Настоящее изобретение относится к соединениям и способам лечения заболеваний, опосредованных простагландинами, и к их определенным фармацевтическим композициям. Более конкретно соединения по настоящему изобретению отличаются по своей структуре от стероидов, антигистаминных агентов или агонистов адренергических рецепторов и представляют собой антагонисты застойных назальных и легочных эффектов простагландинов D-типа.
Две обзорные статьи описывают свойства и терапевтическое значение простаноидных рецепторов, а также наиболее часто применяющиеся селективные агонисты и антагонисты: Eicosanoids: From Biotechnology to Therapeutic Applications, Folco, Samuelsson, Maclouf, and Velo eds., Plenum Press, New York, 1996, глава 14, 137-154, и Journal of Lipid Mediators and Cell Signalling, 1996, 14, 83-87. В статье T. Tsuri et al., опубликованной в 1997 г. в Journal of Medicinal Chemistry, том 40, стр. 3504-3507, указывается, что "PGD2 считают важным медиатором при различных аллергических заболеваниях, таких как аллергический ринит, атопическая астма, аллергический конъюнктивит и атопический дерматит". Более поздняя статья Matsuoka et al. в Science (2000), 287: 2013-7 описывает PGD2 как ключевой медиатор при аллергической астме. Кроме того, патенты, такие как US 4808608, раскрывают антагонисты простагландинов, пригодные для лечения аллергических заболеваний и, главным образом, для лечения аллергической астмы. Антагонисты PGD2 описаны, например, в европейской патентной заявке 837052 и в РСТ заявке WO98/25919, а также WO99/62555.
Настоящее изобретение относится к новым соединениям, которые являются антагонистами простагландиновых рецепторов, более конкретно они являются антагонистами рецепторов простагландина D2 (рецепторов DP). Соединения по настоящему изобретению являются пригодными для лечения различных заболеваний и нарушений, опосредованных простагландинами, соответственно, настоящее изобретение относится к способу лечения заболеваний, опосредованных простагландинами, с помощью новых соединений, описанных в настоящем документе, а также к содержащим их фармацевтическим композициям.
Настоящее изобретение относится с соединениям формулы I:

и их фармацевтически приемлемым солям и гидратам, где
А выбран из С1-3 алкила, необязательно замещенного одним-четырьмя атомами галогена, О(СН2)1-2 и S(СН2)1-2;
Ar представляет собой арил или гетероарил, каждый необязательно замещенный одной-четырьмя группами, независимо выбранными из Rg;
Q выбран из:
(1) СООН,
(2) CONRaRb,
(3) C(O)NHSO2Rc,
(4) SO2NHRa,
(5) SO3H,
(6) PO3H2 и
(7) тетразолила;
один из Х1, Х2, Х3 или Х4 представляет собой азот, а другие независимо выбраны из СН и С-Rg;
Y1 выбран из -(CRdRe)a-X-(CRdRe)b-, фенилена, С3-6 циклоалкилидена и С3-6 циклоалкилена, где а и b представляют собой целые числа 0-1, таким образом, что сумма а и b равняется 0, 1 или 2, а Х представляет собой связь, O, S, NRa, C(O), CH(ORa), OC(O), C(O)O, C(O)NRa, OC(O)NRa, NRaC(O), CRd=CRe или С≡С;
Y2 выбран из (CRdRe)m- и CRd=CRe;
R1 выбран из H, CN, ORa, S(O)nC1-6 алкила и C1-6 алкила, необязательно замещенного одной-шестью группами, независимо выбранными из галогена, ORa и S(O)nC1-6 алкила;
R2 выбран из H и C1-6 алкила, необязательно замещенного одним-шестью атомами галогена; или
R1 и R2 вместе представляют собой оксо; или
R1 и R2 вместе образуют 3- или 4-членное кольцо, содержащее 0 или 1 гетероатом, выбранный из NRf, S и О, необязательно замещенный одной или двумя группами, выбранными из F, CF3 и СН3;
R3 выбран из H и C1-6 алкила, необязательно замещенного одной-шестью группами, независимо выбранными из ORa и галогена;
Ra и Rb независимо выбраны из H, C1-10 алкила, C2-10 алкенила, C2-10 алкинила, Cy и Cy C1-10 алкила, где указанные алкил, алкенил, алкинил и Cy необязательно замещены одним-шестью заместителями, независимо выбранными из галогена, аминогруппы, карбоксильной группы, C1-4 алкила, C1-4 алкиокси, арила, гетероарила, арил C1-4 алкила, гидроксильной группы, CF3, OC(O)C1-4 алкила, OC(O)NRiRj и арилокси; или Ra и Rb вместе с атомом (атомами), к которому они присоединены, образуют гетероциклическое кольцо из 4-7 членов, содержащее 0-2 дополнительных гетероатома, независимо выбранных из кислорода, серы и N-Rf;
Rс выбран из C1-6 алкила, необязательно замещенного одним-шестью атомами галогена, арилом и гетероарилом, где указанные арил и гетероарил необязательно замещены одной-тремя группами, выбранными из галогена, OC1-6 алкила, О-галогенC1-6 алкила, C1-6 алкила и галогенC1-6 алкила;
Rd и Re независимо представляют собой Н, галоген, арил, гетероарил, C1-6 алкил или галогенC1-6 алкил;
Rf выбран из Н, C1-6 алкила, галогенC1-6 алкила, Cy, C(O)C1-6 алкила, C(O)галогенC1-6 алкила и C(O)-Cy;
Rg выбран из
(1) галогена,
(2) CN,
(3) C1-6 алкила, необязательно замещенного одной-восемью группами, независимо выбранными из арила, гетероарила, галогена, NRaRb, C(O)Ra, C(ORa)RaRb, SRa и ORa, где арил, гетероарил и алкил каждый необязательно замещены одной-шестью группами, независимо выбранными из галогена, CF3 и СООН,
(4) C2-6 алкенила, необязательно замещенного одной-шестью группами, независимо выбранными из галогена и ORa,
(5) Cy,
(6) C(O)Ra,
(7) C(O)ORa,
(8) CONRaRb,
(9) OCONRaRb,
(10) OC1-6 алкила, где алкил необязательно замещен одним-шестью заместителями, выбранными из галогена, арила, гетероарила, ОН и OC(O)Ra,
(11) O-Cy,
(12) S(О)nC1-6 алкила, где алкил необязательно замещен одним-шестью заместителями, выбранными из галогена, арила, гетероарила, ОН и OC(O)Ra,
(13) S(О)n-Cy,
(14) -NRaS(O)nRb,
(15) -NRaRb,
(16) -NRaC(O)Rb,
(17) -NRaC(O)ORb,
(18) -NRaC(O)NRaRb,
(19) S(O)nNRaRb,
(20) NO2,
(21) С5-8 циклоалкенила,
где Cy необязательно замещен одной-восемью группами, независимо выбранными из галогена, C(O)Ra, ORa, С1-3 алкила, арила, гетероарила и CF3;
Ri и Rj независимо выбраны из водорода, C1-10 алкила, Cy и Cy-C1-10 алкила; или Ri и Rj вместе с атомом азота, к которому они присоединены, образуют кольцо из 5-7 членов, содержащее 0-2 дополнительных гетероатома, независимо выбранных из кислорода, серы и N-Rf;
Cy выбран из гетероциклила, арила и гетероарила;
m равен 1, 2 или 3; и
n равен 0, 1 или 2.
Настоящее изобретение относится также к фармацевтическим композициям, содержащим соединение формулы I, и к способам лечения или профилактики заболеваний, опосредованных простагландинами, с использованием соединений формулы I.
В настоящем изобретении используются, если не указано иное, следующие определения.
Термин "галоген" включает F, Cl, Br и I.
Термин "алкил" относится к линейным, разветвленным и циклическим и бициклическим структурам и их комбинациям, содержащим указанное количество атомов. Неограничивающие примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил, ундецил, додецил, тридецил, тетрадецил, пентадецил, эйкозил, 3,7-диэтил-2,2-диметил-4-пропилнонил, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопропилметил, циклопентилэтил, циклопропил, замещенный метилом, циклобутил, замещенный этилом, адамантил, циклододецилметил, 2-этил-1-бицикло[4.4.0]децил и т.п. Например, термин С-6 алкил включает ациклические алкильные группы, имеющие указанное количество атомов углерода, а также -Схалкил-Czциклоалкил, где х равен от 0 до 3, а z равен от 3 до 6, при условии, что x+z = от 3 до 6.
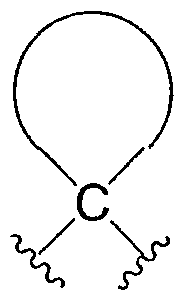
"Циклоалкилиден" относится к следующему двухвалентному радикалу, где точки присоединения находятся на одном и том же атоме углерода:
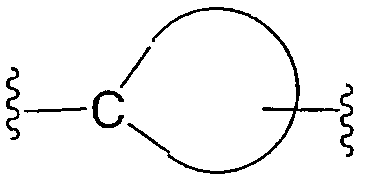
"Циклоалкилен" относится к следующему двухвалентному радикалу, где точки присоединения находятся на разных атомах углерода:
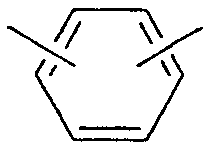
"Фенилен" относится к следующему двухвалентному радикалу и включает 1,2-фенилен, 1,3-фенилен и 1,4-фенилен:
"Галогеналкил" означает алкильную группу, как описано выше, где один или более атомов водорода были заменены на атомы галогена, вплоть до полного замещения всех атомов водорода галогеновыми группами. С1-6 галогеналкил, например, включает -CF3, -CH2CF3, -CF2CF3 и т.п.
"Алкокси" означает алкоксильные группы линейной, разветвленной или циклической конфигурации, имеющие указанное количество атомов углерода. С1-6 алкокси, например, включает метокси, этокси, пропокси, изопропокси и т.п.
"Галогеналкокси" означает алкоксильную группу, как описано выше, в которой один или более атомов водорода были заменены на атомы галогена, вплоть до полного замещения всех атомов водорода галогеновыми группами. С1-6 галогеналкокси, например, включает -OCF3, -OCH2CF3, -OCF2CF3 и т.п.
"Алкенил" означает линейные или разветвленные структуры и их комбинации с указанным количеством атомов углерода, имеющие по меньшей мере одну углерод-углеродную двойную связь, где водород может быть заменен на дополнительную углерод-углеродную двойную связь. С2-6 алкенил, например, включает этенил, пропенил, 1-метилэтенил, бутенил и т.п.
"Гетероциклил" относится к неароматическому кольцу, имеющему от 1 до 4 гетероатомов; указанное кольцо изолировано или конденсировано со вторым кольцом, выбранным из от 3- до 7-членного алициклического кольца, содержащего от 0 до 4 гетероатомов, арила и гетероарила, где указанные гетероатомы независимо выбраны из O, N и S. Неограничивающие примеры гетероциклила включают оксетанил, 1,3-дитиациклопентан, дигидробензофуран и т.п.
"Арил" означает 6-14-членную карбоциклическую ароматическую кольцевую систему, включающую 1-3 бензольных кольца. Если присутствует два или более ароматических колец, то кольца слиты вместе, таким образом, что соседние кольца имеют общую связь. Примеры включают фенил и нафтил.
Термин "гетероарил" (Het), использующийся в настоящем документе, представляет 5-10-членную ароматическую кольцевую систему, содержащую одно кольцо или два конденсированных кольца, 1-4 гетероатома, выбранных из O, S и N. Het включает, без ограничения, тетразолил, бензотиенил, хинолинил, бензотиазолил, фуранил, пиримидинил, пуринил, нафтиридинил, имидазолил, изоксазолил, изотиазолил, оксадиазолил, оксазолил, пиразолил, пиридил, пирролил, тетразинил, тиазолил, тиадизолил, тиенил, триазинил, триазолил, 1Н-пиррол-2,5-дионил, 2-пирон, 4-пирон, пирролопиридин, фуропиридин и тиенопиридин.
"Терапевтически эффективное количество" означает такое количество лекарственного средства или фармацевтического агента, которое будет вызывать биологический или медицинский ответ в ткани, системе, у животного или человека, который требуется исследователю, ветеринару, врачу или другому клиницисту.
Термин "лечение" включает облегчение, улучшение или какое-либо другое уменьшение признаков и симптомов, связанных с заболеванием или нарушением.
Термин "профилактика" означает предотвращение или задержку начала или прогрессирования заболевания или нарушения или признаков и симптомов, связанных с указанным заболеванием или нарушением.
Термин "композиция", как в фармацевтической композиции, относится к продукту, включающему активный ингредиент (ингредиенты) и инертный ингредиент (ингредиенты) (фармацевтически приемлемые наполнители), которые составляют носитель, а также к любому продукту, который получается, прямо или опосредованно, в результате объединения, комплексирования или агрегации любых двух или более ингредиентов, или в результате диссоциации одного или более ингредиентов, или в результате других типов реакций или взаимодействий одного или более ингредиентов. Соответственно, фармацевтические композиции по настоящему изобретению включают любую композицию, изготовленную смешиванием соединения формулы I и фармацевтически приемлемых наполнителей.
Для соединений формулы I примеры А включают, без ограничения, CH2, CH2CH2, CH2CH(CH3), CH(Cl), CH2CF2CH2, CH(Cl)CH2CH(F), OCH2, OCH2CH2, SCH2 и SCH2CH2. Примеры Q включают, без ограничения, CO2H, CONH2, CONHCH3, CONHPh, CON(CH3)2, CON(CH2)4, CONHSO2CH3, SO2NHPh, тетразолил и т.п.
Примеры Y1 включают, без ограничения, CH2, CH2CH2, CH2CH(CH3), CH(Cl), CH(Ph), CH2CH(CF3), CF2CH2, CH(Cl)CH2CH(F), OCH2, OCH2CH2, SCH2, CH2SCH2, S, O, C(O), CH2C(O), CH2C(O)O, CH2C(O)OCH2, NH, NHC(O), CH2NHC(O), CH2NHC(O)CH2, CH=CH, CH2CH=CHCH2, CH2C≡C, 1,4-фенилен, 1,1-циклопропилиден, 1,3-циклогексилен и т.п.
Примеры Ar включают, без ограничения, фенил, 2-, 3-, 4-хлорфенил, 2-, 3-, 4-бромфенил, 2-, 3-, 4-фторфенил, 3,4-дихлорфенил, 2,3-дихлорфенил, 2,4-дихлорфенил, 2,5-дихлорфенил, 2,6-дихлорфенил, 3,5-дихлорфенил, 3-хлор-4-фторфенил, 2-хлор-4-фторфенил, 4-хлор-2-фторфенил, 2-цианофенил, 4-метилфенил, 4-изопропилфенил, 4-трифторметилфенил, бифенил, нафтил, 3-метоксифенил, 3-карбоксифенил, 2-карбоксамидофенил, 4-метоксифенил, 3-феноксифенил, 4-(4-пиридил)фенил, 4-метилсульфонилфенил, 3-диметиламинофенил, 5-тетразолил, 1-метил-5-тетразолил, 2-метил-5-тетразолил, 2-бензотиенил, 2-бензофуранил, 2-индолил, 2-хинолинил, 7-хинолинил, 2-бензотиазолил, 2-бензимидазолил, 1-бензотриазолил, 2-фуранил, 3-фуранил, 2-имидазолил, 5-имидазолил, 5-изоксазолил, 4-изоксазолил, 4-изотиазолил, 1,2,4-оксадиазол-5-ил, 2-оксазолил, 4-оксазолил, 4-пиразолил, 5-пиразолил, 2-пиридил, 3-пиридил, 2-пиразинил, 5-пиримдинил, 2-пирролил, 4-тиазолил, 1,2,4-тиадиазол-3-ил, 1,2,5-тиадиазол-4-ил, 1,2,3-тиадиазол-4-ил, 1,2,5-оксадиазол-4-ил, 1,2,3-оксадиазол-4-ил, 1,2,4-триазол-5-ил, 1,2,3-триазол-4-ил, 3-тиенил, 1,2,4-триазол-5-ил, пирролопиридин, фуро[3,2-b]пиридин-2-ил, тиено[2,3-b]пиридин-2-ил, 5(Н)-2-оксо-4-фуранил, 5(Н)-2-оксо-5-фуранил, (1Н,4Н)-5-оксо-1,2,4-триазол-3-ил, 4-оксо-2-бензопиранил и т.п.
Примеры Y2 включают, без ограничения, CH2, CH2CH2, СН2СН2СН2, CH(Cl), CCl2, CH(CF3), CH(Ph), CH2CHCl, C(Cl)=CH2, C(Cl)=C(Cl), CH=CH, CH=C(CF3) и т.п.
Примеры X1, X2, X3 и X4 включают, без ограничения, N, CH, C-CH3, C-CH(CH3)2, C-Ph, C-Cl, C-Br, C-F, C-CF3, C-C(O)CH3, C-C(O)OH, C-C(O)NH2, C-C(O)N(CH2)2O(CH2)2, C-OCH3, C-OCF3, C-OPh, C-SCH3, C-SOCH3, C-SO2CH3, C-SO2Ph, C-NH2, C-N(CH3)2, C-N(CH3)C(O)CH3, C-N(CH3)C(O)OCH3, C-NHC(O)NHCH3, С-циклопропил, С-циклобутил, С-циклопентил и т.п.
Примеры R1 включают, без ограничения, водород, циано, CH3, CH2CH3, CF3, CH2CH2Cl, циклопропил и т.п.
Примеры R2 включают, без ограничения, водород, CH3, CH2CH3, CF3, CH2CH2Cl, циклопропил и т.п.
Примеры R3 включают, без ограничения, водород, CH3, CH2CH3, CF3, CH2CH2Cl, CH2CH2ОН, циклопропил и т.п.
В одном варианте осуществления формулы I часть A-Q представляет собой CH2CO2H.
Во втором варианте осуществления формулы I присутствуют соединения, в которых часть Y1-Ar представляет собой S-арил или С(О)-арил, где указанный арил представляет собой нафтил или фенил, необязательно замещенный 1-2 группами, выбранными из Rg. В одной их разновидности Y1-Ar представляет собой S-фенил, необязательно замещенный 1-2 группами, выбранными из галогена, C1-6 алкила и трифторметила.
В третьем варианте осуществления формулы I присутствуют соединения, в которых Х1 представляет собой азот, а Х2, Х3 и Х4 независимо выбраны из СН и CRg. В одной их разновидности один из Х2, Х3 и Х4 представляет собой CRg, а остальные представляют собой СН. В другой их разновидности один из Х2, Х3 и Х4 представляет собой CH, а остальные представляют собой CRg.
В четвертом варианте осуществления формулы I присутствуют соединения, в которых Х3 представляет собой азот, а Х1, Х2 и Х4 независимо выбраны из СН и CRg. В одной их разновидности один из Х1, Х2 и Х4 представляет собой CRg, а остальные представляют собой СН. В другой их разновидности один из Х1, Х2 и Х4 представляет собой CH, а остальные представляют собой CRg.
В пятом варианте осуществления формулы I присутствуют соединения, в которых один из Х1, Х2 или Х3 представляет собой азот, а другие представляют собой СН или CRg, а Х4 представляет собой CRg. В одной их разновидности один из Х1, Х2 или Х3 представляет собой азот, а остальные представляют собой СН или С-С1-6 алкил, а Х4 представляет собой C-S(O)n-C1-6 алкил или С-С1-6 алкил, необязательно замещенный ORa.
В шестом варианте осуществления формулы I присутствуют соединения, в которых Y2 выбран из СН2 и СН2СН2.
В седьмом варианте осуществления формулы I присутствуют соединения, в которых каждый из R1, R2 и R3 представляет собой водород или R1 и R2 вместе представляют собой оксо, а R3 представляет собой водород.
Одна группа соединений формулы I представлена формулой Ia:
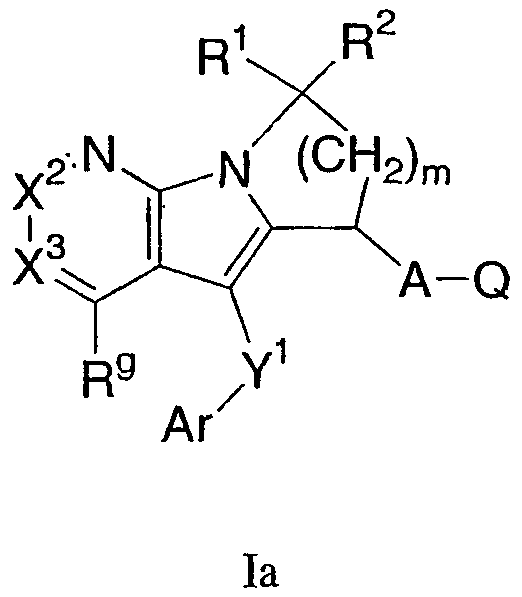
где Х2 и Х3 независимо представляют собой СН или C-Rg, A, Ar, Q, Y1, R1, R2, m и Rg, как определены формулой I. В одном варианте осуществления формулы Ia присутствуют соединения, в которых Х2 и Х3 каждый представляет собой СН. В другом варианте осуществления присутствуют соединения, в которых R1 и R2 каждый представляет собой Н. В еще одном варианте осуществления A-Q представляет собой СН2СО2Н. В еще одном варианте осуществления Y1-Ar представляет собой S-фенил, необязательно замещенный 1-2 группами, независимо выбранными из галогена, С1-6 алкила и трифторметила. В еще одном варианте осуществления Rg выбран из SO2-С1-6 алкила и С1-6 алкила.
Другая группа соединений формулы I представлена формулой Ib:
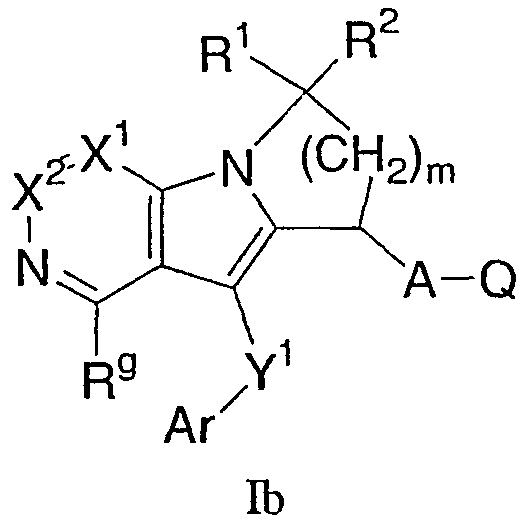
где Х1 и Х2 независимо представляют собой СН или C-Rg, A, Ar, Q, Y1, R1, R2, m и Rg, как определены формулой I. В одном варианте осуществления формулы Ib присутствуют соединения, в которых Х1 и Х2 каждый представляет собой СН. В другом варианте осуществления присутствуют соединения, в которых R1 и R2 каждый представляет собой Н. В еще одном варианте осуществления A-Q представляет собой CH2CO2H. В еще одном варианте осуществления Y1-Ar представляет собой S-фенил, необязательно замещенный 1-2 группами, независимо выбранными из галогена, С1-6 алкила и трифторметила. В еще одном варианте осуществления Rg выбран из SO2-С1-6 алкила и С1-6 алкила.
Другая группа соединений формулы I представлена формулой Ic:
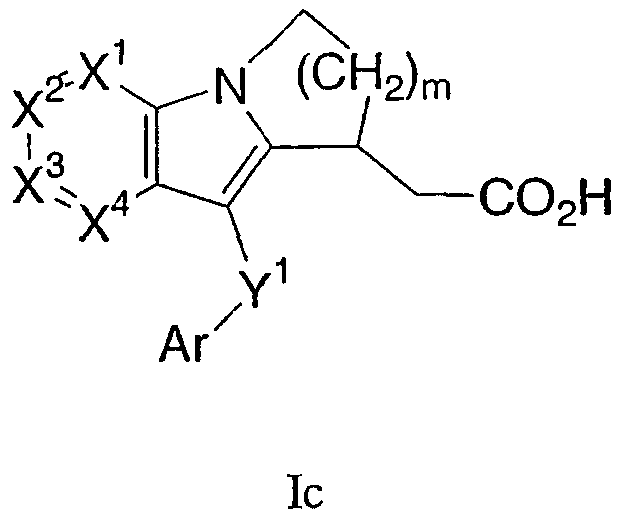
где один из Х1, Х2 и Х3 представляет собой N, а другие представляют собой каждый СН, Х4 представляет собой CRg, m равно 1 или 2, и Ar, Y1 и m определены в формуле I. В одном варианте осуществления Ar представляет собой фенил, необязательно замещенный 1 или 2 группами, независимо выбранными из галогена, С1-3 алкила и трифторметила. В другом варианте осуществления Y1 представляет собой S или С(О). В еще одном варианте осуществления Х4 выбран из С-S(O)n-С1-6 алкила и С-С1-6 алкила, необязательно замещенного ORa.
Показательные примеры соединений формулы I показаны в следующих таблицах:
Таблица 1
Таблица 2
Для целей данного описания следующие сокращения имеют указанное значение:
Ас = ацетил
АсО = ацетат
ВОС = трет-бутилоксикарбонил
CBZ = карбобензокси
CDI = карбонилдиимидазол
DCC= 1,3-дициклогексилкарбодиимид
DCE = 1,2-дихлорэтан
DIBAL = гидрид диизобутилалюминия
DIEA = N,N-диизопропилэтиламин
DMAP = 4-(диметиламино)пиридин
DMF = диметилформамид
EDCI = гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида
EDTA = гидрат тетранатриевой соли этилендиаминтетрауксусной кислоты
FAB = бомбардировка быстрыми атомами
FMOC = 9-флуоренилметоксикарбонил
НМРА = гексаметилфосфорамид
HATU = гексафторфосфат O-(7-азабензотриазол-1-ил)N,N,N',N'-тетраметилурония
HOBt = 1-гидроксибензотриазол
HRMS = масс-спектрометрия высокого разрешения
ICBF = изобутилхлорформиат
KHMDS = гексаметилдисилазан калия
LDA = диизопропиламид лития
MCPBA = метахлорпербензойная кислота
ММРР = гексагидрат монопероксифталата магния
Ms = метансульфонил = мезил
MsO = метансульфонат = мезилат
NBS = N-бромсукцинимид
NMM = 4-метилморфолин
NMP = N-метилпирролидинон
РСС = хлорхромат пиридиния
PDC = дихромат пиридиния
Ph = фенил
PPTS = п-толуолсульфонат пиридиния
pTSA = п-толуолсульфоновая кислота
PyH·Br3 = пиридингидробромидпербромид
r.t./RT = комнатная температура
rac. = рацемический
TFA = трифторуксусная кислота
TfO = трифторметансульфонат = трифлат
THF = тетрагидрофуран
TLC = тонкослойная хроматография
TMSCI = триметилсилилхлорид
Сокращения для алкильных групп:
Me = метил
Et = этил
n-Pr = нормальный пропил
i-Pr = изопропил
c-Pr = циклопропил
n-Bu = нормальный бутил
i-Bu = изобутил
c-Bu = циклобутил
s-Bu = вторичный бутил
t-Bu = третичный бутил
Оптические изомеры - диастереомеры - таутомеры
Соединения формулы I содержат один или более асимметричных центров и могут, таким образом, находиться в форме рацематов и рацемических смесей, одиночных энантиомеров, диастереомерных смесей и отдельных диастереомеров. Настоящее изобретение относится ко всем указанным изомерным формам соединений формулы I.
Некоторые из соединений, описанных в настоящем документе, могут иметь различные точки присоединения водорода; они называются таутомерами. Подобным примером может являться кетон и его енольная форма, известные как кето-енольные таутомеры. Отдельные таутомеры, а также их смеси, охватываются соединениями формулы I.
Соединения формулы I могут быть разделены на диастереоизомерные пары энантиомеров, например, фракционной кристаллизацией из подходящего растворителя, например, метанола или этилацетата или их смеси. Полученная таким способом пара энантиомеров может быть разделена на отдельные стереоизомеры обычными способами, например, использованием оптически активной кислоты или основания в качестве разделяющего агента, или методикой хирального разделения, такой как разделение с помощью ВЭЖХ c использованием хиральной колонки.
Альтернативно любой энантиомер соединения общей формулы I можно получить стереоспецифичным синтезом с использованием оптически чистых исходных материалов или агентов известной конфигурации.
Соли
Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований, включая неорганические основания и органические основания. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и т.п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая натуральные замещенные амины, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
В случае, когда соединение по настоящему изобретению является основным, соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Указанные кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоту и т.п. Особенно предпочтительными являются лимонная, бромистоводородная, хлористоводородная, малеиновая, фосфорная, серная и винная кислоты.
Следует понимать, что, если не указано иное, ссылки на соединение формулы I включают также фармацевтически приемлемые соли.
Полезность
Соединения формулы I являются антагонистами простагландина D2. Способность соединений формулы I взаимодействовать с рецепторами простагландина D2 делает их пригодными для профилактики или снижения нежелательных симптомов, вызванных простагландинами у млекопитающего, особенно у человека. Антагонизм в отношении действий простагландина D2 указывает, что соединения и их фармацевтические композиции являются пригодными для лечения, профилактики или улучшения у млекопитающих и особенно у людей, респираторных состояний, аллергических состояний, боли, воспалительных состояний, нарушений секреции слизи, заболеваний костей, расстройств сна, нарушений фертильности, нарушений свертывания крови, проблем со зрением, а также иммунных и аутоиммунных расстройств. Помимо этого, подобное соединение может ингибировать клеточные неопластические трансформации и метастатический рост опухолей и, следовательно, может быть использовано для лечения злокачественных опухолей. Соединения формулы I можно использовать также для лечения и/или профилактики нарушений пролиферации, опосредованных простагландином D2, таких как нарушения при диабетической ретинопатии и опухолевом ангиогенезе. Соединения формулы I могут также ингибировать простаноид-индуцированное сокращение гладкой мускулатуры путем противодействия контрактильным простаноидам или имитирования действия релаксирующих простаноидов и, следовательно, их можно использовать для лечения дисменореи, преждевременных родов и нарушений, связанных с эозинофилами.
Соответственно, другой аспект настоящего изобретения относится к способу лечения или профилактики заболевания, опосредованного простагландином D2, включающему введение млекопитающему, нуждающемуся в указанном лечении, соединения формулы I в количестве, которое является эффективным для лечения или профилактики указанного заболевания, опосредованного простагландином D2. Заболевания, опосредованные простагландином D2, включают, без ограничения, аллергический ринит, застойные явления в носовой полости, ринорею, круглогодичный насморк, назальное воспаление, астму, включая аллергическую астму, хронические обструктивные заболеваний легких и другие формы воспаления легких; легочную гипотензию; нарушения сна и нарушения цикла сон-бодрствование; простаноид-индуцированное сокращение гладкой мускулатуры, связанное с дисменореей и преждевременными родами; нарушения, связанные с эозинофилами; тромбоз; глаукому и нарушения зрения; окклюзионные заболевания сосудов, такие как, например, атеросклероз; застойную сердечную недостаточность; заболевания или состояния, требующие лечения, предотвращающего свертывание крови, такого как лечение после травмы или оперативного вмешательства; ревматоидный артрит и другие воспалительные заболевания; гангрену; болезнь Рейно; нарушения секреции слизи, включая защиту клеток; боль и мигрень; заболевания, требующие контроля образования и резорбции кости, такие как, например, остеопороз; шок; регуляцию температуры, включая лихорадку; отторжение трансплантированного органа и при хирургическом обходном шунтировании и иммунные нарушения или состояния, при которых желателен иммунорегулирующий эффект. Более конкретно заболевание, подлежащее лечению, представляет собой заболевание, опосредованное простагландином D2, такое как застойные явления в носовой полости, аллергический ринит, застой в легких и астма, включая аллергическую астму.
Пределы доз
Величина профилактической или терапевтической дозы соединения формулы I будет, разумеется, варьироваться в зависимости от характера и тяжести состояния, подвергаемого лечению, и от конкретного соединения формулы I и пути его введения. Она будет также варьироваться в зависимости от ряда факторов, включая возраст, массу тела, общее состояние здоровья, пол, диету, время введения, скорость экскреции, комбинацию лекарственных средств и реакцию отдельного пациента. Обычно суточная доза составляет приблизительно от 0,001 мг до приблизительно 100 мг на кг массы тела млекопитающего, предпочтительно, от 0,01 мг до приблизительно 10 мг на кг. С другой стороны, в некоторых случаях может возникать необходимость использовать дозировки вне указанных пределов.
Количество активного ингредиента, которое можно комбинировать с материалами-носителями для изготовления отдельной лекарственной формы, будет варьироваться в зависимости от пациента, подвергаемого лечению, и конкретного способа введения. Например, композиция, предназначенная для перорального введения человеку, может содержать от 0,05 мг до 5 г активного агента в сочетании с подходящим и удобным количеством материала-носителя, количество которого может варьироваться от приблизительно 5 до приблизительно 99,95 процентов от всей композиции. Дозированные лекарственные формы обычно будут содержать от приблизительно 0,1 мг до приблизительно 0,4 мг активного ингредиента, в типичном случае 0,5 мг, 1 мг, 2 мг, 5 мг, 10 мг, 25 мг, 50 мг, 100 мг, 200 мг или 400 мг.
Фармацевтические композиции
Другой аспект настоящего изобретения относится к фармацевтическим композициям, включающим соединение формулы I с фармацевтически приемлемым носителем. Термин "композиция", как в фармацевтической композиции, относится к продукту, включающему активный ингредиент (ингредиенты) и инертный ингредиент (ингредиенты) (фармацевтически приемлемые наполнители), которые составляют носитель, а также любой продукт, который получается, прямо или опосредованно, в результате объединения, комплексования или агрегации любых двух или более ингредиентов, или в результате диссоциации одного или более ингредиентов, или в результате других типов реакций или взаимодействий одного или более ингредиентов. Соответственно, фармацевтические композиции по настоящему изобретению включают любую композицию, изготовленную смешиванием соединения формулы I, дополнительного активного ингредиента (ингредиентов) и фармацевтически приемлемых наполнителей.
Для лечения любого опосредованного простаноидами заболевания соединения формулы I можно вводить перорально, посредством ингаляционного спрея, местно, парентерально или ректально, в композициях дозированных единиц, содержащих обычные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители. Термин "парентеральный", используемый в настоящем документе, включает подкожные инъекции, внутривенные, внутримышечные, интрастернальные инъекции или инфузии. Помимо лечения теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки, кошки и т.п., соединение по настоящему изобретению является эффективным для лечения людей.
Фармацевтические композиции, содержащие активный ингредиент, могут быть в форме, подходящей для перорального введения, например, в форме таблеток, лепешек, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального введения, могут быть изготовлены с соответствии с любым способом, известным для производства фармацевтических композиций, и указанные композиции могут содержать один или более агентов, выбранных из группы, состоящей из подсластителей, корригентов, окрашивающих агентов и консервантов, с целью изготовления фармацевтически хороших и приятных на вкус препаратов. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые являются подходящими для изготовления таблеток. Указанные наполнители могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие агенты, например, кукурузный крахмал или альгиновая кислота; связывающие агенты, например, крахмал, желатин или аравийская камедь, и смазывающие агенты, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или иметь покрытие, нанесенное известными способами, для задержки распада и всасывания в желудочно-кишечном тракте и, таким образом, обеспечивать продолжительное действие в течение более длительного периода времени. Например, можно применять материал, обеспечивающий задержку времени высвобождения активного агента, такой как глицерилмоностеарат или глицерилдистеарат. На них также можно наносить покрытие с помощью методики, описанной в патентах США №№ 4256108, 4166452 и 4265874, с целью изготовления осмотических терапевтических таблеток с контролируемым высвобождением.
Композиции для перорального введения могут быть также представлены в форме твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в форме мягких желатиновых капсул, в которых активный ингредиент смешан со смешивающимися с водой растворителями, такими как пропиленгликоль, PEG и этанол, или с масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии содержат активный материал в смеси с наполнителями, пригодными для изготовления водных суспензий. Указанные наполнители представляют собой суспендирующие агенты, например, натрий-карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую камедь и аравийскую камедь; диспергирующие или увлажняющие агенты могут представлять собой натуральный фосфатид, например, лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например, полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например, гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и гексита, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из ангидридов жирных кислот и гексита, например, моноолеат полиэтиленсорбитана. Водные суспензии могут также содержать один или более консервантов, например, этил или н-пропил, п-гидроксибензоат, один или более окрашивающих агентов, один или более корригентов и один или более подсластителей, таких как сахароза, сахарин или аспартам.
Масляные суспензии могут быть изготовлены суспендированием активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например, пчелиный воск, твердый парафин или цетиловый спирт. Подсластители, такие, которые перечислены выше, и корригенты можно добавлять для получения приятного на вкус препарата для перорального введения. Указанные композиции можно сохранять путем добавления антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для изготовления водной суспензии путем добавления воды, содержат активный ингредиент в смеси с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или более консервантами. Примеры подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов уже упоминались выше. Могут присутствовать также дополнительные наполнители, например, подсластители, корригенты и окрашивающие агенты.
Фармацевтические композиции по настоящему изобретению могут также быть в форме эмульсии типа "масло-в-воде". Масляная фаза может представлять собой растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смеси. Подходящие эмульгирующие агенты могут представлять собой натуральные фосфатиды, например, соевые фосфатиды, лецитин или сложные эфиры или частичные эфиры, полученные из ангидридов жирных кислот и гексита, например, моноолеат сорбитана, и продукты конденсации указанных частичных сложных эфиров с этиленоксидом, например, моноолеат полиоксиэтиленсорбитана. Эмульсии могут также содержать подсластители и корригенты.
Сиропы и эликсиры можно изготавливать с подсластителями, например глицерином, пропиленгликолем, сорбитом или сахарозой. Указанные композиции могут также содержать уменьшающий раздражение агент, консервант и корригенты и окрашивающие агенты. Фармацевтические композиции могут быть в форме стерильной водной или масляной суспензии для инъекций. Данную суспензию можно изготовить в соответствии с известным уровнем техники с использованием подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов, которые были упомянуты выше. Стерильный препарат для инъекций может также представлять собой стерильный раствор или суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, например, раствор в 1,3-бутандиоле. Приемлемыми носителями и растворителями, которые можно использовать, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Также можно использовать сорастворители, такие как этанол, пропиленгликоль или полиэтиленгликоли. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. Для указанной цели можно использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, при изготовлении препаратов для инъекций используются жирные кислоты, такие как олеиновая кислота.
Соединения формулы I можно также вводить в форме суппозиториев для ректального введения лекарственного средства. Указанные композиции можно изготавливать смешиванием лекарственного средства с подходящим нераздражающим наполнителем, который является твердым при комнатной температуре, но жидким при ректальной температуре, и будет, таким образом, плавиться в прямой кишке для высвобождения лекарственного средства. Указанными материалами являются масло какао и полиэтиленгликоли.
Для местного применения используют кремы, мази, гели, растворы или суспензии и т.п., содержащие соединение формулы I. (Для целей настоящей заявки местное применение будет включать средства для полоскания ротовой полости и горла.) Композиции для местного применения обычно могут включать фармацевтический носитель, сорастворитель, эмульгатор, усилитель проникновения, систему консервантов и смягчающий агент.
Комбинации с другими лекарственными средствами
Для лечения и профилактики заболеваний, опосредованных простагландинами, соединение формулы I можно вводить совместно с другими терапевтическими агентами. Таким образом, в другом аспекте настоящее изобретение относится к фармацевтическим композициям для лечения заболеваний, опосредованных простагландином D2, включающим терапевтически эффективное количество соединения формулы I и один или более других терапевтических агентов. Подходящие терапевтические агенты для комбинированной терапии с соединением формулы I включают: (1) антагонист простагландиновых рецепторов; (2) кортикостероид, такой как триамцинолона ацетонид; (3) β-агонист, такой как сальметерол, формотерол, тербуталин, метапротеренол, альбутерол и т.п.; (4) модификатор лейкотриенов, такой как антагонист лейкотриенов или ингибитор липооксигеназы, такой как монтелукаст, зафирлукаст, пранлукаст или зилеутон; (5) антигистаминный агент (антагонист гистаминовых рецепторов Н1), такой как бромофенирамин, хлорфенирамин, дексхлорфенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин, метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин, фенирамин, пириламин, астемизол, норастемизол, терфенадин, лоратадин, цетиризин, левоцетиризин, фексофенадин, деслоратадин и т.п.; (6) противоотечный агент, включая фенилэфрин, фенилпропаноламин, псевдофедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседрин или леводезоксиэфедрин; (7) агент против кашля, включая кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; (8) другой простагландиновый лиганд, включая агонист простагландина F, такой как латанопрост; мизопростол, энпростил, риопростил, орнопростол или розапростол; (9) диуретик; (10) нестероидные противовоспалительные агенты (NSAID), такие как производные пропионовой кислоты (альминопрофен, беноксапрофен, буклоксиновая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен, пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (индометацин, ацеметацин, алклофенак, клинданак, диклофенак, фенклофенак, фенклозиновая кислота, фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенамовой кислоты (флуфенамовая кислота, меклофенамовая кислота, мефенамовая кислота, нифлуминовая кислота и толфенамовая кислота), производные бифенилкарбоновой кислоты (дифлунизал и флуфенизал), оксикамы (изоксикам, пироксикам, судоксикам и теноксикан), салицилаты (ацетилсалициловая кислота, сульфазалазин) и пиразолоны (апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (11) ингибиторы циклооксигеназы-2 (СОХ-2), такие как целекоксиб и рофекоксиб, эторикоксиб и вальдекоксиб; (12) ингибиторы фосфодиэстеразы типа IV (PDE-IV), например, Ariflo, рофлумиласт; (13) антагонисты хемокиновых рецепторов, особенно CCR-1, CCR-2 и CCR-3; (14) агенты, понижающие уровни холестерина, такие как ингибиторы HMG-CoA-редуктазы (ловастатин, симвастатин и правастатин, флувастатин, аторвастатин и другие статины), секвестранты (холестирамин и колестипол), никотиновая кислота, производные фенофибровой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (15) антидиабетические агенты, такие как инсулин, сульфонилмочевины, бигуаниды (метформин), ингибиторы α-глюкозидазы (акарбоза) и глитазоны (троглитазон, пиоглитазон, энглитазон, розиглитазон и т.п.); (16) препараты интерферона бета (интерферон бета-1а, интерферон бета-1b); (17) антихолинергические агенты, такие как антагонисты мускариновых рецепторов (ипратропия бромид и тиотропия бромид), а также селективные антагонисты мускариновых рецепторов М3; (18) стероиды, такие как беклометазон, метилпреднизолон, бетаметазон, преднизон, дексаметазон и гидрокортизон; (19) триптаны, обычно использующиеся для лечения мигрени, такие как сумитриптан и ризатриптан; (20) алендронат и другие лекарственные средства для лечения остеопороза; (21) другие соединения, такие как 5-аминосалициловая кислота и ее пролекарства, антиметаболиты, такие как азатиоприн и 6-меркаптопурин, цитотоксичные химиотерапевтические агенты для лечения злокачественных опухолей, антагонисты брадикинина (ВК2 или ВК1), антагонисты рецепторов ТР, такие как сератродаст, антагонисты нейрокинина (NK1/NK2), антагонисты VLA-4, такие как те, которые описаны в US 5 510 332, WO97/03094, WO97/02289, WO96/40781, WO96/22966, WO96/20216, WO96/01644, WO96/06108, WO95/15973 и WO96/31206.
Кроме того, настоящее изобретение относится к способу лечения заболеваний, опосредованных простагландином D2, включающему введение пациенту, который нуждается в указанном лечении, терапевтически эффективного количества соединения формулы I, совместно с одним или несколькими ингредиентами, которые перечислены непосредственно выше. Количествами активных ингредиентов являются количества, которые широко используются для каждого активного ингредиента в случае, когда его вводят в отдельности, или, в некоторых случаях, комбинация активных ингредиентов может позволять использование более низкой дозы для одного или более активных ингредиентов.
Способы синтеза
Соединения формулы I по настоящему изобретению можно получить в соответствии со схемами синтеза от А до F и с помощью следующих способов, описанных в примерах, представленных в настоящем документе. Схемы и конкретные примеры, представленные в настоящем документе, служат иллюстративным целям, и специалисту будет понятно, что другие соединения по настоящему изобретению можно получить аналогичным способом, с использованием иллюстративных процедур, или их можно получить из приведенных в примерах соединений посредством процедур интерконверсии функциональных групп, которые известны специалистам, или их можно получить посредством других процедур, известных специалистам в области органического синтеза.
Способ А
Пиридин 1 может быть формилирован с получением альдегида 2 согласно процедуре, описанной в J. Heterocyclic Chem., стр. 81 (1988), Heterocycles, стр. 151, 1993, или в Synthesis, стр. 306 (1999). Замещение галогена тиометоксидом натрия или метоксидом натрия с последующей конденсацией с метилазидоацетатом дает азидоолефин 4, который циклизуется при нагревании с получением индола 5 (см., например, Tetrahedron Lett., 2000, 41:4777-4780). Для получения конденсированного пятичленного кольца индол 5 обрабатывают метилакрилатом в присутствии KOtBu, с последующим декарбоксилированием HCl/EtOH, с получением соединения 6 (m=1). Для получения конденсированного шести- и семичленного кольца индол 5 обрабатывают NaH/DMF и подходящим бромным эфиром с последующей циклизацией KOtBu/THF и, наконец, HCl/EtOH, для воздействия на декарбоксилирование, с получением соединения 6 (m=2,3). Образование сложного эфира 7 осуществляют с использованием условий Реформатского с последующим дезоксигенированием c использованием TMSCl/NaI, или посредством реакции Хорнера-Эммондса с последующим гидрированием над PtO2 или Pd(OH)2. Альтернативно соединение 6 можно превратить в соединение 7 путем реакции восстановления с использованием NaBH4 в этаноле-THF с последующим взаимодействием с дифенилхлорфосфатом с использованием NaHMDS в качестве основания. На полученный фосфат действуют диметилмалонатом и NaHMDS. Бис-эфир нагревают в DMSO с NaCl с получением соединения 7.
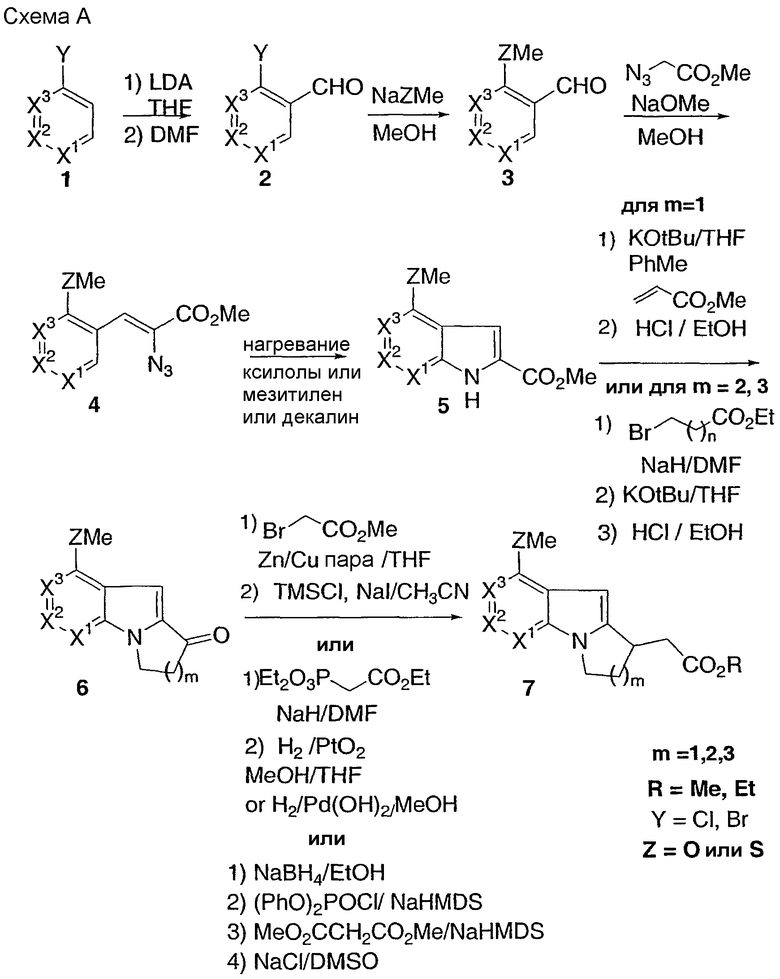
Способ В
Реакция Фриделя-Крафтса соединения 7 с подходящим хлорангидридом в 1,2-дихлорэтане дает соответствующий кетон 8. Последующее расщепление сложного эфира осуществляют с использованием гидроксида натрия с получением кислоты 9. Для получения тиоэфира 11 на подходящий дисульфид действуют SO2Cl2 в 1,2-дихлорэтане с получением соответствующего сульфенилхлорида, который затем взаимодействует с соединением 7 с получением тиоэфирного сложного эфира 10. Гидролиз данного эфира 10 водным раствором гидроксида натрия дает кислоту 11.
aq. - водный.
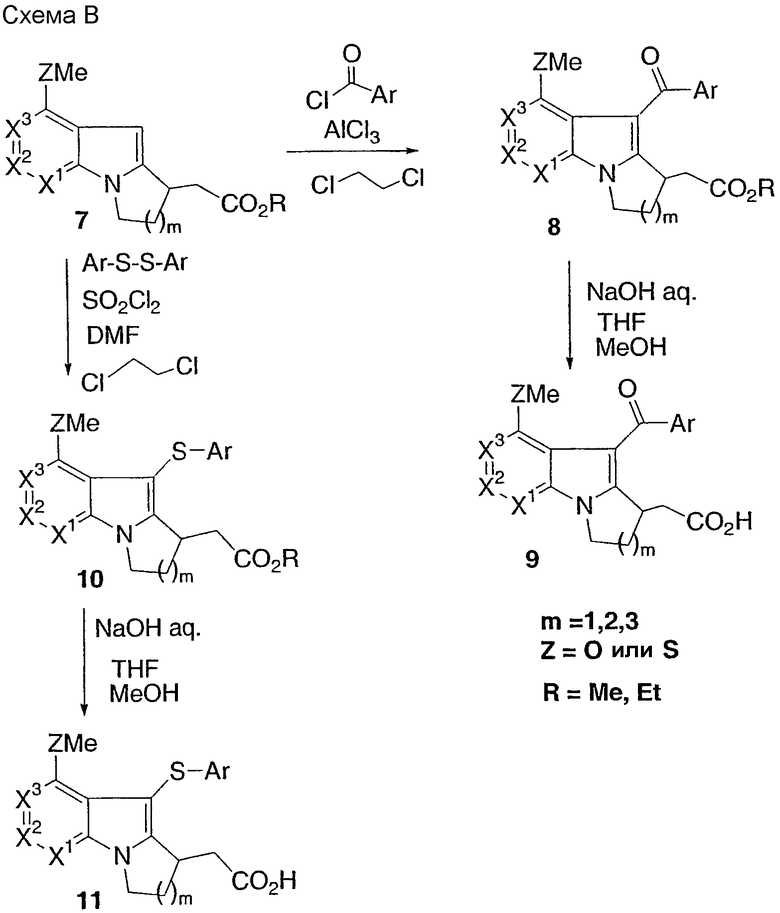
Способ С
Тиоэфирное соединение 8а может быть окислено с использованием Na2WO4/H2O2 для получения соответствующего сульфонового сложного эфира, который после гидролиза дает сульфоновую кислоту 12. Соединение 7а может быть окислено аналогично, а полученный сульфон 13 может быть преобразован в тиоэфир 14 согласно процедурам, описанным в схеме В.
aq. - водный.
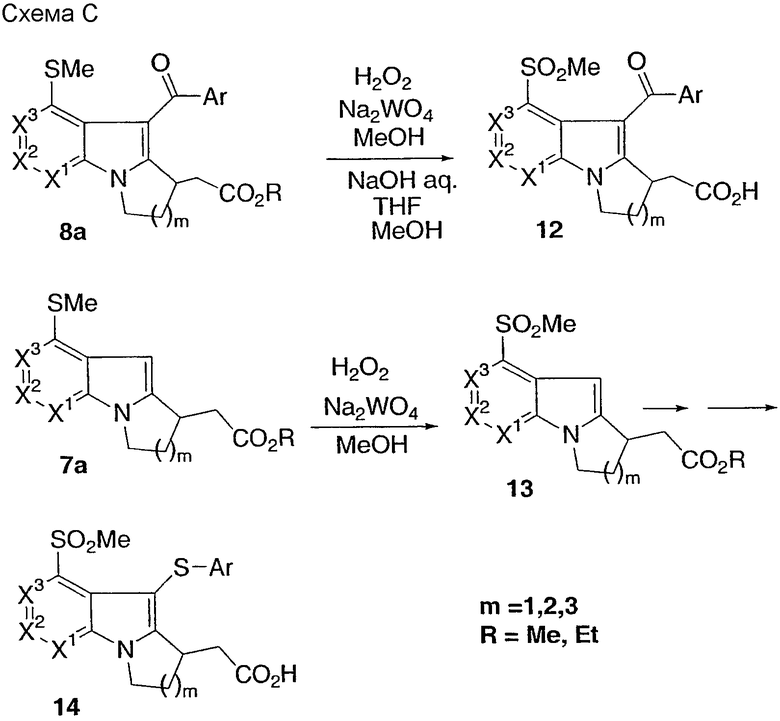
Способ D
Альдегид бромпиридина 2а преобразовывают в соединение 15 с использованием стадий реакции, описанных в способе А. Введение сложноэфирной и изопропильной частей происходит следующим образом. За реакцией Реформатского следует дезоксигенирование c использованием TMSCl/NaI, затем опосредованное палладием сопряжение с 2-бромпропеном, с последующим гидрированием дает соединение 16. Альтернативно реакция Хорнера-Эммондса с последующим опосредованным палладием сопряжением с 2-бромпропеном, и, наконец, гидрирование двух олефинов дает соединение 16, которое преобразовывают в соединение 17, как описано в схеме В. Изопропильную группу также можно ввести на более ранних стадиях синтеза. Соединение 2а можно конвертировать в азаиндольный сложный эфир, как показано в способе А, для получения соединения 5. Изопропил затем вводят, как описано выше, используя опосредованное палладием сопряжение с 2-бромпропеном.
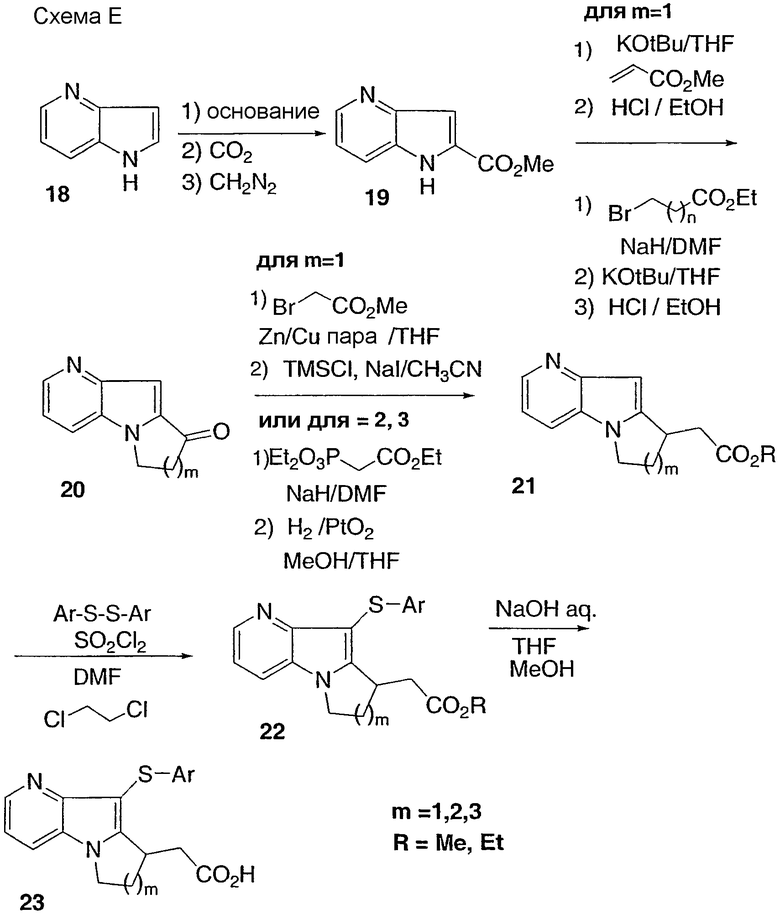
Способ Е
Азаиндол 18 может быть получен согласно процедуре, описанной в J. Heterocyclic Chem. 359 (1992). Обработка соединения 18 основанием, а затем СО2 и диазометаном дает сложный эфир 19, который затем дополнительно функционализируют химическими средствами, описанными в способах А и В, с получением кислоты 23. Альтернативно конденсация 2-пиридинкарбоксальдегида с метилазидоацетатом дает азидоолефин 25, который циклизуют при нагревании, с получением сложного эфира 19, который преобразовывают в кислоту 23.
aq. - водный.
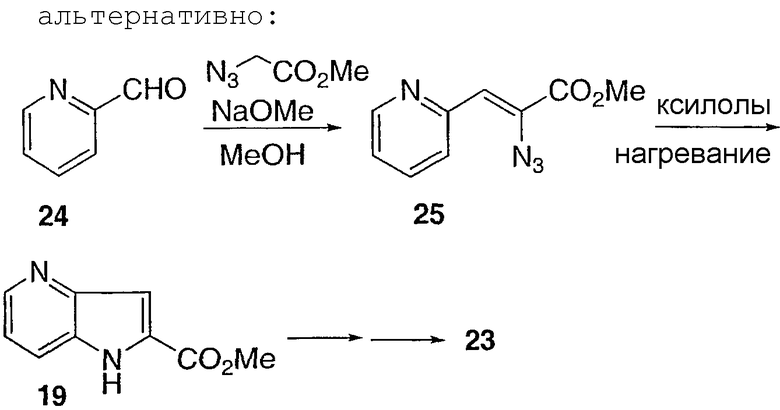
Способ F
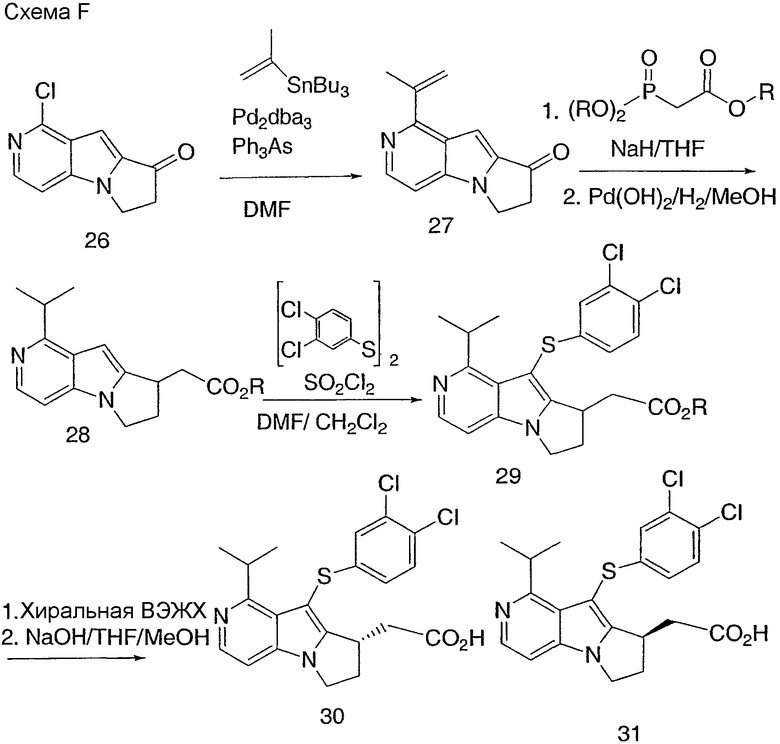
Соединение 26 получают из 2-хлорпиридина согласно общему способу, описанному в способе А. Кетон 26 затем превращают в метилвинильное соединение 27, используя трибутилизопропенилолово (J. Org. Chem. 1988, стр. 3218) и трис(дибензилиденацетон)дипалладия в присутствии трифенилмышьяка. Wittig Horner с кетоном 27 с последующим гидрированием дает сложный эфир 28. Сложный эфир 28 превращают в соединение 29. Энантиомеры разделяют на ОП колонке хиральной ВЭЖХ, с последующим гидролизом, с получением соединений 30 и 31.
Исследования для определения биологической активности
Соединения формулы I можно тестировать с использованием следующих исследований для определения их антагонистической или агонистической активности в отношении простаноидов in vitro и in vivo и их селективности. Продемонстрированная активность в отношении простагландиновых рецепторов представляет собой DP, EP1, EP2, EP3, EP4, FP, IP и TP.
Стабильная экспрессия простаноидных рецепторов в клеточной линии почки человеческого эмбриона (НЕК) 293 (ebna)
кДНК простаноидных рецепторов, соответствующие кодирующим последовательностям полной длины, субклонировали в подходящие сайты экспрессирующих векторов млекопитающих и трансфектировали в клетки НЕК 293 (ebna). Клетки НЕК 293 (ebna), экспрессирующие индивидуальные кДНК, выращивают с селекцией, и отдельные колонии изолируют после 2-3 недель роста с использованием методики клонирующего кольца и впоследствии распространяют в клональные клеточные линии.
Анализы связывания простаноидных рецепторов
Клетки НЕК 293 (ebna) выдерживают в культуре, собирают и получают мембраны путем дифференциального центрифугирования, с последующим лизисом клеток в присутствии ингибиторов протеиназ, для использования в анализах связывания рецепторов. Анализы связывания простаноидных рецепторов осуществляют в 10 мМ MES/KOH (pH 6,0) (EP, FP и ТР) или 10 мМ HEPES/KOH (pH 7,4) (DP и IP), содержащих 1 мМ EDTA, 10 мМ двухвалентный катион и подходящий радиолиганд. Реакцию инициируют добавлением мембранного белка. Лиганды добавляют в диметилсульфоксиде, который поддерживают постоянно на 1% (об./об.) во всех инкубациях. Неспецифичное связывание определяют в присутствии 1 мкМ соответствующего нерадиоактивного простаноида. Инкубации осуществляют в течение 60 минут при комнатной температуре или 30°С и прекращают быстрым фильтрованием. Специфичное связывание рассчитывают вычитанием неспецифичного связывания из общего связывания. Остаточное специфичное связывание при каждой концентрации лиганда рассчитывают и выражают как функцию от концентрации лиганда с целью построения сигмоидальных кривых концентрация-ответ для определения аффинности лиганда.
Изучение агонистов и антагонистов простаноидных рецепторов
Исследования с вторичными мессенджерами цельной клетки, определяющие стимуляцию (EP2, EP4, DP и IP в клетках НЕК 293 (ebna)) или ингибирование (EP3 в клетках эритролейкемии человека (HEL)) аккумуляции внутриклеточного цАМФ или мобилизации внутриклеточного кальция (EP1, FP и ТР в клетках НЕК 293 (ebna), стабильно инфектированных апоэкворином), осуществляли для того, чтобы определить, являются лиганды рецепторов агонистами или антагонистами. Для анализа цАМФ клетки собирают и ресуспендируют в HBSS с содержанием 25 мМ HEPES, рН 7,4. Инкубации содержат 100 мкМ RO-20-1724 (ингибитор фосфодиэстеразы типа IV от компании Biomol), и, в случае анализа ингибирования только ЕР3, 15 мкМ форсколин для стимуляции продукции цАМФ. Образцы инкубируют при 37°С в течение 10 минут, реакцию прекращают, а затем измеряют уровни цАМФ. Для анализов мобилизации кальция клетки нагружают глутатионом и коэлентеразином без кофакторов, собирают и ресуспендируют в среде Хэма F12. Мобилизацию кальция измеряют мониторингом люминесценции, вызываемой связыванием кальция с внутриклеточным фотобелком экворином. Лиганды добавляют в диметилсульфоксиде, который поддерживают постоянно на 1% (об./об.) во всех инкубациях. Для агонистов ответы вторичного мессенджера выражают как функцию от концентрации лиганда, и рассчитывают величины ЕС50 и максимальный ответ по сравнению со стандартом простаноидов. Для антагонистов определяют способность лиганда ингибировать агонистический ответ посредством анализа Шильда, и рассчитывают величины КВ и наклона.
Предотвращение PGD2 или застойных явлений в носовой полости у овец, страдающих аллергией
Подготовка животных: используют здоровых взрослых овец (18-50 кг). Указанных животных отбирают по натуральной положительной кожной реакции на внутрикожное введение экстракта Ascaris suum.
Измерение застойных явлений в носовой полости: эксперимент выполняют на животных, находящихся в сознании. Животных удерживают с помощью тележки в положении лежа на животе с фиксированной головой. Сопротивление назального участка дыхательных путей (NAR) измеряют с помощью методики модифицированной масочной ринометрии. Носовые ходы подвергают местной анестезии (2% лидокаином) для назотрахеальной интубации. Максимальный конец трубки соединяют с пневмотахографом и записывают параметры потока и давления с помощью осциллоскопа, соединенного с компьютером для оперативного расчета NAR. Назальную провокацию осуществляют введением распыленного раствора (10 распылений в ноздрю). Изменения в застое NAR отмечают до и через 60-120 минут после провокации.
Предотвращение PGD2 и индуцированной аллергеном назальной обструкции у обезьян cynomolgus
Подготовка животных: используют здоровых взрослых обезьян cynomolgus мужского пола (4-10 кг). Указанных животных отбирают по натуральной положительной кожной реакции на внутрикожное введение экстракта Ascaris suum. Перед каждым экспериментом обезьяну, отобранную для исследования, не кормили в течение ночи и поили вволю. На следующее утро животное седатируют кетамином (10-15 мг/кг в/м) перед тем, как забирать его из клетки. Его помещают на нагретый стол (36°С) и вводят болюсную дозу (5-12 мг/кг в/в) пропофола. Животное интубируют эндотрахеальной трубкой с манжетой (4-6 мм В.Д.), и анестезию поддерживают непрерывной внутривенной инфузией пропофола (25-30 мг/кг/час). В течение всего эксперимента мониторируют жизненно важные функции (частоту сердечных сокращений, артериальное давление, частоту дыхания, температуру тела).
Измерение застойных явлений в носовой полости: измерение сопротивления дыхательных путей производят с помощью пневмотахографа, соединенного с эндотрахеальной трубкой, чтобы убедиться, что оно в норме. Для оценки застойных явлений в носовой полости используют акустический ринометр Ecovision. Данная методика позволяет получить неинвазивную плоскую эхограмму внутренней поверхности носа. Назальный объем и минимальную площадь поперечного сечения вдоль длины носовой полости рассчитывают за 10 секунд с помощью портативного компьютера, имеющего соответствующую программу (Hood Laboratories, Mass, USA). Назальную провокацию осуществляют введением непосредственно в носовую полость животного (объем 50 мкл). Изменения в застойных явлениях в носовой полости отмечают до и через 60-120 минут после провокации. Если застойные явления в носовой полости имеют место, они преобразовываются в уменьшение назального объема.
Механика внешнего дыхания у тренированных беличьих обезьян, находящихся в сознании
Тест-процедура заключается в помещении тренированных беличьих обезьян в кресла в камеры для воздействия аэрозолем. Для контроля измерения механических параметров внешнего дыхания производят в течение приблизительно 30 минут, чтобы установить нормальные контрольные величины каждой обезьяны на данный день. Для перорального введения соединения растворяют или суспендируют в 1% растворе метоцела (метилцеллюлоза, 65HG, 400 спз) и вводят в объеме 1 мл/кг массы тела. Для аэрозольного введения соединений используют ультразвуковой небулайзер De Vilbiss. Периоды предварительной подготовки варьируют от 5 минут до 4 часов, после чего обезьянам вводят провокационную аэрозольную дозу PGD2 или антигена Ascaris suum, разведение 1:25.
После провокации данные за каждую минуту рассчитывают на компьютере как процент изменения по сравнению с контрольными величинами для каждого параметра дыхания, включая сопротивление дыхательных путей (RL) и динамическое изменение объема легких при колебаниях давления (Cdyn). Результаты для каждого испытуемого соединения впоследствии получают за минимальный период времени 60 минут после провокации, которые затем сравнивают с полученными ранее исходными контрольными величинами для данной обезьяны. Помимо этого, рассчитывают средние значения по всем величинам за 60 минут после провокации для каждой обезьяны (исходные величины и тест-величины) в отдельности и используют их для расчета общего процента ингибирования медиатора или ответа на антиген Ascaris испытуемым соединением. Для статистического анализа используют парный t-критерий. (Ссылки: McFarlane, C.S. et al., Prostaglandins, 28, 173-182 (1984) и McFarlane, C.S. et al., Agents Actions, 22, 63-68 (1987)).
Предотвращение индуцированной бронхоконстрикции у овец, страдающих аллергией
Подготовка животных: используют взрослых овец со средней массой тела 35 кг (пределы от 18 до 50 кг). Все животные отвечают двум критериям: а) они имеют натуральную кожную реакцию на экстракт Ascaris suum в разведении 1:1000 или 1:10 000 (Greer Diagnostics, Lenois, NC) и b) они ранее реагировали на ингаляционную провокацию Ascaris suum острой бронхоконстрикцией и поздней бронхиальной обструкцией (W.M. Abraham et al., Am. Rev. Resp. Dis., 128, 839-44 (1983)).
Измерение механических параметров внешнего дыхания: неседатированных овец удерживают с помощью тележки в положении лежа на животе с фиксированной головой. После местной анестезии носовых ходов 2% раствором лидокаина баллонный катетер продвигают через одну ноздрю в нижний отдел пищевода. Животных затем интубируют эндотрахеальной трубкой с манжетой через другую ноздрю с использованием гибкого фиброоптического бронхоскопа в качестве проводника. Плевральное давление устанавливают с помощью пищеводного баллонного катетера (наполненного одним мл воздуха), который расположен таким образом, что вдох вызывает отклонение отрицательного давления с явно различимыми кардиогенными осцилляциями. Латеральное давление в трахее измеряют с помощью катетера с боковым отверстием (внутреннее измерение 2,5 мм), продвинутого и расположенного дистально по отношению к верхушке назотрахеальной трубки. Транспульмонарное давление, разницу между давлением в трахее и плевральной полости измеряют с помощью дифференциального преобразователя давления (DP45; Validyne Corp., Northridge, CA). Для измерения сопротивления дыхательных путей (RL) максимальный конец назотрахеальной трубки соединяют с пневмотахографом (Fleisch, Dyna Sciences, Blue Bell, PA). Записывают параметры потока и транспульмонарного давления с помощью осциллоскопа (модель DR-12; Electronics for Medicine, White Plains, NY), соединенного с цифровым компьютером PDP-11 (Digital Equipment Corp., Maynard, MA) для оперативного расчета RL по транспульмонарному давлению, дыхательному объему, полученному интегрированием, и потоку. Для определения RL используют анализ 10-15 дыхательных циклов. Объем газа в грудной клетке (Vtg) измеряют с использованием плетизмографа для тела, чтобы получить удельное сопротивление дыхательных путей (SRL = RL·Vtg).
Следующие примеры приводятся для иллюстрации изобретения и не должны толковаться как ограничивающие объем настоящего изобретения. В примерах, если не указано иное:
- все конечные продукты формулы I анализировались с помощью ЯМР, ТСХ и элементного анализа или масс-спектрометрии;
- промежуточные вещества анализировались с помощью ЯМР и ТСХ;
- большинство соединений очищали с помощью флэш-хроматографии на силикагеле, перекристаллизации и/или swish (суспендирования в растворителе с последующим фильтрованием твердого вещества);
- за ходом реакций следили с помощью тонкослойной хроматографии (ТСХ), а время протекания реакции приводится только в целях иллюстрации;
- избыток энантиомеров измеряли с помощью ВЭЖХ с нормальной фазой на хиральной колонке: ChiralPak AD; 250 · 4,6 мм.
ПРИМЕР 1
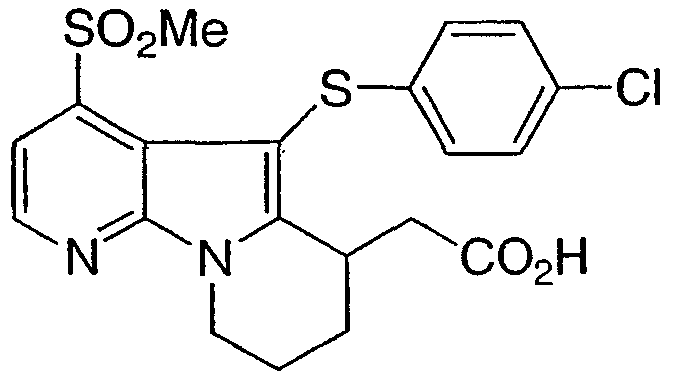
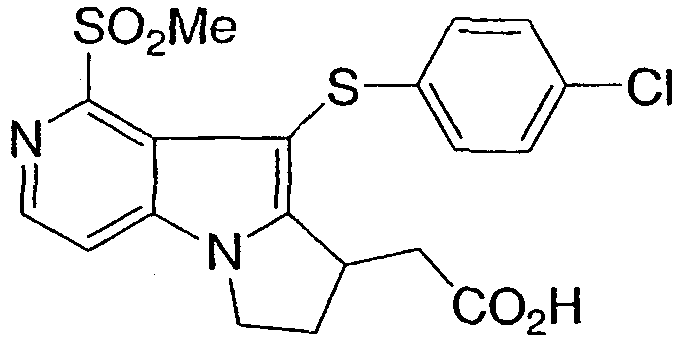
[5-[(4-хлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота
Стадия 14-хлорникотинальдегид
Указанное в заголовке соединение получали, как описано у F. Marsais et al., J. Heterocyclic Chem., 25, 81 (1988).
Стадия 24-(метилтио)никотинальдегид
К раствору NaSMe (9,5 г, 135 ммоль) в МеОН (250 мл) добавляли 4-хлорникотинальдегид (13,5 г, 94,4 ммоль) со стадии 1 в МеОН (250 мл). Температуру реакционной смеси поддерживали на уровне 60°С в течение 15 минут. Реакционную смесь разливали над NH4Cl и EtOAc. Органическую фазу отделяли, промывали Н2О и высушивали над Na2SO4. Соединение затем очищали на силикагеле 50% EtOAc в гексанах с получением указанного в заголовке соединения.
Стадия 3Метил (2Z)-2-азидо-3-[4-(метилтио)пиридин-3-ил]проп-2-еноат
Раствор 4-(метилтио)никотинальдегида (4,8 г, 31 ммоль) и метилазидоацетата (9,0 г, 78 ммоль) в МеОН (50 мл) добавляли к раствору 25% NaOMe в МеОН (16,9 мл, 78 ммоль) при -12°С. Внутреннюю температуру регистрировали и поддерживали при температуре от -10 до -12°С во время добавления в течение 30 минут. Полученную смесь затем перемешивали на бане со льдом в течение нескольких часов, затем держали на бане со льдом в холодной комнате в течение ночи. Суспензию затем выливали в смесь льда и NH4Cl и взвесь фильтровали после перемешивания в течение 10 минут. Продукт промывали холодной Н2О, а затем сушили в условиях вакуума с получением указанного в заголовке соединения в виде твердого вещества бежевого цвета (7,4 г), которое содержало некоторые соли. Соединение затем очищали на силикагеле с использованием EtOAc.
Стадия 4Метил 4-(метилтио)-1Н-пирроло[2,3-b]пиридин-2-карбоксилат
Суспензию соединения со стадии 3 (0,40 г, 1,6 ммоль) в ксилолах (16 мл) медленно нагревали до 140°С. После выдержки при 140°С в течение 15 минут желтый раствор охлаждали до комнатной температуры. Следует предпринимать меры предосторожности, поскольку существует вероятность выделения тепла вследствие образования азота. Суспензию затем охлаждали до 0°С, фильтровали и промывали ксилолом с получением указанного в заголовке соединения.
Стадия 5Этил 4-(метилтио)-6-оксо-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-7-карбоксилат
К раствору соединения со стадии 4 (0,35 г, 1,6 ммоль) в DMF (20 мл) при 0°С добавляли NaH (1,2 экв.). Через 5 минут добавляли нBu4NI (0,10 г) и этил 4-бромбутират (0,40 мл). После периода времени 1 ч при комнатной температуре реакционную смесь выливали в насыщенный раствор NH4Cl и EtOAc. Органическую фазу отделяли, промывали Н2О и высушивали над NaSO4. После выпаривания сырой продукт очищали флэш-хроматографией. Бис-эфир затем растворяли в THF (7,0 мл) и добавляли 1,06 М раствор трет-бутоксида в THF (2,2 мл) при 0°С. После периода времени 1 ч при комнатной температуре реакционную смесь затем выливали в насыщенный раствор NH4Cl и EtOAc. Органическую фазу отделяли, высушивали над Na2SO4 и выпаривали при пониженном давлении с получением указанного в заголовке соединения в виде смеси этилового и метилового эфиров.
Стадия 64-(метилтио)-8,9-дигидропиридо[3,2-b]индолизин-6(7Н)-он
К соединению со стадии 5 (0,32 г) добавляли EtOH (8,0 мл) и концентрированную HCl (2,0 мл). Полученную суспензию кипятили с обратным холодильником в течение 5 ч. Реакционную смесь распределяли между EtOAc и Na2CO3. Органическую фазу отделяли, выпаривали с получением указанного в заголовке соединения.
Стадия 7Этил (2E,2Z)-[4-(метилтио)-8,9-дигидропиридо[3,2-b]индолизин-6(7Н)-илиден]этаноат
К раствору триэтилфосфоноацетата (0,45 г, 2,17 ммоль) в DMF (12 мл) добавляли 80% NaH (0,06 г, 2,00 ммоль) и соединение со стадии 6 (0,22 г, 1,00 ммоль). После 4 ч при 55°С реакционную смесь выливали в насыщенный раствор NH4Cl и EtOAc. Органическую фазу отделяли и выпаривали при пониженном давлении. Сырой продукт очищали флэш-хроматографией с получением указанного в заголовке соединения.
Стадия 8Этил [4-(метилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]ацетат
Соединение со стадии 7 растворяли в MeOH-THF при нагревании. К предварительно охлажденному раствору добавляли при комнатной температуре PtO2 и полученную смесь поддерживали в течение 18 ч при атмосферном давлении водорода. Реакционную смесь осторожно фильтровали через целит с использованием CH2Cl2. Фильтрат выпаривали при пониженном давлении с получением указанного в заголовке соединения. Альтернативно соединение со стадии 7 можно гидрировать с использованием Pd(OH)2 в EtOAc при 40 фунт/дюйм2 Н2 в течение 18 ч.
Стадия 9Этил [4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]ацетат
К соединению со стадии 8 (0,08 г, 0,27 ммоль) в МеОН (3,0 мл) добавляли Na2WO4 (0,10 г) и 30% Н2О2 (600 мкл). Через 1 ч реакционную смесь распределяли между Н2Ои EtOAc. Органическую фазу промывали Н2О,отделяли и выпаривали. Указанное в заголовке соединение очищали флэш-хроматографией.
Стадия 10Этил [5-[(4-хлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]ацетат
К раствору 4,4'-дихлордифенилдисульфида (0,24 г) в 1,2-дихлорэтане (2,0 мл) добавляли SO2Cl2 (50 мкл). К соединению со стадии 9 (0,05 г) в DMF (2,0 мл) добавляли полученную ранее смесь (˜180 мкл). За ходом реакции следили с помощью 1Н ЯМР и поддерживали комнатную температуру до тех пор, пока не осталось исходного материала. Реакционную смесь выливали в насыщенный раствор NaHCO3 и EtOAc. Органическую фазуотделяли, выпаривали и указанное в заголовке соединение очищали флэш-хроматографией.
Стадия 11[5-[(4-хлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота
К соединению со стадии 10, растворенному в смеси 1/1 THF-MeOH, добавляли 1Н NaOH. После 18 ч при комнатной температуре реакционную смесь распределяли между насыщенным NH4Cl и EtOAc. Органическую фазуотделяли, высушивали над Na2SO4 и выпаривали с получением указанного в заголовке соединения.
1Н ЯМР (500 МГц, ацетон-d6) δ 11,00 (ушир.с, 1H), 8,60 (д, 1H), 7,80 (д, 1H), 7,20 (д, 2H), 7,00 (д, 2H), 4,65 (м, 1H), 4,20 (м, 1H), 3,75 (м, 1H), 3,35 (с, 3H), 2,80-2,10 (м, 6H).
ПРИМЕР 2
[5-[(4-хлорфенил)тио]-4-(метилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота

Указанное в заголовке соединение можно получить из соединения примера 1, стадия 8, способом, аналогичным тому, который описан в примере 1, стадия 10 и 11.
m/z 418
ПРИМЕР 3
[5-[(3,4-дихлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота (энантиомер А и энантиомер В)
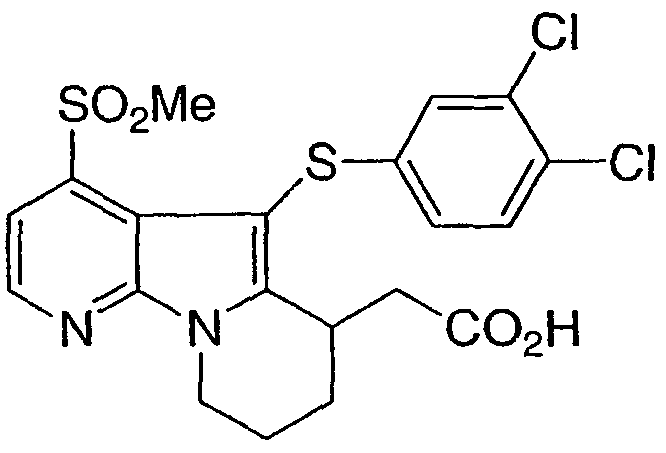
Указанное в заголовке соединение получали, как описано в примере 1, с использованием бис(3,4-дихлорфенил)дисульфида на стадии 10.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,55 (д, 1H), 7,85 (д, 1H), 7,35 (д, 1H), 7,15 (с, 1H), 6,95 (д, 1H), 4,60 (м, 1H), 4,15 (м, 1H), 3,80 (м, 1H), 3,40 (с, 3H), 2,80-2,10 (м, 6H).
Энантиомеры разделяли на колонке Chiralecel OD 25 см · 20 мм, с использованием 30% изопропанола, 7% этанола, 0,2% уксусной кислоты в гексане, скорость потока 8 мл/мин. Чистоту верифицировали на колонке Chiralecel OD 25 см · 4,6 мм с использованием 35% изопропанола, 0,2% уксусной кислоты в гексане, скорость потока 1,0 мл/мин. Более мобильный энантиомер Tr = 9,7 минут, менее мобильный энантиомер Tr = 11,1 минут.
ПРИМЕР 4
[5-(4-хлорбензоил)-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота
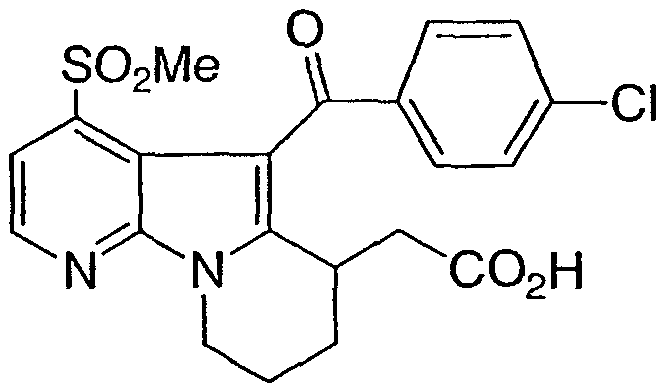
Стадия 1Этил [5-(4-хлорбензоил)-4-(метилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]ацетат
К раствору 4-хлорбензоилхлорида (0,30 г, 1,7 ммоль) в 1,2-дихлорэтане (6,0 мл) добавляли AlCl3 (0,24 г, 1,8 ммоль). Через 5 минут раствор этил [4-(метилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]ацетата примера 1, стадия 8 (0,15 г, 0,47 ммоль) в 1,2-дихлорэтане (6,0 мл) добавляли к полученной ранее смеси. Через 4 ч при 80°С реакционную смесь распределяли между EtOAc и NaHCO3. Органическую фазуотделяли, высушивали над Na2SO4 и выпаривали. Указанное в заголовке соединение очищали флэш-хроматографией.
Стадия 2Этил [5-(4-хлорбензоил)-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]ацетат
К раствору этил [5-(4-хлорбензоил)-4-(метилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]ацетата (0,12 г, 0,27 ммоль) в МеОН (5,0 мл) добавляли Na2WO4 (0,1 г) и 30% Н2О2 (300 мкл). Реакционную смесь перемешивали при 55°С в течение 1 ч. Реакционную смесь затем распределяли между Н2Ои EtOAc. Органическую фазу промывали Н2О,высушивали над Na2SO4 и выпаривали. Указанное в заголовке соединение очищали флэш-хроматографией.
Стадия 3[5-(4-хлорбензоил)-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота
На этил [5-(4-хлорбензоил)-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]ацетат действовали, как описано в примере 1, стадия 11, с получением указанного в заголовке соединения.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,55 (д, 1H), 7,90 (д, 2H), 7,65 (д, 1H), 7,45 (д, 2H), 4,55 (м, 1H), 4,25 (м, 1H), 3,45 (м, 1H), 3,20 (с, 3H), 2,05-3,00 (м, 6H).
ПРИМЕР 5
[5-(4-бромфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота
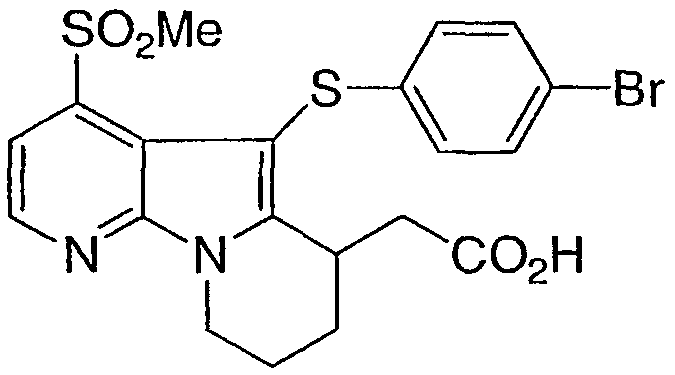
Указанное в заголовке соединение получали, как описано в примере 1, с использованием 4,4'-дибромдифенилдисульфида.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,60 (д, 1H), 7,80 (д, 1H), 7,35 (д, 2H), 7,00 (д, 2H), 4,65 (м, 1H), 4,20 (м, 1H), 3,80 (м, 1H), 3,35 (с, 3H), 2,80-2,10 (м, 6H).
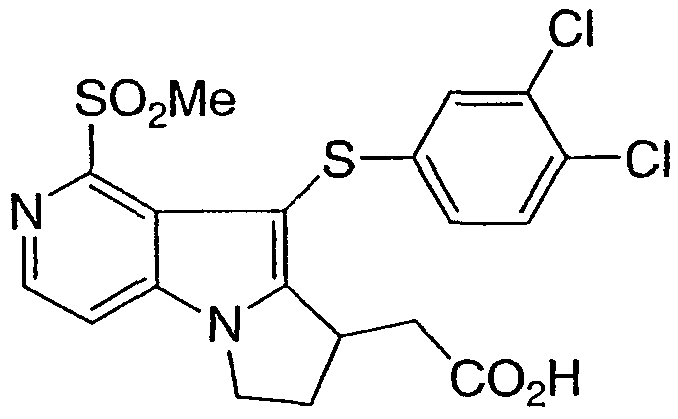
ПРИМЕР 6 СПОСОБ-1
[9-[(3,4-дихлорфенил)тио]-1-(метилсульфонил)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]уксусная кислота
Стадия 12-(метилтио)никотинальдегид
Указанное в заголовке соединение получали из 2-бромникотинальдегида (A. Numata Synthesis 1999, стр. 306), как описано в примере 1, стадия 2, за исключением того, что раствор нагревали до 55°С в течение 2 ч.
Стадия 2Метил (2Z)-2-азидо-3-[2-(метилтио)пиридин-3-ил]проп-2-еноат
Указанное в заголовке соединение получали, как описано в примере 1, стадия 3.
Стадия 3Метил 4-(метилтио)-1Н-пирроло[3,2-c]пиридин-2-карбоксилат
Раствор метил (2Z)-2-азидо-3-[2-(метилтио)пиридин-3-ил]проп-2-еноата (1,00 г, 4,00 ммоль) в мезитилене (50 мл) нагревали до 160°С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры, затем до 0°С, осадок отфильтровывали и промывали холодным мезитиленом с получением указанного в заголовке соединения.
Стадия 4Метил 1-(метилтио)-8-оксо-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-7-карбоксилат
К суспензии метил 4-(метилтио)-1Н-пирроло[3,2-c]пиридин-2-карбоксилата (0,30 г, 1,35 ммоль) в THF (3 мл) - толуоле (12,0 мл) добавляли 1,06 М раствор трет-бутоксида калия (1,42 мл/1,41 ммоль) и метилакрилата (300 мкл) в THF. Полученную смесь нагревали до 80°С в течение 18 ч. Смесь распределяли между EtOAc и NH4Cl, фильтровали через целит. Органическую фазу отделяли,высушивали над Na2SO4 и фильтровали с получением указанного в заголовке соединения.
Стадия 51-(метилтио)-6,7-дигидро-8Н-пиридо[3,4-b]пирролизин-8-он
Метил 1-(метилтио)-8-оксо-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-7-карбоксилат превращали в указанное в заголовке соединение, как описано в примере 1, стадия 6.
Стадия 6Метил [8-гидрокси-1-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]ацетат
Смесь 1-(метилтио)-6,7-дигидро-8Н-пиридо[3,4-b]пирролизин-8-она (0,15 г, 0,68 ммоль), метилбромацетата (0,34 мл), Zn-Cu (0,226 г) в THF (3,0 мл) обрабатывали ультразвуком в течение 2 ч. Смесь затем нагревали до 60°С в течение 5 минут до окончания реакции. Реакционную смесь распределяли между EtOAc и NH4Cl. Органическую фазу отделяли,высушивали над Na2SO4, фильтровали и выпаривали, с получением указанного в заголовке соединения. Соединение очищали флэш-хроматографией.
Стадия 7Метил [1-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]ацетат
К NaI (0,300 г) в CH3CN (3,2 мл) добавляли TMSCI (0,266 мл). Указанную смесь добавляли к суспензии метил [8-гидрокси-1-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]ацетата (0,15 г, 0,515 ммоль) в CH3CN (1,5 мл) на водяной бане. Через 0,5 ч реакционную смесь распределяли между EtOAc и NaHCO3. Органическую фазу отделяли,промывали тиосульфатом натрия, высушивали над MgSO4 и выпаривали. Указанное в заголовке соединение очищали флэш-хроматографией.
Стадия 8Метил [1-(метилсульфонил)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]ацетат
Метил [1-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]ацетат превращали в указанное в заголовке соединение, как описано в примере 1, стадия 9.
Стадия 9[9-[(3,4-дихлорфенил)тио]-1-(метилсульфонил)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]уксусная кислота
Метил [1-(метилсульфонил)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]ацетат превращали в указанное в заголовке соединение, как описано в примере 1, стадии 10 и 11, с использованием бис(3,4-дихлорфенил)дисульфида на стадии 10.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,35 (д, 1H), 7,80 (д, 1H), 7,35 (д, 1H), 7,15 (с, 1H), 6,95 (д, 1H), 4,55 (м, 1H), 4,35 (м, 1H), 3,90 (м, 1H), 3,30 (с, 3H), 3,15 (м, 1H), 3,05 (м, 1H), 2,80 (м, 1H), 2,50 (м, 1H).
ПРИМЕР 6 СПОСОБ-2
[9-[(3,4-дихлорфенил)тио]-1-(метилсульфонил)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]уксусная кислота
Стадия 11-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ол
К суспензии 1-(метилтио)-6,7-дигидро-8Н-пиридо[3,4-b]пирролизин-8-она из примера 6 способ-1, стадия 5 (0,55 г, 2,2 ммоль) в EtOH (10 мл) - THF (1 мл) добавляли NaBH4 (0,10 г, 2,6 мл) при 0°С. Через 30 минут при комнатной температуре реакционную смесь гасили добавлением ацетона. Растворители выпаривали при пониженном давлении и к остатку добавляли EtOAc и Н2О. Органическую фазу отделяли, высушивали над MgSO4 и выпаривали. Указанное в заголовке соединение промывали смесью EtOAc/гексан и фильтровали.
Стадия 2Диметил 2-[1-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]малонат
К суспензии 1-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ола (0,54 г, 2,1 ммоль) в THF (10 мл) при -78°С добавляли 1М NaHMDS в THF (2,35 мл, 2,4 ммоль) и дифенилхлорфосфат (0,53 мл, 2,6 ммоль). Через 30 минут добавляли диметилмалонат (0,73 мл, 6,4 ммоль) и 1М NaHMDS в THF (6,8 мл, 6,8 ммоль). Температуру реакционной смеси доводили до 0°С, а затем до комнатной температуры. Затем смесь распределяли между EtOAc и NaH4Cl. Органическую фазу высушивали над MgSO4, фильтровали и выпаривали. Указанное в заголовке соединение очищали флэш-хроматографией.
Стадия 3Метил [1-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]ацетат
К смеси диметил 2-[1-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]малоната (0,59 г, 2,17 ммоль) и DMSO (4 мл) добавляли NaCl (0,45 г) в Н2О (0,45 мл). Через 18 ч при 150°С реакционную смесь распределяли между EtOAc и Н2О. Органическую фазу отделяли, высушивали над Na2SO4 и выпаривали. Указанное в заголовке соединение очищали флэш-хроматографией.
Стадия 4[9-[(3,4-дихлорфенил)тио]-1-(метилсульфонил)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]уксусная кислота
Указанное в заголовке соединение получали из метил [1-(метилтио)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]ацетата, как описано в примере 6 способ-1, стадия с 8 по 9.
ПРИМЕР 7
[10-[(3,4-дихлорфенил)сульфанил]-1-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,4-b]индолизин-9-ил]уксусная кислота
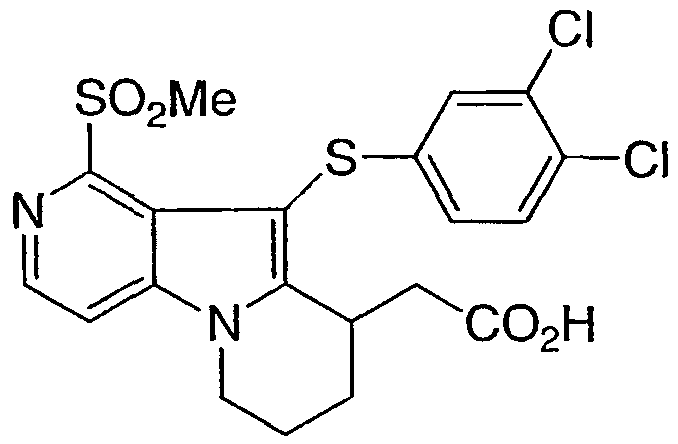
Стадия 1Этил [1-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,4-b]индолизин-9-ил]ацетат
Указанное в заголовке соединение получали из продукта примера 6, стадия 3, таким же способом, который описан в примере 1, стадии с 5 по 9.
Стадия 2[10-[(3,4-дихлорфенил)сульфанил]-1-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,4-b]индолизин-9-ил]уксусная кислота
Продукт со стадии 1 превращали в указанное в заголовке соединение таким же способом, который описан в примере 1, стадии 10-11, с использованием бис(3,4-дихлорфенил)дисульфида на стадии 10.
MS M+1 = 485
ПРИМЕР 8
4-(метилсульфонил)-5-{[4-трифторметил)фенил]тио}-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота
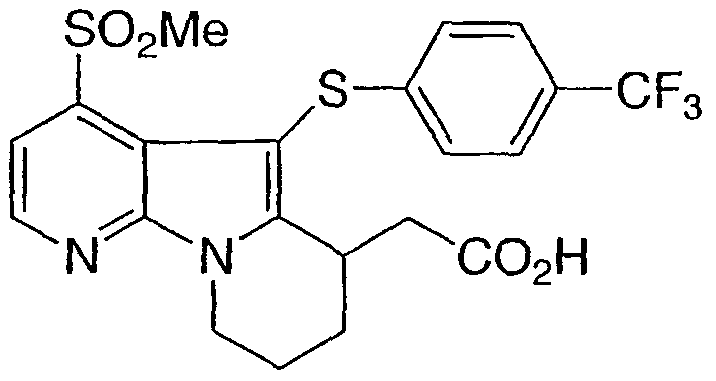
Указанное в заголовке соединение получали, как описано в примере 1, с использованием бис[4-трифторметил)фенил]дисульфида.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,55 (д, 1H), 7,75 (д, 1H), 7,45 (д, 2H), 7,15 (д, 2H), 4,55 (м, 1H), 4,15 (м, 1H), 3,80 (м, 1H), 3,30 (с, 3H), 2,80-2,10 (м, 6H).
ПРИМЕР 9
[5-[(2-хлор-4-фторфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота
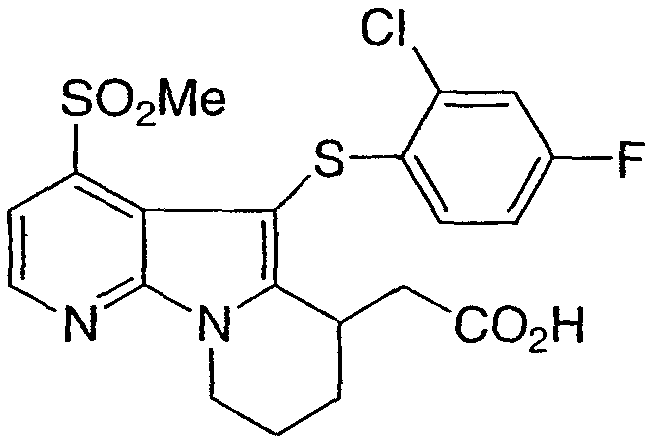
Указанное в заголовке соединение получали, как описано в примере 1, с использованием бис(2-хлор-4-фторфенил)дисульфида.
m/z 469 (M+1)
ПРИМЕР 10
[4-(метилсульфонил)-5-(2-нафтилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота
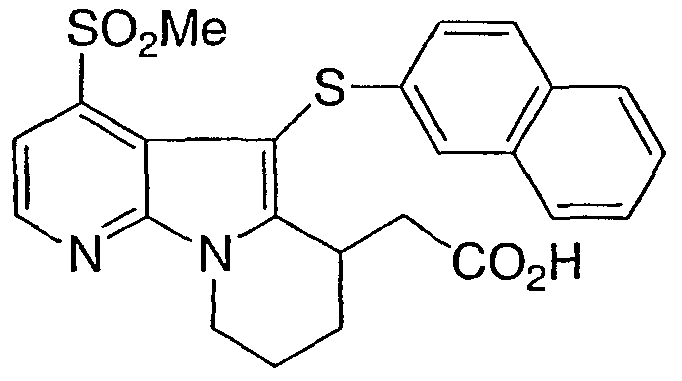
Указанное в заголовке соединение получали, как описано в примере 1, с использованием ди(2-нафтил)дисульфида.
М/z 467 (M+1)
ПРИМЕР 11
[5-[(2,3-дихлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота

Указанное в заголовке соединение получали, как описано в примере 1, с использованием бис(2,3-дихлорфенил)дисульфида.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,85 (д, 1H), 7,80 (д, 1H), 7,30 (д, 1H), 7,00 (т, 1H), 6,60 (д, 1H), 4,60 (м, 1H), 4,20 (м, 1H), 3,80 (м, 1H), 3,40 (с, 3H), 2,80-2,10 (м, 6H).
ПРИМЕР 12
[5-[(4-метилфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота
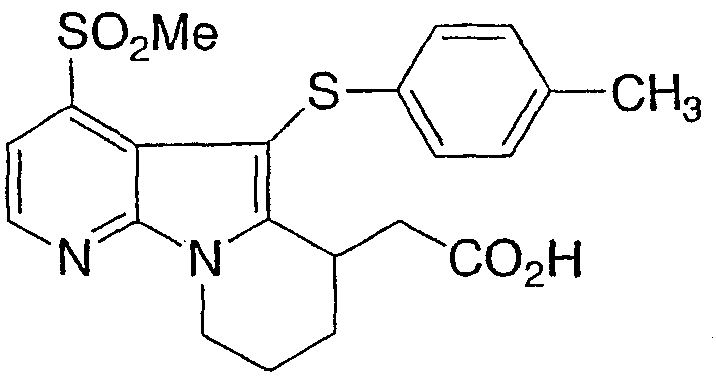
Указанное в заголовке соединение получали, как описано в примере 1, с использованием п-толилдисульфида.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,55 (д, 1H), 7,80 (д, 1H), 6,95 (м, 4H), 4,60 (м, 1H), 4,15 (м, 1H), 3,80 (м, 1H), 3,35 (с, 3H), 2,80-2,10 (м, 6H).
ПРИМЕР 13
[4-(метилсульфонил)-5-(фенилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота
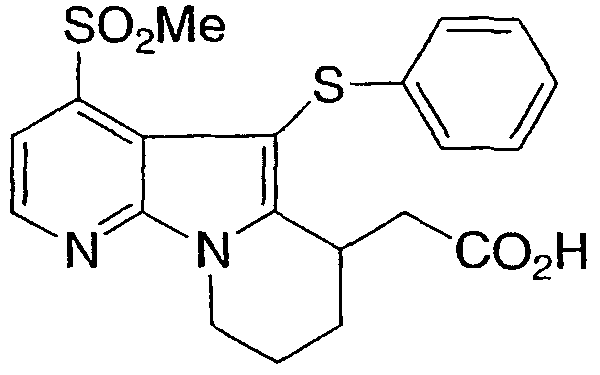
Указанное в заголовке соединение получали, как описано в примере 1, с использованием дифенилдисульфида.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,55 (д, 1H), 7,80 (д, 1H), 7,15-6,90 (м, 5H), 4,60 (м, 1H), 4,15 (м, 1H), 3,75 (м, 1H), 3,30 (s, 3H), 2,80-2,10 (м, 6H).
ПРИМЕР 14
[5-[(2,4-дихлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота
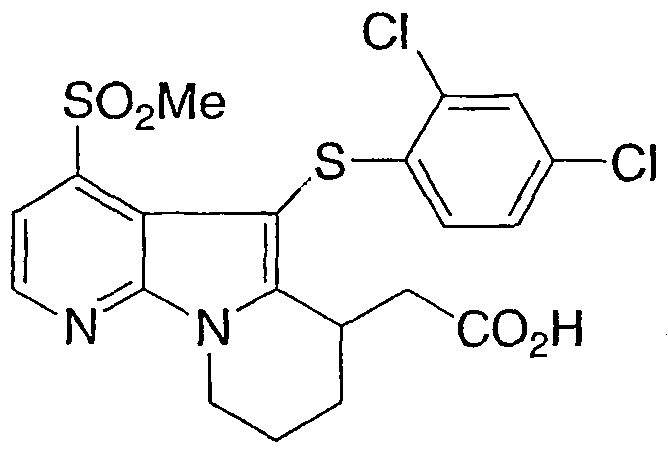
Указанное в заголовке соединение получали, как описано в примере 1, с использованием бис(2,4-дихлорфенил)дисульфида. Дисульфид был получен с использованием Br2 в эфире.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,55 (д, 1H), 7,85 (д, 1H), 7,35 (с, 1H), 7,00 (д, 1H), 6,65 (д, 1H), 4,55 (м, 1H), 4,15 (м, 1H), 3,80 (м, 1H), 3,35 (с, 3H), 2,80-2,10 (м, 6H).
ПРИМЕР 15
[5-[(4-хлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[4,3-b]индолизин-6-ил)уксусная кислота
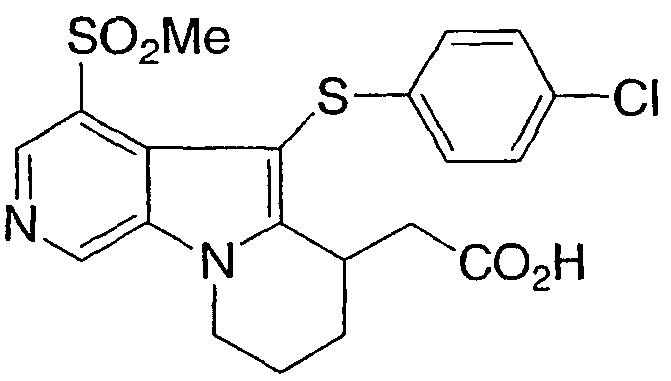
Указанное в заголовке соединение получали, как описано в примере 1, из 3-хлорникотинальдегида (Heterocycles, стр. 151, 1993) за исключением того, что термическая циклизация была осуществлена добавлением азида к декалину при температуре кипячения с обратным холодильником.
1Н ЯМР (500 МГц, ацетон-d6) δ 9,20 (с, 1H), 8,85 (с, 1H), 7,20 (д, 2H), 7,00 (д, 2H), 4,70 (м, 1H), 4,30 (м, 1H), 3,75 (м, 1H), 3,35 (с, 3H), 2,80-2,10 (м, 6H).
ПРИМЕР 16
[9-[(4-хлорфенил)тио]-1-(метилсульфонил)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил)уксусная кислота
Указанное в заголовке соединение получали из продукта примера 6, способ 1, стадия 8, как описано в примере 1. Стадии 10 и 11 с использованием бис(4-хлорфенил)дисульфида на стадии 10.
1Н ЯМР (500 МГц, ацетон-d6) δ 8,25-8,3 (м, 1H), 7,71-7,75 (м, 1H), 7,12-7,17 (м, 2H), 6,97-7,04 (м, 2H), 4,45-4,51 (м, 1H), 4,32-4,39 (м, 1H), 3,73-3,80 (м, 1H), 3,29 (с, 3H), 3,15-3,21 (м, 1H), 2,99-3,08 (м, 1H), 2,66-2,73 (м, 1H), 2,46-2,54 (м, 1H).
ПРИМЕР 17
[9-[(3,4-дихлорфенил)тио]-1-изопропил-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил)уксусная кислота (энантиомер А и энантиомер В)

Стадия 12-хлорникотинальдегид
К раствору диизопропиламина (110 мл, 780 ммоль) в THF (500 мл) добавляли 2,5 М раствор н-BuLi (300 мл, 750 ммоль) в гексанах при -40°С. Через 5 минут реакционную смесь охлаждали до -95°С, затем последовательно добавляли DMPU (15 мл) и 2-хлорпиридин (50 мл, 532 ммоль). Полученную смесь затем подогревали и перемешивали при -78°С в течение 4 ч. Спустя указанный период времени желтую суспензию снова охлаждали до -95°С, после чего добавляли DMF (70 мл). Конечную реакционную смесь затем подогревали до -78°С и перемешивали при указанной температуре в течение 1,5 ч. Реакционную смесь выливали в холодный водный раствор HCl (3н., 800 мл) и перемешивали в течение 5 минут. Добавляли водный концентрированный раствор NH4OH до получения рН 7,5. Водный слой трижды экстрагировали EtOAc. Объединенный органический слой промывали водным раствором NH4Cl и насыщенным солевым раствором, высушивали над безводным Na2SO4, фильтровали и концентрировали. Необработанный материал в дальнейшем очищали слоем силикагеля путем элюирования градиентом от 100% гексанов до 100% EtOAc и продукт кристаллизовали в холодных гексанах, с получением указанного в заголовке соединения в виде твердого вещества бледно-желтого цвета.
Стадия 2Метил (2Z)-2-азидо-3-(2-хлорпиридин-3-ил)проп-2-еноат
Указанное в заголовке соединение получали, как описано в примере 1, стадия 3.
Стадия 3Метил 4-хлор-1Н-пирроло[3,2-c]пиридин-2-карбоксилат
Указанное в заголовке соединение получали, как описано в примере 6, способ-1, стадия 3.
Стадия 4Метил 1-хлор-8-оксо-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-7-карбоксилат
К суспензии метил 4-хлор-1Н-пирроло[3,2-c]пиридин-2-карбоксилата (12,5 г, 59 ммоль) в THF (116 мл) - толуоле (460 мл) добавляли 1,0 М раствор трет-бутоксида калия (64 мл, 64 ммоль) и метилакрилата (55 мл, 611 ммоль) в THF. Полученную смесь нагревали до 100°С в течение 18 ч. После этого суспензию охлаждали до комнатной температуры и выливали ее в смесь насыщенного водного раствора NH4Cl (400 мл) и гексанов (400 мл). Твердые вещества сливали, фильтровали и промывали Н2О и гексанами с получением указанного в заголовке соединения.
Стадия 51-хлор-6,7-дигидро-8Н-пиридо[3,4-b]пирролизин-8-он
Указанное в заголовке соединение получали, как описано в примере 1, стадия 6, с использованием изопропанола вместо этанола и нагревания до 100°С в течение 1 ч.
Стадия 61-изопропенил-6,7-дигидро-8Н-пиридо[3,4-b]пирролизин-8-он
К смеси 1-хлор-6,7-дигидро-8Н-пиридо[3,4-b]пирролизин-8-она (5,0 г, 24,3 ммоль), трис(дибензилиденацетон)дипалладия (0) (1,0 г, 1,09 ммоль) и трифенилмышьяка (2,70 г, 8,82 ммоль) в DMF (100 мл) добавляли трибутилизопропенилолово (9,60 г, 29,00 ммоль). Полученную смесь дегазировали и нагревали до 78°С в течение периода времени 18 ч. Растворитель выпаривали при пониженном давлении. К полученной смеси добавляли CH2Cl2 и целит, а затем фильтровали через целит. Указанное в заголовке соединение очищали флэш-хроматографией (50-100% EtOAc в гексане).
Стадия 7этил (2Е)-1-изопропенил-6,7-дигидро-8Н-пиридо[3,4-b]пирролизин-8-илиден)этаноат
К раствору 1-изопропенил-6,7-дигидро-8Н-пиридо[3,4-b]пирролизин-8-она (0,60 г, 2,8 ммоль) и триэтилфосфоноацетата (1,00 г, 4,46 ммоль) в THF (24 мл) при -78°С добавляли 80% NaH (0,12 г, 4,00 ммоль), реакционную смесь оставляли нагреваться до 0°С, затем до комнатной температуры. Реакционную смесь выливали в насыщенный раствор NH4Cl и EtOAc. Органическую фазу отделяли, высушивали над Na2SO4 и выпаривали. Указанное в заголовке соединение очищали флэш-хроматографией (40% EtOAc в гексане).
Стадия 8Этил (1-изопропил-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил)ацетат
К раствору этил (2Е)-1-изопропенил-6,7-дигидро-8Н-пиридо[3,4-b]пирролизин-8-илиден)этаноата (0,40 г, 1,4 ммоль) в МеОН (20 мл) добавляли Pd(OH)2 (0,20 г). Смесь перемешивали при 1 атм Н2 в течение 3 ч. Смесь фильтровали через целит и выпаривали, с получением указанного в заголовке соединения.
Стадия 9Этил {9-[(3,4-дихлорфенил)тио]-1-изопропил-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил}ацетат
К раствору бис(3,4-дихлорфенил)дисульфида (0,24 г, 0,67 ммоль) в CH2Cl2 (5,6 мл) добавляли SO2Cl2 (0,036 мл). Полученную желтую смесь перемешивали при комнатной температуре в течение 1 ч. Указанный раствор добавляли к раствору этил (1-изопропил-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил)ацетата (0,15 г, 0,52 ммоль) в DMF (5,6 мл) при 0°С. Через 1,5 ч при 0°С реакционную смесь выливали в насыщенный раствор NaHCO3 и EtOAc. Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и выпаривали. Указанное в заголовке соединение очищали флэш-хроматографией (30-40% EtOAc в гексане).
Стадия 10{9-[(3,4-дихлорфенил)тио]-1-изопропил-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил}уксусная кислота
К раствору этил {9-[(3,4-дихлорфенил)тио]-1-изопропил-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил}ацетата (0,23 г, 0,50 ммоль) в THF (5 мл) и МеОН (2,5 мл) добавляли 1,0 М NaOH (1,5 мл, 1,5 ммоль). После перемешивания в течение 18 ч при КТ добавляли НОАс (0,25 мл) и растворитель выпаривали. Остаток помещали в EtOAc/H2O и органический слой промывали Н2О и насыщенным солевым раствором. После сушки (Na2SO4) раствор фильтровали и выпаривали. Остаток перемешивали с 1:1 EtOAc/гексаны с получением после фильтрования указанного в заголовке соединения в виде твердого вещества белого цвета.
1Н ЯМР (MeOH-d4) δ 1,14-1,26 (м, 6H), 2,47-2,56 (м, 1H), 2,56-2,64 (м, 1H), 2,94-3,05 (м, 2H), 3,81-3,89 (м, 1H), 4,22-4,30 (м, 1H), 4,33-4,44 (м, 2H), 6,93-6,99 (м, 1H), 7,14-7,19 (м, 1H), 7,33-7,39 (м, 1H), 7,54-7,59 (м, 1H), 8,16-8,21 (м, 1H).
Продукт со стадии 10 превращали в его метиловый эфир с использованием CH2N2 и эфир подвергали разделению ВЭЖХ с хиральной стационарной фазой (колонка chiralcel OD 2·25 см), с элюированием 12% 2-пропанолом в гексане при скорости потока 6 мл/мин. Энантиомер А (менее полярный) имел время задержки 31,9 минут, а энантиомер В (более полярный) имел время задержки 35,5 минут. И А, и В гидролизовали как в примере 17, стадия 10, с получением энантиомеров А и В указанного в заголовке соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ CRTH2 РЕЦЕПТОРА | 2008 |
|
RU2451019C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
| ПИРИМИДО[5,4-b]ИНДОЛИЗИНОВОЕ ИЛИ ПИРИМИДО[5,4-b]ПИРРОЛИЗИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2721774C1 |
| ПРОИЗВОДНЫЕ ПИРИДОИНДОЛОНА, ЗАМЕЩЕННЫЕ В ПОЛОЖЕНИИ 6, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ | 2005 |
|
RU2364595C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИИ | 2020 |
|
RU2780101C2 |
| ИНГИБИТОРЫ АКТИВНОСТИ АКТ | 2010 |
|
RU2579513C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |
Описываются новые производные пиридопирролизина и пиридоиндолизина общей формулы I
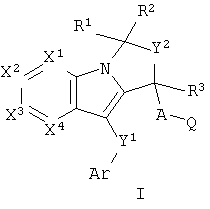
и его фармацевтически приемлемые соли и гидраты, где
А - С1-3алкил;
Ar означает нафтил или фенил, необязательно замещенный одной-двумя группами, выбранными из галогена, С1-6алкила или галоидС1-6алкила;
Q означает СООН;
один из X1, X2, X3 или X4 означает азот, а другие независимо выбраны из СН и C-Rg где Rg означает C1-6алкил или S(O)nС1-6алкил, где n=0, 2;
Y1 означает S или С(O);
Y2 означает (CRdRe)m, где Rd и Re - водород, m - целое число 1 или 2;
R1, R2, R3 означают водород, фармацевтическая композиция, их содержащая, и способ лечения заболеваний, опосредованных простагландином D2. 3 н. и 19 з.п. ф-лы, 2 табл.
и его фармацевтически приемлемые соли и гидраты, где
А выбран из С1-3алкила;
Ar представляет собой нафтил или фенил, необязательно замещенный одной-двумя группами, выбранными из галогена, С1-6алкила или галоидС1-6алкила;
Q означает СООН;
один из Х1, Х2, Х3 или Х4 представляет собой азот, а другие независимо выбраны из СН или C-Rg, где Rg представляет собой C1-6алкил или S(O)nC1-6алкил, где n=0, 2;
Y1 представляет собой S или С(O);
Y2 означает (CRdRe)m, где Rd и Re означают собой водород, m является целым числом 1 или 2;
R1, R2, R3 означает водород.
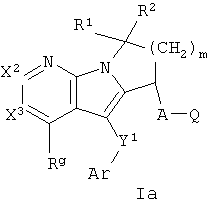
где Х2 и Х3 независимо представляют собой СН или C-Rg, А, Ar, Q, Y1, R1, R2, m и Rg определены в п.1.
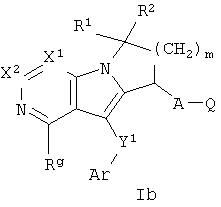
где X1 и X2 независимо представляют собой CH или C-Rg, A, Ar, Q, Y1, R1, R2, m и Rg определены в п.1.
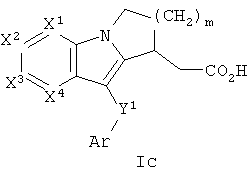
где один из Х1, Х2 и Х3 представляет собой N, а другие представляют собой, каждый, СН, X4 представляет собой CRg, m равно 1 или 2, и Ar, Y1 и m определены в п.1.
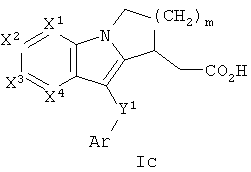
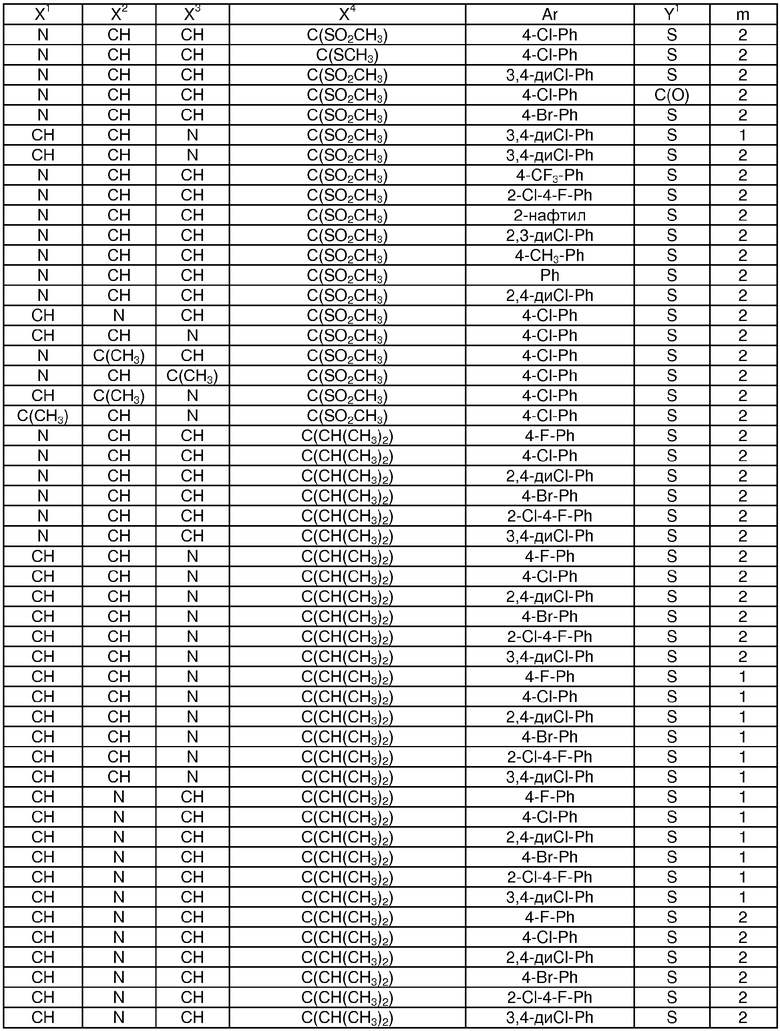
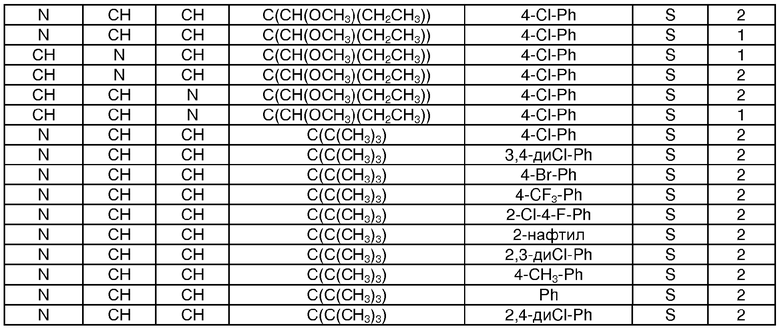
[5-[(4-хлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота;
[5-[(4-хлорфенил)тио]-4-(метилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота;
[5-[(3,4-дихлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота;
[5-(4-хлорбензоил)-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота;
[5-(4-бромфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил]уксусная кислота;
[9-[(3,4-дихлорфенил)тио]-1-(метилсульфонил)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил]уксусная кислота;
[10-[(3,4-дихлорфенил)сульфанил]-1-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,4-b]индолизин-9-ил]уксусная кислота;
4-(метилсульфонил)-5-{[4-трифторметил)фенил]тио}-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота;
[5-[(2-хлор-4-фторфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота;
[4-(метилсульфонил)-5-(2-нафтилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота;
[5-[(2,3-дихлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота;
[5-[(4-метилфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота;
[4-(метилсульфонил)-5-(фенилтио)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота;
[5-[(2,4-дихлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[3,2-b]индолизин-6-ил)уксусная кислота;
[5-[(4-хлорфенил)тио]-4-(метилсульфонил)-6,7,8,9-тетрагидропиридо[4,3-b]индолизин-6-ил)уксусная кислота;
[9-[(4-хлорфенил)тио]-1-(метилсульфонил)-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил)уксусная кислота;
[9-[(3,4-дихлорфенил)тио]-1-изопропил-7,8-дигидро-6Н-пиридо[3,4-b]пирролизин-8-ил)уксусная кислота;
или их фармацевтически приемлемых солей, или гидратов.
| US 5128364 A, 07.07.1992 | |||
| WO 9825919 A, 18.06.1998 | |||
| Логометр | 1971 |
|
SU441517A1 |
| Способ получения незамещенного 4-оксо-1,2,3,3а-тетрагидропирамидо-(1,2,3,3 @ )- @ -карболина или его 5,6-алкил (арил)производных | 1985 |
|
SU1268586A1 |
| ПИРИДО[3,2-Е]ПИРАЗИНОНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРЕПАРАТ НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ПРЕПАРАТА | 1996 |
|
RU2143433C1 |
| НОВЫЕ ТЕТРАЦИКЛИЧЕСКИЕ АНАЛОГИ КАМПТОТЕЦИНОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1998 |
|
RU2220144C2 |

