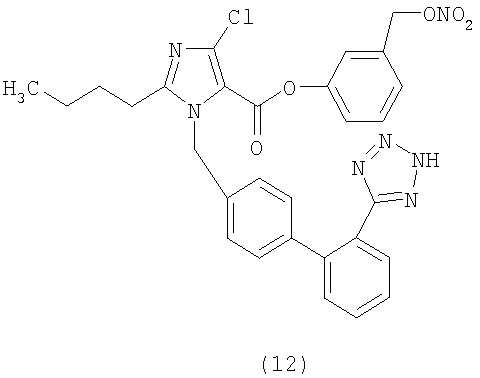
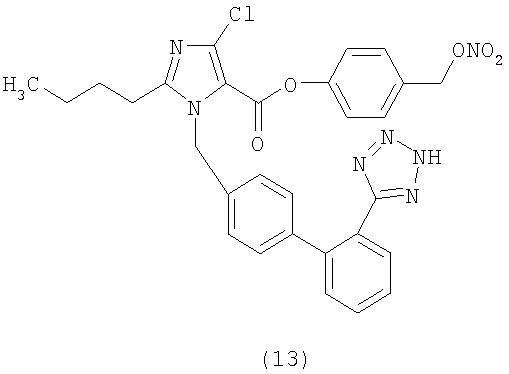
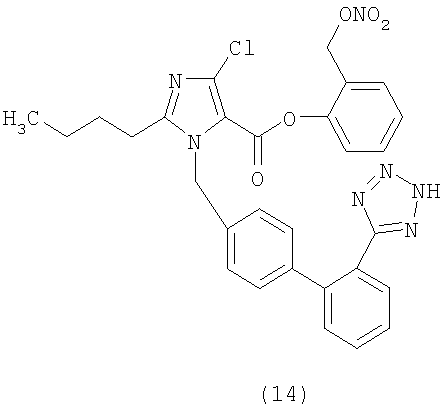
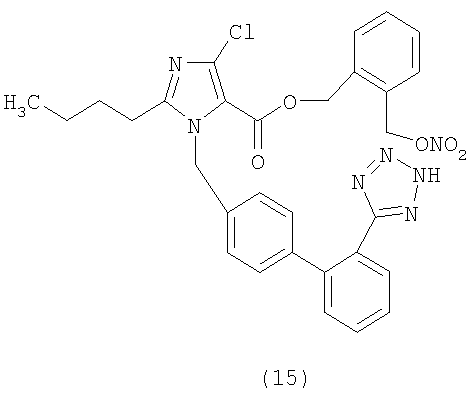
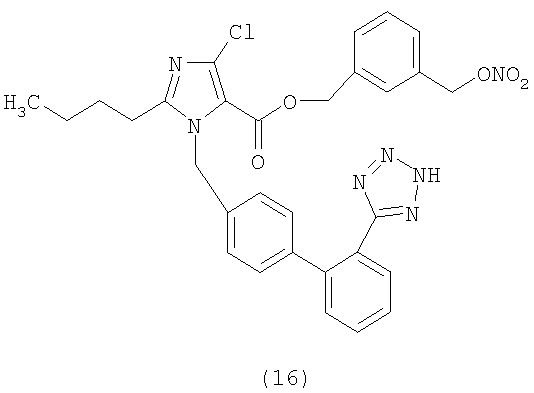
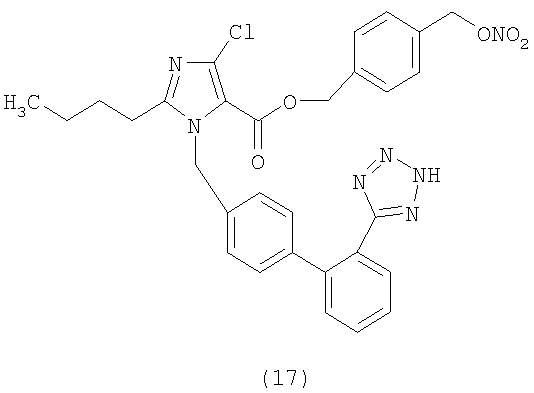
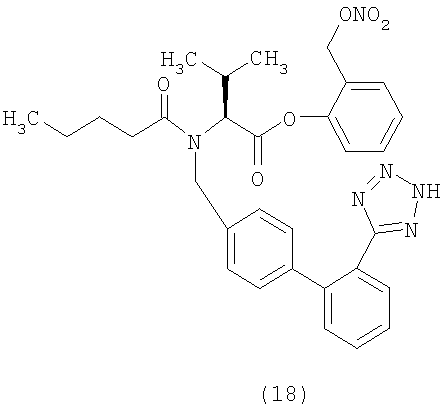
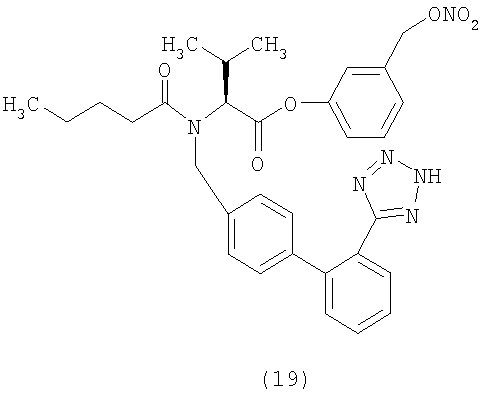
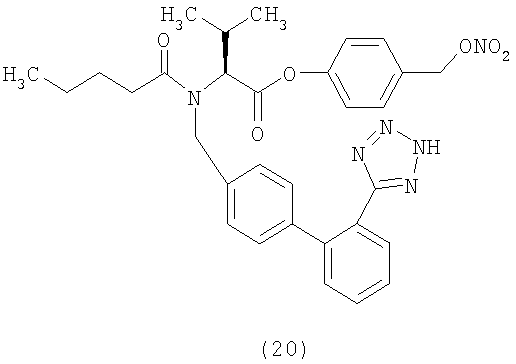
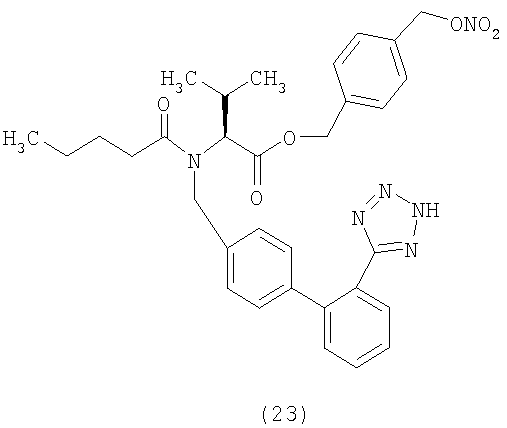
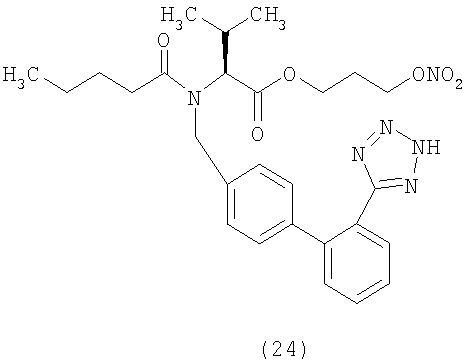
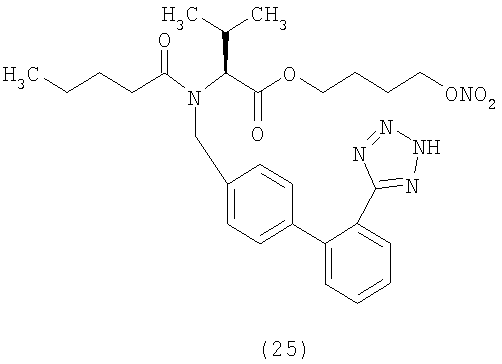
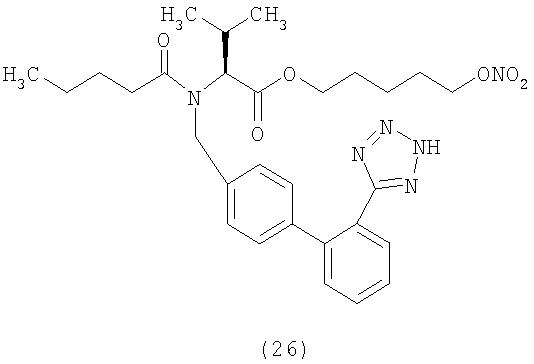


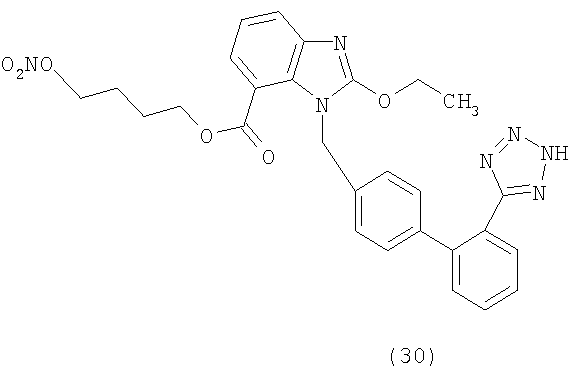
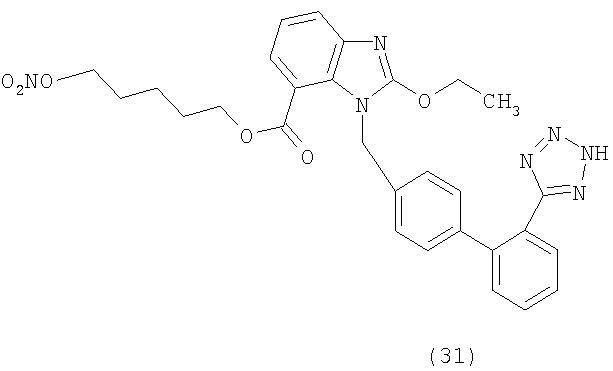

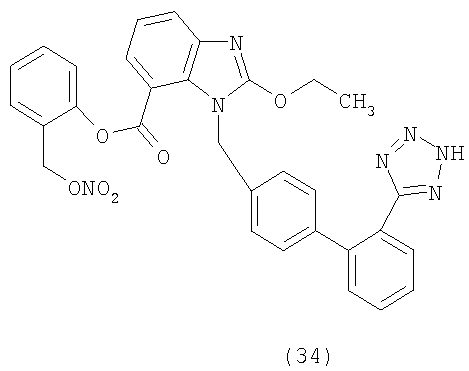
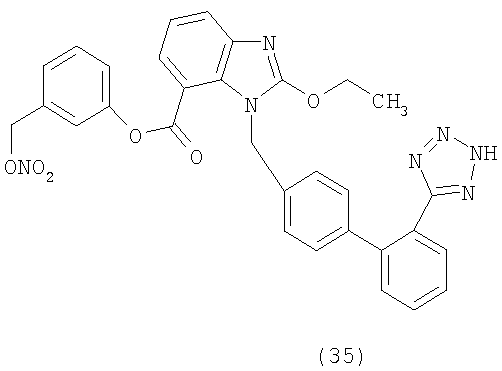
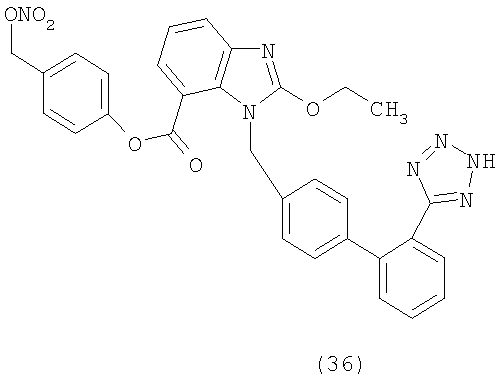
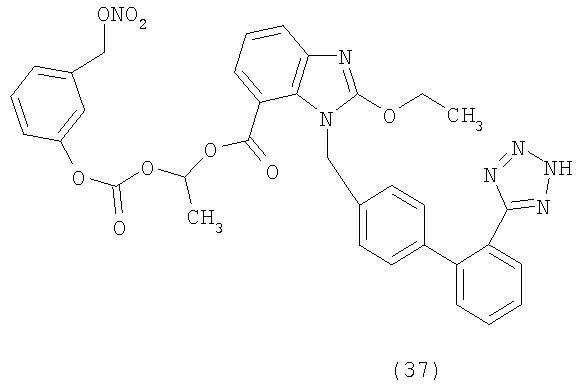
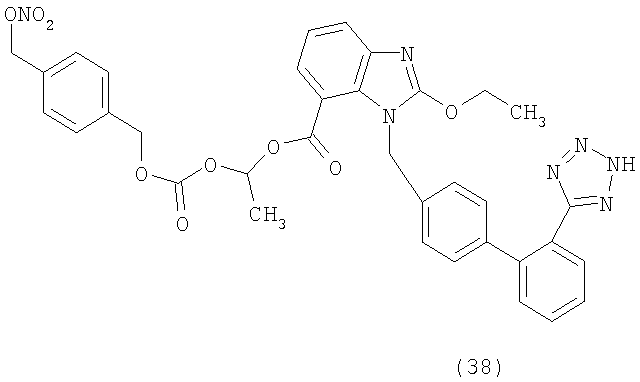
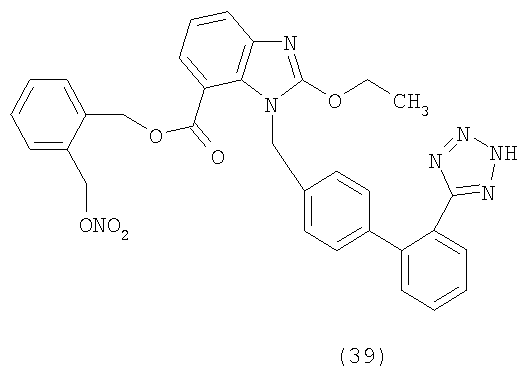
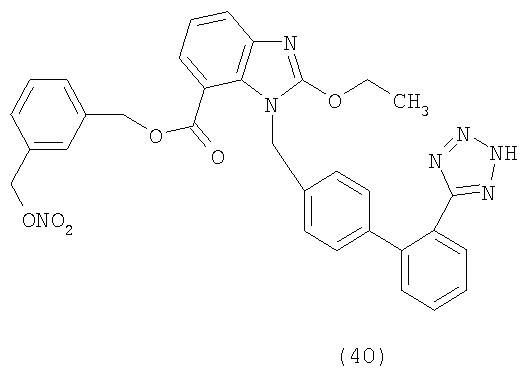

4. Соединения общей формулы (I) по пп.1-3 для применения в качестве лекарственного средства.
5. Соединение общей формулы (1) по пп.1-3 для применения в качестве лекарственного средства, обладающего противовоспалительной, антитромботической и антитромбоцитарной активностью.
6. Соединение общей формулы (1) по пп.1-3 для применения в качестве лекарственного средства, обладающего антигипертензивной активностью.

