Настоящее изобретение относится к способу получения энантиомерно чистого миртазапина, включающему замыкание цикла кислотой.
Миртазапин, 1,2,3,4,10,14b-гексагидро-2-метилпиразино[2,1-а]пиридо[2,3-с][2]бензазепин представляет собой тетрациклическое соединение, имеющее формулу I:
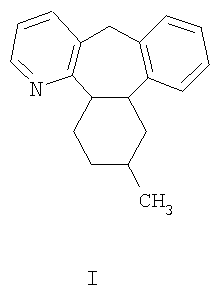
Соединение является хиральным, и рацемическая смесь находит широкое использование в качестве лекарственного средства для лечения депрессии. Сообщалось о других областях использования митразапина в медицине, например WO 99/25356 и WO 01/58453 описывают его использование при лечении расстройств сна и апноэ. Из исследований биологического действия энантиомеров миртазапина (например. О'Connor and Leonard, Neuropharmacology, 1986, vol. 25, pp. 267-270; Kooyman et al., 1994, vol. 33, pp. 501-507; De Boer et al., Neuropharmacology, 1988, vol. 27, pp. 399-408; Gower et al., 1986, vol. 291, pp. 185-201) следует, что данное соединение лучше использовать в его чистых энантиомерных формах, что говорит о необходимости эффективного получения значительных количеств энантиомерно чистого миртазапина. Настоящее изобретение предлагает улучшение в таком способе получения.
Из уровня техники известны многочисленные способы получения миртазапина. US 4062848 описывает варианты внутри четырехстадийной схемы синтеза, посредством чего можно осуществить синтез миртазапина, используя в качестве исходного вещества 2-замещенный никотинонитрил. Дальнейшие модификации различных стадий данного способа были впоследствии описаны в WO 00/62782, WO 01/23345 и US 6376668.
На получение энантиомерно чистого миртазапина были направлены US 4062848, WO 00/62782 и Selditz et al., 1998 (J. Chromatography, 1998, vol. 803, pp. 169-177). Способом, описанным в US 4062848, получают энантиомерно чистый миртазапин дробной кристаллизацией диастереоизомерных солей, полученных взаимодействием рацемического миртазапина с энантиомерно чистой дибензоилвинной кислотой в этаноле, с последующей регенерацией свободного основания обработкой водным аммиаком. Другие методы получения чистого миртазапина перекристаллизацией неочищенного миртазапина описаны в WO 00/62782. Selditz et al. описывают хроматографический метод разделения энантиомеров. В данных методах разделение осуществляется в конце синтеза, приводя к созданию рацемической смеси миртазапина. Поэтому следует, что общий выход каждого полученного энантиомерно чистого соединения является относительно низким и никогда не может превышать 50%. Было бы выгодно иметь более экономичный способ, в котором энантиомерно чистый миртазапин можно получить с общим улучшенным выходом.
Согласно способу, описанному в US 4062848, миртазапин можно получить в результате замыкания цикла соединения формулы (II),
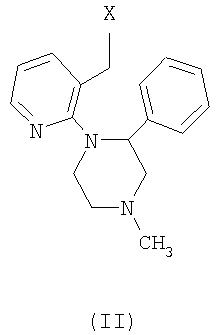
где Х может представлять уходящую группу, такую как гидроксильная группа, этерифицированная в виде простого или сложного эфира, гидроксильная группа или галоген, с использованием множества реагентов для замыкания цикла. Примеры таких реагентов включают кислоты, такие как серная кислота, концентрированная хлористоводородная кислота, пикриновая кислота, трифторуксусная кислота, фосфорная кислота, полифосфорная кислота (РРА), оксихлорид фосфора, триоксид фосфора, пентаоксид фосфора, и кислоты Льюиса, такие как хлорид алюминия, хлорид железа (3), хлорид цинка, хлорид олова, хлорид титана, трифторид бора, хлорид сурьмы (5) и хлорид циркония (4). В US 4062848 получение миртазапина иллюстрируется замыканием цикла с использованием концентрированной серной кислоты. В WO 00/62782 показано, что концентрированная серная кислота является наиболее предпочтительной. В US 4062848 сделано замечание, что чистые энантиомеры миртазапина можно получить синтетически, используя энантиомерно чистое исходное вещество для заключительной стадии замыкания цикла. Однако способ, описанный в WO 00/62782, с концентрированной серной кислотой недостаточно сохраняет оптическую чистоту. По-видимому, данные реакционные условия дают избыточную рацемизацию.
Было обнаружено, что при синтезе энантиомерно чистого миртазапина замыканием цикла энантиомерно чистого соединения формулы (II) стереохимическая чистота исходного материала все-таки может быть сохранена осуществлением специального выбора из вышеуказанных реагентов для замыкания цикла.
Поэтому настоящее изобретение предлагает способ, включающий стадию замыкания цикла соединения формулы (II), где Х представляет собой уходящую группу, причем указанная стадия включает обработку кислотой, где миртазапин с энантиомерным избытком получают замыканием цикла соединения формулы (II) с энантиомерным избытком обработкой подходящей кислотой в отсутствие растворителя или подходящей комбинацией кислоты и органического растворителя. Спирт формулы II, предпочтительно, используют в виде кристаллической соли или сольвата, например в виде оксалатной соли (S)-или (R)-1-(3-гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазина.
Необязательно к реакционной смеси добавляют дикалит, чтобы предотвратить образование крупных кусков.
Термин миртазапин используют в настоящем описании в его общем значении, обычно используемом для ссылки на химическое соединение в виде основания и, в зависимости от контекста, на его соли или сольваты, и дополняется префиксами (R) или (S), и/или (+) или (-) для энантиомеров соединения. Конфигурация (S) вызывает положительное оптическое вращение в обычных растворителях.
Термин энантиомерный избыток в соединении относится к разнице между количеством каждого из энантиомеров, присутствующих в смеси, относительно общего количества соединения в смеси, выраженного в процентах. Например, в 10 г смеси, содержащей 9 г миртазапина (90%), из которых 4 г составляет (R)-миртазапин и 5 г составляет (S)-миртазапин, энантиомерный избыток (S)-энантиомера равен примерно 11%. В сокращенном виде термин миртазапин или соединение с энантиомерным избытком относится к смеси, содержащей миртазапин или соединение с энантиомерным избытком.
Данное изобретение может предоставить энантиомерно чистый миртазапин, если используется энантиомерно чистое исходное вещество, и замыкание цикла осуществляют обработкой подходящей кислотой в отсутствие растворителя или подходящей комбинацией кислоты и органического растворителя.
Энантиомерно чистое соединение представляет собой соединение, включающее менее 20% другого энантиомера, что составляет энантиомерный избыток, равный 60%. В зависимости от конкретных условий заявляемого способа также можно получить энантиомерно чистое соединение, имеющее менее 10% другого энантиомера или менее 1% другого энантиомера. Выходы выделенного энантиомерно чистого миртазапина обычно составляют не менее 50%, но также можно получить выходы не менее 70%.
Уходящая группа представляет собой реакционноспособную функцию на молекуле, которая подвергается замещению в молекуле, когда образуется новая связь, как общеизвестно из уровня техники. Более конкретно - уходящая группа может представлять собой гидроксильную группу, ее активированный сложный эфир, такой как карбоксилат, сульфонат или фосфонат, или галоген. Группы с данной функцией общеизвестны из уровня техники, и список можно дополнительно расширить с помощью обычно имеющихся в распоряжении справочников по органическому синтезу.
Подходящая кислота для способа по настоящему изобретению представляет собой конкретную кислоту или комбинацию кислота/растворитель, как указано в дальнейшем, или кислоту или комбинацию кислота/растворитель, не указанные в дальнейшем, но которые получают, осуществляя тест в отношении пригодности кислоты. Тест состоит в осуществлении замыкания цикла с использованием кислоты, которая является кандидатом кислоты, и исходного вещества, которое представляет собой соединение II, как определено выше, или его соль или сольват, с предварительно определенной энантиомерной чистотой и определении после взаимодействия энантиомерного избытка полученного в результате миртазапина. Количественную степень потери энантиомерной чистоты можно определить простым вычислением и выразить в виде разницы между энантиомерным избытком в исходном веществе до взаимодействия и энантиомерным избытком продукта, представляющего собой миртазапин, после взаимодействия. Если потеря составляет менее 40%, кислота или комбинация кислота/растворитель является подходящей кислотой или комбинацией кислота/растворитель. Более строгий критерий для подходящей кислоты или комбинации кислота/растворитель можно использовать, выбирая соединения, вызывающие потерю менее чем в пределах от 0% до 40%, например 35%, 30%, 25%, 20%, 15%, 10%, 5%, 2%, 1%, 0/5% и 0,3%. Следовательно, один аспект данного изобретения состоит в предложении способа выбора кислоты или комбинации кислота/растворитель, подходящих для стереоспецифичного замыкания кольца, приводящего к энантиомерно чистому миртазапину. Способ включает осуществление реакции замыкания цикла энантиомерно чистого соединения формулы II, или его соли или сольвата, со значением X, как определено ранее, с кислотой или комбинацией кислота/растворитель, которая является кандидатом, определение потери энантиомерного избытка в ходе реакции и идентификацию кислоты и/или комбинации кислота/растворитель в качестве подходящей, если она приводит к потере, составляющей менее 40%. Необязательно, можно использовать более строгий критерий, как указано выше, для более подходящих кислот или комбинаций кислота/растворитель.
Подходящая кислота, используемая в отсутствие растворителя, может представлять собой протонную кислоту или производное протонной кислоты, такое как ангидрид протонной кислоты. Концентрированная серная кислота из способа получения рацемического миртазапина предшествующего уровня техники или треххлористый алюминий не подходят.
Для замыкания цикла, использующего подходящую кислоту в отсутствие растворителя, особенно предпочтительным является использование полифосфорной кислоты или пентаоксида фосфора в фосфорной кислоте. Рекомендуется использовать полифосфорную кислоту или пентаоксид фосфора в фосфорной кислоте в небольшом избытке по отношению к исходному спирту, определенному в виде соединения (II) выше. Взаимодействие будет давать лучший выход и лучшее сохранение энантиомерного избытка, если отношение (мас./мас.) полифосфорной кислоты к спирту (масса основания соединения II) составляет менее чем 10:1 (мас./мас.) или лучше 5:1, еще лучше менее 2,5:1. Когда полифосфорную кислоту вводят в виде пентаоксида фосфора в фосфорной кислоте (возможно, при массовом (мас./мас.) отношении пентаоксида фосфора к фосфорной кислоте от 1:1 до 1:9), навески пентаоксида фосфора и фосфорной кислоты суммируют, чтобы выразить в виде общего количества полифосфорной кислоты.
Подходящая комбинация кислоты и органического растворителя может представлять собой комбинацию протонной кислоты или производного протонной кислоты, такого как ангидрид протонной кислоты, или минеральной кислоты и полярного координирующего растворителя, такого как этанол или высшие спирты, ДМФА, диметиламин (ДМА) или N-метилпирролидинон. Более предпочтительно использовать комбинацию производного протонной кислоты и N-метилпирролидинона, или ДМФА. Особенно предпочтительными являются полифосфорная кислота и N-метилпирролидинон, или ДМФА.
Такие комбинации кислота/растворитель как пентаоксид фосфора или полифосфорная кислота, или серная кислота и ксилол; пентаоксид фосфора или полифосфорная кислота и хлорбензол; пентаоксид фосфора или полифосфорная кислота и толуол, и серная кислота, и дихлорметан не подходят.
В то время как реакция замыкания цикла может происходить при комнатной температуре, протеканию реакции также можно содействовать дополнительным нагреванием. Поэтому дополнительный аспект данного изобретения включает замыкание цикла способами по настоящему изобретению, включающими дополнительное нагревание.
Соединение формулы (II) можно получить синтетическим способом, показанным на схеме I, который описан в US 4062848.
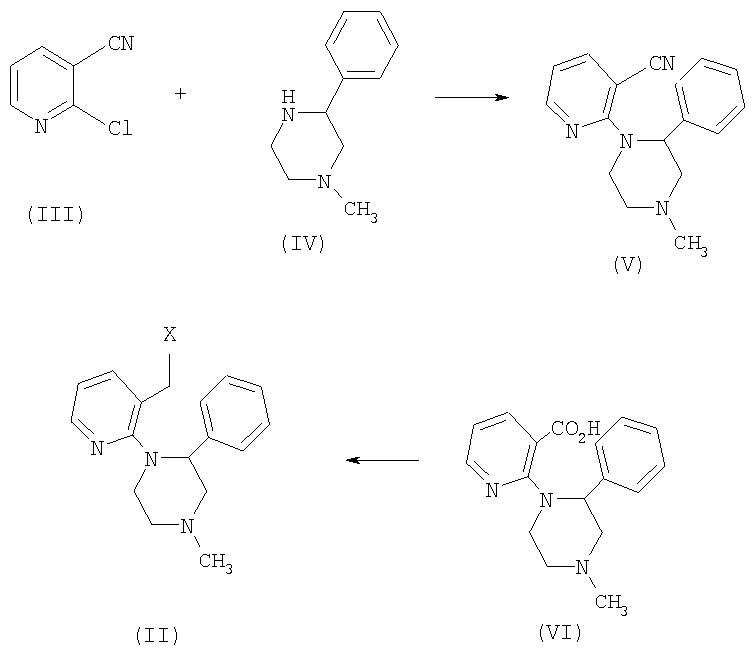
Схема 1
Таким образом, соединение (V) можно получить взаимодействием соединения (IV) с хлорникотинонитрилом (III) в органическом растворителе, таком как тетрагидрофуран или диметилформамид, и в присутствии эквивалента основания, такого как фторид калия. Затем гидролизом соединения (V) можно получить соединение (VI), используя водное основание, такое как гидроксид калия, в спирте, таком как этанол, обычно при температуре кипения. В WO 00/62782 описывается, что молярное отношение основания, используемого для осуществления гидролиза нитрила, можно понизить с 25 молей основания (как описывается в процедуре патента США 4062848) до примерно 12 молей основания. В конечном счете соединение (II) можно получить восстановлением соединения (VI), снова используя гидрид металла, такой как литийалюминий гидрид, в органическом растворителе, таком как тетрагидрофуран. Конверсию спиртовой функциональной группы в другие уходящие группы, такие как карбоксилат и сложные сульфонатные эфиры, и в галогены можно легко осуществить методами, которые хорошо известны из уровня техники.
Затем получения энантиомерно чистого соединения (II) можно достичь, используя способы, хорошо известные из уровня техники, например способы асимметричного синтеза, например синтез с хиральной индукцией, дробная кристаллизация диастереоизомерных солей, полученных при взаимодействии с хиральной кислотой, или разделение хроматографией на хиральной среде методами хроматографии с обычной фазой или обращенной фазой. Такие методы, например, описываются в "Chirality in Industry" (A.N.Collins/ G.N.Sheldrake and J.Crosby, Eds., 1992; John Wiley).
Изобретение также включает энантиомерно чистый миртазапин, полученный способом по настоящему изобретению, и его фармацевтические композиции для использования в терапии. Такие композиции могут включать терапевтически эффективное количество энантиомерно чистого миртазапина в комбинации с фармацевтически приемлемыми носителями и эксципиентами, которые хорошо известны из уровня техники.
Настоящее изобретение иллюстрируется следующими примерами:
Пример 1а
Получение [S]-миртазапина
(S)-1-(3-Гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазин (0,23 г, 1,03 ммоль) растворяют в N-метилпирролидиноне (10 мл). Полученный в результате раствор по каплям при перемешивании добавляют к полифосфорной кислоте (1/46 г) в N-метилпирролидоне (5 мл) при 81°С. Реакционную смесь перемешивают при 100°С в течение 72 часов. Затем ее разбавляют раствором гидроксида натрия и диэтиловым эфиром. Органический слой отделяют и дважды промывают водой. Добавляют сульфат магния, удаляют соль фильтрованием и фильтрат выпаривают. Получают указанное в заголовке соединение (0,19 г, 68%) в виде маслянистого продукта. Энантиомерный избыток (э.и.) продукта составляет 99,2%.
Пример 1b
К смеси полифосфорной кислоты (41,8 г) и N-метилпирролидина (10,5 мл) добавляют раствор (S)-1-(3-гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазина (7,02 г, 24,7 ммоль) в N-метилпирролидиноне (10 мл). Реакционную смесь нагревают при 130°С в течение 1 часа. К реакционной смеси добавляют воду (152 мл), дикалит (8,8 г), толуол (76 мл) и 33% раствор гидроксида натрия (128 мл). Водный слой отделяют и дважды экстрагируют толуолом (76 мл). Объединенные слои толуола три раза промывают водой (76 мл), сушат над MgSO4 и выпаривают. Данная процедура дает 4,63 г [S]-миртазапина (70%) с э.и. 99%.
Пример 2
Получение [S]-миртазапина
(S)-1-(3-Гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазин (0,30 г, 1,0587 ммоль) растворяют в 18,75 мл диметилформамида. К данному раствору добавляют 0,75 г дикалита и 1,5 г полифосфорной кислоты. Реакционную смесь перемешивают в течение одного дня при 100°С. Затем ее разбавляют гидроксидом натрия и экстрагируют диэтиловым эфиром. Органический слой дважды промывают водой, сушат над сульфатом магния, фильтруют, и фильтрат выпаривают. Получают указанное в заголовке соединение (0,19 г, 68%) в виде маслянистого продукта. Э.и. продукта составляет 99,2%.
Пример 3
Получение [S]-миртазапина
(S)-1-(3-Гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазин (0,50 г, 1,76 ммоль) растворяют в N-метилпирролидоне (7,5 мл) и нагревают до 100°С. К данной смеси добавляют дикалит (0,62 г) и пентаоксид фосфора (1,26 г). Через 66 часов реакция завершается. К реакционной смеси добавляют воду. Затем ее фильтруют. Доводят рН до 14, добавляя 4н раствор гидроксида натрия. Водный раствор экстрагируют диэтиловым эфиром. Органический слой сушат над сульфатом магния и выпаривают. Данная процедура дает указанное в заголовке соединение (0,24 г, 51%) с э.и. 99,7%.
Пример 4
Получение [S]-миртазапина
К (S)-1-(3-гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазину (0,5 г, 1,77 ммоль) добавляют полифосфорную кислоту (9,6 г). Реакционную смесь нагревают при 100°С в течение 20 часов. Реакционную смесь разбавляют водой (6,5 мл) и рН доводят до 8, добавляя 4н раствор гидроксида натрия. Водный слой экстрагируют этилацетатом. Органический слой промывают водой, сушат над сульфатом магния и выпаривают. Данная процедура дает указанное в заголовке соединение (0,29 г, 62%) с э.и. 76%.
Пример 5
К (S)-1-(3-гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазину (1,0 г, 3,53 ммоль) добавляют полифосфорную кислоту (2 г). Реакционную смесь нагревают при 130°С в течение 18 часов. Реакционную смесь разбавляют водой (6,5 мл) и рН доводят до 8, добавляя 4н раствор гидроксида натрия. Водный слой экстрагируют этилацетатом. Органический слой промывают водой, сушат над сульфатом магния и выпаривают. Данная процедура дает указанное в заголовке соединение (0,71 г, 76%) с э.и. 98%.
Пример 6
Раствор (S)-1-(3-гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазина (1,0 г, 3,53 ммоль) в дихлорметане добавляют к полифосфорной кислоте, полученной из 85% фосфорной кислоты (2,8 г) и пентаоксида фосфора (1,3 г). Реакционную смесь нагревают при 130°С в течение 18 часов. Реакционную смесь разбавляют водой (6,5 мл) и рН доводят до 8, добавляя 4н раствор гидроксида натрия. Водный слой экстрагируют этилацетатом. Органический слой промывают водой, сушат над сульфатом магния и выпаривают. Данная процедура дает указанное в заголовке соединение (0,79 г, 84%) с э.и. 83%.
Пример 7
К полифосфорной кислоте (20 грамм) добавляют оксалатную соль (S)-1-(3-гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазина (13,2 г, 35,3 ммоль). Реакционную смесь перемешивают при 130°С в течение 18 часов. К реакционной смеси добавляют воду (220 мл), этилацетат (220 мл) и 33% раствор гидроксида натрия (65 мл). Водный слой отделяют и дважды экстрагируют этилацетатом (220 мл). Объединенные органические фракции три раза промывают водой (220 мл) и выпаривают. Данная процедура дает 7,9 г [S]-миртазапина (84%) с э.и. 99,2%.
Пример 8
К полифосфорной кислоте (4 грамма) добавляют иксалатную соль (S)-1-(3-гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазина (1,32 г, 3,53 ммоль). Реакционную смесь перемешивают при 130°С в течение 18 часов. К реакционной смеси добавляют воду (22 мл), этилацетат (22 мл) и 33% раствор гидроксида натрия (6,5 мл). Водный слой отделяют и дважды экстрагируют этилацетатом (22 мл). Объединенные органические фракции три раза промывают водой (22 мл), сушат над MgSO4 и выпаривают. Данная процедура дает 0,79 г [3]-миртазапина (84%) с э.и. 83%.
Пример 9
Получение [S]-миртазапина
К серной кислоте (30,36 мл) при температуре 48°С добавляют раствор (S)-1-(3-гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазина (15,18 г, 51,05 ммоль) в этаноле (30 мл) После 1 ночи добавляют дополнительное количество серной кислоты (30 мл). Через 4 часа реакция завершается. Добавляют воду (195 мл), после чего следует добавление раствора гидроксида натрия (8,3 М) до образования осадка. Водный слой экстрагируют этилацетатом. Затем органический слой промывают раствором гидроксида натрия, затем раствором хлорида натрия, сушат над сульфатом магния и выпаривают. Данная процедура дает указанное в заголовке соединение (7,97 г, 59%) с э.и. 62%.
Пример 10
Дается с целью сравнения с неподходящей комбинацией кислота/растворитель.
Получение [S]-миртазапина
К (S)-1-(3-гидроксиметил-2-пиридил)-4-метил-2-фенилпиперазину (0,29 г, 1,03 ммоль) добавляют концентрированную серную кислоту (2,2 мл). Добавляют дихлорметан, получая прозрачный раствор. Дихлорметан выпаривают при пониженном давлении при 40°С. Реакционную смесь перемешивают при 48°С. Через 4 часа реакция завершается. Добавляют раствор гидроксида натрия (4 н), пока не образуется эмульсия. Водный слой экстрагируют диэтиловым эфиром. Диэтиловый эфир промывают водой, сушат над сульфатом магния и выпаривают. Данная процедура дает указанное в заголовке соединение (0,17 г, 62%) с э.и. 36%.
Изобретение относится к способу получения энантиомера миртазапина, содержащего менее 10% другого энантиомера, который включает реакцию замыкания цикла соединения формулы (II), где X представляет собой уходящую группу, причем указанная стадия включает обработку кислотой, посредством чего миртазапин с энантиомерным избытком получают замыканием цикла R- или S-энантиомера соединения формулы (II) обработкой полифосфорной кислотой в отсутствие растворителя или комбинацией полифосфорной кислоты и N-метилпирролидинона или ДМФА. Техническим результатом является стереохимическая чистота целевого продукта.
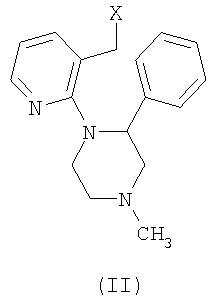
Способ получения энантиомерно чистого миртазапина, содержащего менее 10% другого энантиомера, причем указанный способ включает реакцию замыкания цикла соединения формулы (II)
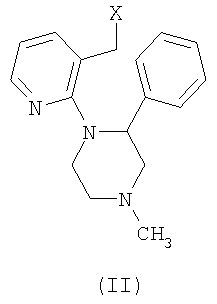
где X представляет собой уходящую группу, причем указанная реакция включает обработку кислотой, отличающийся тем, что миртазапин с энантиомерным избытком получают замыканием цикла R- или S-энантиомера соединения формулы (II) обработкой кислотой или комбинацией кислота/растворитель, выбранных из списка, включающего:
а) полифосфорную кислоту в отсутствии растворителя, причем массовое отношение полифосфорной кислоты к соединению формулы II составляет менее 2,5 к 1;
или
б) полифосфорную кислоту в присутствии N-метилпирролидинона или ДМФА;
или
с) пентоксид фосфора в присутствии N-метилпирролидинона.
| US 4062848 А, 13.12.1977 | |||
| WO 00/62782 А, 26.10.2000 | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| RU 2001128229 A, 2003.07.10. | |||

