Настоящее изобретение относится к 4-[(6-фторспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]морфолинам формулы:
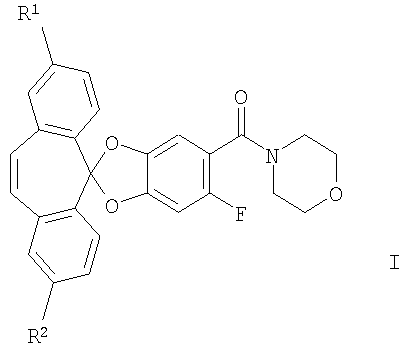
где R1 и R2 каждый независимо представляет собой водород или галоген.
Настоящее изобретение также относится к способу получения соединений формулы I, фармацевтическим композициям, содержащим соединения формулы I, и применению в качестве лекарственного средства, особенно для лечения ожирения и других заболеваний.
Было обнаружено, что соединения формулы I являются модуляторами рецептора CB1.
Были выделены два различных подтипа каннабиноидных рецепторов (CB1 и СВ2) и оба принадлежали к суперсемейству рецептора, связанного с G-белком. Альтернативные родственные формы CB1, CB1A и CB1B также были описаны, но они экспрессируются только в низких уровнях в тестируемых тканях (статьи D.Shire, С.Carrillon, M.Kaghad, В.Calandra, M.Rinaldi-Carmona, G.Le Fur, D.Caput, P.Ferrara, J.Biol. Chem., 1995, 270 (8), сс.3726-31; Е.Ryberg. H.К.Vu, N.Larsson, Т.Groblewski, S.Hjorth, Т.Elebring, S.Sjögren, P.J.Greasley, FEBS Lett., 2005, 579, сс.259-264). CB1 рецептор находится главным образом в головном мозге и в меньшей степени в некоторых периферийных органах, тогда как рецептор CB2 преимущественно находится периферийно в селезенке и клетках иммунной системы (статья S.Munro, K.L.Thomas, M.Abu-Shaar, Nature, 1993, 365, сс.61-61). Следовательно, для устранения побочных эффектов необходимы соединения, селективные по отношению к CB1.
Δ9-тетрагидроканнабинол (Δ9-ТНС) является основным психоактивным соединением индийской конопли (статья Y.Gaoni, R.Mechoulam, J. Am. Chem. Soc., 1964, 86, с.1646), cannabis sativa (марихуана), который используется в медицине очень давно (R.Mechoulam (Ред.) в книге "Cannabinoids as therapeutic Agents", 1986, сс. 1-20, CRC Press). Δ9-ТНС является неселективным агонистом рецептора CB1/2 и доступен в США в виде дронабинола (marinol®) для облегчения рвоты, вызванной раковой химиотерапией (CIE), и устранения потери веса тела у пациентов, страдающих СПИДом, путем стимулирования аппетита. В Великобритании наболинон (LY-109514, Cesamet®), синтетический аналог Δ9-ТНС, используется для CIE (статьи R.G.Pertwee, Pharmaceut. Sci., 1997, 3 (11), сс.539-545, Е.M.Williamson, F.J.Evans, Drugs, 2000. 60 (6), сс.1303-1314).
Анандамид (арахидонилэтаноламид) обнаружен как эндогенный лиганд (агонист) рецептора CB1 (статьи R.G.Pertwee, Curr. Med. Chem., 1999, 6 (8), cc.635-664; W.A.Devane, L.Hanus, A.Breuer, R.G.Pertwee, L.A.Stevenson, G.Griffin, D.Gibson, A.Mandelbaum, A.Etinger, R.Mechoulam, Science, 1992, 258, сс.1946-9). Анандамид и 2-арахидоноилглицерин (2-AG) модулируют в пресинаптическом нерве терминальную отрицательную аденилатциклазу и чувствительные к заряду Са2+ каналы и активируют внутри выпрямляющие К+ каналы (статья V. Di Marzo, D.Melck, T.Bisogno, L. De Petrocellis, Trends in Neuroscience, 1998, 21 (12), сс.521-8), тем самым воздействуя на высвобождение нейротрансмиттеров и/или их действие, что снижает высвобождение нейротрансмиттеров (статья А.С.Porter, С.С.Felder, Pharmacol. Ther., 2001, 90 (1), сс.45-60).
Анандамид как Δ9-ТНС также снижает потребление пищи по механизму, опосредованному рецептором CB1. Селективные антагонисты рецептора CB1 блокируют потребление пищи, связанное с введением анандамида (статьи С.М.Williams, T.C.Kirkham, Psychopharmacology, 1999, 143 (3), сс.315-317; С.С.Felder, E.M.Briley, J. Axelrod, J.T.Simpson, K.Mackie, W.A.Devane, Proc. Natl. Acad. Sci. USA, 1993, 90 (16), сс.7656-60) и подавляют аппетит и вызывают снижение веса (статья G.Colombo, R.Agabio, G.Diaz, С.Lobina, R.Reali, G.L.Gessa, Life Sci., 1998, 63 (8), cc. L113-PL117).
Лептин является первичным сигналом, с помощью которого гипоталамус распознает алиментарный статус и модулирует потребление пищи и энергетический баланс. После временного ограничения пищи мыши с блокированным рецептором CB1 потребляли пищу меньше, чем их сородичи дикого типа, и антагонист CB1 SR141716A снижает потребление пищи у мыши дикого типа, но не у обработанной мыши. Кроме того, нарушенная передача лептина связана с повышенными гипоталамическими, но не церебральными уровнями эндоканнабиноидов у мышей с ожирением db/db и ob/ob и крыс Zucker. Текущее лечение лептином нормальных крыс и мышей ob/ob снижает уровни анандамида и 2-арахидоноилглицерина в гипоталамусе. Это открытие показало, что эндоканнабиноиды в гипоталамусе могут тонически активировать рецепторы CB1 для поддержания потребления пищи и составляют часть нервной цепочки, регулируемой лептином (статья V.Di Marzo, S.K.Goparaju, L.Wang, J.Liu, S.Bitkai, Z.Jarai, F.Fezza, G.I.Miura, R.D.Palmiter, T.Sugiura, G.Kunos, Nature, 410 (6830), cc.822-825).
Сообщалось также, что рецептор CB1 играет роль в регулировании костной массы и потери костной массы в результате недостатка эстрогена. Антагонисты рецепторов СВ1 и СВ2 предотвращают потерю костной массы в результате овариэктомии in vivo и вызывают ингибирование остеокласта in vitro путем промотирования апоптоза остеокласта и ингибирования продукции некоторых жизненных факторов остеокласта (статья A.I.Idris, R.J.van't Hof, I.R.Greig, S.A.Ridge, D.Baker, R.A.Ross, S.H.Wilson, Nature Medicine, 2005, 11 (7), сс.774-779). Антагонисты каннабиноидного рецептора, следовательно, могут быть полезными для лечения остеопороза и других заболеваний костей, таких как рак, связанный с костным заболеванием, и болезнь Паджета костей.
По крайней мере два селективных антагониста / обратных агониста СВ1 (SR-141716 и SLV-319) в настоящее время проходят клинические испытания для лечения ожирения и/или отказа от курения. В двойном слепом испытании с плацебо, в дозах 10 и 20 мг в сутки, SR 141716 существенно снижает вес тела по сравнению с плацебо (статья F.Barth, M.Rinaldi-Carmona, М.Arnone, Н.Heshmati, G.Le Fur, "Cannabinoid antagonists: From research tools to potential new drugs". Сборник докладов, 222nd ACS National Meeting, Chicago, IL, United States, 2001, August, 26-30). SR-141716 снижает массу тела, обхват талии и улучшает метаболические параметры (плазма HDL, чувствительность к триглицеридам и инсулину) в нескольких исследованиях фазы III (RIO-липиды, RIO-Европа и RIO-Северная Америка). Кроме того, как было показано, эффективность SR-141716 в фазе III испытывали при отказе от курения (STRATUS-US). Однако все еще остается потребность в мощных новых низкомолекулярных модуляторах СВ1, которые имели бы фармакокинетические и фармакодинамические свойства, подходящие для применения в качестве фармацевтических средств для человека.
Другими соединениями, которые были предложены в качестве антагонистов рецептора CB1 и соответственно обратных агонистов, являются аминоалкилиндолы (статья AAI; М.Pacheco, S.R.Childers, R.Arnold, F.Casiano, S.J.Ward, J.Pharmacol. Exp. Ther., 1991, 257 (1), сс.170-183), такие как 6-бром-(статья WIN54661; F.М.Casiano, R. Arnold, D.Haycock, J.Kuster, S.J.Ward, NIDA Res. Monogr., 1991, 105, сс.295-6) или 6-йодправадолин (статьи АМ630, К.Hosohata, R.М.Quock, R.M.Hosohata, Т.Н.Burkey, A.Makriyannis, P.Consroe, W.R.Roeske, Н.I.Yamamura, Life Sci., 1997, 61, сс.115-118; R.Pertwee, G.Griffin, S.Fernando, X.Li, A.Hill, A.Makriyannis, Life Sci., 1995, 56 (23-24), сс.1949-55). Арилбензо[b]тиофен и бензо[b]фуран (статья LY320135, С.С.Felder, К.Е.Joyce, E.M.Briley, M.Glass. К.Р.Mackie, К.J.Fahey, G.J.Cullinan, D.С.Hunden, D.W.Johnson, M.O.Chaney, G.A.Koppel, M.Brownstein, J.Pharmacol. Exp. Ther., 1998, 284 (1), сс.291-7) описаны в WO 9602248, US 5596106, 3-алкил-(5,5-дифенил)имидазолидиндионы (статья M.Kanyonyo, S.J.Govaerts, E.Hermans, J.H.Poupaert, D.M.Lambert, Bioorg. Med. Chem. Lett., 1999, 9 (15), сс.2233-2236), а также 3-алкил-5-арилимидазолидиндионы (статья F.Ooms, J.Wouters, О.Oscaro. Т.Happaerts, G.Bouchard, P.-A.Carrupt, B.Testa, D.M.Lambert, J. Med. Chem., 2002, 45 (9), сс.1748-1756), как известно, являются антагонистами рецептора СВ1, и соответственно выступают в качестве обратного агониста рецептора hCB1. WO 0015609 (FR 2783246-A1), WO 0164634 (FR 2805817-A1), WO 0228346, WO 0164632 (FR 2805818-A1), WO 0164633 (FR 2805810-A1) описывают замещенные производные 1-бис(арил)метилазетидинов в качестве антагонистов СВ1. В WO 0170700, WO 02076949 и WO 0276949 A1 описаны производные 4,5-дигидро-1H-пиразола в качестве антагонистов CB1. В нескольких патентах и публикациях описаны мостиковые и немостиковые производные 3-пиразолкарбоксамида в качестве антагонистов/обратных агонистов CB1 (WO 0132663, WO 0046209, WO 9719063, ЕР 658546, ЕР 656354, US 5624941, ЕР 576357, US 3940418, WO 03020217, WO 0335005, статьи J.M.Mussinu и др., Bioorg. Med. Chem., 2003, 11, с.251; S.Ruiu и др., J. Pharm. Expt. Ther., 2003, 306, с.363). Были описаны пиррольные агонисты каннабиноидного рецептора СВ1 в статье G.Tarzia и др., Bioorg. Med. Chem., 2003, 11, с.3965. Фенэтильные амиды были заявлены в качестве антагонистов/обратных агонистов каннабиноидного рецептора CB1 в WO 03077847, WO 03082190, WO 03086288 и WO 03087037. Различные азагетероциклы (имидазолы, триазолы и тиазолы) описаны в WO 0337332, WO 03040107, WO 03063781, WO 03082833 и WO 03078413. Дифенилпиразинкарбоксамиды описаны в WO 03051850, дифенилпиридинкарбоксамиды в WO 03084930 и дифенилбензолкарбоксамиды в WO 03084943.
Целью настоящего изобретения является получение селективных антагонистов рецептора CB1 прямого действия, соответственно обратных агонистам. Такие антагонисты/обратные антагонисты являются полезными в медицине, особенно для лечения и/или профилактики заболеваний, которые связаны с модуляцией рецепторов CB1.
Если не указано иное, следующие определения приведены для иллюстрации и определения значения и объема различных терминов, использующихся для описания настоящего изобретения.
Термин "галоген" обозначает фтор, хлор, бром и йод, предпочтительно хлор и фтор.
Термин "фармацевтически приемлемые соли" включает соли соединений формулы (I) с неорганическими или органическими кислотами, такими как хлористводородная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, малеиновая кислота, уксусная кислота, фумаровая кислота, янтарная кислота, винная кислота, метансульфоновая кислота, салициловая кислота, п-толуолсульфоновая кислота и им подобными, которые являются нетоксичными для живых организмов. Предпочтительными солями с кислотами являются формиаты, малеаты, цитраты, гидрохлориды, гидробромиды и соли с метансульфоновой кислотой, где гидрохлориды являются особенно предпочтительными.
В одном варианте осуществления, настоящее изобретение относится к соединению формулы I как определено выше, которое представляет собой 4-[(6-фторспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]морфолин, соединение формулы:
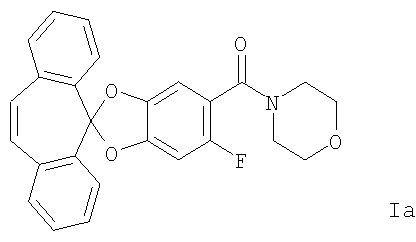
Соединения формулы I настоящего изобретения могут быть получены способами, известными из предшествующего уровня техники, или они могут быть получены способом, как описано далее, который включает:
реакцию 5,5-дихлор-5Н-дибензо[а,d]циклогептена формулы:
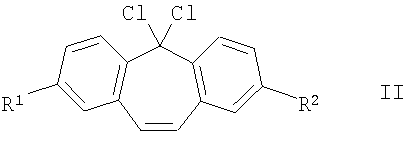
где R1 и R2 каждый независимо представляет собой водород или галоген,
с производным катехола формулы:
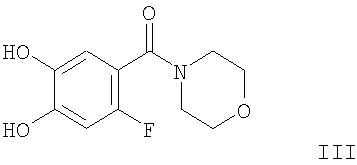
при повышенной температуре с получением соединения формулы:
Повышенная температура обозначает температуру от 100°С до 180°С, предпочтительно температуру от 110 до 130°С.
Так, промежуточное соединение катехола формулы III может быть превращено в кеталь с помощью бисзамещенного производного дихлорметана формулы II в инертном растворителе (например, толуоле или пиридине) или без растворителя, в присутствии или в отсутствии основания (например, пиридина) при повышенной температуре (например, > 100°С) с получением соединения формулы I.
(2-Фтор-4,5-дигидроксифенил)морфолин-4-илметанон (III) может быть легко получен из соответствующего защищенного дифенилметиленкеталя формулы VII обработкой кислотой (например, трифторуксусной кислотой) в подходящем инертном растворителе (например, метиленхлориде) или обработкой кислотой (например, трифторуксусной кислотой) в присутствии подходящего восстанавливающего агента (например, триэтилсилана), без растворителя или в подходящем инертном растворителе (например, метиленхлориде).
Защищенный дифенилметиленкеталь формулы VII получают из 4-фторвератрола способом, описанным на схеме 1. Последовательность реакций описана подробно в примере 1.
Схема 1
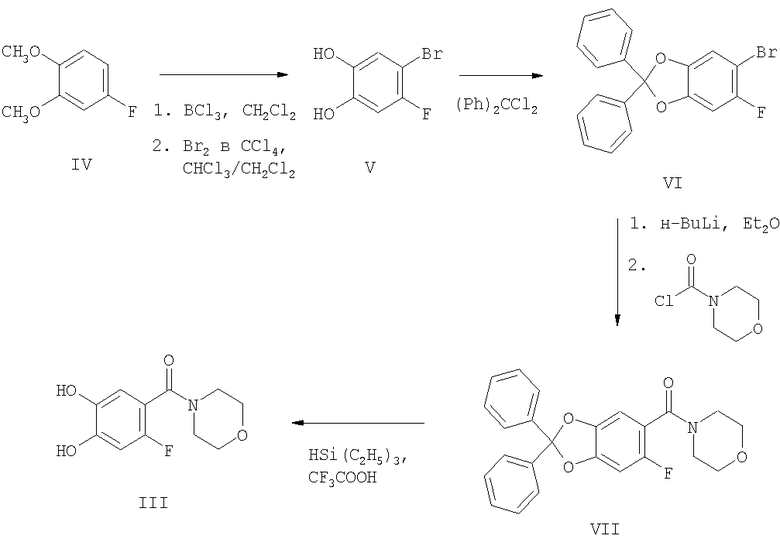
Бисзамещенные производные дихлорметана формулы II могут быть получены способами, известными из предшествующего уровня техники, из соответствующего кетона VIII реакцией с тионилхлоридом в присутствии ДМФА или другого N-формилирующего агента, или реакцией с пентахлоридом фосфора в присутствии или в отсутствии подходящего растворителя, например хлорида оксида фосфора (схема 2).
Схема 2
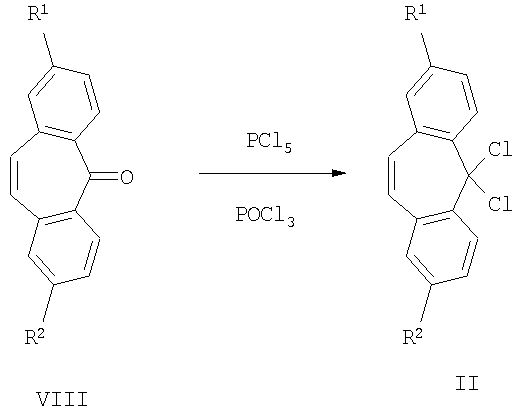
Дибензосубероны формулы VIII являются коммерчески доступными или могут быть получены способами, известными из предшествующего уровня техники.
Некоторые соединения формулы I могут иметь асимметрические центры и поэтому могут существовать более чем в одной стереоизомерной форме. Таким образом, изобретение также относится к соединениям по существу в чистой изомерной форме при одном или нескольких асимметрических центрах, а также к смесям, включая их рацемические смеси. Такие изомеры могут быть получены асимметрическим синтезом, например, с помощью хирального промежуточного соединения, или смеси могут быть разделены обычными методами, например хроматографией (хроматография с использованием хирального адсорбента или элюента), или с использованием расщепляющего агента.
Как описано выше, соединения формулы I могут использоваться в качестве терапевтически активных веществ, особенно в качестве терапевтически активных веществ для лечения и/или профилактики заболеваний, которые связаны с модулированием рецепторов CB1. В одном варианте осуществления, изобретение следовательно относится к соединениям как описано выше для применения в качестве терапевтических активных веществ, особенно в качестве терапевтических активных веществ для лечения и/или профилактики заболеваний, которые связаны с модулированием рецепторов CB1.
Изобретение также относится к фармацевтическим композициям, содержащим соединение как определено выше и фармацевтически приемлемый носитель и/или адъювант.
В другом варианте осуществления, изобретение относится к способу лечения и/или профилактики заболеваний, которые связаны с модулированием рецепторов CB1, который включает введение соединения как определено выше человеку или животному.
Изобретение также относится к применению соединений как определено выше для лечения и/или профилактики заболеваний, которые связаны с модулированием рецепторов CB1.
Кроме того, изобретение относится к применению соединений как определено выше для получения лекарственных средств для лечения и/или профилактики заболеваний, которые связаны с модулированием рецепторов CB1. Такие лекарственные средства включают соединение как определено выше.
В этом контексте, выражение 'заболевания, которые связаны с модулированием рецепторов СВ1'обозначает заболевания, которые могут быть излечены и/или предотвращены модулированием рецепторов СВ1. Такие заболевания включают, но не ограничиваются ими, физические заболевания, особенно беспокойство, психоз, шизофрению, депрессию, зависимость от психотропных средств, например зависимость и/или недостаток веществ, включая алкогольную зависимость и никотиновую зависимость, невропатии, рассеянный склероз, мигрень, стресс, эпилепсию, дискинезии, болезнь Паркинсона, амнезию, когнитивные заболевания, провалы в памяти, старческое слабоумие, болезнь Альцгеймера, заболевания, связанные с приемом пищи, ожирение, диабеты типа II или инсулиннезависимые диабеты (NIDD), желудочно-кишечные заболевания, рвоту, диарею, заболевания мочеиспускания, сердечно-сосудистые заболевания, бесплодие, воспаления, инфекционные заболевания, рак, нейровоспаление, в частности, атеросклероз, или инфекционный полиневрит, вирусный энцефалит, поражения сосудов мозга и черепная травма, а также костные заболевания, такие как остеопороз, особенно остеопороз, связанный с генетической предрасположенностью, гормональным дефицитом или возрастом, костные заболевания, связанные с раком, и болезнь кости Паджета.
В предпочтительном варианте осуществления, выражение 'заболевания, которые связаны с модулированием рецепторов СВ1' относится к заболеваниям, связанным с приемом пищи, ожирению, диабетам типа II или инсулиннезависимым диабетам (NIDD), нейровоспалению, диарее, злоупотреблению и/или зависимости от веществ, включая алкогольную зависимость и никотиновую зависимость. В более предпочтительном варианте осуществления, указанный термин относится к заболеваниям, связанным с приемом пищи, ожирению, диабетам типа II или инсулиннезависимым диабетам (NIDD), злоупотреблению и/или зависимости от веществ, включая алкогольную зависимость и никотиновую зависимость, причем ожирение является особенно предпочтительным.
В другом предпочтительном варианте осуществления, выражение 'заболевания, которые связаны с модулированием рецепторов СВ1' относится к костным заболеваниям, таким как остеопороз, особенно остеопороз, связанный с генетической предрасположенностью, гормональным дефицитом или возрастом, костные заболевания, связанные с раком, и болезнь кости Паджета.
Другим предпочтительным объектом является способ лечения или профилактики ожирения и заболеваний, связанных с ожирением, который включает введение терапевтически эффективного количества соединения формулы I в комбинации или в сочетании с терапевтически эффективным количеством других лекарственных препаратов для лечения ожирения или заболеваний, связанных с приемом пищи, таким образом, чтобы они вместе оказывали эффективный рельеф. Подходящие другие лекарственные препараты включают, но не ограничиваются ими, анорексигенные средства, ингибиторы липазы и селективные ингибиторы захвата серотонина (SSRI). Комбинации или сочетания указанных выше агентов может осуществляться при раздельном, последовательном или одновременном введении.
Предпочтительным ингибитором липазы является тетрагидролипстатин.
Подходящие анорексигенные средства для применения в комбинации с соединением настоящего изобретения включают, но не ограничиваются ими, аминорекс, амфеклорал, амфетамин, бензфетамин, хлорфентермин, клобензорекс, клофорекс, кломинорекс, клортермин, циклекседрин, дексфлерфлурамин, декстроамфетамин, диэтилпропион, дифеметоксидин, N-этиламфетамин, фенбутразат, фенфлурамин, фенизорекс, фенпропорекс, флудорекс, флуминорекс, фурфурилметиламфетамин, левамфетамин, левофацетоперан, мазиндол, мефенорекс, метамфепрамон, метамфетамин, норпсевдоэфедрин, пенторекс, фендиметразин, фенметразин, фентермин, фенилпропаноламин, пицилорекс и сибутрамин и их фармацевтически приемлемые соли.
Наиболее предпочтительными анорексигенными средствами являются сибутрамин и фентермин.
Подходящие селективные ингибиторы захвата серотонина для применения в комбинации с соединением настоящего изобретения включают: флуоксетин, флувоксамин, пароксетин и сертралин, и их фармацевтически приемлемые соли.
Другим предпочтительным объектом является способ лечения или профилактики диабетов типа II (инсулиннезависимые сахарные диабеты (NIDDM) у человека, который включает введение терапевтически эффективного количества соединения в соответствии с формулой I в комбинации или смеси с терапевтически эффективным количеством ингибитора липазы, особенно когда ингибитор липазы представляет собой орлистат. Также объектом изобретения является способ, как описано выше, для одновременного, раздельного или последовательного введения соединения в соответствии с формулой I и ингибитора липазы, особенно тетрагидролипстатина.
Другим предпочтительным объектом является способ лечения или профилактики диабетов типа II (инсулиннезависимые сахарные диабеты (NIDDM) у человека, который включает введение терапевтически эффективного количества соединения в соответствии с формулой I в комбинации или смеси с терапевтически эффективным количеством антидиабетического агента, выбранного из группы, состоящей из 1) агонистов PPARγ, таких как пиоглитазон или розиглитазон и им подобные; 2) бигуанидов, таких как метформин и им подобные; 3) сульфонилмочевин, таких как глибенкламид и им подобные; 4) агонистов PPARα/γ, таких как GW-2331 и им подобные, 5) ингибиторов DPP-IV, таких как LAF-237 (вилдаглиптин) или МК-0431 и им подобные; 6) активаторов глюкокиназы, таких как соединения, описанные, например, в WO 00/58293 А1 и им подобные. Также объектом изобретения является способ как описано выше для одновременного, раздельного или последовательного введения соединения в соответствии с формулой I и терапевтически эффективного количества антидиабетического агента из группы: 1) агонистов PPARγ, таких как пиоглитазон или розиглитазон и им подобные; 2) бигуанидов, таких как метформин и им подобные; 3) сульфонилмочевин, таких как глибенкламид и им подобные; 4) агонистов PPARα/γ, таких как GW-2331 и им подобные, 5) ингибиторов DPP-IV, таких как LAF-237 (вилдаглиптин) или МК-0431 и им подобные; 6) активаторов глюкокиназы, таких как соединения, описанные, например, в WO 00/58293 А1 и им подобные.
Другим предпочтительным объектом является способ лечения или профилактики дислипидемии у человека, который включает введение терапевтически эффективного количества соединения в соответствии с формулой I в комбинации или смеси с терапевтически эффективным количеством агента, снижающего уровень липидов, такого как 1) разрушители желчной кислоты, такие как холестирамин и им подобные; 2) ингибиторы редуктазы HMG-CoA, такие как аторвастатин и им подобные; 3) ингибиторы поглощения холестерина, такие как эзетимиб и им подобные; 4) ингибиторы СЕТР, такие как торцетрапиб, JTT 705 и им подобные; 5) агонисты PPARα, такие как беклофибрат, фенофибрат и им подобные; 6) ингибиторы синтеза липопротеинов, такие как ниацин и им подобные; и 7) агонисты рецептора ниацина. Также объектом изобретения является способ, как описано выше, для одновременного, раздельного или последовательного введения соединения в соответствии с формулой I и терапевтически эффективного количества агента, снижающего уровень липидов, такого как 1) разрушители желчной кислоты, такие как холестирамин и им подобные; 2) ингибиторы редуктазы HMG-CoA, такие как аторвастатин и им подобные; 3) ингибиторы поглощения холестерина, такие как эзетимиб и им подобные; 4) ингибиторы СЕТР, такие как торцетрапиб, JTT 705 и им подобные; 5) агонисты PPARα, такие как беклофибрат, фенофибрат и им подобные; 6) ингибиторы синтеза липопротеинов, такие как ниацин и им подобные; и 7) агонисты рецептора ниацина.
Демонстрация дополнительных видов биологической активности соединений настоящего изобретения может осуществляться в анализах in vitro, ex vivo и in vivo, которые хорошо известны специалистам в данной области техники. Например, для определения эффективности фармацевтического агента для лечения заболеваний, связанных с ожирением, таких как диабеты. Синдром Х или атеросклеротическое заболевание и родственные заболевания, такие как гипертриглицеридемия и гиперхолестеремия, могут использоваться следующие испытания.
Способ измерения уровней глюкозы в крови
У db/db мышей (полученных от Jackson Laboratories, Bar Harbor, ME) брали кровь (через глазную или хвостовую вену) и группировали в соответствии с эквивалентом, обозначающим уровни глюкозы в крови. Им вводили перорально (введением в фармацевтически приемлемом носителе) тестируемое соединение один раз в сутки от 7 до 14 дней. После этого у животных опять брали кровь через глазную или хвостовую вену и определяли уровни глюкозы в крови.
Способ определения уровней триглицеридов
У hApoAl мышей (полученных от Jackson Laboratories, Bar Harbor, ME) брали кровь (через глазную или хвостовую вену) и группировали в соответствии с эквивалентом, обозначающим уровни триглицеридов в сыворотке. Им вводили перорально (введением в фармацевтически приемлемом носителе) тестируемое соединение один раз в сутки от 7 до 14 дней. После этого у животных опять брали кровь через глазную или хвостовую вену и определяли уровни триглицеридов в сыворотке.
Способ определения уровней HDL-холестерина
Для определения уровней HDL-холестерина в плазме у hApoAl мышей брали кровь и группировали в соответствии с эквивалентом, обозначающим уровни HDL-холестерина в плазме. Мышам вводили перорально один раз в сутки носитель или тестируемое соединение в течение от 7 до 14 дней, и затем брали кровь на следующий день. Плазму анализировали на HDL-холестерин.
Кроме того, для демонстрации активности в отношении ЦНС соединений настоящего изобретения, могут использоваться следующие испытания in vivo.
Способ тестирования решения задачи и пространственной памяти
Водяной лабиринт Морриса обычно используется для оценки решения задачи и пространственной памяти (статья Jaspers и др., Neurosci. Lett., 1990, 117, сс.149-153; Morris, J. Neurosci. Methods, 1984, 11, сс.47-60). В этом испытании животных помещают в бассейн, наполненный водой, который разделен на четверти. Одна платформа спрятана в одной из четвертей. Животного помещают в бассейн, наполненный водой, и ждут, пока оно обнаружит спрятанную платформу в течение заранее определенного промежутка времени. В течение ряда тренировочных попыток животное учится находить платформу и выбираться из бассейна. Животному предоставляют несколько попыток в этом эксперименте. Для каждого животного записывают общую пройденную дистанцию, число попыток для обнаружения платформы, время до нахождения платформы и конфигурацию проплытого пути. Способность животного к обучению измеряют длительностью периода или числом попыток, необходимых для нахождения скрытой платформы. Ухудшение или улучшение памяти определяют числом попыток или временем до нахождения платформы за заранее определенную задержку времени после нахождения. Обучение и память может быть измерена количеством раз, за которые животное пересекает четверть, в которой расположена платформа, до момента нахождения.
Способ тестирования наркотической зависимости
Самоуправление у животных является определителем того, что соединение является потенциально опасным для людей. Модификации этой методики также могут использоваться для определения соединений, которые предотвращают или блокируют усиливающие свойства наркотиков, которые являются потенциально опасными. Соединение, которое устраняет самоуправление наркотика, может предотвращать опасность наркотика или зависимость от него (статьи Ranaldi и др., Psychopharmacol., 2002, 161, сс.442-448; Campbell и др., Ехр. Clin. Psychopharmacol., 2000, 8, сс.312-25). В тесте на самоуправление животных помещают в наблюдательные камеры, содержащие как активный, так и неактивный рычаг. Каждое воздействие на активный рычаг производит введение либо тестируемого соединения, либо наркотика, о котором известно, что он приводит к самоуправлению. Нажатие на неактивный рычаг не имеет никакого эффекта, но также записывается. Животные затем обучаются самоуправлению под действием соединения/наркотика в течение ряда периодов времени, имея доступ к наркотику в течение каждой ежедневной сессии. Освещение камеры дневным светом сигнализирует о начале сессии и о доступности соединения/наркотика. Когда сессия заканчивается, освещение дневным светом выключают. Первоначально, введение наркотика имеет место при каждом нажатии на активный рычаг. Как только формируется зависимость от нажатия рычага, число нажатий увеличивается с получением инъекции наркотика. После того как достигнуто устойчивое самоуправление от соединения/наркотика, может оцениваться воздействие второго соединения на поведение, зависимое от наркотика. Введение этого второго соединения до сессии может потенциировать, устранять или не производить никаких изменений в поведении самоуправления.
Следующие тесты были проведены для определения активности соединения формулы I.
Сродство соединений по изобретению по отношению к каннабиноидным рецепторам CB1 определяли с помощью мембранных препаратов из эмбриональных клеток почек человека (НЕК), в которых каннабиноидный рецептор CB1 человека кратковременно трансфицируется, с использованием системы вируса Semliki Forest в сочетании с [3H]-СР-55,940 в качестве радиолиганда. После инкубирования свежеприготовленного клеточного мембранного препарата с [3Н]-лигандом, с или без добавления соединений по изобретению, осуществляли разделение связавшегося и свободного лиганда фильтрацией через стекловолоконные фильтры. Радиоактивность фильтра определяли с помощью измерения сцинтилляции в жидкости.
Сродство соединений по изобретению по отношению к каннабиноидным рецепторам СВ2 определяли с помощью мембранных препаратов из эмбриональных клеток почек человека (НЕК), в которых каннабиноидный рецептор СВ2 человека кратковременно трансфицируется, с использованием системы вируса Semliki Forest в сочетании с [3H]-СР-55,940 в качестве радиолиганда. После инкубирования свежеприготовленного клеточного мембранного препарата с [3H]-лигандом, с или без добавления соединений по изобретению, осуществляли разделение связавшегося и свободного лиганда фильтрацией через стекловолоконные фильтры. Радиоактивность фильтра определяли с помощью измерения сцинтилляции в жидкости.
Каннабиноидную антагонистическую активность в отношении CB1 соединений по изобретению определяли функциональными исследованиями, используя СНО клетки, в которых каннабиноидные рецепторы CB1 человека стабильно экспрессируются (см. статью М. Rinaldi-Carmona и др., J. Pharmacol. Exp. Ther., 1996, 278, с.871). Стабильную экспрессию каннабиноидного рецептора человека в клеточных системах впервые описали в статьях Nature, 1990, 346, сс.561-564 (CB1) и Nature, 1993, 365, cc.61-65 (СВ2) соответственно. Аденилилциклазу стимулировали с помощью форсколина и количественно измеряли количество аккумулированного циклического АМФ. Сопутствующее активирование рецепторов CB1 агонистами рецептора CB1 (например, СР-55,940 или (R)-WIN-55212-2) может смягчаться вызванным форсколином накоплением цАМФ в зависимости от концентрации. Этот обусловленный рецептором CB1 отклик может быть антагонизирован антагонистами рецептора CB1, такими как соединения по изобретению.
Соединения формулы I показывают отличное сродство по отношению к рецептору CB1, определенное в экспериментальных условиях, описанных в статье Devane и др. Mol. Pharmacol., 1988, 34, сс.605-613. Соединения настоящего изобретения или их фармацевтически приемлемые соли или сольваты являются антагонистами и селективны в отношении рецептора CB1 со сродством менее IC50=1 мкМ, предпочтительно менее 0,100 мкМ. Они проявляют по крайней мере 10-кратную селективность в отношении рецептора СВ2.
Действие антагониста/обратного агониста по отношению к рецептору CBI на СР 55,940-индуцированной гипотермии у NMRI мышей
Животные
Самцов NMRI мыши использовали в этом исследовании и получали их от Research Consulting Company Ltd (RCC) Füllinsdorf (Switzerland). Мышей с весом 30-31 г использовали в этом исследовании. Окружающая температура составляла примерно 20-21°С и относительная влажность составляла 55-65%. 12-часовой цикл светового дня обеспечивали в комнатах, в которых осуществляли все тесты, в течение светлой фазы. Доступ к поилке и еде являлся неограниченным.
Способ
Все измерения проводили между 12:00 am и 5:00 pm. Мышей помещали в эту среду и они привыкали по меньшей мере в течение двух часов до начала эксперимента. Они всегда имели свободный доступ к еде и воде. Для каждой дозы использовали 8 мышей. Измерения ректальной температуры тела записывали с помощью ректальной пробы (RET2 Physitemp) и цифрового термометра (Digi-sense n°8528-20 Cole Parmer, Chicago USA). Пробник помещали на глубину 3,5 см в каждой мыши.
Температуру тела измеряли за 15 мин до введения либо носителя, либо антагониста/обратного агониста рецептора CB1. Через 30 или 90 мин после внутрибрюшинного или перорального введения этого соединения, соответственно, ректальную температуру тела записывали для оценки любого воздействия самого соединения. Агонист рецептора СВ СР 55,940 (0,3 мг/кг) немедленно вводили внутривенно, и через 20 мин после внутривенного введения СР 55940 вновь замеряли температуру тела.
Активность in vivo соединений формулы (1) оценивали по их способности регулировать аппетит, записывая поглощение пищи у животных, лишенных пищи.
Крыс тренировали получать пищу в течение 2 ч в день и отбирали пищу на 22 ч. Когда они привыкали к этой схеме, количество выданной пищи каждый день в течение этих 2 ч сессии поступления пищи было постоянным в последующие дни.
Для тестирования способности соединений формулы (1) уменьшать потребление пищи, 8 животных использовали в перекрестном анализе. Крыс индивидуально помещали в боксы из Plexiglas с решеткой на полу и под клеткой помещали бумагу для сбора любых выделений. Кормушку (стакан) наполняли предварительно взвешенным количеством пищи и предоставляли им на 2 ч. В конце сеанса кормления крыс возвращали в их боксы. Каждую крысу взвешивали до начала эксперимента и записывали количество пищи, поглощенное в течение этого 2 ч сеанса. За 60 мин до 2 ч сеанса питания перорально вводили либо различные дозы тестируемого соединения, либо носитель. В эксперимент включали положительный контроль римонабанта (SR141716). Тест Anova с повторными измерениями использовали после теста Student Neumann-Keuls. * Р<0,05 по сравнению с крысами, обработанными физиологическим раствором.
Кроме того, применимость соединений формулы I для лечения болезней или заболеваний может быть продемонстрирована на моделях заболеваний животных, которые описаны в предшествующем уровне техники. Далее приведены примеры таких моделей заболеваний животных: а) уменьшение поглощения сладкой пищи мармозетками (статья Behavioural Pharm., 1998, 9, сс.179-181); б) уменьшение поглощения сахарозы и этанола мышами (статья Psychopharm., 1997, 132, сс.104-106); в) уменьшение двигательной активности и пространственных условий у крыс (статьи Psychopharm., 1998, 135, сс.324-332; Psychopharmacol, 2000, 151, сс.25-30); г) спонтанная локомоторная активность у мышей (статья J. Pharm. Exp. Ther., 1996, 277, сс. 586-594); д) уменьшение опиатного самоуправления у мышей (статья Sci., 1999, 283, сс.401-404).
Соединения формулы I и/или их фармацевтически приемлемые соли могут использоваться в качестве лекарственных средств, например, в форме фармацевтических препаратов для энтерального, парентерального или местного введения. Они могут вводиться, например, перорально, например, в форме таблеток, покрытых таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий, ректально, например, в форме суппозиториев, парентерально, например, в форме инъекционных растворов или инфузионных растворов, или местно, например, в форме мазей, кремов или масел. Пероральное введение является предпочтительным.
Получение фармацевтических препаратов может осуществляться способом, известным специалисту в данной области техники, путем введения описанных соединений формулы (I) и/или их фармацевтически приемлемых солей, необязательно в комбинации с другими терапевтически ценными веществами, в галеновую форму для введения вместе с подходящими, нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими материалами носителя и, при необходимости, обычными фармацевтическими адъювантами.
Подходящими материалами носителя являются не только неорганические материалы носителя, но также органические материалы носителя. Так, например, лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли могут использоваться в качестве материалов носителя для таблеток, покрытых таблеток, драже и твердых желатиновых капсул. Подходящими материалами носителя для мягких желатиновых капсул являются, например, растительные масла, воски, жиры и полутвердые и жидкие полиолы (в зависимости от природы активного ингредиента, однако носитель может не использоваться для мягких желатиновых капсул). Подходящими материалами носителя для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертированный сахар и им подобные. Подходящими материалами носителя для инъекционных растворов являются, например, вода, спирты, полиолы, глицерин и растительные масла. Подходящими материалами носителя для суппозиториев являются, например, природные или отвержденные масла, воски, жиры и полужидкие или жидкие полиолы. Подходящими материалами носителя для местных препаратов являются глицериды, полусинтетические и синтетические глицериды, гидрированные масла, жидкие воски, жидкие парафины, жидкие жирные спирты, стеролы, полиэтиленгликоли и производные целлюлозы.
Обычные стабилизаторы, консерванты, увлажняющие и эмульгирующие агенты, агенты, улучшающие консистенцию, агенты, улучшающие вкус, соли для регулирования осмотического давления, буферные вещества, солюбилизаторы, красители и маскирующие агенты и антиоксиданты могут рассматриваться в качестве фармацевтических адъювантов.
Дозировка соединений формулы I может изменяться в широких пределах в зависимости от контролируемого заболевания, возраста и индивидуального состояния пациента и способа введения, и будет, конечно, подбираться по индивидуальным требованиям в каждом отдельном случае. Для взрослых пациентов суточная доза составляет от около 1 до 1000 мг, особенно от около 1 до 100 мг. В зависимости от степени тяжести заболевания и точности фармакокинетического профиля соединение может вводиться одной или несколькими суточными дозированными единицами, например в 1-3 дозированных единицах.
Фармацевтические препараты обычно содержат около 1-500 мг, предпочтительно 1-100 мг соединения формулы I.
Следующие примеры служат для более подробной иллюстрации настоящего изобретения. Однако они не должны рассматриваться как ограничение его объема.
Примеры
MS = масс-спектрометрия, EI = захват электрона, ISP = ионный спрей (положительный ион). Все эксперименты проводили в инертной атмосфере (азот или аргон).
Пример 1
Получение (2-фтор-4,5-дигидроксифенил)морфолин-4-илметанона
а) Получение 4-бром-5-фторбензол-1,2-диола
К охлажденному (-78°С) раствору 4-фторвератрола (5,0 г, 32 ммоля) в дихлорметане (106 мл) медленно добавляли раствор трихлорида бора в дихлорметане (1М, 96 мл, 96 ммолей, 3,0 эквив.). Реакционную смесь нагревали до 20°С и перемешивали в течение ночи. Реакционную смесь выливали в ледяную воду, экстрагировали этилацетатом (3 раза). Объединенные органические слои промывали водным раствором бикарбоната натрия, высушивали сульфатом натрия и отфильтровывали. Летучие примеси удаляли в вакууме. Коричневое твердое вещество разбавляли хлороформом (50 мл) и дихлорметаном (10 мл). Медленно добавляли раствор брома в тетрахлориде углерода (5 мл). После перемешивания в течение 3 ч при комнатной температуре летучие примеси удаляли в вакууме. Очистка ускоренной хроматографией приводила к получению указанного в заголовке соединения (6,51 г, 98%) в виде коричневого твердого вещества.
ISP MS: m/e=207,9 ([М+Н]+).
б) Получение 5-бром-6-фтор-2,2-дифенилбензо[1,3]диоксола
Смесь 4-бром-5-фторбензол-1,2-диола (12 г, 58,0 ммолей) и дифенилдихлорметана (1,2 эквив., 16,50 г) перемешивали при комнатной температуре до прекращения выделения газа. Смесь нагревали при перемешивании при 180°С в течение 20 мин. Реакционной смеси позволяли охладиться до комнатной температуры, разбавляли метанолом (50 мл) и осторожно перемешивали. Осадившийся продукт собирали фильтрацией и растворяли в толуоле (50 мл). Добавляли метанол (100 мл) и смесь перемешивали в течение 30 мин при комнатной температуре. Осадившийся продукт собирали фильтрацией (выход 10,3 г, 48%), другую партию (6,4 г, 30%) извлекали из маточника.
ISP MS: m/e=370,0 ([М+Н+]).
в) Получение (6-фтор-2,2-дифенилбензо[1,3]диоксол-5-ил)морфолин-4-илметанона
К охлажденному (-78°С) раствору 5-бром-6-фтор-2,2-дифенилбензо[1,3]диоксола (17,59 г, 47,4 ммоля) в диэтиловом эфире (300 мл) медленно добавляли раствор н-бутиллития в гексане (1,6М, 30 мл, 48 ммолей, 1,0 эквив.). Реакционную смесь перемешивали в течение 1 ч при -78°С перед добавлением 4-морфолинкарбонилхлорида (8,5 г, 56,9 ммоля, 1,2 эквив.). Реакционной смеси позволяли нагреться до 20°С и выливали в водный раствор бикарбоната натрия. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором хлорида натрия. Летучие примеси удаляли в вакууме. Очистка ускоренной хроматографией приводила к получению соединения (6-фтор-2,2-дифенилбензо[1,3]диоксол-5-ил)морфолин-4-илметанона (13,0 г, 68%) в виде светло-желтого твердого вещества.
ISP MS: m/e=406,2 ([М+Н]+).
г) Получение (2-фтор-4,5-дигидроксифенил)морфолин-4-илметанона
К охлажденному (на ледяной бане) раствору (6-фтор-2,2-дифенилбензо[1,3]диоксол-5-ил)морфолин-4-илметанона (5,70 г, 14,06 ммоля) в трифторуксусной кислоте (60 мл) добавляли триэтилсилан (2,1 эквив., 4,7 мл) в течение 10 мин. Смесь перемешивали в течение 20 мин при 0°С и в течение 4 ч при комнатной температуре. Летучие примеси удаляли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (2:1 этилацетат/гептан-этилацетат- 10:1 этилацетат/метанол) с получением (2-фтор-4,5-дигидроксифенил)морфолин-4-илметанона в виде светло-коричневого твердого вещества (3,19 г, 94%).
ISP MS: m/e=242,2 ([М+Н]+).
Пример 2
Получение 5,5-дихлор-5Н-дибензо[a,d]циклогептена
5,5-Дихлор-5Н-дибензо[а,d]циклогептен получали в соответствии с методикой, описанной в статье J.J. Looker, J. Org. Chem., 1966, 31, с.3599: 5-Дибензосуберенон (2 г, 9,7 ммоля) растворяли в оксихлориде фосфора (4,5 мл) и добавляли пентахлорид фосфора (3,13 г, 15,03 ммоля). Смесь нагревали в течение 4 ч при 120°С. Смеси позволяли охладиться до комнатной температуры и растворитель удаляли при пониженном давлении. Сырой продукт использовали без дополнительной очистки.
Пример 3
Получение 4-[(6-фторспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]морфолина
5,5-Дихлор-5Н-дибензо[а,d]циклогептен (0,783 г, 3,0 ммоля) растворяли в толуоле (8 мл) и нагревали до 120°С. По каплям добавляли раствор 4-(2-фтор-4,5-дигидроксибензоил)морфолина (0,36 г, 1,5 ммоля) в толуоле (4 мл) в течение 20 мин. После окончания добавления смесь нагревали в течение 1 ч при 120°С. Смесь охлаждали до комнатной температуры и упаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент от 1:0 до 10:1 дихлорметан/этилацетат) с получением указанного в заголовке соединения в виде беловатой пены.
MS: m/e 430,4 [(М+Н)+]
NMR: δ (CDCl3) 3,33 (br s, 2H), 3,60 (br s, 2H), 3,74 (br s, 4H), 6,54, (d, 1H, J=8,8HZ), 6,87 (d, 1H, J=5,2Hz), 7,17, (s, 2H), 7,44 (m, 4H), 7,50 (m, 2H), 7,93 (m, 2H) ppm.
Примеры галеновых форм
Пример А
Покрытые пленкой таблетки, содержащие следующие ингредиенты, могут быть получены следующим способом:
Активный ингредиент просеивали и смешивали с микрокристаллической целлюлозой и смесь гранулировали с раствором поливинилпирролидона в воде. Гранулят смешивали с натриевой солью гликолята крахмала и стеаратом магния и спрессовывали с получением ядер по 120 или 350 мг соответственно. Ядра покрывали водным раствором/суспензией указанной выше пленочной оболочки.
Пример Б
Капсулы, содержащие следующие ингредиенты, могут быть получены обычным образом:
Компоненты просеивали и смешивали и наполняли ими капсулы размера 2.
Пример В
Инъекционные растворы могут иметь следующий состав:
Активный ингредиент растворяли в смеси полиэтиленгликоля 400 и воды для инъекций (часть). Значение рН доводили до 5,0 уксусной кислотой. Объем доводили до 1,0 мл добавлением оставшегося количества воды. Раствор отфильтровывали, наполняли им сосуды, используя подходящее устройство, и стерилизовали.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СПИРОПЕНТАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2360909C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРА DPP-IV ДЛЯ СНИЖЕНИЯ ПРИСТУПОВ ГЛИКЕМИИ | 2006 |
|
RU2440143C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА СВ1 | 2012 |
|
RU2593751C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ РКС ПРИ ОСЛОЖНЕНИЯХ, ВЫЗВАННЫХ ДИАБЕТОМ | 2007 |
|
RU2447892C2 |
| 2-АМИНОБЕНЗОТИАЗОЛЫ В КАЧЕСТВЕ ОБРАТНЫХ АГОНИСТОВ РЕЦЕПТОРОВ CB | 2004 |
|
RU2344132C2 |
| ПРИМЕНЕНИЕ АНТАГОНИСТОВ РЕЦЕПТОРА СВ1 ДЛЯ ПОЛУЧЕНИЯ КОМПОЗИЦИИ, ПРИГОДНОЙ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ПЕЧЕНИ | 2005 |
|
RU2402328C2 |
| НОВОЕ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ГАСТРОЭЗОФАГЕАЛЬНОЙ РЕФЛЮКСНОЙ БОЛЕЗНИ | 2002 |
|
RU2280443C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛИНА, ОБЛАДАЮЩИЕ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2357958C2 |
| 5-(3,4-ДИХЛОРФЕНИЛ)-N-(2-ГИДРОКСИЦИКЛОГЕКСИЛ)-6-(2,2,2-ТРИФТОРЭТОКСИ)НИКОТИНАМИД И ЕГО СОЛИ В КАЧЕСТВЕ СРЕДСТВ, ПОВЫШАЮЩИХ КОНЦЕНТРАЦИЮ ЛВП ХОЛЕСТЕРИНА | 2010 |
|
RU2541475C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ЗАВИСИМОСТЕЙ | 2008 |
|
RU2492858C2 |
Настоящее изобретение относится к новым производным дибензосуберона формулы (I):
где R1 и R2 каждый независимо представляет собой водород или галоген. Эти соединения используются для лечения и/или профилактики заболеваний, которые связаны с модулированием рецепторов CB1. Изобретение относится также к фармацевтической композиции на основе этих соединений. 2 н. и 3 з.п. ф-лы.
1. Соединения общей формулы
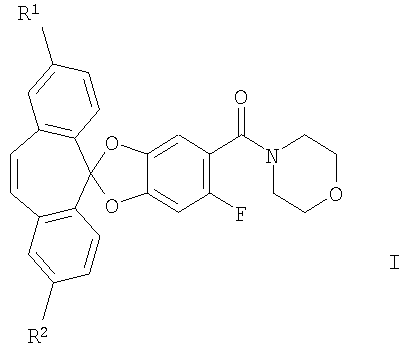
где R1 и R2 каждый независимо представляет собой водород или галоген.
2. Соединение по п.1, где R1 и R2 представляют собой водород.
3. Соединение по п.1 или 2, которое представляет собой 4-[(6-фторспиро[1,3-бензодиоксол-2,5'-[5Н]дибензо[а,d]циклогептен]-5-ил)карбонил]морфолин.
4. Фармацевтическая композиция для лечения и/или профилактики заболеваний, которые связаны с модулированием рецептора CB1, содержащая соединение по любому из пп.1-3 и фармацевтически приемлемый носитель и/или адъювант.
5. Соединения по любому из пп.1-3, предназначенные для использования в качестве терапевтически активных веществ для лечения и/или профилактики заболеваний, которые связаны с модулированием рецептора CB1.
| WO 2004013120 A1, 12.02.2004 | |||
| ЭЛЕКТРОМЕХАНИЧЕСКАЯ ВИНТОВАЯ СИЛОВАЯ ГОЛОВКА | 0 |
|
SU217890A1 |
| Способ получения производных спиро (дибензо (а-д) циклогептади(или три)-ен-5:2"-(4"-аминометилдиоксолана-1",3")) | 1973 |
|
SU504488A3 |

