Настоящее изобретение относится к нейропатической боли. В частности, настоящее изобретение относится к лечению нейропатической боли путем селективного взаимодействия с подтипами мускариновых рецепторов.
У многих больных повреждение сенсорных нервов сопровождается болью различной интенсивности. Восприятие может варьировать от умеренного увеличения тактильной или температурной чувствительности до мучительной боли. Такой вид боли назван нейропатической болью, так как считается, что она вызвана функциональным нарушением нервной системы или изменением структуры нервной системы. Нейропатическая боль чрезвычайно трудно поддается лечению, обычно является хронической, и не отвечает на стандартные анальгетические вмешательства.
Приблизительно 1,5% населения США может страдать от нейропатической боли того или иного вида. Эта группа оказывается шире, если в нее включают больных с различными формами боли в спине неврогенного происхождения. Таким образом, нейропатическая боль может быть связана с повреждениями нерва, вызванными травмой, с такими заболеваниями, как диабет, опоясывающий лишай, синдром раздраженной кишки, рак последней стадии, или химическим повреждением (например, неблагоприятным последствием лекарственной терапии, включая антивирусные препараты).
Важно отметить, что препараты, эффективные при лечении воспалительной и острой боли (такие как опиаты и нестероидные противовоспалительные средства), обычно не эффективны при лечении нейропатической боли). Наоборот, соединения, которые облегчают нейропатическую боль, могут быть не эффективны для лечения острой боли (например, гапапентин, трициклические антидепрессанты). Предлагаемая в настоящее время терапия нейропатической боли явно не разработана для этого вида боли и не удивительно, что эти препараты не имеют высокой эффективности и при этом воздействуют не на всех пациентов. Таким образом, существует актуальная потребность в более эффективном и более толерантном лечении нейропатической боли.
Существует один класс молекул, перспективный для терапии нейропатической боли, - это молекулы, которые прямо или опосредованно взаимодействуют с мускариновыми рецепторами. Например, блокада активности ацетилхолинэстеразы (ACHE-I) повышает уровень ацетилхолина, предотвращая его расщепление, что, в свою очередь, приводит к одновременной активации всех холинергических рецепторов.
Препараты, которые у человека ингибируют активность холинэстеразы, являются эффективными анальгетиками. Например, ACHE-I физостигмин вызывает кратко действующую аналгезию у хирургических больных при послеоперационном применении. Спинальное введение другого химически родственного препарата ACHE-I неостигмина уменьшает острую послеоперационную боль, хроническую нейропатическую боль и потенцирует болеутоляющую активность опиатов при их спинальном применении. Чтобы добиться антиноцицептивной и аллодинической реакции ингибиторов холинэстеразы, из различных холинергических рецепторов были предложены как мускариновые, так и никотиновые рецепторы, Однако антиаллодинические эффекты физостигмина блокировались антагонистами мускариновых рецепторов, но не антагонистами никотиновых рецепторов. Предполагается, что эффекты холинэстеразного ингибирования этой формы боли достигается путем активации мускариновых, а не никотиновых рецепторов.
В различных моделях острой боли на животных прямое действие агонистов мускариновых рецепторов является антиноцицептивным (Bartolini и другие, 1992; Brodie и Proudfit, 1984; Capone et al., 1999; Hartvig et al., 1989; Pedigo и et al., 1975; Przewlocka et al., 1999; Shannon et al., 1997; Sheardown et al., 1997). Эти эффекты могут быть блокированы мускариновыми антагонистами (Bartolini et al., 1992; Hwang et al., 1999; Naguib и Yaksh, 1997; Sheardown et al. 1997). Эти данные далее подтверждают роль активации мускариновых рецепторов в регуляции состояния острой боли.
Некоторые исследования изучали роль активации мускариновых рецепторов при хронической или нейропатической боли. В этих исследованиях прямое и непрямое повышение холинергического тонуса демонстрировало облегчение тактильной аллодинии после спинального введения у крыс моделей нейропатической боли с лигированным спинным мозгом, и эти эффекты были обратимы применением мускариновых антагонистов (Hwang et al., 1999; и Lee et al., 2002). Таким образом, прямая или косвенная активация мускариновых рецепторов проявляла и высокую анальгетическую активность и облегчала нейропатическую боль. Мускариновые агонисты и ACHE-I вследствие их склонности вызывать множество побочных эффектов при применении у людей клинически не нашли широкого применения. Нежелательные побочные эффекты включают повышенное слюноотделение и потоотделение, увеличение моторики желудочно-кишечного тракта и брадикардию, а также другие побочные эффекты. Эти побочные эффекты связаны с повсеместной экспрессией в организме семейства мускариновых рецепторов.
Открытие в середине 1980-х годов 5 генетически уникальных мускариновых рецепторов М(1)-M(5) с их дифференцированным распределением в организме позволило задуматься о разработке молекул, которые селективно взаимодействуют только с одним из этих подтипов рецепторов, а не с другими. Предполагалось, что разработка селективных молекул даст возможность модуляции, например, мускариновых рецепторов, регулирующих функции центральной нервной системы без одновременного активирования мускариновых рецепторов, регулирующих кардиальные, желудочно-кишечные или секреторные функции. Несмотря на огромные усилия, не было разработано никаких препаратов с такой ожидаемой селективностью, что обусловлено главным образом структурным сходством важных центров активации у этих 5 подтипов рецепторов.
Также не известно, какой из 5 подтипов мускариновых рецепторов вызывает эффекты мускариновых соединений при различных болевых состояниях. В действительности возможно, что в регулирование боли может быть вовлечена активация более чем одного подтипа мускаринового рецептора или что активация различных подтипов мускариновых рецепторов может вызывать различные виды боли. Например, М(2) рецепторы в высокой степени выражены в ганглии дорсального корешка в малых и средних нейронах, в дорсальном роге спинного мозга и таламуса, и предполагается, что активация М(2) рецепторов может участвовать в модуляции трансдукции токсичных стимулов от периферии через спинной мозг к головному мозгу. Эта гипотеза подтвердилась тем, что удаление М(2) рецепторов у мышей уменьшает высокую антиноцицептивную активность мускариновых агонистов. К тому же, основываясь на удалении других подтипов мускариновых рецепторов у мышей, только М(2) и, возможно, М(4) рецепторы по причине их меньшей распространенности проявляют высокую анальгетическую активность мускариновых агонистов. В других случаях были получены подобные выводы: "Эти данные обеспечивают однозначные доказательства, что мускариновая аналгезия опосредована комбинацией исключительно М(2) и М(4) мускариновых рецепторов как на спинальном, так и супраспинальном уровнях" (Duttaroy A, et al., 2002). Тем не менее, другие исследователи в дальнейшем отметили: «Однако активность М(1) подтипа рецептора не является необходимой для антиноцицептивного действия» (Sheardown, et al., 1997).
Несмотря на эти данные, терапевтическая применимость соединения, действующего непосредственно на М(2) рецепторы, ограничена. Это обусловлено тем, что М(2) рецепторы также очень распространены в сердце и желудочно-кишечном тракте. Предполагается, что желудочно-кишечные расстройства и побочные эффекты мускариновых рецепторов со стороны сердечно-сосудистой системы также опосредованы этими рецепторами. Аналогично это предположение было подтверждено на мышах с удалением М(2) рецепторов. Таким образом, средства, которые прямо или опосредованно активируют М(2) мускариновые рецепторы, не могли бы быть полезны для лечения даже острой боли вследствие нежелательных и потенциально опасных побочных эффектов.
Подобное научное руководство не применимо для нейропатической боли. Точный подтип мускаринового рецептора, связанного с активностью прямых и косвенных мускариновых агонистов при нейропатической боли, достоверно не известен. В медицине существует насущная потребность определить подтип(ы) мускариновых рецепторов, вовлеченных в облегчение нейропатической боли, и разработать препараты, селективно активирующие эти рецепторы.
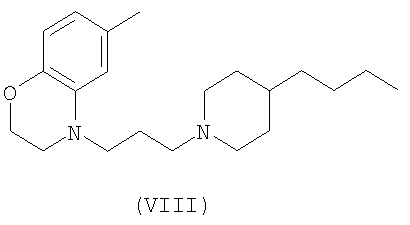
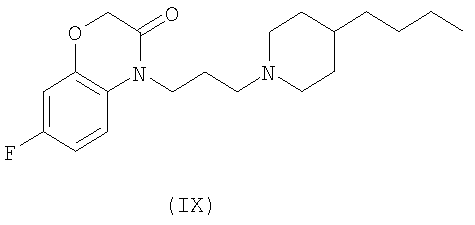
Способ лечения нейропатической боли, раскрытый в настоящем изобретении, включает идентификацию субъекта, нуждающегося в таком лечении, и предоставление субъекту эффективного количества, по меньшей мере, одного соединения, которое селективно активирует М(1) подтип рецепторов, посредством чего частично снимаются один или более симптомов нейропатической боли. В некоторых вариантах осуществления изобретения у субъекта проявляется гипералгезия. В некоторых вариантах осуществления изобретения у субъекта проявляется аллодиния. В некоторых вариантах осуществления изобретения нейропатическая боль связана с диабетом, вирусной инфекцией, синдромом раздраженной кишки, ампутацией, раком или химическим повреждением. В некоторых вариантах осуществления изобретения соединение, селективно активирующее М(1) подтип рецептора, не облегчает острую боль. В некоторых вариантах осуществления изобретения соединение выбирается из группы, состоящей из соединений Формул VII, VIII и IX:

Изобретение также относится к способу идентификации соединения, частично снимающего гипералгезию или аллодинию у субъекта, который включает предоставление субъекту, по меньшей мере, одного тестированного в отношении мускариновых рецепторов соединения и определение того, облегчает ли, по меньшей мере, одно тестированное соединение гипералгезию или аллодинию у субъекта. В некоторых вариантах осуществления изобретения, по меньшей мере, одно тестированное соединение является селективным для М(1) или М(4), но не для М(2) или М(3) рецепторов. В некоторых вариантах осуществления изобретения, по меньшей мере, одно тестированное соединение является селективным для М(1) рецептора. В некоторых вариантах осуществления изобретения гипералгезией является тепловая гипералгезия. В некоторых вариантах осуществления изобретения аллодинией является тактильная аллодиния.
Изобретение относится к фармацевтической композиции, содержащей эффективное количество, по меньшей мере, одного соединения, которое селективно активирует рецепторы подтипа М(1) в количестве, эффективном для снижения одного или более симптомов нейропатической боли. В некоторых вариантах осуществления изобретения соединения выбирают из группы, состоящей из соединений Формул VII, VIII и IX.
Краткое описание чертежей
Фиг.1 показывает химические структуры примеров соединений Формулы (VI).
Фиг.2 показывает эффект лечения соединением Формулы IX на тактильную чувствительность после частичного лигирования седалищного нерва.
Фиг.3 показывает эффект введения соединения Формулы IX интрасеребровентикулярно на тактильную чувствительность после частичного лигирования седалищного нерва.
Подробное описание предпочтительного варианта осуществления
Разработаны соединения с беспрецедентной селективностью для М(1) рецепторов относительно других подтипов мускариновых рецепторов (Spalding ТА, Trotter C, Skjaerbaek N, Messier TL, Currier EA, Burstein ES, Li D, Hacksell U, Brann MR. Discovery of an ectopic activation site on the M(1) muscarinic receptor. Mol. Pharmacol, 61(6): 1297-302, 2002; U.S. Appl. No. 10/262,517 (publication number 20030100545), entitled, "Benzimidazolidinone Derivatives as Muscarinic Agents"; U.S. Patent No. 6,627,645, entitled, "Muscarinic Agonists"; U.S. Patent No. 6,528,529, entitled, "Compounds with Activity on Muscarinic Receptors"; U.S. Appl. No. 10/338,937 (publication number 20030144285), entitled, "Compounds with Activity on Muscarinic Receptors"; U.S. Appl. No. 10/329,455 (publication number 20030176418), entitled, "Tetrahydroisoquinoline Analogues as Muscarinic Agonists"; and U.S. Provisional No. 60/432,692, entitled, "Piperidinyl Dimers as Muscarinic Agents".
Обнаружено, что соединения с относительной селективностью для М(1) мускариновых рецепторов были очень эффективны в облегчении тепловой гипералгезии и тактильной аллодинии на моделях нейропатической боли при системном применении у грызунов. Поскольку эти соединения также не активируют другие подтипы мускариновых рецепторов, эти М(1) агонисты не вызывают, как предшествующие неселективные агонисты, нежелательных и несовместимых с жизнью действий. Поэтому М(1) селективные агонисты являются особенно привлекательными в качестве препаратов для лечения хронической нейропатической боли. Напротив, в отличие от неселективных мускариновых агонистов, которые взаимодействуют с М(2) и всеми другими подтипами мускариновых рецепторов, эти М(1) селективные агонисты не эффективны для уменьшения острой боли. Таким образом, на грызунах показано, что М(1) селективные агонисты имеют особенно привлекательный профиль. Они блокируют нейропатическую боль, но не меняют реакцию на другие формы боли. При длительном применении эти средства должны позволить пациентам обычным образом реагировать на острую боль, в то же время блокируя хроническую нейропатическую боль.
Используемый в описании термин "селективный" определен как свойство соединения, при этом количество соединения достаточно, чтобы вызывать желательную реакцию специфического типа, подтипа, класса или подкласса рецепторов с значительно меньшим или существенно малым или отсутствием всякого эффекта на активность других типов рецепторов. Например, селективное соединение может иметь, по меньшей мере, 10-кратный эффект на активность желательного рецептора, чем на другие типы рецепторов. В некоторых случаях селективное соединение может иметь, по меньшей мере, 20-кратный эффект на активность желательного рецептора, чем на другие типы рецепторов, или, по меньшей мере, 50-кратный эффект, или, по меньшей мере, 100-кратный эффект, или, по меньшей мере, 1000-кратный эффект, или, по крайней мере, 10000-кратным большим эффектом, или по, меньшей мере, 100000-кратный эффект, или более чем 100000-кратный эффект.
Участок действия М(1) агонистов на нейропатическую боль требует объяснения. Тем не менее, показано, что уменьшающее нейропатическую боль действие М(1) селективных агонистов блокируется мускариновым антагонистом скополамина гидрохлоридом, проникающим в центральную нервную систему, но не блокируется мускариновым антагонистом метилскополамина гидрохлоридом, обладающим главным образом периферическим действием. Предполагается, что уменьшающее нейропатическую боль действие М(1) селективных агонистов осуществляется через воздействие на центральную нервную систему. К тому же, эти М(1) селективные агонисты не эффективны в облегчении нейропатической боли при спинальном применении, но эффективно облегчают эту форму боли при интрацеребровентрикулярном применении. Предполагается, что активация уменьшающего нейропатическую боль действия М(1) рецептора осуществляется супраспинально и спинальная локализация действия не обязательна.
Соединения, которые взаимодействуют с М(1) подтипом рецептора, обладают прежде недооцененной аналгетической активностью и являются эффективными при лечении нейропатической боли. Эти наблюдения имеют практическое применение, которые поддерживают использование М(1) агонистов для лечения нейропатической боли, вызванной травмой и такими заболеваниями, как диабет, опоясывающий лишай, синдром раздраженной кишки, рак последней стадии, или химическим повреждением (например, неблагоприятным последствием лекарственной терапии, включая антивирусные препараты).
Таким образом, в некоторых вариантах осуществления настоящего изобретения нейропатическая боль в организме лечится путем введения объекту фармакологически активной дозы соединения, которое взаимодействует с М(1) подтипом рецепторов, с целью регуляции боли, не вызывая нежелательных и ограничивающих ее полезность побочных эффектов.
В некоторых вариантах осуществления изобретения соединения в соответствии с настоящим изобретением селективно взаимодействуют с М(1) подтипами рецепторов.
В некоторых вариантах осуществления изобретения соединения в соответствии с настоящим изобретением описаны в U.S. Patent Application No. 10/262,517 (publication number 20030100545) и имеют структуру Формулы (I)
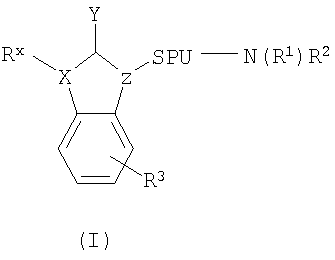
в которой X выбран из от группы, состоящей из C, O, N и S;
Z выбран из от группы, состоящей из CH и N;
Y выбран из от группы, состоящей из =O, =N и =S или их таутомеров, таких как Y-алкилированные таутомеры;
SPU представляет единицу - спейсер, обеспечивающую расстояние d между Z и N, в котором -SPU- представляет бирадикал, выбранный из группы, состоящей из -(CR6R7)n-A- и -C3-8-циклоалкил-, в котором n находится в диапазоне от 1 до 5, такого типа, как 1, 2, 3, 4, или 5, и А отсутствует или произвольно замещен на -C3-8-циклоалкил-;
N вместе с R1 и R2 образуют гетероциклическое кольцо, в котором гетероциклическое кольцо выбрано из группы, состоящей из пергидроазоцина, пергидроазепина, пиперидина, пирролидина, азетидина, азиридина и 8-азабицикло[3,2,1]октана, и в котором гетероциклическое кольцо замещено одним или более заместителями R4, выбранными из группы, состоящей из гидрокси, галогена, С1-8-алкила, C3-8-циклоалкила, C1-8-алкокси, C1-8-алкилкарбонила, C1-8-алкилидена, C2-8-алкенила, C2-8-алкинила, C1-6-алкилоксиимино и C1-6-алкилоксиамино, каждый из которых может быть необязательно замещен заместителем R5 и в котором, по меньшей мере, одним из упомянутых заместителей R4 является R4′, выбранный из группы, состоящей из С1-8-алкила, C3-8-циклоалкила, C1-8-алкокси, C1-8-алкилкарбонила, C1-8-алкилидена, C1-8-алкилоксиимино и C1-8-алкилоксиамино, каждый из которых может быть необязательно замещен заместителем R5;
R5 выбран из группы, состоящей из водорода, галогена, гидрокси, С1-8-алкила, C1-8-алкокси, C3-8-циклоалкила, C3-8-гетероциклила, C1-8-алкилкарбонила, C1-8-алкилидена, C2-8-алкенила и C2-8-алкинила;
Rx может отсутствовать или выбираться из группы, состоящей из водорода, необязательно замещенного С1-8-алкила, необязательно замещенного C3-8-циклоалкила, необязательно замещенного C2-8-алкенила, необязательно замещенного C2-8-алкинила, необязательно замещенного арила, необязательно замещенного гетероарила CH2N(R5)(R5), CH2-OR5, CH2-SR5, CH2-O-C(=O)R5, CH2-O-C(=S)R5;
R3 может присутствовать 0-4 раза и выбираться из группы, состоящей из галогена, гидрокси, необязательно замещенного С1-8-алкила, С1-8-алкокси, необязательно замещенного C1-8-алкилидена, необязательно замещенного C2-8-алкенила, необязательно замещенного C2-8-алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного C3-8-циклоалкила, необязательно замещенного C3-8-гетероциклила и необязательно замещенного C1-8-алкилкарбонила; и
каждый R6 и каждый R7 независимо выбирается из группы, состоящей из водорода, галогена, гидрокси, необязательно замещенного С1-8-алкила, С1-8-алкокси, необязательно замещенного C1-8-алкилидена, необязательно замещенного C2-8-алкенила, необязательно замещенного C2-8-алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного C3-8-циклоалкила, необязательно замещенного C3-8-гетероциклила, и необязательно замещенного C1-8-алкилкарбонила.
В некоторых вариантах осуществления изобретения соединения, использующиеся в настоящем изобретении, описаны в U.S. Patent No. 6627645 и имеют структуру Формулы (II):
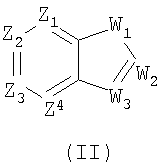
в которой Z1 представляет CR1 или N, Z2 представляет CR2 или N, Z3 представляет CR3 или N и Z4 представляет CR4 или N, где не больше чем два из Z1, Z2, Z3 и Z4 являются N;
W1 представляет O, S или NR5, один из W2 и W3 представляет N или CR6, а другой из W2 и W3 представляет CG; W1 является NG, W2 представляет CR5 или N, и W3 представляет CR6 или N; или W1 и W3 представляет N и W2 представляет NG;
G имеет формулу (III):
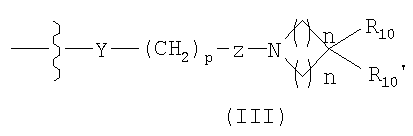
Y представляет O, S, CHOH, -NHC(O)-, -C(O)NH-, -C(O)-, OC(O)-, -(O)СО-, -NR7-, -CH=N- или отсутствует;
р = 1, 2, 3, 4 или 5;
Z представляет CR8R9 или отсутствует;
каждое t = 1, 2 или 3;
каждое R1, R2, R3 и R4 независимо является Н, амино, гидроксилом, галогеном или прямой или разветвленной цепью С1-6алкил, C2-6алкенил, C2-6алкинил, C1-6гетероалкил, C1-6галогеналкил, -CN, -CF3-OR11, -COR11, -NO2, -SR11, -NHC(O)R1, -C(O)NR12R13, -NR12R3, -NR11C(O)NR12R13, -SO2NR12R13, -OC(O)R11, -O(CH2)qNR12R13 или -(CH2)qNR12R13, где q - целое число от 2 до 6, или R1 и R2 вместе образуют -NH-N=N- или R3 и R4 вместе образуют -NH-N=N-;
каждое R5, R6 и R7, независимо, является Н, С1-6алкилом; формилом; C3-6циклоалкилом; C5-6арилом, необязательно замещенным галогеном или С1-6алкилом; или C5-6гетероарилом, необязательно замещенным галогеном или С1-6алкилом; каждое R8 и R9, независимо, является Н или прямой или разветвленной цепью С1-8алкила;
R10 представляет прямую или разветвленную цепь С1-8алкила; C2-8алкенил, C2-8алкинил, C1-8алкилиден, C1-8алкокси, C1-8гетероалкил, C1-8аминоалкил, C1-8галогеналкил, C1-8алкоксикарбонил, C1-8гидроксиалкокси, C1-8гидроксиалкил, -SH, C1-8алкилтио, -O-CH2-C5-6арил, -C(O)-C5-6арил, замещенный С1-3алкилом или галогеном, C5-6арил, C5-6циклоалкил, C5-6гетероарил, C5-6гетероциклоалкил, -NR12R13, -C(O)NR12R13, -NR11C(O)NR12R13, -CR11R12R13, -OC(O)R11, -(O)(CH2)SNRI2R13 или -(CH2)SNR12R13, где s является целым числом от 2 до 8;
R10' является Н, прямой или разветвленной цепью С1-8алкила, C2-8алкенилом, C2-8алкинилом, C1-8алкилиденом, C1-8алкокси, C1-8гетероалкилом, C1-8аминоалкилом, C1-8галогеналкилом, C1-8алкоксикарбонилом, C1-8гидроксиалкокси, C1-8гидроксиалкилом или C1-8алкилтио; каждый R11, независимо, является Н, прямой или разветвленной цепью С1-8алкила, C2-8алкенилом, C2-8алкинилом, C2-8гетероалкилом, C2-8аминоалкилом, C2-8 галогеналкилом, C1-8алкоксикарбонилом, C2-8гидроксиалкилом, -C(O)-C5-6арилом, замещенным C1-3алкилом, или галогеном, C5-6арилом, C5-6гетероарилом, C5-6циклоалкилом, C5-6гетероциклоалкилом, -C(O)NR12R13, -CR5R12R13, -(CH2)tNR12R13, где t является целым числом от 2 до 8; и
каждый R12 и R13, независимо, являются Н, С1-6алкилом; C3-6циклоалкилом; C5-6арилом, необязательно замещенным галогеном или С1-6алкилом; или C5-6гетероарилом, необязательно замещенным галогеном или С1-6алкилом; или R12 и R13 вместе образуют циклическую структуру; или фармацевтически приемлемой солью, сложным эфиром или его пролекарством.
В некоторых вариантах осуществления изобретения соединения, используемые в настоящем изобретении, описаны в US Patent No. 6528529 и имеют структуру Формулы (IV):
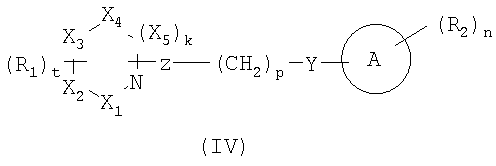
в которой Х1, X2, X3, X4 и X5 выбраны из C, N и O;
k является 0 или 1;
t является 0, 1 или 2;
R1 представляет прямую или разветвленную цепь С1-8алкила, C2-8алкенил, C2-8алкинил, C1-8алкилиден, C1-8алкокси, C1-8гетероалкил, C1-8аминоалкил, C1-8галогеналкил, C1-8алкоксикарбонил, C1-8гидроксиалкокси, C1-8гидроксиалкил, -SH, C1-8алкилтио, -O-CH2-C5-6арил, -C(O)-C5-6арил, замещенный С1-3алкилом или галогеном; C5-6арил или C5-6циклоалкил необязательно содержащий 1 или более гетероатомов, выбранных из N, S и O; -C(O)NR3R4, -NR3R4, -NR3C(O)NR4R5, -CR3R4, -OC(O)R3, -(O)(CH2)SNR3R4 или -(CH2)SNR3R4;
где R3, R4 и R5 являются одинаковыми или различными, каждый независимо выбирается из Н, С1-6алкила; C5-6арила, необязательно содержащим 1 или более гетероатомов, выбранных из N, O и S, и необязательно замещенным галогеном или С1-6алкилом; C3-6циклоалкила; или R3 и R4 вместе с атомом N, если он присутствует, образуют циклическую кольцевую структуру, содержащую 5-6 атомов, выбранных из C, N, S и O; и
s является целым числом от 0 до 8;
А представляет C5-12арил или C5-7циклоалкил, каждый из которых необязательно содержит 1 или более гетероатомов, выбранных из N, S и O;
R2 представляет Н, амино, гидроксил, галоген или прямую или разветвленную цепь С1-6алкила, C2-6алкенил, C2-6алкинил, C1-6алкокси, C1-6гетероалкил, C1-6аминоалкил, C1-6галогеналкил, C1-6алкилтио, C1-6алкилкарбонил, -CN, -CF3, -OR3, -COR3, NO2, -NHR3, -NHC(O)R3, -C(O)NR3R4, -NR3R4, -NR3C(O)NR4R5, -OC(O)R3, -C(O)R3R4, -O(CH2)qNR3, -CNR3R4 или -(CH2)qNR3R4;
где q = целое число от 1 до 6;
n=0, 1, 2, 3 или 4, группы R2, при n>1, являются одинаковыми или различными;
р=0 или целое число от 1 до 5;
Y представляет O, S, CHOH, -NHC(O)-, -C(O)NH-, -C(O)-, -OC(O)-, NR7 или -CH=N-, и
R7 представляет Н или С1-4алкил; или отсутствует; и
Z представляет CR8R9, в котором R8 и R9 независимо выбраны из Н, и прямой или разветвленной цепи С1-8алкила; или фармацевтически приемлемой соли, сложного эфира или его пролекарства.
В некоторых вариантах осуществления изобретения соединения, используемые в настоящем изобретении, описаны в US Patent Application No. 10/329455 (publication number 20030176418) и имеют структуру Формулы (V):
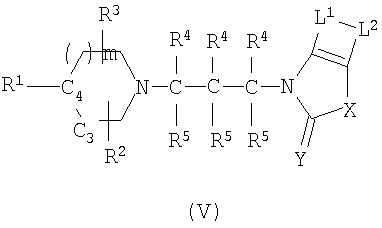
в которой R1 представляет монорадикал, выбранный из группы, состоящей из необязательно замещенного С1-6алкила, необязательно замещенного C2-6-алкилидена, необязательно замещенного C2-6-алкенила, необязательно замещенного C2-6-алкинила, необязательно замещенного O-С1-6алкила, необязательно замещенного O-C2-6-алкенила, необязательно замещенного O-C2-6-алкинила; необязательно замещенного S-С1-6алкила, необязательно замещенного S-C2-6-алкенила, необязательно замещенного S-C2-6-алкинила;
m=0, 1 или 2;
C3-C4 представляет CH2-CH или CH=C, или C4 представляет CH, и C3 отсутствует;
R2 и R3 независимо выбран из группы, состоящей из водорода, необязательно замещенного С1-6алкила, необязательно замещенного O-С1-6алкила, галогена, гидрокси или выбраны так, что R2 и R3 вместе образуют кольцевую систему;
каждый R4 и R5 независимо выбран из группы, состоящей из водорода, галогена, гидрокси, необязательно замещенного С1-6алкила, необязательно замещенного O-С1-6алкила, необязательно замещенного арил-С1-6алкила, и необязательно замещенного арилгетероалкила;
L1 и L2 представляет бирадикалы, независимо выбранные из группы, состоящей из -C(R6)=C(R7), -C(R6)=N-, -N=C(R6)-, -S-, -NH- и -O-; причем только один из L1 и L2 может быть выбран из группы, состоящей из -S-, -NH- и -O-;
Y выбран из группы, состоящей из O, S и H2;
X представляет бирадикал, выбранный из группы, состоящей из -C(R6)(R7)-C(R6)(R7)-, -C(R6)=C(R7)-, -O-C(R6)(R7)-, C(R6)(R7)-O-, -S-C(R6)(R7)-, -С(R6)(R7)-S-, -N(RN)-C(R6)(R7)-, -C(R6)(R7)-N(RN)-, -C(R6)(R7)-C(R6)(R7)-C(R6)(R7)-, -O-C(R6)(R7)-C(R6)(R7)-, S-C(R6)(R7)-C(R6)(R7)-, N(RN)-C(R6)(R7)-C(R6)(R7)-, -C(R6)(R7)-C(R6)(R7)-O, -C(R6)(R7)-C(R6)(R7)-S, -C(R6)(R7)-C(R6)(R7)-N(RN)-, -C(R6)(R7)-C(R6)=C(R7)- и -C(R6)=C(R7)-C(R6)(R7),
в котором R6 и R7 независимо выбираются из группы, состоящей из водорода, галогена, гидрокси, нитро, циано, NRNRN, N(RN)-C(O)N(RN), необязательно замещенного С1-6алкила, C2-6-алкенила, C2-6-алкинила, необязательно замещенного O-C1-6-алкила, необязательно замещенного O-арила, необязательно замещенного O-C2-6-алкенила, необязательно замещенного O-C2-6-алкинила,
в котором RN выбран из группы, состоящей из водорода, и необязательно замещенного C1-6-алкила.
В некоторых вариантах осуществления изобретения соединения настоящего изобретения описаны в US Provisional Application No. 60/432692, и имеют структуру Формулы (VI):
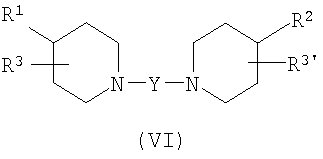
в которой Y представляет бирадикал (CR4R5)m-Z-C(R4R5)n;
в котором сумма m+n составляет от 1 до 7;
Z выбран из группы, состоящей из C(R4R5), C(O), O, N(R6), S, O-C(O), N(R6)C(O), C(O)-O и P; и
R4 и R5 независимо выбраны из группы, состоящей из водорода, галогена, гидрокси, нитро, NR6N6', необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного C3-8-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного C1-6-алкила, необязательно замещенного C1-6-алкокси, необязательно замещенного фенокси, необязательно замещенного C2-8-алкенила и необязательно замещенного C2-8-алкинила; и
в которой R1 и R2 независимо выбраны из группы, состоящей из необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного C3-8-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного C1-6-алкила, необязательно замещенного C1-6-алкокси, необязательно замещенного C2-8-алкенила и необязательно замещенного C2-8-алкинила;
в которой R3 и R3' независимо выбраны из группы, состоящей из водорода, галогена, гидрокси, нитро, NR6N6', необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного C3-8-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного C1-6-алкила, необязательно замещенного C1-6-алкокси, необязательно замещенного C2-8-алкенила и необязательно замещенного C2-8-алкинила; и
R6 и R6' независимо выбраны из группы, состоящей из водорода, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного C3-8-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного C1-6-алкила, необязательно замещенного C1-6-алкокси, необязательно замещенного C2-8-алкенила и необязательно замещенного C2-8-алкинила.
Химические структуры, показывающие определенные примеры соединений Формулы (VI), изображены на Фиг.1. Ниже представлены примеры, показывающие синтез этих соединений:
1,2-бис(4-(2-оксобензимидазолин-1-ил)пиперидино)этан (55LH-4-1A)
В пробирку помещали 4-(2-оксобензимидазолин-1-ил)пиперидин (0,27 г, 1,25 ммоль), 1-хлоро-2-йодоэтан (95 мг, 0,5 ммоль), K2CO3 (0,17 г, 1,25 ммоль) и этанол (2 мл) и смесь взбалтывали при 60°C в течение ночи. Добавляли воду и этилацетат, фильтровали и сушили с получением названного соединения в количестве 113 мг.
1Н ЯМР (ДМСО-d6) δ 1,59-1,66 (м, 4Н), 2,06-2,15 (м, 4Н), 2,27-2,40 (м, 4Н), 2,45 (арр с, 4Н), 2,99-3,06 (м, 4Н), 4,07-4,18 (м, 2Н), 6,92-7,00 (арр с, 6Н), 7,16-7,21 (м, 2Н); 13С ЯМР (ДМСО-d6) δ 29,4, 50,9, 53,9, 56,3, 109,3, 109,5, 121,1, 121,1, 129,0, 129,9, 154,4. LC-МС[M-H]+ 461,4.
1,4-бис(4-(2-оксобензимидазолин-1-ил)пиперидино)бутан трифторацетат (55-LH-25A)
В пробирку помещали 4-(2-оксобензимидазолин-1-ил)пиперидин (1,1 г, 5,0 ммоль), 4-бромо-1-бутанол (0,92 мг, 6,0 ммоль), K2CO3 (0,86 г, 6,25 ммоль) и этанол (3 мл) и смесь взбалтывали при 60°C в течение девяти дней. Добавляли воду и этилацетат, органический слой высушивали (Na2SO4), фильтровали и концентрировали. Остаток очищали на хроматографической колонке [(SiO2, 5% NH4OH в MeOH/EtOAc (1:9)] с получением 0,22 мг 4-(4-(2-оксобензимидазолин-1-ил)пиперидино)бутанола (55-LH-10), который использовали на следующем этапе без дальнейшего определения характеристик. LC-MC[M-H]+290,1.
Смесь (55-LH-10) (0,22 г, 0,78 ммоль), диметилсульфоксида (DMSO) (66 мкл, 0,93 ммоль) и дихлорметана (1 мл) охлаждали до -78°C и перемешивали в течение 0,5 часа. Добавляли оксалихлорид (73 мкл, 0,85 ммоль) и смесь выдерживали дополнительные 0,5 часа при -78°C. Добавляли триэтиламин (0,54 мл, 3,9 ммоль) и реакционную смесь доводили до комнатной температуры. Добавляли воду и дихлорметан, органический слой отделяли и промывали насыщенным солевым раствором, высушивали (Na2SO4), фильтровали и выпаривали. Полученный альдегид растворяли в MeOH (2,5 мл) и добавляли 4-(2-оксобензимидазолин-1-ил)пиперидин (0,17 г, 0,78 ммоль) с последующим добавлением HOAc до достижения pH 4-5. Добавляли свежеприготовленный раствор NaCNBH3 (54 мг, 0,85 ммоль) в MeOH (1 мл), и смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду и этилацетат, органический слой высушивали (Na2SO4), фильтровали и концентрировали. Остаток растворяли в водном HCl (1N) и очищали препаративной ВЭЖХ [Luna колонка (21,2×250 мм, 15 мкм C18(2), 0,1% TFA (трифторуксусная кислота) в H2O/0,1% TFA в CH3CN/H2O (8:2) (9:1 градиент к 0:100)]. Очищенное соединение выпадало из воды в виде трифторацетатной соли (24 мг).
1Н ЯМР (CD3OD) δ 1,86-1,96 (м, 4Н), 2,06-2,14 (м, 4Н), 2,79-2,93 (м, 4Н), 3,09-3,32 (м, 8Н), 3,73-3,82 (м, 4Н), 4,55-4,65 (м, 2Н), 7,05-7,15 (м, 6Н), 7,28-7,33 (м, 2Н); LC-МС[M-H]+ 489,2.
5-(4-(2-Оксобензимидазолин-1-ил)пиперидино)пентанол (55-LH-27A)
Соединение 55-LH-27 приготавливали согласно процедуре, используемой для приготовления 55-LH-10 с использованием 5-бромо-1-пентанола (1,0 г, 6,0 ммоль). После 10 дней при 60°C добавляли воду, продукт фильтровали с получением 0,79 г названного соединения.
1Н ЯМР (CD3OD) δ 1,35-1,50 (м, 2Н), 1,55-1,65 (м, 4Н), 1,70-1,85 (м, 2Н), 2,10-2,25 (м, 2Н), 2,40-2,60 (м, 4Н), 3,05-3,15 (м, 2Н), 3,50-3,60 (м, 2Н), 4,25-4,40 (м, 1Н), 7,05-7,15 (м, 3Н), 7,35-7,45 (м, 1Н); 13С ЯМР (CD3OD) δ 23,8, 26,5, 28,4, 32,3, 50,7, 53,1, 58,4, 61,6, 109,4, 109,6, 121,0, 121,3, 128,5, 129,1, 155,1; LC-МС[M-H]+ 304,3.
1,5-бис(4-(2-оксобензимидазолин-1-ил)пиперидино)пентан (55-LH-31A)
Соединение (55-LH-31A) приготавливали согласно процедуре, используемой для приготовления 55-LH-25А с использованием 55-LH-27А (0,30 г, 1,0 ммоль). Остаток очищали на препаративной ВЭЖХ [Luna колонка (21,2×250 мм, 15 мкм C18(2), 0,1% TFA в H2O/0,1% TFA в CH3CN/H2O (8:2) 9:1 градиент к 0:100)]. Испаряли растворитель, и остаток растворяли в воде и дихлорметане. Добавляли гидроокись аммония до pH 10 и высушивали органический слой (Na2SO4), фильтровали и концентрировали. Остаток растворяли в MeOH и добавляли трифторуксусную кислоту (5 мкл). Трифторацетат очищали на препаративной ВЭЖХ [Luna колонка (21,2×250 мм, 15 мкм C18(2), 0,1% TFA в H2O/0,1% TFA в CH3CN/H2O (8:2) (9:1 градиент к 0:100)]. Растворитель испаряли и к водному раствору добавляли NH4OH до pH 10. Продукт фильтровали и высушивали с получением 47 мг названного соединения.
1Н ЯМР (CD3OD) δ 1,37-1,46 (м, 2Н), 1,59-1,68 (м, 4Н), 1,74-1,82 (м, 4Н), 2,16-2,25 (м, 4Н), 2,44-2,60 (м, 8Н), 3,12-3,20 (м, 4Н), 4,28-4,38 (м, 2Н), 7,02-7,08 (м, 6Н), 7,36-7,41 (м, 2Н); 13С ЯМР (CD3OD) δ 25,6, 26,6, 28,4, 50,7, 53,1, 58,3, 109,4, 109,6, 121,0, 121,3, 128,5, 129,1, 155,1; LC-МС[M-H]+ 503,1.
1,3-бис(4-(2-оксобензимидазолин-1-ил)пиперидино)пропан (55-LH-3B)
В пробирку помещали 4-(2-оксобензимидазолин-1-ил)пиперидин (1,09 г, 5 ммоль), 1-хлоро-3-йодопропан (250 мкл, 2 ммоль), K2CO3 (0,69 г, 5 ммоль) и этанол (10 мл) и взбалтывали при 60°C в течение шести дней. Добавляли воду, этилацетат и MeOH. Органический слой испаряли и остаток очищали на хроматографической колонке [(SiO2, 5% NH4OH в MeOH/этилацетате (1:9)] и затем путем препаративной ВЭЖХ [Luna колонка (21,2×250 мм, 15 мкм C18(2), 0,1% TFA в H2O/0,1% TFA в CH3CN/H2O (8:2) (9:1 градиент к 0:100)]. Растворитель испаряли и к водному раствору добавляли NH4OH до pH 10. Продукт отфильтровывали, промывали водой и высушивали с получением 235 мг названного соединения.
1Н ЯМР (CD3OD) δ 1,76-1,88 (м, 6Н), 2,20-2,28 (м, 4Н), 2,48-2,62 (м, 8Н), 3,14-3,22 (м, 4Н), 4,28-4,38 (м, 2Н), 7,02-7,09 (м, 6Н), 7,35-7,40 (м, 2Н); 13С ЯМР (CD3OD) δ 24,0, 28,4, 50,7, 53,1, 56,3, 109,4, 109,5, 121,1, 121,3, 128,5, 128,2, 155,1; LC-МС[M-H]+ 475,4.
1,3-бис(1-фенил-4-оксо-1,3,8-триазаспиро[4,5]декан-8-ил)пропан (55-LH4-3A)
В пробирку помещали 1-фенил-1,3,8-триазаспиро[4,5]декан-4-он (0,29 г, 1,25 ммоль), 1-хлоро-3-йодопропан (0,10 г, 0,5 ммоль), K2CO3 (0,17 г, 1,25 ммоль) и этанол (2 мл) и взбалтывали при 60°C в течение ночи. Добавляли воду и этилацетат. Продукт отфильтровывали и высушивали до получения 154 мг названного соединения.
1Н ЯМР (CD3OD) δ 1,69-1,83 (м, 6Н), 2,43-2,49 (м, 4Н), 2,57-2,67 (м, 4Н), 2,84-2,90 (м, 8Н), 4,68 (с, 4Н), 6,82-6,87 (м, 2Н), 6,99-7,04 (м, 4Н), 7,22-7,27 (м, 4Н); 13С ЯМР (CD3OD) δ 23,9, 28,8, 49,5, 56,5, 59,4, 59,7, 116,5, 119,4, 128,9, 143,6, 178,2; LC-МС[M-H]+ 503,4.
3-[4-(2-Оксобензимидазолин-1-ил)пиперидино]-1-(4-бутилпиперидино)пропан (55-LH-11C)
В пробирку помещали 4-(2-оксобензимидазолин-1-ил)пиперидин (0,13 г, 0,6 ммоль), 1-хлоро-3-йодопропан (64 мкл, 0,6 ммоль), K2CO3 (0,173 г, 1,25 ммоль) и этанол (2 мл) и взбалтывали при 60°C в течение пяти дней. Добавляли 4-Бутилпиперидин (0,85 г, 0,6 ммоль) и смесь взбалтывали при 60°C дополнительно в течение двух дней. Добавляли воду и этилацетат. Органический слой высушивали (Na2SO4), отфильтровывали и концентрировали. Остаток очищали на хроматографической колонке [(SiO)2, 5% NH4OH в MeOH/этилацетате (1:9)], препаративная жидкостная хроматография - масс-спектрометрия LC-MS [Waters симметрия C18 (19×50 мм, частицы 5 мкм), 0,15% TFA в H2O/0,15% TFA в CH3CN/H2O (95:5) (9:1 градиент к 0:100)] и путем препаративной ВЭЖХ [Luna колонка (21,2×250 мм, 15 мкм C18 (2), 0,1 % TFA в H2O/0,1 % TFA в CH3CN/H2O (8:2) (9:1 градиент к 0:100)]. Растворитель испаряли и к водному раствору добавляли NH4OH до pH 10. Органический слой высушивали (Na2SO4), фильтровали и выпаривали с получением 11,4 мг названного соединения.
1Н ЯМР (CD3OD) δ 0,88-0,93 (м, 3Н), 1,18-1,34 (м, 9Н), 1,68-1,83 (м, 6Н), 1,97-2,06 (м, 2Н), 2,15-2,24 (м, 2Н), 2,38-2,58 (м, 6Н), 2,94-3,01 (м, 2Н), 3,10-3,17 (м, 2Н), 4,26-4,36 (м, 1Н), 7,02-7,08 (м, 3Н), 7,36-7,39 (м, 1Н); 13С ЯМР (CD3OD) δ 13,2, 22,8, 23,7, 28,4, 28,9, 29,7, 35,6, 36,2, 50,8, 53,1, 53,9, 56,4, 56,9, 109,4, 109,5, 121,0, 121,3, 128,5, 129,2, 155,1; LC-МС[M-H]+ 399,3.
1,3-бис(4-бутилпиперидино)пропан (40-LH-67)
В пробирку помещали 4-бутилпиперидин (0,13 г, 0,9 ммоль), 1-хлоро-3-йодпропан (107 мкл, 1,0 ммоль), K2CO3 (0,35 г, 2,5 ммоль) и этанол (4 мл) и смесь взбалтывали при 60°C в течение ночи. Добавляли воду и этилацетат. Органический слой выпаривали и остаток очищали препаративной жидкостной хроматографией - масс-спектрометрией LC-MS [Waters симметрия C18 (19×50 мм, частицы 5 мкм), 0,15% TFA в H2O/0,15% TFA в CH3CN/H2O (95:5) (9:1 градиент к 0:100)] с получением 6,4 мг названного соединения.
1Н ЯМР (CDCl3) δ 0,84-1,10 (м, 6Н), 1,16-1,32 (м, 18Н), 1,62-1,74 (м, 6Н), 1,82-1,91 (м, 4Н), 2,26-2,32 (м, 4Н), 2,86-2,92 (м, 4Н); 13С ЯМР (CDCl3) δ 14,3, 23,1, 25,0, 29,3, 32,7, 36,1, 36,6, 54,4, 57,6; LC-МС[M-H]+ 323,4.
1,3-бис[4-(2-оксобензимидазолин-1-ил)пиперидино]-2-пропанол (55-LH-30B)
В пробирку помещали 4-(2-оксобензимидазолин-1-ил)пиперидин (0,44 г, 2 ммоль), эпихлоргидрин (78 мкл, 1 ммоль), K2CO3 (0,35 г, 2,5 ммоль) и этанол (3 мл) и смесь взбалтывали при 60°C в течение 19 дней. Добавляли воду и продукт фильтровали до получения 400 мг сырого продукта, 150 мг которого очищали путем препаративной ВЭЖХ [Luna колонка (21,2×250 мм, 15 мкм C18(2), 0,1% TFA в H2O/0,1% TFA в CH3CN/H2O (8:2) (9:1 градиент к 0:100)], с получением 50 мг названного соединения.
1Н ЯМР (CD3OD) δ 1,76-1,84 (м, 4Н), 2,32-2,66 (м, 12Н), 3,20-3,28 (м, 4Н), 4,01-4,08 (м, 1Н), 4,28-4,38 (м, 2Н), 7,02-7,09 (м, 6Н), 7,35-7,40 (м, 2Н); 13С ЯМР (CD3OD) δ 28,4, 28,4, 50,7, 53,2, 54,2, 62,6, 65,4, 109,4, 109,5, 121,1, 121,3, 128,5, 128,2, 155,1; LC-МС[M-H]+ 491,0.
1,3-бис(4-фенил-1-пиперазинил)пропан (55-LH-15)
В пробирку помещали 4-фенилпиперазин (191 мкл, 1,25 ммоль), 1-хлоро-3-йодопропан (54 мкл, 0,5 ммоль), K2CO3 (0,17 г, 1,25 ммоль) и этанол (3 мл) и смесь взбалтывали при 60°C в течение пяти дней. Добавляли воду, продукт фильтровали и высушивали с получением 145 мг названного соединения.
1Н ЯМР (CD3OD) δ 1,76-1,86 (м, 2Н), 2,44-2,51 (м, 4Н), 2,63-2,69 (м, 8Н), 3,17-3,22 (м, 8Н), 6,81-6,86 (м, 2Н), 6,94-6,99 (м, 4Н), 7,20-7,26 (м, 4Н); 13С ЯМР (CD3OD) δ 23,4, 49,1, 53,1, 56,5, 116,3, 120,0, 128,9, 151,5; LC-МС[M-H]+ 365,2.
1,3-бис(4-(2-нитро-4-трифторметилфенил)-1-пиперазинил)пропан (55-LH-16B)
В пробирку помещали 4-(2-нитро-4-трифторметилфенил) пиперазин (0,34 г, 1,25 ммоль), 1-хлоро-3-йодопропан (54 мкл, 0,5 ммоль), K2CO3 (0,17 г, 1,25 ммоль) и этанол (3 мл) и смесь взбалтывали при 60°C в течение пяти дней. Добавляли воду, продукт фильтровали и высушивали. В результате перекристаллизации (из 2-пропанола) получали 226 мг названного соединения.
1Н ЯМР (CD3OD) δ 1,74-1,83 (м, 2Н), 2,46-2,52 (м, 4Н), 2,61-2,66 (м, 8Н), 3,18-3,23 (м, 8Н), 7,37-7,42 (м, 2Н), 7,76-7,79 (м, 2Н), 8,04-8,07 (м, 2Н); 13С ЯМР (CD3OD) δ 23,4, 50,4, 52,7, 56,2, 121,3, 121,9, 123,5, 123,8, 129,9, 141,2, 148,0;
LC-МС[M-H]+ 591,2.
1,3-бис(4-(2-бензотиазолил)пиперидино)пропан (55-LH-46)
В пробирку помещали (4-(2-бензотиазолил)пиперидин (0,15 г, 0,69 ммоль), 1-хлоро-3-йодопропан (36 мкл, 0,34 ммоль), K2CO3 (97 мг, 0,70 ммоль) и этанол (2 мл) и смесь взбалтывали при 60°C в течение пяти дней. Добавляли воду, продукт фильтровали и высушивали с получением 138 мг названного соединения.
1Н ЯМР (CD3OD) δ 1,74-1,84 (м, 2Н), 1,90-2,03 (м, 4Н), 2,14-2,26 (м, 8Н), 2,41-2,48 (м, 4Н), 3,04-3,20 (м, 6Н), 7,36-7,42 (м, 2Н), 7,44-7,51 (м, 2Н), 7,89-7,96 (м, 4Н); 13С ЯМР (CD3OD) δ 23,6, 32,0, 41,2, 53,2, 56,6, 121,7, 122,0, 125,0, 126,1, 134,4, 152,8, 176,8; LC-МС[M-H]+ 477,1.
1,3-бис(4-(2-бензотиазолил)пиперидино)-2-пропанол (55-LH-47)
В пробирку помещали 4-(2-бензотиазолил)пиперидин (0,15 г, 0,69 ммоль), эпихлорогидрин (27 мкл, 0,34 ммоль), K2CO3 (97 мг, 0,70 моль) и этанол (2 мл) и взбалтывали при 60°C в течение пяти дней. Добавляли воду, продукт фильтровали и высушивали с получением 140 мг названного соединения.
1Н ЯМР (CD3OD) δ 1,90-2,05 (м, 4Н), 2,10-2,20 (м, 4Н), 2,21-2,52 (м, 8Н), 3,07-3,18 (м, 6Н), 3,96-4,04 (м, 1Н), 7,35-7,42 (м, 2Н), 7,44-7,51 (м, 2Н), 7,88-7,96 (м, 4Н); 13С ЯМР (CD3OD) δ 32,2, 32,2, 41,2, 53,4, 54,2, 63,2, 65,7, 121,7, 122,0, 125,0, 126,1, 134,4, 152,8, 177,1; LC-МС[M-H]+ 493,1.
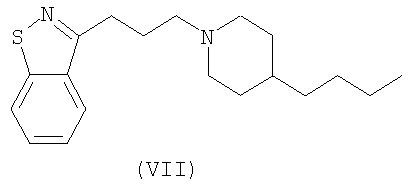
В некоторых вариантах осуществления изобретения соединения настоящего изобретения включают соединение Формулы VII, описанное в US Patent No. 6627645
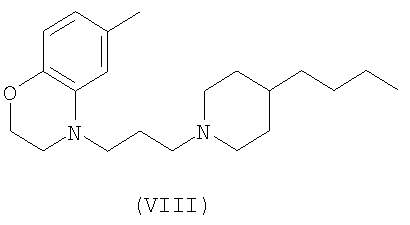
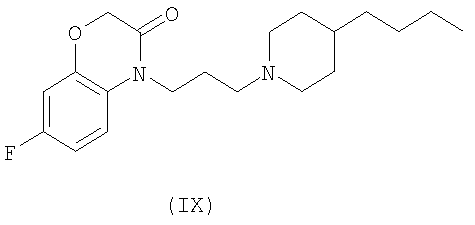
и соединения Формул VIII и IX, которые описаны в US Appl. No. 10/329455 (publication number 20030176418)
Некоторые соединения настоящего изобретения могут существовать как стереоизомеры, включая в себя оптические изомеры. Изобретение включает все стереоизомеры - как рацемические смеси таких стереоизомеров, так и отдельные энантиомеры, которые могут быть отделены согласно способам, хорошо известным специалистам в данной области техники.
Примеры фармацевтически приемлемых аддитивных солей включают соли присоединения неорганических и органических кислот, такие как гидрохлорид, гидробромид, фосфат, сульфат, ацетат, цитрат, лактат, тартрат, малеат, фумарат, манделат и оксалат; и соли присоединения неорганических и органических оснований, такие как гидроокись натрия, и Три(гидроксиметил)аминометан (ТРИС, трометан).
В дополнение к применению соединения в качестве химического сырья, соединения изобретения могут применяться как часть фармацевтических препаратов, содержащих подходящие фармацевтически приемлемые носители, включающие наполнители и добавки, которые обеспечивают переработку соединений в препараты для использования в фармации. Предпочтительно, чтобы препараты, особенно те препараты, которые могут применяться перорально или местно и которые могут использоваться для предпочтительного введения, такие как таблетки, драже, пастилки длительного действия и капсулы, полоскания и лосьоны для полости рта, гели, жидкие суспензии, ополаскиватели для волос, гели для волос, шампуни и также препараты, которые могут применяться ректально, такие как суппозитории, так же как и растворы, подходящие для инъекций и для местного или перорального применения, содержали около 0,01 до 99 процентов, предпочтительно около 0,25 до 75 процентов активного соединения (активных соединений) вместе с наполнителем.
Нетоксичные фармацевтически приемлемые соли соединений настоящего изобретения также включены в объем настоящего изобретения. Аддитивные соли кислот получают, смешивая раствор агонистов М1 рецепторов, описанных в этом документе, с раствором фармацевтически приемлемой нетоксичной кислоты, такой как соляная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, лимонная кислота, винная кислота, угольная кислота, фосфорная кислота, щавелевая кислота и т.п. Основные соли получают, смешивая раствор М1 селективных рецепторов, описанных в настоящем изобретении, с раствором фармацевтически приемлемого нетоксичного основания, такого как гидроксид натрия, гидроксид калия, гидроксид холина, карбонат натрия Трис и т.п.
Фармацевтические композиции настоящего изобретения могут вводиться любому животному, на которое соединения изобретения оказывают благоприятное влияние. Прежде всего среди животных рассматриваются млекопитающие, например люди, хотя настоящее изобретения не предназначается быть настолько ограниченным.
Агонисты М1 рецептора и его фармацевтические композиции могут быть введены любым способом, которое достигает предназначенной цели. Например, введение может быть парентеральным, подкожным, внутривенным, внутримышечным, интраперитонеальным, трансдермальным, спинальным, внутричерепным, интраназальным или местным. Альтернативно или одновременно введение может быть пероральным. Вводимая дозировка будет зависеть от возраста, состояния здоровья и веса получающего назначение, вида параллельного лечения, если таковое имеется, частоты лечения и характера желаемого эффекта.
Фармацевтические препараты агонистов М1 рецепторов, описанных в этом документе, изготовлены известным способом, например посредством обычного смешивания, гранулирования, дражирования, процессов растворения или лиофилизации. Таким образом, фармацевтические препараты для перорального применения, если желательно или необходимо получить форму таблеток или драже, могут быть получены путем смешивания активных соединений с твердыми наполнителями, необязательно измельчения получившейся смеси и переработки смеси гранул после добавления подходящих добавок.
Пригодными наполнителями являются, в частности, наполнители, такие как сахариды, например лактоза или сахароза, маннит или сорбит, препараты целлюлозы и/или фосфаты кальция, например трикальций фосфат или кислый фосфат кальция, так же как и связующие вещества типа крахмального клейстера, использование, например, крахмала кукурузы, крахмала пшеницы, крахмала риса, картофельного крахмала, желатина, траганта, метилцеллюлозы, гидроксипропилметилцеллюлозы, натрия карбоксиметилцеллюлозы и/или поливинилпирролидона. По желанию могут быть добавлены такие расщепляющие средства как вышеупомянутые крахмалы, а также карбоксиметилкрахмал, сшитый поливинилпирролидон, агар, или альгиновая кислота или их соли, такие как альгинат натрия. Добавки - это прежде всего средства, регулирующие скорость потока, и смазывающие вещества, например кварц, тальк, стеариновая кислота или их соли, такие как стеарат магния или стеарат кальция, и/или полиэтиленгликоль. Ядро драже покрывается подходящими покрытиями, которые, при необходимости, могут быть устойчивыми к желудочному соку. Для этой цели могут использоваться концентрированные растворы сахаридов, которые могут необязательно содержать, гумми-арабик тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, лакирующие растворы и подходящие органические растворители или смеси растворителей. Для получения покрытия, устойчивого к желудочному соку, используются растворы подходящих препаратов целлюлозы, такие как ацетилцеллюлоза фталат или гидроксипропилметилцеллюлоза фталат. К покрытиям таблеток или драже могут быть добавлены красящие вещества или пигменты, например, для идентификации или для того, чтобы отличать комбинации доз активного соединения.
Другие фармацевтические препараты для перорального применения, включают плотнонасаженные капсулы из желатина, а также мягкие, герметичные капсулы из желатина и пластификатора, такого, как глицерин или сорбит. Плотнонасаженные капсулы могут содержать активные соединения в форме гранул, которые могут смешиваться с наполнителями, такими как лактоза, связующими веществами типа крахмалов, и/или смазывающими веществами типа талька или стеарата магния и, необязательно, стабилизаторами. В мягких капсулах активные соединения предпочтительно растворять или в подходящих жидкостях, таких как жирные масла или вазелиновое масло. Кроме того, могут добавляться стабилизаторы.
Возможные фармацевтические препараты для ректального применения включают в себя, например, клизмы или суппозитории, которые состоят из комбинации одного или большего числа активных соединений в основе суппозитория. Подходящей для суппозитория основой, являются, например, натуральные или синтетические триглицериды или парафиновые углеводороды. Кроме того, также возможно использовать желатиновые ректальные капсулы, которые состоят из комбинации активных соединений с основой. Возможные материалы основы включают, например, жидкие триглицериды, полиэтиленгликоль или парафиновые углеводороды.
Подходящие лекарственные формы для парентерального введения включают в себя водные растворы активных соединений в водорастворимой форме, например водорастворимые соли и щелочные растворы. Кроме того, могут назначаться суспензии активных соединений как адекватные масляным суспензиям для инъекций. Подходящие липофильные растворители или носители включают жирные масла, например кунжутное масло или синтетические сложные эфиры жирных кислот, например этилолеат или триглицериды или полиэтиленгликоль-400 (соединения растворимы в ПЭГ-400). Водные суспензии для инъекций могут содержать вещества, которые увеличивают вязкость суспензии, включающие, например, натриевую соль карбоксиметилцеллюлозы, сорбит и/или декстран. Необязательно суспензия может содержать стабилизаторы.
Композиции в объеме настоящего изобретения включают все композиции, в которых соединения, описанные в настоящей заявке, содержатся в количестве, эффективном для достижения предназначенной цели. В то время как отдельные потребности могут меняться, определение оптимальных диапазонов эффективных количеств каждого компонента находится в пределах компетентности в данной области техники. Как правило, соединения могут вводиться млекопитающим, например людям, перорально в дозе от 0,0025 до 50 мг/кг в день или эквивалентного количества фармацевтически приемлемой соли этого соединения, в расчете на вес тела млекопитающего, подвергаемого лечению. Предпочтительно перорально вводить около 0,01 до около 10 мг/кг. Для внутримышечного введения доза в большинстве случаев составляет половину пероральной дозы.
Однократная пероральная доза может содержать около 0,01 до около 50 мг, предпочтительно около 0,1 до около 10 мг соединения. Однократная доза может применяться один или более раз в день в количестве одной или более таблеток, каждая из которых будет содержать около 0,1 до около 10, удобной дозой будет около 0,25 до 50 мг соединения или его сольватов.
В лекарственной форме для наружного применения соединение может находиться в концентрации около 0,01 до 100 мг на грамм носителя. В предпочтительных вариантах осуществления изобретения соединение находится в концентрации около 0,07-1,0 мг/мл, более предпочтительно около 0,1-0,5 мг/мл, наиболее предпочтительно около 0,4 мг/мл.
Следующие примеры изложены для обеспечения рядовых специалистов в данной области техники полным раскрытием и описанием того, как изготавливать и использовать настоящее изобретение, и не рассматриваются как ограничивающие объем настоящего изобретения, и при этом ниже перечисленные эксперименты не рассматриваются как единственно выполненные эксперименты.
Пример 1
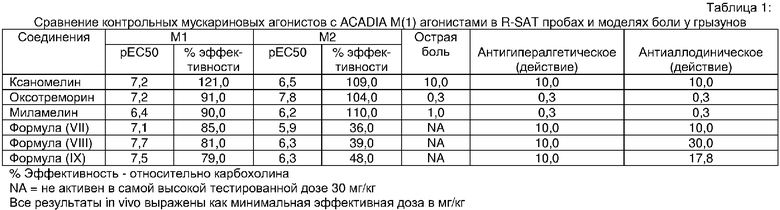
Проба функциональных рецепторов, Receptor Selection and Amplification Technology (R-SAT), по существу, как раскрыто в US Patent Nos. 5,707,798, 5,912,132 and 5,955,281), использовалась для исследования фармакологических свойств известных и новых мускариновых агонистов. Соответственно были тестированы ксаномелин, оксотреморин, миламелин и соединения формул VII, VIII и IX.
Эти эксперименты обеспечили молекулярный профиль или фингерпринт, для каждого из этих средств по наиболее значащим рецепторам, М(1) и М(2) подтипам мускариновых рецепторов. Как показано в Таблице 1, три контрольных средства: ксаномелин, оксотреморин, миламелин являются мощными и эффективными полными агонистами и М(1) и М(2) подтипов рецепторов. Напротив, соединения Формул VII, VIII, и IX являются мощными и эффективными М(1) агонистами, но в отношении М(2) рецепторов они являются только слабыми частичными агонистами.
CCI/Термальная гипералгезия
В асептических условиях и согретом состоянии крысам вводилась анестезия комбинацией 1,6 мл кетамина (100 мг/мл) и 1,6 мл ксилазина (100 мг/мл) в 6,8 мл 0,9% физиологического раствора в объеме 0,1 мл/100 г. Левая четырехглавая мышца была выбрита и полностью обработана раствором йода. Седалищный нерв был открыт на уровне седалищной вырезки дистальнее седалищной трифуркации. Нерв был очень тщательно выделен из подлежащей мышцы и соединительной ткани, чтобы не вызвать его непосредственную травму. В качестве шовного материала использовался 4-0 хромированный кетгут, наложены четыре полусвободных лигатуры вокруг седалищного нерва, начиная от максимально проксимального уровня близко от седалищной вырезки, с интервалами примерно 1 мм друг от друга и заканчивая проксимальнее седалищной трифуркации. Увеличивали натяжение лигатур до появления у животных небольшого подергивания левой лапы или мускулатуры, окружающей нерв. Мышечный разрез закрывался шелковым шовным материалам 4-0, на кожу накладывались зажимы для раны. За животными внимательно наблюдали до их полного восстановления после анестезии. Ту же самую операцию проводили для экспериментов с аллодинией и гипералгезией.
Для тестов с гипералгезией крысы помещались в тонированную пластмассовую коробку с прозрачным стеклянным полом с регулируемой температурой, поддерживаемой на уровне 31±1°C. Пол содержал фокальный излучатель высокой температуры (проекционная галогеновая лампа CXL/CXP, 50 W, 8v, USHIO, Tokyo). Излучатель высокой температуры на нижней стороне стекла был подвижным и имел излучаемый поток приблизительно 3 мм в диаметре, который можно было расположить под плантарной поверхностью задней лапы крысы.
До начала теста крыс помещали в тонированные пластмассовые коробки, где им позволяли освоиться в новой обстановке 10-20 минут. После этого излучатель высокой температуры помещали под плантарной поверхностью задней лапы. С активацией излучателя высокой температуры одновременно запускался таймер. С рефлекторным движением задней лапы активизировался датчик движения, останавливая таймер и инактивируя излучатель высокой температуры. Термальный излучатель был отрегулирован так, чтобы среднее латентное время реакции для неоперированного животного было не более 20 секунд. У каждой крысы в течение двух дней провели предоперационные исходные измерения латентного времени, при этом измерения на плантарной поверхности левой задней лапы сделаны от трех до четырех раз. Были проведены от двух до трех послеоперационных исходных измерений латентного времени слева, до начала и после применения лечения. Измерения на 2 и 4 послеоперационные дни показали самую большую степень гипералгезии и таким образом использовались в этой пробе. Каждое животное было тестировано дважды, по меньшей мере, с 48 часами перерыва между каждым испытанием.
Термальная гипералгезия, проявленная на оперированной левой лапе, доказывается уменьшением латентного периода отдергивания при термальном раздражении. Максимальная гипералгезия наблюдалась со 2 по 4 послеоперационные дни. Латентные периоды отдергивания лапы на оперированной левой стороне постепенно возвращались к исходным уровням в течение от 5 до 12 дней послеоперационного периода. На неоперированную правую лапу проведенная операция не оказывала значительного воздействия, что доказано одинаковым латентным периодом отдергивания лапы в течение 12 дней тестирования.
Введение носителя в каждой группе не влияло на термальную гипералгезию. Напротив, контрольная доза мускариновых агонистов зависимо инвертировала термальную гипералгезию (Таблица 1). Ксаномелин инвертировал термальную гипералгезию [F(2,15)=57,43, p<0,001]. Последующее сравнение Dunnett's показало, что ксаномелин инвертировал термальную гипералгезию при 10 мг/кг (p<0,001), но не при 3 мг/кг (p>0,05) относительно носителя. Оксотреморин также инвертировал термальную гипералгезию [F(2,11)=13,74, p=0,0018]. Последующее сравнение показало, что латентный период отдергивания лапы после применения оксотреморина в дозе 1 мг/кг (18,468±1,532 с; p<0,001) и 0,3 мг/кг (13,683 с ± 1,36; p<0,05) статистически отличались от наполнителя. Значительная антигипералгезия также наблюдалась в исследованиях с миламелином, [F(2,14)=106,9, p<0,0001], при дозах 1 мг/кг p (p<0,001) и 0,3 мг/кг (p<0,0001) значительно увеличивающих латентный период отдергивания лапы. В сравнении морфин [F(3,20)=15,55, p<0,0001] вызывал значительную антигипералгезию в дозах 1 мг/кг (16,856 с ± 1,05, p<0,01) и 3 мг/кг (16,817 с ± 1,6, p<0,01).
Подобно контрольным мускариновым агонистам соединения Формул VII, VIII, и IX дозозависимо инвертировали термальную гипералгезию: Формула VII, F(4,29)=13,2, p<0,0001; Формула VIII, F(2,23)=6,066, p=0,0041; Формула IX, [F(4,24)=14,51, p<0,0001]. Последующее сравнение Dunnett's показало, что соединения Формул VII, VIII и IX инвертировали термальную гипералгезию в дозе 10 мг/кг (p<0,001).
CCI/Тактильная аллодиния
Начало и продолжительность выраженной механической послеоперационной аллодинии наступает приблизительно через 10-14 дней и длится в течение примерно двух месяцев. В пределах этого периода аллодинии и для каждого конкретного эксперимента по аллодинии, перед и после введения лекарственного средства проводили измерения с использованием семи волосков von Frey, которые обозначались в журнале (10* силы, требуемой для сгибания волоска, мг) и варьировались от 2 до 26 граммов ((#'s 4,31-5,46). На каждый волосок над серединой плантарной поверхности левой поврежденной задней лапы нажимали перпендикулярно с достаточной для небольшого изгиба силой, и нажим удерживался в течение 6-8 секунд, начиная с самого тонкого калиброванного волоска и продолжая до самого толстого. Регистрировалась положительная реакция, когда поврежденная лапа резко отдергивалась, и эта реакция подтверждалась как положительная в тесте со следующим, более толстым калиброванным волоском и той же самой реакцией. В расчет принималась только дважды отмеченная реакция. Если максимальная сила в 26 граммов не достигала реакции, это считалось границей пороговой чувствительности для аллодинического поведения и данные регистрировались. Животные считались аллодиническими, когда послеоперационные исходные измерения составляли 6 граммов и ниже. Проводились два исходных дня измерений с одним циклом испытаний в день. В день тестирования препаратов проводился один цикл исходных измерений, делали адекватную i.p. (внутрибрюшинную) премедикацию, и регистрировался второй цикл измерений. Каждое животное использовалось в многократных экспериментах, с одним лечебным воздействием за эксперимент и адекватным периодом выведения между экспериментами.
Выраженная тактильная аллодиния отмечалась, начиная с 8 дня и продолжаясь в течение 35 дней послеоперационного периода. Оценка тактильной чувствительности после данных мускариновых агонистов проводилась в пределах этих послеоперационных отсчетов времени. В группе, у которой применялся носитель, послеоперационные предварительные показатели статистически незначительно отличались от исходного уровня, [F(2,95)=1,275, p>0,05]. Три контрольных мускариновых агониста также дозозависимо инвертировали тактильную аллодинию. Ксаномелин инвертировал тактильную аллодинию, (F(3,22)=12,58, p<0,0001] в дозах 10,0 и 30 мг/кг (p<0,01). Оксотреморин также инвертировал тактильную аллодинию [F(3,19)=32,49, p<0,0001] в дозе 0,3 мг/кг (p<0,05) и 1 мг/кг (p<0,01). Результаты для CI-979 были сходны с результатами других мускариновых агонистов, [F(2,14)=24,38, p<0,0001]. В дозах 0,3 мг/кг (p<0,05) и 1 мг/кг (p<0,01), CI-979 увеличивал пороги тактильной чувствительности. Морфин проявлял антиаллодинию подобно этим мускариновым агонистам, [F(2,17)=6,257, p=0,0106].
Вместе с тем, подобно контрольным мускариновым агонистам соединения Формул VII, VIII и IX дозозависимо инвертировали тактильную аллодинию: Формула VII, F(3,20)=29,11, p<0,0001; Формула VIII, F(3,23)=11,764, p<0,0001; Формула IX, F(4,28)=7,569, p=0,0004. Последующее сравнение Dunnett's показало, что Формула VII инвертировала тактильную аллодинию в дозе 10 мг/кг (p<0,001), Формула VIII инвертировала тактильную аллодинию в дозе 30 мг/кг (p=0,08) и Формула IX инвертировала тактильную аллодинию в дозе 17,8 мг/кг (p<0,001).
Острая Термальная Аналгезия
Вода нагревалась и поддерживалась при 55°C ± 1°C с помощью горячей пластины, регулируемой датчиком. Самкам крыс, весом приблизительно 200 г - 250 г, за несколько дней давали освоиться, помещая и доставая их из пластмассовой клетки для фиксации крыс. В день эксперимента каждая крыса была помещена в клетку для фиксации за 1 минуту до самого испытания. Примерно один дюйм хвоста погружали в воду после включения таймера. Как только хвост полностью извлекался из воды, таймер останавливали и зарегистрировали время. Если животное не реагировало в пределах 10 секунд, экспериментатор удалял хвост из нагретой воды и регистрировал это как максимальный показатель. Был получен один цикл исходных измерений. Применяли исследуемое соединение и после адекватного предварительного интервала процедуру повторяли. Каждое животное использовалось в многократных экспериментах, с одним лечебным воздействием за эксперимент и адекватным периодом выведения, по меньшей мере, 48 часов между экспериментами. Результаты тестирования соединений на острой ноцицепции показаны в Таблице 1. Предварительный исходный латентный период отдергивания хвоста был 2,281 с ± 0,25. Применение носителя не изменяло латентного периода отдергивания хвоста со средним временем ожидания 3,16 с ± 0,21. Ксаномелин [F(2,16)=4,952, p<0,05], оксотреморин [F(2,17)=20,50, p<0,05], и миламелин [F(2,17)=19,25, p<0,05] произвели значительный антиноцицептивный эффект. При этом ксаномелин был активен в дозе 10,0 мг/кг, оксотреморин в дозе 0,3 мг/кг и дозе 1,0 мг/кг и миламелин в дозе 1,0 мг/кг. Морфин [F(3,23)=5,903, p<0,01] был антиноцицептивен в дозе 10 мг/кг.
Поразительно, но соединения Формул VII, VIII и IX выявили отсутствие активности в облегчении острой термальной боли (Таблица 1). Таким образом, соединения Формул VII, VIII и IX инвертируют хроническую нейропатическую боль, но не обладают острой антиноцицептивностью.
Пример 2
Мускариновые побочные эффекты
Все из тестированных эталонных мускариновых агонистов рецепторов оказывали холинергические побочные эффекты, как показано в Таблице 2. Количество животных, у которых наблюдался каждый побочный эффект в каждой дозе показано по сравнению с количеством животных в тесте (N). Ксаномелин в дозе 30 мг/кг вызывал диарею, слюноотделение и сонливость у всех тестированных с этой дозой животных, принимая во внимание, что более низкая доза 10 мг/кг вызывала только диарею у 2 из 11 тестированных животных. Оксотреморин в дозе 1 мг/кг вызывал все пять из измеренных мускариновых побочных эффектов у большинства крыс, тогда как 0,3 мг/кг вызывала только диарею, слюноотделение и сонливость. Миламелин в дозе 1 мг/кг подобно оксотреморину вызывал четыре из измеренных побочных эффекта, кроме тремора, тогда как более низкая доза 0,3 мг/кг вызывала преимущественно диарею. Напротив, ни одно из соединений Формул VII, VIII или IX в дозах от 3,0 мг/кг до 30 мг/кг не вызывало ни один из этих побочных эффектов. Таким образом, контрольные мускариновые агонисты вызывают тяжелые мускариновые побочные эффекты в дозах, сходных с теми, что требуются для достижения эффективности у этих моделей боли, принимая во внимание, что соединения Формул VII, VIII и IX не вызывают эти побочные эффекты в дозах, которые эффективны в моделях нейропатической боли.
Профиль побочных эффектов эталонных мускариновых агонистов
Пример 3
Операция - частичное седалищное лигирование (PSL) / Тактильная аллодиния
В асептических условиях и согретом состоянии самцам мышей (C57B1/6) проводилась ингаляционная анестезия 1 % изофлураном (1 Lpm - 1 литр в минуту). Левая четырехглавая мышца была выбрита и полностью обработана раствором йода. Пальпировалась седалищная вырезка и проводился разрез от вырезки до середины четырехглавой мышцы. Седалищный нерв был открыт на уровне седалищной вырезки дистальнее седалищной трифуркации. Нерв был очень тщательно выделен из подлежащей мышцы и соединительной ткани, чтобы не вызвать его непосредственную травму. При необходимости к открытым тканям прикладывали стерильный солевой раствор, чтобы предотвратить высыхание. Седалищный нерв выводился непосредственно дистальнее седалищной вырезки и, используя 10-0 шовную полипропиленовую синюю мононить, накладывалась лигатура, пережимающая от 1/3 до 1/2 седалищного нерва. Увеличивали натяжение лигатур до появления у животных небольшого подергивания левой лапы. Мышечный разрез закрывался при необходимости полипропиленовым шовным материалам 7-0, на кожу накладывались зажимы для раны. После операции применялся бупринекс в дозе 0,075 мг/кг SC. За животными внимательно наблюдали до их полного восстановления после анестезии.
Выраженная тактильная аллодиния начинает проявляться на 4-6 день после PSL операции и продолжается в течение примерно одного месяца. В пределах времени аллодинии и для каждого конкретного эксперимента с аллодинией проводились измерения предварительные и после применения препаратов с восемью волосками von Frey, которые регистрировались в журнале (10* силы, требуемой для сгибания волоска, мг) и варьировались от 0,07-4 граммов. На каждый волосок над серединой плантарной поверхности левой поврежденной задней лапы нажимали перпендикулярно с достаточной для небольшого изгиба силой, и нажим удерживался в течение 6-8 секунд, начиная с самого тонкого калиброванного волоска и продолжая до самого толстого. Регистрировалась положительная реакция, когда поврежденная лапа резко отдергивалась, и эта реакция подтверждалась как положительная, в тесте со следующим, более толстым калиброванным волоском и той же самой реакцией. В расчет принималась только дважды отмеченная реакция. Если максимальная сила в 10 граммов не достигала реакции, это считалось границей пороговой чувствительности для аллодинического поведения и данные регистрировались. Животные считались аллодиническими, когда послеоперационные исходные измерения составляли ~60% от предоперационных исходных измерений. Проводились два исходных дня измерений с одним циклом испытаний в день. В день тестирования препаратов проводился один цикл исходных измерений, делали адекватную i.p. (внутрибрюшинную) или sc (подкожную) премедикацию, и регистрировался второй цикл измерений. Каждое животное использовалось в многократных экспериментах, с одним лечебным воздействием за эксперимент и адекватным периодом выведения между экспериментами.
Трансгенные мыши (KO) с М(1) мускариновыми рецепторами не отличаются от дикого типа мышей (WT) ни в отношении предоперационной тактильной чувствительности (t=1,094, df=15, p=0,2913), ни в отношении послеоперационной аллодинии (t=0,2338, df=15, p=0,8183). Как М(1) KO (t=5,765, df=7, p=0,0007), так и WT (t=3,551, df=8, p=0,0075) мыши развивали разумную тактильную аллодинию в след за PSL-операцией. Несмотря на значительное ослабление тактильной аллодинии у мышей WT соединениями Формулы IX в дозе 30 мг/кг, эффекты соединения Формулы IX полностью отсутствовали у М(1) KO мышей, подтверждая роль М(1) рецепторов в нейропатической боли in vivo. Контрольная тактильная чувствительность перед PSL-операцией и после PSL-операции показана в Фиг.2 для сравнения с чувствительностью после применения соединений Формулы IX у дикого типа (+/+) и трансгенных (-/-) М(1) рецепторных мышей.
Далее, как изображено в Фиг.3, соединения Формулы IX значительно инвертируют тактильную аллодинию у мышей с PSL нейропатическим повреждением после интрацеребровентрикулярного (i.c.v.) применения, что предполагает супраспинальный механизм действия, согласующийся с распределений М(1) рецепторов.
Источники информации
Bartolini A., Ghelardini C., Fantetti L., Malcangio M., Malmberg-Aiello P., Giotti A. Role of muscarinic receptor subtypes in central antinociception. Br. J. Pharmacol. 105:77-82, 1992.
Brodie M.S. and Proudfit H.K. Hypoalgesia induced by the local injection of carbachol into the nucleus raphe magnus. Brain Research 291:337-342, 1984.
Capone F., Aloisi A.M., Carli G., Sacerdote P., Pavone F. Oxotremorine-induced modifications of the behavioral and neuroendocrine responses to formalin pain in male rats. Brain Res. 830:292-300, 1999.
Duttaroy A, Gomeza J, Gan JW, Siddiqui N, Basile AS, Harman WD, Smith PL, Felder CC, Levey AI, Wess J. Evaluation of muscarinic agonist-induced analgesia in muscarinic acetylcholine receptor knockout mice. Mol. Pharmacol. 62:1084-93, 2002.
Hartvig P., Gillberg P.G., Gordh T. Jr., Post C. Cholinergic mechanisms in pain and analgesia. Trends Pharmacol. Sci. Dec. Suppl.:75-79, 1989.
Hwang J.-H, Hwang K.-S, Leem J.-K., Park P.-H., Han S.-M., Lee D.-M. The antiallodynic effects of intrathecal cholinesterase inhibitors in a rat model of neuropathic pain. Anesthesiology 90:492-494, 1999.
Lee E.J., Sim J.Y, Park J.Y., Hwang J.H., Park P.H., Han S.M. Intrathecal carbachol and clonidine produce a synergistic antiallodynic effect in rats with a nerve ligation injury. Can J Anaesth 49:178-84, 2002.
Naguib M. and Yaksh T.L. Characterization of muscarinic receptor subtypes that mediate antinociception in the rat spinal cord. Anesth. Analg. 85:847-853, 1997.
Pedigo N.W., Dewey W.L. and Harris L.S. Determination and characterization of the antinociceptive avtivity if intraventricularly administered acetylcholine in mice. J. Pharmacol. Exp. Ther. 193: 845-852, 1975.
Prezewlocka В., Mika J., Capone F., Machelska H., Pavone F. Intrathecal oxotremorine affects formalin-induced behavior and spinal nitric oxide synthase immunoreactivity in rats. Pharmacol. Biochem. Behav. 62:531-536, 1999.
Shannon H.E., Womer D.E., Bymaster P.P., Calligaro D.O., DeLapp N.C., Mitch C.H., Ward J.S., Whitesitt C.A., Swedberg M.D.B., Sheardown M.J., Fink-Jensen A., Olesen P.H., Rimvall K., Sauerberg P. In vivo pharmacology of butylthio[2.2.2.] (LY297802/NNC11-1053), an orally acting antinociceptive muscarinic agonist. Life Sci. 60:969-976, 1997.
Sheardown M.J., Shannon H.E., Swedberg M.D.B., Suzdak P.D., Bymaster F.P., Olesen P.H., Mitch C.H., Ward J.S., Sauerberg P. M1 receptor agonist activity is not a requirement for muscarinic antinociception. J. Pharmacol. Exp. Ther. 281:868-875, 1997.
| название | год | авторы | номер документа |
|---|---|---|---|
| АГОНИСТЫ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2678835C2 |
| Производные гомопиперазинил- и гомопиперидинил-хиназолин-4(3H)-она, обладающие мультимодальной активностью в отношении боли | 2020 |
|
RU2833258C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ МУСКАРИНОВЫЕ АГОНИСТЫ И КОМПОЗИЦИИ, ИХ ПРИМЕНЕНИЕ И СПОСОБЫ ЛЕЧЕНИЯ | 2002 |
|
RU2292346C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, ХАРАКТЕРИЗУЮЩИЕСЯ МУЛЬТИМОДАЛЬНОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ БОЛИ | 2014 |
|
RU2709482C1 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛИДИНОНА В КАЧЕСТВЕ АГЕНТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2288919C2 |
| НОВЫЙ КЛАСС СЕЛЕКТИВНЫХ АГОНИСТОВ СОМАТОСТАТИНОВЫХ РЕЦЕПТОРОВ | 2014 |
|
RU2603962C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИЗОХИНОЛИНА, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СИНАПТИЧЕСКОГО ЗАХВАТА ДОПАМИНА И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2293728C2 |
| АЗОЛЬНЫЕ И ТИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2436779C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| ПИПЕРИДИНИЛ-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛОНА КАК ИНГИБИТОРЫ Rho-КИНАЗЫ | 2006 |
|
RU2414467C2 |
Изобретение относится к медицине, а именно к неврологии, и может быть использовано для лечения нейропатической боли. Изобретение заключается в том, что для лечения нейропатической боли вводят соединение, которое селективно активирует мускариновые рецепторы подтипа М(1), но при этом не облегчает острую боль. Использование изобретения позволяет лечить нейропатическую боль, не ослабляя способности воспринимать обычные раздражители. 9 з.п. ф-лы, 3 ил., 2 табл.
1. Способ лечения нейропатической боли, не ослабляющий острую боль, включающий идентификацию субъекта, нуждающегося в таком лечении, и предоставление субъекту эффективного количества, по меньшей мере, одного соединения, которое селективно активирует мускариновые рецепторы подтипа М(1).
2. Способ по п.1, в котором субъект проявляет гипералгезию.
3. Способ по п.1, в котором субъект проявляет аллодинию.
4. Способ по п.1, в котором нейропатическая боль связана с диабетом, вирусной инфекцией, синдромом раздраженной кишки, ампутацией, раком или химическим повреждением.
5. Способ по п.1, в котором соединение выбирается из группы, состоящей из соединений формул VII, VIII и IX
6. Способ по п.1, где соединение имеет структуру формулы (I)
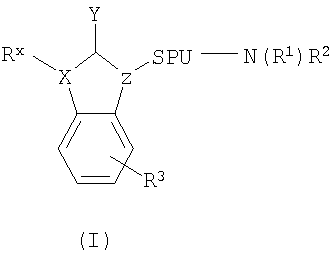
в которой X выбран из группы, состоящей из С, О, N и S; Z выбран из группы, состоящей из СН и N; Y выбран из группы, состоящей из =O, =N и =S или их таутомеров, таких как Y-алкилированные таутомеры; SPU представляет единицу - спейсер, обеспечивающую расстояние d между Z и N, в котором -SPU- представляет бирадикал, выбранный из группы, состоящей из -(CR6R7)n-A- и -С3-8-циклоалкил-, в котором n находится в диапазоне от 1 до 5, такого типа, как 1, 2, 3, 4, или 5, и А отсутствует или произвольно замещен на -С3-8-циклоалкил-; N вместе с R1 и R2 образуют гетероциклическое кольцо, в котором гетероциклическое кольцо выбрано из группы, состоящей из пергидроазоцина, пергидроазепина, пиперидина, пирролидина, азетидина, азиридина и 8-азабицикло[3,2,1]октана, и в котором гетероциклическое кольцо замещено одним или более заместителями R4, выбранными из группы, состоящей из гидрокси, галогена, C1-8-алкила, С3-8-циклоалкила, С1-8-алкокси, C1-8-алкилкарбонила, C1-8-алкилидена, С2-8-алкенила, С2-8-алкинила, C1-6-алкилоксиимино и С1-6-алкилоксиамино, каждый из, которых может быть необязательно замещен заместителем R5 и в котором, по меньшей мере, одним из упомянутых заместителей R4 является R4′, выбранный из группы, состоящей из C1-8-алкила, С3-8-циклоалкил, С1-8-алкокси, C1-8-алкилкарбонила, C1-8-алкилидена, C1-8-алкилоксиимино, и C1-8-алкилоксиамино, каждый из которых может быть необязательно замещен заместителем R5; R5 выбран из группы, состоящей из водорода, галогена, гидрокси, C1-8-алкила, C1-8-алкокси, С3-8-циклоалкила, С3-8-гетероциклила, С1-8-алкилкарбонила, C1-8-алкилидена, C2-8-алкенила и С2-8-алкинила; Rx может отсутствовать или выбираться из группы, состоящей из водорода, необязательно замещенного C1-8-алкила, необязательно замещенного С3-8-циклоалкила, необязательно замещенного C2-8-алкенила, необязательно замещенного С2-8-алкинила, необязательно замещенного арила, необязательно замещенного гетероарила CH2N(R5)(R5), CH2-OR5, СН2-SR5, CH2-O-C(=O)R5, CH2-O-C(=S)R5; R3 может присутствовать 0-4 раза и выбираться из группы, состоящей из галогена, гидрокси, необязательно замещенного C1-8-алкила, C1-8-алкокси, необязательно замещенного C1-8-алкилидена, необязательно замещенного C2-8-алкенила, необязательно замещенного С2-8-алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного С3-8-циклоалкила, необязательно замещенного С3-8-гетероциклила, и необязательно замещенного C1-8-алкилкарбонила; и каждый R6 и каждый R7 независимо выбирается из группы, состоящей из водорода, галогена, гидрокси, необязательно замещенного C1-8-алкила, C1-8-алкокси, необязательно замещенного C1-8-алкилидена, необязательно замещенного С2-8-алкенила, необязательно замещенного С2-8-алкинила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного С3-8-циклоалкила, необязательно замещенного С3-8-гетероциклила, и необязательно замещенного С1-8-алкилкарбонила.
7. Способ по п.1, где соединение имеет структуру формулы (II)

в которой Z1 представляет CR1 или N, Z2 представляет CR2 или N, Z3 представляет CR3 или N, и Z4 представляет CR4 или N, где не больше, чем два из Z1, Z2, Z3 и Z4 являются N; W1 представляет О, S или NR5, один из W2 и W3 представляет N или CR6, а другой из W2 и W3 представляет CG; W1 является NG, W2 представляет CR5 или N, и W3 представляет CR6, или N; или W1 и W3 представляет N, и W2 представляет NG; G имеет формулу (III)
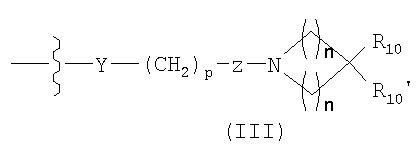
Y представляет О, S, CHOH, -NHC(O)-, -C(O)NH-, -С(O)-, ОС(O)-, -(О)СО-, -NR7-, -CH=N-, или отсутствует; р=1, 2, 3, 4 или 5; Z представляет CR8R9 или отсутствует; каждое t=1, 2, или 3; каждое R1, R2, R3 и R4, независимо, является Н, амино, гидроксилом, галогеном или прямой или разветвленной цепью C1-6алкил, С2-6алкенил, С2-6алкинил, С1-6гетероалкил, С1-6галогеналкил, -CN, -CF3-OR11, -COR11, -NO2, -SR11, -NHC(O)R1, -C(O)NR12R13, -NR12R3, -NR11C(O)NR12R13, -SO2NR12R13, -OC(O)R11, -O(CH2)qNR12R13, или -(CH2)qNR12R13, где q - целое число от 2 до 6, или R1 и R2 вместе образуют -NH-N=N-, или R3 и R4 вместе образуют -NH-N=N-; каждое R5, R6 и R7, независимо, является Н, C1-6алкилом; формилом; С3-6циклоалкилом; С5-6арилом, необязательно замещенным галогеном или С1-6алкилом; или C5-6 гетероарилом, необязательно замещенным галогеном или C1-6алкилом; каждое R8 и R9, независимо, является Н или прямой или разветвленной цепью C1-8алкила; R10 представляет прямую или разветвленную цепь C1-8алкила; С2-8алкенил, С2-8алкинил, C1-8алкилиден, C1-8алкокси, С1-8гетероалкил, C1-8аминоалкил, С1-8галогеналкил, С1-8алкоксикарбонил, С1-8гидроксиалкокси, C1-8гидроксиалкил, -SH, C1-8алкилтио, -О-СН2-С5-6арил, -С(O)-С5-6арил, замещенный С1-3алкилом или галогеном, С5-6арил, С5-6циклоалкил, С5-6гетероарил, С5-6гетероциклоалкил, -NR12R13, -C(O)NR12R13, -NR11C(O)NR12R13, -CR11R12R13, -OC(O)R11, -(O)(CH2)sNR12R13 или -(CH2)sNR12R13, где s является целым числом от 2 до 8; R10' является Н, прямой или разветвленной цепью C1-8алкила, C2-8алкенилом, С2-8алкинилом, C1-8алкилиденом, C1-8алкокси, C1-8гетероалкилом, С1-8аминоалкилом, C1-8галогеналкилом, С1-8алкоксикарбонилом, C1-8гидроксиалкокси, C1-8гидроксиалкилом, или C1-8алкилтио; каждый R11, независимо, является Н, прямой или разветвленной цепью C1-8алкила, С2-8алкенилом, C2-8алкинилом, С2-8гетероалкилом, С2-8аминоалкилом, С2-8галогеналкилом, С1-8алкоксикарбонилом, С2-8гидроксиалкилом, -С(O)-С5-6арилом, замещенным С1-3алкилом, или галогеном,
С5-6арилом, С5-6гетероарилом, C5-6 циклоалкилом, С5-6гетероциклоалкилом,
-C(O)NR12R13, -CR5R12R13, -(CH2)tNR12R13, где t является целым числом от 2 до 8; и каждый R12 и
R13, независимо, являются Н, С1-6алкилом; С3-6циклоалкилом; С5-6арилом, необязательно замещенным галогеном или C1-6алкилом; или С5-6гетероарилом, необязательно замещенным галогеном или C1-6алкилом; или R12 и R13 вместе образуют циклическую структуру; или фармацевтически приемлемой солью, сложным эфиром или его пролекарством.
8. Способ по п.1, где соединение имеет структуру формулы (IV)
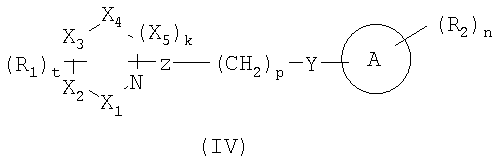
в которой X1, Х2, Х3, Х4 и Х5 выбраны из С, N и О; k является 0 или 1; t является 0, 1 или 2; R1 представляет прямую или разветвленную цепь С1-8алкила, С2-8алкенил, С2-8алкинил, C1-8алкилиден, C1-8алкокси, C1-8гетероалкил, С1-8аминоалкил, C1-8галогеналкил, C1-8алкоксикарбонил, С1-8 гидроксиалкокси, С1-8гидроксиалкил, -SH, C1-8алкилтио, -O-CH2-C5-6арил, -С(O)-С5-6арил, замещенный С1-3алкилом или галогеном; С5-6арил или C5-6циклоалкил необязательно содержащий 1 или более гетероатомов, выбранных из N, S и О; -C(O)NR3R4, -NR3R4, -NR3С(O)NR4R5, -CR3R4, -ОС(O)R3, -(O)(CH2)sNR3R4 или -(СН2)sNR3R4; где R3, R4 и R5 являются одинаковыми или различными, каждый независимо выбирается из Н, C1-6алкила; C5-6арила, необязательно содержащим 1 или более гетероатомов, выбранных из N, О и S, и необязательно замещенным галогеном или C1-6алкилом; С3-6циклоалкила; или R3 и R4 вместе с атомом N, если он присутствует, образуют циклическую кольцевую структуру, содержащую 5-6 атомов, выбранных из С, N, S и О; и s является целым числом от 0 до 8; А представляет C5-12арил или C5-7циклоалкил, каждый из которых необязательно содержит 1 или более гетероатомов, выбранных из N, S и О; R2 представляет Н, амино, гидроксил, галоген, или прямую или разветвленную цепь C1-6алкила, С2-6алкенил, С2-6алкинил, С1-6алкокси, C1-6гетероалкил, C1-6аминоалкил, С1-6галогеналкил, C1-6алкилтио, C1-6алкилкарбонил, -CN, -CF3, -OR3, -COR3, NO2, -NHR3, -NHC(O)R3, -С(O)NR3R4, -NR3R4, -NR3C(O)NR4R5, -ОС(O)R3, -С(O)R3R4, -O(CH2)qNR3, -CNR3R4 или -(СН2)qNR3R4; где q = целое число от 1 до 6; n=0, 1, 2, 3 или 4, группы R2, при n>1, являются одинаковыми или различными; р=0 или целое число от 1 до 5; Y представляет О, S, СНОН, -NHC(O)-, -C(O)NH-, -C(O)-, -OC(O)-, NR7 или -CH=N-, и R7 представляет Н или С1-4алкил; или отсутствует; и Z представляет CR8R9, в котором R8 и R9 независимо выбраны из Н, и прямой или разветвленной цепи C1-8алкила; или фармацевтически приемлемой соли, сложного эфира или его пролекарства.
9. Способ по п.1, где соединение имеет структуру формулы (V)
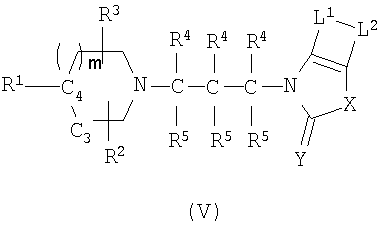
в которой R1 представляет монорадикал, выбранный из группы, состоящей из необязательно замещенного C1-6алкила, необязательно замещенного С2-6-алкилидена, необязательно замещенного С2-6-алкенила, необязательно замещенного С2-6-алкинила, необязательно замещенного O-C1-6алкила, необязательно замещенного O-С2-6-алкенила, необязательно замещенного O-С2-6-алкинила; необязательно замещенного S-C1-6алкила, необязательно замещенного S-С2-6-алкенила, необязательно замещенного S-С2-6-алкинила; m=0, 1 или 2; С3-С4 представляет СН2-СН или СН=С, или C4 представляет СН, и С3 отсутствует; R2 и R3 независимо выбраны из группы, состоящей из водорода, необязательно замещенного C1-6алкила, необязательно замещенного О-C1-6алкила, галогена, гидрокси или выбраны так, что R2 и R3 вместе образуют кольцевую систему; каждый R4 и R5 независимо выбраны из группы, состоящей из водорода, галогена, гидрокси, необязательно замещенного C1-6 алкила, необязательно замещенного O-C1-6 алкила, необязательно замещенного арил-С1-6 алкила, и необязательно замещенного арилгетероалкила; L1 и L2 представляет бирадикалы, независимо выбранные из группы, состоящей из -C(R6)=C(R7), -C(R6)=N-, -N=C(R6)-, -S-, -NH- и -O-; причем только один из L1 и L2 может быть выбран из группы, состоящей из -S-, -NH- и -O-; Y выбран из группы, состоящей из О, S и Н2; Х представляет бирадикал, выбранный из группы, состоящей из -C(R6)(R7)-C(R6)(R7)-, -C(R6)=C(R7)-, -O-C(R6)(R7)-, C(R6)(R7)-O-, -S-C(R6)(R7)-, -C(R6)(R7)-S-, -N(RN)=C(R6)-(R7)-, -C(R6)(R7)-N(RN)-, -C(R6)(R7)-C(R6)(R7)-C(R6)(R7)-, -O-C(R6)(R7)-C(R6)(R7)-, S-C(R6)(R7)-C(R6)(R7)-, N(RN)-С(R6)(R7)-С(R6)(R7)-, -C(R6)(R7)-C(R6)(R7)-O, -C(R6)(R7)-C(R6)(R7)-S, -C(R6)(R7)-C(R6)(R7)-N(RN)-, -C(R6)(R7)-C(R6)=C(R7)- и -C(R6)=C(R7)-C(R6)(R7), в котором R6 и R7 независимо выбираются из группы, состоящей из водорода, галогена, гидрокси, нитро, циано, NRNRN, N(RN)-С(O)N(RN), необязательно замещенного C1-6алкила, С2-6-алкенила, С2-6-алкинила, необязательно замещенного O-С1-6-алкила, необязательно замещенного O-арила, необязательно замещенного О-С2-6-алкенила, необязательно замещенного О-С2-6-алкинила, в котором RN выбран из группы, состоящей из водорода, и необязательно замещенного С1-6-алкила.
10. Способ по п.1, где соединение имеет структуру формулы (VI)
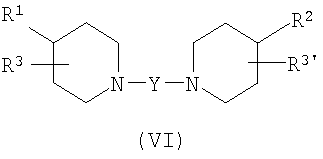
в которой Y представляет бирадикал (CR4R5)m-Z-C(R4R5)n; в котором сумма m+n составляет от 1 до 7; Z выбран из группы, состоящей из C(R4R5), C(O), О, N(R6), S, O-C(O), N(R6)C(O), С(O)-O и Р; и R4 и R5 независимо выбраны из группы, состоящей из водорода, галогена, гидрокси, нитро, NR6N6′, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного С3-8-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного C1-6-алкила, необязательно замещенного С1-6-алкокси, необязательно замещенного фенокси, необязательно замещенного С2-8-алкенила и необязательно замещенного С2-8-алкинила; и в которой R1 и R2 независимо выбраны из группы, состоящей из необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного С3-8-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного С1-6-алкила, необязательно замещенного С1-6-алкокси, необязательно замещенного С2-8-алкенила и необязательно замещенного С2-8-алкинила; в которой R3 и R3′ независимо выбраны из группы, состоящей из водорода, галогена, гидрокси, нитро, NR6N6′, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного С3-8-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного С1-6-алкила, необязательно замещенного C1-6-алкокси, необязательно замещенного С2-8-алкенила и необязательно замещенного С2-8-алкинила; и R6 и R6′ независимо выбраны из группы, состоящей из водорода, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного С3-8-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного C1-6-алкила, необязательно замещенного С1-6-алкокси, необязательно замещенного С2-8-алкенила и необязательно замещенного С2-8-алкинила.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 6271196, 07.08.2001 | |||
| ПРИМЕНЕНИЕ БЕНЗОНАФТАЛИНОВЫХ ПРОИЗВОДНЫХ ДЛЯ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ, ПРЕДНАЗНАЧЕННЫХ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ НЕРВНОЙ СИСТЕМЫ | 1997 |
|
RU2160098C2 |
| HWANG J.H | |||
| et al | |||
| The antiallodynic effects of intrathecal cholinesterase inhibitors in a rat model of neuropathic pain | |||
| Anesthesiology | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |

