ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к низкомолекулярным ингибиторам, направленным на киназу BTK, и, в частности, к пиримидо[5,4-b]индолизиновому или пиримидо[5,4-b]пирролизиновому соединению, способу его получения и применению.
УРОВЕНЬ ТЕХНИКИ
Тирозинкиназа Брутона (BTK) представляет собой класс нерецепторных тирозинкиназ и принадлежит к семейству тирозинкиназ ТЕС, включающему Тес, Bmx, BTK , Itk и Txk. BTK в настоящее время является клинически подтвержденной лекарственной мишенью в семействе ТЕС.
BTK, главным образом, экспрессируется в клетках, относящихся к гемопоэтической системе, таких как В-клетки, мастоциты, макрофаги и т.д. В-клетки вырабатывают разнообразные физиологические сигналы посредством сигнальной трансдукции через В-клеточный рецептор (BCR). Сигнальный путь BCR играет ключевую роль в развитии и мутации В-клеток. Нарушение и дисфункция сигнального пути BCR часто приводят к тяжелому иммунодефициту и воспалительному ответу, а также сигнальный путь BCR тесно связан с появлением и развитием различных опухолей, связанных с В-клетками. BTK является ключевым регулятором образования ранних В-клеток и выживания зрелых В-клеток и является посредником в сигнальном пути-предшественнике пути BCR. Кроме того, BTK также тесно связана с пролиферацией и апоптозом клеток. После стимуляции иммунного ответа в организме BCR распознает антиген и активирует экспрессию факторов транскрипции NF-κB/Rel через опосредованный BTK сигнальный путь BCR для регулирования пролиферации В-клеток. С другой стороны, активированная BTK также может регулировать транскрипционную активность BAP-135/TFII-I, а впоследствии регулировать экспрессию белков, связанных с апоптозом, таких как VpreB, CD5 и Bcl-2. Таким образом, ингибиторы BTK можно применять для лечения определенных гематологических опухолей и аутоиммунных заболеваний.
Ибрутиниб, разработанный Pharmacyclics Inc., является единственным коммерчески доступным в настоящее время низкомолекулярным ингибитором, направленным на киназу BTK. Это лекарственное средство получило одобрение FDA для реализации на рынке в ноябре 2013 года в качестве принципиально нового терапевтического средства для клинического лечения рецидивирующей или рефрактерной мантийноклеточной лимфомы (МКЛ). Ибрутиниб обеспечивает общий уровень улучшения состояния 65,8%, где медианная продолжительность улучшения состояния составляет 1,9 месяца. Тем не менее, ибрутиниб имеет относительно тяжелые токсичные и побочные эффекты, так как он эффективно ингибирует киназы, такие как Tek и EGFR.
Таким образом, сохраняется необходимость в дальнейшей разработке других низкомолекулярных ингибиторов, направленных на киназу BTK.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено 5-арил-пиримидоиндолизиновое или 5-арил-пиримидопирролизиновое соединение, такое как показано на формуле I, или его фармацевтически приемлемая соль
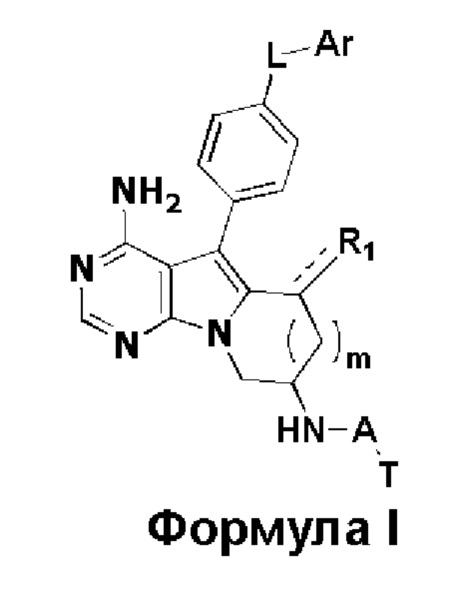
где:
R1 представляет собой Н, метил или метилиден;
L представляет собой -О-, -C(=O)NH- или -C(=O)NHCHR2-, причем R2 представляет собой водород или замещенный или незамещенный С1-С3 алкил, и заместитель в R2 представляет собой галоген или С1-С3 алкокси;
m равен 0 или 1;
Ar представляет собой замещенный или незамещенный С6-С20 арил, замещенный или незамещенный 5-20-членный гетероарил, содержащий один или более гетероатомов, выбранных из группы, состоящей из О, N и S, предпочтительно замещенный или незамещенный С6-С10 арил, замещенный или незамещенный 5-10-членный гетероарил, содержащий один или более гетероатомов, выбранных из группы, состоящей из О, N и S, более предпочтительно замещенный или незамещенный фенил, замещенный или незамещенный 5-6-членный гетероарил (в частности, пиридил), где заместитель в Ar представляет собой галоген, С1-С6 алкил, С1-С6 алкокси, трифторметил или трифторметокси, предпочтительно галоген, С1-С3 алкил, С1-С3 алкокси, трифторметил или трифторметокси, более предпочтительно галоген, С1-С3 алкил, С1-С3 алкокси, трифторметил или трифторметокси;
А представляет собой карбонил, сульфонил или 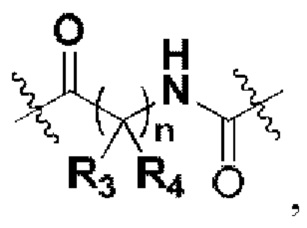 где R3 и R4 независимо могут представлять собой водород или С1-С3 алкил, или R3, R4 совместно с присоединенным к ним атомом углерода образуют С3-С5 циклоалкил;
где R3 и R4 независимо могут представлять собой водород или С1-С3 алкил, или R3, R4 совместно с присоединенным к ним атомом углерода образуют С3-С5 циклоалкил;
n равен 0, 1 или 2, предпочтительно 0 или 1;
Т представляет собой 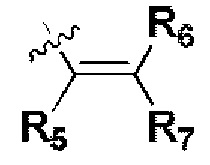 или
или 
R5, R6 и R7 независимо могут быть выбраны из группы, состоящей из водорода, циано, галогена и замещенного или незамещенного С1-С20 алкила, предпочтительно независимо выбраны из группы, состоящей из водорода, циано, галогена и замещенного или незамещенного С1-С10 алкила, более предпочтительно независимо выбраны из группы, состоящей из водорода, циано, галогена и замещенного или незамещенного С1-С5 алкила; заместитель в R5, R6 или R7 представляет собой диметиламино, С1-С10 алкокси или 3-10-членный гетероциклил, содержащий один или более гетероатомов, выбранных из группы, состоящей из О, N и S; предпочтительно диметиламино, С1-С6 алкокси или 3-10-членный гетероциклил, содержащий один или более гетероатомов, выбранных из группы, состоящей из О, N и S; более предпочтительно диметиламино, С1-С5 алкокси или 3-6-членный гетероциклил, содержащий от 1 до 3 гетероатомов, выбранных из группы, состоящей из О, N и S;
R8 выбран из группы, состоящей из водорода и С1-С10 алкила, предпочтительно выбран из группы, состоящей из водорода и С1-С5 алкила, более предпочтительно выбран из группы, состоящей из водорода и С1-С3 алкила.
В одном из вариантов реализации L представляет собой -О- или -C(=O)NH-.
В одном из вариантов реализации Ar представляет собой фенил, 4-фторфенил, 4-трифторметилфенил, пиридин-2-ил, 3-фторпиридин-6-ил или 4-трифторметилпиридин-6-ил.
В одном из вариантов реализации R3 представляет собой водород.
В одном из вариантов реализации R4 представляет собой Н или метил.
В одном из вариантов реализации R3, R4 совместно с присоединенным к ним атомом углерода образуют циклопропил.
В одном из вариантов реализации n равен 0.
В одном из вариантов реализации R5 представляет собой Н, циано или метил.
В одном из вариантов реализации R6 представляет собой Н.
В одном из вариантов реализации R7 представляет собой Н, трет-бутил или диметиламинометил.
В одном из вариантов реализации R8 представляет собой Н или метил.
В одном из вариантов реализации Т представляет собой винил или пропинил.
В настоящем изобретении галоген включает фтор, хлор, бром и йод.
В настоящем изобретении С6-С20 арил обозначает 6-20-членную моноциклическую или полициклическую ароматическую группу, кольцо в которой состоит только из атомов углерода, такую как фенил, нафтил и т.д. С6-С10 арил имеет схожее значение.
В настоящем изобретении 5-20-членный гетероарил обозначает 5-20-членную моноциклическую или полициклическую ароматическую группу, кольцо в которой содержит один или более гетероатомов, выбранных из группы, состоящей из О, N и S, например, фуранил, пирролил, пиридил, пиримидинил и т.д. 5-10- и 5-6-членный гетероарил имеют схожие значения.
В настоящем изобретении 3-10-членный гетероциклил обозначает 3-10-членную моноциклическую или полициклическую неароматическую группу, кольцо в которой содержит один или более гетероатомов, выбранных из группы, состоящей из О, N и S, например, азиридинил, оксиранил, азетидинил, оксетанил, тетрагидрофуранил, азациклопентанил, пиперидинил и т.д. 3-6-членный гетероциклил имеет схожее значение.
Более предпочтительно соединение выбрано из группы, состоящей из соединений, приведенных в следующей таблице:

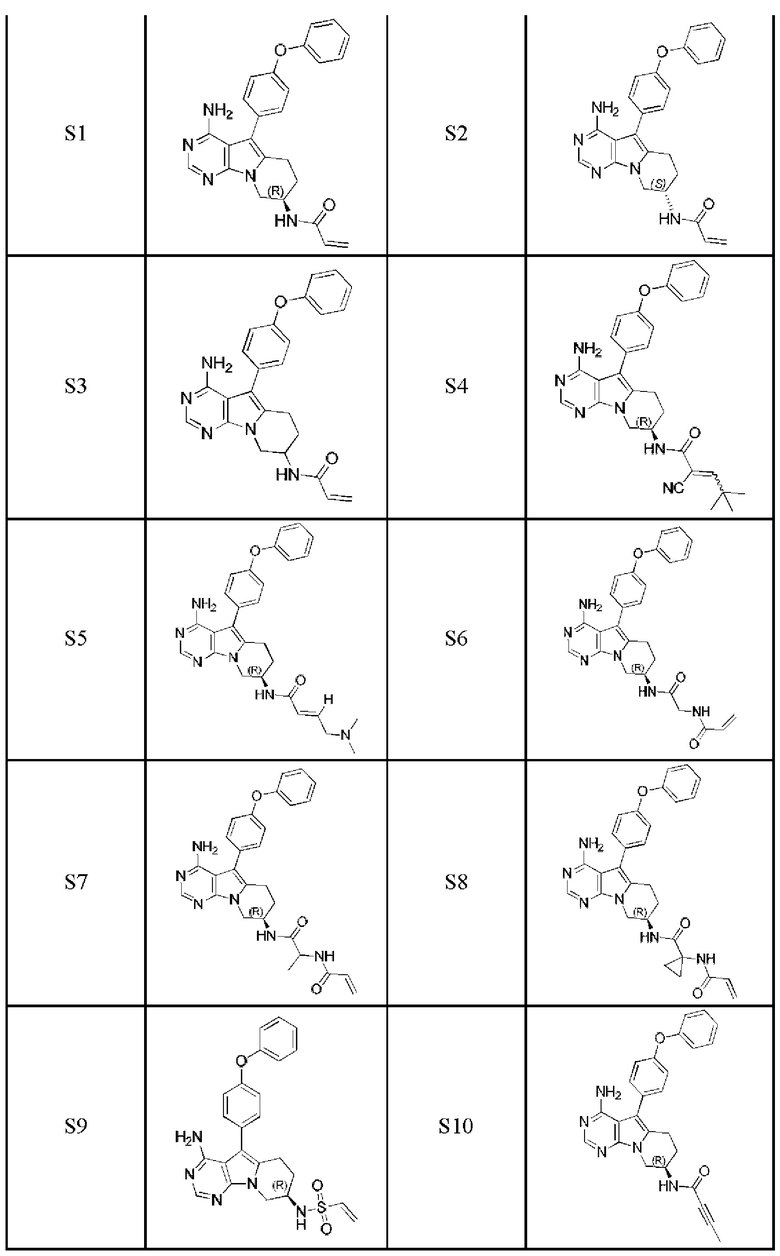
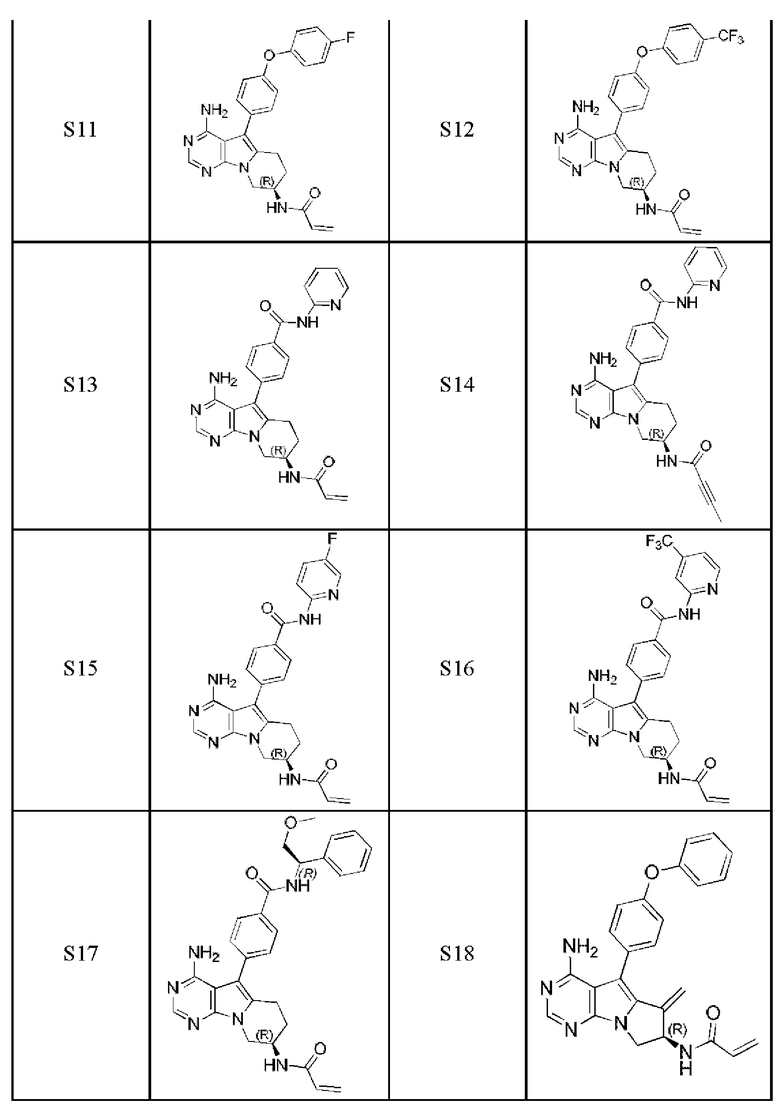
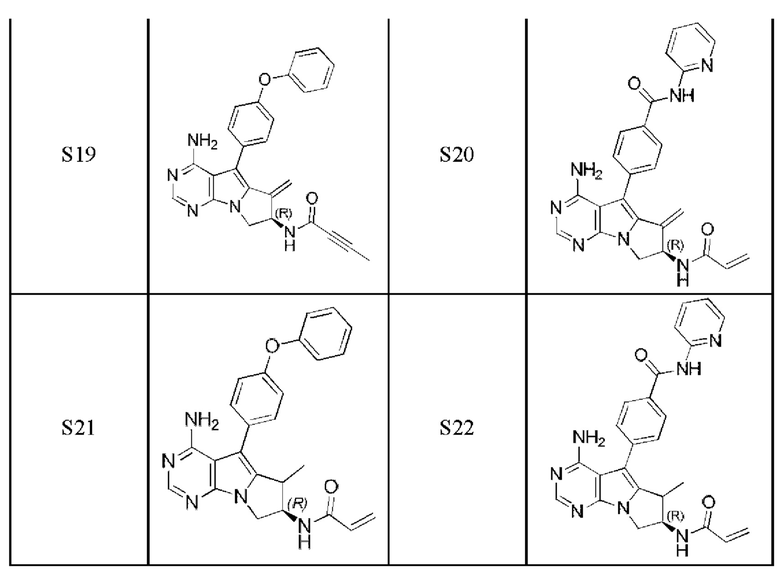
В настоящем изобретении также предложен способ получения 5-арил-пиримидоиндолизинового или 5-арил-пиримидопирролизинового соединения. Сокращения и условные обозначения, используемые при описании способа получения (а также в других фрагментах настоящего описания) имеют следующие значения:
Сокращения и условные обозначения
9-BBN: 9-бор-бицикло[3.3.1]нонан
DCM: дихлорметан
DIAD: диизопропилазодикарбоксилат
DIPEA: диизопропилэтиламин
DMF: N,N-диметилформамид
ЕА: этилацетат
HATU: гексафторфосфат 2-(7-оксибензотриазол)-N,N,N',N'-тетраметилмочевины
NBS: N-бромсукцинимид
NIS: N-йодсукцинимид
PdCl2(dppf): дихлорид [1,1'-бис(дифенилфосфинил)ферроцен]палладия
РЕ: петролейный эфир
THF: тетрагидрофуран
Способ получения соединения включает следующие стадии:
Схема 1:
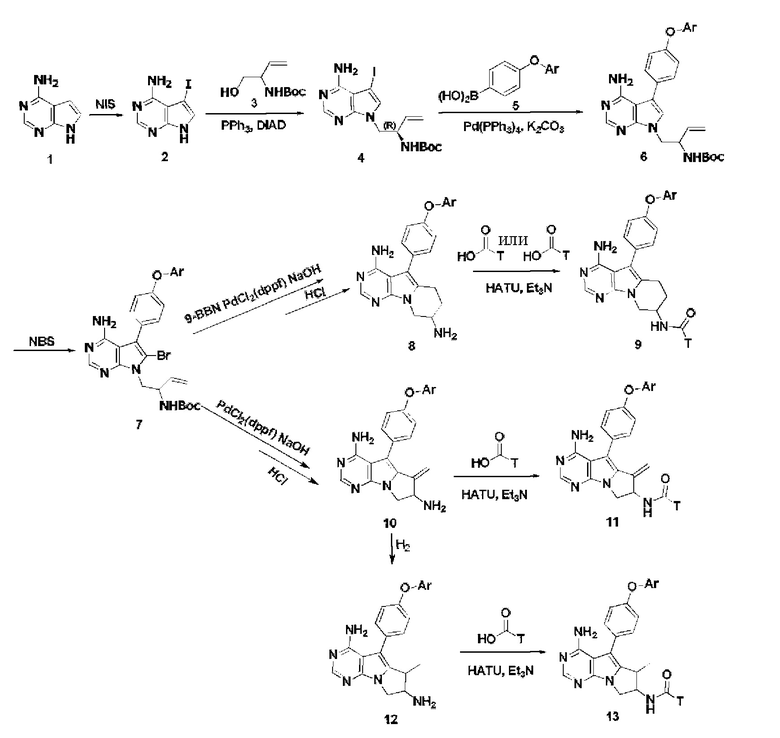
Схема 1а:
В коммерчески доступном 4-амино-пирроло[2,3-d]пиримидине, т.е. в соединении 1, в качестве исходного вещества проводят замещение йодом с использованием N-йодсукцинимида (NIS) по положению 5 с получением соединения 2,
В соединение 2 встраивают фрагмент 3 по реакции Мицунобу с получением соединения 4,
Проводят сочетание соединения 4 с замещенной фенилбороновой кислотой или боратом с получением соединения 6,
Вводят бром по положению 6 в соединение 6 с использованием N-бромсукцинимида (NBS) с получением соединения 7,
Проводят реакцию соединения 7 с 9-бор-бицикло[3.3.1]нонаном (9-BBN) в безводном тетрагидрофуране (THF), затем образуется шестичленное кольцо в результате реакции внутреннего сочетания Сузуки-Мияура в присутствии дихлорида [1,1'-бис(дифенилфосфинил)ферроцен]палладия (PdCl2(dppf)), и удаляют защитные группы с получением соединения 8, имеющего ядро тетрагидропиридо [5,4-b] индола,
Проводят конденсацию соединения 8 с карбоновой кислотой или сульфокислотой с получением соединения 9,
или
Схема 1b:
Из соединения 7 получают пятичленное циклическое соединение по внутримолекулярной реакции Хека в присутствии PdCl2(dppf), и удаляют защитные группы с получением соединения 10, из которого путем конденсации с замещенной карбоновой кислотой получают соединение 11,
или
Схема 1с:
Соединение 10 гидрируют в присутствии катализатора с получением соединения 12,
Проводят конденсацию соединения 12 с кислотой с получением соединения 13;
или
Схема 2:
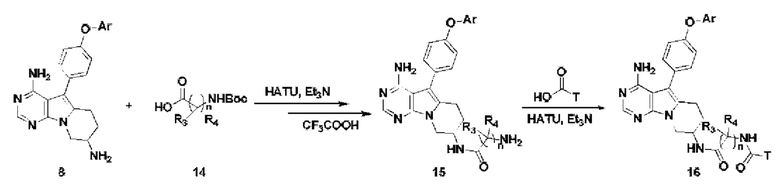
Проводят конденсацию соединения 8 с N-Boc-защищенной аминокислотой 14 и удаляют защитные группы с получением соединения 15,
Проводят конденсацию соединения 15 с замещенной карбоновой кислотой с получением соединения 16;
или
Схема 3:
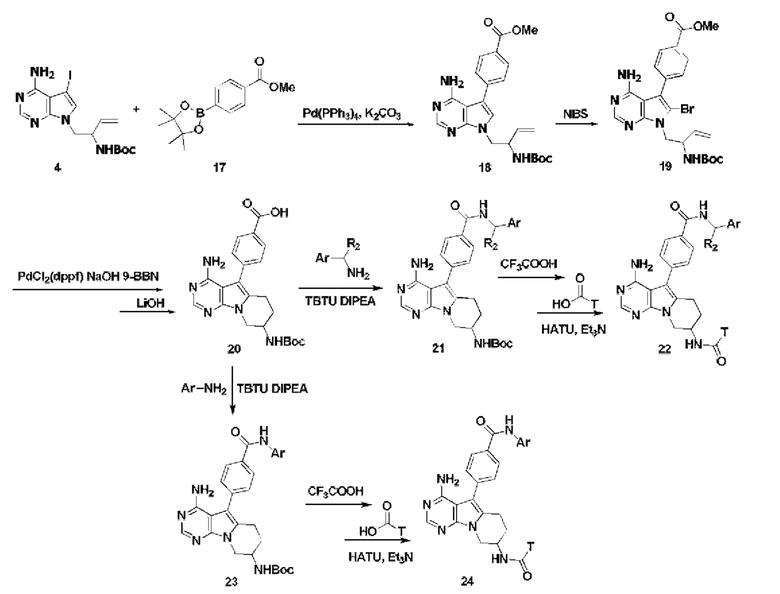
Схема 3а:
Проводят сочетание соединения 4 с замещенной фенилбороновой кислотой или боратом 17 с получением соединения 18,
Вводят бром по положению 6 в соединение 18 с использованием NBS с получением бромированного соединения 19,
Проводят реакцию соединения 19 с 9-BBN в безводном THF, образуется шестичленное кольцо путем реакции внутреннего сочетания Сузуки-Мияура в присутствии PdCl2(dppf), затем проводят гидролиз в присутствии гидроксида лития с получением соединения 20,
Проводят конденсацию соединения 20 с замещенным алкиламином с получением соединения 21,
Удаляют защитные группы в соединении 21 и проводят конденсацию с карбоновой кислотой с получением соединения 22, или
Схема 3b:
Проводят конденсацию соединения 20 с ариламином с получением соединения 23,
Удаляют защитные группы в соединении 23 и проводят конденсацию с карбоновой кислотой с получением соединения 24;
или
Схема 4:
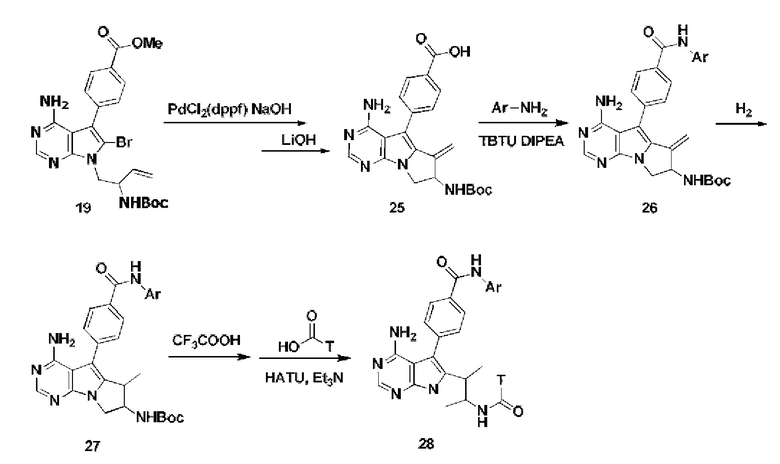
Из бромированного соединения 19 получают пятичленное циклическое соединение по внутримолекулярной реакции Хека в присутствии PdCl2(dppf), и обрабатывают LiOH с получением карбоновой кислоты 25,
Проводят конденсацию карбоновой кислоты 25 с ариламином с получением амида 26,
Амид 26 гидрируют в присутствии катализатора с получением соединения 27,
Удаляют защитные группы в соединении 27 с использованием CF3COOH для удаления Вос-защиты и проводят конденсацию с карбоновой кислотой с получением соединения 28.
В указанных выше реакциях:
R2 представляет собой водород или замещенный или незамещенный С1-С3 алкил, где заместитель в R2 представляет собой галоген или С1-С3 алкокси;
Ar представляет собой замещенный или незамещенный С6-С20 арил, замещенный или незамещенный 5-20-членный гетероарил, содержащий один или более гетероатомов, выбранных из группы, состоящей из О, N и S, предпочтительно замещенный или незамещенный С6-С10 арил, замещенный или незамещенный 5-10-членный гетероарил, содержащий один или более гетероатомов, выбранных из группы, состоящей из О, N и S, более предпочтительно замещенный или незамещенный фенил, замещенный или незамещенный 5-6-членный гетероарил (в частности, пиридил), где заместитель в Ar представляет собой галоген, С1-С6 алкил, С1-С6 алкокси, трифторметил или трифторметокси, предпочтительно галоген, С1-С3 алкил, С1-С3 алкокси, трифторметил или трифторметокси, более предпочтительно галоген, С1-С3 алкил, С1-С3 алкокси, трифторметил или трифторметокси;
R3 и R4 независимо могут представлять собой водород или С1-С3 алкил, или R3, R4 совместно с присоединенным к ним атомом углерода образуют С3-С5 циклоалкил;
n равен 0, 1 или 2, предпочтительно 0 или 1;
Т представляет собой 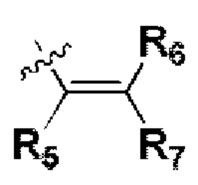 или
или 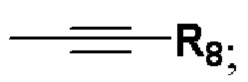
R5, R6 и R7 независимо могут быть выбраны из группы, состоящей из водорода, циано, галогена и замещенного или незамещенного С1-С20 алкила, предпочтительно независимо выбраны из группы, состоящей из водорода, циано, галогена и замещенного или незамещенного С1-С10 алкила, более предпочтительно независимо выбраны из группы, состоящей из водорода, циано, галогена и замещенного или незамещенного С1-С5 алкила; заместитель в R5, R6 или R7 представляет собой диметиламино, С1-С10 алкокси или 3-10-членный гетероциклил, содержащий один или более гетероатомов, выбранных из группы, состоящей из О, N и S; предпочтительно диметиламино, С1-С6 алкокси или 3-10-членный гетероциклил, содержащий один или более гетероатомов, выбранных из группы, состоящей из О, N и S; более предпочтительно диметиламино, С1-С5 алкокси или 3-6-членный гетероциклил, содержащий от 1 до 3 гетероатомов, выбранных из группы, состоящей из О, N и S;
R8 выбран из группы, состоящей из водорода и С1-С10 алкила, предпочтительно выбран из группы, состоящей из водорода и С1-С5 алкила, более предпочтительно выбран из группы, состоящей из водорода и С1-С3 алкила.
В одном из вариантов реализации Ar представляет собой фенил, 4-фторфенил, 4-трифторметилфенил, пиридин-2-ил, 3-фторпиридин-6-ил или 4-трифторметилпиридин-6-ил.
В одном из вариантов реализации R3 представляет собой водород.
В одном из вариантов реализации R4 представляет собой Н или метил.
В одном из вариантов реализации R3, R4 совместно с присоединенным к ним атомом углерода образуют циклопропил.
В одном из вариантов реализации n равен 0.
В одном из вариантов реализации R5 представляет собой Н, циано или метил.
В одном из вариантов реализации R6 представляет собой Н.
В одном из вариантов реализации R7 представляет собой Н, трет-бутил или диметиламинометил.
В одном из вариантов реализации R8 представляет собой Н или метил.
В одном из вариантов реализации Т представляет собой винил или пропинил.
Соединения согласно изобретению могут содержать асимметрические атомы, оси хиральности и плоскости хиральности и могут существовать в виде энантиомеров, диастереомеров, рацематов и их смесей.
Фармацевтически приемлемая соль соединения формулы I может представлять собой традиционную нетоксичную соль, образованную в результате реакции соединения формулы I с неорганической или органической кислотой. Например, традиционная нетоксичная соль может быть получена путем реакции соединения формулы I с неорганической или органической кислотой. Неорганические кислоты включают хлороводородную кислоту, бромоводородную кислоту, серную кислоту, азотную кислоту, аминосульфокислоту, фосфорную кислоту и т.д. Органические кислоты включают лимонную кислоту, винную кислоту, молочную кислоту, виноградную кислоту, уксусную кислоту, бензолсульфокислоту, n-толуолсульфокислоту, метансульфокислоту, нафталинсульфокислоту, этансульфокислоту, нафталиндисульфокислоту, малеиновую кислоту, яблочную кислоту, малоновую кислоту, фумаровую кислоту, янтарную кислоту, пропановую кислоту, щавелевую кислоту, трифторуксусную кислоту, стеариновую кислоту, памовую кислоту, гидроксималеиновую кислоту, фенилуксусную кислоту, бензойную кислоту, салициловую кислоту, глутаминовую кислоту, аскорбиновую кислоту, п-аминобензолсульфокислоту, 2-ацетоксибензойную кислоту, гидроксиэтансульфокислоту и т.д. В качестве альтернативы, фармацевтически приемлемая соль соединения формулы I может представлять собой соль натрия, соль калия, соль кальция, соль алюминия или соль аммония, которая образуется в результате реакции соединения формулы I с пропановой кислотой, щавелевой кислотой, малоновой кислотой, янтарной кислотой, фумаровой кислотой, малеиновой кислотой, молочной кислотой, яблочной кислотой, винной кислотой, лимонной кислотой, аспарагиновой кислотой или глутаминовой кислотой для получения сложного эфира и последующей реакции с неорганическим основанием; или соль метиламина, соль этиламина или соль этаноламина, образующуюся в результате реакции соединения формулы I с органическим основанием; или соль неорганической кислоты, которая образуется в результате реакции соединения формулы I с лизином, аргинином или орнитином для получения сложного эфира и последующей реакции с хлороводородной кислотой, бромоводородной кислотой, фтороводородной кислотой, серной кислотой, азотной кислотой или фосфорной кислотой; или соль органической кислоты, которая образуется в результате реакции соединения формулы I с лизином, аргинином или орнитином для получения сложного эфира и последующей реакции с муравьиной кислотой, уксусной кислотой, пикриновой кислотой, метансульфокислотой и этансульфокислотой.
В настоящем изобретении предложено применение соединения описанной выше формулы I для получения лекарственного средства для лечения заболевания, связанного с путем сигнальной трансдукции киназы BTK.
В изобретении также предложено применение соединения описанной выше формулы I в лечении заболевания, связанного с путем сигнальной трансдукции киназы BTK.
В изобретении также предложен способ лечения заболевания, связанного с путем сигнальной трансдукции BTK, включающий введение терапевтически эффективного количества одного или более соединений описанной выше формулы I или их фармацевтически приемлемых солей субъекту.
Заболевание, связанное с путем сигнальной трансдукции киназы BTK, выбрано из группы, состоящей из, например, рака, гиперплазии, рестеноза, иммунных нарушений и воспаления.
В изобретении также предложено применение соединения описанной выше формулы I в получении лекарственного средства для ингибирования киназы BTK.
В изобретении также предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения описанной выше формулы I или его фармацевтически приемлемой соли и один или более фармацевтически приемлемых носителей. Фармацевтическая композиция может дополнительно содержать фармацевтическое вспомогательное вещество, такое как отдушка, ароматизатор и т.д.
Фармацевтическая композиция согласно настоящему изобретению предпочтительно содержит активный ингредиент в массовом отношении от 1 до 99%. Предпочтительно соединение формулы I присутствует в отношении от 65% до 99% по массе в качестве активного ингредиента, а оставшуюся часть составляют фармацевтически приемлемые носители, такие как разбавитель или растворитель или солевой раствор.
Соединения и композиции согласно изобретению также подходят для лечения, предупреждения или модуляции раковых клеток и метастатических раковых опухолей. Соответственно, в настоящем изобретении также предложено применение соединения описанной выше формулы I в получении лекарственного средства для лечения, предупреждения или модуляции раковых клеток и метастатических раковых опухолей.
Рак согласно настоящему изобретению включает, но не ограничивается ими, гистиоцитарную лимфому, рак яичников, плоскоклеточный рак головы и шеи, рак желудка, рак молочной железы, детскую печеночноклеточную карциному, колоректальный рак, рак шейки матки, рак легкого, саркому, носоглоточную карциному, рак поджелудочной железы, глиобластому, рак предстательной железы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, множественную миелому, рак щитовидной железы, рак яичек, рак шейки матки, рак эндометрия, рак пищевода, лейкоз, почечноклеточную карциному, рак мочевого пузыря, рак печени, астроцитому и т.д.; более предпочтительно плоскоклеточный рак головы и шеи, гистиоцитарную лимфому, аденокарциному легкого, мелкоклеточный рак легкого, немелкоклеточный рак легкого, рак поджелудочной железы, папиллярный почечноклеточный рак, рак печени, рак желудка, рак толстой кишки, множественную миелому и глиобластому; предпочтительно рак представляет собой лимфому.
Соединение и фармацевтическая композиция согласно настоящему изобретению могут входить в различные составы, такие как таблетки, капсулы, порошки, сиропы, растворы, суспензии, аэрозоли и т.д., и они могут содержаться в подходящем твердом или жидком носителе или разбавителе и в подходящем стерилизующем устройстве для инъекции или капельницы.
Различные лекарственные формы фармацевтической композиции согласно настоящему изобретению можно получать согласно способам получения, общепринятым в области фармацевтики. Стандартная дозировка состава содержит от 0,05 мг до 200 мг соединения формулы I, предпочтительно стандартная дозировка состава содержит от 0,1 мг до 100 мг соединения формулы I.
Соединение и фармацевтическую композицию согласно настоящему изобретению можно клинически вводить млекопитающему, включая человека и животных, способом, таким как пероральный, через нос, на кожу, в легкие или желудочно-кишечный тракт, наиболее предпочтительно пероральным способом. Наиболее предпочтительная дневная доза составляет от 0,01 до 200 мг/кг массы тела за один раз или от 0,01 до 100 мг/кг массы тела за несколько раз. Выбор оптимальной дозировки для индивидуума должен основываться на конкретном режиме лечения независимо от способа введения. Как правило, наиболее подходящую дозу определяют, начиная с небольшой дозы, а затем постепенно увеличивая дозу до достижения оптимального значения.
Согласно экспериментам, приведенным в настоящем изобретении, соединение согласно настоящему изобретению имеет хорошую ингибирующую активность в отношении BTK на молекулярном и клеточном уровне. Важно отметить, что соединение согласно настоящему изобретению имеет низкую активность в отношении нормальных клеток Рамоса при В-клеточной лимфоме человека и высокую активность в отношении восприимчивых к BTK клеток диффузной крупноклеточной В-клеточной лимфомы человека TMD8, это указывает на то, что данный тип соединений с новой структурой обладает высокой селективностью и низкой нецелевой активностью, а также низкими побочными эффектами, и, таким образом, представляет собой класс селективных ингибиторов BTK, имеющий потенциал для разработки.
НАИЛУЧШИЙ ВАРИАНТ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
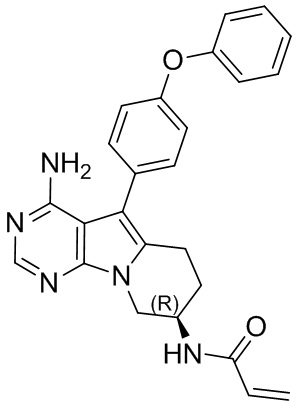
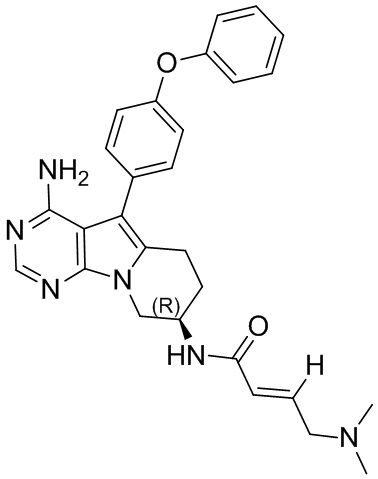
Пример 1: S1:
(R)-N-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид (схема 1)
Стадия 1: Синтез 4-амино-5-йод-7H-пирроло[2,3-d]пиримидина
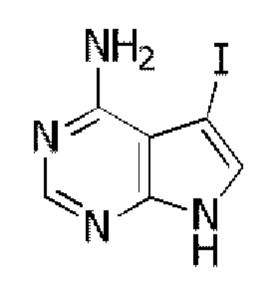
4-амино-7H-пирроло[2,3-d]пиримидин (2,7 г, 20 ммоль) растворяли в 60 мл хлороформа, добавляли NIS (4,5 г, 20 ммоль). Реакционный раствор кипятили с обратным холодильником в течение 2 часов и собирали нерастворившиеся вещества путем фильтрования. Неочищенный продукт очищали с помощью колоночной хроматографии с использованием смеси CHCl3/МеОН=20/1 с получением 4,2 г целевого продукта, выход 81%.
Стадия 2: Синтез трет-бутил-(R)-(1-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
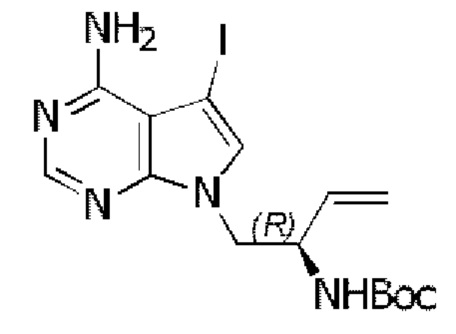
В реакционную колбу добавляли трифенилфосфин (787 мг, 3 ммоль), затем добавляли 15 мл безводного THF в качестве растворителя и охлаждали полученную смесь до 0°С. По каплям добавляли диизопропилазодикарбоксилат (DIAD) (606 мг, 3 ммоль) и перемешивали при указанной температуре в течение 15 минут, затем добавляли трет-бутил-(R)-(1-гидроксибут-3-ен-2-ил)карбамат (562 мг, 3 ммоль), а после него 4-амино-5-йод-7H-пирроло[2,3-d]пиримидин (520 мг, 2 ммоль). Через 30 минут нагревали реакционную смесь до комнатной температуры и перемешивали в течение 12 часов. Отфильтровывали нерастворившиеся вещества, концентрировали фильтрат и очищали неочищенный продукт путем колоночной хроматографии с использованием смеси CHCl3/МеОН=30/1 с получением 308 мг целевого продукта, выход 35,9%.
Стадия 3: Синтез трет-бутил-(R)-(1-(4-амино-5-(4-(феноксифенил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
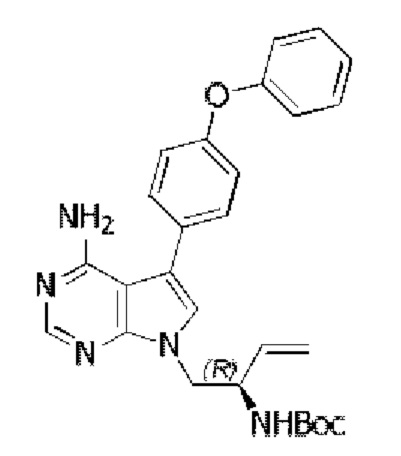
В запаянную ампулу добавляли трет-бутил-(R)-(1-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат (859 мг, 2 ммоль), 4-феноксифенилбороновую кислоту (642 мг, 3 ммоль), Pd(PPh3)4 (116 мг, 0,1 ммоль) и K2CO3 (553 мг, 4 ммоль), в качестве растворителя добавляли 25 мл 1,4-диоксана и 5 мл воды, реакцию проводили в запаянной ампуле в атмосфере N2 при 90°С на масляной бане в течение 2 часов. После завершения реакции экстрагировали полученную смесь этилацетатом и сушили над безводным Na2SO4. Неочищенный продукт очищали путем колоночной хроматографии с использованием смеси РЕ/ЕА=2/1 с получением 493 мг целевого продукта, выход 52,3%.
Стадия 4: Синтез трет-бутил-(R)-(1-(4-амино-5-(4-(феноксифенил)-6-бром-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата
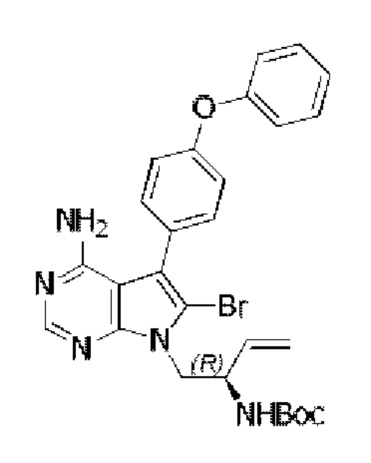
В 50 мл DMF добавляли трет-бутил-(R)-(1-(4-амино-5-(4-(феноксифенил)-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат (943 мг, 2 ммоль), медленно добавляли NBS (356 мг, 2 ммоль), перемешивали реакционную смесь при комнатной температуре в течение 8 часов. После завершения реакции экстрагировали полученную смесь этилацетатом и сушили над безводным Na2SO4. Неочищенный продукт очищали путем колоночной хроматографии с использованием смеси РЕ/ЕА=2/1 с получением 1,05 г целевого продукта, выход 95,4%.
Стадия 5: Синтез трет-бутил-(R)-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо [5,4-b]индолизин-8-ил)карбамата
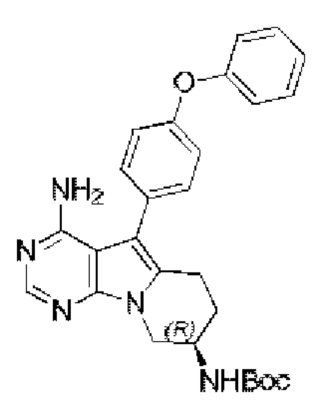
В запаянную ампулу добавляли трет-бутил-(R)-(1-(4-амино-5-(4-(феноксифенил)-6-бром-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат (550 мг, 1 ммоль), добавляли 10 мл безводного THF в атмосфере N2 и охлаждали до 0°С. По каплям добавляли раствор 9-BBN в тетрагидрофуране (12 мл, 0,5 М) и перемешивали смесь при 0°С в течение 10 минут, а затем при комнатной температуре в течение 5 часов. Последовательно добавляли водный раствор NaOH (4,7 мл, 3 М) и PdCl2(dppf) (190 мг, 0,25 ммоль), проводили реакцию при 80°С в запаянной ампуле в течение 15 часов. После завершения реакции экстрагировали полученную смесь этилацетатом и сушили над безводным Na2SO4. Неочищенный продукт очищали путем колоночной хроматографии с использованием ЕА с получением 316 мг целевого продукта, выход 67,0%.
Стадия 6: Синтез (R)-N-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламида (S1)
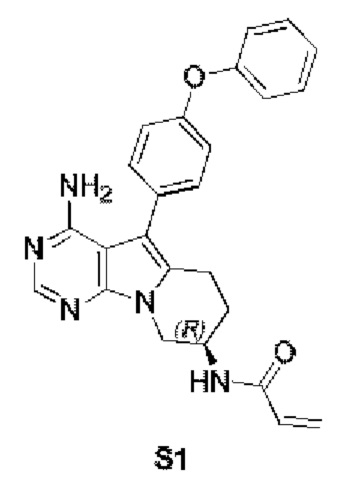
Трет-бутил-(R)-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат (472 мг, 1 ммоль) растворяли в 15 мл метанола, по каплям добавляли раствор HCl в метаноле (5 мл, 2 М) при 0°С, нагревали реакционную смесь до комнатной температуры и перемешивали в течение 8 часов. После концентрирования реакционного раствора досуха на роторном испарителе немедленно добавляли 20 мл DCM, затем по каплям добавляли Et3N (280 мкл, 2 ммоль) и акриловую кислоту (75 мкл, 1,1 ммоль), HATU (418 мг, 1,1 ммоль) и проводили реакцию при комнатной температуре в течение 2 часов. После завершения реакции экстрагировали полученную смесь этилацетатом и сушили над безводным Na2SO4. Неочищенный продукт очищали путем колоночной хроматографии с использованием смеси CHCl3/МеОН=30/1 с получением 129 мг целевого продукта, выход 30,3%.
1H ЯМР (300 МГц, CDCl3) δ 8,12 (s, 1H), 7,38 (dd, J=14,2, 7,3 Гц, 4Н), 7,16 (t, J=7,2 Гц, 1H), 7,09 (d, J=8,1 Гц, 4Н), 6,78 (d, J=6,7 Гц, 1H), 6,38 (d, J=16,4 Гц, 1H), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,69 (d, J=9,8 Гц, 1Н), 5,04 (s, 2Н), 4,72 (шир., 1H), 4,36 (dd, J=12,7, 4,6 Гц, 1H), 4,16 (dd, J=12,7, 5,7 Гц, 1H), 2,97 (t, J=6,2 Гц, 2Н), 2,16-2,04 (m, 2Н).
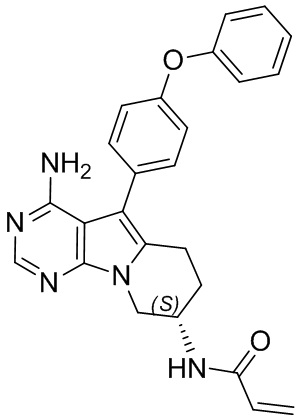
Пример 2: S2:
(S)-N-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид
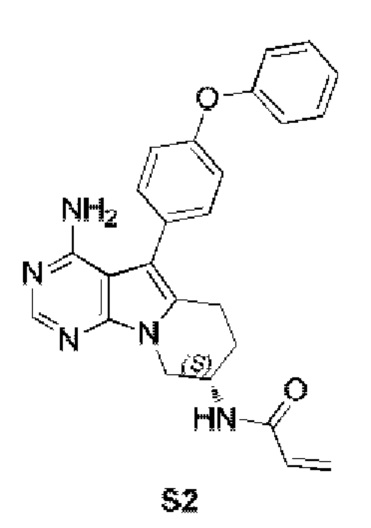
S2 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 2 трет-бутил-(S)-(1-гидроксибут-3-ен-2-ил)карбамат использовали вместо трет-бутил-(R)-(1-гидроксибут-3-ен-2-ил)карбамата.
1H ЯМР (300 МГц, CDCl3) δ 8,11 (s, 1H), 7,38 (dd, J=14,2, 7,3 Гц, 4Н), 7,15 (t, J=7,2 Гц, 1H), 7,09 (d, J=8,1 Гц, 4Н), 6,78 (d, J=6,7 Гц, 1H), 6,37 (d, J=16,4 Гц, 1H), 6,23 (dd, J=16,9, 10,0 Гц, 1H), 5,67 (d, J=9,8 Гц, 1Н), 5,08 (s, 2H), 4,70 (шир., 1H), 4,35 (dd, J=12,7, 4,6 Гц, 1H), 4,16 (dd, J=12,7, 5,7 Гц, 1H), 2,95 (t, J=6,2 Гц, 2H), 2,15-2,02 (m, 2H).
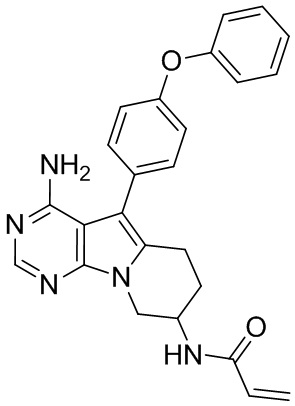
Пример 3: S3:
N-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид
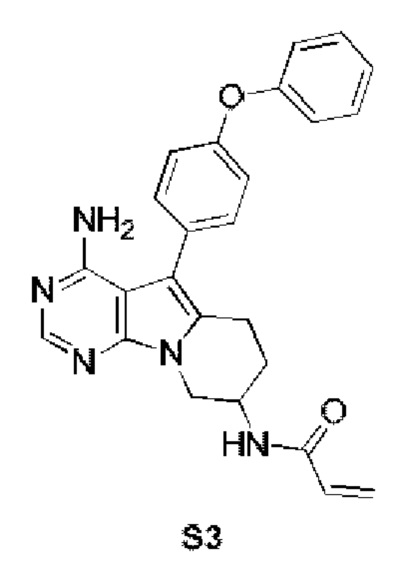
S3 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 2 рацемат трет-бутил-(1-гидроксибут-3-ен-2-ил)карбамата использовали вместо трет-бутил-(R)-(1-гидроксибут-3-ен-2-ил)карбамата.
1H ЯМР (300 МГц, CDCl3) δ 8,11 (s, 1H), 7,38 (dd, J=14,2, 7,3 Гц, 4Н), 7,15 (t, J=7,2 Гц, 1H), 7,08 (d, J=8,1 Гц, 4Н), 6,77 (d, J=6,7 Гц, 1H), 6,36 (d, J=16,4 Гц, 1H), 6,23 (dd, J=16,9, 10,0 Гц, 1H), 5,69 (d, J=9,8 Гц, 1H), 5,10 (s, 2H), 4,72 (s, шир., 1H), 4,35 (dd, J=12,7, 4,6 Гц, 1H), 4,15 (dd, J=12,7, 5,7 Гц, 1H), 2,96 (t, J=6,2 Гц, 2H), 2,16-2,03 (m, 2H).
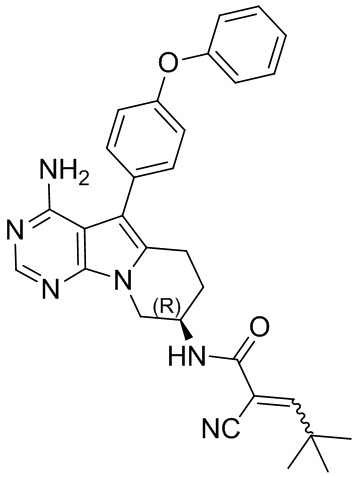
Пример 4: S4:
(R)-N-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-2-циано-4,4-диметилпент-2-енамид
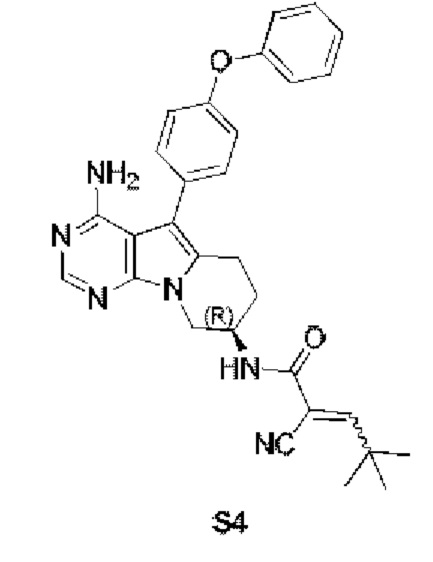
S4 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 6 2-циано-4,4-диметил-2-пентеновую кислоту использовали вместо акриловой кислоты.
1H ЯМР (300 МГц, CDCl3) δ 8,28 (s, 1H), 7,43-7,29 (m, 4H), 7,16 (t, J=7,3 Гц, 1H), 7,09 (d, J=8,3 Гц, 4H), 5,05 (s, 2H), 4,72 (dd, J=12,6, 5,7 Гц, 1H), 4,58-4,40 (m, 1H), 3,79 (d, J=2,9 Гц, 2H), 3,65 (d, J=2,9 Гц, 1H), 3,11 (dt, J=17,3, 4,3 Гц, 1H), 2,86 (ddd, J=16,7, 11,3, 5,2 Гц, 1H), 2,45 (ddd, J=16,5, 12,0, 5,4 Гц, 1H), 2,23 (dd,J=12,4, 4,0 Гц, 1H), 1,97 (s, 1H), 1,09 (s, 9H).
Пример 5: S5:
(R,E)-N-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)-4-(диметиламино)бут-2-енамид
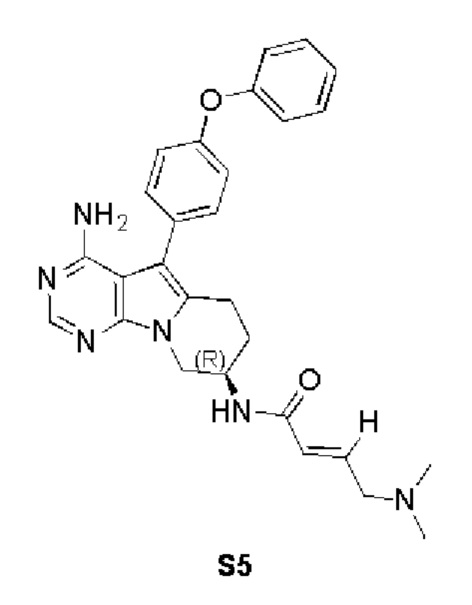
S5 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 6 (E)-4-диметиламино-2-бутеновую кислоту использовали вместо акриловой кислоты.
1H ЯМР (300 МГц, CDCl3) δ 8,10 (s, 1H), 7,42-7,28 (m, 5H), 7,15 (t, J=7,5 Гц, 1H), 7,07 (dd, J=7,5, 5,2 Гц, 4H), 6,94 6,80 (m, 1H), 6,29 (d, J=15,2 Гц, 1H), 5,24 (s, 2H), 4,65 (dd, J=10,4, 5,6 Гц, 1H), 4,36 (dd, J=11,9, 4,3 Гц, 1H), 4,11 (dd, J=12,9, 5,6 Гц, 1H), 3,36 (d, J=6,1 Гц, 2H), 3,04-2,88 (m, 2H), 2,49 (s, 6H), 2,07 (d, J=4,7 Гц, 2H).
Пример 6: S6:
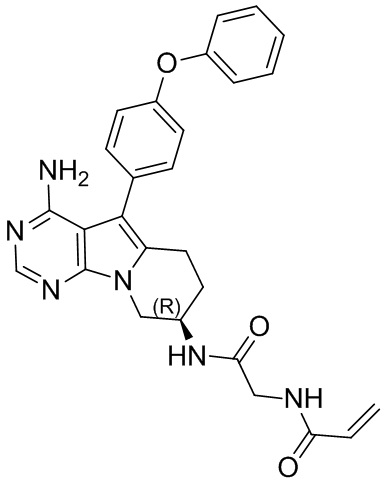
(R)-N-(2-((4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)амино)-2-оксоэтил)акриламид
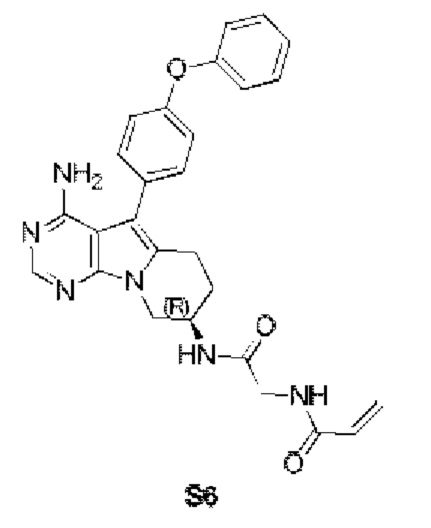
S6 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 6 2-акриламидоуксусную кислоту использовали вместо акриловой кислоты.
1H ЯМР (300 МГц, CDCl3) δ 8,12 (s, 1Н), 7,44-7,30 (m, 4H), 7,16 (t, J=7,3 Гц, 1H), 7,13-7,01 (m, 4Н), 6,36-6,07 (m, 2Н), 5,67 (d, J=9,8 Гц, 1H), 5,41 (s, 2H), 4,58-4,46 (m, 1H), 4,37 (dd, J=12,7, 4,8 Гц, 1H), 4,02 (dd, J=12,7, 6,1 Гц, 3H), 3,11-2,82 (m, 2H), 2,17-1,90 (m, 2H).
Пример 7: S7:
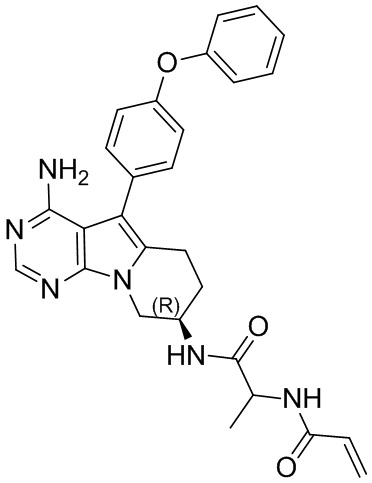
N-(1-(((R)-4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)имино)-1-оксопроп-2-ил)акриламид
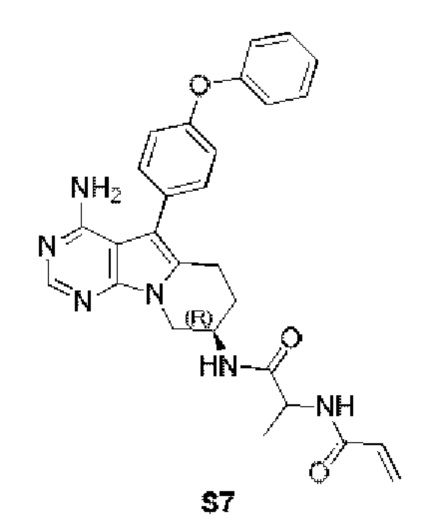
S7 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 6 2-акриламидопропановую кислоту использовали вместо акриловой кислоты.
1H ЯМР (300 МГц, CDCl3) δ 8,17 (s, 1H), 7,49-7,35 (m, 4H), 7,21 (t, J=7,3 Гц, 1H), 7,17-7,04 (m, 4H), 6,41-6,12 (m, 2H), 5,70 (d, J=9,8 Гц, 1H), 5,26 (s, 2H), 4,62-4,46 (m, 2H), 4,39 (dd, J=12,7, 4,8 Гц, 1H), 4,03 (dd, J=12,7, 5,7 Гц, 1H), 3,16-2,87 (m, 2H), 2,21-1,94 (m, 2H), 1,48 (d, J=4,5 Гц, 3H).
Пример 8: S8:
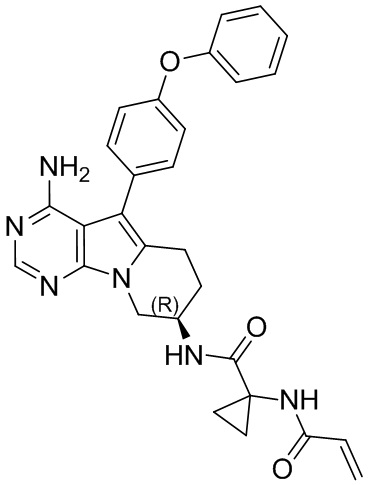
(R)-1-акриламидо-N-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)циклопропанкарбоксамид
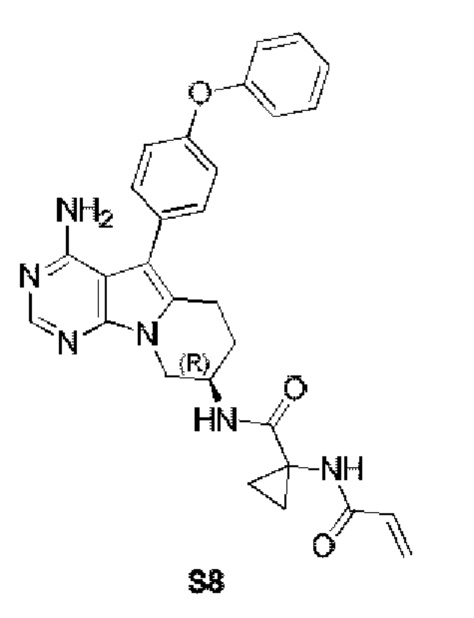
S8 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 6 1-акриламидоциклопропанкарбоновую кислоту использовали вместо акриловой кислоты.
1H ЯМР (300 МГц, CDCl3) δ 8,12 (s, 1H), 7,44-7,30 (m, 4H), 7,16 (t, J=7,3 Гц, 1H), 7,13-7,01 (m, 4H), 6,36-6,07 (m, 2H), 5,67 (d, J=9,8 Гц, 1H), 5,41 (s, 2H), 4,58-4,46 (m, 1H), 4,37 (dd, J=12,7, 4,8 Гц, 1H), 4,02 (dd, J=12,7, 6,1 Гц, 3H), 3,11-2,82 (m, 2H), 2,17-1,90 (m, 2H), 1,26-1,18 (m, 2H), 1,00-0,88 (m, 2H).
Пример 9: S9:
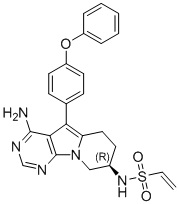
(R)-N-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)винилсульфамид
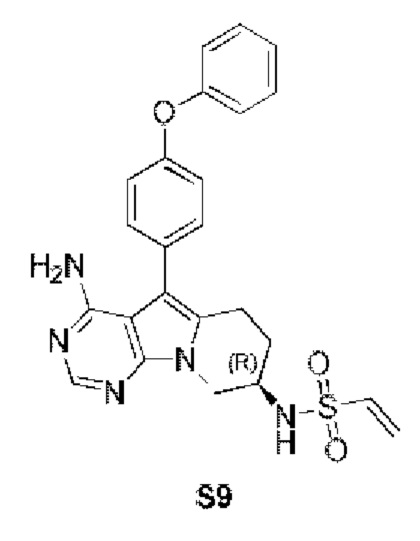
Трет-бутил-(R)-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамат (472 мг, 1 ммоль) (получали согласно стадии 1-5 в примере 1) растворяли в 15 мл метанола, по каплям добавляли раствор HCl в метаноле (5 мл, 2 М) при 0°С, нагревали реакционную смесь до комнатной температуры при перемешивании в течение 8 часов. После концентрирования реакционного раствора досуха на роторном испарителе немедленно добавляли 20 мл DCM, затем добавляли Et3N (280 мкл, 2 ммоль) и винилсульфонилхлорид (138 мг, 1,1 ммоль) и проводили реакцию при комнатной температуре в течение 2 часов. После завершения реакции экстрагировали полученную смесь этилацетатом и сушили над безводным Na2SO4. Неочищенный продукт очищали путем колоночной хроматографии с использованием смеси CHCl3/МеОН=30/1 с получением 115 мг целевого продукта, выход 24,9%.
1H ЯМР (300 МГц, CDCl3) δ 8,12 (s, 1H), 7,38 (dd, J=14,2, 7,3 Гц, 4Н), 7,16 (t, J=7,2 Гц, 1H), 7,09 (d, J=8,1 Гц, 4Н), 6,78 (d, J=6,7 Гц, 1H), 6,38 (d, J=16,4 Гц, 1H), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,69 (d, J=9,8 Гц, 1Н), 5,04 (s, 2H), 4,72 (шир., 1H), 4,36 (dd, J=12,7, 4,6 Гц, 1H), 4,16 (dd, J=12,7, 5,7 Гц, 1Н), 2,97 (t, J=6,2 Гц, 2H), 2,16-2,04 (m, 2H).
Пример 10: S10:
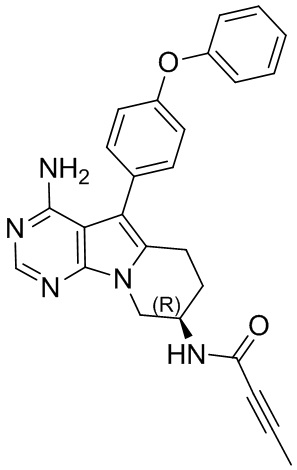
(R)-N-(4-амино-5-(4-феноксифенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)бут-2-иниламид
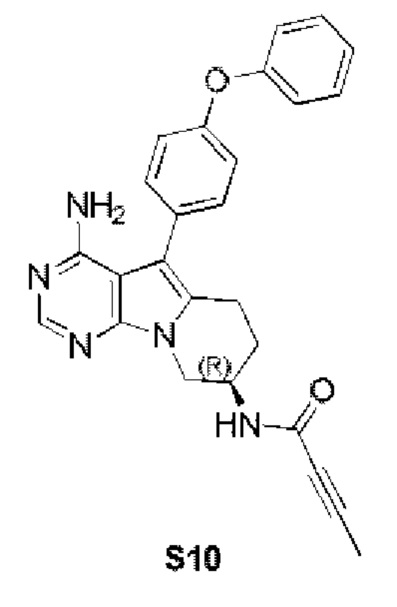
S10 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 6 бут-2-иновую кислоту использовали вместо акриловой кислоты.
1Н ЯМР (300 МГц, CDCl3) δ 8,14 (s, 1H), 7,31 (dd, J=16,2, 8,4 Гц, 4H), 7,10 (t, J=7,8 Гц, 1H), 7,02 (d, J=8,1 Гц, 4H), 6,50 (d, J=7,5 Гц, 1H), 4,94 (s, 2H), 4,58 (dd, J=12,4, 5,3 Гц, 1H), 4,31 (dd, J=13,0, 4,7 Гц, 1H), 4,04 (dd, J=12,9, 5,8 Гц, 1H), 2,91 (t, J=6,2 Гц, 2H), 2,01 (dd, J=10,6, 5,0 Гц, 2H), 1,87 (s, 3H).
Пример 11: S11:
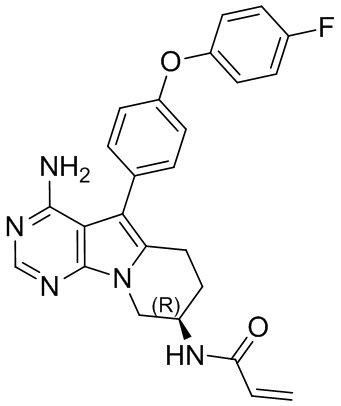
(R)-N-(4-амино-5-(4-(4-фторфенокси)фенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид
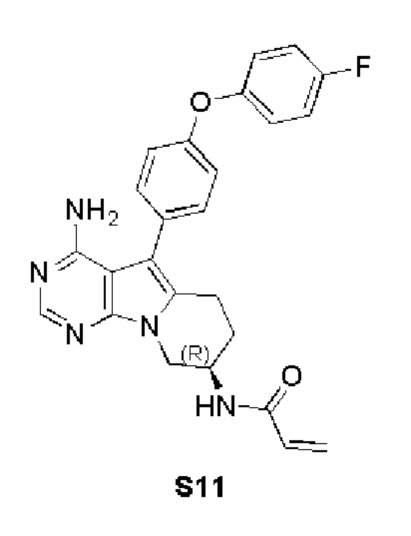
S11 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 3 4-(4-фторфенокси)фенилбороновую кислоту использовали вместо 4-феноксифенилбороновой кислоты.
1H ЯМР (300 МГц, CDCl3) δ 8,12 (s, 1H), 7,38 (dd, J=14,2, 7,3 Гц, 4Н), 7,16 (t, J=7,2 Гц, 1H), 7,09 (d, J=8,1 Гц, 4Н), 6,78 (d, J=6,7 Гц, 1H), 6,38 (d, J=16,4 Гц, 1H), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,69 (d, J=9,8 Гц, 1H), 5,04 (s, 2H), 4,72 (шир., 1H), 4,36 (dd, J=12,7, 4,6 Гц, 1H), 4,16 (dd, J=12,7, 5,7 Гц, 1Н), 2,97 (t, J=6,2 Гц, 2H), 2,16-2,04 (m, 2H).
Пример 12: S12:
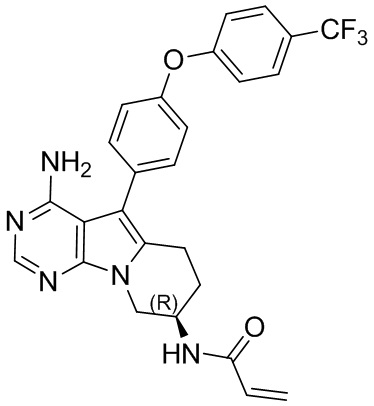
(R)-N-(4-амино-5-(4-(4-(трифторметил)фенокси)фенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)акриламид
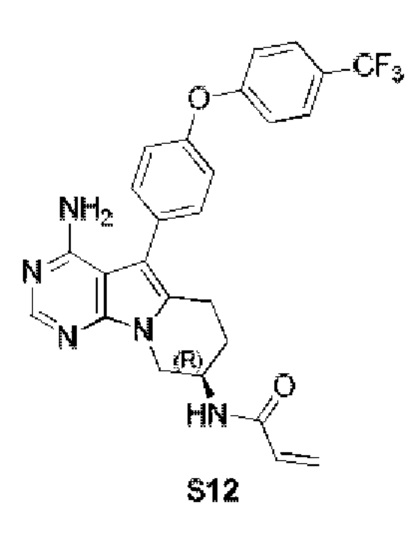
S12 синтезировали согласно стадиям синтеза S1 в примере 1 с тем исключением, что на стадии 3 4-(4-трифторметилфенокси)фенилбороновую кислоту использовали вместо 4-феноксифенилбороновой кислоты.
1H ЯМР (300 МГц, CDCl3) δ 8,15 (s, 1H), 7,38 (dd, J=14,2, 7,3 Гц, 4Н), 7,21 (d, J=8,1 Гц, 4Н), 6,78 (d, J=6,7 Гц, 1H), 6,38 (d, J=16,4 Гц, 1Н), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,69 (d, J=9,8 Гц, 1H), 5,04 (s, 2H), 4,72 (шир., 1H), 4,36 (dd, J=12,7, 4,6 Гц, 1H), 4,16 (dd, J=12,7, 5,7 Гц, 1H), 2,97 (t, J=6,2 Гц, 2H), 2,16-2,04 (m, 2H).
Пример 13: S13:
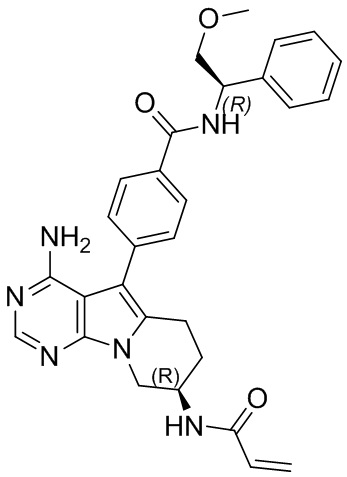
(R)-4-(8-акриламидо-4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)-N-(пиридин-2-ил)бензамид (схема 3b)
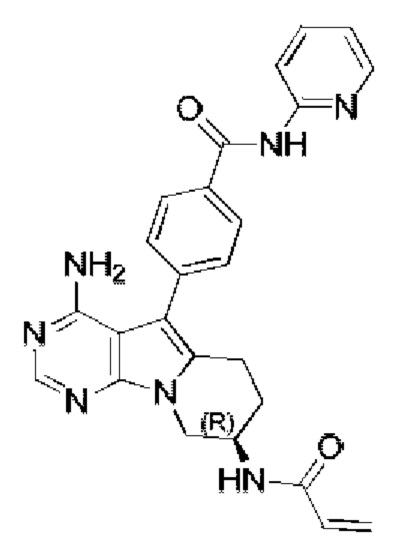
Стадия 1: Синтез метил-(R)-4-(4-амино-7-(2-((трет-бутоксикарбонил)-амино)бут-3-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)бензоата
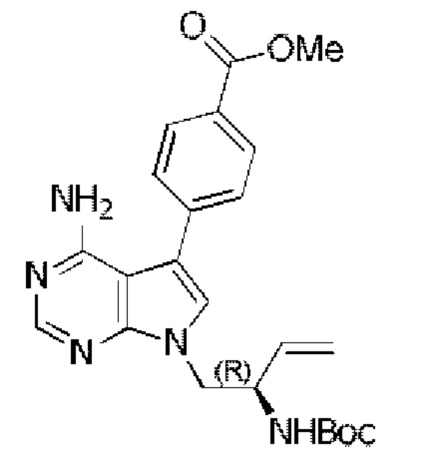
Метил-(R)-4-(4-амино-7-(2-((трет-бутоксикарбонил)амино)бут-3-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)бензоат синтезировали согласно стадии 3 в примере 1 с тем исключением, что 4-метоксикарбонилфенилбороновую кислоту использовали вместо 4-феноксифенилбороновой кислоты.
Стадия 2: Синтез метил-(R)-4-(4-амино-6-бром-7-(2-((трет-бутоксикарбонил)амино)бут-3-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)бензоата
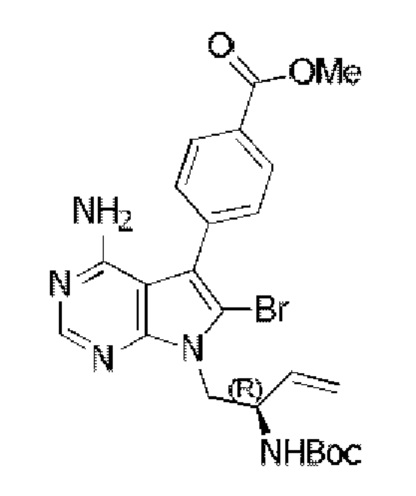
Метил-(R)-4-(4-амино-6-бром-7-(2-((трет-бутоксикарбонил)амино)бут-3-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)бензоат синтезировали согласно стадии 4 в примере 1 с тем исключением, что соединение, полученное на стадии 1 в данном примере 13, использовали вместо соответствующего соединения на стадии 4 в примере 1.
Стадия 3: Синтез метил-(R)-4-(4-амино-8-((трет-бутоксикарбонил)амино)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)бензоата
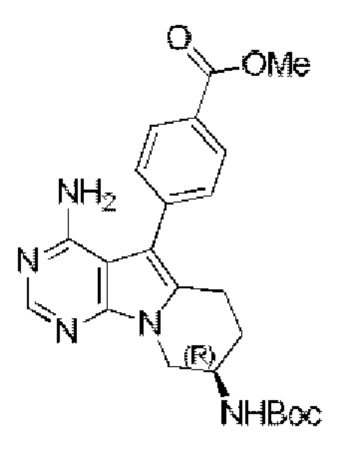
Метил-(R)-4-(4-амино-8-((трет-бутоксикарбонил)амино)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)бензоат синтезировали согласно стадии 5 в примере 1 с тем исключением, что соединение, полученное на стадии 2 в данном примере 13, использовали вместо соответствующего соединения на стадии 5 в примере 1.
Стадия 4: Синтез (R)-4-(4-амино-8-((трет-бутоксикарбонил)амино)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)бензойной кислоты
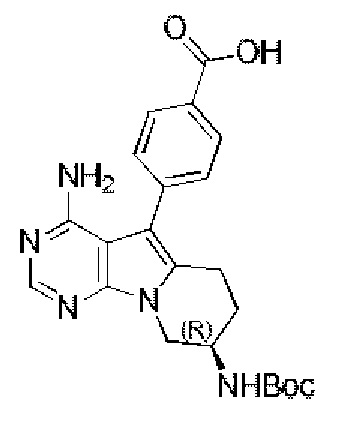
В смесь 10 мл метанола/10 мл воды добавляли метил-(R)-4-(4-амино-8-((трет-бутоксикарбонил)амино)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)бензоат (437 мг, 1 ммоль), добавляли гидроксид лития (72 мг, 3 ммоль), перемешивали реакционную смесь при комнатной температуре в течение 12 часов. После завершения реакции добавляли разбавленную хлороводородную кислоту и доводили рН до нейтрального, получали 330 мг неочищенного продукта путем фильтрования, выход 77,9%.
Стадия 5: Синтез трет-бутил-(R)-(4-амино-5-(4-(пиридин-2-илкарбамоил)-фенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-8-ил)карбамата
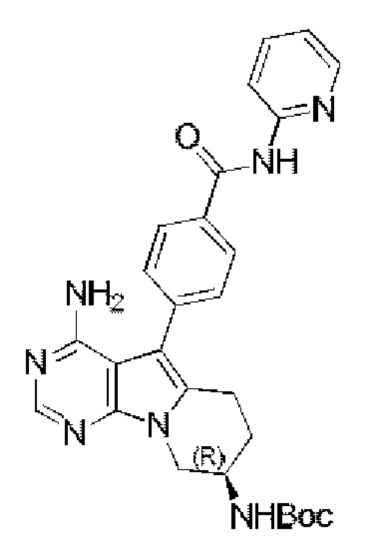
(R)-4-(4-амино-8-((трет-бутоксикарбонил)амино)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)бензойную кислоту (423 мг, 1 ммоль) добавляли в 20 мл DCM, используемого в качестве растворителя, затем последовательно добавляли по каплям Et3N (280 мкл, 2 ммоль) и 2-аминопиридин (104 мг, 1,1 ммоль), добавляли гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилмочевины (HATU) (418 мг, 1,1 ммоль), проводили реакцию при комнатной температуре в течение 8 часов. После завершения реакции экстрагировали полученную смесь ЕА и сушили над безводным Na2SO4. Неочищенный продукт очищали путем колоночной хроматографии с использованием смеси CHCl3/МеОН=30/1 с получением 264 мг целевого продукта, выход 52,8%.
Стадия 6: Синтез (R)-4-(8-акриламидо-4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)-N-(пиридин-2-ил)бензамида (S13)
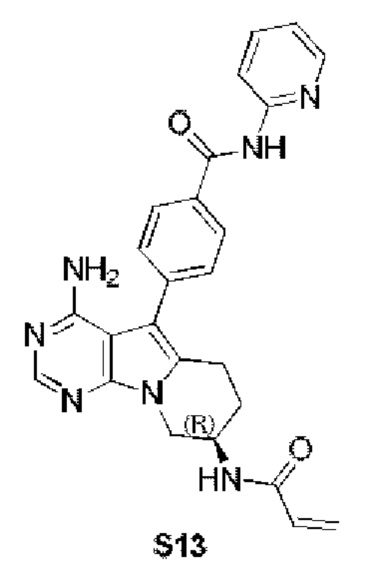
S13 синтезировали согласно стадии 6 в примере 1 с тем исключением, что соединение, полученное на стадии 5 в данном примере 13, использовали вместо соответствующего соединения на стадии 6 в примере 1.
1Н ЯМР (300 МГц, CDCl3) δ 9,15 (s, 1H), 8,38 (d, J=8,4 Гц, 1H), 8,28 (d, шир., 1H), 8,20 (s, 1H), 7,77 (t, шир., 1H), 7,44-7,30 (m, 4H), 7,08 (ddd, J=7,3, 4,9,0,9 Гц, 1H), 6,78 (s, 1H), 6,38 (d, J=16,4 Гц, 1H), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,69 (d, J=9,8 Гц, 1H), 5,20 (s, 2H), 4,72 (шир., 1H), 4,36 (dd,J=12,7, 4,6 Гц, 1H), 4,16 (dd, J=12,7, 5,7 Гц, 1H), 2,97 (t, J=6,2 Гц, 2H), 2,16-2,04 (m, 2H).
Пример 14: S14:
(R)-4-(4-амино-8-(бут-2-иниламид)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)-N-(пиридин-2-ил)бензамид
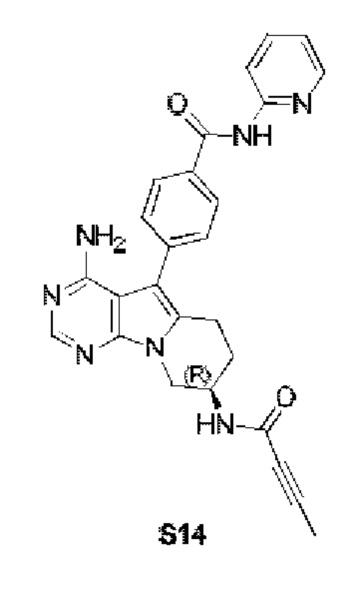
S14 синтезировали согласно стадии 6 в примере 1 с тем исключением, что соединение, полученное на стадии 5 в примере 13, использовали вместо соответствующего соединения на стадии 6 в примере 1 и бут-2-иновую кислоту использовали вместо акриловой кислоты на стадии 6 в примере 1.
1Н ЯМР (300 МГц, CDCl3) δ 9,20 (s, 1Н), 8,38 (d, J=8,4 Гц, 1H), 8,27 (d, шир., 1H), 8,19 (s, 1H), 7,75 (t, шир., 1H), 7,44-7,30 (m, 4Н), 7,08 (ddd, J=7,3, 4,9,0,9 Гц, 1H), 6,70 (s, 1H), 5,03 (s, 2Н), 4,57 (dd, J=12,4, 5,3 Гц, 1H), 4,30 (dd, J=13,0, 4,7 Гц, 1H), 4,03 (dd, J=12,9, 5,8 Гц, 1Н), 2,91 (t, J=6,2 Гц, 2H), 2,02 (dd, J=10,6, 5,0 Гц, 2H), 1,86 (s, 3Н).
Пример 15: S15:
(R)-4-(8-акриламидо-4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)-N-(5-фторпиридин-2-ил)бензамид
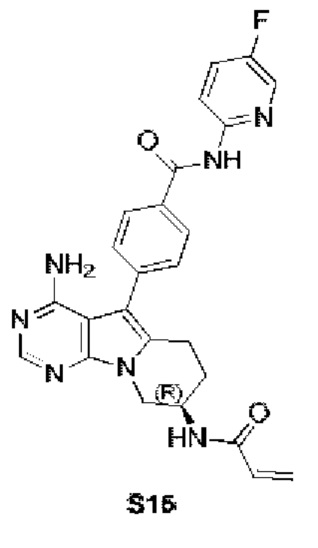
S15 синтезировали согласно стадиям синтеза S13 в примере 13 с тем исключением, что 2-амино-5-фторпиридин использовали вместо 2-аминопиридина на стадии 5 в примере 13.
1H ЯМР (300 МГц, CDCl3) δ 8,90 (s, 1H), 8,28 (d, J=8,0 Гц, 1H), 8,17 (s, 1H), 8,04 (s, 1H), 7,85 (d, J=8,0 Гц, 2H), 7,65 (d, J=8,0 Гц, 2H), 7,59 (d, J=8,0 Гц, 1H), 6,78 (s, 1H), 6,38 (d, J=16,4 Гц, 1H), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,69 (d, J=9,8 Гц, 1H), 5,20 (s, 2H), 4,72 (шир., 1H), 4,36 (dd, J=12,7, 4,6 Гц, 1H), 4,16 (dd, J=12,7, 5,7 Гц, 1H), 2,97 (t, J=6,2 Гц, 2H), 2,16-2,04 (m, 2H).
Пример 16: S16:
(R)-4-(8-акриламидо-4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
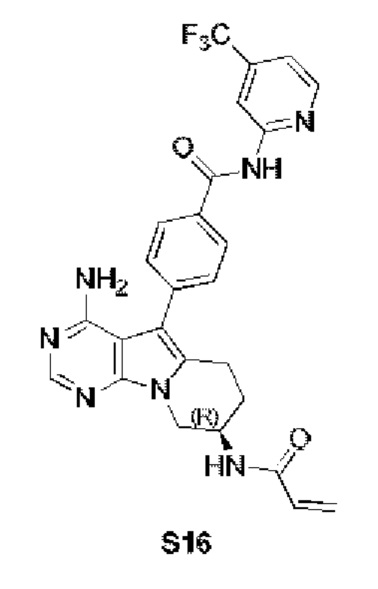
S16 синтезировали согласно стадиям синтеза S13 в примере 13 с тем исключением, что 2-амино-4-трифторметилпиридин использовали вместо 2-аминопиридина на стадии 5 в примере 13.
1Н ЯМР (300 МГц, CDCl3) δ 8,80 (s, 1H), 8,25 (s, 1H), 8,21 (s, 1H), 8,07 (d,J=5,1 Гц, 1H), 7,86 (dd, J=6,8, 1,8 Гц, 2H), 7,45 (dd, J=6,8, 1,8 Гц, 2H), 6,89 (d, J=5 Гц, 1H), 6,78 (s, 1H),6,38(d, J=16,4 Гц, 1H), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,69 (d, J=9,8 Гц, 1H), 5,20 (s, 2H), 4,72 (шир., 1H), 4,36 (dd, J=12,7, 4,6 Гц, 1H), 4,16 (dd, J=12,7, 5,7 Гц, 1H), 2,97 (t, J=6,2 Гц, 2H), 2,16-2,04 (m, 2H).
Пример 17: S17:
4-((R)-8-акриламидо-4-амино-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-5-ил)-N-((R)-2-метокси-1-фенилэтил)бензамид
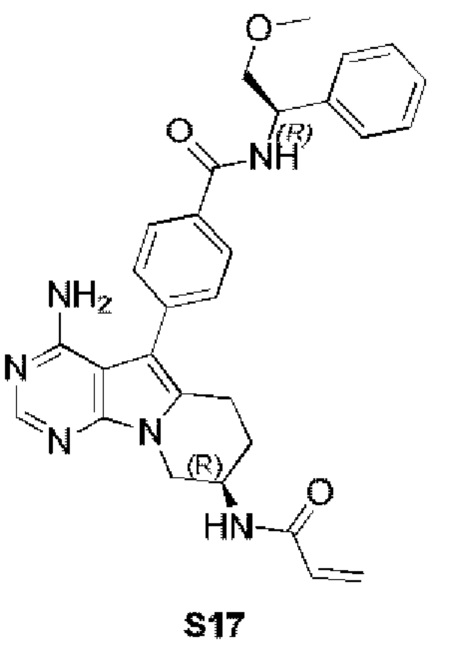
S17 синтезировали согласно стадиям синтеза S13 в примере 13 с тем исключением, что (R)-2-метокси-1-фенилэтиламин использовали вместо 2-аминопиридина на стадии 5 в примере 13.
1H ЯМР (300 МГц, CDCl3) δ 8,40 (s, 1H), 7,99 (d, J=7,9 Гц, 2Н), 7,78 (d, J=7,8 Гц, 2Н), 7,49-7,28 (m, 5Н), 6,99 (d, J=7,0 Гц, 1H), 6,78 (d, J=6,7 Гц, 1H), 6,38 (d, J=16,4 Гц, 1H), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,69 (d, J=9,8 Гц, 1H), 5,42-5,35 (m, 1H), 5,04 (s, 2H), 4,72 (шир., 1H), 4,36 (dd, J=12,7, 4,6 Гц, 1H), 4,16 (dd, J=12,7, 5,7 Гц, 1H), 4,09 (s, 1H), 3,84 (s, 1H), 3,42 (s, 3H), 2,97 (t, J=6,2 Гц, 2H), 2,16-2,04 (m, 2H).
Пример 18: S18:
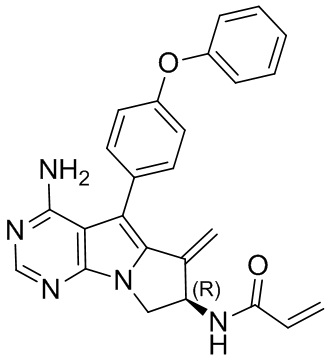
(R)-N-(4-амино-6-метилиден-5-(4-феноксифенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-7-ил)акриламид (схема 1b)
Стадия 1: Синтез трет-бутил-(R)-(4-амино-6-метилиден-5-(4-феноксифенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-7-ил)карбамата
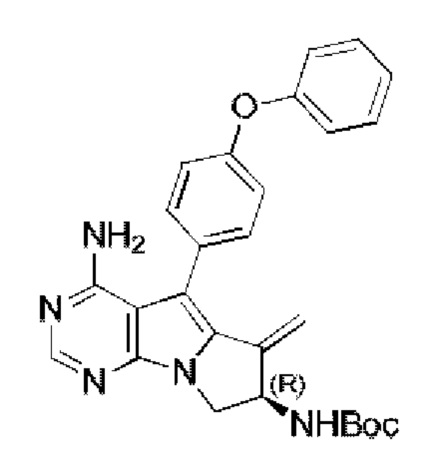
В запаянную ампулу добавляли трет-бутил-(R)-(1-(4-амино-5-(4-феноксифенил)-6-бром-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамат (550 мг, 1 ммоль) (соединение, которое синтезировали на стадии 4 в примере 1) в атмосфере N2. Добавляли 10 мл безводного THF, затем последовательно добавляли водный раствор NaOH (4,7 мл, 3 М) и PdCl2(dppf) (190 мг, 0,25 ммоль), проводили реакцию при 80°С в течение 15 часов. После завершения реакции экстрагировали полученную смесь этилацетатом и сушили над безводным Na2SO4. Неочищенный продукт очищали путем колоночной хроматографии с использованием ЕА с получением 245 мг целевого продукта, выход 52,0%.
Стадия 2: Синтез (R)-N-(4-амино-6-метилиден-5-(4-феноксифенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-7-ил)акриламида (S18)
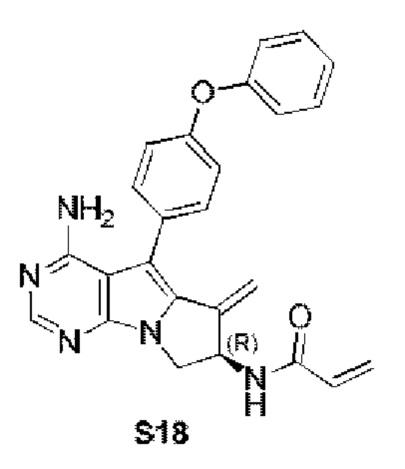
S18 синтезировали согласно стадии 6 в примере 1 с тем исключением, что соединение, полученное на стадии 1 в примере 18, использовали вместо соответствующего соединения на стадии 6 в примере 1.
1Н ЯМР (300 МГц, CDCl3) δ 8,25 (s, 1H), 7,50-7,36 (m, 4H), 7,18 (t, J=7,4 Гц, 1H), 7,10 (d, J=8,4 Гц, 4H), 6,40 (d, J=16,9 Гц, 1H), 6,28-6,08 (m, 2H), 5,75 (d, J=10,4 Гц, 1H), 5,73-5,66 (m, 1H), 5,53 (s, 1H), 5,19 (s, 1H), 5,09 (s, 2H), 4,75-4,62 (m, 1H), 3,99 (dd, J=11,5, 4,7 Гц, 1H).
Пример 19: S19:
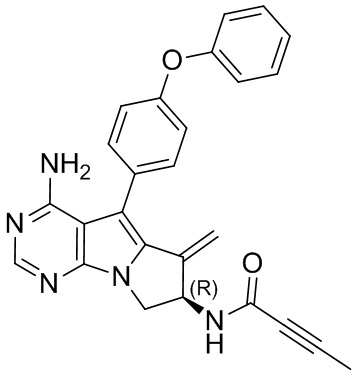
(R)-N-(4-амино-6-метилиден-5-(4-феноксифенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-7-ил)бут-2-иниламид
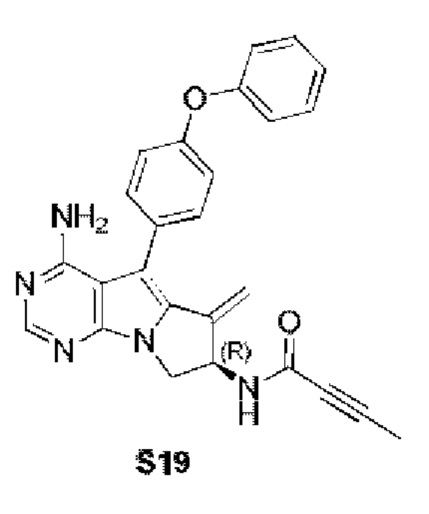
S19 синтезировали согласно стадии синтеза S18 в примере 18 с тем исключением, что соединение, полученное на стадии 1 в примере 18, использовали вместо соответствующего соединения на стадии 6 в примере 1 и бут-2-иновую кислоту использовали вместо акриловой кислоты на стадии 6 в примере 1.
1H ЯМР (300 МГц, CDCl3) δ 8,21 (s, 1H), 7,45-7,29 (m, 4H), 7,12 (t, J=7,2 Гц, 1H), 7,05 (d, J=8,1 Гц, 4H), 6,19-6,07 (m, 1H), 5,63-5,52 (m, 1H), 5,47 (s, 1H), 5,14 (s, 1H), 4,97 (s, 2H), 4,62 (dd, J=11,4, 7,8 Гц, 1H), 3,92 (dd, J=11,5, 4,7 Гц, 1H), 1,91 (s, 3H).
Пример 20: S20:
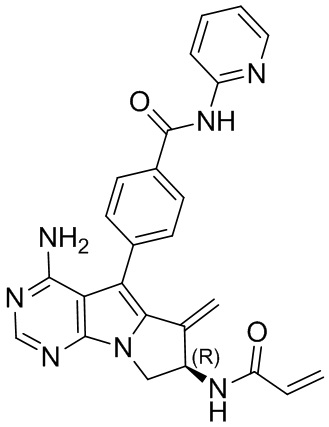
(R)-4-(7-акриламидо-4-амино-6-метилиден-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-5-ил)-N-(пиридин-2-ил)бензамид (схема 3b)
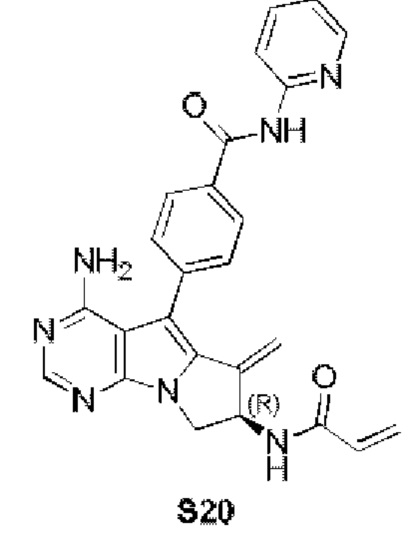
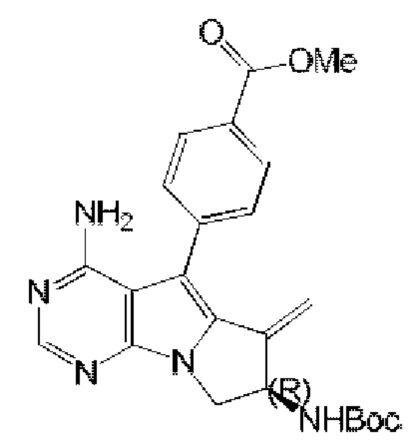
синтезировали согласно стадии 1 в примере 18 с тем исключением, что метил-(R)-4-(4-амино-6-бром-7-(2-((трет-бутоксикарбонил)-амино)бут-3-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)бензоат (соединение, полученное на стадии 2 в примере 13) использовали вместо трет-бутил-(R)-(1-(4-амино-5-(4-феноксифенил)-6-бром-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (550 мг, 1 ммоль) (соединение, которое синтезировали на стадии 4 в примере 1). S20 синтезировали согласно стадиям 3-6 в примере 13 с тем исключением, что
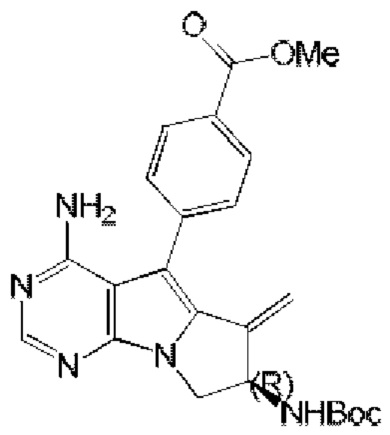
использовали вместо соединения, полученного на стадии 2 в примере 13.
1Н ЯМР (300 МГц, CDCl3) δ 9,15 (s, 1Н), 8,38 (d, J=8,4 Гц, 1H), 8,28 (d, шир., 1H), 8,20 (s, 1H), 7,77 (t, шир., 1H), 7,44-7,30 (m, 4Н), 7,08 (ddd, J=7,3, 4,9,0,9 Гц, 1H), 6,78 (s, 1Н), 6,40 (d, J=16,9 Гц, 1H), 6,28-6,08 (m, 2H), 5,73-5,66 (m, 1H), 5,53 (s, 1H), 5,19 (s, 1H), 5,09 (s, 2H), 4,75-4,62 (m, 1H), 3,99 (dd,J=11,5, 4,7 Гц, 1H).
Пример 21: S21:
N-((7R)-4-амино-6-метил-5-(4-феноксифенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-7-ил)акриламид (схема 1с)
Стадия 1: Синтез трет-бутил-((7R)-(4-амино-6-метил-5-(4-феноксифенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-7-ил)карбамата
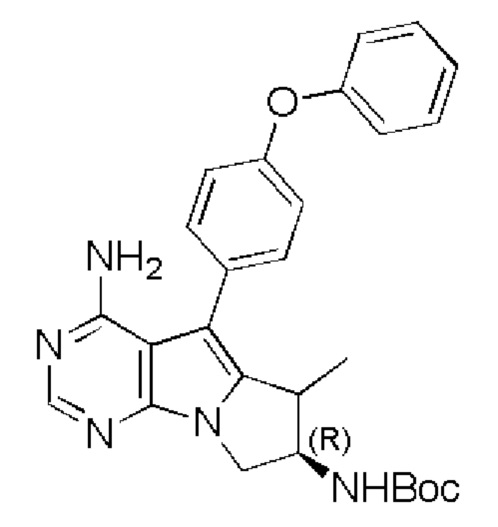
В 20 мл метанола добавляли трет-бутил-(R)-(4-амино-6-метилиден-5-(4-феноксифенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-7-ил)карбамат (соединение, полученное на стадии 1 в примере 18) (470 мг, 1 ммоль), добавляли Pd/C (213 мг, 0,2 ммоль, 10%), перемешивали реакционную смесь, нагнетая при этом H2, в течение 6 часов. После завершения реакции удаляли Pd/C путем фильтрования. Отделяли реакционный раствор и очищали путем колоночной хроматографии с использованием смеси CHCl3/МеОН=20/1 с получением 102 мг целевого продукта, выход 21,6%.
Стадия 2: Синтез N-((7R)-4-амино-6-метил-5-(4-феноксифенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-7-ил)акриламида (S21)
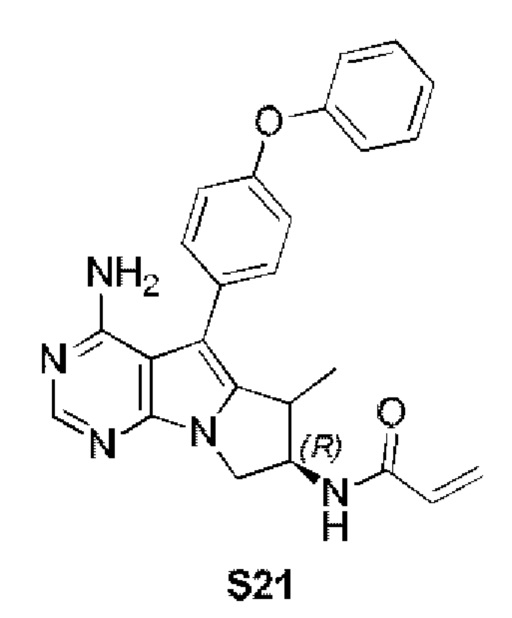
S21 синтезировали согласно стадии 6 в примере 1 с тем исключением, что соединение, полученное на стадии 1 в данном примере 21, использовали вместо соответствующего соединения на стадии 6 в примере 1.
1H ЯМР (300 МГц, CDCl3) δ 8,17 (s, 1Н), 7,44-7,35 (m, 4H), 7,16 (t,J=7,4 Гц, 1H), 7,08 (dd, J=7,7, 3,7 Гц, 4H), 6,78 (s, 1H), 6,38 (d, J=16,4 Гц, 1H), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,85 (s, 2Н), 5,69 (d, J=9,8 Гц, 1H), 4,41 (dd, J=11,0, 6,7 Гц, 1H), 4,20 (dd, J=12,3, 5,8 Гц, 1H), 3,85 (dd, J=10,7, 6,2 Гц, 1H), 3,50-3,33 (m, 1H), 1,15 (d, J=7,2 Гц, 3Н).
Пример 22: S22:
4-((7R)-7-акриламидо-4-амино-6-метил-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-5-ил)-N-(пиридин-2-ил)бензамид (схема 4)
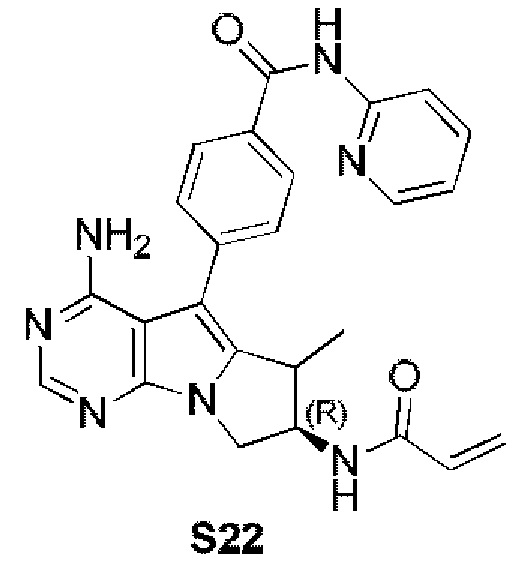
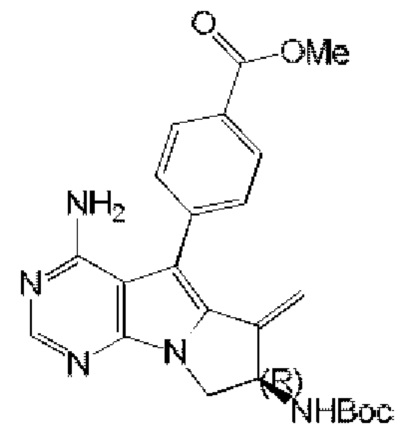
синтезировали согласно стадии 1 в примере 18 с тем исключением, что метил-(R)-4-(4-амино-6-бром-7-(2-((трет-бутоксикарбонил)-амино)бут-3-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)бензоат (соединение, полученное на стадии 2 в примере 13) использовали вместо трет-бутил-(R)-(1-(4-амино-5-(4-феноксифенил)-6-бром-7H-пирроло[2,3-d]пиримидин-7-ил)бут-3-ен-2-ил)карбамата (550 мг, 1 ммоль) (соединение, которое синтезировали на стадии 4 в примере 1). S22 синтезировали согласно стадиям 1-2 в примере 21 с тем исключением, что
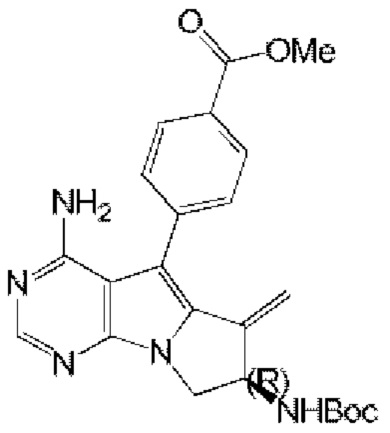
использовали вместо трет-бутил-(R)-(4-амино-6-метилиден-5-(4-феноксифенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-7-ил)карбамата (соединение, полученное на стадии 1 в примере 18).
1H ЯМР (300 МГц, CDCl3) δ 9,15 (s, 1Н), 8,38 (d, J=8,4 Гц, 1H), 8,28 (d, шир., 1H), 8,20 (s, 1H), 7,77 (t, шир., 1H), 7,44-7,30 (m, 4Н), 7,08 (ddd, J=7,3, 4,9,0,9 Гц, 1H), 6,78 (s, 1H), 6,38 (d, J=16,4 Гц, 1H), 6,24 (dd, J=16,9, 10,0 Гц, 1H), 5,85 (s, 2H), 5,69 (d, J=9,8 Гц, 1H), 4,41 (dd, J=11,0, 6,7 Гц, 1H), 4,20 (dd, J=12,3, 5,8 Гц, 1H), 3,85 (dd, J=10,7, 6,2 Гц, 1H), 3,50-3,33 (m, 1H), 1,15 (d, J=7,2 Гц, 3H).
Экспериментальный пример 1: Оценка ингибирующей активности в отношении киназы Брутона (BTK) на молекулярном уровне
1. Экспериментальный способ
В качестве субстрата для катализируемой ферментом реакции поли-(Glu, Tyr) 4:1 разбавляли в не содержащем калий PBS (10 мМ буфер фосфата натрия, 150 мМ NaCl, рН 7,2-7,4) до концентрации 20 мкг/мл на нанесения покрытия в планшете для ELISA. Планшет выдерживали при 37°С в течение 12-16 часов, трижды промывали 200 мкл/лунка T-PBS (PBS, содержащий 0,1% Tween-20) и сушили в печи при 37°С в течение 1-2 часов. Сначала в описанный выше планшет для ELISA с покрытием субстрата добавляли раствор АТФ, разбавленный в реакционном буфере (50 мМ HEPES, рН 7,4, 50 мМ MgCl2, 5 мМ MnCl2, 0,2 мМ Na3VO4, 1 мМ DTT) в концентрации 50 мкл/лунка (концентрация 10 мкМ). Затем добавляли исследуемое соединение, разбавленное в 1% ДМСО до подходящей концентрации (10 мкл/лунка), отдельно готовили лунки с отрицательным контролем и положительным контролем. Наконец, запускали реакцию путем добавления белка тирозинкиназы BTK, разбавленного в 40 мкл реакционного буфера.
Полученную выше реакционную систему помещали на шейкер (100 об./мин) при 37°С на 1 час, затем трижды промывали T-PBS, добавляли первичное антитело PY99 (Cell Signaling Technology) в количестве 100 мкл/лунка и проводили реакцию на шейкере при 37°С в течение 0,5 часа. После промывки планшета Т-РВS добавляли вторичное антитело, меченный пероксидазой хрена козий антимышиный IgG, в количестве 100 мкл/лунка и проводили реакцию на шейкере при 37°С в течение 0,5 часа. После промывки планшета Т-РВS добавляли 2 мг/мл проявляющего раствора OPD в количестве 100 мкл/лунка и проводили реакцию без доступа света при 25°С в течение 1-10 минут. Затем гасили реакцию путем добавления 2 М H2SO4 в количестве 50 мкл/лунка. Данные получали на анализаторе микропланшетов с настраиваемой длиной волны ELISA SPECTRA MAX 190 при длине волны 490 нм.
Уровень ингибирования для соединения вычисляли при помощи следующего уравнения: уровень ингибирования (%)=(ОПконтрольная лунка - ОПисследуемая лунка)/ОПконтрольная лунка × 100%. IC50 вычисляли путем подстановки в кривую ингибирования согласно четырехпараметрической методике.
2. Результаты эксперимента
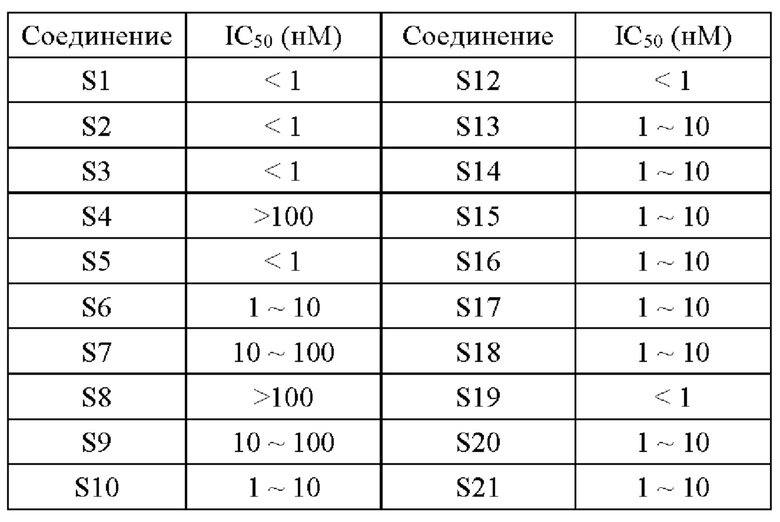

Экспериментальный пример 2: Исследование активности ингибирования пролиферации in vitro клеток Рамоса В-клеточной лимфомы человека и клеток диффузной крупноклеточной В-клеточной лимфомы человека TMD8 предложенными соединениями
1. Экспериментальный способ:
Клетки высеивали в количестве 10000-15000/лунка в 96-луночный планшет для клеточных культур, в каждую лунку добавляли исследуемое соединение в различных концентрациях (3 повтора для каждой концентрации), также готовили пустой контроль, положительный контроль и отрицательный контроль. Через 48 часов после введения лекарственных средств добавляли 20 мл МТТ (5 мг/мл) и инкубировали планшет при 37°С в течение 4 часов, а затем добавляли 100 мл трехкомпонентного раствора (10% SDS, 5% изобутанол, 0,01 М HCl). Полученную смесь выдерживали при 37°С в течение ночи и измеряли значение ОП при длине волны 570 нм на анализаторе микропланшетов с настраиваемой длиной волны ELISA SPECTRA MAX 190. Уровень ингибирования для соединения вычисляли при помощи следующего уравнения: уровень ингибирования (%)=(ОПконтрольная лунка - ОПисследуемая лунка)/ОПконтрольная лунка × 100%. Значение IC50 вычисляли путем подстановки в кривую ингибирования согласно четырехпараметрической методике. Эксперимент независимо повторяли 3 раза.
2. Результаты эксперимента
(1) Активность ингибирования пролиферации клеток Рамоса В-клеточной лимфомы человека предложенными соединениями
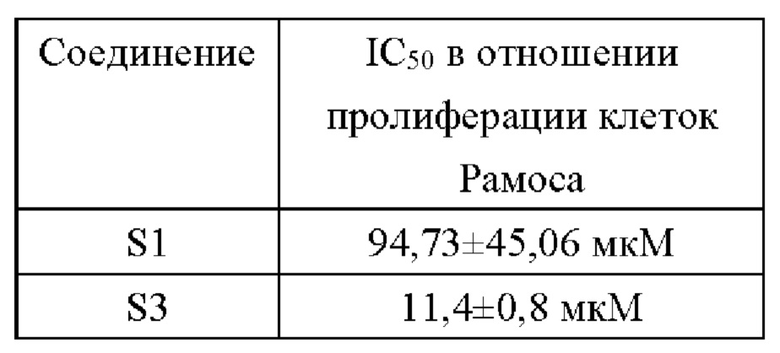
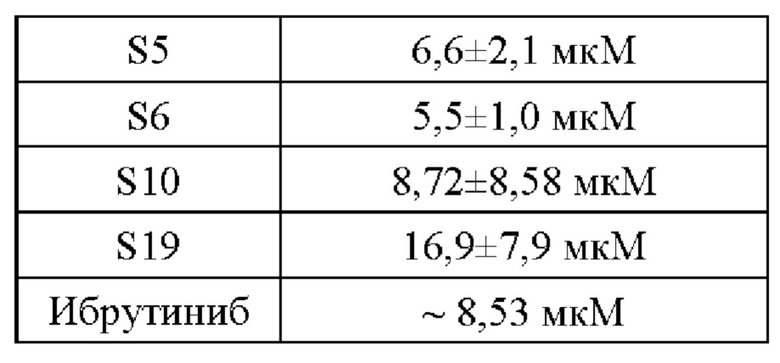
(2) Активность ингибирования пролиферации клеток диффузной крупноклеточной В-клеточной лимфомы человека TMD8 предложенными соединениями
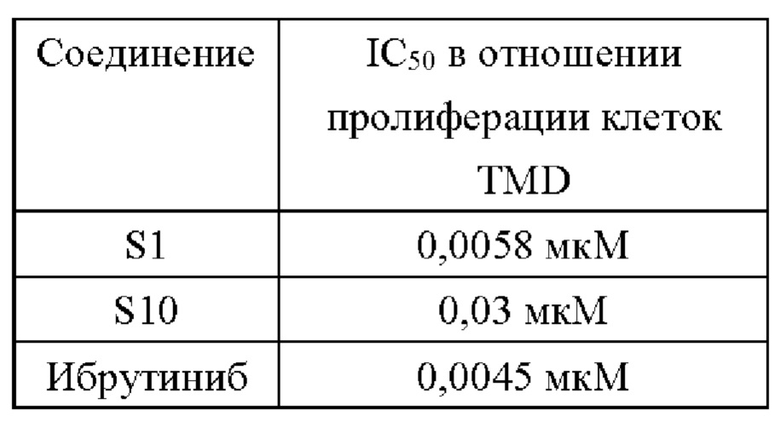
В настоящем изобретении предложена группа 5-арил пиримидопирролизиновых или 5-арил-пиримидоиндолизиновых трициклических соединений. Данная группа производных включает ингибиторы киназы Брутона, имеющие новую структуру, и некоторые соединения имею значительную ингибирующую активность в отношении ферментов BTK значительную ингибирующую активность в отношении BTK-зависимых клеток TMD8 на молекулярном уровне. И, что более важно, некоторые соединения (например, S1) имеют низкую клеточную активность в отношении клеток Рамос В-клеточной лимфомы человека и хорошую селективность к BTK и, таким образом, они представляют собой многообещающий класс селективных ингибиторов BTK.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИНОЛИЛПИРРОЛПИРИМИДИЛЬНОЕ КОНДЕНСИРОВАННОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2581039C1 |
| СПОСОБ СИНТЕЗИРОВАНИЯ НОВОГО ХИРАЛЬНОГО ЛИГАНДА, ХЕЛАТА МЕТАЛЛА, РАЗЛИЧНЫХ НЕПРИРОДНЫХ АМИНОКИСЛОТ, МАРАВИРОКА И ЕГО ОСНОВНЫХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2017 |
|
RU2727723C1 |
| Соединения на основе триазолопиримидина и их соли, композиции на их основе и пути их применения | 2020 |
|
RU2809821C2 |
| ПИРИДИНЫ, ПИРИМИДИНЫ И ПИРАЗИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ БРУТОНА И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2712220C2 |
| ФТОРИРОВАННОЕ СОЕДИНЕНИЕ ЦИКЛОПРОПИЛАМИНА, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ИСПОЛЬЗОВАНИЕ | 2017 |
|
RU2746323C2 |
| 5-АРОМАТИЧЕСКОЕ АЛКИНИЛЗАМЕЩЕННОЕ БЕНЗАМИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2695371C2 |
| СОЕДИНЕНИЯ ПУРИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2655388C2 |
| СОЕДИНЕНИЕ ИЗОИНДОЛИН, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2813232C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ BTK ИНГИБИТОРОВ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2743040C2 |
| НОВОЕ ХИНОЛИН-ЗАМЕЩЕННОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2689158C2 |
Изобретение относится к 5-арил-пиримидоиндолизиновому или 5-арил-пиримидопирролизиновому соединению формулы I, в которой R1 представляет собой H, метил или метилиден; L представляет собой -O-, -C(=O)NH- или -C(=O)NHCHR2-, причем R2 представляет собой замещенный C1-C3 алкил и заместитель в R2 представляет собой C1-C3 алкокси; m равен 0 или 1; Ar представляет собой замещенный или незамещенный C6 арил, замещенный или незамещенный 6-членный гетероарил, содержащий один гетероатом N, где заместитель в Ar представляет собой галоген или трифторметил, A представляет собой карбонил, сульфонил или 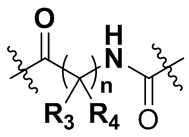 , где R3 и R4 независимо представляют собой водород или C1-C3 алкил, или R3, R4 совместно с присоединенным к ним атомом углерода образуют C3-C5 циклоалкил; n равен 1; T представляет собой
, где R3 и R4 независимо представляют собой водород или C1-C3 алкил, или R3, R4 совместно с присоединенным к ним атомом углерода образуют C3-C5 циклоалкил; n равен 1; T представляет собой 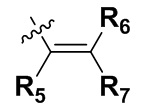 или
или 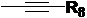 ; R5, R6 и R7 независимо выбраны из группы, состоящей из водорода, циано и замещенного или незамещенного C1-C5 алкила; заместитель в R5, R6 или R7 представляет собой диметиламино; R8 представляет собой метил. Изобретение также относится к способу получения, к фармацевтической композиции и к применению 5-арил-пиримидоиндолизинового или 5-арил-пиримидопирролизинового соединения для получения лекарственного средства для лечения лимфомы человека, связанной с путём сигнальной трансдукции киназы ВТК. Технический результат: получены новые 5-арил-пиримидоиндолизиновое и 5-арил-пиримидопирролизиновое соединения формулы I, обладающие ингибирующей активностью в отношении BTK. 4 н. и 4 з.п. ф-лы, 24 пр.
; R5, R6 и R7 независимо выбраны из группы, состоящей из водорода, циано и замещенного или незамещенного C1-C5 алкила; заместитель в R5, R6 или R7 представляет собой диметиламино; R8 представляет собой метил. Изобретение также относится к способу получения, к фармацевтической композиции и к применению 5-арил-пиримидоиндолизинового или 5-арил-пиримидопирролизинового соединения для получения лекарственного средства для лечения лимфомы человека, связанной с путём сигнальной трансдукции киназы ВТК. Технический результат: получены новые 5-арил-пиримидоиндолизиновое и 5-арил-пиримидопирролизиновое соединения формулы I, обладающие ингибирующей активностью в отношении BTK. 4 н. и 4 з.п. ф-лы, 24 пр.
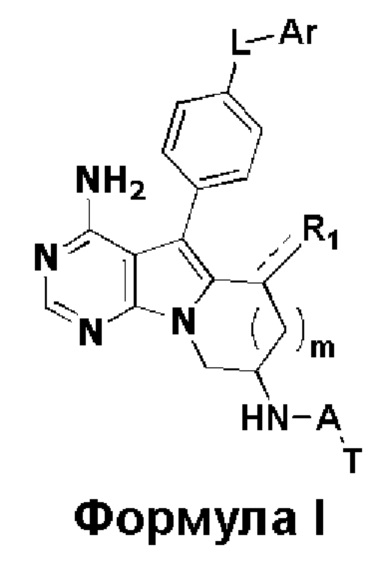
1. 5-арил-пиримидоиндолизиновое или 5-арил-пиримидопирролизиновое соединение формулы I;
Формула I
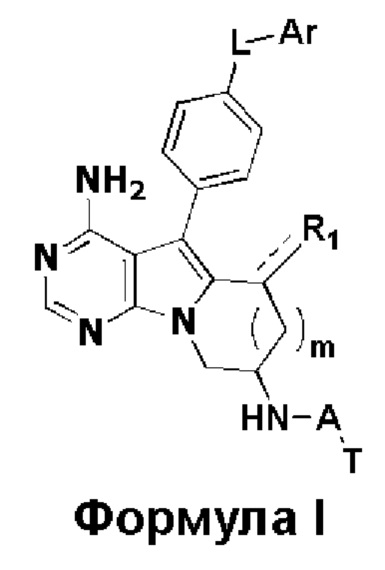
где:
R1 представляет собой H, метил или метилиден;
L представляет собой -O-, -C(=O)NH- или -C(=O)NHCHR2-, причем R2 представляет собой замещенный C1-C3 алкил и заместитель в R2 представляет собой C1-C3 алкокси;
m равен 0 или 1;
Ar представляет собой замещенный или незамещенный C6 арил, замещенный или незамещенный 6-членный гетероарил, содержащий один гетероатом N, где заместитель в Ar представляет собой галоген или трифторметил,
A представляет собой карбонил, сульфонил или 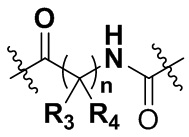 , где R3 и R4 независимо представляют собой водород или C1-C3 алкил, или R3, R4 совместно с присоединенным к ним атомом углерода образуют C3-C5 циклоалкил;
, где R3 и R4 независимо представляют собой водород или C1-C3 алкил, или R3, R4 совместно с присоединенным к ним атомом углерода образуют C3-C5 циклоалкил;
n равен 1;
T представляет собой 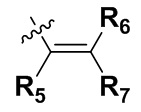 или
или 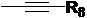 ;
;
R5, R6 и R7 независимо выбраны из группы, состоящей из водорода, циано и замещенного или незамещенного C1-C5 алкила; заместитель в R5, R6 или R7 представляет собой диметиламино;
R8 представляет собой метил.
2. 5-арил-пиримидоиндолизиновое или 5-арил-пиримидопирролизиновое соединение по п.1,
в котором Ar представляет собой замещенный или незамещенный C6 арил, замещенный или незамещенный 6-членный гетероарил, содержащий один гетероатом N, где заместитель в Ar представляет собой галоген или трифторметил;
n равен 1;
T представляет собой или ;
R5, R6 и R7 независимо выбраны из группы, состоящей из водорода, циано, и замещенного или незамещенного C1-С5 алкила; заместитель в R5, R6 или R7 представляет собой диметиламино;
R8 представляет собой метил.
3. 5-арил-пиримидоиндолизиновое или 5-арил-пиримидопирролизиновое соединение по п.1,
в котором Ar представляет собой замещенный или незамещенный фенил, замещенный или незамещенный 6-членный гетероарил, где заместитель в Ar представляет собой галоген или трифторметил;
n равен 1;
T представляет собой или ;
R5, R6 и R7 независимо выбраны из группы, состоящей из водорода, циано, и замещенного или незамещенного C1-C5 алкила; заместитель в R5, R6 или R7 представляет собой диметиламино;
R8 представляет собой метил.
4. 5-арил-пиримидоиндолизиновое или 5-арил-пиримидопирролизиновое соединение по п.1,
в котором L представляет собой -O-, -C(=O)NH-,
в котором Ar представляет собой фенил, 4-фторфенил, 4-трифторметилфенил, пиридин-2-ил, 3-фторпиридин-6-ил или 4-трифторметилпиридин-6-ил;
в котором A представляет собой карбонил, сульфонил или , n равен 1, R3 представляет собой водород, R4 представляет собой H или метил, или R3, R4 совместно с присоединенным к ним атомом углерода образуют циклопропил;
в котором T представляет собой или ; R5 представляет собой H, циано или метил, R6 представляет собой H, R7 представляет собой H, трет-бутил или диметиламинометил, R8 представляет собой метил.
5. 5-арил-пиримидоиндолизиновое или 5-арил-пиримидопирролизиновое соединение по п. 1, выбранное из группы, состоящей из соединений, приведенных в следующей таблице:
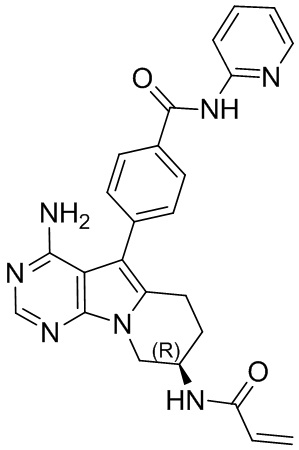
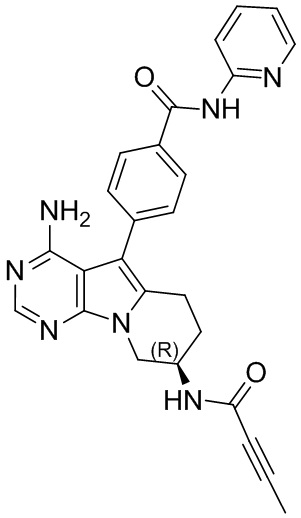
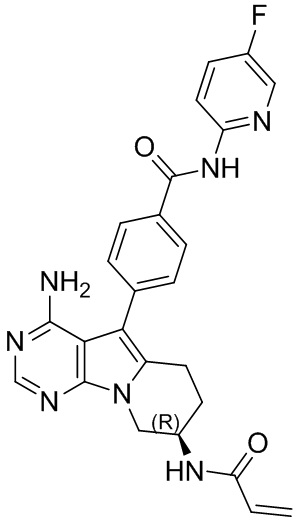
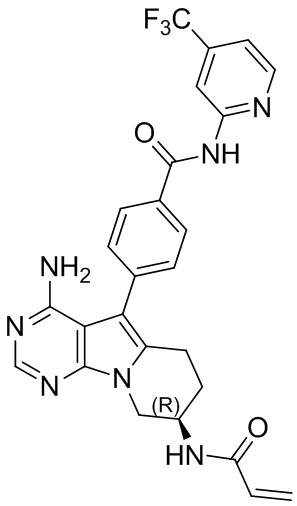
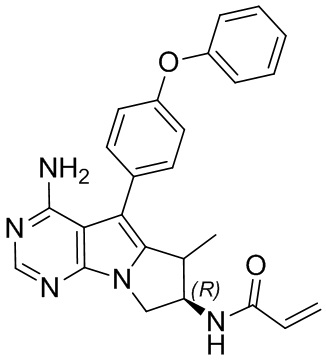
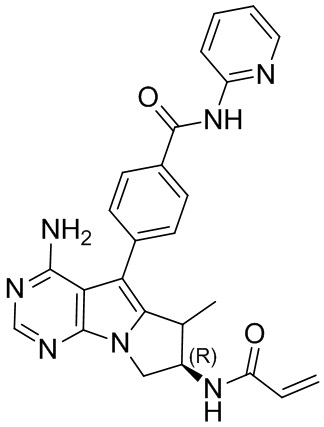 .
.
6. Способ получения 5-арил-пиримидоиндолизинового или 5-арил-пиримидопирролизинового соединения, включающий следующие стадии:
Схема 1a:
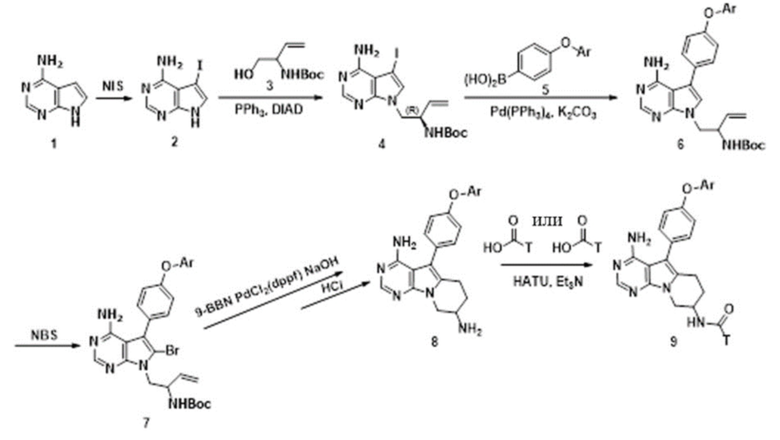
4-амино-пирроло[2,3-d]пиримидин, т.е. соединение 1, в качестве исходного вещества растворяют в хлороформе и проводят замещение йодом с использованием N-йодсукцинимида (NIS) по положению 5 при кипячении с обратным холодильником с получением соединения 2,
в соединение 2 встраивают фрагмент 3 (трет-бутил-(R)-(1-гидроксибут-3-ен-2-ил)карбамат) по реакции Мицунобу с получением соединения 4, при этом к трифенилфосфину добавляют безводный тетрагидрофуран (THF), и охлаждают до 0 °С, по каплям добавляют диизопропилазодикарбоксилат (DIAD), затем добавляют соединение 2, через 30 минут нагревают реакционную смесь до комнатной температуры и перемешивают,
проводят сочетание соединения 4 с замещенной фенилбороновой кислотой или боратом в присутствии Pd(PPh3)4 и карбоната калия в 1,4-диоксане в качестве растворителя в атмосфере азота при 90 °С с получением соединения 6,
вводят бром по положению 6 в соединение 6 с использованием N-бромсукцинимида (NBS) в N,N-диметилформамиде с получением соединения 7,
проводят реакцию соединения 7 с 9-бор-бицикло[3.3.1]нонаном (9-BBN) в безводном тетрагидрофуране (THF) в атмосфере азота с образованием шестичленного кольца в результате реакции внутреннего сочетания Сузуки-Мияура в присутствии дихлорида [1,1'-бис(дифенилфосфинил)ферроцен]палладия (PdCl2(dppf)) и гидроксида натрия при нагревании и удаляют защитные группы с получением соединения 8,
проводят конденсацию соединения 8 с карбоновой кислотой или сульфокислотой с получением соединения 9 в среде дихлорметана в присутствии гексафторфосфата 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилмочевины и триэтиламина, при этом
Ar представляет собой замещенный или незамещенный C6 арил, где заместитель в Ar представляет собой галоген или трифторметил;
T представляет собой или ;
R5, R6 и R7 каждый независимо выбран из группы, состоящей из водорода, циано, и замещенного или незамещенного C1-C5 алкила; заместитель в R5, R6 или R7 представляет собой диметиламино;
R8 представляет собой метил.
7. Фармацевтическая композиция, содержащая терапевтически эффективное количество 5-арил-пиримидоиндолизинового или 5-арил-пиримидопирролизинового соединения по любому из пп. 1-5 для ингибирования киназы BTK и один или более фармацевтически приемлемых носителей для переноса указанного 5-арил-пиримидоиндолизинового или 5-арил-пиримидопирролизинового соединения.
8. Применение 5-арил-пиримидоиндолизинового или 5-арил-пиримидопирролизинового соединения по любому из пп. 1-5 для получения лекарственного средства для лечения лимфомы человека, связанной с путем сигнальной трансдукции киназы BTK.
| Прибор для взятия пробы газа | 1929 |
|
SU18573A1 |
| КОНВЕЙЕРНАЯ УСТАНОВКА | 1994 |
|
RU2081039C1 |
| EP 2960241 A1, 30.12.2015 | |||
| EP 3037424 A1, 29.06.2016. | |||

