Настоящее изобретение относится к новым спиропентациклическим соединениям, их получению, содержащим их фармацевтическим композициям и к их применению в качестве лекарственных средств. Соединения, предлагаемые в настоящем изобретении, применимы для лечения ожирения и других нарушений.
В частности, настоящее изобретение относится к соединениям формулы (I)
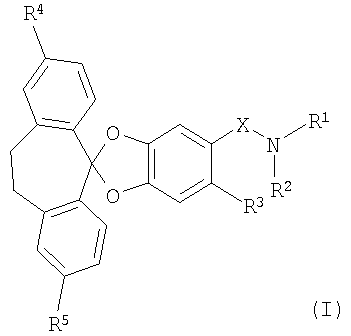
в которой
R1 и R2 совместно с атомом азота, к которому они присоединены, образуют 4-, 5-, 6- или 7-членное моноциклическое насыщенное гетероциклическое кольцо, которое необязательно может содержать 1 или 2 дополнительных гетероатома, независимо выбранных из О, N и S;
R3 обозначает водород, галоген, низш. алкил или цианогруппу;
R4 и R5 каждый независимо обозначает галоген; и
Х обозначает -С(O)- или -SO2-;
или к их фармацевтически приемлемой соли.
Выделены 2 разных подтипа каннабиноидных рецепторов (СВ1 и СВ2), и они относятся к подгруппе рецепторов, связанных с белком G. Также описана альтернативная сплайсированная форма СВ1, СВ1А, но она не отличается от СВ1 по способности связывать лиганд и активировать рецептор (D.Shire, С.Carrillon, M.Kaghad, В.Calandra, M.Rinaldi-Carmona, G.Le Fur, D.Caput, P.Fenara, J.Biol. Chem. 270 (8) (1995) 3726-31). Рецептор СВ1 расположен в основном в головном мозге, тогда как рецептор СВ2 преимущественно распределен на периферии и расположен в основном в селезенке и клетках иммунной системы (S.Munro, K.L.Thomas, M.AbuShaar, Nature 365 (1993) 61-61). Поэтому для исключения побочных эффектов необходимо соединение, селективное по отношению к СВ1.
Δ9-Тетрагидроканнабинол (Δ9-ТГК) является главным психотропным соединением индийской конопли (Y.Gaoni, R.Mechoulam, J.Am.Chem. Soc., 86 (1964) 1646), canabis sativa (марихуана), которое длительное время применяется в медицине (R.Mechoulam (Ed.) in "Cannabinoids as therapeutic Agents", 1986, pp.1-20, CRC Press). Δ9-ТГК является неселективным агонистом рецептора СВ1/2 и в США выпускается под названием дронабинол (маринол®) для ослабления рвоты, вызванной химиотерапией (РВХ), и для устранения потери массы у страдающих СПИД пациентов путем стимулирования аппетита. В Великобритании наболинон (LY-109514, Cesamet®), синтетический аналог Δ9-ТГК, применяется для РВХ (R.G.Pertwee, Pharmaceut. Sci. 3 (11) (1997) 539-545, E.M.Williamson, F.J.Evans, Drugs 60 (6) (2000) 1303-1314).
Установлено, что анандамид (арахидонилэтаноламин) является эндогенным лигандом (агонистом) рецептора СВ1 (R.G.Pertwee, Curr. Med. Chem., 6 (8) (1999) 635-664; W.A.Devane, L.Hanus, A.Breuer, R.G.Pertwee, L.A.Stevenson, G.Griffin, D.Gibson, A.Mandelbaum, A.Etinger, R.Mechoulam, Science 258 (1992) 1946-9). Анандамид и 2-арахидоноилглицерин (2-AG) на пресинапсическом нервном окончании негативно модулируют аденилатциклазу и потенциалочувствительные Са2+ каналы и активируют пропускающий внутрь К+ канал (V.Di Marzo, D.Melck, Т.Bisogno, L.De Petrocellis, Trends in Neuroscience 21 (12) (1998) 521-8), тем самым влияя на высвобождение и/или действие нейротрансмиттера, что уменьшает высвобождение нейротрансмиттера (А.С.Porter, C.C.Felder, Pharmacol. Ther., 90 (1) (2001) 45-60).
Анандамид в качестве Δ9-ТГК усиливает питание по опосредуемому рецептором СВ1 механизму. Селективные антагонисты рецептора СВ1 блокируют усиление питания, связанное с введением анандамида (С.М.Williams, Т.С.Kirkham. Psychopharmacology 143 (3) (1999) 315-317; C.C.Felder, E.M.Briley, J.Axelrod, J.T.Simpson, K.Mackie, W.A.Devane, Proc. Natl. Acad. Sci. U.S.A. 90 (16) (1993) 7656-60) и приводят к подавлению аппетита и снижению массы (G.Colombo, R.Agabio, G.Diaz, C.Lobina, R.Reali, G.L.Gessa, Life Sci. 63 (8) (1998) L113-PL117).
Лептин является первичным сигналом, с помощью которого гипоталамус воспринимает состояние питания и изменяет потребление пищи и энергетический баланс. После временного ограничения количества пищи мыши с нарушенным рецептором СВ1 потребляют меньше корма, чем однопометные животные дикого типа, и антагонист рецептора СВ1, SR141716A, уменьшает потребление корма у мышей дикого типа с ненарушенным СВ1. Кроме того, нарушение сигналов с участием лептина связывают с повышенными уровнями эндоканнабиоидов в гипоталамусе, но не в мозжечке у страдающих ожирением мышей db/db и ob/ob и крыс Zucker. Введение больших доз лептина нормальным крысам и мышам ob/ob снижает уровни анандамида и 2-арахидоноилглицерина в гипоталамусе. Эти данные показывают, что в гипоталамусе эндоканнабиоиды могут тонизирующим образом активировать рецепторы СВ1 для поддержания потребления пищи и образуют часть нейронной цепи, регулируемой лептином (V.Di Marzo, S.K.Goparaju, L.Wang, J.Liu, S.Bitkai, Z.Jarai, F.Fezza, G.I.Miura, R.D.Palmiter, T.Sugiura, G.Kunos, Nature 410 (6830) 822-825).
SR-141716A, селективный антагонист/обратный агонист СВ1, в настоящее время проходит фазу III клинического исследования, посвященного лечению ожирения. В двойном слепом исследовании при контроле с помощью плацебо при дозах, равных 5, 10 и 20 мг в сутки, SR 141716 значительно снижает массу тела по сравнению с плацебо (F.Barth, M.Rinaldi-Carmona, M.Amone, Н.Heshmati, G.Le Fur, "Cannabinoid antagonists: From research tools to potential new drugs." Abstracts of Papers, 222nd ACS National Meeting, Chicago, IL, United States, August 26-30, 2001).
Другие соединения, предложенные в качестве антагонистов, соответственно обратных агонистов рецептора СВ1, представляют собой аминоалкилиндолы (ААИ; M.Pacheco, S.R.Childers, R.Arnold, F.Casiano, S.J.Ward, J.Pharmacol. Exp. Ther. 257 (1) (1991) 170-183), такие как 6-бром-(WIN54661; F.M.Casiano, R.Arnold, D.Haycock, J.Kuster, S.J.Ward, NIDA Res. Monogr. 105 (1991) 295-6) и 6-йодправадолин (АМ630, К.Hosohata, R.M.Quock, R.M.Hosohata, T.Н.Burkey, A.Makriyannis, P.Consroe, W.R.Roeske, Н.I.Yamamura, Life Sci. 61 (1997) 115-118; R.Pertwee, G.Griffin, S.Femando, X.Li, A.Hill, A.Makriyannis, Life Sci. 56 (23-24) (1995) 1949-55). Арилбензо[b]тиофен и бензо[b]фуран (LY320135, С.С.Felder, К.Е.Joyce, E.M.Briley, M.Glass, K.P.Mackie, K.J.Fahey, G.J.Cullinan, D.С.Hunden, D.W.Johnson, M.O.Chaney, G.A.Koppel, M.Brownstein, J.Pharmacol. Exp. Ther. 284 (1) (1998) 291-7) раскрыты BW09602248, US 5596106, 3-алкил-(5,5-дифенил)имидазолидиндионы (M.Kanyonyo, S.J.Govaerts, E.Hermans, J.Н.Poupaert, D.M.Lambert, Bioorg. Med. Chem. Lett. 9 (15) (1999) 2233-2236.), а также 3-алкил-5-арилимидазолидиндионы (F.Ooms, J.Wouters, O.Oscaro. T.Happaerts, G.Bouchard, P.-A.Carrupt, B.Testa, D.M.Lambert, J.Med.Chem. 45 (9) (2002) 1748-1756), известные как антагонисты рецептора СВ1, действуют на рецептор hCBl, как обратные агонисты. В WO 0015609 (FR 2783246-A1), WO 0164634 (FR 2805817-A1), WO 0228346, WO 0164632 (FR 2805818-A1), WO 0164633 (FR 2805810-A1) раскрыты замещенные производные 1-бис(арил)метилазетидина как антагонисты СВ1. В WO 0170700, WO 02076949 и WO 0276949 A1 производные 4,5-дигидро-1Н-пиразола описаны как антагонисты СВ1. В нескольких патентах и публикациях мостиковые и немостиковые производные 3-пиразолкарбоксамида раскрыты как антагонисты/обратные агонисты (W00132663, WO 0046209, WO 9719063, ЕР 658546, ЕР 656354, US 5624941, ЕР 576357, US 3940418, WO 03020217, WO 0335005, J.M.Mussinu et al., Bioorg. Med. Chem. 2003,11,251; S.Ruiu et al., J.Pharm.Expt.Ther., 2003, 306, 363). Пирроловые антагонисты каннабиноидного рецептора СВ1 описаны в публикации G.Tarzia et al., Bioorg. Med. Chem. 2003, 11, 3965. Фенетиламиды заявлены в качестве антагонистов/обратных агонистов каннабиноидного рецептора СВ1 в WO 03077847, WO 03082190, WO 03086288 и WO 03087037. Различные азагетероциклы (имидазолы, триазолы и тиазолы) описаны в W00337332, WO 03040107, WO 03063781, WO 03082833 и WO 03078413. Дифенилпиразинкарбоксамиды описаны в WO 03051850, дифенилпиридинкарбоксамиды - в WO 03084930 и дифенилбензолкарбоксамиды - в WO 03084943.
Объектом настоящего изобретения являются селективные, обладающие прямым действием антагонисты соответственно обратные агонисты рецептора СВ1. Такие антагонисты/обратные агонисты применимы при медикаментозном лечении, предпочтительно - при лечении и/или предупреждении заболеваний, которые связаны с модуляцией рецепторов СВ1.
Если не указано иное, то приведенные ниже определения предназначены для иллюстрации и определения значений и объема различных терминов, использованных для описания настоящего изобретения.
В настоящем описании "низш." означает группу, содержащую от 1 до 8, предпочтительно - от 1 до 4 атома (атомов) углерода.
Термин "алкил", по отдельности или в комбинации с другими группами, означает обладающий разветвленной или линейной цепью одновалентный насыщенный алифатический углеводородный радикал, содержащий от 1 до 20 атомов углерода, предпочтительно - от 1 до 16 атомов углерода, более предпочтительно - от 1 до 10 атомов углерода.
Термин "низш. алкил", по отдельности или в комбинации с другими группами, означает обладающий разветвленной или линейной цепью одновалентный алкильный радикал, содержащий от 1 до 8 атомов углерода, предпочтительно - от 1 до 4 атомов углерода. Для этого термина примерами также являются такие радикалы, как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, 3-метилбутил, н-гексил, 2-этилбутил и т.п.
Термин "галоген" означает фтор, хлор, бром или йод, предпочтительно - хлор и фтор.
Термин "фармацевтически приемлемые соли" включает соли соединений формулы (I) с неорганическими или органическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, малеиновая кислота, уксусная кислота, фумаровая кислота, янтарная кислота, виннокаменная кислота, метансульфоновая кислота, салициловая кислота, п-толуолсульфоновая кислота и т.п., которые нетоксичны для живых организмов. Предпочтительными солями с кислотами являются формиаты, малеаты, цитраты, гидрохлориды, гидробромиды и соли метансульфоновой кислоты, а особенно предпочтительными являются гидрохлориды.
В одном варианте осуществления настоящее изобретение относится к соединению формулы (I), определенному выше, в которой R1 и R2 совместно с атомом азота, к которому они присоединены, образуют 4-, 5-, 6- или 7-членное моноциклическое насыщенное гетероциклическое кольцо, которое необязательно может содержать 1 или 2 дополнительных гетероатома, независимо выбранных из группы, включающей О, N и S. В предпочтительном варианте осуществления R1 и R2 совместно с атомом азота, к которому они присоединены, образуют 6-членное моноциклическое насыщенное гетероциклическое кольцо, которое необязательно может содержать в цикле еще один атом кислорода. Наиболее предпочтительными гетероциклическими кольцами, образованными с помощью R1 и R2 совместно с атомом азота, к которому они присоединены, являются пиперидинильное и морфолиновое.
В другом варианте осуществления настоящее изобретение относится к соединению формулы (I), определенному выше, в которой R3 обозначает водород, галоген, низш. алкил или цианогруппу. Предпочтительным низш. алкил остатком R3 является метил. Предпочтительными галогенидными остатками R3 являются фтор и хлор, а фтор является особенно предпочтительным. Предпочтительно, если R3 обозначает водород или галоген, такой как фтор. В другом варианте осуществления настоящее изобретение относится к соединению формулы (I), определенному выше, в которой R4 и R5 каждый независимо обозначает галоген. Предпочтительно, если R4 и R5 оба обозначают фтор или R4 и R5 оба обозначают хлор. Наиболее предпочтительно, если R4 и R5 обозначают фтор.
В другом варианте осуществления настоящее изобретение относится к соединению формулы (I), определенному выше, в которой Х обозначает -С(O)- или -SO2-. Предпочтительно, если Х обозначает -С(O)-.
Предпочтительными соединениями общей формулы (I) являются соединения, независимо выбранные из группы, включающей:
4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]-морфолин,
4-[(2',8'-дихлор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5H]дибензо[а,d]циклогептен]-5-ил)карбонил]-пиперидин,
4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-
[5H]дибензо [a,d]циклогептен]-5-ил)карбонил]-пиперидин,
4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-
[5H]дибензо [a,d]циклогептен]-5-ил)сульфонил]-морфолин,
4-[(2',6,8'-трифтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5H]дибензо[а,d]циклогептен]-5-ил)карбонил]-морфолин,
4-[(2',6,8'-трифтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5H]дибензо[a,d]циклогептен]-5-ил)карбонил]-пиперидин,
и фармацевтически их приемлемые соли.
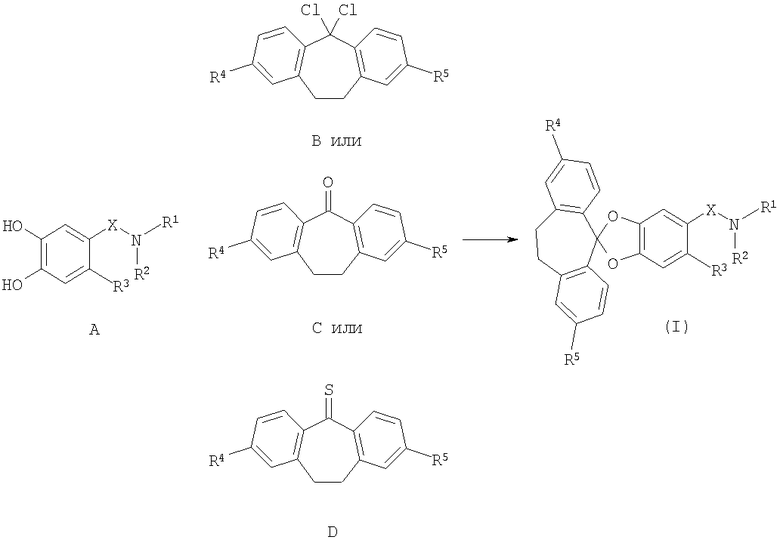
Способы получения соединений формулы I также являются объектом настоящего изобретения. Таким образом, настоящее изобретение также относится к способу получения соединений формулы (I), и этот способ включает:
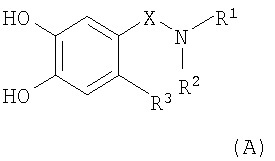
(а) кетализацию соединения формулы (А)
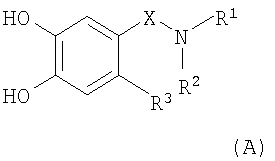 ,
,
в которой R1, R2, R3 и Х являются такими, как определено выше в настоящем изобретении;
с соединением формулы (В)
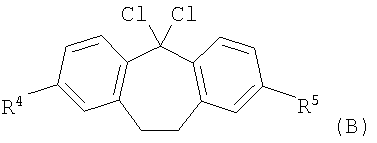 ,
,
в которой R4 и R5 являются такими, как определено выше в настоящем изобретении; или
(b) взаимодействие соединения формулы (А)
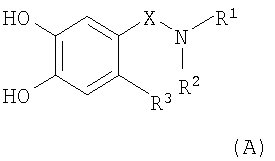 ,
,
в которой R1, R2, R3 и Х являются такими, как определено в п.1 формулы изобретения;
с соединением формулы (С)
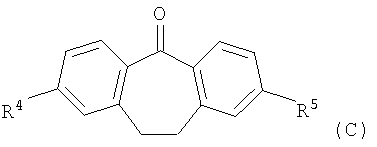 ,
,
в которой R4 и R5 являются такими, как определено выше в настоящем изобретении; или
(с) взаимодействие соединения формулы (А)
 ,
,
в которой R1, R2, R3 и Х являются такими, как определено выше в настоящем изобретении;
с соединением формулы (D)
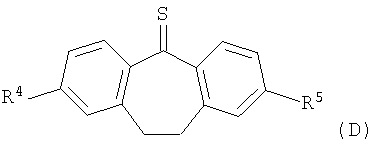
в которой R4 и R5 являются такими, как определено выше в настоящем изобретении.
При подробном описании соединения формулы (I), в которой R1-R5 и Х являются такими, как определено выше, можно получить по общим методикам, представленным на схеме 1 и обсужденным ниже.
В соответствии со схемой 1 промежуточный катехин формулы (А) можно кетализировать с помощью бис-замещенного производного дихлорметана формулы (В) в инертном растворителе (например, толуоле или пиридине) или в неразбавленном виде, в присутствии основания (например, пиридина) или без него, при повышенной температуре (например, >100°С) с получением продукта I. Альтернативно соединение формулы (I) можно получить по реакции промежуточного катехина формулы (А) с кетоном формулы (С) при повышенной температуре (например, >150°С) неразбавленного или в инертном растворителе (например, толуоле) без удаления воды или с ее с удалением путем отгонки или азеотропной отгонки или прибавления осушающих агентов (например, молекулярных сит или 2,2-диметоксипропана) по методикам, известным в данной области техники (см., например, Т.R.Kelly, A.Szabados, Y.-J.Lee, J.Org.Chem. 62 (2) (1997) 428). Альтернативно соединение формулы (I) можно получить по реакции промежуточного катехина формулы (А) с тиокетоном формулы (D) неразбавленным или в инертном растворителе (например, ацетонитриле) в присутствии основания (например, триэтиламина) или без него, с солью металла (например, AgI) по методикам, известным в данной области техники (см., например, I.Shibuya, E.Katoh, Y.Gama, A.Oishi, Y.Taguchi and Т.Tsuchiya, Heterocycles, 43 (1996) 851).
Схема 1:
Альтернативно соединение формулы (I) можно получить по реакции диазосоединения формулы (Е) с 1,2-дикетоном формулы (F) (схема 2) по методикам, известным в данной области техники (см., например, J.M.Fox, N.R.Goldberg and T.J.Katz, J.Org.Chem., 1998, 63, 7456). Диазосоединения формулы (Е) можно получить из соответствующих гидразонов по методикам, известным в данной области техники; 1,2-дикетоны формулы (F) можно получить из катехинов формулы (А) по методикам, известным в данной области техники.
Схема 2:
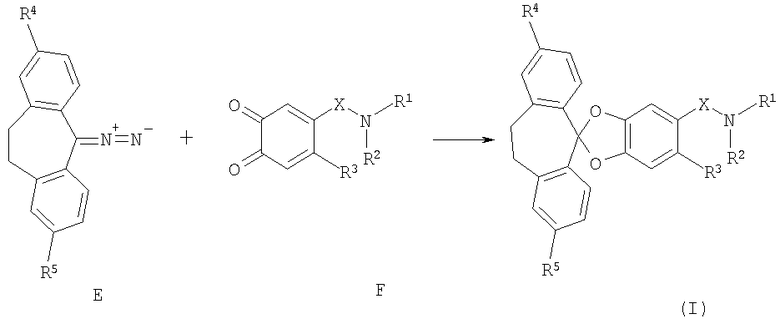
Бис-замещенные производные дихлорметана формулы (В) можно получить по методикам, известным в данной области техники, из соответствующего кетона по реакции с тионилхлоридом в присутствии ДМФ (диметилформамид) или другого N-формилирующего реагента, по реакции с пентахлоридом фосфора в присутствии подходящего растворителя или без него, например оксихлорида фосфора (схема 3).
Кетоны формулы (С) можно получить с помощью электрофильного ароматического замещения производного (G) оксалилхлоридом в присутствии кислоты Льюиса (например, трихлорида алюминия) в инертном растворителе (например, сероуглероде) (см., например, M.R. Pavia et al., Journal of Medicinal Chemistry 1992, 35, 4238-48), с помощью внутримолекулярной циклизации производного карбоновой кислоты формулы (Н) в присутствии кислоты Льюиса (например, полифосфорной кислоты) в присутствии инертного растворителя (например, 1,2-дихлорэтана) или без него (см., например, WO 9854180).
Тиокетоны формулы (D) можно легко получить из соответствующих дикетонов (С) по реакции с подходящим тионилирующим реагентом (например, пентасульфидом фосфора), по методикам, известным в данной области техники, в присутствии подходящего растворителя (например, toluene) в присутствии силана (например, гексаметилдисилоксана) или без него (см., например, T.J.Curphey, J.Org.Chem., 2002, 67, 6461).
Схема 3:
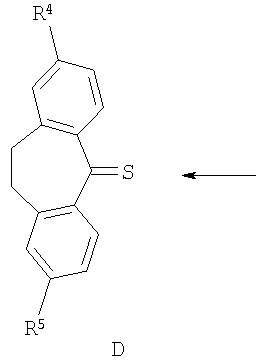
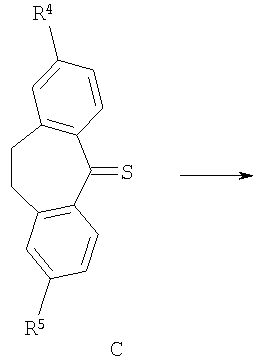
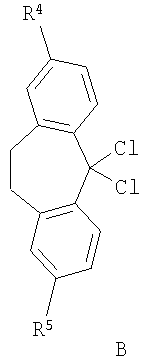
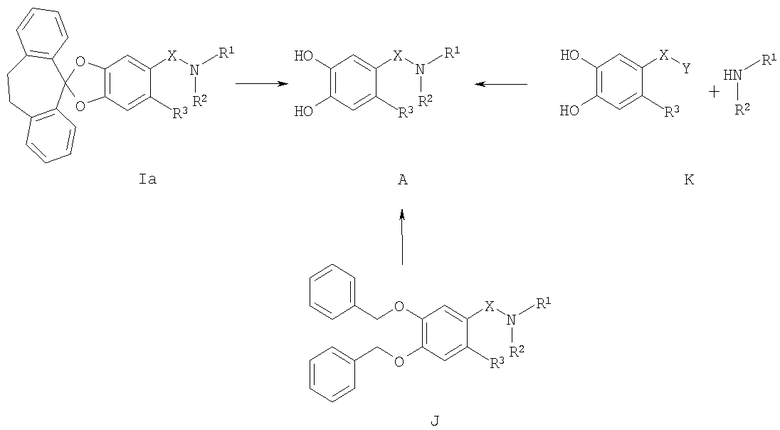
Катехины формулы А можно легко получить из соответствующих защищенных дифенилметиленовой группой кеталей формулы (Ia) путем обработки кислотой (например, трифторуксусной кислотой) в подходящем инертном растворителе (например, метиленхлориде) или путем обработки кислотой (например, трифторуксусной кислотой) в присутствии подходящего восстановительного реагента (например, триэтилсилана), неразбавленных или в подходящем инертном растворителе (например, метиленхлориде). Альтернативно катехин формулы А можно легко получить из соответствующего защищенного бис-бензильной группой катехина формулы (J) путем восстановления (например, гидрирования в присутствии подходящего катализатора, например палладия на угле, по методикам, известным в данной области техники). Альтернативно производное катехина формулы (К) можно ввести в реакцию сочетания с подходящим амином в подходящем инертном растворителе (например, ДМФ, метиленхлориде, пиридине или ТГФ (тетрагидрофуран)) в присутствии основания (например, триэтиламина). Любой из соответствующих хлорангидридов кислот (Х=СО, Z=Cl) или соответствующих сульфонилхлоридов (X=SO2, Z=Cl), или соответствующих карбоновых кислот (Х=СО, Z=OH) после активации подходящим реагентом реакции сочетания (например, карбонил диимидазолом) используют для получения катехинов формулы (А) по методикам, известным в данной области техники (схема 4). Защищенные дифенилметиленовой группой кетали формулы (Ia), защищенные бис-бензильной группой катехины формулы (J) и производное катехина формулы (K) можно получить по методикам, аналогичным описанным в литературе.
Схема 4:
Некоторые соединения формулы (I) могут содержать асимметрические центры и поэтому могут существовать более, чем в одной стереоизомерной форме. Поэтому настоящее изобретение также относится к соединениям, находящимся в основном в чистой изомерной форме по одному или большему количеству асимметрических центров, а также к их смесям, включая рацемические смеси. Такие изомеры можно получить с помощью асимметрического синтеза, например, с использованием хирального промежуточного продукта, или смеси можно разделить с помощью обычных методик, например, хроматографии (хроматографии использованием хирального сорбента или элюента), или и использованием разделяющего реагента.
Следует понимать, что соединения общей формулы (I), предлагаемые в настоящем изобретении, можно превратить в производные по функциональным группам, которые способны к обратному превращению в исходные соединения in vivo.
Как отмечено выше, соединения формулы (I) или их фармацевтически приемлемые соли можно применять в качестве терапевтически активных веществ, предпочтительно - в качестве терапевтически активных веществ, предназначенных для лечения и/или профилактики заболеваний, которые связаны с модуляцией рецепторов СВ1. Поэтому в одном варианте осуществления настоящее изобретение относится к соединениям, определенным выше, предназначенным для применения в качестве терапевтически активных веществ, предпочтительно - в качестве терапевтически активных веществ, предназначенных для лечения и/или профилактики заболеваний, которые связаны с модуляцией рецепторов СВ1.
Настоящее изобретение также относится к фармацевтическим композициям, включающим соединение, определенное выше, и фармацевтически приемлемый носитель и/или вспомогательное вещество.
В другом варианте осуществления настоящее изобретение относится к способу лечения и/или профилактики заболеваний, которые связаны с модуляцией рецепторов СВ1, включающем введение соединения, определенного выше, человеку или животному.
Настоящее изобретение также относится к применению соединений, определенных выше, для лечения и/или профилактики заболеваний, которые связаны с модуляцией рецепторов СВ1.
Кроме того, настоящее изобретение относится к применению соединений, определенных выше, для приготовления лекарственных средств, предназначенных для лечения и/или профилактики заболеваний, которые связаны с модуляцией рецепторов СВ1. Такие лекарственные средства включают соединение, определенное выше.
В этом контексте выражение "заболевания, связанные с модуляцией рецепторов СВ1" означает заболевания, которые можно лечить и/или предупреждать путем модуляции рецепторов СВ1. Такие заболевания включают, но не ограничиваются только ими, психические нарушения, в особенности тревогу и связанные с тревогой нарушения, психоз, шизофрению, депрессию, нарушения, связанные со злоупотреблением веществами, включая зависимость от психотропных веществ, например злоупотребление веществами и/или зависимость от веществ, включая алкогольную зависимость и никотиновую зависимость, невропатии, мигрень, стресс, эпилепсию, дискинезии, болезнь Паркинсона, амнезию, нарушения памяти и познавательной способности, старческое слабоумие, болезнь Альцгеймера, нарушения питания, ожирение, диабет типа II или инсулиннезависимый диабет (ИННД), желудочно-кишечные заболевания, рвоту, диарею, нарушения мочевых путей, сердечно-сосудистые нарушения, бесплодие, воспаления, инфекции, рак, нарушения, связанные с разрушением миелинового слоя, нейровоспаления, в частности, при атеросклерозе, и синдром Гийена-Барре, вирусный энцефалит, нарушения, связанные с сосудами головного мозга, и черепно-мозговую травму.
В предпочтительном варианте осуществления выражение "заболевания, связанные с модуляцией рецепторов СВ1'' включает нарушения питания, ожирение, диабет типа II или инсулиннезависимый диабет (ИННД), нейровоспаление, диарею, злоупотребление веществами и/или зависимость от веществ, включая алкогольную зависимость и никотиновую зависимость. В более предпочтительном варианте осуществления указанное выражение включает нарушения питания, ожирение, диабет типа II или инсулиннезависимый диабет (ИННД), злоупотребление веществами и/или зависимость от веществ, включая алкогольную зависимость и никотиновую зависимость, и особенно предпочтительным является ожирение.
Другим предпочтительным объектом является лечение или предупреждение диабета типа II (инсулиннезависимого сахарного диабета (ИННСД) у человека, которое включает введение терапевтически эффективного количества соединения формулы (I) в комбинации или совместно с терапевтически эффективным количеством ингибитора липазы, в особенности если ингибитором липазы является орлистат. Объектом настоящего изобретения также является описанный выше способ одновременного, раздельного или последовательного введения соединения формулы (I) и ингибитора липазы, предпочтительно - тетрагидролипстатина.
Другим предпочтительным объектом является лечение или предупреждение ожирения и связанных с ожирением нарушений, которое включает введение терапевтически эффективного количества соединения формулы (I) в комбинации или совместно с терапевтически эффективным количеством других лекарственных препаратов, предназначенных для лечения ожирения или нарушений питания, такое, что совместно они приводят к эффективному ослаблению заболевания. Подходящие другие лекарственные препараты включают, но не ограничиваются только ими, анорексические средства, ингибиторы липазы и селективные ингибиторы повторного всасывания серотонина (СИПС). Комбинации или совместные препараты указанных выше средств можно вводить по отдельности, последовательно или одновременно.
Предпочтительным ингибитором липазы является тетрагидролипстатин.
Анорексические средства, подходящие для применения в комбинации с соединением, предлагаемым в настоящем изобретении, включают, но не ограничиваются только ими, аминорекс, амфехлорал, амфетамин, бензфетамин, хлорфентермин, клобензорекс, клофорекс, кломинорекс, клортермин, циклекседрин, дексфенфлурамин, декстроамфетамин, диэтилпропион, дифеметоксидин, N-этиламфетамин, фенбутразат, фенфлурамин, фенизорекс, фенпропорекс, флудорекс, флуминорекс, фурфурилметиламфетамин, левамфетамин, левофацетоперан, мазиндол, мефенорекс, метамфепрамон, метамфетамин, норпсевдоэфедрин, пенторекс, фендиметразин, фенметразин, фентермин, фенилпропаноламин, пицилорекс и сибутрамин и их фармацевтически приемлемые соли.
Наиболее предпочтительными анорексическими средствами являются сибутрамин и фентермин.
Ингибиторы повторного всасывания серотонина, подходящие для применения в комбинации с соединением, предлагаемым в настоящем изобретении, включают флуоксетин, флувоксамин, пароксетин и сертралин и их фармацевтически приемлемые соли.
Продемонстрировать дополнительную биологическую активность соединений, предлагаемых в настоящем изобретении, можно с помощью анализов, проводимых in vitro, ex vivo и in vivo, которые хорошо известны в данной области техники. Например, чтобы продемонстрировать эффективность фармацевтического препарата для лечения связанных с ожирением нарушений, таких как диабет, синдром Х и атеросклеротических и родственных нарушений, таких как гипертриглицеридемия и гиперхолестеринемия, можно провести указанные ниже анализы.
Методика измерения уровней глюкозы в крови
У мышей db/db (получены от фирмы Jackson Laboratories, Bar Harbor, ME) берут кровь для анализа (из глазной или хвостовой вены) и их разделяют на группы в соответствии со средними эквивалентными уровнями глюкозы в крови. Им перорально (путем искусственного питания в фармацевтически приемлемом разбавителе) вводят исследуемые соединения один раз в сутки в течение от 7 до 14 дней. Затем у животных из глазной или хвостовой вены повторно берут кровь для анализа и определяют уровни глюкозы в крови.
Методика измерения уровней триглицеридов
У мышей hApoAl (получены от фирмы Jackson Laboratories, Bar Harbor, ME) берут кровь для анализа (из глазной или хвостовой вены) и их разделяют на группы в соответствии со средними эквивалентными уровнями триглицеридов в сыворотке. Им перорально (путем искусственного питания в фармацевтически приемлемом разбавителе) вводят исследуемые соединения один раз в сутки в течение от 7 до 14 дней. Затем у животных из глазной или хвостовой вены повторно берут кровь для анализа и определяют уровни триглицеридов в сыворотке.
Методика измерения уровней ЛВП-холестерина
Для определения уровней ЛВП-холестерина (ЛВП - липопротеины высокой плотности) у мышей hApoAl берут кровь для анализа и их разделяют на группы в соответствии со средними эквивалентными уровнями ЛВП-холестерина в плазме. Мышам перорально один раз в сутки в течение от 7 до 14 дней вводят разбавитель или исследуемое соединение, и на следующий день у них берут кровь для анализа. Плазму анализируют на содержание ЛВП-холестерина.
Кроме того, чтобы показать активность соединений, предлагаемых в настоящем изобретении, по отношению к центральной нервной системе, можно провести следующие исследования in vivo.
Методика исследования обучения заданиям и пространственной памяти
Для оценки обучения заданиям и пространственной памяти обычно используют водный лабиринт Морриса (Jaspers et al., Neurosci. Lett. 117:149-153, 1990; Morris, J.Neurosci. Methods 11:47-60, 1984). В этом исследовании животных помещают в бассейн с водой, разделенный на 4 части. В одной из частей скрыта платформа. Животных помещают в бассейн с водой и в течение заранее заданного времени ждут, найдут ли они скрытую платформу. После выполнения некоторого количества попыток животное находит место расположения платформы и выходит из бассейна. Животным предоставляют возможность совершить множество попыток решения этой задачи. Для каждого животного регистрируют суммарное пройденное расстояние, количество попыток для обнаружения платформы, время нахождения платформы и траекторию плавания. Способность животных к обучению оценивают по количеству времени или количеству попыток, необходимых для нахождения скрытой платформы. Ухудшение или улучшение памяти оценивают по количеству попыток или времени, необходимому для нахождения платформы, после заранее заданного времени выдерживания после приобретения навыков. Обучение и память можно оценить по тому, сколько раз животное пересекает четвертую часть бассейна, когда платформа размещена на стадии обучения.
Методика исследования наркотической зависимости
Самовведение животными является средством предсказания возможности злоупотребления соединением у людей. Модификации этой методики также можно использовать для выявления соединений, которые блокируют усиление воздействия наркотиков, характеризующихся возможностью к злоупотреблению. Соединение, которое препятствует самовведению наркотика, может предупредить злоупотребление наркотиком или возникновение зависимости от него. (Ranaldi et al., Psychopharmacol. 161:442-448, 2002; Campbell et al., Exp. Clin. Psychopharmacol. 8:312-25, 2000). Рот исследовании самовведения животных помещают в оперантную камеру, содержащую активный и неактивный рычаги. Каждое нажатие на активный рычаг приводит к вливанию исследуемого соединения или наркотика, пригодного для самовведения. Нажатие на неактивный рычаг не приводит к какому-либо воздействию, но также регистрируется. Затем животных обучают самовведению соединения/наркотика в течение установленного периода времени, предоставляя им доступ к наркотику в течение каждого ежедневного сеанса. Включение освещения камеры сигнализирует о начале сеанса и доступности соединения/наркотика. По окончании сеанса освещение выключается. Вначале вливание наркотика происходит при каждом нажатии на активный рычаг. После возникновения зависимости от нажатия на рычаг увеличивают количество нажатий, необходимых для вливания наркотика. После установления стабильного режима самовведения соединения/наркотика можно оценить влияние второго соединения на поведение, приводящее к увеличению количества наркотика. Введение этого второго соединения до сеанса может усилить, ослабить склонность к самовведению или не привести к изменениям.
Для определения активности соединений формулы (I) проведены следующие исследования.
Сродство соединений, предлагаемых в настоящем изобретении, к каннабиноидным рецепторам СВ1 определяли с использованием препаратов мембран клеток почек эмбриона человека (ПЭЧ), при этом каннабиноидный рецептор человека СВ1 временно трансфицировали с использованием вируса лихорадка леса Семлики совместно с [3Н]-СР-55940 в качестве радиолиганда. После инкубации свежеприготовленного препарата мембран клеток с лигандом [3Н] с прибавлением или без прибавления соединений, предлагаемых в настоящем изобретении, разделение связанного и свободного лиганда проводили путем фильтрования через стекловолоконные фильтры. Радиоактивность фильтра измеряли с помощью жидкостного сцинтилляционного счетчика.
Сродство соединений, предлагаемых в настоящем изобретении, к каннабиноидным рецепторам СВ2 определяли с использованием препаратов мембран клеток почек эмбриона человека (ПЭЧ), при этом каннабиноидный рецептор человека СВ2 временно трансфицировали с использованием вируса лихорадка леса Семлики совместно с [3Н]-СР-55940 в качестве радиолиганда. После инкубации свежеприготовленного препарата мембран клеток с лигандом [3Н] с прибавлением или без прибавления соединений, предлагаемых в настоящем изобретении, разделение связанного и свободного лиганда проводили путем фильтрования через стекловолоконные фильтры. Радиоактивность фильтра измеряли с помощью жидкостного сцинтилляционного счетчика.
Антагонистическую активность соединений, предлагаемых в настоящем изобретении, к каннабиноидным рецепторам СВ1 определяли с помощью функционального исследования с использованием клеток яичников китайского хомячка, в которые стабильно экспрессированы каннабиноидные рецепторы человека СВ1 (см. М.Rinaldi-Carmona et. al., J.Pharmacol. Exp. Ther. 278 (1996) 871). Стабильное экспрессирование каннабиноидного рецептора человека в клеточных системах впервые описано в публикациях Nature 1990, 346, 561-564 (СВ1) и Nature 1993, 365, 61-65 (CB2). Аденилилциклазу стимулировали с помощью форсколина и определяли путем количественного определения количества накопившегося циклического аденозинмонофосфата (цАМФ).
Происходящая при этом активация рецептора СВ1 агонистами рецептора СВ1 (например, СР-55940 или (R)-WIN-55212-2) может ослабить вызванное форсколином накопление цАМФ зависящим от концентрации образом. Этой опосредуемой рецептором СВ1 реакции можно противодействовать с помощью антагонистов рецептора СВ1, таких как соединения, предлагаемые в настоящем изобретении.
Соединения формулы (I) обнаруживают превосходное сродство к рецептору СВ1, которое определяли при условиях проведения эксперимента, описанных в публикации Devane et.al. Mol. Pharmacol. 34 (1988) 605-613. Соединения, предлагаемые в настоящем изобретении, или их фармацевтически приемлемые соли или сольваты являются антагонистами рецептора СВ1 и селективны по отношению к рецептору СВ1 при значениях сродства, составляющих менее IC50=5 мкМ, предпочтительно - менее IC50=2 мкМ. Они обнаруживают примерно в 10 раз более значительную селективность, чем селективность по отношению к рецептору CB2.
Влияние антагониста/обратного агониста рецептора СВ1 на вызванную СР 55940 гипотермию у мышей NMRI
Животные
В этом исследовании использовали самцов мышей NMRI, полученных от фирмы Research Consulting Company Ltd (RCC) of Füllinsdorf (Switzerland). В этом исследовании использовали мышей массой 30-31 г. Температура окружающей среды составляла примерно 20-21°С и относительная влажность составляла 55-65%. В помещениях поддерживали 12-часовой цикл света-темноты и все исследования проводили во время освещения. Предоставлялся неограниченный доступ к питьевой воде и корму.
Методика
Все исследования проводили в период от 12:00 до 17:00. Мышей помещали в эту среду и давали им привыкнуть к среде в течение не менее 2 ч до начала эксперимента. Им также предоставлялся свободный доступ к питьевой воде и корму. Для каждой дозы использовали по 8 мышей. Измерения ректальной температуры тела проводили с помощью ректального зонда (RET2, выпускающегося фирмой Physitemp) и цифрового термометра (Digi-sense n°8528-20, выпускающегося фирмой Cole Farmer, Chicago USA). Каждой мыши зонд вводили на глубину, равную примерно 3,5 см.
Температуру тела измеряли за 15 мин до введения разбавителя или антагониста/обратного агониста рецептора СВ1. Для оценки влияния самого соединения через 30 или 90 мин после внутрибрюшинного или перорального введения этого соединения соответственно регистрировали ректальную температуру тела. Сразу же внутривенно вводили агонист рецептора СВ, СР 55940 (0,3 мг/кг), затем, через 20 мин после внутривенного введения СР 55940, повторно измеряли температуру тела.
Активность in vivo соединений формулы (I) оценивали по их способности регулировать поведение при кормлении путем регистрации потребления корма предварительно лишенными корма животными.
Крыс подготавливали путем предоставления им доступа к корму в течение 2 ч в сутки и затем их лишали корма в течение 22 ч. При подготовке в таком режиме количество корма, съеденного каждый день в течение этого 2-часового сеанса, в последующие дни было постоянным.
Для исследования способности соединений формулы (I) уменьшать потребление корма проводили перекрестное исследование с использованием 8 животных. Крыс по отдельности помещали в клетки из плексигласа с решеткой на дне и под пол клетки помещали бумагу для сбора рассыпавшегося корма. Им на 2 ч предоставляли дозатор корма (стакан), содержащий взвешенное количество корма. В конце сеанса кормления крыс возвращали в свои собственные клетки. Каждую крысу взвешивали до начала эксперимента и регистрировали количество корма, съеденного в течение этого 2-часового сеанса. Различные дозы исследуемого соединения или разбавителя вводили перорально за 60 мин до 2-часового сеанса кормления. В качестве положительного контроля в этот эксперимент включали римонабант (SR 141716). Результаты многократных исследований обрабатывали с помощью дисперсионного анализа, а затем использовали методику Неймана-Кеулса с критерием Стьюдента. * Р<0,05 по сравнению с крысами, которым давали физиологический раствор.
Кроме того, применимость соединений формулы (I) в случае заболеваний или нарушений можно продемонстрировать с помощью моделей заболеваний на подопытных животных, которые описаны в литературе. Ниже приведены примеры таких моделей заболеваний на подопытных животных: а) уменьшение потребления сладкого корма мартышками (Behavioural Pharm, 1998, 9,179-181); b) уменьшение потребления сахарозы и этанола мышами (Psychopharm. 1997, 132, 104-106); с) улучшение двигательной активности и обустройства местообитания у крыс (Psychopharm. 1998, 135, 324-332; Psychopharmacol 2000, 151: 25-30); d) самопроизвольная локомоторная активность у мышей (J. Pharm. Exp. Ther. 1996, 277, 586-594); е) уменьшение самовведения опиатов у мышей (Sci. 1999, 283, 401-404).
Соединения формулы (I) и/или их фармацевтически приемлемые соли можно использовать в качестве лекарственных средств, например, в форме фармацевтических препаратов, предназначенных для энтерального, парентерального или местного введения. Их можно вводить, например, перорально, например, в виде таблеток, таблеток с покрытием, драже, капсул из твердого и мягкого желатина, растворов, эмульсий или суспензий, ректально, например, в виде суппозиториев, парентерально, например, в виде растворов для инъекций или растворов для вливания, или местно, например, в виде мазей, кремов или масел. Предпочтительным является пероральное введение.
Изготовление фармацевтических препаратов можно проводить способами, которые должны быть известны любому специалисту в данной области техники, путем внесения описанных соединений формулы (I) и/или их фармацевтически приемлемых солей, необязательно в комбинации с другими терапевтически ценными веществами, в галеновы вводимые формы совместно с подходящими нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими материалами носителей и при желании с обычными фармацевтическими вспомогательными веществами.
Подходящими материалами носителей являются не только неорганические материалы носителей, но и органические материалы носителей. Так, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли можно использовать в качестве материалов носителей для таблеток, таблеток с покрытием, драже и капсул из твердого желатина. Подходящими материалами носителей для капсул из мягкого желатина являются, например, растительные масла, воска, жиры и полужидкие и жидкие полиолы (однако в случае капсул из мягкого желатина в зависимости от природы активного ингредиента носители могут не потребоваться). Подходящими материалами носителей для приготовления растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар и т.п. Подходящими материалами носителей для растворов для инъекций являются, например, вода, спирты, полиолы, глицерин и растительные масла. Подходящими материалами носителей для суппозиториев являются, например, натуральные или затвердевшие масла, воска, жиры и полужидкие и жидкие полиолы. Подходящими материалами носителей для препаратов местного действия являются глицериды, полусинтетические и синтетические глицериды, гидрированные масла, жидкие воска, жидкие парафины, жидкие жирные спирты, стерины, полиэтиленгликоли и производные целлюлозы.
В качестве фармацевтических вспомогательных веществ рассматриваются обычные стабилизаторы, консерванты, смачивающие и эмульгирующие агенты, агенты, улучшающие консистенцию, агенты, улучшающие вкус, соли для регулирования осмотического давления, буферные вещества, солюбилизаторы, красители и маскирующие агенты и антиоксиданты.
Дозировка соединений формулы (I) может меняться в широких пределах в зависимости от подвергающегося лечению заболевания, возраста и индивидуального состояния пациента и способа введения, и, разумеется, в каждом конкретном случае подбирается в соответствии с индивидуальной потребностью. Для взрослых пациентов рассматривается суточная доза, составляющая примерно от 1 до 1000 мг, предпочтительно - примерно от 1 до 100 мг. В зависимости от тяжести заболевания и точного фармакокинетического профиля соединение следует вводить в виде одной или нескольких дозировочных единиц в сутки, например от 1 до 3 дозировочных единиц.
Фармацевтические препараты обычно содержат примерно 1-500 мг, предпочтительно - 1-100 мг соединения формулы (I).
Приведенные ниже примеры предназначены для более подробной иллюстрации настоящего изобретения. Однако они не предназначены для какого-либо ограничения его объема.
Примеры
МС = масс-спектрометрия, ЭУ = электронный удар, ИРП = ионное распыление (положительный ион). Все эксперименты проводили в инертной атмосфере (азота или аргона).
Пример 1
Получение 4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5H]дибензо[а,d]циклогептен]-5-ил)карбонил]-морфолина
а) Получение 4-бром-5-фтор-бензол-1,2-диола
При охлаждении (-78°С) к раствору 4-фторвератрола (5,0 г, 32 ммоль) в дихлорметане (106 мл) медленно прибавляли раствор трихлорида бора в дихлорметане (1 М, 96 мл, 96 ммоль, 3,0 экв.). Реакционную смесь нагревали до 20°С и перемешивали в течение ночи. Реакционную смесь выливали в воду со льдом, экстрагировали этилацетатом (3 раза). Объединенные органические слои промывали водным раствором бикарбоната натрия, сушили над сульфатом натрия и фильтровали. Летучие вещества удаляли в вакууме. Коричневое твердое вещество разбавляли хлороформом (50 мл) и дихлорметаном (10 мл). Медленно прибавляли раствор брома в тетрахлориде углерода (5 мл). После перемешивания в течение 3 ч при комнатной температуре летучие вещества удаляли в вакууме. Очистка с помощью флэш-хроматографии давала искомое соединение (6,51 г, 98%) в виде коричневого твердого вещества.
ИРП МС: m/e=207,9 ([М+Н]+).
b) Получение 5-бром-6-фтор-2,2-дифенилбензо[1,3]диоксола
Смесь 4-бром-5-фтор-бензол-1,2-диола (12 г, 58,0 ммоль) и дифенилдихлорметана (1,2 экв., 16,50 г) перемешивали при комнатной температуре до прекращения выделения газа. Смесь нагревали при перемешивании при 180°С в течение 20 мин. Реакционной смеси давали охладиться до комнатной температуры, разбавляли метанолом (50 мл) и энергично перемешивали. Осадившийся продукт собирали фильтрованием и растворяли в толуоле (50 мл). Прибавляли метанол (100 мл) и смесь перемешивали 30 мин при комнатной температуре. Осадившийся продукт собирали фильтрованием (выход 10,3 г, 48%), еще одну порцию (6,4 г, 30%) извлекали из маточного раствора.
ИРП МС: m/e=370,0 ([М+Н+]).
c) Получение (6-фтор-2,2-дифенилбензо[1,3]диоксол-5-ил)-морфолин-4-илметанона
К охлаждаемому раствору (-78°С) раствору 5-бром-6-фтор-2,2-дифенилбензо[1,3]диоксола (17,59 г, 47,4 ммоль) в диэтиловом эфире (300 мл) медленно прибавляли раствор н-бутиллития в гексанах (1,6 М, 30 мл, 48 ммоль, 1,0 экв.). Реакционную смесь перемешивали в течение 1 ч при -78°С и затем прибавляли 4-морфолинкарбонилхлорид (8,5 г, 56,9 ммоль, 1,2 экв.). Реакционной смеси давали нагреться до 20°С и ее выливали в водный раствор бикарбоната натрия. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали рассолом. Летучие вещества удаляли в вакууме. Очистка с помощью флэш-хроматографии давала (6-фтор-2,2-дифенилбензо[1,3]диоксол-5-ил)-морфолин-4-илметанон (13,0 г, 68%) в виде светло-желтого твердого вещества.
ИРП МС: m/e=406,2 ([М+Н]+).
d) Получение (2-фтор-4,5-дигидроксифенил)-морфолин-4-илметанона
К охлаждаемому (в бане со льдом) раствору (6-фтор-2,2-дифенилбензо[1,3]диоксол-5-ил)-морфолин-4-илметанона (5,70 г, 14,06 ммоль) в трифторуксусной кислоте (60 мл) в течение 10 мин прибавляли триэтилсилан (2,1 экв., 4,7 мл). Смесь перемешивали в течение 20 мин при 0°С и в течение 4 ч при комнатной температуре. Летучие вещества удаляли при пониженном давлении и остаток очищали с помощью хроматографии на колонке с силикагелем (2:1 этилацетат/гептан-этилацетат - 10:1 этилацетат/метанол) и получали (2-фтор-4,5-дигидроксифенил)-морфолин-4-илметанон в виде светло-коричневого твердого вещества (3,19 г, 94%).
ИРП МС: m/е=242.2 ([М+Н]+).
e) Получение 2,8-дихлор-10,11-дигидродибензо[a,d]циклогептен-5-тиона
При перемешивании к раствору 2,8-дихлор-10,11-дигидродибензо[а,d]циклогептен-5-она (0,70 г, 2,53 ммоль) в толуоле (20 мл) прибавляли пентасульфид фосфора (1 экв., 561 мг) и гексаметилдисилоксан (1 экв., 0,54 мл). Смесь 3 ч кипятили с обратным холодильником и ей давали охладиться до комнатной температуры. Смесь фильтровали через слой силикагеля (70 г) и промывали смесью 8:1 гептан/этилацетат. Выпаривание растворителя при пониженном давлении давало 2,8-дихлор-10,11-дигидродибензо[а,d]циклогептен-5-тион в виде голубого твердого вещества (710 мг, 95%), которое использовали без дополнительной очистки.
ЭУ МС: m/е=292,0 ([М]+).
f) Получение 4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'[5H]дибензо[a,d]циклогептен]-5-ил)карбонил)морфолина
К раствору (3,4-дигидроксифенил)-морфолин-4-илметанона (360 мг, 1,49 ммоль) в ацетонитриле (14 мл) прибавляли 2,8-дихлор-10,11-дигидродибензо[а,d]циклогептен-5-тион (1,5 экв., 657 мг), трифторацетат серебра (2,5 экв., 824 мг) и хинуклидин (4 экв., 667 мг). Смесь 4 ч кипятили с обратным холодильником, охлаждали до комнатной температуры, фильтровали через диатомовую землю (дикалит), промывали этилацетатом и выпаривали при пониженном давлении. Остаток очищали с помощью хроматографии на колонке с силикагелем (от 1:0 до 10:1 дихлорметан/этилацетат) и получали продукт в виде почти белого вспененного вещества (396 мг, 53%).
ЭУ МС: m/е=499,1 ([М]+).
Пример 2
Получение 4-[(2',8'-дихлор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5H]дибензо[а,d]циклогептен]-5-ил)карбонил]-пиперидина
Искомое соединение получали по общей методике, приведенной в примере 1f, из (3,4-дигидроксифенил)-пиперидин-4-илметанона и 2,8-дихлор-10,11-дигидродибензо[а,d]циклогептен-5-тиона (пример 1е). Желтое твердое вещество.
ЭУ МС: m/е=478,2 ([М]+).
Пример 3
Получение 4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро [1,3-бензодиоксол-2,5'-[5H]дибензо[а,d]циклогептен]-5-ил)карбонил]-пиперидина
a) Получение (6-фтор-2,2-дифенилбензо[1,3]диоксол-5-ил)-пиперидин-1-илметанона
Искомое соединение получали по общей методике, приведенной в примере 1с, из 5-бром-6-фтор-2,2-дифенилбензо[1,3]диоксола (пример 1b) и 1-пиперидинкарбонилхлорида. Бесцветное полужидкое вещество.
МС: m/е=404,3 ([М+Н]+).
b) Получение (2-фтор-4,5-дигидроксифенил)-пиперидин-1-илметанона
Искомое соединение получали по общей методике, приведенной в примере 1d, из (6-фтор-2,2-дифенилбензо[1,3]диоксол-5-ил)-пиперидин-1-илметанона. Бесцветное полужидкое вещество.
МС: m/е=240,2 ([М+Н]+).
c) Получение 4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5H]дибензо[a,d]циклогептен]-5-ил)карбонил]-пиперидина
Искомое соединение получали по общей методике, приведенной в примере 1f, из (2-фтор-4,5-дигидроксифенил)-пиперидин-1-илметанона и 2,8-дихлор-10,11-дигидродибензо[а,d]циклогептен-5-тиона (пример 1е). Светло-желтая смола.
ИРП МС: m/е=498,2 ([М+Н]+).
Пример 4
Получение 4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро [1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)сульфонил]-морфолина
а) Получение 2-фтор-4,5-диметоксибензолсульфонилхлорида
К суспензии комплекса триоксида серы с N,N-диметилформамидом (4,108 г, 27 ммоль) в 1,2-дихлорэтане по каплям прибавляли 4-фторвератрол (3,49 г, 22 ммоль). Смесь медленно нагревали до 85°С на масляной бане. Через 2,5 ч твердые вещества растворялись и образовывался золотисто-желтый раствор. Все еще содержались следы исходного вещества и нагревание продолжали в течение еще 4,5 ч. Масляную баню удаляли и по каплям прибавляли тионилхлорид (1,95 мл, 27 ммоль). Смесь нагревали в течение 4 ч при 85°C и ей давали охладиться до комнатной температуры. Раствор выливали в воду и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали водой, сушили над сульфатом магния и выпаривали. Оставшиеся следы N,N-диметилформамида удаляли путем азеотропной отгонки с толуолом и получали продукт в виде почти белого твердого вещества, которое использовали без дополнительной очистки.
ЭУ МС: m/е=254,0 ([М]+).
b) Получение 1-(2-фтор-4,5-диметоксибензолсульфонил)-морфолина
К раствору 2-фтор-4,5-диметоксибензолсульфонилхлорида (5,30 г, 20,81 ммоль) в дихлорметане (30 мл) по каплям прибавляли морфолин (2,1 экв., 3,81 мл) (экзотермическая реакция). Смесь перемешивали в течение ночи при комнатной температуре и фильтровали через слой силикагеля и элюировали этилацетатом. Растворитель выпаривали при пониженном давлении и получали 1-(2-фтор-4,5-диметоксибензолсульфонил)-морфолин в виде бледно-желтого твердого вещества, которое использовали без дополнительной очистки.
ЭУ МС: m/е=305,1 ([М]+).
c) Получение 4-фтор-5-(морфолин-1-сульфонил)-бензол-1,2-диола
К охлаждаемому (в бане со льдом) раствору 1-(2-фтор-4,5-диметоксибензолсульфонил)-морфолина (837 мг, 2,74 ммоль) в дихлорметане (25 мл) прибавляли 1 М раствор трибромида бора в дихлорметане (2 экв., 5,48 мл). Смесь перемешивали в течение ночи при комнатной температуре и выливали в 1 М водный раствор дигидрофосфата калия в смеси со льдом. После перемешивания в течение 1 ч фазы разделяли и водную фазу экстрагировали этилацетатом. Объединенные органические фазы промывали рассолом, сушили над сульфатом магния и выпаривали. Остаток очищали с помощью хроматографии на колонке с силикагелем (элюент - смесь 19:1 хлороформ/этанол) и получали 4-фтор-5-(морфолин-1-сульфонил)-бензол-1,2-диол в виде коричневого масла (564 мг, 74%), которое при выдерживании в холодильнике затвердевало.
МС: m/е=276,0 ([М-Н]+).
d) Получение 4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5H]дибензо[a,d]циклогептен]-5-ил)сульфонил]-морфолина
Искомое соединение получали по общей методике, приведенной в примере 1f, из 4-фтор-5-(морфолин-1-сульфонил)-бензол-1,2-диола и 2,8-дихлор-10,11-дигидродибензо[а,d]циклогептен-5-тиона (пример 1е).
ЭУ МС: m/е=535,1 ([М]+).
Пример 5
Получение 4-[(2',6,8'-трифтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5H]дибензо[а,d]пиклогептен]-5-ил)карбонил]-морфолина
a) Получение 2,8-дифтор-10,11-дигидродибензо[а,d]циклогептен-5-она
К охлаждаемой (в бане твердый диоксид углерода/ацетон) суспензии безводного трихлорида алюминия (5 экв., 45,82 г, 0,34 моль) в сероуглероде (450 мл) по каплям прибавляли оксалилхлорид (4 экв., 23,58 мл). Смеси давали нагреться до комнатной температуры, а затем медленно прибавляли 1,1'-(1,2-этандиил)бис[3-фторбензол] (1 экв., 15,00 г) в виде раствора в сероуглероде (20 мл). Смесь нагревали в течение ночи при 35°С и разбавляли хлороформом (400 мл). Смесь охлаждали (в бане со льдом) и медленно прибавляли воду (400 мл). Фазы разделяли, водную фазу экстрагировали хлороформом и объединенные органические фазы сушили над сульфатом магния и выпаривали при пониженном давлении. Остаток очищали с помощью хроматографии на колонке с силикагелем (15:1 гептан/этилацетат) и получали 2,8-дифтор-10,11-дигидродибензо[а,d]циклогептен-5-он в виде желтого твердого вещества (4,98 г, 29%).
ЭУ МС: m/е=244,1 ([M]+).
b) Получение 2,8-дифтор-10,11-дигидродибензо[а,d]циклогептен-5-тиона
Искомое соединение получали по общей методике, приведенной в примере 1е, из 2,8-дифтор-10,11-дигидродибензо[а,d]циклогептен-5-она. Голубое твердое вещество.
ЭУ МС: m/е=260,0 ([М]+).
с) Получение 4-[(2',6,8'-трифтор-10',11'-дигидроспиро [1,3-бензодиоксол-2,5'-[5H]дибензо[а,d]циклогептен]-5-ил)карбонил]-морфолина
Искомое соединение получали по общей методике, приведенной в примере 1f, из (2-фтор-4,5-дигидроксифенил)-морфолин-4-илметанона (пример 1d) и 2,8-дифтор-10,11-дигидродибензо[а,d]циклогептен-5-тиона. Почти белое вспененное вещество.
ЭУ МС: m/е=467,1 ([М]+).
Пример 6
Получение 4-[(2',6,8'-трифтор-10',11'-дигидроспиро [1,3-бензодиоксол-2,5'-[5H]дибензо[a,d]циклогептен]-5-ил)карбонил]-пиперидина
Искомое соединение получали по общей методике, приведенной в примере 1f, из (2-фтор-4,5-дигидроксифенил)-пиперидин-4-илметанона (пример 3b) и 2,8-дифтор-10,11-дигидродибензо[a,d]циклогептен-5-тиона (пример 5b). Почти белое вспененное вещество.
ИРП МС: m/е=466,4 ([M+H]+).
Примеры галеновых форм
Пример А
Таблетки с пленочным покрытием, содержащие указанные ниже ингредиенты, можно изготовить обычным образом:
Активный ингредиент просеивают и смешивают с микрокристаллической целлюлозой, и смесь гранулируют с использованием водного раствора поливинилпирролидона. Гранулят смешивают с натриевой солью гликолята крахмала и стеаратом магния и прессуют с получением ядер таблеток массой 120 или 350 мг соответственно. Ядра лакируют водным раствором/суспензией указанного выше пленочного покрытия.
Пример В
Капсулы, содержащие указанные ниже ингредиенты, можно изготовить обычным образом:
Компоненты просеивают и смешивают и помещают в капсулы размера 2.
Пример С
Растворы для инъекций могут обладать следующим составом:
Активный ингредиент растворяют в смеси полиэтиленгликоля 400 с водой для инъекций (частью). Значение рН доводят до 5,0 путем прибавления уксусной кислоты. Объем доводят до 1,0 мл путем прибавления оставшегося количества воды. Раствор фильтруют, помещают во флаконы в соответствующем избытке и стерилизуют.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ДИБЕНЗОСУБЕРОНА | 2005 |
|
RU2391337C2 |
| НОВЫЕ БЕНЗОДИОКСОЛЫ | 2003 |
|
RU2304580C2 |
| АМИНОЗАМЕЩЕННЫЕ АНАЛОГИ ДИАРИЛ [a,d] ЦИКЛОГЕПТЕНА В КАЧЕСТВЕ МУСКАРИНОВЫХ АГОНИСТОВ И СПОСОБЫ ЛЕЧЕНИЯ ПСИХОНЕВРОЛОГИЧЕСКИХ РАССТРОЙСТВ | 2004 |
|
RU2394030C2 |
| ПРОИЗВОДНЫЕ 3-ЗАМЕЩЕННОГО 1,5-ДИФЕНИЛПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ СВ1 МОДУЛЯТОРОВ | 2005 |
|
RU2375349C2 |
| (3-ЦИКЛОАЛКИЛ-2,3,4,5-ТЕТРАГИДРО-1Н-БЕНЗО[d]АЗЕПИН-7-ИЛОКСИ)ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ Н3 РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ | 2003 |
|
RU2388752C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА | 2005 |
|
RU2391338C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ1 | 2005 |
|
RU2377238C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
| НОВЫЕ ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗ | 2008 |
|
RU2495043C2 |
| N-ЗАМЕЩЕННЫЕ АЗАГЕТЕРОЦИКЛИЧЕСКИЕ КАРБОНОВЫЕ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2186769C2 |
Изобретение относится к новым спиропентациклическим соединениям формулы (I)
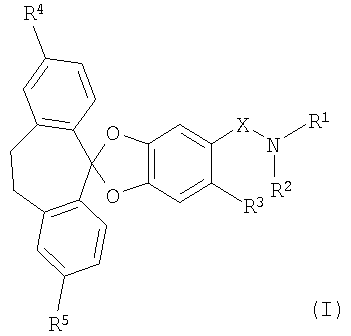
в которой R1 и R2 совместно с атомом азота, к которому они присоединены, образуют 6- членное моноциклическое насыщенное гетероциклическое кольцо, которое необязательно может содержать 1 дополнительный О или N;
R3 обозначает водород, галоген или низш. алкил;
R4 и R5 каждый независимо обозначает галоген;
и Х обозначает -C(O)- или -SO2-;
или их фармацевтически приемлемым солям, предназначенным для применения в качестве терапевтически активных веществ для лечения и/или профилактики заболеваний, которые связаны с модуляцией рецепторов СВ1, и также фармацевтической композиции и на основе этих соединений. 2 н. и 10 з.п. ф-лы, 1 табл.
1. Соединения формулы (I)
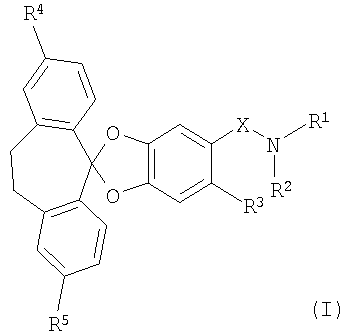
в которой
R1 и R2 совместно с атомом азота, к которому они присоединены, образуют 6- членное моноциклическое насыщенное гетероциклическое кольцо, которое необязательно может содержать один дополнительный гетероатом О или N;
R3 обозначает водород, галоген или низш. алкил;
R4 и R5 каждый независимо обозначает галоген; и Х обозначает -С(O)- или -SO2-;
или их фармацевтически приемлемая соль.
2. Соединения по п.1, в которых R1 и R2 совместно с атомом азота, к которому они присоединены, образуют 6-членное моноциклическое насыщенное гетероциклическое кольцо, которое необязательно может содержать в цикле еще один атом кислорода.
3. Соединения по п.1, в которых R1 и R2 совместно с атомом азота, к которому они присоединены, выбраны из пиперидинила и морфолина.
4. Соединения по п.1, в которых R3 обозначает водород.
5. Соединения по п.1, в которых R3 обозначает галоген.
6. Соединения по п.1, в которых R4 и R5 обозначают хлор.
7. Соединения по п.1, в которых R4 и R5 обозначают фтор.
8. Соединения по п.,1, в которых Х обозначает -C(O)-.
9. Соединения по п.1, выбранные из группы, включающей
4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]-морфолин,
4-[(2',8'-дихлор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]-пиперидин,
4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]-пиперидин,
4-[(2',8'-дихлор-6-фтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)сульфонил]-морфолин,
4-[(2',6,8'-трифтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]-морфолин,
4-[(2',6,8'-трифтор-10',11'-дигидроспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]-пиперидин,
и их фармацевтически приемлемые соли.
10. Фармацевтическая композиция, обладающая антагонистическим действием в отношении рецептора СВ1, включающая соединение по любому из пп.1-9 и фармацевтически приемлемый носитель и/или вспомогательное вещество.
11. Соединения по п.1, обладающие антагонистическим действием в отношении рецептора СВ1.
12. Соединения по п.1, предназначенные для применения в качестве терапевтически активных веществ для лечения и/или профилактики заболеваний, которые связаны с модуляцией рецепторов СВ 1.
| OOMS F | |||
| et al | |||
| J.Med | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Керамическая масса для изготовления химически стойких изделий | 1989 |
|
SU1707000A1 |
| WO 03084930 A1, 16.10.2003 | |||
| РЕГУЛЯТОР РОСТА И РАЗВИТИЯ РАСТЕНИЙ | 1992 |
|
RU2072998C1 |
| RU 2001117199 А, 10.07.2003. | |||

