Изобретение относится к промежуточным соединениям и к способу синтеза соединений, ингибирующих протеазу, кодированную вирусом иммунодефицита человека (ВИЧ), а в частности, к способу синтеза соединений L-735524 или их фармацевтически приемлемых солей. Эти соединения могут быть использованы для предупреждения инфицирования вирусом ВИЧ, лечения ВИЧ-инфекций и лечения вызываемого ВИЧ-инфекцией синдрома приобретенного иммунодефицита (СПИД).
Более конкретно, способ настоящего изобретения предусматривает проведение реакции аминового нуклеофила, такого как пиперазиновое производное, с активированным глицидиловым производным, таким как 2(S)-глицидилнозилат, с получением эпоксидного промежуточного соединения, которое может быть использовано для получения соединения, ингибирующего протеазу ВИЧ, включая соединение L-735524. Настоящее изобретение также относится к улучшенному способу синтеза специфических диалкиламинов, используемых в синтезе ингибиторов протеазы ВИЧ.
Ретровирус, называемый вирусом иммунодефицита человека (ВИЧ), является этиологическим фактором, вызывающим комплексное заболевание, характеризующееся прогрессирующей диструкцией иммунной системы (синдром приобретенного иммунодефицита; СПИД) и дегенерацией центральной и периферической нервной системы. Ранее этот вирус был известен как вирус, вызывающий лимфаденопатию (LAV), Т-лимфотропный вирус человека, тип III (HTLV-III), или ретровирус, вызывающий СПИД (ARV ). Общим признаком репликации ретровирусов является экстенсивный посттрансляционный процессинг полипротенов-предшественников с помощью протеазы, кодируемой вирусным геномом, в результате чего образуются зрелые вирусные белки, необходимые для сборки и функционирования вируса. Ингибирование описанного процессинга предотвращает продуцирование нормального инфекционного вируса. Так, например. Kohl N.E. и др. ( Proc. Natl. Acad. Sci. 85, 4686 (1988)) показали, что генетическая инактивация протеазы, кодируемой ВИЧ, приводит к продуцированию незрелых, неинфицированных вирусных частиц. Эти результаты свидетельствуют о том, что ингибирование ВИЧ-протеазы представляет собой практически осуществимый способ лечения СПИДа и предупреждения или лечения ВИЧ-инфекций.
Нуклеотидная последовательность ВИЧ имеет ген po1 в одной открытой рамке считывания (Patner L. et al., Nature, 313, 277 (1985)). Гомология аминокислотной последовательности свидетельствует о том, что последовательность po1 кодирует обратную транскриптазу, эндонуклеазу и ВИЧ-протеазу (Toh H. et al., , EMBO J. 4, 1267 (1985); Power M.D. et al., Science, 231, 1567 (1986); Pearl L.H. et al., Nature 329, 351 (1987) ).
Соединения конечных продуктов, включая соединение L -735524, которое показано ниже в Примере 11, являются ингибиторами ВИЧ-протеазы и могут быть получены из новых промежуточных соединений в соответствии со способами настоящего изобретения, причем указанные соединения раскрываются в EPO 541168, опубликованном 12 мая 1993 г.
Ранее синтез соединения L-735524 и родственных соединений осуществляли 12-ступенчатым методом, предусматривающим использование гидрокси-защищенного дигидро-5(S)-гидроксиметил-3(2H)-фуранона, который был алкилирован; и замену спиртовой уходящей группы на алкилированный фуранон с пиперидиновой частью. Затем продукт реакции присоединения гидролизовали для размыкания кольца с образованием гидрокси-кислотной группы, после чего, на конечном этапе, добавляли кислоту с получением 2(R)-гидрокси-1(S)-аминоиндана. Этот способ описан в EPO 541168. Этот чрезмерно продолжительный процесс (12 стадий) требует слишком много времени и является весьма трудоемким; при этом он требует использования множества дорогостоящих реагентов и дорогостоящего исходного продукта. А поэтому было бы желательно найти более экономически выгодный и менее трудоемкий способ, для которого потребовалось бы меньшее число реакционных стадий и меньшее количество реагентов.
Модифицированный способ получения L-735524 и родственных соединений был также описан в EPO 541168 и заключался в диастереоселективном алкилировании энолята, происходящего от N-(2(R)-гидрокси-1(S)-индан-H,O-изопропилиден-ил)-3-фенилпропанамида, где в качестве аллильной группы вводят C3-C5-трехуглеродный элемент, с последующим окислением. Этот способ имеет несколько недостатков, а именно: (a) для осуществления введения трехуглеродного глицидилового фрагмента требуются четыре стадии; (b) в этом способе используется высокотоксичный OSO4 и (c) в стадии гидроксилирования имеет место низкая диастереоселективность. Таким образом, желательно, чтобы в этом способе осуществлялось непосредственное введение трехуглеродного элемента в соответствующей хиральной окисленной форме.
Кроме того, синтез хирального пиперазинового промежуточного соединения был осуществлен из 2- пиразинкарбоновой кислоты 6-стадийным методом, который требовал использования таких дорогостоящих реагентов, как BOC-ON и EDC. В связи с этим очевидно, что было бы желательно найти более короткий путь к получению пиперазинового промежуточного соединения, который также не требовал бы использования дорогостоящих реагентов.
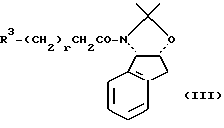
В литературе описано несколько примеров конденсации стабилизированных карбанионов с глицидолом и его производными (активированными или неактивированными); однако пока еще не описаны методы прямого продуцирования нового эпоксида с хорошим выходом. См., например, Hanson R.M. Chem. Rev., 1991, 91, 437-435. В случае активированных глицидоловых производных и углеродных нуклеофилов отсутствие таких методов обусловлено прежде всего преждевременным и нежелательным "двойным" присоединением нуклеофила к эпоксидному продукту. Конденсация стабилизированных карбанионов с активированными нерацимическими глицидоловыми производными показала, что анион малоната был связан с нерацемическим эпихлорогидрином I и нерацемическим глицидилтрифталатом II, в результате чего образовывался циклопропил-лактон III. См., например, Pirrung М. С. и др. Helvetica Chimica Acta 1989, 72, 1301- 1310, и Burgess K. и др., J. Org. Chem. 1992, 57, 5931-5936. В этом случае промежуточный эпоксид подвергается дальнейшей реакции с образованием циклопропиловой кольцевой системы. В случае I начальная реакция с анионом малоната происходит у эпоксидного конца (C3), тогда как в случае II начальная реакция происходит у C1-конца трифталата.
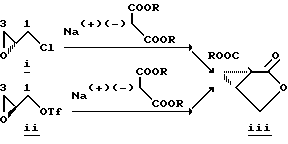
Аналогичным примером является реакция сульфон- стабилизированного карбаниона, полученного из соединения v, с глицидилтозилатом iv с образованием гидрокси-тозилата vi. См., например, Baldwin J.E. и др., J. Chem. Soc. Chem. Commun. 1992, 1249-1251. Хотя в этом случае "двойное" присоединение карбаниона не имеет большого значения, однако для превращения промежуточного соединения гидрокси-тозилата VI в нужный эпоксид VII необходимо проведение дополнительной стадии. - 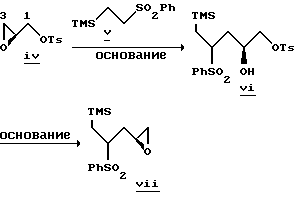
Аналогично, неизвестным в литературе и неожиданным является тот факт, что азотные нуклеофилы могут быть селективно присоединены к активированным глицидоловым производным с хорошим выходом.
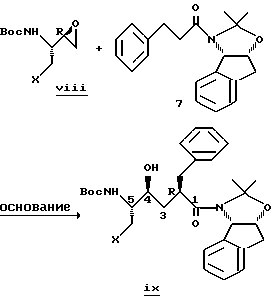
Специалистам также известна реакция конденсации энолятов, полученных из N-(2(R)-гидрокси-1(S)-индан-N, O-изопропилиден-ил)-3-фенилпропанамида 7, с защищенными альфа-аминоэпоксидами VIII, в результате которой получают нужные гидроксиэтилен-дипептидные изостерные промежуточные соединения IX с высокой степенью стерического контроля для C2-(R)-стереоцентра. См., например, Askin D. и др., J. Org. Chem., 1992, 57, 2771-2773, и Askin D. и др., патент США 5169952. После гидролиза получают деблокированные гидроксиэтилен-дипептидные изостерные ингибиторы.
Выделение 2-пиперазинкарбоновой кислоты с помощью (+) -CSA является известным методом. См. , например, Felder E. и др. Helvetica, Chim. Acta, 1960, 43, 888. Однако примеры выделения пиперазинамидов в литературе пока не описаны.
В настоящем изобретении раскрывается способ получения ингибиторов ВИЧ-протеазы, обладающий большими преимуществами, чем способы, описанные ранее. Этот способ занимает меньше времени, является в высокой степени диастереоселективным, позволяет осуществлять синтез соединений, раскрытых в EPO 541168, а в частности соединения L-735524, с более высоким выходом и не предусматривает использования токсичных реагентов, таких как тетраокись осмия, или дорогостоящих реагентов, таких как (S)-(+)-дигидро-5-(гидрокси-метил)-2(3H)-фуранон.
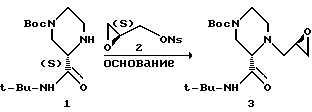
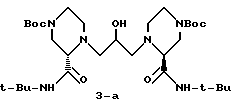
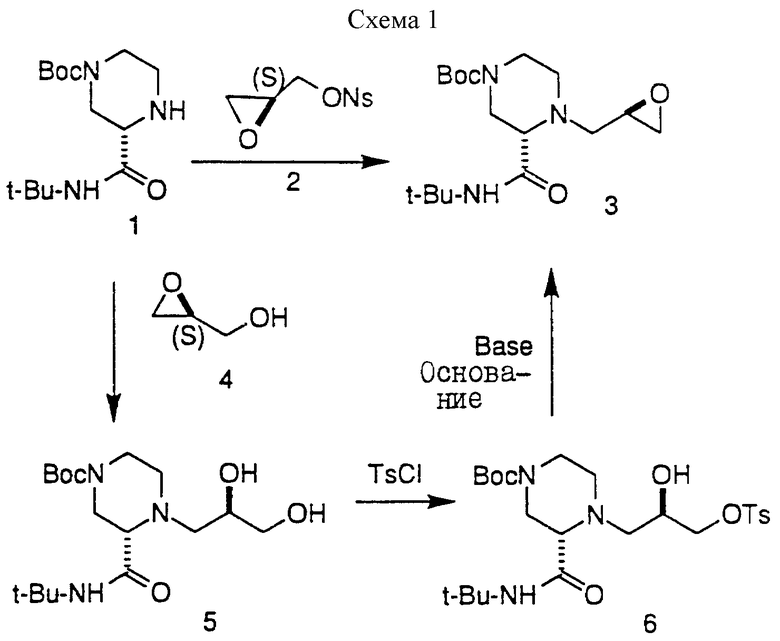
Настоящее изобретение относится к новым способам синтеза амино-эпоксидных промежуточных соединений, таких как 3, которые могут быть использованы для получения ингибиторов ВИЧ-протеазы. Один из вариантов настоящего изобретения предусматривает осуществление реакции аминового нуклеофила, такого как 1, с активированным глицидоловым производным, таким как 2(S)-глицидилнозилат 2, с получением эпоксидного продукта, такого как 3, с хорошим выходом. Результат этой реакции оказался неожиданным, поскольку предполагалось, что в результате дальнейшей реакции эпоксида 3 в условиях присоединения будет образовываться большое количество димерного продукта 3-а, что приведет тем самым к низкому выходу 3.
Другой вариант настоящего изобретения предусматривает осуществление реакции аминового нуклеофила с нерацемическим глицидолом с последующей обработкой тозилхлоридом (TSCI), а затем основанием и получение в результате аминоэпоксидного промежуточного соединения.
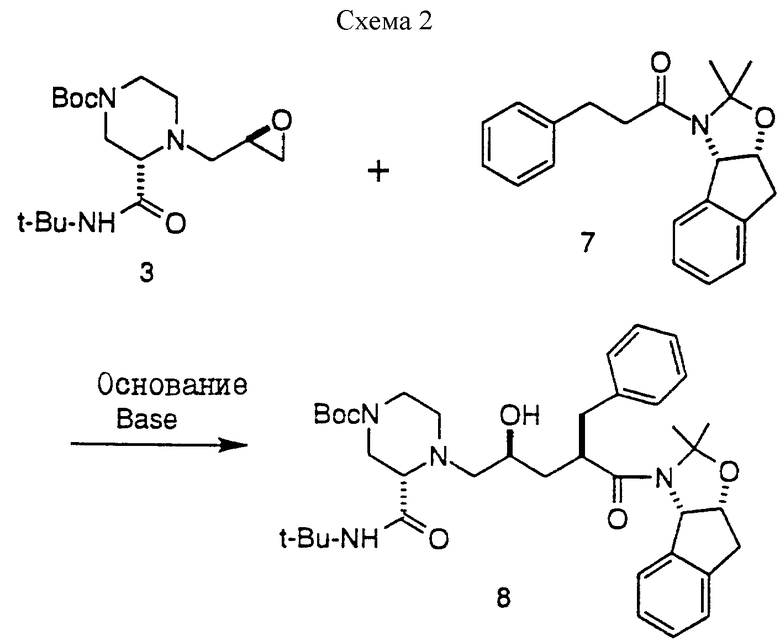
Следующий вариант настоящего изобретения предусматривает осуществление реакции амино-эпоксидного промежуточного соединения с амидом формулы VIII, определенным ниже, и получение промежуточных соединений формулы I, определенных ниже.
Еще один вариант настоящего изобретения относится к новым способам получения соединения I в нужной хиральной конфигурации. Этот способ может быть осуществлен с высокой степенью эффективности и высоким выходом путем обработки рацемического 2-трет-бутил-карбоксамидпиперизина (+)-камфорсульфоновой кислотой (CSA) или L-пироглутаминовой кислотой (PGA) с последующей кристаллизацией хирального продукта, который затем защищают ди-трет-бутилдикарбонатом (BOC2O). Альтернативно, соединение 1 получают кинетическими методами.
В настоящей заявке могут встретиться следующие сокращения:
Обозначение - Защитная группа
BOC (Boc) - т-бутилоксикарбонил
CBZ (Сbz) - бензилоксикарбонил (карбобензокси)
TBS (TBDMs) - т-бутил-диметилсилил
Обозначение - Активирующая группа
Ts или тозил, или тозилат - п-толуолсульфонил
Ns или нозил, или нозилат - 3-нитробензолсульфонил
Ts или трифлил, или трифлат - трифторометансульфонил
Ms или мезил, или мезилат - метансульфонил
Обозначение - Связующий реагент
BOP-реагент - бензотриазол-1-илокситрис (диметиламино) фосфония гексафторофосфат
BOP-C1 - хлорангидрид бис (2-оксо-3-оксазо-лидинил)фосфиновой кислоты
EDC - 1-этил-3-(3-диметиламинопропил)-карбодиимида гидрохлорид
Обозначение - Другие соединения
BOC-ON - 2-(трет-бутилкарбонилоксиимино)-2-фенилацетонитрил
(BOC)2(BOC2O или Boc2O) - ди-трет-бутилдикарбонат
n-Bu4N+F- - фторид тетрабутиламмония
n BuLi (n-Buli) - н-бутиллитий
(S)-CSA - (1S)-(+)-10-камфорсульфоновая кислота
DIEA или DIPEA - диизопропилэтиламин
DMAP - диметиламинопиридин
DME - диметоксиэтан
ДМФ (DMF) - диметилформамид
Et3N - триэтиламин
EtOAc - этилацетат
h - час (ы)
IPA - 2-пропанол
LDA - диизопропиламид лития
L-PGA - (L)-пироглутаминовая кислота
TFA - трифторуксусная кислота
ТГФ (THF) - тетрагидрофуран
ТСХ (TLC) - тонкослойная хроматография
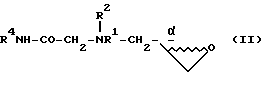
Настоящее изобретение относится к новому способу получения промежуточных соединений формул I и II, которые могут быть использованы для синтеза ингибиторов ВИЧ-протеазы, а в частности соединений, раскрытых в EPO 541168. Способ получения промежуточных соединений формулы I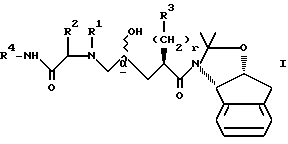
включает в себя следующие стадии:
(1) получение соединения формулы II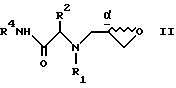
посредством реакции амина формулы III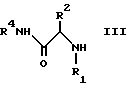
с простым эфиром: (а) глицидолом формулы IV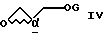
в присутствии основания
или (b) с глицидолом формулы V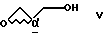
с получением соединения формулы VI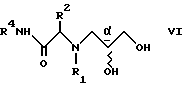
и последующей обработкой соединения VI активирующим агентом, выбранным из группы, состоящей из п-толуолсульфонилхлорида (также известного как тозилхлорид или TSCl), метансульфонилхлорида (также известного как мезилхлорид или MsCl), ангидрида трифторометансульфоновой кислоты (также известного как ангидрид трифторометансульфоновой кислоты или Tf2O и PBr3 c получением соединения VII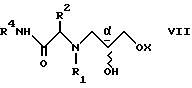
и обработкой этого соединения VII сильным основанием с получением соединения II;
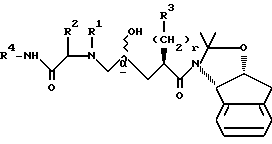
(2) осуществление реакции соединения II с амидом формулы VIII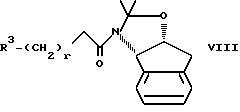
в присутствии сильного основания при низкой температуре, где стереоцентр 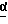 находится либо в R-конфигурации, либо в S-конфигурации, либо является рацемическим;
находится либо в R-конфигурации, либо в S-конфигурации, либо является рацемическим;
r является целым числом от 0 до 5 включительно;
G представляет собой группу, выбранную из 3-нитробензолсульфонила и трифторометансульфонила;
X представляет собой группу, выбранную из п-толуолсульфонила, метансульфонила и трифторометансульфонила;
R1 и R2 в каждом случае независимо выбирают из группы, включающей в себя:
1) водород;
2) C1-4-алкил, незамещенный или замещенный одним или несколькими из следующих заместителей:
a) гидрокси,
b) C1-3-алкокси,
c) арил, незамещенный или замещенный одним или несколькими заместителями, выбранными из C1-4-алкила, гидрокси или арила;
d) -W-арил или -W-бензил, где W является -O- или - S-;
e) 5-7-членная циклоалкильная группа, незамещенная или замещенная одним или несколькими из следующих заместителей:
i) гидрокси
ii) C1-3-алкокси или
iii) арил,
f) гетероцикл, незамещенный или замещенный одним или несколькими заместителями, выбранными из гидрокси: C1-4-алкила, C1-4-алкила, замещенного гидрокси-группой или Boc,
g) -NH-COOC1-3-алкил,
h) -NH-CO-C1-3-алкил,
i) -NH-SO2C1-3-алкил,
j) -COOR или
k) -((CH2)mO)nR, или
3) арил, незамещенный или замещенный одним или несколькими из следующих заместителей:
a) галоген,
b) гидрокси,
с) -NO2 или -N(R)2,
d) C1-4-алкил,
е) C1-3-алкокси-группа, незамещенная или замещенная одним или обоими из - OH и C1-3-алкокси,
f) -COOR,
g) -CON(R)2,
h) -CH2N(R)2,
i) - CH2NHCOR,
j) -CN,
k) -CF3
1) -NHCOR,
m) арил C1-3-алкокси,
n) арил,
o) -NRSO2R,
p) -OP(O)(ORx)2 или
q) -R5, определенный выше; или
R1 и R2, взятые вместе с атомами азота, с которыми связан R1, и атомом углерода, с которым связан R2, образуют 3-10-членную моноциклическую или бициклическую насыщенную кольцевую систему, состоящую из атома азота, с которым связан R1, и из 2-9 атомов углерода, например, такую как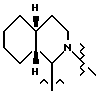
и незамещенную или замещенную одним или несколькими заместителями, такими как
1) гидрокси,
2) C1-4-алкил, незамещенный или замещенный одним или несколькими заместителями, такими как
а) галоген,
b) гидрокси,
с) C1-3-алкокси,
d) арил,
е) циклоалкильная группа с 5-7 членами, незамещенная или замещенная одним или несколькими заместителями, такими как
i) гидрокси,
ii) C1-3-алкокси или
iii) арил, или
f) гетероцикл;
3) C1-3-алкокси,
4) -NH-COOC1-3-алкил,
5) -NH-CO-C1-3-алкил,
6) -NH-SO2C1-3-алкил,
7) гетероцикл,
8) -W-арил или
9) -W-CO-арил,
где W определен выше; или
R1 и R2, взятые вместе с атомами азота, с которым связан R1, и с атомом углерода, с которым связан R2, образуют 3-10-членную моноциклическую или бициклическую насыщенную кольцевую систему, состоящую из атома азота, с которым связан R1, из 1-8 атомов углерода и из одного или нескольких незамещенных или замещенных гетероатомов, выбранных из
1) 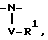
где V отсутствует или является -CO-Q-, или -SO2-Q-R1 определен выше для случая, когда R1 является независимым от R2 и не связан с ним; и где Q либо отсутствует, либо является -O-, -NR- или гетероциклом, необязательно замещенным -C1-4-алкилом;
2) 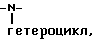
3) 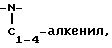 незамещенный или замещенный арилом,
незамещенный или замещенный арилом,
4) 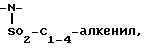 незамещенный или замещенный арилом,
незамещенный или замещенный арилом,
5) -S(O)p-,
где p=0, 1 или 2, или
6) -O-,
например, такую как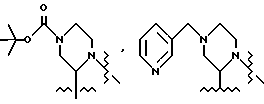
или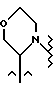
R3 выбирают из группы, включающей в себя
1) водород,
2) -C1-4-алкил,
3) C5-C10-циклоалкил, необязательно замещенный гидрокси-группой,
4) C6-C10-арил, незамещенный или замещенный одним или несколькими заместителями, такими как
a) галоген,
b) гидрокси,
с) -NO2 или -N(R)2,
d) C1-4-алкил,
е) C1-3-алкокси-группа, незамещенная или замещенная одним или двумя из -OH или C1-3-алкокси,
f) -COOR,
g) -CON(R)2,
h) -CH2N(R)2,
i) -CH2NHCOR,
j) -CN,
k) -CF3,
1) -NHCOR,
m) арил C1-3-алкокси,
n) арил,
о) -NRSO2R,
p) -OP(O)(ORx)2 или
q) -R5, определенный ниже; или
5) моноциклический или бициклический гетероцикл, который содержит от 1 до 3 гетероатомов, выбранных из группы, состоящей из N, O и S, например, такой как 2-пиридил, 3-пиридил или 4-пиридил; и который является незамещенным или замещенным R5 и необязательно одним или несколькими заместителями, такими как
а) галоген,
b) C1-4-алкил или
с) C1-3-алкокси;
m равно 2, 3, 4 или 5;
n равно 0, 1, 2 или 3;
R представляет собой водород или C1-4-алкил;
Rx представляет собой H или арил;
R4 представляет собой C1-5-алкил с прямой или разветвленной цепью и
R5 представляет собой:
1) -W-(CH2)m-NR6R7, где W и m является такими, как они были определены выше, а R6 и R7 в каждом случае независимо выбирают из:
а) водорода,
b) C1-6-алкила, незамещенного или замещенного одним или несколькими заместителями из:
i) C1-3-алкокси,
ii) -OH или
iii) -N(R)2;
с) ароматического гетероцикла, незамещенного или замещенного одним или несколькими заместителями из:
i) C1-4-алкила или
ii) -N(R)2;
d) либо R6 и R7, взятые вместе с атомом азота, с которым они связаны, образуют гетероцикл с 5-7 членами, такой как морфолино, содержащий до 2 дополнительных гетероатомов, выбранных из -N(R), -O-, - S-, -S(O)- или -S(O)2-; причем указанный гетероцикл необязательно замещен C1-4-алкилом,
2) -(CH2)q-NR6R7, где q является целым числом от 1 до 5, a R6 и R7 являются такими, как они были определены выше, за исключением того, что R6 или R7 не являются H или незамещенным C1-6-алкилом; или
3) бензофурил, индолил, азациклоалкил, азабицикло C7-11-циклоалкил, или бензопиперидинил, незамещенный или замещенный C1-4-алкилом.
Этот способ проиллюстрирован нижеприведенными Схемами 1 и 2. Однако при этом следует отметить, что этот способ не ограничивается конкретными заместителями, используемыми в указанных схемах, которые приводятся лишь в иллюстративных целях.
Для активации глицидола IV может быть использована подходящая группа, например такая, как 3-нитробензолсульфонил или трифторометансульфонил, а предпочтительно 3-нитробензолсульфонил. Для проведения реакции соединения III с соединением IV может быть использован любой подходящий полярный растворитель, например, такой как диметилформамид (ДМФ), N-метилпирролидинон, ацетон, ацетонитрил, трет-бутиловый спирт, трет-амиловый спирт, 2-пропанол, N-этилпирролидинон, 1,1,3,3-тетраметилмочевина, диметилсульфоксид, 1,3-диметил-3,4, 5,6-тетрагидро-2(1H)-пиримидинон, тетраметилсульфон, тетрагидрофуран (ТГФ), 1,4-диоксан, пиридин и вода, а также их комбинации. При этом предпочтительными полярными растворителями являются ДМФ, N-метилпирролидинон, ацетон, 2-бутанон и ацетонитрил; а наиболее предпочтительным является ДМФ. Для осуществления реакции соединения III с соединением IV может быть использовано любое подходящее основание, например такое, как диизопропилэтиламин (DIEA), карбонат калия, карбонат натрия, бикарбонат натрия, триэтиламин, пиридин и диметиланилин; при этом предпочтительными являются DIEA и карбонат калия, а наиболее предпочтительным является DIEA. В этой реакционной стадии предпочтительно использовать молярные эквиваленты III:IV = 1:1. Реакцию осуществляют предпочтительно при повышенной температуре, например при температуре от около 50oC до 70oC, а наиболее предпочтительно от около 60oC до 65oC.
Реакцию глицидола V с амином III осуществляют при молярном соотношении 1 - 3 эквивалента V на один моль II, а предпочтительно при молярном соотношении около 1,5 : IV : III, в соответствующем растворителе. Подходящими растворителями являются, например, углеводороды; простые эфиры; такие как диэтиловый эфир; спирты, такие как метанол, этанол или изопропанол; нитрилы, такие как ацетонитрил; и сложные эфиры, такие как этилацетат; или их комбинации, при этом предпочтительными растворителями являются спирты. Эта реакция может быть проведена при температуре в пределах от комнатной температуры до температуры перегонки используемого растворителя, а предпочтительно при повышенных температурах. Наиболее предпочтительно, если реакция протекает примерно при 85oC, а растворителем является изопропанол.
Активацию гидрокси-группы соединения VI с получением соединения VII осуществляют с использованием стандартной техники, известной специалистам. В этих целях могут быть использованы от около 1 до 3 молярных эквивалентов TsCl, MsCl или Ts2O на один молярный эквивалент VI, а предпочтительно, если отношение молярных эквивалентов активирующего агента и VI составляло 1,5:1 соответственно. Для этой стадии предпочтительно, чтобы X представлял собой п-толуолсульфонил, а поэтому предпочтительно использовать TsCl. Реакцию проводят предпочтительно при комнатной температуре, т.е. при 25oC, но могут быть использованы и более низкие или более высокие температуры, например в пределах от -20 до 80oC. В этой стадии может быть использован любой подходящий растворитель, известный специалистам, например такой, как углеводороды, простые эфиры, нитрилы, сложные эфиры, и амины, такие как пиридин, или их комбинации. Наиболее предпочтительным растворителем является пиридин. При этом предпочтительно, если не используется спиртовой растворитель. И наконец, активированное промежуточное соединение VII обрабатывают сильным основанием предпочтительно при комнатной температуре и получают в результате соединение II; причем эта реакция может быть осуществлена при более низких и при более высоких температурах, например в пределах от 0oC до температуры перегонки растворителя. Подходящими сильными основаниями являются NaH, KOC(CH3)3, KOC(CH3)2CH2CH3, NaOC(CH3)2CH2CH3, диизопропиламид лития (LDA), н-бутиллитий (n-BuLi), бис(триметилсилил)амид лития или аналогичные сильные основания, известные специалистам, при этом предпочтительным основанием является NaH. В этой реакции обычно используют около 1 - 3 молярных эквивалента основания на один молярный эквивалент соединения VII, при этом предпочтительное отношение молярных эквивалентов основания и VII составляет 1,5:1 соответственно. В этой стадии могут быть использованы любые подходящие растворители, например такие, как углеводороды, простые эфиры, нитрилы, сложные эфиры, или их комбинации, при этом предпочтительным растворителем является ТГФ. Кроме того, предпочтительно не использовать спиртовой растворитель.
Реакцию взаимодействия промежуточных соединений VIII и II осуществляют с использованием сильного основания в эфирном растворителе. Это сильное основание должно быть металлсодержащим основанием. Кроме того, это сильное основание может присутствовать, а может и не присутствовать в инертном безводном органическом растворителе, таком как циклические или ациклические углеводороды, включая гексан, пентан, циклогексан и т.п. Подходящими сильными основаниями являются н-бутиллитий (n-BuLi), S-BuLi, t-BuLi, диизопропиламид лития (LDA), изопропилциклогексиламид лития, пирролид лития, тетраметилпиперид лития, фениллитий, хлорид изопропилмагния, хлорид изобутилмагния и другие аналогичные сильные основания, хорошо известные специалистам. Предпочтительными сильными основаниями являются n-BuLi, S-BuLi и LDA, а наиболее предпочтительным основанием является n-BuLi. В этой реакции может быть использовано около 1-2 эквивалентов основания на один молярный эквивалент соединения VII, предпочтительно 1,05-1,2, а наиболее предпочтительно 1,15 молярных эквивалентов основания на один молярный эквивалент соединения VII. Реакция соединения VIII с соединением II может быть осуществлена путем объединения VIII и II в одном сосуде с последующим добавлением сильного основания либо она может быть осуществлена последовательно, т.е., амин VIII может быть сначала обработан основанием, а затем добавлено соединение II.
Сильное основание осуществляет металлирование амида VIII в положении альфа по отношению к карбонильной группе, в результате чего образуется реактивный амидоенолят металла, который затем размыкает кольцо эпоксида II в концевом положении, образуя соединение I. В изостерном продукте 1 новый центр асимметрии создается во 2-положении.
Эта реакция протекает предпочтительно при низкой температуре, например в пределах приблизительно -82oC - 0oC. Для осуществления металлирования амида VIII температуру предпочтительно поддерживать в пределах приблизительно от -82oC до -40oC, а наиболее предпочтительно примерно от -50oC до -45oC. Для осуществления реакции между металлированным амидным производным VIII и глицидоловым производным II с образованием соединения I предпочтительно поддерживать температуру в пределах приблизительно от -50oC до -10oC, а наиболее предпочтительно в пределах приблизительно от -30oC до -20oC, в течение около 4 - 5 часов, хотя продолжительность может варьироваться в зависимости от масштаба реакции и других факторов, хорошо известных специалистам.
В качестве эфирных растворителей могут быть использованы любые растворители, подходящие для такой реакции, например тетрагидрофуран (ТГФ), 1,2- диметоксиэтан, диэтиловый эфир и метил-трет-бутиловый эфир, при этом предпочтительным растворителем является ТГФ.
Активированные глицидолы формулы IV могут быть получены известными методами, например методами, описанными J. Klunder и др. в J.Org. Chem., 1989, 54, 1295-1304, и в работах, цитируемых в указанной работе.
Соединения формулы VII могут быть получены в соответствии со стандартными способами, хорошо известными специалистам, например способом, описанным в Примере 1, с использованием соответствующих исходных соединений.
В целях настоящего изобретения, там, где это необходимо, могут быть использованы защитные группы, такие как азотзащитные группы. Например, азот в 4-положении 2-трет-бутилкарбоксамидпиперазина может быть защищен такой группой, как BOC, CBZ, бензил, 4-метокси-бензил, 2,4- диметоксибензил, трифтороацетамид, триалкилсилил, или другими группами, хорошо известными специалистам.
Конечные продукты, а именно ингибиторы ВИЧ-протеазы, получают из соединений формулы I путем удаления любых оставшихся защитных групп в соответствии с методами деблокирования, хорошо известными специалистам. Например, кетальная защитная группа может быть удалена путем обработки соединения I кислотой в присутствии метанола или водной кислотой, или 1 н. HCl в ТГФ с получением конечных продуктов ингибиторов ВИЧ-протеазы. Соединения формулы I могут быть также, кроме того, замещены методами, известными специалистам.
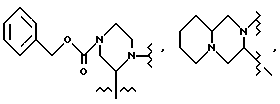
В одном из вариантов настоящего изобретения стереоцентр 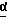 имеет S-конфигурацию; r = 1; G представляет собой 3-нитробензолсульфонил; X представляет собой п-толуолсульфонил; R1 и R2, взятые вместе, образуют циклическую структуру, выбранную из группы, состоящей из
имеет S-конфигурацию; r = 1; G представляет собой 3-нитробензолсульфонил; X представляет собой п-толуолсульфонил; R1 и R2, взятые вместе, образуют циклическую структуру, выбранную из группы, состоящей из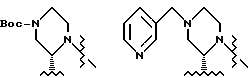
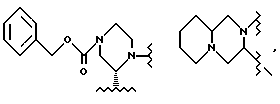

R3 выбирают из фенила,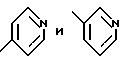
а R4 представляет собой трет-бутил.
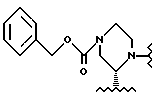
В этом варианте предпочтительным типом соединений формулы II является промежуточное соединение формулы IIa: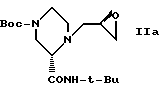
Кроме того, в этом варианте предпочтительным типом соединений формулы I являются промежуточные соединения формул I-a и I-b: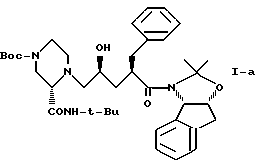
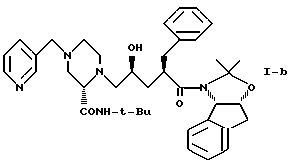
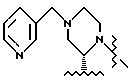
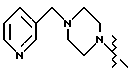
Соединение I-b может быть получено путем непосредственного взаимодействия метилпиридилового соединения II-а с соответствующими соединениями формулы VII. Конечный продукт L-735524 получают предпочтительно путем деблокирования и пиколилирования соединения I-а, как описано в Примерах 10 и 11.
В другом варианте настоящего изобретения предусматривается получение хирального промежуточного соединения (S)-2-трет-бутилкарбоксамида-4-трет- бутоксикарбонилпиперазина I. Соединение I получают путем обработки солевой формулы (S)-2-трет-бутилкарбоксамидопиперазина основанием, а затем Boc2O. При этом помимо Boc для защиты азота могут быть также использованы и другие группы, такие как CBZ, бензил и т.п.
Получение и выделение солевой формы (S)-2-трет- бутилкарбоксамидпиперазина осуществляли новым способом, включающим в себя следующие стадии:
(a) контактирование раствора (S) (R)-2- трет-бутилкарбоксамидпиперазина с кислотой в водно-органической смеси растворителей, где кислоту выбирают из группы, состоящей из (+) или (-) винной кислоты, (+) или (-) миндальной кислоты, (+) или (-) дибензоилвинной кислоты, D- или L-пироглутаминовой кислоты (также известной как (+) или (-) 2-пирролидин-5-карбоновая кислота), (+) или (-) ди-O,O'-п-толуол-винной кислоты, (+) или (-) яблочной кислоты, (+) или (-)-10-камфорсульфоновой кислоты, (+) или (-)-3-бромо-10- камфорсульфоновой кислоты и (-) или (-)-хлоро-10- камфорсульфоновой кислоты;
(b) нагревание смеси для растворения любых образующихся твердых веществ;
(с) охлаждение смеси;
(d) выделение осажденных кристаллов из маточного раствора и,
(е) если маточный раствор состоит преимущественно из (S)-антипода, то удаление из этого раствора растворителя.
Органическими растворителями, подходящими для данного способа выделения, являются несмешивающиеся с водой растворители, такие как ТГФ, 1,4-диоксан, ацетонитрил, ДМФ, 1-метил-2-пирролидинон, диметоксиэтан, этилацетат, C1-4-спирты, такие как метанол, этанол, 1-пропанол, изопропанол, н-бутанол и втор-бутанол, и их комбинации. Предпочтительным органическим растворителем является C1-4-спирт или смесь C1-4-спирта и ацетонитрила, а более предпочтительно, если спирт выбирают из 1-пропанола и этанола. Хотя количество воды в водно-органической смеси растворителей может варьироваться, однако предпочтительно, если процент объема воды в смеси составляет 15% или менее, а более предпочтительно 5% или менее.
В этом процессе предпочтительно использовать около 1-3 молярных эквивалента кислоты на один молярный эквивалент рацемического пиперазинового производного; причем это количество является предпочтительным для образования соли бис-кислоты. Предпочтительными кислотами являются (1S)-(+)-10-камфорсульфоновая кислота и (L)-пироглутаминовая кислота, а наиболее предпочтительной является (L)- пироглутаминовая кислота.
Температуры при осуществлении стадий (b) и (с) могут варьироваться в зависимости от используемых методов, хорошо известных специалистам. В основном для стадии (b) необходима температура, достаточная для растворения любых твердых веществ, и для этих целей может быть использована температура в пределах от около 70oC до температуры перегонки используемого растворителя. Нагретый раствор оставляют для медленного охлаждения, предпочтительно для естественного охлаждения до температуры окружающей среды, а затем он может быть дополнительно охлажден примерно до 20 - 23oC для стадии (с). Необязательно в этот раствор может быть внесена затравка в виде соответствующей соли либо (S), либо (R)-2-трет-бутилкарбоксамидопиперазина для стимуляции кристаллизации.
После выделения кристаллического осадка из маточного раствора можно определить, содержит ли данный осадок преимущественно R- или S-антипод, используя при этом стандартную технику, известную специалистам, например хиральный ВРЖХ-анализ. Термин "преимущественно" означает энантиомерный избыток (э. и.), составляющий 90% или более. S-антипод может быть затем выделен из осадка или из маточного раствора соответственно. Например, когда получают соль CSA, то L-энантиомер кристаллизуется из раствора, а когда образуется соль L-PGA, то S-энантиомер остается в маточном растворе, а R-энантиомер выпадает в осадок в виде кристаллов.
Способы и промежуточные соединения настоящего изобретения предназначены для получения конечных продуктов, которые могут быть использованы для ингибирования ВИЧ-протеазы, для предупреждения или лечения инфекций, вызываемых вирусом иммунодефицита человека (ВИЧ), и для лечения последующих патологических состояний, таких как СПИД. В настоящей заявке лечение СПИДа либо предупреждение или лечение ВИЧ-инфекций рассматривается как частный случай и не ограничивает более широкий спектр возможных состояний, вызываемых ВИЧ-инфекцией, а именно СПИД, ARC (СПИД-ассоциированный комплекс), как симптоматический, так и бессимптомный, а также фактическая или потенциальная угроза заражения вирусом ВИЧ. Например, конечные продукты, которые могут быть получены с помощью способов и промежуточных соединений настоящего изобретения, являются подходящими для лечения подозреваемых ВИЧ-инфекций, которые могли быть вызваны попаданием в организм ВИЧ, например, при переливании крови, трансплантации органов, обмена физиологических жидкостей, укусах, случайном уколе иглой или во время хирургической операции.
Конечные продукты, ингибирующие ВИЧ-протеазу, могут быть также использованы для получения и проведения скринирующих анализов на противовирусные соединения. Например, эти конечные продукты могут быть использованы для выделения ферментных мутантов, которые являются прекрасным инструментом для поиска более сильных противовирусных соединений. Кроме того, эти продукты могут быть использованы для установления или определения сайтов связывания других противовирусных агентов с ВИЧ-протеазой, например, путем конкурентного ингибирования. Таким образом, конечные продукты, которые могут быть получены с использованием методов и промежуточных соединений настоящего изобретения, являются коммерческими продуктами, предназначенными для продажи в целях использования как указано выше.
Соединения-ингибиторы ВИЧ-протеазы, которые могут быть получены с использованием методов и промежуточных соединений настоящего изобретения, раскрываются в EPO 541164. Соединения, ингибирующие ВИЧ-протеазу, могут быть введены пациентам, нуждающимся в таком лечении, в виде фармацевтических композиций, содержащих фармацевтически приемлемый носитель и терапевтически эффективные количества данного соединения или его фармацевтически приемлемой соли. B EPO 541164 раскрываются подходящие фармацевтические композиции, способы их введения, а также солевые формы и дозы соединений-ингибиторов.
Соединения настоящего изобретения могут иметь асимметрические центры и присутствовать в виде рацематов, рацемических смесей, а также в виде отдельных диастереомеров или энантиомеров, причем все указанные формы входят в объем настоящего изобретения.
Если в какой-либо составляющей части или в формулах I-VIII одновременно присутствует более чем одно переменное (например, арил, гетероцикл, R, R1, R2, n, X и т.п.), то его определение в каждом случае не зависит от определения других переменных. Кроме того, комбинации заместителей и/или переменных являются допустимыми только в том случае, если такие комбинации приводят к получению стабильных соединений. Если это не оговорено особо, то используемый в настоящем описании термин "алкил" означает насыщенные алифатические углеводородные группы с разветвленной или прямой цепью, имеющие определенное число атомов углерода (Me - метил, Et - этил, Pr - пропил, Bu - бутил, t-Bu - трет-бутил); термин "алкокси" означает алкильную группу с указанным числом атомов углерода, связанных посредством кислородного мостика; а термин "циклоалкил" означает насыщенные кольцевые группы, такие как циклопропил, циклобутил, циклопентил, циклогексил (Cyh) и циклогептил. Термин "алкенил" означает углеводородные группы прямой или разветвленной конфигурации с одной или несколькими углерод-углеродными двойными связями, которые могут присутствовать в любой стабильной точке цепи, например такие, как этенил, пропенил, бутенил, пентенил и т.п. Термин "алкинил" означает углеводородные группы прямой или разветвленной конфигурации с одной или несколькими углерод-углеродными тройными связями, которые могут присутствовать в любой стабильной точке цепи, например такие, как этинил, пропинил, бутинил, пентинил и т.п. Термин "галоген", используемый в настоящем описании, означает фтор, хлор, бром и йод. Используемый в настоящем описании термин "арил" означает фенил (Ph) или нафтил.
Используемые в настоящем описании термины "гетероцикл" или "гетероциклический" относятся, если это не оговорено особо, к стабильной 5 - 7-членной моно- или бициклической или 7-10-членной бициклической гетероциклической кольцевой системе, в которой любое кольцо может быть насыщенным или ненасыщенным и которая состоит из атомов углерода и 1-3 гетероатомов, выбранных из N, O и S, где гетероатомы азота и серы могут быть, но необязательно, окислены, в гетероатом азота может быть, но необязательно, кватернизирован; причем указанная система включает в себя бициклическую группу, в которой любое из вышеуказанных гетероциклических колец конденсировано с бензольным кольцом. Гетероциклическое кольцо может быть присоединено в любом гетероатоме или атоме углерода, который способствует образованию стабильной структуры. Примерами таких гетероциклических элементов являются пиперидинил, пиперазинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролодинил, 2-оксоазепинил, азепинил, пирролил, 4-пиперидонил, пирролидинил, пиразолил, пиразолидинил, имидазолил, имидазолинил, имидазолидинил, пиридил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, хинуклидинил, изотиазолидинил, индолил, хинолинил, изохинолинил, бензимидазолил, тиадиазоил, бензопиранил, бензотиазолил, бензоксазолил, фурил, тетрагидрофурил, тетрагидропиранил, тиенил, бензотиенил, тиаморфолинил, тиаморфолинилсульфоксид, тиаморфолинилсульфон и оксадиазолил. Морфолино - это то же самое, что и морфолинил.
Ниже подробно описана типичная методика экспериментов с использованием нового способа. Эта методика описана лишь в качестве иллюстрации метода настоящего изобретения и не должна рассматриваться как некое ограничение изобретения.
Пример 1. Получение амида 7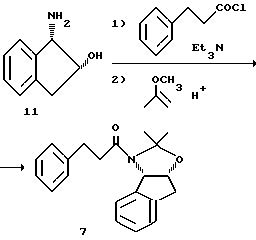
Раствор (-)-цис-1-аминоиндан-2-ола II (884 г, 5,93 М) в 17,8 л сухого тетрагидрофурана (KF = 55 мг/мл) и триэтиламина (868 мл, 6,22 М) в 50-литровой круглодонной колбе, снабженной термопарой, механической мешалкой, устройством для впуска азота и барботером, охлаждали до температуры 15oC. Затем в течение 75 минут к раствору добавляли 3-фенилпропионилхлорид (1000 г, 5,93 М), поддерживая при этом внутреннюю температуру 14-24oC с помощью охлаждающей бани из льда/воды. После завершения добавления смесь выдерживали в течение 30 минут при температуре 18-20oC, а затем анализировали с помощью ВРЖХ-анализа на исчезновение соединения II.
Ход реакции прослеживали с помощью жидкостной хроматографии высокого разрешения (ВРЖХ-анализ): 25-смколонка Dupont C8-RX; ацетонитрил/10 мМ (КH2PO4/К2HPO4) (60: 40) при 1,0 мл/мин.; объем впрыска 20 мл; детекция 200 нм; приготовление образцов 500 X-разведение. Приблизительное время удерживания: 4,1 мин гидроксиамид; 6,3 мин - цис-аминоинданол.
Реакционную смесь обрабатывали пара-толуолсульфонатом пиридиния (241 г, 0,96 М, 0,16 экв.) и перемешивали в течение 10 минут (pH смеси после разведения 1 мл образца равным объемом воды составлял 4,3-4,6). После добавления 2-метоксипропена (1,27 л, 13,24 М, 2,2 эквивалента) реакционную смесь нагревали до 38-40oC в течение 2 часов. После этого реакционную смесь охлаждали до температуры 20oC и распределяли между этилацетатом (12 л) и 5%-ным водным раствором бикарбоната натрия (10 л). Полученную смесь перемешивали, а слои отделяли. Этилацетатный экстракт промывали 5%-ным водным раствором бикарбоната натрия (10 л) и водой (4 л). Затем этилацетатный экстракт осушали путем дистилляции при атмосферном давлении, а растворитель меняли на циклогексан (полный объем около 30 л). По окончании дистилляции и концентрирования (20%-ный объем от объема экстракции этилацетатом) горячий циклогексановый раствор медленно охлаждали до 25oC для кристаллизации продукта. Затем суспензию охлаждали до температуры 10oC и выдерживали в течение одного часа. Продукт выделяли путем фильтрации, а сырой осадок на фильтре промывали холодным (10oC) циклогексаном (2 х 800 мл). Промытый осадок на фильтре осушали в вакууме (26 мм рт.ст.) при температуре 40oC и получали 1,65 кг ацетонида 7 (86,4%, 98% площадь (ВРЖХ-анализ)). 1H-ЯМР (300,13 МГц, CDCI3, главный ротамер) δ: 7,36-7,14 (м, 9H), 5,03 (д, J=44, 1H), 4,66 (м, 1H), 3,15 (м, 2H), 3,06 (шир. с., 2H), 2,97 (м, 2H), 1,62 (с, 3H), 1,37 (с, 3H); 13C-ЯМР (75,5 МГц, CDCl3, главный ротамер) δC: 168,8, 140,9, 140,8, 140,6, 128,6, 128,5, 128,4, 127,1, 126,3, 125,8, 124,1, 96,5, 78,6, 65,9, 38,4, 36,2, 31,9, 26,5, 24,1. Анализ для C11H23NO2: Вычислено: C 78,47; H 7,21; N 4,36; Найдено: C 78,65; H 7,24; N 4,40.
Пример 2. Пиразин-2-трет-бутилкарбоксамид 13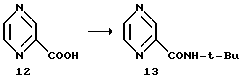
2-Пиразинкарбоновая кислота, кг - 3,35 (27 М)
Оксалилхлорид, кг - 3,46 (27,2 М)
трет-Бутиламин (КF = 460 мкг/мл), л - 9,36 (89 М)
EtOAc (КF = 56 мкг/мл), л - 27
ДМФ, мл - 120
1-Пропанол, л - 30
Карбоновую кислоту 12 суспендировали в 27 л этилацетата и 120 мл ДМФ в 72-литровой трехгорловой колбе, снабженной механической мешалкой, в атмосфере азота, а затем суспензию охлаждали до температуры 2oC. К полученной суспензии добавляли оксалилхлорид, поддерживая при этом температуру 5-8oC.
Добавление завершали через 5 часов. В процессе экзотермического добавления выделялся CO и CO2. Образовавшаяся соляная кислота присутствовала в растворе в большом количестве. Присутствующим осадком, вероятно, являлась гидрохлоридная соль хлорангидрида пиразина. Анализ на образование хлорангидрида осуществляли путем гашения безводного образца этой реакции с использованием т-бутиламина. После завершения реакции оставалось < 0,7% кислоты 12.
Анализ на завершение образования хлорангидрида является важным, поскольку незавершенная реакция приводит к образованию бис-трет-бутилоксамидных примесей.
Ход реакции может быть прослежен с помощью ВРЖХ: 25-см колонка Zorbax Dupont RXC8 при скорости потока 1 мл/мин и детекции при 250 нм; линейный градиент: от 98% 0,1% водного H3PO4 и 2% CH3CN до 50% водного H3PO4 и 50% CH3CN при 30 мин. Время удерживания: кислота 12 = 10,7 минут, амид 13 = 28,1 мин.
Реакционную смесь выдерживали в течение одного часа при температуре 5oC. Полученную суспензию охлаждали до температуры 0oC и добавляли трет-бутиламин, так чтобы внутренняя температура поддерживалась ниже 20oC.
Поскольку реакция была очень экзотермичной, то для добавления требовалось 6 часов. Небольшая часть образованного гидрохлорида трет-бутиламмония выпадала из реакции в виде хлопьеобразного белого твердого вещества.
После этого смесь выдерживали при температуре 18oC в течение еще 30 минут. Осажденные соли аммония удаляли путем фильтрации. Образовавшийся осадок на фильтре промывали 12 литрами этилацетата. Объединенные органические фазы промывали 6 литрами 3%-ного бикарбоната натрия и насыщенным водным раствором хлорида натрия (2 х 2 л). Органическую фазу обрабатывали 200 граммами углерода G60 Darco, фильтровали через Solka Floc и образовавшийся осадок на фильтре промывали 4 литрами этилацетата. Обработка углеродом эффективно удаляла слабый пурпурный оттенок продукта.
Этилацетатный раствор 13 концентрировали при 10 мбар до 25% первоначального объема. После добавления 30 л 1-пропанола дистилляцию продолжали до получения конечного объема 20 л.
На этой стадии EtOAc был ниже границы детекции при 1H-ЯМР (< 1%). Внутренняя температура при замене этого растворителя составляла <30oC. Раствор 1- пропанола/этилацетата (13) был стабильным до температуры перегонки при атмосферном давлении в течение нескольких дней.
В результате выпаривания аликвот получали твердое вещество коричневатого цвета, т. пл. 87-88oC; 13C-ЯМР (75 МГц, CDCl3, м.д.): 161,8, 146,8, 145,0, 143,8, 142,1, 51,0, 28,5.
Пример 3. рац.-2-трет-Бутил-карбоксамид-пиперазин 14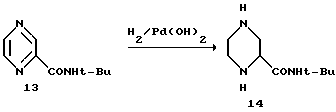
Материалы:
Пиразин-2-трет-бутилкарбоксамид 13 (2,4 кг, 13,4 М) в 1-пропаноловом растворе 12 л 20% Pd(OH)2/C и 144 г 16 мас.% воды.
Раствор пиразин-2-трет-бутилкарбоксамида 13 в 1-пропаноле помещали в автоклав емкостью 5 галлонов (≈18,9 млтров (америк.) После добавления катализатора смесь гидрировали под давлением водорода 40 фунт/кв.дюйм (2,812 кг/см2) (3 атмосферы) при температуре 65oC.
Через 24 ч реакции было израсходовано теоретическое количество водорода, а газовая хроматография (ГХ) указывала на присутствие < 1% соединения 13. Затем смесь охлаждали, продували азотом, а катализатор удаляли путем фильтрации через Solka Floc. После этого катализатор промывали 2 литрами теплого 1-пропанола.
Было обнаружено, что использование теплого 1- пропанола на стадии промывания осадка на фильтре улучшает фильтрацию и снижает потери продукта на этом осадке.
Ход реакции прослеживали с помощью газовой хроматографии (ГХ): 30-м колонка Megabore от 100oC до 160oC при 10oC/мин., выдерживание 5 минут, а затем при 10oC/мин до 250oC; время удерживания: соединение 13 = 7,0 мин, соединение 14 = 9,4 мин. Ход реакции может быть также прослежен с помощью тонкослойной хроматографии (ТСХ) с использованием в качестве растворителя EtOAc/MeOH (50:50), а в качестве проявляющего растворителя нингидрина.
Выпаривание аликвот указывало на то, что выход после амидирования и гидрирования составлял 88%, а концентрация соединения 14 составляла 133 г/л.
После выпаривания аликвот было получено соединение 14 в виде белого твердого вещества, т.пл. 150-151oC; 13C-ЯМР (75 МГц, D2O, млн.д.): 173,5, 59,8, 52,0, 48,7, 45,0, 44,8, 28,7.
Пример 4. Соль (S)-2-трет-бутил-карбоксамид-пиперазин-бис(S) - камфарсульфоновой кислоты (S)-15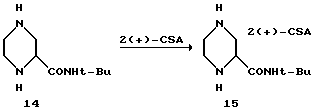
Материалы:
Раствор рацемич. - 2-трет-бутил-карбоксамид-пиперазина 14 в 1-пропаноле (в 25,5 кг растворителя), кг - 14,10 (22,12 M)
(S)-(+)-10-Камфарсульфоновая кислота, кг - 10,0 (43,2 M)
1-Пропанол, л - 12
Ацетонитрил, л - 39
Вода, л - 2,4
Раствор амина 14 в 1-пропаноле загружали в 100-литровую колбу, снабженную концентратором периодического действия. Полученный раствор концентрировали при 10 мбар (104 Па) и температуре < 25oC до объема, приблизительно равного 12 л.
На этой стадии продукт осаждался из раствора, но снова поступал в раствор, когда смесь нагревалась до температуры 50oC.
Анализ гомогенных аликвот указывал на то, что концентрация амина 14 составляла 341 г/л. Концентрацию определяли с помощью ВРЖХ: 25-см колонка RXC8 Zorbax Dupont при скорости потока 1,5 мл/мин и детекции при 210 нм; изократическая смесь (98/2) CH3CN/0,1% водного H3PO4; время удерживания амина 14: 2,5 минут.
После добавления ацетонитрила (39 л) и воды (2,4 л) получали прозрачный, слегка коричневатый раствор.
Определение содержания воды путем титрования Карла Фишера и отношения CH3CN/1-пропанола путем 1H-ЯМР-суммирования показало, что отношение CH3CN/1-пропанол/H2O составляло 26/8/1,6. Концентрация в растворе составляла 72,2 г/л.
После этого в течение 30 минут четырьмя порциями загружали (S)-10-камфарсульфоновую кислоту при 20oC. После добавления CSA температуру повышали до 40oC. Через несколько минут образовывался густой белый осадок. Эту белую суспензию нагревали до температуры 76oC для растворения всех твердых веществ, а затем слегка коричневатый раствор оставляли на 8 часов для охлаждения до 21oC.
Полученный продукт осаждали при температуре 62oC. Затем продукт фильтровали без отстаивания при температуре 21oC, а образовавшийся осадок на фильтре промывали 5 литрами смеси растворителей из CH3CN/1-пропанола/H2O (26/8/1,6). После осушки при температуре 35oC в вакуумной печи с продувкой азотом получали 5,6 кг (39%) соединения 15 в виде белого кристаллического твердого вещества с т. пл. 288-290oC (с разложением). [α]
Энантиомерный избыток (э. и.) вещества составлял 95%, в соответствии с нижеследующим хиральным ВРЖХ-анализом: аликвоту соединения 15 (33 мг) суспендировали в 4 мл EtOH и 1 мл триэтиламина. После добавления Boc2O (11 мг), реакционную смесь выдерживали в течение одного часа. Растворитель полностью удаляли в вакууме, а остаток растворяли приблизительно в 1 мл этилацетата и фильтровали через Пастеровскую пипетку с двуокисью кремния, используя в качестве элюента этилацетат. Выпаренный продукт разделяли на фракции и снова растворяли в гексане (≈1 мг/мл). Энантиомеры отделяли на колонке Daicel Chiracell AS, используя систему растворителей из гексана/1PA (97:3), при скорости потока 1 мл/мин и детекции при 228 нм. Время удерживания: энантиомер S=7,4 мин, R = 9,7 мин.
Пример 5. (S)-2-трет-Бутилкарбоксамид-4-трет-бутилоксикарбонил-пиперазин 1 из соли 15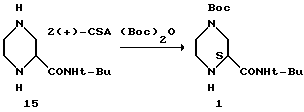
Материалы:
(S)-2-трет-Бутил-карбоксамид-пиперазина бис (S)-(+)-CSA-соль 15, 95% э. и., кг - 5,54 (8,53 М)
ди-трет-Бутилкарбонат, кг - 1,86 (8,53 М)
Lacamas Et3N, л - 5,95 (42,6 М)
Aldrich
-EtOH (абсолютный этиловый спирт), л - 55
EtOAc, л - 2
К соли (S)-камфарсульфоновой кислоты (22) в 100-литровой трехгорлой колбе, снабженной капельной воронкой, в атмосфере азота добавляли EtOH, а затем триэтиламин при температуре 25oC. Твердое вещество растворяли сразу после добавления триэтиламина. Boc3O растворяли в этилацетате и загружали в капельную воронку. Затем добавляли раствор Boc2O в EtOAc таким образом, чтобы температура поддерживалась ниже 25oC. Добавление проводили в течение 3 часов. После завершения добавления раствора Boc2O реакционную смесь выдерживали в течение 1 часа.
Ход реакции может быть прослежен с помощью ВРЖХ: 25-см колонка PXC8 Zorbax Dupont при скорости потока 1 мл/мин и детекции при 228 нм; изократическая смесь (50/50) CH3CN/0,1 М KH2PO4, доведенная до pH 6,8 путем добавления гидроксида натрия. Время удерживания соединения 1 = 7,2 мин. Хиральный анализ осуществляли с использованием той же самой системы, описанной в предыдущей стадии. Ход этой реакции также может быть прослежен с помощью ТСХ с использованием в качестве растворителя 100% этилацетата (Rf=0,7).
Раствор концентрировали приблизительно до 10 литров при внутренней температуре < 20oC в концентраторе периодического действия в вакууме при 10 мбар (104 Па). Переключение растворителя осуществляли путем медленного продувания в 20 литрах этилацетата и повторного концентрирования приблизительно до 10 литров. Реакционную смесь промывали в экстракторе 60 литрами этилацетата. Органическую фазу промывали 5% водным раствором Na2CO3 (16 л), дистиллированной водой (2х10 л) и насыщенным водным раствором хлорида натрия (2х6 л). Объединенные водные промывки подвергали обратному экстрагированию 20 литрами этилацетата, а органическую фазу промывали водой (2х3 л) и насыщенным водным раствором хлорида натрия (2х4 л). Объединенные этилацетатные экстракты концентрировали в вакууме при 10 мбар (104 Па) приблизительно до 8 литров в 100-литровом концентраторе периодического действия с внутренней температурой <20oC. Растворитель переключали на циклогексан путем медленного продувания приблизительно в 20 л циклогексана и повторного концентрирования приблизительно до 8 л. К суспензии добавляли 5 л циклогексана и 280 мл этилацетата, и эту смесь нагревали до температуры перегонки, когда все вещества были растворены в растворе. Полученный раствор охлаждали, а затем добавляли затравку (10 г) при 58oC. Эту суспензию охлаждали до 22oC в течение 4 часов, и продукт выделяли путем фильтрации после выдерживания в течение 1 часа при температуре 22oC. Осадок на фильтре промывали 1,8 литрами циклогексана и осушали в вакуумной печи при температуре 35oC, используя продувку азотом, в результате чего получали 1,87 кг (77%, > 99,9% площадь (ВРЖХ-анализ), R-изомер ниже уровня детекции) соединения I в виде слегка коричневатого порошка. [α]
Пример 6. (S)-2-трет-Бутил-карбоксамид-пиперазин-бис (L)-пироглутаминовая кислота 16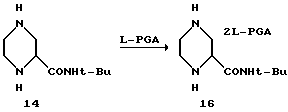
Материалы;
рац-2-трет-Бутил-карбоксамид-пиперазин 14 в 1-пропаноле, мл - 155, анализ=21,1 г (0,11 М)
L-пироглутаминовая кислота, г - 28 (0,21 М)
Вода, мл - 5
Раствор рацемич.2-трет-бутил-карбоксамид-пиперазина 14 в 1-пропаноле загружали в 500-миллилитровую круглодонную колбу, снабженную обратным холодильником, механической мешалкой и устройством для впуска азота. После добавления воды вместе с L-пироглутаминовой кислотой полученную суспензию нагревали до температуры перегонки. Гомогенный желтый раствор охлаждали до 50oC и в качестве затравки добавляли кристаллы соли бис-(L)-пироглутаминовой кислоты R-амина (50 мг). После этого сразу начиналось образование твердых веществ. Раствор охлаждали до 25oC и выдерживали в течение 16 часов. Твердые вещества фильтровали при температуре 22oC, а осадок на фильтре промывали 35 миллилитрами холодного 1-пропанола/-1%-ной воды. Затем осадок на фильтре осушали при температуре 35oC в вакуумной печи с продувкой азотом, в результате чего получали 23,74 г (48%) (R)-2-трет-бутил-карбоксамид-пиперазин-бис (L)-пироглутаминовой кислоты. Энантиомерный избыток вещества составлял 98% в соответствии с хиральным ВРЖХ-анализом, описанным выше. Желтые маточные растворы содержали 22,6 г (46%) соли (S)-2-трет-бутил-карбоксамид-пиперазин-бис (L)-пироглутаминовой кислоты 16, а энантиомерный избыток составлял 95% в соответствии с хиральным ВРЖХ-анализом. После этого маточные растворы выпаривали и использовали непосредственно в стадии защиты.
(S)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонил- пиперазин 1 из соли (S)-2-трет-бутил-карбоксамид-пиперазин- бис-(L)-пироглутаминовой кислоты 16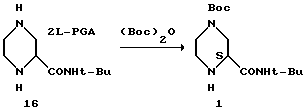
Материалы:
Соль (S)-2-трет-бутил-карбоксамид-пиперазин-бис (L)-пироглутаминовой кислоты, 95% э.и., г - 22,6 (50,1 мМ)
Ди-трет-бутилдикарбонат, г - 11,1 (50,1 мМ)
Et3N, мл - 35,5 (0,254 М)
1-Пропанол, мл - 226
EtOAc, мл - 24
К соли (S)-2-трет-бутил-карбоксамид-пиперазин-бис-(L)- пироглутаминовой кислоты в 500-миллилитровую трехгорловую колбу, снабженную капельной воронкой, в атмосфере азота добавляли 1-пропанол. Камедеобразное желтое твердое вещество растворялось сразу после добавления триэтиламина. Раствор Boc2O в EtOAc добавляли в течение 2 часов при температуре 22oC. После завершения добавления реакционную смесь выдерживали в течение одного часа.
Ход реакции может быть прослежен с помощью ВРЖХ (высокоразрешающей жидкостной хроматографии) и ТСХ (тонкослойной хроматографии) с использованием той же самой методики, описанной в способе превращения соединения 15 в соединение I.
После этого раствор концентрировали, а растворитель переключали на этилацетат (200 мл). Реакционную смесь промывали 7% водным раствором Na2CO3 (50 мл) и водой (2 х 30 мл), а затем осушали сульфатом натрия и фильтровали. Этилацетатный раствор концентрировали, а растворитель переключали на циклогексан (60 мл). После добавления этилацетата (1 мл) смесь нагревали до температуры перегонки для растворения всех твердых веществ. Полученную смесь охлаждали и добавляли затравку (50 мг) при температуре 52oC. Образовавшуюся суспензию охлаждали до 22oC в течение 2 часов, и после выдерживания в течение одного часа при температуре 22oC продукт выделяли путем фильтрации. Осадок на фильтре промывали 8 миллилитрами циклогексана и осушали в вакуумной печи при температуре 35oC, продувая азотом, в результате чего получали 10,8 г (74%, > 99,9% площадь (ВРЖХ-анализ), R-изомер ниже уровня детекции) соединение I в виде беловатого порошка.
Пример 7. 1-((R)-2', 3'-Эпоксипропил)-(S)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонил-пиперазин 3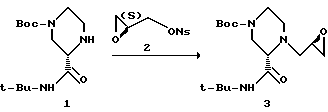
Материалы:
(S)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонил-пиперазин 1, г - 11,0 (38,4 мМ)
(2S)-(+)-Глицидил-З-нитробензолсульфонат, г - 9,96 (38,4 мМ)
Диизопропилэтиламин, мл - 5,5 (42,2 мМ)
ДМФ, мл - 38
Пиперазин 1 и (2S)-(+)-глицидил-3-нитробензолсульфонат 2 в 250-миллилитровой колбе, снабженной магнитной мешалкой, в атмосфере азота растворяли в ДМФ и DIEA. Полученный гомогенный раствор нагревали в течение 9 часов до температуры 60-62oC.
ТСХ (элюент:100%-ный EtOAc; окрашивание нингидрином) указывала на полное израсходование пиперазина 1.
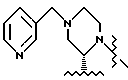
Реакцию гасили путем добавления 30 мл 5%-ного водного раствора NaHCO3. Реакционную смесь экстрагировали 400 миллилитрами изопропилацетата. Органическую фазу промывали водой (3 х 50 мл) и солевым раствором (1 х 50 мл), а затем осушали сульфатом натрия и выпаривали с получением желтого маслообразного вещества. После флеш-хроматографии (4 см х 20-см колонка; SiO2, элюирование градиентом EtOAc (30:70):гексана --> EtOAc:гексана (60:40)) и выпаривания продукта, содержащего фракции, получали 9,24 г (выход 71%) соединения 3 в виде маслообразного вещества. [α]
Пример 8. Получение эпоксида 3 из пиперазина 1 и (S)-глицидола 4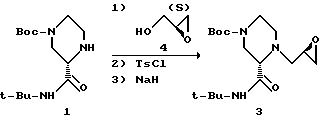
Пиперазин 1 (2,00 г, 7,00 мМ) и (3)-глицидол 4 (930 мкл, 14,0 мМ) нагревали с обратным холодильником в 19 мл изопропанола в течение 17 часов, а затем смесь распределяли между 100 мл этилацетата и 50 мл воды. После этого слои отделяли, а этилацетатный слой промывали насыщенным хлоридом натрия, осушали сульфатом магния и концентрировали с получением 2,4 г камеди. Часть этой камеди (241 мг) обрабатывали 2 миллилитрами пиридина и пара-толуолсульфонилхлоридом (130 мг, 0,68 мМ) в течение ночи, а затем концентрировали с получением маслообразного вещества. Это маслообразное вещество распределяли между 25 мл этилацетата и 10 мл воды. Этилацетатный слой промывали солевым раствором, осушали сульфатом магния и концентрировали с получением маслообразного вещества. Неочищенное маслообразное вещество растворяли в 2 мл ТГФ и обрабатывали 100 миллиграммами гидрида натрия (60% дисперсия в масле). Через 1 час смесь распределяли между 50 мл этилацетата и 10 мл воды. Этилацетатный слой осушали сульфатом магния и концентрировали, в результате чего получали целевой эпоксид 3 (см. спектральные данные предыдущих экспериментов).
Пример 9. Получение продукта 8 реакции взаимодействия амида 7 с эпоксидом 3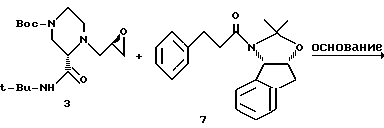
Раствор ацетонида 7 (216 мг, 0,67 мМ) и эпоксида N-Boc- пиперазина 3 (228 г, 0,67 мМ, 1,0 экв.) в 3,5 мл ТГФ (КF= 22 мкг/мл) (KF означает титрование Карла Фишера для воды) в 100-миллилитровой круглодонной колбе, снабженной термопарой и магнитной мешалкой, в атмосфере азота охлаждали до температуры -78oC. Затем добавляли раствор н-бутиллития в гексане (0,9 мл, 1,6 М, 2,1 экв. ), поддерживая при этом внутреннюю температуру от -78oC до - 73oC. Реакционную смесь перемешивали в течение одного часа при температуре -76oC, а затем нагревали в течение одного часа до температуры -25oC. Полученную смесь перемешивали в течение 2,5 часа при температуре от -25oC до -22oC. После этого реакционную смесь гасили 5 миллилитрами дистиллированной воды при температуре -15oC и распределяли с этилацетатом (20 мл). Затем смесь перемешивали, а слои разделяли. Этилацетатный экстракт промывали 10 миллилитрами насыщенного хлорида натрия и концентрировали при пониженном давлении (28 мм рт.ст.) с получением неочищенного продукта, который хроматографировали на колонках с силикагелем, используя в качестве элюента этилацетат/гексан (3: 2), в результате чего был получен целевой продукт 8 (84 мг, 20%) в виде бледно-желтого сиропообразного вещества: 13C-ЯМР (CDCI3, 75,4 МГц) δ: 172,6, 170,2, 164,6, 140,8, 140,4, 139,6, 129,5, 128,8, 128,1, 127,2, 126,8, 125,6, 124,1, 96,7, 80,4, 79,2, 65,9, 65,8, 62,2, 51,3, 50,1, 45,3, 43,5, 39,5, 39,1, 36,2, 28,8, 28,4, 26,5, 24,2.
Пример 10. Получение пенултимата 9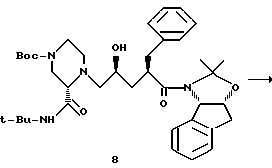
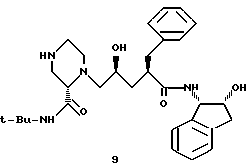
К раствору соединения 8 (5,79 г, 8,73 мМ) в 25,5 мл изопропанола при 0oC добавляли 20 мл 6 н. водного раствора соляной кислоты, а через 15 минут добавляли еще 10 мл концентрированной соляной кислоты. Через 1 час смесь нагревали до температуры 20oC и выдерживали в течение 4 часов. Полученную смесь охлаждали до 0oC, а pH доводили до 12,5 путем добавления 13 мл 50%-ного водного NaOH, поддерживая при этом температуру ≤29oC. Эту смесь экстрагировали этилацетатом (2 х 80 мл), после чего экстракты осушали сульфатом магния и концентрировали, в результате чего получали 5,46 г продукта 9 в виде бесцветной пены. 13C-ЯМР (75,4 Мгц, CDCl3) δ: 175,2, 170,5, 140,8, 140,5, 139,9, 129,1, 128,5, 127,9, 126,8, 126,5, 125,2, 124,2, 73,0, 66,0, 64,8, 62,2, 57,5, 49,5, 47,9, 46,4, 45,3, 39,6, 39,3, 38,2, 28,9.
Пример 11. Получение L-735.524-моногидрата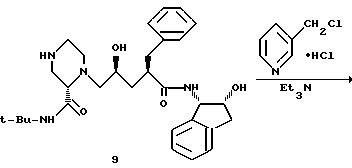
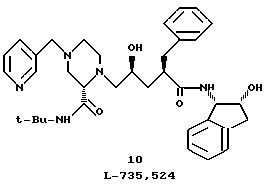
К раствору соединения 9 в EtOAc (10,5 л, KF = 10 мг/мл), полученному в предыдущей стадии, добавляли 20 литров осушенного на ситах диметилформамида (KF < 30 мг/л), и эту смесь нагревали на паровой бане под вакуумом 30'' рт. ст. для отгонки главным образом воды и/или любого остаточного изопропанолового или этилацетатного растворителя. Конечный объем концентрата составлял 13,5 л (KF = 1,8 мг/мл). К раствору (25oC) добавляли триэтиламин (2,86 л, 20,51 М), а затем гидрохлорид 3-пиколилхлорида (96%, 1287 г, 7,84 М). Полученную суспензию нагревали до 68oC.
Ход реакции прослеживали с помощью ВРЖХ-анализа, используя те же самые условия, что и в предыдущей стадии. Приблизительное время удерживания, мин: 2,7 - ДМФ; 4,2 - 3-пиколилхлорид; 4,8 - L-735,524; 9,1 - Пенултимат 9.
Полученную смесь выдерживали при температуре 68oC до тех пор, пока остаточное пенултиматное соединение 9 не составило <0,3% площадь (ВРЖХ-анализ). ВРЖХ-условия: 25-см колонка C8-RX Dupont; ацетонитрил/10 мМ (KH2PO4/K2HPO4) (60:40) при 1,0 мл/мин и детекция = 220 нм.
Затем смесь перемешивали в течение 4 часов при температуре 68oC и охлаждали до 25oC, после чего распределяли между этилацетатом (80 л) и смесью из 24 литров насыщенного водного раствора бикарбоната натрия и 14 литров дистиллированной воды. Полученную смесь перемешивали при температуре 55oC, а слои отделяли. Этилацетатный слой промывали три раза водой (20 л) при 55oC. Промытый этилацетатный слой концентрировали при атмосферном давлении до конечного объема 30 литров. По окончании концентрирования при атмосферном давлении к горячему раствору добавляли воду (560 мл), смесь охлаждали до 55oC и в качестве затравки добавляли кристаллы моногидрата L -735,524. Эту смесь охлаждали до температуры 4oC и фильтровали с получением продукта. Этот продукт промывали холодным этилацетатом (2 х 3 л) и осушали в "бытовом" вакууме при температуре 25oC, в результате чего получали 2905 г (70,7%) L-735,524 в виде белого твердого вещества.
Пример 12. Кинетическое разделение (S/R)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонил-пиперазина 17 с получением соединения I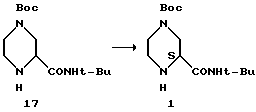
Материалы:
Неочищенный (S/R)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонил-пиперазин 17, г - 1,40
(S)-2-трет-Бутилкарбоксамид-4-трет-бутокси- карбонилпиперазин 1 (>99,5% э.и.), г - 4x0,14
Метилциклогексан с 2% (об/об) EtOAc, мл - 14
Неочищенный камедеобразный продукт 17 растворяли в 14 мл смеси растворителей путем нагревания до температуры 90oC. Этот раствор оставляли для охлаждения и при интервалах в 10oC к нему в качестве затравки добавляли 0,14 г соединения I (>99,5% э.и.). При температуре 55oC четыре 0,14-граммовые партии затравок больше не растворялись, и после дополнительного медленного охлаждения до комнатной температуры образовывали белую кристаллическую массу. Реакционную смесь фильтровали, промывали 3 миллилитрами смеси метилциклогексанового/этилацетатного растворителя и осушали в вакуумной печи с продувкой азотом, в результате чего получали 0,95 г белого твердого вещества. Определение энантиомерной чистоты с помощью колонки Chiracell AS указало на 93% энантиомерного избытка (э.и.).
Пример 13. Получение транс-3-(4-пиридил)акриловой кислоты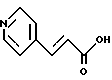
К раствору карбоксальдегида 4-пиридина (36,7 мл, 0,384 М) и малоновой кислоты (40 г, 0,384 М) в 31 мл пиридина добавляли пиперидин (0,12 мл), и эту смесь нагревали до температуры 100oC. Осторожно: выделяется большое количество CO2. Через 0,5 часа реакционную смесь охлаждали до комнатной температуры, и раствор отверждали. Этот раствор растирали с 240 мл воды и фильтровали, после чего дважды промывали 50-миллилитровыми порциями воды. Полученное твердое вещество осушали в течение ночи при температуре 42oC под вакуумом (10 мм рт.ст.), в результате чего получали 37,1 г белого твердого продукта, т. пл. 295- 297oC.
Пример 14. Получение N-(2-(R)-гидрокси-1(S)-инданил)-транс-3-(4-пиридил)акриламида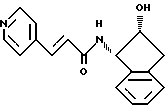
К суспензии, состоящей из транс-3-(4-пиридил)акриловой кислоты (10,0 г, 0,067 М) и 500 мл ТГФ, добавляли триэтиламин (10,29 мл, 0,0738 М), и полученный раствор охлаждали до 0oC. После добавления триметилацетилхлорида (8,68 мл, 0,0704 М) реакционную смесь перемешивали в течение получаса. К этой смеси через канюлю добавляли 2(R)-гидрокси-1(S)-индан (10,0 г, 0667 М), растворенный в 260 мл тетрагидрофурана. Через 2 часа реакционную смесь нагревали до комнатной температуры и перемешивали еще 15 часов. Растворитель удаляли в вакууме, а полученное твердое вещество растирали с холодным этилацетатом (150 мл) и фильтровали. Затем это вещество осушали в течение ночи под вакуумом (0,5 мм рт.ст.) и получали 18,5 г белого твердого продукта, т. пл. 205-207oC.
Пример 15. Получение N-(1,2-N,O-изопропилиден-2(R)-гидрокси-1(S)-инданил)-транс-3-(4-пиридил)акриламида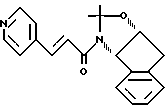
К суспензии N-(2(R)-гидрокси-1(S)-инданил)-транс-3-(4-пиридил)акриламида (18,5 г, 0,066 М) в 700 мл метиленхлорида добавляли диметоксипропан (49,0 мл, 0,402 М), а затем (+/-)-камфарсульфоновую кислоту (46,8 г, 0,201 М). Через 20 минут реакционная смесь становилась гомогенной. После этого смесь перемешивали в течение 3 часов и промывали насыщенным бикарбонатом натрия (2 х 150 мл). Водный слой экстрагировали метиленхлоридом (3 х 200 мл), а объединенный органический слой осушали сульфатом магния, фильтровали и концентрировали с получением маслообразного вещества. B результате очистки с помощью колоночной флеш-хроматографии (100 х 150-мм колонка с силикагелем; градиентное элюирование смесью (1:30:69, 2:30:68, 3:30:67, 5:30:65) NeOH: CHCl3, насыщенной NH3:CH2Cl2)) получали 16,0 г белой пены. (Rf=0,46 в смеси (5:30:65) MeOH:CHCl3, насыщенной NH3:CH2Cl2)).
Пример 16 Получение N-(1,2-N,O-изопропилиден-ен-2(R)-гидрокси-1(S)-инданил) -3-(4-пиридил)пропиламида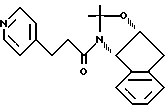
К N-(1,2-N, 0-изопропилиденен-2(R)-гидрокси-1(S)- инданил)транс-3-(4-пиридил)акриламиду (16,0 г, 0,0499 M), растворенному в этаноле (200 мл) и ТГФ (200 мл), добавляли 14,0 г Pd(OH)2 на угле (20% по массе). Затем в колбу загружали H2 и содержимое перемешивали в течение 9 часов. Полученный раствор продували аргоном, фильтровали через пробку из целита и промывали этанолом (100 мл). После удаления растворителя в вакууме продукт очищали с помощью колоночной флеш-хроматографии (100 х 150-мм колонка с силикагелем; градиентное элюирование смесью (1:30:69, 2:30:68, 3:30:67, 5:30:65) MeOH:CHCl3, насыщенной NH3:CH2Cl2)), в результате чего получали 13,8 г белой пены. (Rf= 0,5 в смеси (5:30:65) MeOH:CHCl3, насыщенной NH3:CH2Cl2)).
Пример 17. Получение N-(2(R)-гидрокси-1(S)-инданил)-транс-3-(3-пиридил)акриламида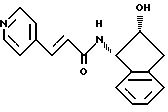
Целевое соединение было получено с использованием методики, в основном аналогичной методике, описанной для получения N-(2(R)-гидрокси-1(S)-инданил)-транс-3-(4-пиридил)-акриламида, за исключением того, что были использованы другие соответствующие исходные соединения. Физические данные этого соединения: т. пл. 119-120oC. Анализ для C17H16N2O2•0,65 H2O: Вычислено: C 69,92; H 5,97; N 9,59; Найдено: C 69,94, H 5,74, N 9,84.
Пример 18. Получение N-(1, 2-N,O-изопропилиденен-2(R)-гидрокси-1(S)-инданил)-транс-3-(3-пиридил)акриламида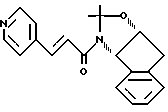
Целевое соединение было получено с использованием методики, в основном аналогичной методике, описанной для получения N-(1,2-N,O-изопропилиденен-2(R)-гидрокси-1(S)- инданил)-транс-3-(4-пиридил)акриламида, за исключением того, что было использовано другое соответствующее исходное соединение. Физические данные целевого соединения: т. пл. 134-136oC. Анализ для C20H20N2O2•0,25 H2O: Вычислено: C 73,94; H 6,36, N 8,62; Найдено: C 73,95, H 6,18, N 8,70.
Пример 19. Получение N-(1,2-N,O-изопропилиденен-2(R) -гидрокси-1(S)-инданил)-3-(3-пиридил)пропиламида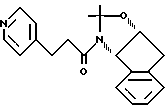
Целевое соединение было получено с использованием методики, в основном аналогичной методике, описанной для получения N-(1,2-N,O-изопропилиденен-2 (R)-гидрокси-1(S)-инданил)-3-(4-пиридил)пропиламида, за исключением того, что был использован другой соответствующий исходный продукт.
Хотя в вышеприведенном описании изложены принципы настоящего изобретения, проиллюстрированные соответствующими примерами, однако следует отметить, что в него могут быть внесены различные изменения, варианты и модификации, не выходящие за рамки нижеследующей формулы изобретения.
Соединение формулы I, где стереоцентр  имеет R- или S-конфигурацию либо является рацемическим; r = 0 - 5, целое число, R1, R2 - водород, алкил или R1 и R2 вместе с атомом азота, с которым связан R1, и атомом углерода, с которым связан R2, образуют 3-10-членную моноциклическую насыщенную кольцевую систему, включающую азот, 1-8 атомов углерода и один гетероатом формулы =N-V-R1, где V отсутствует или представляет -СОО-, или =Н-СОО-бензил, или = N-СН2-пиридил; R3 - Н, C1-4-алкил, С2-10-циклоалкил, С6-10-незамещенный арил и пиридил, R4 - С1-5-алкил, получают взаимодействием соединения формулы II и формулы III в присутствии сильного основания при низкой температуре. Способ занимает меньше времени, является в высокой степени диастереоселективным и не предусматривает использование токсичных реагентов. 5 с. и 24 з. п. ф-лы.
имеет R- или S-конфигурацию либо является рацемическим; r = 0 - 5, целое число, R1, R2 - водород, алкил или R1 и R2 вместе с атомом азота, с которым связан R1, и атомом углерода, с которым связан R2, образуют 3-10-членную моноциклическую насыщенную кольцевую систему, включающую азот, 1-8 атомов углерода и один гетероатом формулы =N-V-R1, где V отсутствует или представляет -СОО-, или =Н-СОО-бензил, или = N-СН2-пиридил; R3 - Н, C1-4-алкил, С2-10-циклоалкил, С6-10-незамещенный арил и пиридил, R4 - С1-5-алкил, получают взаимодействием соединения формулы II и формулы III в присутствии сильного основания при низкой температуре. Способ занимает меньше времени, является в высокой степени диастереоселективным и не предусматривает использование токсичных реагентов. 5 с. и 24 з. п. ф-лы.
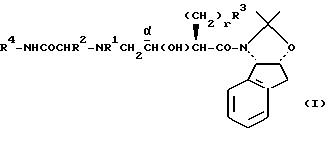
где стереоцентр 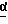 имеет либо R-, либо S-конфигурацию либо является рацемическим;
имеет либо R-, либо S-конфигурацию либо является рацемическим;
r = 0 - 5, целое число;
R1 и R2 независимо выбраны из группы, состоящей из водорода и незамещенного C1 - C4-алкила, или R1 и R2, объединенные вместе с атомом азота, с которым связан R1, и атомом углерода, с которым связан R2, образуют 3 - 10-членную моноциклическую насыщенную кольцевую систему, состоящую из азота, с которым связан R1, 1 - 8 атомов углерода и одного гетероатома, имеющую формулу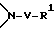
где V отсутствует или представляет -COO-;
R1 определен выше,
или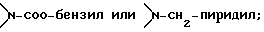
R3 выбран из группы, состоящей из водорода, C1 - C4-алкила, C5 - C10-циклоалкила, C6 - C10-незамещенного арила и пиридила;
R4 - C1 - C5-алкил с прямой или разветвленной цепью,
отличающийся тем, что соединение формулы II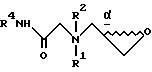
где R1, R2, R4 и  имеют указанные значения,
имеют указанные значения,
подвергают реакции с амидом формулы VIII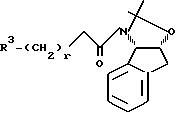
где R3 и r имеют указанные значения,
в присутствии сильного основания при низкой температуре. имеет S-конфигурацию, r = 1, R1 и R2, взятые вместе, образуют
имеет S-конфигурацию, r = 1, R1 и R2, взятые вместе, образуют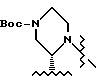
или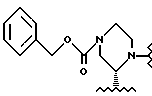
R3 выбирают из группы, состоящей из фенила,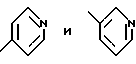
R4 - трет-бутил.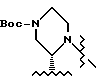
6. Способ по п.1, включающий, кроме того, стадию получения соединения формулы II посредством реакции амина формулы III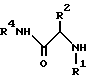
с глицидолом формулы IV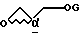
в присутствии основания,
где G является защитной группой, выбранной из 3-нитробензолсульфонила и трифторометансульфонила.
(a) взаимодействие раствора (S)(R)-2-трет-бутилкарбоксамидпиперазина с кислотой в водно-органической смеси растворителей, где кислоту выбирают из группы, включающей (+) или (-) винную кислоту, (+) или (-) миндальную кислоту, (+) или (-) дибензоилвинную кислоту, D- или L-пироглутаминовую кислоту, (+) или (-) ди-0,0'-п-толуолвинную кислоту, (+) или (-) яблочную кислоту, (+) или (-) 10-камфорсульфоновую кислоту, (+) или (-) 3-бромо-10-камфорсульфоновую кислоту и (+) или (-)-3-хлоро-10-камфорсульфоновую кислоту;
(b) нагревание смеси для растворения любых образующихся твердых веществ;
(с) охлаждение смеси;
(d) выделение (S)-2-трет-бутилкарбоксамид-пиперазина в виде осажденных кристаллов или из маточного раствора и
(e) обработку выделенного (S)-энантиомера основанием, а затем Boc2O;
с продуцированием (S)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонилпиперазина.
a) проведение реакции амина формулы III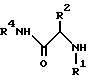
с глицидолом формулы V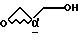
с получением соединения формулы VI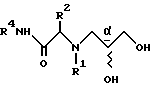
(b) обработку соединения VI активирующим агентом, выбранным из группы, включающей в себя п-толуолсульфонилхлорид, метансульфонилхлорид и ангидрид трифторометансульфоновой кислоты, с получением соединения VII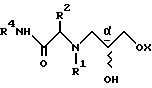
(c) обработку соединения VII сильным основанием с получением соединения формулы II, где X является группой, выбранной из п-толуолсульфонила, метансульфонила и трифторометансульфонила.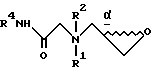
где стереоцентр 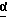 имеет либо R-, либо S-конфигурацию либо является рацемическим;
имеет либо R-, либо S-конфигурацию либо является рацемическим;
R1 и R2 независимо выбраны из группы, состоящей из водорода и незамещенного C1 - C4-алкила, или R1 и R2, объединенные вместе с атомом азота, с которым связан R1, и атомом углерода, с которым связан R2, образуют 3 - 10-членную моноциклическую насыщенную кольцевую систему, состоящую из азота, с которым связан R1, 1 - 8 атомов углерода и одного гетероатома, имеющую формулу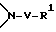
где V отсутствует или представляет -COO-;
R1 определен выше, или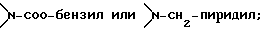
R4 - C1 - C5-алкил с прямой или разветвленной цепью,
отличающийся тем, что осуществляют реакцию амина формулы III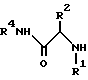
где R1, R2 и R4 имеют указанные значения,
с глицидолом формулы IV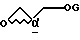
где G является защитной группой, выбранной из 3-нитробензолсульфонила и трифторметансульфонила,
в присутствии основания.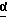 имеет S-конфигурацию, G - 3-нитробензолсульфонил; R1 и R2, взятые вместе, образуют циклическую структуру, выбранную из группы, состоящей из
имеет S-конфигурацию, G - 3-нитробензолсульфонил; R1 и R2, взятые вместе, образуют циклическую структуру, выбранную из группы, состоящей из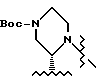
или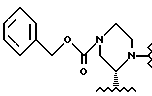
а R4 - трет-бутил.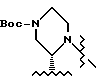
19. Производные α-аминоэпоксидов формулы II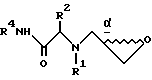
где стереоцентр 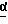 имеет либо R-, либо S-конфигурацию либо является рацемическим;
имеет либо R-, либо S-конфигурацию либо является рацемическим;
R1 и R2 независимо выбраны из группы, состоящей из водорода и незамещенного C1 - C4-алкила, или R1 и R2, объединенные вместе с атомом азота, с которым связан R1, и атомами углерода, с которым связан R2, образуют 3 - 10-членную моноциклическую насыщенную кольцевую систему, состоящую из азота, с которым связан R1, 1 - 8 атомов углерода и одного гетероатома, имеющую формулу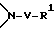
где V отсутствует или представляет -COO-;
R1 определен выше, или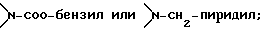
R4 - C1 - C5-алкил с прямой или разветвленной цепью.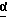 имеет S-конфигурацию, R4 - трет-бутил, R1 и R2, взятые вместе, образуют циклическую структуру, выбранную из группы, состоящей из
имеет S-конфигурацию, R4 - трет-бутил, R1 и R2, взятые вместе, образуют циклическую структуру, выбранную из группы, состоящей из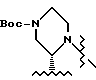
21. 1-((R)-2', 3'-Эпоксипропил)-(S)-2-трет-бутилкарбоксамид-4-третбутоксикарбонилпиперазин формулы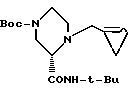
22. Способ получения и выделения (S)-2-трет-бутилкарбоксамидпиперазина, включающий в себя следующие стадии:
(a) взаимодействие раствора (S)(R)-2-трет-бутилкарбоксамид-пиперазина с кислотой в водно-органической смеси растворителей, где кислоту выбирают из группы, включающей (+)- или (-)-винную кислоту, (+)- или (-)-миндальную кислоту, (+)- или (-)-дибензоилвинную кислоту, D- или L-пироглутаминовую кислоту, или (+) или (-)-ди-0,0'-п-толуол-винную кислоту, (+)- или (-)-яблочную кислоту, (+)- или (-)-10-камфорсульфоновую кислоту, (+)- или (-)-3-бромо-10-камфорсульфоновую кислоту и (+)- или (-)-3-хлоро-10-камфорсульфоновую кислоту;
(b) нагревание смеси для растворения любых образующихся твердых веществ;
(c) охлаждение смеси;
(d) в случае, если осажденные кристаллы состоят преимущественно из (S)-антипода, то выделение осажденных кристаллов из маточного раствора, и
(e) в случае, если маточный раствор состоит преимущественно из (S)-антипода, то выделение осажденных кристаллов из этого маточного раствора и выделение (S)-антипода.
| Люминофоры в сине-фиолетовой области спектра | 1977 |
|
SU699001A1 |
| SU 1155599 A, 1985 | |||
| Тетраглицидиловый эфир 3,3 -дихлор4,4 -диаминотрифенилметана для получения хемоустойчивых эпоксидных полимеров | 1976 |
|
SU667553A1 |
| 1-Пиперидино-4-(1-метил-2,2-гем.дихлорциклопропил)-2,3-эпоксибутан в качестве противозадирной и противоизносной присадки к смазочным маслам | 1985 |
|
SU1273359A1 |
| Устройство для возведения двоичных чисел в степень | 1974 |
|
SU541168A1 |
| EP 521686 A1, 07.01.93 | |||
| СПОСОБ ПРЕССОВАНИЯ ЛИСТОВ ИЛИ ПЛЕНОК | 0 |
|
SU346847A1 |

