Настоящее изобретение относится к соединениям, которые применимы в качестве ингибиторов активности одной или нескольких изоформ серин/треонин-киназы, AKT. Настоящее изобретение также относится к фармацевтическим композициям, содержащим указанные соединения, а также к способам использования указанных соединений при лечении злокачественной опухоли и к способам лечения злокачественной опухоли.
ПРЕДПОСЫЛКИ СОЗДАНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Семейство белков AKT, известное также как протеинкиназы В (PKB), как известно, участвует в широком спектре биологических процессов, включая клеточную пролиферацию, дифференцировку, апоптоз, онкогенез, а также синтез гликогена и поглощение глюкозы. Указанные ферменты являются представителями семейства серин/треонин-специфичных протеинкиназ.
Путь PKB/AKT был определен как важный регулятор передачи сигнала в клетках, опосредующего клеточное выживание и апоптоз. Считается, что передача сигнала посредством целого ряда рецепторов факторов роста, включая тромбоцитарный фактор роста, инсулиновый фактор роста и фактор роста нервов, приводит к активации фосфатидилинозитол-3-ОН-киназы (PI3K). Эта активация, в свою очередь, приводит к образованию фосфатидилинозитол-(3,4,5)-трифосфата (PIP3). Активированный PIP3 связывается с ферментом PDK-1, основным активатором AKT, через свой домен гомологии к плекстрину и, в свою очередь, фосфорилирует его. Активированный PDK-1 отвечает за фосфорилирование Thr308 в AKT, что индуцирует конформационное изменение, которое облегчает дальнейшее фосфорилирование Ser473 в AKT посредством PDK-2.
PDK-1 фосфорилирование киназ, расположенных ниже в цепи передачи сигнала, не является уникальным для AKT, поскольку сообщалось об активации ею p70 S6 киназы и протеинкиназы С.
Активация AKT влияет на множество событий в клетке, включая ингибирование апоптоза, прогрессию клеточного цикла, выживание клеток, метаболизм, ангиогенез и устойчивость к гормонам.
На настоящий момент идентифицировано три представителя семейства AKT - AKT 1, AKT 2 и AKT 3 (также известные как PKBα, PKBβ и PKBγ). Представители семейства характеризуются 80% гомологией аминокислотных последовательностей, и все сохраняют подобную структуру участков. Они содержат С-концевой домен гомологии к плекстрину (PH), каталитический домен, короткий α-спиральный линкерный участок и карбоксильный концевой домен. Домен PH обеспечивает связывание белков с клеточной мембраной посредством взаимодействия с фосфолипидами. Каталитический домен представителей семейства AKT содержит два остатка, необходимые для активации киназы, а именно Thr308 и Ser473. В свою очередь, AKT может фосфорилировать любой белок, содержащий мотив RXRXXS/T-B, где X представляет собой любую аминокислоту, а В представляет собой объемистые гидрофобные остатки.
Что касается клеточной функции AKT, то гиперактивация AKT была связана с ингибированием апоптоза клеток вследствие фосфорилирования и с негативной регуляцией семейства транскрипционных факторов forkhead, которые регулируют различные гены, ответственные за инициацию процессов гибели, включая FKHR, FKHRL1 и AFX. Напротив, сообщалось, что AKT активирует гены, которые, как известно, являются антиапоптическими, включая IKK и CREB. Именно это сочетание положительной и негативной регуляции подчеркивает важность AKT в регуляции апоптоза. AKT способствует выживанию нежелательных клеток посредством фосфорилирования ею нескольких ключевых апоптических белков, включая Bad и про-каспазу 9, что тем самым делает их неактивными и предотвращает передачу сигнала через этот путь. AKT активирует и ингибирует множество механизмов, которые играют важную роль в прогрессии клеточного цикла, что в конечном итоге приводит к пролиферации клеток. Регуляция лучше всего охарактеризованных белков-регуляторов клеточного цикла и белков-супрессоров опухолевого роста p53 может быть нарушена посредством фосфорилирования AKT и активации основного негативного регулятора р53 - MDM2. Фосфорилированный MDM2 перемещается в ядро, где он предотвращает транскрипцию p53. Ингибирование p53 способствует аберрантной пролиферации клеток и переходу их в доброкачественное состояние.
Аналогичным образом, AKT также может фосфорилировать p27kip1 и p21 - два основных ингибитора прогрессии клеточного цикла, что приводит к утрате ими функции, в результате чего происходит неконтролируемая прогрессия клеточного цикла и избыточная пролиферация.
Активация AKT вызывает увеличение скорости гликолиза за счет увеличения скорости метаболизма глюкозы. Также сообщалось, что активированная AKT стимулирует транспорт аминокислот и поддерживает mTOR-зависимое усиление трансляции белков. Сообщалось, что проангиогенные факторы, такие как фактор роста эндотелия сосудов (VEGF), активируют AKT, что в конечном итоге приводит к ингибированию апоптоза эндотелия, а также к активации эндотелиальной синтазы окиси азота (eNOS). Суммарным результатом этого является быстрая неоваскуляризация и миграция клеток.
Запускаемый гипоксией ангиогенез, опосредованный, главным образом, индуцируемым гипоксией фактором (HIF 1а), может приводить к индукции нескольких белков, включая VEGF. Усиленная активация AKT, как сообщалось, усиливает экспрессию HIF-1α, что приводит к усилению ангиогенеза, независимого от гипоксической среды. Последние данные показали, что активность HIF-1α при инвазивной злокачественной опухоли молочной железы коррелирует с усиленным фосфорилированием, активированным AKT1.
Ингибиторы рецептора эстрогенов (ER) и рецептора андрогенов (AR), разработанные для ингибирования клеточной передачи сигнала и индукции апоптоза, являются важными инструментами в способах терапии злокачественных опухолей. Частота возникновения устойчивости к указанным лекарственным средствам быстро растет при злокачественных опухолях, включая злокачественные опухоли предстательной железы, молочной железы и яичника. Сообщалось, что AKT фосфорилирует рецепторы андрогенов, что приводит к ингибированию активности AR и блокаде нормальной апоптотической передачи сигнала при злокачественной опухоли предстательной железы, индуцированной андрогенами.
Аналогичным образом, активация AKT приводит к фосфорилированию ERα, в результате чего происходит ингибирование опосредованного тамоксифеном апоптоза или регрессии опухоли в сочетании с образованием эстроген-независимого пути передачи сигнала. Активированная AKT2 была идентифицирована как промотор транскрипции ERα в присутствии или отсутствии эстрогена, увеличивая скорость пролиферации клеток злокачественной опухоли молочной железы.
Сообщалось о гиперактивированной по сравнению с нормальными тканями AKT при ряде злокачественных опухолей, включая злокачественные опухоли молочной железы, легкого, предстательной железы, желудка, яичника, поджелудочной железы, щитовидной железы, глиобластому и при гематологических злокачественных опухолях. Фосфорилирование AKT также было ассоциировано с клиническими показателями, включающими повышенную стадию и степень опухоли, и более неблагоприятный прогноз. Активация AKT может возникать в результате целого ряда различных генетических мутаций в сигнальном пути AKT/PI3K.
О соматических мутациях в гене PI3KCA неоднократно сообщалось при самых различных опухолях, включая опухоли молочной железы, предстательной железы и головы и шеи. Большое число таких мутаций увеличивает число копий этого гена, что приводит к усилению активности PI3K. В недавнем исследовании была выявлена мутация PI3K, которая селективно фосфорилирует AKT при злокачественной опухоли толстой кишки, что приводит к усилению клеточной пролиферации и инвазии.
Любая мутация, которая повышает активность сигнального пути PI3K, в конечном итоге приводит к повышенной активации AKT. Амплификации генов являются распространенными явлениями при злокачественной опухоли. Амплификации AKT2 были отмечены при карциноме яичника, поджелудочной железы, молочной железы и плоскоклеточной карциноме головы и шеи. На настоящий момент об амплификациях или мутациях в AKT3 не сообщалось, хотя сообщалось о делеционных мутациях, приводящих к гиперактивации и амплификации мутаций, ассоциированных с AKT1. Одна мутация, E17K, приводит к патологической локализации AKT1 в клеточной мембране, что индуцирует ее активацию и приводит к нисходящей передаче сигнала и к клеточной трансформации. Было показано in vivo, что указанная мутация индуцирует развитие лейкоза у мышей.
Делетированный ген фосфатазы и гомолога тензина в хромосоме 10 (PTEN) представляет собой ген-супрессор опухоли, известный негативной регуляцией функции AKT. При злокачественной опухоли утрата функции PTEN приводит к конститутивному фосфорилированию AKT и других нисходящих эффекторов пути PI3K. Утрата PTEN вследствие делеционных мутаций или метилирования промотора отмечалась при ряде различных злокачественных опухолей, включая глиобластому, злокачественные опухоли эндометрия, легких, молочной железы, предстательной железы и щитовидной железы. Такая утрата обычно ассоциирована с гиперактивацией AKT. В недавних исследованиях было показано, что утрата гетерозиготности (LOH) по гену PTEN прямо коррелирует с повышенной активацией AKT и устойчивостью к химиотерапии при карциноме желудка, а также с пониженной экспрессией рецептора прогестерона при карциноме молочной железы.
Активация AKT обычно инициируется на клеточной поверхности посредством передачи сигнала на рецепторе, как правило, одной из семейства тирозинкиназ. Двумя тирозинкиназными рецепторами, которые обычно амплифицируются или сверхэкспрессируются при злокачественной опухоли, являются HER2 и EGFR. При опухолях со сверхэкспрессией HER2 часто происходит гиперактивация AKT, что отмечалось при злокачественных опухолях яичника, желудка и мочевого пузыря. Аналогичным образом, при опухолях со сверхэкспрессией EGFR, в частности при активирующей EGFRvIII мутации, сообщалось о селективной активации AKT при целом ряде злокачественных опухолей, включая немелкоклеточный рак легких, злокачественную опухоль молочной железы, яичника и, наиболее часто, глиому высокой степени злокачественности.
Примеры ингибиторов AKT представлены в WO 2008/070134, WO 2008/070016 и WO 2008/070041. В указанных документах раскрыты конкретные нафтиридиновые соединения, конденсированные с пятичленным гетероциклом. Другие ингибиторы AKT могут быть найдены, например, в WO 2009/148887 и WO 2009/148916.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В первом аспекте настоящее изобретение относится к соединению в соответствии с формулой (I):
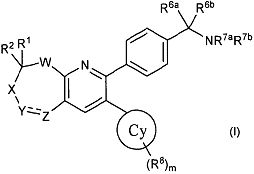 ,
,
в которой:
каждый R1 и R2 независимо выбирают из водорода, арила, C1-C10алкила, CN, CHO, CO2H, CONH2, CONHR3, CONR3aR3b, COR3, CO2R3, NH2, NHR3, NR3aR3b, NHCOR3, NHSO2R3, NR3aCOR3b, NR3aSO2R3b, OH, OR3, SH, SR3, SOR3, SO2R3, SO2NHR3, SO2NR3aR3b, F, Cl, Br и I, где каждый R3, R3a и R3b независимо выбирают из C1-C10алкила, включая случаи, когда R3a и R3b вместе с атомом азота, к которому они прикреплены, образуют гетероцикл;
где отдельные R1 и R2 вместе с атомом углерода, к которому они присоединены, могут образовывать необязательно замещенный и необязательно насыщенный гетероцикл или карбоцикл; или R1 и R2 вместе представляют собой оксо или необязательно C1-C10алкила O-замещенный оксим;
W представляет собой O, S, SO, SO2, NR' или CRaRb, где R' представляет собой либо водород, либо C1-C10алкил, и где каждый Ra и Rb независимо выбирают из представителей представленной выше группы, из которой выбирают R1 и R2;
X либо отсутствует, либо представляет собой CR4R5, где каждый R4 и R5 независимо выбирают из представителей представленной выше группы, из которой выбирают R1 и R2;
Y и Z независимо представляют собой либо замещенный или незамещенный азот или углерод, и где углерод замещен заместителями, независимо выбранными из представителей представленной выше группы, из которой выбирают R1 и R2, либо в случае азота заместитель выбирают из арила, C1-C10алкила, SO2R3, CONHR3, CONR3aR3b, COR3 и CO2R3, где значения R3, R3a и R3b определены выше, или где Y и Z вместе образуют необязательно замещенный гетероциклил или карбоциклическую группу, или где Y представляет собой SO2;
R7a и R7b независимо выбирают из H и алкила, включая случаи, когда R7a и R7b вместе с атомом азота, к которому они присоединены, образуют гетероцикл; и
R6a и R6b независимо выбирают из H, (C1-C6)алкила, (C1-C6)алкенила и (C1-C6)алкинила, где упомянутый алкил необязательно замещен не более чем тремя заместителями, выбранными из OH или галогена; или R6a и R6b могут быть объединены с образованием моноциклического или бициклического карбо- или гетероцикла с 3-7 атомами в каждом кольце, причем упомянутый гетероцикл содержит один или несколько гетероатомов, выбранных из N, O и S, и упомянутый карбо- или гетероцикл необязательно замещен одним или несколькими заместителями, выбранными из (C1-C6)алкила, (C3-C6)циклоалкила, (C1-C6)алкокси, CO2H, галогена, OH, CN и NR3aR3b, и упомянутые алкил, циклоалкил и алкокси необязательно замещены одним или несколькими заместителями, выбранными из галогена, CN, OH и NR3aR3b; и
кольцо Cy выбирают из (C3-C8)циклоалкила, алкилциклоалкила, гетероциклоалкила, гетероарила и арила, где m равен 0, 1, 2, 3, 4 или 5, и каждый R8 независимо выбирают из алкила, CN, CHO, CO2H, CONH2, CONHR9, CONHR9aR9b, COR9, CO2R9, NH2, NHR9, NR9aR9b, NHCOR9, NHSO2R9, NR9aCOR9b, NR9aSO2R9b, OH, OR9, SH, SR9, F, Cl, Br и I, где каждый R9, R9a и R9b независимо выбирают из алкила, включая случаи, когда R9a и R9b вместе с атомом азота, к которому они присоединены, образуют гетероцикл, или Cy может представлять собой йод;
и к его фармацевтически приемлемым солям, стереоизомерам и таутомерам.
Один аспект настоящего изобретения также относится к соединениям, выбранным из следующей группы структур:
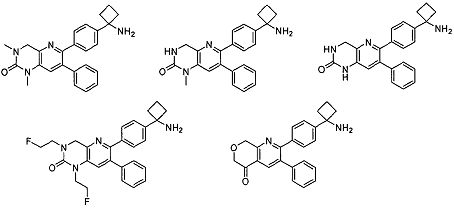 ,
,
и к их фармацевтически приемлемым солям, стереоизомерам и таутомерам.
В предпочтительных вариантах осуществления X отсутствует.
В предпочтительных вариантах осуществления R6a и R6b вместе образуют  , то есть предпочтительно они образуют циклобутан. В более предпочтительных вариантах осуществления группа, связанная с фенильным кольцом в структуре I, представляет собой 1-аминоциклобутил.
, то есть предпочтительно они образуют циклобутан. В более предпочтительных вариантах осуществления группа, связанная с фенильным кольцом в структуре I, представляет собой 1-аминоциклобутил.
В предпочтительных вариантах осуществления R6a и R6b вместе образуют  или
или 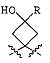 , где R представляет собой алкильную группу, предпочтительно C1-C6алкильную группу.
, где R представляет собой алкильную группу, предпочтительно C1-C6алкильную группу.
В предпочтительных вариантах осуществления кольцо Cy представляет собой незамещенный C6арил, то есть фенил.
В дополнительном варианте осуществления соединения согласно настоящему изобретению фенильное кольцо, представленное в формуле (I), замещено пиридиновым кольцом.
Связь между Y и Z необязательно представляет собой либо одинарную, либо двойную связь. В предпочтительных вариантах осуществления связь между Y и Z представляет собой одинарную связь.
В предпочтительных вариантах осуществления X отсутствует, Y представляет собой карбонил, и Z представляет собой необязательно замещенный амино, или X отсутствует, Y представляет собой необязательно замещенный амино, и Z представляет собой карбонил. В указанных вариантах осуществления W предпочтительно представляет собой CH2 или O, более предпочтительно O. В указанных же вариантах осуществления R1 и R2 предпочтительно представляют собой водород. Если амино является замещенным в указанных вариантах осуществления, то предпочтительно он является метил- или ацетамидо-замещенным.
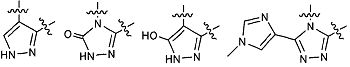
В предпочтительных вариантах осуществления заместители, присоединенные к Y и Z, и собственно Y и Z вместе образуют 5- или 6-членное необязательно замещенное карбоциклическое или гетероциклическое кольцо.
В особо предпочтительных вариантах осуществления заместители, присоединенные к Y и Z, и собственно Y и Z вместе образуют фрагмент, выбранный из группы:
 и
и  ,
,
где R представляет собой  или
или  .
.
В одном особо предпочтительном варианте осуществления соединения согласно настоящему изобретению кольцо Cy заменено атомом йода.
В предпочтительных вариантах осуществления, если Cy представляет собой алкилциклоалкил, то Cy представляет собой метилциклопропил.
В предпочтительных вариантах осуществления W представляет собой либо CH2, либо O. В одном особо предпочтительном варианте осуществления W представляет собой CH2. В другом особо предпочтительном варианте осуществления W представляет собой О.
Если соединение согласно настоящему изобретению содержит R6a и R6b группы, взаимодействующие таким образом, что вместе образуют 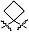 , то есть предпочтительно они образуют циклобутан, который дополнительно содержит полярный заместитель в дистальном положении относительно бензольного кольца, к которому присоединено циклобутановое кольцо, то полярный заместитель предпочтительно находится в транс-положении относительно атома азота, который имеет присоединенные к нему боковые группы R7a и R7b.
, то есть предпочтительно они образуют циклобутан, который дополнительно содержит полярный заместитель в дистальном положении относительно бензольного кольца, к которому присоединено циклобутановое кольцо, то полярный заместитель предпочтительно находится в транс-положении относительно атома азота, который имеет присоединенные к нему боковые группы R7a и R7b.
Другие предпочтительные варианты осуществления соединений согласно настоящему изобретению представлены на всем протяжении подробного описания и, в частности, в примерах. Особо предпочтительными являются такие упомянутые соединения, которые обладают большей активностью по результатам испытаний. Соединения, обладающие более высокой активностью, являются более предпочтительными по сравнению с теми, которые обладают меньшей активностью.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Термин «алкильная группа» относится к алифатической группе, содержащей, по меньшей мере, углерод и водород и содержащей от 1 до 15 атомов углерода, как, например, 1-10 атомов углерода. Присоединение к алкильной группе происходит через атом углерода.
Термин «Cnалкильная» группа относится к алифатической группе, содержащей n атомов углерода. Например, C1-C10алкильная группа содержит 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода.
Алкильная группа может быть неразветвленной, или она может быть разветвленной.
Алкильная группа может не содержать кольцевые структуры или может содержать одно или несколько колец.
Например, «циклоалкильная» группа содержит, по меньшей мере, одно кольцо. При этом понимается, что присоединение к циклоалкильной группе происходит через кольцо циклоалкильной группы. Каждое кольцо может содержать от 3 до 10 атомов, как, например, от 4 до 8 или от 5 до 7 атомов. Каждое кольцо может быть независимо выбрано как содержащее только атомы углерода или как содержащее и атомы углерода и от 1 до 4 гетероатомов, выбранных из O, N и S. Для циклогетероалкильных групп (т.е., циклоалкильных групп, которые содержат один или несколько гетероатомов) присоединение к циклоалкильной группе может происходить или через атом углерода, или присоединение также может происходить через содержащийся в кольце гетероатом, если в кольце содержатся один или несколько гетероатомов.
Например, циклоалкильная группа может быть моноциклической или бициклической.
Таким образом, «Cnциклоалкильная» группа содержит n атомов углерода. Все n атомы углерода могут содержаться в составе кольца(ец) циклоалкильной группы, или один или несколько атомов углерода могут не содержаться в составе кольца(ец) и вместо этого могут образовывать одно или несколько ответвлений от кольца. Если Cnалкильная группа присоединена к отдельной Cmалкильной группе, содержащей m атомов углерода, с образованием, например, гетероцикла, то две алкильные группы содержат общее число m+n атомов углерода.
Алкильная группа может быть насыщенной или ненасыщенной. Таким образом, алкильная группа может представлять собой алкенильную группу (например, содержащую двойную углерод-углеродную связь) и/или алкинильную группу (например, содержащую тройную углерод-углеродную связь). Если алкильная группа является ненасыщенной, то она может содержать, по меньшей мере, 2 атома углерода. При этом понимается, что любые ненасыщенные части алкильной группы являются неароматическими (ароматические группы подпадают под объем определения «арил»). Любая часть алкильной группы может быть ненасыщенной, например, неразветвленная, разветвленная или циклическая часть алкильной группы может содержать двойную углерод-углеродную связь или тройную углерод-углеродную связь. Присоединение к ненасыщенной алкильной группе может происходить через ненасыщенную часть алкильной группы или может происходить через ненасыщенную часть группы.
Например, ненасыщенная алкильная группа может содержать от 1 до 4 двойных углерод-углеродных связей или от 1 до 3 тройных углерод-углеродных связей, или от 1 до 4 сочетаний двойных углерод-углеродных связей и тройных углерод-углеродных связей.
Алкильная группа может быть замещена одним или несколькими гетероатомами или она может быть незамещенной (т.е., не содержать какие-либо гетероатомы). Если присутствует более чем один гетерозаместитель, то заместители выбирают относительно независимо друг друга, кроме случаев, когда они образуют часть конкретной функциональной группы (например, амидогруппы).
В свою очередь, заместители гетероатома могут быть замещены дополнительными углеродсодержащими группами. В этом случае префикс Cn или Cm, который определяет замещенную алкильную группу, относится к общему числу углеродов, содержащихся в группе, т.е. включая атомы углерода, содержащиеся в любых замещенных гетероатомных группах, и алкильная группа содержит всего от 1 до 15 атомов углерода, как определено ранее.
Соответственно, если алкильная группа является замещенной, то она, например, может содержать одну или несколько из CN, CO2H, CONH2, CONHR, CONRaRb, CO2R, NH2, NHR, NRaRb, OH, OR, SH, SR, F, Cl, Br и I, где каждый R, Ra и Rb независимо представляет собой выбранные группы (например, алкильные/арильные группы), присоединенные к атому, к которому группа присоединена через атом углерода каждой группы, включая случаи, когда Ra и Rb образуют гетероцикл, содержащий гетероатом, к которому они присоединены. Группа, содержащая два Cm-Cnалкильных фрагмента, которые образуют цикл, который содержит, например, гетероатом, к которому они присоединены, может содержать от C2m до C2n атомов углерода.
Примеры незамещенных насыщенных алкильных групп, не содержащих циклические структуры, включают метил, этил, н-пропил, втор-пропил, н-бутил, втор-бутил, трет-бутил, пентил (разветвленный или неразветвленный), гексил (разветвленный или неразветвленный), гептил (разветвленный или неразветвленный), октил (разветвленный или неразветвленный), нонил (разветвленный или неразветвленный) и децил (разветвленный или неразветвленный).
Примеры незамещенных насыщенных циклических алкильных групп включают циклопропил, циклобутил, циклопентил и циклогексил.
Примеры ненасыщенных алкильных групп включают этенил, пропенил, бутенил, 2-метилбутенил и циклогексенил.
Термин «арильная группа» относится к группе, содержащей, по меньшей мере, одно кольцо, которое является ароматическим и содержит от 1 до 15 атомов углерода, как, например, от 1 до 10 атомов углерода. В случае, когда арильная группа указана как замещенная в конкретном положении, положение присоединения к арильной группе расположено собственно на ароматическом кольце арильной группы, а не на какой-либо неароматической боковой цепи арильной группы. Например, если R1 представляет собой арильную группу в CR1, то C присоединен к ароматической части арильной группы.
Каждое кольцо может быть независимо выбрано как содержащее только атомы углерода или как содержащее и атомы углерода и от 1 до 4 гетероатомов, выбранных из O, N и S. Для гетероарильных групп (например, арильные группы содержащие один или несколько гетероатомов) присоединение к арильной группе может происходить либо посредством атома углерода, либо, если в кольце содержится один или несколько гетероатомов, присоединение может также происходить посредством гетероатома, содержащегося в кольце.
Констатируется, что содержащиеся в кольце гетероарильной группы гетероатомы могут быть замещены, например, с образованием N-оксида.
Например, ароматическая группа может быть моноциклической или бициклической, где одно или оба кольца бициклической системы являются ароматическими.
Примеры арильных групп включают акридинил, фенил, карбазолил, циннолинил, хиноксалинил, пирразолил, бензотриазолил, фуранил, нафтил, тиенил, тиазолил, бензотиенил, бензофуранил, хинолинил, изохинолинил, оксазолил, изоксазолил, индолил, пиразинил, пиридазинил, пиридинил, пиримидинил, пирролил, тетрагидрохинолин, бензимидазолил и меламинил.
Констатируется, что под объем термина «гетероцикл» подпадают и циклоалкильные группы, содержащие один или несколько гетероатомов в кольцевой системе, и арильные группы, содержащие один или несколько гетероатомов в кольцевой системе.
Гетероциклические группы могут представлять собой любой из различных пиразолов, имидазолов и триазолов и могут включать кислородсодержащие и/или серосодержащие аналоги различных пиразолов, имидазолов и триазолов, то есть оксазолы, изоксазолы, тиазолы и изотиазолы и производные.
Термин «галоген» относится к группе, выбранной из хлора, фтора, брома и йода.
Далее будет описано настоящее изобретение. В последующем, различные аспекты настоящего изобретения определены более подробно. Каждый определенный таким образом аспект может быть объединен с любым другим аспектом или аспектами, кроме тех, которые четко указаны как имеющие противоположный смысл. В частности, любая характерная черта, указанная в качестве предпочтительной или благоприятной, может быть сочетана с любой другой характерной чертой или характерными чертами, указанными в качестве предпочтительных или благоприятных.
Для полноты, также констатируется, что определенные химические формулы, используемые в настоящем документе, обозначают делокализованные системы. Это определение известно в данной области техники как определение ароматичности и может указывать на присутствие, например, моно-, ди- или трициклических систем, содержащих (4n+2) электронов, где n представляет собой целое число. Другими словами, эти системы могут отображать ароматичность Хюккеля.
В каком бы то ни было аспекте, соединения согласно настоящему изобретению могут обладать некоторым аспектом стереохимии. Например, соединения могут обладать хиральными центрами и/или плоскостями и/или осями. Например, соединения могут быть представлены в виде отдельных стереоизомеров, отдельных диастереомеров, смесей стереоизомеров или в виде рацемических смесей. Стереоизомеры известны в данной области техники как молекулы, которые имеют одинаковую молекулярную формулу и последовательность связанных атомов, но отличаются пространственной ориентацией своих атомов и/или групп.
Кроме того, соединения согласно настоящему изобретению могут обладать таутомерией. Предполагается, что каждая таутомерная форма подпадает под объем настоящего изобретения.
Кроме того, соединения согласно настоящему изобретению могут быть представлены в виде пролекарства. Пролекарства преобразуются, главным образом in vivo, из одной формы в активные формы лекарственных средств, описанных в этом документе. Например, пролекарство может быть образовано в виде физиологически гидролизуемого амида путем защиты амина присоединением циклобутана. В качестве альтернативы или дополнения, если Y или Z или присоединенные к ним любые фрагменты представляют собой NH, то один или несколько из них могут быть защищены в виде гидролизуемого в физиологических условиях амида.
В тех соединениях согласно настоящему изобретению, в которых X «отсутствует», вместо него присутствует одинарная ковалентная связь, так что W и Y связаны с одним и тем же атомом углерода, содержащим заместители R1 и R2.
Кроме того, соединения согласно настоящему изобретению могут быть представлены в виде их фармацевтически приемлемых солей или в виде сокристаллов. Например, соединения могут быть содержать протонированные аминогруппы.
Термин «фармацевтически приемлемая соль» относится к ионным соединениям, образованным путем добавления кислоты к основанию. Этот термин относится к таким солям, которые рассматриваются в данной области техники как подходящие для применения в контакте с пациентом, например, in vivo, и обычно фармацевтически приемлемые соли выбирают исходя из их нетоксичных и не вызывающих раздражения свойств.
Термин «сокристалл» относится к многокомпонентному молекулярному кристаллу, который может иметь в своем составе неионные взаимодействия.
Фармацевтически приемлемые соли и сокристаллы могут быть получены путем ионообменной хроматографии или путем осуществления взаимодействия свободной основной или кислотной формы соединения со стехиометрическими количествами или с избытком желаемой солеобразующей кислоты или основания в одном или нескольких подходящих растворителях, или путем смешивания соединения с другим фармацевтически приемлемым соединением, способным к образованию сокристалла.
Соли, известные в данной области техники в качестве обычно подходящих для применения в контакте с пациентом, включают соли, полученные из неорганических и/или органических кислот, включая гидробромид, гидрохлорид, сульфат, бисульфат, нитрат, ацетат, оксалат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат и тартрат. Они могут содержать катионы щелочных и щелочноземельных металлов, таких как натрий, калий, кальций и магний, а также аммоний, тетраметиламмоний, тетраэтиламмоний. Дополнительно дается ссылка на целый ряд литературных источников, в которых представлен обзор подходящих фармацевтически приемлемых солей, например, Handbook of pharmaceutical salts, опубликованный IUPAC.
Кроме того, соединения согласно настоящему изобретению могут иногда существовать в виде цвиттерионов, которые рассматриваются как часть настоящего изобретения.
Соединения согласно настоящему изобретению применимы для лечения медицинских состояний, ассоциированных с нарушенным клеточным ростом, включая без ограничения злокачественной опухоли, в частности злокачественных опухолей, ассоциированных с гиперактивностью AKT, происходящей либо в результате прямого изменения в самой киназе, такого, которое может произойти в результате мутации в какой-либо из ее субъединиц, либо в результате повышенной активности вверх по цепочке передачи сигнала, включая без ограничения повышенную активность PI3K или PDK. Повышение активности PI3K может происходить вследствие утраты супрессора опухоли PTEN.
Например, злокачественные опухоли включают злокачественные опухоли сердца, злокачественные опухоли легких, злокачественные опухоли желудочно-кишечного тракта, злокачественные опухоли мочеполовых путей, злокачественные опухоли печени, злокачественные опухоли костей, злокачественные опухоли нервной системы, гинекологические злокачественные опухоли, гематологические злокачественные опухоли, злокачественные опухоли кожи и злокачественные опухоли надпочечников.
Например, злокачественные опухоли включают опухоли надпочечников, желчного протока, мочевого пузыря, крови, костной и соединительной тканей, головного мозга и центральной нервной системы, молочной железы, шейки матки, ободочной и прямой кишки (колоректальная), эндометрия, пищевода, желчного пузыря, головы и шеи, лимфому Ходжкина, гипофарингеальную, почки, гортани, лейкозы, печени, легких, лимфому, средостенные опухоли, меланому (злокачественную меланому), мезотелиому, множественную миелому, носовой полости, носоглотки, нейроэндокринные опухоли, неходжкинскую лимфому, ротовой полости, пищевода, ротоглоточной области, яичника, поджелудочной железы, придаточной пазухи носа, паращитовидной железы, полового члена, опухоли гипофиза, предстательной железы, слюнной железы, саркому, кожи, позвоночника, желудка, яичек, щитовидной железы, мочеиспускательного канала, матки, влагалища и вульвы.
Соединения согласно настоящему изобретению также применимы при получении лекарственного средства, которое применимо при лечении описанных выше заболеваний, в частности, злокачественной опухоли.
Соединения согласно настоящему изобретению могут селективно ингибировать активность одну или две из изоформ семейства белков AKT по сравнению с другой(ими) изоформой(ами) AKT. Например, эти соединения могут селективно ингибировать одну или две из AKT1, AKT2 или AKT3 по сравнению с другой(ими) изоформой(ами) AKT.
Например, соединения согласно настоящему изобретению могут ингибировать, по меньшей мере, AKT1 и/или AKT2. Например, эти соединения могут селективно ингибировать AKT1 и/или AKT2 по сравнению с AKT3.
Настоящее изобретение, кроме того, относится к способу ингибирования активности AKT, который включает введение фармацевтически эффективного количества соединения согласно настоящему изобретению нуждающемуся в этом млекопитающему.
Соединения согласно настоящему изобретению могут быть введены млекопитающим, включая людей, либо отдельно, либо в сочетании с фармацевтически приемлемыми носителями, наполнителями или разбавителями, в фармацевтической композиции, в соответствии со стандартной фармацевтической практикой. Соединения могут быть введены перорально или парентерально, включая внутривенный, внутримышечный, интраперитонеальный, подкожный, ректальный и местный пути введения.
Под объем настоящего изобретения также подпадает применение соединений согласно настоящему изобретению в сочетании со вторым лекарственным средством при лечении злокачественной опухоли. Второе лекарственное средство может представлять собой такое, которое уже известно в области техники лечения злокачественной опухоли. Настоящее изобретение также включает применение соединений согласно настоящему изобретению в схеме лечения, включающей стадию лучевой терапии.
В частности, злокачественные опухоли часто становятся резистентными к терапии. Развитие резистентности может быть отложено или преодолено путем введения сочетания лекарственных средств, которые включают соединения согласно настоящему изобретению.
Например, лекарственные средства, которые могут быть использованы в сочетании с соединениями согласно настоящему изобретению, могут быть направлены на тот же или сходный биологический путь, на который направлены и соединения согласно настоящему изобретению, или могут воздействовать на другой или несвязанный путь.
В зависимости от подлежащего лечению заболевания вместе с соединениями согласно настоящему изобретению может быть совместно введен целый ряд партнеров сочетания. Второй активный ингредиент может включать без ограничения: алкилирующие агенты, включая циклофосфамид, ифосфамид, тиотепа, мелфалан, хлорэтилнитрозомочевину и бендамустин; производные платины, включая цисплатин, оксалиплатин, карбоплатин и сатраплатин; антимитотические средства, включая алкалоиды барвинка (винкристин, винорелбин, винбластин), таксаны (паклитаксел, доцетаксел), эпотилоны и ингибиторы митотических киназ, включая киназы семейств aurora и polo; ингибиторы топоизомераз, включая антрациклины, эпиподофиллотоксины, камптотецин и аналоги камтотецина; антиметаболиты, включая 5-фторурацил, капецитабин, цитарабин, гемцитабин, 6-меркаптопурин, 6-тиогуанин, флударабин, метотрексат и преметрексед; ингибиторы протенкиназ, включая иматиниб, гефитиниб, сорафениб, сунитиниб, эрлотиниб, дазатиниб и лапатиниб; ингибиторы протеосом, включая бортезомиб; ингибиторы гистондеацетилаз, включая вальпроат и SAHA; антиангиогенные лекарственные средства, включая бевацизумаб; моноклональные антитела, включая трастузумаб, ритуксимаб, алемтузумаб, тозитумомаб, цетуксимаб, панитумумаб, конъюгаты миоклональных антител, включая гемтузумаб озогамицин, ибритумомаб тиуксетан, гормональные терапии, включая антиэстрогены (тамоксифен, ралоксифен, анастразол, летрозол, эксаместан), антиандрогены (флутамид, биклутамид) и аналоги или антагонисты лютеинизирующего гормона.
Что касается сочетанной терапии, то соединения согласно настоящему изобретению могут быть введены отдельно, последовательно, одновременно, параллельно или могут быть хронологически разнесены с одним или несколькими стандартными терапевтическими средствами, такими как любое из средств, упомянутых выше.
Настоящее изобретение также относится к фармацевтической композиции, подходящей для клинического применения.
В частности, фармацевтическая композиция может содержать фармацевтический носитель и распределенное в нем терапевтически эффективное количество соединений согласно настоящему изобретению. Композиция может быть твердой или жидкой. Фармацевтический носитель обычно выбирают, основываясь на используемом способе введения, и фармацевтический носитель может быть, например, твердым или жидким. Соединения согласно настоящему изобретению могут быть в одинаковой фазе с фармацевтическим носителем или в разных фазах.
Фармацевтические композиции могут быть составлены в соответствии с их конкретным использованием и назначением путем смешивания, например, наполнителя, связующего вещества, смазки, разрыхлителя, покровного вещества, эмульгатора, суспендирующего агента, растворителя, стабилизатора, усилителя абсорбции и/или мазевой основы. Композиция может быть подходящей для перорального, инъекционного, ректального или местного введения.
Например, фармацевтическая композиция может быть введена перорально, например, в форме таблеток, таблеток с покрытием, твердых или мягких желатиновых капсул, растворов, эмульсий или суспензий. Введение может также проводиться ректально, например, с использованием суппозиториев, местно или подкожно, например, с использованием мазей, кремов, гелей или растворов, или парентерально, например, с использованием инъекционных растворов.
Для приготовления таблеток, таблеток с покрытием или твердых желатиновых капсул, соединения согласно настоящему изобретению могут быть смешаны с фармацевтически инертными, неорганическими или органическими наполнителями. Примеры подходящих наполнителей включают лактозу, кукурузный крахмал или его производные, тальк или стеариновую кислоту или ее соли. Подходящие наполнители для использования с мягкими желатиновыми капсулами включают, например, растительные масла, воски, жиры и полутвердые или жидкие полиолы.
Наполнители для приготовления растворов и сиропов включают, например, воду, полиолы, сахарозу, инвертный сахар и глюкозу.
Наполнители для инъекционных растворов включают, например, воду, спирты, полиолы, глицерин или растительное масло.
Наполнители для суппозиториев и для местного и подкожного применения включают, например, натуральные или гидрогенизированные масла, воски, жиры и полутвердые или жидкие полиолы.
Фармацевтические композиции могут также содержать консерванты, солюбилизаторы, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, ароматизаторы, буферы, покровные вещества и/или антиоксиданты.
Второе лекарство для сочетанной терапии может находиться в фармацевтической композиции с настоящим изобретением или может быть представлено отдельно.
Таким образом, фармацевтический состав для перорального введения может представлять собой, например, гранулу, таблетку, покрытую сахаром таблетку, капсулу, пилюлю, суспензию или эмульсию. Для парентеральных инъекций, например, для внутривенного, внутримышечного или подкожного применения, может быть представлен стерильный водный раствор, который может содержать другие вещества, включая, например, соли и/или глюкозу для создания изотонического раствора. Противоопухолевое средство также может быть введено в форме суппозитория или вагинального суппозитория, или может быть применено местно в форме лосьона, раствора, крема, мази или присыпки.
Настоящее изобретение также относится к способам лечения или профилактики злокачественной опухоли, включающим введение нуждающемуся в этом субъекту соединений согласно настоящему изобретению, описанных в настоящем документе. В предпочтительных вариантах осуществления способ включает использование одного или нескольких соединений согласно настоящему изобретению в схеме лечения, включающей стадию лучевой терапии.
ПРИМЕРЫ
Далее, настоящее изобретение будет описано в отношении нескольких примеров.
Соединения примеров 1-153 синтезировали в соответствии со способами, описанными далее. Затем их значения IC50 определяли, как описано ниже, и представляли в виде последующей таблицы, в которой номера соединений соответствуют номерам примеров.






Условные обозначения
+ IC50>10 мкМ
++ 1 мкΜ<IC50≤10 мкМ
+++ 0,1 мкМ<IC50≤1 мкМ
++++ IC50≤0,1 мкМ
Кроме того, методом ELISA исследовали статус фосфорилирования различных участников цепи передачи сигнала AKT/PI3K. Например, соединение примера 4 обуславливает фосфорилирование GSK3β со значением IC50<0,1 мкМ, измеренным через 48 ч.
В таблице можно увидеть, что соединения некоторых примеров обладают селективностью в отношении одной или нескольких изоформ AKT по сравнению с другой(ими) изоформой(ами). Например, в отношении AKT1 и/или AKT2 наблюдается большая активность по сравнению с AKT3.
Кроме того, исследовали активность соединений в методах анализа клеточной пролиферации in vitro. Для типичных примеров (например, соединения примеров 4, 28 и 31) показано ингибирование пролиферации клеточных линий PC3 и/или LnCaP со значением IC50<10 мкМ.
Сокращения
AcOH: уксусная кислота; nBuLi: н-бутиллитий; BINAP: 2,2'-бис(дифенилфосфино)-1,1'-бинафтил; DBU: 1,8-диазабицикло[5.4.0]ундец-7-ен; DCM: дихлорметан; DIPEA: диизопропилэтиламин; DMA: N,N-диметилацетамид; DMAP: 4-диметиламинопиридин; DME: диметоксиэтан; DMF: N,N-диметилформамид; DMSO: диметилсульфоксид; EDCI: 1-этил-3-(3'-диметиламинопропил)карбодиимид; EtOAc: этилацетат; ч: час(ы); HATU: O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; HCl: соляная кислота; HOBt: 1-гидроксибензотриазол; ЖХВД: жидкостная хроматография высокого давления; IMS: технические метилированные спирты: M: молярная концентрация (М); MeOH: метанол; NMP: N-метил-2-пирролидон; ЯМР: ядерный магнитный резонанс; мин: минуты; к.т.: комнатная температура; SCX: сильный катионообменный силикагель SCX; TBAF: тетра-н-бутиламмония фторид; TEA: триэтиламин; TFA: трифторуксусная кислота; THF: тетрагидрофуран; TMSCl: триметилсилилхлорид
Общие условия проведения эксперимента
1H-ЯМР спектры регистрировали при комнатной температуре с использованием спектрометра Varian Unity Inova (400 МГц) с 5-мм зондом тройного резонанса, спектрометра Bruker Avance DRX (400 МГц) с 5-мм TXI датчиком тройного резонанса с инверсным детектированием, спектрометра Bruker Avance (500 МГц) с 5-мм QNP датчиком или спектрометра Bruker Avance DPX (300 МГц) со стандартным 5-мм датчиком двойного резонанса. Химические сдвиги выражены в м.д. относительно тетраметилсилана.
Эксперименты с жидкостной хроматографией высокого давления/масс-спектрометрией (ЖХ/МС) для определения значений времени удерживания (RT) и ассоциированных масс ионов проводили с использованием одного из последующих методов.
Метод A: Система состоит из масс-спектрометра ThermoFinnigan LCQ Advantage с ЖХ-системой Surveyor и автодозатором на 200 образцов. Спектрометр оснащен системой электрораспыления, работающей в режиме определения положительных и отрицательных ионов. ЖХ-МС определения проводили на каждом образце с использованием следующих условий: ЖХ колонка - Luna 3 мкм C18 50×2 мм; подвижная фаза: A) вода с 0,1% муравьиной кислоты, B) ацетонитрил с 0,1% муравьиной кислоты.
Деление потока - 100 мкл/мин к ESI источнику с детекцией на встроенном DAD Surveyor
Детекция - МС, УФ
Метод МС ионизации - электрораспылением (положительные и отрицательные ионы)
Общее время эксперимента - 11 мин (приблизительно)
Метод B: Система состоит из квадрупольного масс-спектрометра Waters ZMD, соединенного с ЖХ-системой Waters 1525 с диодно-матричным детектором Waters 996. Введение образца осуществляли при помощи автодозатора Waters 2700. Спектрометр оснащен системой электрораспыления, работающей в режиме определения положительных и отрицательных ионов. Дополнительно проводили детекцию с использованием испарительного детектора светорассеяния Sedex 85. ЖХ-МС определения проводили на каждом образце с использованием следующих условий: ЖХ колонка - Luna 3 мкм C18(2) 30×4,6 мм или эквивалент. Подвижная фаза: A) вода с 0,1% муравьиной кислоты, B) метанол с 0,1% муравьиной кислоты.
Деление потока - 200 мкл/мин к ESI источнику с детекцией на встроенном DAD Waters 996.
Детекция - МС, испарительное светорассеяние, УФ
Метод МС ионизации - электрораспылением (положительные и отрицательные ионы)
Общее время эксперимента - 6 мин (приблизительно)
Метод C: Система состоит из квадрупольного масс-спектрометра Waters Micromass ZQ2000, соединенного с UPLC-системой Waters Acquity с диодно-матричным (PDA) УФ-детектором. Спектрометр оснащен системой электрораспыления, работающей в режиме определения положительных и отрицательных ионов. ЖХ-МС определения проводили на каждом образце с использованием следующих условий: ЖХ колонка - Acquity BEH C18 1,7 мкм 100×2,1 мм, термостатирование при 40°C, или Acquity BEH Shield RP18 1,7 мкм 100×2,1 мм, термостатирование при 40°C. Подвижная фаза: A) вода с 0,1% муравьиной кислоты, B) ацетонитрил с 0,1% муравьиной кислоты.
Детекция - МС, PDA-УФ
Метод МС ионизации - электрораспылением (положительные и отрицательные ионы)
Метод D: Система состоит из квадрупольного масс-спектрометра Agilent Technologies 6140, соединенного с ЖХ-системой Agilent Technologies 1290, оснащенной диодно-матричным УФ-детектором и автодозатором. Спектрометр оснащен мультимодальной системой ионизации (электрораспылением и химическая ионизация при атмосферном давлении (APCI)), работающей в режиме определения положительных и отрицательных ионов. ЖХ-МС определения проводили на каждом образце с использованием следующих условий: ЖХ колонка - Zorbax Eclipse Plus C18 RRHD 1,8 мкм 50×2,1 мм, термостатирование при 40°C. Подвижная фаза: A) вода с 0,1% муравьиной кислоты, B) ацетонитрил с 0,1% муравьиной кислоты.
Детекция - МС, УФ
Метод МС ионизации - мультимодальный (положительные и отрицательные ионы)
Общее время эксперимента - 2,50 мин (приблизительно)
Метод E: Система состоит из квадрупольного масс-спектрометра Agilent Technologies 6120, соединенного с препаративной ЖХ-системой Agilent Technologies 1200, оснащенной многоволновым детектором и автодозатором. Спектрометр оснащен мультимодальной системой ионизации (электрораспылением и химическая ионизация при атмосферном давлении), работающей в режиме определения положительных и отрицательных ионов. Сбор фракций является масс-инициированным. Очистку каждого образца проводили с использованием следующих условий: ЖХ колонка - Waters XBridge™ Prep C18 5 мкм OBD™ 19×50 мм, при комнатной температуре. Подвижная фаза: A) вода с 0,1% гидроксида аммония, B) ацетонитрил/вода 95/5 с 0,1% гидроксида аммония.
Детекция - МС, УФ
Метод МС ионизации - мультимодальный (положительные и отрицательные ионы)
Общее время эксперимента - 10 мин (приблизительно)
Метод F: Система состоит из квадрупольного масс-спектрометра Agilent Technologies 6120, соединенного с препаративной ЖХ-системой Agilent Technologies 1200, оснащенной многоволновым детектором и автодозатором. Спектрометр оснащен мультимодальной системой ионизации (электрораспылением и химическая ионизация при атмосферном давлении), работающей в режиме определения положительных и отрицательных ионов. Сбор фракций является масс-инициированным. Очистку каждого образца проводили с использованием следующих условий: ЖХ колонка - Waters XBridge™Prep C18 5 мкм OBD™ 30×100 мм, при комнатной температуре. Подвижная фаза: A) вода с 0,1% гидроксида аммония, B) ацетонитрил/вода 95/5 с 0,1% гидроксида аммония.
Детекция - МС, УФ
Метод МС ионизации - мультимодальный (положительные и отрицательные ионы)
Общее время эксперимента - 18 мин (приблизительно)
Метод G: Система состоит из квадрупольного ЖХ/МС масс-спектрометра Agilent 6110, соединенного с ЖХ-системой Agilent 1200, оснащенной диодно-матричным детектором Agilent 1200. Введение образца осуществляли при помощи автодозатора Agilent 1200. Спектрометр оснащен системой ионизации электрораспылением и APCI, работающей в режиме определения положительных и отрицательных ионов. Очистку каждого образца проводили с использованием следующих условий: ЖХ колонка - Sunfire 5 мкм C18(2) 15×4,6 мм или эквивалент. Подвижная фаза: A) вода с 0,1% муравьиной кислоты, B) ACN с 0,1% муравьиной кислоты.
Деление потока - 40/мин к ESI/APCI источнику
Детекция - МС, УФ
Метод МС ионизации - электрораспылением и APCI (положительные и отрицательные ионы)
Общее время эксперимента - 6 или 20 мин (приблизительно)
Эксперименты с использованием микроволн проводили с использованием аппаратов CEM Explorer™ или Biotage Initator™. Могут быть достигнуты температуры 60-300°C, и значения давления до 20 бар.
Если иное не указано особо, то использовали номенклатуру структур «структура-название» производства ChemBioDraw 11 (CambridgeSoft). Некоторым соединения, получившие названия с использованием упомянутого выше программного обеспечения, содержат стереоцентры, обозначенные как 'S' и 'R', тогда как обычно они должны были бы быть обозначены как 'цис' или 'транс'. Для таких соединений представленные структуры являются типичными для полученных соединений.
Если иное не указано особо, то исходные вещества и промежуточные вещества приобретали из коммерческих источников, получали в соответствии с изложенными в литературе методиками (например, WO 2008/070016, WO 2009/148887 и WO 2009/148916) или путем стандартных преобразований, очевидных специалисту в данной области техники.
Пример 1: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-он
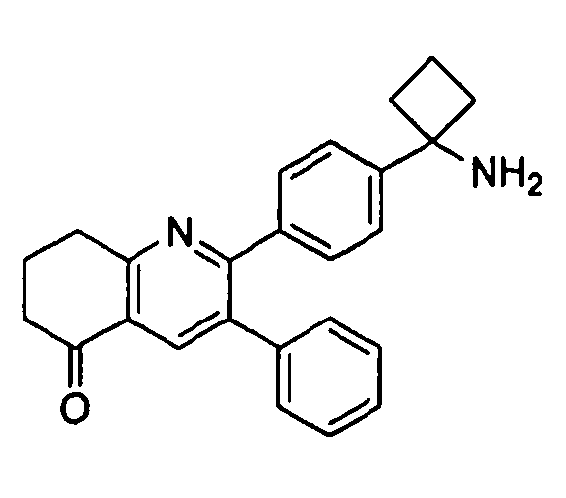
Стадия 1: трет-бутил-(1-(4-(3-(диметиламино)-2-фенилакрилоил)фенил)циклобутил)карбамат
Раствор трет-бутил-(1-(4-(2-фенилацетил)фенил)циклобутил)карбамата (25,0 г, 68 ммоль) в Ν,Ν-диметилформамида диметилацетале (150 мл) нагревали до 80°C в атмосфере азота в течение 18 часов. Полученный коричневый раствор оставляли охлаждаться до комнатной температуры и концентрировали досуха в условиях пониженного давления с получением целевого продукта в виде желто-коричневого твердого вещества (30,6 г, количественный выход).
1H-ЯМР (500 МГц, CDCl3) δ 7,14-7,48 (10H, м), 5,09 (1H, ушир. с), 2,74 (6H, с), 2,45-2,60 (4H, м), 2,04-2,16 (1H, м), 1,80-1,90 (1H, м), 1,10-1,52 (9H, ушир. м).
Стадия 2: трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат
К уксусной кислоте (120 мл) добавляли молекулярные сита (5Å, 1,50 г), трет-бутил-(1-(4-(3-(диметиламино)-2-фенилакрилоил)фенил)циклобутил)карбамат (8,00 г, 19,0 ммоль) и 3-амино-2-циклогексен-1-он (3,20 г, 28,8 ммоль). Реакционную смесь нагревали до 100°C в атмосфере азота в течение 2 часов. Смесь оставляли охлаждаться до комнатной температуры и концентрировали в условиях пониженного давления. Остаток распределяли между водой (160 мл) и дихлорметаном (160 мл) и декантировали с молекулярных сит. Слои разделяли, и экстрагировали водную фазу введением в дихлорметан (2×160 мл). Объединенные органические фазы промывали насыщенным раствором NaHCO3 (2×160 мл), солевым раствором (160 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желто-коричневого твердого вещества. Это вещество очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде желтого твердого вещества (3,30 г, выход 37%).
1H-ЯМР (500 МГц, CDCl3) δ 8,30 (1H, с), 7,37 (2H, д), 7,30 (2H, д), 7,24-7,27 (3H, м), 7,15-7,20 (2H, м), 5,01 (1H, ушир. с), 3,26 (2H, т), 2,75 (2H, т), 2,42-2,55 (4H, м), 2,26 (2H, p), 2,00-2,10 (1H, м), 1,73-1,85 (1H, м), 1,12-1,44 (9H, ушир. м).
ЖХ/МС (способ A) RT=7,54 мин, M+H+=469,15.
Стадия 3: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-он
трет-Бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат (11 мг, 0,023 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления, и сушили с получением целевого продукта в виде не совсем белого твердого вещества (10 мг, выход 91%).
1H-ЯМР (500 МГц, CD3OD) δ 8,29 (1H, с), 7,51 (2H, д), 7,45 (2H, д), 7,31 (3H, т), 7,21 (2H, дд), 3,26 (2H, т), 2,80 (2H, т), 2,72-2,81 (2H, м), 2,53-2,62 (2H, м), 2,30 (2H, т), 2,20-2,30 (1H, м), 1,92-2,01 (1H, м).
ЖХ/МС (способ A) RT=4,09 мин, M+H+=368,91.
Пример 2: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она оксим
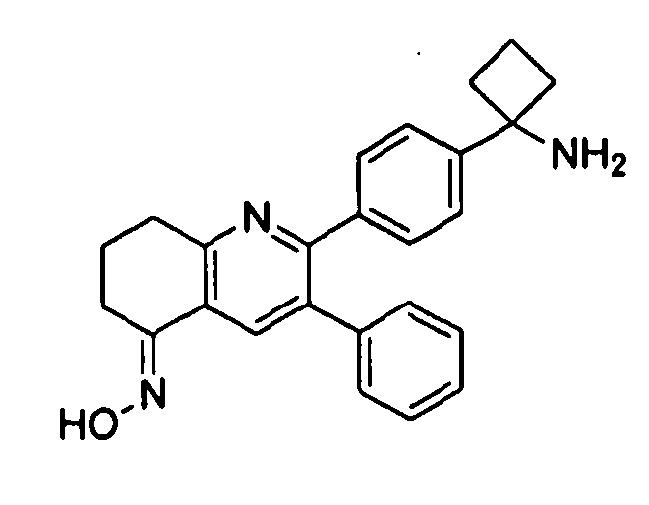
Стадия 1: трет-бутил-(1-(4-(5-(гидроксиимино)-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она (80 мг, 0,17 ммоль) в безводном этаноле (2,0 мл) добавляли гидроксиламина гидрохлорид (65 мг, 0,9 ммоль) и пиридин (75 мкл, 0,9 ммоль). Полученный желтый раствор нагревали до 90°C в течение 2 часов в атмосфере азота. Реакционную смесь оставляли охлаждаться до комнатной температуры и концентрировали досуха в условиях пониженного давления. Остаток смешивали с водой (2,0 мл) и экстрагировали введением в дихлорметан (3×2,0 мл). Объединенные органические фазы промывали солевым раствором (2×4,0 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Остаток очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 95/5→60/40) с получением целевого продукта в виде белого твердого вещества (47 мг, выход 56%).
1H ЯМР (500 МГц, CH3OD) 8,30 (с, 1H), 7,30 (д, 2H), 7,26 (м, 5H), 7,19 (д, 2H), 3,19 (м, 2H), 2,87 (м, 2H), 2,45-2,50 (м, 4H), 2,0-2,1 (м, 1H), 1,90-1,99 (м, 2H), 1,80-1,86 (м, 1H), 1,2-1,4 (ушир. с, 9H).
Стадия 2: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она оксим
трет-Бутил-(1-(4-(5-(гидроксиимино)-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат (7 мг, 0,017 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления, и сушили с получением целевого продукта в виде не совсем белого твердого вещества (3 мг, выход 35%).
1H ЯМР (500 МГц, CH3OD) 8,45 (с, 1H), 7,46 (м, 5H), 7,34 (д, 2H), 7,21 (д, 2H), 3,10 (т, 2H), 2,90 (т, 2H), 2,74-2,78 (м, 2H), 2,56-2,6 (м, 2H), 2,20-2,28 (м, 1H), 2,02-2,10 (м, 2H), 1,93-1,98 (м, 1H).
Пример 3: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она O-метилоксим
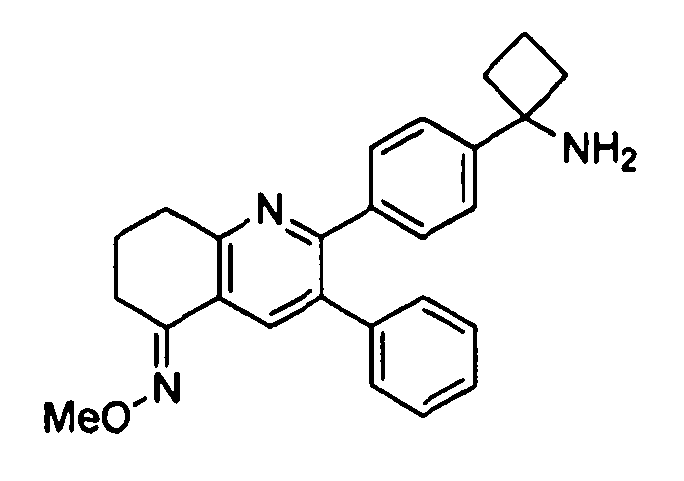
Стадия 1: трет-бутил-(1-(4-(5-(метоксиимино)-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (90 мг, 0,19 ммоль) в безводном этаноле (2,0 мл) добавляли O-метилгидроксиламина гидрохлорид (80 мг, 0,96 ммоль) и пиридин (80 мкл, 0,99 ммоль). Полученный желтый раствор нагревали до 90°C в атмосфере азота. Реакционную смесь оставляли охлаждаться до комнатной температуры и концентрировали досуха в условиях пониженного давления. Остаток смешивали с водой (2,0 мл) и экстрагировали введением в этилацетат (3×2,0 мл). Объединенные органические фазы промывали солевым раствором (2×4,0 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Остаток очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 95/5→60/40) с получением целевого продукта в виде белого твердого вещества (44 мг, выход 46%).
1H-ЯМР (500 МГц, CD3OD) δ 8,29 (1H, с), 7,35 (2H, д), 7,26-7,32 (5H, м), 7,16-7,21 (2H, м), 4,01 (3H, с), 3,02 (2H, т), 2,83 (2H, т), 2,36-2,52 (4H, м), 2,04-2,16 (1H, м), 2,01 (2H, p), 1,81-1,92 (1H, м), 1,15- 1,50 (9H, ушир. м).
ЖХ/МС (способ A) RT=8,28 мин, M+H+=498,10.
Стадия 2: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она O-метилоксим
трет-Бутил-(1-(4-(5-(метоксиимино)-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат (44 мг, 0,088 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (32 мг, выход 73%).
1H-ЯМР (500 МГц, CD3OD) δ 8,35 (1Н, с), 7,48 (2Н, д), 7,43 (2Н, д), 7,30 (3Н, т), 7,18-7,23 (2Н, м), 4,02 (3Н, с), 3,05 (2Н, т), 2,85 (2Н, т), 2,73-2,81 (2H, м), 2,54-2,63 (2H, м), 2,19-2,30 (1H, м), 2,03 (2H, p), 1,91-2,03 (1H, м).
ЖХ/МС (способ A) RT=4,57 мин, M+H+=398,10.
Пример 4: 1-(4-(8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
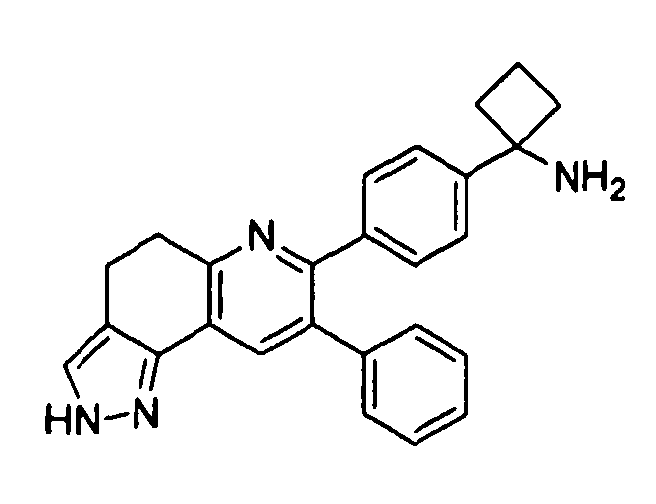
Стадия 1: трет-бутил-1-(4-(6-((диметиламино)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидронафталин-2-ил)фенил)циклобутил)карбамат
трет-Бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат (500 мг, 1,06 ммоль) растворяли в 5 мл безводного Ν,Ν-диметилформамида диметилацеталя. Полученную смесь нагревали до 100°C в атмосфере азота в течение 2,5 часов. Анализ по методам ТСХ и ЖХ/МС обнаруживал полное расходование исходного вещества. Реакционную смесь концентрировали досуха в условиях пониженного давления. Неочищенный остаток суспендировали в н-гексане (10 мл) в течение 1 часа. Суспензию фильтровали, и сушили твердое вещество до постоянного веса с получением целевого продукта в виде светло-желтого твердого вещества (420 мг, выход 76%). ЖХ/МС (способ A): RT=6,89 мин, M+H+=524; RT=8,37 мин, M+H+=497.
Стадия 2: трет-бутил-(1-(4-(8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-1-(4-(6-((диметиламино)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидронафталин-2-ил)фенил)циклобутил)карбамата (40 мг, 0,07 ммоль) в безводном этаноле (1500 мкл) добавляли гидразина моногидрат (15 мкл, 0,23 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 2,5 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления, очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 95/5→60/40) и суспендировали в диэтиловом эфире (1 мл) с получением целевого продукта в виде белого твердого вещества (6 мг, выход 16%).
1H ЯМР (500 МГц, CH3OD) 8,15 (с, 1H), 7,22-7,37 (м, 9Н), 3,22 (т, 2Н), 3,00 (т, 2Н), 2,45-2,51 (м, 4Н), 2,03-2,09 (м, 1H), 1,86-1,89 (м, 1H), 1,3-1,5 (ушир. с, 9H).
Стадия 3: 1-(4-(8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (6 мг, 0,012 ммоль) растворяли в TFA (0,8 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (3 мг, выход 50%).
1H ЯМР (500 МГц, CH3OD) 8,16 (с, 1H), 7,58 (с, 1H), 7,49 (д, 2H), 7,43 (д, 2H), 7,30 (м, 3H), 3,24 (т, 2H), 3,02 (т, 2H), 2,78 (м, 2H), 2,59 (м, 2H), 2,23 (м, 1H), 1,98 (м, 1H).
Пример 5: 1-(4-(2-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
Пример 6: 1-(4-(1-метил-8-фенил-4,5-дигидро-1H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
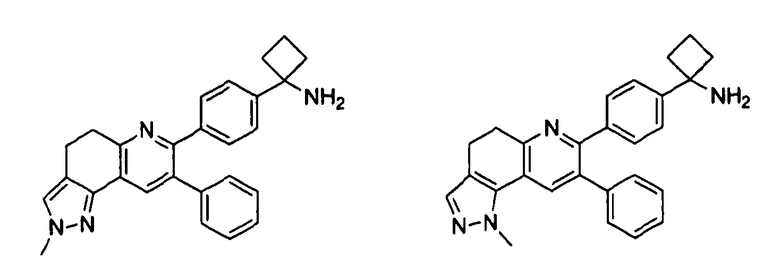
Стадия 1: трет-бутил-(1-(4-(2-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат и трет-бутил-(1-(4-(1-метил-8-фенил-4,5-дигидро-1H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-1-(4-(6-((диметиламино)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидронафталин-2-ил)фенил)циклобутил)карбамата (100 мг, 0,19 ммоль) в безводном этаноле (2 мл) добавляли метилгидразин (30 мкл, 0,57 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 2 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления, очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 99/1→70/30) с получением целевого продукта трет-бутил-(1-(4-(2-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамата в виде белого твердого вещества (14 мг, выход 15%).
1H ЯМР (500 МГц, CH3OD) 8,10 (с, 1H), 7,36 (д, 2H), 7,29 (д, 2H), 7,20-7,22 (м, 6H), 4,99 (с, 1H), 3,94 (с, 3H), 3,20-3,36 (ушир. с, 2H), 2,86-3,01 (м, 4H), 2,43-2,55 (м, 2H), 1,99-2,10 (м, 1H), 1,74-1,85 (м, 1H), 1,49-1,68 (ушир. с, 9H).
1H-1H NOESY (500 МГц, CH3OD) положительная корреляция между 7,22 (с, 1H) и 3,94 (с, 3H).
Смешанные фракции с колонки (45 мг) очищали методом препаративной ЖХВД (метод G, градиент 5→95% 0,1% FA/ACN в 0,1%FA/H2O, прогон 20 минут) с получением целевого продукта трет-бутил-(1-(4-(1-метил-8-фенил-4,5-дигидро-1H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамата в виде белого твердого вещества (9 мг, выход 10%).
1H ЯМР (500 МГц, CH3OD) 7,83 (с, 1H), 7,37 (м, 3H), 7,29 (м, 5H), 7,22 (м, 2H), 5,00 (с, 1H), 4,16 (с, 3H), 3,22-3,33 (ушир. м, 2H), 2,87-2,94 (м, 2H), 2,41-2,55 (м, 4H), 2,04 (м, 1H), 1,57 (м, 1H), 1,50-1,62 (ушир. с, 9H).
ЖХ/МС (способ A): RT=7,36 мин, M+H+=507.
Стадия 2A: 1-(4-(2-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
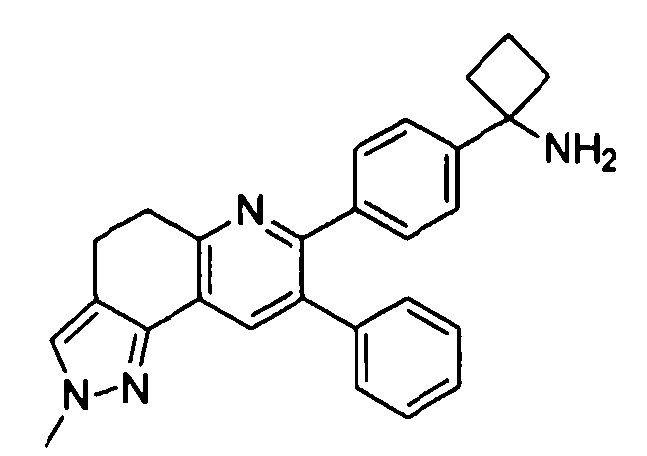
трет-Бутил-(1-(4-(2-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (14 мг, 0,027 ммоль) растворяли в TFA (0,5 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~1 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (7 мг, выход 50%).
1H ЯМР (500 МГц, CH3OD) 8,20 (с, 1H), 7,50 (с, 1H), 7,47 (д, 2H), 7,43 (д, 2H), 7,28 (м, 3H), 7,22 (м, 2H), 3,94 (с, 1H), 3,24 (т, 2H), 2,97 (т, 2H), 2,70-2,80 (ушир. м, 2H), 2,52-2,61 (ушир. м, 2H), 2,15-2,29(ушир. м, 1H), 1,88-2,00 (ушир. м, 1H).
Стадия 2B. 1-(4-(1-метил-8-фенил-4,5-дигидро-1H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
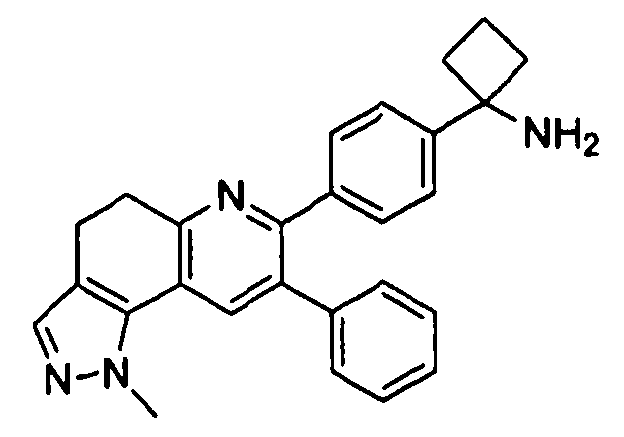
трет-Бутил-(1-(4-(1-метил-8-фенил-4,5-дигидро-1H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (3 мг, 0,006 ммоль) растворяли в TFA (0,5 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~1 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (0,65 мг, выход 22%).
1H ЯМР (500 МГц, CH3OD) 8,06 (с, 1H), 7,50 (д, 2H), 7,43 (д, 3H), 7,27-7,36 (ушир. м, 5H), 4,19 (с, 1H), 3,22 (т, 2H), 2,93 (т, 2H), 2,73-2,83 (ушир. м, 2H), 2,54-2,66 (ушир. м, 2H), 2,18-2,33 (ушир. м, 1H), 1,92-2,03 (ушир. м, 1H).
ЖХ/МС (способ A): RT=4,21 мин, M+H+=407.
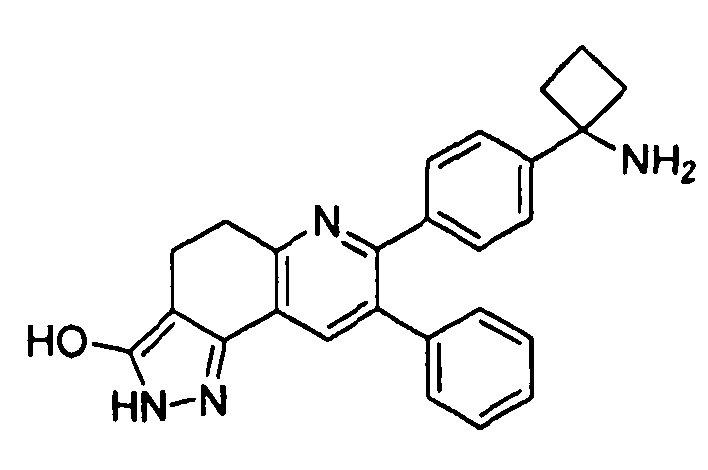
Пример 7: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-3-ол
Стадия 1: трет-бутил-(1-(4-(6-(бис(метилтио)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (1,00 г, 2,13 ммоль) в безводном тетрагидрофуране (10 мл) по каплям в течение 10 мин добавляли трет-бутоксид калия (1M в THF, 4,69 мл, 4,69 ммоль) с получением темно-красной суспензии. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 15 мин. К этой смеси по каплям в течение 5 мин добавляли дисульфид углерода (142 мкл, 2,35 ммоль) с получением темно-красного раствора. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 30 мин. Добавляли метилйодид (294 мкл, 4,69 ммоль) и перемешивали смесь при комнатной температуре в течение 90 мин. Реакционную смесь разбавляли насыщенным раствором бикарбоната натрия (40 мл) и экстрагировали этилацетатом (3×40 мл). Объединенные органические фазы промывали водой/солевым раствором (50/50, 40 мл), а затем солевым раствором (40 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением целевого продукта в виде светло-желтого твердого вещества (1,21 г, выход 99%).
1H-ЯМР (500 МГц, CDCl3) δ 8,40 (1H, с), 7,37 (2H, д), 7,31 (2H, д), 7,23-7,28 (3Н, м), 7,17-7,21 (2H, м), 3,40 (2H, т), 3,31 (2H, ушир. с), 2,50 (3Н, с), 2,46 (3Н, с), 2,42-2,57 (4H, м), 2,00-2,12 (1H, м), 1,75-1,87 (1H, м), 1,12-1,47 (9H, ушир. м).
ЖХ/МС (способ A) RT=8,64 мин, M+H+=573,13.
Стадия 2: метил-2-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-6-карбоксилат
К перемешанному раствору трет-бутил-(1-(4-(6-(бис(метилтио)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (140 мг, 0,24 ммоль) в безводном тетрагидрофуране (2,8 мл) добавляли раствор гидроксида натрия (96 мг, 2,4 ммоль) в метаноле (2,2 мл). Реакционную смесь нагревали до 50°C в атмосфере азота в течение 2 часов. После охлаждения до комнатной температуры смесь разбавляли 1M раствором HCl (3 мл) и экстрагировали введением в этилацетат (3×6 мл). Объединенные органические фазы промывали солевым раствором (3×6 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением целевого продукта в виде коричневого твердого вещества (126 мг, количественный выход). ЖХ/МС (способ A) RT=7,56 мин, M+H+=527,14.
Стадия 3: трет-бутил-(1-(4-(3-гидрокси-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору метил-2-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-6-карбоксилата (60 мг, 0,11 ммоль) в безводном этаноле (1500 мкл) добавляли гидразина моногидрат (9 мкл, 0,9 ммоль). Реакционную смесь нагревали до температуры возгонки в атмосфере азота в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали досуха в условиях пониженного давления, очищали методом хроматографии (силикагель Biotage, дихлорметан/метанол, элюирование градиентом 99/1→90/10) и суспендировали в диэтиловом эфире (1 мл) с получением целевого продукта в виде белого твердого вещества (19 мг, выход 33%).
1H-ЯМР (500 МГц, CD3OD) δ 7,94 (1H, с), 7,32 (2H, д), 7,21-7,29 (5H, м), 7,14-7,21 (2H, м), 3,18 (2H, т), 2,81 (2H, т), 2,31-2,50 (4H, м), 2,00-2,12 (1, ушир. м), 1,76-1,91 (1H, ушир. м), 1,09-1,48 (ушир. м).
ЖХ/МС (способ A) RT=5,57 мин, M+H+=509,17.
Стадия 4: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-3-ол
трет-Бутил-(1-(4-(3-гидрокси-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (19 мг, 0,037 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (3 мг, выход 17%).
1H-ЯМР (500 МГц, CD3OD) δ 8,05 (1H, с), 7,49 (2H, д), 7,44 (2H, д), 7,27-7,34 (3Н, м), 7,20-7,27 (2H, м), 3,25 (2H, т), 2,87 (2H, т), 2,72-2,82 (2H, м), 2,54-2,65 (2H, м), 2,18-2,30 (1H, м), 1,90-2,03 (1H, м).
ЖХ/МС (способ A) RT=3,40 мин, M+H+=409,19.
Пример 8: 7-(4-(1-аминоциклобутил)фенил)-3a-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-3(3aH)-он
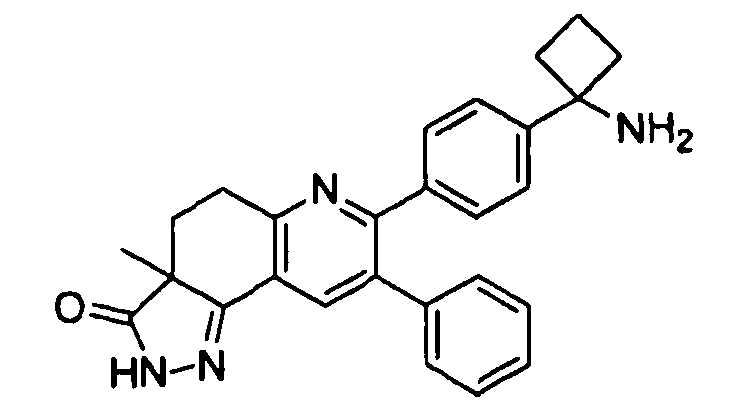
7-(4-(1-аминоциклобутил)фенил)-3a-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-3(3aH)-он
Стадия 1: метил-2-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-6-метил-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-6-карбоксилат
Раствор метил-2-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-6-карбоксилата (65 мг, 0,12 ммоль) в безводном THF (1,0 мл) охлаждали до 0°C в атмосфере азота, а затем по каплям добавляли раствор трет-бутоксида калия (1M в THF, 150 мкл, 0,15 ммоль). Смесь перемешивали при 0°C в течение 15 минут, а затем добавляли метилйодид (10 мкл, 0,16 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали еще в течение 4 часов. Смесь разбавляли этилацетатом (2,0 мл), промывали 1M раствором HCl (2,0 мл), солевым раствором (2×2,0 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением целевого продукта в виде коричневого твердого вещества (54 мг, 83% выход). ЖХ/МС (способ A) RT=7,92 мин, M+H+=541,18.
Стадия 2: трет-бутил-(1-(4-(3a-метил-3-оксо-8-фенил-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору метил-2-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-6-метил-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-6-карбоксилата (54 мг, 0,10 ммоль) в безводном этаноле (1300 мкл) добавляли гидразина моногидрат (8 мкл, 0,16 ммоль). Реакционную смесь нагревали до температуры возгонки в атмосфере азота в течение 5 часов, дополнительно добавляли гидразина моногидрат (40 мкл, 0,82 ммоль), и нагревали смесь с обратным холодильником в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали досуха в условиях пониженного давления, очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 88/12→0/100) с получением целевого продукта в виде желтого твердого вещества (12 мг, выход 23%).
1H-ЯМР (500 МГц, CDCl3) δ 8,72 (1H, с), 8,09 (1H, с), 7,34 (2H, д), 7,30 (2H, д), 7,23-7,28 (3Н, м), 7,16-7,22 (2H, м), 5,05 (1H, ушир. с), 3,32-3,44 (1H, м), 3,19-3,32 (1H, ушир. м), 2,41-2,60 (4H, ушир. м), 2,26-2,35 (1H, дд), 1,97-2,11 (2H, м), 1,74-1,86 (1 H, м), 1,45 (3H, с), 1,14-1,44 (9H, ушир. м).
ЖХ/МС (способ A) RT=6,60 мин, M+H+=523,17.
Стадия 3: 7-(4-(1-аминоциклобутил)фенил)-3a-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-3(3aH)-он
трет-Бутил-(1-(4-(3a-метил-3-оксо-8-фенил-3,3a,4,5-тетрагидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (12 мг, 0,023 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (9 мг, выход 60%).
1H-ЯМР (500 МГц, CD3OD) δ 8,15 (1H, с), 7,49 (2H, д), 7,44 (2H, д), 7,29-7,36 (3Н, м), 7,20-7,26 (2H, м), 3,35-3,45 (1H, м), 3,16-3,26 (1H, дд), 2,72-2,84 (2H, м), 2,54-2,66 (2H, м), 2,19-2,39 (2H, м), 1,92-2,09 (2H, м), 1,43 (3Н, с).
ЖХ/МС (способ A) RT=3,76 мин, M+H+=423,16.
Пример 9: 1-(4-(3-(метилтио)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
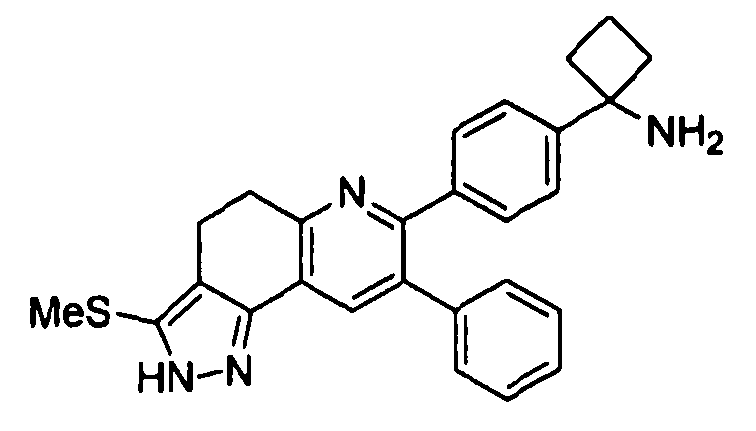
Стадия 1: трет-бутил-(1-(4-(3-(метилтиоV8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(6-(бис(метилтио)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (200 мг, 0,35 ммоль) в безводном этаноле (1,7 мл) добавляли гидразина моногидрат (25 мкл, 0,52 ммоль). Реакционную смесь нагревали до 80°C в атмосфере азота в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали досуха в условиях пониженного давления, очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 90/10→80/20) с получением целевого продукта в виде желтого твердого вещества (53 мг, выход 28%).
1H-ЯМР (500 МГц, CDCl3) δ 7,94 (1H, с), 7,16-7,24 (4H, м), 7,11-7,16 (3Н, м), 7,06-7,10 (2Н, м), 3,16 (2Н, т), 2,83 (2Н, т), 2,35 (3Н, с), 2,15-2,45 (4Н, ушир. м), 1,88-2,01 (1H, м), 1,64-1,77 (1H, м), 1,00-1,35 (9H, ушир. м).
ЖХ/МС (способ A) RT=6,94 мин, M+H+=539,15.
Стадия 2: 1-(4-(3-(метилтио)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(3-(метилтио)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (53 мг, 0,098 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и лиофилизировали с получением целевого продукта в виде не совсем белого твердого вещества (24 мг, выход 37%).
1H-ЯМР (500 МГц, CD3OD) δ 8,14 (1H, ушир. с), 7,48 (2H, д), 7,43 (2H, д), 7,27-7,34 (3H, м), 7,21-7,27 (2H, м), 3,26 (2H, т), 2,97 (2H, ушир. т), 2,72-2,82 (2H, м), 2,53-2,63 (2H, м), 2,49 (3H, с), 2,19-2,30 (1H, м), 1,91-2,02 (1H, м).
ЖХ/МС (способ A) RT=4,06 мин, M+H+=439,14.
Пример 10: 1-(4-(3-(метилсульфонил)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
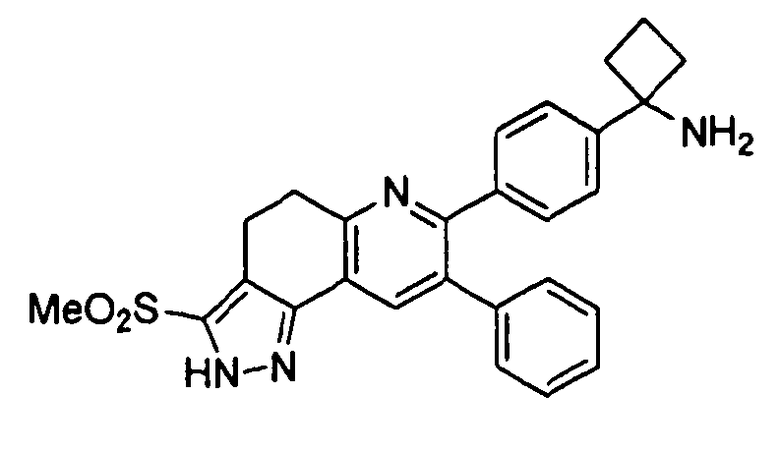
Стадия 1: трет-бутил-(1-(4-(3-(метилсульфонил)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-(метилтио)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамата (158 мг, 0,29 ммоль) в смеси THF (2 мл) и метанола (2 мл) по каплям добавляли раствор Oxone® (моноперсульфатное соединение, 1,08 г, 1,76 ммоль) в воде (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем гасили путем добавления по каплям насыщенного раствора NaHCO3 (4 мл) и экстрагировали введением в этилацетат (3×8 мл). Объединенные органические фазы промывали насыщенным раствором NaHCO3 (8 мл), солевым раствором (8 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 92/12→100/0) с получением целевого продукта в виде белого твердого вещества (44 мг, выход 26%).
1H-ЯМР (500 МГц, CDCl3) δ 7,85 (1H, с), 7,27 (2H, д), 7,17-7,25 (5H, м), 7,11-7,16 (2H, м), 3,24 (2H, т), 3,14 (2H, т), 3,15 (3Н, с), 2,18-2,51 (4H, ушир. м), 1,96-2,10 (1H, ушир. м), 1,68-1,80 (1H, ушир. м), 1,09-1,38 (9H, ушир. м).
ЖХ/МС (способ A) RT=6,59 мин, M+H+=571,11.
Стадия 2: 1-(4-(3-(метилсульфонил)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(3-(метилсульфонил)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (40 мг, 0,070 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и лиофилизировали с получением целевого продукта в виде не совсем белого твердого вещества (18 мг, выход 37%).
1H-ЯМР (500 МГц, CD3OD) δ 8,10 (1H, с), 7,50 (2H, д), 7,44 (2H, д), 7,31-7,35 (3Н, м), 7,24-7,29 (2H, м), 3,32 (2H, т), 3,28 (3Н, с), 3,24 (2H, т), 2,74-2,82 (2H, м), 2,54-2,64 (2H, м), 2,19-2,30 (1H, м), 1,92-2,03 (1H, м).
ЖХ/МС (способ A) RT=3,81 мин, M+H+=471,16.
Пример 11: 7-(4-(1-аминоциклобутил)фенил)-N-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-3-амин
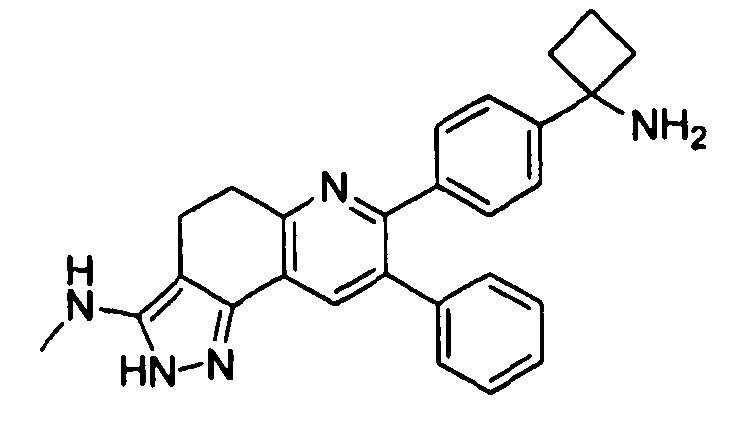
Стадия 1: трет-бутил-(1-(4-(3-(метиламино)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(6-(бис(метилтио)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (100 мг, 0,18 ммоль) в этаноле (873 мкл) добавляли метиламин (2M в THF, 87 мкл, 0,18 ммоль) и нагревали смесь до 80°C в атмосфере азота в течение 2 часов. Дополнительно добавляли метиламин (2M в THF, 873 мкл, 1,746 ммоль) и нагревали смесь до 80°C в атмосфере азота в течение 2 часов. Добавляли гидразина моногидрат (25,4 мкл, 0,52 ммоль) и нагревали смесь до 80°C в течение 18 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии (силикагель Biotage, DCM/MeOH, элюирование градиентом 100/0→90/10) с получением бесцветного твердого вещества (47 мг, выход 52%).
1H-ЯМР (500 МГц, CDCL3) δ 7,63 (1H, с), 7,32 (2H, д), 7,27 (2H, д), 7,16-7,22 (3Н, м), 7,09-7,16 (2H, м), 5,06 (1H, ушир. с), 3,10 (2H, т), 2,68 (3Н, с), 2,22-2,60 (6H, ушир. м), 1,97-2,09 (1H, м), 1,72-1,84 (1H, м), 1,03-1,53 (9H, ушир. м).
ЖХ/МС (способ A) RT=5,34 мин, M+H+=522,19.
Стадия 2: 7-(4-(1-аминоциклобутил)фенил)-N-метил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-3-амин
трет-Бутил-(1-(4-(3-(метиламино)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (47 мг, 0,090 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и лиофилизировали с получением целевого продукта в виде не совсем белого твердого вещества (31 мг, выход 53%).
1H-ЯМР (500 МГц, CD3OD) δ 8,08 (1H, с), 7,50 (2H, д), 7,44 (2H, д), 7,30-7,50 (3Н, м), 7,23-7,27 (2H, м), 3,30 (2H, т), 2,98 (3Н, с), 2,87 (2H, т), 2,73-2,81 (2H, м), 2,54-2,64 (2H, м), 2,19-2,31 (1H, м), 1,92-2,04 (1H, м).
ЖХ/МС (способ A) RT=3,28 мин, M+H+=422,18.
Пример 12: 2-((7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-3-ил)амино)этанол
Стадия 1: трет-бутил-(1-(4-(3-((2-гидроксиэтил)амино)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(6-(бис(метилтио)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (100 мг, 0,18 ммоль) в этаноле (873 мкл) добавляли этаноламин (10,6 мкл, 0,18 ммоль) и нагревали смесь до 80°C в атмосфере азота в течение 2 часов. Дополнительно добавляли этаноламин (106 мкл, 1,75 ммоль) и нагревали смесь до 80°C в атмосфере азота в течение 18 часов. Добавляли гидразина моногидрат (25,4 мкл, 0,52 ммоль) и нагревали смесь до 80°C в течение 3 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии (силикагель Biotage, DCM/MeOH, элюирование градиентом 100/0→90/10) с получением бесцветного твердого вещества (51 мг, выход 53%).
1H-ЯМР (500 МГц, CDCl3) δ 7,67 (1H, с), 7,18-7,28 (4H, ушир. м), 7,10-7,16 (3Н, м), 7,01-7,08 (2Н, м), 5,11 (1Н, ушир. с), 3,74 (2H, т), 3,35 (2H, т), 3,15 (2H, т), 2,66 (2H, т), 2,19-2,58 (4H, ушир. м), 1,95-2,08 (1H, м), 1,69-1,81 (1H, м), 1,03-1,49 (9H, ушир. м).
ЖХ/МС (способ A) RT=5,20 мин, M+H+=552,24.
Стадия 2: 2-((7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-3-ил)амино)этанол
трет-Бутил-(1-(4-(3-((2-гидроксиэтил)амино)-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (25 мг, 0,045 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и лиофилизировали с получением целевого продукта в виде не совсем белого твердого вещества (22 мг, выход 71%).
1H-ЯМР (500 МГц, CD3OD) δ 8,06 (1H, с), 7,50 (2H, д), 7,44 (2H, д), 7,29-7,34 (3Н, м), 7,22-7,28 (2H, м), 3,78 (2H, т), 3,41 (2H, т), 3,30 (2H, т), 2,90 (2H, т), 2,71-2,82 (2H, м), 2,54-2,66 (2H, м), 2,19-2,30 (1H, м), 1,91-2,03 (1H, м).
ЖХ/МС (способ A) RT=3,30 мин, M+H+=452,17.
Пример 13: 1-(4-(3-морфолино-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(3-морфолино-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(6-(бис(метилтио)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (100 мг, 0,18 ммоль) в этаноле (873 мкл) добавляли морфолин (76 мкл, 0,87 ммоль) и нагревали смесь до 80°C в атмосфере азота в течение 2 часов. Добавляли гидразина моногидрат (12,7 мкл, 0,26 ммоль) и нагревали смесь до 80°C в течение 1 часа. Дополнительно добавляли гидразина моногидрат (72 мкл, 1,48 ммоль) и нагревали смесь до 80ºC в течение 18 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии (силикагель Biotage, циклогексан/этилацетат, элюирование градиентом 92/2→100/0) с получением белого твердого вещества (30 мг, выход 30%).
1H-ЯМР (500 МГц, CDCl3) δ 7,67 (1H, с), 7,31 (2H, д), 7,23-7,28 (2H, м), 7,18-7,24 (3Н, м), 7,07-7,14 (2H, м), 4,95-5,44 (1H, ушир. м), 3,82 (4H, т), 3,24 (2H, т), 3,21 (4H, т), 2,91 (2H, т), 2,19-2,64 (4H, ушир. м), 1,97-2,09 (1H, м), 1,73-1,84 (1H, м), 1,10-1,46 (9H, ушир. м).
ЖХ/МС (способ A) RT=6,18 мин, M+H+=578,24.
Стадия 2: 1-(4-(3-морфолино-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(3-морфолино-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (30 мг, 0,052 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и лиофилизировали с получением целевого продукта в виде не совсем белого твердого вещества (23 мг, выход 63%).
1H-ЯМР (500 МГц, CD3OD) δ 8,08 (1H, с), 7,49 (2H, д), 7,44 (2H, д), 7,29-7,33 (3Н, м), 7,22-7,27 (2H, м), 3,86 (4H, т), 3,26 (2H, т), 3,22 (4H, т), 2,98 (2H, т), 2,73-2,82 (2H, м), 2,54-2,64 (2H, м), 2,19-2,30 (1H, м), 1,92-2,03 (1H, м).
ЖХ/МС (способ A) RT=3,74 мин, M+H+=478,23.
Пример 14: 7-(4-(1-аминоциклобутил)фенил)-2-метил-8-фенил-4,5-дигидро-3H-имидазо[4,5-f]хинолин-3-ол
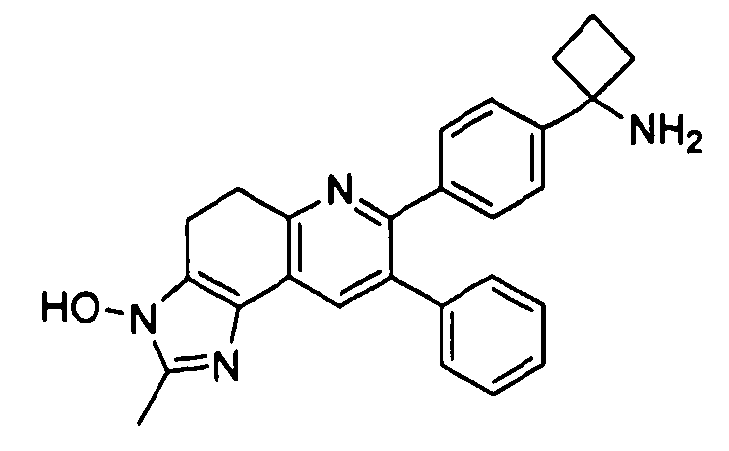
Стадия 1: трет-бутил-(1-(4-(6-(гидроксиимино)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (1,00 г, 2,134 ммоль) в безводном THF (30 мл) по каплям в течение 10 мин добавляли трет-бутоксид калия (1 М в THF, 2,56 мл, 2,56 ммоль) с получением темно-красного раствора. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 30 мин. К этой смеси по каплям в течение 5 мин добавляли изопентилнитрит (0,286 мл, 2,134 ммоль) с получением красного раствора. Реакционную смесь перемешивали при 50°C в атмосфере азота в течение 1 часа, затем оставляли охлаждаться до комнатной температуры и концентрировали досуха в условиях пониженного давления. Остаток повторно растворяли в воде (5 мл), подкисляли добавлением 1M раствора HCl (5 мл) и экстрагировали введением в этилацетат (3×10 мл). Объединенные органические фазы промывали водой (20 мл), солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 88/12→0/100) с получением целевого продукта в виде коричневого твердого вещества (436 мг, выход 41%).
1H-ЯМР (500 МГц, CDCl3) δ 8,40 (1H, с), 7,40 (2H, д), 7,32 (2H, д), 7,25-7,29 (3H, м), 7,18-7,22 (2H, м), 5,04 (1H, ушир. с), 3,38 (2H, т), 3,29 (2H, т), 2,26-2,59 (4H, ушир. м), 2,01-2,12 (1H, м), 1,76-1,86 (1H, м), 1,15-1,45 (9H, ушир. м).
ЖХ/МС (способ A) RT=6,74 мин, M+H+=498,13.
Стадия 2: трет-бутил-(1-(4-(3-гидрокси-2-метил-8-фенил-4,5-дигидро-3H-имидазо[4,5-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(6-(гидроксиимино)-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (300 мг, 0,603 ммоль) в этаноле (3,0 мл) добавляли ацетальдегид (68,1 мкл, 1,206 ммоль), а затем гидроксид аммония (329 мкл, 2,53 ммоль). Реакционную смесь нагревали при 80°C в атмосфере азота в течение 2 часов, затем оставляли охлаждаться до комнатной температуры и концентрировали досуха в условиях пониженного давления, и сушили с получением неочищенного продукта в виде коричневого твердого вещества (306 мг, выход 97%).
Порцию этого продукта массой 50 мг дополнительно очищали методом хроматографии (силикагель Biotage, DCM/MeOH, элюирование градиентом 100/0→50/50) с получением целевого продукта в виде коричневого твердого вещества (10 мг).
1H-ЯМР (500 МГц, CDCl3) δ 7,70 (1H, с), 7,15-7,24 (7H, м), 7,09-7,14 (2H, ушир. м), 3,19 (2H, т), 2,91 (2H, т), 2,20-2,52 (4H, ушир. м), 2,30 (3H, с), 1,93-2,06 (1H, м), 1,69-1,79 (1H, м), 1,06-1,45 (9H, ушир. м).
ЖХ/МС (способ A) RT=5,00 мин, M+H+=523,19.
Стадия 3: 7-(4-(1-аминоциклобутил)фенил)-2-метил-8-фенил-4,5-дигидро-3H-имидазо[4,5-f]хинолин-3-ол
трет-Бутил-(1-(4-(3-гидрокси-2-метил-8-фенил-4,5-дигидро-3H-имидазо[4,5-f]хинолин-7-ил)фенил)циклобутил)карбамат (10 мг, 0,019 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и лиофилизировали с получением целевого продукта в виде бледно-коричневого твердого вещества (10 мг, выход 80%).
1H-ЯМР (500 МГц, CD3OD) δ 7,92 (1H, с), 7,48 (2H, д), 7,43 (2H, д), 7,30-7,34 (3Н, м), 7,22-7,26 (2H, м), 3,42 (2H, т), 3,18 (2H, т), 2,72-2,81 (2H, м), 2,63 (3H, с), 2,54-2,64 (2H, м), 2,18-2,29 (1H, м), 1,91-2,02 (1H, м).
ЖХ/МС (способ A) RT=3,14 мин, M+H+=423,12.
Пример 15: 1-(4-(2-метил-8-фенил-4,5-дигидро-3H-имидазо[4,5-f]хинолин-7-ил)фенил)циклобутанамин
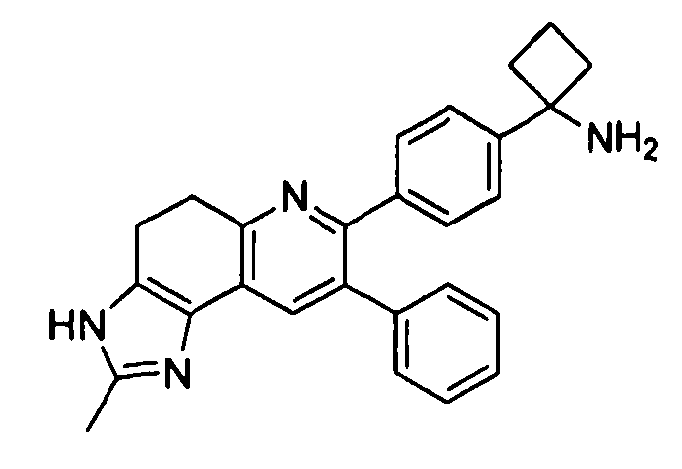
Стадия 1: трет-бутил-(1-(4-(2-метил-8-фенил-4,5-дигидро-3H-имидазо[4,5-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-гидрокси-2-метил-8-фенил-4,5-дигидро-3H-имидазо[4,5-f]хинолин-7-ил)фенил)циклобутил)карбамата (276 мг, 0,528 ммоль) в безводном DMF (1,4 мл) добавляли триэтилфосфит (181 мкл, 1,056 ммоль) и нагревали до 100°C в атмосфере азота в течение ночи. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли водой/солевым раствором (50/50, 10 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы промывали водой/солевым раствором (50/50, 2×10 мл), солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии (силикагель Biotage, DCM/MeOH, элюирование градиентом 100/0→90/10) с получением целевого продукта в виде бледно-коричневого твердого вещества (100 мг, выход 37%).
1H-ЯМР (500 МГц, CDCl3) δ 7,74 (1H, с), 7,28 (2H, д), 7,23 (2H, д), 7,13-7,17 (3Н, м), 7,06-7,11 (2H, м), 5,09 (1H, ушир. с), 3,25 (2H, т), 2,92 (2H, т), 2,16-2,59 (4H, ушир. м), 2,41 (3H, с), 1,95-2,08 (1H, м), 1,70-1,82 (1H, м), 1,05-1,47 (9H, ушир. м).
ЖХ/МС (способ A) RT=4,66 мин, M+H+=507,20.
Стадия 2: 1-(4-(2-метил-8-фенил-4,5-дигидро-3H-имидазо[4,5-f]хинолин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(2-метил-8-фенил-4,5-дигидро-3H-имидазо[4,5-f]хинолин-7-ил)фенил)циклобутил)карбамат (25 мг, 0,049 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и лиофилизировали с получением целевого продукта в виде не совсем белого твердого вещества (23 мг, выход 74%).
1H-ЯМР (500 МГц, CD3OD) δ 7,90 (1H, с), 7,49 (2H, д), 7,44 (2H, д), 7,31-7,35 (3Н, м), 7,23-7,27 (2H, м), 3,44 (2H, т), 3,20 (2H, т), 2,73-2,81 (2H, м), 2,72 (3H, с), 2,55-2,65 (2H, м), 2,19-2,30 (1H, м), 1,92-2,03 (1H, м).
ЖХ/МС (способ A) RT=3,02 мин, M+H+=407,14.
Пример 16: 1-(4-(9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-8-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-8-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-1-(4-(6-((диметиламино)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидронафталин-2-ил)фенил)циклобутил)карбамата (50 мг, 0,09 ммоль) в безводном этаноле (1000 мкл) добавляли раствор этоксида натрия (21% в этаноле, 150 мкл, 0,38 ммоль) и формамидина ацетат (20 мг, 0,19 ммоль). Реакционную смесь нагревали до 80°C в атмосфере азота в течение 2 часов. Смесь концентрировали досуха в условиях пониженного давления, очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 95/5→60/40) и суспендировали в диэтиловом эфире (1 мл) с получением целевого продукта в виде не совсем белого твердого вещества (12 мг, выход 26%).
1H ЯМР (500 МГц, CH3OD) 9,03 (с, 1H), 8,66 (с, 1H), 8,59 (с, 1H), 7,31 (ушир. с, 4H), 7,25 (дд, 3H), 7,21 (м, 2H), 3,23 (м, 2H), 3,16 (м, 2H), 2,31-2,45 (м, 4H), 1,92-1,97 (м, 1H), 1,72-1,74 (м, 1H), 1,2-1,5 (ушир. с, 9H).
ЖХ/МС (способ A): RT=7,64 мин, M+H+=505.
Стадия 2: 1-(4-(9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-8-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-8-ил)фенил)циклобутил)карбамат (12 мг, 0,023 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (8 мг, выход 70%).
1H ЯМР (500 МГц, CH3OD) 9,09 (с, 1H), 8,74 (с, 1H), 8,67 (с, 1H), 7,52 (д, 2H), 7,43 (д, 2H), 7,32 (м, 3H), 7,26 (м, 2H), 3,22 (м, 2H), 2,75 (м, 2H), 2,59 (м, 2H), 2,23 (м, 1H), 1,98 (м, 1H).
ЖХ/МС (способ A): RT=4,26 мин, M+2H+=406.
Пример 17: 8-(4-(1-аминоциклобутил)фенил)-9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-2-амин
Стадия 1: трет-бутил-(1-(4-(2-амино-9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-8-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-1-(4-(6-((диметиламино)метилен)-5-оксо-3-фенил-5,6,7,8-тетрагидронафталин-2-ил)фенил)циклобутил)карбамата (45 мг, 0,085 ммоль) в безводном этаноле (1500 мкл) добавляли раствор этоксида натрия (21% в этаноле, 130 мкл, 0,34 ммоль) и гуанидина нитрат (22 мг, 0,17 ммоль). Реакционную смесь нагревали до 80°C в атмосфере азота в течение 1 часа. Смесь концентрировали досуха в условиях пониженного давления, очищали методом хроматографии (силикагель Biotage, дихлорметан/метанол, элюирование градиентом 99/1→90/10) и суспендировали в диэтиловом эфире (1 мл) с получением целевого продукта в виде не совсем белого твердого вещества (10 мг, выход 23%).
1H ЯМР (500 МГц, d6-DMSO) 8,36 (с, 1H), 8,25 (с, 1H), 7,31 (м, 5H), 7,25 (м, 4H), 6,56 (ушир. с, 1H), 3,11 (т, 2H), 2,91 (т, 2H), 2,33 (ушир. м, 4H), 1,93-2,04 (м, 1H), 1,70-1,82 (м, 1H), 1,1-1,41 (ушир. с, 9Н).
ЖХ/МС (способ A): RT=6,23 мин, M+H+=520.
Стадия 2: 8-(4-(9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-2-амин
трет-Бутил-(1-(4-(2-амино-9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-8-ил)фенил)циклобутил)карбамат (10 мг, 0,019 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (2 мг, выход 20%).
1H ЯМР (500 МГц, d6-DMSO) 8,04 (ушир. с, 2H), 8,31 (с, 1H), 8,23 (с, 1H), 7,40 (д, 2H), 7,35 (д, 2H), 7,29 (м, 3H), 7,20 (м, 2H), 3,13 (т, 2H), 2,94 (т, 2H), 2,56 (м, 4H), 2,07-2,19 (ушир. м, 1H), 1,74-1,85 (ушир. м, 1H).
ЖХ/МС (способ A): RT=3,58 мин, M+H+=420.
Пример 18: 2-(4-(1-аминоциклобутил)фенил)-7,7-диметил-3-фенил-7,8-дигидрохинолин-5(6H)-он
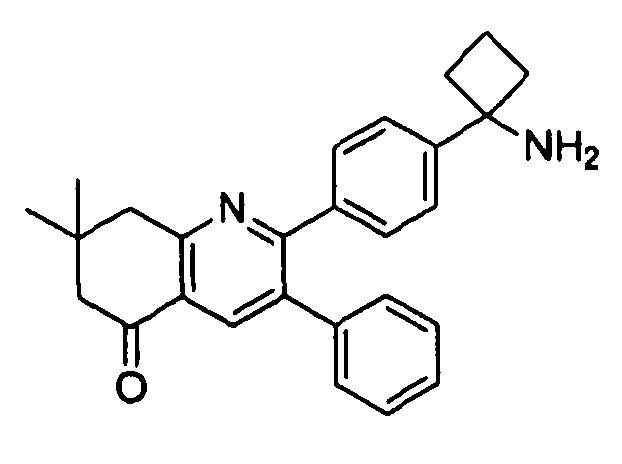
Стадия 1: трет-бутил-(1-(4-(7,7-диметил-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-(диметиламино)-2-фенилакрилоил)фенил)циклобутил)карбамата (166 мг, 0,40 ммоль) в уксусной кислоте (1 мл) в атмосфере азота добавляли молекулярные сита (10 мг) и 3-амино-5,5-диметилциклогекс-2-енон (82 мг, 0,59 ммоль). Полученную смесь нагревали при 100°C в течение 2 ч. После охлаждения до комнатной температуры, смесь распределяли между водой (10 мл) и дихлорметаном (10 мл). Слои разделяли, и экстрагировали водную фазу введением в дихлорметан (2×10 мл). Объединенные органические фазы промывали насыщенным раствором NaHCO3 (20 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→20% EtOAc в гексане) с получением указанного в заголовке соединения (80 мг, 41%).
1H ЯМР (500 МГц, CDCl3): 8,27 (1H, с), 7,37 (2H, д), 7,30 (2H, д), 7,25-7,24 (3H, м), 7,19-7,17 (2H, м), 5,06 (1H, ушир. с), 3,13 (2H, с), 2,59 (2H, с), 2,55-2,25 (4H, м), 2,09-2,01 (1H, м), 1,82-1,77 (1H, м), 1,44-1,15 (9H, ушир.), 1,17 (6H, с).
Стадия 2: 2-(4-(1-аминоциклобутил)фенил)-7,7-диметил-3-фенил-7,8-дигидрохинолин-5(6H)-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(7,7-диметил-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат (30 мг, 0,06 ммоль) с получением указанного в заголовке соединения в виде белого твердого вещества (27 мг, 87%). ЖХ/МС (способ A): RT=4,48 мин, M-NH2=380.
1H ЯМР (500 МГц, MeOD): 8,26 (1H, с), 7,50 (2H, д), 7,43 (2H, д), 7,31-7,29 (3H, м), 7,21-7,19 (2H, м), 3,15 (2H, с), 2,78-2,72 (2H, м), 2,66 (2H, с), 2,60-2,54 (2H, м), 2,27-2,18 (1H, м), 2,00-1,91 (1H, м), 1,17 (6H, с).
Пример 19: 1-(4-(4,4-диметил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
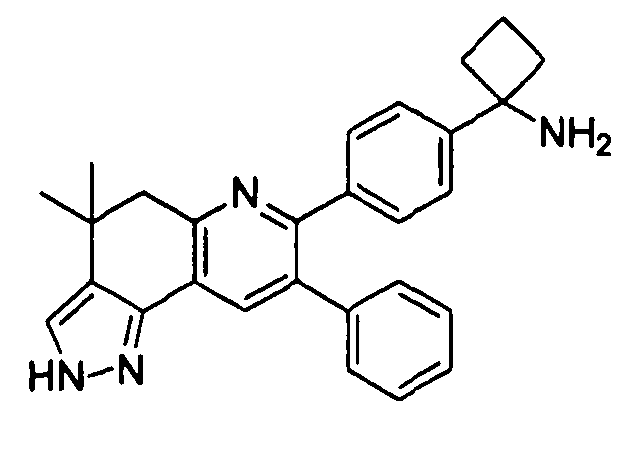
Стадия 1: трет-бутил-(1-(4-(6-((диметиламино)метилен)-7,7-диметил-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат
трет-Бутил-(1-(4-(7,7-диметил-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат (1 г, 2 ммоль) растворяли в 10 мл безводного N,N-диметилформамида диметилацеталя. Полученную смесь нагревали до 100°C в атмосфере азота в течение 48 часов. Анализ по методам ТСХ и ЖХ/МС обнаруживал полное расходование исходного вещества. Реакционную смесь концентрировали досуха в условиях пониженного давления. Неочищенный остаток суспендировали в н-гексане (10 мл) в течение 1 часа. Суспензию фильтровали, и сушили твердое вещество до постоянного веса с получением целевого продукта в виде светло-желтого твердого вещества (420 мг, выход 76%). ЖХ/МС (способ A): RT=8,51 мин, M+H+=552; RT=8,81 мин, M+H+=525 (трет-бутил-(1-(4-(6-(гидроксиметилен)-7,7-диметил-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамат).
Стадия 2: трет-бутил-(1-(4-(4,4-диметил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(6-((диметиламино)метилен)-7,7-диметил-5-оксо-3-фенил-5,6,7,8-тетрагидрохинолин-2-ил)фенил)циклобутил)карбамата (180 мг, 0,33 ммоль) в безводном этаноле (4 мл) добавляли гидразина гидрат (48 мкл, 1 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 2 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления, очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 99/1→60/40) и препаративной ЖХВД (способ A) с получением целевого продукта в виде белого твердого вещества (8 мг, выход 5%).
1H ЯМР (500 МГц, CDCl3) 8,12 (с, 1H), 7,27-7,48 (ушир. м, 5H), 7,25-7,18 (ушир. м, 5H), 5,03 (ушир. с, 1H), 3,16 (с, 2H), 2,23-2,68 (ушир. м, 4H), 1,97-2,12 (м, 1H), 1,75-1,88 (м, 1H), 1,11-1,57 (ушир. с, 9H+6H).
ЖХ/МС (способ A): RT=6,62 мин, M+H+=521.
Стадия 3: 1-(4-(4,4-диметил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(4,4-диметил-8-фенил-4,5-дигидро-2H-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутил)карбамат (8 мг, 0,015 ммоль) растворяли в TFA (0,5 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~1 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (4 мг, выход 50%).
1H ЯМР (500 МГц, CH3OD) 8,18 (с, 1H), 7,63 (с, 1H), 7,47 (д, 2H), 7,41 (д, 2H), 7,19-7,33 (ушир. м, 5H), 3,08 (с, 2H), 2,68-2,79 (ушир. м, 2H), 2,51-2,64 (ушир. м, 2H), 2,15-2,30 (ушир. м, 1H), 1,87-2,01 (ушир. м, 1H), 1,35 (с, 6H).
ЖХ/МС (способ A): RT=3,03 мин, M+H+=421.
Пример 20: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-5-он
Стадия 1: трет-бутил-(1-(4-(5-оксо-3-фенил-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-(диметиламино)-2-фенилакрилоил)фенил)циклобутил)карбамата (119 мг, 0,28 ммоль) в уксусной кислоте (1 мл) в атмосфере азота добавляли молекулярные сита (10 мг) и циклогептан-1,3-дион (36 мкл, 0,42 ммоль). Полученную смесь нагревали при 100°C в течение 2 ч. После охлаждения до комнатной температуры смесь распределяли между водой (10 мл) и дихлорметаном (10 мл). Слои разделяли, и экстрагировали водную фазу введением в дихлорметан (2×10 мл). Объединенные органические фазы промывали насыщенным раствором NaHCO3 (20 мл), солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→20% EtOAc в гексане) с получением указанного в заголовке соединения (32 мг, 24%).
1H ЯМР (500 МГц, CDCl3): 8,09 (1H, с), 7,38 (2H, д), 7,30 (2H, д), 7,25-7,24 (3H, м), 7,19-7,17 (2H, м), 5,04 (1H, ушир. с), 3,29 (2H, т), 2,87 (2H, т), 2,60-2,25 (4H, м), 2,07-2,01 (3H, м), 1,99-1,94 (2H, м), 1,84-1,75 (1H, м), 1,42-1,15 (9H, ушир.).
Стадия 2: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-5-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(5-оксо-3-фенил-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-2-ил)фенил)циклобутил)карбамат (14 мг, 0,03 ммоль) с получением указанного в заголовке соединения в виде белого твердого вещества (14 мг, 100%). ЖХ/МС (способ A): RT=4,26 мин, M-NH2=366.
1H ЯМР (500 МГц, MeOD): 8,06 (1H, с), 7,50 (2H, д), 7,42 (2H, д), 7,29-7,28 (3H, м), 7,20-7,18 (2H, м), 3,30 (2H, т), 2,89 (2H, т), 2,78-2,72 (2H, м), 2,59-2,54 (2H, м), 2,25-2,19 (1H, м), 2,06-2,01 (2H, м), 1,98-1,92 (3H, м).
Пример 21: 1-(4-(9-фенил-2,4,5,6-тетрагидропиразоло[3',4':3,4]циклогепта[1,2-b]пиридин-8-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(6-((диметиламино)метилен)-5-оксо-3-фенил-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-2-ил)фенил)циклобутил)карбамат
трет-Бутил-(1-(4-(5-оксо-3-фенил-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-2-ил)фенил)циклобутил)карбамат (270 мг, 0,56 ммоль) растворяли в 3 мл безводного Ν,Ν-диметилформамида диметилацеталя. Полученную смесь нагревали до 100°C в атмосфере азота в течение 18 часов. Анализ по методам ТСХ и ЖХ/МС обнаруживал полное расходование исходного вещества. Реакционную смесь концентрировали досуха в условиях пониженного давления. Неочищенный остаток суспендировали в н-гексане (5 мл) в течение 1 часа. Суспензию фильтровали, и сушили твердое вещество до постоянного веса с получением целевого продукта в виде светло-желтого твердого вещества (190 мг, выход 63%). ЖХ/МС (способ A): RT=6,55 мин, M+H+=538; RT=8,43 мин, M+H+=511 (трет-бутил-(1-(4-(6-(гидроксиметилен)-5-оксо-3-фенил-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-2-ил)циклобутил)карбамата).
Стадия 2: трет-бутил-(1-(4-(9-фенил-2,4,5,6-тетрагидропиразоло[3',4':3,4]циклогепта[1,2-b]пиридин-8-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(6-((диметиламино)метилен)-5-оксо-3-фенил-6,7,8,9-тетрагидро-5H-циклогепта[b]пиридин-2-ил)фенил)циклобутил)карбамата (90 мг, 0,17 ммоль) в безводном этаноле (2 мл) добавляли гидразина гидрат (25 мкл, 0,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 18 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления, очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 99/1→60/40) и препаративной ХЖВД (способ G, градиент 5→95% 0,1% FA/ACN в 0,1% FA/H2O, прогон 20 минут) с получением целевого продукта в виде белого твердого вещества (50 мг, выход 58%).
1H ЯМР (500 МГц, CDCl3) 7,49 (с, 1H), 7,38 (д, 2H), 7,29 (д, 2H), 7,21-7,25 (ушир. м, 5H+1Н), 3,25 (м, 2Н), 2,94 (т, 2H), 2,53-2,61 (ушир. м, 1H), 2,42-2,52 (м, 2H), 2,31-2,41 (ушир. м, 1H), 2,14-2,21 (м, 2H), 1,98-2,10 (м, 1H), 1,74-1,85 (м, 1H), 1,30-1,43 (ушир. с, 9H).
ЖХ/МС (способ A): RT=6,09 мин, M+H+=507.
Стадия 3: 1-(4-(9-фенил-2,4,5,6-тетрагидропиразоло[3',4':3,4]циклогепта[1,2-b]пиридин-8-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(9-фенил-2,4,5,6-тетрагидропиразоло[3',4':3,4]циклогепта[1,2-b]пиридин-8-ил)фенил)циклобутил)карбамат (10 мг, 0,019 ммоль) растворяли в TFA (0,5 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~1 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (2 мг, выход 20%).
1H ЯМР (500 МГц, CH3OD) 8,69 (с, 1H), 7,66 (с, 1H), 7,56 (д, 2H), 7,2 (д, 2H), 7,34 (м, 3H), 7,29 (м, 2H), 3,47-3,53 (м, 1H), 3,03 (м, 2H), 2,74-2,82 (ушир. м, 2H), 2,56-2,67 (ушир. м, 2H), 2,19-2,34 (ушир. м, 3H), 1,91-2,03 (м, 1H).
ЖХ/МС (способ A): RT=3,03 мин, M+H+=407.
Пример 22: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6H)-он
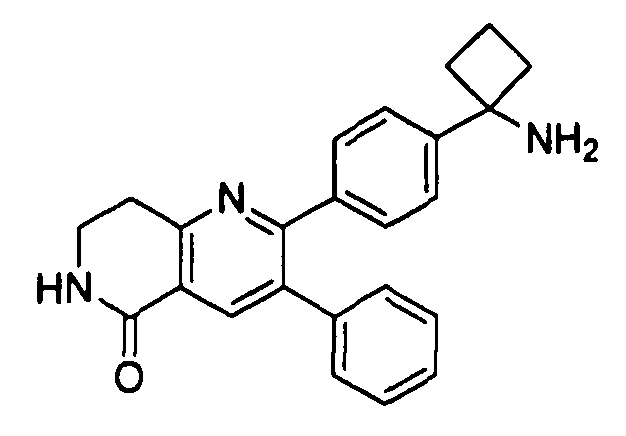
Стадия 1: трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-(диметиламино)-2-фенилакрилоил)фенил)циклобутил)карбамата (952 мг, 2,27 ммоль) в уксусной кислоте (10 мл) в атмосфере азота добавляли молекулярные сита (100 мг), ацетат аммония (524 мг, 6,80 ммоль) и пиперидин-2,4-дион (385 мг, 3,40 ммоль). Полученную смесь нагревали при 100°C в течение 2 ч. После охлаждения до комнатной температуры смесь распределяли между водой (100 мл) и дихлорметаном (100 мл). Слои разделяли, и экстрагировали водную фазу введением в дихлорметан (2×50 мл). Объединенные органические фазы промывали насыщенным раствором NaHCO3 (100 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→100% EtOAc в гексане) с получением указанного в заголовке соединения в виде желтого твердого вещества (353 мг, 33%).
1H ЯМР (500 МГц, MeOD): 8,28 (1H, с), 7,38-7,34 (4H, м), 7,32-7,29 (3H, м), 7,23-7,21 (2H, м), 3,69 (2H, т), 3,25 (2H, т), 2,51-2,40 (4H, м), 2,13-2,05 (1H, м), 1,91-1,84 (1H, м), 1,44-1,18 (9H, ушир.)
Стадия 2: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6H)-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат (10 мг, 0,021 ммоль) с получением указанного в заголовке соединения (7 мг, 69%). ЖХ/МС (способ A): RT=3,62 мин, M+H+=370.
1H ЯМР (500 МГц, MeOD) 8,30 (1H, с), 7,51 (2H, д), 7,45 (2H, д), 7,31 (3H, м), 7,22 (2H, м), 3,70 (2H, т), 3,27 (2H, т), 2,85-2,70 (2H, м), 2,65-2,50 (2H, м), 2,30-2,20 (1H, м), 2,05-1,90 (1H, м).
Пример 23: 2-(4-(1-аминоциклобутил)фенил)-6-метил-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6H)-он
Стадия 1: трет-бутил-(1-(4-(6-метил-5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамата (30 мг, 0,06 ммоль) в безводном DMF (1 мл) при 0°C в атмосфере азота добавляли гидрид натрия (3 мг, 0,07 ммоль). Спустя 1 ч при 0°C добавляли йодметан (17 мкл, 0,19 ммоль) и перемешивали полученную смесь при 0°C еще в течение 10 мин. Добавляли насыщенный раствор NH4Cl и экстрагировали смесь дихлорметаном (3×) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (градиент 0→50% EtOAc в гексане) с получением указанного в заголовке соединения (22 мг, 71%).
1H ЯМР (500 МГц, CDCl3): 8,35 (1H, с), 7,34 (2H, д), 7,29 (2H, д), 7,24-7,22 (3H, м), 7,19-7,17 (2H, м), 5,06 (1H, ушир. с), 3,70 (2H, т), 3,29 (2H, т), 3,19 (3H, с), 2,54-2,30 (4H, м), 2,08-2,00 (1H, м), 1,83-1,74 (1H, м), 1,44-1,18 (9H, ушир.).
Стадия 2: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6H)-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(6-метил-5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат (29 мг, 0,06 ммоль) с получением указанного в заголовке соединения (32 мг, 87%). ЖХ/МС (способ A): RT=3,76 мин, M-NH2=367.
1H ЯМР (500 МГц, MeOD): 8,29 (1H, с), 7,49 (2H, д), 7,42 (2H, д), 7,30-7,29 (3H, м), 7,21-7,19 (м, 2H), 3,80 (2H, т), 3,30 (2H, т), 3,20 (3H, с), 2,78-2,72 (м, 2H), 2,60-2,54 (м, 2H), 2,27-2,18 (м, 1H), 2,00-1,91 (м, 1H).
Пример 24: 2-(2-(4-(1-аминоциклобутил)фенил)-5-оксо-3-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)ацетонитрил
Стадия 1: трет-бутил-(1-(4-(6-(цианометил)-5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамата (31 мг, 0,07 ммоль) в безводном DMF (1 мл) при 0°C в атмосфере азота добавляли гидрид натрия (3 мг, 0,07 ммоль). Спустя 1 ч при 0°C добавляли 2-бромацетонитрил (14 мкл, 0,20 ммоль) и полученную смесь перемешивали при 0°C еще в течение 30 мин. Добавляли насыщенный раствор NH4Cl и экстрагировали смесь дихлорметаном (3×) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (градиент 0→40% EtOAc в гексане) с получением указанного в заголовке соединения (10 мг, 29%).
1H ЯМР (500 МГц, CDCl3): 8,36 (1H, с), 7,35 (2H, д), 7,30 (2H, д), 7,27-7,26 (3H, м), 7,20-7,18 (2H, м), 5,05 (1H, ушир. с), 4,59 (2H, с), 3,86 (2H, т), 3,40 (2Н, т), 2,55-2,25 (4Н, м), 2,10-2,02 (1H, м), 1,85-1,76 (1H, м), 1,44-1,18 (9H, ушир.).
Стадия 2: 2-(2-(4-(1-аминоциклобутил)фенил)-5-оксо-3-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)ацетонитрил
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(6-(цианометил)-5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат (17 мг, 0,03 ммоль) с получением указанного в заголовке соединения (15 мг, 71%). ЖХ/МС (способ A): RT=3,80 мин, M-NH2=392.
1H ЯМР (500 МГц, MeOD): 8,33 (1H, с), 7,50 (2H, д), 7,42 (2H, д), 7,31-7,29 (3H, м), 7,22-7,20 (м, 2H), 4,66 (2H, с), 3,92 (2H, т), 3,38 (2H, т), 2,78-2,72 (м, 2H), 2,60-2,54 (м, 2H), 2,27-2,18 (м, 1H), 2,00-1,91 (м, 1H).
Пример 25: 2-(4-(1-аминоциклобутил)фенил)-6-(2-(диметиламино)этил)-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6H)-он
Стадия 1: трет-бутил-(1-(4-(6-(2-(диметиламино)этил)-5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамата (25 мг, 0,05 ммоль) в безводном DMF (1 мл) при 0°C в атмосфере азота добавляли гидрид натрия (5 мг, 0,106 ммоль). Спустя 1 ч при 0°C добавляли раствор 2-хлор-N,N-диметилэтанамина гидрохлорида (9 мг, 0,064 ммоль) и карбоната калия (9 мг, 0,064) в 1 мл DMF и перемешивали полученную смесь при 40°C в течение 1 часа. Реакционную смесь охлаждали до 0°C, и добавляли при перемешивании насыщенный раствор NH4Cl, поддерживая температуру ниже 10°C. Полученную смесь экстрагировали дихлорметаном (3×) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом препаративной ЖХВД (метод G) с получением целевого продукта в виде белого твердого вещества (5 мг, выход 17%).
1H ЯМР (500 МГц, CDCl3): 8,35 (1H, с), 7,34 (2H, д), 7,29 (2H, д), 7,23-7,25 (3H, м), 7,17-7,20 (2H, м), 5,01 (1H, с), 3,77 (4H, м) 3,28 (2H, т), 2,63 (2H, т), 2,47 (2H+1H, м), 2,35 (6H, с), 2,24-2,32 (2H, м), 1,98-2,12 (1H, м), 1,67-1,89 (9H, ушир. с).
Стадия 2: 2-(4-(1-аминоциклобутил)фенил)-6-(2-(диметиламино)этил)-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6H)-он
трет-Бутил-(1-(4-(6-(2-(диметиламино)этил)-5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат (5 мг, 0,01 ммоль) растворяли в TFA (0,5 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~1 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (1,66 мг, выход 35%).
1H ЯМР (500 МГц, CH3OD) 8,22 (с, 1H), 7,50 (д, 2H), 7,39 (д, 2H), 7,33 (д, 2H), 7,18-7,22 (м, 3H), 7,08-7,13 (м, 2H), 3,90 (т, 2H), 3,76 (т, 2H), 3,39 (т, 2H), 3,24 (т, 2H), 2,94 (с, 6H), 2,60-2,70 (ушир. м, 2H), 2,43-2,52 (ушир. м, 2H), 2,09-2,19 (ушир. м, 1H), 1,80-1,95 (ушир. м, 1H).
ЖХ/МС (способ A): RT=3,03 мин, M+H+=441.
Пример 26: 8-(4-(1-аминоциклобутил)фенил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-3(2H)-он
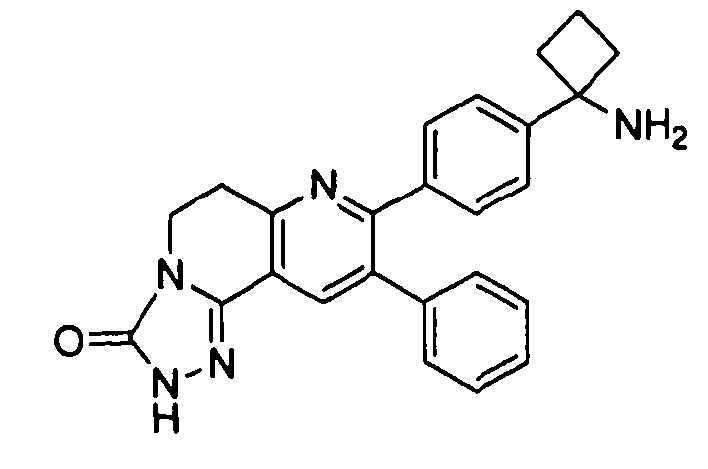
Стадия 1: трет-бутил-(1-(4-(3-фенил-5-тиоксо-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-оксо-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамата (223 мг, 0,47 ммоль) в толуоле (15 мл) в атмосфере азота добавляли реагент Лауссона (192 мг, 0,47 ммоль). Полученную смесь нагревали с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→1% MeOH в дихлорметане) с получением указанного в заголовке соединения в виде желтого твердого вещества (231 мг, количественный выход).
1H ЯМР (500 МГц, MeOD): 8,76 (1H, с), 7,37-7,33 (4H, м), 7,31-7,28 (3H, м), 7,22-7,20 (2H, м), 3,69 (2H, т), 3,25 (2H, т), 2,49-2,39 (4H, м), 2,15-2,05 (1H, м), 1,91-1,80 (1H, м), 1,45-1,15 (9H, ушир.).
Стадия 2: трет-бутил-(1-(4-(3-оксо-9-фенил-2,3,5,6-тетрагидро-[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-фенил-5-тиоксо-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамата (55 мг, 0,113 ммоль) в THF (7 мл) в атмосфере азота добавляли этилкарбозат (23,6 мг, 0,226 ммоль) и ацетат ртути (54,1 мг, 0,17 ммоль). Полученную смесь перемешивали при к.т. в течение 2 ч. Смесь концентрировали досуха в условиях пониженного давления и распределяли между DCM (15 мл) и водой (15 мл). Органическую фазу разделяли и концентрировали досуха в условиях пониженного давления. Полученный остаток суспендировали в толуоле (7 мл). Полученную смесь нагревали с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→4% MeOH в дихлорметане) с получением трет-бутил-(1-(4-(3-оксо-9-фенил-2,3,5,6-тетрагидро-[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутил)карбамата (32 мг, 56%). ЖХ/МС (способ A): RT=6,29 мин, M+1=510.
1H ЯМР (500 МГц, CDCl3): 9,83 (1H, с), 8,10 (1H, с), 7,29 (2H, д), 7,22 (2H, д), 7,19-7,18 (3H, м), 7,13-7,11 (2H, м), 5,1 (1H, ушир.), 4,01 (2H, т), 3,36 (2H, т), 2,43-2,28 (4H, м), 2,02-1,97 (1H, м), 1,76-1,70 (1H, м), 1,30-1,14 (9H, ушир.).
Стадия 3: 8-(4-(1-аминоциклобутил)фенил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-3(2H)-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(3-оксо-9-фенил-2,3,5,6-тетрагидро-[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутил)карбамат (10 мг, 0,019 ммоль) с получением указанного в заголовке соединения (6 мг, 58%). ЖХ/МС (способ A): RT=3,51 мин, M+2=411,2.
1H ЯМР (500 МГц, MeOD): 8,23 (1H, с), 7,52 (2H, д), 7,43 (2H, д), 7,34-7,32 (3H, м), 7,26-7,24 (2H, м), 4,09 (2H, т), 3,46 (2H, т), 2,81-2,75 (2H, м), 2,62-2,56 (2H, м), 2,28-2,22 (1H, м), 2,01-1,95 (1H, м).
Пример 27: 1-(4-(6-этил-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(6-этил-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-фенил-5-тиоксо-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамата (41 мг, 0,08 ммоль) в безводном этаноле (1 мл) в атмосфере азота добавляли никель Ренея (1 мл). Полученную смесь нагревали с обратным холодильником в течение 4 ч. После охлаждения до комнатной температуры, смесь фильтровали на целите (несколько раз промывали этанолом) и концентрировали досуха в условиях пониженного давления с получением указанного в заголовке соединения (28 мг, 74%).
1H ЯМР (500 МГц, CDCl3): 7,29 (1H, с), 7,22-7,18 (4H, м), 7,15-7,14 (3H, м), 7,07-7,06 (2H, м), 4,98 (1H, ушир. с), 3,64 (2H, с), 3,09 (2H, т), 2,83 (2H, т), 2,58 (2H, кв), 2,50-2,20 (4H, м), 2,00-1,91 (1H, м), 1,74-1,66 (1H, м), 1,40-1,10 (9H, ушир.), 1,15 (3H, т).
Стадия 2: 1-(4-(6-этил-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутанамин
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(6-этил-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат (14 мг, 0,03 ммоль) с получением указанного в заголовке соединения (18 мг, количественный выход). ЖХ/МС (способ A): RT=0,44 мин, M-NH2=367.
1H ЯМР (500 МГц, MeOD) 7,76 (1H, с), 7,45 (2H, д), 7,42 (2H, д), 7,30-7,29 (3H, м), 7,19-7,18 (2H, м), 3,47 (2H, кв), 3,46 (2H, т), 3,39 (2H, ушир. с), 3,32 (2H, м), 2,77-2,71 (2H, м), 2,60-2,54 (2H, м), 2,27-2,18 (1H, м), 1,99-1,90 (1H, м), 1,50 (3H, т).
Пример 28: 1-(4-(9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(5-гидразоно-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-фенил-5-тиоксо-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамата (127 мг, 0,26 ммоль) в THF (2 мл) в атмосфере азота добавляли гидразин·H2O (63 мкл, 1,31 ммоль) и ацетат ртути (125 мг, 0,39 ммоль) с получением черного раствора. Полученную смесь перемешивали при комнатной температуре в течение 2 ч. После охлаждения до комнатной температуры, смесь фильтровали на целите (промывали дихлорметаном) и концентрировали досуха в условиях пониженного давления. Полученное темно-желтое масло использовали непосредственно на следующей стадии без дополнительной очистки. ЖХ/МС (способ A): RT=4,77 мин, M+H+=484.
Стадия 2: трет-бутил-(1-(4-(9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутил)карбамат
Раствор трет-бутил-(1-(4-(5-гидразоно-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамата (40 мг, 0,08 ммоль) в триэтилортоформиате (1 мл) нагревали при 150°C в течение 1,5 ч в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом препаративной ЖХВД (метод G, градиент 5→95% 0,1% FA/ACN в 0,1% FA/H2O) с получением указанного в заголовке соединения (9 мг, 22%).
1H ЯМР (500 МГц, CDCl3): 8,47 (1H, с), 8,29 (1H, с), 7,37 (2H, д), 7,31 (2H, д), 7,28-7,26 (3H, м), 7,23-7,21 (2H, м), 5,07 (1H, ушир. с), 4,42 (2H, т), 3,50 (2H, т), 2,55-2,25 (4H, м), 2,10-2,02 (1H, м), 1,85-1,76 (1H, м), 1,45-1,20 (9H, ушир.).
Стадия 3: 1-(4-(9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутанамин
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутил)карбамат (9 мг, 0,02 ммоль) с получением указанного в заголовке соединения (10 мг, количественный выход). ЖХ/МС (способ A): RT=3,53 мин, M-NH2=377.
1H ЯМР (500 МГц, MeOD) 8,70 (1H, с), 8,41 (1H, с), 7,52 (2H, д), 7,43 (2H, д), 7,33-7,31 (3H, м), 7,29-7,27 (2H, м), 4,57 (2H, т), 3,51 (2H, т), 2,79-2,73 (2H, м), 2,60-2,54 (2H, м), 2,27-2,20 (1H, м), 1,99-1,92 (1H, м).
Пример 29: 1-(4-(3-(1-метил-1H-имидазол-4-ил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(3-(1-метил-1H-имидазол-4-ил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-гидразоно-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутил)карбамата (22 мг, 0,04 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли EDCI (11 мг, 0,06 ммоль), HOBt·H2O (8 мг, 0,06 ммоль) и 1-метил-1H-имидазол-4-карбоновую кислоту (8 мг, 0,06 ммоль). Реакционную смесь нагревали при 60°C в течение ночи. После охлаждения до комнатной температуры, смесь распределяли между насыщенным раствором NaHCO3 и этилацетатом. Слои разделяли, органическую фазу промывали водой и солевым раствором, сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом препаративной ЖХВД (метод G, градиент 5→95% 0,1% FA/ACN в 0,1% FA/H2O) с получением указанного в заголовке соединения (1 мг, 5%).
1H ЯМР (500 МГц, CDCl3): 8,49 (1H, с), 7,74 (1H, с), 7,54 (1H, с), 7,38 (2H, д), 7,31 (2H, д), 7,28-7,23 (5H, м), 5,01 (1H, ушир. с), 4,94 (2H, т), 3,81 (3H, с), 3,49 (2H, т), 2,60-2,30 (4H, м), 2,10-2,01 (1H, м), 1,85-1,76 (1H, м), 1,45-1,20 (9H, ушир.).
Стадия 2: 1-(4-(3-(1-метил-1H-имидазол-4-ил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутанамин
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(3-(1-метил-1H-имидазол-4-ил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутил)карбамат (9 мг, 0,02 ммоль) с получением указанного в заголовке соединения (10 мг, количественный выход). ЖХ/МС (способ A): RT=3,53 мин, M-NH2=377.
1H ЯМР (500 МГц, MeOD): 8,43 (1H, с), 7,99 (1H, ушир. с), 7,91 (1H, ушир. с), 7,52 (2H, д), 7,44 (2H, д), 7,33-7,27 (5H, м), 3,89 (3H, с), 3,54 (2H, т), 3,32 (2H, т), 2,79-2,73 (2H, м), 2,61-2,55 (2H, м), 2,27-2,20 (1H, м), 2,00-1,92 (1H,м).
Пример 30: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
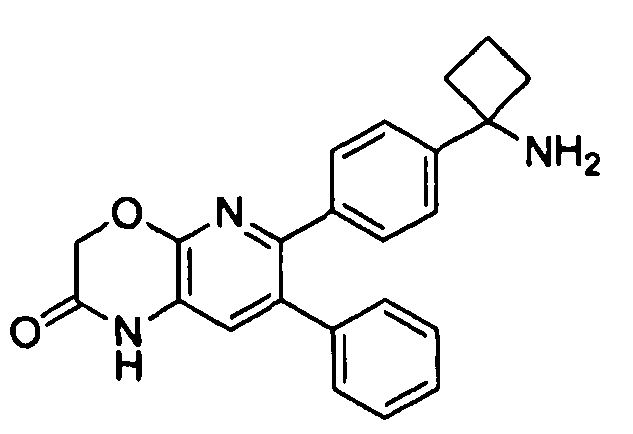
Стадия 1: трет-бутил-(1-(4-(5-циан-6-гидрокси-3-фенилпиридин-2-ил)фенил)циклобутил)карбамат
К суспензии гидрида натрия (3,58 г, 0,089 моль) в DMF (150 мл) при 0°C добавляли раствор 2-цианоацетамида (3,48 г, 0,041 моль) и (E)-трет-бутил-(1-(4-(3-(диметиламино)-2-фенилакрилоил)фенил)циклобутил)карбамата (15 г, 0,0357 моль) в метаноле (5,5 мл) и DMF (150 мл). Полученную смесь нагревали при 40°C в течение 16 ч. После охлаждения до комнатной температуры, смесь медленно добавляли к раствору уксусной кислоты (50 мл) в воде (300 мл). Полученный осадок фильтровали, и распределяли осадок на фильтре между водой (250 мл) и дихлорметаном (150 мл). Слои разделяли, и экстрагировали водную фазу введением в дихлорметан (150 мл). Объединенные органические фазы промывали водой (200 мл) и солевым раствором (150 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением указанного в заголовке соединения в виде желтого твердого вещества (14,6 г, 93%). ЖХ/МС (способ A): RT=6,59 мин, M+1=442.
1H ЯМР (500 МГц, CDCl3): 7,89 (1H, с), 7,37-7,32 (2H, м), 7,22-7,16 (5H, м), 7,03-6,98 (2H, м), 2,50-2,37 (4H, м), 2,06-1,97 (1H, м), 1,82-1,77 (1H, м), 1,45-1,05 (9H, ушир.).
Стадия 2: трет-бутил-(1-(4-(5-карбамоил-6-гидрокси-3-фенилпиридин-2-ил)фенил)циклобутил)карбамат
К суспензии трет-бутил-(1-(4-(5-циано-6-гидрокси-3-фенилпиридин-2-ил)фенил)циклобутил)карбамата (14,6 г, 3,58 г, 0,033 моль) в этаноле (112 мл) и воде (112 мл) при комнатной температуре добавляли раствор гидроксида натрия (6 M, 54,4 мл) и пероксид водорода (30%, 21,9 мл). Полученную смесь нагревали при 50°C в течение 16 ч. После охлаждения до 0°C, к смеси добавляли уксусную кислоту для корректировки значения pH до 4. Полученный осадок фильтровали, и сушили осадок на фильтре с получением указанного в заголовке соединения в виде белого твердого вещества (10,8 г, 72%). ЖХ/МС (способ A): RT=5,88 мин, M+1=460.
1H ЯМР (500 МГц, DMSO-d6): 9,65 (1H, ушир.), 8,10 (1H, с), 7,25-7,05 (7H, м), 6,98 (2H, м), 2,45-2,25 (4H, м), 2,0-1,95 (1H, м), 1,79-1,75 (1H, м), 1,3-1,0 (9H, ушир.).
Стадия 3: трет-бутил-(1-(4-(5-амино-6-гидрокси-3-фенилпиридин-2-ил)фенил)циклобутил)карбамат
К раствору брома (5,99 г, 0,037 моль) в воде (350 мл) добавляли раствор гидроксида натрия (15,02 г, 0,37 моль) в воде (350 мл). Полученный раствор перемешивали при 0°C в течение 10 мин. К этому раствору добавляли трет-бутил-(1-(4-(5-карбамоил-6-гидрокси-3-фенилпиридин-2-ил)фенил)циклобутил)карбамат (10,8 г, 0,0235 моль). Полученную смесь нагревали при 65°C в течение 24 ч. После охлаждения до комнатной температуры, смесь экстрагировали этилацетатом (200 мл). Слои разделяли, и экстрагировали водную фазу введением в этилацетат (2×200 мл). Объединенные органические фазы промывали водой (400 мл) и солевым раствором (300 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→5% метанол в DCM) с получением указанного в заголовке соединения (1,8 г, 18%). ЖХ/МС (способ A): RT=6,13 мин, M+1=432.
1H ЯМР (500 МГц, CDCl3): 7,45-6,9 (10H, м), 7,37 (2H, д), 5,0 (1H, ушир.), 2,4-2,1 (4H, м), 2,09-1,95 (1H, м), 1,85-1,7 (1H, м), 1,4-1,0 (9H, ушир.).
Стадия 4: трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-амино-6-гидрокси-3-фенилпиридин-2-ил)фенил)циклобутил)карбамата (1,8 г, 4,17 ммоль), DIPEA (3,63 мл, 20 ммоль) в DCM (100 мл) при 0°C добавляли раствор хлорацетилхлорида (0,465 мл) в DCM (20 мл). Полученную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь концентрировали досуха в условиях пониженного давления. К остатку добавляли MIBK (120 мл) и насыщенный бикарбонат натрия (120 мл). Полученную смесь нагревали при 115°C в течение 16 ч. После охлаждения до комнатной температуры MIBK удаляли в условиях пониженного давления. Остаток экстрагировали этилацетатом (75 мл). Слои разделяли, и экстрагировали водную фазу введением в этилацетат (2×50 мл). Объединенные органические фазы промывали водой (150 мл) и солевым раствором (150 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→2% метанол в DCM) с получением указанного в заголовке соединения (0,82 г, 42%). ЖХ/МС (способ A): RT=6,55 мин, M+1=472.
1H ЯМР (500 МГц, CDCl3): 7,3-7,0 (10H, м), 4,96 (1H, ушир.), 4,82 (2H, с), 2,55-2,25 (4H, м), 2,1-1,9 (1H, м), 1,8-1,65 (1H, м), 1,35-1,1 (9H, ушир.).
Стадия 5: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (8 мг, 0,017 ммоль) с получением указанного в заголовке соединения (6 мг, 73%). ЖХ/МС (способ A): RT=3,59 мин, M+1=373.
1H ЯМР (500 МГц, MeOD): 7,40 (2H, д), 7,37 (2H, д), 7,33 (1H, с), 7,32-7,28 (3H, м), 7,19-7,17 (2H, м), 4,93 (2H, с), 2,78-2,72 (2H, м), 2,60-2,54 (2H, м), 2,27-2,19 (1H, м), 2,00-1,91 (1H, м).
Пример 31: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Стадия 1: трет-бутил-(1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (60 мг, 0,13 ммоль) в безводном DMF (1 мл) при 0°C в атмосфере азота добавляли гидрид натрия (6 мг, 0,14 ммоль). Спустя 1 ч при 0°C, добавляли йодметан (9 мкл, 0,14 ммоль) и перемешивали полученную смесь при 0°C еще в течение 10 мин. Добавляли насыщенный раствор NH4Cl и экстрагировали смесь дихлорметаном (3×) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→30% EtOAc в циклогексане) с получением указанного в заголовке соединения в виде белого твердого вещества (38 мг, 60%).
1H ЯМР (500 МГц, CDCl3): 7,31-7,24 (8H, м), 7,21-7,19 (2H, м), 5,01 (1H, ушир. с), 4,90 (2H, с), 3,39 (3H, с), 2,55-2,25 (4H, м), 2,09-2,01 (1H, м), 1,84-1,76 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (37 мг, 0,08 ммоль) с получением указанного в заголовке соединения (25 мг, 78%). ЖХ/МС (способ A): RT=3,90 мин, M+2H+=387.
1H ЯМР (500 МГц, MeOD): 7,54 (1H, с), 7,40 (2H, д), 7,37 (2H, д), 7,29-7,27 (3H, м), 7,23-7,21 (м, 2H), 4,95 (2H, с), 3,42 (3H, с), 2,77-2,71 (м, 2H), 2,59-2,53 (м, 2H), 2,26-2,17 (м, 1H), 1,99-1,90 (м, 1H).
Пример 32: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(проп-2-ин-1-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Стадия 1: трет-бутил-(1-(4-(2-оксо-7-фенил-1-(проп-2-ин-1-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (23 мг, 0,05 ммоль) в безводном DMF (1 мл) при 0°C в атмосфере азота добавляли гидрид натрия (3 мг, 0,05 ммоль). Спустя 1 ч при 0°C добавляли пропаргилбромид (16 мкл, 0,15 ммоль) и перемешивали полученную смесь при 0°C еще в течение 1 ч. Добавляли насыщенный раствор NH4Cl и экстрагировали смесь дихлорметаном (3×) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (градиент 0→30% EtOAc в циклогексане) с получением указанного в заголовке соединения (17 мг, 68%).
1H ЯМР (500 МГц, CDCl3): 7,50 (1H, с), 7,31-7,24 (7H, м), 7,22-7,20 (2H, м), 5,01 (1H, ушир. с), 4,92 (2H, с), 4,73 (2H, д), 2,55-2,25 (4H, м), 2,32 (1H, т), 2,09-2,01 (1H, м), 1,85-1,76 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(проп-2-ин-1-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(2-оксо-7-фенил-1-(проп-2-ин-1-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (6 мг, 0,01 ммоль) с получением указанного в заголовке соединения (3 мг, 40%). ЖХ/МС (способ A): RT=4,12 мин, M+2H+=411.
1H ЯМР (500 МГц, MeOD): 7,69 (1H, с), 7,41 (2H, д), 7,38 (2H, д), 7,30-7,29 (3H, м), 7,23-7,21 (м, 2H), 4,98 (2H, с), 4,85 (2H, с), 2,80 (1H, т), 2,77-2,71 (м, 2H), 2,59-2,53 (м, 2H), 2,26-2,17 (м, 1H), 1,97-1,92 (м, 1H).
Пример 33: 2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
Стадия 1: трет-бутил-(1-(4-(1-(цианометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (14 мг, 0,03 ммоль) в безводном DMF (1 мл) при 0°C в атмосфере азота добавляли гидрид натрия (2 мг, 0,05 ммоль). Спустя 1 ч при 0°C добавляли 2-бромацетонитрил (7 мкл, 0,09 ммоль) и перемешивали полученную смесь при комнатной температуре еще в течение 4 ч. Добавляли воду и экстрагировали смесь этилацетатом (3×). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (градиент 0→40% EtOAc в гексане) с получением указанного в заголовке соединения (6 мг, 40%).
1H ЯМР (500 МГц, CDCl3): 7,36 (1H, с), 7,30-7,26 (7H, м), 7,22-7,20 (2H, м), 5,01 (1H, ушир. с), 4,96 (2H, с), 4,88 (2H, с), 2,55-2,25 (4H, м), 2,10-2,02 (1H, м), 1,85-1,76 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(1-(цианометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (6 мг, 0,02 ммоль) с получением указанного в заголовке соединения (6 мг, количественный выход). ЖХ/МС (способ A): RT=3,95 мин, Μ+2Η+=412.
1H ЯМР (500 МГц, MeOD): 7,69 (1H, с), 7,43 (2H, д), 7,38 (2H, д), 7,31-7,30 (3H, м), 7,25-7,24 (м, 2H), 5,13 (2H, с), 5,03 (2H, с), 2,77-2,72 (м, 2H), 2,59-2,53 (м, 2H), 2,26-2,17 (м, 1H), 1,99-1,92 (м, 1H).
Пример 34: 6-(4-(1-аминоциклобутил)фенил)-1-(2-(диметиламино)этил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
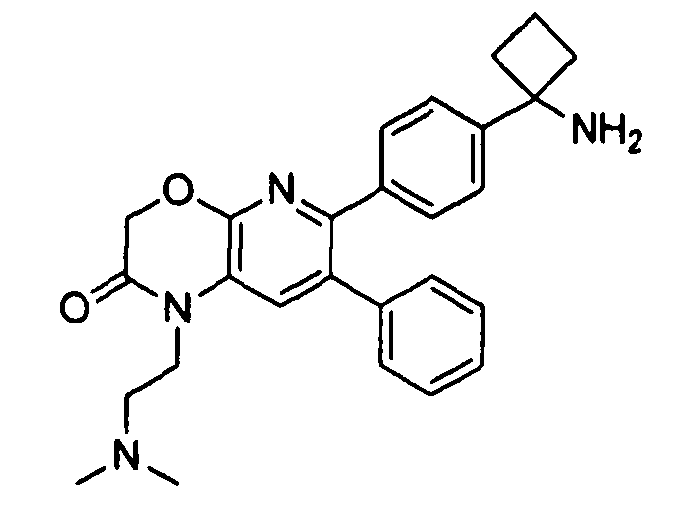
Стадия 1: трет-бутил-(1-(4-(1-(2-(диметиламино)этил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (19 мг, 0,04 ммоль) в безводном DMF (1 мл) при 0°C в атмосфере азота добавляли гидрид натрия (8 мг, 0,18 ммоль). Спустя 1 ч при 0°C добавляли 2-хлор-N,N-диметилэтанамина гидрохлорид (18 мг, 0,12 ммоль) и перемешивали полученную смесь при 40°C еще в течение 5 ч. Добавляли воду и экстрагировали смесь этилацетатом (3×). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (градиент 0→3% MeOH в дихлорметане) с получением указанного в заголовке соединения (7 мг, 32%).
1H ЯМР (500 МГц, CDCl3): 7,38 (1H, с), 7,31-7,24 (7H, м), 7,20-7,18 (2H, м), 5,01 (1H, ушир. с), 4,88 (2H, с), 4,08 (2H, т), 2,60 (2H, т), 2,55-2,25 (4H, м), 2,33 (6H, с), 2,09-2,01 (1H, м), 1,85-1,76 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(2-(диметиламино)этил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(1-(2-(диметиламино)этил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (7 мг, 0,01 ммоль) с получением указанного в заголовке соединения (7 мг, количественный выход). ЖХ/МС (способ A): RT=2,92 мин, M+H+=443.
1H ЯМР (500 МГц, MeOD): 7,63 (1H, с), 7,41 (2H, д), 7,38 (2H, д), 7,31-7,29 (3H, м), 7,25-7,23 (м, 2H), 5,00 (2H, с), 4,45 (2H, т), 3,48 (2H, т), 3,02 (6H, с), 2,77-2,72 (м, 2H), 2,59-2,53 (м, 2H), 2,25-2,19 (м, 1H), 1,99-1,92 (м, 1H).
Пример 35: 2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетамид
Стадия 1: трет-бутил-(1-(4-(1-(2-амино-2-оксоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (19 мг, 0,04 ммоль) в безводном DMF (1 мл) при комнатной температуре в атмосфере азота добавляли карбонат калия (12 мг, 0,08 ммоль) и 2-хлорацетамид (12 мг, 0,12 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (градиент 0→80% AcOEt в гексане) с получением указанного в заголовке соединения (14 мг, 67%).
1H ЯМР (500 МГц, CDCl3): 7,32 (1H, с), 7,28-7,23 (7H, м), 7,16-7,14 (2H, м), 6,22 (1H, ушир. с), 5,65 (1H, ушир. с), 5,09 (1H, ушир. с), 4,94 (2H, с), 4,52 (2H, с), 2,55-2,25 (4H, м), 2,09-2,01 (1H, м), 1,85-1,76 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетамид
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(1-(2-амино-2-оксоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (10 мг, 0,02 ммоль) с получением указанного в заголовке соединения (10 мг, количественный выход). ЖХ/МС (способ A): RT=3,43 мин, M-NH2=412.
1H ЯМР (500 МГц, MeOD): 7,39 (2H, д), 7,36 (2H, д), 7,34 (1H, с), 7,28-7,26 (3H, м), 7,18-7,17 (м, 2H), 5,01 (2H, с), 4,72 (2H, с), 2,76-2,70 (м, 2H), 2,57-2,51 (м, 2H), 2,25-2,16 (м, 1H), 1,98-1,89 (м, 1H).
Пример 36: 6-(4-(1-аминоциклобутил)фенил)-1-(2-оксопропил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он

Стадия 1: трет-бутил-(1-(4-(1-(2-оксо-1-(2-оксопропил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-1-(2-оксипропил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (60 мг, 0,13 ммоль) в безводном DMF (2,5 мл) при 0°C в атмосфере азота добавляли гидрид натрия (5 мг, 0,14 ммоль). Спустя 1 ч при 0°C добавляли хлорацетон (11 мкл, 0,14 ммоль) и перемешивали полученную смесь при 0°C в течение 2 часов. При перемешивании добавляли насыщенный раствор NH4Cl, поддерживая температуру ниже 10°C. Полученную смесь экстрагировали дихлорметаном (3×) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии (силикагель Biotage, гексан/этилацетат, элюирование градиентом 100/0→55/45) с получением целевого продукта в виде белого твердого вещества (41 мг, выход 60%).
1H ЯМР (500 МГц, CDCl3): 7,12-7,28 (9H, м), 6,82 (1H, с), 4,95 (2H, с), 4,54 (2H, с), 2,36-2,52 (4H, м), 2,23 (3H, с), 1,98-2,12 (1H, м), 1,69-1,75 (1H, м), 1,47 (9H, ушир. с).
ЖХ/МС (способ A): RT=6,87 мин, M+H+=528.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(2-оксопропил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-(1-(4-(1-(2-оксо-1-(2-оксопропил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (5 мг, 0,009 ммоль) растворяли в TFA (0,5 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~1 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (0,5 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (2,2 мг, выход 46%).
1H ЯМР (500 МГц, CH3OD): 7,15-7,50 (м, 10H), 5,01 (с, 2H), 4,98 (с, 2H), 2,71-2,81 (м, 2H), 2,53-2,63 (м, 2H), 2,30 (с, 3H), 2,18-2,27 (ушир. м, 1H), 1,92-2,03 (ушир. м, 1H).
ЖХ/МС (способ A): RT=3,03 мин, M+H+=428.
Пример 37: 1-(4-(7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (143 мг, 0,42 ммоль) в безводном THF (4 мл) при 0°C в атмосфере азота добавляли трифторида бора диэтилэфират (91 мкл, 0,73 ммоль) и борогидрид натрия (27 мг, 0,73 ммоль). Полученную смесь перемешивали в течение 4 ч при 0°C. Добавляли AcOEt (5 мл) и насыщенный раствор NaHCO3 (10 мл). Водную фазу экстрагировали дихлорметаном (3×) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→2% MeOH в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (66 мг, 48%).
1H ЯМР (500 МГц, CDCl3): 7,21-7,26 (7H, м), 7,14-7,12 (2H, м), 6,97 (1H, с), 4,48 (2H, т), 3,48 (2H, ушир. с),2,55-2,25 (4H, м), 2,06-1,98 (1H, м), 1,82-1,73 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 1-(4-(7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (15 мг, 0,03 ммоль) с получением указанного в заголовке соединения (15 мг, количественный выход). ЖХ/МС (способ A): RT=3,80 мин, M-NH2=341.
1H ЯМР (500 МГц, MeOD): 7,31 (4H, с), 7,22-7,21 (3H, м), 7,12-7,11 (2H, м), 7,01 (1H, с), 4,45 (2H, т), 3,45 (2H, т), 2,75-2,70 (м, 2H), 2,56-2,50 (м, 2H), 2,24-2,15 (м, 1H), 1,97-1,88 (м, 1H).
Пример 38: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он
Стадия 1: трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (211 мг, 0,45 ммоль) в толуоле (15 мл) в атмосфере азота добавляли реагент Лауссона (181 мг, 0,45 ммоль). Полученную смесь нагревали с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→5% MeOH в дихлорметане) с получением указанного в заголовке соединения в виде желтого твердого вещества (124 мг, 57%).
1H ЯМР (500 МГц, CDCl3): 9,47 (1H, ушир. с), 7,30-7,24 (7H, м), 7,18 (1H, с), 7,16-7,14 (2H, м), 5,17 (2H, с), 5,04 (1H, ушир. с), 2,55-2,25 (4H, м), 2,10-2,01 (1H, м), 1,85-1,76 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: этил-2-(6-(4-(1-(трет-бутоксикарбониламино)циклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-илиден)гидразинкарбоксилат
К раствору трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (20 мг, 0,04 ммоль) в безводном THF (1 мл) в атмосфере азота добавляли этилгидразинкарбоксилат (21 мг, 0,20 ммоль) и ацетат ртути (21 мг, 0,06 ммоль). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре, фильтровали на целите и концентрировали досуха в условиях пониженного давления с получением указанного в заголовке соединения, которое использовали без очистки на следующей стадии.
Стадия 3: трет-бутил-(1-(4-(1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Раствор этил-2-(6-(4-(1-(трет-бутоксикарбониламино)циклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-илиден)гидразинкарбоксилата (24 мг, 0,04 ммоль) в безводном DMF (1 мл) нагревали при 200°C в течение 10 мин в условиях обработки микроволновым излучением. Реакционную смесь разбавляли дихлорметаном, фильтровали на целите и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом препаративной ЖХВД (метод G, градиент 5→95% 0,1% FA/ACN в 0,1% FA/H2O) с получением указанного в заголовке соединения (7 мг, 32%).
1H ЯМР (500 МГц, CDCl3): 9,18 (1H, ушир. с), 8,54 (1H, с), 7,32 (2H, д), 7,28-7,26 (5H, м), 7,22-7,20 (2H, м), 5,32 (2H, с), 5,07 (1H, ушир. с), 2,55-2,25 (4H, м), 2,10-2,03 (1H, м), 1,86-1,78 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 4: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (7 мг, 0,01 ммоль) с получением указанного в заголовке соединения (7 мг, количественный выход). ЖХ/МС (способ A): RT=3,54 мин, M+2H+=413.
1H ЯМР (500 МГц, MeOD): 8,56 (1H, с), 7,44 (2H, д), 7,38 (2H, д), 7,30-7,29 (3H, м), 7,23-7,21 (м, 2H), 5,41 (2H, с), 2,78-2,72 (м, 2H), 2,59-2,53 (м, 2H), 2,25-2,18 (м, 1H), 1,99-1,91 (м, 1H).
Пример 39: 1-(4-(8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
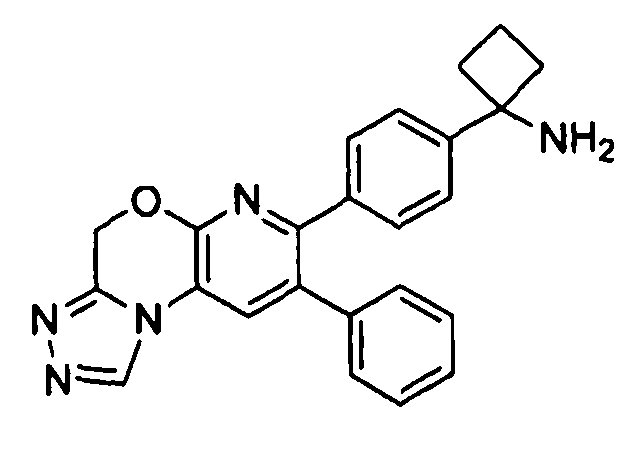
Стадия 1: трет-бутил-(1-(4-(2-гидразоно-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (20 мг, 0,04 ммоль) в THF (2 мл) в атмосфере азота добавляли гидразин·H2O (10 мкл, 0,20 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1,5 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях пониженного давления с получением указанного в заголовке соединения, которое использовали непосредственно на следующей стадии без какой-либо очистки.
Стадия 2: трет-бутил-(1-(4-(8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Раствор трет-бутил-(1-(4-(2-гидразоно-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (20 мг, 0,04 ммоль) в триэтилортоформиате (1 мл) нагревали при 150°C в течение 10 мин в условиях обработки микроволновым излучением. Реакционную смесь концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (градиент 0→2% MeOH в дихлорметане) с получением указанного в заголовке соединения (3 мг, 15%).
1H ЯМР (500 МГц, CDCl3): 8,68 (1H, с), 7,75 (1H, с), 7,34-7,26 (7H, м), 7,22-7,20 (2H, м), 5,70 (2H, с), 5,02 (1H, ушир. с), 2,55-2,25 (4H, м), 2,10-2,03 (1H, м), 1,86-1,78 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 3: 1-(4-(8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Следуя методике получения 2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6H)-она, в реакции использовали трет-бутил-(1-(4-(8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (2 мг, 0,004 ммоль) с получением указанного в заголовке соединения (2 мг, количественный выход). ЖХ/МС (способ A): RT=3,60 мин, M+2H+=397.
1H ЯМР (500 МГц, MeOD): 9,31 (1H, с), 8,30 (1H, с), 7,44 (2H, д), 7,38 (2H, д), 7,32-7,31 (3H, м), 7,27-7,25 (м, 2H), 5,77 (2H, с), 2,78-2,72 (м, 2H), 2,59-2,53 (м, 2H), 2,25-2,18 (м, 1H), 1,99-1,91 (м, 1H).
Пример 40: 1-(4-(1-метил-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин
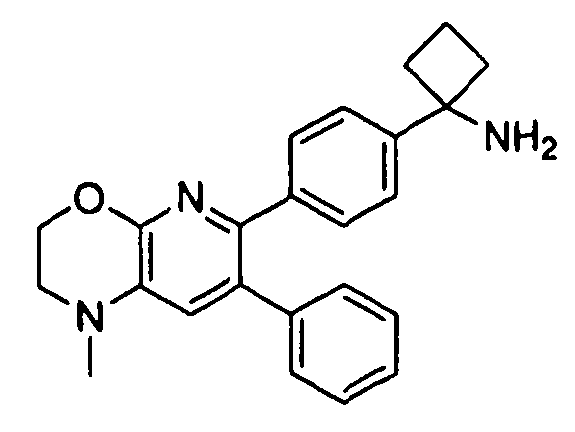
Стадия 1: трет-бутил-(1-(4-(1-метил-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (20 мг, 0,04 ммоль) в безводном DMF (1 мл) при 0°C в атмосфере азота добавляли гидрид натрия (2 мг, 0,05 ммоль) и метилйодид (5 мкл, 0,05 ммоль). Полученную смесь перемешивали в течение 30 минут при 0°C. Добавляли насыщенный раствор NH4Cl и экстрагировали смесь DCM (3×10 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→20% EtOAc в циклогексане) с получением указанного в заголовке соединения (8 мг, 44%).
1H ЯМР (500 МГц, CDCl3): 7,39-7,19 (9H, м), 6,85 (1H, с), 4,51-4,50 (2H, д), 3,45-3,43 (2H, д), 2,80-2,70 (2H, м), 2,61-2,51 (4H, м), 2,01-1,88 (1H, м), 2,00 (3H, с), 1,40-1,10 (9H, ушир.).
Стадия 2: 1-(4-(1-метил-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали 2-((1r,3r)-1-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион (21 мг, 0,04 ммоль) с получением указанного в заголовке соединения (3 мг, количественный выход). ЖХ/МС (способ A): RT=4,21 мин, M+1=355.
1H ЯМР (500 МГц, MeOD): 7,36-7,32 (4H, м), 7,29-7,20 (3H, м), 7,19-7,15 (2H, м), 7,00 (1H, с), 4,45 (2H, д), 3,45 (2H, д), 2,88 (3H, с), 2,75-2,70 (2H, м), 2,61-2,51 (2H, м), 2,28-2,17 (1H, м), 2,01-1,88 (1H, м).
Пример 41: 1-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)этанон
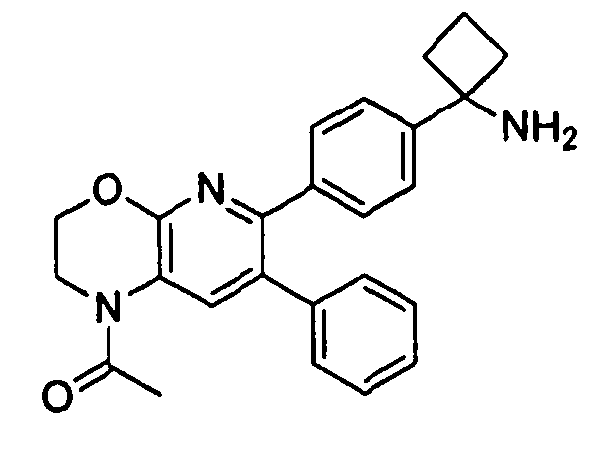
Стадия 1: трет-бутил-(1-(4-(1-ацетил-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (22 мг, 0,05 ммоль) в безводном DCM (1 мл) в атмосфере азота добавляли триэтиламин (10 мкл, 0,07 ммоль) и ацетилхлорид (5 мкл, 0,07 ммоль). Полученную смесь перемешивали в течение 1 ч при комнатной температуре. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь дихлорметаном (3×10 мл) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→80% EtOAc в циклогексане) с получением указанного в заголовке соединения (20 мг, 83%).
1H ЯМР (500 МГц, CDCl3): 8,50 (1H, ушир. с), 7,25 (2H, д), 7,19-7,17 (5H, м), 7,11-7,10 (2H, м), 4,95 (1H, с), 4,43 (2H, т), 3,94 (2H, ушир. с), 2,55-2,25 (7H, м), 2,06-1,98 (1H, м), 1,82-1,73 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 1-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)этанон
трет-Бутил-(1-(4-(1-ацетил-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (20 мг, 0,04 ммоль) растворяли в 2M HCl в Et2O (1 мл). Полученную смесь перемешивали в течение ночи при комнатной температуре и упаривали в условиях пониженного давления. Соединение со снятой защитой дважды снова вводили в диэтиловый эфир. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением указанного в заголовке соединения (21 мг, количественный выход). ЖХ/МС (способ A): RT=3,87 мин, M-16=383.
1H ЯМР (500 МГц, MeOD): 8,70 (1H, ушир. с), 7,44-7,43 (4H, м), 7,30-7,28 (3H, м), 7,21-7,19 (2H, м), 4,63 (2H, т), 4,11 (2H, т), 2,75-2,70 (2H, м), 2,56-2,50 (2H, м), 2,42 (3H, с), 2,24-2,15 (1H, м), 1,97-1,88 (1H, м).
Пример 42: 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
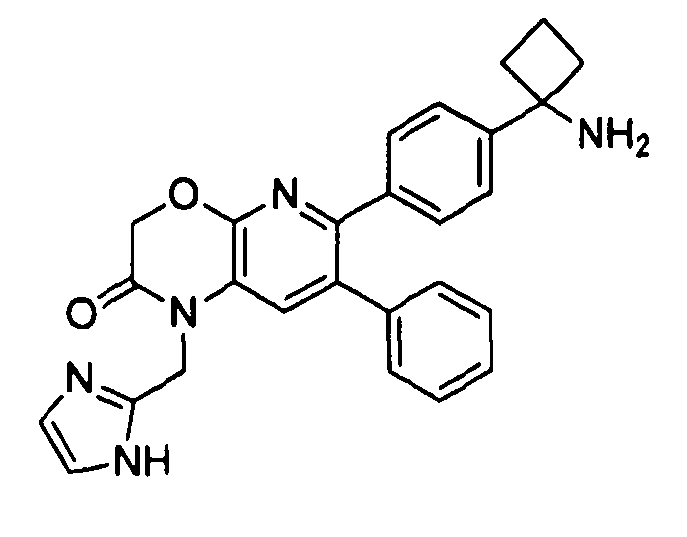
Стадия 1: трет-бутил-(1-(4-(1-((1H-имидазол-2-ил)метил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (26 мг, 0,05 ммоль) в безводном DMF (1 мл) при 0°C в атмосфере азота добавляли гидрид натрия (5 мг, 0,13 ммоль). Спустя 1 час при 0°C добавляли 2-(хлорметил)-1H-имидазол·HCl (10 мг, 0,07 ммоль) и перемешивали полученную смесь еще в течение 1 ч при комнатной температуре. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь дихлорметаном (3×10 мл) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом препаративной ЖХВД (метод G, градиент 5→95% 0,1% FA/ACN в 0,1% FA/H2O) с получением указанного в заголовке соединения (5 мг, 17%).
1H ЯМР (500 МГц, CDCl3): 7,83 (1H, с), 7,22-7,15 (7H, м), 7,14-7,13 (2H, м), 6,94 (2H, с), 6,22 (1H, ушир. с), 5,14 (2H, с), 4,98 (1H, ушир. с), 4,83 (2H, с), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,75-1,70 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-(1-(4-(1-((1H-имидазол-2-ил)метил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (5 мг, 0,001 ммоль) растворяли в TFA (1 мл). Полученную смесь перемешивали в течение 30 секунд при комнатной температуре и упаривали в условиях пониженного давления. Соединение со снятой защитой дважды снова вводили в диэтиловый эфир, и дважды промывали образовавшееся твердое вещество DCM. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением указанного в заголовке соединения (5 мг, количественный выход). ЖХ/МС (способ A): RT=2,80 мин, M-NH2=435.
1H ЯМР (500 МГц, MeOD): 7,44 (1H, с), 7,41 (2H, с), 7,31-7,26 (4H, м), 7,18-7,16 (3H, м), 7,06-7,05 (2H, м), 5,44 (2H, с), 4,97 (2H, с), 2,66-2,60 (2H, м), 2,49-2,43 (2H, м), 2,14-2,10 (1H, м), 1,89-1,82 (1H, м).
Пример 43: 2-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
Пример 44: 2-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетамид
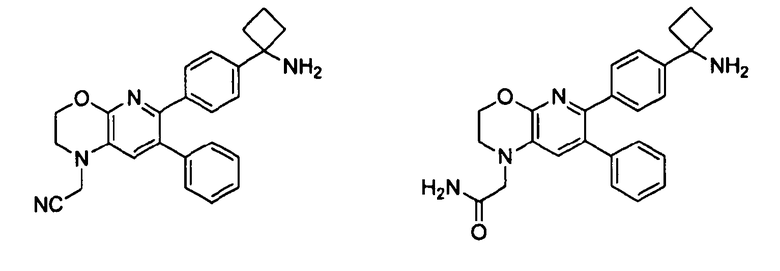
Стадия 1: трет-бутил-(1-(4-(1-(цианометил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (24 мг, 0,05 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли карбонат калия (93 мг, 1,57 ммоль) и бромацетонитрил (110 мкл, 1,57 ммоль). Полученную смесь перемешивали в течение 4 часов при 80°C. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь дихлорметаном (3×10 мл) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→60% EtOAc в циклогексане) с получением указанного в заголовке соединения (5 мг, 19%).
1H ЯМР (500 МГц, CDCl3): 7,22-7,11 (9H, м), 6,93 (1H, с), 4,92 (1H, с), 4,51-4,49 (2H, м), 4,15 (2H, с), 3,39-3,37 (2H, м), 2,55-2,25 (4H, м), 1,99-1,94 (1H, м), 1,75-1,63 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 2-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
2-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетамид
Следуя методике получения 1-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)этанона и 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1-(цианометил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (5 мг, 0,01 ммоль) с получением указанных в заголовке соединений (1 мг, 25% и 2 мг, 41%). ЖХ/МС: RT=3,99 мин, M-16=380.
1H ЯМР (500 МГц, MeOD): 7,20-7,13 (8H, м), 7,09-7,07 (2H, м), 4,46 (2H, т), 3,54 (2H, с), 3,22 (2H, т), 2,46-2,40 (2H, м), 2,17-2,11 (2H, м), 1,97-1,94 (1H, м), 1,66-1,62 (1H, м).
ЖХ/МС (способ A): RT=3,99 мин, M-16=398.
1H ЯМР (500 МГц, D2O): 8,39 (2H, с), 7,27-7,22 (7H, м), 7,11-7,09 (2H, м), 6,89 (1H, с), 4,78 (2H, с), 4,48 (2H, т), 4,00 (2H, с), 3,50 (2H, т), 2,65-2,60 (2H, м), 2,53-2,47 (2H, м), 2,09-2,05 (1H, м), 1,86-1,79 (1H, м).
Пример 45: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-4-илметил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Стадия 1: трет-бутил-(1-(4-(2-оксо-7-фенил-1-(пиридин-4-илметил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (35 мг, 0,07 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли гидрид натрия (7 мг, 0,18 ммоль) и 4-(бромметил)пиридин·HBr (23 мг, 0,09 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь дихлорметаном (3×10 мл) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→80% EtOAc в циклогексане) с получением указанного в заголовке соединения (21 мг, 50%).
1H ЯМР (500 МГц, CDCl3): 8,54 (2H, с), 7,19-7,12 (9H, м), 6,97 (1H, с), 6,93-6,91 (2H, м), 5,11 (2H, с), 4,98 (1H, ушир. с), 4,95 (2H, с), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,75-1,70 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-4-илметил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(2-оксо-7-фенил-1-(пиридин-4-илметил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (21 мг, 0,04 ммоль) с получением указанного в заголовке соединения (26 мг, количественный выход). ЖХ/МС (способ A): RT=3,25 мин, M+1=464.
1H ЯМР (500 МГц, MeOD): 8,57 (2H, с), 7,67 (2H, с), 7,28-7,24 (4H, м), 7,23 (1H, с), 7,14-7,10 (3H, м), 6,96-6,94 (2H, м), 5,37 (2H, с), 5,03 (2H, с), 2,66-2,60 (2H, м), 2,49-2,43 (2H, м), 2,14-2,10 (1H, м), 1,89-1,82 (1H, м).
Пример 46: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-3-илметил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Стадия 1: трет-бутил-(1-(4-(2-оксо-7-фенил-1-(пиридин-3-илметил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (51 мг, 0,11 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли гидрид натрия (11 мг, 0,26 ммоль) и 3-(бромметил)пиридин·HBr (33 мг, 0,13 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь дихлорметаном (3×10 мл) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→80% EtOAc в циклогексане) с получением указанного в заголовке соединения (23 мг, 38%).
1H ЯМР (500 МГц, CDCl3): 8,60-8,50 (2H, ушир. с), 7,60 (2H, д), 7,28 (1H, с), 7,19-7,15 (6H, м), 7,12 (1H, с), 6,97 (2H, д), 5,13 (2H, с), 4,93 (3H, с), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,75-1,70 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-3-илметил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(2-оксо-7-фенил-1-(пиридин-3-илметил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (23 мг, 0,04 ммоль) с получением указанного в заголовке соединения (27 мг, количественный выход). ЖХ/МС (способ A): RT=3,48 мин, M+1=464.
1H ЯМР (500 МГц, MeOD): 8,75 (1H, ушир. с), 8,60 (1H, ушир. с), 8,12 (1H, д), 7,67 (1H, с), 7,49 (1H, с), 7,40-7,36 (4H, м), 7,28-7,24 (3H, м), 7,09 (2H, д), 5,42 (2H, с), 5,12 (2H, с), 2,77-2,72 (2H, м), 2,59-2,53 (2H, м), 2,25-2,21 (1H, м), 1,98-1,94 (1H, м).
Пример 47: 1-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (5 г, 10,60 ммоль) в толуоле (380 мл) добавляли реагент Лауссона (4,29 г, 10,60 ммоль). Полученную смесь нагревали с обратным холодильником в течение 5 часов. После охлаждения смеси до комнатной температуры, смесь концентрировали досуха в условиях пониженного давления. Полученный остаток растворяли в минимальном количестве дихлорметана (500 мл) и обрабатывали диизопропиловым эфиром (800 мл). Отжимали желтое твердое вещество. Твердое вещество фильтровали и сушили до постоянного веса (4,7 г, 91%). ЖХ/МС (способ D): RT=1,526 мин, M+1=488.
Стадия 2: (E)-трет-бутил-(1-(4-(2-гидразоно-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (2 г, 2,05 ммоль) в THF (350 мл) добавляли гидразина моногидрат (645 мкл, 20,51 ммоль) с получением желтого раствора. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь фильтровали для удаления нерастворимых черных частиц и концентрировали досуха в условиях пониженного давления. Полученное желтое твердое вещество использовали непосредственно на следующей стадии в виде неочищенного вещества без какой-либо очистки. ЖХ/МС (способ D): RT=1,04 мин, M+1=486.
Стадия 3: трет-бутил-(1-(4-(9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутил)карбамат
Раствор (E)-трет-бутил-(1-(4-(2-гидразоно-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (50 мг, 0,103 ммоль) в триэтилортопропионате (1 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→100% этилацетат в н-гексанах) с получением указанного в заголовке соединения (30 мг, 55%). ЖХ/МС (способ D): RT=1,43 мин, M+1=524.
1H ЯМР (500 МГц, CDCl3): 7,80 (1H, с), 7,27-7,26 (5H, м), 7,22 (2H, д), 7,15 (2H, м), 5,56 (2H, с), 3,22 (2H, кв.), 2,50-2,33 (4H, м), 2,07-1,94 (1H, м), 1,81-1,68 (1H, м), 1,52 (3H, т), 1,38-1,11 (9H, ушир.).
Стадия 4: 1-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутил)карбамат (30 мг, 0,057 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления, и сушили остаток с получением целевого продукта в виде не совсем белого твердого вещества (15 мг, 61% выход). ЖХ/МС (способ A) RT=3,78 мин, M+1=425.
1H-ЯМР (500 МГц, CD3OD): δ 8,10 (1H, с), 7,49 (2H, д), 7,35 (3H, м), 7,29 (4H, м), 5,66 (2H, с), 3,26 (2H, т), 2,84-2,71 (2H, м), 2,64-2,54 (2H, м), 2,31-2,14 (1H, м), 2,04-1,93 (1H, м), 1,51 (3H, т).
Пример 48: 6-(4-(1-аминоциклобутил)фенил)-1-(циклобутилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
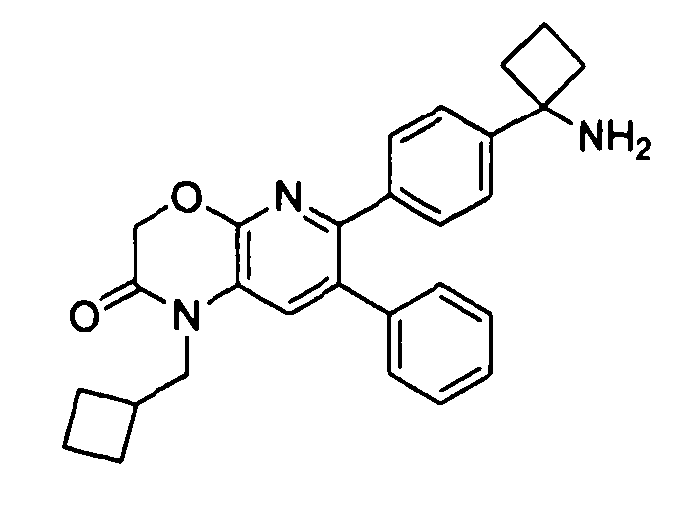
Стадия 1: трет-бутил-(1-(4-(1-(циклобутилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (44 мг, 0,09 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли гидрид натрия (9 мг, 0,22 ммоль) и (бромметил)циклобутан (13 мкл, 0,11 ммоль). Полученную смесь перемешивали в течение 1 ч при 80°C. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь дихлорметаном (3×10 мл) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→40% EtOAc в циклогексане) с получением указанного в заголовке соединения (8 мг, 16%).
1H ЯМР (500 МГц, CDCl3): 7,23-7,17 (8H, м), 7,13-7,11 (2H, м), 4,95 (1H, с), 4,80 (2H, с), 3,96 (2H, д), 2,68-2,64 (1H, м), 2,52-2,20 (4H, м), 2,00-1,95 (3H, м), 1,86-1,50 (5H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(циклобутилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1-(циклобутилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (8 мг, 0,02 ммоль) с получением указанного в заголовке соединения (8 мг, 97%). ЖХ/МС (способ A): RT=4,65 мин, M-16=423.
1H ЯМР (500 МГц, MeOD): 7,56 (1H, с), 7,42-7,37 (4H, м), 7,32-7,30 (3H, м), 7,23-7,21 (2H, м), 4,97 (1H, с), 4,59 (2H, с), 4,14 (2H, д), 2,80-2,72 (3H, м), 2,59-2,53 (2H, м), 2,24-2,20 (1H, м), 2,10-2,06 (2H, м), 1,98-1,87 (5H, м).
Пример 49: этил-2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетат
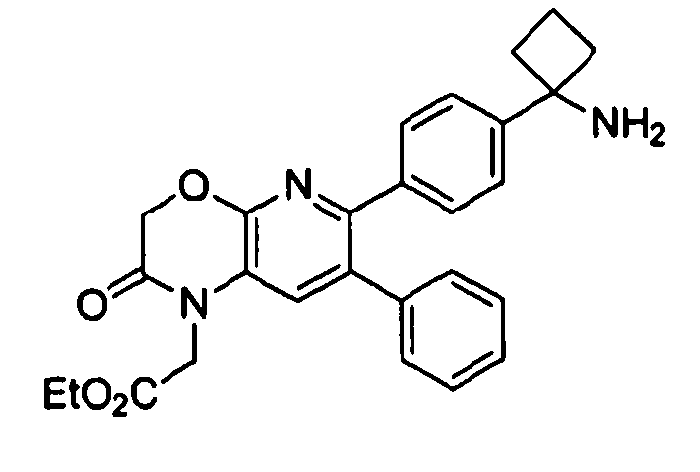
Стадия 1: этил-2-(6-(4-(1-(трет-бутоксикарбониламино)циклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (50 мг, 0,106 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением красного раствора, а затем охлаждали до 0°C в атмосфере азота. Добавляли гидрид натрия (60% в масле, 6,4 мг, 0,159 ммоль) и перемешивали смесь при 0°C в течение 15 минут. Добавляли этил-2-бромацетат (0,035 мл, 0,318 ммоль), перемешивали смесь при 0°C в течение 30 минут, оставляли нагреваться до комнатной температуры, а затем перемешивали в течение 1 часа. Реакционную смесь гасили добавлением насыщенного раствора хлорида аммония (5 мл) и экстрагировали введением в этилацетат (3×3 мл). Органическую фазу промывали водой/солевым раствором (50/50, 2×5 мл), а затем солевым раствором (5 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 88/12→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (22 мг, выход 39%).
1H-ЯМР (500 МГц, CDCl3) δ 7,13-7,28 (8H, м), 7,37-7,43 (2H, м), 5,24 (1H, ушир. с), 5,18 (2H, с), 4,89 (2H, с), 4,49 (2H, кв.), 2,40-2,90 (4H, м), 2,21-2,35 (1H, м), 1,90-2,10 (1H, м), 1,20-1,75 (9H, ушир. м), 1,53 (3H, т).
ЖХ/МС (способ A) RT=7,20 мин, M+H+=558,13.
Стадия 2: этил-2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетат
Этил-2-(6-(4-(1-(трет-бутоксикарбониламино)циклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетат (22 мг, 0,039 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (22 мг, выход 98%).
1H-ЯМР (500 МГц, MeOD) δ 7,37-7,48 (5H, м), 7,25-7,37 (3H, м), 7,15-7,25 (2H, м), 5,01 (2H, с), 4,85 (2H, с), 4,26 (2H, кв.), 2,70-2,83 (2H, м), 2,48-2,62 (2H, м), 2,15-2,30 (1H, м), 1,90-2,05 (1H, м), 1,28 (3H, т),
ЖХ/МС (способ A) RT=4,08 мин, M+2H+=459,20.
Пример 50: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-амин
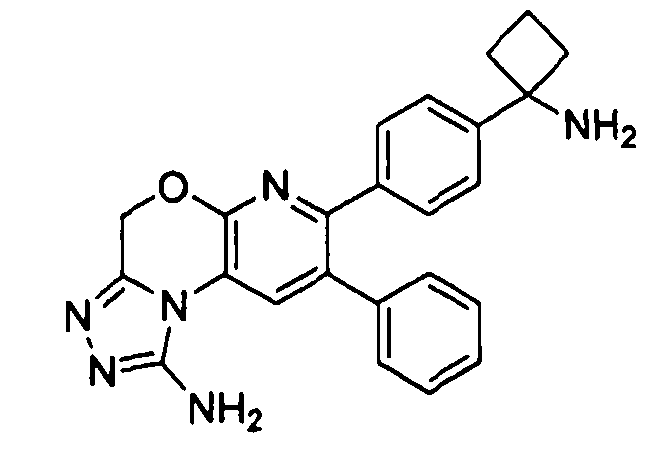
Стадия 1: трет-бутил-(1-(4-(1-амино-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[3,4-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Раствор (E)-трет-бутил-(1-(4-(2-гидразоно-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (50 мг, 0,103 ммоль), цианогена бромида (33 мг, 0,309 ммоль) и карбоната натрия (22 мг, 0,206 ммоль) в смеси этанола (1 мл) и 1,4-диоксана (1 мл) перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 100%→94/6 дихлорметан в метаноле) с получением указанного в заголовке соединения (15 мг, 30%). ЖХ/МС (способ A): RT=5,73 мин, M+1=511.
1H ЯМР (500 МГц, CDCl3): 7,80 (1H, с), 7,36-7,28 (7H, м), 7,23-7,19 (2H, м), 5,48 (2H, с), 2,56-2,41 (4H, м), 2,13-2,02 (1H, м), 1,89-1,77 (1H, м), 1,46-1,21 (9H, ушир. с).
Стадия 2: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-амин
трет-Бутил-(1-(4-(1-амино-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[3,4-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (15 мг, 0,029 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления, и сушили остаток с получением целевого продукта в виде не совсем белого твердого вещества (10 мг, выход 80%). ЖХ/МС (способ A) RT=3,40 мин, M+1=412.
1H-ЯМР (500 МГц, CD3OD) δ 8,21 (1H, с), 7,41-7,36 (2H, д), 7,35-7,30 (2H, д), 7,27-7,19 (5Н, м), 5,44 (2Н, с), 2,72-2,62 (2Н, м), 2,53-2,43 (2Н, м), 2,20-2,09 (1H, м), 1,92-1,82 (1H, м).
Пример 51: 6-(4-(1-аминоциклобутил)фенил)-1-(2-метоксиэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
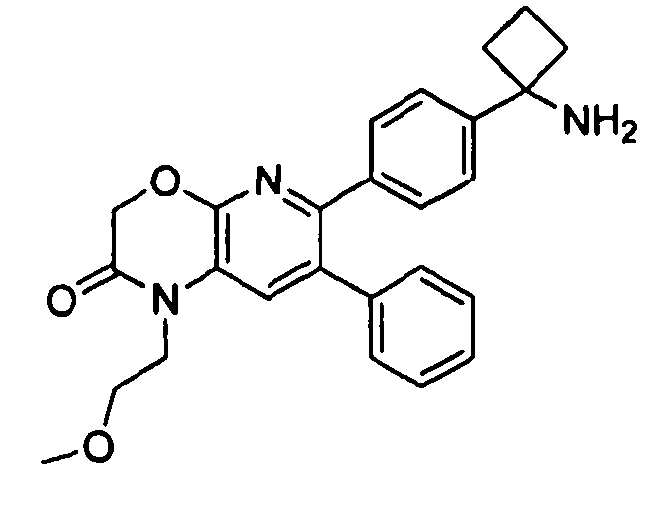
Стадия 1: трет-бутил-(1-(4-(1-(2-метоксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (48 мг, 0,10 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли гидрид натрия (16 мг, 0,41 ммоль) и 1-бром-2-метоксиэтан (28 мг, 0,20 ммоль). Полученную смесь перемешивали в течение 2 часов при 80°C. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь дихлорметаном (3×10 мл) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→40% EtOAc в циклогексане) с получением указанного в заголовке соединения (10 мг, 18%).
1H ЯМР (500 МГц, CDCl3): 7,49 (1H, с), 7,23-7,17 (7H, м), 7,12-7,11 (2H, м), 4,95 (1H, с), 4,82 (2H, с), 4,05 (2H, т), 3,61 (2H, т), 3,29 (3H, с), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,79-1,65 (1H, м), 1,40-1,10 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(2-метоксиэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1-(2-метоксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (10 мг, 0,02 ммоль) с получением указанного в заголовке соединения (7 мг, 68%). ЖХ/МС (способ D): RT=4,11 мин, M-16=423.
1H ЯМР (500 МГц, MeOD): 7,77 (1H, с), 7,43-7,38 (4H, м), 7,33-7,28 (3H, м), 7,23-7,21 (2H, м), 4,96 (2H, с), 4,24-4,22 (2H, т), 3,72-3,70 (2H, т), 3,36 (3H, с), 2,77-2,72 (2H, м), 2,59-2,53 (2H, м), 2,25-2,21 (1H, м), 1,98-1,94 (1H, м).
Пример 52: 6-(4-(1-аминоциклобутил)фенил)-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
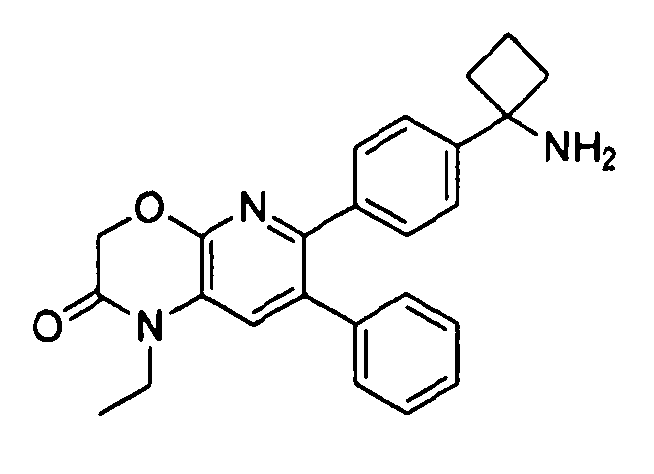
Стадия 1: трет-бутил-1-(4-(1-этил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (50 мг, 0,106 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением желтого раствора, а затем охлаждали до 0°C в атмосфере азота. Добавляли гидрид натрия (60% в масле, 10 мг, 0,254 ммоль), перемешивали смесь при 0°C в течение 15 минут. Добавляли йодэтан (10 мкл, 0,127 ммоль), смесь перемешивали при 0°C в течение 30 минут, оставляли нагреваться до комнатной температуры, а затем перемешивали в течение 1 часа. Дополнительно добавляли гидрид натрия (60% в масле, 10,18 мг, 0,254 ммоль) и нагревали реакционную смесь до 40°C в атмосфере азота в течение 1 часа. Дополнительно добавляли йодэтан (10 мкл, 0,127 ммоль) и перемешивали смесь при комнатной температуре в течение 30 минут. Реакционную смесь гасили путем добавления насыщенного раствора бикарбоната натрия (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Органическую фазу промывали водой/солевым раствором (50/50, 2×10 мл), а затем солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 88/12→0/100) с получением целевого продукта в виде бледно-желтого твердого вещества (7 мг, выход 13%).
1H-ЯМР (500 МГц, CDCl3) δ 7,10-7,28 (10Н, м), 4,93 (1H, ушир. с), 4,81 (2H, с), 3,94 (2H, кв.), 2,10-2,55 (4H, м), 1,90-2,08 (1H, м), 1,65-1,80 (1H, м), 1,00-1,40 (9H, ушир. м), 1,14 (3H, т).
ЖХ/МС (способ A) RT=7,29 мин, M+H+=500,12.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(1-этил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (7 мг, 0,014 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (6 мг, выход 83%).
1H-ЯМР (500 МГц, MeOD) δ 7,57 (1H, с), 7,42 (2H, д), 7,38 (2H, д), 7,28-7,35 (3H, м), 7,20-7,28 (2H, м), 4,95 (2H, с), 4,10 (2H, кв.), 2,68-2,85 (2H, м), 2,50-2,65 (2H, м), 2,15-2,30 (1H, м), 1,90-2,05 (1H, м), 1,30 (3H, т).
ЖХ/МС (способ A) RT=4,04 мин, M+2H+=401,18.
Пример 53: 1-((1H-имидазол-5-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Стадия 1: трет-бутил-(1-(4-(1-((1H-имидазол-5-ил)метил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
В круглодонную колбу добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (150 мг, 0,318 ммоль) в DMF (объем: 8 мл) с получением коричневого раствора. Добавляли гидрид натрия (30,5 мг, 0,763 ммоль) и 5-(хлорметил)-1H-имидазола гидрохлорид (58,4 мг, 0,501 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Дополнительно добавляли гидрид натрия (15,2 мг) и имидазол (29 мг) и перемешивали полученную смесь еще в течение 1 часа. Реакционную смесь концентрировали досуха. Остаток распределяли между этилацетатом (35 мл) и водой (30 мл). Органическую фазу разделяли и промывали водой (2×25 мл), солевым раствором и концентрировали. Остаток очищали на колонке (силикагель Biotage, 25 г) с получением продукта (14 мг). ЖХ/МС (способ A) RT=4,58 мин, M+H+=552,1.
Стадия 2: 1-((1H-имидазол-5-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-(1-(4-(1-((1H-имидазол-5-ил)метил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (12 мг, 0,022 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения (7,5 мг).
1H-ЯМР (500 МГц, MeOD) δ 8,81 (1H, с), 7,62 (2H, ушир.), 7,39-7,41 (м, 4H), 7,28-7,30 (3H, м), 7,16-7,18 (2H, м), 5,39 (2H, с), 5,07 (2H, с), 2,7-2,8 (2H, м), 2,55-2,65 (2H, м), 2,2-2,3 (1H, м), 1,9-2,05 (1H, м).
ЖХ/МС (способ A) RT=2,83 мин, M+H+=453,1.
Пример 54: 2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрил
Стадия 1: трет-бутил-(1-(4-(1-(1-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
В круглодонную колбу объемом 50 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (50 мг, 0,106 ммоль) в DMF (объем: 4 мл) с получением коричневого раствора. Добавляли гидрид натрия (10,18 мг, 0,254 ммоль) и 2-бромпропаннитрил (17,05 мг, 0,127 ммоль). Реакционную смесь перемешивали при 50°C в течение 1 ч, а затем при комнатной температуре в течение ночи. Реакционную смесь распределяли между DCM (20 мл) и водой (20 мл). Органическую фазу разделяли и концентрировали. Остаток очищали на колонке (силикагель Biotage, 25 г), элюируя этилацетатом/циклогексаном (0-50%), с получением продукта (19 мг). ЖХ/МС (способ A): RT=7,14 мин, M+H+=525,0.
Стадия 2: 2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрил
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(1-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (7 мг, 0,013 ммоль) с получением указанного в заголовке соединения (5,5 мг).
1H ЯМР (500 МГц, CH3OD) 7,77 (1H, с), 7,44 (2H, д), 7,40 (2H, д), 7,30-7,34 (3H, м), 7,25-7,28 (2H, м), 6,03 (1H, кв.), 4,99 (2H, с), 2,7-2,85 (2H, м), 2,55-2,65 (2H, м), 2,2-2,3 (1H, м), 1,9-2,1 (1H, м), 1,81 (3H, д).
ЖХ/МС (способ A): RT=4,11 мин, M+H+=426,1.
Пример 55: 6-(4-(1-аминоциклобутил)фенил)-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он

Стадия 1: трет-бутил-1-(4-(1-(циклопропилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (50 мг, 0,106 ммоль), карбонат калия (44 мг, 0,318 ммоль) и (бромметил)циклопропан (0,031 мл, 0,318 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением оранжевой суспензии. Реакционную смесь перемешивали при 80°C в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в дихлорметан (3×3 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде не совсем белого твердого вещества (40 мг, выход 72%).
1H-ЯМР (500 МГц, CDCl3) δ 7,32 (1H, с), 7,10-7,28 (9H, м), 4,98 (1H, ушир. с), 4,82 (2H, с), 3,79 (2H, д), 2,05-2,60 (4H, м), 1,90-2,05 (1H, м), 1,65-1,79 (1H, м), 1,00-1,45 (10H, ушир. м), 0,45-0,57 (2H, м), 0,35-0,45 (2H, м).
ЖХ/МС (способ A) RT=7,79 мин, M+H+=526,18.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(1-(циклопропилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (40 мг, 0,076 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (30 мг, выход 73%).
1H-ЯМР (500 МГц, MeOD) δ 7,67 (1H, с), 7,43 (2H, д), 7,40 (2H, д), 7,28-7,36 (3H, м), 7,20-7,25 (2H, м), 4,96 (2H, с), 3,98 (2H, д), 2,70-2,80 (2H, м), 2,52-2,63 (2H, м), 2,18-2,30 (1H, м), 1,90-2,03 (1H, м), 1,17-1,30 (1H, м), 0,55-0,65 (2H, м), 0,42-0,50 (2H, м).
ЖХ/МС (способ A) RT=4,44 мин, M+2H+=427,20.
Пример 56: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-2-илметил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он

Стадия 1: трет-бутил-(1-(4-(2-оксо-7-фенил-1-(пиридин-2-илметил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (75 мг, 0,16 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли гидрид натрия (15 мг, 0,38 ммоль) и 2-(бромметил)пиридин.HBr (48 мг, 0,19 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь дихлорметаном (3×10 мл) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→40% EtOAc в циклогексане) с получением указанного в заголовке соединения (11 мг, 12%).
1H ЯМР (500 МГц, CDCl3): 8,49 (1H, д), 7,63 (1H, т), 7,42 (1H, с), 7,27 (1H, д), 7,19-7,13 (8H, м), 7,01-6,98 (2H, д), 5,23 (2H, с), 4,92 (2H, с), 4,91 (1H, с), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,75-1,70 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-2-илметил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(2-оксо-7-фенил-1-(пиридин-2-илметил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (16 мг, 0,03 ммоль) с получением указанного в заголовке соединения (17 мг, 87%). ЖХ/МС (способ A): RT=4,11 мин, M-16=446.
1H ЯМР (500 МГц, MeOD): 8,54 (1H, д), 7,91-7,87 (1H, т), 7,53 (1H, д), 7,42-7,35 (6H, м), 7,26-7,21 (3H, м), 7,04 (2H, д), 5,39 (2H, с), 5,11 (2H, с), 2,77-2,72 (2H, м), 2,59-2,53 (2H, м), 2,25-2,21 (1H, м), 1,98-1,94 (1H, м).
Пример 57; 6-(4-(1-аминоциклобутил)фенил)-1-изопропил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Стадия 1: трет-бутил-1-(4-(1-изопропил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (25 мг, 0,053 ммоль), карбонат калия (22 мг, 0,159 ммоль) и 2-йодпропан (16 мкл, 0,159 ммоль) в безводном N,N-диметилформамиде (500 мкл) с получением оранжевой суспензии. Реакционную смесь перемешивали при 80°C в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в дихлорметан (3×3 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде не совсем белого твердого вещества (13 мг, выход 48%).
1H-ЯМР (500 МГц, CDCl3) δ 7,37 (1H, с), 7,10-7,28 (9H, м), 4,95 (1H, ушир. с), 4,65-4,77 (3H, м), 2,05-2,60 (4H, м), 1,92-2,06 (1H, м), 1,65-1,80 (1H, м), 1,50 (6H, д), 1,00-1,45 (9H, ушир. м).
ЖХ/МС (способ A) RT=7,73 мин, M+H+=514,15.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-изопропил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(1-изопропил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (7 мг, 0,014 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (7 мг, выход 97%).
1H-ЯМР (500 МГц, MeOD) δ 7,64 (1H, с), 7,41 (2H, д), 7,36 (2H, д), 7,26-7,32 (3H, м), 7,19-7,26 (2H, м), 4,80 (2H, с), 4,75 (1H, септ.), 2,68-2,80 (2H, м), 2,50-2,62 (2H, м), 2,13-2,29 (1H, м), 1,89-2,00 (1H, м), 1,48 (6H, д).
ЖХ/МС (способ A) RT=4,20 мин, M+2H+=415,19.
Пример 58: 6-(4-(1-аминоциклобутил)фенил)-1-циклопентил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
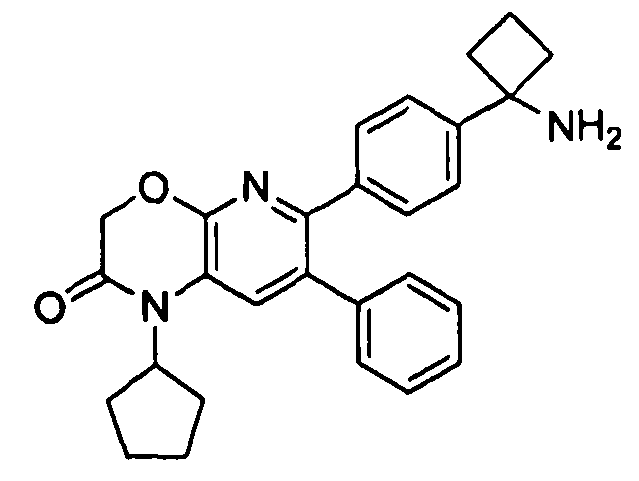
Стадия 1: трет-бутил-1-(4-(1-циклопентил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (50 мг, 0,106 ммоль), карбонат калия (44 мг, 0,318 ммоль) и бромциклопентан (0,034 мл, 0,318 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением оранжевой суспензии. Реакционную смесь перемешивали при 80°C в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в дихлорметан (3×3 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде не совсем белого твердого вещества (12 мг, выход 21%).
1H-ЯМР (500 МГц, CDCl3) δ 7,32 (1H, с), 7,17-7,28 (7H, м), 7,10-7,17 (2H, м), 4,94 (1H, ушир. с), 4,83 (1H, квинт.), 4,72 (2H, с), 2,05-2,60 (6H, м), 1,83-2,03 (5H, м), 1,54-1,82 (3H, м), 1,00-1,45 (9H, ушир. м).
ЖХ/МС (способ A) RT=8,25 мин, M+H+=540,19.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-циклопентил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(1-циклопентил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (12 мг, 0,022 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (6 мг, выход 49%).
1H-ЯМР (500 МГц, MeOD) δ 7,60 (1H, с), 7,40 (2H, д), 7,36 (2H, д), 7,25-7,36 (3H, м), 7,18-7,25 (2H, м), 4,91 (1H, квинт.), 4,84 (2H, с), 2,66-2,31 (2H, м), 2,47-2,61 (2H, м), 2,12-2,31 (3H, м), 1,83-2,12 (5H, м), 1,63-1,81 (2H, м).
ЖХ/МС (способ A) RT=4,55 мин, M+2H+=441,19.
Пример 59: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(тиазол-4-илметил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
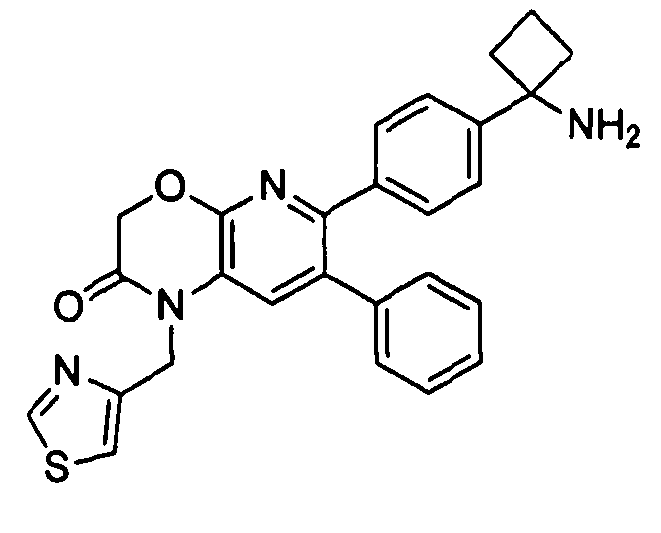
Стадия 1: трет-бутил-1-(4-(2-оксо-7-фенил-1-(тиазол-4-илметил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (50 мг, 0,106 ммоль), карбонат калия (59 мг, 0,424 ммоль) и 4-(хлорметил)тиазол·HCl (54 мг, 0,318 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением оранжевой суспензии. Реакционную смесь перемешивали при 80°C в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в дихлорметан (3×3 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (54 мг, выход 90%).
1H-ЯМР (500 МГц, CDCl3) δ 8,70 (1H, с), 7,61 (1H, с), 7,28 (1H, с), 7,12-7,27 (7H, м), 7,03-7,12 (2H, м), 5,23 (2H, с), 4,99 (1H, ушир. с), 4,86 (2H, с), 2,10-2,60 (4H, м), 2,41-2,06 (1H, м), 1,65-1,79(1H, м), 1,00-1,45 (9H, ушир. м).
ЖХ/МС (способ A) RT=7,11 мин, M+H+=569,14.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(тиазол-4-илметил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(2-оксо-7-фенил-1-(тиазол-4-илметил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (54 мг, 0,095 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (30 мг, выход 54%).
1H-ЯМР (500 МГц, MeOD) δ 8,97 (1H, с), 7,63 (1H, с), 7,60 (1H, с), 7,32-7,41 (4H, м), 7,20-7,29 (3H, м), 7,05-7,15 (2H, м), 5,39 (2H, с), 5,03 (2H, с), 2,66-2,88 (2H, м), 2,47-2,38 (2H, м), 2,13-2,28 (1H, м), 1,85-1,98 (1H, м).
ЖХ/МС (способ A) RT=3,81 мин, M+2H+=470,18.
Пример 60: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
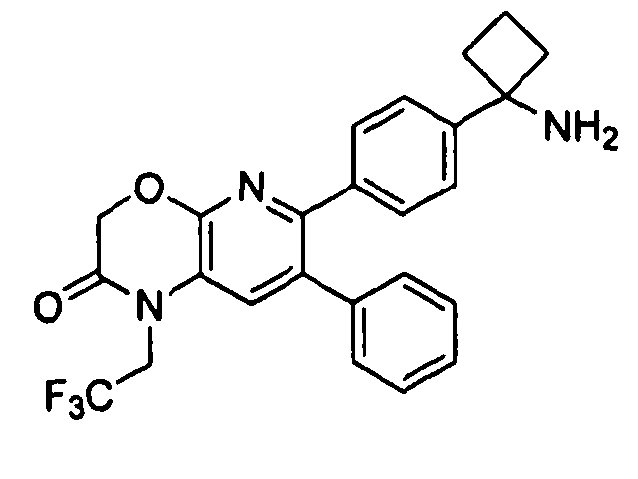
Стадия 1: трет-бутил-1-(4-(2-оксо-7-фенил-1-(2,2,2-трифторэтил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (50 мг, 0,106 ммоль), карбонат калия (44 мг, 0,318 ммоль) и 2-бром-1,1,1-трифторэтан (0,193 мл, 2,121 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением оранжевой суспензии. Реакционную смесь перемешивали при 80°C в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в дихлорметан (3×3 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде не совсем белого твердого вещества (31 мг, выход 53%).
1H-ЯМР (500 МГц, CDCl3) δ 7,38 (1H, с), 7,25-7,37 (7H, м), 7,17-7,23 (2H, м), 5,05 (1H, ушир. с), 4,97 (2H, с), 4,63 (2H, кв.), 2,20-2,58 (4H, м), 2,00-2,15 (1H, м), 1,75-1,88 (1H, м), 1,10-1,50 (9H, ушир. м).
ЖХ/МС (способ A) RT=7,65 мин, M+H+=554,05.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(2-оксо-7-фенил-1-(2,2,2-трифторэтил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (31 мг, 0,056 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (6 мг, выход 19%).
1H-ЯМР (500 МГц, MeOD) δ 7,74 (1H, с), 7,41 (2H, д), 7,37 (2H, д), 7,25-7,33 (3H, м), 7,17-7,25 (2H, м), 5,01 (2H, с), 4,90 (2H, кв.), 2,65-2,82 (2H, м), 2,50-2,63 (2H, м), 2,13-2,30 (1H, м), 1,85-2,02 (1H, м).
ЖХ/МС (способ A) RT=4,22 мин, M+2H+=455,19.
Пример 61: 6-(4-(1-аминоциклобутил)фенил)-1-циклобутил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
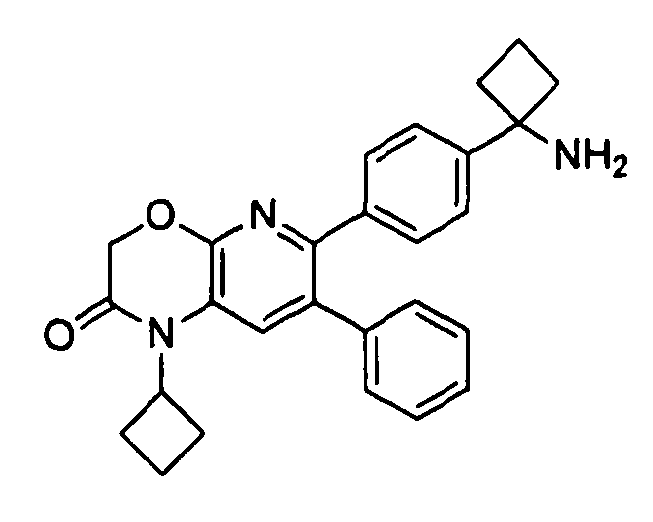
Стадия 1. трет-бутил-1-(4-(1-циклобутил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (50 мг, 0,106 ммоль), карбонат калия (44 мг, 0,318 ммоль) и бромциклобутан (0,030 мл, 0,318 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением оранжевой суспензии. Реакционную смесь перемешивали при 80°C в течение 1 часа, а затем при 100°C в течение 2 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в дихлорметан (3×3 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде не совсем белого твердого вещества (15 мг, выход 27%).
1H-ЯМР (500 МГц, CDCl3) δ 7,25-7,37 (8H, м), 7,16-7,25 (2H, м), 5,03 (1H, ушир. с), 4,78 (2H, с), 4,53 (1H, квинт.), 2,30-2,67 (8H, м), 2,02-2,14 (1H, м), 1,76-1,97 (3H, м), 1,10-1,50 (9H, ушир. м).
ЖХ/МС (способ A) RT=7,76 мин, M+H+=526,16.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-циклобутил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(1-циклобутил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (31 мг, 0,059 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (11 мг, выход 35%).
1H-ЯМР (500 МГц, MeOD) δ 7,35-7,58 (5H, м), 7,25-7,35 (3Н, м), 7,10-7,25 (2H, м), 4,80 (2H, с), 4,59 (1H, квинт.), 2,65-2,83 (2H, м), 2,40-2,65 (6Н, м), 2,13-2,29 (1Н, м), 1,79-2,02 (3Н, м).
ЖХ/МС (способ A) RT=4,58 мин, M+2H+=427,21.
Пример 62: 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрил
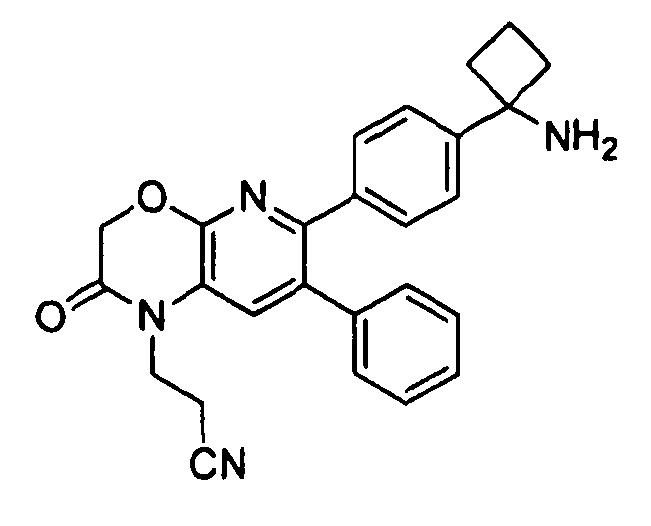
Стадия 1: трет-бутил-(1-(4-(1-(2-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Смесь трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (100 мг, 0,212 ммоль), 3-бромпропаннитрила (85 мг, 0,636 ммоль) и карбоната калия (88 мг) в DMF (4 мл) нагревали при 80°C в течение 16 ч. Реакционную смесь распределяли между дихлорметаном (20 мл) и водой (20 мл). Органическую фазу разделяли с использованием фазового сепаратора и концентрировали. Неочищенный продукт очищали на колонке (Biotage, 25 г), элюируя этилацетатом/циклогексаном, с получением продукта (45 мг).
1H ЯМР (500 МГц, CDCl3): 7,39 (1H, с), 7,16-7,39 (9H, м), 5,1 (1H, ушир.), 4,91 (2H, с), 4,25 (2H, т), 2,83 (2H, т), 2,3-2,5 (4H, м), 2,04-2,09 (1H, м), 1,79-1,84 (1H, м), 1,27-1,36 (9H, ушир.).
Стадия 2: 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрил
трет-Бутил-(1-(4-(1-(2-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (15 мг, 0,029 ммоль) растворяли в TFA (0,5 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (1 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (0,5 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (12 мг).
1H ЯМР (500 МГц, CH3OD) 7,77 (1H, с), 7,43 (2H, д), 7,39 (2H, д), 7,3 (3H, м), 7,27 (2H, м), 4,99 (2H, с), 4,37 (2H, т), 2,94 (2H, т), 2,73-2,79 (2H, м), 2,55-2,61 (2H, м), 2,2-2,27 (1H, м), 1,93-1,99 (1Н, м)
ЖХ/МС (способ A): RT=3,73 мин, M+H+=425.
Пример 63: 1-(4-(1-метил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
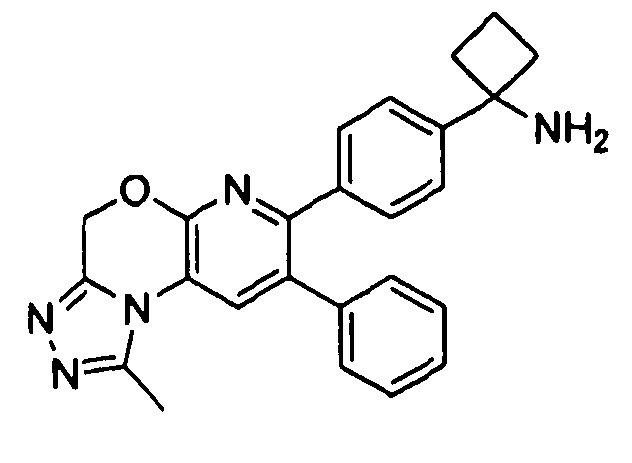
Стадия 1: трет-бутил-(1-(4-(1-метил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Раствор (E)-трет-бутил-(1-(4-(2-гидразоно-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (50 мг, 0,103 ммоль) в 1,1,1-триметоксиэтане (1 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→100% этилацетат в н-гексанах) с получением указанного в заголовке соединения (24 мг, 47%). ЖХ/МС (способ A): RT=6,71 мин, M+1=510.
1H ЯМР (500 МГц, CDCl3): 7,84 (1H, с), 7,37-7,33 (5H, м), 7,30-7,27 (2H, м), 7,24-7,19 (2H, м), 5,59 (2H, с), 2,87 (3H, с), 2,54-2,40 (4H, м), 2,13-2,02 (1H, м), 1,87-1,78 (1H, м), 1,45-1,27 (9H, ушир. с).
Стадия 2: 1-(4-(1-метил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(1-метил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (11 мг, 0,022 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления, и сушили остаток с получением целевого продукта в виде не совсем белого твердого вещества (5 мг, выход 55%). ЖХ/МС (способ A): RT=3,71 мин, M+1=411.
1H-ЯМР (500 МГц, CD3OD) δ 8,11 (1H, с), 7,49-7,44 (2H, д), 7,42-7,37 (2H, д), 7,35-7,30 (3Н, м), 7,29-7,25 (2Н, м), 5,63 (2Н, с), 2,84 (3Н, с), 2,80-2,70 (2H, м), 2,61-2,51 (2H, м), 2,28-2,17 (1H, м), 2,01-1,88 (1H, м).
Пример 64: 4-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)бутаннитрил
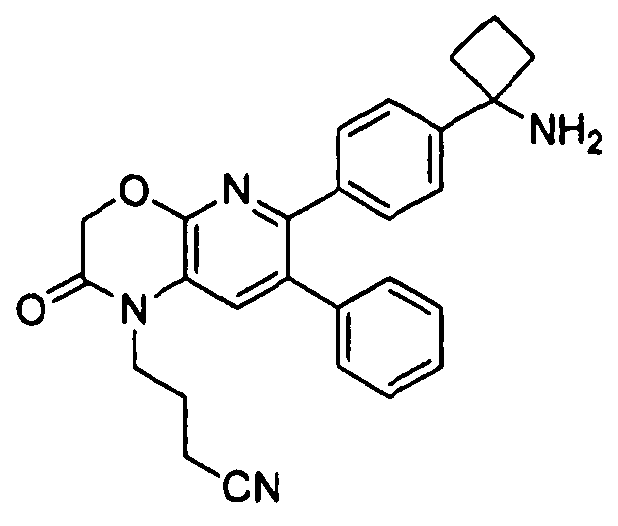
Стадия 1: трет-бутил-(1-(4-(1-(3-цианопропил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Следуя методике получения трет-бутил-(1-(4-(1-(2-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата, осуществляли взаимодействие трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (100 мг, 0,212 ммоль) с 4-бромбутаннитрилом (94 мг, 0,636 ммоль) с получением указанного в заголовке соединения (68 мг).
1H ЯМР (500 МГц, CDCl3): 7,20-7,32 (10H, м), 5,1 (1H, ушир.), 4,90 (2H, с), 4,11 (2H, м), 2,44-2,50 (6H, м), 2,05-2,13 (3H, м), 1,79-1,84 (1H, м), 1,27-1,36 (9H, ушир.).
Стадия 2: 4-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)бутаннитрил
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(3-цианопропил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (35 мг, 0,065 ммоль) с получением указанного в заголовке соединения (28 мг).
1H ЯМР (500 МГц, CH3OD) 7,62 (1H, с), 7,41 (2H, д), 7,39 (2H, д), 7,3 (3H, м), 7,25 (2H, м), 4,98 (2H, с), 4,19 (2H, т), 2,73-2,79 (2H, м), 2,55-2,61 (4H, м), 2,22-2,25 (1H, м), 2,10-2,25 (2H, м), 1,95-1,99 (1H, м).
ЖХ/МС (способ A): RT=4,01 мин, M+H+=440,2.
Пример 65: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(тетрагидро-2H-пиран-4-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
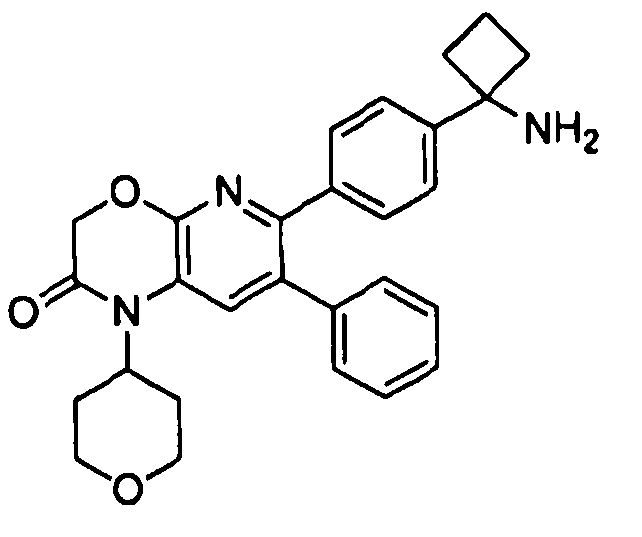
Стадия 1: трет-бутил-(1-(4-(6-гидрокси-3-фенил-5-((тетрагидро-2H-пиран-4-ил)амино)пиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-амино-6-гидрокси-3-фенилпиридин-2-ил)фенил)циклобутил)карбамата (200 мг, 0,46 ммоль) в безводном DCE (2 мл) в атмосфере азота добавляли дигидро-2H-пиран-4(3H)-он (88 мг, 0,88 ммоль), AcOH (0,16 мл, 2,78 ммоль), триацетоксиборгидрид натрия (275 мг, 1,30 ммоль). Полученную смесь перемешивали в течение 3 ч при комнатной температуре. Добавляли насыщенный раствор NaHCO3, и экстрагировали смесь AcOEt (3×10 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток использовали на следующей стадии без какой-либо очистки.
Стадия 2: трет-бутил-(1-(4-(2-оксо-7-фенил-1-(тетрагидро-2H-пиран-4-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(6-гидрокси-3-фенил-5-((тетрагидро-2H-пиран-4-ил)амино)пиридин-2-ил)фенил)циклобутил)карбамата (240 мг, 0,46 ммоль) в безводном THF (11 мл) добавляли N,N-диизопропилэтиламин (0,41 мл, 2,32 ммоль). При 0°C по каплям добавляли 2-хлорацетилхлорид (0,19 мл, 2,32 ммоль). Полученную смесь перемешивали в течение ночи при комнатной температуре. Добавляли насыщенный раствор NaHCO3 (8 мл) и перемешивали смесь с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры, органическую фазу разделяли, и экстрагировали водную фазу AcOEt (3×10 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→50% EtOAc в циклогексане) с получением указанного в заголовке соединения (154 мг, 59%).
1H ЯМР (500 МГц, CDCl3): 7,45 (1H, с), 7,24-7,22 (5H, м), 7,19-7,17 (2H, м), 7,14-7,12 (2H, м), 4,94 (1H, с), 4,71 (2H, с), 4,60-4,55 (1H, м), 4,07-4,03 (2H, м), 3,45 (2H, т), 2,63-2,54 (2H, м), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,79-1,65 (3Н, м), 1,40-1,10 (9H, ушир.).
Стадия 3: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(тетрагидро-2H-пиран-4-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(2-оксо-7-фенил-1-(тетрагидро-2H-пиран-4-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (55 мг, 0,10 ммоль) с получением указанного в заголовке соединения (53 мг, 93%). ЖХ/МС (способ A): RT=4,19 мин, M+1=457.
1H ЯМР (500 МГц, MeOD): 7,76 (1H, с), 7,44-7,38 (4H, м), 7,33-7,31 (3H, м), 7,27-7,25 (2H, м), 4,90 (2H, с), 4,59-4,53 (1H, м), 4,09-4,06 (2H, м), 3,60 (2H, т), 2,80-2,72 (4H, м), 2,59-2,53 (2H, м), 2,25-2,21 (1H, м), 1,98-1,94 (1H, м), 1,81-1,79 (2H, м).
Пример 66: 5-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пентаннитрил
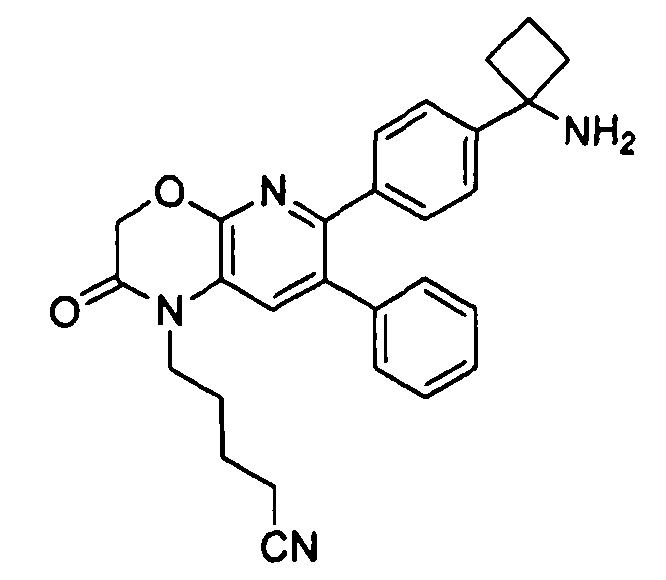
Стадия 1: трет-бутил-(1-(4-(1-(4-цианобутил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Следуя методике получения трет-бутил-(1-(4-(1-(2-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата, осуществляли взаимодействие трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (100 мг, 0,212 ммоль) с 5-бромпентаннитрилом (103 мг, 0,636 ммоль) с получением указанного в заголовке соединения (82 мг).
1H ЯМР (500 МГц, CDCl3): 7,20-7,33 (10H, м), 5,03 (1H, ушир.), 4,91 (2H, с), 4,05 (2H, т), 2,45-2,51 (6H, м), 2,05-2,10 (1H, м), 1,88-1,94 (2H, м), 1,67-1,85 (3H, м), 1,27-1,38 (9H, ушир.).
Стадия 2: 5-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пентаннитрил
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(4-цианобутил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (25 мг, 0,045 ммоль) с получением указанного в заголовке соединения (14,5 мг).
1H ЯМР (500 МГц, CH3OD) 7,60 (1H, с), 7,42 (2H, д), 7,39 (2H, д), 7,31 (3H, м), 7,25 (2H, м), 4,97 (2H, с), 4,11 (2H, т), 2,73-2,79 (2H, м), 2,54-2,61 (4H, м), 2,22-2,25 (1H, м), 1,95-1,97 (1H, м), 1,84-1,89 (2H, м), 1,74-1,78 (2H, м).
ЖХ/МС (способ A): RT=4,19 мин, M+H+=453,5.
Пример 67: 1-аллил-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
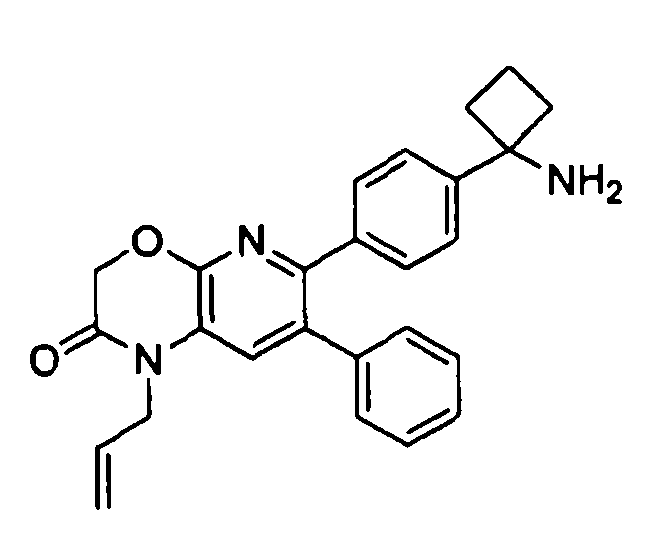
Стадия 1: трет-бутил-1-(4-(1-аллил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В герметично закрытую пробирку добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (100 мг, 0,212 ммоль) в DMF с получением коричневого раствора. Добавляли бромциклопропан (76,9652 мг, 0,636 ммоль) и карбонат калия. Реакционную смесь нагревали при 80°C в течение 16 ч. Затем реакционную смесь нагревали в условиях обработки микроволнами при 100°C в течение 2 ч и при 130°C в течение 8 ч. Реакционную смесь распределяли между дихлорметаном (20 мл) и водой (20 мл). Органическую фазу разделяли с использованием фазового сепаратора и концентрировали. Неочищенный продукт очищали на колонке (силикагель Biotage, 25 г), элюируя этилацетатом/циклогексаном с получением продукта (12 мг).
1H ЯМР (500 МГц, CDCl3) 7,17-7,23 (м, 8H), 7,07-7,10 (м, 2H), 5,77-5,84 (м, 1H), 5,18-5,24 (м, 2H), 4,51 (д, 2H), 2,2-2,5 (м, 4H), 1,9-2,0 (м, 1H), 1,69-1,79 (м, 1H), 1,1-1,35 (ушир., 9H).
ЖХ/МС (способ A): RT=7,9 мин, M+H+=512,17.
Стадия 2: 1-аллил-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В круглодонную колбу, содержащую трет-бутил-1-(4-(1-аллил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (6,5 мг, 0,013 ммоль), добавляли трифторуксусную кислоту (0,5 мл). Полученный раствор перемешивали в течение 60 секунд, а затем концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (1 мл) и трижды концентрировали досуха с получением трифторуксуснокислой соли продукта (3,55 мг).
1H ЯМР (500 МГц, CD3OD) 7,49 (с, 1H), 7,38-7,43 (м, 4H), 7,29-7,30 (м, 3H), 7,18-7,20 (м, 2H), 5,9-6,0 (м, 1H), 5,25-5,35 (м, 2H), 5,01 (с, 2H), 4,68 (д, 2H), 2,7-2,8 (м, 2H), 2,55-2,65 (м, 2H), 2,21-2,27 (м, 1H), 1,9-2,0 (м, 1H).
ЖХ/МС (способ A): RT=4,38 мин, M+H+=413,18.
Пример 68: 6-(4-(1-аминоциклобутил)фенил)-1-(2-гидроксиэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
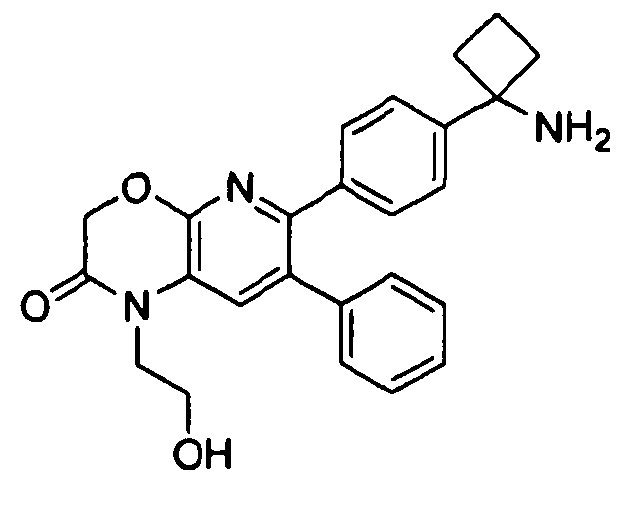
Стадия 1: трет-бутил-(1-(4-(1-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Следуя методике получения трет-бутил-(1-(4-(1-(2-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата, осуществляли взаимодействие трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (100 мг, 0,212 ммоль) с 2-бромэтанолом (80 мг, 0,636 ммоль) с получением указанного в заголовке соединения (75 мг).
1H ЯМР (500 МГц, CDCl3): 7,47 (с, 1H), 7,23-7,29 (м, 7H), 7,16-7,19 (м, 2H), 5,04 (ушир., 1H), 4,89 (с, 2H), 4,12 (м, 2H), 3,94 (м, 2H), 2,37-2,48 (м, 4H), 2,37 (т, 1H), 2,05 (м, 1H), 1,81 (м, 1H), 1,2-1,43 (ушир., 9H).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(2-гидроксиэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В круглодонную колбу, содержащую трет-бутил-1-(4-(1-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (25 мг, 0,048 ммоль), добавляли дихлорметан (2,5 мл), а затем соляную кислоту (2,5 мл, 2M в диэтиловом эфире). Полученный раствор перемешивали в течение 16 ч. Осадок фильтровали и промывали эфиром (2×10 мл) и сушили с получением продукта (15,5 мг).
1H ЯМР (500 МГц, CH3OD) 7,78 (с, 1H), 7,42 (д, 2H), 7,38 (д, 2H), 7,30 (м, 3H), 7,22 (м, 2H), 4,96 (с, 2H), 4,16 (т, 2H), 3,86 (т, 2H), 2,73-2,79 (м, 2H), 2,54-2,60 (м, 2H), 2,21-2,26 (м, 1H), 1,94-1,99 (м, 1H).
ЖХ/МС (способ D): RT=1,99 мин, M+H+=416,2.
Пример 69: 1-(4-(8-фенил-1-пропил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин

Стадия 1: трет-бутил-(1-(4-(8-фенил-1-пропил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Раствор (E)-трет-бутил-(1-(4-(2-гидразоно-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (60 мг, 0,124 ммоль) в 1,1,1-триметоксибутане (1 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→100% этилацетат в н-гексанах) с получением указанного в заголовке соединения (30 мг, 45%). ЖХ/МС (способ A): RT=7,34 мин, M+1=538.
1H ЯМР (500 МГц, CDCl3): 7,80 (1H, с), 7,36-7,32 (5H, м), 7,31-7,27 (2H, м), 7,23-7,19 (2H, м), 5,57 (2H, с), 3,08 (2H, т), 2,55-2,40 (4H, м), 2,15-2,03 (1H, м), 2,00 (2H, кв.), 1,89-1,77 (1H, м), 1,41-1,29 (9H, ушир.), 1,12 (3H, т).
Стадия 2: 1-(4-(8-фенил-1-пропил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(8-фенил-1-пропил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (39 мг, 0,073 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли в условиях пониженного давления, и сушили остаток с получением целевого продукта в виде не совсем белого твердого вещества (10 мг, выход 30%). ЖХ/МС (способ A): RT=3,85 мин, M+1=439.
1H-ЯМР (500 МГц, CD3OD) δ 8,09 (1H, с), 7,50 (2H, д), 7,43 (2H, д), 7,37-7,34 (3Н, м), 7,31-7,27 (2H, м), 5,66 (2H, с), 3,2 (2H, т), 2,83-2,71 (2H, м), 2,64-2,54 (2H, м), 2,32-2,19 (1H, м), 2,02-1,90 (1H, м+2H, т), 1,16-1,09 (3Н, т).
Пример 70: 1-(4-(1-(метилсульфонил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин
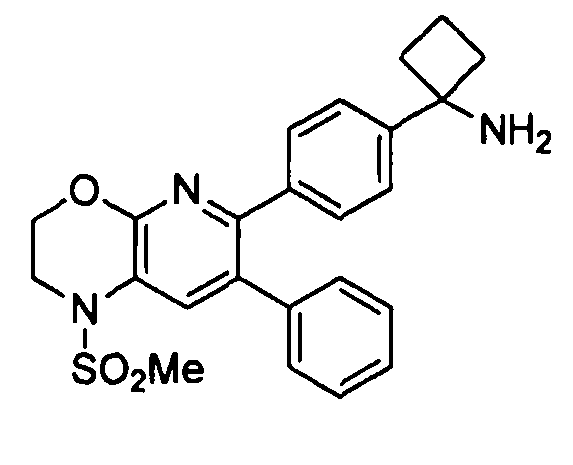
Стадия 1: трет-бутил-(1-(4-(1-(метилсульфонил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (50 мг, 0,11 ммоль) в безводном DCM (1 мл) в атмосфере азота добавляли триэтиламин (46 мкл, 0,33 ммоль) и метансульфонилхлорид (26 мкл, 0,33 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь дихлорметаном (3×10 мл) с использованием фазового сепаратора (Isolute® SPE). Объединенные органические фазы концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→50% EtOAc в циклогексане) с получением указанного в заголовке соединения (45 мг, 77%).
1H ЯМР (500 МГц, CDCl3): 8,00 (1H, с), 7,25 (2H, д), 7,19-7,17 (5H, м), 7,11-7,10 (2H, м), 4,95 (1H, с), 4,43 (2H, т), 3,90 (2H, т), 2,55-2,25 (7H, м), 2,06-1,98 (1H, м), 1,82-1,73 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 1-(4-(1-(метилсульфонил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1-(метилсульфонил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (45 мг, 0,08 ммоль) с получением указанного в заголовке соединения (44 мг, 95%). ЖХ/МС (способ A): RT=2,34 мин, M+1=437.
1H ЯМР (500 МГц, MeOD): 8,15 (1H, с), 7,43-7,38 (4H, м), 7,30-7,28 (3H, м), 7,21-7,19 (2H, м), 4,57 (2H, т), 4,01 (2H, т), 3,19 (3H, с), 2,79-2,73 (2H, м), 2,60-2,54 (2H, м), 2,24-2,21 (1H, м), 1,99-1,94 (1H, м).
Пример 71: 6-(4-(1-аминоциклобутил)фенил)-3-(2-гидроксиэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
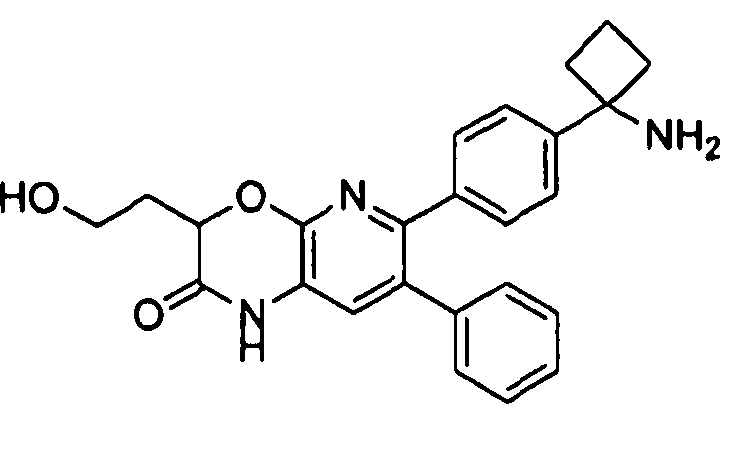
Стадия 1: трет-бутил-(1-(4-(3-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
В круглодонную колбу объемом 100 мл добавляли трет-бутил-1-(4-(5-амино-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутилкарбамат (0,5 г, 1,159 ммоль) в THF (объем: 25 мл) с получением красной суспензии, а затем добавляли DIPEA (1,0 мл). При 0°C по каплям добавляли 2,4-дибромбутаноилхлорид (0,735 г, 2,78 ммоль) в THF (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь переносили в круглодонную колбу объемом 250 мл, и добавляли насыщенный раствор бикарбоната натрия (40 мл) и бикарбонат натрия (1 г). Полученную смесь нагревали до температуры возгонки в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры. Органическую фазу разделяли. Водную фазу экстрагировали этилацетатом (45 мл). Органические фазы объединяли и промывали водой (2×30 мл), солевым раствором (25 мл), а затем концентрировали в условиях пониженного давления. Остаток суспендировали в DCM (10 мл). Осадок собирали путем фильтрования и промывали DCM (5 мл) с получением неочищенного продукта, который очищали на колонке (силикагель Biotage, 25 г) с получением трет-бутил-1-(4-(3-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (45 мг) и трет-бутил-1-(4-(7-фенил-3,3a-дигидро-2H-фуро[2,3-e]пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (0,35 г).
1H ЯМР (500 МГц, CD3OD): 7,50 (с, 1H), 7,31 (м, 2H), 7,26 (м, 5H), 7,16 (м, 2H), 5,38 (дд, 1H), 4,72 (т, 1H), 4,55 (м, 1H), 2,92 (м, 1H), 2,67 (м, 1H), 2,43-2,47 (м, 4H), 2,07 (м, 1H), 1,85 (м, 1H), 1,23-1,39 (ушир., 9H).
ЖХ/МС (способ A): RT=6,89 мин, M+H+=498,12.
Раствор трет-бутил-1-(4-(7-фенил-3,3a-дигидро-2H-фуро[2,3-e]пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (0,35 г, 0,703 ммоль) в DCM (15 мл) и уксусной кислоте (10 мл) перемешивали при комнатной температуре в течение 48 ч. Реакционную смесь концентрировали и распределяли между этилацетатом (40 мл) и водой (30 мл). Отделенную органическую фазу промывали водой (2×25 мл), солевым раствором и концентрировали с получением продукта (0,36 г, 45 мг).
1H ЯМР (500 МГц, CD3OD): 7,23-7,31 (м, 8H), 7,16 (м, 2H), 5,05 (дд, 1H), 3,91 (м, 1H), 3,85 (м, 1H), 2,45 (м, 4H), 2,26 (м, 1H), 2,15 (м, 1H), 2,07 (м, 1H), 1,86 (м, 1H), 1,23-1,39 (ушир., 9H).
ЖХ/МС (способ A): RT=5,92 мин, M+H+=516,04.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-3-(2-гидроксиэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(4-цианобутил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (15 мг, 0,029 ммоль) с получением указанного в заголовке соединения (8,5 мг).
1H ЯМР (500 МГц, CD3OD): 7,28-7,42 (м, 8H), 7,18 (м, 2H), 5,08 (дд, 1H), 3,84-3,93 (м, 2H), 2,73-2,79 (м, 2H), 2,55-2,61 (м, 2H), 2,25-2,29 (м, 2H), 2,15-2,24 (м, 1H), 1,95-1,97 (м, 1H).
ЖХ/МС (способ A): RT=3,49 мин, M+H+=416,18.
Пример 72: 6-(4-(1-аминоциклобутил)фенил)-4-метил-7-фенил-3,4-дигидропиридо[2,3-b]пиразин-2(1H)-он
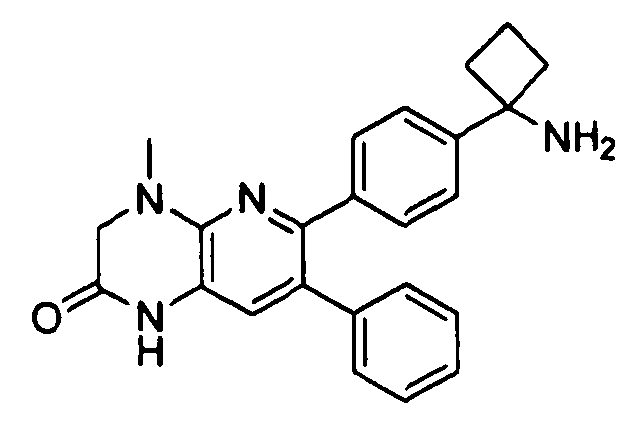
Стадия 1: метил-2-((6-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-3-нитро-5-фенилпиридин-2-ил)(метил)амино)ацетат
В пробирку для микроволновой обработки добавляли трет-бутил-(1-(4-(6-хлор-5-нитро-3-фенилпиридин-2-ил)фенил)циклобутил)карбамат (200 мг, 0,417 ммоль), метил-2-(метиламино)ацетата гидрохлорид (58 мг, 0,417 ммоль) и триэтиламин (58 мкл, 0,47 ммоль) в метаноле (2 мл). Раствор нагревали до 80°C в условиях обработки микроволновым излучением в течение 1 часа. Реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 100/0→0/100) с получением указанного в заголовке соединения (103 мг, 65%).
1H ЯМР (500 МГц, CDCl3): 8,29 (1H, с), 7,25-7,38 (7H, м), 7,15-7,23 (2H, м), 5,05 (1H, ушир. с), 4,39 (2H, с), 3,83 (3H, с), 3,11 (3H, с), 2,25-2,65 (4H, м), 2,05-2,20 (1H, м), 1,80-1,95 (1H, м), 1,10-1,50 (9H, ушир.).
Стадия 2: трет-бутил-(1-(4-(4-метил-2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[2,3-b]пиразин-6-ил)фенил)циклобутил)карбамат
В стеклянный автоклав добавляли метил-2-((6-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-3-нитро-5-фенилпиридин-2-ил)(метил)амино)ацетат (800 мг, 1,46 ммоль) и 10% палладированный уголь (300 мг, 0,28 ммоль) в THF (100 мл). Раствор гидрировали при комнатной температуре водородом под давлением 1,5 атм. в течение 20 часов. Реакционную смесь фильтровали через целит, а затем концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 100/0→0/100) с получением указанного в заголовке соединения (270 мг, 38%).
1H ЯМР (500 МГц, CDCl3): 7,35 (2H, д), 7,20-7,33 (5H, м), 7,15 (2H, д), 6,98 (1H, с), 5,05 (1H, ушир. с), 4,11 (2H, с), 3,18 (3H, с), 2,20-2,70 (4H, ушир. м), 2,00-2,14 (1H, м), 1,77-1,87 (1H, м), 1,10-1,50 (9H, ушир.).
Стадия 3: 6-(4-(1-аминоциклобутил)фенил)-4-метил-7-фенил-3,4-дигидропиридо[2,3-b]пиразин-2(1H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-1-(4-(4-метил-2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[2,3-b]пиразин-6-ил)фенил)циклобутилкарбамат (29 мг, 0,060 ммоль) с получением указанного в заголовке соединения (29 мг, 97%). ЖХ/МС (способ A): RT=3,69 мин, M+2H+=386.
1H ЯМР (500 МГц, MeOD): 7,44 (2H, д), 7,32 (2H, д), 7,20-7,28 (3H, м), 7,12 (2H, д), 7,02 (1H, с), 4,08 (2H, с), 3,11 (3H, с), 2,70-2,80 (2H, м), 2,50-2,63 (2H, м), 2,13-2,27 (1H, м), 1,88-2,02 (1H, м).
Пример 73: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-2,2-диоксид
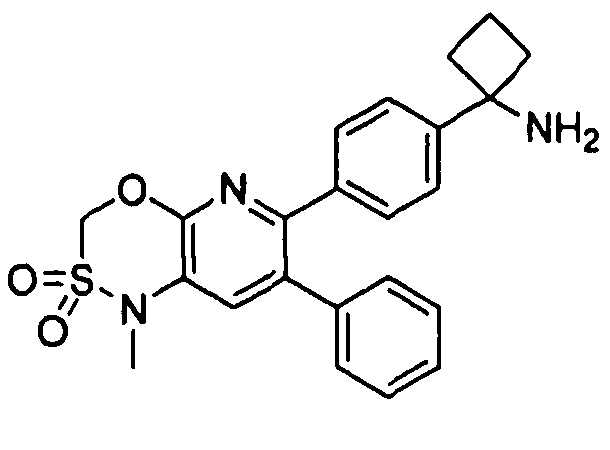
Стадия 1: трет-бутил-(1-(4-(2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат
В реакционную пробирку объемом 40 мл добавляли трет-бутил-1-(4-(5-амино-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутилкарбамат (500 мг, 1,159 ммоль) в безводном пиридин (5 мл) с получением желтого раствора. После перемешивания при комнатной температуре в течение 15 минут, добавляли хлорметансульфонилхлорид (126 мкл, 1,390 ммоль) и перемешивали полученный раствор при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления и повторно растворяли в безводном метаноле (5 мл), а затем добавляли карбонат калия (641 мг, 4,63 ммоль) и нагревали до 60°C в течение ночи. Реакционную смесь оставляли охлаждаться до комнатной температуры и концентрировали досуха в условиях пониженного давления. Остаток повторно растворяли в этилацетате (5 мл) и промывали водой/солевым раствором (50/50, 2×5 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением коричневого масла. Это масло очищали методом хроматографии на силикагеле Biotage (элюирование градиентом циклогексан/этилацетат/дихлорметан/метанол 80/10/10/0→0/90/10/0→0/90/10) с получением целевого продукта в виде не совсем белого твердого вещества (330 мг, выход 56%).
1H-ЯМР (500 МГц, d6-ацетон) δ 7,35 (1H, с), 7,23-7,32 (7H, м), 7,16-7,32 (2H, м), 5,39 (2Н, с), 2,30-2,60 (4H, м), 2,00-2,12 (1H, м), 1,75-1,88 (1H, м), 1,05-1,40 (9H, ушир. м).
ЖХ/МС (способ A) RT=6,68 мин, M+H+=508,01.
Стадия 2: трет-бутил-(1-(4-(1-метил-2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-(1-(4-(2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат (50 мг, 0,099 ммоль), карбонат калия (41 мг, 0,296 ммоль) и йодметан (7,5 мкл, 0,118 ммоль) в безводном Ν,Ν-диметилформамиде (1 мл) с получением оранжевой суспензии. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в дихлорметан (3×3 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде бесцветного масла (50 мг, выход 97%).
1H-ЯМР (500 МГц, CDCl3) δ 7,24-7,47 (8H, м), 7,16-7,23 (2H, м), 5,23 (2H, с), 5,05 (1Н, ушир. с), 3,36 (3Н, с), 2,20-2,65 (4H, м), 2,01-2,15 (1H, м), 1,77-1,90 (1H, м), 1,10-1,50 (9H, ушир. м).
ЖХ/МС (способ A) RT=7,16 мин, M+H+=522,01.
Стадия 3: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-2,2-диоксид
трет-Бутил-(1-(4-(1-метил-2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат (50 мг, 0,096 ммоль) растворяли в TFA (3 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~3 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (32 мг, выход 62%).
1H-ЯМР (500 МГц, MeOD) δ 7,58 (1H, с), 7,41 (2H, д), 7,37 (2H, д), 7,25-7,33 (3Н, м), 7,17-7,25 (2H, м), 5,45 (2H, с), 3,35 (3H, с), 2,66-2,78 (2H, м), 2,46-2,57 (2H, м), 2,12-2,25 (1H, м), 1,85-1,97 (1H, м).
ЖХ/МС (способ A) RT=3,98 мин, M+2H+=423,13.
Пример 74: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-3-ил)-1H-пиридо[3,2-e][1,3,4]оксатиазин-2,2-диоксид
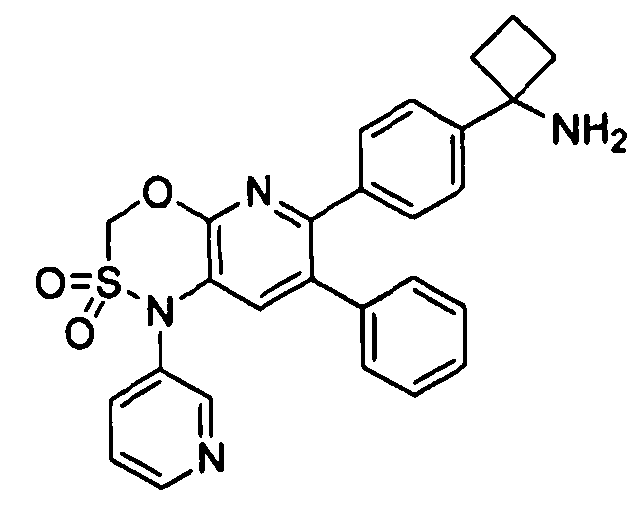
Стадия 1: трет-бутил-(1-(4-(2,2-диоксидо-7-фенил-1-(пиридин-3-ил)-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-(1-(4-(2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат (50 мг, 0,099 ммоль), пиридин-3-илбороновую кислоту (24 мг, 0,197 ммоль), ацетат меди(II) (27 мг, 0,148 ммоль) и 5Å молекулярные сита (30 мг) в безводном дихлорметане (1 мл). К этой смеси добавляли безводный пиридин (16 мкл, 0,197 ммоль) и перемешивали на воздухе полученную черную суспензию при комнатной температуре в течение 24 часов. Дополнительно добавляли пиридин (1 мл) и перемешивали полученную черную суспензию при комнатной температуре в течение 24 часов. Дополнительно добавляли пиридин-3-илбороновую кислоту (24 мг, 0,197 ммоль) и ацетат меди(II) (27 мг, 0,148 ммоль) и перемешивали смесь при комнатной температуре в течение 72 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления, и очищали остаток методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 88/12→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (40 мг, выход 70%).
1H-ЯМР (500 МГц, CDCl3) δ 8,67-8,76 (2H, м), 7,89 (1H, д), 7,54 (1H, дд), 7,20-7,35 (7H, м), 7,06 (2H, д), 7,03 (1H, с), 5,38 (2H, с), 5,07 (1H, ушир. с), 2,20-2,60 (4H, м), 2,00-2,15 (1H, м), 1,78-1,90 (1H, м), 1,10-1,50 (9H, ушир. с).
ЖХ/МС (способ A) RT=6,98 мин, M+H+=584,99.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-3-ил)-1H-пиридо[3,2-e][1,3,4]оксатиазин-2,2-диоксид
трет-Бутил-(1-(4-(2,2-диоксидо-7-фенил-1-(пиридин-3-ил)-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат (40 мг, 0,068 ммоль) растворяли в TFA (3 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~3 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (23 мг, выход 56%).
1H-ЯМР (500 МГц, MeOD) δ 8,70 (1H, д), 8,65 (1H, дд), 8,00 (1H, дд), 7,61 (1H, дд), 7,44 (2H, д), 7,38 (2H, д), 7,17-7,25 (3H, м), 7,09 (1H, с), 7,06 (2H, д), 5,67 (2H, с), 2,68-2,77 (2H, м), 2,50-2,59 (2H, м), 2,14-2,25 (1H, м), 1,87-2,00 (1H, м).
ЖХ/МС (способ A) RT=4,00 мин, M+2H+=486,07.
Пример 75: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-3-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
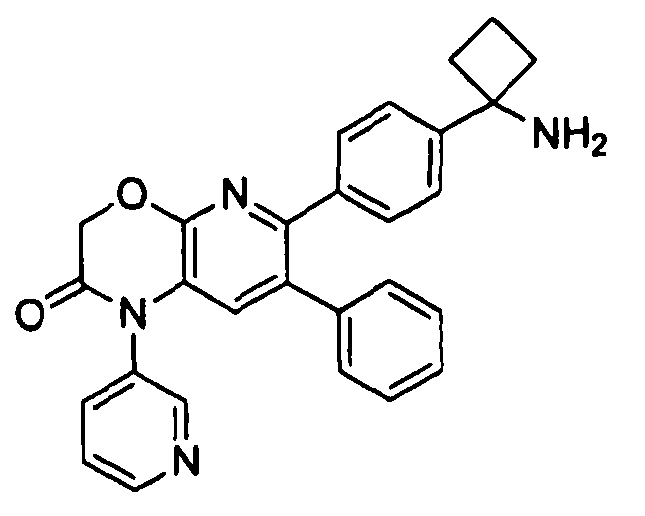
Стадия 1: трет-бутил-1-(4-(2-оксо-7-фенил-1-(пиридин-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (100 мг, 0,212 ммоль), пиридин-3-илбороновую кислоту (52 мг, 0,424 ммоль), ацетат меди(II) (58 мг, 0,318 ммоль) и 5Å молекулярные сита (60 мг) в безводном дихлорметане (2 мл). К этой смеси добавляли безводный пиридин (34 мкл, 0,424 ммоль) и перемешивали на воздухе полученную голубую суспензию при комнатной температуре в течение 24 часов. Дополнительно добавляли пиридин (1 мл) и перемешивали полученную голубую суспензию при комнатной температуре в течение 24 часов. Дополнительно добавляли пиридин-3-илбороновую кислоту (52 мг, 0,424 ммоль) и ацетат меди(II) (58 мг, 0,318 ммоль) и перемешивали смесь при комнатной температуре в течение 72 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления, и очищали остаток методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 88/12→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (32 мг, выход 28%).
1H-ЯМР (500 МГц, CDCl3) δ 8,50-8,80 (2H, ушир. м), 7,71 (1H, д), 7,50 (1H, ушир. с), 7,09-7,26 (7H, м), 6,92-7,02 (2H, д), 6,71 (1H, с), 5,00 (3H, с), 2,10-2,55 (4H, м), 1,92-2,05 (1H, м), 1,66-1,80 (1H, м), 1,00-1,45 (9H, ушир. м).
ЖХ/МС (способ A) RT=6,61 мин, M+H+=549,09.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-3-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(2-оксо-7-фенил-1-(пиридин-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (30 мг, 0,055 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (15 мг, выход 49%).
1H-ЯМР (500 МГц, MeOD) δ 8,62-8,77 (2H, м), 7,98 (1H, д), 7,68 (1H, дд), 7,36 (2H, д), 7,39 (2H, д), 7,15-7,24 (3H, м), 6,97-7,05 (2H, м), 6,77 (1H, с), 5,14 (2H, с), 2,66-2,77 (2H, м), 2,46-2,58 (2H, м), 2,13-2,25 (1H, м), 1,86-1,97 (1H, м).
ЖХ/МС (способ A) RT=3,71 мин, M+2H+=450,20.
Пример 76: 2-(6-(4-(1-аминоциклобутил)фенил)-2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-1-ил)ацетонитрил
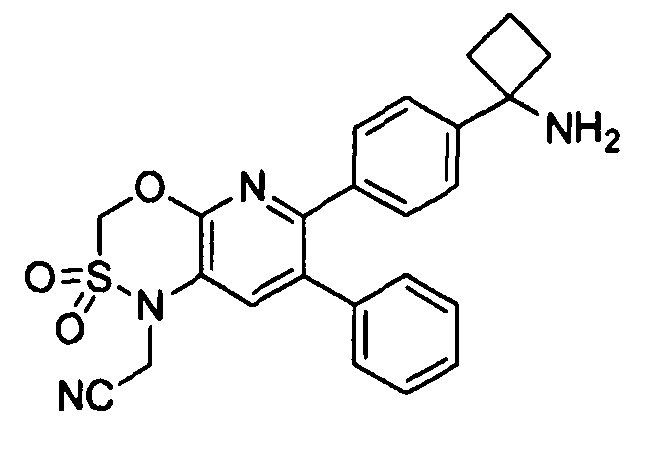
Стадия 1: трет-бутил-(1-(4-(1-(цианометил)-2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-(1-(4-(2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат (50 мг, 0,099 ммоль), карбонат калия (41 мг, 0,296 ммоль) и 2-бромацетонитрил (0,021 мл, 0,296 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением желтой суспензии. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в дихлорметан (3×3 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде бесцветного масла (32 мг, выход 59%).
1H-ЯМР (500 МГц, CDCl3) δ 8,04 (1Н, с), 7,25-7,38 (7H, м), 7,17-7,25 (2H, м), 5,32 (2H, с), 5,05 (1H, ушир. с), 4,73 (2H, с), 2,20-2,65 (4Н, м), 2,00-2,18 (1Н, м), 1,77-1,90 (1H, м), 1,05-1,55 (9Н, ушир. м).
ЖХ/МС (способ D) RT=1,60 мин, M+H+=547,10.
Стадия 2: 2-(6-(4-(1-аминоциклобутил)фенил)-2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-1-ил)ацетонитрил
трет-Бутил-(1-(4-(1-(цианометил)-2,2-диоксидо-7-фенил-1H-пиридо[3,2-e][1,3,4]оксатиазин-6-ил)фенил)циклобутил)карбамат (32 мг, 0,059 ммоль) растворяли в TFA (3 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~3 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (16 мг, выход 49%).
1H-ЯМР (500 МГц, MeOD) δ 7,83 (1H, с), 7,44 (2H, д), 7,39 (2H, д), 7,26-7,35 (3Н, м), 7,20-7,26 (2H, м), 5,53 (2H, с), 5,01 (2H, с), 2,68-2,78 (2H, м), 2,50-2,60 (2H, м), 2,14-2,27 (1H, м), 1,87-1,99 (1H, м).
ЖХ/МС (способ A) RT=4,00 мин, M+2H+=448,12.
Пример 77: 6-(4-(1-аминоциклобутил)фенил)-3-(2-гидроксиэтил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
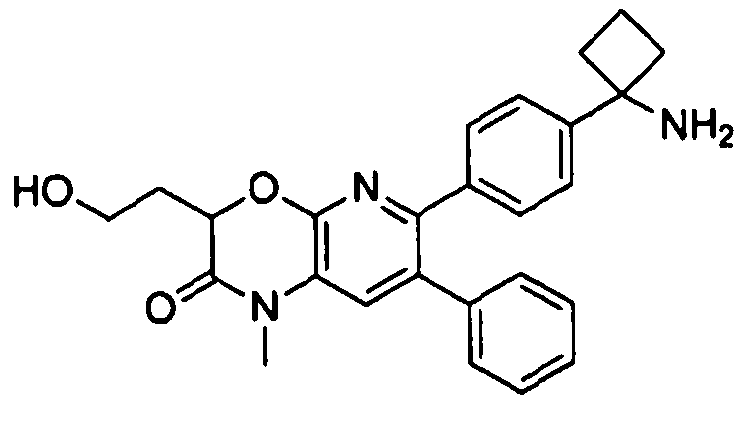
Стадия 1: трет-бутил-(1-(4-(3-(2-гидроксиэтил)-1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-1-(4-(3-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (40 мг, 0,078 ммоль) в DMF (1 мл) добавляли карбонат калия (32 мг) и метийодид (13,2 мг, 0,093 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь распределяли между дихлорметаном (20 мл) и водой (20 мл). Органическую фазу разделяли с использованием фазового сепаратора и концентрировали. Неочищенный продукт очищали на колонке (силикагель Biotage, размер колонки: 25 г), элюируя этилацетатом/циклогексаном, с получением продукта (18 мг).
1H ЯМР (500 МГц, CDCl3): 7,25-7,30 (м, 8H), 7,19 (м, 2H), 5,04 (дд, 1H), 5,01 (ушир., 1H), 3,95 (м, 2H), 3,40 (с, 3H), 2,45 (м, 4H), 2,45 (м, 2H), 2,02 (м, 1H), 1,79 (м, 1H), 1,15-1,35 (м, 9H).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-3-(2-гидроксиэтил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(3-(2-гидроксиэтил)-1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (12,5 мг, 0,024 ммоль) с получением указанного в заголовке соединения (5,5 мг).
1H ЯМР (500 МГц, CH3OD) 7,64 (с, 1H), 7,41 (м, 4H), 7,28 (м, 3H), 7,18 (м, 2H), 5,09 (дд, 1H), 3,80-3,92 (м, 2H), 3,41 (с, 1H), 2,72 (м, 2H), 2,54 (м, 2H), 2,24 (м, 2H), 2,13 (м, 1H), 1,94 (м, 1H).
ЖХ/МС (способ A): RT=3,76 мин, M+H+=413,16.
Пример 78: 2-(6-(4-(1-аминоциклобутил)фенил)-3-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
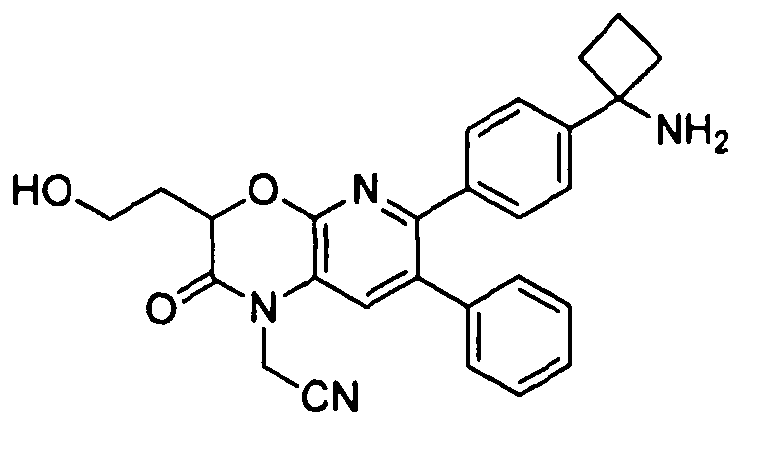
Стадия 1: трет-бутил-(1-(4-(1-(цианометил)-3-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-1-(4-(3-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (51 мг, 0,099 ммоль) в DMF (2 мл) добавляли карбонат калия (41 мг) и 2-бромацетонитрил (35,5 мг, 0,297 ммоль). Реакционную смесь перемешивали при 50°C в течение 3 ч. Реакционную смесь распределяли между дихлорметаном (20 мл) и водой (20 мл). Органическую фазу разделяли с использованием фазового сепаратора и концентрировали. Неочищенный продукт очищали на колонке (силикагель Biotage, размер колонки: 25 г), элюируя этилацетатом/циклогексаном, с получением продукта (18 мг). ЖХ/МС (способ A): RT=6,45 мин, M+H: 555,0.
Стадия 2: 2-(6-(4-(1-аминоциклобутил)фенил)-3-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(цианометил)-3-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (15 мг, 0,027 ммоль) с получением указанного в заголовке соединения (7,5 мг).
1H ЯМР (500 МГц, CH3OD) 7,67 (с, 1H), 7,41 (д, 2H), 7,38 (д, 2H), 7,30 (м, 3H), 7,24 (м, 2H), 5,18 (дд, 1H), 5,11 (с, 2H), 3,81-3,89 (м, 2H), 2,71-2,77 (м, 2H), 2,52-2,58 (м, 2H), 2,27-2,30 (м, 1H), 2,16-2,23 (м, 2H), 1,93-2,10 (м, 1H).
ЖХ/МС (способ A): RT=3,78 мин, M+H+=456,2.
Пример 79: 7-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он
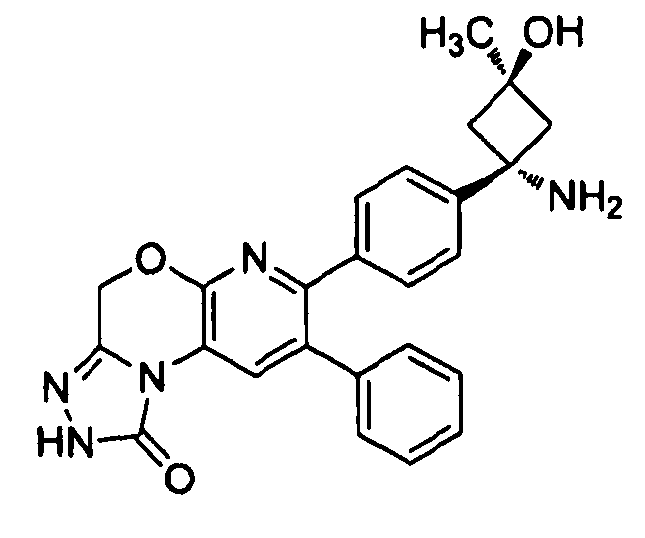
Стадия 1: 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)изоиндолин-1,3-дион
В круглодонную колбу объемом 15 мл добавляли 7-бром-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он (30 мг, 0,087 ммоль), 2-((1r,3r)-1-(4-(5,5-диметил-1,3,2-диоксаборинан-2-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион (30,4 мг, 0,072 ммоль) и карбонат цезия (118 мг, 0,362 ммоль) в 1,4-диоксане (1,55 мл) и воде (0,5 мл) с получением желтого раствора. Реакционную смесь дегазировали в течение 15 минут, затем добавляли аддукт PdCl2(dppf)-CH2Cl2 (20 мг) и дегазировали еще в течение 15 минут. Реакционную смесь нагревали в атмосфере азота до 50°C в течение ночи. Реакционную смесь оставляли охлаждаться до комнатной температуры и добавляли воду (7 мл). Реакционную смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические слои концентрировали в условиях пониженного давления. Неочищенный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0%→100% этилацетат в н-гексанах) с получением указанного в заголовке соединения (5 мг, 12%). ЖХ/МС (способ D): RT=1,167 мин, M+1=572.
1H ЯМР (500 МГц, CD3OD): 8,53 (1H, с), 7,84-7,78 (4H, м), 7,71-7,64 (2H, м), 7,51-7,45 (3Н, м), 7,30-7,22 (4Н, м), 5,38 (2Н, с), 3,50-3,45 (2Н, м), 3,36 (3Н, с), 3,02-2,96 (2Н, м).
Стадия 2: 7-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он
В пробирку для микроволновой обработки объемом 10 мл добавляли 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)изоиндолин-1,3-дион (5 мг, 8,75 мкмоль), гидразина моногидрат (2,75 мкл, 0,087 ммоль) в 1,4-диоксане (0,3 мл) и этанол (0,1 мл) с получением бесцветного раствора. Реакционную смесь нагревали до 120°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления твердого вещества. Это вещество тщательно промывали этанолом. Фильтраты объединяли и концентрировали в условиях пониженного давления. Неочищенный остаток (5 мг) очищали методом препаративной ЖХВД. Объединенные содержащие продукт фракции концентрировали досуха в условиях пониженного давления с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (3 мг, 78%). ЖХ/МС (способ D) RT=0,578 мин, M+1=442.
1H-ЯМР (500 МГц, CD3OD) δ 8,43 (1H, с), 7,27-7,17 (7H, м), 7,15-7,10 (2H, м), 5,29 (2H, с), 2,59-2,52 (2H+3Н, м), 2,32-2,24 (2Н, м).
Пример 80: 6-(4-(1-аминоциклобутил)фенил)-3-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
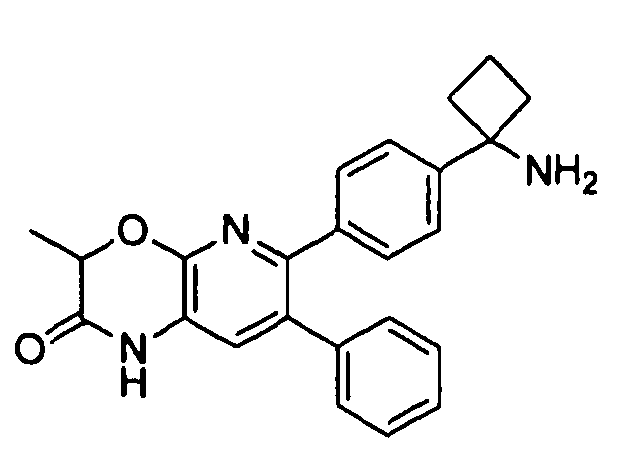
Стадия 1: трет-бутил-(1-(4-(3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Раствор трет-бутил-1-(4-(5-амино-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутилкарбамат (0,5 г, 1,159 ммоль) в THF (объем: 20 мл) добавляли DIPEA (1,01 мл), а затем при 0°C по каплям в течение 1 ч добавляли 2-хлорпропаноилхлорид (0,235 г, 1,854 ммоль) в THF (5 мл). Полученную смесь перемешивали при к.т. в течение ночи. Добавляли насыщенный раствор бикарбоната натрия (30 мл). Полученную смесь нагревали до температуры возгонки в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры. Органическую фазу разделяли. Водную фазу экстрагировали этилацетатом (40 мл). Органическую фазу объединяли и промывали водой (2×30 мл), солевым раствором (25 мл), а затем концентрировали в условиях пониженного давления. Остаток суспендировали в DCM (10 мл). Осадок собирали путем фильтрования и промывали DCM (5 мл) с получением продукта (439 мг).
1H ЯМР (500 МГц, CD3OD): 7,20-7,29 (м, 8H), 7,13-7,15 (м, 2H), 4,98 (кв., 1H), 2,41-2,47 (м, 4H), 2,05 (м, 1H), 1,86 (м, 1H), 1,64 (д, 3H), 1,21-1,36 (ушир., 9H).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-3-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (25 мг, 0,051 ммоль) с получением указанного в заголовке соединения (18,5 мг).
1H ЯМР (500 МГц, CD3OD): 7,37-7,42 (м, 4H), 7,34 (с, 1H), 7,28-7,30 (м, 3H), 7,18-7,20 (м, 2H), 5,03 (кв., 1H), 2,73-2,79 (м, 2H), 2,56-2,60 (м, 2H), 2,20-2,26 (м, 1H), 1,96-1,99 (м, 1H), 1,65 (д, 3H).
ЖХ/МС (способ D): RT=0,73 мин, M+H+=386,2.
Пример 81: 2-(6-(4-(1-аминоциклобутил)фенил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
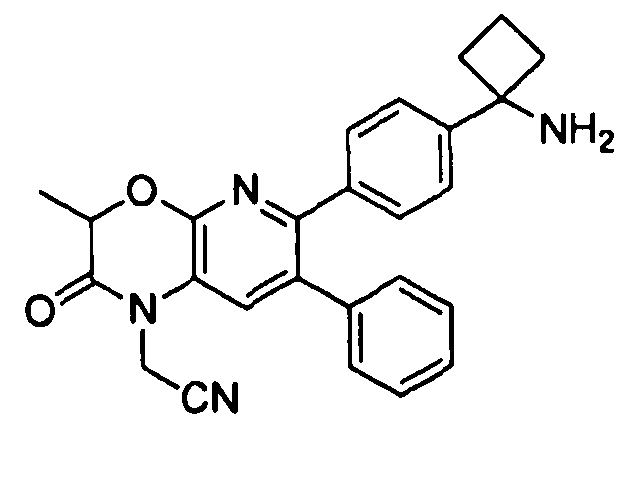
Стадия 1: трет-бутил-(1-(4-(1-(цианометил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-1-(4-(3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (50 мг, 0,103 ммоль) в DMF (2 мл) добавляли карбонат калия (42,7 мг) и 2-бромацетонитрил (22 мкл, 0,309 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 48 ч, а затем разбавляли этилацетатом (20 мл). Промывали водой, солевым раствором, сушили с Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали на колонке (силикагель Biotage, 25 г), элюируя этилацетатом и циклогексаном, с получением продукта (45 мг).
'H ЯМР (500 МГц, CDCl3): 7,35 (с, 1H), 7,20-7,32 (м, 9H), 4,98 (кв., 1H), 4,87 (дд, 2H), 2,44-2,49 (м, 4H), 2,06 (м, 1H), 1,80 (м, 1H), 1,75 (д, 3H), 1,24-1,36 (ушир., 9H),
Стадия 2: 2-(6-(4-(1-аминоциклобутил)фенил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(цианометил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (25 мг, 0,048 ммоль) с получением указанного в заголовке соединения (20 мг).
1H ЯМР (500 МГц, CH3OD) 7,71 (с, 1H), 7,45 (д, 2H), 7,39 (д, 2H), 7,32 (м, 3H), 7,26 (м, 2H), 5,14 (с, 2H), 5,10 (кв., 1H), 2,74-2,80 (м, 2H), 2,55-2,61 (м, 2H), 2,22-2,26 (м, 1H), 1,96-1,99 (м, 1H), 1,71 (д, 3H).
ЖХ/МС (способ D): RT=0,81 мин, M+H+=425,2.
Пример 82: 6-(4-(1-аминоциклобутил)фенил)-1-(метилсульфонилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
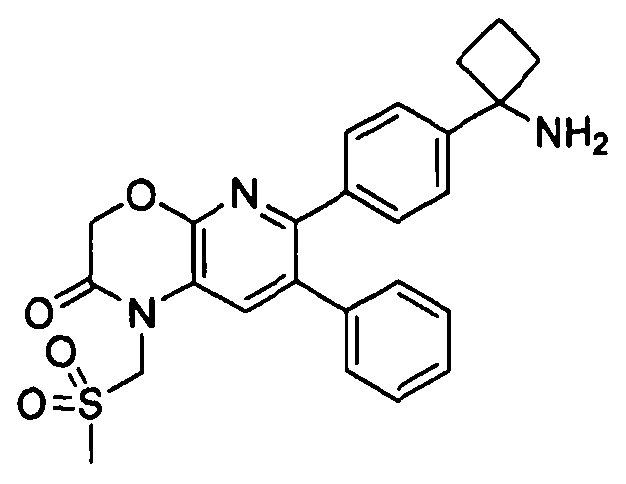
Стадия 1: трет-бутил-1-(4-(1-(метилтиометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (150 мг, 0,318 ммоль), карбонат калия (132 мг, 0,954 ммоль) и хлорметилметилсульфид (0,079 мл, 0,954 ммоль) в безводном N,N-диметилформамиде (2 мл) с получением желтой суспензии. Реакционную смесь нагревали до 80°C в атмосфере азота в течение 2 часов, а затем оставляли охлаждаться до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия (10 мл) и экстрагировали введением в дихлорметан (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→80:20) с получением целевого продукта в виде белого твердого вещества (75 мг, выход 44%).
1H-ЯМР (500 МГц, CDCl3) δ 7,43 (1H, с), 7,23-7,35 (7H, м), 7,19-7,23 (2H, м), 5,09 (3Н, ушир. с), 4,94 (2H, с), 2,30-2,55 (4H, м), 2,26 (3H, с), 2,00-2,15 (1H, м), 1,75-1,90 (1H, м), 1,10-1,50 (9H, ушир. с).
ЖХ/МС (способ A) RT=7,42 мин, M+H+=532,14.
Стадия 2: трет-бутил-1-(4-(1-(метилсульфонилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(1-(метилтиометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (50 мг, 0,094 ммоль) в смеси метанола (1500 мкл) и тетрагидрофурана (1500 мкл) с получением желтого раствора. К этому раствору по каплям добавляли раствор Oxone® (моноперсульфатное соединение, 463 мг, 0,752 ммоль) в воде (1500 мкл) и перемешивали полученную суспензию при комнатной температуре в течение ночи. Реакционную смесь гасили путем осторожного добавления насыщенного раствора бикарбоната натрия (3 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы промывали насыщенным раствором бикарбоната натрия (5 мл), солевым раствором (5 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением не совсем белого твердого вещества. Это вещество очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 88/12→0/100) с получением целевого продукта в виде белого твердого вещества (30 мг, 57%).
1H-ЯМР (500 МГц, CDCl3) δ 7,67 (1H, с), 7,25-7,34 (7H, м), 7,17-7,23 (2H, м), 5,21 (2H, ушир. с), 5,08 (1H, ушир. с), 4,99 (2H, с), 3,08 (3H, с), 2,20-2,60 (4H, м), 2,00-2,15 (1H, м), 1,75-1,90 (1H, м), 1,10-1,50 (9H, ушир. м).
ЖХ/МС (способ D) RT=1,44 мин, M+H+=564,00.
Стадия 3: 6-(4-(1-аминоциклобутил)фенил)-1-(метилсульфонилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(1-(метилсульфонилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (28 мг, 0,050 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~3 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и лиофилизировали в течение выходных дней с получением целевого продукта в виде не совсем белого твердого вещества (28 мг, выход 98%).
1H-ЯМР (500 МГц, MeOD) δ 7,78 (1H, с), 7,32 (2H, д), 7,28 (2H, д), 7,13-7,23 (3Н, м), 7,08-7,13 (2H, м), 5,43 (2H, с), 5,01 (2H, с), 2,86 (3Н, с), 2,58-2,70 (2H, м), 2,40-2,52 (2H, м), 2,03-2,17 (1H, м), 1,76-1,90 (1H, м).
ЖХ/МС (способ D) RT=0,87 мин, M+H+=464,00.
Пример 83: 6-(4-(1-аминоциклобутил)фенил)-1-(1-метил-1H-пиразол-4-ил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
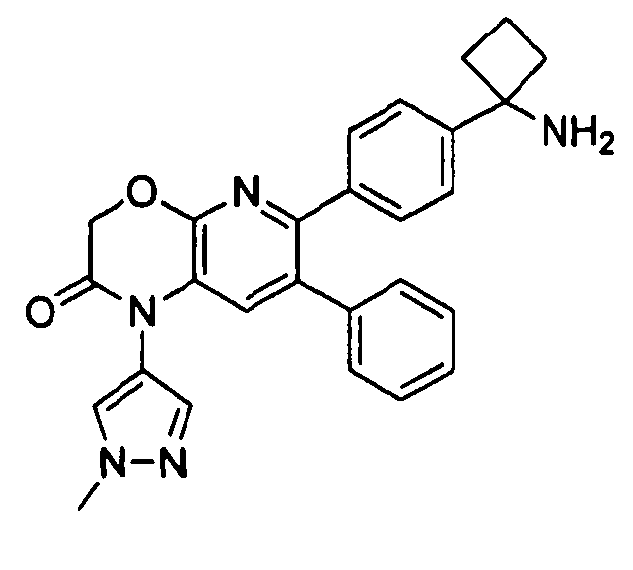
Стадия 1: трет-бутил-1-(4-(1-(1-метил-1H-пиразол-4-ил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (100 мг, 0,212 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (44 мг, 0,212 ммоль), ацетат меди(II) (58 мг, 0,318 ммоль) и 5Å молекулярные сита (60 мг) в безводном дихлорметане (2 мл). К этой смеси добавляли безводный пиридин (1 мл, 12,36 ммоль) и перемешивали полученный темно-пурпурный раствор на воздухе при комнатной температуре в течение 48 часов. Дополнительно добавляли 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (44 мг, 0,212 ммоль) и ацетат меди(II) (58 мг, 0,318 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 72 часов, а затем нагревали до 40°C в течение ночи. К реакционной смеси дополнительно добавляли 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (44 мг, 0,212 ммоль) и ацетат меди(II) (58 мг, 0,318 ммоль) и нагревали при 40°C в течение 6 суток. Реакционную смесь оставляли охлаждаться до комнатной температуры и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 88/12→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (30 мг, выход 51%).
1H-ЯМР (500 МГц, CDCl3) δ 7,53 (1H, с), 7,49 (1H, с), 7,12-7,27 (7H, м), 6,98-7,08 (3Н, м), 4,93 (2H, с), 3,90 (3Н, с), 2,10-2,50 (4H, м), 1,92-2,04 (1H, м), 1,65-1,79 (1H, м), 1,00-1,45 (9H, ушир. м).
ЖХ/МС (способ D) RT=1,45 мин, M+H+=552,20.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(1-метил-1H-пиразол-4-ил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-1-(4-(1-(1-метил-1H-пиразол-4-ил)-2-оксо-7-фенил-2)3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (30 мг, 0,054 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (29 мг, выход 94%).
1H-ЯМР (500 МГц, MeOD) δ 7,89 (1H, с), 7,62 (1H, с), 7,42-7,51 (4H, м), 7,40 (2H, д), 7,37 (2H, д), 7,18-7,27 (3Н, м), 7,03-7,12 (3Н, м), 5,06 (2H, с), 3,96 (3Н, с), 2,66-2,78 (2H, м), 2,48-2,58 (2H, м), 2,13-2,26 (1H, м), 1,85-1,98 (1H, м).
ЖХ/МС (способ D) RT=0,86 мин, M+H+=452,20.
Пример 84: 6-(4-(1-аминоциклобутил)фенил)-1,3-диметил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
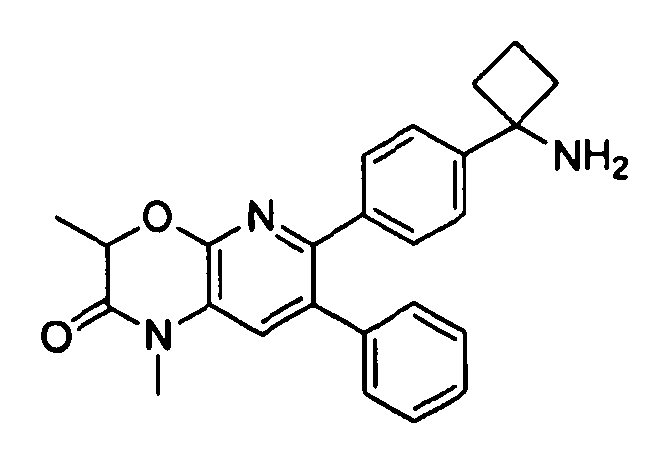
Стадия 1: трет-бутил-(1-(4-(1,3-диметил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-1-(4-(3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (50 мг, 0,103 ммоль) в DMF (2 мл) добавляли карбонат калия (42,7 мг) и йодметан (7,69 мкл, 0,124 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 48 ч, а затем разбавляли этилацетатом (20 мл). Промывали водой, солевым раствором и сушили с Na2SO4, фильтровали и концентрировали с получением продукта (43 мг).
1H ЯМР (500 МГц, CDCl3): 7,19-7,32 (м, 10H), 4,96 (кв., 1H), 3,39 (с, 3H), 2,43-2,48 (м, 4H), 2,04 (м, 1H), 1,79 (м, 1H), 1,70 (д, 3H), 1,24-1,36 (м, 9H).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1,3-диметил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1,3-диметил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (40 мг, 0,08 ммоль) с получением указанного в заголовке соединения (34,5 мг).
1H ЯМР (500 МГц, CH3OD) 7,56 (с, 1H), 7,42 (д, 2H), 7,38 (д, 2H), 7,31 (м, 3H), 7,24 (м, 2H), 5,06 (кв., 1H), 3,44 (с, 3H), 2,73-2,77 (м, 2H), 2,53-2,59 (м, 2H), 2,20-2,26 (м, 1H), 1,95-1,97 (м, 1H), 1,67 (д, 3H).
ЖХ/МС (способ D): RT=0,85 мин, M+H+=400,2.
Пример 85: 6-(4-(1-аминоциклобутил)фенил)-1-(2,2-дифторэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
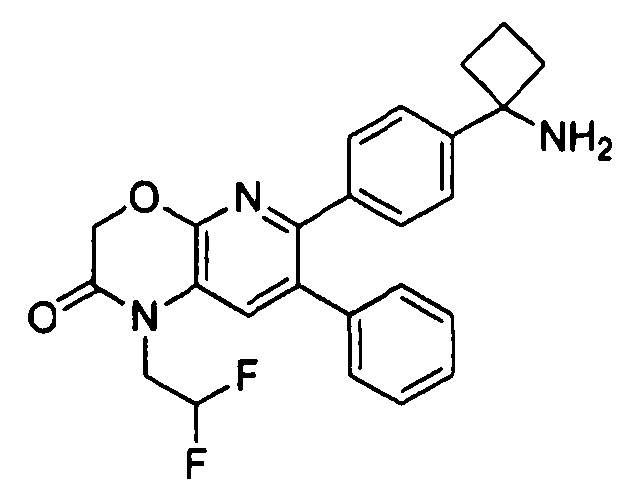
Стадия 1: трет-бутил-(1-(4-(1-(2,2-дифторэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Следуя методике получения трет-бутил-(1-(4-(1-(2-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата, осуществляли взаимодействие трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (100 мг, 0,212 ммоль) с 1,1-дифтор-2-йодэтаном (122 мг, 0,636 ммоль) с получением указанного в заголовке соединения (85 мг).
1H ЯМР (500 МГц, CDCl3): 7,42 (1H, с), 7,24-7,30 (7H, м), 7,17-7,19 (2H, м), 6,11 (1H, тт), 5,05 (1H, ушир.), 4,92 (с, 2H), 4,28 (дт, 2H), 2,44-2,49 (м, 4H), 2,05 (м, 1H), 1,81 (м, 1H), 1,2-1,43 (ушир., 9H).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(2,2-дифторэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(2,2-дифторэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (20 мг, 0,037 ммоль) с получением указанного в заголовке соединения (16 мг).
1H ЯМР (500 МГц, CH3OD) 7,71 (с, 1H), 7,42 (д, 2Н), 7,38 (д, 2Н), 7,31 (м, 3Н), 7,22 (м, 2Н), 6,21 (тт, 1H), 5,01 (с, 2H), 4,50 (дт, 2H), 2,73-2,78 (м, 2H), 2,54-2,59 (м, 2H), 2,21-2,25 (м, 1H), 1,93-1,99 (м, 1H).
ЖХ/МС (способ D): RT=0,84 мин, M+H+=436.
Пример 86: 6-(4-(1-аминоциклобутил)фенил)-1-(2-фторэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
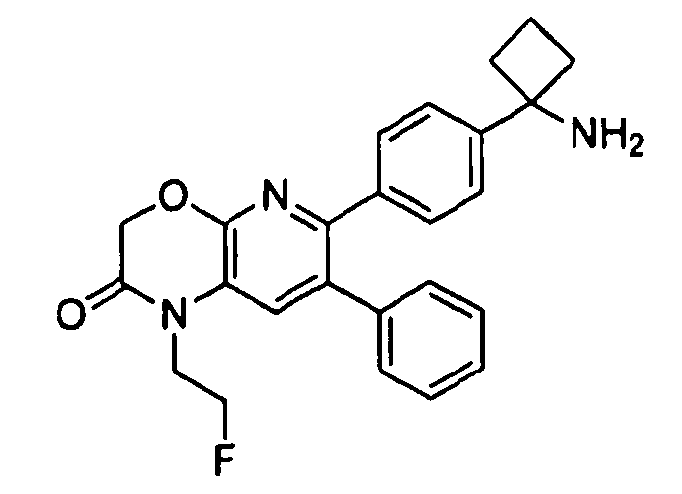
Стадия 1: трет-бутил-(1-(4-(1-(2-фторэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Следуя методике получения трет-бутил-(1-(4-(1-(2-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата, осуществляли взаимодействие трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (100 мг, 0,212 ммоль) с 1-фтор-2-йодэтаном (111 мг, 0,636 ммоль) с получением указанного в заголовке соединения (85 мг).
1H ЯМР (500 МГц, CDCl3): 7,45 (1H, с), 7,24-7,31 (7H, м), 7,17-7,20 (2H, м), 5,01 (1H, с), 4,94 (2H, с), 4,80 (1H, т), 4,70 (1H, т), 4,27 (1H, т), 4,22 (1H, т), 2,44-2,49 (4H, м), 2,06 (1H, м), 1,86 (1H, м), 1,2-1,43 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(2-фторэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(2-фторэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (60 мг, 0,116 ммоль) с получением указанного в заголовке соединения (50,5 мг).
1H ЯМР (500 МГц, CH3OD) 7,69 (1H, с), 7,42 (2H, д), 7,38 (2H, д), 7,30 (3H, м), 7,22 (2H, м), 4,99 (2H, с), 4,80 (1H, т), 4,70 (1H, т), 4,40 (1H, т), 4,35 (1H, т), 2,73-2,79 (2H, м), 2,54-2,60 (2H, м), 2,20-2,25 (1H, м), 1,95-1,97 (1H, м).
ЖХ/МС (способ D): RT=0,83 мин, M+H+=418.
Пример 87: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1H)-он
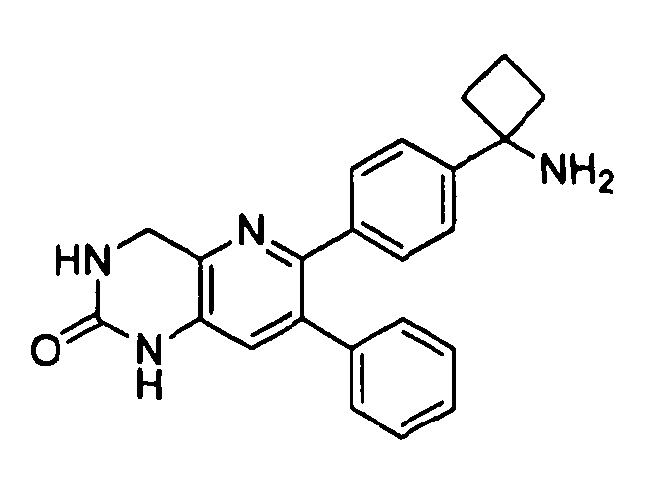
Стадия 1: трет-бутил-(1-(4-(5-нитро-6-(нитрометил)-3-фенилпиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутоксида калия (468 мг, 4,17 ммоль) в безводном DMSO (20 мл) в атмосфере азота медленно добавляли нитрометан (225 мкл, 4,17 ммоль). Полученную смесь перемешивали в течение 5 мин при комнатной температуре, после чего добавляли трет-бутил-(1-(4-(6-хлор-5-нитро-3-фенилпиридин-2-ил)фенил)циклобутил)карбамат (1 г, 2,08 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли насыщенный раствор NH4Cl и экстрагировали смесь AcOEt (3×50 мл). Объединенные органические фазы промывали водой (3×20 мл), сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток использовали непосредственно без очистки.
1H ЯМР (500 МГц, CDCl3): 8,60 (1H, с), 7,44-7,35 (7H, м), 7,30-7,28 (2H, м), 6,21 (2H, с), 5,09 (1H, с), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,79-1,65 (1H, м), 1,40-1,10 (9H, ушир.).
Стадия 2: трет-бутил-(1-(4-(5-амино-6-(аминометил)-3-фенилпиридин-2-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-нитро-6-(нитрометил)-3-фенилпиридин-2-ил)фенил)циклобутил)карбамата (74 мг, 0,15 ммоль) в EtOH (50 мл) добавляли никель Ренея (17 мг, 0,15 ммоль). Полученную смесь продували азотом и H2, а затем перемешивали при комнатной температуре в атмосфере H2 (1,5 бар) в течение ночи. Черную смесь фильтровали через целит, несколько раз промывали EtOH (каждый раз убеждаясь, что осадок на фильтре оставался влажным), сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток использовали непосредственно без очистки.
Стадия 3: трет-бутил-(1-(4-(2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(5-амино-6-(аминометил)-3-фенилпиридин-2-ил)фенил)циклобутил)карбамата (113 мг, 0,25 ммоль) в безводном THF (2 мл) в атмосфере азота добавляли CDI (41 мг, 0,25 ммоль). Полученную смесь перемешивали в течение 1 ч при 60°C и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→5% MeOH в DCM) с получением указанного в заголовке соединения (71 мг, 59%).
1H ЯМР (500 МГц, CDCl3): 8,91 (1H, ушир. с), 7,19-7,11 (7H, м), 7,00-6,97 (3H, м), 5,73 (1H, с), 4,93 (1H, с), 4,65 (2H, с), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,79-1,65 (1H, м), 1,40-1,10 (9H, ушир.).
Стадия 4: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутил)карбамат (12 мг, 0,03 ммоль) с получением указанного в заголовке соединения (12 мг, 97%). ЖХ/МС (способ D): RT=0,684 мин, M+1=371.
1H ЯМР (500 МГц, MeOD): 7,42-7,38 (4H, м), 7,31-7,28 (3H, м), 7,23 (1H, с), 7,21-7,18 (2H, м), 4,66 (2H, с), 2,81-2,75 (2H, м), 2,63-2,57 (2H, м), 2,26-2,24 (1H, м), 1,99-1,97 (1H, м).
Пример 88: 6-(4-(1-аминоциклобутил)фенил)-N-этил-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-карбоксамид
Стадия 1: трет-Бутил-(1-(4-(этилкарбамоил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
трет-Бутил-1-(4-(7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (50 мг, 0,109 ммоль) и изоцианатоэтан (40 мл, 0,546 ммоль) в THF (1,5 мл) нагревали до 50°C в течение 24 ч. Полученную реакционную смесь концентрировали в условиях пониженного давления. Полученное неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 100%→80% н-гексан в этилацетате) с получением указанного в заголовке соединения (40 мг, 70%).
1H ЯМР (500 МГц, CDCl3): 7,83 (1H, с), 7,33-7,29 (2H, д), 7,7-7,21 (4H, м), 7,18-7,13 (4H, м), 4,47 (2H, т), 3,93 (3H, т), 3,38 (2H, кв.), 2,53-2,38 (4H, м), 2,12-1,99 (1H, м), 1,96-1,86 (1H, м), 1,48-1,28 (9H, ушир.), 1,29 (3H, т).
ЖХ/МС (способ D): RT=1,508 мин, +1=529.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-N-этил-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-карбоксамид
трет-Бутил-(1-(4-(этилкарбамоил)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (45 мг, 0,093 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (13 мг, выход 53%).
1H-ЯМР (500 МГц, CD3OD) δ 8,24 (1H, с), 7,41 (2H, д), 7,38 (2H, д), 7,29-7,25 (3Н, м), 7,23-7,18 (2H, м), 4,50 (2H, т), 3,89 (2H, т), 3,30 (2H, кв.), 2,81-2,71 (2H, м), 2,63-2,52 (2H, м), 2,29-2,19 (1H, м), 2,02-1,90 (1H, м), 1,21 (3H, т).
ЖХ/МС (способ D) RT=0,779 мин, M+1=429.
Пример 89: 6-(4-(1-аминоциклобутил)фенил)-1,3-диметил-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1H)-он
Стадия 1: трет-бутил-(1-(4-(1,3-диметил-2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутил)карбамата (46 мг, 0,10 ммоль) в безводном DMF (1 мл) в атмосфере азота при -78°C добавляли гидрид натрия (3,9 мг, 0,10 ммоль) и метилйодид (6,1 мкл, 0,10 ммоль). Баню удаляли, и перемешивали полученную смесь в течение 1 ч при комнатной температуре. Смесь охлаждали до -78°C. В атмосфере азота добавляли гидрид натрия (3,9 мг, 0,10 ммоль) и метилйодид (6,1 мкл, 0,10 ммоль). Баню удаляли, и перемешивали полученную смесь в течение 1 ч при комнатной температуре. Добавляли воду и экстрагировали смесь EtOAc (3×10 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→50% EtOAc в циклогексане) с получением указанного в заголовке соединения (30 мг, 61%).
1H ЯМР (500 МГц, CDCl3): 7,20-7,19 (7H, м), 7,13-7,11 (2H, м), 7,02 (1H, с), 5,00 (1H, ушир. с), 4,56 (2H, с), 3,27 (3H, с), 3,03 (3H, с), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,79-1,65 (1H, м), 1,40-1,10 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1,3-диметил-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1,3-диметил-2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутил)карбамат (30 мг, 0,06 ммоль) с получением указанного в заголовке соединения (28 мг, 91%). ЖХ/МС (способ D): RT=0,798 мин, M-NH2=382.
1H ЯМР (500 МГц, MeOD): 7,45-7,39 (4H, м), 7,34 (1H, с), 7,32-7,30 (3H, м), 7,26-7,24 (2H, м), 4,67 (2H, с), 3,36 (3H, с), 3,11 (3H, с), 2,81-2,75 (2H, м), 2,63-2,57 (2H, м), 2,26-2,24 (1H, м), 1,99-1,97 (1H, м).
Пример 90: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она O-метилоксим
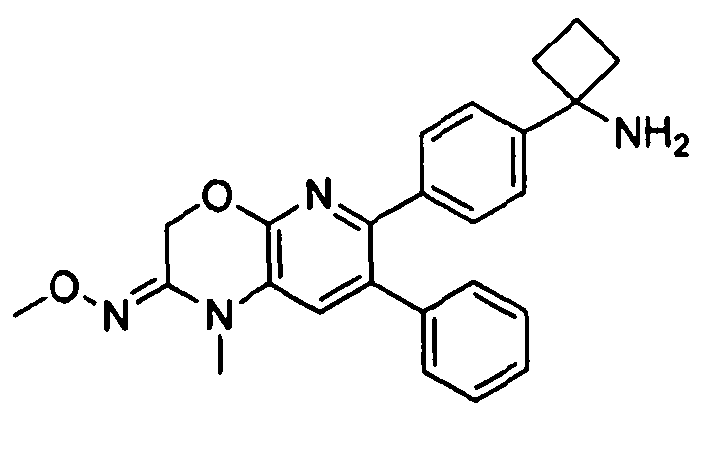
Стадия 1: трет-бутил-(1-(4-(2-(метоксиимино)-1-метил-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
В круглодонную колбу добавляли трет-бутил-1-(4-(1-метил-7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (0,1 г, 0,199 ммоль) и орто-метилгидроксиламина гидрохлорид (0,067 г, 0,797 ммоль) в пиридине (объем: 2 мл). Полученный раствор перемешивали и нагревали при 70°C в течение 18 ч. Реакционную смесь концентрировали досуха и распределяли между этилацетатом (20 мл) и водой (20 мл). Органическую фазу разделяли и промывали водой (2×15 мл), солевым раствором (15 мл) и концентрировали с получением неочищенного продукта, который очищали на колонке (силикагель Biotage, 25 г), элюируя этилацетатом/циклогексаном (0→50%), с получением продукта (21 мг).
1H ЯМР (500 МГц, CDCl3): 7,20-7,29 (м, 9H), 7,10 (с, 1H), 5,19 (с, 2H), 4,95 (с, 1H), 3,81 (с, 3H), 3,31 (с, 3H), 2,2-2,6 (м, 4H), 1,95-2,15 (м, 1H), 1,75-1,85 (м, 1H), 1,2-1,4 (ушир., 9H).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она O-метилоксим
В круглодонную колбу, содержащую трет-бутил-1-(4-(2-(метоксиимино)-1-метил-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (28 мг, 0,054 ммоль), добавляли трифторуксусную кислоту (1 мл). Полученный раствор перемешивали в течение 60 секунд, а затем концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (1 мл) и трижды концентрировали досуха с получением трифторуксуснокислой соли продукта (14,5 мг).
1H ЯМР (500 МГц, CD3OD): 7,49 (с, 1H), 7,35-7,39 (м, 5H), 7,28-7,30 (м, 3H), 7,21-7,23 (м, 2H), 5,24 (с, 2H), 3,83 (с, 3H), 3,35 (с, 3H), 2,72-2,78 (м, 2H), 2,54-2,60 (м, 2H), 2,2-2,3 (м, 1H), 1,9-2,0 (м, 1H).
ЖХ/МС (способ D): RT=0,96 мин, M+H+=415,2.
Пример 91: 1-(4-(7-фенил-1-(пиридин-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин
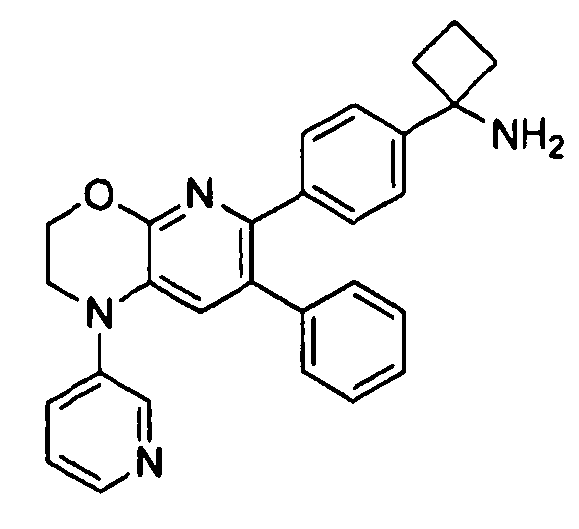
Стадия 1: трет-бутил-1-(4-(7-фенил-1-(пиридин-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 15 мл добавляли трет-бутил-1-(4-(2-оксо-7-фенил-1-(пиридин-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (90 мг, 0,164 ммоль) в безводном тетрагидрофуране (3 мл) с получением бесцветного раствора. Этот раствор охлаждали до 0°C, а затем добавляли боргидрид натрия (15 мг, 0,394 ммоль) и трифторида бора диэтилэфират (50 мкл, 0,394 ммоль). Реакционную смесь перемешивали при 0°C в атмосфере азота в течение 3 часов, а затем нагревали до 40°C в течение ночи. Реакционную смесь разбавляли этилацетатом (3 мл) и гасили путем промывания насыщенным раствором хлорида аммония (2×3 мл). Органическую фазу сушили над Na2SO4, фильтровали, концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 88/12→0/100), затем методом хроматографии на силикагеле Biotage (дихлорметан/метанол, элюирование градиентом 99/1→90/10), затем методом хроматографии на силикагеле Biotage (дихлорметан/этилацетат, элюирование градиентом 88/12→0/100), с получением целевого продукта в виде желтого масла (30 мг, выход 34%).
1H-ЯМР (500 МГц, CDCl3): δ 8,60 (1H, ушир. с), 8,24 (1H, ушир. с), 8,12 (1H, д), 7,65 (1H, ушир. с), 7,36 (1H, с), 7,13-7,28 (7H, м), 7,02-7,08 (2H, м), 4,94 (1H, ушир. с), 4,51 (2H, т), 3,86 (2H, т), 2,20-2,50 (4H, м), 1,95-2,05 (1H, м), 1,70-1,80 (1H, м), 1,00-1,38 (9H, ушир. м).
ЖХ/МС (способ D) RT=1,45 мин, M+H+=535,20.
Стадия 2: 1-(4-(7-фенил-1-(пиридин-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин
трет-Бутил-1-(4-(7-фенил-1-(пиридин-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (10 мг, 0,019 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (4 мг, выход 32%).
1H-ЯМР (500 МГц, MeOD): δ 8,57 (1H, ушир. с), 8,28 (1H, ушир. с), 7,80 (1H, д), 7,43 (1H, ушир. с), 7,26 (2H, д), 7,24 (2H, д), 7,04-7,16 (4H, м), 6,90-7,00 (2H, м), 4,51 (2H, т), 3,80 (2H, т), 2,56-2,70 (2H, м), 2,37-2,52 (2H, м), 2,02-2,16 (1H, м), 1,74-1,90 (1H, м).
ЖХ/МС (способ D) RT=0,77 мин, M+H+=435,20.
Пример 92: 1-(4-(1-бром-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
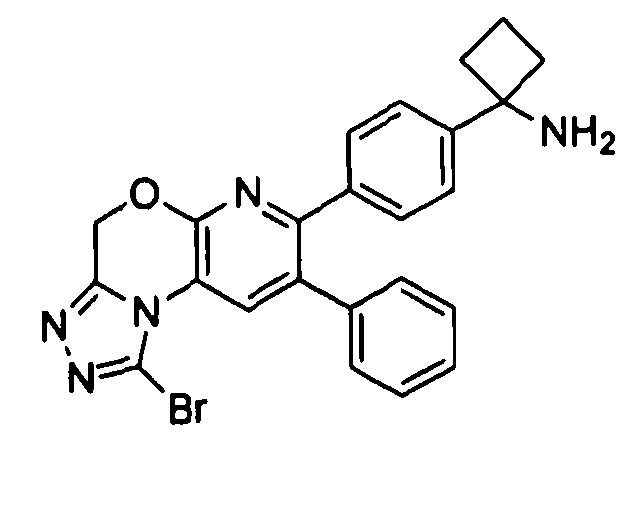
Стадия 1: трет-бутил-(1-(4-(1-бром-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Смесь трет-бутил-1-(4-(8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутилкарбамата (200 мг, 0,404 ммоль) и 1-бромпирролидин-2,5-диона (114,9275 мг, 0,646 ммоль) в DCM (12 мл) и CCL4 (18 мл) нагревали при 50°C в течение 24 ч. Смесь концентрировали, и распределяли остаток между этилацетатом (40 мл) и водой (40 мл). Органическую фазу разделяли, и экстрагировали водную фазу этилацетатом (30 мл). Объединенную органическую фазу промывали водой (50 мл), солевым раствором (30 мл) и концентрировали с получением продукта (159 мг).
1H-ЯМР (500 МГц, CDCl3): 8,55 (с, 1H), 7,22-7,35 (м, 9H), 5,58 (с, 2H), 5,1 (ушир., 1H), 2,4-2,5 (м, 4H), 2,01-2,11 (м, 1H), 1,79-1,89 (м, 1H), 1,24-1,43 (ушир., 9H).
Стадия 2: 1-(4-(1-бром-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(цианометил)-3-(метоксиметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (18 мг, 0,031 ммоль) с получением указанного в заголовке соединения (4,5 мг).
1H-ЯМР (500 МГц, CH3OD) 8,71 (с, 1H), 7,50 (д, 2H), 7,43 (д, 2H), 7,34-7,36 (м, 3H), 7,28-7,31 (м, 2H), 5,70 (с, 2H), 2,76-2,80 (м, 2H), 2,57-2,61 (м, 2H), 2,2-2,27 (м, 1H), 1,9-2,0 (м, 1H).
ЖХ/МС (способ D): RT=0,774 мин, M+=474,2, M+2=476,2.
Пример 93: (R)-2-(6-(4-(1-аминоциклобутил)фенил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
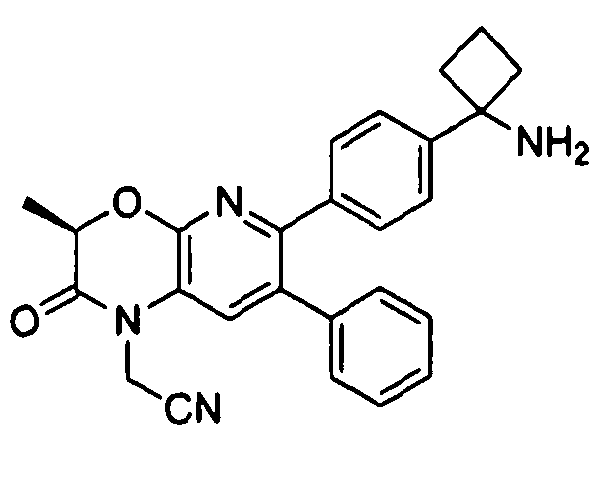
Стадия 1: (R)-трет-бутил-(1-(4-(1-(цианометил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору (R)-трет-бутил-1-(4-(3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (100 мг, 0,206 ммоль) в DMF (2 мл) добавляли карбонат калия (85 мг) и 2-бромацетонитрил (44 мкл, 0,618 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 48 ч, а затем разбавляли этилацетатом (20 мл). Промывали водой, солевым раствором, сушили с Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали на колонке (силикагель Biotage, 25 г), элюируя этилацетатом и циклогексаном, с получением продукта (55 мг).
1H-ЯМР (500 МГц, CDCl3): 7,35 (с, 1H), 7,20-7,32 (м, 9H), 4,98 (кв., 1H), 4,87 (дд, 2H), 2,44-2,49 (м, 4H), 2,06 (м, 1H), 1,80 (м, 1H), 1,75 (д, 3H), 1,24-1,36 (ушир., 9H).
Стадия 2: (R)-2-(6-(4-(1-аминоциклобутил)фенил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали (R)-трет-бутил-(1-(4-(1-(цианометил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (41 мг, 0,078 ммоль) с получением указанного в заголовке соединения (22 мг).
1H-ЯМР (500 МГц, CH3OD) 7,71 (с, 1H), 7,45 (д, 2H), 7,39 (д, 2H), 7,32 (м, 3H), 7,26 (м, 2H), 5,14 (с, 2H), 5,10 (кв., 1H), 2,74-2,80 (м, 2H), 2,55-2,61 (м, 2H), 2,22-2,26 (м, 1H), 1,96-1,99 (м, 1H), 1,71 (д, 3H).
ЖХ/МС (способ D): RT=0,81 мин, M+H+=425,2.
Пример 94: (R)-6-4-(1-аминоциклобутил)фенил)-1,3-диметил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
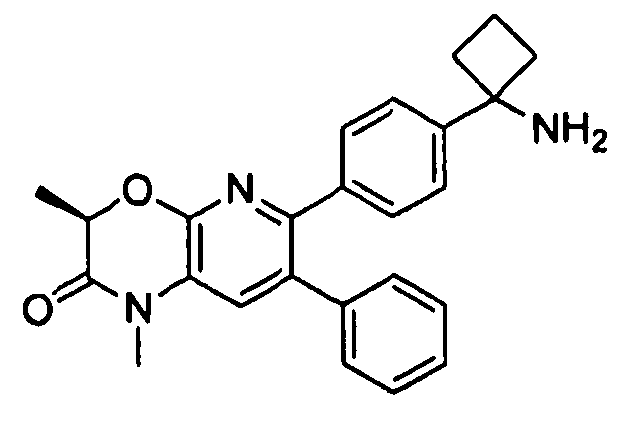
Стадия 1: (R)-трет-бутил-(1-(4-(3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
(S)-2-хлорпропановую кислоту (0,20 г, 1,854 ммоль) в круглодонной колбе перемешивали с тионилхлоридом (0,149 мл, 2,039 ммоль). Добавляли каплю DMF и перемешивали смесь в течение 0,5 ч при комнатной температуре, а затем при 80°C в течение 2 ч. Полученную светло-желтую жидкость разбавляли THF (5 мл). При 0°C в круглодонную колбу объемом 100 мл добавляли трет-бутил-1-(4-(5-амино-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутилкарбамат (0,5 г, 1,159 ммоль) и N-этил-N-изопропилпропан-2-амин (1,015 мл, 5,79 ммоль) в THF (25 мл) с получением желтой суспензии. К суспензии в течение 30 мин по каплям добавляли указанный выше хлорангидрид в THF. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь переносили в круглодонную колбу объемом 250 мл. Добавляли насыщенный раствор бикарбоната натрия (30 мл) и бикарбонат натрия (1 г). Полученную смесь нагревали до температуры возгонки в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры. Органическую фазу разделяли. Водную фазу экстрагировали этилацетатом (40 мл). Органическую фазу объединяли и промывали водой (2×30 мл), солевым раствором (25 мл), а затем концентрировали в условиях пониженного давления. Остаток суспендировали в DCM (10 мл). Осадок собирали путем фильтрования и промывали DCM (5 мл) с получением продукта (0,23 г).
1H-ЯМР (500 МГц, CD3OD): 7,20-7,32 (м, 8H), 7,13-7,15 (м, 2H), 4,98 (кв., 1H), 2,41-2,47 (м, 4H), 2,05 (м, 1H), 1,86 (м, 1H), 1,64 (д, 3H), 1,21-1,36 (ушир., 9H).
Стадия 2: (R)-трет-бутил-(1-(4-(1,3-диметил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору (R)-трет-бутил-1-(4-(3-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (100 мг, 0,206 ммоль) в DMF (2 мл) добавляли карбонат калия (85 мг) и йодметан (15 мкл, 0,247 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, а затем разбавляли этилацетатом (20 мл). Промывали водой, солевым раствором, сушили с Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали на колонке (силикагель Biotage, размер: 25 г), элюируя этилацетатом/циклогексаном (0-50%) с получением продукта (33 мг).
1H-ЯМР (500 МГц, CDCl3): 7,19-7,32 (м, 10H), 4,96 (кв., 1H), 3,39 (с, 3H), 2,43-2,48 (м, 4H), 2,04 (м, 1H), 1,80 (м, 1H), 1,70 (д, 3H), 1,24-1,36 (м, 9H).
Стадия 3: 6-(4-(1-аминоциклобутил)фенил)-1,3-диметил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали (R)-трет-бутил-(1-(4-(1,3-диметил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (16 мг, 0,032 ммоль) с получением указанного в заголовке соединения (6,5 мг).
1H-ЯМР (500 МГц, CH3OD) 7,56 (с, 1H), 7,42 (д, 2H), 7,38 (д, 2H), 7,31 (м, 3H), 7,24 (м, 2H), 5,06 (кв., 1H), 3,44 (с, 3H), 2,73-2,77 (м, 2H), 2,53-2,59 (м, 2H), 2,20-2,26 (м, 1H), 1,95-1,97 (м, 1H), 1,67 (д, 3H).
ЖХ/МС (способ D): RT=0,85 мин, M+H+=400,2.
Пример 95: 2-(6-(4-(1-аминоциклобутил)фенил)-3-(метоксиметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
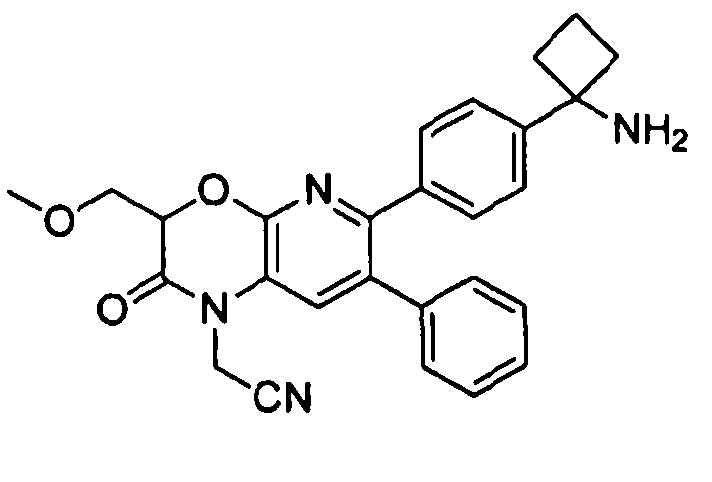
Стадия 1: трет-бутил-(1-(4-(1-(цианометил)-3-(метоксиметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-(метоксиметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (60 мг, 0,116 ммоль) в DMF (2 мл) добавляли карбонат калия (48 мг) и 2-бромацетонитрил (41,9 мг, 0,349 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, а затем разбавляли этилацетатом (20 мл). Промывали водой, солевым раствором, сушили с Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали на колонке (силикагель Biotage, 25 г), элюируя этилацетатом и циклогексаном, с получением продукта (35 мг).
1Н-ЯМР (500 МГц, CDCl3): 7,20-7,33 (м, 10H), 5,10 (т, 1H), 4,97 (с, 1H), 4,82 (дд, 2H), 4,03 (дд, 2H), 3,36 (с, 3H), 2,44-2,50 (м, 4H), 2,06 (м, 1H), 1,80 (м, 1H), 1,24-1,36 (ушир., 9H).
Стадия 2: 2-(6-(4-(1-аминоциклобутил)фенил)-3-(метоксиметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(цианометил)-3-(метоксиметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (35 мг, 0,063 ммоль) с получением указанного в заголовке соединения (18,5 мг).
1Н-ЯМР (500 МГц, CH3OD) 7,63 (с, 1H), 7,45 (д, 2H), 7,39 (д, 2H), 7,32 (м, 3H), 7,26 (м, 2H), 5,28 (т, 1H), 5,16 (дд, 2H), 4,01 (дд, 2H), 2,74-2,79 (м, 2H), 2,55-2,61 (м, 2H), 2,22-2,26 (м, 1H), 1,96-1,99 (м, 1H).
ЖХ/МС (способ D): RT=0,78 мин, M+H+=456,2.
Пример 96: 6-(4-(1-аминоциклобутил)фенил)-3-(метоксиметил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
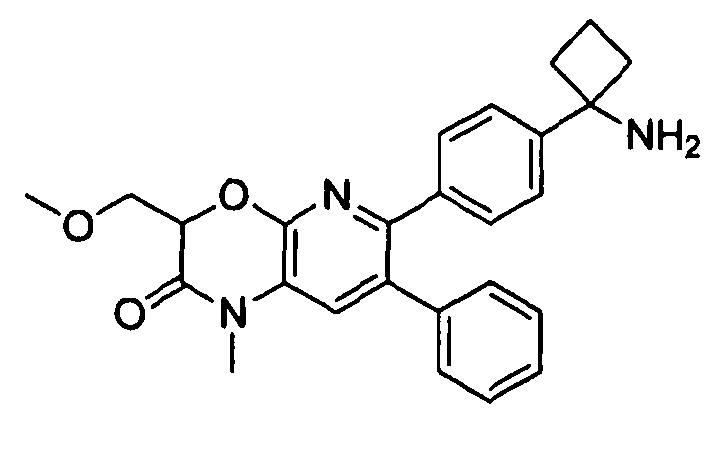
Стадия 1: трет-бутил-(1-(4-(3-(метоксиметил)-1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
В круглодонной колбе 2-хлор-3-метоксипропановую кислоту (0,257 г, 1,854 ммоль) смешивали с тионилхлоридом (0,149 мл, 2,039 ммоль). Добавляли каплю DMF и перемешивали смесь в течение 0,5 ч при комнатной температуре, а затем при 80°C в течение 2 ч. Полученную светло-желтую жидкость разбавляли THF (5 мл). При 0°C в круглодонную колбу объемом 100 мл добавляли трет-бутил-1-(4-(5-амино-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутилкарбамат (0,5 г, 1,159 ммоль) и N-этил-N-изопропилпропан-2-амин (1,015 мл, 5,79 ммоль) в THF (25 мл) с получением желтой суспензии. К суспензии в течение 30 мин по каплям добавляли указанный выше хлорангидрид в THF. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь переносили в круглодонную колбу объемом 250 мл. Добавляли насыщенный раствор бикарбоната натрия (30 мл) и бикарбонат натрия (1 г). Полученную смесь нагревали до температуры возгонки в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры. Органическую фазу разделяли. Водную фазу экстрагировали этилацетатом (40 мл). Органическую фазу объединяли и промывали водой (2×30 мл), солевым раствором (25 мл), а затем концентрировали в условиях пониженного давления. Остаток суспендировали в DCM (10 мл). Осадок собирали путем фильтрования и промывали DCM (5 мл) с получением продукта (0,26 г).
1Н-ЯМР (500 МГц, CDCl3): 8,11 (с, 1H), 7,23-7,31 (м, 6H), 7,15 (м, 2H), 7,07 (с, 1H), 5,06 (т, 1H), 5,01 (ушир., 1H), 4,08 (дд, 1H), 3,95 (дд, 1H), 3,39 (с, 3H), 2,41-2,47 (м, 4H), 2,05 (м, 1H), 1,80 (м, 1H), 1,21-1,36 (ушир., 9H).
Стадия 2: трет-бутил-(1-(4-(3-(метоксиметил)-1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(3-(метоксиметил)-1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (60 мг, 0,116 ммоль) в DMF (1 мл) добавляли карбонат калия (48 мг) и йодметан (8,7 мкл, 0,14 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, а затем разбавляли этилацетатом (20 мл). Промывали водой, солевым раствором, сушили с Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали на колонке (силикагель Biotage, размер: 25 г), элюируя этилацетатом/циклогексаном (0-50%), с получением продукта (27 мг).
1Н-ЯМР (500 МГц, CDCl3): 7,19-7,32 (м, 10H), 5,05 (т, 1H), 4,97 (ушир., 1H), 4,06 (дд, 1H), 3,94 (дд, 1H), 3,40 (с, 3H), 3,37 (с, 3H), 2,43-2,48 (м, 4H), 2,04 (м, 1H), 1,79 (м, 1H), 1,24-1,36 (м, 9H).
Стадия 3: 6-(4-(1-аминоциклобутил)фенил)-3-(метоксиметил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(3-(метоксиметил)-1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (35 мг, 0,066 ммоль) с получением указанного в заголовке соединения (28 мг).
1Н-ЯМР (500 МГц, CH3OD) 7,49 (с, 1H), 7,42 (д, 2H), 7,38 (д, 2H), 7,31 (м, 3H), 7,25 (м, 2H), 5,19 (т, 1H), 4,02 (дд, 1H), 3,92 (дд, 1H), 3,44 (с, 3H), 2,74-2,79 (м, 2H), 2,56-2,60 (м, 2H), 2,21-2,26 (м, 1H), 1,94-1,99 (м, 1H).
ЖХ/МС (способ D): RT=0,79 мин, M+H+=430,2.
Пример 97: 6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
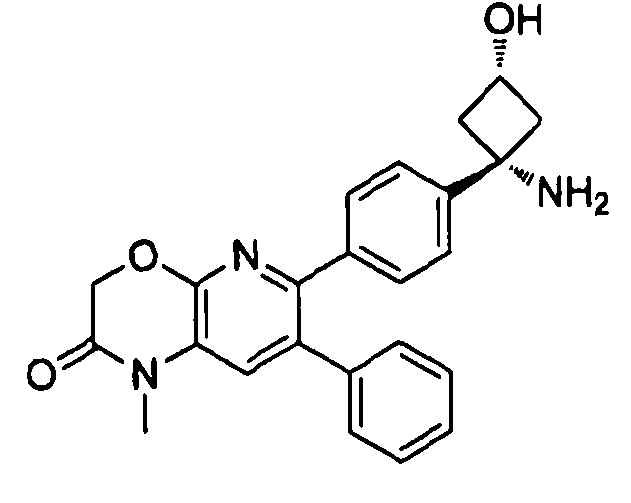
Стадия 1: трет-бутил-(1s,3s)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутилкарбамат
В реакционную пробирку объемом 40 мл добавляли трет-бутил-(1s,3s)-1-(4-бромфенил)-3-гидроксициклобутилкарбамат (0,25 г, 0,731 ммоль) в безводном тетрагидрофуране (14 мл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 20 минут, а затем добавляли аддукт [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и дихлорметана (60 мг, 0,073 ммоль). После барботирования азотом еще в течение 15 минут, добавляли ацетат калия (143 мг, 1,461 ммоль) и бис(пинаколато)дибор (223 мг, 0,877 ммоль). Реакционную смесь нагревали до температуры возгонки в течение ночи, затем концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 88/12→0/100) с получением целевого продукта в виде бесцветного масла, которое отверждалось при отстаивании (240 мг, выход 84%).
1H-ЯМР (500 МГц, CDCl3) δ 7,71 (2H, д), 7,44 (2H, д), 4,15 (1H, ушир. с), 2,87-2,98 (2H, м), 2,27-2,44 (2H, м), 1,22-1,49 (21H, ушир. м).
Стадия 2: трет-бутил-(1s,3s)-3-гидрокси-1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В пробирку для микроволновой обработки объемом 10 мл добавляли 6-бром-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (50 мг, 0,157 ммоль), трет-бутил-(1s,3s)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутилкарбамат (73 мг, 0,188 ммоль) и трет-бутил-(1s,3s)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутилкарбамат (73 мг, 0,188 ммоль) в смеси 1,4-диоксана (3,6 мл) и воды (1,2 мл) с получением желтого раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем нагревали до 100°C в условиях обработки микроволнами в течение 15 минут. Реакционную смесь разбавляли водой (10 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 100/0→0/100), а затем методом хроматографии на силикагеле Biotage (дихлорметан/метанол, элюирование градиентом 100/0→90/10), с получением целевого продукта в виде белого твердого вещества (8 мг, выход 10%).
1H-ЯМР (500 МГц, CDCl3) δ 7,18-7,36 (10Н, м), 4,91 (3Н, с), 4,06 (1Н, ушир. с), 3,40 (3Н, с), 3,00 (2H, ушир. с), 2,77 (2H, ушир. с), 1,12-1,52 (9H, ушир. м).
ЖХ/МС (способ D) RT=1,19 мин, M+H+=502,20.
Стадия 3: 6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В круглодонную колбу объемом 10 мл добавляли трет-бутил-(1s,3s)-3-гидрокси-1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (5 мг, 9,97 мкмоль) в 1,4-диоксане (1 мл) с получением бесцветного раствора. К этому раствору по каплям добавляли 4M HCl в 1,4-диоксане (1 мл, 4,00 ммоль), и перемешивали полученный раствор при комнатной температуре в течение 6 часов. Реакционную смесь разбавляли путем добавления по каплям диэтилового эфира (2 мл) и перемешивали в течение 10 минут с получением белого осадка. Слои оставляли отстаиваться, и удаляли надосадочную жидкость. Твердое вещество дважды промывали диэтиловым эфиром (2 мл), оставляли отстаиваться, каждый раз удаляя надосадочную жидкость. Удаляли оставшийся растворитель путем концентрирования досуха в условиях пониженного давления, а затем лиофилизировали в течение выходных с получением целевого продукта в виде белого твердого вещества (4 мг, выход 92%).
1H-ЯМР (500 МГц, MeOD) δ 7,53 (1H, с), 7,38-7,45 (4H, м), 7,24-7,32 (3Н, м), 7,18-7,24 (2H, м), 4,93 (2H, с), 4,05 (1H, квинт.), 3,42 (3Н, с), 3,00-3,12 (2H, м), 2,37-2,48 (2H, м).
ЖХ/МС (способ D) RT=0,71 мин, M+H+=402,20.
Пример 98: 1-(4-(1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
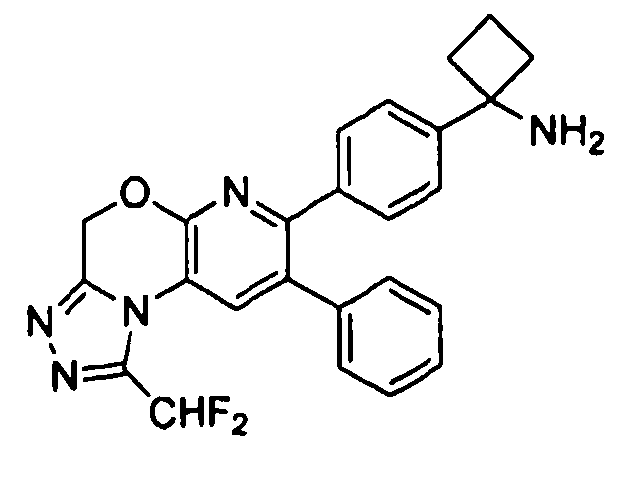
Стадия 1: (E)-трет-бутил-(1-(4-(2-(2-(2,2-дифторацетил)гидразоно)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Этил-2,2-дифторацетат (0,43 мл, 4,10 ммоль) и гидразина моногидрат нагревали до 60°C в N.N-диметилформамиде (1 мл) в течение 30 минут с получением бесцветного раствора. К полученному раствору при комнатной температуре добавляли трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (100 мг, 0,205 ммоль). Полученную смесь нагревали до 70°C в течение 20 часов. Реакционную смесь концентрировали в условиях пониженного давления, и использовали неочищенное вещество (100 мг, количественный выход) на следующей стадии без дополнительной очистки. ЖХ/МС (способ D): RT=1,37 мин, M+1=564.
Стадия 2: трет-бутил-(1-(4-(1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Раствор (E)-трет-бутил-(1-(4-(2-(2-(2,2-дифторацетил)гидразоно)-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (80 мг, 0,142 ммоль) в пара-ксилоле (1 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→2% метанол в дихлорметане) с получением указанного в заголовке соединения (15 мг, 20%). ЖХ/МС (способ D): RT=1,529 мин, M+1=546.
Стадия 3: 1-(4-(1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (15 мг, 0,073 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли в условиях пониженного давления, и сушили остаток с получением целевого продукта в виде не совсем белого твердого вещества (8 мг, выход 65%). ЖХ/МС (способ D): RT=0,803 мин, M+1=446.
1H-ЯМР (500 МГц, CD3OD) δ 8,20 (1H, с), 7,53-7,47 (3Н, м), 7,46-7,41 (2H, м), 7,39-7,33 (3Н, м), 7,30-7,23 (2H, м), 5,77 (2H, с), 2,83-2,70 (2H, м), 2,63-2,53 (2H, м), 2,30-2,19 (1H, м), 2,06-1,90 (1H, м).
Пример 99: 7-(4-(1-аминоциклобутил)фенил)-2-метил-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он
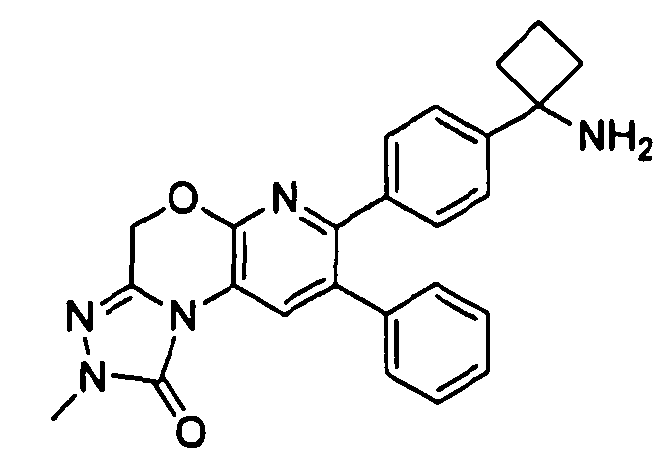
Стадия 1: трет-бутил-(1-(4-(2-метил-1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамата (26 мг, 0,05 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли карбонат калия (21 мг, 0,15 ммоль) и метилйодид (10 мкл, 0,15 ммоль). Полученную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и экстрагировали смесь EtOAc (3×5 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→40% EtOAc в циклогексане) с получением указанного в заголовке соединения (22 мг, 82%).
1Н-ЯМР (500 МГц, CDCl3): 8,50 (1H, с), 7,25 (2H, д), 7,20-7,19 (5H, м), 7,15-7,12 (2H, м), 5,21 (2H, с), 5,00 (1H, ушир. с), 3,46 (3H, с), 2,55-2,25 (4H, м), 2,10-2,03 (1H, м), 1,86-1,78 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 7-(4-(1-аминоциклобутил)фенил)-2-метил-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(2-метил-1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (26 мг, 0,05 ммоль) с получением указанного в заголовке соединения (18 мг, 56%). ЖХ/МС (метод D): RT=0,764 мин, M-NH2=409.
1Н-ЯМР (500 МГц, MeOD): 8,56 (1H, с), 7,45 (2H, д), 7,41 (2H, д), 7,33-7,31 (3H, м), 7,24-7,23 (2H, м), 5,43 (2H, с), 3,53 (3H, с), 2,78-2,72 (2H, м), 2,59-2,53 (2H, м), 2,25-2,18 (1H, м), 1,99-1,91 (1H, м).
Пример 100: 1-(4-(8-фенил-1-(трифторметил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
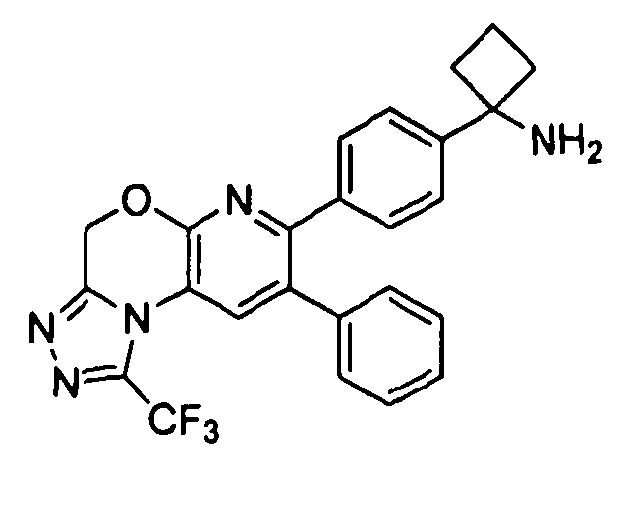
Стадия 1: трет-бутил-(1-(4-(8-фенил-1-(трифторметил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Раствор трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (70 мг, 0,144 ммоль) и 2,2,2-трифторацетогидразида (7,3 мг, 0,057 ммоль) в пара-ксилоле (1 мл) нагревали до 150°C в течение 30 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в н-гексанах) с получением указанного в заголовке соединения (7 мг, 20%). ЖХ/МС (способ D): RT=1,633 мин, M+1=564.
1Н-ЯМР (500 МГц, CDCl3): 7,96 (1H, с), 7,36-7,27 (7H, м), 7,21-7,17 (2H, м), 5,63 (2H, с), 2,55-2,40 (4H, м), 2,14-1,98 (1H, м), 1,88-1,78 (1H, м), 1,37-1,20 (9H, ушир. с).
Стадия 2: 1-(4-(8-фенил-1-(трифторметил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(8-фенил-1-(трифторметил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (7 мг, 0,012 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли в условиях пониженного давления, и сушили остаток с получением целевого продукта в виде не совсем белого твердого вещества (1,5 мг, выход 26%). ЖХ/МС (способ D): RT=0,877 мин, M+1=465.
1H-ЯМР (500 МГц, CD3OD) δ 8,02 (1H, с), 7,50 (2H, д), 7,43 (2H, д), 7,38-7,36 (3Н, м), 7,25 (2H, м), 5,77 (2H, с), 2,75-2,69 (2H, м), 2,55-2,42 (2H, м), 2,29-2,17 (1H, м), 2,05-1,90 (1H, м).
Пример 101: 2-(7-(4-(1-аминоциклобутил)фенил)-1-оксо-8-фенил-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-2(4H)-ил)ацетонитрил
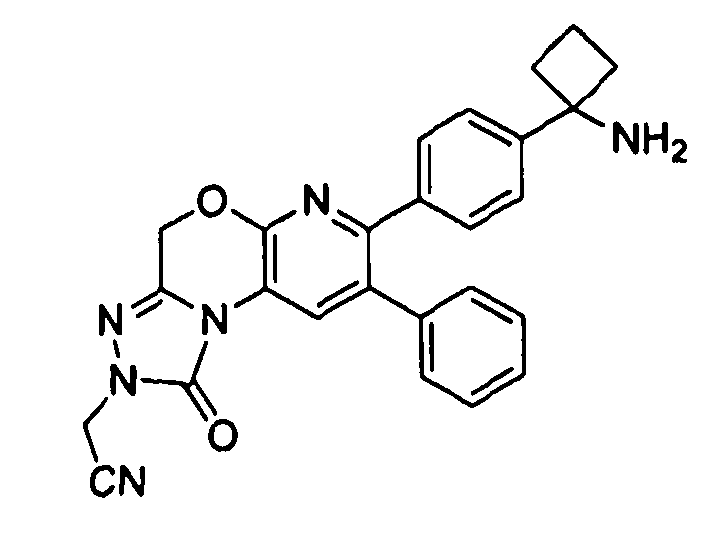
Стадия 1: трет-бутил-(1-(4-(2-(цианометил)-1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамата (70 мг, 0,14 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли карбонат калия (57 мг, 0,41 ммоль) и бромацетонитрил (29 мкл, 0,41 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли воду и экстрагировали смесь EtOAc (3×10 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→35% EtOAc в циклогексане) с получением указанного в заголовке соединения (45 мг, 60%).
1Н-ЯМР (500 МГц, CDCl3): 8,43 (1H, с), 7,26-7,19 (7H, м), 7,14-7,12 (2H, м), 5,25 (2H, с), 4,96 (1H, ушир. с), 4,72 (2H, с), 2,55-2,25 (4H, м), 2,10-2,03 (1H, м), 1,86-1,78 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 2-(7-(4-(1-аминоциклобутил)фенил)-1-оксо-8-фенил-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-2(4H)-ил)ацетонитрил
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(2-(цианометил)-1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (45 мг, 0,08 ммоль) с получением указанного в заголовке соединения (34 мг, 61%). ЖХ/МС (способ D): RT=0,777 мин, M-NH2=434.
1Н-ЯМР (500 МГц, MeOD): 8,54 (1H, с), 7,45 (2H, д), 7,41 (2H, д), 7,33-7,31 (3H, м), 7,24-7,23 (м, 2H), 5,47 (2H, с), 5,01 (2H, с), 2,78-2,72 (м, 2H), 2,59-2,53 (м, 2H), 2,25-2,18 (м, 1H), 1,99-1,91 (м, 1H).
Пример 102: 6-(4-(1-аминоциклобутил)фенил)-1-изобутил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
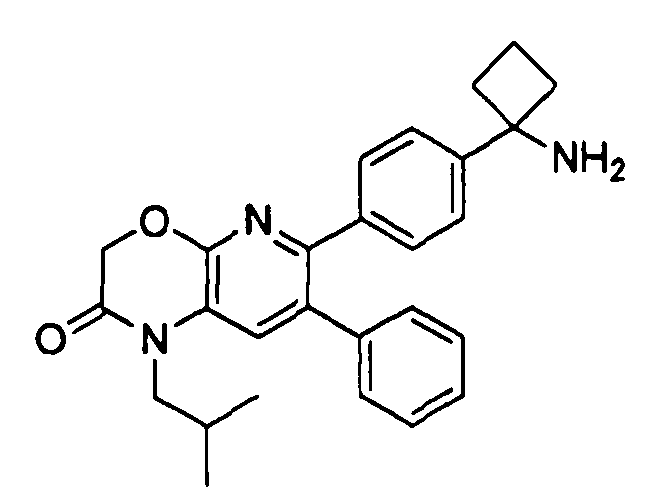
Стадия 1: трет-бутил-(1-(4-(1-изобутил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Следуя методике получения трет-бутил-(1-(4-(1-(2-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата, осуществляли взаимодействие трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (100 мг, 0,212 ммоль) с 1-бром-2-метилпропаном (87 мг, 0,636 ммоль) с получением указанного в заголовке соединения (78 мг). ЖХ/МС (метод D) RT=1,73 мин, M+H=528,2.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-изобутил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(2-оксо-7-фенил-1-пропил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (50 мг, 0,078 ммоль) с получением указанного в заголовке соединения (34,5 мг).
1Н-ЯМР (500 МГц, CH3OD) 7,57 (1H, с), 7,38-7,42 (4H, м), 7,30-32 (3H, м), 7,21-7,2 (2H, м), 4,97 (2H, с), 3,92 (2H, д), 2,73-2,79 (2H, м), 2,54-2,60 (2H, м), 2,22-2,25 (1H, м), 2,13-2,16 (1H, м), 1,95-1,99 (1H, м), 1,0 (6H, д).
ЖХ/МС (метод D) RT: 0,93, M+H+=428,2.
Пример 103: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-2-(2,2,2-трифторэтил)-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он

Стадия 1: трет-бутил-(1-(4-(1-оксо-8-фенил-2-(2,2,2-трифторэтил)-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамата (98 мг, 0,19 ммоль) в безводном DMF (2 мл) в атмосфере азота добавляли карбонат калия (79 мг, 0,57 ммоль) и 2-бром-1,1,1-трифторэтан (94 мг, 0,57 ммоль). Полученную смесь перемешивали в течение 1 часа при 80°C. Добавляли воду и экстрагировали смесь EtOAc (3×). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→27% EtOAc в циклогексане) с получением указанного в заголовке соединения (45 мг, 40%).
1Н-ЯМР (500 МГц, CDCl3): 8,47 (1H, с), 7,26-7,20 (7H, м), 7,14-7,12 (2H, м), 5,25 (2H, с), 4,96 (1H, ушир. с), 4,38 (2H, кв.), 2,55-2,25 (4H, м), 2,10-2,03 (1H, м), 1,86-1,78 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-2-(2,2,2-трифторэтил)-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1-оксо-8-фенил-2-(2,2,2-трифторэтил)-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (45 мг, 0,08 ммоль) с получением указанного в заголовке соединения (34 мг, 61%). ЖХ/МС (способ D): RT=0,923 мин, M-NH2=477.
1Н-ЯМР (500 МГц, MeOD): 8,43 (1H, с), 7,34-7,28 (4Н, м), 7,21-7,20 (3H, м), 7,13-7,12 (2H, м), 5,34 (2H, с), 4,49 (2H, кв.), 2,70-2,62 (2H, м), 2,49-2,43 (2H, м), 2,15-2,08 (1H, м), 1,91-1,77 (1H,м).
Пример 104: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-1H-пиридо[2,3-b][1,4]диазепин-2(3H)-он
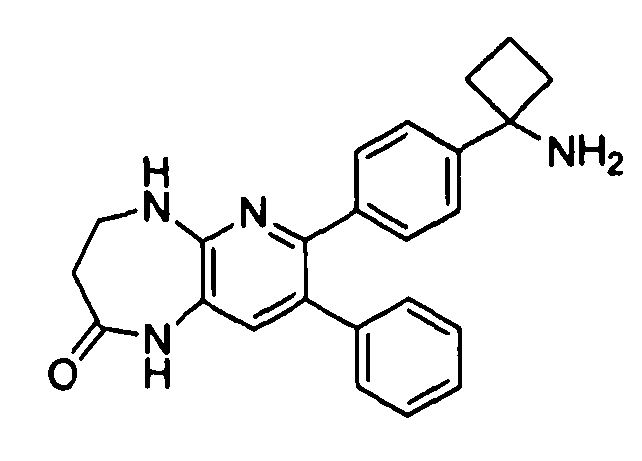
Стадия 1: этил-3-((6-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-3-нитро-5-фенилпиридин-2-ил)амино)пропаноат
К раствору трет-бутил-(1-(4-(6-хлор-5-нитро-3-фенилпиридин-2-ил)фенил)циклобутил)карбамата (1,03 г, 2,14 ммоль) в EtOH (8 мл) в атмосфере азота добавляли триэтиламин (1,49 мл, 10,72 ммоль) и этил-3-аминопропаноат·HCl (1,65 г, 10,72 ммоль). Полученную смесь перемешивали в течение 1,5 часов при 80°C в условиях обработки микроволновым излучением. Добавляли воду, и экстрагировали смесь AcOEt (3×15 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→30% EtOAc в циклогексане) с получением указанного в заголовке соединения (1,2 г, количественный выход).
1Н-ЯМР (500 МГц, CDCl3): 8,50 (1H, с), 8,38 (1H, с), 7,32 (2H, д), 7,23-7,18 (5H, м), 7,09-7,07 (2H, м), 5,00 (1H, ушир. с), 4,12 (2H, кв.), 3,98 (2H, кв.), 2,71-2,68 (2H, т), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,79-1,65 (1H, м), 1,40-1,10 (9H, ушир.), 1,20 (3H, т).
Стадия 2: 3-((6-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-3-нитро-5-фенилпиридин-2-ил)амино)пропановая кислота
К раствору этил-3-((6-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-3-нитро-5-фенилпиридин-2-ил)амино)пропаноата (558 мг, 0,99 ммоль) в THF (4 мл) добавляли 1M NaOH (4 мл, 4 ммоль). Полученную смесь перемешивали в течение ночи при 50°C. Органическую фазу разделяли и концентрировали досуха в условиях пониженного давления. Полученный остаток использовали непосредственно без очистки.
Стадия 3: 3-((3-амино-6-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-5-фенилпиридин-2-ил)амино)пропановая кислота
К раствору 3-((6-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-3-нитро-5-фенилпиридин-2-ил)амино)пропановой кислоты (200 мг, 0,38 ммоль) в THF (100 мл) добавляли Pd/C (4 мг, 0,04 ммоль). Полученную смесь продували азотом и H2, а затем перемешивали в атмосфере H2 (2 бар) в течение ночи при комнатной температуре. Черную смесь фильтровали через целит, несколько раз промывали THF (каждый раз убеждаясь, что осадок на фильтре оставался влажным), сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток использовали непосредственно без очистки.
Стадия 4: трет-бутил-(1-(4-(2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамат
К раствору 3-((3-амино-6-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-5-фенилпиридин-2-ил)амино)пропановой кислоты (100 мг, 0,20 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли EDC (153 мг, 0,80 ммоль) и HOBT (122 мг, 0,796 ммоль). Полученную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и экстрагировали смесь EtOAc (3×10 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→55% EtOAc в циклогексане) с получением указанного в заголовке соединения (68 мг, 71%).
1Н-ЯМР (500 МГц, CDCl3): 8,38 (1H, ушир. с), 7,94 (1H, ушир. с), 7,72-7,12 (8H, м), 7,01-6,99 (2H, м), 5,00 (1H, с), 3,68-3,66 (2H, м), 2,82-2,80 (2H, м), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,79-1,65 (1H, м), 1,40-1,10 (9H, ушир.).
Стадия 5: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-4l5-дигидро-1H-пиридо[2,3-b][1,4]диазепин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамат (27 мг, 0,06 ммоль) с получением указанного в заголовке соединения (25 мг, 73%). ЖХ/МС (способ D): RT=0,644 мин, M+1=385.
1Н-ЯМР (500 МГц, MeOD): 7,47-7,43 (5H, м), 7,27-7,25 (3H, м), 7,16-7,14 (2H, м), 3,78-3,76 (2H, м), 2,90-2,88 (2H, м), 2,79-2,73 (2H, м), 2,62-2,56 (2H, м), 2,26-2,22 (1H, м), 1,99-1,95 (1H, м).
Пример 105: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-пропил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
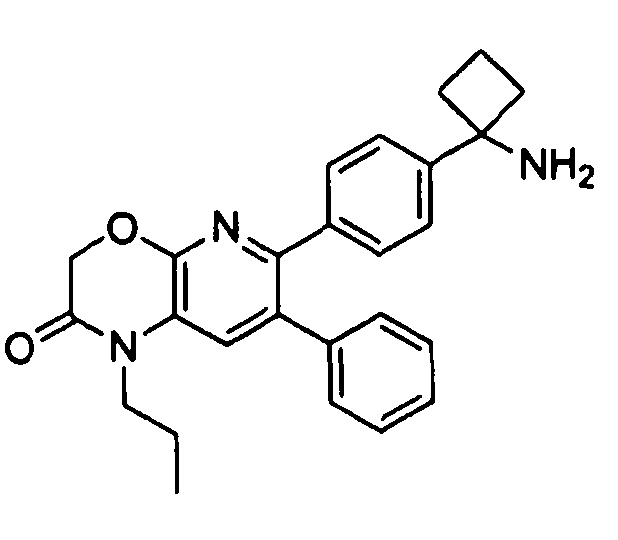
Стадия 1: трет-бутил-(1-(4-(2-оксо-7-фенил-1-пропил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Следуя методике получения трет-бутил-(1-(4-(1-(2-цианоэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата, осуществляли взаимодействие трет-бутил-1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (60 мг, 0,127 ммоль) с 1-йодпропаном (32,4 мг, 0,191 ммоль) с получением указанного в заголовке соединения (43 мг).
1Н-ЯМР (500 МГц, CD3OD): 7,53 (1H, с), 7,21-7,32 (9H, м), 4,94 (2H, с), 4,02 (2H, т), 2,44-2,47 (4H, м), 2,05-2,10 (1H, м), 1,85-1,87 (1H, м), 1,73-1,77 (2H, м), 1,02-1,48 (9H, ушир.), 1,01 (3H, т).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-пропил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(2-оксо-7-фенил-1-пропил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (40 мг, 0,078 ммоль) с получением указанного в заголовке соединения (16,5 мг).
1Н-ЯМР (500 МГц, CH3OD) 7,55 (1H, с), 7,41 (2H, д), 7,38 (2H, д), 7,32 (3H, м), 7,24 (2H, м), 4,96 (2H, с), 4,02 (2H, т), 2,73-2,78 (2H, м), 2,54-2,60 (2H, м), 2,20-2,28 (1H, м), 1,91-1,99 (1H, м), 1,71-1,79 (2H, м), 1,01 (3H, т).
ЖХ/МС (способ D): RT=0,79 мин, M+H+=414,2.
Пример 106: 6-(4-(1-аминоциклобутил)фенил)-1-(3-гидрокси-2-(гидроксиметил)-2-метилпропил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
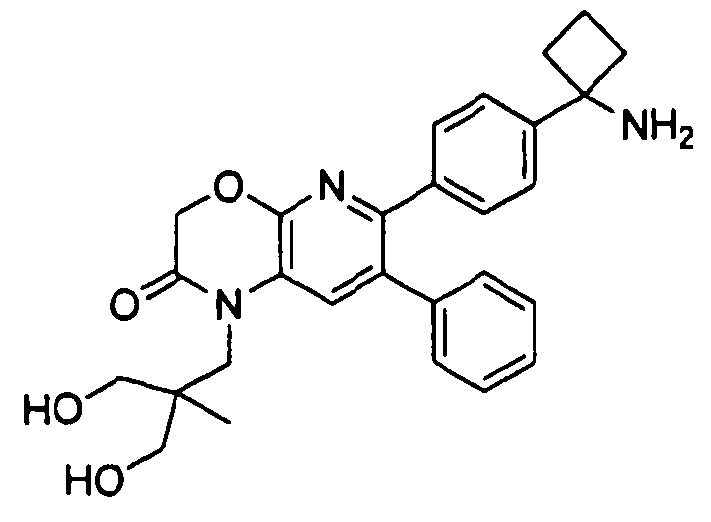
Стадия 1: трет-бутил-(1-(4-(1-((3-метилоксетан-3-ил)метил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (50 мг, 0,11 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли карбонат калия (44 мг, 0,32 ммоль) и 3-(хлорметил)-3-метилоксетан (38 мг, 0,32 ммоль). Полученную смесь перемешивали в течение 2,5 часов при 80°C. Добавляли насыщенный раствор NaHCO3 и экстрагировали смесь AcOEt (3×). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→40% EtOAc в циклогексане) с получением указанного в заголовке соединения (21 мг, 36%).
1Н-ЯМР (500 МГц, CDCl3): 7,23-7,17 (8H, м), 7,09-7,07 (2H, м), 4,94 (2H, с), 4,83 (2H, с), 4,64 (2H, д), 4,23 (2H, д), 4,05 (2H, с), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,79-1,65 (1H, м), 1,36 (3H, с), 1,40-1,10 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-(3-гидрокси-2-(гидроксиметил)-2-метилпропил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1-((3-метилоксетан-3-ил)метил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (16 мг, 0,03 ммоль) с получением указанного в заголовке соединения (16 мг, 79%). ЖХ/МС (способ D): RT=0,721 мин, M+1=474.
1Н-ЯМР (500 МГц, MeOD): 8,03 (1H, с), 7,44-7,38 (4H, м), 7,30-7,24 (5H, м), 4,96 (2H, с), 4,11 (2H, с), 3,47-3,41 (4H, м), 2,77-2,72 (2H, м), 2,59-2,53 (2H, м), 2,25-2,21 (1H, м), 1,98-1,94 (1H, м), 0,92 (3H, с).
Пример 107: ((7-(4-(1-аминоциклобутил)фенил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-ил)фенил)метанол
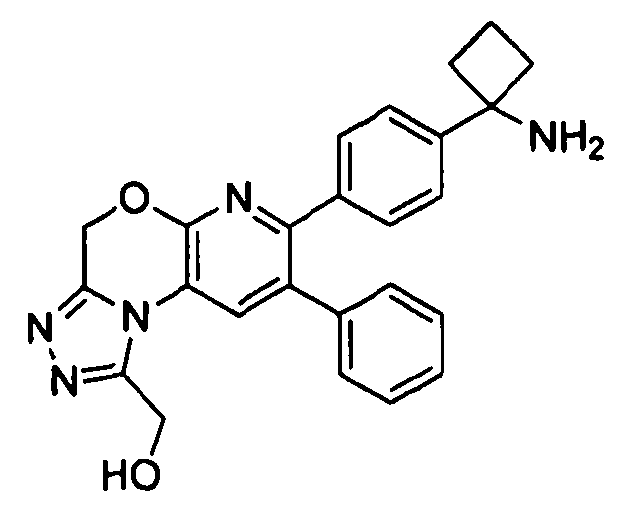
Стадия 1: 2-гидроксиацетогидразид
Этилгликолят (5 г, 0,047 моль) и гидразина моногидрат (3,6 г, 0,071 моль) нагревали до температуры возгонки в течение 15 часов. Реакционную смесь концентрировали в условиях пониженного давления, и обрабатывали остаток толуолом (3×50 мл) для азеотропной отгонки избытка гидразина. Указанную процедуру повторяли трижды. Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
Стадия 2: трет-бутил-(1-(4-(1-(гидроксиметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (250 мг, 0,513 ммоль) и 2-гидроксиацетогидразида (230 мг, 2,56 ммоль) в пара-ксилоле (1,5 мл) нагревали до 150°C в течение 30 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в н-гексанах) с получением указанного в заголовке соединения (40 мг, 15%). ЖХ/МС (способ D): RT=1,297 мин, M+1=526.
1Н-ЯМР (500 МГц, CDCl3) δ 8,33 (1H, с), 7,37-7,32 (2H, м), 7,31-7,27 (5H, м), 7,25-7,20 (2Н, м), 5,59 (2Н, с), 5,06 (2Н, ушир. с), 5,02 (1H, ушир. с), 2,53-2,43 (4H, м), 2,14-2,02 (1H, м), 1,88-1,76 (1H, м), 1,45-1,27 (9H, ушир. с).
Стадия 3: ((7-(4-(1-аминоциклобутил)фенил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-ил)фенил)метанол
трет-Бутил-(1-(4-(1-(гидроксиметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (5 мг, 9,51 мкмоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (2,5 мг, выход 60%). ЖХ/МС (способ D) RT=0,747 мин, M+1=436.
1H-ЯМР (500 МГц, CD3OD) δ 8,49 (1H, с), 7,42-7,36 (2H, д), 7,34-7,29 (2H, д), 7,26-7,20 (3H, м), 7,19-7,14 (2H, м), 5,60 (2H, с), 4,90 (2H, с), 2,72-2,61 (2H, м), 2,53-2,43 (2H, м), 2,20-2,08 (1H, м), 1,93-1,82 (1H, м), 2,01-1,92 (1H, м).
Пример 108: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1H)-он
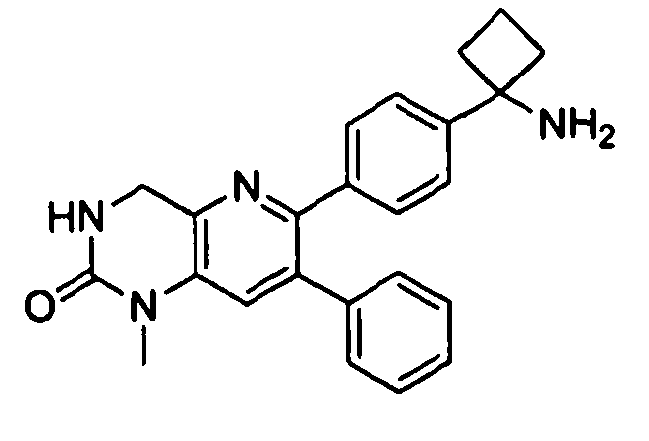
Стадия 1: трет-бутил-1-(4-(3-метил-2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[2,3-b]пиразин-6-ил)фенил)циклобутилкарбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутил)карбамата (71 мг, 0,15 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли карбонат калия (21 мг, 0,15 ммоль) и метилйодид (9,4 мкл, 0,15 ммоль). Полученную смесь перемешивали в течение ночи при комнатной температуре. В атмосфере азота добавляли карбонат калия (42 мг, 0,30 ммоль) и метилйодид (18,8 мкл, 0,30 ммоль). Полученную смесь перемешивали в течение 1 ч при комнатной температуре. В атмосфере азота добавляли карбонат калия (146 мг, 1,05 ммоль) и метилйодид (66 мкл, 1,05 ммоль). Полученную смесь перемешивали в течение 4 ч при 60°C. Добавляли насыщенный раствор бикарбоната натрия и экстрагировали смесь этилацетатом (3×10 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→100% EtOAc в циклогексане) с получением указанного в заголовке соединения (31 мг, 42%). ЖХ/МС (способ D): RT=1,437 мин, M+H+=485.
1Н-ЯМР (500 МГц, CDCl3): 7,25-7,38 (7H, м), 7,20-7,25 (2H, м), 7,16 (1H, с), 5,47 (1H, ушир. с), 5,10 (1H, ушир. с), 4,73 (2H, с), 3,35 (3H, с), 2,25-2,70 (4H, ушир. м), 2,00-2,13 (1H, м), 1,75-1,85 (1H, м), 1,10-1,40 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-1-(4-(1-метил-2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутилкарбамат (31 мг, 0,064 ммоль) с получением указанного в заголовке соединения (5 мг, 20%). ЖХ/МС (способ D): RT=0,763 мин, M-NH2=368.
1Н-ЯМР (500 МГц, MeOD): 7,20-7,40 (10H, м), 4,62 (2H, с), 3,30 (3H, с), 2,48-2,13 (2H, м), 2,20-2,35 (2H, м), 2,00-2,13 (1H, м), 1,70-1,80 (1H, м).
Пример 109: 6-(4-(1-аминоциклобутил)фенил)-1,3-бис(2-фторэтил)-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1H)-он
Стадия 1: трет-бутил-(1-(4-(1,3-бис(2-фторэтил)-2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутил)карбамата (41 мг, 0,09 ммоль) в безводном DMF (1 мл) при комнатной температуре в атмосфере азота добавляли гидрид натрия (3,5 мг, 0,09 ммоль) и 1-фтор-2-йодэтан (7,6 мкл, 0,09 ммоль). Полученную смесь перемешивали в течение 1 ч при комнатной температуре. В атмосфере азота добавляли гидрид натрия (3,5 мг, 0,09 ммоль) и 1-фтор-2-йодэтан (7,6 мкл, 0,09 ммоль). Полученную смесь перемешивали в течение 1 ч при комнатной температуре. Добавляли воду и экстрагировали смесь EtOAc (3×5 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→35% EtOAc в циклогексане) с получением указанного в заголовке соединения (30 мг, 61%).
1Н-ЯМР (500 МГц, CDCl3): 7,24 (1H, с), 7,20-7,17 (7H, м), 7,12-7,10 (2H, м), 4,96 (1H, ушир. с), 4,74 (2H, с), 4,72 (2H, дт), 4,62 (2H, дт), 4,12 (2H, дт), 3,71 (2H, дт), 2,52-2,20 (4H, м), 2,00-1,95 (1H, м), 1,79-1,65 (1H, м), 1,40-1,10 (9H, ушир.).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1,3-бис(2-фторэтил)-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1,3-бис(2-фторэтил)-2-оксо-7-фенил-1,2,3,4-тетрагидропиридо[3,2-d]пиримидин-6-ил)фенил)циклобутил)карбамат (15 мг, 0,03 ммоль) с получением указанного в заголовке соединения (5 мг, 27%). ЖХ/МС (способ D): RT=0,872 мин, M-NH2=446.
1Н-ЯМР (500 МГц, MeOD): 7,52 (1H, с), 7,45-7,39 (4H, м), 7,32-7,30 (3H, м), 7,24-7,22 (2H, м), 4,81-4,77 (4H, м), 4,68 (2H, дт), 4,28 (2H, дт), 3,83 (2H, дт), 2,81-2,75 (2H, м), 2,63-2,57 (2H, м), 2,26-2,24 (1H, м), 1,99-1,97 (1H, м).
Пример 110: 1-(4-(1-циклопропил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(1-циклопропил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (150 мг, 0,308 ммоль) и циклопропанкарбогидразида (41 мг, 0,410 ммоль) в пара-ксилоле (1,5 мл) нагревали до 150°C в течение 20 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в н-гексанах) с получением указанного в заголовке соединения (36 мг, 32%). ЖХ/МС (способ D): RT=1,457 мин, M+1=536.
1Н-ЯМР (500 МГц, CDCl3): 8,24 (1H, с), 7,37-7,27 (7H, м), 7,24-7,19 (2H, м), 5,57 (2H, с), 2,55-2,40 (4H, м), 2,12-2,05 (1H, м), 1,87-1,78 (1H, м), 1,43-1,31 (9H, ушир. с), 1,34-1,29 (3H, м), 1,23-1,17 (2H, м).
Стадия 2: 1-(4-(1-циклопропил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(1-циклопропил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (36 мг, 0,012 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли в условиях пониженного давления, и сушили остаток с получением целевого продукта в виде не совсем белого твердого вещества (24 мг, 82% выход). ЖХ/МС (способ D) RT=0,747 мин, M+1=436.
1H-ЯМР (500 МГц, CD3OD) δ 8,48 (1H, с), 7,53-7,47 (2H, м), 7,45-7,41 (2H, д), 7,38-7,33 (3Н, м), 7,32-7,26 (2H, м), 5,66 (2H, с), 2,82-2,72 (2H, м), 2,63-2,55 (2H, м), 2,44-2,36 (1H, м), 2,30-2,19 (1H, м), 2,01-1,92 (1H, м), 1,28-1,17 (4H, перекрывающийся ддд).
Пример 111: 7-(4-(1-аминоциклобутил)фенил)-1,5-диметил-8-фенил-4,5-дигидро-1H-пиридо[2,3-b][1,4]диазепин-2(3H)-он
Стадия 1: трет-бутил-(1-(4-(1,5-диметил-2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамата (41 мг, 0,09 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли карбонат калия (117 мг, 0,85 ммоль) и метилйодид (53 мкл, 0,85 ммоль). Полученную смесь перемешивали в течение 1 часа при 80°C. Добавляли воду и экстрагировали смесь EtOAc (3×15 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→60% EtOAc в циклогексане) с получением указанного в заголовке соединения (15 мг, 35%).
1Н-ЯМР (500 МГц, CDCl3): 7,32-7,28 (3H, м), 7,21-7,18 (5H, м), 7,12-7,10 (2H, м), 4,94 (1H, ушир. с), 3,65 (2H, т), 3,28 (3H, с), 2,96 (3H, с), 2,61-2,59 (2H, т), 2,55-2,25 (4H, м), 2,10-2,03 (1H, м), 1,86-1,78 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 7-(4-(1-аминоциклобутил)фенил)-1,5-диметил-8-фенил-4,5-дигидро-1H-пиридо[2,3-b][1,4]диазепин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1,5-диметил-2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамат (15 мг, 0,03 ммоль) с получением указанного в заголовке соединения (12 мг, 64%). ЖХ/МС (способ D): RT=0,880 мин, M+1=413.
1Н-ЯМР (500 МГц, MeOD): 7,65 (1H, с), 7,53 (2H, д), 7,39 (2H, д), 7,31-7,28 (3H, м), 7,24-7,22 (2H, м), 3,73 (2H, т), 3,37 (3H, с), 3,02 (3H, с), 2,81-2,75 (2H, м), 2,68 (2H, т), 2,61-2,55 (2H, м), 2,25-2,18 (1H, м), 1,99-1,91 (1H, м).
Пример 112: 1-(4-(1-изопропил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
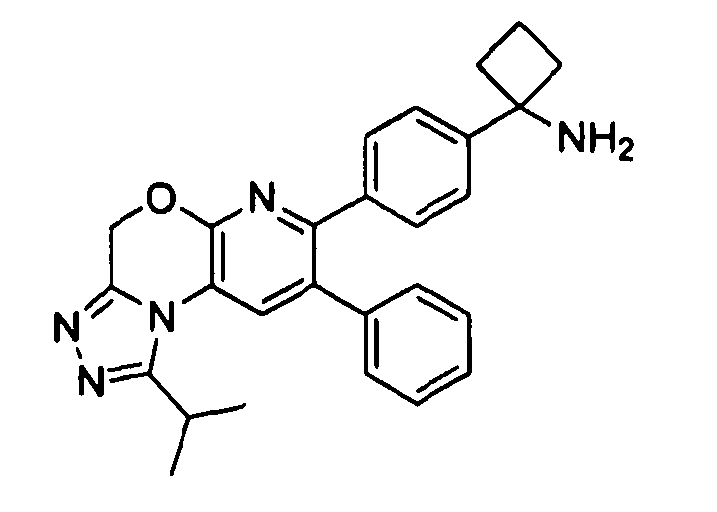
Стадия 1: изобутирогидразид
Этилизобутират (5 г, 0,043 моль) и гидразина моногидрат (3,2 г, 0,065 моль) нагревали до температуры возгонки в течение 15 часов. Реакционную смесь концентрировали в условиях пониженного давления, и обрабатывали остаток толуолом (3×100 мл) для азеотропной отгонки избытка гидразина. Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
Стадия 2: трет-бутил-(1-(4-(1-изопропил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (150 мг, 0,308 ммоль) и изобутирогидразида (28 мг, 0,308 ммоль) в пара-ксилоле (1,5 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления. Неочищенный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в н-гексанах) с получением указанного в заголовке соединения (28 мг, 17%). ЖХ/МС (способ D): RT=1,497 мин, M+1=538.
1Н-ЯМР (500 МГц, CDCl3) δ 7,85 (1H, с), 7,36-7,31 (5H, м), 7,28 (2H, м), 7,24-7,19 (2H, м), 5,56 (2H, с), 3,46-3,36 (1H, м), 2,57-2,39 (4H, м), 2,13-2,02 (1H, м), 1,88-1,76 (1H, м), 1,57-1,53 (6H, д), 1,44-1,29 (9H, ушир. с).
Стадия 3: 1-(4-(1-изопропил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(1-изопропил-8-фенил-4H-пиридо[2>3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (24 мг, 0,045 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (18 мг, выход 92%). ЖХ/МС (способ D) RT=0,815 мин, M+1=438.
1H-ЯМР (500 МГц, CD3OD) δ 8,18 (1H, с), 7,52-7,47 (2H, м), 7,45-7,41 (2H, д), 7,38-7,34 (3H, м), 7,31-7,26 (2H, м), 5,64 (2H, с), 2,82-2,72 (2H, м), 2,63-2,54 (2H, м), 2,30-2,19 (1H, м), 2,02-1,92 (1H, м), 1,51 (6H, д).
Пример 113: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Стадия 1: этил-2-((5-бром-3-нитропиридин-2-ил)окси)ацетат
К суспензии гидрид натрия (5,31 г, 133 ммоль) в 1,4-диоксане (250 мл) по каплям в течение 30 минут добавляли этилгликолят (12,56 мл, 133 ммоль), будучи уверенным в поддержании температуры ниже 30°C. Полученную густую суспензию перемешивали при комнатной температуре в течение 15 минут. В отдельную круглодонную колбу объемом 1 л добавляли 5-бром-2-хлор-3-нитропиридин (21 г, 88 ммоль) в 1,4-диоксане (150 мл) с получением коричневого раствора. При 0°C в течение 30 минут по каплям добавляли суспензию гидрида натрия и этилгликолята. Полученную реакционную смесь нагревали до 80°C в течение ночи. Реакционную смесь концентрировали в условиях пониженного давления, и очищали неочищенный остаток методом хроматографии на силикагеле Biotage (градиент 0%→10% этилацетат в н-гексанах) с получением указанного в заголовке соединения (11,8 г, 44%).
1Н-ЯМР (500 МГц, CDCl3): 8,48 (1H, с), 8,42 (1H, c), 5,07 (2H, с), 4,28-4,24 (2H, кв.), 1,31-1,28 (3H, т).
Стадия 2: этил-2-((3-нитро-5-фенилпиридин-2-ил)окси)ацетат
В круглодонную колбу объемом 1 л добавляли этил-2-(5-бром-3-нитропиридин-2-илокси)ацетат (18,33 г, 60,1 ммоль), фенилбороновую кислоту (10,99 г, 90 ммоль), трифенилфосфин (4,73 г, 18,02 ммоль) и фторид цезия (45,6 г, 300 ммоль) в 1,2-диметоксиэтане (300 мл) с получением желтого раствора. Реакционную смесь дегазировали путем барботирования азотом в течение 30 минут. Добавляли ацетат палладия(II) (2,023 г, 9,01 ммоль) и нагревали смесь до 75°C в атмосфере азота в течение ночи. Реакционную смесь оставляли охлаждаться до комнатной температуры и концентрировали досуха в условиях пониженного давления с получением коричневого твердого вещества. Это твердое вещество повторно растворяли в дихлорметане, фильтровали и концентрировали досуха в условиях пониженного давления с получением коричневого твердого вещества. Неочищенный остаток очищали методом хроматографии на силикагеле Biotage (градиент 5%→60% этилацетат в н-гексанах) с получением указанного в заголовке соединения (6,9 г, 38%).
1Н-ЯМР (500 МГц, CDCl3) δ 8,58 (1H, с), 8,56 (1H, c), 7,59-7,52 (2H, м), 7,48-7,46 (2H, м), 7,45-7,43 (1H, м), 5,13 (2H, с), 4,30-4,26 (2H, кв.), 1,33-1,30 (3Н, т).
Стадия 3: 7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В круглодонную колбу объемом 500 мл добавляли этил-2-(3-нитро-5-фенилпиридин-2-илокси)ацетат (4,6 г, 15,22 ммоль) в соляной кислоте (37%, 40 мл) с получением желтой суспензии. Смесь охлаждали до 0-5°C, а затем порциями добавляли порошковое олово (9,94 г, 84 ммоль). Добавление сопровождается экзотермической реакцией. В процессе добавления следует соблюдать осторожность. Затем смесь перемешивали при комнатной температуре еще в течение 30 минут до окончания пенообразования. Реакционную смесь нагревали до 80°C в атмосфере азота в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (800 мл). Белый осадок отделяли путем фильтрования, промывали водой (100 мл) и сушили отсасыванием с получением белого твердого вещества. Твердое вещество подвергали азеотропной перегонке с толуолом (3×30 мл) с получением указанного в заголовке соединения в виде белого твердого вещества (2,6 г, 77%).
1Н-ЯМР (500 МГц, (CD3)2SO) δ 10,41 (1H, с), 8,10 (1H, с), 7,59 (2H, д), 7,49-7,42 (2H, т), 7,39-7-38 (1H, д), 4,83 (2H, с).
Стадия 4: 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В пробирку для микроволновой обработки объемом 10 мл добавляли 7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (50 мг, 0,221 ммоль) и N-бромсукцинимид (78,6 мг, 0,441 ммоль) в диметилформаиде (1 мл). Реакционную смесь нагревали до 80°C в условиях обработки микроволновым излучением в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (10 мл). Органический раствор промывали водой (2×10 мл) и солевым раствором (2×10 мл). Органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали в условиях пониженного давления. Неочищенный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0%→5% метанол в дихлорметане) с получением указанного в заголовке соединения в виде желтого твердого вещества (61 мг, 90%).
1Н-ЯМР (500 МГц, CD3OD) δ 7,48-7,32 (5H, м), 7,12 (1H, с), 4,82 (2H, с).
Стадия 5: 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)изоиндолин-1,3-дион
В пробирку для микроволновой обработки объемом 10 мл добавляли 6-бром-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (32 мг, 0,100 ммоль), 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-дион (46 мг, 0,105 ммоль) и карбонат натрия (21 мг, 0,201 ммоль) в смеси 1,4-диоксана (3,50 мл) и воды (1,20 мл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 10 минут, а затем добавляли тетракис(трифенилфосфин)палладий(0) (12 мг, 10,03 мкмоль). Реакционную смесь нагревали до 100°C в условиях обработки микроволновым излучением в течение 15 минут. Дополнительно добавляли 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-дион (24 мг, 0,055 ммоль) и нагревали смесь до 100°C в условиях обработки микроволновым излучением еще в течение 15 минут. Реакционную смесь разбавляли этилацетатом (12 мл), промывали водой (10 мл), солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого масла. Это масло очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (18 мг, выход 33%).
1H-ЯМР (500 МГц, CDCl3) δ 7,74 (2H, дд), 7,66 (2H, дд), 7,51 (2H, д), 7,31 (2H, д), 7,22-7,29 (4H, м), 7,15-7,20 (2H, м), 4,87 (2H, с), 3,37 (3H, с), 3,32 (2H, д), 3,09 (2H, д), 1,40 (3H, с).
ЖХ/МС (способ D) RT=1,30 мин, M+H+=546,00.
Стадия 6: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В реакционную пробирку объемом 15 мл добавляли 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)изоиндолин-1,3-дион (18 мг, 0,033 ммоль) и гидразина моногидрат (8 мкл, 0,165 ммоль) в этаноле (1650 мкл) с получением желтого раствора. Этот раствор нагревали до 90°C в атмосфере азота в течение 5 часов, а затем оставляли охлаждаться до комнатной температуры. Дополнительно добавляли гидразина моногидрат (16 мкл, 0,330 ммоль) и нагревали реакционную смесь до 100°C в условиях обработки микроволновым излучением в течение 10 минут, а затем до 120°C в условиях обработки микроволновым излучением в течение 20 минут. Дополнительно добавляли гидразина моногидрат (16 мкл, 0,330 ммоль) и нагревали смесь до 120°C в условиях обработки микроволновым излучением в течение 60 минут. Полученный осадок удаляли путем фильтрования и хорошо промывали этанолом. Объединенные фильтраты концентрировали досуха в условиях пониженного давления с получением белого твердого вещества. Это вещество очищали на SCX catch-and-release колонке и лиофилизировали с получением целевого продукта в виде белого твердого вещества (10 мг, выход 73%).
1H-ЯМР (500 МГц, MeOD) δ 7,52 (1H, с), 7,20-7,35 (9H, м), 4,94 (2H, с), 3,42 (3H, с), 2,66 (2H, д), 2,39 (2H, д), 1,55 (3H, с).
ЖХ/МС (способ D) RT=0,67 мин, M+H+=416,20.
Пример 114: 7-(4-(1-аминоциклобутил)фенил)-1-метил-8-фенил-4f5-дигидро-1H-пиридо[2,3-b][1,4]диазепин-2(3H)-он
Стадия 1: трет-бутил-(1-(4-(1-метил-2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамата (53 мг, 0,11 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли гидрид натрия (4,4 мг, 0,11 ммоль) и метилйодид (6,9 мкл, 0,11 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли воду и экстрагировали смесь EtOAc (3×15 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→57% EtOAc в циклогексане) с получением указанного в заголовке соединения (21 мг, 38%).
1Н-ЯМР (500 МГц, CDCl3): 7,39 (1H, с), 7,27-7,23 (7H, м), 7,16-7,14 (2H, м), 5,04 (1H, с), 4,91 (1H, ушир. с), 3,88 (2H, ушир. с), 3,39 (3H, с), 2,74 (2H, т), 2,55-2,25 (4H, м), 2,10-2,03 (1H, м), 1,86-1,78 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 7-(4-(1-аминоциклобутил)фенил)-1-метил-8-фенил-4,5-дигидро-1H-пиридо[2,3-b][1,4]диазепин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1-метил-2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамат (21 мг, 0,04 ммоль) с получением указанного в заголовке соединения (19 мг, 72%). ЖХ/МС (способ D): RT=0,806 мин, M+1=399.
1Н-ЯМР (500 МГц, MeOD): 7,65 (1H, с), 7,47 (2H, д), 7,41 (2H, д), 7,29-7,25 (3H, м), 7,20-7,19 (2H, м), 3,86-3,84 (4H, м), 3,41 (3H, с), 2,79-2,73 (4H, м), 2,61-2,55 (2H, м), 2,25-2,18 (1H, м), 1,99-1,91 (1H, м).
Пример 115: 1-(4-(1-изобутил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Стадия 1: 3-метилбутангидразид
Этилвалерат (5 г, 0,038 моль) и гидразина моногидрат (2,8 г, 0,057 моль) нагревали до температуры возгонки в течение 15 часов. Реакционную смесь концентрировали в условиях пониженного давления, и обрабатывали остаток толуолом (3×100 мл) для азеотропной отгонки избытка гидразина. Указанную процедуру повторяли трижды. Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
Стадия 2: трет-бутил-(1-(4-(1-изобутил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (200 мг, 0,410 ммоль) и 3-метилбутангидразида (47 мг, 0,410 ммоль) в пара-ксилоле (1,5 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в н-гексанах) с получением указанного в заголовке соединения (15 мг, 5%). ЖХ/МС (способ D): RT=1,5687 мин, M+1=552.
1Н-ЯМР (500 МГц, CDCl3): 7,80 (1H, с), 7,36-7,31 (5H, м), 7,30-7,27 (2H, м), 7,23-7,19 (2H, м), 5,57 (2H, с), 2,98 (2H, д), 2,54-2,41 (4H, м), 2,33-2,24 (1H, м), 2,13-2,01 (1H, м), 1,88-1,76 (1H, м), 1,44-1,26 (9H, ушир. с), 1,11 (6H, д).
Стадия 3: 1-(4-(1-изобутил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(1-изобутил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (7 мг, 0,013 ммоль) растворяли в дихлорметане (1 мл), при комнатной температуре добавляли TFA (1 мл) и перемешивали полученную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (4 мг, выход 68%). ЖХ/МС (способ D): RT=0,844 мин, M+1=452.
1H-ЯМР (500 МГц, CD3OD) δ 8,07 (1H, с), 7,52-7,45 (2H, д), 7,44-7,40 (2H, д), 7,38-7,33 (3H, м), 7,31-7,26 (2H, м), 5,66 (2H, с), 3,11 (2H, д), 2,81-2,68 (2H, м), 2,61-2,45 (2H, м), 2,32-2,17 (1H+1H, 2 м), 2,02-1,87 (1H, м), 1,10 (6H, д).
Пример 116: 7-(4-(1-аминоциклобутил)фенил)-N,N-диметил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-амин
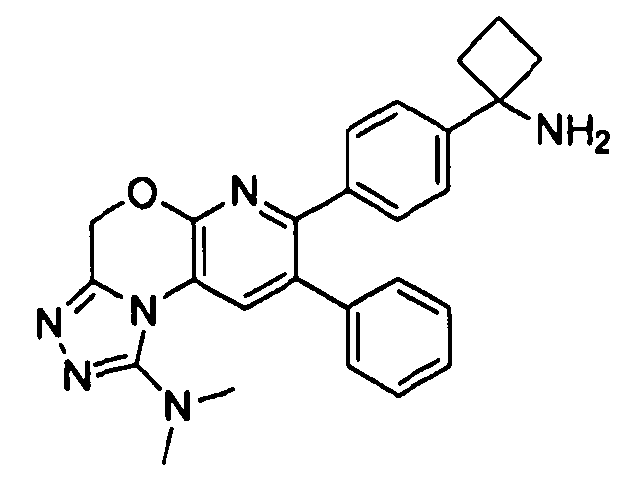
Стадия 1: трет-бутил-(1-(4-(1-(диметиламино)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Смесь трет-бутил-1-(4-(1-бром-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутилкарбамата (41 мг, 0,071 ммоль), диметиламина гидрохлорида (58,2 мг, 0,714 ммоль) и триэтиламина (0,099 мл, 0,714 ммоль) в NMP (1 мл) нагревали при 130°C в течение 50 мин в условиях обработки микроволнами. Смесь разбавляли DCM (8 мл) и водой (8 мл). Органическую фазу промывали водой (3×8 мл) и солевым раствором (5 мл), концентрировали и очищали на колонке, элюируя MeOH/DCM (0-4%), с получением продукта (8 мг).
1Н-ЯМР (500 МГц, CDCl3) 8,15 (с, 1H), 7,15-7,34 (м, 9H), 5,51 (с, 2H), 5,01 (с, 1H), 2,95 (с, 6H), 2,4-2,5 (м, 4H), 2,0-2,15 (м, 1H), 1,75-1,85 (м, 1H), 1,2-1,35 (м, 9H).
Стадия 2: 7-(4-(1-аминоциклобутил)фенил)-N,N-диметил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-амин
Следуя методике получения 3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрила, в реакции использовали трет-бутил-(1-(4-(1-(цианометил)-3-(метоксиметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (8 мг, 0,015 ммоль) с получением указанного в заголовке соединения (5,5 мг).
1Н-ЯМР (500 МГц, CH3OD) 8,26 (с, 1H), 7,48 (д, 2H), 7,42 (д, 2H), 7,35 (м, 3H), 7,26 (м, 2H), 5,6 (с, 2H), 2,96 (с, 6H), 2,7-2,8 (м, 2H), 2,55-2,65 (м, 2H), 2,2-2,3 (м, 1H), 1,9-2,0 (м, 1H).
ЖХ/МС (способ D): RT=0,77 мин, M+H+=439,2.
Пример 117: 7-(4-(1-аминоциклобутил)фенил)-1-(2-фторэтил)-8-фенил-4,5-дигидро-1H-пиридо[2,3-b][1,4]диазепин-2(3H)-он
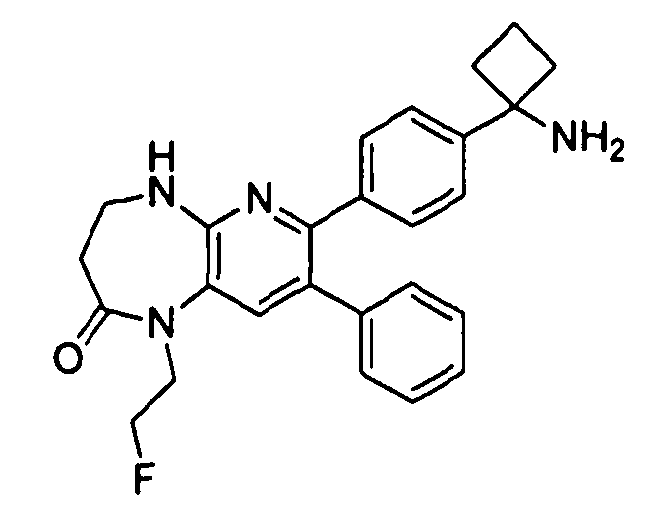
Стадия 1: трет-бутил-(1-(4-(1-(2-фторэтил)-2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамат
К раствору трет-бутил-(1-(4-(2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамата (101 мг, 0,21 ммоль) в безводном DMF (1 мл) в атмосфере азота добавляли гидрид натрия (12 мг, 0,32 ммоль) и 1-фтор-2-йодэтан (27 мкл, 0,32 ммоль). Полученную смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли воду и экстрагировали смесь EtOAc (3×10 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали досуха в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0→100% EtOAc в циклогексане) с получением указанного в заголовке соединения (52 мг, 47%).
1Н-ЯМР (500 МГц, CDCl3): 7,62 (1H, с), 7,31-7,22 (7H, м), 7,16-7,14 (2H, м), 5,01 (1H, с), 4,75 (1H, ушир. с), 4,74 (2H, дт), 4,07 (2H, дт), 3,92-3,88 (2H, м), 2,73 (2H, т), 2,55-2,25 (4H, м), 2,10-2,03 (1H, м), 1,86-1,78 (1H, м), 1,44-1,15 (9H, ушир.).
Стадия 2: 7-(4-(1-аминоциклобутил)фенил)-1-(2-фторэтил)-8-фенил-4,5-дигидро-1H-пиридо[2,3-b][1,4]диазепин-2(3H)-он
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали трет-бутил-(1-(4-(1-(2-фторэтил)-2-оксо-8-фенил-2,3,4,5-тетрагидро-1H-пиридо[2,3-b][1,4]диазепин-7-ил)фенил)циклобутил)карбамат (15 мг, 0,03 ммоль) с получением указанного в заголовке соединения (12 мг, 64%). ЖХ/МС (способ D): RT=0,808 мин, M+1=431.
1Н-ЯМР (500 МГц, MeOD): 7,76 (1H, с), 7,48 (2H, д), 7,41 (2H, д), 7,29-7,25 (3H, м), 7,20-7,18 (2H, м), 4,67 (2H, дт), 4,16 (2H, дт), 3,88-3,86 (2H, м), 2,79-2,73 (4H, м), 2,61-2,55 (2H, м), 2,25-2,18 (1H, м), 1,99-1,91 (1H, м).
Пример 118: 1-(4-(8-фенил-1-(2,2,2-трифторэтил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
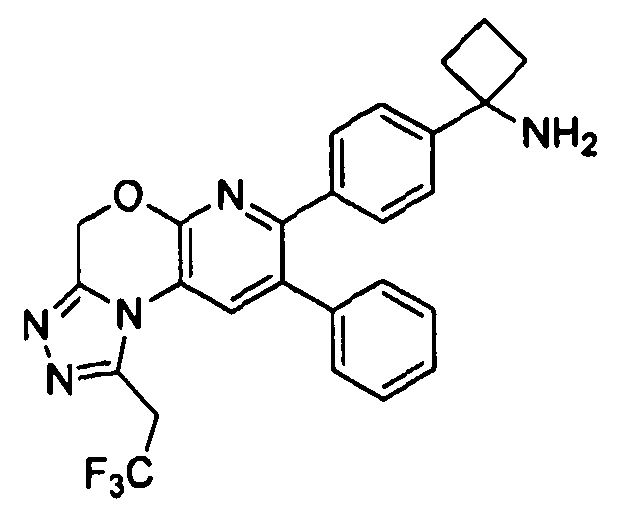
Стадия 1: 3,3,3-трифторпропангидразид
Этил-3,3,3-трифторпропаноат (2,5 г, 0,016 моль) и гидразина моногидрат (1,2 г, 0,024 моль) нагревали до температуры возгонки в течение 1 часа. Реакционную смесь концентрировали в условиях пониженного давления, и обрабатывали остаток толуолом (3×100 мл) для азеотропной отгонки избытка гидразина. Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
Стадия 2: трет-бутил-(1-(4-(8-фенил-1-(2,2,2-трифторэтил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (250 мг, 0,513 ммоль) и 3,3,3-трифторпропангидразида (145 мг, 1,205 ммоль) в пара-ксилоле (2 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в н-гексанах) с получением указанного в заголовке соединения (38 мг, 13%). ЖХ/МС (способ D): RT=1,499 мин, M+1=578.
1Н-ЯМР (500 МГц, CDCl3): 7,84 (1H, с), 7,36-7,32 (5H, м), 7,31-7,28 (2H, м), 7,23-7,18 (2H, м), 5,60 (2H, с), 4,12 (2H, кв.), 2,52-2,42 (4H, м), 2,12-2,01 (1H, м), 1,87-1,78 (1H, м), 1,44-1,26 (9H, ушир. с).
Стадия 3: 1-(4-(8-фенил-1-(2,2,2-трифторэтил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(8-фенил-1-(2,2,2-трифторэтил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (25 мг, 0,043 ммоль) растворяли в дихлорметане (1 мл), при комнатной температуре добавляли TFA (1 мл) и перемешивали полученную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (16 мг, выход 77%). ЖХ/МС (способ D): RT=0,791 мин, M+1=478.
1H-ЯМР (500 МГц, CD3OD) δ 8,19 (1H, с), 7,52-7,48 (2H, д), 7,45-7,42 (2H, д), 7,38-7,34 (3Н, м), 7,31-7,27 (2H, м), 5,69 (2H, с), 4,47 (2H, кв.), 2,82-2,73 (2H, м), 2,64-2,54 (2H, м), 2,31-2,19 (1H, м), 2,02-1,92 (1H, м).
Пример 119: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-карбоксамид
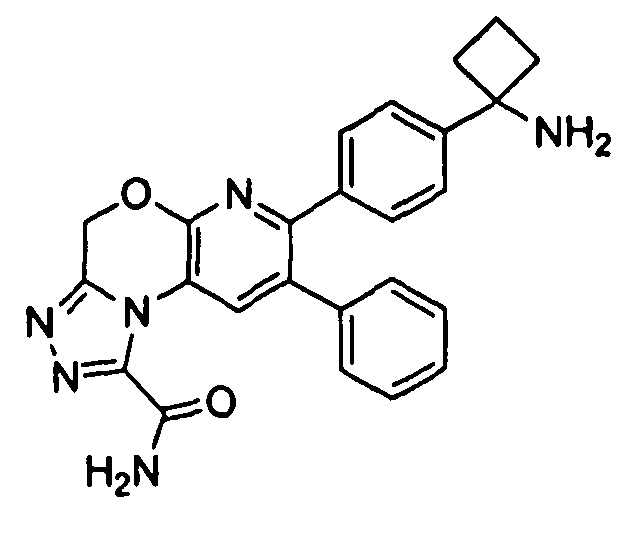
Стадия 1: этилгидразина карбоксилат
Диэтилоксалат (5 г, 0,042 моль) и гидразина моногидрат (2,8 г, 0,057 моль) в этаноле (3 мл) нагревали до температуры возгонки в течение 15 часов. Реакционную смесь концентрировали в условиях пониженного давления, и обрабатывали остаток толуолом (3×100 мл) для азеотропной отгонки избытка гидразина. Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
Стадия 2: этил-7-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-карбоксилат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (250 мг, 0,513 ммоль) и этилгидразина карбоксилата (271 мг, 2,05 ммоль) в пара-ксилоле (2 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→30% этилацетат в н-гексанах) с получением указанного в заголовке соединения (38 мг, 13%). ЖХ/МС (способ D): RT=1,531 мин, M+1=568.
Стадия 3: трет-бутил-(1-(4-(1-карбамоил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию этил-7-(4-(1-((трет-бутоксикарбонил)амино)циклобутил)фенил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-карбоксилата (70 мг, 0,123 ммоль) и хлорида аммония (10 мг, 0,19 ммоль) в гидроксиде аммония (1 мл) нагревали до 100°C в течение 10 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→10% этилацетат в циклогексане) с получением указанного в заголовке соединения (38 мг, 13%). ЖХ/МС (способ D): RT=1,347 мин, M+1=539.
1H-ЯМР (500 МГц, CD3OD) δ 9,10 (1Н, с), 7,47-7,40 (1Н, ушир. с), 7,40-7,37 (2Н, д), 7,32-7,27 (6Н, м), 5,59 (2Н, с), 2,59-2,39 (4H, м), 2,14-2,01 (1H, м), 1,88-1,76 (1H, м), 1,44-1,33 (9H, ушир. с).
Стадия 4: 7-(4-(1-аминоциклобутил)фенил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-карбоксамид
трет-Бутил-(1-(4-(1-карбамоил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (10 мг, 0,019 ммоль) растворяли в дихлорметане (0,5 мл), этот раствор добавляли к SCX-2 catch and release картридж (1 г), предварительно уравновешенный метанолом (15 мл) и дихлорметаном (15 мл). После связывания в течение 15 часов, колонку промывали метанолом (6 мл), а затем элюировали продукт 7M аммиаком в метаноле (2×4 мл). Объединенные содержащие продукт фракции концентрировали досуха в условиях пониженного давления с получением целевого продукта в виде белого твердого вещества (5 мг, выход 61%). ЖХ/МС (способ D) RT=0,695 мин, M+1=440.
1H-ЯМР (500 МГц, CD3OD) δ 8,80 (1H, с), 7,27-7,24 (4H, ушир. с), 7,22-7,17 (3Н, м), 7,16-7,13 (2H, м), 5,57 (2H, с), 2,49-2,38 (2H, м), 2,19-2,10 (2H, м), 2,03-1,92 (1H, м), 1,71-1,60 (1H, м).
Пример 120: 1-(4-(1-(фторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
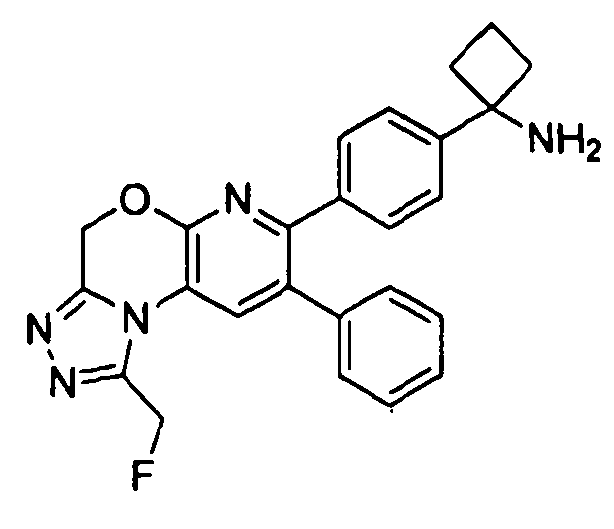
Стадия 1: трет-бутил-(1-(4-(1-(фторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Раствор трет-бутил-(1-(4-(1-(гидроксиметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамата (40 мг, 0,019 ммоль), триэтиламина (2,65 мл, 0,019 ммоль) и DeoxoFluor® (50% в THF, 21,05 мг, 0,048 ммоль) в дихлорметане (1 мл) нагревали до 30°C в течение 15 часов. Полученную реакционную смесь гасили насыщенным раствором бикарбоната натрия и экстрагировали дихлорметаном (3×10 мл). Объединенные органические слои концентрировали досуха в условиях пониженного давления. Неочищенный остаток очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в циклогексане) с получением указанного в заголовке соединения (38 мг, 13%). ЖХ/МС (способ D): RT=1,439 мин, M+1=528.
1Н-ЯМР (500 МГц, CDCl3): 8,01 (1H, с), 7,37-7,27 (7H, м), 7,24-7,20 (2H, дд), 5,83 (2H, с), 5,73 (1H, с), 5,64 (1H, с), 2,54-2,41 (4H, м), 2,12-2,02 (1H, м), 1,88-1,76 (1H, м), 1,43-1,30 (9H, ушир. с).
Стадия 2: 1-(4-(1-(фторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(1-(фторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (3 мг, 5,69 мкмоль) растворяли в дихлорметане (1 мл), при комнатной температуре добавляли TFA (0,5 мл) и перемешивали полученную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (1,5 мг, выход 61%). ЖХ/МС (способ D) RT=0,774 мин, M+1=428.
1H-ЯМР (500 МГц, CD3OD) δ 8,03 (1H, с), 7,41-7,35 (2H, д), 7,34-7,29 (2H, д), 7,27-7,23 (3Н, м), 7,21-7,15 (2Н, м), 5,86 (1Н, с), 5,75 (1Н, с), 5,64 (2H, с), 2,71-2,58 (2H, м), 2,49-2,37 (2H, м), 2,18-2,07 (1H, м), 1,91-1,79 (1H, м).
Пример 121: 1-(4-(1-(2-метоксиэтил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(1-(2-метоксиэтил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (100 мг, 0,205 ммоль) и 3-метоксипропангидразида (48 мг, 0,410 ммоль) в пара-ксилоле (2 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в циклогексане) с получением указанного в заголовке соединения (33 мг, 30%). ЖХ/МС (способ D): RT=1,414 мин, M+1=554.
1Н-ЯМР (500 МГц, CDCl3) δ 8,16 (1H, с), 7,27-7,36 (7H, м), 7,23-7,18 (2H, м), 5,60 (2H, с), 3,99 (2H, т), 3,43 (3H, с), 3,38 (2H, т), 2,57-2,40 (4H, м), 2,12-2,01 (1H, м), 1,87-1,77 (1H, м), 1,49-1,27 (9H, ушир. с).
Стадия 2: 1-(4-(1-(2-метоксиэтил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(1-(2-метоксиэтил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (12 мг, 0,022 ммоль) растворяли в дихлорметане (0,5 мл), при комнатной температуре добавляли TFA (0,5 мл) и перемешивали полученную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (5 мг, выход 50%). ЖХ/МС (способ D) RT=0,741 мин, M+1=454.
1H-ЯМР (500 МГц, CD3OD) δ 8,40 (1H, с), 7,52-7,47 (2H, д), 7,46-7,41 (2H, д), 7,38-7,34 (3Н, м), 7,32-7,26 (2H, дд), 5,65 (2H, с), 3,94 (2H, т), 3,49 (2H, т), 3,35 (3H, с), 2,82-2,74 (2H, м), 2,63-2,54 (2H, м), 2,30-2,19 (1H, м), 2,04-1,93 (1H, м).
Пример 122: 1-(4-(8-фенил-1-(пиридин-3-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(8-фенил-1-(пиридин-3-ил)-4H-пиридо[2,3-b][1,2,4]]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (100 мг, 0,205 ммоль) и никотиновой кислоты гидразид (84 мг, 0,308 ммоль) в пара-ксилоле (2 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в циклогексане) с получением указанного в заголовке соединения (33 мг, 25%). ЖХ/МС (способ D): RT=1,364 мин, M+1=573.
1Н-ЯМР (500 МГц, CDCl3): 9,00 (1H, д), 8,87 (1H, дд), 8,10 (1H, дт), 7,56 (1H, дд), 7,36-7,27 (5H, м), 7,24-7,17 (3H, м), 6,99-6,93 (2H, м), 5,67 (2H, с), 2,56-2,39 (4H, м), 2,13-2,01 (1H, м), 1,88-1,75 (1H, м), 1,49-1,26 (9H, ушир. с).
Стадия 2: 1-(4-(8-фенил-1-(пиридин-3-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(8-фенил-1-(пиридин-2-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (15 мг, 0,026 ммоль) растворяли в дихлорметане (1 мл), при комнатной температуре добавляли TFA (0,5 мл) и перемешивали полученную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (8 мг, 56% выход). ЖХ/МС (способ A) RT=3,73 мин, M+1=474.
1Н-ЯМР (500 МГц, CDCl3): 8,99 (1H, д), 8,86 (1H, дд), 8,30 (1H, дт), 7,75 (1H, дд), 7,48 (2H, д), 7,42 (2H, д), 7,37 (1H, с), 7,31-7,20 (3H, м), 7,01 (2H, д), 5,78 (2H, с), 2,83-2,72 (2H, м), 2,63-2,54 (2H, м), 2,30-2,17 (1H, м), 2,05-1,90 (1H, м).
Пример 123: 1-(4-(8-фенил-1-(пиридин-4-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(8-фенил-1-(пиридин-4-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (100 мг, 0,205 ммоль) и изоникотиновой кислоты гидразида (127 мг, 0,923 ммоль) в пара-ксилоле (2 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в н-гексанах) с получением указанного в заголовке соединения (14 мг, 12%). ЖХ/МС (способ A): RT=6,28 мин, M+1=573.
1Н-ЯМР (500 МГц, CDCl3): 8,90 (2H, д), 7,70 (2H, д), 7,38 (1H, с), 7,36-7,32 (2H, д), 7,31-7,27 (2H, д), 7,25-7,18 (3Н, м), 6,98-6,93 (2H, дд), 5,66 (2H, с), 2,55-2,41 (4H, м), 2,14-2,01 (1H, м), 1,88-1,78 (1H, м), 1,46-1,24 (9H, ушир. с).
Стадия 2: 1-(4-(8-фенил-1-(пиридин-4-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(8-фенил-1-(пиридин-4-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (14 мг, 0,024 ммоль) растворяли в дихлорметане (1 мл), при комнатной температуре добавляли TFA (0,5 мл) и перемешивали полученную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (10 мг, 87% выход). ЖХ/МС (способ A) RT=3,66 мин, M+H+=474.
1Н-ЯМР (500 МГц, CDCl3): 8,77 (2H, д), 8,79 (2H, д), 7,41 (2H, д), 7,33 (1H, с), 7,32 (2H, д), 7,18-7,13 (3H, м), 6,96 (2H, дд), 5,66 (2H, с), 2,70-2,61 (2H, м), 2,52-2,43 (2H, м), 2,19-2,08 (1H, м), 1,92-1,82 (1H, м).
Пример 124: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-(пиридин-3-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
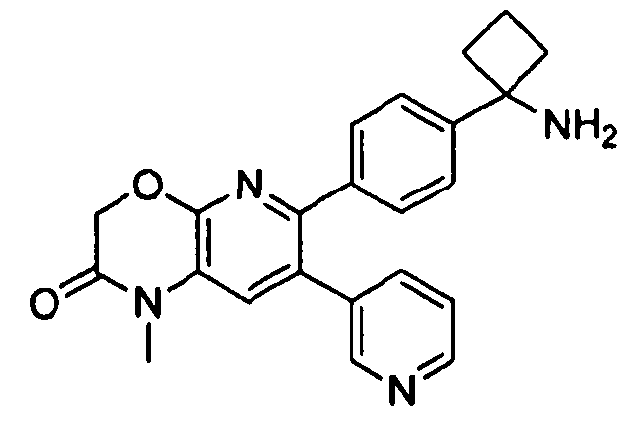
Стадия 1: 6,7-дибром-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
7-Бром-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (1,5 г, 6,55 ммоль) и NBS (5,83, 32,7 ммоль) нагревали до 80°C в DMF (30 мл) в течение 2 часов. Полученную реакционную смесь разбавляли этилацетатом (30 мл) и водой (30 мл). Водную фазу экстрагировали свежим этилацетатом (3×50 мл), объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением указанного в заголовке соединения (1,6 г, 79%). Это соединение использовали на следующей стадии без дополнительной очистки. ЖХ/МС (способ D): RT=0,91 мин, M+1=309.
Стадия 2: 6,7-дибром-1-метил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
6,7-Дибром-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (1,5 мг, 4,88 ммоль), карбонат калия (2,02 г, 14,61 ммоль) и метилйодид (2,07, 14,61 ммоль) перемешивали в DMF (35 мл) при комнатной температуре в течение 1 часа. Полученную реакционную смесь гасили добавлением воды (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали водой (100 мл), солевым раствором (100 мл) и сушили над безводным Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением указанного в заголовке соединения (1,3 г, 83%). Это соединение использовали на следующей стадии без дополнительной очистки. ЖХ/МС (способ D) RT=1,053 мин, M+1=322.
1H-ЯМР (500 МГц, CD3OD) δ 7,41 (1H, с), 4,85 (2H, с), 3,35 (3Н, с).
Стадия 3: трет-бутил-(1-(4-(триметилстаннил)фенил)циклобутил)карбамат
трет-Бутил-(1-(4-бромфенил)циклобутил)карбамат (5,45 мг, 13,36 ммоль) растворяли в безводном толуоле (250 мл). Раствор дегазировали путем барботирования азотом в течение 15 минут. Добавляли тетракис(трифенилфосфин)палладий(0) (3,09 г, 2,67 ммоль) и дегазировали полученную реакционную смесь путем барботирования азотом в течение 15 минут. К полученной смеси добавляли гексаметилдиолово (6,57 г, 4,19 мл, 20,05 ммоль), заранее растворенное в безводном толуоле (30 мл). Полученную реакционную смесь дегазировали путем барботирования азотом в течение 10 минут и нагревали до температуры возгонки в течение 2 часов. Реакционную смесь охлаждали и фильтровали через целит, используя свежий этилацетат для промывки осадка на фильтре. Фильтрат концентрировали досуха в условиях пониженного давления. Полученное неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→90% этилацетат в н-гексанах; пластины ТСХ проявляли KMnO4) с получением указанного в заголовке соединения (1,2 г, 22%).
1Н-ЯМР (500 МГц, CDCl3): 7,53-7,36 (4H, м), 2,60-2,47 (4H, м), 2,13-2,03 (1H, м), 1,90-1,78 (1H, м), 1,47-1,27 (9H, ушир.).
Стадия 4: трет-бутил-(1-(4-(7-бром-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
6,7-Дибром-1-метил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (0,628 г, 1,951 ммоль) и трет-бутил-1-(4-(триметилстаннил)фенил)циклобутилкарбамат (1,2 г, 2,93 ммоль) растворяли в DMF (10 мл) с получением желтого раствора. Реакционную смесь дегазировали путем барботирования азотом в течение 30 минут. При комнатной температуре добавляли хлорид бис(диметилфосфино)палладия(IV) (0,137 г, 0,195 ммоль). Полученную реакционную смесь дегазировали и нагревали до 80°C. Реакционную смесь гасили добавлением воды (80 мл); водную фазу экстрагировали этилацетатом (3×80 мл); объединенные органические слои промывали водой (2×80 мл) и солевым раствором (2×80 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→70% этилацетат в н-гексанах) с получением указанного в заголовке соединения (400 мг, 42%).
1 1Н-ЯМР (500 МГц, CDCl3): 7,73-7,56 (2H, м), 7,56-7,46 (3H, м), 4,88 (2H, с), 3,33 (3H, с), 2,68-2,49 (4H, м), 2,20-2,04 (1H, м), 1,94-1,80 (1H, м), 1,47-1,27 (9H, ушир. с).
1 Это соединение представляло собой смесь двух региоизомеров (в соотношении 7/3).
Стадия 5: трет-бутил-(1-(4-(1-метил-2-оксо-7-(пиридин-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
трет-Бутил-(1-(4-(7-бром-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (57 мг, 0,117 ммоль), пиридин-3-илбороновую кислоту (21,52 мг, 0,175 ммоль), трифенилфосфин (9,18 мг, 0,035 ммоль), фторид цезия (89 мг, 0,584 ммоль) растворяли в 1,2-диметоксиэтане (2 мл) с получением оранжевого раствора. Реакционную смесь дегазировали путем барботирования азотом в течение 10 минут. При комнатной температуре добавляли ацетат палладия(II) (3,93 мг, 0,018 ммоль). Полученную реакционную смесь дегазировали и нагревали до 80°C в течение 18 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления. Неочищенное вещество очищали методом препаративной ЖХВД с получением указанного в заголовке соединения (12 мг, 21%). ЖХ/МС (способ D) RT=1,158 мин, M+1=487.
1Н-ЯМР (500 МГц, CDCl3): 7,64-7,52 (1H, м), 7,37-7,21 (8H, м), 4,94 (2H, с), 3,43 (3H, с), 2,59-2,36 (4H, м), 2,15-2,01 (1H, м), 1,91-1,77 (1H, м), 1,47-1,20 (9H, ушир. с).
Стадия 6: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-(пиридин-3-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-(1-(4-(1-метил-2-оксо-7-(пиридин-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (12 мг, 0,025 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (14 мг, выход 78%). ЖХ/МС (способ D) RT=0,546 мин, M+1=387.
1H-ЯМР (500 МГц, CD3OD) δ 8,57-8,49 (1H, ушир. с), 8,45-8,36 (1H, ушир. с), 7,90 (1H, д), 7,67 (1H, с), 7,61-7,48 (2H, д), 7,48-7,36 (4H, м), 5,00 (2H, с), 3,45 (3H, с), 2,84-2,72 (2H, м), 2,65-2,54 (2H, м), 2,32-2,16 (1H, м), 2,06-1,90 (1H, м).
Пример 125: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-(тиофен-3-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
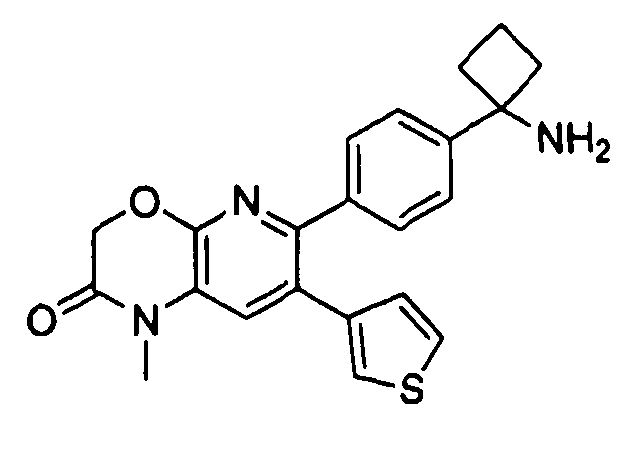
Стадия 1: трет-бутил-(1-(4-(1-метил-2-оксо-7-(тиофен-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
трет-Бутил-(1-(4-(7-бром-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (70 мг, 0,143 ммоль), тиофен-3-илбороновуюкислоту (30 мг, 0,215 ммоль), трифенилфосфин (11 мг, 0,043 ммоль), фторид цезия (109 мг, 0,717 ммоль) растворяли в 1,2-диметоксиэтане (3 мл) с получением оранжевого раствора. Реакционную смесь дегазировали путем барботирования азотом в течение 10 минут. При комнатной температуре добавляли ацетат палладия(II) (5 мг, 0,021 ммоль). Полученную реакционную смесь дегазировали и нагревали до 80°C в течение 18 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления. Неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→70% этилацетат в н-гексанах) с получением указанного в заголовке соединения в виде смеси двух региоизомеров (37 мг, 52%, 7/3) 3.
1Н-ЯМР (500 МГц, CDCl3): 7,39-7,30 (5H, м), 7,24-7,20 (1H, дд), 7,15-7,12 (1H, дд), 6,82-6,78 (1H, дд), 4,81 (2H, с), 3,39 (3H, с), 2,68-2,33 (4H, м), 2,15-2,05 (1H, м), 1,90-1,78 (1H, м), 1,47-1,20 (9H, ушир. с).
ЖХ/МС (способ D): RT=1,514 мин, M+1=492.
3 Это соединение представляло собой смесь двух региоизомеров (в соотношении 7/3).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-(тиофен-3-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-(1-(4-(1-метил-2-оксо-7-(тиофен-3-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (25 мг, 0,051 ммоль) растворяли в дихлорметане (0,5 мл), этот раствор добавляли на SCX-2 catch and release картридж (5 г), предварительно уравновешенный метанолом (15 мл) и дихлорметаном (15 мл). После связывания в течение 12 часов, колонку промывали метанолом (6 мл), а затем продукт элюировали 7M аммиаком в метаноле (4×2 мл). Объединенные содержащие продукт фракции концентрировали досуха в условиях пониженного давления с получением целевого продукта в виде белого твердого вещества (5,8 мг, выход 30%). ЖХ/МС (способ D): RT=0,777 мин, M+1=393.
1H-ЯМР (500 МГц, CD3OD) δ 7,35-7,29 (3H, м), 7,28-7,25 (1H, дд), 7,21-7,18 (1H, с), 7,17-7,14 (1H, дд), 7,07-7,03 (1H, дд), 6,71-6,67 (1H, дд), 4,80 (2H, с), 3,32 (3H, с), 2,51-2,37 (4H, м), 2,25-2,13 (1H,м).
Пример 126: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-(тиофен-2-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
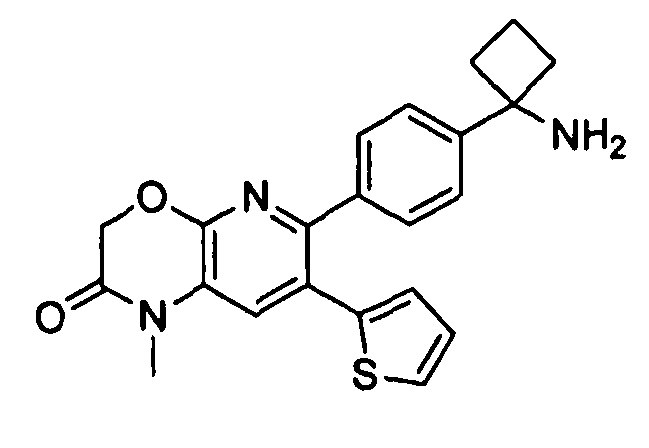
Стадия 1: трет-бутил-(1-(4-(1-метил-2-оксо-7-(тиофен-2-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
трет-Бутил-(1-(4-(7-бром-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (70 мг, 0,143 ммоль), тиофен-2-илбороновую кислоту (30 мг, 0,215 ммоль), трифенилфосфин (11 мг, 0,043 ммоль), фторид цезия (109 мг, 0,717 ммоль) растворяли в 1,2-диметоксиэтане (3 мл) с получением оранжевого раствора. Реакционную смесь дегазировали путем барботирования азотом в течение 10 минут. При к.т. добавляли ацетат палладия(II) (4 мг, 0,018 ммоль). Полученную реакционную смесь дегазировали, барботируя N2, и нагревали до 80°C в течение 18 часов. Реакционную смесь концентрировали досуха в условиях пониженного давления. Неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→70% этилацетат в н-гексанах) с получением указанного в заголовке соединения в виде смеси двух региоизомеров (15 мг, 22%, 7/3). 2 ЖХ/МС (способ D): RT=1,524 мин, M+1=492.
1Н-ЯМР (500 МГц, CDCl3): 7,69-7,32 (7H, м), 6,81-6,76 (1H, м), 4,93 (2H, с), 3,43 (3H, с), 2,58-2,48 (4H, м), 2,20-2,07 (1H, м), 1,96-1,81 (1H, м), 1,46-1,20 (9H, ушир. с).
2 Это соединение представляло собой смесь двух региоизомеров (в соотношении 7/3).
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-1-метил-7-(тиофен-2-ил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-(1-(4-(1-метил-2-оксо-7-(тиофен-2-ил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (12 мг, 0,024 ммоль) растворяли в дихлорметане (1,5 мл). При комнатной температуре добавляли TFA (1 мл) и перемешивали реакционную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (14 мг, выход 78%). ЖХ/МС (способ D): RT=0,781 мин, M+1=393.
1H-ЯМР (500 МГц, CD3OD) δ 7,66 (1H, с), 7,53-7,49 (2H, дд), 7,48-7,44 (2H, дд), 7,42-7,39 (1H, дд), 7,01-7,97 (1H, дд), 6,97-6,95 (1H, дд), 4,97 (2H, с), 3,44 (3H, с), 2,84-2,74 (2H, м), 2,64-2,55 (2H, м), 2,31-2,21 (1H, м), 2,07-1,93 (1H, м).
Пример 127: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-6,8-дигидро-5H-пирано[3,4-b]пиридин-5-он
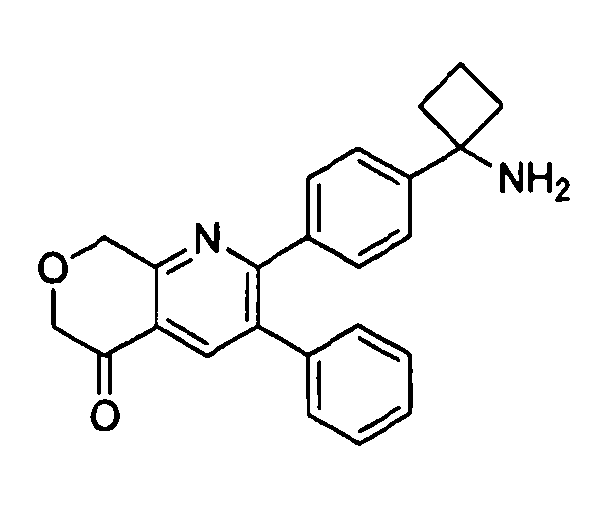
Стадия 1: трет-бутил-(1-(4-(5-оксо-3-фенил-6,8-дигидро-5H-пирано[3,4-b]пиридин-2-ил)фенил)циклобутил)карбамат
В реакционную пробирку объемом 5 мл добавляли уксусную кислоту (5 мл), а затем (E)-трет-бутил-1-(4-(3-(диметиламино)-2-фенилакрилоил)фенил)циклобутилкарбамат (750 мг, 1,783 ммоль), 2H-пиран-3,5(4H,6H)-дион (305 мг, 2,68 ммоль), ацетат аммония (412 мг, 5,35 ммоль) и молекулярные сита (100 мг) с получением коричневой суспензии. Реакционную смесь перемешивали при 100°C в атмосфере азота в течение 2 часов, а затем оставляли охлаждаться до комнатной температуры и концентрировали в условиях пониженного давления. Остаток распределяли между водой (10 мл) и дихлорметаном (10 мл) и декантировали с молекулярных сит. Слои разделяли, и водную фазу экстрагировали введением в дихлорметан (2×10 мл). Объединенные органические фазы промывали насыщенным раствором бикарбоната натрия (2×10 мл), солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желто-коричневого твердого вещества. Это вещество дважды очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 93/7→60/40), а затем методом препаративной ЖХВД (способ F) с получением целевого продукта в виде белого твердого вещества (12 мг, выход 1,4%).
1H-ЯМР (500 МГц, CDCl3) δ 8,29 (1H, с), 7,16-7,41 (9H, м), 5,05 (2H, с), 5,02 (1H, ушир. с), 4,44 (2H, с), 2,21-2,66 (4H, ушир. м), 2,01-2,16 (1H, м), 1,74-1,88 (1H, м), 1,10-1,50 (9H, ушир. м).
Стадия 2: 2-(4-(1-аминоциклобутил)фенил)-3-фенил-6,8-дигидро-5H-пирано[3,4-b]пиридин-5-он
трет-Бутил-(1-(4-(5-оксо-3-фенил-6,8-дигидро-5H-пирано[3,4-b]пиридин-2-ил)фенил)циклобутил)карбамат (12 мг, 0,026 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~3 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (10 мг, выход 77%).
1H-ЯМР (500 МГц, MeOD) δ 8,19 (1H, с), 7,41 (2H, д), 7,32 (2H, д), 7,20-7,24 (3H, м), 7,11-7,15 (2H, м), 4,94 (2H, с), 4,37 (2H, с), 2,59-2,68 (2H, м), 2,39-2,49 (2H, м), 2,06-2,16 (1H, м), 1,79-1,95 (1H, м).
ЖХ/МС (способ D) RT=0,86 мин, M+H+=371,00.
Пример 128: (1r,3r)-3-амино-3-(4-(1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-1-метилциклобутанол
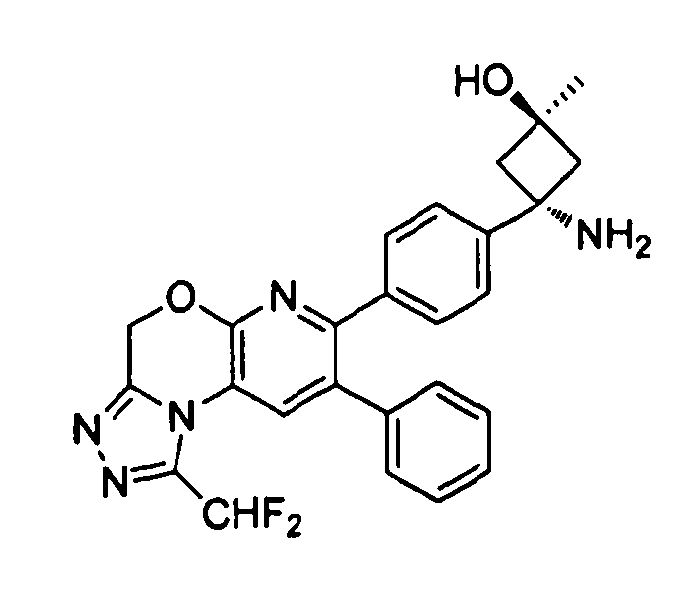
Стадия 1: 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-тион
Смесь 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она (0,26 г, 0,852 ммоль) и реагента Лауссона (0,34 г, 0,852 ммоль) в толуоле (10 мл) нагревали при 120°C в течение 16 ч. Реакционную смесь концентрировали досуха и добавляли DCM (5 мл) и диизопропиловый эфир (20 мл). Осадок фильтровали с получением продукта (0,18 г).
1Н-ЯМР (500 МГц, DMSO-d6): 12,9 (ушир., 1H), 7,35-7,55 (м, 5H), 7,32 (с, 1H), 5,16 (с, 2H).
Стадия 2: 7-бром-1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин
Смесь 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-тиона (0,21 г, 0,654 ммоль) и 2,2-дифторацетогидразида (0,14 г, 1,3 ммоль) в ксилоле (6 мл) нагревали при 150°C в условиях обработки микроволнами в течение 60 мин. Реакционный раствор концентрировали, и распределяли остаток между этилацетатом (10 мл) и водой (10 мл). Органическую фазу разделяли, и экстрагировали водную фазу этилацетатом (10 мл). Объединенную органическую фазу концентрировали и очищали на колонке (силикагель Biotage, 25 г), элюируя этилацетатом/циклогексаном (0-60%), с получением продукта (55 мг).
1Н-ЯМР (500 МГц, CDCl3) 8,09 (с, 1H), 7,4-7,55 (м, 5H), 7,06 (т, 1H), 5,66 (с, 2H).
Стадия 3: 2-((1r,3r)-1-(4-(1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион
Смесь 7-бром-1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазина (30 мг, 0,079 ммоль), 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-диона (46 мг, 0,106 ммоль) и карбоната натрия (16,7721 мг, 0,158 ммоль) в диоксане и воде (соотношение: 4/1, объем: 4 мл/1 мл) с получением желтой суспензии. Суспензию дегазировали в течение 5 мин. Добавляли Pd(PPh3)4 (6,4614 мг, 7,91 мкмоль) и нагревали смесь в условиях обработки микроволнами при 100°C в течение 20 мин. Реакционную смесь концентрировали досуха. Остаток распределяли между этилацетатом (15 мл) и водой (15 мл). Органическую фазу разделяли. Водную фазу экстрагировали этилацетатом (2×15 мл). Объединенную органическую фазу промывали водой (2×30 мл), солевым раствором (25 мл), а затем концентрировали. Неочищенный продукт очищали на колонке (силикагель Biotage, 10 г), элюируя этилацетатом/циклогексаном (0-50%), с получением продукта (10 мг).
1Н-ЯМР (500 МГц, CDCl3): 8,14 (с, 1H), 7,74 (м, 2H), 7,69 (м, 2H), 7,54 (д, 2H), 7,33 (д, 2H), 7,27-7,31 (м, 3H), 7,05-7,19 (м, 3H), 5,61 (с, 2H), 3,35 (д, 2H), 3,09 (д, 2H), 1,4 (с, 3H).
Стадия 4: (1r,3r)-3-амино-3-(4-(1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-1-метилциклобутанол
Смесь 2-((1r,3r)-1-(4-(1-(дифторметил)-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-диона (10 мг, 0,017 ммоль) и гидразина гидрата (0,1 мл, 3,19 ммоль) в этаноле (3 мл) нагревали в условиях обработки микроволновым излучением при 120°C в течение 30 мин. Смесь концентрировали досуха. Остаток распределяли между этилацетатом (10 мл) и водой (10 мл). Органическую фазу разделяли. Водную фазу экстрагировали этилацетатом (2×10 мл). Объединенную органическую фазу промывали водой (2×15 мл), солевым раствором (15 мл) и концентрировали. Неочищенный продукт очищали на SCX-2 колонке (2 г), элюируя аммиаком в метаноле (2M) с получением продукта (3 мг).
1Н-ЯМР (500 МГц, CD3OD): 8,05 (с, 1H), 7,4 (т, 1H), 7,2-7,3 (м, 7H), 7,11-7,15 (м, 2H), 5,63 (с, 2H), 2,59 (д, 3H), 2,34 (д, 2H), 1,43 (с, 3H).
ЖХ/МС (способ D): RT=0,78 мин, M+H+=476,0.
Пример 129: 1-(4-(1,8-дифенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Стадия 1: трет-бутил-(1-(4-(1,8-дифенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (100 мг, 0,205 ммоль) и бензойной кислоты гидразида (84 мг, 0,615 ммоль) в пара-ксилоле (2 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в н-гексане) с получением указанного в заголовке соединения (46 мг, 40%). ЖХ/МС (способ D): RT=1,546 мин, M+1=572.
1Н-ЯМР (500 МГц, CDCl3): 7,77-7,69 (2H, м), 7,64-7,57 (2H, м), 7,56-7,50 (1H, т), 7,49-7,41 (2H, т), 7,34-7,30 (2H, м), 7,28 (1H, с), 7,22-7,14 (3H, м), 6,95-6,90 (2H, дд), 5,65 (2H, с), 2,55-2,40 (3H, м), 2,15-2,01 (2H, м), 1,89-1,77 (1H, м), 1,46-1,24 (9H, ушир. с).
Стадия 2: 1-(4-(1,8-дифенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(1,8-дифенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (15 мг, 0,026 ммоль) растворяли в дихлорметане (1 мл), при комнатной температуре добавляли TFA (0,5 мл) и перемешивали полученную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексанах (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением целевого продукта в виде не совсем белого твердого вещества (5 мг, выход 40%). ЖХ/МС (способ D) RT=0,846 мин, M+1=472.
1Н-ЯМР (500 МГц, CDCl3): 7,91-7,86 (2H, дд), 7,83-7,76 (3H, м), 7,59-7,54 (2H, д), 7,53-7,48 (2H, д), 7,42 (1H, с), 7,37-7,28 (3H, м), 7,09-7,03 (2H, дд), 5,86 (2H, с), 2,91-2,81 (2H, м), 2,74-2,63 (2H, м), 2,40-2,29 (1H, м), 2,13-2,02 (1H, м).
Пример 130: 1-(4-(8-фенил-1-(пиридин-2-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
Стадия 1: 2-пиколинилгидразид
Этил-2-пиколинат (5 г, 33,1 ммоль) и гидразина моногидрат (5,3 г, 165 ммоль) в этаноле (70 мл) нагревали до температуры возгонки в течение 15 часов. Реакционную смесь концентрировали в условиях пониженного давления, и обрабатывали остаток толуолом для азеотропной отгонки избытка гидразина. Указанную процедуру повторяли трижды. Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
Стадия 2: трет-бутил-(1-(4-(8-фенил-1-(пиридин-4-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
Суспензию трет-бутил-(1-(4-(7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамата (100 мг, 0,205 ммоль) и 2-пиколинилгидразида (56,2 мг, 0,410 ммоль) в пара-ксилоле (1 мл) нагревали до 150°C в течение 15 минут в условиях обработки микроволновым излучением. Полученную реакционную смесь концентрировали досуха в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→4% метанол в дихлорметане) с получением указанного в заголовке соединения (15 мг, 13%). ЖХ/МС (способ A): RT=6,95 мин, M+1=573.
1Н-ЯМР (500 МГц, CDCl3): 8,78-8,74 (1H, дд), 8,45 (1H, с), 8,27-8,23 (1H, дд), 7,96-7,91 (1H, дт), 7,53-7,48 (1H, дд), 7,38-7,33 (2H, дд), 7,31-7,27 (3H, м), 7,25-7,23 (2H, м), 7,19-7,13 (2H, дд), 5,63 (2H, с), 2,56-2,41 (4H, м), 2,14-2,01 (1H, м), 1,88-1,75 (1H, м), 1,46-1,21 (9H, ушир. с).
Стадия 3: 1-(4-(8-фенил-1-(пиридин-2-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин
трет-Бутил-(1-(4-(8-фенил-1-(пиридин-2-ил)-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (15 мг, 0,026 ммоль) растворяли в дихлорметане (1 мл), при комнатной температуре добавляли TFA (0,5 мл) и перемешивали полученную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Затем остаток суспендировали в н-гексане (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Удаляли оставшийся растворитель в условиях пониженного давления. Полученный остаток очищали методом препаративной ЖХВД с получением целевого продукта в виде белого твердого вещества (8 мг, выход 64%). ЖХ/МС (способ D) RT=0,825 мин, M+H+=474.
1Н-ЯМР (500 МГц, d6-DMSO): 8,79 (1H, д), 8,44 (1H, ушир. с), 8,13 (1H, с), 8,11-8,05 (1H, м), 7,62-7,59 (1H, м), 7,41 (2H, с), 7,42-7,38 (2H, д), 7,25-7,27 (3H, м), 7,10 (2H, м), 5,71 (2H, с), 2,60-2,51 (4H, м), 2,27-2,19 (1H, м), 1,88-1,76 (1H, м).
Пример 131: 6-(4-(аминометил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Стадия 1: трет-бутил-4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат
В реакционную пробирку объемом 15 мл добавляли 6-бром-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (40 мг, 0,125 ммоль) и трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбамат (50 мг, 0,150 ммоль) в 1,4-диоксане (2 мл), а затем раствор карбоната натрия (40 мг, 0,3 ммоль) в воде (0,5 мл), с получением белой суспензии. Эту суспензию дегазировали путем барботирования азотом в течение 10 минут, а затем добавляли аддукт [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и дихлорметана (9 мг, 0,013 ммоль). Реакционную смесь нагревали до 80°C в течение ночи. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли солевым раствором (4 мл) и экстрагировали введением в этилацетат (3×4 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде белого твердого вещества (40 мг, выход 72%).
1H-ЯМР (500 МГц, CDCl3) δ 7,27-7,32 (5H, м), 7,25 (1H, с), 7,16-7,20 (2H, м), 7,10 (2H, д), 4,89 (2H, с), 4,81 (1H, ушир. с), 4,26 (2H, ушир. с), 3,39 (3H, с), 1,23 (9H, ушир. с).
ЖХ/МС (способ D): RT=1,40 мин, M+H+=446,20.
Стадия 2: 6-(4-(аминометил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат (40 мг, 0,090 ммоль) растворяли в TFA (2 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~2 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (2 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде белого твердого вещества (35 мг, выход 85%).
1H-ЯМР (500 МГц, MeOD) δ 7,54 (1Н, с), 7,36 (2Н, д), 7,31 (2Н, д), 7,25-7,29 (3Н, м), 7,18-7,23 (2Н, м), 4,95 (2Н, с), 4,07 (2Н, с), 3,41 (3Н, с).
ЖХ/МС (способ D): RT=0,69 мин, M+H+=346,20.
Пример 132: 6-(4-(1-аминоциклобутил)фенил)-7-(4-фторфенил)-1-метил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
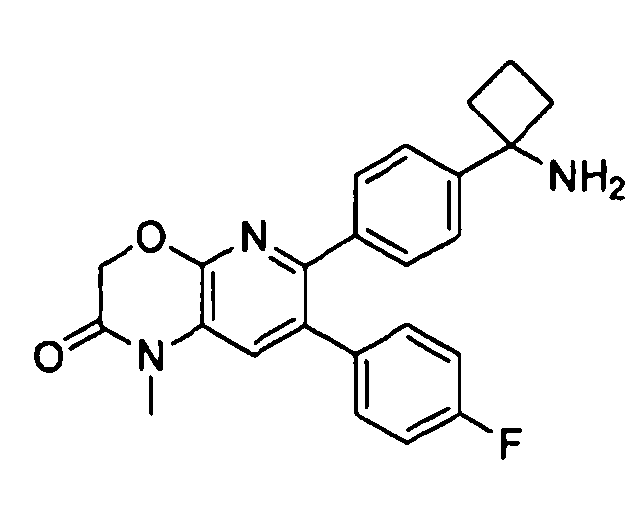
Стадия 1: трет-бутил-1-(4-(7-(4-фторфенил)-1-метил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат
В герметично закрытую пробирку добавляли трет-бутил-1-(4-(7-бром-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамат (82 мг, 0,168 ммоль), 4-фторфенилбороновую кислоту (35,2 мг, 0,252 ммоль), трифенилфосфин (13,21 мг, 0,05 ммоль) и фторид цезия (128 мг, 0,84 ммоль) в 1,2-диметоксиэтане (2 мл). Реакционную смесь дегазировали путем барботирования азотом в течение 10 мин, добавляли ацетат палладия (5,65 мг, 0,025 ммоль) и нагревали смесь при 80°C в течение ночи. Реакционную смесь концентрировали, остаток разбавляли этилацетатом, и пропускали смесь через силикагель. Фильтрат концентрировали, и добавляли к остатку метанол (2,5 мл) с образованием суспензии. Твердое вещество фильтровали и сушили с получением трет-бутил-1-(4-(7-(4-фторфенил)-1-метил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутилкарбамата (19 мг). ЖХ/МС (способ D): RT=1,54 мин, M+H+=504.
Стадия 2: 6-(4-(1-аминоциклобутил)фенил)-7-(4-фторфенил)-1-метил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В круглодонной колбе (5 мл) к трет-бутил-(1-(4-(7-(4-фторфенил)-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамату (16 мг, 0,032 ммоль) добавляли трифторуксусную кислоту (0,5 мл, 6,49 ммоль). Полученный раствор перемешивали в течение 60 секунд, а затем концентрировали досуха в условиях пониженного давления. Остаток суспендировали в диэтиловом эфире (1 мл) и трижды концентрировали досуха с получением трифторуксуснокислой соли продукта (7 мг). ЖХ/МС (способ D): RT=0,78 мин, M+H+=404.
1Н-ЯМР (500 МГц, CD3OD) 7,44 (с, 1H), 7,29 (м, 4H), 7,12-7,15 (м, 2H), 6,91-6,95 (м, 2H), 4,85 (с, 2H), 3,31 (с, 3H), 2,62-2,69 (м, 2H), 2,43-2,49 (м, 2H), 2,10-2,15 (м, 1H), 1,80-1,86 (м, 1H).
Пример 133: 6-(4-(1-аминоциклобутил)фенил)-7-(циклопропилметил)-1-метил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
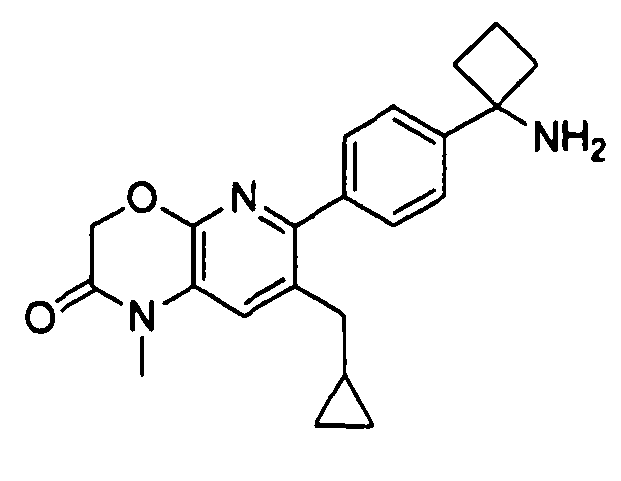
Стадия 1: трет-бутил-(1-(4-(7-аллил-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
трет-Бутил-(1-(4-(7-бром-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (100 мг, 0,205 ммоль) и аллилтрибутилстаннан растворяли в безводном диметилформамиде (1 мл) с получением белого раствора. Реакционную смесь дегазировали путем барботирования азотом в течение 30 минут. При комнатной температуре добавляли дихлорид бис(трифенилфосфин)палладия(II) (15 мг, 0,020 ммоль). Полученную реакционную смесь дегазировали и нагревали до 80°C в течение 2 часов. Реакционную смесь разбавляли этилацетатом (3 мл), промывали водой (5 мл) и солевым раствором (5 мл), и концентрировали органическую фазу досуха в условиях пониженного давления. Неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→20% этилацетат в н-гексанах) с получением указанного в заголовке соединения (52 мг, 56%). ЖХ/МС (способ A) RT=6,83 мин, M+1=450.
1Н-ЯМР (500 МГц, CDCl3): 7,57-7,40 (4H, м), 7,15 (1H, с), 6,04-5,88 (1H, м), 5,20-5,13 (1H, м), 5,11-4,99 (2H, м), 4,85 (2H, с), 3,52-3,41 (1H, дд), 3,37 (3H, с), 2,67-2,41 (4H, м), 2,15-2,04 (1H, м), 1,90-1,80 (1H, м), 1,47-1,31 (9H, ушир. с).
Стадия 2: трет-бутил-(1-(4-(7-(циклопропилметил)-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
В трехгорлую круглодонную колбу объемом 50 мл при комнатной температуре в атмосфере азота добавляли трет-бутил-(1-(4-(7-аллил-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (52 мг, 0,116 ммоль), ацетат палладия (32 мг, 0,180 ммоль) в диэтиловом эфире (5 мл) с получением желтого раствора. Реакционную смесь охлаждали до 0°C, и пластиковым шприцем добавляли триметилсилилдиазометан (2 мл). Реакционную смесь оставляли перемешиваться при 0°C в течение 10 минут и нагреваться до комнатной температуры в течение ночи. Реакционную смесь гасили добавлением водной уксусной кислоты (3 мл). Затем реакционную смесь разбавляли этилацетатом (5 мл) и концентрировали в условиях пониженного давления. Неочищенный остаток растворяли в этилацетате (10 мл) и фильтровали через целит. Фильтрат концентрировали в условиях пониженного давления. Неочищенный остаток очищали методом препаративной ЖХВД (способ E). Содержащие продукт фракции объединяли и концентрировали в условиях пониженного давления с получением указанного в заголовке соединения (3 мг, 5%). ЖХ/МС (способ D) RT=1,561 мин, M+1=464.
1Н-ЯМР (500 МГц, CDCl3): 7,58-7,39 (4H, м), 7,36 (1H, с), 4,85 (2H, с), 3,41 (3H, с), 2,64-2,60 (2H, м), 2,59-2,52 (4H, м), 2,15-2,04 (1H, м), 1,91-1,84 (2H, м), 1,46-1,35 (9H, ушир. с), 1,33-1,27 (1H, м), 0,95-0,84 (2H, м), 0,17-0,10 (1H, м).
Стадия 3: 6-(4-(1-аминоциклобутил)фенил)-7-(циклопропилметил)-1-метил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-(1-(4-(7-(циклопропилметил)-1-метил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (0,5 мг, 1,79 мкмоль) растворяли в дихлорметане (1 мл). При комнатной температуре добавляли TFA (0,5 мл) и перемешивали реакционную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~1 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли дважды. Затем остаток суспендировали в н-гексане (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли дважды. Остаток, содержащий растворитель, сушили с получением целевого продукта в виде не совсем белого твердого вещества в виде указанного в заголовке соединения (0,3 мг, выход 77%). ЖХ/МС (способ D) RT=0,77 мин, M+1=364.
1H-ЯМР (500 МГц, CD3OD) δ 7,66-7,50 (4H, м), 7,47 (1H, с), 4,48 (2H, с), 4,15-4,09 (2H, м), 3,06 (3Н, с), 2,75-2,65 (2H, м), 2,51-2,42 (2H, м), 2,28-2,20 (1H, м), 1,97-1,90 (1H, м), 1,76-1,71 (1H, м), 1,66-1,55 (2H, м), 1,38-1,31 (2H, м).
Пример 134: 6-(4-((1s,3s)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
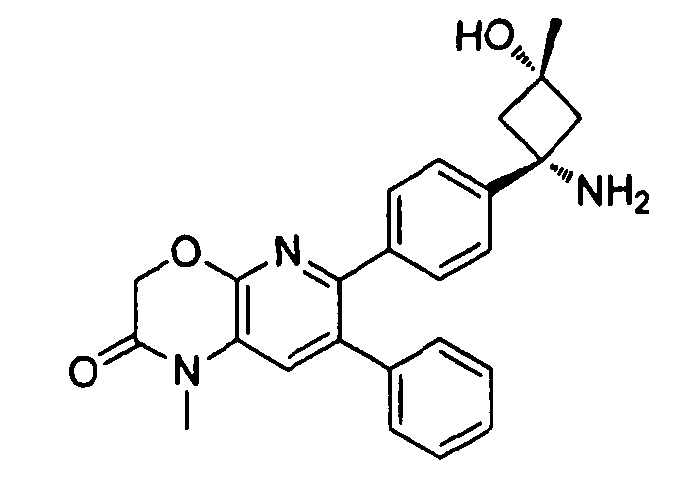
Стадия 1: 2-((1s,3s)-3-гидрокси-3-метил-1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)изоиндолин-1,3-дион
В реакционную пробирку объемом 15 мл добавляли 6-бром-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (50 мг, 0,157 ммоль), 2-((1s,3s)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-дион (56,6 мг, 0,131 ммоль) и карбонат цезия (213 мг, 0,652 ммоль) в смеси 1,4-диоксана (2 мл) и воды (667 мкл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт PdCl2(dppf)-CH2Cl2 и дегазировали еще в течение 15 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1,5 часов. Реакционную смесь охлаждали до комнатной температуры, и добавляли воду (7 мл). Полученную смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в условиях пониженного давления. Неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→30% этилацетат в н-гексанах) с получением указанного в заголовке соединения (35 мг, 50%).
1Н-ЯМР (500 МГц, CDCl3): 7,75-7,70 (2H, м), 7,66-7,63 (2H, м), 7,58-7,54 (2H, м), 7,32-7,29 (2H+1H, м), 7,25-7,21 (2H+1H, м), 7,18-7,14 (2H, м), 4,90 (2H, с), 3,37 (3H, с), 3,34-3,25 (2H, м), 3,10-3,01 (2H, м), 1,09 (3H, с).
ЖХ/МС (способ D) RT=1,252 мин, M+1=546.
Стадия 2: 6-(4-((1s,3s)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В пробирку для микроволновой обработки объемом 10 мл добавляли 2-((1s,3s)-3-гидрокси-3-метил-1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)изоиндолин-1,3-дион (35 мг, 0,064 ммоль) и гидразина моногидрат (0,040 мл, 1,2 ммоль) в этаноле (1,5 мл) и 1,4-диоксане (0,5 мл) с получением желтого раствора. Реакционную смесь нагревали до 120°C в течение 1 ч в условиях обработки микроволнами. Реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления твердого вещества. Это вещество тщательно промывали этанолом. Фильтраты объединяли и концентрировали в условиях пониженного давления. Неочищенный остаток (20 мг) растворяли в 1 мл раствора метанол/дихлорметан (1/1) и добавляли на SCX-2 catch-and-release картридж (5 г). После связывания в течение 15 минут, колонку промывали метанолом (4×2 мл), а затем элюировали продукт 2M аммиаком в метаноле (4×2 мл). Объединенные содержащие продукт фракции концентрировали досуха в условиях пониженного давления с получением не совсем белого твердого вещества (15 мг). Это вещество повторно растворяли в метаноле/воде (2 мл, 1/3) и лиофилизировали в течение ночи с получением указанного в заголовке соединения (13 мг, 50%).
1H-ЯМР (500 МГц, CD3OD) δ 7,54 (1H, с), 7,39-7,18 (4H, м), 4,95 (2H, с), 3,43 (3H, с), 2,76-2,68 (2H, м), 2,46-2,33 (2H, м), 1,14 (3H, с).
ЖХ/МС (способ D) RT=0,747 мин, M+1=416.
Пример 135: 2-(6-(4-(аминометил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
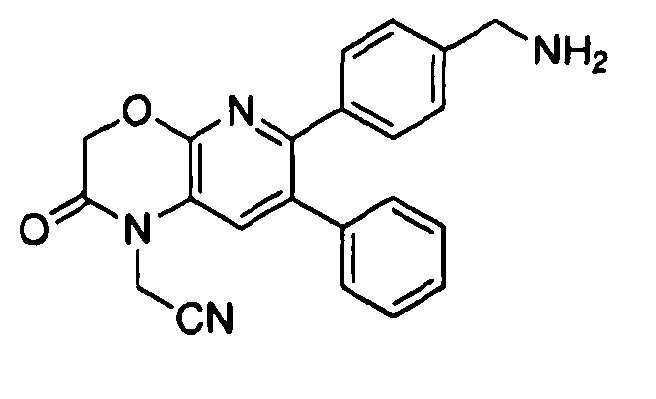
Стадия 1: трет-бутил-4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат
В реакционную пробирку добавляли 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (0,19 г, 0,623 ммоль) и трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбамат (0,25 г, 0,750 ммоль) в 1,4-диоксане (10 мл), а затем раствор карбоната натрия (0,198 г, 1,868 ммоль) в воде (2,5 мл) с получением суспензии. Эту суспензию дегазировали, а затем добавляли аддукт PdCl2(dppf)-CH2Cl2 (0,051 г, 0,062 ммоль). Полученную смесь нагревали при 80°C в течение 20 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали водой, солевым раствором и концентрировали с получением продукта (0,21 г), который использовали на следующей стадии без дополнительной очистки. ЖХ/МС (способ D): RT=1,25 мин, M+H+=433,2.
Стадия 2: трет-бутил-4-(1-(цианометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат
Смесь трет-бутил-4-(2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамата (70 мг, 0,162 ммоль), 2-бромацетонитрила (58,4 мг, 0,487 ммоль) и карбоната калия (67,3 мг, 0,487 ммоль) в DMF (2 мл) перемешивали при 40°C в течение 16 ч. Реакционную смесь охлаждали, разбавляли водой (10 мл) и экстрагировали DCM (3×8 мл). Объединенную органическую фазу концентрировали, и очищали остаток на колонке (силикагель Biotage, 10 г), элюируя этилацетатом/циклогексаном 0-30%, с получением продукта (12 мг).
1Н-ЯМР (500 МГц, CDCl3) 7,29 (с, 1H), 7,19-7,26 (м, 5H), 7,13 (м, 2H), 7,05 (д, 2H), 4,88 (с, 2H), 4,81 (с, 2H), 4,73 (ушир., 1H), 4,21 (д, 2H), 1,38 (ушир., 9H).
ЖХ/МС (способ D): RT=1,35 мин, M+H+=472,2.
Стадия 3: 2-(6-(4-(аминометил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
В круглодонную колбу, содержащую трет-бутил-4-(1-(цианометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат (12 мг, 0,026 ммоль), добавляли TFA (0,5 мл, 6,49 ммоль). Полученный раствор перемешивали в течение 60 секунд, а затем концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (1 мл) и трижды концентрировали досуха с получением трифторуксуснокислой соли продукта (9,5 мг).
1Н-ЯМР (500 МГц, CD3OD) 7,58 (с, 1H), 7,29 (м, 2H), 7,19-7,23 (м, 5H), 7,13 (м, 2H), 5,03 (с, 2H), 4,93 (с, 2H), 3,98 (с, 2H).
ЖХ/МС (способ D): RT=0,65 мин, M+H+=372,2.
Пример 136: (1s,3s)-3-амино-3-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанол
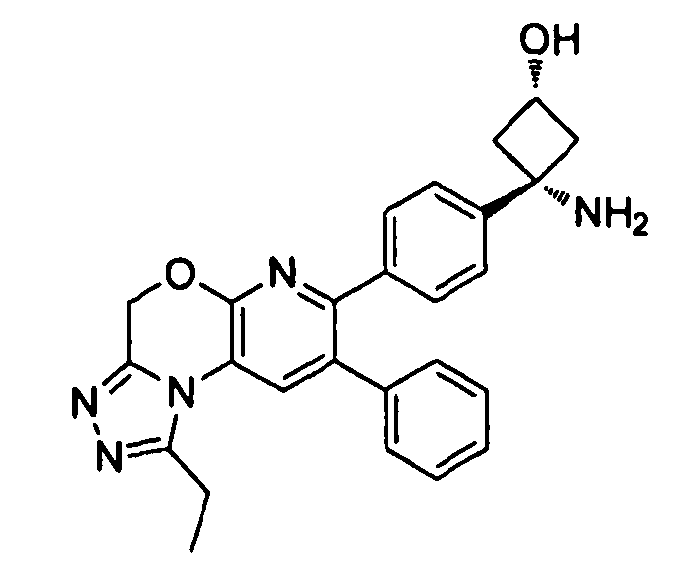
Стадия 1: 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-тион
В круглодонную колбу объемом 500 мл добавляли 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (2 г, 6,55 ммоль) и реагент Лауссона (2,65 г, 6,55 ммоль) в толуоле (150 мл) с получением оранжевой суспензии. Реакционную смесь нагревали до температуры возгонки в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, и удаляли толуол в условиях пониженного давления. Затем неочищенный остаток растворяли в минимальном количестве дихлорметана. Добавляли диизопропиловый эфир, и образовывался осадок. Осадок фильтровали и сушили до постоянного веса с получением указанного в заголовке соединения (1,5 г, 71%). Это соединение использовали на следующей стадии без дополнительной очистки. ЖХ/МС (способ D): RT=1,274 мин, M+1=323.
Стадия 2: (E)-6-бром-2-гидразоно-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин
В пробирку объемом 15 мл добавляли 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-тион (100 мг, 0,311 ммоль) и гидразина моногидрат (0,049 мл, 1,557 ммоль) в тетрагидрофуране (5 мл) с получением коричневого раствора. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали в условиях пониженного давления, и суспендировали неочищенный остаток в диэтиловом эфире. Надосадочную жидкость удаляли, и сушили коричневое твердое вещество до постоянного веса с получением указанного в заголовке соединения (90 мг, 90%). Это соединение использовали на следующей стадии без дополнительной очистки. ЖХ/МС (способ D): RT=0,770 мин, M+1=321.
Стадия 3: 7-бром-1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин
В пробирку для микроволновой обработки объемом 10 мл добавляли (E)-6-бром-2-гидразоно-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин (90 мг, 0,282 ммоль) в 1,1,1-триэтоксипропане с получением желтой суспензии. Реакционную смесь нагревали до 120°C в течение 20 минут. Неочищенный остаток концентрировали в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 100%→100% н-гексан в этилацетате) с получением указанного в заголовке соединения (41 мг, 40%).
1Н-ЯМР (500 МГц, CDCl3): 7,73 (1H, с), 7,54-7,48 (3H, м), 7,45-7,41 (2H, м), 5,56 (2H, с), 3,10-3,00 (2H, кв.), 1,53-1,48 (3H, т).
ЖХ/МС (способ D) RT=1,117 мин, M+1=358.
Стадия 4: трет-бутил-((1s,3s)-1-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-3-гидроксициклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли 7-бром-1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин (40 мг, 0,112 ммоль), трет-бутил-((1s,3s)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)карбамат (36,3 мг, 0,093 ммоль) и карбонат цезия (152 мг, 0,467 ммоль) в 1,4-диоксане (1,8 мл) и воде (0,6 мл) с получением оранжевой суспензии. Реакционную смесь дегазировали путем барботирования азотом в течение 15 минут. Добавляли аддукт PdCl2(dppf)-CH2Cl2 (15,24 мг, 0,019 ммоль), и дегазировали реакционную смесь путем барботирования азотом еще в течение 15 минут. Реакционную смесь нагревали до 50°C в течение 1 ч. Реакционную смесь оставляли охлаждаться до к.т. и добавляли воду (7 мл). Реакционную смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические слои концентрировали в условиях пониженного давления. Неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→5% метанол в дихлорметане) с получением указанного в заголовке соединения (27 мг, 53%). ЖХ/МС (способ D) RT=1,091 мин, M+1=530.
1Н-ЯМР (500 МГц, CDCl3): 7,82 (1H, с), 7,39-7,32 (5H, м), 7,25-7,19 (4H, м), 5,57 (2H, с), 3,53-3,46 (2H, д), 3,18-3,09 (2H, кв.), 3,08-2,96 (2H, ушир. с), 2,19 (1H, с), 1,82-1,75 (3H, т), 1,59-1,53 (9H, ушир. с).
Стадия 5: (1s,3s)-3-амино-3-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанол
трет-Бутил-((1s,3s)-1-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-3-гидроксициклобутил)карбамат (34 мг, 0,063 ммоль) растворяли в дихлорметане (1 мл). При комнатной температуре добавляли TFA (1 мл) и перемешивали реакционную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~1 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли дважды. Затем остаток суспендировали в н-гексане (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли дважды. Удаляли оставшийся растворитель в условиях пониженного давления, и сушили остаток до постоянного веса. Затем остаток очищали методом препаративной ЖХВД с получением целевого продукта в виде не совсем белого твердого вещества (7 мг, выход 25%). ЖХ/МС (способ D) RT=0,658 мин, M+1=440.
1H-ЯМР (500 МГц, CD3OD) δ 7,95 (1H, с), 7,31-7,13 (8H, м), 5,52 (2H, с), 3,84-3,76 (1H, м), 3,17-3,10 (2H, кв.), 2,75-2,65 (2H, м), 2,86-2,77 (2H, м), 2,13-2,02 (1H, м), 1,43-1,38 (3H, т).
Пример 137: 6-(4-((1r,3r)-1-амино-3-гидроксициклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
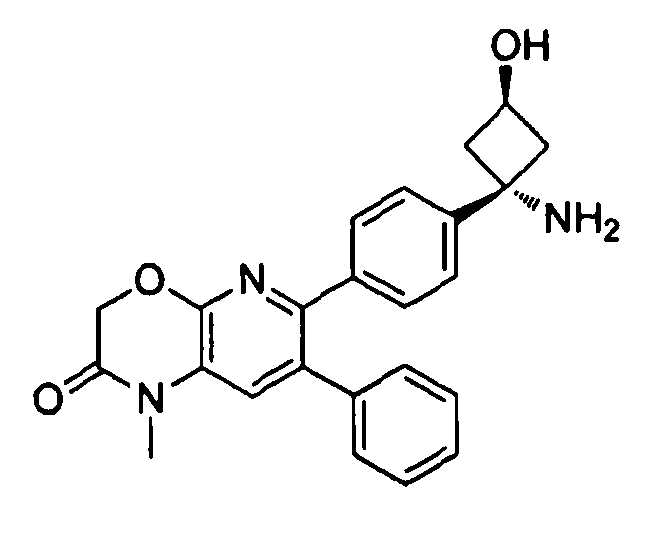
Стадия 1: 1-(4-бромфенил)-3,3-диметоксициклобутанкарбонитрил
Суспензию гидрида натрия (2 г, 50,0 ммоль) в DMF (20 мл) охлаждали до 0°C, после чего медленно добавляли 2-(4-бромфенил)ацетонитрил (4 г, 20,40 ммоль). Суспензию перемешивали при 0°C еще в течение 10 мин, после чего добавляли 1,3-дибром-2,2-диметоксипропан (2,62 г, 10,00 ммоль). Реакционную смесь перемешивали при 60°C в течение 20 ч, после чего охлаждали до комнатной температуры, вливали в воду (75 мл) и экстрагировали этилацетатом (2×50 мл). Объединенную органическую фазу промывали водой (75 мл), солевым раствором (50 мл) и концентрировали. Неочищенный продукт очищали на колонке (100 г, силикагель Biotage), элюируя этилацетатом/циклогексаном (0-10%), с получением продукта (1,7 г, выход 59%).
1Н-ЯМР (500 МГц, DMSO-d6): 7,46 (д, 2H), 7,29 (д, 2H), 3,21 (с, 3H), 3,11 (с, 3H), 3,03 (д, 2H), 2,62 (д, 2H).
Стадия 2: 1-(4-бромфенил)-3,3-диметоксициклобутанкарбоксамид
К перемешанной смеси 1-(4-бромфенил)-3,3-диметоксициклобутанкарбонитрила (3,18 г, 10,74 ммоль) и карбоната калия (0,297 г, 2,15 ммоль) в DMSO (11 мл) при 40°C в атмосфере азота в течение 2 ч добавляли пероксид водорода (2,05 мл, 20,07 ммоль). После добавления реакционную смесь нагревали до 80°C в течение 3 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и добавляли воду (30 мл). Реакционную смесь экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали водой (2×30 мл), солевым раствором (30 мл) и концентрировали с получением светло-желтого масла (3,3 г).
1Н-ЯМР (500 МГц, CDCl3) 7,40 (д, 2H), 7,11 (д, 2H), 5,89 (ушир., 1H), 5,58 (ушир., 1H), 3,12 (с, 3H), 3,02 (с, 3H), 2,97 (д, 2H), 2,52 (д, 2H).
Стадия 3: 1-(4-бромфенил)-3,3-диметоксициклобутанамин
К раствору 1-(4-бромфенил)-3,3-диметоксициклобутанкарбоксамида (2,3 г, 7,32 ммоль) а ацетонитриле (9 мл) и воде (9,00 мл) добавляли бис(трифторацетат)фенилйод (4,73 г, 11,00 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 18 ч. Реакционную смесь медленно вливали в насыщенный раствор бикарбоната натрия (~45 мл) и экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали водой, солевым раствором и концентрировали с получением неочищенного продукта (3,4 г). Неочищенный продукт очищали на колонке (силикагель Biotage, 50 г), элюируя 0-5% DCM/метанолом (смесь 500 мл метанола и 60 мл 2M NH4OH в метаноле) с получением продукта, 1-(4-бромфенил)-3,3-диметоксициклобутанамина (1,06 г).
1Н-ЯМР (500 МГц, CDCl3): 7,48 (д, 2H), 7,32 (д, 2H), 3,27 (д, 3H), 3,17 (д, 3H), 2,66 (д, 2H), 2,44 (д, 2H).
Стадия 4: 2-(1-(4-бромфенил)-3,3-диметоксициклобутил)изоиндолин-1,3-дион
Смесь 1-(4-бромфенил)-3,3-диметоксициклобутанамина (0,66 г, 2,306 ммоль), этил-1,3-диоксоизоиндолин-2-карбоксилата (0,56 г, 2,55 ммоль) и триэтиламина (1,3 мл, 9,33 ммоль) в CHCl3 (15 мл) нагревали при 70°C в течение 16 ч. Реакционную смесь концентрировали до максимально возможно сухого состояния. Добавляли метанол (~5 мл). Раствор концентрировали досуха. Снова добавляли метанол (~5 мл), и собирали полученное твердое вещество путем фильтрования с получением продукта в виде белого твердого вещества (0,39 г).
1Н-ЯМР (500 МГц, CDCl3): 7,70 (дд, 2H), 7,60 (дд, 2H), 7,44 (дд, 2H), 7,37 (дд, 2H), 3,18 (м, 4H), 3,10 (с, 3H), 3,02 (с, 3H).
Стадия 5: 2-(1-(4-бромфенил)-3-оксоциклобутил)изоиндолин-1,3-дион
К раствору 2-(1-(4-бромфенил)-3,3-диметоксициклобутил)изоиндолин-1,3-диона (0,3 г, 0,72 ммоль) в ацетоне (30 мл) добавляли 6M HCl (3 мл) и перемешивали полученный раствор при 50°C в течение 2 ч. Реакционную смесь концентрировали с получением продукта (0,26 г).
1Н-ЯМР (500 МГц, CDCl3): 7,76 (дд, 2H), 7,67 (дд, 2H), 7,41 (м, 4H), 4,21 (м, 2H), 3,87 (м, 2H).
Стадия 6: 2-((1r,3r)-1-(4-бромфенил)-3-гидроксициклобутил)изоиндолин-1,3-дион
К раствору 2-(1-(4-бромфенил)-3-оксоциклобутил)изоиндолин-1,3-диона (194 мг, 0,524 ммоль) в THF (5 мл) при -78°C добавляли L-селектрид (0,524 мл, 0,524 ммоль, 1M раствор в THF) с получением желтого раствора и перемешивали при -78°C в течение 1 ч. Реакционную смесь гасили добавлением насыщенного раствора бикарбоната натрия при -78°C и экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу концентрировали и очищали на колонке (силикагель Biotage, 25 г) с получением продукта (99 мг).
1Н-ЯМР (500 МГц, CDCl3): 7,76-7,79 (м, 2H), 7,67-7,71 (м, 2H), 7,42-7,50 (м, 4H), 4,5-4,6 (м, 1H), 3,73-3,84 (м, 2H), 2,76-2,83 (м, 2H).
Стадия 7: 2-((1r,3r)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-дион
Из смеси 2-((1r,3r)-1-(4-бромфенил)-3-гидроксициклобутил)изоиндолин-1,3-диона (99 мг, 0,266 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (67,5 мг, 0,266 ммоль) и ацетата калия (131 мг, 1,330 ммоль) в диоксане (10 мл) откачивали воздух и трижды продували ее азотом. Добавляли Pd(dppf)Cl2·CH2Cl2 (21,72 мг, 0,027 ммоль), из полученной смеси откачивали воздух, и трижды продували ее азотом. Затем смесь нагревали при 90°C в течение 22 ч. Реакционную смесь концентрировали, и остаток очищали методом хроматографии (силикагель Biotage, 25 г), элюируя этилацетатом/циклогексаном 0-70%, с получением продукта (45 мг).
1Н-ЯМР (500 МГц, CDCl3): 7,73-7,78 (м, 4H), 7,65 (м, 2H), 7,57-7,59 (м, 2H), 4,45-4,55 (м, 1H), 3,81-3,85 (м, 2H), 2,79-2,82 (м, 2H), 1,30 (с, 12H).
Стадия 8: 2-((1r,3r)-3-гидрокси-1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)изоиндолин-1,3-дион
Смесь 6-бром-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она (45 мг, 0,141 ммоль), 2-((1r,3r)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-диона (49,3 мг, 0,117 ммоль) и карбоната цезия (191 мг, 0,587 ммоль) в диоксане (3 мл) и воде (1 мл) дегазировали. Добавляли PdCl2(dppf)·CH2Cl2 (19,19 мг, 0,023 ммоль). Реакционную смесь снова дегазировали и нагревали при 50°C при перемешивании в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (15 мл) и экстрагировали этилацетатом (3×15 мл). Объединенную органическую фазу промывали водой, сушили над сульфатом натрия и концентрировали с получением неочищенного продукта, который очищали методом колоночной хроматографии (силикагель Biotage 10 г), элюируя этилацетатом/циклогексаном 0-80%, с получением продукта (26 мг).
1Н-ЯМР (500 МГц, CDCl3): 7,74-7,76 (м, 2H), 7,66-7,68 (м, 2H), 7,40-7,42 (м, 2H), 7,23-7,30 (м, 6H), 7,17-7,19 (м, 2H), 4,87 (с, 2H), 4,45-4,55 (м, 1H), 3,79-3,82 (м, 2H), 3,38 (с, 3H), 2,74-2,78 (м, 2H).
ЖХ/МС (способ D): RT=1,22 мин, M+H+=533,0.
Стадия 9: 6-(4-((1r,3r)-1-амино-3-гидроксициклобутил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
К смеси 2-((1r,3r)-3-гидрокси-1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)изоиндолин-1,3-диона (26 мг, 0,049 ммоль) в диоксане (1 мл) и MeOH (1,0 мл) добавляли гидразина моногидрат (0,25 мл, 7,97 ммоль). Реакционную смесь нагревали при 120°C в течение 30 мин в условиях обработки микроволнами. Реакционную смесь разбавляли насыщенным раствором бикарбоната (10 мл) и экстрагировали DCM (3×10 мл). Объединенную органическую фазу промывали водой, солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали методом препаративной ЖХВД (метод F) с получением продукта (5 мг).
1Н-ЯМР (500 МГц, CD3OD): 7,44 (с, 1H), 7,28-7,29 (м, 2H), 7,18-7,20 (м, 5H), 7,11-7,13 (м, 2H), 4,85 (с, 2H), 4,52-4,54 (м, 1H), 3,32 (с, 3H), 32,83-2,87 (м, 2H), 2,46-2,51 (м, 2H).
ЖХ/МС (способ D): RT=0,67 мин, M+H+=403,2.
Пример 138: (1r,3r)-3-амино-3-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-1-метилциклобутанол
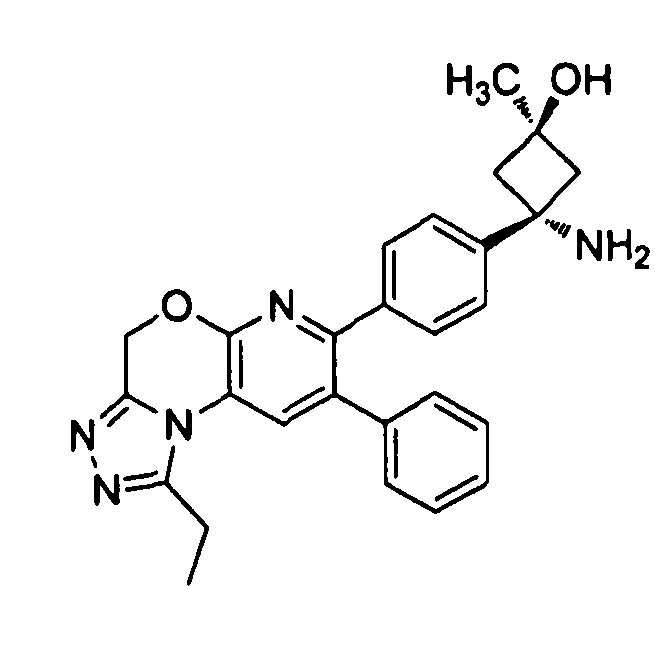
Стадия 1: 2-((1r,3r)-1-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион
В реакционную пробирку объемом 15 мл добавляли 7-бром-1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин (62 мг, 0,174 ммоль), 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-дион (62,7 мг, 0,145 ммоль) и карбонат цезия (236 мг, 0,723 ммоль) в 1,4-диоксане (2,5 мл) с получением желтого раствора. Реакционную смесь дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт PdCl2(dppf)-CH2Cl2. Реакционную смесь нагревали до 50°C в течение 1 ч. Реакционную смесь оставляли охлаждаться до к.т. и добавляли воду (7 мл). Реакционную смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические слои концентрировали в условиях пониженного давления. Неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→100% этилацетат в н-гексанах, а затем 0%→3% метанол в дихлорметане) с получением указанного в заголовке соединения (65 мг, 77%). ЖХ/МС (способ D) RT=1,212 мин, M+1=584.
1Н-ЯМР (500 МГц, CDCl3): 7,79 (1H, с), 7,77-7,72 (2H, м), 7,71-7,64 (2H, м), 7,58-7,51 (2H, м), 7,38-7,28 (5H, м), 7,22-7,16 (1H, м), 5,54 (2H, с), 3,39-3,29 (2H, м), 3,16-3,06 (2H+2H, м), 1,52-1,50 (3H, т), 1,42 (3H, с).
Стадия 2: (1r,3r)-3-амино-3-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-1-метилциклобутанол
В пробирку для микроволновой обработки объемом 10 мл добавляли 2-((1r,3r)-1-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион (30 мг, 0,051 ммоль) и гидразина моногидрат (0,016 мл, 0,514 ммоль) в 1,4-диоксане (1,5 мл) и этаноле (0,5 мл) с получением желтого раствора. Реакционную смесь нагревали до 140°C в течение 3 часов в условиях обработки микроволнами. Реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления твердого вещества, которое образовывалось в процессе реакции. Это твердое вещество тщательно промывали этанолом. Фильтраты объединяли и концентрировали в условиях пониженного давления. Неочищенный остаток (20 мг) растворяли в 1 мл раствора метанол/дихлорметан (1/1) и наносили на картридж SCX-2 catch-and-release (5 г). После связывания в течение 15 минут, колонку промывали метанолом (4×2 мл), а затем элюировали продукт 2M аммиаком в метаноле (4×2 мл). Объединенные содержащие продукт фракции концентрировали досуха в условиях пониженного давления с получением не совсем белого твердого вещества (15 мг). Это твердое вещество повторно растворяли в метаноле/воде и лиофилизировали в течение ночи с получением указанного в заголовке соединения (15 мг, 64%). ЖХ/МС (способ D) RT=0,630 мин, M+1=454.
1H-ЯМР (500 МГц, CD3OD) δ 7,95 (1H, с), 7,26-7,23 (3Н, м), 7,22-7,20 (3Н, м), 7,18-7,13 (3Н, м), 5,52 (2Н, с), 3,15-3,09 (2H, кв.), 2,59-2,52 (2H, м), 2,33-2,24 (2H, м), 1,43 (3Н, с), 1,41-1,36 (3Н, т).
Пример 139: 6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
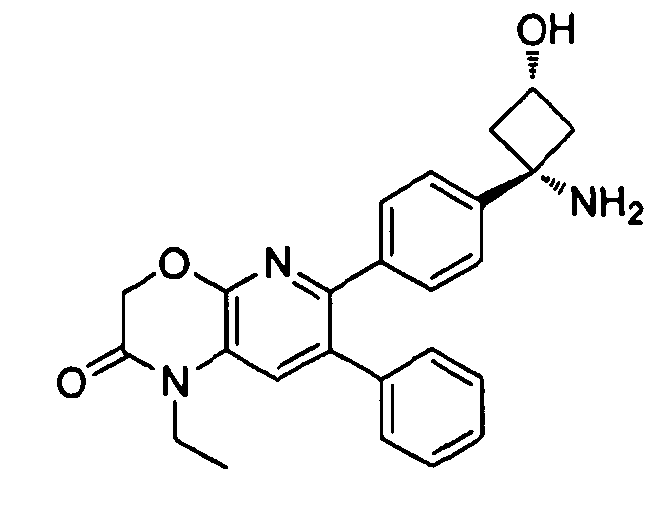
Стадия 1: трет-бутил-((1s,3s)-1-(4-(1-этил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидроксициклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли 6-бром-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (50 мг, 0,150 ммоль), трет-бутил-((1s,3s)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)карбамат (49 мг, 0,125 ммоль) и карбонат цезия (204 мг, 0,625 ммоль) в смеси 1,4-диоксана (2,3 мл) и воды (0,8 мл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и дихлорметана (20 мг, 0,025 ммоль) и дегазировали еще в течение 5 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1 часа, затем оставляли охлаждаться до комнатной температуры, разбавляли водой (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (45 мг, выход 70%).
1H-ЯМР (500 МГц, CDCl3) δ 7,29-7,35 (5H, м), 7,28 (1H, с), 7,18-7,24 (4H, м), 4,96 (1H, ушир. с), 4,88 (2H, с), 4,05 (1H, ушир. с), 4,01 (2H, кв.), 2,98 (2H, ушир. с), 2,75 (2H, ушир. с), 1,20-1,51 (9H, ушир. м), 1,32 (3H, т).
ЖХ/МС (способ D) RT=1,25 мин, M+H+=516,20.
Стадия 2: 6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-((1s,3s)-1-(4-(1-этил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидроксициклобутил)карбамат (45 мг, 0,087 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~3 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде не совсем белого твердого вещества (33 мг, выход 71%).
1H-ЯМР (500 МГц, MeOD) δ 7,55 (1H, с), 7,39-7,42 (4H, м), 7,27-7,31 (3Н, м), 7,20-7,24 (2H, м), 4,93 (2H, с), 4,01-4,11 (3Н, м), 3,03-3,11 (2H, м), 2,42-2,50 (2H, м), 1,28 (3Н, т).
ЖХ/МС (способ D) RT=0,74 мин, M+H+=416,20.
Пример 140: 6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
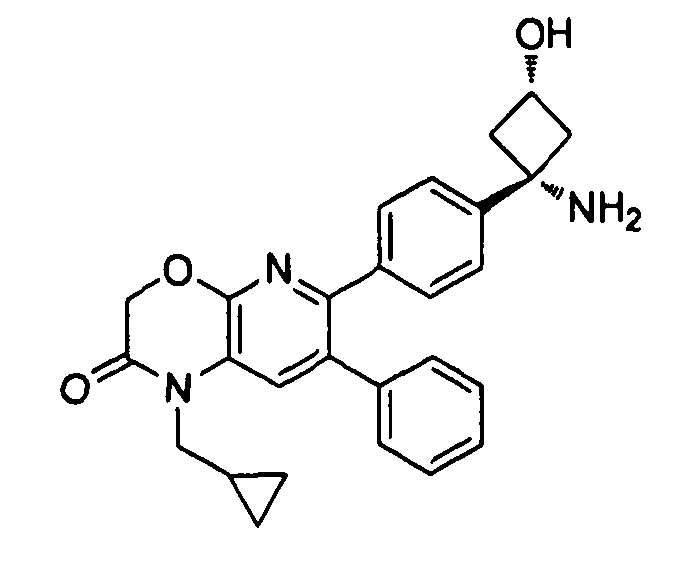
Стадия 1: трет-бутил-((1s,3s)-1-(4-(1-(циклопропилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидроксициклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли 6-бром-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (50 мг, 0,139 ммоль), трет-бутил-((1s,3s)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)карбамат (45 мг, 0,116 ммоль) и карбонат цезия (189 мг, 0,580 ммоль) в смеси 1,4-диоксана (2,2 мл) и воды (0,7 мл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, затем добавляли аддукт [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и дихлорметана (19 мг, 0,023 ммоль) и дегазировали еще в течение 5 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1 часа, затем оставляли охлаждаться до комнатной температуры, разбавляли водой (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (43 мг, выход 68%).
1H-ЯМР (500 МГц, CDCl3) δ 7,40 (1H, с), 7,28-7,34 (5H, м), 7,18-7,24 (4H, м), 4,96 (1H, ушир. с), 4,90 (2H, с), 4,05 (1H, ушир. с), 3,87 (2H, д), 2,99 (2H, ушир. с), 2,75 (2H, ушир. с), 1,14-1,54 (9H, ушир. м), 1,11-1,22 (1H, м), 0,54-0,62 (2H, м), 0,43-0,50 (2H, м).
ЖХ/МС (способ D) RT=1,36 мин, M+H+=542,20.
Стадия 2: 6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-((1s,3s)-1-(4-(1-(циклопропилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидроксициклобутил)карбамат (43 мг, 0,079 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~3 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде желтого твердого вещества (31 мг, выход 70%).
1H-ЯМР (500 МГц, MeOD) δ 7,65 (1H, с), 3,60 (4H, с), 7,28-7,31 (3H, м), 7,20-7,24 (2H, м), 4,94 (2H, с), 4,06 (1H, квинт.), 3,96 (2H, д), 3,02-3,12 (2H, м), 2,40-2,51 (2H, м), 1,14-1,25 (1H, м), 0,53-0,60 (2H, м), 0,40-0,47 (2H, м).
ЖХ/МС (способ D) RT=0,83 мин, M+H+=442,20.
Пример 141: 7-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он
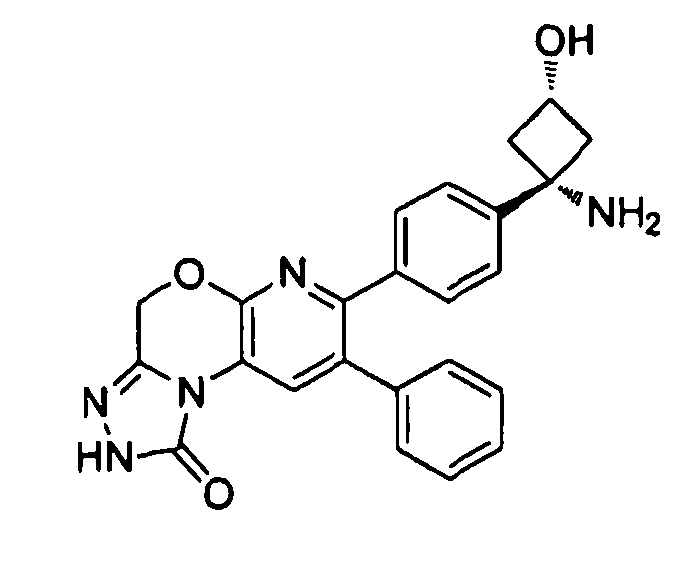
Стадия 1: 7-бром-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он
В пробирку для микроволновой обработки объемом 10 мл добавляли 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-тион (1 г, 3,11 ммоль) и этилгидразина карбоксилат (1,2 г, 12,44 ммоль) в пара-ксилоле (10 мл) с получением желтой суспензии. Реакционную смесь нагревали до 180°C в течение 45 минут в условиях обработки микроволнами. Неочищенный остаток концентрировали в условиях пониженного давления и очищали методом хроматографии на силикагеле Biotage (градиент 0%→100% этилацетат в н-гексане) с получением указанного в заголовке соединения (293 мг, 27%). ЖХ/МС (способ D) RT=1,069 мин, M+1=346.
Стадия 2: трет-бутил-((1s,3s)-3-гидрокси-1-(4-(1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли 7-бром-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он (70 мг, 0,203 ммоль), трет-бутил-((1s,3s)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)карбамат (65,8 мг, 0,169 ммоль) и карбонат цезия (275 мг, 0,845 ммоль) в 1,4-диоксане (3 мл) и воде (0,733 мл) с получением желтого раствора. Реакционную смесь дегазировали путем барботирования азотом в течение 15 минут, затем добавляли аддукт PdCl2(dppf)-CH2Cl2 (20 мг) и дегазировали еще в течение 15 минут. Реакционную смесь нагревали до 70°C в течение 96 часов в атмосфере азота. Реакционную смесь оставляли охлаждаться до комнатной температуры, и добавляли воду (7 мл). Реакционную смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические слои концентрировали в условиях пониженного давления. Неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→100% этилацетат в н-гексанах) с получением указанного в заголовке соединения (15 мг, 16%). ЖХ/МС (способ D) RT=1,034 мин, M+1=528.
1Н-ЯМР (500 МГц, CDCl3): 8,57 (1H, с), 7,48-7,27 (5H, м), 7,25-7,19 (4H, м), 5,30 (2H, с), 3,09-2,94 (2H, м), 2,84-2,70 (2H, м), 2,04 (1H, с), 1,47-1,32 (9H, ушир. с).
Стадия 3: 7-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]]триазоло[4,3-d][1,4]оксазин-1-он
трет-Бутил-((1s,3s)-3-гидрокси-1-(4-(1-оксо-8-фенил-2,4-дигидро-1H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутил)карбамат (15 мг, 0,028 ммоль) растворяли в дихлорметане (1 мл). При комнатной температуре добавляли TFA (1 мл) и перемешивали реакционную смесь в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~1 мл) и концентрировали досуха в условиях пониженного давления. Указанную процедуру повторяли трижды. Затем остаток суспендировали в диэтиловом эфире (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли дважды. Затем остаток суспендировали в н-гексанах (1 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли дважды. Удаляли оставшийся растворитель в условиях пониженного давления, и остаток сушили до постоянного веса. Затем остаток очищали методом препаративной ЖХВД с получением целевого продукта в виде не совсем белого твердого вещества (4 мг, выход 33%). ЖХ/МС (способ D) RT=0,613 мин, M-NH2=410.
1H-ЯМР (500 МГц, CD3OD) δ 8,56 (1H, с), 7,42-7,37 (2H, м), 7,36-7,28 (5H, м), 7,25-7,20 (2H, м), 5,41 (2H, с), 3,96-3,87 (1H, м), 3,00-2,88 (2H, м), 2,28-2,13 (2H, м).
Пример 142: (4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)метанамин
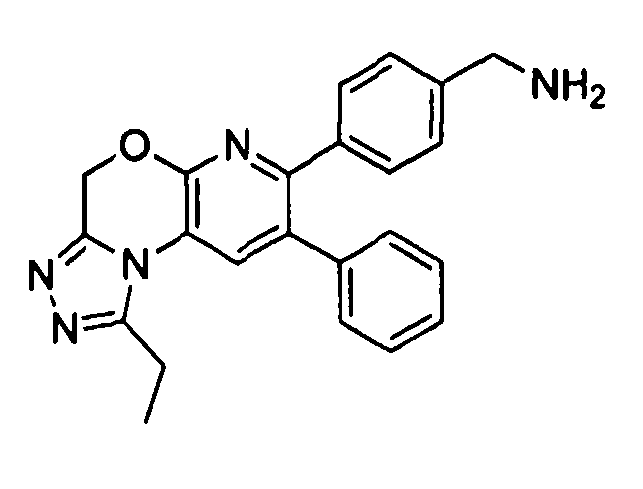
Стадия 1: 2-((1r,3r)-1-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион
В реакционную пробирку объемом 15 мл добавляли 7-бром-1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин (75 мг, 0,210 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбамат (84 мг, 0,252 ммоль) и карбонат цезия (342 мг, 1,050 ммоль) в 1,4-диоксане (3 мл) и воде (1 мл) с получением желтого раствора. Реакционную смесь дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт PdCl2(dppf)-CH2Cl2 (34 мг). Реакционную смесь нагревали до 50°C в течение 1 часа. Реакционную смесь оставляли охлаждаться до к.т. и добавляли воду (7 мл). Реакционную смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические слои концентрировали в условиях пониженного давления. Неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→100% этилацетат в циклогексане) с получением указанного в заголовке соединения (51 мг, 50%). ЖХ/МС (способ B) RT=1,292 мин, M+1=484.
1Н-ЯМР (500 МГц, CDCl3): 7,81 (1H, с), 7,38-7,30 (5H, м), 7,23-7,18 (2H, м), 7,16-7,12 (2H, м), 5,57 (2H, с), 4,29 (2H, ушир. с), 3,19-3,09 (2H, кв.), 1,58-1,50 (3H, т), 1,48-1,39 (9H, ушир. с).
Стадия 2: (4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)метанамин
Следуя методике получения 1-((1H-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она, в реакции использовали 2-((1r,3r)-1-(4-(1-этил-8-фенил-4H-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион (21 мг, 0,04 ммоль) с получением указанного в заголовке соединения (26 мг, количественный выход). ЖХ/МС: RT=0,669 мин, M+1=384.
1Н-ЯМР (500 МГц, MeOD): 7,99 (1H, с), 7,36-7,32 (2H, м), 7,29-7,20 (5H, м), 7,19-7,15 (2H, м), 5,55 (2H, с), 4,00 (2H, с), 3,25-3,16 (2H, кв.), 1,69-1,43 (3H, кв.).
Пример 143: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
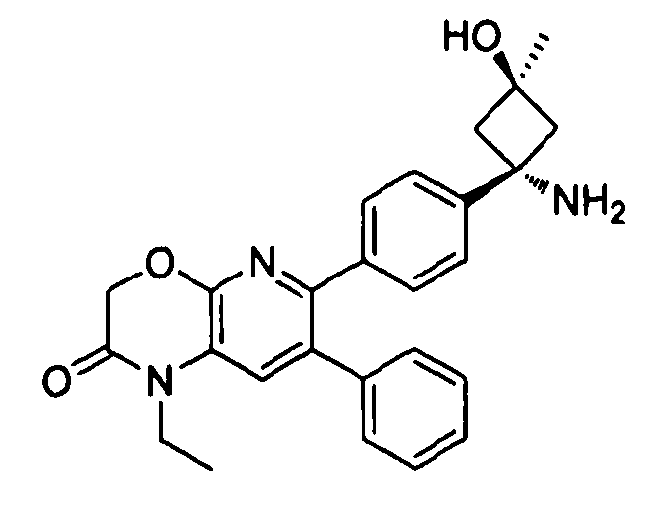
Стадия 1: 2-((1r,3r)-1-(4-(1-этил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион
В реакционную пробирку объемом 15 мл добавляли 6-бром-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (40 мг, 0,120 ммоль), 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-дион (43 мг, 0,100 ммоль) и карбонат цезия (163 мг, 0,500 ммоль) в смеси 1,4-диоксана (1,9 мл) и воды (0,6 мл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и дихлорметана (16 мг, 0,020 ммоль) и дегазировали еще в течение 5 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли водой (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (40 мг, выход 71%).
1H-ЯМР (500 МГц, CDCl3) δ 7,71-7,77 (2H, м), 7,63-7,69 (2H, м), 7,50 (2H, д), 7,23-7,33 (6H, м), 7,15-7,20 (2H, м), 4,84 (2H, с), 3,99 (2H, кв.), 3,32 (2H, д), 3,08 (2H, д), 1,40 (3H, с), 1,30 (3H, кв.).
ЖХ/МС (способ D) RT=1,36 мин, M+H+=560,20.
Стадия 2: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
К раствору 2-((1r,3r)-1-(4-(1-этил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-диона (40 мг, 0,071 ммоль) в смеси метанола (1 мл) и 1,4-диоксана (1 мл) добавляли гидразина моногидрат (0,25 мл, 5,15 ммоль) и нагревали до 120°C в условиях обработки микроволновым излучением в течение 30 минут. Реакционную смесь разбавляли дихлорметаном (8 мл) и промывали насыщенным раствором бикарбоната натрия (8 мл), солевым раствором (8 мл) и водой (8 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток повторно растворяли в 1,4-диоксане (1 мл), а затем по каплям добавляли 4M HCl в 1,4-диоксане (0,054 мл, 0,214 ммоль). Полученную суспензию перемешивали при комнатной температуре в течение 15 минут, а затем разбавляли диэтиловым (6 мл) и суспендировали в течение 10 минут. После отстаивания пипеткой удаляли надосадочную жидкость. Твердое вещество суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли дважды. Оставшийся растворитель удаляли путем концентрирования в условиях пониженного давления и лиофилизировали в течение ночи с получением целевого соединения в виде белого твердого вещества (27 мг, выход 81%).
1H-ЯМР (500 МГц, MeOD) δ 7,56 (1H, с), 7,37-7,44 (4H, м), 7,27-7,31 (3H, м), 7,20-7,25 (2H, м), 4,93 (2H, с), 4,07 (2H, кв.), 2,85 (2H, д), 2,67 (2H, д), 1,49 (3H, с), 1,28 (3H, кв.).
ЖХ/МС (способ D) RT=0,72 мин, M+H+=430,20.
Пример 144: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
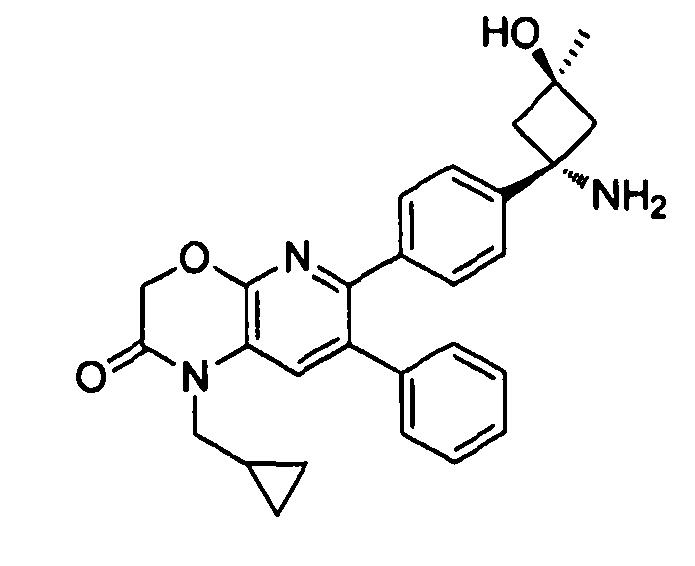
Стадия 1: 2-((1r,3r)-1-(4-(1-(циклопропилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион
В реакционную пробирку объемом 15 мл добавляли 6-бром-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (40 мг, 0,111 ммоль), 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-дион (40 мг, 0,093 ммоль) и карбонат цезия (151 мг, 0,464 ммоль) в смеси 1,4-диоксана (1,7 мл) и воды (0,6 мл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и дихлорметана (15 мг, 0,019 ммоль) и дегазировали еще в течение 5 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли водой (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→0/100), а затем методом хроматографии на силикагеле Biotage (дихлорметан/метанол, элюирование градиентом 100/0→90/10) с получением целевого продукта в виде не совсем белого твердого вещества (20 мг, выход 37%).
1H-ЯМР (500 МГц, CDCl3) δ 7,71-7,76 (2H, м), 7,63-7,69 (2H, м), 7,50 (2H, д), 7,36 (1H, с), 7,24-7,33 (5H, м), 7,15-7,19 (2H, м), 4,85 (2H, с), 3,84 (2H, д), 3,32 (2H, д), 3,08 (2H, д), 1,40 (3H, с), 1,09-1,19 (1H, м), 0,52-0,59 (2H, м), 0,42-0,48 (2H, м).
ЖХ/МС (способ D) RT=1,45 мин, M+H+=586,20.
Стадия 2: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
К раствору 2-((1r,3r)-1-(4-(1-(циклопропилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-диона (20 мг, 0,034 ммоль) в смеси метанола (1 мл) и 1,4-диоксана (1 мл) добавляли гидразина моногидрат (0,25 мл, 5,15 ммоль) и нагревали до 120°C в условиях обработки микроволновым излучением в течение 30 минут. Реакционную смесь разбавляли дихлорметаном (8 мл) и промывали насыщенным раствором бикарбоната натрия (8 мл), солевым раствором (8 мл) и водой (8 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток повторно растворяли в 1,4-диоксане (1 мл), а затем по каплям добавляли 4M HCl в 1,4-диоксане (0,026 мл, 0,102 ммоль). Полученную суспензию перемешивали при комнатной температуре в течение 15 минут, а затем разбавляли диэтиловым эфиром (6 мл) и суспендировали в течение 10 минут. После отстаивания пипеткой удаляли надосадочную жидкость. Твердое вещество суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли дважды. Удаляли оставшийся растворитель путем концентрирования в условиях пониженного давления и лиофилизировали в течение ночи с получением целевого соединения в виде белого твердого вещества (18 мг, количественный выход).
1H-ЯМР (500 МГц, MeOD) δ 7,65 (1H, с), 7,39-7,45 (4H, м), 7,27-7,31 (3H, м), 7,20-7,25 (2Н, м), 4,94 (2Н, с), 3,96 (2Н, д), 2,85 (2H, д), 2,68 (2H, д), 1,49 (3H, с), 1,14-1,26 (1H, м), 0,54-0,61 (2H, м), 0,40-0,46 (2H, м).
ЖХ/МС (способ D) RT=0,81 мин, M+H+=456,20.
Пример 145: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-(2-фторэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
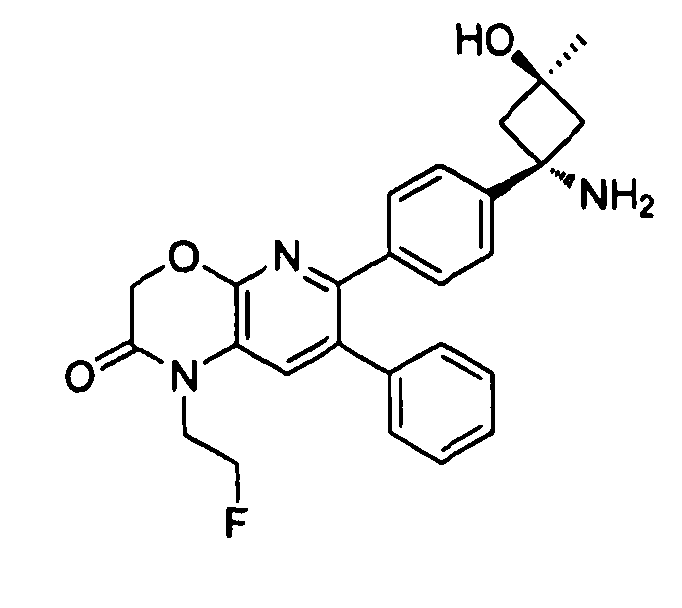
Стадия 1: 6-бром-1-(2-фторэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Смесь 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она (400 мг, 1,311 ммоль), 1-фтор-2-йодэтана (684 мг, 3,93 ммоль) и карбоната калия (544 мг, 3,93 ммоль) в DMF (4 мл) нагревали при перемешивании в течение 3 ч. Реакционную смесь охлаждали и разбавляли водой (20 мл), полученную смесь экстрагировали DCM (3×20 мл). Объединенную органическую фазу промывали водой, солевым раствором, сушили над сульфатом натрия и концентрировали с получением неочищенного материала, который очищали методом колоночной хроматографии, элюируя этилацетатом/циклогексаном 0-40%, с получением продукта (0,21 г).
1Н-ЯМР (500 МГц, CDCl3): 7,39-7,48 (м, 5H), 4,89 (с, 2H), 4,77 (т, 1H), 4,67 (т, 1H), 4,21 (т, 1H), 4,16 (т, 1H).
ЖХ/МС (способ D): RT=1,24 мин, M+H+=353,0.
Стадия 2: 2-((1r,3r)-1-(4-(1-(2-фторэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион
Смесь 6-бром-1-(2-фторэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она (38,9 мг, 0,111 ммоль), 2-((1r,3r)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)изоиндолин-1,3-диона (40 мг, 0,092 ммоль) и карбоната цезия (150 мг, 0,462 ммоль) в диоксане (3 мл) и воде (1 мл) дегазировали. После дегазирования добавляли Pd(dppf)Cl2·CH2Cl2 (15,08 мг, 0,018 ммоль) и нагревали реакционную смесь при 50°C при перемешивании в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (20 мл), и экстрагировали полученную смесь этилацетатом (3×20 мл). Объединенную органическую фазу промывали водой (30 мл) и сушили с сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали методом хроматографии (силикагель Biotage, 10 г), элюируя этилацетатом/циклогексаном 0-60%, с получением продукта (31 мг). ЖХ/МС (способ D): RT=1,31 мин, M+H+=578,2.
Стадия 3: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-(2-фторэтил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
К смеси 2-((1r,3r)-1-(4-(1-(2-фторэтил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-диона (31 мг, 0,054 ммоль) в диоксане (1 мл) и метаноле (1,000 мл) добавляли гидразина моногидрат (0,25 мл, 7,97 ммоль). Реакционную смесь нагревали при 120°C в течение 30 мин в условиях обработки микроволнами. Реакционную смесь разбавляли насыщенным раствором бикарбоната (10 мл) и экстрагировали DCM (3×10 мл). Объединенную органическую фазу промывали водой, солевым раствором и сушили над сульфатом натрия, фильтровали и концентрировали с получением продукта (13 мг).
1Н-ЯМР (500 МГц, CD3OD) 7,54 (с, 1H), 7,20-7,22 (м, 2H), 7,15-7,17 (м, 5H), 7,07-7,09 (м, 2H), 4,86 (с, 2H), 4,66 (т, 1H), 4,57 (т, 1H), 4,27 (т, 1H), 4,22 (т, 1H), 2,53 (д, 2H), 2,56 (д, 2H).
ЖХ/МС (способ D): RT=0,63 мин, M+H+=448,2.
Пример 146: 6-(4-(аминометил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
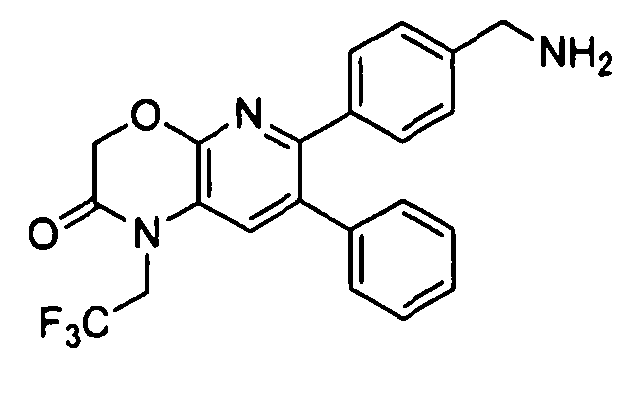
Стадия 1: трет-бутил-4-(2-оксо-7-фенил-1-(2,2,2-трифторэтил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат
В реакционную пробирку объемом 15 мл добавляли 6-бром-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (40 мг, 0,103 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбамат (41,3 мг, 0,124 ммоль) и карбонат цезия (168 мг, 0,517 ммоль) в смеси 1,4-диоксана (1937 мкл) и воды (646 мкл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт PdCl2(dppf)-CH2Cl2 (16,87 мг, 0,021 ммоль) и дегазировали еще в течение 5 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1 часа. Методом ЖХ/МС регистрировали завершение реакции. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли водой (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением коричневого твердого вещества. Полученный остаток очищали на силикагеле Biotage (0→45% циклогексан/EtOAc; колонка 10 г) с получением указанного в заголовке соединения (45 мг, 85%) в виде не совсем белого твердого вещества.
1Н-ЯМР (500 МГц, CDCl3): 7,36 (1H, с), 7,32-7,26 (5H, м), 7,17-7,13 (4H, м), 4,96 (2H, с), 4,77 (1H, ушир.), 4,63-4,58 (2H, кв.), 4,28 (2H, с), 1,48-1,45 (9H, ушир.).
Стадия 2: 6-(4-(аминометил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-4-(2-оксо-7-фенил-1-(2,2,2-трифторэтил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат (45 мг, 0,088 ммоль) растворяли в TFA (1 мл). Полученную смесь перемешивали в течение 30 секунд при комнатной температуре и упаривали в условиях пониженного давления. Соединение со снятой защитой дважды снова вводили в диэтиловый эфир, и дважды промывали полученное твердое вещество диэтиловым эфиром. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением указанного в заголовке соединения (34 мг, 94%) в виде не совсем белого твердого вещества. ЖХ/МС (способ D): RT=0,82 мин, M+H+=414.
1Н-ЯМР (500 МГц, CDCl3): 8,25 (2H, ушир.), 7,47 (1H, с), 7,28-7,26 (4H, м), 7,20-7,18 (3H,м), 7,10-7,08 (2H, м), 4,88 (2H, с), 4,64-4,59 (2H, кв.), 3,92 (2H, с).
Пример 147: 2-(6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
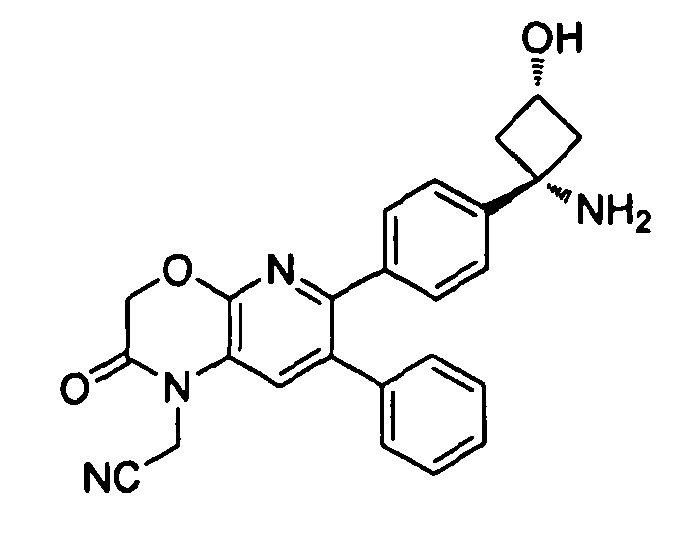
Стадия 1: 2-(6-бром-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
Смесь 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она (0,4 г, 1,311 ммоль), 2-бромацетонитрила (0,472 г, 3,93 ммоль) и карбоната калия (0,544 г, 3,93 ммоль) в DMF (4 мл) перемешивали при 50°C в течение 2 ч. Реакционную смесь разбавляли водой (20 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенную органическую фазу промывали водой, солевым раствором, сушили над сульфатом натрия и концентрировали с получением неочищенного продукта, который очищали методом колоночной хроматографии (силикагель Biotage, 25 г), элюируя 0-50% этилацетатом/циклогексаном, с получением продукта (0,19 г).
1Н-ЯМР (500 МГц, CDCl3) 7,45-7,50 (м, 3H), 7,4-7,43 (м, 2H), 7,31 (с, 1H), 4,93 (с, 2H), 4,83 (с, 2H).
Стадия 2: трет-бутил-((1s,3s)-1-(4-(1-(цианометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидроксициклобутил)карбамат
Смесь 2-(6-бром-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрила (42,4 мг, 0,123 ммоль), трет-бутил-((1s,3s)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)карбамата (40 мг, 0,103 ммоль) и карбоната цезия (167 мг, 0,514 ммоль) в диоксане (3 мл) и воде (1 мл) дегазировали. После дегазирования добавляли Pd(dppf)Cl2·CH2Cl2 (16,78 мг, 0,021 ммоль) и нагревали реакционную смесь при 50°C при перемешивании в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (20 мл) и экстрагировали полученную смесь этилацетатом (3×20 мл). Объединенную органическую фазу промывали водой (30 мл) и сушили с сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали методом хроматографии (силикагель Biotage, 10 г), элюируя этилацетатом/циклогексаном 0-70%, с получением продукта (21 мг). ЖХ/МС (способ D): RT=1,15 мин, M+H+=527,2.
Стадия 3: 2-(6-(4-(аминометил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
В круглодонную колбу, содержащую трет-бутил-((1s,3s)-1-(4-(1-(цианометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидроксициклобутил)карбамат (18 мг, 0,034 ммоль), добавляли трифторуксусную кислоту (0,5 мл, 6,49 ммоль). Полученный раствор перемешивали в течение 60 секунд, а затем концентрировали досуха в условиях пониженного давления. Остаток суспендировали в диэтиловом эфире (1 мл) и трижды концентрировали досуха с получением трифторуксуснокислой соли продукта (8,5 мг).
1Н-ЯМР (500 МГц, CD3OD) 7,58 (с, 1H), 7,31-7,35 (м, 4H), 7,19-7,22 (м, 3H), 7,13-7,15 (м, 2H), 5,02 (с, 2H), 4,92 (с, 2H), 3,92-3,96 (м, 1H), 2,95-2,99 (м, 2H), 2,35-2,39 (м, 2H).
ЖХ/МС (способ A): RT=3,53 мин, M+H+=428,11.
Пример 148: 6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
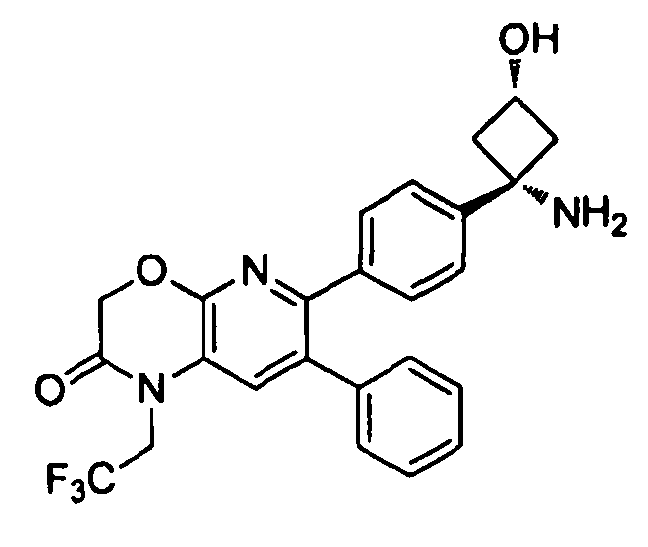
Стадия 1: 6-бром-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Смесь 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она (0,4 г, 1,311 ммоль), карбоната калия (0,544 г, 3,93 ммоль) и 1,1,1-трифтор-2-йодэтана (0,826 г, 3,93 ммоль) в DMF (2 мл) нагревали при 70°C при перемешивании в течение 18 ч. Реакционную смесь охлаждали, разбавляли водой (15 мл) и экстрагировали DCM (3×15 мл). Объединенную органическую фазу промывали водой (25 мл) и сушили с Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии (силикагель Biotage, 25 г), элюируя этилацетатом/циклогексаном 0-60%, с получением продукта 6-бром-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она (0,17 г).
1Н-ЯМР (500 МГц, CDCl3) 7,45-7,50 (м, 3H), 7,38-7,40 (м, 2H), 7,28 (с, 1H), 4,94 (с, 2H), 4,55 (кв., 2H).
Стадия 2: трет-бутил-((1s,3s)-3-гидрокси-1-(4-(2-оксо-7-фенил-1-(2,2,2-трифторэтил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
Смесь 6-бром-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-она (47,7 мг, 0,123 ммоль), трет-бутил-((1s,3s)-3-гидрокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)карбамата (40 мг, 0,103 ммоль) и карбоната цезия (167 мг, 0,514 ммоль) в диоксане (3 мл) и воде (1 мл) дегазировали. После дегазирования добавляли Pd(dppf)Cl2·CH2Cl2 (16,78 мг, 0,021 ммоль) и нагревали реакционную смесь при 50°C при перемешивании в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (20 мл), и экстрагировали полученную смесь этилацетатом (3×20 мл). Объединенную органическую фазу промывали водой (30 мл), сушили с сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали методом хроматографии (силикагель Biotage, 10 г), элюируя этилацетатом/циклогексаном 0-70%, с получением продукта (32 мг).
1Н-ЯМР (500 МГц, CDCl3) 7,31-7,37 (м, 5H), 7,16-7,22 (м, 5H), 4,96 (с, 2H), 4,95 (ушир., 1H), 4,61 (кв., 2H), 4,09 (м, 1H), 2,95-3,05 (м, 2H), 2,7-2,8 (м, 2H), 1,40 (ушир., 9H).
ЖХ/МС (способ D): RT=1,31 мин, M+H+=571,0.
Стадия 3: 6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В круглодонную колбу, содержащую трет-бутил-((1s,3s)-3-гидрокси-1-(4-(2-оксо-7-фенил-1-(2,2,2-трифторэтил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (30 мг, 0,053 ммоль), добавляли трифторуксусную кислоту (0,5 мл, 6,49 ммоль). Полученный раствор перемешивали в течение 60 секунд, а затем концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (1 мл) и трижды концентрировали досуха с получением трифторуксуснокислой соли продукта (21 мг).
1Н-ЯМР (500 МГц, CD3OD) 7,76 (с, 1H), 7,45 (м, 4H), 7,31-7,32 (м, 3H), 7,21-7,23 (м, 2H), 5,03 (с, 2H), 4,92 (кв., 2H), 4,06-4,09 (м, 1H), 3,08-3,11 (м, 2H), 2,46-2,50 (м, 2H).
ЖХ/МС (способ A): RT=3,94 мин, M+H+=471,04.
Пример 149: 6-(4-(1-аминоциклопропил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
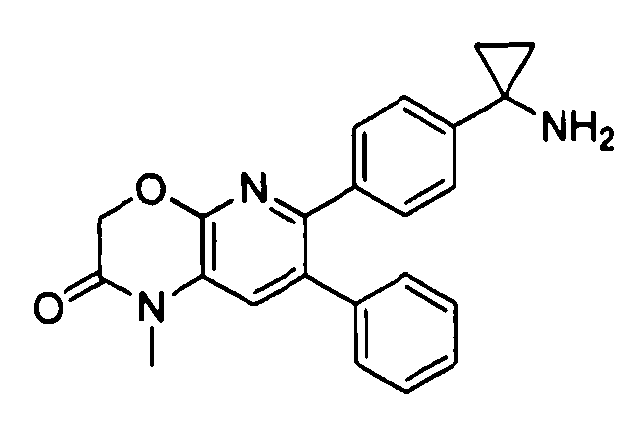
Стадия 1: 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклопропанамин
В реакционную пробирку объемом 40 мл добавляли 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензонитрил (500 мг, 2,18 ммоль) и изопропоксид титана(IV) (710 мкл, 2,40 ммоль) в безводном тетрагидрофуране (11 мл) с получением бесцветного раствора. Реакционную смесь охлаждали до -70°C в атмосфере азота, а затем по каплям в течение 30 минут добавляли этилмагния бромид (1M в THF, 4,80 мл, 4,80 ммоль). Полученный оранжевый раствор перемешивали при -78°C в течение 10 минут, а затем нагревали до комнатной температуры и перемешивали в течение 60 минут. После этого добавляли трифторида бора диэтилэфират (540 мкл, 4,37 ммоль) и перемешивали смесь при комнатной температуре в течение 60 минут. Затем к реакционной смеси добавляли 1M раствор HCl (6 мл) и перемешивали в течение 15 минут, после чего подщелачивали добавлением 2M раствора NaOH. Органическую фазу разделяли, и промывали водную фазу этилацетатом (3×4 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (70 мг, выход 12%).
1H-ЯМР (500 МГц, CDCl3) δ 7,76 (2H, д), 7,29 (2H, д), 2,32 (2H, ушир. с), 1,34 (12H, с), 1,12 (2H, дд), 1,01 (2H, дд).
ЖХ/МС RT=0,68 мин, M+H+=260,20.
Стадия 2: трет-бутил-(1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклопропил)карбамат
К раствору 1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклопропанамина (70 мг, 0,270 ммоль) в безводном THF (1,35 мл) добавляли триэтиламин (83 мкл, 0,594 ммоль) и ди-трет-бутилдикарбонат (69 мкл, 0,297 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 3 часов, а затем концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→40:60) с получением целевого продукта в виде белого твердого вещества (54 мг, выход 56%).
1H-ЯМР (500 МГц, CDCl3) δ 7,73 (2H, д), 7,19 (2H, д), 1,39-1,59 (9H, ушир. м), 1,33 (12H, с), 1,30 (2H, ушир. с), 1,25 (2H, ушир. с).
Стадия 3: трет-бутил-(1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклопропил)карбамат
В реакционную пробирку объемом 15 мл добавляли 6-бром-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (40 мг, 0,125 ммоль) и трет-бутил-(1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклопропил)карбамат (54 мг, 0,150 ммоль) в 1,4-диоксане (1 мл), а затем раствор карбоната натрия (40 мг, 0,3 ммоль) в воде (0,25 мл) с получением белой суспензии. Эту суспензию дегазировали путем барботирования азотом в течение 10 минут, а затем добавляли аддукт [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и дихлорметана (9 мг, 0,013 ммоль). Реакционную смесь нагревали до 80°C в течение ночи. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли солевым раствором (4 мл) и экстрагировали введением в этилацетат (3×4 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением целевого продукта в виде белого твердого вещества (60 мг, количественный выход).
1H-ЯМР (500 МГц, CDCl3) δ 7,14-7,26 (6H, м), 7,08-7,14 (2H, м), 6,93 (2H, д), 4,99-5,27 (1H, ушир. м), 4,80 (2H, с), 3,31 (3H, с), 1,35 (9H, ушир. с), 1,25 (2H, ушир. с), 1,03 (2H, ушир. с).
ЖХ/МС (способ D): RT=1,44 мин, M+H+=472,20.
Стадия 4: 6-(4-(1-аминоциклопропил)фенил)-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
К раствору трет-бутил-(1-(4-(1-метил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклопропил)карбамата (60 мг, 0,127 ммоль) в 1,4-диоксане (1 мл) по каплям добавляли 4M HCl в 1,4-диоксане (0,5 мл, 2,00 ммоль) и перемешивали полученный раствор при комнатной температуре в течение 1 часа. Реакционную смесь по каплям разбавляли диэтиловым эфиром (6 мл) и перемешивали в течение 15 минут с получением белого осадка. Слои оставляли отстаиваться, и удаляли надосадочную жидкость. Твердое вещество еще дважды промывали диэтиловым эфиром (6 мл) и оставляли отстаиваться, каждый раз удаляя надосадочную жидкость. Удаляли оставшийся растворитель путем концентрирования досуха в условиях пониженного давления, а затем лиофилизировали в течение выходных с получением целевого продукта в виде не совсем белого твердого вещества (27 мг, выход 52%).
1H-ЯМР (500 МГц, MeOD) δ 7,53 (1H, с), 7,34 (2H, д), 7,31 (2H, д), 7,25-7,29 (3H, м), 7,18-7,23 (2H, м), 4,94 (2H, с), 3,41 (3H, с), 1,29-1,35 (2H, м), 1,24-1,29 (2H, м).
ЖХ/МС (способ D): RT=0,75 мин, M+H+=372,10.
Пример 150: 2-(6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитриле
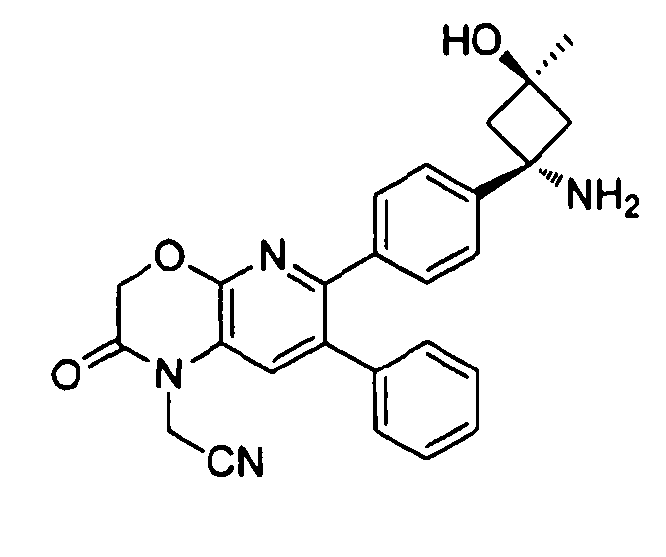
Стадия 1: трет-бутил-((1r,3r)-1-(4-(1-(цианометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидрокси-3-метилциклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли 2-(6-бром-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил (40 мг, 0,116 ммоль), трет-бутил-((1r,3r)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)карбамат (39 мг, 0,097 ммоль) и карбонат цезия (158 мг, 0,484 ммоль) в смеси 1,4-диоксана (1,8 мл) и воды (0,6 мл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и дихлорметана (16 мг, 0,019 ммоль) и дегазировали еще в течение 5 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли водой (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток дважды очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (30 мг, выход 57%).
1H-ЯМР (500 МГц, d6-ацетон) δ 7,67 (1H, с), 7,23-7,35 (9H, м), 6,55 (1H, ушир. с), 5,21 (2Н, с), 5,04 (2Н, с), 2,70-2,78 (2Н, м), 2,45-2,60 (2Н, м), 1,49 (3, с), 1,15-1,40 (ушир. м, 9H).
ЖХ/МС (способ D) RT=1,22 мин, M+H+=541,15.
Стадия 2: 2-(6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил
трет-Бутил-((1r,3r)-1-(4-(1-(цианометил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)-3-гидрокси-3-метилциклобутил)карбамат (30 мг, 0,055 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~3 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде белого твердого вещества (18 мг, выход 59%).
1H-ЯМР (500 МГц, MeOD) δ 7,68 (1H, с), 7,40-7,45 (4H, м), 7,28-7,31 (3H, м), 7,22-7,26 (2H, м), 5,12 (2H, с), 5,00 (2H, с), 2,85 (2H, д), 2,68 (2H, д), 1,48 (3H, с).
ЖХ/МС (способ D) RT=0,64 мин, M+H+=441,10.
Пример 151: 6-(4-(аминометил)фенил)-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
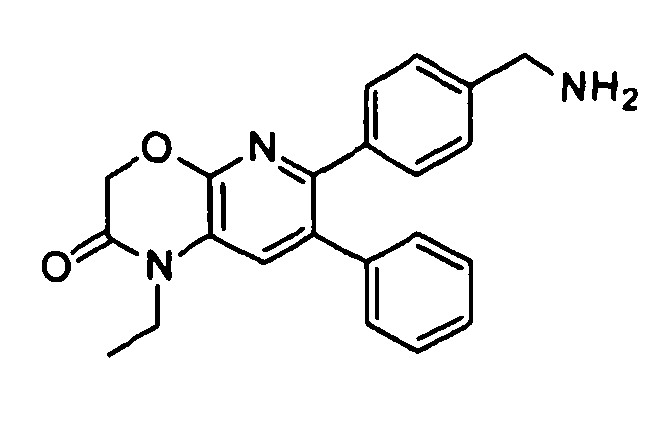
Стадия 1: трет-бутил-4-(1-этил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат
В реакционную пробирку объемом 15 мл добавляли 6-бром-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (50 мг, 0,150 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбамат (60,0 мг, 0,180 ммоль) и карбонат цезия (244 мг, 0,750 ммоль) в смеси 1,4-диоксана (2814 мкл) и воды (938 мкл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт PdCl2(dppf)-CH2Cl2 (24,51 мг, 0,030 ммоль) и дегазировали еще в течение 5 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1 часа. Методом ЖХ/МС регистрировали завершение реакции. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли водой (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением не совсем белого твердого вещества. Полученный остаток очищали на силикагеле Biotage (15-85% циклогексан/EtOAc; колонка 10 г) с получением указанного в заголовке соединения (45 мг, 65,3%) в виде не совсем белого твердого вещества.
1Н-ЯМР (500 МГц, CDCl3): 7,31-7,27 (6H, м), 7,19-7,18 (2H, д), 7,11-7,10 (2H, д), 4,87 (2H, с), 4,84 (1H, ушир.), 4,27 (2H, с), 4,03-3,98 (2H, кв.), 1,45 (9H, ушир.), 1,32-1,30 (3H, т).
Стадия 2: 6-(4-(аминометил)фенил)-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-4-(1-этил-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат (45 мг, 0,098 ммоль) растворяли в TFA (1 мл). Полученную смесь перемешивали в течение 30 секунд при комнатной температуре и упаривали в условиях пониженного давления. Соединение со снятой защитой еще дважды вводили в диэтиловый эфир, и дважды промывали образовавшееся твердое вещество диэтиловым эфиром. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением указанного в заголовке соединения (35 мг, 99%) в виде белого твердого вещества. ЖХ/МС: RT=0,75 мин, M+H+=360.
'Н-ЯМР (500 МГц, CDCl3): 8,34 (2H, ушир.), 7,30-7,27 (5H, м), 7,18 (3H, м), 7,13-7,11 (2H, м), 4,80 (2H, с), 4,03-3,99 (2H, кв.), 3,96 (2H, с), 1,32-1,30 (3H, т).
Пример 152: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
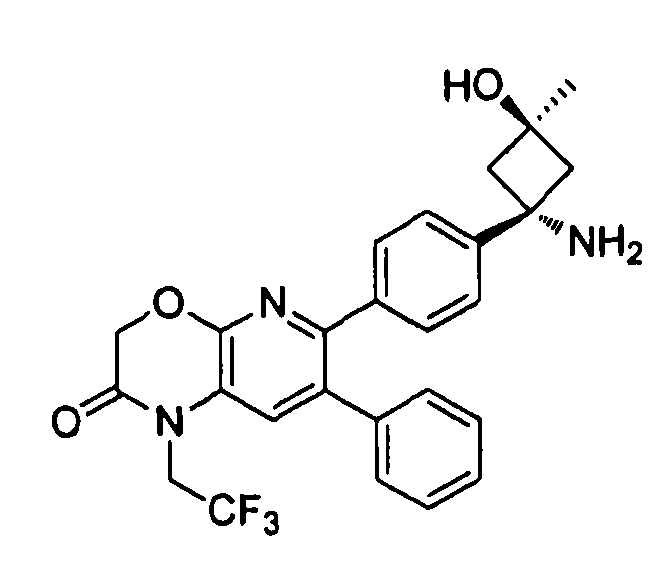
Стадия 1: трет-бутил-((1r,3r)-3-гидрокси-3-метил-1-(4-(2-оксо-7-фенил-1-(2,2,2-трифторэтил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли 6-бром-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (45 мг, 0,116 ммоль), трет-бутил-((1r,3r)-3-гидрокси-3-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)циклобутил)карбамат (39 мг, 0,097 ммоль) и карбонат цезия (158 мг, 0,484 ммоль) в смеси 1,4-диоксана (1,8 мл) и воды (0,6 мл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и дихлорметана (16 мг, 0,019 ммоль) и дегазировали еще в течение 5 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли водой (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Остаток дважды очищали методом хроматографии на силикагеле Biotage (циклогексан/этилацетат, элюирование градиентом 90/10→0/100) с получением целевого продукта в виде не совсем белого твердого вещества (25 мг, выход 44%).
1H-ЯМР (500 МГц, CDCl3) δ 7,35 (1H, с), 7,26-7,32 (5H, м), 7,21-7,25 (2H, м), 7,14-7,18 (2H, м), 4,94 (2H, с), 2,93 (1H, ушир. с), 4,60 (2H, кв.), 2,40-2,68 (4H, м), 1,55 (3H, с), 1,20-1,40 (9H, ушир. м).
ЖХ/МС (способ D) RT=1,38 мин, M+H+=584,10.
Стадия 2: 6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-((1r,3r)-3-гидрокси-3-метил-1-(4-(2-оксо-7-фенил-1-(2,2,2-трифторэтил)-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (25 мг, 0,043 ммоль) растворяли в TFA (1 мл) и перемешивали в течение 30 секунд. Раствор немедленно концентрировали досуха в условиях пониженного давления. Остаток растворяли в диэтиловом эфире (~3 мл) и трижды концентрировали досуха в условиях пониженного давления. Затем остаток суспендировали в диэтиловом эфире (3 мл), и после отстаивания удаляли пипеткой надосадочную жидкость. Указанную процедуру повторяли трижды. Оставшийся растворитель удаляли путем лиофилизации в течение ночи с получением целевого соединения в виде белого твердого вещества (8 мг, выход 31%).
1H-ЯМР (500 МГц, MeOD) δ 7,73 (1H, с), 7,39-7,44 (4H, м), 7,26-7,32 (3H, м), 7,17-7,23 (2H, м), 5,01 (2H, с), 4,89 (2H, кв.), 2,85 (2H, д), 2,66 (2H, д), 1,49 (3H, с).
ЖХ/МС (способ D) RT=0,80 мин, M+H+=484,05.
Пример 153: 6-(4-(аминометил)фенил)-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
Стадия 1: трет-бутил-4-(1-(циклопропилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат
В реакционную пробирку объемом 15 мл добавляли 6-бром-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (80 мг, 0,223 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилкарбамат (89 мг, 0,267 ммоль) и карбонат цезия (363 мг, 1,114 ммоль) в смеси 1,4-диоксана (3341 мкл) и воды (1114 мкл) с получением бесцветного раствора. Этот раствор дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт PdCl2(dppf)-CH2Cl2 (36,4 мг, 0,045 ммоль) и дегазировали еще в течение 5 минут. Реакционную смесь нагревали до 50°C в атмосфере азота в течение 1 часа. Методом ЖХ/МС регистрировали завершение реакции. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли водой (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением не совсем белого твердого вещества. Полученный остаток очищали на силикагеле Biotage (15-85% циклогексан/EtOAc; колонка 10 г) с получением указанного в заголовке соединения (80 мг, 74%).
1Н-ЯМР (500 МГц, CDCl3): 7,39 (1H, с), 7,32-7,26 (5H, м), 7,20-7,18 (2H, дд), 7,12-7,10 (2H, д), 4,89 (2H, с), 4,80 (1H, ушир.), 4,27 (2H, с), 3,87-3,86 (2H, д), 1,48-1,45 (9H, ушир.), 1,18-1,15 (1H, м), 0,60-0,56 (2H, м), 0,48-0,45 (2H, м).
Стадия 2: 6-(4-(аминометил)фенил)-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
трет-Бутил-4-(1-(циклопропилметил)-2-оксо-7-фенил-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)бензилкарбамат (80 мг, 0,165 ммоль) растворяли в TFA (1 мл). Полученную смесь перемешивали в течение 30 секунд при комнатной температуре и упаривали в условиях пониженного давления. Соединение со снятой защитой еще дважды вводили в диэтиловый эфир, и дважды промывали образовавшееся твердое вещество диэтиловым эфиром. Удаляли оставшийся растворитель в условиях пониженного давления и сушили с получением указанного в заголовке соединения (45 мг, 70,9%) в виде белого твердого вещества. ЖХ/МС: RT=0,84 мин, M+H+=386.
1Н-ЯМР (500 МГц, CDCl3): 8,33 (2H, ушир.), 7,41 (1H, с), 7,27-7,26 (4H, м), 7,18 (3H, м), 7,13-7,12 (2H, м), 4,82 (2H, с), 3,94 (2H, с), 3,87-3,85 (2H, д), 1,16-1,14 (1H, м), 0,60-0,57 (2H, м), 0,48-0,45 (2H, м).
трет-Бутил-1-(4-(6-хлор-5-нитро-3-фенилпиридин-2-ил)фенил)циклобутилкарбамат
Стадия 1: трет-бутил-(1-(4-(5-нитро-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутил)карбамат
Раствор (E)-трет-бутил-1-(4-(3-(диметиламино)-2-фенилакрилоил)фенил)циклобутилкарбамата (95 г, 214 ммоль) и 2-нитроацетамида (20 г, 192 ммоль) в уксусной кислоте (600 мл) перемешивали при 45°C в течение 3 часов, а затем при комнатной температуре в течение 18 часов. Полученный осадок фильтровали, промывали уксусной кислотой (150 мл), диэтиловым эфиром (400 мл) и сушили при комнатной температуре с получением указанного в заголовке соединения в виде желтого твердого вещества (41 г). Фильтрат концентрировали в условиях пониженного давления. Остаток поглощали диэтиловым эфиром. Полученный осадок фильтровали и сушили при комнатной температуре с получением второй порции продукта (9 г). Общий выход указанного в заголовке соединения составил 48%.
1Н-ЯМР (500 МГц, d6-DMSO): 13,02 (1H, ушир. с), 8,42 (1H, с), 7,6-7,1 (9H, м), 2,35 (4H, м), 1,98 (1H, м), 1,76 (1H, м), 1,3-1,1 (9H, м).
Стадия 2: трет-бутил-1-(4-(6-хлор-5-нитро-3-фенилпиридин-2-ил)фенил)циклобутилкарбамат
Раствор трет-бутил-1-(4-(6-гидрокси-5-нитро-3-фенилпиридин-2-ил)фенил)циклобутилкарбамата (50 г, 2,16 ммоль) и 10% Pd/C (0,2 г) в THF (2 л) гидрировали в автоклаве (2 бар) в течение 15 часов при комнатной температуре. Затем раствор фильтровали через целит, и промывали целит THF (0,5 л). Объединенные фильтраты концентрировали в условиях пониженного давления. Остаток растирали с диэтиловым эфиром. Полученный белый осадок фильтровали и сушили при комнатной температуре с получением указанного в заголовке соединения (50 г, 91%). Это соединение использовали на следующей стадии без дополнительной очистки.
Стадия 3: трет-бутил-1-(4-(6-хлор-5-нитро-3-фенилпиридин-2-ил)фенил)циклобутилкарбамат
В пробирку Шленка добавляли трет-бутил-1-(4-(6-хлор-5-нитро-3-фенилпиридин-2-ил)фенил)циклобутилкарбамат (10 г, 21,67 ммоль), трифенилфосфин (17,05 г, 65 ммоль), DCE (200 мл) и тетрахлорид углерода (2,19 мл, 22,75 ммоль). Полученный гомогенный раствор нагревали при 100°C в течение 35 минут. Затем его концентрировали в условиях пониженного давления. Остаток очищали методом хроматографии на силикагеле Biotage (градиент 0%→3% метанол в дихлорметане) с получением указанного в заголовке соединения в виде белого твердого вещества (6 г, 57%).
1Н-ЯМР (500 МГц, CDCl3): 8,30 (1H, с), 7,4-7,2 (9H, м), 2,51 (4H, м), 2,10 (1H, м), 1,88 (1H, м), 1,3-1,1 (9H, м).
трет-Бутил-(1-(4-(5-амино-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутил)карбамат
Стадия 1: трет-бутил-(1-(4-(5-нитро-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутил)карбамат
(E)-трет-бутил-1-(4-(3-(диметиламино)фенил)циклобутилкарбамат (10 г, 23,78 ммоль) и 2-нитроацетамид (2,45 г, 23,78 ммоль) растворяли в уксусной кислоте (125 мл) с получением желтого раствора. Эту реакционную смесь нагревали до 50°C в течение 2 часов, а затем перемешивали ее при комнатной температуре в течение 16 часов. Полученный осадок фильтровали, промывали диэтиловым эфиром и сушили до постоянного веса с получением указанного в заголовке соединения в виде желтого твердого вещества (4,1 г, 37,4%).
Стадия 2: трет-бутил-(1-(4-(5-амино-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутил)карбамат
трет-Бутил-(1-(4-(5-нитро-6-оксо-3-фенил-1,6-дигидропиридин-2-ил)фенил)циклобутил)карбамат (10,95 г, 23,77 ммоль) растворяли в уксусной кислоте (250 мл) при комнатной температуре. При 30°C добавляли порошковый цинк (10 г, 153 ммоль) и перемешивали полученную суспензию при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали в условиях пониженного давления, и распределяли полученный осадок между водой (250 мл) и этилацетатом (250 мл). Водную фазу экстрагировали свежим этилацетатом (3×200 мл), объединенные органические слои промывали водой (200 мл), солевым раствором (200 мл), сушили над безводным Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления. Полученное неочищенное вещество очищали методом хроматографии на силикагеле Biotage (градиент 0%→3% метанол в дихлорметане) с получением указанного в заголовке соединения (4,3 г, 43%). Это соединение использовали на следующей стадии без дополнительной очистки. ЖХ/МС (способ A) RT=5,96 мин, M+1=432.
трет-Бутил-(1-(4-(1-метил-7-фенил-2-тиоксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат
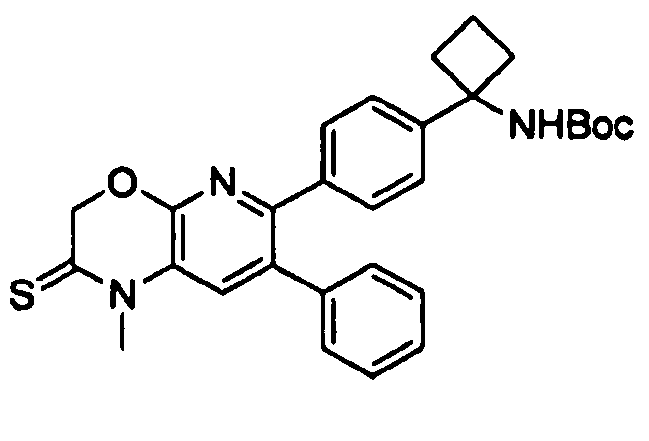
трет-Бутил-(1-(4-(1-метил-7-фенил-2-оксо-2,3-дигидро-1H-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутил)карбамат (200 мг, 0,412 ммоль) суспендировали в толуоле (5 мл). При комнатной температуре добавляли реагент Лауссона (125 мг, 0,309 ммоль). Полученную смесь нагревали с обратным холодильником в течение 5 часов. После ее охлаждения до комнатной температуры, смесь концентрировали досуха в условиях пониженного давления. Полученный остаток растворяли в минимальном количестве дихлорметана (1 мл) и обрабатывали диизопропиловым эфиром (7 мл). Отжимали желтое твердое вещество. Это твердое вещество фильтровали и сушили до постоянного веса (200 мг, 96%). ЖХ/МС (способ A): RT=7,08 мин, M+1=502.
6-Бром-1-метил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В реакционную пробирку объемом 15 мл добавляли 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (1,00 г, 3,28 ммоль) в безводном N,N-диметилформамиде (5 мл), карбонат калия (1,359 г, 9,83 ммоль) и йодметан (0,246 мл, 3,93 ммоль) с получением коричневой суспензии. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 1 часа. Реакционную смесь разбавляли насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали введением в дихлорметан (3×15 мл). Объединенные органические фазы сушили над безводным Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением коричневого масла. Остаток очищали методом хроматографии на силикагеле Biotage (колонка 50 г, циклогексан/этилацетат, элюирование градиентом 88/12→0/100) с получением бледно-желтого твердого вещества (780 мг, выход 74,6%).
1Н-ЯМР (500 МГц, CDCl3): 7,51-7,44 (5H, м), 7,20 (1H, с), 4,91 (2H, с), 3,36 (3H, с).
7-Бром-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В круглодонную колбу объемом 500 мл добавляли этил-2-(5-бром-3-нитропиридин-2-илокси)ацетат (5,2 г, 17,04 ммоль) в соляной кислоте (37%, 50 мл) с получением желтой суспензии. Смесь охлаждали до 0-5°C, а затем порциями добавляли порошковое олово (10,1168 г, 85 ммоль). В процессе добавления следует соблюдать осторожность, поскольку реакция является резко экзотермической. Смесь перемешивали при комнатной температуре еще в течение 30 минут после добавления. Реакционную смесь нагревали до 80°C в атмосфере азота в течение 3 часов. Реакционную смесь разбавляли водой (250 мл). Осадок фильтровали, промывали водой (200 мл) и диэтиловым эфиром (200 мл) и сушили до постоянного веса с получением указанного в заголовке соединения (3,2 г, 82%).
1Н-ЯМР (500 МГц, d6-DMSO): 10,41 (2H, с), 7,88 (2H, с), 7,34 (2H, с), 4,85 (2H, с).
Этил-2-((5-бром-3-нитропиридин-2-ил)окси)ацетат
К суспензии гидрида натрия (5,31 г, 133 ммоль) в 1,4-диоксане (250 мл) по каплям в течение 30 минут добавляли этилгликолят (12,56 мл, 133 ммоль), убеждаясь в поддержании температуры ниже 30°C. Полученную вязкую суспензию перемешивали при комнатной температуре в течение 15 минут. В отдельную круглодонную колбу объемом 1 л добавляли 5-бром-2-хлор-3-нитропиридин (21 г, 88 ммоль) в 1,4-диоксане (150 мл) с получением коричневого раствора. По каплям в течение 30 минут добавляли суспензию гидрида натрия/этилгликолята. Полученную реакционную смесь нагревали до 80°C в течение ночи. Реакционную смесь концентрировали в условиях пониженного давления, и очищали остаток методом хроматографии на силикагеле Biotage (колонка 340 г, элюирование градиентом 0%→10% этилацетат в циклогексане) с получением бледно-желтого твердого вещества (11,8 г, выход 43%).
1Н-ЯМР (500 МГц, CDCl3): 8,48 (1H, д), 8,42 (1H, д), 5,07 (2H, с), 4,25 (2H, кв.), 1,30 (3H, с).
6-Бром-1-этил-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В реакционную пробирку объемом 15 мл добавляли 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (300 мг, 0,983 ммоль), йодэтан (0,095 мл, 1,180 ммоль) и карбонат калия (408 мг, 2,95 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением коричневой суспензии. Эту суспензию перемешивали при 50°C в атмосфере азота в течение 60 минут. Реакционную смесь разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы промывали водой/солевым раствором (50/50, 3×5 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением коричневого твердого вещества. Это вещество очищали методом хроматографии на силикагеле Biotage (картридж с 25 г силикагеля, циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением указанного в заголовке соединения в виде бежевого твердого вещества (160 мг, выход 48,8%).
1Н-ЯМР (500 МГц, CDCl3): 7,58-7,37 (5H, м), 7,21 (1H, с), 4,86 (2H, с), 3,96 (2H, кв.), 1,27 (3H, т).
ЖХ/МС (способ D) RT=1,293 мин, M+1=334.
6-Бром-1-(циклопропилметил)-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он
В реакционную пробирку объемом 15 мл добавляли 6-бром-7-фенил-1H-пиридо[2,3-b][1,4]оксазин-2(3H)-он (300 мг, 0,983 ммоль), (бромметил)циклопропан (0,114 мл, 1,180 ммоль) и карбонат калия (408 мг, 2,95 ммоль) в безводном N,N-диметилформамиде (1 мл) с получением коричневой суспензии. Эту суспензию перемешивали при 50°C в атмосфере азота в течение 60 минут. Реакционную смесь разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали введением в этилацетат (3×5 мл). Объединенные органические фазы промывали водой/солевым раствором (50/50, 3×5 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением коричневого твердого вещества. Это вещество очищали методом хроматографии на силикагеле Biotage (картридж с 25 г силикагеля, циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением указанного в заголовке соединения в виде бежевого твердого вещества (160 мг, 0,445 ммоль, выход 45,3%).
1Н-ЯМР (500 МГц, CDCl3): 7,58-7,37 (5H, м), 7,32 (1H, с), 4,88 (2H, с), 3,81 (2H, д), 1,14-1,01 (1H, м), 0,60-0,53 (2H, м), 0,47-0,40 (2H, м).
трет-Бутил-((1r,3r)-1-(4-(5,5-диметил-1,3,2-диоксаборинан-2-ил)фенил)-3-гидрокси-3-метилциклобутил)карбамат
Стадия 1: 2-((1r,3r)-1-(4-(5,5-диметил-1,3,2-диоксаборинан-2-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион
В круглодонную колбу объемом 100 мл добавляли 2-((1r,3r)-1-(4-бромфенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион (0,710 г, 1,838 ммоль), 5,5,5',5'-тетраметил-2,2'-би(1,3,2-диоксаборинан) (0,700 г, 3,10 ммоль) и ацетат калия (0,361 г, 3,68 ммоль) в безводном DMSO (35,4 мл) с получением бесцветной суспензии. Эту суспензию дегазировали путем барботирования азотом в течение 15 минут, а затем добавляли аддукт PdCl2(dppf)·CH2Cl2 (0,150 г, 0,184 ммоль). Полученную суспензию нагревали до 80°C в атмосфере азота в течение ночи. Реакционную смесь оставляли охлаждаться до комнатной температуры, разбавляли этилацетатом (180 мл), фильтровали через целит, промывали водой (2×180 мл), а затем солевым раствором (180 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением коричневого масла. Это масло очищали методом хроматографии на силикагеле Biotage (картридж с 50 г силикагеля, циклогексан/этилацетат, элюирование градиентом 90/10→0/100) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (263 мг, 0,627 ммоль, выход 34,1%).
1Н-ЯМР (500 МГц, CDCl3): 7,82-7,72 (5H, м), 7,70-7,63 (4H, м), 3,73 (3H, с), 3,36 (2H, д), 3,19 (2H, д).
ЖХ/МС (способ D) RT=0,890 мин, M+1=420.
Стадия 2: трет-бутил-((1r,3r)-1-(4-(5,5-диметил-1,3,2-диоксаборинан-2-ил)фенил)-3-гидрокси-3-метилциклобутил)карбамат
В реакционную пробирку объемом 15 мл добавляли 2-((1r,3r)-1-(4-(5,5-диметил-1,3,2-диоксаборинан-2-ил)фенил)-3-гидрокси-3-метилциклобутил)изоиндолин-1,3-дион (220 мг, 0,525 ммоль) и гидразина моногидрат (0,2 мл, 4,12 ммоль) в этаноле (4 мл) с получением бледно-желтого раствора. Этот раствор нагревали до 80°C в течение ночи с получением белой суспензии. Реакционную смесь фильтровали, и концентрировали фильтрат досуха в условиях пониженного давления. Остаток повторно растворяли в метаноле (4 мл), а затем добавляли ди-трет-бутилдикарбонат (0,244 мл, 1,049 ммоль) и триэтиламин (0,183 мл, 1,312 ммоль). Реакционную смесь нагревали до 50°C в течение ночи. Реакционную смесь разбавляли этилацетатом (16 мл) и промывали водой (8 мл), сушили над Na2SO4, фильтровали и концентрировали досуха в условиях пониженного давления с получением желтого твердого вещества. Это вещество очищали методом хроматографии на силикагеле Biotage (картридж с 25 г силикагеля, циклогексан/этилацетат, элюирование градиентом 90/10→20/80) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (88 мг, 0,226 ммоль, выход 43,1%).
1H-ЯМР (500 МГц, CD3Cl3) δ 7,75 (2H, м), 7,38 (2H, с), 3,76 (3Н, с), 2,80-2,63 (4H, м), 2,60-2,45 (2H, м), 2,16-2,03 (2H, м), 1,49-1,29 (9H, м), 1,01 (6H, с).
ЖХ/МС (способ D) RT=0,814 мин, M+1=390.
Метод анализа активности AKT киназы
Тестирование соединений проводили с использованием метода анализа активности AKT киназы.
Активированные изоформы AKT 1, 2 и 3 анализировали с использованием 5' FAM Crosstide (SEQ GRPRTSSFAEG-OH). Степень фосфорилирования киназы определяли по поляризации флуоресценции с использованием связывающего реагента iMAP Progressive, в котором применяются связывающие гранулы, позволяющие реагенту специфически связываться с фосфатными остатками посредством ковалентных координационных комплексных связей.
Связывающий раствор с iMAP останавливает взаимодействие Crosstide и киназы и специфически связывает фосфорилированные субстраты. Степень фосфорилирования определяют по поляризации флуоресценции (возбуждение при 485 нм; эмиссия при 528 нм) или по снижению скорости вращения возбужденного субстрата.
В анализе использовали следующие вещества:
a) активированные изоформы AKT (SignalChem.), растворенные в полном реакционном буфере в предварительно определенной концентрации, подобранной так, что метод анализа проводили в линейном диапазоне;
b) пептид-субстрат AKT: Crosstide FAM (R7110) производства Molecular Devices, разбавленный полным реакционным буфером;
c) экспресс-набор для iMAP-скрининга (R8127) производства Molecular Devices;
d) полный реакционный буфер, содержащий 0,1% BSA, 10 мМ Tris-HCl, 10 мМ MgCl2, 0,05% NaN3 и 0,01% BSA, не содержащий фосфатов, 1 мМ DTT;
e) связывающий раствор, содержащий 75% буфера А, 25% буфера В и небольшой объем связывающего реагента, который содержит связываемый субъект для данного метода анализа;
f) ATP, разбавленную в полном реакционном буфере;
g) 384-луночные аналитические планшеты из черного полистирола (Nunc);
h) гибридный планшет-ридер Biotek Synergy 4.
В DMSO (Sigma Aldrich) растворяли 5 мкл тестируемого соединения, проводили серийные разведения полным реакционным буфером до снижения эффекта дозы на четырнадцать с половиной порядков и помещали в черные 384-луночные планшеты. Соединение инкубировали с активированной изоформой AKT (5 мкл) в предварительно определенной концентрации в течение 45 минут при комнатной температуре.
В каждую лунку добавляли 2,5 мкл раствора ATP, смешанного с 2,5 мкл пептида-субстрата AKT (Crosstide FAM (R7110) производства Molecular Devices), и центрифугировали планшет при 1000 об/мин в течение 20 секунд для обеспечения однородного перемешивания реагентов. Реакционную смесь инкубировали в темноте в течение одного часа при комнатной температуре.
Киназную реакцию останавливали добавлением связывающего раствора, и оставляли смесь уравновешиваться в течение одного часа в темноте при комнатной температуре.
Поляризацию флуоресценции в каждой лунке определяли с использованием гибридного планшет-ридера Biotek Synergy 4. Вкратце, возбуждение флуоресценции в каждом реакционном растворе проводили при 485 нм, и измеряли эмиссию при 528 нм как в параллельном, так и в перпендикулярном направлениях.
Значение поляризации в каждой лунке рассчитывали с помощью программного обеспечения Gen5 (Biotek), а процент ингибирования активности киназы по сравнению с носителем в качестве контроля рассчитывали с помощью GraphPad Prism. Значения IC50 для каждого соединения рассчитывали с помощью нелинейного регрессионного анализа с использованием программного обеспечения Prism.
Все планшеты подвергали внутреннему контролю двумя способами. Во-первых, путем расчета соотношения сигнал/шум на основании поляризации в присутствии киназы без ингибитора и поляризации в полном реакционном буфере в отсутствие активированной киназы. Во-вторых, путем определения значений IC50, получаемых с известными ингибиторами изоформ AKT.
Тестирование соединений также проводили с использованием методов анализа пролиферации клеток in vitro.
Cell Titre Glo (Promega) представляет собой высокочувствительный однородный реагент, используемый для определения жизнеспособности клеток. В указанном реагенте стабильная форма люциферазы используется для измерения количества ATP в качестве показателя жизнеспособности. Полученные при анализе значения люминесценции прямо пропорциональны числу жизнеспособных клеток в данном методе анализа.
Были использованы следующие вещества: белые 96-луночные аналитические планшеты с прозрачным дном (Costar); реагенты Cell titre Glo; клетки LnCaP (ЕСАСС), выращенные в среде RPMI (Invitrogen), дополненной 10 мМ HEPES (Invitrogen), 1 мМ пирувата натрия (Invitrogen), 2 мМ L-глутамина (Invitrogen) и 10% эмбриональной бычьей сыворотки (Invitrogen); клетки PC3 (ЕСАСС), выращенные в среде RPMI, дополненной 10% эмбриональной бычьей сыворотки (Invitrogen); трипсином (Invitrogen), PBS (Invitrogen); гибридный планшет-ридер Biotek Synergy 4; шейкер для 96-луночных планшетов (Stuart SSL5); настольная центрифуга Eppendorf 5414; устройство для подсчета клеток Beckman Coulter Z1 с установкой одного порогового значения.
Линии клеток предстательной железы PC3 и LNCaP промывали, снимали и повторно суспендировали в соответствующих свежих средах. Клетки осаждали центрифугированием (Eppendorf 5414), а надосадочную жидкость удаляли. Клетки повторно суспендировали перемешиванием на вортексе, подсчитывали и высевали в белые 96-луночные планшеты с прозрачным дном при плотности 5000 клеток на лунку. Клетки инкубировали (Sanyo) в течение ночи при 37°C (95% О2/5% СО2) и на следующий день обрабатывали возрастающими концентрациями тестируемого соединения, приготовленного в свежей среде. Планшеты возвращали в инкубатор на 72 часа.
Cell Titre Glo (Promega) получали путем смешивания поставляемых реагентов в соответствии с инструкциями изготовителя и оставляли при комнатной температуре. Планшеты с клетками извлекали из инкубатора, и в каждую лунку добавляли 80 мкл раствора Cell Titre Glo. Планшет встряхивали в течение пяти минут для обеспечения однородного перемешивания реагентов и клеток, а затем оставляли отстаиваться в течение десяти минут при комнатной температуре.
Жизнеспособность клеток после обработки соединением определяли по интенсивности люминесценции, испускаемой из обработанных лекарственным средством лунок планшета. Вкратце, в гибридный планшет-ридер Synergy Biotex 4 помещали аналитический планшет, и регистрировали люминесценцию в каждой лунке. Обработанные соединением лунки сравнивали с лунками, обработанными носителем, и рассчитывали процент ингибирования жизнеспособности клеток.
Данные анализировали с использованием GraphPad Prism со значениями IC50, полученными с использованием нелинейного регрессионного анализа набора данных.
Анализ эффектов соединения на пути передачи сигнала AKT
Статус фосфорилирования различных участников пути передачи сигнала AKT/PI3K исследовали методом вестерн-блоттинга.
Вещества, требуемые для указанного метода анализа: 20-кратный подвижный буфер (Invitrogen); шкала маркеров Rainbow (GE Healthcare); восстанавливающий буфер (Invitrogen); 20-кратный гибридизационный буфер (Invitrogen); 4-12% гели Bis-Tris (Invitrogen); фильтровальная бумага (Whatman); нитроцеллюлоза (Amersham); реагенты для выявления ECL plus (GE Healthcare); рентгенографическая пленка (Kodak); реагенты для определения белка Biorad (Biorad); антитела пути передачи сигнала AKT (Cell Signalling).
Клеточные линии LNCaP и PC3 промывали, снимали и повторно суспендировали в свежей среде. Их высевали в 90-мм2 чашки и инкубировали в течение ночи (95% О2/5% СО2), давая возможность им прикрепиться. Когда конфлуэнтность клеток достигала 60%, среду удаляли и заменяли ее средой, дополненной соединением или носителем. Планшеты инкубировали в течение некоторого диапазона времени.
Среду удаляли, а клетки помещали на лед и промывали PBS. В чашку добавляли 300 мкл лизирующего буфера и оставляли на несколько минут, после чего клетки счищали в раствор и переносили с помощью пипетки в центрифужную пробирку. Пробирку помещали на лед на 10 минут, а затем встряхивали на вортексе для лизиса клеток. Образец центрифугировали (в настольной ультрацентрифуге Eppendorf) при 13200 об/мин в течение 10 минут при 4°C. Полученную надосадочную жидкость анализировали на содержание белка с использованием метода Брэдфорда (Biorad), и равные согласно расчету количества белка нагревали в восстанавливающем буфере до 95°C в течение десяти минут.
Образцы прогоняли на гелях с 4-12% Bis-Tris (Invitrogen), переносили на нитроцеллюлозную мембрану (Amersham) и блокировали 5% раствором обезжиренного молока.
Мембраны использовали для определения различия в общей AKT, AKT pSer473, pGSK3β и общей GSK3β (все производства Cell Signalling). Соответствующие первичные антитела разводили в блокирующем растворе 1% обезжиренного молока и инкубировали на мембранах при 4°С в течение ночи. Мембраны трижды промывали PBS, инкубировали в течение двух часов с соответствующими вторичными антителами (Sigma Aldrich), и определяли белки с использованием реагентов для выявления ECL plus (GE Healthcare).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ С ПОМОЩЬЮ СОЕДИНЕНИЙ 2,4-ПИРИМИДИНДИАМИНА | 2013 |
|
RU2659777C2 |
| СОЕДИНЕНИЯ 2, 4-ПИРИМИДИНДИАМИНА, ОБЛАДАЮЩИЕ ВОЗДЕЙСТВИЕМ ПРИ АУТОИММУННЫХ РАССТРОЙСТВАХ | 2004 |
|
RU2356901C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ И ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 2007 |
|
RU2434868C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ПОВЫШЕНИЯ АКТИВНОСТИ CFTR | 2015 |
|
RU2767460C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |

Изобретение относится к соединениям формулы (I), где каждый R1 и R2 независимо выбирают из водорода, C1-С10алкила, необязательно замещенного ОН и OR3, где R3 представляет собой С1-С10алкил, или R1 и R2 вместе представляют собой оксо или необязательно C1-С10алкила О-замещенный оксим; W представляет собой О, NR′ или CRaRb, где R′ представляет собой либо водород, либо C1-С10алкил; X либо отсутствует, либо представляет собой CR4R5, где каждый Ra, Rb, R4 и R5 независимо выбирают из заместителей, указанных для R1 и R2; Y и Z независимо представляют собой замещенный или незамещенный азот или углерод (заместители для которых указаны в п.1 формулы изобретения), или Y и Z вместе образуют необязательно замещенный пиразолил, имидазолил, пиримидинил, 1,2,4-триазолил (заместители для которых указаны в п.1 формулы изобретения); R7a и R7b представляют собой Н; R6a и R6b представляют собой Н или R6a и R6b могут быть объединены с образованием С3-С6циклоалкила, необязательно замещенного одним или двумя заместителями, выбранными из С1-С6алкила и ОН; Су выбирают из C1-6алкил(С3-С8)циклоалкила, фенила, пиридинила и тиенила; m равен 0 или 1; R8 представляет собой F. Также изобретение относится к конкретным соединениям, указанным в п.1 формулы изобретения, к фармацевтической композиции и способу лечения злокачественной опухоли. Технический результат - новые ингибиторы AKT. 3 н. и 16 з.п. ф-лы, 1 ил., 153 пр.

1. Соединение формулы (I):
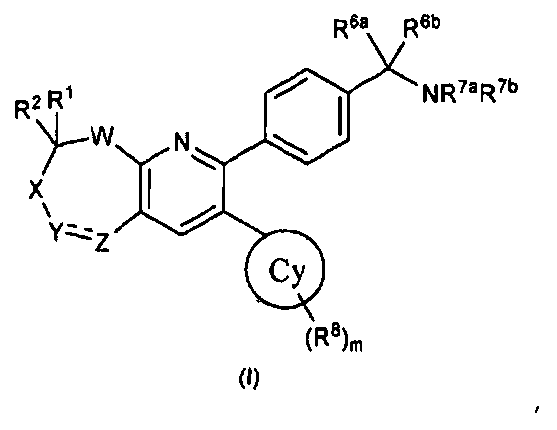
в которой:
каждый R1 и R2 независимо выбирают из водорода, C1-С10алкила, необязательно замещенного ОН и OR3, где каждый R3 представляет собой С1-С10алкил или
R1 и R2 вместе представляют собой оксо или необязательно C1-С10алкила О-замещенный оксим;
W представляет собой О, NR′ или CRaRb, где R′ представляет собой либо водород, либо C1-С10алкил и где каждый Ra и Rb независимо выбирают из представителей представленной выше группы, из которой выбирают R1 и R2;
X либо отсутствует, либо представляет собой CR4R5, где каждый R4 и R5 независимо выбирают из представителей представленной выше группы, из которой выбирают R1 и R2;
Y и Z независимо представляют собой или замещенный или незамещенный азот или углерод и углерод замещен заместителями, независимо выбранными из представителей представленной выше группы, из которой выбирают R1 и R2, или в случае азота заместитель представляет собой тетрагидропиранил, пиридинил или 1-метилпиразолил, C1-С10алкил, необязательно замещенный одним-тремя заместителями, выбранными из CN, NR3aR3b, CONH2, COR3, CO2R3, OR3, F, ОН, С3-С6циклоалкила, гетероарила, который представляет собой тиазолил, имидазолил или пиридинил, SO2R3, CONHR3, где значения R3 определены выше, R3a и R3b представляют собой С1-С10алкил, или где Y и Z вместе образуют необязательно замещенный гетероарил, выбранный из пиразолила, имидазолила, пиримидинила, 1,2,4-триазолила, необязательно замещенных C1-С10алкилом, необязательно замещенного F, CN, ОН, OR3; ОН, Br, оксо, SR3, SO2R3, NHR3, NH2, NR3, CONH2, С3-С6циклоалкилом, -NHCH2CH2OH, 1-метил-1Н-имидазолилом, пиридинилом и морфолинилом, где R3 определен выше, или где Y представляет собой SO2;
R7a и R7b независимо выбирают из Н; и
R6a и R6b независимо представляют собой Н или R6a и R6b могут быть объединены с образованием С3-С6циклоалкила, необязательно замещенного одним или двумя заместителями, выбранными из С1-С6алкила и ОН; и
кольцо Су выбирают из C1-6алкил(С3-С8)циклоалкила, фенила и гетероарила, выбранного из пиридинила и тиенила;
где m равен 0 или 1;
R8 представляет собой F; или соединение, выбранное из группы:
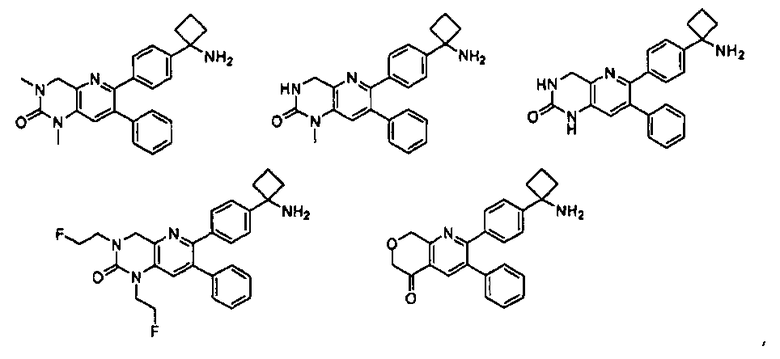
и его фармацевтически приемлемые соли, стереоизомеры и таутомеры.
2. Соединение по п. 1, где X отсутствует и W представляет собой СН2 или О.
3. Соединение по п. 1 или 2, где R6a и R6b вместе образуют  , то есть предпочтительно они образуют циклобутан.
, то есть предпочтительно они образуют циклобутан.
4. Соединение по п. 3, где группа, связанная с фенильным кольцом в структуре I, представляет собой 1-аминоциклобутил.
5. Соединение по любому из предшествующих пунктов, где связь между Y и Z представляет собой одинарную связь.
6. Соединение по п. 2, где Y представляет собой карбонил и Z представляет собой необязательно замещенный амино, или Z представляет собой карбонил и Y представляет собой необязательно замещенный амино.
7. Соединение по п. 6, где W представляет собой О и R1 и R2 предпочтительно представляют собой водород.
8. Соединение по п. 6 или 7, где если амино является замещенным, то предпочтительно он является метил- или ацетамидозамещенным.
9. Соединение по п. 1, где заместители, присоединенные к Y и Z, и собственно Y и Z вместе образуют 5- или 6-членное необязательно замещенное гетероарильное кольцо.
10. Соединение по п. 1, где заместители, присоединенные к Y и Z, и собственно Y и Z вместе образуют фрагмент, выбранный из группы:
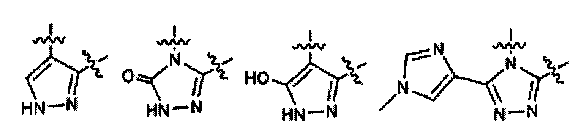 и
и  ,
,
где R представляет собой 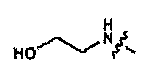 или
или  .
.
11. Соединение по п. 1, где Cy представляет собой фенил и m равен 0.
12. Соединение по п. 1, представляющее собой
2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6Н)-он,
2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6Н)-она оксим,
2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидрохинолин-5(6Н)-она О-метилоксим,
1-(4-(8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин,
1-(4-(2-метил-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин,
1-(4-(1-метил-8-фенил-4,5-дигидро-1Н-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин,
7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-3-ол,
7-(4-(1-аминоциклобутил)фенил)-3а-метил-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-3(3аН)-он,
1-(4-(3-(метилтио)-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин,
1-(4-(3-(метилсульфонил)-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин,
7-(4-(1-аминоциклобутил)фенил)-N-метил-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-3-амин,
2-((7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-3-ил)амино)этанол,
1-(4-(3-морфолино-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин,
7-(4-(1-аминоциклобутил)фенил)-2-метил-8-фенил-4,5-дигидро-3Н-имидазо[4,5-f]хинолин-3-ол,
1-(4-(2-метил-8-фенил-4,5-дигидро-3Н-имидазо[4,5-f]хинолин-7-ил)фенил)циклобутанамин,
1-(4-(9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-8-ил)фенил)циклобутанамин,
8-(4-(1-аминоциклобутил)фенил)-9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-2-амин,
2-(4-(1-аминоциклобутил)фенил)-7,7-диметил-3-фенил-7,8-дигидрохинолин-5(6Н)-он,
1-(4-(4,4-диметил-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин,
2-(4-(1-аминоциклобутил)фенил)-3-фенил-6,7,8,9-тетрагидро-5Н-циклогепта[b]пиридин-5-он,
1-(4-(9-фенил-2,4,5,6-тетрагидропиразоло[3′,4′:3,4]циклогепта[1,2-b]пиридин-8-ил)фенил)циклобутанамин,
2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6Н)-он,
2-(4-(1-аминоциклобутил)фенил)-6-метил-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6Н)-он,
2-(2-(4-(1-аминоциклобутил)фенил)-5-оксо-3-фенил-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил)ацетонитрил,
2-(4-(1-аминоциклобутил)фенил)-6-(2-(диметиламино)этил)-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6Н)-он,
8-(4-(1-аминоциклобутил)фенил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-3(2Н)-он,
1-(4-(6-этил-3-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)фенил)циклобутанамин,
1-(4-(9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутанамин,
1-(4-(3-(1-метил-1Н-имидазол-4-ил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутанамин,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(проп-2-ин-1-ил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
6-(4-(1-аминоциклобутил)фенил)-1-(2-(диметиламино)этил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетамид,
6-(4-(1-аминоциклобутил)фенил)-1-(2-оксипропил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
1-(4-(7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин,
7-(4-(1-аминоциклобутил)фенил)-8-фенил-2,4-дигидро-1Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он,
1-(4-(8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
1-(4-(1-метил-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин,
1-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)этанон,
1-((1Н-имидазол-2-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
2-(6-(4-(1-аминоциклобутил)фенил)-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетамид,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-4-илметил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-3-илметил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
1-(4-(1-этил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
6-(4-(1-аминоциклобутил)фенил)-1-(циклобутилметил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
этил-2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетат,
7-(4-(1-аминоциклобутил)фенил)-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-амин,
6-(4-(1-аминоциклобутил)фенил)-1-(2-метоксиэтил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-этил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
1-((1Н-имидазол-5-ил)метил)-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрил,
6-(4-(1-аминоциклобутил)фенил)-1-(циклопропилметил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-2-илметил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-изопропил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-циклопентил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(тиазол-4-илметил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-циклобутил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
3-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)пропаннитрил,
1-(4-(1-метил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
4-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)бутаннитрил,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
5-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)пентаннитрил,
1-аллил-6-(4-(1-аминоциклобутил)фенил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-(2-гидроксиэтил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
1-(4-(8-фенил-1-пропил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
1-(4-(1-(метилсульфонил)-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин,
6-(4-(1-аминоциклобутил)фенил)-3-(2-гидроксиэтил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-4-метил-7-фенил-3,4-дигидропиридо[2,3-b]пиразин-2(1Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1Н-пиридо[3,2-е][1,3,4]оксатиазин-2,2-диоксид,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-3-ил)-1Н-пиридо[3,2-е][1,3,4]оксатиазин-2,2-диоксид,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(пиридин-3-ил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-(1-аминоциклобутил)фенил)-2,2-диоксидо-7-фенил-1Н-пиридо[3,2-е][1,3,4]оксатиазин-1-ил)ацетонитрил
6-(4-(1-аминоциклобутил)фенил)-3-(2-гидроксиэтил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-(1-аминоциклобутил)фенил)-3-(2-гидроксиэтил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
6-(4-(1-аминоциклобутил)фенил)-3-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-(1-аминоциклобутил)фенил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
6-(4-(1-аминоциклобутил)фенил)-1-(метилсульфонилметил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-(1-метил-1Н-пиразол-4-ил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1,3-диметил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-(2,2-дифторэтил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-(2-фторэтил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1Н)-он,
6-(4-(1-аминоциклобутил)фенил)-N-этил-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-карбоксамид,
6-(4-(1-аминоциклобутил)фенил)-1,3-диметил-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-она О-метилоксим,
1-(4-(7-фенил-1-(пиридин-3-ил)-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-6-ил)фенил)циклобутанамин,
1-(4-(1-бром-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
(R)-2-(6-(4-(1-аминоциклобутил)фенил)-3-метил-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
2-(6-(4-(1-аминоциклобутил)фенил)-3-(метоксиметил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
6-(4-(1-аминоциклобутил)фенил)-3-(метоксиметил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
1-(4-(1-(дифторметил)-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
7-(4-(1-аминоциклобутил)фенил)-2-метил-8-фенил-2,4-дигидро-1Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он,
1-(4-(8-фенил-1-(трифторметил)-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
2-(7-(4-(1-аминоциклобутил)фенил)-1-оксо-8-фенил-1Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-2(4Н)-ил)ацетонитрил,
6-(4-(1-аминоциклобутил)фенил)-1-изобутил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
7-(4-(1-аминоциклобутил)фенил)-8-фенил-2-(2,2,2-трифторэтил)-2,4-дигидро-1Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он,
7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-1Н-пиридо[2,3-b][1,4]диазепин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-пропил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-(3-гидрокси-2-(гидроксиметил)-2-метилпропил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
((7-(4-(1-аминоциклобутил)фенил)-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-ил)фенил)метанол,
6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1,3-бис(2-фторэтил)-7-фенил-3,4-дигидропиридо[3,2-d]пиримидин-2(1Н)-он,
1-(4-(1-циклопропил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
7-(4-(1-аминоциклобутил)фенил)-1,5-диметил-8-фенил-4,5-дигидро-1Н-пиридо[2,3-b][1,4]диазепин-2(3Н)-он,
1-(4-(1-изопропил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
7-(4-(1-аминоциклобутил)фенил)-1-метил-8-фенил-4,5-дигидро-1Н-пиридо[2,3-b][1,4]диазепин-2(3Н)-он,
1-(4-(1-изобутил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
7-(4-(1-аминоциклобутил)фенил)-N,N-диметил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-амин,
7-(4-(1-аминоциклобутил)фенил)-1-(2-фторэтил)-8-фенил-4,5-дигидро-1Н-пиридо[2,3-b][1,4]диазепин-2(3Н)-он,
1-(4-(8-фенил-1-(2,2,2-трифторэтил)-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
7-(4-(1-аминоциклобутил)фенил)-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-карбоксамид,
1-(4-(1-(фторметил)-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
1-(4-(1-(2-метоксиэтил)-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
1-(4-(8-фенил-1-(пиридин-3-ил)-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d)[1,4]оксазин-7-ил)фенил)циклобутанамин,
1-(4-(8-фенил-1-(пиридин-4-ил)-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
6-(4-(1-аминоциклобутил)фенил)-1-метил-7-(пиридин-3-ил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-метил-7-(тиофен-3-ил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-метил-7-(тиофен-2-ил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(4-(1-аминоциклобутил)фенил)-3-фенил-6,8-дигидро-5Н-пирано[3,4-b]пиридин-5-он,
(1r,3r)-3-амино-3-(4-(1-(дифторметил)-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-1-метилциклобутанол,
1-(4-(1,8-дифенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
1-(4-(8-фенил-1-(пиридин-2-ил)-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
6-(4-(аминометил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-(4-фторфенил)-1-метил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-(циклопропилметил)-1-метил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-((1s,3s)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-(аминометил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
(1s,3s)-3-амино-3-(4-(1-этил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанол,
6-(4-((1r,3r)-1-амино-3-гидроксициклобутил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
(1r,3r)-3-амино-3-(4-(1-этил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-1-метилциклобутанол,
6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-1-этил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-1-(циклопропилметил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
7-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-8-фенил-2,4-дигидро-1Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он,
(4-(1-этил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)метанамин,
6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-этил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-(циклопропилметил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-(2-фторэтил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(аминометил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
6-(4-((1s,3s)-1-амино-3-гидроксициклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклопропил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
6-(4-(аминометил)фенил)-1-этил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(аминометил)фенил)-1-(циклопропилметил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
7-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-8-фенил-2,4-дигидро-1Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-1-он,
и его фармацевтически приемлемые соли, стереоизомеры и таутомеры.
13. Соединение по п. 1, представляющее собой
1-(4-(8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-7-ил)фенил)циклобутанамин,
7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-3-ол,
2-((7-(4-(1-аминоциклобутил)фенил)-8-фенил-4,5-дигидро-2Н-пиразоло[3,4-f]хинолин-3-ил)амино)этанол,
1-(4-(9-фенил-5,6-дигидропиридо[2,3-h]хиназолин-8-ил)фенил)-циклобутанамин,
2-(4-(1-аминоциклобутил)фенил)-3-фенил-7,8-дигидро-1,6-нафтиридин-5(6Н)-он,
8-(4-(1-аминоциклобутил)фенил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-3(2Н)-он,
1-(4-(3-(1-метил-1Н-имидазол-4-ил)-9-фенил-5,6-дигидро[1,2,4]триазоло[3,4-f][1,6]нафтиридин-8-ил)фенил)циклобутанамин,
6-(4-(1-аминоциклобутил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
и его фармацевтически приемлемые соли, стереоизомеры и таутомеры.
14. Соединение по п. 1, представляющее собой
2-(6-(4-(1-аминоциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
и его фармацевтически приемлемые соли, стереоизомеры и таутомеры.
15. Соединение по п. 1, представляющее собой
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(проп-2-ин-1-ил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
1-(4-(1-этил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
6-(4-(1-аминоциклобутил)фенил)-1-этил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-(циклопропилметил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-(1-аминоциклобутил)фенил)-1-(2-фторэтил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
1-(4-(1-(дифторметил)-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
1-(4-(1-(фторметил)-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)циклобутанамин,
6-(4-((1r,3r)-1-амино-3-гидроксициклобутил)фенил)-1-метил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
(1r,3r)-3-амино-3-(4-(1-этил-8-фенил-4Н-пиридо[2,3-b][1,2,4]триазоло[4,3-d][1,4]оксазин-7-ил)фенил)-1-метилциклобутанол,
6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-этил-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-(циклопропилметил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-1-(2-фторэтил)-7-фенил-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
2-(6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-2-оксо-7-фенил-2,3-дигидро-1Н-пиридо[2,3-b][1,4]оксазин-1-ил)ацетонитрил,
6-(4-((1r,3r)-1-амино-3-гидрокси-3-метилциклобутил)фенил)-7-фенил-1-(2,2,2-трифторэтил)-1Н-пиридо[2,3-b][1,4]оксазин-2(3Н)-он,
и его фармацевтически приемлемые соли, стереоизомеры и таутомеры.
16. Фармацевтическая композиция, ингибирующая АКТ активность, содержащая фармацевтический носитель и диспергированное в нем соединение по любому из пп. 1, 12, 13, 14 или 15 в эффективном количестве.
17. Соединение по любому из пп. 1, 12, 13, 14 или 15 для использования в лечении или предотвращении состояния, обусловленного АКТ активностью.
18. Соединение по любому из пп. 1, 12, 13, 14 или 15 для использования при лечении или профилактике злокачественной опухоли.
19. Способ лечения или профилактики злокачественной опухоли, включающий введение соединения по любому из пп. 1, 12, 13, 14 или 15 нуждающемуся в этом субъекту, необязательно вместе с лучевой терапией.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Wu Z | |||
| et al., Rapid assembly of diverse and potent allosteric Akt inhibitors, Bioorganic & Medicinal Chemistry Letters, 2008, vol.18, no.6, p.2211-2214 | |||
| ПРИБОР ДЛЯ ОПРЕДЕЛЕНИЯ КАЧЕСТВА МУКИ | 1926 |
|
SU12112A1 |
| ПРОИЗВОДНЫЕ 6-АРИЛПИРИДО[2,3-D]-ПИРИМИДИНЫ И -НАФТИРИДИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ КЛЕТОЧНОЙ ПРОЛИФЕРАЦИИ, ВЫЗЫВАЕМОЙ ПРОТЕИНОВОЙ ТИРОЗИНКИНАЗОЙ, И СПОСОБ ИНГИБИРОВАНИЯ КЛЕТОЧНОЙ ПРОЛИФЕРАЦИИ | 1995 |
|
RU2191188C2 |

