Данная заявка утверждает приоритет предварительной заявки США № 60/612458, поданной 22 сентября 2004 г., и предварительной заявки США № 60/683006, поданной 19 мая 2005 г., полное содержание которых включено здесь в виде ссылки.
Область техники, к которой относится изобретение
Данное изобретение главным образом относится к применению 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-она в качестве химического сенсибилизирующего вещества, которое повышает эффективность цитотоксических дозированных средств или лучевой терапии. Данное изобретение относится к фармацевтическим комбинациям 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-она, или его фармацевтически приемлемых солей и, по меньшей мере, одного дополнительного терапевтического средства, наборам, содержащим такие комбинации и способам применения таких комбинаций при лечении субъектов, страдающих такими заболеваниями как рак.
Предпосылки к созданию изобретения
Соединение 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-он, представленное формулой 1
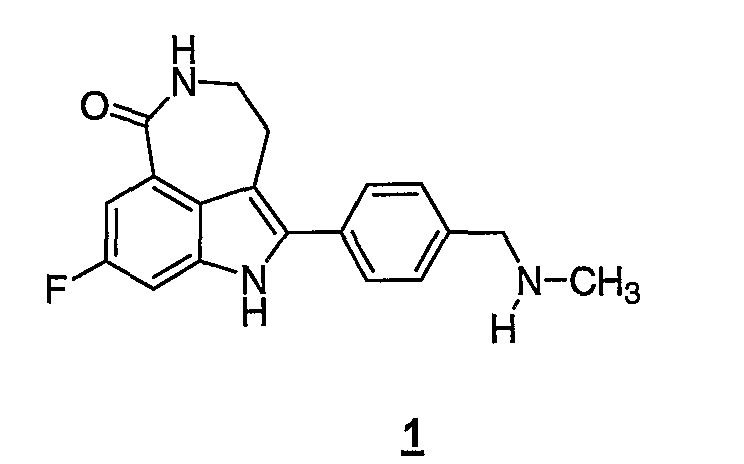
является синтетическим ингибитором поли-(AДФ-рибоза) полимеразы (PARP). Соединение формулы 1 и его соли, можно получить, как описано в патенте США №6495541; заявке PCT № PCT/IB2004/000915, международной публикации № WO 2004/087713; предварительных заявках на выдачу патента США №№60/612457, 60/612459 и 60/679296, раскрытия которых включены здесь в полном объеме в виде ссылки.
В настоящее время в семействе PARP, посредством гомологии ДНК последовательностей, выявлено восемнадцать ферментов, и изучены химические и ферментативные свойства семи из них: PARP-1 и PARP-2 стимулируются разрывами нитей ДНК, PARP-3 взаимодействует с PARP-1 и центросомой, PARP-4, также известный как PARP-запасник (VPARP), является самым большим PARP и связан с цитоплазматическими хранилищами, танкираза 1 и 2 (PARP-5a и 5b) связаны с теломерными белками, и функция PARP-7 (TiPARP) в настоящее время не ясна, но он может быть задействован в функции Т-клеток и может поли(АДФ-рибозил)ировать гистоны (Ame JC, Splenlehauer C and de Murcia G. The PARP Superfamily. Bioessays 26 882-893 (2004)). Фармакологические исследования показали, что соединение формулы 1 является ингибитором PARP-1 (Ki=1,4 нМ) и PARP-2 (Ki=0,17 нМ). Исходя из структурного подобия аминокислотных последовательностей среди ферментов PARP, соединение формулы 1, возможно, также связывается с высокой аффинностью с другими членами этого семейства.
Опосредованное ферментом восстановление одно- или двунитевых разрывов ДНК является потенциальным механизмом устойчивости к лучевой терапии или воздействию цитотоксических дозированных средств, чей механизм действия обусловлен повреждением ДНК. Таким образом, стратегией усиления действия этих средств является ингибирование ДНК-репаративных ферментов. PARP-1, наиболее изученный член семейства PARP, представляет собой ядерный фермент, регулируемый повреждением ДНК, опосредует передачу фрагментов АДФ-рибозы от NAD+ к ряду акцепторных белков. В зависимости от степени повреждения ДНК, активирование PARP-1 и последующее поли(АДФ-рибозил)ирование опосредуют восстановление поврежденной ДНК или индуцируют клеточную гибель. При умеренных повреждениях ДНК, PARP-1 играет значительную роль в процессе восстановления ДНК. С другой стороны, в случае массивных повреждений ДНК, избыточная активация PARP-1 истощает запасы АТФ (при восполнении NAD+), что в итоге приводит к гибели клеток в результате некроза (Tentori L, Portarena I, Graziani G. Potential applications of poly(ADP-ribose) polymerase (PARP) inhibitors. Pharmacol Res 2002, 45, 73-85). Это активирование PARP может также привести к высвобождению АИФ (апоптоз-индуцирующий фактор), который запускает независимый от каспазы путь апоптоза. (Hong SJ, Dawson TM and Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF. Trends in Pharmacological Sciences 25 259-264 (2004)).
В результате двойного назначения PARP-1 ингибиторы этого фермента, такие как 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-он, представленный формулой 1, могут действовать как химические сенсибилизирущие агенты (предотвращая восстановление ДНК, например, после противоопухолевой терапии), или в качестве дозированных средств при различных заболеваниях и токсических состояниях, которые предполагают окислительный или индуцированный оксидом азота стресс и последующую гиперактивацию PARP. Такие состояния включают неврологические и нейродегенеративные заболевания (например, болезнь Паркинсона, болезнь Альцгеймера) (Love S, Barber R, Wilcock GK. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain 1999; 122:247-53; Mandir AS, Przedborski S, Jackson-Lewis V, et al. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci USA 1999; 96:5774-9); сердечно-сосудистые заболевания (например, инфаркт миокарда, ишемическо-реперфузионное повреждение) (Pieper AA, Walles T, Wei G, et al. Myocardial postischemic injury is reduced by poly(ADP-ribose) polymerase-1 gene disruption. J MoI Med 2000; 6:271-82; Szabó G, Bährle S, Stumpf N, et al. Poly(ADP-ribose) polymerase inhibition reduces reperfusion injury after heart transplantation. Circ Res 2002; 90:100-6; патент США 6423705); воспалительные заболевания, (Szabó C, Dawson V. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. TIPS 1998; 19:287-98); сосудистые нарушения при диабете (Soriano FG, Virág L, Szabó C. Diabetic endothelial dysfunction: role of reactive oxygen and nitrogen species production and poly(ADP-ribose) polymerase activation. J Mol Med 2001; 79: 437-48); артриты (Szabó C, Virág L, Cuzzocrea S, et al. Protection against peroxynitrite-induced fibroblast injury and arthritis development by inhibition of poly(ADP-ribose) synthase. Proc Natl Acad Sci USA 1998, vol.95, pp. 3867-72); и цисплатин-индуцированную нефротоксичность (Racz et al. «BGP-15 - a novel poly(ADP-ribose) polymerase inhibitor - protects against nephrotoxicity of cisplatin without compromising its antitumor activity» Biochem Pharmacol 2002; 63: 1099-111). Более того, показано, что опухолевые клетки, лишенные BRCA2, резко чувствительны к ингибиторам PARP, применяемым отдельно. (Bryant et al. «Specific killing of BRCA2 deficient tumors with inhibitors of poly(ADP-ribose)polymerase» Nature, 2005, vol. 434, pp. 913-917; Farmer et al. «Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy» Nature, 2005, vol. 434, pp. 917-921). Ингибиторы PARP также участвуют в усилении индукции экспрессии гена Reg в β-клетках и гена HGF и, соответственно, способствуют пролиферации панкреатических β-клеток в островках Лангерганса и тормозят клеточный апоптоз (публикация патентной заявки США 2004/0091453; публикация PCT № WO 02/00665). Дополнительно, ингибиторы PARP также используются в косметических препаратах, главным образом в лосьонах после загара (публикация PCT № WO 01/82877). В настоящее время отсутствуют реализуемые на рынке ингибиторы PARP.
Рак остается заболеванием, при котором медицинские потребности являются крайне неудовлетворенными. Цитотоксическая химиотерапия остается основой системной терапии большинства злокачественных новообразований, особенно на поздней стадии заболевания. Однако у пациентов с прогрессирующей или метастазирующей опухолью немногие цитотоксические химиотерапевтические средства или режимы эффективны в повышении общей выживаемости. Кроме того, маленькое терапевтическое окно, связанное с цитотоксическими веществами, приводит к сильной токсичности в сочетании с субоптимальной эффективностью. Таким образом, химическое сенсибилизирующее вещество, которое повышает эффективность цитотоксических дозированных средств в хорошо переносимых дозах может удовлетворять критической необходимости онкологических больных.
Лучевая терапия является эффективным способом лечения рака, применяемым при большинстве типов опухолей для ограниченного контроля заболевания. Более 50% всех онкологических больных будут получать лучевую терапию в течение их болезни (Foroudi F. et al. An evidence-based estimate of appropriate radiotherapy utilization rate for breast cancer. Int J Radiat Oncol Biol Phys. 2002, 53: 1240-53; Foroudi F. et al. An evidence-based estimate of the appropriate radiotherapy utilization rate for colorectal cancer. Int J Radiat Oncol Biol Phys. 2003, 56: 1295-307; Foroudi F. et al. Evidence-based estimate of appropriate radiotherapy utilization rate for prostate cancer. Int J Radiat Oncol Biol Phys. 2003, 55: 51-63; Barbera L. et al. Estimating the benefit and cost of radiotherapy for lung cancer. Int J Technol Assess Health Care. 2004, 20: 545-51). Однако даже в первой линии терапии раков, при которых лучевую терапию применяют в лечебных целях (например, рак головы и шеи, саркома мягких тканей и карцинома шейки матки), не все пациенты реагируют удовлетворительно. Таким образом, существует необходимость в способах лечения, которые увеличат реакцию у всех пациентов. Обычно стандартную химиотерапию проводят перед или после лучевой терапии. Альтернативным подходом является объединение лучевой терапии с новейшими противоопухолевыми средствами, которые специально разработаны для усиления эффективности лучевой терапии. Такие средства воздействуют на пять ключевых факторов, которые определяют реакцию опухоли на облучение («Cell survival as a determinant of tumor response». Basic clinical radiobiology 3rd Edition. Steel GG (Ed.). Arnold Press UK, pp. 52-63, 2002). К ним относятся способность к восстановлению повреждений ДНК, вызванных лучевой терапией; перераспределение клеток во время клеточного цикла после лучевой терапии (так что опухолевые клетки, которые были в устойчивой фазе при первой дозе облучения, могут прогрессировать к более чувствительной фазе посредством следующей фракции облучения); репопуляция, в результате чего выжившие клетки продолжают делиться, и таким образом увеличивается опухолевая масса между фракциями облучения; реоксигенация клеток, выживших в начальном цикле лучевой терапии, в результате являются более слабо оксигенированы, и наконец, врожденная радиочувствительность конкретной ткани. Среди этих факторов повышенное восстановление и репопуляция приводят к радиоустойчивости, тогда как перераспределение, реоксигенация и врожденная радиочувствительность могут делать опухоль более отзывчивой на лучевую терапию. Очевидно, что использование агентов, которые уменьшают способность к репарации ДНК, совместно с лучевой терапией имеют потенциальную возможность усилить радиотерапевтический результат. Активирование PARP-1 и последующее поли-(ADP-рибозил)ирование отмечены в ответ на повреждение ДНК, вызванное радиацией (Satoh MS & Lindahl T. «Role of poly(ADP-ribose) formation in DNA repair» Nature. 1992, 356: 356-358). Кроме того, клеточные линии и нокаут-мыши, выведенные с дефектом экспрессии и активности PARP-1, показывают сильную радиочувствительность, подтверждая, что PARP-1 является выгодной мишенью для потенцирования действием излучения (Wang et al. «Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease». Genes Dev. 1995, 9: 509-20; de Murcia et al. «Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells» Proc Natl Acad Sci USA. 1997, 94: 7303-7; Masutani et al. «Function of poly(ADP-ribose) polymerase in response to DNA damage: gene-disruption study in mice» Mol Cell Biochem. 1999, 193: 149-52). В дополнение к прямым эффектам на репарацию ДНК, семейство ингибиторов PARP-1 детально является вазоактивным и по существу увеличивает потенциальные возможности для реоксигенации опухоли между фракциями облучения, что может далее способствовать усиленной реакции на облучение (Calabrese et al. «Anticancer chemo- and radio-sensitisation in vitro and in vivo by a potent novel poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor, AG14361». J. Natl. Cancer Inst. 2004, 96: 56-67).
Краткое изложение сущности изобретения
В одном варианте осуществления настоящее изобретение относится к дозированной форме для применения у млекопитающих, где данная дозированная форма содержит соединение формулы 1:
его фармацевтически приемлемую соль или сольват, или их смесь, в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 часов после введения млекопитающему.
В другом варианте осуществления данное изобретение относится к дозированной форме для введения млекопитающему, где дозированная форма содержит соединение формулы 1, его фармацевтически приемлемую соль или сольват, или их смесь, в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 10 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 часов после введения млекопитающему.
В другом варианте осуществления данное изобретение относится к дозированной форме для введения млекопитающему, где дозированная форма содержит соединение формулы 1, его фармацевтически приемлемую соль или сольват, или их смесь, в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 часов после введения млекопитающему, где дозированная форма представляет собой лиофилизированный порошок для инъекций.
В другом варианте осуществления данное изобретение относится к дозированной форме для введения млекопитающему, где дозированная форма содержит соединение формулы 1, фармацевтически приемлемую соль или сольват, или их смесь, в количестве, эффективном для ингибирования в лимфоцитах перифирической крови фермента поли-(ADP-рибозил) полимераза, по меньшей мере, на 50% в течение, по меньшей мере, 24 часов после введения млекопитающему.
В другом варианте осуществления данное изобретение относится к дозированной форме для введения млекопитающему, где дозированная форма содержит соединение формулы 1, фармацевтически приемлемую соль или сольват, или их смесь, в количестве, эффективном для ингибирования в лимфоцитах периферической крови фермента поли-(ADP-рибозил) полимераза, по меньшей мере, на 50%, в течение, по меньшей мере, 24 часов после введения млекопитающему, где дозированная форма представляет собой лиофилизированный порошок для инъекций.
В другом варианте осуществления данное изобретение относится к дозированной форме для введения млекопитающему, где дозированная форма содержит соединение формулы 1, его фармацевтически приемлемую соль или сольват, или их смесь, в количестве от 1 до 48 мг/м2, выраженном как эквивалентная масса свободного основания соединения формулы 1.
В другом варианте осуществления данное изобретение относится к дозированной форме для введения млекопитающему, где дозированная форма содержит соединение формулы 1, его фармацевтически приемлемую соль или сольват, или их смесь, в количестве от 1 до 48 мг/м2, выраженном как эквивалентная масса свободного основания соединения формулы 1, где дозированная форма представляет собой лиофилизированный порошок для инъекций.
В другом варианте осуществления данное изобретение относится к дозированной форме для введения млекопитающему, где дозированная форма содержит соединение формулы 1, его фармацевтически приемлемую соль или сольват, или их смесь, в количестве от 2 до 96 мг, выраженном как эквивалентная масса свободного основания соединения формулы 1.
В другом варианте осуществления данное изобретение относится к дозированной форме для введения млекопитающему, где дозированная форма содержит соединение формулы 1, его фармацевтически приемлемую соль или сольват, или их смесь, в количестве от 2 до 96 мг, выраженном как эквивалентная масса свободного основания соединения формулы 1, где дозированная форма представляет собой лиофилизированный порошок для инъекций.
В другом варианте осуществления данное изобретение относится к способу лечения рака у млекопитающего, включающему введение млекопитающему
(a) соединения формулы 1, его фармацевтически приемлемой соли или сольвата, или их смеси в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 часов после введения млекопитающему; и
(б) терапевтически эффективного количества, по меньшей мере, одного противоопухолевого средства.
В другом варианте осуществления данное изобретение относится к способу лечения рака у млекопитающего, включающему введение млекопитающему
(a) соединения формулы 1, его фармацевтически приемлемой соли или сольвата, или их смеси в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1, по меньшей мере, в течение 24 часов после введения млекопитающему; и
(б) терапевтически эффективного количества, по меньшей мере, одного противоопухолевого средства,
где противоопухолевое средство применяют в течение 1 часа после применения соединения формулы 1.
В другом варианте осуществления, данное изобретение относится к способу лечения рака у млекопитающего, включающему введение млекопитающему
(a) соединения формулы 1, его фармацевтически приемлемой соли или сольвата, или их смеси в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 часов после введения млекопитающему; и
(б) терапевтически эффективного количества, по меньшей мере, одного противоопухолевого средства,
где рак выбран из рака легких, рака костей, рака поджелудочной железы, рака кожи, рака головы или шеи, кожной или внутриглазной меланомы, рака матки, рака яичников, рака прямой кишки, рака анальной области, рака желудка, рака толстой кишки, рака молочной железы, карциномы фаллопиевых труб, карциномы эндометрия, карциномы шейки матки, карциномы влагалища, карциномы вульвы, болезни Ходжкина, рака пищевода, рака тонкой кишки, рака эндокринной системы, рака щитовидной железы, рака паращитовидной железы, рака надпочечников, саркомы мягких тканей, рака уретры, рака пениса, рака предстательной железы, хронического или острого лейкоза, лимфоцитарной лимфомы, рака мочевого пузыря, рака почки или мочеточника, почечноклеточного рака, карциномы почечной лоханки, новообразования центральной нервной системы (ЦНС), первичной лимфомы ЦНС, опухолей позвоночника, глиомы ствола мозга, аденомы гипофиза, и их сочетаний.
В другом варианте осуществления, данное изобретение обеспечивает набор для лечения рака у млекопитающего, где набор содержит:
(a) количество соединения формулы 1, его фармацевтически приемлемой соли или сольвата, или их смеси, и фармацевтически приемлемый носитель или разбавитель в первой единичной дозированной форме;
(б) количество, по меньшей мере, одного противоопухолевого средства и фармацевтически приемлемый носитель или разбавитель по меньшей мере во второй единичной дозированной форме; и
(c) контейнер для содержания первой и, по меньшей мере, второй дозированных форм;
где количество соединения формулы 1 является эффективным для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 часов после введения млекопитающему.
В другом варианте осуществления данное изобретение обеспечивает способ лечения рака, где способ включает применение у млекопитающего
(a) соединения формулы 1, его фармацевтически приемлемой соли или сольвата, или их смеси, в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 часов после введения млекопитающему; и
(б) комбинации иринотекана, 5-фторурацила и лейковорина.
В другом варианте осуществления данное изобретение обеспечивает способ лечения рака, где способ включает применение у млекопитающего
(a) соединения формулы 1, его фармацевтически приемлемой соли или сольвата, или их смеси, в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 часов после введения млекопитающему; и
(б) дозы излучения, эффективной для уничтожения рака.
Определения и аббревиатуры терминов
Термин «соединение I» относится к фосфатной соли 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-она. Термин «соединение формулы 1» означает 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-он, свободное основание.
«Аномальный клеточный рост», как используется здесь, если не указано особо, означает клеточный рост, который не зависит от нормальных регуляторных механизмов (например, нарушение контактного ингибирования).
Термин «лечение», как используется здесь, если не оговорено особо, означает реверсирование, облегчение, торможение прогрессирования, или предотвращение заболевания или состояния, к которому этот термин применяется, или одного или нескольких симптомов такого заболевания или состояния. Термин «лечить», как используется здесь, если не оговорено особо, относится к процессу лечения как «лечение» определено непосредственно ранее.
Термин «радиосенсибилизирующее вещество», как используется здесь, означает дозированное средство, которое делает опухолевые клетки более чувствительными к лучевой терапии.
Термин «лучевая терапия», как используется здесь, включает наружную лучевую радиотерапию (XBRT) или дистанционную лучевую терапию, брахитерапию или радиотерапию с использованием закрытых источников излучения, или радиотерапию с использованием негерметизированного источника излучения. Различия между этими тремя основными направлениями лучевой терапии относятся к положению источника радиоактивного излучения; наружный находится вне тела, в то время как при радиотерапии с применением закрытого и негерметизированного источника излучения радиоактивный материал доставляется внутрь организма. Наружная лучевая радиотерапия является наиболее принятой формой лучевой терапии, при которой пациент лежит на кушетке, и внешний источник рентгеновских лучей направлен на конкретную область тела. Ионизирующее излучение взаимодействует с клетками и поглощается, разрушая ДНК клеток. Брахитерапия представляет собой применение лучевой терапии с использованием закрытых источников, которые помещаются как можно ближе к области, подвергающейся обработке. Этот вид лучевой терапии применяют при лечении опухолей, когда источник излучения может быть помещен в полость тела, например в пищевод или бронх, или когда опухоль достигаема для иглы или катетера, помещенных в опухоль, например, области головы и шеи и кожа. Брахитерапию возможно применять при большинстве локализаций опухоли. Ее можно применять в качестве первичного лечения или в комбинации с наружной лучевой радиотерапией. Лучевая терапия с использованием негерметизированного источника излучения относится к применению растворимых форм радиоактивных веществ, которые парентерально вводят в организм. У всех этих веществ есть одна общая особенность, а именно нерадиоактивная роль исходного вещества. Протонная терапия является частным случаем наружной лучевой радиотерапии, где частицы являются протонами.
Термин «радиоиммунотерапия», как используется здесь, означает лучевую терапию, при которой цитотоксические радионуклиды связаны с антителами с целью доставки токсинов прямо к опухолям-мишеням. Терапия с использованием направленного излучения по сравнению с использованием антител, сцепленных с токсинами (иммунотоксины), имеет преимущество, заключающееся в том, что прилегающие опухолевые клетки, не имеющие соответствующих антигенных детерминант, могут быть разрушены посредством перекрестного облучения. Радиоиммунотерапию иногда называют направленной лучевой терапией, но последний термин может также означать радионуклиды, связанные с неиммунными молекулами (лучевая терапия).
Фраза «фармацевтически приемлемая соль (соли)», как используется здесь, если не оговорено особым образом, включает соли кислотных или основных групп, которые могут присутствовать в соединении. Соединения, являющиеся по природе основаниями, способны образовывать большое разнообразие солей с различными неорганическими и органическими кислотами. Кислотами, которые можно использовать для получения фармацевтически приемлемых кислотных аддитивных солей таких основных соединений, являются те, которые образуют нетоксичные кислотные аддитивные соли, а именно соли, содержащие в себе фармацевтически приемлемые анионы, например ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, кальциевая соль этилендиаминтетрауксусной кислоты, камсилат, карбонат, хлорид, клавуланат, цитрат, дихлоргидрат, соль этилендиаминтетрауксусной кислоты, эдизилат, эстолат, эзилат, этилсукцинат, фумарат, глюцептат, глуконат, глутамат, гликолильарсанилат, гексилрезорцинат, гидрабамин, бромгидрат, хлоргидрат, иодид, изотионад, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилсульфат, мукат, напсилат, нитрат, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и соли валериановой кислоты. В частности предпочтительные соли включают фосфаты и глюконаты.
Данное изобретение также включает меченные изотопами соединения, которые являются идентичными соединению, указанному в формуле 1, если не считать того, что один или несколько атомов замещены атомом, чья атомная масса или массовое число отличается от атомной массы или массового числа, обычно встречающегося в природе. Примеры изотопов, которые могут быть введены в соединение по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, например 2H, 3H, 11C, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F, и 36Cl, соответственно. Соединения по настоящему изобретению и фармацевтически приемлемые соли вышеуказанных соединений, которые содержат в себе вышеупомянутые изотопы и/или другие изотопы других элементов, находятся в пределах объема данного изобретения. Некоторые меченные изотопами соединения по настоящему изобретению, например те, в которые включены такие радиоактивные изотопы, как 3H, 14C, 11C или 18F, используются в анализе распределения в тканях дозированного средства и/или субстрата. Изотопы тритий, то есть 3H, и углерод -14, то есть 14C, чрезвычайно предпочтительны вследствие их легкого получения и выявления, и 11C и 18F для применения в позиционно-эмиссионной томографии. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, то есть 2H, может оказать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например увеличение полужизни in vivo, или уменьшение требуемой дозировки, и таким образом, могут быть предпочтительны в некоторых случаях. Меченное изотопом соединение формулы 1 по настоящему изобретению обычно может быть получено в результате проведения процедур, описанных для немеченных соединений, с заменой немеченного изотопом реагента на легко доступный меченный изотопом реагент.
ADP - аденозиндифосфат.
AE - нежелательное явление.
ALT аланинаминотрансфераза.
ANC - абсолютное число нейтрофилов.
AST - аспартатаминотрансфераза.
AUC - область (площадь) под кривой зависимости концентрация-время.
AUC(0-24) - область (площадь) под кривой зависимости концентрация-время от момента времени 0 до 24 часов.
AUC(0-tlast) - область (площадь) под кривой зависимости концентрация-время от начального момента времени до последнего зарегистрированного наблюдения.
BLD - ниже уровня обнаружения.
BSA - площадь поверхности тела.
BUN - азот мочевины крови.
C0 - исходная концентрация.
CL - клиренс.
Cmax - максимальная концентрация в плазме.
CRC - колоректальный рак.
CTCAEv3 - общие номенклатурные критерии для нежелательных явлений, версия 3.
CV - сердечно-сосудистый.
DLT - предельная токсичность дозы.
DNA - дезоксирибонуклеиновая кислота.
EC50 - концентрация, обуславливающая 50% от максимального эффекта.
ECG - электрокардиограмма.
FcR - рецептор Fc.
5-FU - 5-фторурацил.
GI - желудочно-кишечный.
GIST - желудочно-кишечная стромальная опухоль.
GLP - апробированный лабораторный метод.
HCT - гематокрит.
HERG - ген человека ERG.
hERG-Ikr - ген человека hERG, блокирующий калиевый канал.
HGB - гемоглобин.
GI50 - концентрация, при которой подавляется рост 50% клеток.
IC50 - концентрация, при которой подавляется ферментативная активность на 50%.
IGF - инсулиноподобный фактор роста.
IGF-1 R - рецептор инсулиноподобного фактора роста, тип 1.
IL - интерлейкин.
IP - внутрибрюшинный.
IV - внутривенный.
LLN - нижняя граница нормы.
LLOQ - нижний предел количественных определений.
LV - лейковорин.
MMNG - N-метил-N'-нитро-N-нитрозогуанидин.
MTD - максимально переносимая доза.
NAD - никотинамидадениндинуклеотид.
NOAEL - уровень без отрицательных действий.
PARP - поли(АДФ-рибоза) полимераза.
PBMCs - моноциты периферической крови.
PD - фармакодинамика.
PID - доза, ингибирующая PARP.
PK - фармакокинетика.
PO - перорально.
RBC - эритроциты.
RECIST - критерии оценки ответной реакции солидных опхолей.
QC - контроль качества.
SAE - тяжелые неблагоприятные явления.
SWFI/SWI - стерильная вода для инъекций.
T1/2 - видимый конечный период полувыведения.
Tmax - время достижения Cmax.
ULN - верхняя граница нормы.
Vdss - объем распределения в стационарном состоянии.
WFI - вода для инъекций.
Краткое описание чертежей
На фиг.1 представлены данные эффективности темозоломида в комбинации с 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-оном в виде фосфатной соли в отношении ксенотрансплантата SW620.
На фиг.2 представлены данные эффективности темозоломида в комбинации с 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-оном в виде соли глюкуроновой кислоты в отношении ксенотрансплантата SW620.
На фиг.3 представлены средние профили зависимости концентрации в плазме от времени 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-оном на День-7 (фосфатная соль 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-оном, отдельно) и День 1 и 4 (фосфатная соль 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-оном плюс темозоломид), где фосфатную соль вводили посредством 30-минутной внутривенной инфузии и темозоломид давали
перорально в дозе 100 мг/м2.
На фиг.4 представлена медианная активность PARP в лимфоцитах периферической крови после введение фосфатной соли 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-оном.
Подробное описание изобретения
I. Фармацевтические составы, содержащие 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-он
Соединение формулы 1 и его соли могут быть получены, как описано в патенте США № 6495541; заявке PCT № PCT/IB2004/000915; предварительная заявка на выдачу патента США № 60/612457; и предварительная заявка на выдачу патента США № 60/612459, полные содержания которых включены здесь в виде ссылки. Некоторые исходные вещества могут быть получены в соответствии с методами, известными специалисту в уровне техники и некоторые синтетические модификации могут быть проведены в соответствии с методами, известными специалисту в уровне техники.
Соединение формулы 1 способно образовывать большое разнообразие различных солей с разными неорганическими и органическими кислотами. Несмотря на то, что такие соли должны быть фармацевтически приемлемыми для применения у млекопитающих, зачастую целесообразно на практике первоначально выделить соединение формулы 1 из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнюю в свободное основание соединения посредством обработки щелочным реагентом, и затем превратить последнее свободное основание в фармацевтически приемлемую кислотную аддитивную соль. Кислотные аддитивные соли основных соединений по настоящему изобретению легко получить посредством обработки основного соединения по существу эквивалентным количеством выбранной неорганической или органической кислоты в водорастворимой среде или в подходящем органическом растворителе, например метиловом или этиловом спирте. После тщательного выпаривания растворителя быстро получают необходимую твердую соль. Необходимую кислую соль также можно осадить из раствора свободного основания в органическом растворителе посредством добавления в раствор соответствующей неорганической или органической кислоты. Конкретные примеры получения предпочтительной соли, фосфатной соли, можно найти в заявке PCT № PCT/IB2004/000915; предварительной заявке на выдачу патента США № 60/612457; и предварительной заявке на выдачу патента США № 60/612459, полные содержания которых включены здесь в виде ссылки.
Применение соединения формулы 1 может быть осуществлено посредством любого способа, позволяющего доставить соединение до места его действия. Эти способы включают пероральные пути введения, парентеральные инъекции (в том числе внутривенные, подкожные, внутримышечные, внутрисосудистые или вливания), местное или ректальное введение.
Это соединение, например, может быть представлено в такой форме, подходящей для перорального применения, как таблетка, капсула, пилюля, порошок, состав с замедленным высвобождением, раствор, суспензия, для парентерального введения в виде стерильного раствора, суспензии или эмульсии, для местного применения в виде мази или крема, или для ректального введения в виде суппозитории.
Данное соединение может быть представленным в виде единичных дозированных форм, пригодных для однократного введения определенных доз. Предпочтительно, дозированные формы включают подходящий фармацевтический носитель или эксципиент и соединение формулы 1 в качестве активного ингредиента. Дополнительно, дозированные формы могут включать другие дозированные или фармацевтические вещества, носители, адъюванты и тому подобное.
Примеры форм для парентерального введения включают растворы или суспензии в стерильных водных растворах, например водные растворы пропиленгликоля или декстрозы. Такие дозированные формы при желании могут быть соответствующим образом забуферены.
Приемлемые фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители. Данная фармацевтическая композиция, при необходимости, может содержать в себе дополнительные ингредиенты, такие как ароматизаторы, связующие вещества, эксципиенты и тому подобное. Так при пероральном применении, таблетки, содержащие различные эксципиенты, такие как лимонная кислота, можно использовать вместе с различными дезинтегрирующими веществами, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, и со связывающими веществами, такими как сахароза, желатин и камедь. Часто для получения таблеток дополнительно используют смазывающие вещества, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции схожего типа также могут использоваться в мягких и твердых наполненных желатиновых капсулах. Предпочтительные вещества для этого включают лактозу или молочный сахар, и полиэтиленгликоли с высокой молекулярной массой. В случае, когда для перорального применения желательны водные суспензии или эликсиры, действующее вещество в них может сочетаться с различными подслащивающими или ароматизирующими веществами, красящими веществами или красителями и, при желании, эмульгирующими веществами или суспендирующими веществами, вместе с такими разбавителями как вода, этанол, пропиленгликоль, глицерин или их сочетанием.
В предпочтительных вариантах осуществления дозированных форм по настоящему изобретению, дозированная форма представляет собой дозированную форму для перорального применения, более предпочтительно, таблетку или капсулу.
В предпочтительных вариантах осуществления способов по настоящему изобретению, соединение формулы 1 применяют парентерально, например, используя лиофилизированный порошок. Получение лиофилизированного порошка для инъекций для клинического использования описано в предварительной заявке на выдачу патента США № 60/612459, полное содержание которой включено здесь в виде ссылки.
Например, фосфатная соль соединения формулы 1 может быть приготовлена и поставлена в виде лиофилизированного порошка для инъекции, 12 мг/ампула (в виде свободного основания), в 10 мл/20 мм ампуле из оранжевого стекла, тип I. Готовая дозированная форма композиции фосфатной соли соединения формулы 1 может содержать в себе фосфатную соль соединения формулы 1, маннит, воду для инъекций и азот. Полученная готовая дозированная форма может представлять собой спекшуюся массу цвета от грязно-белого до желтого. Каждая готовая дозированная форма в ампуле может быть восстановлена с помощью 6 мл стерильной воды для инъекций для получения 2,02 мг/мл (округленно до 2 мг/мл) соединения формулы 1 в виде свободного основания.
В предпочтительных вариантах осуществления, концентрация соединения формулы 1 в плазме поддерживается на уровне 5,9 нг/мл или выше. Это значение обусловлено прицельным действием (IC89) для подавления истощения
клеточного NAD+ и образования полимера поли АДФ-рибозы, скорректированного с учетом связывания с белком. В частности, как показано в примере 4, 5 нМ соединения формулы 1 (темозоломид PF50=1,3), значительно снижает истощение клеточного NAD+, индуцированное MNNG и ингибирование образования клеточной поли-АДФ-рибозы на 89% в клетках A549. Корректируя конечный результат связывания белков человека под действием 5 нМ соединения формулы 1 (27,4% среднее значение несвязанных для соединения формулы 1 концентрации от 0,05 до 25 нМ) получали концентрацию в плазме 5,9 нг/мл:
5 нМ × 323,37=5,9 нг/мл
0,274×1000
II. Фармацевтические комбинации по настоящему изобретению и их применение
В одном варианте осуществления настоящего изобретения соединение формулы используют для увеличения эффективности цитотоксических средств, чей механизм действия основан на повреждении ДНК. Эти дозированные средства включают в себя, но не ограничиваются, темозоломид (SCHERING), иринотекан (PFIZER), топотекан (GLAXO SMITHKLINE), цисплатин (BRISTOL MEYERS SQUIBB; AM PHARM PARTNERS; BEDFORD; GENSIA SICOR PHARMS; PHARMACHEMIE), и доксорубицина гидрохлорид (AM PHARM PARTNERS; BEDFORD; GENSIA; SICOR PHARMS; PHARMACHEMIE; ADRIA; ALZA).
Терапевтически эффективные количества веществ по данному изобретению можно применять, обычно в форме фармацевтической композиции, для лечения заболеваний, опосредованных модуляцией или регуляцией PARP. Термин «эффективное количество» означает, что количества агента, вводимого в организм млекопитающего, в том числе человека, нуждающегося в таком лечении, достаточно для обеспечения лечения заболевания, опосредованного действием одного или нескольких ферментов PARP. Таким образом, терапевтически эффективное количество соединения по данному изобретению количественно достаточно для модулирования, регулирования или ингибирования действия одного или нескольких ферментов PARP, таким образом, что болезненное состояние, опосредованное таким действием, улучшается или смягчается. Эффективное количество применяемого соединения будет изменяться в зависимости от таких факторов, как вид болезненного состояния и его тяжесть, и индивидуальных особенностей и состояния (например, масса тела) млекопитающего, нуждающегося в таком лечении, но обычно может быть определено, используя стандартные методики, специалистом в уровне техники. Термин «лечение» означает, по меньшей мере, облегчение болезненного состояния у млекопитающего, в том числе у человека, вызванного, по меньшей мере, частично, действием одного или нескольких ферментов PARP, и включает в себя: профилактику появления болезненного состояния у млекопитающего, в частности, когда обнаружено, что данное млекопитающее предрасположено иметь болезненное состояние, но оно еще не диагностировано; модулирование и/или ингибирование болезненного состояния; и/или облегчение болезненного состояния. Типичным болезненным состоянием является рак.
Активность соединения формулы 1 в качестве модулятора активности PARP можно измерить, используя любые способы, доступные специалисту в уровне техники, в том числе методы in vivo и/или in vitro. Примеры подходящих способов определения активности включают те, которые описаны в патенте США № 6495541, и конкретные примеры настоящего изобретения.
Настоящее изобретение относится к способам лечения болезненного состояния, опосредованного действием PARP, например, рака и ряда заболеваний и токсических состояний, которые приводят к окислительному или индуцированному оксидом азота стрессу и последующей гиперактивации PARP. Такие состояния включают, но не ограничиваются, неврологические и нейродегенеративные заболевания (например, болезнь Паркинсона, болезнь Альцгеймера), сердечно-сосудистые заболевания (например, инфаркт миокарда, ишемическо-реперфузионное повреждение), сосудистые нарушения при диабете, цисплатин-индуцированную нефротоксичность. Способы лечения по настоящему изобретению включают введение в организм млекопитающего, нуждающегося в таком лечении, терапевтически эффективное количество фармацевтической композиции, которая содержит в себе любую из полиморфных форм, или фармацевтических композиций, обсуждавшихся выше.
Данное изобретение также относится к способу воздействия на аномальный клеточный рост у млекопитающего, в том числе у человека, включающему введение в организм указанного млекопитающего такого количества соединения формулы 1, как определено выше, или его фармацевтически приемлемой соли или сольвата, которое является эффективным при воздействии на аномальный клеточный рост.
В одном варианте осуществление этого способа аномальный клеточный рост представляет собой рак, включающий, но не ограниченно, мезотелиому, гепатобиллиарный (печеночный и желчный проток), первичную или вторичную опухоль ЦНС, первичную или вторичную опухоль головного мозга, рак легких (NSCLC и SCLC), рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожную или внутриглазную меланому, рак яичников, рак толстой кишки, рак прямой кишки, рак анальной области, рак желудка, гастроинтестинальный (желудочный, колоректальный и дуоденальный), рак молочной железы, рак матки, карциному фаллопиевых труб, карциному эндометрия, карциному шейки матки, карциному влагалища, карциному вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечников, саркому мягких тканей, рак уретры, рак пениса, рак предстательной железы, рак яичка, хронический или острый лейкоз, хроническую миелоидную лейкемию, лимфоцитарую лимфому, рак мочевого пузыря, рак почки или мочеточника, почечноклеточный рак, карциному почечной лоханки, новообразования центральной нервной системы (ЦНС), первичную лимфому ЦНС, неходжкинские лимфомы, опухоли позвоночника, глиому ствола мозга, аденому гипофиза, рак коры надпочечников, рак желчного пузыря, множественную миелому, холангиокарциному, фибросаркому, нейробластому, ретинобластому, или сочетание одного или нескольких вышеупомянутых типов рака.
В другом варианте осуществления вышеуказанного способа вышеупомянутый аномальный клеточный рост является доброкачественным пролиферативным заболеванием, включающим, но не ограниченно, псориаз, доброкачественную гипертрофию предстательной железы или рестеноз.
Данное изобретение также относится к способу воздействия на аномальный клеточный рост у млекопитающего, который включает введение в организм вышеупомянутого млекопитающего такого количества соединения формулы 1, или его фармацевтически приемлемой соли или сольвата, которое является эффективным при воздействии на аномальный клеточный рост в комбинации с противоопухолевым агентом, выбранным из группы, включающей ингибиторы митоза, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологической реакции, антитела, цитотоксические агенты, антигормональные агенты и антиандрогенные агенты.
Данное изобретение также относится к фармацевтической композиции для воздействия на аномальный клеточный рост у млекопитающего, в том числе у человека, включающей такое количество соединения формулы 1, как определено выше, или его фармацевтически приемлемой соли или сольвата, которое эффективно при воздействии на аномальный клеточный рост, и фармацевтически приемлемый носитель.
В одном варианте осуществления вышеуказанной композиции вышеупомянутый аномальный клеточный рост представляет собой рак, включающий, но не ограниченно, мезотелиому, гепатобиллиарный (печеночный и желчный проток), первичную или вторичную опухоль ЦНС, первичную или вторичную опухоль головного мозга, рак легких (NSCLC и SCLC), рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожную или внутриглазную меланому, рак яичников, рак толстой кишки, рак прямой кишки, рак анальной области, рак желудка, гастроинтестинальный (желудочный, колоректальный и дуоденальный), рак молочной железы, рак матки, карциному фаллопиевых труб, карциному эндометрия, карциному шейки матки, карциному влагалища, карциному вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечников, саркому мягких тканей, рак уретры, рак пениса, рак предстательной железы, рак яичка, хронический или острый лейкоз, хронический миелоидный лейкоз, лимфоцитарую лимфому, рак мочевого пузыря, рак почки или мочеточника, почечноклеточный рак, карциному почечной лоханки, новообразования центральной нервной системы (ЦНС), первичную лимфому ЦНС, неходжкинские лимфомы, опухоли позвоночника, глиому ствола мозга, аденому гипофиза, рак коры надпочечников, рак желчного пузыря, множественную миелому, холангиокарциному, фибросаркому, нейробластому, ретинобластому, или сочетание одного или нескольких вышеупомянутых типов рака.
В другом варианте осуществления вышеназванной композиции вышеуказанный аномальный клеточный рост представляет собой доброкачественное пролиферативное заболевание, в том числе, но не ограниченно, псориаз, доброкачественную гипертрофию предстательной железы и рестеноз.
Данное изобретение также относится к фармацевтической композиции для воздействия на аномальный клеточный рост у млекопитающего, в том числе у человека, которая содержит такое количество соединения формулы 1, которое определено выше, или его фармацевтически приемлемой соли или сольвата, которое является эффективным при воздействии на аномальный клеточный рост, в комбинации с фармацевтически приемлемым носителем и противоопухолевым агентом, выбранным из группы, включающей ингибиторы митоза, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологической реакции, антигормоны и антиандрогены.
Данное изобретение также относится к способу лечения гиперпролиферативного заболевания у млекопитающего, который включает введение в организм указанного млекопитающего терапевтически эффективного количества соединения формулы 1, или его фармацевтически приемлемой соли или гидрата, в комбинации с противоопухолевым агентом, выбранным из группы, включающей антипролиферативные агенты, ингибиторы киназы, ингибиторы ангиотензина, ингибиторы фактора роста, ингибиторы COX-I, ингибиторы COX-II, ингибиторы митоза, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, радиацию, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологической реакции, антитела, цитотоксины, антигормоны, статины и антиандрогены.
Данное изобретение также относится к комбинации терапевтических способов лечения болезненного состояния, опосредованного действием PARP, которая включает введение в организм млекопитающего, нуждающегося в таком лечении, терапевтически эффективного количества фармацевтической композиции, которая содержит в себе любую из полиморфных форм, или фармацевтических композиций обсуждавшихся выше, в комбинации с терапевтически эффективным количеством одного или нескольких веществ, выбранных из противоопухолевых агентов, антиангиогенных агентов, ингибиторов сигнальной трансдукции и антипролиферативных агентов. Такие вещества включают те, которые раскрыты в публикациях PCT №№ WO 00/38715, WO 00/38716, WO 00/38717, WO 00/38718, WO 00/38719, WO 00/38730, WO 00/38665, WO 00/37107 и WO 00/38786, полные описания которых включены здесь в виде ссылки.
Примеры противоопухолевых агентов включают темозоломид (SCHERING), иринотекан (PFIZER), топотекан (GLAXO SMITHKLINE), цисплатин (BRISTOL MEYERS SQUIBB; AM PHARM PARTNERS; BEDFORD; GENSIA SICOR PHARMS; PHARMACHEMIE), и доксорубицина гидрохлорид (AM PHARM PARTNERS; BEDFORD; GENSIA; SICOR PHARMS; PHARMACHEMIE; ADRIA; ALZA).
Комбинация терапевтических способов включает применение соединения формулы 1 и противоопухолевого агента, используя любую желаемую схему введения/дозирования лекарственного средства. Например, режимы могут зависеть от комбинации агентов как указано далее:
(a) соединение формулы 1, его фармацевтически приемлемая соль или сольват, или их смесь, может быть введено в организм в объеме от 1 до 48 мг/м2, выраженном как эквивалентная масса свободного основания соединения формулы 1, ежедневно, 5 дней, каждые 28 дней за 1 час перед приемом 25-200 мг/м2 темозоломида, предпочтительно, 100-200 мг/м2 темозоломида;
(б) соединение формулы 1, его фармацевтически приемлемая соль или сольват, или их смесь, может быть введено в организм в объеме от 1 до 48 мг/м2, выраженном как эквивалентная масса свободного основания соединения формулы 1, за 1 час до введения дозы иринотекана и спустя 24 часа.
Диапазон доз для иринотекана:
62-125 мг/м2 еженедельно, 4 недели каждые 6 недель;
175-350 мг/м2 каждые 3 недели;
90-180 мг/м2 каждые 2 недели.
(в) соединение формулы 1, его фармацевтически приемлемая соль или сольват, или их смесь, может быть введено в организм в объеме от 1 до 48 мг/м2, выраженном как эквивалентная масса свободного основания соединения формулы 1, ежедневно, 5 дней, каждые 21 день, за 1 час до введения дозы топотекана.
Диапазон доз для топотекана:
0,75-1,5 мг/м2 ежедневно, 5 дней, каждые 21 день.
(г) соединение формулы 1, его фармацевтически приемлемая соль или сольват, или их смесь, может быть введено в организм в объеме от 1 до 48 мг/м2, выраженном как эквивалентная масса свободного основания соединения формулы 1, либо однократно каждые 3-4 недели, либо ежедневно в течение 3-5 дней каждые 3-4 недели, за 1 час до введения дозы цисплатина.
Диапазон доз для цисплатина:
10-100 мг/м2 каждые 3-4 недели;
10-40 мг/м2 ежедневно в течение 3-5 дней каждые 3-4 недели.
(д) соединение формулы 1, его фармацевтически приемлемая соль или сольват, или их смесь, может быть введено в организм в объеме от 1 до 48 мг/м2, выраженном как эквивалентная масса свободного основания соединения формулы 1, за 1 час до введения дозы доксорубицина и через 24 часа.
Диапазон доз для доксорубицина:
20-75 мг/м2 каждые 21-28 дней.
Комбинация терапевтических способов по настоящему изобретению может включать применение соединения формулы 1, его фармацевтически приемлемой соли или сольвата, или их смеси, в количестве от 1 дo 48 мг/м2, выраженном как эквивалентная масса свободного основания соединения формулы 1, и противоопухолевого агента (агентов), используя, например, схемы приема дозированного средства, представленные в таблице 1.
Режим Saltz
LV 20 мг/м2 внутривенный болюс, день 1, 8, 15, 22
5-FU 500 мг/м2 внутривенный болюс, день 1, 8, 15, 22
Повторять каждые 6 недель
LV 200 мг/м2 внутривенный болюс, за 2 часа до 5-FU, день 1 и 2
5-FU 400 мг/м2 внутривенный болюс, затем 600 мг/м2 продолжительная инфузия в течение 22 часов, день 1и 2
Повторять каждые 2 недели
LV 200 мг/м2 в течение 2-часовой инфузии во время приема иринотекана
5-FU болюс 400 мг/м2, затем 2,4-3 г/м2 продолжительная инфузия в течение 46 часов, день 1и 2
Повторять каждые 2 недели
Tournigand et al. J Clin Oncol. 2004; 23: 229-237
Иринотекан 100 мг/м2, день 1 и 8
Повторять каждые 22 дня
Капецитабин 1000 мг/м2 перорально 2 раза в день, вечер день 1-утро день 15
Повторять каждые 3 недели
Оксалиплатин 85 мг/м2 внутривенно в течение 2 часов, день 1
Повторять каждые 3 недели
LV 20 мг/м2 внутривенно, день 1, 8, 15, 22
5-FU 500 мг/м2, внутривенно, день 1, 8, 15, 22
Повторять каждые 6 недель
Бевацизумаб 5 мг/кг внутривенно в течение 90 минут* после хемотерапии, день 1
Повторять каждые 2 недели
*если первое вливание переносится хорошо, то последующие вливания можно осуществлять в течение 60 минут, а затем 30 минут.
Схемы приема дозированного препарата, указанные в таблице 1, могут быть модифицированы. Например, ириноректан можно применять в дозе 50-350 мг/м2; 5-FU можно применять в дозе 370 мг/м2 - 3,0 г. LV можно применять в дозе 20-500 мг/м2.
Комбинацию терапевтических способов по настоящему изобретению, которая включает применение соединения формулы 1, его фармацевтически приемлемой соли или сольвата, или их смеси, в количестве от 1 до 48 мг/м2, выраженном как эквивалентная масса свободного основания соединения формулы 1, и противоопухолевого агента (агентов), можно применять, например, у пациентов, у которых, например, лечение с использованием схем применения дозированного средства, представленных в таблице 2, было неэффективным.
LV 200 мг/м2 внутривенно в течение 2 часов, день 1 и 2
5-FU 400 мг/м2 внутривенный болюс, затем 600 мг/м2 внутривенно в течение 22 часов, день 1 и 2
Повторять каждые 2 недели
Rothenberg et al. J Clin Oncol. 2003; 21: 2059-2069.
LV 200 мг/м2 в течение 2 часов, день 1
5-FU 400 мг/м2 внутривенный болюс, затем 2,4-3 г/м2 в течение 46 часов, продолжительная инфузия
Повторять каждые 2 недели
Tournigand et al. J Clin Oncol. 2004; 23: 229-237
LV 175 мг/м2 внутривенно в течение 2 часов, день 1
5-FU 400 мг/м2 внутривенный болюс, затем 2,4-3 г/м2 в течение 46 часов, продолжительная инфузия
Повторять каждые 2 недели
LV 400 мг/м2 внутривенно в течение 2 часов
5-FU 2400 мг/м2 внутривенно, в течение 46 часов, продолжительная инфузия
Повторять каждые 2 недели 6 циклов
LV 500 мг/м2 внутривенно в течение 2 часов, день 1, 8, 15, 22, 29, 36
5-FU 500 мг/м2 внутривенный болюс, после начала приема LV
, день 1, 8, 15, 22, 29, 36
Повторять каждые 8 недель, 3 цикла
LV 500 мг/м2 внутривенно в течение 2 часов, день 1, 8, 15, 22
5-FU 2,6 г/м2 внутривенно в течение 24 часов, продолжительная инфузия, день 1, 8, 15, 22
Повторять каждые 36 дней
LV 20 мг/м2 внутривенно в течение 10-20 минут, день 1, 8, 15
5-FU 500 мг/м2 внутривенный болюс, день 1, 8, 15
Повторять каждые 28 дней
LV 200 мг/м2 внутривенно в течение 2 часов, день 1 и 2
5-FU 400 мг/м2 внутривенный болюс, затем 600 мг/м2 в течение 22 часов, день 1 и 2
Бевацизумаб 10 мг/кг внутривенно в течение 90 минут, * день 1
Повторять каждые 2 недели
LV 200 мг/м2 внутривенно в течение 2 часов, день 1 и 2
5-FU 400 мг/м2 внутривенный болюс, затем 600 мг/м2 в течение 22 часов, день 1 и 2
Повторять каждые 2 недели
Цетуксимаб 400 мг/м2 внутривенно в течение 2 часов 1 неделю, затем 250 мг/м2 внутривенно в течение 60 минут еженедельно
Единицы дозировки выражены в мг на м2 BSA. Например, для определения BSA применимы формула Мостеллера, формула Дюбуа и Дюбуа, формула Хэйкока, формула Гехана-Джорджа, формула Бойда (Mosteller RD: Simplified Calculation of Body Surface Area. N Engl J Med 1987 Oct 22; 317(17): 1098; DuBois D; DuBois EF: A formula to estimate the approximate surface area if height and weight be known. Arch Int Med 1916 17: 863-71; Haycock G.B., Schwartz G.J., Wisotsky D.H. Geometric method for measuring body surface area: A height weight formula validated in infants, children and adults. The Journal of Pediatrics 1978 93: 1: 62-66; Gehan EA, George SL, Estimation of human body surface area from height and weight. Cancer Chemother Rep 1970 54: 225-35; Boyd E, The growth of the surface area of the human body. Minneapolis: university of Minnesota Press, 1935; Lam TK, Leung DT: More on simplified calculation of body-surface area. N Engl J Med 1988 Apr 28; 318(17): 1130).
Дополнительные примеры противоопухолевых агентов включают антипролиферативные агенты, ингибиторы киназы, ингибиторы ангиогенеза, ингибиторы фактора роста, ингибиторы COX-I, ингибиторы COX-II, ингибиторы митоза, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, радиацию, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологической реакции, антитела, цитотоксины, антигормоны, статины, и антиандрогены.
В одном варианте осуществления настоящего изобретения противоопухолевый агент, используемый в сочетании с соединением формулы 1 и фармацевтическими композициями, описанными здесь, представляет собой антиангиогеный агент, ингибитор киназы, ингибитор пан-киназы или игибитор фактора роста.
Предпочтительные ингибиторы пан-киназы включают SU-11248, описанный в патенте США № 6573293 (Pfizer, Inc, NY, USA).
Антиангиогенные агенты включают в себя, но не ограничены, следующие агенты, такие как ингибитор EGF, ингибиторы EGFR, ингибиторы VEGF, ингибиторы VEGFR, ингибиторы TIE2, ингибиторы IGF1R, ингибиторы COX-II (циклооксигеназа II), ингибиторы MMP-2 (металлопротеиназа 2 матрикса), и ингибиторы MMP-9 (металлопротеиназа 9 матрикса).
Предпочтительные ингибиторы VEGF, включают в себя, например, Авастин (бевацизумаб), моноклональные тела против VEGF фирмы Genentech, Inc. of South San Francisco, California.
Дополнительные ингибиторы VEGF включают CP-547632 (Pfizer Inc., NY, USA), AG13736 (Pfizer Inc.), ZD-6474 (AstraZeneca), AEE788 (Novartis), AZD-2171), VEGF Trap (Regeneron/Aventis), Ваталаниб (также известный как PTK-787, ZK-222584: Novartis & Schering AG), Макуген (пегаптаниб октанатрия, NX-1838, EYE-001, Pfizer Inc/Gilead/Eyetech), IM862 (Cytran Inc. of Kirkland, Washington, USA); и ангиозим, синтетический рибозим, полученный от компаний Ribozyme (Boulder, Colorado) и Chiron (Emeryville, California), и их комбинации. Ингибиторы VEGF, используемые в практическом применении настоящего изобретения, раскрыты в патентах США №№6534524 и 6235764, содержания которых включены здесь в полном объеме в виде ссылки для любых целей. В частности, предпочтительные ингибиторы VEGF включают CP-547632, AG13736, Ваталаниб, Макуген и их комбинации.
Дополнительные ингибиторы VEGF описаны, например, в международной публикации WO 99/24440 (опубликована 20 мая 1999 г.), международной заявке PCT PCT/IB99/00797 (подана 3 мая, 1999 г.), международной публикации WO 95/21613 (опубликована 17 августа, 1995 г.), международной публикации WO 99/61422 (опубликована 2 декабря, 1999 г.), патенте США 6534524 (раскрывает AG13736), патенте США 5834504 (выдан 10 ноября, 1998 г.), международной публикации WO 98/50356 (опубликована 12 ноября, 1998 г.), патенте США 5883113 (выдан 16 марта, 1999 г.), патенте США 5886020 (выдан 23 марта, 1999 г.), патенте США 5792783 (выдан 11 августа, 1998 г.), патенте США № 6653308 (выдан 25 ноября, 2003 г.), международной публикации WO 99/10349 (опубликована 4 марта, 1999 г.), международной публикации WO 97/32856 (опубликована 12 сентября, 1997 г.), международной публикации WO 97/22596 (опубликована 26 июня, 1997 г.), международной публикации WO 98/54093 (опубликована 3 декабря, 1998 г.), международной публикации WO 98/02438 (опубликована 22 января, 1998 г.), международной публикации WO 99/16755 (опубликована 8 апреля, 1999 г.), и международной публикации WO 98/02437 (опубликована 22 января, 1998 г.), полные описания всех включены здесь в виде ссылки.
Другие антипролиферативные агенты, которые можно использовать с соединением по настоящему изобретению, включают ингибиторы фермента фарнезил-протеин трансферазы и ингибиторы рецептора тирозин киназы PDGFr, включая соединения, раскрытые и заявленные в следующих патентных заявках США: 09/221946 (подана 28 декабря, 1998 г.); 09/454058 (подана 2 декабря, 1999 г.); 09/501163 (подана 9 февраля, 2000 г.); 09/539930 (подана 31 марта, 2000 г.); 09/202796 (подана 22 мая, 1997 г.); 09/384339 (подана 26 августа, 1999 г.); и 09/383755 (подана 26 августа, 1999 г.); и соединения, раскрытые и заявленные в следующих предварительных заявках на выдачу патента: 60/168207 (подана 30 ноября, 1999 г.); 60/170119 (подана 10 декабря, 1999 г.); 60/177718 (подана 21 января, 2000 г.); 60/168217 (подана 30 ноября, 1999 г.), и 60/200834 (подана 1 мая, 2000 г.). Описание каждой из перечисленных выше патентных заявок и предварительных заявок на выдачу патента включено здесь в полном объеме в виде ссылки.
Ингибиторы PDGRr включают в себя, но не ограничены, те, которые раскрыты в публикации международной патентной заявки WO01/40217, опубликованной 7 июля, 2001 г. и публикации международной патентной заявки WO2004/020431, опубликованной 11 марта, 2004 г., содержания которых включены здесь в полном объеме в виде ссылки для любых целей.
Предпочтительные ингибиторы PDGFr включают продукты компании Pfizer CP-673451 и CP-868596 и их фармацевтически приемлемые соли.
Предпочтительные ингибиторы GARF включают продукт компании Pfizer AG-2037 (пелитрексол) и его фармацевтически приемлемые соли. Ингибиторы GARF, используемые при практическом применении настоящего изобретения, раскрыты в патенте США № 5608082, описание которого включено здесь в полном объеме в виде ссылки для любых целей.
Примеры приемлемых ингибиторов COX-II, которые можно использовать в сочетании с соединением формулы 1 и фармацевтическими композициями, описанными здесь, включают CELEBREXTM (целекоксиб), парекоксиб, деракоксиб, ABT-963, MK-663 (эторикоксиб), COX-189 (люмиракоксиб), BMS 347070, RS 57067, NS-398, Бекстра (валдекоксиб), паракоксиб, Виокс (рофекоксиб), SD-8381, 4-метил-2-(3,4-диметилфенил)-1-(4-сульфамоил-фенил)-1H-пиррол, 2-(4-этоксифенил)-4-метил-1-(4-сульфамоилфенил)-1H-пиррол, T-614, JTE-522, S-2474, SVT-2016, CT-3, SC-58125 и Аркоксия (эторикоксиб). Дополнительно, ингибиторы COX-II раскрыты в патентных заявках США №№ 10/801446 и 10/801429, содержания которых включены здесь в полном объеме в виде ссылки для любых целей.
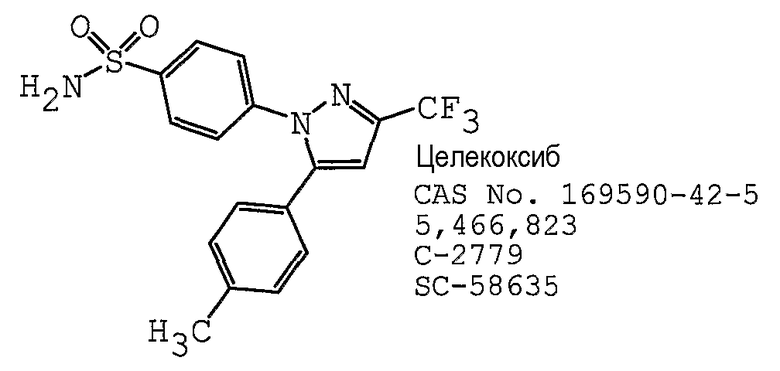
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой целекоксиб, как раскрыто в патенте США № 5466823, содержание которого включено здесь в полном объеме в виде ссылки для любых целей. Структурная формула Целекоксиба представлена ниже:
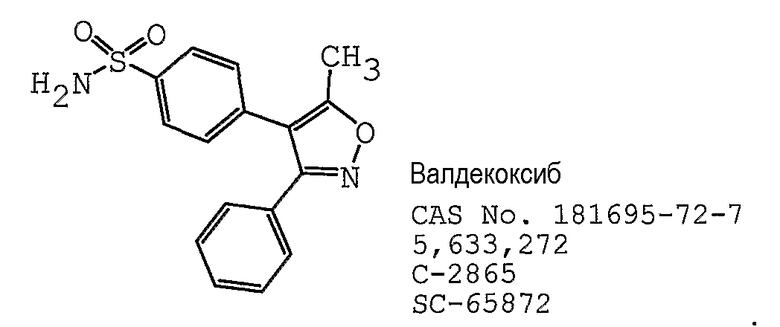
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой валдекоксиб, как раскрыто в патенте США № 5633272, содержание которого включено здесь в полном объеме в виде ссылки для любых целей. Структурная формула валдекоксиба представлена ниже:
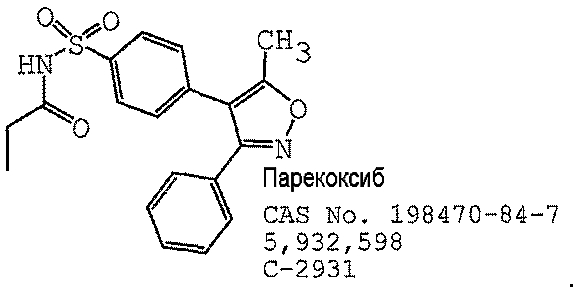
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой парекоксиб, как раскрыто в патенте США № 5932598, содержание которого включено здесь в полном объеме в виде ссылки для любых целей. Структурная формула парекоксиба представлена ниже:
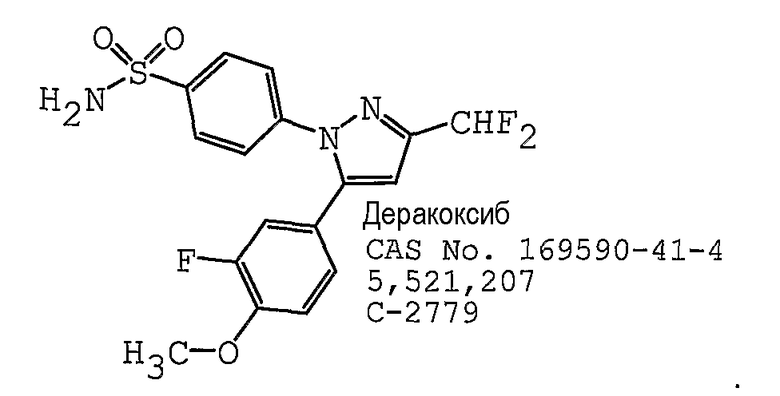
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой деракоксиб, как раскрыто в патенте США № 5521207, содержание которого включено здесь в полном объеме в виде ссылки для любых целей. Структурная формула деракоксиба представлена ниже:
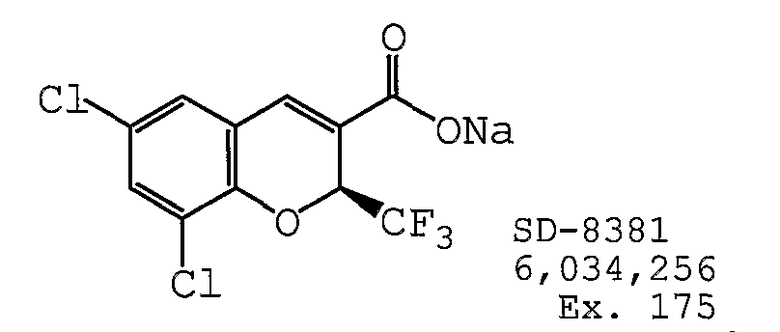
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой SD-8381, как раскрыто в патенте США № 6034256, содержание которого включено здесь в полном объеме в виде ссылки для любых целей. Структурная формула SD-8381 представлена ниже:
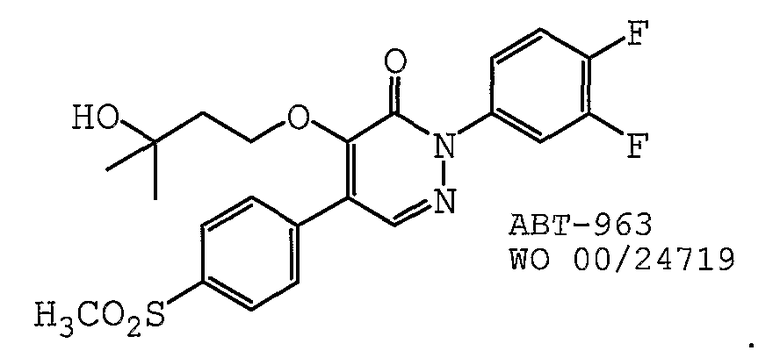
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой ABT-963, как раскрыто в международной публикации № WO 2002/24719, содержание которой включено здесь в полном объеме в виде ссылки для любых целей. Структурная формула ABT-963 представлена ниже:
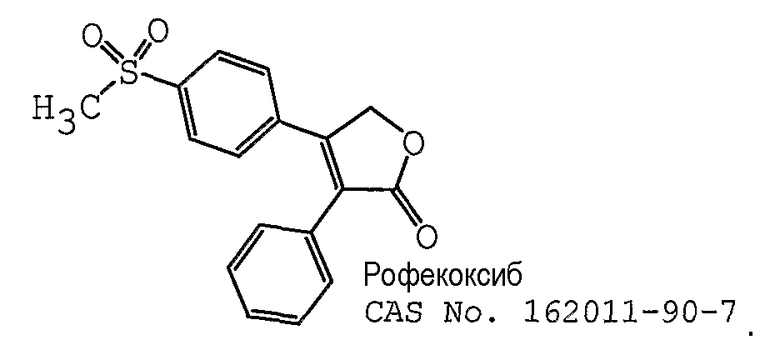
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой рофекоксиб, который представлен ниже:
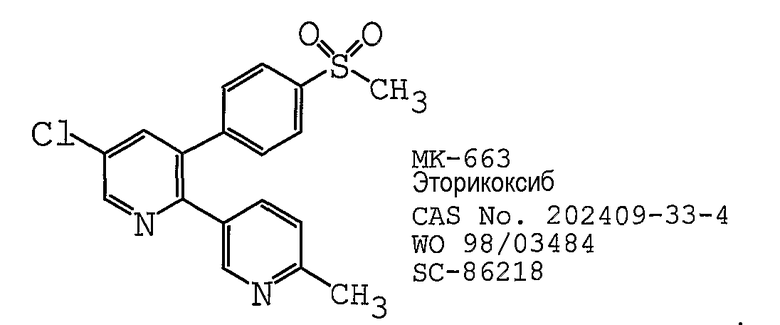
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой MK-663 (эторикоксиб), как раскрыто в международной публикации № WO 1998/03484, содержание которой включено здесь в полном объеме в виде ссылки для любых целей. Структурная формула эторикоксиба представлена ниже:
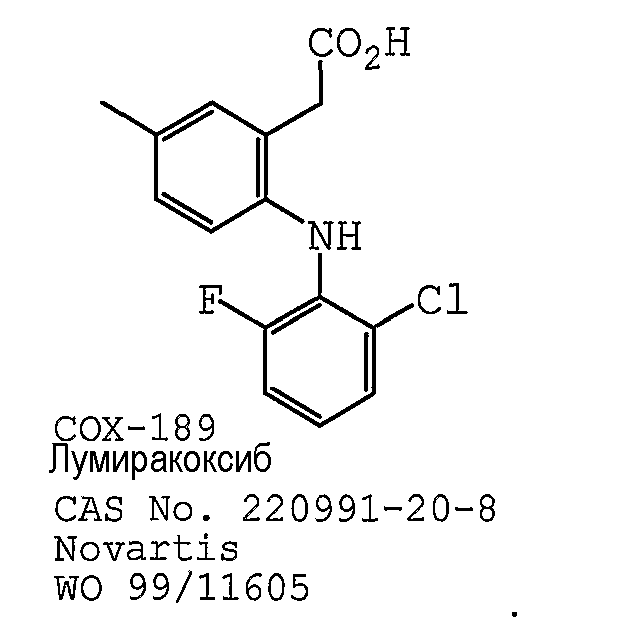
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой COX-189 (лумиракоксиб), как раскрыто в международной публикации № WO 1999/11605, содержание которой включено здесь в полном объеме в виде ссылки для любых целей. Структурная формула лумиракоксиба представлена ниже:
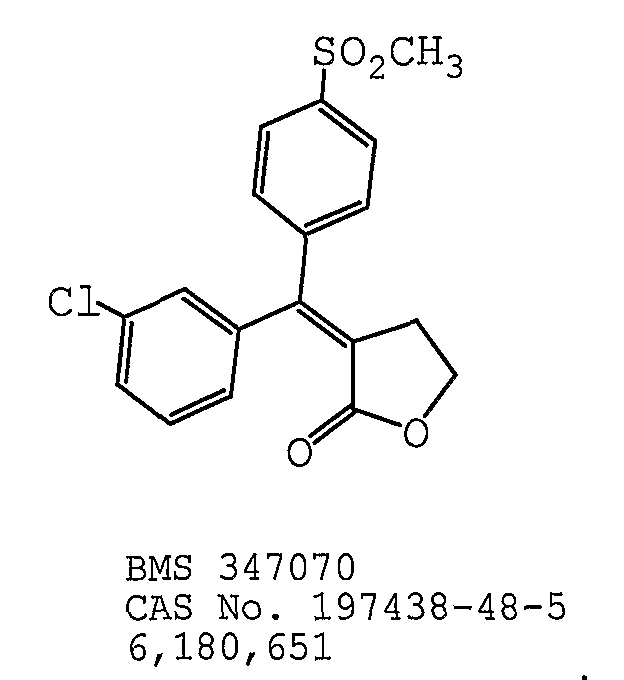
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой BMS-347070, как раскрыто в патенте США № 6180651, содержание которого включено здесь в полном объеме в виде ссылки для любых целей. Структурная формула BMS-347070 представлена ниже:
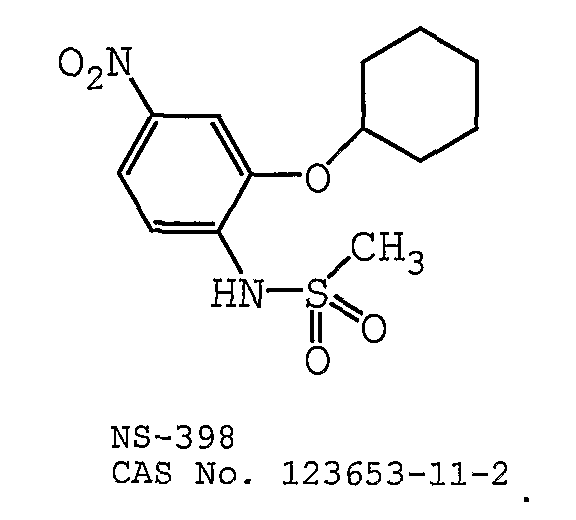
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой NS-398 (CAS 123653-11-2). Структурная формула NS-398 (CAS 123653-11-2) представлена ниже:
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой RS 57067 (CAS 17932-91-3). Структурная формула RS-57067 (CAS 17932-91-3) представлена ниже:
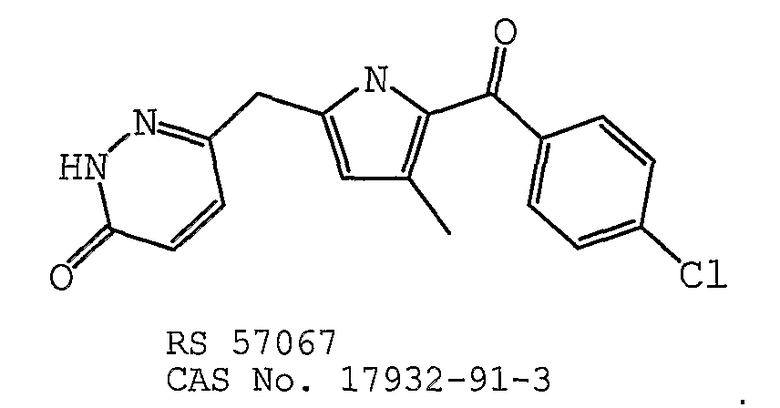
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой 4-метил-2-(3,4-диметилфенил)-1-(4-сульфамоилфенил)-1H-пиррол. Структурная формула 4-метил-2-(3,4-диметилфенил)-1-(4-сульфамоилфенил)-1H-пиррола представлена ниже:
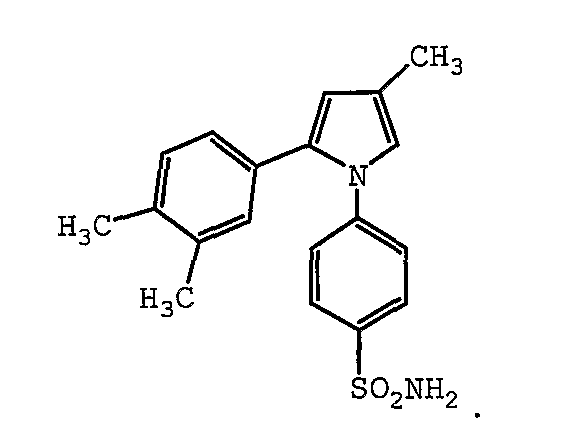
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой 2-(4-этоксифенил)-4-метил-1-(4-сульфамоилфенил)-1H-пиррол. Структурная формула 2-(4-этоксифенил)-4-метил-1-(4-сульфамоилфенил)-1H-пиррола представлена ниже:
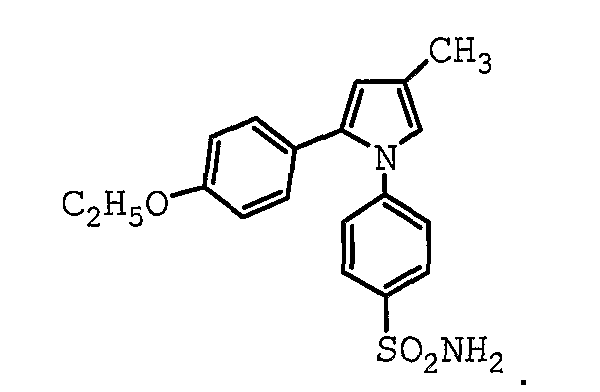
В одном предпочтительном варианте осуществления противоопухолевый агент представляет собой мелоксикам. Структурная формула мелоксикама представлена ниже:
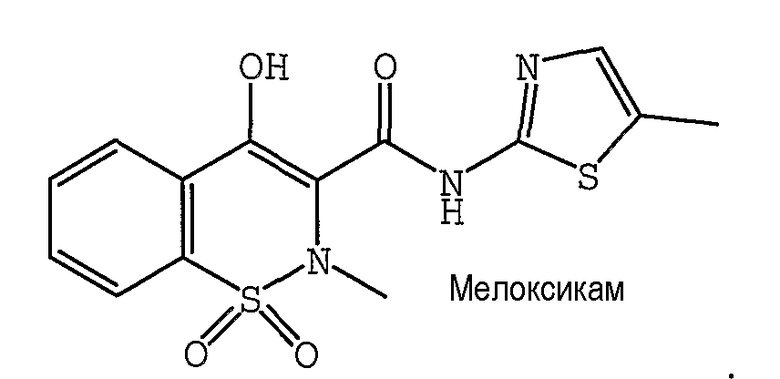
Другие ингибиторы, эффективные в качестве противоопухолевых агентов, используемые в сочетании с соединением формулы 1 и фармацевтическими композициями, описанными здесь, включают аспирин и нестероидные противовоспалительные дозированные средства (НПВС), которые ингибируют фермент, который производит простагландины (циклооксигеназы I и II), что приводит к снижению уровней простагландинов, в том числе, но не ограниченно, Салсалат (Амиджезик), Дифлюнисал (Долобид), Ибупрофен (Мотрин), Кетопрофен (Орудис), Набуметон (Релафен), Пироксикам (Фелден), Напроксен (Алев, Напросин), Диклофенак (Вольтарен), Индометацин (Индоцин), Сулиндак (Клинорил), Толметин (Толектин), Этодолак (Лодин), Кеторолак (Торадол), Оксапрозин (Дэйпро) и их комбинации.
Предпочтительные ингибиторы COX-I включают ибупрофен (Мотрин), нуприн, напроксен (Алев), индометацин (Индоцин), набуметон (Релафен) и их комбинации.
Целевые средства, используемые в сочетании с соединением формулы 1 и фармацевтическими композициями, описанными здесь, включают такие ингибиторы EGF, как Иресса (гефитиниб, AstraZeneca), Тарцева (эрлотиниб или OSI-774, OSI Pharmaceuticals Inc.), Эрбитукс (цетуксимаб, Imclone Pharmaceuticals, Inc.), EMD-7200 (Merck AG), ABX-EGF (Amgen Inc. and Abgenix Inc.), HR3 (правительство Кубы), IgA антитела (University of Erlangen-Nuremberg), TP-38 (IVAX), гибридный белок EGFR, вакцина EGF, иммунолипосомы против EGFr (Hermes Biosciences Inc.) и их комбинации.
Предпочтительными ингибиторами EGFr являются Иресса, Эрбитукс, Тарцева и их комбинации.
Настоящее изобретение также относится к противоопухолевым агентам, выбранным из ингибиторов пан erb рецептора или ингибиторов рецептора ErbB2, например CP-724714 (Pfizer, Inc.), CI-1033 (канертиниб, Pfizer, Inc.), Херцептин (трастузумаб, Genentech Inc.), Омитарг (2С4, петузумаб, Genentech Inc.), TAK-165 (Takeda), GW-572016 (лонафарниб, GlaxoSmithKline), GW-282974 (GlaxoSmithKline), EKB-569 (Wyeth), PKI-166 (Novartis), dHER2 (вакцина HER2, Corixa and GlaxoSmithKline), APC8024 (вакцина HER2, Dendreon), биспецифические антитела против HER2/neu (Decof Cancer Center), B7.her2.IgG3 (Agensys), AS HER2 (Research Institute for Rad Biology & Medicine), трифункциональные биспецифические антитела (University of Munich) и mAB AR-209 (Aronex Pharmaceuticals Inc) и mAB 2B-1 (Chiron) и их комбинации.
Предпочтительные противоопухолевые агенты селективные для erb включают Херцептин, TAK-165, CP-724714, ABX-EGF, HER3 и их комбинации.
Предпочтительные ингибиторы пан erbb рецептора включают GW572016, CI-1033, EKB-569, и Омитарг и их комбинации.
Дополнительные ингибиторы erbB2 включают те, которые описаны в международных публикациях WO 98/02434 (опубликована 22 января, 1998 г.), WO 99/35146 (опубликована 15 июля, 1999 г.), WO 99/35132 (опубликована 15 июля, 1999 г.), WO 98/02437 (опубликована 22 января, 1998 г.), WO 97/13760 (опубликована 17 апреля, 1997 г.), WO 95/19970 (опубликована 27 июля, 1995 г.), патенте США 5587458 (выдан 24 декабря, 1996 г.), и патенте США 5877305 (выдан 2 марта, 1999 г.), описания которых включены здесь в полном объеме в виде ссылки. Ингибиторы рецептора ErbB2, используемые в настоящем изобретении, также описаны в патентах США №№ 6465449 и 6284764, и международной заявке WO 2001/98277, описания которых включены здесь в полном объеме в виде ссылки.
Дополнительно, другие противоопухолевые агенты могут быть выбраны из следующих агентов, BAY-43-9006 (Onyx Pharmaceuticals Inc.), Генасенс (аугмеросен, Genta), Панитумумаб (Abgenix/Amgen), Зевалин (Schering), Бекссар (Corixa/GlaxoSmithKline), Абареликс, Алимта, EPO 906 (Novartis), дискодермолид (XAA-296), ABT-510 (Abbott), Неовастат (Aeterna), энзастаурин (Eli Lilly), Комбрестатин A4P (Oxigene), ZD-6126 (AstraZeneca), флавопиридол (Aventis), CYC-202 (Cyclacel), AVE-8062 (Aventis), DMXAA (Roche/Antisoma), Тимитаг (Eximias), Темодар (темозоломид, Schering Plough) и Ревилимд (Celegene) и их комбинации.
Другие противоопухолевые агенты могут быть выбраны из следующих агентов, CyPat (ципротерона ацетат), Хистерелин (хистерелина ацетат), Пленаиксис (абареликс депот), Атразентан (ABT-627), Сатраплатин (JM-216), Таломид (Талидомид), Тератоп, Темилифен (DPPE), ABI-007 (паклитаксел), Эвиста (ралоксифен), Атаместан (Biomed-777), Ксиотакс (полиглутамат паклитаксел), Таргетин (бексаротин) и их комбинации.
Дополнительно, другие противоопухолевые агенты могут быть выбраны из следующих агентов: Тризаон (тирапазамин), Апозин (эксисулинд), Невастат (AE-941), Цеплен (гистамина гидрохлорид), Оратецин (рубитекан), Вирулизин, Гастриммун (G17DT), DX-8951f (экзатекан мезилат), Онконаза (ранпирназа), BEC2 (митумоаб), Кситрин (мотексафина гадолин) и их комбинации.
Кроме того, противоопухолевые агенты могут быть выбраны из следующих агентов: ЦеаВак (CEA), НеуТрексин (триметрезат глюкуронат) и их комбинации.
Дополнительные противоопухолевые агенты могут быть выбраны из следующих агентов: ОваРекс (ореговомаб), Осидем (IDM-1), и их комбинации.
Дополнительные противоопухолевые агенты могут быть выбраны из следующих агентов: Адвексин (ING 201), Тиразон (тирапазамин), и их комбинации.
Дополнительные противоопухолевые агенты могут быть выбраны из следующих агентов: RSR13 (эфапроксипал), Котара (1311 chTNT 1/b), NBI-3001 (IL-4) и их комбинации.
Дополнительные противоопухолевые агенты могут быть выбраны из следующих агентов: Канваксин, вакцина GMK, PEG интерон A, Таксопрексин (DHA/паксилтаксел) и их комбинации.
Другие предпочтительные противоопухолевые агенты включают продукцию фирмы Pfizer MEK1/2-ингибитор PD325901, продукцию фирмы Array Biopharm MEK-ингибитор ARRY-142886, продукцию фирмы Bristol Myers CDK2-ингибитор BMS-387032, продукцию фирмы Pfizer CDK-ингибитор PD0332991 и продукцию фирмы AstraZeneca AXD-5438 и их комбинации.
Дополнительно, также можно использовать такие ингибиторы mTOR, как CCI-779 (Wyeth) и производные рапамицина RAD001 (Novartis) и AP-23573 (Ariad), HDAC-ингибиторы SAHA (Merck Inc/Aton Pharmaceuticals) и их комбинации.
Дополнительные противоопухолевые агенты включают ингибитор aurora 2-киназы VX-680 (Vertex), Chk1/2-ингибитор XL844 (Exilixis).
Следующие цитотоксические агенты, например один или несколько, выбранные из группы, содержащей в себе эпирубицин (Элленс), доцетаксел (Таксотер), паклитаксел, Зинекард (дексразоксан), ритуксимаб (Ритуксан) иматиниба мезилат (Гливек), и их комбинации, могут быть использованы в сочетании с соединением формулы 1 и фармацевтическими композициями, описанными здесь.
Данное изобретение также относится к применению соединений по настоящему изобретению в сочетании с гормональной терапией, включающей, но не ограниченно, экземестан (Аромазин, Pfizer Inc.), леупрорелин (Лупрон или Леуплин, TAP/Abbott/Takeda), анастрозол (Аримидекс, Astrazeneca), Госрелин (Золадекс, AstraZeneca), доксеркальциферол, фадрозол, форместан, тамоксифена цитрат (тамоксифен, Нолвадекс, AstraZeneca), Казодекс (AstraZeneca), Абареликс (Praecis), Трелстар, и их комбинации.
Данное изобретение также относится к таким средствам гормональной терапии как антиэстрогены, включающие, но не ограниченно, фулвестрант, торемифен, ралоксифен, лазофоксифен, летрозол (Фемара, Novartis), такие антиандрогены, как бикалутамид, флутамид, мифепристон, нилутамид, Casodex® (4'-циано-3-(4-фторфенилсульфонил)-2-гидрокси-2-метил-3'-(трифторметил)пропионанилид, бикалутамид) и их комбинации.
Кроме того, данное изобретение относится к соединению по настоящему изобретению самостоятельно или в сочетании с одним или несколькими средствами заместительной терапии, например, средство выбрано из группы, содержащей в себе Филграстим (Неупоген), ондансетрон (Зофран), Фрагмин, Прокрит, Алокси, Эменд или их комбинации.
Конкретные предпочтительные цитотоксические агенты включают Камптосар, Эрбитукс, Ирессу, Гливек, Таксотер или их комбинации.
Следующие ингибиторы топоизомеразы I могут быть использованы в качестве противоопухолевых агентов: камптотецин, иринотекан HCl (Камптосар), эдотекарин, оратецин (Supergen), экзатекан (Daiichi), BN-80915 (Roche) и их комбинации.
Конкретные предпочтительные ингибиторы топоизомеразы II включают эпирубицин (Элленс).
Соединения по настоящему изобретению могут быть использованы в сочетании с противоопухолевыми агентами, алкилирующими агентами, антиметаболитами, антибиотиками, противоопухолевыми агентами растительного происхождения, производными камптотецина, ингибиторами тирозин киназы, антителами, интерферонами, и/или модификаторами биологических реакций.
Алкилирующие агенты включают, но не ограничиваются, N-оксид азотистого иприта, циклофосфамид, ифосфамид, мелфалан, бусульфан, митобронитол, карбоквон, тиотепа, ранимустин, нимустин, темозоломид, AMD-473, альтретамин, AP-5280, апазиквон, бросталлицин, бендамустин, кармустин, эстрамустин, фотемустин, глюфосфамид, ифосфамид, KW-2170, мафосфамид, и митолактол; алкилирующие соединения на основе координационных соединений платины включают, но не ограничиваются, цисплатин, Параплатин (карбоплатин), эптаплатин, лобаплатин, недаплатин, Элоксатин (оксалиплатин, Sanofi) или сатрплатин и их комбинации. Конкретные предпочтительные алкилирующие агенты включают Элоксатин (оксалиплатин).
Антиметаболиты включают, но не ограничиваются, метотрексат, 6-меркаптопурин рибозид, меркаптопурин, 5-фторурацил (5-FU) отдельно или в комбинации с лейковорином, тегафуром, UFT, доксифлуридином, кармофуром, цитарабином, цитарабина окфосфатом, эноцитабином, S-1, Алимтой (пеметрексед динатрий, LY231514, MTA), Гемзаром (гемцитабин, EIi Lilly), флударабином, 5-азацитидином, капецитадином, кладрибином, клофарабином, децитабином, эфлорнитином, этинилцитидином, цитозид арабинозидом, гидроксимочевиной, TS-1, мелфаланом, неларабином, нолатрекседом, окфосфат, пеметрексед динатрием, пентостатином, пелитрексолом, ралтитрекседом, триапином, триметрексатом, видарабином, винкристином, винорелбином; или, например, одним из предпочтительных антиметаболитов, раскрытых в европейской патентной заявке № 239362, например N-(5-[N-(3,4-дигидро-2-метил-4-оксоквиназолин-6-илметил)-N-метиламино]-2-теноил)-L-глутаминовая кислота и их комбинации.
Антибиотики включают интеркалирующие антибиотики, но не ограничиваются: акларубицин, актиномицин D, амрибицин, аннамицин, адриамицин, блеомицин, даунорубицин, доксорубицин, элсамитруцин, эпирубицин, галарубицин, идарубицин, митомицин С, неморубицин, неокарциностатин, пеломицин, пирарубицин, ребеккамицин, стималамер, стрептозоцин, валрубицин, зиностатин и их комбинации.
Противовоспалительные вещества растительного происхождения включают, например, те, которые выбраны из ингибиторов митоза, например, винбластин, доцетаксел (Таксотер), паклитаксел и их комбинации.
Цитотоксические агенты, ингибирующие топоизомеразу, включают один или несколько агентов, выбранных из группы, содержащей в себе акларубицин, амонафид, белотекан, камптотецин, 10-гидроксикамптотецин, 9-аминокамптотецин, дифломотекан, иринотекан HCl (Камптосар), эдотекарин, эпирубицин (Элленс), этопозид, эксатекан, гиматекан, луртотекан, митоксантрон, пирарубицин, пиксантрон, рибитекан, собузоксан, SN-38, тафлупозид, топотекан и их комбинации.
Предпочтительные цитотоксические агенты, ингибирующие топоизомеразу, включают один или несколько агентов, выбранных из группы, содержащей в себе камптотецин, 10-гидроксикамптотецин, 9-аминокамптотецин, иринотекан HCI (Камптосар), эдотекарин, эпирубицин (Элленс), этопозид, SN-38, топотекан, и их комбинации.
Иммунологические средства включают интерфероны и многочисленные другие усиливающие иммунитет агенты. Интерфероны включают интерферон альфа, интерферон альфа-2а, интерферон альфа-2b, интерферон бета, интерферон гамма-1a, интерферон гамма-1b (Актиммун), или интерферон гамма-n1 и их комбинации. Другие агенты включают филграстим, лентинан, сизофилан, ТераЦис, убенимекс, WF-10, алдеслейкин, алемтузумаб, BAM-002, дакарбазин, даклизумаб, денилейкин, гемтузумаб озогамицин, ибритумомаб, имиквимод, ленограстим, лентинан, вакцину против меланомы (Corixa), молграмостин, OncoVAX-CL, сарграмостин, тазонермин, теклейкин, тималазин, тозитумомаб, Вирулизин, Z-100, эпратузумаб, митомомаб, ореговомам, пемтимомаб (Y-muHMFG1), Провенг (Дендреон) и их комбинации.
Модификаторы биологической реакции представляют собой агенты, которые изменяют защитные механизмы живых организмов или биологические реакции, такие как выживаемость, рост, или дифференцировка клеток тканей, побуждают их иметь противоопухолевое действие. Такие агенты включают крестин, лентинан, сизофиран, пицибанил, убенимекс и их комбинации.
Другие противоопухолевые препараты включают алитретиноин, амплиген, атрасентан бексаротен, бортезомиб. Бозентан, кальцитриол, эксисулинд, финастерид, фотемустин, ибандроновая кислота, милтефозин, митоксантрон, I-аспарагиназа, прокарбазин, дакарбазин, гидроксикарбамид, пегаспаргаза, пентостатин, тазаротен, Телцита (TLK-286, Telik Inc.), Велкад (бортемазиб, Millenium), третиноин и их комбинации.
Другие антиангиогенные соединения включают ацитретин, фенретинид, талидомид, золедроновую кислоту, ангиостатин, аплидин, циленгтид, комбретастатин A-4, эндостатин, халофугинон, ребимастат, ремоваб, ревлимид, скваламин, украин, витаксин и их комбинции.
Координационные соединения платины включают, но не ограничены, цисплатин, карбоплатин, недаплатин, оксалиплатин и их комбинации.
Производные камптотецина включают, но не ограничены, камптотецин, 10-гидроксикамптотецин, 9-аминокамптотецин, иринотекан, SN-38, эдотекарин, топотекан и их комбинации.
Другие противоопухолевые агенты включают митоксантрон, I-аспарагиназу, прокарбазин, дакарбазин, гидроксикарбамид, пентостатин, третиноин и их комбинации.
Противоопухолевые агенты, способные усиливать противоопухолевые иммунные реакции, такие как антитела против CTLA4 (цитотоксический лимфоцитарный антиген 4), и другие агенты, способные блокировать CTLA4, также могут быть использованы, например MDX-010 (Medarex) и соединения CTLA4, раскрытые в патенте США № 6682736; и такие антипролиферативные агенты, как другие ингибиторы фарнезил-протеин трансферазы, например, ингибиторы фарнезил-протеин трансферазы. Дополнительно, специфические антитела против CTLA4, которые могут быть использованы в настоящем изобретении, включают те, которые описаны в предварительной заявке на выдачу патента США № 60/113647 (подана 23 декабря, 1998), патенте США № 6682736, описания которых включены здесь в полном объеме в виде ссылки.
Антитела, специфичные к IGF1R, которые могут быть использованы в настоящем изобретении, включают те, которые описаны в международной патентной заявке WO 2002/053596, которая включена здесь в полном объеме в виде ссылки.
Антитела, специфичные к CD40, которые могут быть использованы в настоящем изобретении, включают те, которые описаны в международной патентной заявке WO 2003/040170, которая включена здесь в полном объеме в виде ссылки.
Также могут быть использованы в качестве противоопухолевых агентов такие средства генотерапии, как, например TNFерад (GeneVec), которые экспрессируют TNF альфа в ответ на лучевую терапию.
В одном варианте осуществления настоящего изобретения статины могут быть использованы в сочетании с соединением формулы 1 и фармацевтическими композициями. Статины (ингибиторы HMG-CoA редуктазы) могут быть выбраны из группы, содержащей в себе Аторвастатин (Липитор, Pfizer Inc.), Правастатин (Правахол, Bristol-Myers Squibb), Ловастатин (Мевакор, Merck Inc.), Симвастатин (Зокор, Merck Inc.), Флувастатин (Лескол, Novartis), Церивастатин (Байкол, Bayer), Розувастатин (Крестор, AstraZeneca), Ловостатин и Ниацин (Адвикор, Kos Pharmaceuticals), их производные и комбинации.
В предпочтительном варианте осуществления статин выбран из группы, содержащей в себе Аторвастатин и Ловастатин, их производные и комбинации.
Другие средства, применимые в качестве противоопухолевых средств, включают Кадуэт.
Способы включают применение соединения формулы 1, используя любую желаемую схему применения дозированного средства.
В одном конкретном варианте осуществления, соединение вводят в организм один раз в день, хотя более или менее частое введение находится в рамках данного изобретения. Соединение формулы 1 можно применять в таком же режиме, как и цитотоксическое средство, с которым оно применяется вместе. В случаях, когда время полужизни цитотоксического агента длинное (а именно > 10 часов) также может быть рассмотрено применение соединения формулы 1 на следующий день, после введения цитотоксического агента. Соединение формулы 1 можно вводить в организм млекопитающего, в том числе человека, предпочтительно посредством внутривенного вливания в течение 30 минут.
В другом варианте осуществления настоящего изобретения соединение формулы 1 используют в качестве радиосенсибилизирующего средства, которое увеличивает эффективность лучевой терапии. В соответствии с настоящим изобретением соединение формулы 1 можно использовать в сочетании с любым типом лучевой терапии, в том числе наружной дистанционной лучевой терапией (XBRT) или телетерапией, брахитерапией или радиотерапией с использованием закрытого источника излучения, радиотерапией с использованием негерметизированного источника излучение и радиоиммунотерапией. В соответствии с настоящим изобретением для обеспечения максимального клинического канцерогенного эффекта, облучение обычно дают в ежедневной дозе 2-4 Гр до суммарной дозы 50-60 Гр. Специалисту в уровне техники понятно, что точная схема будет различной в зависимости от локализации заболевания и того, применяется радиация в лечебных целях или является средством паллиативного лечения. Дополнительную информацию, относящуюся к различным видам лучевой терапии, можно найти, например, в «Absorbed Dose Determination in External Beam Radiotherapy» International Atomic Energy Agency, Vienna, 2000, Technical Reports Series № 398; «Principles and Practice of Brachytherapy: Using Afterloading Systems,» Joslin et al. (Eds.), Arnold Publishers, 1st Edition, 2001; «Proton Therapy and Radiosurgery» Smit et al. (Eds.), Springer-Verlag Telos, 1st Edition, 2000; Greig et al. «Treatment with unsealed radioisotopes» Br. Med. Bull., 1973, 29 (1): 63-68; «Radioimmunotherapy of Cancer» Abrams et al. (Eds.), Marcel Dekker, 1st Edition, 2000. В патенте США № 6649645 приводятся сведения о комбинированной терапии радиацией и ингибитором циклооксигеназы-2 при лечении неопластических заболеваний.
В другом варианте осуществления настоящего изобретения соединение формулы 1 используется в сочетании с лучевой терапией и, по меньшей мере, одним противоопухолевым агентом.
В другом варианте осуществления настоящего изобретения соединение формулы 1 используется в сочетании с лучевой терапией и, по меньшей мере, одним радиопотенцирующим агентом, например антагонистами рецептора фактора роста.
Способы и композиции по настоящему изобретению обеспечивают одно или несколько преимуществ. Комбинацию соединения формулы 1 с химиотерапией или лучевой терапией по настоящему изобретению можно применять в низкой дозе, то есть дозе, ниже, чем обычно используется в клинических ситуациях для каждого отдельного компонента, применяемого самостоятельно. Преимущество снижения дозы химиотерапии или лучевой терапии по настоящему изобретению, применяемой у млекопитающего, включает снижение частоты возникновения отрицательных эффектов, вызванных более высокими дозами. Посредством снижения частоты возникновения отрицательных эффектов ожидается улучшение качества жизни пациента, получающего терапию в отношении рака. Дополнительные преимущества снижения частоты возникновения отрицательных эффектов включают улучшение соблюдения больным режима и схемы лечения и уменьшение количества госпитализаций, необходимых для устранения отрицательных эффектов.
Альтернативно, способы и комбинации по настоящему изобретению также могут обеспечить максимальный терапевтический эффект при более высоких дозах.
Примеры
Примеры и препаративные формы, представленные ниже, дополнительно иллюстрируют и приводят примеры комбинаций, дозированных форм и способов по настоящему изобретению. Понятно, что объем данного изобретения не ограничивается, никаким образом, объемом следующих примеров.
Материалы
Все химические вещества, если не оговорено иначе, получены от фирмы Sigma (Poole, Dorset, UK). Физиологический раствор, забуференный фосфатом по Дульбекко (PBS) получен от фирмы Gibco (Paisley, UK), сахароза, гидроксид натрия и хлорид калия поставлялись фирмой BDH (Lutterworth, UK) и дигитонин получен от фирмы Boehringer Mannheim (Roche Diagnostics, Lewes, UK). Набор реактивов BCA protein (Pierce, Perbio Science, Rockford, IL, USA) использовали для определения концентрации белка. Молочный порошок получен от фирмы Marvel Premier Brands UK Ltd (Spalding, UK), и наборы ECL Western Blot Detection получены от фирмы Amersham (Little Chalfont, UK). Nycomed® Lymphoprep получен от фирмы Axis-Shield (Oslo, Norway)
и пробирки с ЭДТА для сбора крови производства BD Vacutainer (Plymouth, UK). Мышиные моноклональные первичные антитела 10H были великодушно предоставлены профессором Alexander Burkle, и вторичные антимышиные антитела козы (HRP-конъюгированные) получены от фирмы DAKO (Ely, UK). Олигонуклеотиды, используемые для стимуляции активности PARP, первоначально синтезированы доктором J Lunec (Northern Institute for Cancer Research, Newcastle), и последующие поставки получены от фирмы Invitrogen (Glasgow, UK). Очищенный полимер поли(АДФ рибоза) (PAR) получен от фирмы BIOMOL Research Lab (Plymouth, PA, USA).
Культура тканей клеток SW620 и L1210 (контроль качества)
Клетки подращивали в среде RPMI 1640 (Sigma), дополненной 10% (об/об) фетальной сыворотки теленка (Invitrogen) и 1 Ед/мл раствора пенициллин-стрептомицин (Sigma), в инкубаторе Hereus (Fischer Scientific, Manchester, UK) подращивали при 37°C во влажной атмосфере с 5% содержанием CO2 в воздухе. Используемые клетки L1210 получали от ATCC (American Type Culture Collection, Manassas, VA) и выращивали в виде суспензии до плотности приблизительно 6×105/мл при получении, для обеспечения экспоненциальной фазы роста. Аликвоты 1×106 клеток для использования в качестве контроля качества образцов ресуспендировали в 1 мл среды плюс 10% (об/об) DMSO и 10% (об/об) фетальной сыворотки теленка и замораживали при -80°C.
Получение образцов опухолевых ксенотрансплантатов
Опухоли удаляли и мгновенно замораживали в жидком азоте и хранили при -80°С до момента приготовления гомогенатов для анализа. Образцы размораживали на льду и регистрировали сырую массу. Ткань гомогенизировали с использованием инструмента Pro 2000 (Pro Scientific Inc, Monroe, CT, USA) в 3 объемах (то есть 1 мг плюс 3 мкл изотонического буфера - 7 мМ Hepes, 26 мМ KCl, 0,1 мМ декстрана, 0,4 мM EGTA, 0,5 мM MgCl2, 45 мM сахарозы, pH 7,8), получая разведение гомогената 1 к 4. Гомогенат хранили на льду на протяжении всей процедуры, и гомогенизирование проводили 10-секундными сериями для предотвращения излишнего нагревания образца. Перед проведением анализа образцы дополнительно разбавляли изотоническим буфером в случае необходимости получить конечное разведение 1 к 40 для исследования инкорпорирования [32P]NAD, или 1 к 1000 для иммуноблот-анализа.
Получение PBL и образцов опухоли
Цельную кровь собирали в вакуумные пробирки с EDTA и получали лимфоциты периферической крови человека в результате процесса выделения лимфоцитов в соответствии с рекомендациями фирмы-изготовителя. Биопсии опухолей получали из операционной в стерильном контейнере и немедленно помещали на лед. В срок не более 30 минут образцы опухоли мгновенно замораживали в жидком азоте и хранили при -80°C до момента приготовления гомогената для анализа. Образец размораживали на льду и регистрировали сырую массу. Образцы массой более 100 мг гомогенизировали с применением инструмента Pro 2000 (Pro Scientific Inc, Monroe, CT, USA) в 3 объемах (то есть 1 мг плюс 3 мкл изотонического буфера - 7 мМ Hepes, 26 мМ KCl, 0,1 мМ декстрана, 0,4 мM EGTA, 0,5 мM MgCl2, 45 мM сахарозы, pH 7,8), получая разведение гомогената 1 к 4. В случае получения образцов с меньшей массой их гомогенизировали в 99 или 999 объемах, получая конечные разведения 1 к 100, и 1 к 1000, соответственно. Гомогенат хранили на льду в течение всей процедуры, гомогенизирование проводили 10-секундными сериями для предотвращения излишнего нагревания образца. Если образец не подвергали анализу в день получения гомогената, образцы повторно замораживали до -80°C и хранили при этой температуре до момента проведения анализа. Перед проведением анализа образцы дополнительно разбавляли изотоническим буфером, когда необходимо получить конечное разведение 1 к 1000.
Анализ PARP с использованием инкорпорирования [ 32 P]NAD
Как описано в: Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, Durkacz BW, Hostomsky Z, Kumpf RA, Kyle S, Li J, Maegley K, Newell DR, North M, Notarianni E, Stratford IJ, Skalitzky D, Thomas HD, Wang L-Z, Webber SE, Williams KJ and Curtin NJ. Preclinical evaluation of a novel poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor, AG14361, with significant anticancer chemo- and radio-sensitization activity. JNCI 96 56-67 (2004) and Bowman KJ, Newell DR, Calvert AH and Curtin NJ. Differential effects of the poly(ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity. Br. J Cancer 84 106-112 (2001). На основании ранее опубликованных методов (Halldorsson H., Gray D.A., and Shall S. (1978). Poly(ADP-ribose)polymerase activity in nucleotide permeable cells. FEBS Letters 85: 349-352, Grube, K., Kupper, J.H. & Burkle, A. Direct stimulation of poly(ADP-ribose) polymerase in permeabilised cells by double-stranded DNA oligomers. Anal. Biochemistry. 1991; 193: 236-239).
Ингибирование PARP определяли в клетках, с нарушенной дигитонином (0,15 мг/мл) проницаемостью мембраны (8×105-1×106/реакция), стимулированных добавленным извне 12-мерным двухцепочечным ДНК олигонуклеотидом с тупыми концами (2,5 мкг/мл), посредством измерения ингибирования 75 мкМ NAD++[32P]NAD+ (Amersham), инкорпорирования внутрь клеточных макромолекул в течение 6-минутного инкубирования при 25°С, затем осажденные добавлением ледяного 10% ТСА, 10% NaPPi (мас./об.), как описано ранее. Вкратце, клетки в течение 30 минут суспендировали на льду в гипотоническом буфере (9 мM HEPES, pH 7,8, 4,5% (об./об.) декстран, 4,5 мM MgCl2 и 5 мM DTT) из расчета плотности 1,5×107/мл, затем добавляли 9 объемов изотонического буфера (40 мM HEPES pH 7,8, 130 мM KCl, 4% (об./об.) декстран, 2 мM EGTA, 2,3 мM MgCl2, 225 мM сахароза и 2,5 мM DTT). Реакцию начинали посредством добавления 300 мкл клеток к 100 мкл 300 мкМ NAD+, содержащего [32P]-NAD+ (Amersham, UK), и останавливали посредством добавления 2 мл ледяной 10% (мас./об.) TCA+10% (мас./об.) пирофосфата натрия. После 30 минут преципитации на льду 32Р-меченные полимеры АДФ-рибоза фильтровали на фильтрах Whatman GC/C (Whatman International Ltd, Kent, UK), отмывали 5 раз с использованием 1% (об./об.) TCA/1% (об./об.) пирофосфат натрия, высушивали и подсчитывали. Значения IC50 ингибирования PARP высчитывали по кривым, построенным по экспериментальным точкам с помощью компьютера. (GraphPad Software, Inc., San Diego, CA).
Опухолевые гомогенаты анализировали схожим образом; однако процедура гомогенизирования вносит достаточное разрушение ДНК для максимального стимулирования PARP, таким образом, не требуется применение олигонуклеотида. Результаты выражены в пересчете на пмоль PAR на мг опухоли.
Анализ PARP с применением моноклональных антител
Как описано у Plummer ER, Middleton MR, Jones C, Olsen A, Hickson I, McHugh P, Margison G, McGown G, Thomcroft M, Watson AJ, Boddy AV, Calvert AH, Harris AL, Newell DR, Curtin NJ. Temozolomide pharmacodynamics in patients with metastatic melanoma: DNA damage and activity of repair enzymes ATase and PARP-1. Clinical Cancer Research. 11 3402-3409 (2005) на основании модификации ранее опубликованных методов (Pfieffer R, Brabeck C, Burkle A: Quantitative nonisotopic immuno-dot-blot method for the assessment of cellular poly(ADP-ribosyl)ation capacity. Analytical Biochemistry 1999; 275: 118-122).
Культивируемые клетки или быстро размороженные препараты лимфоцитов дважды отмывали ледяным PBS. Дебрис ресуспендировали в 0,15 мг/мл дигитонина до получения плотности приблизительно 1-2×106 клеток на мл в течение 5 минут для нарушения проницаемости клеток, с последующим добавлением 9 объемов ледяного буфера (7 мM HEPES, 26 мM KCl, 0,1 мM декстран, 0,4 мM EGTA, 0,5 мM MgCl2, 45 мM сахароза, pH 7,8) и образцы помещали на лед. Подсчитывали плотность клеток с нарушенной проницаемостью мембраны (то есть окрашенные трипановым синим), и клеточную суспензию при необходимости разбавляли вышеуказанным буфером для получения такой плотности клеток, которая позволит добавить в каждую реакционную пробирку 20000 клеток с нарушенной проницаемостью. В этом исследовании максимально стимулированную активность PARP определяли под действием олигонуклеотида с «тупыми» концами в присутствии NAD+ субстрата [25] при 26°С на водяной бане с вибрационным устройством. Смешивали пять мкл 7 мM NAD+ и 5 мкл 200 мкг/мл палиндромного олигонуклеотида (CGGAATTCCG) с клетками с нарушенной проницаемостью и реакционным буфером (100 мM Tris HCl, 120 мM MgCl2, pH 7,8) до конечного объема 100 мкл. Реакцию останавливали через 6 минут добавлением избытка ингибитора PARP (400 мкл 12,5 мкM соединения I) и клетки блоттировали на нитроцеллюлозную мембрану (Hybond N, Amersham), используя 24-луночный манифолд, для обеспечения возможности количественного анализа, калибровочную кривую очищенного PAR нагружали на каждую мембрану (0-25 пмол эквивалент мономера). После инкубирования с первичными антителами (1 к 500 в PBS-MT (PBS плюс 0,05% Tween 20 плюс 5% молочный порошок) в течение ночи при 4°C 2 раза отмывали с помощью PBS-T (PBS плюс 0,05% Tween 20) и затем инкубировали со вторичными антителами (1 к 1000 в PBS-MT) в течение 1 часа при комнатной температуре. Инкубированную мембрану многократно отмывали с помощью PBS в течение одного часа, затем в течение одной минуты подвергали действию реакционного раствора ECL, поставляемого фирмой-изготовителем. Хемилюминесценцию, выявленную в течение 5 минутной экспозиции, измеряли с использованием УФ-иллюминатора Fuji LAS3000 (Raytek, Sheffield, UK) и оцифровывали с использованием программного обеспечения для обработки изображений (Fuji LAS Image версия 1.1, Raytek). Сканированное изображение анализировали с помощью анализатора Aida Image Analyser (версия 3.28.001) и результаты выражали в LAU/мм2. Измеряли три фоновые области на блоте, подверженном воздействию, и среднее значение фонового сигнала мембраны вычитали из всех результатов. Калибровочную кривую полимера PAR анализировали с помощью нелинейной регрессионной модели невзвешенного одностороннего связывания и таким образом выводили неизвестные величины калибровочной кривой. Затем результаты были выражены относительно количества нагруженных клеток. Тройные образцы QC клеток 5000 L1210 применяли в каждом исследовании, все образцы, полученные от одного больного, анализировали на одном и том же блоте.
Опухолевые гомогенаты анализировали в схожей манере; однако процедура гомогенизирования вносит достаточное разрушение ДНК для максимального стимулирования PARP, таким образом, не требуется применение олигонуклеотида. Концентрацию белка в гомогенате измеряли с использованием BCA белкового анализа и планшет-ридера Titertek Multiscan MCC/340. Результаты могут быть выражены в пересчете на пмоль PAR на мг белка или на мг опухоли.
Анализ активности PARP в моноцитах периферической крови (PBMC) основан на методе Boulton et al. («Potentiation of temozolomide-induced cytotoxicity: a comparative study of the biological effects of poly(ADP-ribose) polymerase inhibitors». 1995. British J. Cancer 72, 849-856). Все стадии проводят при 0-4°C.
Получение PBMC
1. Собрать 5 мл крови в пробирку с литиевым гепарином и осторожно перемешать.
2. Разбавить гепаринизированную кровь с помощью PBS в пропорции 1:1 в 30-миллилитровой одноразовой универсальной пробирке (конечный объем 10 мл).
3. Осторожно наслоить разбавленную кровь на 8-10 мл предварительно охлажденного Lymphoprep в 30-миллилитровой одноразовой универсальной пробирке. Принять меры, чтобы кровь не смешивалась с разделительной жидкостью.
4. Центрифугировать образцы в течение 15 минут на центрифуге с поворотно-откидным ротором (центрифуга Mistral) при 800×G, 4°C, скорость торможения 0.
5. После центрифугирования слой лейкоцитов должен быть виден на поверхности. Этот слой клеток должен быть собран с помощью стеклянной пастеровской пипетки и помещен в 30-миллилитровую одноразовую универсальную пробирку.
6. Разбавить суспензию лимфоцитов с помощью 20 мл ледяного PBS и центрифугировать клетки в течение 10 минут при 500×G, 4°C.
7. Удалить супернатант.
8. Ресуспендировать осадок в 20 мл ледяного PBS и центрифугировать при 333×G, 4°C в течение 5 минут.
9. Удалить супернатант и ресуспендировать клетки в 500 мкл предварительно охлажденной среды (RPMI плюс 10% фетальная сыворотка теленка), с добавленной 10% DMSO
10. Перенести в маркированную пробирку Eppendorf с закручивающейся крышкой и заморозить.
11. Хранить при -70°C.
Анализ PARP в PBMC
1. В день проведения эксперимента готовили свежий раствор 32P 600 мкM NAD, как подробно описано выше. Исходный олигонуклеотид перемещали из хранилища и размораживали.
2. Водяную баню нагревали до 26°C и устанавливали встряхивающий режим на 70 колебаний в минуту.
3. Реакционные пробирки подготавливали к работе следующим образом.
4. Каждый образец и стандарт QC анализировали трижды, по три пробирки с TO, + олиго и - олиго образцами (всего 9 пробирок на образец).
5. Подсчитывали клеточную плотность в каждой суспензии. 10 мкл образца каждой клеточной суспензии разбавляли трипановым синим в пропорции 1:1 и в счетной камере подсчитывали количество клеток с нарушенной проницаемостью мембраны в мл.
6. Реакционные пробирки и суспензию клеток с нарушенной проницаемостью мембраны прогревали на водяной бане до 26°С в течение 7 минут.
7. Суспензию клеток с нарушенной проницаемостью мембраны в течение короткого времени встряхивали и начинали реакцию посредством добавления 300 мкл (приблизительно 1×106 клеток) этой суспензии в каждую реакционную пробирку.
8. Реакцию останавливали ровно через 6 минут после добавления клеток, посредством добавления 2 мл ледяной 10% TCA+10% NaPPi и интенсивного перемешивания.
9. Затем пробирку, перед фильтрацией, инкубировали на льду в течение, по меньшей мере, одного часа (на этом этапе анализа должна произойти преципитации в течение, по меньшей мере, одного часа, реакционные пробирки можно оставить на ночь, если температура поддерживается на уровне ≤4°C).
10. Перед добавлением клеток с нарушенной проницаемостью мембраны в пробирки с TO добавляли 2 мл ледяной 10% TCA+10% NaPPi для внесения поправки на неспецифическое связывание радиоактивной метки с фильтром.
Получение образцов ткани опухоли
1. Взвешивали замороженные образцы опухоли.
2. 3 объема (то есть 3 мкл раствора, добавленные на каждый 1 мг ткани) изотонического буфера плюс DTT добавляли к каждому образцу опухоли. Хранили на льду до и во время гомогенизирования.
3. Образец гомогенизировали на льду внутри бокса класса II 10-секундными сериями до тех пор, пока не исчезнут макроскопические кусочки ткани.
4. Достаточный объем гомогената разводили в соотношении 1 к 10 изотоническим буфером плюс DTT для обеспечения итогового разведения исходного образца 1 к 40. Конечный объем 3 мл достаточен для тройного исследования и последующего белкового анализа.
5. Разбавленный гомогенат хранили на льду и анализировали в срок не позднее одного часа как описано ниже.
Анализ PARP в образцах опухолевой ткани
1. В день проведения эксперимента готовили свежий раствор 600 мкM [32P] NAD, как подробно описано выше. Исходный олигонуклеотид перемещали из хранилища и размораживали.
2. Водяную баню нагревали до 26°C и устанавливали встряхивающий режим на 70 колебаний в минуту.
3. Реакционные пробирки подготавливали к работе как указано в таблице А для образцов QC и как указано в таблице Б для гомогената.
4. Каждый образец QC анализировали трижды, по три пробирки с TO, + олиго и - олиго образцами (всего 9 пробирок на образец, если количество клеток небольшое, то образцы - олиго пропускают). Гомогенаты также анализируют трижды, по три пробирки с TO и реакционными образцами (всего 6 пробирок на образец).
5. Перед добавлением гомогената или клеток в пробирки с TO добавляли 2 мл ледяной 10% TCA+10% NaPPi для внесения поправки на неспецифическое связывание радиоактивной метки с фильтром.
6. Реакционные пробирки с гомогенатами и клетками QC прогревали на водяной бане до 26°С в течение 7 минут.
7. Каждый препарат в течение короткого времени интенсивно встряхивали и начинали реакцию посредством добавления 300 мкл его в каждую реакционную пробирку.
8. Реакцию останавливали ровно через 6 минут после добавления гомогената посредством добавления 2 мл ледяной 10% TCA+10% NaPPi и интенсивного перемешивания.
9. Затем пробирку, перед фильтрацией, инкубировали на льду в течение, по меньшей мере, одного часа.
10. 10 мкл образца суспензии QC разбавляли в соотношении 1:1 трипановым синим и в счетной камере подсчитывали количество клеток с нарушенной проницаемостью мембраны в мл.
11. Оставшиеся гомогенаты центрифугировали при 500×G в течение 5 минут при 4°C, отбирали 200 мкл супернатанта и для измерения белка помещали в маркированную микропробирку с закручивающейся крышкой. Если анализ не проводят сразу же, образцы супернатанта можно хранить, по меньшей мере, в течение одного месяца при -20°C.
Пример 1. Ингибирование поли-АДФ-рибоза полимеразы
Кристаллографический анализ соединения формулы 1, связанного с ингибированным ферментом-мишенью, показал, что данное дозированное средство связывается с активным участком PARP-1, образуя 3 водородные связи. Действие соединения формулы 1, ингибирующее фермент PARP, испытывали, как описано в патенте США 6495541. Ki, определенная с использованием 32P-NAD+, инкорпорированного в полимер с помощью очищенного полноразмерного PARP-1 человека, составляет 1,4 нM (Таблица 3). Соединение формулы 1 также является мощным ингибитором PARP-2 (Ki=0,17 нM) и, на основании значительного структурного подобия аминокислотных последовательностей среди различных ферментов семейства PARP (танкираза, V-PARP), фосфатная соль соединения формулы 1 (Соединение I) вероятно будет связываться с высокой аффинностью также и с этими ферментами.
Кинетические константы для взаимодействия соединения формулы 1 с PARP
*SD= стандартное отклонение
Пример 2. Ингибирование клеточного роста
Свойственную соединению формулы 1 рост-ингибиторную активность после 5-дневного непрерывного воздействия (Таблица 4) определяли в клеточных линиях A549, LoVo и SW620, как описано в патенте США 6495541. Значения GI50 (концентрация необходимая для ингибирования роста на 50%) распределены от 7 до 12 мкM. Схожим образом определяли способность 0,4 мкМ соединения формулы 1 (то есть, <5% от IC50) увеличивать мощность темозоломида и топотекана ингибировать рост (Таблица 2). Фактор потенцирования при концентрации IC50; PF50, рассчитывали следующим образом: GI50 темозоломида или топотекана по отдельности/GI50 темозоломида или топотекана+0,4 мкМ соединения формулы 1. Наблюдали 8-кратное уменьшение GI50 темозоломида в клетках LoVo и 3,5-кратное уменьшение GI50 темозоломида в клетках А549 после добавления 0,4 мкМ соединения формулы 1. Наблюдали 1,6-кратное уменьшение GI50 топотекана в клетках LoVo и 2,6-кратное уменьшение IC50 топотекана и в клетках A549, и в клетках SW620 после добавления 0,4 мкМ соединения формулы 1.
Ингибирование клеточного роста под воздействием соединения формулы 1 и потенцирование темозоломида и топотекана с помощью 0,4 мкM соединения формулы 1
Пример 3. Хемосенсибилизирование стандартных химеотерапевтических агентов соединением I.
Исследования In vitro опухолевых клеточных линий человека, проводимые согласно способу, описанному в патенте США 6495541, показали, что в сумбикромолярных концентрациях соединение формулы 1 увеличивает чувствительность клеток к темозоломиду и ингибиторов топоизомеразы типа I, топотекану и SN-38 (активный метаболит иринотекана) в отношении клеток Н460 немелкоклеточного рака легких человека (NSCLC) (Таблица 5).
Действие соли глюкуроновой кислоты соединения формулы 1 на эффективность in vitro стандартных химиотерапевтических агентов в клетках H460 NSCLC человека
бСоединение I (фосфатная соль соединения формулы 1) использовали вместо соли глюкуроновой кислоты соединения формулы 1 в этих экспериментах.
Пример 4. Ингибирование истощения клеточного NAD и образование полимера поли-АДФ-рибозы посредством соединения формулы 1
Полимеры поли-(АДФ-рибоза) и NAD+ количественно определяли, как описано Abou-Ela et.al (Anal Biochem. (1988), 174: 239-250) с незначительными модификациями как указано далее. Клетки A549 (ATCC, Rockville, MD.) высевали в 35 мм чашки для культивирования и позволяли расти до слияния. Среду удаляли и заменяли свежей средой, содержащей в себе 20-50 мкКи мл-1 [3H] аденина. Клетки метили в течение 16 ч при 37°С. За 45 минут до экспериментальной процедуры среду заменяли свежей средой. После экспериментальной процедуры среду удаляли и клетки промывали ледяным забуференным фосфатом физиологическим раствором, рН 7,2, и собирали посредством добавления 1 мл 20% ледяной трихлоруксусной кислоты. Нерастворимое кислотой вещество удаляли с чашки посредством соскабливания. Чашки отмывали один раз 1 мл 20% трихлоруксусной кислотой и образцы подвергали центрифугированию. Супернатант сохраняли для определения NAD+. Осадок растворяли в 0,2 мл ледяной 98% муравьиной кислоты, затем разбавляли до 10 мл ледяной деионизированной H2O. Для облегчения преципитации добавляли двести микролитров 10 мг/мл альбумина бычьей сыворотки. Концентрацию трихлоруксусной кислоты доводили до 20% путем добавления 2,55 мл 100% трихлоруксусной кислоты. Нерастворимую кислотой фракцию собирали посредством центрифугирования.
Определение NAD +. Трихлоруксусный супернатант разбавляли до 10 мл 250 мМ муравьинокислым аммонием, рН 8,6, и доводили рН до значения 8,6 концентрированным гидроксидом аммония. Образец наносили на 0,5 мл колонку с DHB-сефарозой, которая была предварительно отмыта 10 мл 250 мМ муравьинокислого аммония, рН 8,6. Данную колонку отмывали 10 мл 250 мМ муравьинокислого аммония, рН 8,6 и 2 мл H2O. NAD+ элюировали с помощью 4 мл 250 мМ муравьинокислого аммония, pH 4,5.
Определение полимеров АДФ-рибозы. Нерастворимый в кислоте осадок растворяли в 1 мл гуанидинхлорида, 250 мМ ацетата аммония, 10 мМ EDTA, pH 6,0; и 1 мл 1 M KOH, 100 мM EDTA. Образец инкубировали при 37°С в течение 2 ч. Образец разводили до 10 мл с помощью 1М гуанидинхлорида, 250 мM ацетата аммония, 10 мM EDTA, pH 9,0 (буфер A), доводили pH до 9,0 и наносили на 0,5 мл колонку DHBB (Bio Rad), которая предварительно была отмыта 5 мл H2O и 10 мл буфера A. После нанесения колонку отмывают 25 мл буфера А, после чего 10 мл 1М бикарбоната аммония, 1 мM EDTA, pH 9,0. Поли(АДФ-рибозу) элюировали с помощью 0,5 мл H2O. Образец лиофилизировали до сухого состояния, затем суспендировали в 2 мл 50 мМ MOPS, 5 мM McGiII, pH 7,5. В течение 3 ч при 37°C суспензию переваривали, посредством добавления 1 единицы фосфодиэстеразы 1 змеиного яда (Worthington Biochemicals) и 1 единицы BAP.
Анализ методом ВЭЖХ: Анализ осуществляли посредством ВЭЖХ на 5 мкм колонке для обращенно-фазовой хроматографии Beckman C18 ODS, с 7 мM муравьинокислого аммония, подвижная фаза 7% метанол, при скорости потока 1 мл/мин. Каждый образец совместно впрыскивали с 10 нмоль и аденозина, и дезоксиаденозина. Собирали один миллилитр фракции, прошедшей через колонку и включали в 5 мл сцинтилляционной жидкости.
Потенциирование In vitro темозоломида соединением формулы 1 коррелируется с ингибированием истощения клеточного NAD, индуцированого алкилирующим агентом, и блокированием формирования полимера поли-АДФ-рибозы, с эффективным диапазоном от 5 до 400 нМ. После повреждения ДНК клеточный NAD быстро встраивается в полимер поли-АДФ-рибозы. 5 нМ соединения формулы 1 (PF50=1,3), значительно снижает потребление клеточного NAD, индуцированного MNNG, и ингибирует образование клеточной поли-АДФ-рибозы на 89% (Таблица 6). Соединение формулы 1 повышает эффективность темозоломида в клетках 549, по меньшей мере, в 2 раза, уже при концентрациях 50 нМ, соответствуя ингибированию истощения NAD, индуцированного MNNG, на 93% и ингибированию образования полимера поли-АДФ-рибозы на 95%. Соединение формулы 1 также ингибирует каталитическую активность PARP, измеренную как ингибирование поглощения клеточного NAD в мышиных лейкозных клетках P388 и мышиных лимфоцитах периферической крови, после активации PARP с помощью MNNG, перекиси водорода, или гамма-облучения (данные не представлены).
In Vitro Ингибирование активности PARP соединением формулы 1 в клеточной линии A549
бMNNG только контроль.
Пример 5. Исследования In Vivo противоопухолевой эффективности соединения формулы 1 - темозоломида
Эксперименты In vivo выполняли, как описано Calabrese et. al (JNCI (2004), 96: 56-67).
Для этих исследований вычисление дозы Соединения I (фосфатная соль) и дозы соли глюкуроновой кислоты проводили по свободному основанию.
В этих исследованиях Соединение I не проявляло противоопухолевых эффектов при применении отдельно. В комбинированных исследованиях Соединение I повышало эффективность дозы гамма-облучения, иринотекана и темозоломида. При испытании однократной дозы соединение формулы 1 усиливало противоопухолевые эффекты 200 мг/кг темозоломида более чем в 10 раз диапазона нетоксических дозировок в отношении ксенотрансплантата карциномы толстой кишки SW620 человека у мышей (Таблица 7).
Действие in vivo соединения формулы 1 в комбинации с однократной дозой темозоломида в отношении ксенотрансплантата карциномы толстой кишки SW620 человека
бдозы соединения формулы 1, введенные посредством внутрибрюшинной инъекции.
вn=5 для всех групп.
г%усиления=100×(время задержки темозоломида+соединение формулы 1 - время задержки только темозоломида)/время задержки только темозоломида. Время задержки рассчитывали как время для RTV4 (относительный объем опухоли) в группе, в которой проводилось лечение - время для RTV4 в контрольных группах, где RTV4 представляет собой объем опухоли, эквивалентный 4-кратному объему опухоли в начале лечения.
дзначительно отличается от отдельно применяемого агента темозоломид (критерий Манна-Уитни)
е2 смертельных исхода в результате токсичности.
При испытании повторной дозы (QD×5 для каждого агента) в отношении ксенотрансплантата SW620 соединение формулы 1 (в виде соли глюкуроновой кислоты) при 0,05, 0,15, и 0,5 мг/кг в комбинации с 68 мг/кг темозоломида увеличивало активность темозоломида во всех 3 группах сочетаний (темозоломид 68 мг/кг + 0,05, 0,15, или 0,5 мг/кг соли глюкуроновой кислоты соединения формулы 1. Полная 100 % ремиссия наблюдалась в случае сочетания групп с 0,15 или 0,5 мг/кг соли глюкуроновой кислоты с соединением формулы 1. Не наблюдали потери массы тела при комбинации доз соли глюкуроновой кислоты соединения формулы 1 (темозоломид 68 мг/кг + 0,05 или 0,15 мг/кг соли глюкуроновой кислоты соединения формулы). Наблюдали один летальный исход в результате токсичности в группе комбинации высокой дозы (темозоломид 68 мг/кг + 0,5 мг/кг соли глюкуроновой кислоты соединения формулы 1). В подобном эксперименте в отношении ксенотрансплантатов LoVo соль глюкуроновой кислоты соединения формулы 1 (0,5 мг/кг) усиливала противоопухолевую активность темозоломида (68 мг/кг) на 67% (Таблица 8, фиг.1). Ни в одной группе доз не наблюдали потерю массы тела в эксперименте с LoVo. Ни в одном исследовании комбинации не наблюдали антагонизма.
Эффективность темозоломида в комбинации с солью глюкуроновой кислоты соединения формулы 1 в отношении ксенотрансплантатов SW620 и LoVo
бдозы соединения формулы 1, введенные посредством внутрибрюшинной инъекции.
всоединение формулы 1 (эквивалент свободного основания), дозированное в виде соли глюкуроновой кислоты
гn=5 для всех групп.
д%усиления=100×(время задержки темозоломида+соединение формулы 1 - время задержки только темозоломида)/время задержки только темозоломида. Время задержки рассчитывали как время для RTV (относительный объем опухоли)4 в группе, в которой проводилось лечение - время для RTV4 в контрольных группах, где RTV4 представляет собой объем опухоли, эквивалентный 4-кратному объему опухоли в начале лечения.
езначительно отличается от отдельно применяемого агента темозоломид (критерий Манна-Уитни)
ж1 смертельный исход в результате токсичности.
Пример 7. Исследования In Vivo противоопухолевой эффективности соединения формулы 1 - иринотекана
Эксперименты in vivo выполняли, как описано Calabrese et.al (J. Natl. Cancer Inst. (2004), 96:56-67).
В виде отдельно применяемого агента ингибитор топоизомеразы I, иринотекан (25 мг/кг, QW×3 IP), существенно не ингибировал рост опухоли SW620. Комбинация 25 мг/кг иринотекана с соединением формулы 1 в виде соли глюкуроновой кислоты приводила к существенному противоопухолевому эффекту действия иринотекана во всех группах комбинаций (иринотекан 25 мг/кг + 0,05, 0,15, или 0,5 мг/кг соединения формулы 1 (Таблица 9)). Не наблюдали значительной токсичности в группах, где иринотекан был единственным агентом, или в группах, где иринотекан сочетался с ингибитором PARP. Ингибирование роста опухоли (процент усиления) увеличивалось с увеличением доз соединения формулы 1. В подобном эксперименте в отношении ксенотрансплантатов LoVo соединение формулы 1 (0,5 мг/кг) усиливало противоопухолевую активность иринотекана (25 мг/кг) на 86% (Таблица 9). Ни в одном исследовании комбинации не наблюдали антагонизма.
Эффективность иринотекана в комбинации с солью глюкуроновой кислоты соединения формулы 1 в отношении ксенотрансплантатов SW620 и LoVo
бдозы соли глюкуроновой кислоты соединения формулы 1, введенные посредством внутрибрюшинной инъекции.
вn=5 для всех групп.
г%усиления=100×(время задержки иринотекана+соединение формулы 1 - время задержки только иринотекана)/время задержки только иринотекана. Время задержки рассчитывали как время для RTV4 (относительный объем опухоли) в группе, в которой проводилось лечение - время для RTV4 в контрольных группах, где RTV4 представляет собой объем опухоли, эквивалентный 4-кратному объему опухоли в начале лечения.
дсоединение формулы 1 (эквивалент свободного основания), дозированное в виде соли глюкуроновой кислоты
езначительно отличается от отдельно применяемого агента иринотекан (критерий Манна-Уитни).
Пример 8. Фармакодинамика соединения формулы 1 - темозоломид
Лечение мышей, несущих ксенотрансплантат карциномы толстой кишки SW620 человека, с применением только 10 мг/кг соединения формулы 1 (QD×5) не приводило к замедлению роста опухоли и было нетоксическим. В таблице 10(a) представлены значения концентрации соединения формулы 1 в плазме и опухоли после внутрибрюшинного введения фосфатной соли (Соединение I). При испытании повторной дозы (QD×5 для каждого агента) в отношении ксенотрансплантата SW620, 0,1 мг/кг соединения формулы 1 усиливало противоопухолевый эффект 68 мг/кг темозоломида на 28% более чем отдельное применение 68 или 136 мг/кг темозоломида (Таблица 10(б)). Увеличение дозы соединения формулы 1 до 1 мг/кг увеличивало противоопухолевый эффект темозоломида (68 мг/кг) на 100%. Комбинация 10 мг/кг соединения формулы 1 и 68 мг/кг темозоломида была токсической.
В параллельном исследовании уровни соединения формулы 1 в плазме и опухоли измеряли посредством ВЭЖХ/МС анализа. Дополнительно, степень ингибирования каталитической активности опухолевой PARP оценивали посредством инкорпорирования Р32-NAD в полимер поли-АДФ-рибозы в гомогенатах опухоли SW620, полученных от животных, подвергавшихся лечению. При эффективной дозе соединения формулы 1 (1,0 мг/кг) концентрации соединения формулы 1 в плазме были едва определимы через 6 часов, но в опухоли уровни от 40 до 60 нг/мл были определены через 6 и 24 ч после инъекции. Каталитическая активность PARP была ингибирована на 50 % через 6 ч и на 25 % через 24 ч.
При токсической дозировке соединения формулы 1 (10 мг/кг), концентрации соединения формулы 1 в плазме были 30 нг/мл через 6 ч, но едва определимы через 24 ч. Уровни соединения формулы 1 в опухоли >200 нг/мл были определимы в любое время вплоть до 24 ч после введения 10 мг/кг соединения формулы 1, и каталитическая активность PARP была ингибирована на 90 % через 6 ч и на 75 % через 24 ч.
Концентрация соединения формулы 1 в плазме крови и опухоли после внутрибрюшинного введения фосфатной соли (соединение I)
формулы 1а,б (мг/кг)
бсоединение формулы 1 (эквивалент свободного основания), дозированное в виде фосфатной соли.
вBLQ: ниже уровня подсчета
Эффективность и фармакодинамика соли глюкуроновой кислоты соединения формулы 1 в комбинации с темозоломидом в отношении ксенотрансплантата карциномы толстой кишки SW620 человека
формулы 1а,б,г (мг/кг)
бсоединение формулы 1 (эквивалент свободного основания), дозированное в виде соли глюкуроновой кислоты
вдозы темозоломида, введенные перорально посредством зонда.
гдозы соединения формулы 1, введенные в/б, в комбинации с 68 мг/кг темозоломида, введенного п/о.
дSD=стандартное отклонение.
еусиление, вычисленное как ((время задержки (комбинация)/время задержки (темозоломид отдельно))×100-100)
жзначительно отличается от отдельно применяемого агента темозоломид (критерий Манна-Уитни).
зN/A, неприменимо, 5/5 случаев со смертельным исходом в результате токсичности.
Пример 9. Фармакокинетические исследования на животных
Фармакокинетику соединения формулы 1 (дозированная субстанция в виде свободного основания), после внутривенного введения соли соединения формулы 1, оценивали у мышей CD-1, крыс линии Вистар, собак породы бигль и человекообразных обезьян, результаты приведены в таблице 11. Внутривенное введение дозы у всех видов привело к клиренсу от умеренного до быстрого (от 34 до 136 мл/мин/кг) и большому объему распределения (от 7 до 15 л/кг), свидетельствуя о том, что это соединение равномерно распределяется в организме. Конечный период полувыведения был от относительно короткого до среднего (от 2 до 5 часов). Комбинированные исследования соединения I (фосфатная соль) с темозоломидом проводили на мышах и крысах для изучения потенциального воздействия этого цитотоксического агента на фармакокинетику соединения формулы 1. В комбинированном исследовании мышей одна группа из 8 мышей получала однократно внутривенную дозу 6,5 мг/кг соединения I (эквивалентно 5 мг/кг соединения формулы 1), в то время как вторая группа из 8 мышей получала однократно внутривенную дозу 6,5 мг/кг соединения I и однократно пероральную дозу 200 мг/кг темозоломида. Каждая из групп, получавших лечение, была разделена на 2 когортные группы по 4 мыши. Повод для когортного забора крови является результатом ограниченного объема циркулирующей крови у мышей. Забор крови проводили в каждой когорте в каждый момент времени определения фармакокинетических параметров. В комбинированных исследованиях крыс одна группа из 2 мышей получала однократно внутривенную дозу 6,5 мг/кг соединения I (5 мг/кг), в то время как вторая группа из 2 крыс получала и внутривенную дозу 6,5 мг/кг соединения I и пероральную дозу 50 мг/кг темозоломида. Результаты комбинированных исследований соединения I и темозоломида у мышей и крыс показали, что этот цитотоксический агент имеет только незначительный эффект на фармакокинетический профиль соединения формулы 1 (Таблица 12 и Таблица 13). Аналогично показано, что соединение I имеет только незначительный эффект на фармакокинетический профиль темозоломида (данные не представлены). Дополнительно, комбинированные исследования соединения I и иринотекана проводили у самцов мышей линии CD-1 и самцов крыс линии Вистар. В комбинированном исследовании у мышей одна группа из 15 мышей получала однократно внутривенную дозу 6,5 мг/кг соединения I (эквивалентно 5 мг/кг соединения формулы 1), в то время как вторая группа получала и внутривенную дозу 6,5 мг/кг соединения I и внутривенную дозу 45 мг/кг иринотекана. В каждый момент времени для забора образца в каждой группе умерщвляли по три мыши. В комбинированном исследовании у крыс одна группа получала внутривенную дозу 6,5 мг/кг соединения I, в то время как вторая группа получала и внутривенную дозу 6,5 мг/кг соединения I и внутривенную дозу 45 мг/кг иринотекана. Кровь брали у каждой крысы в каждый момент времени для забора образца. Результаты этого исследования предполагают, что в применяемых дозах не существуют взаимодействия лекарство-лекарство между соединением I и иринотеканом, которое приводит к нарушению фармакокинетики (Таблица 14 и Таблица 15).
Средние значения фармакокинетических параметров однократной внутривенной доза соединения формулы 1 у мышей, крыс, собак и обезьян
аданные комбинированных исследований с темозоломидом у мышей и крыс
бсоединение формулы 1 (эквивалент свободного основания), дозированное в виде соединения I (фосфатная соль)
всоединение формулы 1 (эквивалент свободного основания), дозированное в виде соли глюкуроновой кислоты
Групповое среднее фармакокинетических параметров соединения формулы 1 у мышей, получавших соединение I (фосфатная соль) отдельно или в комбинации с темозоломидом
асоединение I (фосфатная соль соединения формулы 1); дозы скорректированы с учетом соли.
Среднее значение фармакокинетических параметров соединения формулы 1 у крыс, получавших соединение I (фосфатная соль) отдельно или в комбинации с темозоломидом
асоединение I (фосфатная соль соединения формулы 1); дозы скорректированы с учетом соли.
Групповое среднее фармакокинетических параметров соединения формулы 1, введенного внутривенно, у мышей, получавших соединение I (фосфатная соль) отдельно или в комбинации с иринотеканом
асоединение I (фосфатная соль соединения формулы 1); дозы скорректированы с учетом соли.
Среднее значение (SD) фармакокинетических параметров соединения формулы 1, введенного внутривенно, у крыс, получавших соединение I (фосфатная соль) отдельно или в комбинации с иринотеканом
асоединение I (фосфатная соль соединения формулы 1); дозы скорректированы с учетом соли.
Пример 10. Эффекты на организм человека: первый этап испытания внутривенного введения соединения I как ингибитора PARP в комбинации с пятидневным пероральным применением темозоломида, получаемом каждые четыре недели
Открытое многоцентровое исследование с повышением дозы, проводимое в 2 частях.
Часть 1 данного исследования проводят у пациентов с запущенными опухолями. После пробной дозы отдельно взятого агента соединения I, введенной на День-7, соединение I применяли ежедневно в течение 5 дней в виде внутривенной инфузии с темозоломидом (100 мг/м2/доза). В последовательных когортах пациентов дозы соединения I повышали вплоть до дозы, ингибирующей PARP (PID, см. раздел D ниже), установленной с помощью фармакодинамических и фармакокинетических данных. Установленная PID составляла 12 мг/м2. Повышение дозы соединения I в следующей когорте становится возможным после того, как безопасность высших доз была установлена в предыдущей когорте.
Часть 2 испытания проводят среди пациентов с метастатической меланомой. Последовательные когорты пациентов получают PID соединения I в дополнение к повышенным дозам темозоломида до тех пор, пока максимально переносимая доза комбинированных дозированных средств не установится или доза темозоломида достигнет максимума 200 мг/м2. Пациенты, вошедшие в часть 2 исследования, обязаны дать согласие на проведение биопсии опухоли до и после лечения для измерения ингибирования PARP. Повышение дозы темозоломида в следующей когорте становится возможным после того, как безопасность высших доз была установлена в предыдущей когорте.
Клинические результаты у 17 пациентов, давших согласия и получавших лечение в части 1 исследования. В таблице 16 представлены демографические данные этих пациентов.
демографические данные пациентов, вошедших в часть 1 фазы 1 исследования
% белых
(1,32-1,82)
(1,86-2,44)
(1,71-2,01)
(1,82-2,10)
(2,04-2,26)
(1,32-2,44)
Первичный рак у этих пациентов, у всех запущенное заболевание, был раком грудной железы (1), толстого кишечника (2), почек (1), печени (1), поджелудочной железы (2), предстательной железы (1), прямой кишки (1), меланомой (3), саркомой мягких тканей (3), и желудка (2). Двенадцать (71%) пациентов предварительно получали хемотерапию, 3 (18%) пациента не получали, и о 2 (12%) нет информации.
A. Фармакокинетика и метаболический продукт в организме человека
Фармакокинетику соединения формулы 1 оценивали на этапе 1 открытого испытания с повышением дозы внутривенного введения соединения I в комбинации с темозоломидом. В части 1 испытания (повышение дозы соединения I) серии образцов крови для определения соединения формулы 1 собирали в следующие моменты времени:
Цикл 1, День-7 (C1D-7, соединение I, однократная доза).
Цикл 1, День 1 (C1D1, соединение I плюс темозоломид, однократная доза).
Цикл 1, День 4 (C1D4, соединение I плюс темозоломид, многократная доза).
Анализ фармакокинетики проводили по предварительным промежуточным данным используя расчетные времена забора.
В. Анализ фармакокинетики во всех циклах вплоть до Дня 4
Определение соединения формулы 1 в плазме крови человека проводили посредством экстракции белкового осадка, с последующим определением посредством ВЭЖХ с обращенной фазой и тандемной масс-спектрометрией. Использовались следующие условия хроматографии.
Аналитическая колонка: Thermo Hypersil Keystone Betabasic C8, 5 мкм, 100×2,1 мм (внутренний диаметр).
Композиция подвижной фазы A: 0,1% муравьиной кислоты в воде.
Композиция подвижной фазы В: 0,1% муравьиной кислоты в ацетонитриле.
Скорость потока: 200 мкл/мин.
Объем инъекции: 10 мкл.
Промывка иглы автоматического пробоотборника:вода:ацетонитрил:муравьиная кислота (500:500:1, об.:об.:об.).
Типичное время удерживания*: соединение формулы 1:1,5 минуты;
d6 - соединение формулы 1:1,5 минуты;
*время удерживания приблизительно, и может различаться в пределах аналитических серий.
Следующие типичные параметры масс спектрометрии могут изменяться в зависимости от прибора для получения эквивалентного ответа.
Масс-спектрометр: Sciex API 365.
Ионизация: Sciex Turbo Ion Spray.
Турбоионораспылитель: режим положительно зараженных ионов.
Вольтаж ионораспыления: 4000 V.
Температура турбонагревателя: 450°C.
Преобразования масс (номинально): соединение формулы 1: отношение массы к заряду=324,4 -> отношение массы к заряду 293,2.
d6 - соединение формулы 1: отношение массы к заряду=330,3→ отношение массы к заряду=299,1.
Время выдержки: соединение формулы 1: 350 мс.
d6-соединение формулы 1: 150 мс.
Давление газа в распылителе: 6.
Параметр газовой заслонки: 8.
CAD: 2.
Обобщенные предварительные параметры фармакокинетики у всех 17 пациентов представлены в таблице 17 и среднее значение концентрации в плазме крови в зависимости от времени соединения формулы 1 для каждой категории доз представлены на фиг.3.
Таблица 17
Обобщенные предварительные фармакокинетические (среднее (CV%)) соединения формулы 1 после 30-минутной внутривенной инфузии соединения I отдельно (C1 D-7), или соединения I плюс темозоломид перорально 100 мг/м2 (C1 D1 и C1 D4)
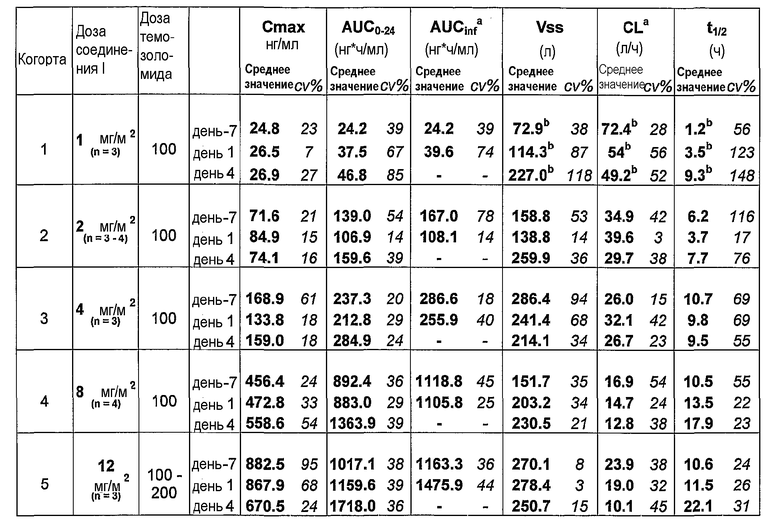
аAUC0-inf и CL могут и не отражать точно экстраполирование AUC0-inf был >20% от AUC0-24 для некоторых пациентов.
бне включено в статистический анализ. Значение может быть некорректно оценено по причине недостаточности данных.
В.1. Анализ фармакокинетики в цикле 1, день -7 (C1D-7)
После внутривенной инъекции отдельно соединения I в течение 30 минут (C1 D-7) концентрации соединения формулы 1 в плазме уменьшались в мультиэкспоненциальной манере со средним значением конечного периода полувыведения приблизительно 6,2-10,7 часов. При введении соединения I между 2 и 12 мг/м2 в виде внутривенной инфузии в течение 30 минут наблюдалась линейная пропорциональность доз в AUC(0-24) и Cmax. Значение AUC0-24 при 1 мг/м2 не включали в оценку пропорциональности дозы в силу того, что концентрации были ниже предела аналитического исследования (LLOQ=2 нг/мл) у всех пациентов после 3 часов введения дозированного средства. Среднее значение общего клиренса организма составляло 27 л/ч (C1D-7), что составляет приблизительно 30% печеночного кровотока. Среднее значения распределения объема в стационарном состоянии составляло 197 л (C1D-7).
В.2. Анализ фармакокинетики в цикле 1, день 1 (C1D1)
После однократного приема пероральной дозы 100 мг/м2 темозоломида и однократных доз от 2 до 12 мг/м2 соединения I концентрации соединения формулы 1 были схожи с концентрациями соединения I, применяемого
отдельно. Значение AUC0-24 соединения формулы 1 на C1D-7 (соединение I отдельно) было сравнимо с таковым на C1D1 (соединение I плюс темозоломид) при всех дозах.
В.3. Анализ фармакокинетики в цикле 1, день 4 (C1D4)
После 4 дней ежедневного приема соединения I плюс темозоломид наблюдалась минимальная аккумуляция соединения формулы 1 в плазме, исходя из визуального контроля индивидуальных значений концентрации в плазме крови в зависимости от времени. Однако имелась тенденция к увеличению (диапазон: 50% до 75%) значения AUC(0-24) соединения формулы 1 между дозой в день 4, цикл 1 и дозой в день 1, цикл 1 (Таблица 17).
В.4. Вариабельность между пациентами из разных когорт и у пациентов внутри одной когорты
Вариабельность AUC(0-24) для соединения формулы 1 между пациентами из разных когорт составляла от 14% до 85%, а вариабельность Cmax составляла от 7% до 95%. Однако вариабельность между пациентами в пределах каждой когорты в целом была <60% и для AUC(0-24) и для Cmax (Таблица 17). Значения вариабельности AUC(0-24) и Cmax у пациентов оценивали посредством сравнения приема соединения I отдельно на C1D-7 с приемом соединения I + темозоломид на C1D1. Вариабельность у пациентов внутри когорты колебалась от 7% до 47 % для AUC(0-24) и от 3% до 44% для Cmax.
C. Определение дозы, ингибирующей PARP: методика исследования
При фармакологическом исследовании активирования и ингибирования PARP использовали моноклональные антитела для определения количества полимера PAR, которое образуется при установленных условиях в лимфоцитах периферической крови с нарушенной проницаемостью мембраны, и в образцах гомогенизированной опухоли. Это количество образовавшегося полимера может быть использовано для установления соотношения активности PARP, таким образом, уменьшение образование полимера коррелирует со степенью ингибирования PARP. Активность PARP выражена как процентное содержание исходного уровня, и она вычисляется путем деления количества полимера PAR, образовавшегося после инфузии, на количество, образовавшееся до инфузии. Осуществимость данного метода была успешно протестирована на 12 пациентах на этапе 2 исследования темозоломида в качестве единственного агента среди пациентов с метастатической меланомой. Это исследование показало, что темозоломид в качестве единственного агента не ингибирует активность PARP ни в лимфоцитах периферической крови, ни в пунктатах опухоли.
D. Фармакодинамика, определенная на этапе 1 клинического исследования
На этапе 1 открытого исследования с повышением дозы внутривенного введения соединения I в комбинации с темозоломидом одной из первоочередных задач было определение PID соединения I; данная PID была определена как доза, при которой активность PARP в лимфоцитах периферической крови была снижена до менее 50% от исходного уровня, и имелось плато (+/- 10 абсолютных %) степени ингибирования PARP между 2 уровнями доз соединения I. Оценка базировалась на активности PARP, наблюдаемой через 24 часа после введения соединения I в День 1.
Используя фармакодинамическое исследование активности PARP, раскрытое выше в разделе С, на этапе 1 клинического испытания было определено ингибирование PARP
в лимфоцитах периферической крови и ткани опухоли. Как указано выше, количество пациентов, принимавших участие в части 2 этапа 1 испытания, было ограничено пациентами, имеющими метастатическую меланому с возможностью взятия биопсии. От всех пациентов требовалось информированное согласие на проведение биопсии до и после лечения, так что активность PARP в опухоли могла быть оценена.
На этапе 1 исследования образцы цельной крови всех пациентов собирали в день, когда вводили пробную дозу соединения I (обычно День -7), и в День 1 и 4. Время момента забора крови выбирали до инфузии соединения I, в конце инфузии, 4-6 часов после инфузии и 24 часа после инфузии (перед инфузией следующего дня). Соединение I вводили посредством внутривенной инфузии в течение 30 минут. Лимфоциты периферической крови получали из образцов крови, и по возможности, образцы анализировали трижды.
В таблице 18 суммированы активности PARP в лимфоцитах периферической крови после применения соединения I в День 1. Заметное ингибирование PARP (по меньшей мере, уменьшение на 50% медианной активности PARP) показано у всех пациентов вне зависимости от дозы после завершения 30-минутной инфузии соединения I в День 1. В зависимости от пациента, максимальное ингибирование наблюдали в момент времени 0,5 или 4-6 часов. Длительное ингибирование активности PARP показано у всех пациентов в когортах 4 и 5, где наблюдали >50% уменьшение медианной активности PARP через 24 часа после введения дозы соединения I в День 1.
Как показано в таблице 19, активность PARP была ингибирована на 50%-93% в образцах опухоли, полученных от 6 пациентов, входящих в часть 2 исследования, через 4-6 часов после введения соединения I в дозе 12 мг/м2 в День 1, 4 или 5 первого цикла лечения.
Активность PARP через 0,5, 4-6, и 24 часов после введения соединения I в День 1
4-6
24
2
2
57
77
BLD-114
40-114
4-6
24
3
2
27
36
15-62
22-48
4-6
24
3
3
28
108
22-47
50-264
4-6
24
3
3
16
9
5-30
7-31
4-6
24
12
12
13
29
BLD-114
3-73
4-6
24
6
6
7,5
14
0-21
0-34
атолько для первой когорты, образцы не были собраны в День 1. Данные, полученные с использованием образцов, собранных после пробной дозы соединения I (обычно День-7).
Ингибирование PARP в опухолях
Пример 11. Эффекты на организм человека: фазы 1/2 испытания внутривенного введения соединения I ингибитора PARP в комбинации с режимом «FOLFIRI» у пациентов с запущенным колоректальным раком, у которых применение режима «FOLFOX» на первой линии терапии метастатической стадии было неэффективным.
В водной части фазы 1 исследования устанавливают дозу соединения I в комбинации с иринотеканом, 5-FU и лейковорином, которую использовали на этапе фазы 2. Этап фазы 2 представляет собой открытое мультицентровое исследование соединения I, применяемого в комбинации с режимом FOLFIRI у пациентов, которые ранее получали режим FOLFOX химиотерапии на первой линии терапии метастатического колоректального рака.
Этап фазы 2 данного испытания разделен на 2 части. Часть 1 представляет собой открытое исследование с повышением дозы, в котором оценивается безопасность и иммунность комбинации соединения I с иринотеканом (см. таблицу 20 - часть 1). В части 2 к комбинации, уже созданной в части 1, добавляют 5-FU + лейковорин (см. таблицу 20 - часть 2). Пациентам вводили дозы 2-недельными циклами для облегчения перехода к схеме дозировки FOLFIRI фазы 2. Пациенты имели гистологически или цитологически подтвержденное наличие колоректального рака, который был невосприимчив к или для которого было неэффективным применение режима FOLFOX в первой линии терапии метастатической стадии, возраст, по меньшей мере, 18 лет, были работоспособны (ВОЗ 0 или 1), имели подходящие функции костного мозга, печени и почек, как определено в стандартных исследованиях крови, дали информированное согласие, а также удовлетворяли некоторым другим критериям включения в исследование. Пациенты получали соединение I в дозе, ингибирующей PARP (как определено ранее в фазе 1 исследования и части 1 этого испытания) и FOLFIRI.
В фазе 2 количество циклов относится к каждому 2-недельному циклу FOLFIRI, который применяется в стандартном режиме. Иринотекан (доза исходит из фазы 1) применяется внутривенно в течение 90 минут в День 1. Инфузию лейковорина (LV 200 мг/м2) начинают одновременно с иринотеканом и продолжают в течение 2 часов в День 1. Болюс 5-FU (400 мг/м2) и 46-часовую инфузию 5-FU (2400 мг/м2) начинали немедленно после инфузии лейковорина. Соединение I добавляли к иринотекану в возрастающей дозе в серийных когортах пациентов, как показано в таблице 20. Соединение I сначала вводили в начальной дозе 12 мг/м2, вводимой посредством 30-минутной внутривенной инфузии за 1 час до каждой дозы иринотекана и еще раз 24 часа спустя. Начальная доза иринотекана составляет 150 мг/м2 (около 80% от полной дозы иринотекана, применяемой в режиме FOLFIRI). Образцы крови собирали в цикле 1 для определения профилей фармакокинетики соединения I, иринотекана и SN-38.
Предельная токсичность дозы (DLT) используется для определения максимальной переносимой дозы (MTD) и оценивается в первые 4 недели. Первоначально по 3 пациента были включены в каждый уровень дозировки. Если DLT наблюдали у 1 из первых 3 пациентов в любой когорте, вводились дополнительные 3 пациента. MTD определяется как наивысший уровень дозы, при котором ≤1/6 пациентов испытывают DLT в течение первых 4 недель. Дозу соединения I не поднимали выше 18 мг/м2. Как только 4 недели лечения 6 пациентов с использованием MTD заканчиваются, начинается стадия фазы 2. MTD определяется как доза, ниже которой более чем 30% (2 из 6 пациентов) когорты испытывает предельную токсичность дозы в результате комбинации дозированных средств в течение первых 21 дня лечения. Заменяют пациентов, которые не выдерживают время, необходимое для определения DLT по любой причине, кроме связанной с токсичностью лечения. MTD является рекомендованной начальной дозой в фазе 2 испытаний.
Дозы фазы 1
Объективным показателем эффективности дозированного средства является первичная критическая точка в фазе 2 исследования. Канцерогенный эффект оценивается каждые 3 цикла FOLFIRI. Объективный показатель эффективности (RR) комбинации соединения I и FOLFIRI определяется с помощью критериев оценки ответной реакции солидных опухолей (RECIST). Therasse et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst, 2000, v. 92, pp. 205-216.
Пример 12. Радиосенсибилизация посредством соединения формулы 1
Предрасполагающим фактором к устойчивости к радиации in vivo является способность покоящихся клеток восстанавливаться после потенциально летального повреждения (PLD) (Weichselbaum, R.R. and Little, J.B. The differential response of human tumors to fractionated radiation may be due to a post-irradiation repair process. Br. J. Cancer 1982; 46: 532-537). В модели in vitro измеряют повышение выживаемости облученных с подавленным ростом клеток после отсроченного посева для колониеобразования. Восстановление после потенциально летального повреждения измеряли in vitro, используя клетки LoVo, у которых был подавлена фаза G1, посредством увеличения слияния для имитирования популяции радиорезистентных покоящихся клеток в опухолях. Клетки подвергали воздействию γ-облучения в дозе 8 Гр (Gammacel 1000 Elite, Nordian International Inc. Canada), и либо немедленно собирали и высевали для анализа колониеобразования, либо поддерживали в виде конфлюэнтных культур с подавленным ростом в течение 24-часового восстановительного периода перед сбором и высеванием для анализа колониеобразования. Где указано, 0,4 мкM соединения формулы 1 добавляли за 30 минут до облучения и имелось в наличии на протяжении восстановительного инкубирования. Как показано в таблице 21, выживаемость клеток увеличивалась приблизительно в 7 раз после 24 ч восстановления в регулирующей среде. Инкубирование с соединением формулы 1 в течение восстановительного периода ингибировало восстановление PLD на 64,9%.
Ингибирование восстановления после потенциально летального повреждения (PLDR)
б% ингибирование восстановления подсчитан как 100-((PLDR в присутствии соединения формулы 1/PLDR контроля)×100)
всреднее значение в 3 независимых экспериментах
Эффективность in vivo соединения формулы 1 в качестве радиопотенцирующего агента оценивали, используя два независимых подхода: ex-vivo клоногенный анализ и анализ отсроченного опухолевого роста. Для первого подхода адаптированные LoVo ксенотрансплантанты обрабатывали соединением формулы 1 (15 или 30 мг/кг; исходное соединение) 30 минут перед опухоль-ограниченным облучением в дозе 5 Гр. Через 24 часа опухоль удаляли, размельчали для получения одноклеточных суспензий и высевали на чашку Петри для анализа колониеобразования. Как показано в таблице 22, выжившая фракция (SF) опухолевых клеток, обработанных соединением формулы 1 и 5 Гр, была увеличена по сравнению с применением только облучения. SF для 15 мг/кг и 30 мг/кг IR комбинаций эквивалентна той, которую можно было бы достичь, применяя дозы облучения 8 Гр или 9 Гр, допускающие факторы изменения дозы (DMF) 1,6 и 1,9, соответственно.
Ex-vivo радиопотенцирование посредством соединения формулы 1
бвыжившая фракция: эффективность колониеобразования (CFE) в качестве функции необработанных контрольных опухолей
вфактор изменения дозы: кратность увеличения дозы облучения, которая требовалась бы для обеспечения такого же уровня клоногенной выживаемости, как комбинация формулы 1 и облучения.
При исследовании отсроченного опухолевого роста LoVo ксенотрансплантаты объемом приблизительно 250 мм3 обрабатывали дозой облучения 10 Гр, применяемой дробно по 2 Гр один раз в день в течение 5 дней. В комбинированных группах, соединение формулы 1 вводили за 30 минут перед каждой обработкой/дозой 2 Гр либо в дозе 15 мг/кг, либо в дозе 30 мг/кг (снова использовали исходное соединение). Экспериментальная конечная точка определялась как время, необходимое для увеличения относительного объема опухоли до четырехкратного объема, определенного перед началом обработки (RTV4). Задержки роста высчитывали по разнице во времени, потребовавшемся для достижения RTV4 (дни) между опухолями, обработанными IR/формула 1 и необработанными контролями. Как показано в таблице 23, обе дозировки соединения формулы 1 приводили к значительному (36%) повышению действия облучения в отношении LoVo ксенотрансплантатов.
Эффективность облучения рентгеновскими лучами в комбинации с соединением формулы 1 в отношении LoVo ксенотрансплантатов
бдозы соединения формулы, доставленные посредством внутривенной инъекции.
вn=5 во всех группах.
гувеличение, подсчитанное как % увеличения=100×(задержка роста при использовании IR+соединение формулы 1-задержка роста при использовании IR отдельно)/задержка роста при использовании IR отдельно.
дзначительно отличающийся от применения IR отдельно p=0,015 (критерий Манна-Уитни)
езначительно отличающийся от применения IR отдельно p=0,009 (критерий Манна-Уитни)
Описания всех цитируемых здесь документов включены здесь в виде ссылки в их полном объеме.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2018 |
|
RU2767664C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2018 |
|
RU2757373C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2011 |
|
RU2743643C2 |
| КОМБИНИРОВАННАЯ ПРОТИВОРАКОВАЯ ТЕРАПИЯ | 2011 |
|
RU2607596C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ПРОТИВООПУХОЛЕВЫМ АЛКАЛОИДОМ | 2011 |
|
RU2605335C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ПРИМЕНЕНИЕМ ЛИПОСОМАЛЬНОГО ИРИНОТЕКАНА И ИНГИБИТОРА PARP ДЛЯ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2760185C2 |
| ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ 2-{4-[(3S)-ПИПЕРИДИН-3-ИЛ]ФЕНИЛ}-2Н-ИНДАЗОЛ-7-КАРБОКСАМИДА | 2009 |
|
RU2495035C2 |
| ПРОТИВООПУХОЛЕВЫЕ КОМБИНАЦИИ С ИНГИБИТОРАМИ MTOR, ГЕРЦЕПТИНОМ И/ИЛИ HKI-272 | 2006 |
|
RU2451524C2 |
| ЛЕЧЕНИЕ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ С ПОМОЩЬЮ СОЕДИНЕНИЯ 4-ИОД-3-НИТРОБЕНЗАМИД В КОМБИНАЦИИ С ПРОТИВООПУХОЛЕВЫМИ СРЕДСТВАМИ | 2008 |
|
RU2480211C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ ИНГИБИТОРА ГИСТОНДИАЦЕТИЛАЗЫ В КОМБИНАЦИИ С БЕНДАМУТИНОМ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2609833C2 |
Изобретение относится к лекарственным средствам и касается дозированной формы для введения млекопитающему, предназначенной для лечения или профилактики болезненного состояния у млекопитающего, опосредованного действием PARP, где дозированная форма содержит соединение формулы 1, его фармацевтически приемлемую соль или сольват, или их смеси, в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 часов после введения в организм млекопитающего, которая является концентрацией ингибитора, подавляющей на 89% (IC89) потребление клеточного NAD+и образование полимера поли АДФ-рибозы, и скорректирована с учетом связывания с белком. Предложенная дозированная форма обеспечивает стабильную величину концентрации в плазме 5,9 нг/мл AG-014699 в течение, по меньшей мере, 24 часов после введения в организм млекопитающего. 5 н. и 9 з.п. ф-лы, 4 ил., 23 табл.
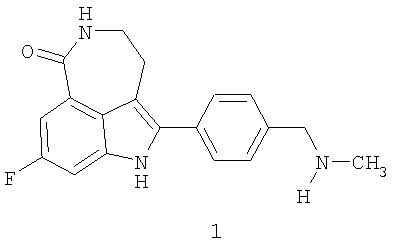
1. Дозированная форма для введения млекопитающему, предназначенная для лечения или профилактики болезненного состояния у млекопитающего, опосредованного действием PARP, где дозированная форма содержит соединение формулы 1:
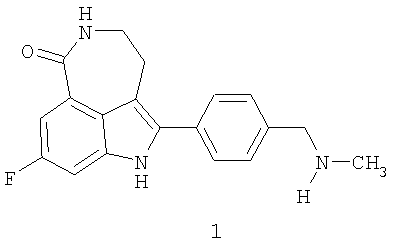
его фармацевтически приемлемую соль или сольват, или их смеси, в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 ч после введения в организм млекопитающего, которая является концентрацией ингибитора, подавляющей на 89% (IC89) потребление клеточного NAD+ и образование полимера полиАДФ-рибозы, и скорректирована с учетом связывания с белком.
2. Дозированная форма по п.1, где дозированная форма представляет собой лиофилизированный порошок для инъекций.
3. Дозированная форма по п.1, где фармацевтически приемлемой солью является фосфатная соль.
4. Дозированная форма для введения млекопитающему, предназначенная для лечения или профилактики болезненного состояния у млекопитающего, опосредованного действием PARP, где дозированная форма содержит соединение формулы 1:
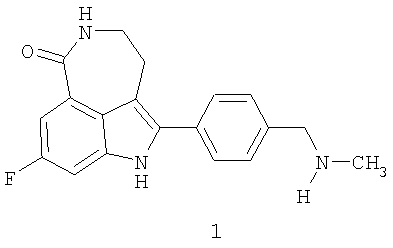
его фармацевтически приемлемую соль или сольват, или их смесь, в количестве, эффективном для ингибирования в лимфоцитах периферической крови фермента поли(АДФ-рибоза)полимеразы, по меньшей мере, на 50% в течение, по меньшей мере, 24 ч, по результатам исследования ингибирования PARP, после введения в организм млекопитающего.
5. Дозированная форма по п.4, где ингибирование фермента поли(АДФ-рибоза)полимеразы составляет, по меньшей мере, на 75%, по результатам исследования ингибирования PARP.
6. Дозированная форма по п.4, где дозированная форма представляет собой лиофилизированный порошок для инъекций.
7. Дозированная форма по п.4, где фармацевтически приемлемой солью является фосфатная соль.
8. Способ лечения рака у млекопитающего, где способ включает введение млекопитающему
(а) соединения формулы 1:
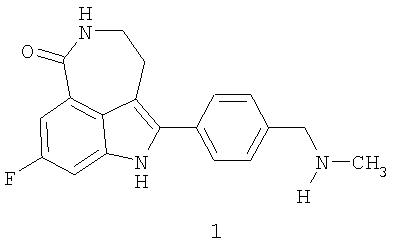
его фармацевтически приемлемой соли или сольвата, или их смеси в количестве, эффективном для обеспечения стабильной величины концентрации соединения формулы 1 в плазме, по меньшей мере, 5,9 нг/мл в течение, по меньшей мере, 24 ч после введения млекопитающему, которая является концентрацией ингибитора, подавляющей на 89% (IC89) потребление клеточного NAD+ и образование полимера полиАДФ-рибозы, и скорректирована с учетом связывания с белком; и
(б) терапевтически эффективного количества одного противоопухолевого средства, выбранного из группы, состоящей из темозоломида, иринотекана, топотекана, цисплатина, карбоплатина, доксорубицина и 5-фторурацила.
9. Способ по п.8, где противоопухолевое средство вводят в пределах одного часа после введения соединения формулы 1.
10. Способ по п.8, где терапевтически эффективное количество темозоломида составляет от 3 до 6 мг/кг.
11. Набор для лечения рака у млекопитающего, включающий:
(а) количество соединения формулы 1
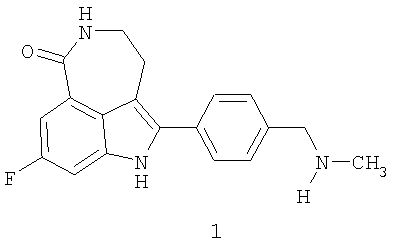
его фармацевтически приемлемой соли или сольвата, или их комбинации, и фармацевтически приемлемый носитель или разбавитель в первой единичной дозированной форме;
(б) количество одного противоопухолевого средства, выбранного из группы, состоящей из темозоломида, иринотекана, топотекана, цисплатина, карбоплатина, доксорубицина и 5-фторурацила, и фармацевтически приемлемый носитель или разбавитель в, по меньшей мере, второй единичной дозированной форме; и
(с) контейнер для содержания первой и второй дозированных форм;
где количество соединения формулы 1 соответствует количеству, эффективному для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 ч после введения в организм млекопитающего.
12. Набор по п.11, где дозированная форма в первой единичной дозированной форме представляет собой лиофилизированный порошок для инъекций.
13. Способ по п.8, где стадия (б) включает введение комбинации иринотекана, 5-фторурацила и лейковорина.
14. Способ лечения рака у млекопитающего, где способ включает введение млекопитающему
(а) соединения формулы 1:
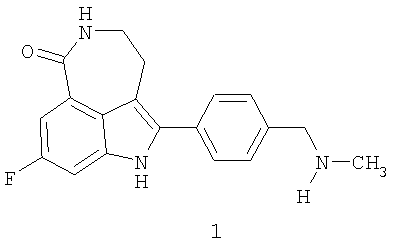
его фармацевтически приемлемой соли или сольвата, или их смеси в количестве, эффективном для обеспечения стабильной величины концентрации в плазме, по меньшей мере, 5,9 нг/мл соединения формулы 1 в течение, по меньшей мере, 24 ч после введения млекопитающему, которая является концентрацией ингибитора, подавляющей на 89% (IC89) потребление клеточного NAD+ и образование полимера полиАДФ-рибозы, и скорректирована с учетом связывания с белком; и
(б) дозы облучения, эффективной для уничтожения рака.
| ПОДДОННИК ДЛЯ ФИЛЬТР-ПРЕССОВ | 1934 |
|
SU42040A1 |
| Гидравлический рулевой привод для транспортной машины | 1970 |
|
SU525584A1 |
| WO 9959975 A1, 25.11.1999. | |||

