ЗАЯВКА НА ПРИОРИТЕТ
Настоящая заявка заявляет приоритет согласно патенту США № 61/895472, поданного 25 октября 2013, и патенту США № 61/927782, поданного 15 января 2014, каждый из которых включен в данное описание посредством ссылки во всей своей полноте.
УРОВЕНЬ ТЕХНИКИ
Рецептор 4 фактора роста фибробластов (FGFR-4) представляет собой белок, который у людей кодируется геном FGFR-4. Этот белок является членом семейства рецепторов фактора роста фибробластов, где аминокислотная последовательность членов в ходе эволюции оставалась высококонсервативной. Члены 1-4 семейства FGFR отличаются друг от друга сродством их лигандов и распределением в тканях. Типичный непроцессированный белок состоит из внеклеточной области, состоящей из трех иммуноглобулин-подобных доменов, одного гидрофобного трансмембранного сегмента и цитоплазматического домена тирозинкиназы. Внеклеточная часть белка взаимодействует с факторами роста фибробластов, приводя в движение каскад нисходящих сигналов, в конечном счете, влияющих на митогенез и дифференциацию. Геномная организация гена FGFR4 охватывает 18 экзонов. Несмотря на то, что наблюдается альтернативный сплайсинг, нет никаких доказательств того, что С-концевая половина домена IgIII этого белка варьируется между тремя альтернативными формами, как указано для FGFR 1-3.
У крыс, получавших ингибитор FGFR-1, была обнаружена эктопическая минерализация, характеризующаяся неадекватным фосфорно-кальциевым отложением в мягких тканях (Brown, AP et al. (2005), Toxicol. Pathol., p. 449-455). Это говорит о том, что селективное ингибирование FGFR-4 без ингибирования других изоформ FGFR, в том числе FGFR-1, может быть желательным для того, чтобы избежать некоторых токсичностей. FGFR-4 преимущественно связывает фактор роста фибробластов 19 (FGF19) и в последнее время связывается с прогрессированием некоторых сарком, почечно-клеточного рака, рака молочной железы и рака печени.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фигура 1 представляет собой график, отражающий ингибирование роста групп, получающих Соединение 27, против ксенотрансплантатных опухолей Hep3B у бестимусных мышей.
Фигура 2 представляет собой график, отражающий изменение массы тела (%) несущих Нер3В бестимусных мышей в течение периода исследования.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение описывает ингибиторы FGFR-4. Данное изобретение дополнительно описывает фармацевтические составы, содержащие ингибитор FGFR-4.
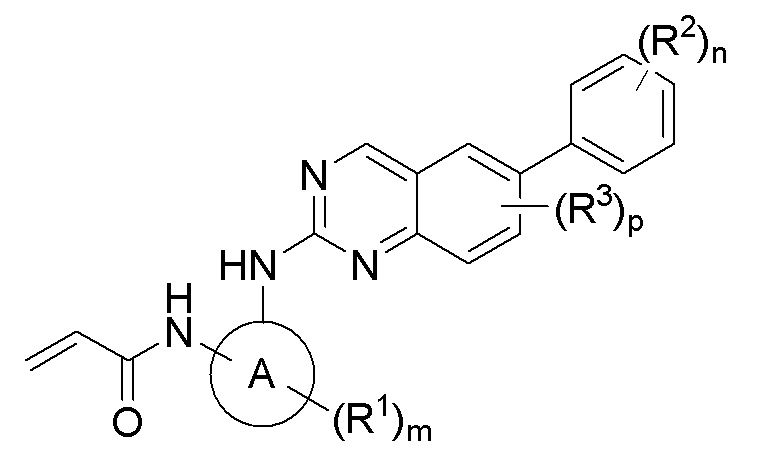
В одном аспекте данное изобретение предлагает соединение Формулы I или его фармацевтически приемлемую соль:

Формула I
где
головная часть представляет собой функциональную группу, способную образовывать ковалентную связь с нуклеофилом;
кольцо A представляет собой 3-8-членный моноциклический или бициклический циклоалкил или гетероциклил;
каждый из R1 и R2 независимо представляет собой галоген, циано, C1-6 алкокси, гидрокси, оксо, амино, амидо, сульфонил, сульфонамидо, сложный эфир, алкилмочевину, C1-6 алкил, -C(O)O-, -C(O)-C1-6 алкил, -C(O)-C1-6 алкиламино, C1-6 гетероалкил, гетероциклил или гетероциклилалкил, где каждый из C1-6 алкокси, амино, амидо, сульфонамидо, сложного эфира, алкилмочевины, C1-6 алкила, C1-6 гетероалкила, гетероциклила или гетероциклилалкила независимо замещен 0-5 случаями R4;
R3 представляет собой галоген;
каждый R4 независимо выбран из C1-6 алкила, C1-6 алкокси, галогена, гидрокси, оксо, амино, циано, циклоалкила и гетероциклила;
m равен 0-3;
n равен 0-4; и
p равен 0-2.
В некоторых вариантах реализации изобретения кольцо А представляет собой моноциклический циклоалкил. В некоторых вариантах реализации изобретения кольцо А представляет собой циклобутил, циклопентил или циклогексил. В некоторых вариантах реализации изобретения R3 независимо представляет собой галоген.
В некоторых вариантах реализации изобретения кольцо А представляет собой бициклический циклоалкил.
В некоторых вариантах реализации изобретения кольцо А представляет собой гетероциклил. В некоторых вариантах реализации изобретения кольцо А представляет собой пирролидинил, пиперидинил, тетрагидрофуранил или тетрагидропиранил. В некоторых вариантах реализации изобретения R3 независимо представляет собой галоген.
В другом аспекте, данное изобретение предлагает соединение Формулы II или его фармацевтически приемлемую соль:
Формула II
где
кольцо A представляет собой 3-6-членный циклоалкил или гетероциклил;
R1 независимо представляет собой галоген, циано, C1-6 алкокси, гидрокси, оксо, амино, амидо, сульфонил, сульфонамидо, сложный эфир, алкилмочевину, C1-6 алкил, -C(O)O-, -C(O)-C1-6 алкил, -C(O)-C1-6 алкиламино или C1-6 гетероалкил;
R2 представляет собой галоген или C1-6 алкокси;
R3 представляет собой галоген; и
m равен 0-1; n равен 0-4; и p равен 0-1.
В некоторых вариантах реализации изобретения кольцо А представляет собой циклоалкил.
В некоторых вариантах реализации изобретения кольцо А представляет собой гетероциклил. В некоторых вариантах реализации изобретения R3 независимо представляет собой галоген.
В некоторых вариантах реализации изобретения кольцо А представляет собой циклобутил, циклопентил, циклогексил, пирролидинил, пиперидинил, тетрагидрофуранил или тетрагидропиранил.
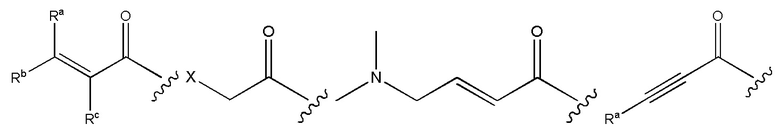
В соединениях, описанных в данном документе, головная часть представляет собой функциональную группу, которая вступает в реакцию с нуклеофилом, например, способную образовывать ковалентную связь с нуклеофилом. Примеры головных частей включают, но не ограничиваясь этим, галоидные алкилы, алкилсульфонаты, гетероарильные галиды, эпоксиды, галогенацетамиды, малеимиды, эфиры сульфокислоты, альфа-бета-ненасыщенные кетоны, альфа-бета-ненасыщенные сложные эфиры, винилсульфоны, пропаргиловые амиды, акриламиды. В некоторых из этих случаев, например, акриламида и пропаргилового амида, в формулах, приведенных выше, N головной части является соседним по отношению к N. Структуры типовых головных частей приведены ниже:


где X представляет собой уходящую группу, такую как галоген, или активированную гидроксильную функциональную группу (например, трифлат); и каждый из Ra, Rb и Rc независимо представляет собой H, замещенный или незамещенный C1-4 алкил, замещенный или незамещенный C1-4 циклоалкил или циано.
В формулах, приведенных выше, головные части, как правило, присоединены к атому N на ингибиторе. В других вариантах реализации изобретения головная часть в качестве альтернативы может быть присоединена к атому, отличному от N. Примеры типовых головных частей включают, но не ограничиваясь этим,


Другие примеры головных частей могут быть обнаружены, например, в WO 2010/028236 и WO 2011/034907.
В некоторых вариантах реализации изобретения ингибиторы FGFR-4 в соответствии с данным изобретением ингибируют активность FGFR-4 более эффективно, чем они ингибируют активность FGFR-1. Например, ингибиторы FGFR-4 в соответствии с данным изобретением могут ингибировать активность FGFR-4 в по меньшей мере 10 раз, по меньшей мере 50 раз, по меньшей мере 100 раз, по меньшей мере 200 раз или по меньшей мере 500 раз более эффективно, чем они ингибируют активность FGFR-1.
В одном аспекте, селективность измеряют путем сравнения ингибирования FGFR-1 и FGFR-4, вызванного соединением по данному изобретению, в том же типе анализа. В одном варианте реализации изобретения анализы, применяемые для измерения ингибирования FGFR-1 и FGFR-4, представляют собой любые описанные в данном документе анализы. Как правило, ингибирование выражают в виде IC50 (концентрация ингибитора, при которой ингибируется 50% активности фермента) и, таким образом, кратную селективность измеряют с помощью уравнения:
(IC50 FGFR-1)/ (IC50 FGFR-4). Те же измерения и расчеты могут быть также применены для измерения селективности по FGFR-2 и FGFR-3.
Для определения относительного ингибирования FGFR-1 и FGFR-4 с помощью соединений по данному изобретению могут быть применены любые другие анализы активности FGFR, поскольку такие анализы применяют то, что специалист в данной области техники может полагать, что параметры являются такими же, что и при измерении активности FGFR.
В другом аспекте, изобретение предлагает фармацевтическую композицию, содержащую фармацевтически приемлемый носитель и соединение, описанное в данном документе.
В другом аспекте изобретение предлагает способ лечения состояния, медиированного с помощью FGFR-4, состояния, характеризующегося чрезмерной экспрессией FGFR-4, состояния, характеризующегося амплификацией FGFR4, состояния, медиированного с помощью FGF19, состояния, характеризующегося амплификацией FGF-19, или состояния, характеризующегося чрезмерной экспрессией FGF19, любой из этих способов включает введение субъекту терапевтически эффективного количества соединения, описанного в данном документе.
В другом аспекте, изобретение предлагает способ лечения любого из следующих состояний с помощью введения субъекту терапевтически эффективного количества соединения, описанного в данном документе: гепатоклеточной карциномы, рака молочной железы, рака яичника, рака легких, рака печени, саркомы или гиперлипидемии.
Изобретение включает все возможные комбинации вариантов реализации изобретения, описанных выше и ниже.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Соединения, описанные ниже, могут образовывать ковалентную связь с белком FGFR4; например, соединения могут образовывать ковалентную связь с остатком цистеина FGFR4, например, цистеина в остатке 552. FGFR1-3 не содержат этот цистеин. Возможность образовывать ковалентную связь между соединением и FGFR4, следовательно, является важным фактором в селективности соединений, описанных в данном документе в случае FGFR4.
Подробности устройства и размещения компонентов, изложенные в следующем описании или проиллюстрированные на фигурах, не являются ограничивающими. Другие варианты реализации изобретения и различные пути практического применения изобретения включены явным образом. Кроме того, фразеология и терминология, применяемые в настоящем документе, приведены с целью описания и не должны интерпретироваться как ограничивающие. Применение в данном документе слов «включающий», «включает», «включать», «содержит» или «имеющий», «содержащий», «принимающий участие» и их вариаций обозначает включение элементов последующего списка и их эквивалентов, а также дополнительных элементов.
Определения
«Алифатическая группа», как применяют в данном документе, обозначает неразветвленную, разветвленную или циклическую углеводородную группу и содержит замещенные и незамещенные группы, такие как алкильная группа, алкильная группа и алкинильная группа.
«Алкенил», как применяют в данном документе, обозначает алифатическую группу, содержащую по меньшей мере одну двойную связь.
«Алкоксил» или «алкокси», как применяют в данном документе, обозначает алкильную группу, имеющую присоединенный к ней кислородный радикал. Типичные алкоксильные группы включают метокси, этокси, пропилокси, трет-бутокси и тому подобное.
«Алкил» обозначает моновалентный радикал замещенного неразветвленного или разветвленного углеводорода, такого как неразветвленная или разветвленная группа из 1 12, 1 10 или 1 6 атомов углерода, упоминаемая в данном документе как C1 C12 алкил, C1 C10 алкил и C1 C6 алкил, соответственно. Типовые алкил группы включают, но не ограничиваясь этим, метил, этил, пропил, изопропил, 2 метил 1 пропил, 2 метил 2 пропил, 2 метил 1 бутил, 3 метил 1 бутил, 2 метил 3 бутил, 2,2 диметил 1 пропил, 2 метил 1 пентил, 3 метил 1 пентил, 4 метил 1 пентил, 2 метил 2 пентил, 3 метил 2 пентил, 4 метил 2 пентил, 2,2 диметил 1 бутил, 3,3 диметил 1 бутил, 2 этил 1 бутил, бутил, изобутил, трет бутил, пентил, изопентил, неопентил, гексил, гептил, октил и т.д.
«Алкилен» обозначает дивалентный радикал алкильной группы, например, -CH2-, -CH2CH2- и CH2CH2CH2-.
«Алкинил» обозначает разветвленную или разветвленную углеводородную цепь, содержащую 2-12 атомов углерода и отличающуюся тем, что имеет одну или несколько тройных связей. Примеры алкинильных групп включают, но не ограничиваясь этим, этинил, пропаргил и 3-гексинил. Один из углеродов тройной связи необязательно может быть точкой присоединения алкильного заместителя.
«Алкинилен» обозначает алкинил, имеющий две точки присоединения. Например, «этинилен» представляет группу -C≡C-. Алкиниленовые группы также могут быть в незамещенной форме или замещенной форме с одним или более заместителями.
«Алкилтио», как применяют в данном документе, обозначает гидрокарбильную группу, имеющую присоединенный к нему радикал, содержащий серу. В некоторых вариантах реализации изобретения "алкилтио" функциональная группа представлена одним из -S-алкила, -S-алкенила или -S-алкинила. Типичные алкилтио группы включают метилтио, этилтио и тому подобное.
"Амидо", как применяют в данном документе, обозначает -C(=O)-N(R1)( R2) или
-N(R1)-C(=O)-R2, где каждый из R1 и R2 представляет собой H, алкил, циклоалкил, алкокси или гидрокси.
«Амино», как применяют в данном документе, обозначает -NH2, -NH(алкил) или -N(алкил)(алкил).
«Амплифицированный», как применяют в данном документе, означает дополнительные копии гена или сегмента хромосомы, получаемые в раковых клетках, что может обеспечить преимущество роста или выживания.
«Арилалкил» или «аралкил», как применяют в данном документе, обозначает алкильную группу, замещенную арильной группой (например, ароматической или гетероароматической группой). Аралкил включает группы, в которых более чем один атом водорода замещен арильной группой. Примеры «арилалкила» или «аралкила» включают бензильную, 2-фенилэтильную, 3-фенилпропильную, 9-флуоренильную, бензгидрильную и тритильную группы.
«Арил», как применяют в данном документе, обозначает 5-, 6- и 7-членные однокольцевые ароматические группы, которые могут содержать от ноля до четырех гетероатомов, например, фенил, пирролил, фуранил, тиофенил, имидазолил, оксазолил, тиазолил, триазолил, пиразолил, пиридинил, пиразинил, пиридазинил и пиримидинил и тому подобное. Те арильные группы, которые содержат гетероатомы в кольцевой структуре, могут также называться «арильными гетероциклами» или "гетероароматическими соединениями." Ароматическое кольцо может быть замещено по одной или более позициям в кольце такими заместителями, как описано выше, например, галогеном, азидом, алкилом, аралкилом, алкенилом, алкинилом, циклоалкилом, полициклилом, гидроксилом, алкоксилом, амино, нитро, сульфгидрилом, имино, амидо, фосфатом, фосфонатом, фосфинатом, карбонилом, карбоксилом, силилом, простым эфиром, алкилтио, сульфонилом, сульфонамидо, кетоном, альдегидом, сложным эфиром, гетероциклилом, ароматическими и гетероароматическими функциональными группами, -CF3, -CN или тому подобным. Термин «арил» также включает полициклические кольцевые системы, содержащие два или более циклических колец, в которых два или более углеродов являются общими для двух смежных колец (кольца являются «слитыми кольцами»), причем по меньшей мере одно из колец является ароматическим, например, другие кольца могут быть циклоалкилами, циклоалкенилами, циклоалкинилами, арилами и/или гетероциклилами. Каждое кольцо может содержать, например, 5-7 членов.
«Карбоциклическая кольцевая система», как применяют в данном документе, обозначает моноциклическую, бициклическую или полициклическую углеводородную кольцевую систему, причем каждое кольцо является либо полностью замещенным, либо содержит одно или более ненасыщенных звеньев, но где отсутствует ароматическое кольцо.
«Карбоциклил», как применяют в данном документе, обозначает моновалентный радикал карбоциклической кольцевой системы. Типичные карбоциклильные группы включают циклоалкильные группы (например, циклопентил, циклобутил, циклопентил, циклогексил и тому подобное) и циклоалкенильные группы (например, циклопентенил, циклогексенил, циклопентадиенил и тому подобное).
«Циклоалкил», как применяют в данном документе, обозначает циклическую, бициклическую, трициклическую или полициклическую неароматические углеводородные группы, имеющие 3-12 углеродов. Любой замещаемый атом в кольце может быть замещен (например, одним или более заместителями). Циклоалкильные группы могут содержать слитые или имеющие один общий атом кольца. Слитые кольца представляют собой кольца, которые имеют общий атом углерода. Примеры циклоалкильных функциональных групп содержат, но не ограничиваясь этим, циклопропил, циклогексил, метилциклогексил, адамантил и норборнил.
«Циклоалкилалкил», как применяют в данном документе, обозначает радикал
-(циклоалкил)-алкил, где циклоалкил и алкил являются такими, как описано в данном документе. «Циклоалкилалкил» связан с исходной молекулярной структурой посредством циклоалкильной группы.
«Циано», как применяют в данном документе, обозначает -CN.
«Ковалентный ингибитор», как применяют в данном документе, означает ингибитор, который может образовать ковалентную связь с белком.
«Сложный эфир», как применяют в данном документе, обозначает -C(=O)-O(R1) или -O-C(=O)-R1, R1 представляет собой H или алкил.
«FGFR-4» или «белок FGFR-4» обозначает любую форму белка FGFR-4, в том числе дикого типа и все вариантные формы (в том числе, без ограничения, мутантные формы и сплайс-варианты). Белок FGFR-4 представляет собой продукт гена FGFR-4 и белок FGFR-4, следовательно, содержит любой белок, кодируемый любой формой гена FGFR4, в том числе все аберрации, например, точечные мутации, инсерционно-делеционные мутации, транслокационные слияния и фокальные амплификации.
«Гетероароматическая кольцевая система» является принятой в данной области техники и обозначает моноциклическую, бициклическую или полициклическую кольцевую систему, причем по меньшей мере одно кольцо является и ароматическим, и содержит по меньшей мере один гетероатом (например, N, O или S); и при этом другие кольца не являются гетероциклилом (как определено ниже). В некоторых случаях, кольцо, которое является ароматическим и содержит гетероатом, содержит в таком кольце 1, 2, 3 или 4 кольцевые гетероатома.
«Гетероарил» обозначает моновалентный радикал гетероароматической кольцевой системы. Типичные гетероарильные группы включают кольцевые системы, где (i) каждое кольцо содержит гетероатом и является ароматическим, например, имидазолил, оксазолил, тиазолил, триазолил, пирролил, фуранил, тиофенил пиразолил, пиридинил, пиразинил, пиридазинил, пиримидинил, индолизинил, пуринил, нафтиридинил, пиридо[2,3-d]пиримидин и птеридинил; (ii) каждое кольцо является ароматическим или карбоциклилом, по меньшей мере одно ароматическое кольцо содержит гетероатом и по меньшей мере одно отличное кольцо представляет собой углеводородное кольцо или например, индолил, изоиндолил, бензотиенил, бензофуранил, дибензофуранил, индазолил, бензимидазолил, бензтиазолил, хинолил, изохинолил, циннолинил, фталазилин, хиназолинил, хиноксалинил, карбазолил, акридинил, феназинил, фенотиазинил, феноксазинил, пиридо[2,3 b] 1,4 оксазин 3-(4H) он, 5,6,7,8 тетрагидрохинолинил и 5,6,7,8 тетрагидроизохинолинил; и (iii) каждое кольцо является ароматическим или карбоциклилом и по меньшей мере одно ароматическое кольцо имеет общий мостиковый гетероатом с другим ароматическим кольцом, например, 4H хинолизинилом.
«Гетероциклическая кольцевая система» обозначает моноциклическую, бициклическую и полициклическую кольцевые системы, где по меньшей мере одно кольцо является замещенным или частично незамещенным (но не ароматическим) и содержит по меньшей мере один гетероатом. Гетероциклическая кольцевая система может быть присоединена к ее боковой группе на любом гетероатоме или атоме углерода, что приводит к стабильной структуре, а любой из атомов кольца может быть необязательно замещенным.
«Гетероциклил» обозначает моновалентный радикал гетероциклической кольцевой системы. Типичные гетероциклилы включают кольцевые системы, в которых (i) каждое кольцо является неароматическим и по меньшей мере одно кольцо содержит гетероатом, например, тетрагидрофуранил, тетрагидропиранил, тетрагидротиенил, пирролидинил, пиранил, тианил, пирролидонил, пиперидинил, пирролинил, декагидрохинолинил, оксазолидинил, пиперазинил, диоксанил, диоксоланил, диазепинил, оксазепинил, тиазепинил, морфолинил и хинуклидинил; (ii) по меньшей мере одно кольцо является неароматическим и содержит гетероатом и по меньшей мере одно другое кольцо представляет собой ароматическое углеродное кольцо, например, 1,2,3,4 тетрагидрохинолинил, 1,2,3,4 тетрагидроизохинолинил; и (iii) по меньшей мере одно кольцо является неароматическим и содержит гетероатом и по меньшей мере одно другое кольцо является ароматическим и содержит гетероатом, например, 3,4 дигидро 1H пирано[4,3 c]пиридин и 1,2,3,4 тетрагидро 2,6 нафтиридин. В некоторых вариантах реализации изобретения гетероциклил может содержать:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и  .
.
«Гетероциклилалкил», как применяют в данном документе, обозначает алкильную группу, замещенную гетероциклической группой.
«Гетероарилалкил», как применяют в данном документе, обозначает алкильную группу, замещенную гетероарильной группой.
«Гидрокси» или «гидроксид», как применяют в данном документе, обозначает -OH.
«Ингибитор», как применяют в данном документе, обозначает соединение, которое ингибирует фермент таким образом, что можно наблюдать снижение активности фермента, например, в биохимическом анализе. В некоторых вариантах реализации изобретения ингибитор имеет IC50 менее около 1 мкмоль, менее около 500 нмоль, менее около 250 нмоль, менее около 100 нмоль, менее около 50 нмоль или менее около 10 нмоль. Ингибитор FGFR-4 обозначает соединение, которое ингибирует FGFR-4.
«Нитро», как применяют в данном документе, обозначает -NO2.
«Нуклеофил», как применяют в данном документе, обозначает соединения, которые отдают электронную пару электрофилу с образованием химической связи в реакции. В некоторых вариантах реализации изобретения нуклеофил может быть нуклеофилом, содержащим кислород, например, водой или гидроксилом, нуклеофилом, содержащим азот, например, амин, или нуклеофилом, содержащим серу, например, тиол, такой как, например, тиол в боковой цепи остатка цистеина.
«Сверхэкспрессированный», как применяют в данном документе, означает, что существует получение продукта гена в образце, которое является значительно более высоким, чем это наблюдалось в совокупности контрольных образцов (например, нормальной ткани).
«Селективное» обозначает соединение, которое ингибирует активность белка-мишени, например, FGFR-4, более эффективно, нежели он ингибирует активность других белков. В этом случае, все изоформы FGFR-1, FGFR-2, FGFR-3 и FGFR-4 считают различными белками. В некоторых вариантах реализации изобретения соединение может ингибировать активность белка-мишени, например, FGFR-4, в по меньшей мере 1,5, по меньшей мере 2, по меньшей мере 5, по меньшей мере 10, по меньшей мере 20, по меньшей мере 30, по меньшей мере 40, по меньшей мере 50, по меньшей мере 60, по меньшей мере 70, по меньшей мере 80, по меньшей мере 90, по меньшей мере 100, по меньшей мере 200, по меньшей мере 500 или по меньшей мере 1000 или более раз эффективнее, чем оно ингибирует активность не являющегося мишенью белка.
«Замещенный», либо с предшествующим термином «необязательно», либо без него, относится в данном документе к функциональным группам, имеющим заместители, замещающие водород на одном или более атомах углерода остова. Следует понимать, что «замещение» или «замещенный на» включает косвенное условие, что такое замещение находится в соответствии с допустимой валентностью замещенного атома и заместителя и что замещение приводит к образованию стабильного соединения, например, которое самопроизвольно не претерпевает такую трансформацию, как перегруппировка, циклизация, отщепление и т.д. Как применяют в данном документе, термин «замещенный», как предполагают, включает все допустимые заместители органических соединений. В широком аспекте, допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Допустимых заместителей может быть один или более и в случае соответствующих органических соединений они могут быть одинаковыми или различными. Для целей данного изобретения, гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанные в данном документе, которые заполняют валентности гетероатомов. Заместители могут содержать любые заместители, описанные в данном документе, например, галоген, гидроксил, карбонил (такой как карбоксил, алкоксикарбонил, формил или ацил), тиокарбонил (такой как сложный тиоэфир, тиоацетат или тиоформиат), алкоксил, фосфорил, фосфат, фосфонат, фосфинат, амино, амидо, амидин, имин, циано, нитро, азидо, сульфгидрил, алкилтио, сульфат, сульфонат, сульфамоил, сульфонамидо, сульфонил, гетероциклил, аралкил или ароматическую или гетероароматическую функциональную группу. Специалистам в данной области техники должно быть понятно, что функциональные группы, замещенные по углеводородной цепи, при необходимости, могут быть сами замещены. К примеру, заместители замещенного алкила могут содержать замещенные и незамещенные формы амино, азидо, имино, амидо, фосфорильные (в том числе фосфонатные и фосфинатные), сульфонильные (в том числе сульфатные, сульфонамидо, сульфамоильные и сульфонатные) и силильные группы, также как простые эфиры, алкилтио, карбонилы (в том числе кетоны, альдегиды, карбоксилаты и сложные эфиры), -CF3, -CN и тому подобное. Типовые замещенные алкилы описаны ниже. Циклоалкилы могут быть дополнительно замещены алкилами, алкенилами, алкокси, алкилтио, аминоалкилами, карбонил-замещенными алкилами, -CF3, -CN и тому подобными. Аналогичные замены могут быть сделаны в алкенильных и алкинильных группах для получения, например, аминоалкенилов, аминоалкинилов, амидоалкенилов, амидоалкинилов, иминоалкенилов, иминоалкинилов, тиоалкенилов, тиоалкинилов, карбонил-замещенных алкенилов или алкинилов.
Как применяют в данном документе, определение каждого выражения, например, алкил, m, n и т.д., если оно встречается более одного раза в любой структуре, предназначено для того, чтобы не зависеть от его определения в другом месте той же структуры.
«Сульфонил», как применяют в данном документе, обозначает -SO2-.
«Сульфонамидо», как применяют в данном документе, обозначает -S(=O)-N(R1)(R2) или -N(R1)-S(=O)-R2, где каждый из R1 и R2 независимо представляет собой H или алкил.
«Головная функциональная группа» или «головная группа» обозначает функциональную группу ингибитора, которая принимает участие, либо обратимо, либо необратимо, в реакции донора, например, белка, с субстратом. Головные группы могут, например, образовывать ковалентные связи с белком, либо могут создавать стабильные переходные состояния, либо быть обратимым или необратимым алкилирующим средством. Например, головная функциональная группа может представлять собой функциональную группу на ингибиторе, которая может принимать участие в реакции образования связи, в котором новая ковалентная связь образуется между частью головной группы и донором, например, аминокислотным остатком белка. Головная часть представляет собой электрофил, а "донор" представляет собой нуклеофил, такой как боковой цепи остатка цистеина. Примеры подходящих головных групп включают, но не ограничиваясь этим, группы, приведенные ниже:

где X представляет собой уходящую группу, такую как галоген, или активированную гидроксильную функциональную группу (например, трифлат); и каждый из Ra, Rb и Rc независимо представляет собой H, замещенный или незамещенный C1-4 алкил, замещенный или незамещенный C1-4 циклоалкил или циано.
Соединения, описанные в данном документе, могут содержать неестественные пропорции атомных изотопов одного или более атомов, которые составляют подобные соединения. Например, соединения может быть радиоактивно меченными радиоактивными изотопами, такими как, например, тритий (3H) или углерод-14 (14C). Все изотопные варианты соединений, описанных в данном документе, либо радиоактивные, либо нет, охватываются объемом данного изобретения. Например, дейтерированные соединения или соединения, содержащие 13C, охватываются объемом данного изобретения.
Определенные соединения могут существовать в различных таутомерных формах, а все возможные таутомерные формы всех соединений, описанных в данном документе, охватываются объемом данного изобретения.
«Энантиомерный избыток» или «% энантиомерного избытка» композиции могут быть рассчитаны с применением уравнения, приведенного ниже. В примере, приведенном ниже, композиция содержит 90% одного энантиомера, например, S-энантиомера, и 10% другого энантиомера, т.е., R-энантиомера.
э.и.=(90-10)/100=80%.
Таким образом, композиция, содержащая 90% одного энантиомера и 10% другого энантиомера, как говорят, имеет энантиомерный избыток в 80%. Некоторые композиции, описанные в данном документе, содержат энантиомерный избыток по меньшей мере 50%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 99% Соединения 1 (S-энантиомера). Другими словами, композиции содержат энантиомерный избыток S-энантиомера по отношению к R-энантиомеру.
Если не указано иное, структуры, изображенные в данном документе, также предназначены для включения всех изомерных (например, энантиомерных, диастереомерных и геометрических (или конформационных)) форм структуры; например, R и S конфигурации каждого асимметрического центра, Z и E изомеров двойных связей и Z и E конформационных изомеров. Следовательно, отдельные стереохимические изомеры, такие как энантиомерные, диастереомерные и геометрические (или конформационные) смеси данного соединения, находятся в пределах объема изобретения. Если не указано иное, все таутомерные формы соединений в соответствии с данным изобретением находятся в пределах объема изобретения.
Соединения, описанные в данном документе, могут быть пригодными в качестве свободного основания или в виде соли. Типичные соли включают гидробромидные, гидрохлоридные, сульфатные, бисульфатные, фосфатные, нитратные, ацетатные, валератные, олеатные, пальмитатные, стеаратные, лауратные, бензоатные, лактатные, фосфатные, тозилатные, цитратные, малеатные, фумаратные, сукцинатные, тартратные, нафтилатные, мезилатные, глюкогептонатные, лактобионатные, лаурилсульфонатные соли и тому подобное. (Смотри, например, Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66:1-19.)
Некоторые соединения, описанные в данном документе, могут существовать как в нерастворимых формах, так и в сольватированных формах, в том числе в гидратированных формах. В общем, сольватированные формы эквивалентны несольватированным формам и находятся в пределах объема данного изобретения. Некоторые соединения, описанные в данном документе, могут существовать во многих кристаллических или аморфных формах. В общем, все физические формы эквивалентны для применений, предусматриваемых данным изобретением, и находятся в пределах объема данного изобретения.
Фармацевтические композиции
Если соединение, описанное в данном документе, может быть введено отдельно, предпочтительно вводить соединение в виде фармацевтического состава, в котором соединение объединяют с одним или более фармацевтически приемлемыми вспомогательными веществами или носителями. Соединения, описанные в данном документе, могут быть составлены для введения любым удобным способом для применения в медицине или ветеринарии. В некоторых вариантах реализации изобретения соединение, содержащееся в фармацевтическом препарате, может быть активным само по себе, или может быть пролекарством, например, способным превращаться в активное соединение в физиологической обстановке. В некоторых вариантах реализации изобретения соединения, предлагаемые в данном документе, содержат их гидраты.
Фразу "фармацевтически приемлемые" применяют в данном документе для обозначения таких соединений, материалов, композиций и/или лекарственных форм, которые, с медицинской точки зрения, являются подходящими для применения в контакте с тканями человеческих существ и животных без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, пропорционально разумному соотношению польза/риск.
Примеры фармацевтически приемлемых солей соединения, описанного в данном документе, включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих кислых солей включают ацетат, адипат, бензоат, бензолсульфонат, бутират, цитрат, диглюконат, додецилсульфат, формиат, фумарат, гликолят, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, лактат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, пальмоат, фосфат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тозилат и ундеканоат. Соли, полученные из подходящих оснований, включают соли щелочного металла (например, натрия), щелочно-земельного металла (например, магния), аммония и N-(алкила)4+. Данное изобретение также предусматривает кватернизацию любых основных азотсодержащих групп соединений, описанных в данном документе. Вода или жирорастворимые или диспергируемые продукты могут быть получены с помощью подобной кватернизации.
Примеры фармацевтически приемлемых носителей включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) вспомогательные вещества, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) многоатомные спирты, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и метиллаурат; (13) агар; (14) буферные средства, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатные буферные растворы; (21) циклодекстрины, такие как Captisol®; таргетированные лиганды, прикрепленные к наночастицами, такие как Accurins™; и (22) другие нетоксичные совместимые вещества, такие как композиции на основе полимера, применяемые в фармацевтических составах.
Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и тому подобное; (2) жирорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (BHA),бутилированный гидрокситолуол (BHT), лецитин, пропилгаллат, альфа-токоферол и тому подобное; и (3) средства, образующие комплексные соединения с металлом, такие как лимонная кислота, этилендиаминтетрауксусная кислота (ЭДТА), сорбитол, винная кислота, фосфорная кислота и тому подобное.
Твердые лекарственные формы (например, капсулы, таблетки, пилюли, драже, порошки, гранулы и тому подобное) могут содержать один или более фармацевтически приемлемых носителей, таких как цитрат натрия или дикальцийфосфат и/или любой из следующих: (1) наполнителей или разбавителей, таких как крахмалы, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; (2) связывающих средств, таких как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или камедь; (3) увлажнителей, таких как глицерин; (4) разрыхлителей, таких как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) замедляющих растворение средств, таких как парафин; (6) ускорителей абсорбции, таких как четвертичные аммониевые соединения; (7) смачивающих средств, таких как, например, цетиловый спирт и глицеролмоностеарат; (8) абсорбентов, таких как каолин и бентонитовая глина; (9) смазывающих средств, таких как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси; и (10) красящих средств.
Жидкие лекарственные формы могут содержать фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активному ингредиенту, жидкие лекарственные формы могут содержать инертные разбавители, обычно применяемые в уровне техники, такие как, например, вода или другие растворители, солюбилизирующие средства и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (в частности, хлопковое, арахисовое, кукурузное, масло зародышей пшеницы, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и жирнокислотные эфиры сорбитана, а также их смеси.
Суспензии, в дополнение к активным соединениям, могут содержать суспендирующие средства, такие как этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также их смеси.
Мази, пасты, кремы и гели могут содержать, в дополнение к активному соединению, вспомогательные вещества, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевая кислота, тальк и оксид цинка или их смеси.
Порошки и спреи могут содержать, в дополнение к активному соединению, вспомогательные вещества, такие как лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция и полиамидный порошок, или смесь этих веществ. Спреи могут дополнительно содержать обычные пропелленты, такие как хлорфторуглеводороды и летучие незамещенные углеводороды, такие как бутан и пропан.
В целях удобства составы могут быть представлены в единичной лекарственной форме и могут быть получены по любому из способов, хорошо известных в области фармации. Количество активного ингредиента, которое может быть объединено с материалом носителя для получения единой лекарственной формы, будет варьироваться в зависимости от хозяина, подлежащего лечению, конкретного способа введения. Количество активного ингредиента, которое может быть объединено с материалом носителя для получения единой лекарственной формы, как правило, будет представлять собой такое количество соединения, которое обеспечивает терапевтический эффект.
Лекарственные формы для местного применения или трансдермального введения соединения по данному описанию, включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и средства для ингаляций. Активный компонент может быть смешан в стерильных условиях с фармацевтически приемлемым носителем и любыми консервантами, буферными средствами или пропеллентами, которые могут потребоваться.
При условии, что соединения, описанные в данном документе, людям и животным вводят в виде фармацевтического препарата, они могут быть предоставлены per se или в виде фармацевтической композиции, содержащей, например, от 0,1 до 99,5% (более предпочтительно, от 0,5 до 90%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
Составы могут быть введены местно, перорально, трансдермально, ректально, вагинально, парентерально, интраназально, внутрилегочно, внутриглазно, внутривенно, внутримышечно, внутрисуставно, внутриартериально, интратекально, внутрикапсульно, внутрикожно, внутрибрюшинно, подкожно, субкутикулярно или путем ингаляции.
Показания
FGFR-4 регулирует пролиферацию, выживание и секрецию альфа-фетопротеина во время прогрессирования гепатоклеточной карциномы (HCC); ингибиторы FGFR-4, следовательно, являются перспективными потенциальными терапевтическими средствами для этой неудовлетворенной медицинской потребности (Ho et al., Journal of Hepatology, 2009, 50:118-27). HCC ежегодно затрагивает более 550000 человек во всем мире и имеет один из худших коэффициентов однолетней выживаемости по сравнению с любым типом рака.
Дополнительное подтверждение связи между FGFR-4 и HCC проявляется посредством вовлечения FGF19, члена семейства факторов роста фибробластов (FGF), которое состоит из гормонов, регулирующих глюкозу, жир и энергетический гомеостаз. У FGF19 трансгенных мышей наблюдались повышенная пролиферация гепатоцитов и образование опухоли печени. FGF19 активирует FGFR-4, его преобладающий рецептор в печени, и считается, что активация FGFR-4 представляет собой механизм, посредством которого FGF19 может повышать пролиферацию гепатоцитов и индуцировать образование опухоли печени (Wu et al., J Biol Chem (2010) 285(8):5165-5170). В другом случае FGF19 также был идентифицирован как драйверный ген в HCC (Sawey et al., Cancer Cell (2011) 19: 347-358). Поэтому полагают, что соединения, описанные в данном документе, являющиеся сильнодействующими и селективными ингибиторами FGFR-4, могут быть применены для лечения HCC и иных видов рака печени.
Онкогенный скрининг определил активирующую мутацию Y367C рецептора 4 фактора роста фибробластов (FGFR-4)в клеточной линии MDA-MB-453 рака молочной железы человека. Эта мутация, как было показано, вызывает конститутивное фосфорилирование, что приводит к активации каскада митоген-активируемой протеинкиназы. Соответственно, было высказано предположение, что FGFR-4 может быть драйвером роста опухоли рака молочной железы (Roidl et al., Oncogene (2010) 29(10):1543-1552). Поэтому полагают, что соединения, описанные в данном документе, которые являются сильнодействующими и селективными ингибиторами FGFR-4, могут быть применены для лечения рака молочной железы, модулированного с помощью FGFR-4.
Молекулярные изменения (например, транслокации) в генах против хода транскрипции от FGFR-4 могут привести к активации/сверхэкспрессии FGFR-4. Например, транслокация/слияние генов PAX3-FKHR может привести к сверхэкспрессии FGFR-4. Сверхэкспрессия FGFR-4, в соответствии с этим механизмом, связана с рабдомиосаркомой (RMS) (Cao et al., Cancer Res (2010) 70(16): 6497-6508). Мутации в самом FGFR-4 (например, мутации домена киназы) могут привести к чрезмерной активации белка; этот механизм связан с субпопуляцией RMS (Taylor et al., J Clin Invest (2009) 119: 3395-3407). Поэтому полагают, что соединения, описанные в данном документе, которые являются сильнодействующими и селективными ингибиторами FGFR-4, могут быть применены для лечения RMS, модулированной с помощью FGFR-4, и других сарком.
Другие заболевания были связаны с изменениями в генах против хода транскрипции от FGFR-4 или с мутациями в самом FGFR-4. Например, мутации в домене киназы FGFR-4 приводят к сверхактивации, которая связана с аденокарциномой легкого (Ding et al., Nature (2008) 455(7216): 1069-1075). Амплификация EGFR-4 была связана с такими состояниями, как почечно-клеточная карцинома (предварительные данные TCGA). Кроме того, сайленсинг FGFR4 и ингибирование лиганд-рецепторного связывания значительно снижают рост опухоли яичников, предполагая, что ингибиторы FGFR4 могут быть полезны при лечении рака яичников (Zaid et al., Clin. Cancer Res. (2013) 809).
Патогенные повышения уровней желчных кислот были связаны с изменениями в уровнях FGF19 (Vergnes et al., Cell Metabolism (2013) 17, 916-28). Поэтому снижение уровня FGF19 может быть полезным в промотировании синтеза желчной кислоты и, таким образом, при лечении гиперлипидемии.
Величины дозы
Фактические уровни дозировки активных ингредиентов в фармацевтических композициях по данному изобретению могут варьироваться таким образом, чтобы получать количество активного ингредиента, являющееся эффективным для достижения заданного терапевтического ответа в случае конкретного пациента, композиции и способа введения, не будучи токсичным для пациента.
Выбранный уровень дозировки будет зависеть от множества факторов, в том числе активности конкретного применяемого соединения, описанного в данном изобретении, или его сложного эфира, соли или амида, пути введения, времени введения, скорости экскреции конкретного применяемого соединения, длительности лечения, других лекарственных средств, соединений и/или материалов, применяемых в комбинации с конкретным применяемым соединением, возраста, пола, массы, состояния, общего состояния здоровья и медицинского анамнеза пациента, подлежащего лечению, и подобных факторов, хорошо известных из уровня техники в области медицины.
Врач или ветеринар, имеющие обычную квалификацию в данной области техники, могут легко определить и прописать эффективное количество необходимой фармацевтической композиции. Например, врач или ветеринар может начинать дозирование соединений, применяемых в фармацевтической композиции в соответствии с данным изобретением, в меньшем количестве, чем это требуется для достижения желаемого терапевтического эффекта, и постепенно увеличивать дозировку, пока не будет достигнут желаемый эффект.
В общем, подходящая суточная доза соединения в соответствии с данным изобретением будет представлять собой такое количество соединения, которое является самой низкой дозой, эффективной для получения терапевтического эффекта. Такая эффективная доза обычно зависит от факторов, описанных выше. Как правило, дозы соединений по данному изобретению для пациента будут варьироваться от около 0,0001 до около 100 мг на килограмм массы тела в сутки. Например, доза может составлять от 10 до 2000 мг в день. В альтернативном варианте, доза может составлять от 100 до 1000 мг в день или от 200 до 600 мг в день. При необходимости, эффективная суточная доза активного соединения может быть введена в виде одной, двух, трех, четырех или более частей дозы, вводимых раздельно через соответствующие промежутки времени в течение дня, необязательно, в единичных лекарственных формах.
Комбинированная и целевая терапия
Введение ингибиторов FGFR-4, описанных в данном документе, может быть объединено с другими способами лечения рака. Например, ингибиторы могут быть введены в комбинации с хирургическими вмешательствами, облучением или другими терапевтическими средствами, такими как антитела, другими селективными ингибиторами киназы или химиотерапевтическими средствами. Ингибиторы могут быть введены в комбинации с терапией с применением RNAi или антисмысловой терапией. Ингибиторы FGFR-4, описанные в данном документе, могут быть объединены с одним, двумя или несколькими другими терапевтическими средствами. В примерах, описанных ниже, следует понимать, что «второе терапевтическое средство» также включает в себя более чем одно терапевтическое средство, отличное от ингибитора FGFR4. К примеру, соединения, описанные в данном документе, могут быть объединены с таким средством, как сорафениб. Ингибитор FGFR-4, описанный в данном документе, может быть введен с одним, двумя или более другими терапевтическими средствами.
Ингибиторы FGFR-4, описанные в данном документе, и второе терапевтическое средство не должны быть введены в одной и той же фармацевтической композиции, и, возможно, ввиду различных физических и химических характеристик, могут быть введены различными способами. Например, ингибитор FGFR-4 может быть введен перорально, в то время как второе терапевтическое средство вводят внутривенно. Определение способа введения и целесообразности введения, где это возможно, в одной и той же фармацевтической композиции, находится в пределах знаний квалифицированного врача. Начальное введение может быть осуществлено согласно установленным протоколам, известным в данной области, а затем, на основании наблюдаемых эффектов, дозировки, способов введения и количеств введения, может быть модифицировано квалифицированным врачом.
Ингибитор FGFR-4 и второе терапевтическое средство могут вводиться одновременно (например, одновременно, по существу одновременно или в пределах того же протокола лечения) или последовательно (т.е. последовательно одно за другим, с оптимальным временным интервалом между ними), в зависимости от природы пролиферативного заболевания, состояния пациента и фактического выбора второго терапевтического средства, подлежащего введению.
Кроме того, ингибиторы FGFR-4, описанные в данном документе, могут быть введены в качестве части конъюгата антитело-лекарственное средство, где ингибитор FGFR-4 представляет собой «полезную» часть конъюгата.
Соединения
В приведенной ниже таблице показаны структуры соединений, описанных в данном документе.
Номер





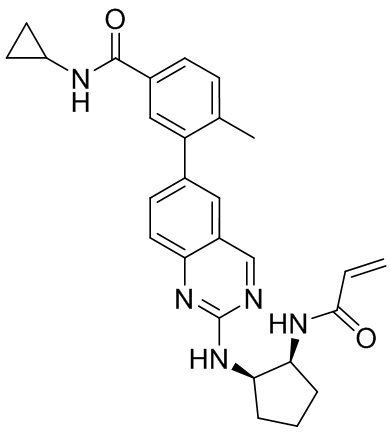

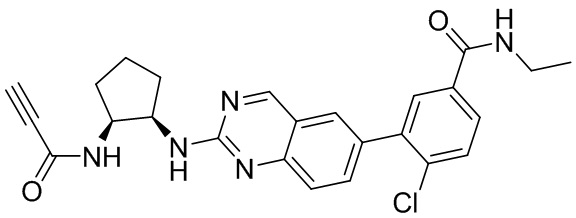

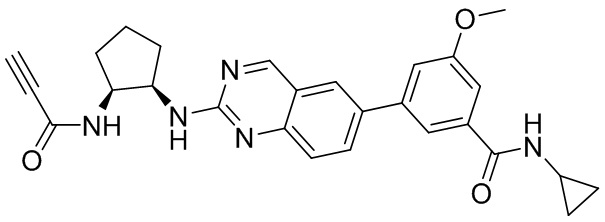


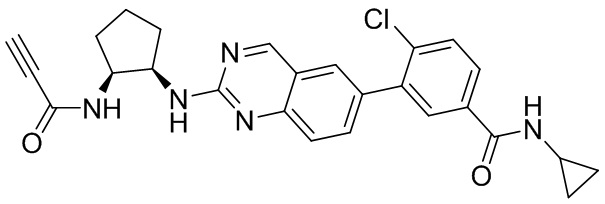

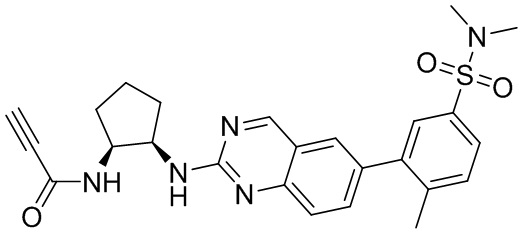


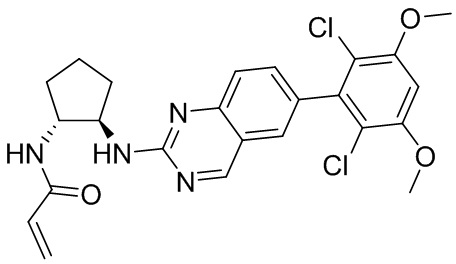
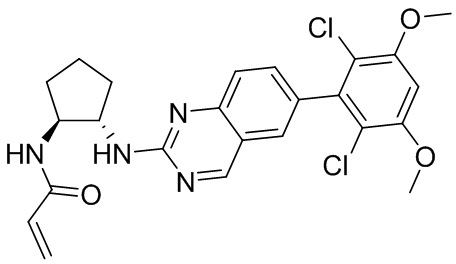

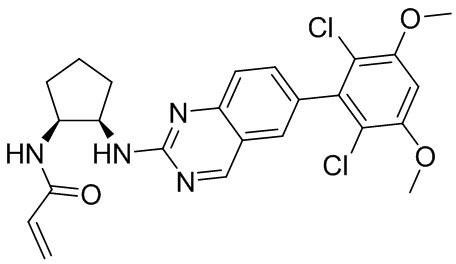





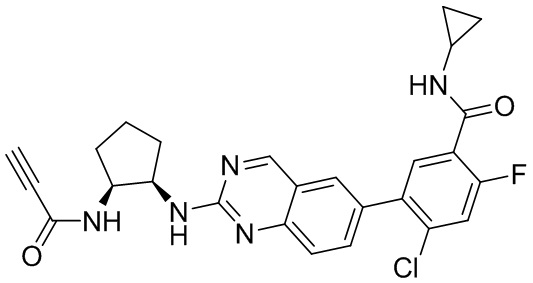

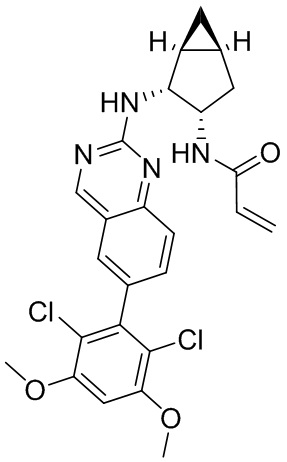


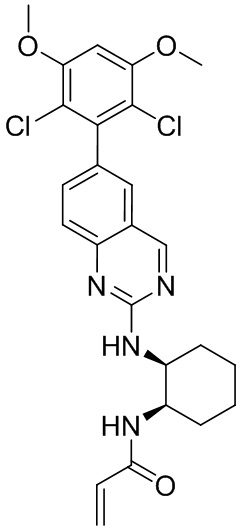






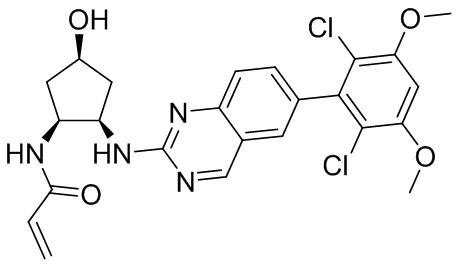

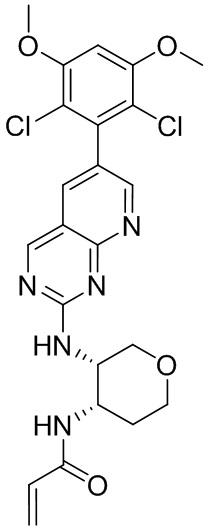
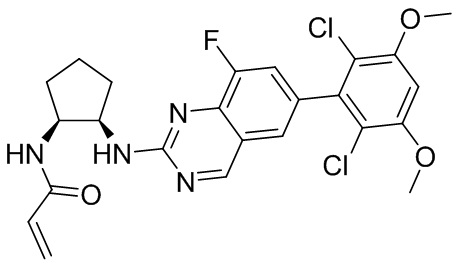






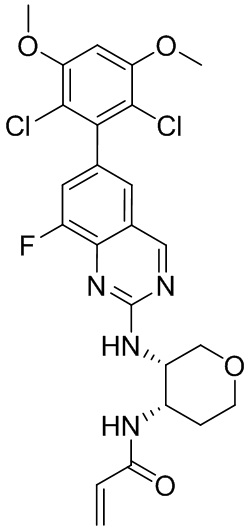


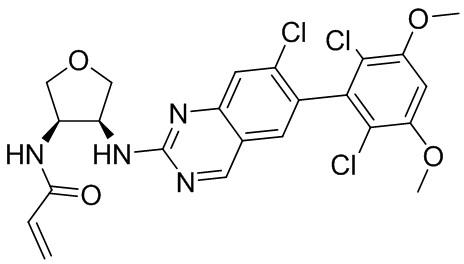

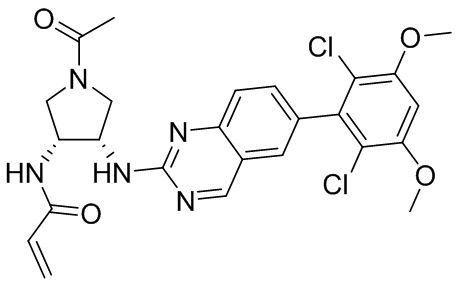
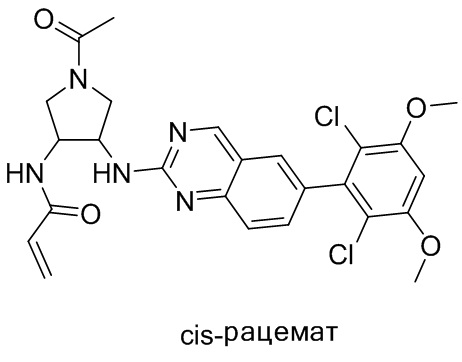



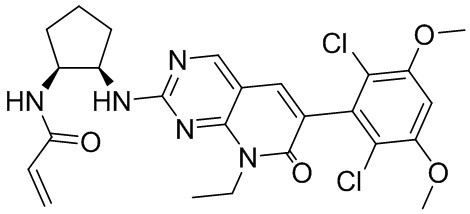








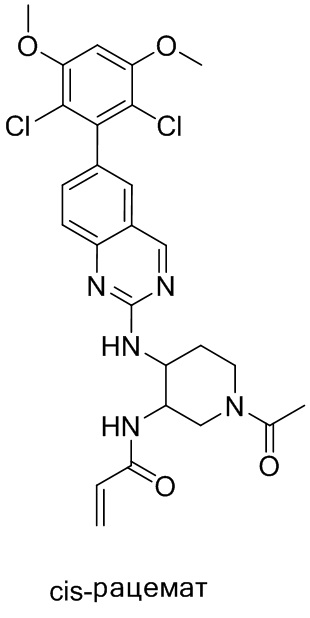

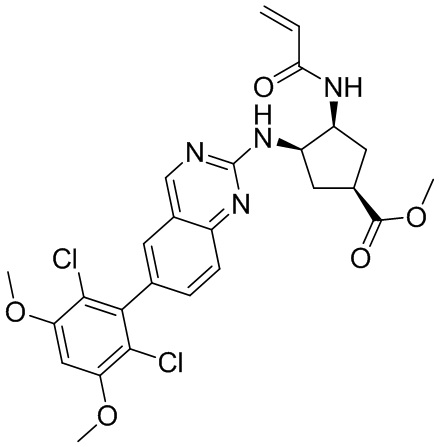



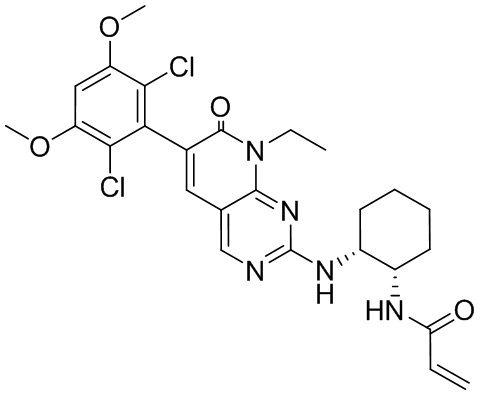
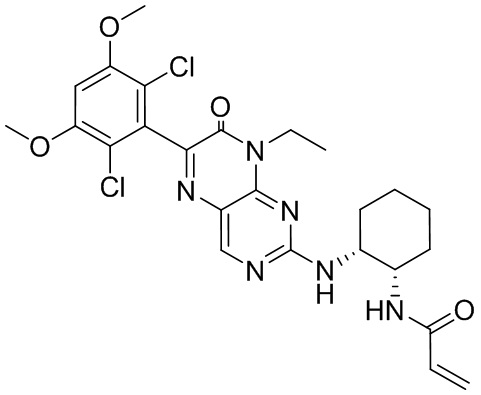
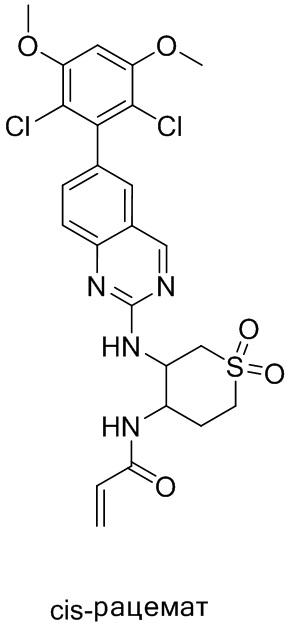


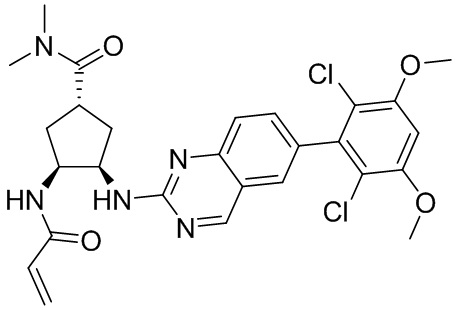

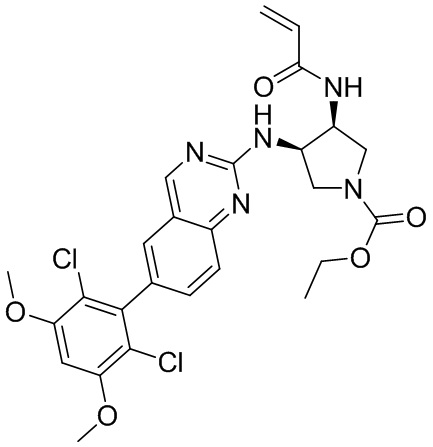

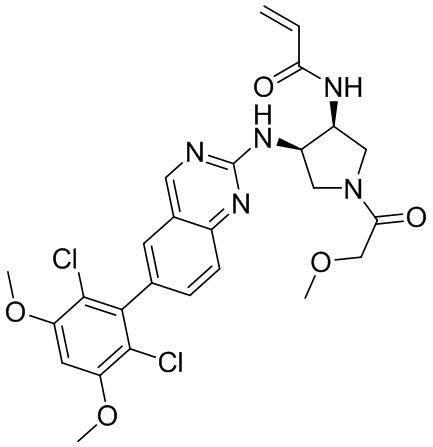
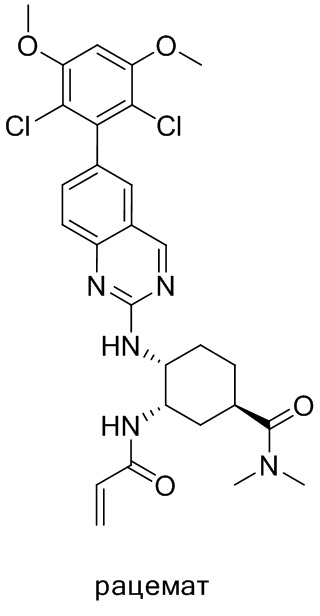




Синтез
Соединения в соответствии с данным изобретением, в том числе их соли и N-оксиды, могут быть получены с применением известных технологий органического синтеза и могут быть синтезированы в соответствии с любым из многочисленных возможных путей синтеза, таких как приведены на Схемах ниже. Реакции для получения соединений в соответствии с данным изобретением могут быть выполнены в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящие растворители могут быть по большей части не реагирующими с исходными материалами (реагентами), интермедиатами или продуктами при температурах, при которых осуществляют реакцию, например, температурах, которые могут изменяться от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может быть осуществлена в одном растворителе или в смеси более чем одного растворителя. В зависимости от конкретного этапа реакции, специалистом в данной области техники могут быть выбраны подходящие для конкретного этапа реакции растворители.
Получение соединений в соответствии с данным изобретением может включать защиту и снятие защиты с различных химических групп. Необходимость защиты или снятия защиты, а также выбор соответствующих защитных групп, могут быть легко определены специалистом в данной области техники. Химия защитных групп могут быть обнаружена, например, в Wuts and Greene, Protective Groups in Organic Synthesis, 4th ed., John Wiley & Sons: New Jersey, (2006), включенной в данный документ в полном объеме посредством ссылки.
Реакции возможно контролировать в соответствии с любым подходящим известным в данной области техники способом. Например, образование продукта можно контролировать спектроскопическими способами, такими как спектроскопия ядерного магнитного резонанса (ЯМР) (например, H1 или C13), инфракрасная спектроскопия (ИК), спектрофотометрия (например, УФ-видимая), масс-спектрометрия (МС) или с помощью хроматографических способов, таких как высокоэффективная жидкостная хроматография (ВЭЖХ) или тонкослойная хроматография (ТСХ). Аналитические устройства и способы для определения характеристик соединения:
ЖХ-МС: Если не указано иное, все данные жидкостной хроматографии-масс-спектрометрии (ЖХ-МС) (образец анализируют на чистоту и идентичность) получают с помощью ЖХ системы Agilent model-1260 с применением масс-спектрометра Agilent model 6120, применяя ES-API ионизацию, оснащенного обращенно-фазовой колонкой Agilent Poroshel 120 (EC-C18, размер частиц 2,7 мкм, размеры 3,0×50 мм) при 22,4 градусов Цельсия. Подвижная фаза состоит из смеси растворителя 0,1% муравьиной кислоты в воде и 0,1% муравьиной кислоты в ацетонитриле. Применяют постоянный градиент от 95% водной/5% органической до 5% водной/95% органической подвижной фазы в течение 4 минут. Скорость потока являлась постоянной при 1 мл/мин.
Преп. ЖХ-МС: Препаративную ВЭЖХ осуществляют на Shimadzu Discovery VP® Preparative system, оснащенной обращенно-фазовой колонкой Luna 5u C18(2) 100A, AXIA packed, 250×21,2 мм при 22,4 градусов Цельсия. Подвижная фаза состоит из смеси растворителя 0,1% муравьиной кислоты в воде и 0,1% муравьиной кислоты в ацетонитриле. Применяют постоянный градиент от 95% водной/5% органической до 5% водной/95% органической подвижной фазы в течение 25 минут. Скорость потока являлась постоянной при 20 мл/мин. Реакции, осуществляемые в микроволновой печи, выполняют в микроволновом блоке Biotage Initiator.
Хроматография на силикагеле: Хроматографию на силикагеле осуществляют либо на блоке Teledyne Isco CombiFlash® Rf, либо на блоке Biotage® Isolera Four.
Протонный ЯМР: Если не указано иное, все спектры ЯМР Н1 получают с помощью ЯМР устройства Varian 400 МГц Unity Inova 400 МГц (время регистрации=3,5 секунд с задержкой 1 секунда; 16-64 сканирований). Там, где это определено, все протоны регистрируют в растворителе ДМСО-d6 в виде частей на миллион (ч./млн.) по отношению к остаточному ДМСО (2,50 ч./млн.).
ПРИМЕРЫ
Следующие примеры предназначены для иллюстрации и не ограничивают объем изобретения каким-либо образом.
Приведенные ниже схемы предназначены для обеспечения общего руководства по вопросу получения соединений в соответствии с данным изобретением. Специалисту в данной области техники будет понятно, что препараты, показанные на Схемах, могут быть изменены или оптимизированы с применением общих знаний по органической химии для получения различных соединений в соответствии с данным изобретением.
Протокол Синтеза 1

6-Бром-2-хлорхиназолин может быть замещен 1,2-монозащищенным циклоалкилдиамином при условиях реакции нуклеофильного замещения в ароматическом ядре с применением основания, такого как диизопропилэтиламин (DIPEA) или триэтиламин (TEA) в полярном растворителе, таком как диоксан, для получения диамин-замещенного хиназолина. 6-Бромхиназолин могут быть соединен с арильным, гетероарильным реагентом бора, олова или цинка посредством медиированной палладием реакции конденсации, например, реакции Сузуки, Стилла, Негиши, для получения интермедиата, с которого затем снимают защиту, чтобы открыть амин. Аминный заместитель циклоалкана может вступать в реакцию с пропионовой кислотой с применением условий реакции конденсации амида или вступать в реакцию с акрилоилхлоридом для получения акриламида. Как приведено ниже, Соединения 2 и 6 получали с применением Протокола Синтеза 1.
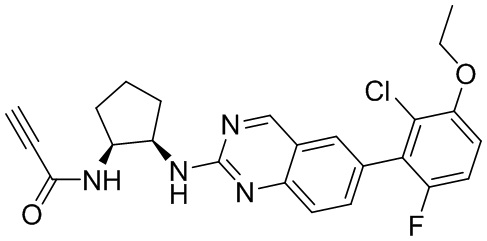
Пример 1: Синтез N-((1S,1R)-2-((6-(2,6,-дифтор-3-метоксифенил)хиназолин-2-ил)амино)циклопентил)пропиоламида (Соединение 2)
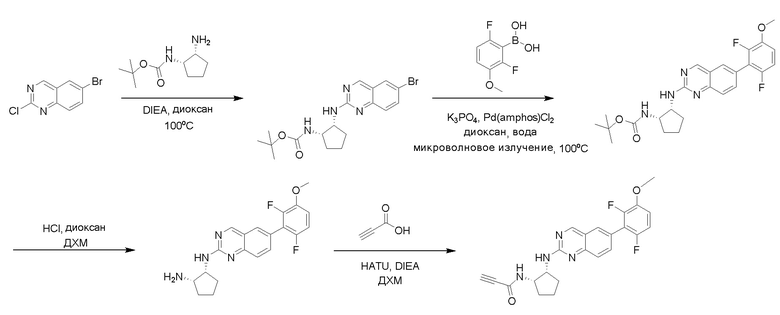
Этап 1: Синтез трет-бутил((1S,2R)-2-((6-бромхиназолин-2-ил)амино)циклопентил)карбамата

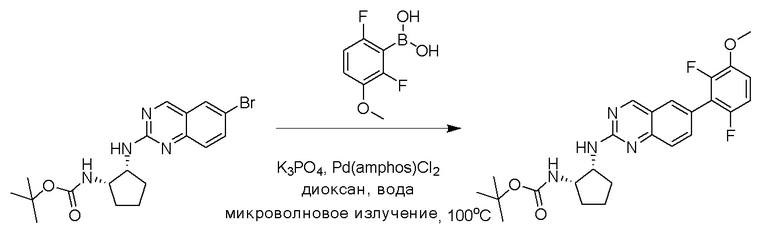
Смесь трет-бутил((1S,2R)-2-((6-бромхиназолин-2-ил)амино)циклопентил)карбамата (25 мг, 0,06 ммоль), (2,6-дифтор-3-метоксифенил)бороновой кислоты (24 мг, 0,12 ммоль), бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладия(II) (3 мг, 0,003 ммоль) и фосфата калия (40 мг, 0,19 ммоль) в 1,4-диоксане/воде (1 мл/0,2 мл) дегазировали азотом в течение 5 минут и перемешивали при 100°C в течение 30 минут под микроволновым излучением. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, промывали насыщенным раствором хлорида аммония и высушивали с помощью сульфата натрия. Остаток очищали с помощью колоночной хроматографии на силикагеле, чтобы обеспечить трет-бутил((1S,2R)-2-((6-(2,6-дифтор-3-метоксифенил)хиназолин-2-ил)амино)циклопентил)карбамат (21 мг, 37%). МС (ЭР+) C26H30N4O5 требует: 470, обнаружено: 471 [M+H]+.
Этап 3: Синтез (1R,2S)-N1-(6-(2,6-дифтор-3-метоксифенил)хиназолин-2-ил)циклопентан-1,2-диамина
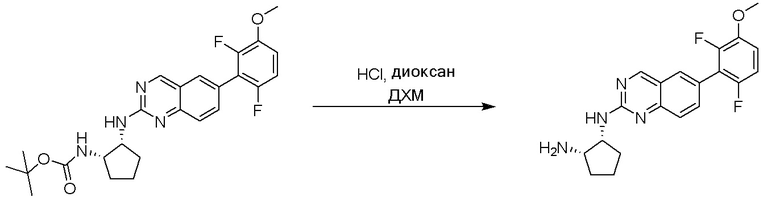
Смесь трет-бутил((1S,2R)-2-((6-(2,6-дифтор-3-метоксифенил)хиназолин-2-ил)амино)циклопентил)карбамата (21 мг, 0,045 ммоль) и 4 M HCl в диоксане (0,5 мл) в дихлорметане (1 мл) перемешивали при комнатной температуре в течение 16 ч. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь концентрировали и применяли на следующем этапе без дополнительной очистки.
Этап 4: Синтез N-((1S,2R)-2-((6-(2,6-дифтор-3-метоксифенил)хиназолин-2-ил)амино)циклопентил)пропиоламида

Смесь (1R,2S)-N1-(6-(2,6-дифтор-3-метоксифенил)хиназолин-2-ил)циклопентан-1,2-диамина (0,045 ммоль), пропионовой кислоты (0,004 мл, 0,067 ммоль), HATU (25 мг, 0,067 ммоль) и DIEA (0,023 мл, 0,135 ммоль) в дихлорметане (1 мл) перемешивали при комнатной температуре в течение 60 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением N-((1S,2R)-2-((6-(2,6-дифтор-3-метоксифенил)хиназолин-2-ил)амино)циклопентил)пропиоламида (Соединение 2) (13 мг, 68%). МС (ЭР+) C27H27N5O3 требует: 422, обнаружено: 423 [M+H]+.
Пример 2: Синтез N-((1S,2R)-2-((6-(2-хлор-3-этокси-6-фторфенил)хиназолин-2-ил)амино)циклопентил)пропиоламида (Соединение 6)

Этап 1: Синтез трет-бутил((1S,2R)-2-((6-бромхиназолин-2-ил)амино)циклопентил)карбамата
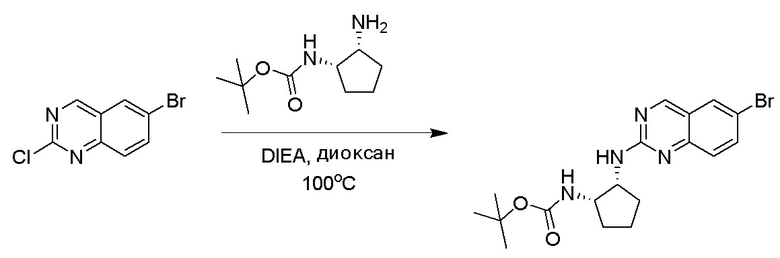
Смесь 6-бром-2-хлорхиназолина (1 г, 4,14 ммоль) и трет-бутил((1S,2R)-2-аминоциклопентил)карбамата (0,826 г, 4,14 ммоль) перемешивали при 100°C в диоксане (10 мл) в течение 48 часов. Реакционную смесь охлаждали до комнатной температуры, концентрировали и очищали остаток с помощью колоночной хроматографии на силикагеле, чтобы обеспечить трет-бутил((1S,2R)-2-((6-бромхиназолин-2-ил)амино)циклопентил)карбамат (1 г, 59%). МС (ЭР+) C18H23BrN4O2 требует: 406, обнаружено: 407 [M+H]+.
Этап 2: Синтез трет-бутил((1S,2R)-2-((6-(2-хлор-3-этокси-6-фторфенил)хиназолин-2-ил)амино)циклопентил)карбамата
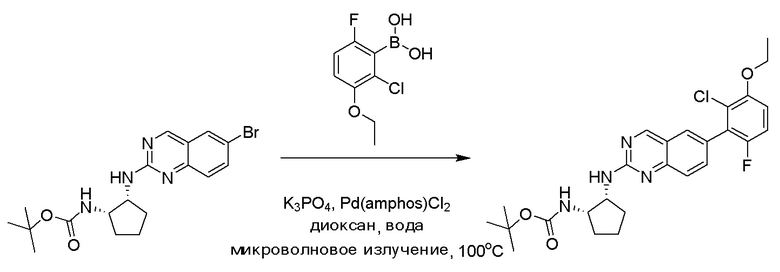
Смесь трет-бутил((1S,2R)-2-((6-бромхиназолин-2-ил)амино)циклопентил)карбамата (50 мг, 0,12 ммоль), (2-хлор-3-этокси-6-фторфенил)бороновой кислоты (40 мг, 0,18 ммоль), бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладия(II) (4 мг, 0,005 ммоль) и фосфата калия (78 мг, 0,37 ммоль) в 1,4-диоксане/воде (1,15 мл/0,15 мл) дегазировали азотом в течение 5 минут и перемешивали при 100°C в течение 30 минут под микроволновым излучением. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, промывали насыщенным раствором хлорида аммония и высушивали с помощью сульфата натрия. Остаток очищали с помощью колоночной хроматографии на силикагеле, чтобы обеспечить трет-бутил((1S,2R)-2-((6-(2-хлор-3-этокси-6-фторфенил)хиназолин-2-ил)амино)циклопентил)карбамат (51 мг, 83%). МС (ЭР+) C26H30ClFN4O3 требует: 500, обнаружено: 501 [M+H]+.
Этап 3: Синтез (1R,2S)-N1-(6-(2-хлор-3-этокси-6-фторфенил)хиназолин-2-ил)циклопентан-1,2-диамина

Смесь трет-бутил((1S,2R)-2-((6-(2-хлор-3-этокси-6-фторфенил)хиназолин-2-ил)амино)циклопентил)карбамата (51 мг, 0,1 ммоль) и 4 M HCl в диоксане (0,5 мл) в дихлорметане (1 мл) перемешивали при комнатной температуре в течение 2 ч. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь концентрировали и применяли на следующем этапе без дополнительной очистки.
Этап 4: Синтез N-((1S,2R)-2-((6-(2-хлор-3-этокси-6-фторфенил)хиназолин-2-ил)амино)циклопентил)пропиоламида

Смесь (1R,2S)-N1-(6-(2-хлор-3-этокси-6-фторфенил)хиназолин-2-ил)циклопентан-1,2-диамина (0,1 ммоль), пропионовой кислоты (0,007 мл, 0,12 ммоль), HATU (57 мг, 0,15 ммоль) и DIEA (0,052 мл, 0,3 ммоль) в дихлорметане (1 мл) перемешивали при комнатной температуре в течение 40 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением N-((1S,2R)-2-((6-(2-хлор-3-этокси-6-фторфенил)хиназолин-2-ил)амино)циклопентил)пропиоламида (Соединение 6) (35 мг, 76%). МС (ЭР+) C24H22ClFN4O2 требует: 452, обнаружено: 453 [M+H]+.
Протокол Синтеза 2
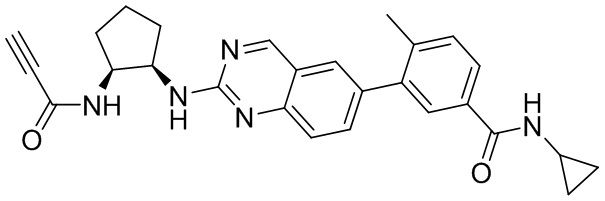
 6-Бром-2-хлорхиназолин может быть замещен 1,2-монозащищенным циклоалкилдиамином при условиях реакции нуклеофильного замещения в ароматическом ядре с применением основания, такого как диизопропилэтиламин (DIPEA) или триэтиламин (TEA) в полярном растворителе, таком как диоксан, для получения диамин-замещенного хиназолина. 6-Бромхиназолин может быть соединен с арильным, гетероарильным карбоксильным или сложноэфирным реагентом бора, олова или цинка посредством медиированной палладием реакции конденсации, например, реакции Сузуки, Стилла, Негиши. Карбоновая кислота может затем вступать в реакцию с амином с применением условий реакции конденсации амида (такого как HATU и диизопропилэтиламин) для обеспечения интермедиата, с которого затем снимают защиту, чтобы открыть аминный заместитель циклоалкана. Амин может вступать в реакцию с пропионовой кислотой с применением условий реакции конденсации амида или вступать в реакцию с акрилоилхлоридом для получения акриламида. Как приведено ниже, Соединение 13 получали с применением Протокола Синтеза 2.
6-Бром-2-хлорхиназолин может быть замещен 1,2-монозащищенным циклоалкилдиамином при условиях реакции нуклеофильного замещения в ароматическом ядре с применением основания, такого как диизопропилэтиламин (DIPEA) или триэтиламин (TEA) в полярном растворителе, таком как диоксан, для получения диамин-замещенного хиназолина. 6-Бромхиназолин может быть соединен с арильным, гетероарильным карбоксильным или сложноэфирным реагентом бора, олова или цинка посредством медиированной палладием реакции конденсации, например, реакции Сузуки, Стилла, Негиши. Карбоновая кислота может затем вступать в реакцию с амином с применением условий реакции конденсации амида (такого как HATU и диизопропилэтиламин) для обеспечения интермедиата, с которого затем снимают защиту, чтобы открыть аминный заместитель циклоалкана. Амин может вступать в реакцию с пропионовой кислотой с применением условий реакции конденсации амида или вступать в реакцию с акрилоилхлоридом для получения акриламида. Как приведено ниже, Соединение 13 получали с применением Протокола Синтеза 2.
Соединение 13
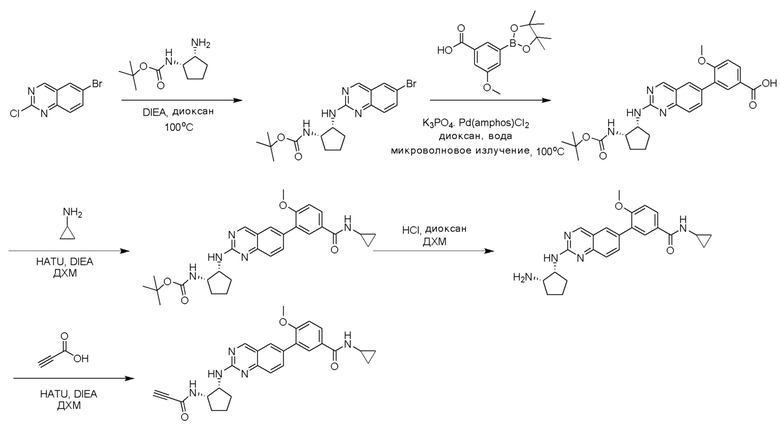 Этап 1: Синтез трет-бутил((1S,2R)-2-((6-бромхиназолин-2-ил)амино)циклопентил)карбамата
Этап 1: Синтез трет-бутил((1S,2R)-2-((6-бромхиназолин-2-ил)амино)циклопентил)карбамата

Смесь 6-бром-2-хлорхиназолина (1 г, 4,14 ммоль) и трет-бутил((1S,2R)-2-аминоциклопентил)карбамата (0,826 г, 4,14 ммоль) перемешивали при 100°C в диоксане (10 мл) в течение 48 часов. Реакционную смесь охлаждали до комнатной температуры, концентрировали и очищали остаток с помощью колоночной хроматографии на силикагеле, чтобы обеспечить трет-бутил((1S,2R)-2-((6-бромхиназолин-2-ил)амино)циклопентил)карбамат (1 г, 59%). МС (ЭР+) C18H23BrN4O2 требует: 406, обнаружено: 407 [M+H]+.
Этап 2: Синтез 4-(2-(((1R,2S)-2-((трет-бутоксикарбонил)амино)циклопентил)амино)хиназолин-6-ил)-3-метоксибензойной кислоты

Смесь трет-бутил((1S,2R)-2-((6-бромхиназолин-2-ил)амино)циклопентил)карбамата (100 мг, 0,25 ммоль), 3-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензойной кислоты (82 мг, 0,29 ммоль), бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладия(II) (9 мг, 0,01 ммоль) и фосфата калия (157 мг, 0,74 ммоль) в 1,4-диоксане/воде (2,5 мл/0,25 мл) дегазировали азотом в течение 5 минут и перемешивали при 100°C в течение 30 минут под микроволновым излучением. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, промывали насыщенным раствором хлорида аммония и высушивали с помощью сульфата натрия. Остаток очищали с помощью колоночной хроматографии на силикагеле, чтобы обеспечить метил 4-(2-(((1R,2S)-2-((трет-бутоксикарбонил)амино)циклопентил)амино)хиназолин-6-ил)-3-метоксибензойную кислоту (114 мг, 96%). МС (ЭР+) C26H30N4O5 требует: 478, обнаружено: 479 [M+H]+.
Этап 3: Синтез трет-бутил((1S,2R)-2-((6-(4-(циклопропилкарбамоил)-2-метоксифенил)хиназолин-2-ил)амино)циклопентил)карбамата
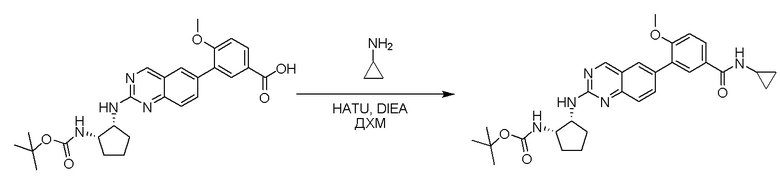
Смесь 4-(2-(((1R,2S)-2-((трет-бутоксикарбонил)амино)циклопентил)амино)хиназолин-6-ил)-3-метоксибензойной кислоты (57 мг, 0,12 ммоль), циклопропиламина (0,012 мл, 0,18 ммоль), HATU (68 мг, 0,18 ммоль) и DIEA (0,052 мл, 0,30 ммоль) в дихлорметане (1,5 мл) перемешивали при комнатной температуре в течение 30 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением трет-бутил((1S,2R)-2-((6-(4-(циклопропилкарбамоил)-2-метоксифенил)хиназолин-2-ил)амино)циклопентил)карбамата (58 мг, 93%). МС (ЭР+) C29H35N5O4 требует: 517, обнаружено: 518 [M+H]+.
Этап 4: Синтез 4-(2-(((1R,2S)-2-аминоциклопентил)амино)хиназолин-6-ил)-N-циклопропил-3-метоксибензамида

Смесь трет-бутил((1S,2R)-2-((6-(4-(циклопропилкарбамоил)-2-метоксифенил)хиназолин-2-ил)амино)циклопентил)карбамата (58 мг, 0,11 ммоль) и 4 M HCl в диоксане (0,8 мл) в дихлорметане (1,5 мл) перемешивали при комнатной температуре в течение 120 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь концентрировали и применяли на следующем этапе без дополнительной очистки.
Этап 5: Синтез N-циклопропил-3-метокси-4-(2-(((1R,2S)-2-пропиоламидоциклопентил)амино)хиназолин-6-ил)бензамида
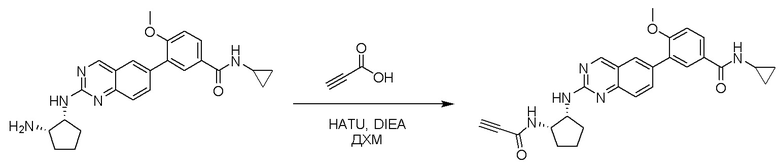
Смесь 4-(2-(((1R,2S)-2-аминоциклопентил)амино)хиназолин-6-ил)-N-циклопропил-3-метоксибензамида (0,11 ммоль), пропионовой кислоты (0,010 мл, 0,17 ммоль), HATU (64 мг, 0,17 ммоль) и DIEA (0,06 мл, 0,34 ммоль) в дихлорметане (1,5 мл) перемешивали при комнатной температуре в течение 45 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением N-циклопропил-3-метокси-4-(2-(((1R,2S)-2-пропиоламидоциклопентил)амино)хиназолин-6-ил)бензамида (Соединение 13) (35 мг, 69%). МС (ЭР+) C27H27N5O3 требует: 469, обнаружено: 470 [M+H]+.
Протокол Синтеза 3

2-Хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (описанный в WO 2014011900) может быть замещен 1,2-монозащищенным циклоалкилдиамином при различных условиях реакции нуклеофильного замещения в ароматическом ядре с применением основания (такого как диизопропилэтиламин (DIPEA), DBU или NaHCO3) в полярном растворителе (таком как диоксан, CH3CN или NMP) или посредством медиированной палладием реакции конденсации Бухвальда для получения диамин-замещенного хиназолина. Защитную группу на амине удаляют, чтобы открыть аминный заместитель циклоалкана. Амин может вступать в реакцию с пропионовой кислотой с применением условий реакции конденсации амида или вступать в реакцию с акрилоилхлоридом для получения акриламида. Как приведено ниже, Соединения 27, 32, 34, 36 и 40 получали с применением Протокола Синтеза 3.
Соединение 27
Синтез N-[(3R,4S)-4-{[6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил]амино}оксолан-3-ил]проп-2-енамида

Этап 1: Синтез трет-бутил((3R,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)тетрагидрофуран-3-ил)карбамата в виде светло-желтой пены

Смесь 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина (1,02 г, 2,76 ммоль), трет-бутил((3R,4S)-4-аминотетрагидрофуран-3-ил)карбамата (0,85 г, 4,20 ммоль) и бикарбоната натрия (0,58 г, 6,90 ммоль) перемешивали в NMP (5,5 мл, 0,5 M) при 95°C в течение 12 часов.
Реакционную смесь удаляли с масляной бани и при охлаждении до комнатной температуры обрабатывали с помощью около 90 мл воды, а затем обрабатывали ультразвуком и перемешивали в течение 20 минут. Желто-оранжевое твердое вещество выделяли с помощью фильтрования, промывали несколько раз небольшими количествами воды и высушивали под вакуумом в течение почти 1 часа с получением 3,35 г неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 1,10 г (74,5% выхода) трет-бутил((3R,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)тетрагидрофуран-3-ил)карбамата в виде светло-желтой пены. МС (ЭР+) C25H28Cl2N4O5 требует: 534, обнаружено: 535 [M+H]+.
Этап 2: Синтез (3S,4R)-N3-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)тетрагидрофуран-3,4-диамина
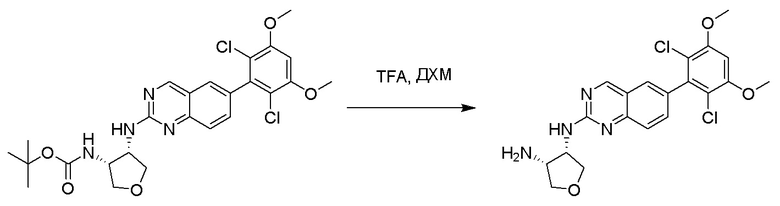
Раствор трет-бутил((3R,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)тетрагидрофуран-3-ил)карбамата (1,097 г, 2,049 ммоль) в ДХМ (15 мл, 0,137 M) и TFA (11,7 г, 102 ммоль) перемешивали около 40 минут при комнатной температуре. Избыточные растворители удаляли при пониженном давлении. Желтое масло растворяли в ДХМ (~60 мл) и промывали водным 1N NaOH (~30 мл). Водный слой затем разбавляли насыщенным раствором соли (~15 мл) и экстрагировали с помощью свежего ДХМ (3×30 мл). Объединенные органические слои высушивали над сульфатом натрия, отфильтровывали, сгущали и высушивали с получением (3S,4R)-N3-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)тетрагидрофуран-3,4-диамина в виде очень светло-желтой пены(0,879 г, 99%).
Этап 3: Синтез N-[(3R,4S)-4-{[6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил]амино}оксолан-3-ил]проп-2-енамида

К раствору (3S,4R)-N3-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)тетрагидрофуран-3,4-диамина (0,94 г, 2,1 ммоль) в дихлорметане (25 мл) при 0°C добавляли DIEA (0,37 мл, 2,1 ммоль) и акрилоилхлорид (0,17 мл, 2,1 ммоль) и перемешивали реакционную смесь в течение 3 ч. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением N-((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)акриламида (Соединение 27) (0,8 г, 76%). МС (ЭР+) C23H22Cl2N4O4 требует: 488, обнаружено: 489
Соединение 32
Синтез N-((1S,2R,3S,5S)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)бицикло[3.1.0]гексан-3-ил)акриламида

Этап 1: Синтез 2-(триметилсилил)этил(1S,2R,3S,5S)-2-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-иламино)бицикло[3.1.0]гексан-3-илкарбамата

Раствор 2-(триметилсилил)этил(1S,2R,3S,5S)-2-аминобицикло[3.1.0]гексан-3-илкарбамата (250 мг, 0,977 ммоль), 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина (300 мг, 0,814 ммоль) и бикарбоната натрия (205 мг, 2,442 ммоль) в N-метил-2-пирролидоне (10 мл) перемешивали при 100°C в течение ночи. Реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом (100 мл) и промывали водой (восемь раз) и соляным раствором (50 мл). Органический слой высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить неочищенный продукт, который очищали с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=4:2), чтобы обеспечить указанное в названии соединение (300 мг, 52%) в виде желтого твердого вещества. МС (ЭР+) C28H34Cl2N4O4Si требует: 588, 590, обнаружено: 589, 591 [M+H]+.
Этап 2: Синтез (1S,2R,3S,5S)-N2-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)бицикло[3.1.0]гексан-2,3-диамина

К раствору 2-(триметилсилил)этил(1S,2R,3S,5S)-2-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-иламино)бицикло[3.1.0]гексан-3-илкарбамата (200 мг, 340 ммоль) в диоксане (10 мл) добавляли 12 M конц. HCl (1 мл) при комнатной температуре. Получаемую смесь перемешивали в течение ночи, затем гасили водой (50 мл) и доводили pH раствора до pH=8-9 с помощью насыщенного раствора карбоната натрия. Смесь растворов экстрагировали этилацетатом (3 × 50 мл) и промывали объединенные слои насыщенным раствором соли (50 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии (преп-ТСХ) (дихлорметан:метанол=15:1) и затем дополнительно очищали с помощью колоночной хроматографии на силикагеле (дихлорметан:метанол=20:1), чтобы обеспечить указанное в названии соединение (70 мг, 46%) в виде белого твердого вещества. МС (ЭР+) C22H22Cl2N4O2 требует: 444, 446, обнаружено: 445, 447 [M+H]+.
Этап 3: Синтез N-((1S,2R,3S,5S)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)бицикло[3.1.0]гексан-3-ил)акриламида

К раствору (1S,2R,3S,5S)-N2-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)бицикло[3.1.0]гексан-2,3-диамина (42 мг, 0,094 ммоль) в дихлорметане (1,9 мл) при 0°C добавляли DIEA (0,025 мл, 0,14 ммоль) и акрилоилхлорид (0,009 мл, 0,11 ммоль) и перемешивали реакционную смесь в течение 1 ч. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением N-((1S,2R,3S,5S)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)бицикло[3.1.0]гексан-3-ил)акриламида (36 мг, 76%) в виде бледно-желтого твердого вещества. МС (ЭР+) C25H24Cl2N4O3 требует: 498, обнаружено: 499
Соединение 34
Синтез N-((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)акриламида
 Этап 1: Синтез трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)карбамата:
Этап 1: Синтез трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)карбамата:
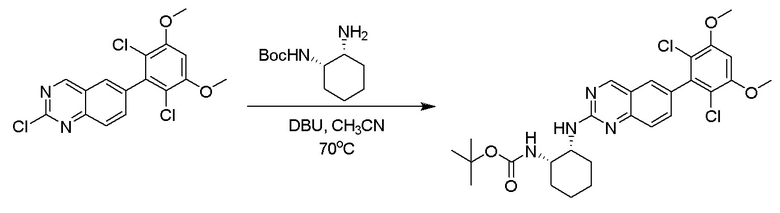
Смесь 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина (0,95 г, 2,6 ммоль), трет-бутил((1S,2R)-2-аминоциклогексил)карбамата (1,1 г, 5,14 ммоль) и DBU (0,77 мл, 5,14 ммоль) в ацетонитриле (9 мл) дегазировали с помощью N2 в течение 5 минут и нагревали при 70°C в течение 16 ч. Смесь охлаждали до комнатной температуры, концентрировали и очищали остаток с помощью колоночной хроматографии на силикагеле, чтобы обеспечить трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)карбамат (1,1 г, 81%). МС (ЭР+) C27H32Cl2N4O4 требует: 546, обнаружено: 547 [M+H]+.
Этап 2: Синтез (1R,2S)-N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)циклогексан-1,2-диамина
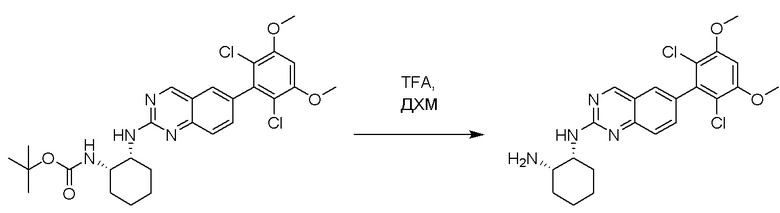
Смесь трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)карбамата (1,14 г, 2,1 ммоль) и 4N HCl в диоксане (5,2 мл) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 30 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь концентрировали до получения (1R,2S)-N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)циклогексан-1,2-диамина (0,94 г, 100%), который применяли на следующем этапе без дополнительной очистки.
Этап 3: Синтез N-((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)акриламида

К раствору (1R,2S)-N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)циклогексан-1,2-диамина (0,94 г, 2,1 ммоль) в дихлорметане (25 мл) при 0°C добавляли DIEA (0,37 мл, 2,1 ммоль) и акрилоилхлорид (0,17 мл, 2,1 ммоль) и перемешивали реакционную смесь в течение 3 ч. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением N-((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)акриламида (0,8 г, 76%). МС (ЭР+) C25H26Cl2N4O3 требует: 500, обнаружено: 501.
Соединение 36
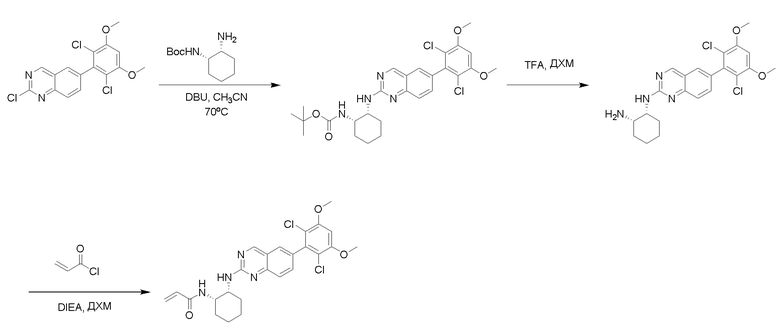
Синтез N-((1S,2S)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)акриламида

Этап 1: Синтез трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)карбамата
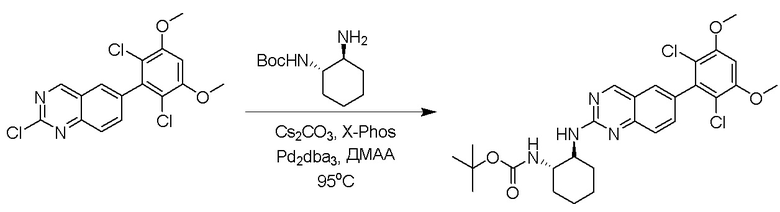
Смесь 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина (0,1 г, 0,27 ммоль), трет-бутил((1S,2S)-2-аминоциклогексил)карбамата (75 мг, 0,35 ммоль), Cs2CO3 (176 мг, 0,54 ммоль), X-Phos (13 мг, 0,027 ммоль) и Pd2dba3 (12,5 мг, 0,013 ммоль) в DMA (1,8 мл) дегазировали с помощью N2 в течение 5 минут и нагревали в микроволновом реакторе при 125°C в течение 30 минут. Смесь охлаждали до комнатной температуры, отфильтровывали сквозь целит и промывали водой, а затем насыщенным солевым раствором. Остаток очищали с помощью колоночной хроматографии на силикагеле, чтобы обеспечить трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)карбамат (67 мг, 45%). МС (ЭР+) C27H32Cl2N4O4 требует: 546, обнаружено: 547 [M+H]+.
Этап 2: Синтез (1R,2S)-N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)циклогексан-1,2-диамина

Смесь трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)карбамата (67 мг, 0,12 ммоль) и TFA (0,6 мл) в дихлорметане (0,6 мл) перемешивали при комнатной температуре в течение 60 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь разбавляли насыщенным NaHCO3, а затем экстрагировали с помощью дихлорметана. Объединенные органические слои высушивали над Na2SO4, отфильтровывали, концентрировали до получения (1R,2S)-N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)циклогексан-1,2-диамина, который применяли на следующем этапе без дополнительной очистки.
Этап 3: Синтез N-((1S,2S)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)акриламида
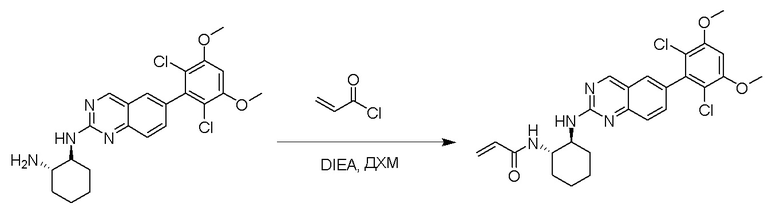
К раствору (1R,2S)-N1-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)циклогексан-1,2-диамина (0,12 ммоль) в дихлорметане (1,3 мл) при 0°C добавляли DIEA (0,004 мл, 0,02 ммоль) и акрилоилхлорид (0,012 мл, 0,15 ммоль) и перемешивали реакционную смесь в течение 1 ч. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением N-((1S,2S)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклогексил)акриламида (35 мг, 58%). МС (ЭР+) C25H26Cl2N4O3 требует: 500, обнаружено: 501.
Соединение 40
Синтез N-((3S,4S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)тетрагидро-2H-пиран-4-ил)акриламида

Этап 1: Синтез N-((3S,4S)-4-азидотетрагидро-2H-пиран-3-ил)-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-амина

(3S,4S)-4-Азидотетрагидро-2H-пиран-3-амин, HCl (0,200 г, 1,120 ммоль) и 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (0,318 г, 0,861 ммоль) помещали в NMP (2 мл) и добавляли карбонат натрия (0,217 г, 2,58 ммоль). Реакционную смесь нагревали до 100°C в течение ночи. После охлаждения до температуры окружающей среды реакционную смесь выливали в 5 мл воды и перемешивали в течение 30 минут. Твердый слой фильтровали, промывали водой и дополнительно высушивали под высоким вакуумом до получения N-((3S,4S)-4-азидотетрагидро-2H-пиран-3-ил)-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-амина (0,300 г, 0,631 ммоль, 73,3% выхода). МС (ЭР+) C21H20Cl2N6O3 требует: 474, обнаружено: 475 [M+H]+.
Этап 2: Синтез (3S,4S)-N3-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)тетрагидро-2H-пиран-3,4-диамина

N-((3S,4S)-4-Азидотетрагидро-2H-пиран-3-ил)-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-амин (0,063 г, 0,133 ммоль) помещали в метанол (7 мл) и EtOAc (7,00 мл), добавляли Pd-C (0,014 г, 0,133 ммоль) и перемешивали в емкости под атмосферой H2 в течение 1 часа. После того, как реакция была завершена, его отфильтровывали сквозь целит и удаляли растворитель. (3S,4S)-N3-(6-(2,6-Дихлор-3,5-диметоксифенил)хиназолин-2-ил)тетрагидро-2H-пиран-3,4-диамин (0,060 г, 0,134 ммоль, 101% выхода) извлекали в виде желтого твердого вещества, которое применяли без дополнительной очистки. МС (ЭР+) C21H22Cl2N4O3 требует: 448, обнаружено: 449 [M+H]+.
Этап 3: Синтез N-((3S,4S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)тетрагидро-2H-пиран-4-ил)акриламида
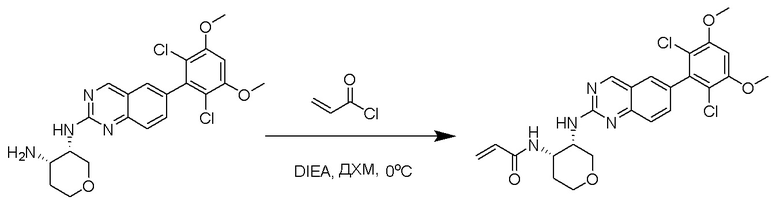
(3S,4S)-N3-(6-(2,6-Дихлор-3,5-диметоксифенил)хиназолин-2-ил)тетрагидро-2H-пиран-3,4-диамин (0,060 г, 0,134 ммоль) помещали в CH2Cl2 (2 мл) и охлаждали до 0°C, после чего добавляли DIEA (0,023 мл, 0,134 ммоль), а затем медленно акрилоилхлорид (0,012 мл, 0,147 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 минут, затем смесь сразу загружали на силикагель и очищали с помощью флэш-хроматографии, с применением 0-10% CH2Cl2/MeOH. N-((3S,4S)-3-((6-(2,6-Дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)тетрагидро-2H-пиран-4-ил)акриламид (0,041 г, 0,081 ммоль, 61% выхода) извлекали в виде белесоватого твердого вещества. МС (ЭР+) C24H24Cl2N4O4 требует: 502, обнаружено: 503 [M+H]+.
Протокол Синтеза 4
 2-Хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (описанный в WO 2014011900) может быть замещен 1,2-монозащищенным пирролидин диамином при условиях реакции нуклеофильного замещения в ароматическом ядре с применением основания (такого как NaHCO3) в полярном растворителе (таком как NMP) для получения диамин-замещенного хиназолина. Защитную группу на амине удаляют при соответствующих условиях, чтобы открыть аминный заместитель пирролидина. Амин может вступать в реакцию с акрилоилхлоридом для получения акриламида. Как приведено ниже, Соединения 56 и 83 получали с применением Протокола Синтеза 4.
2-Хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (описанный в WO 2014011900) может быть замещен 1,2-монозащищенным пирролидин диамином при условиях реакции нуклеофильного замещения в ароматическом ядре с применением основания (такого как NaHCO3) в полярном растворителе (таком как NMP) для получения диамин-замещенного хиназолина. Защитную группу на амине удаляют при соответствующих условиях, чтобы открыть аминный заместитель пирролидина. Амин может вступать в реакцию с акрилоилхлоридом для получения акриламида. Как приведено ниже, Соединения 56 и 83 получали с применением Протокола Синтеза 4.
Соединение 56
Синтез N-((3S,4R)-1-ацетил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-3-ил)акриламида
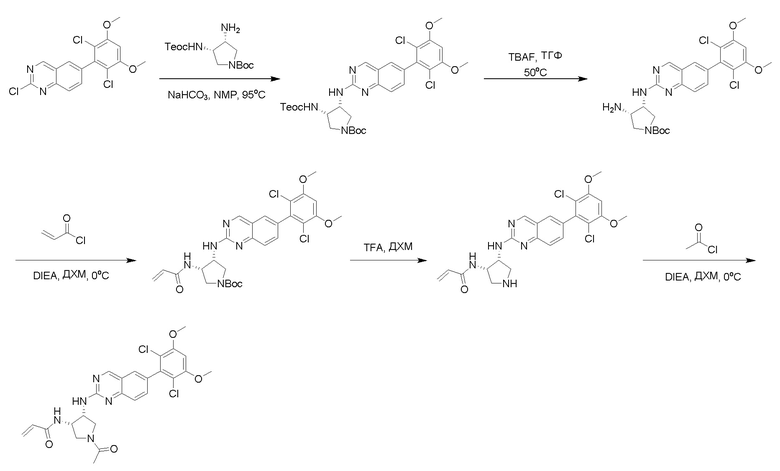
Этап 1: Синтез трет-бутил(3R,4S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(((2-(триметилсилил)этокси)карбонил)амино)пирролидин-1-карбоксилата

Смесь 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина (2,65 г, 7,17 ммоль), трет-бутил(3R,4S)-3-амино-4-(((2,2,2-трихлорэтокси)карбонил)амино)пирролидин-1-карбоксилата (2,97 г, 8,6 ммоль) и бикарбоната натрия (2,41 г, 28,7 ммоль) перемешивали в NMP (40 мл) при 95°C в течение 16 часов. Реакционную смесь удаляли с масляной бани, охлаждали до комнатной температуры и добавляли к 300 мл воды. Желто-оранжевое твердое вещество выделяли с помощью фильтрования, промывали несколько раз небольшими количествами воды и высушивали под вакуумом в течение почти 1 часа с получением 5 г неочищенного продукта, который очищали с помощью хроматографии на силикагеле с получением 2,82 г (58% выхода) трет-бутил(3R,4S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(((2-(триметилсилил)этокси)карбонил)амино)пирролидин-1-карбоксилата. МС (ЭР+) C31H41Cl2N5O6Si требует: 677, обнаружено: 678 [M+H]+.
Этап 2: Синтез трет-бутил(3S,4R)-3-амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
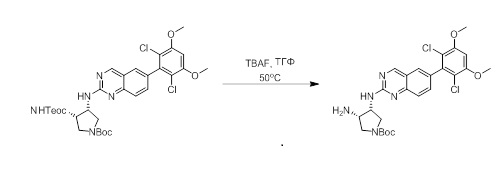
Смесь трет-бутил(3R,4S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(((2-(триметилсилил)этокси)карбонил)амино)пирролидин-1-карбоксилата (2,77 г, 4,1 ммоль) и 1 M TBAF в ТГФ (6,1 мл, 6,1 ммоль) перемешивали при в ТГФ (27 мл) при 50°C в течение 4 ч., а затем 16 ч. при комнатной температуре. Реакционную смесь разбавляли с помощью 10% метанола в дихлорметане (100 мл) и промывали водой (50 мл). Водный слой затем экстрагировали с помощью свежего дихлорметана (3×20 мл). Объединенные органические слои промывали насыщенным солевым раствором, высушивали над сульфатом натрия, отфильтровывали, сгущали и высушивали с получением трет-бутил(3S,4R)-3-амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата в виде желтого твердого вещества (2,1 г, 94%).
Этап 3: Синтез трет-бутил(3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
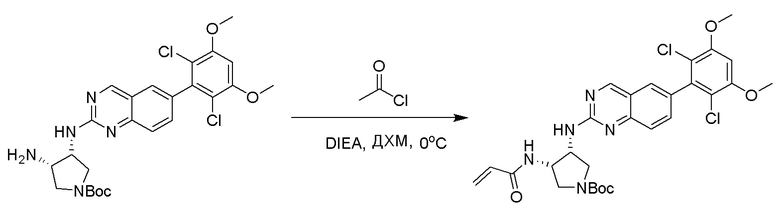
К раствору (3S,4R)-3-амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (2,1 г, 4,1 ммоль) в дихлорметане (82 мл) при 0°C добавляли DIEA (1,07 мл, 6,1 ммоль) и акрилоилхлорид (0,36 мл, 4,5 ммоль) и перемешивали реакционную смесь в течение 30 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением трет-бутил(3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (1,26 г, 52%). МС (ЭР+) C28H31Cl2N5O5 требует: 587, обнаружено: 588.
Этап 4: Синтез N-((3S,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-3-ил)акриламида

Раствор трет-бутил(3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (1,26 г, 2,14 ммоль) в ДХМ (8 мл) и TFA (3 мл, 39 ммоль) перемешивали 3 ч. при комнатной температуре. Избыточные растворители удаляли при пониженном давлении. Желтое масло растворяли в ДХМ (~100 мл) и промывали водным насыщенным раствором бикарбоната натрия (~50 мл). Водный слой затем экстрагировали с помощью свежего ДХМ (3×30 мл). Объединенные органические слои высушивали над сульфатом натрия, отфильтровывали, сгущали и высушивали с получением N-((3S,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-3-ил)акриламида, который применяли на следующем этапе без дополнительной очистки.
Этап 5: Синтез N-((3S,4R)-1-ацетил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-3-ил)акриламида

К раствору N-((3S,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-3-ил)акриламида (0,37 г, 0,76 ммоль) в дихлорметане (15 мл) при 0°C добавляли DIEA (0,16 мл, 0,92 ммоль) и ацетилхлорид (0,054 мл, 0,76 ммоль) и перемешивали реакционную смесь в течение 60 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением N-((3S,4R)-1-ацетил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-3-ил)акриламида (0,207 г, 51%). МС (ЭР+) C25H25Cl2N5O4 требует: 529, обнаружено: 530.
Соединение 83
Синтез (3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N-этилпирролидин-1-карбоксамида

К раствору N-((3S,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-3-ил)акриламида (0,040 г, 0,082 ммоль) в дихлорметане (1,5 мл) при 0°C добавляли TEA (0,014 мл, 0,098 ммоль) и этилизоцианат (0,008 мл, 0,098 ммоль) и перемешивали реакционную смесь в течение 45 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением (3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N-этилпирролидин-1-карбоксамида (0,035 г, 76%). МС (ЭР+) C26H28Cl2N6O4 требует: 558, обнаружено: 559.
Протокол Синтеза 5

2-Cl гетероцикл (описанный в WO 2014/011900) может быть замещен 1,2-монозащищенным диамином посредством медиированной палладием реакции конденсации Бухвальда для получения диамин-замещенного гетероцикла. Защитную группу на амине затем удаляют, чтобы открыть аминный заместитель циклоалкана. Амин может вступать в реакцию с пропионовой кислотой с применением условий реакции конденсации амида, чтобы обеспечить пропаргиловый амид или вступать в реакцию с акрилоилхлоридом для получения акриламида. Как приведено ниже, Соединение 62 получали с применением Протокола Синтеза 5.
Соединение 62
Синтез N-((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-этил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)циклопентил)акриламида
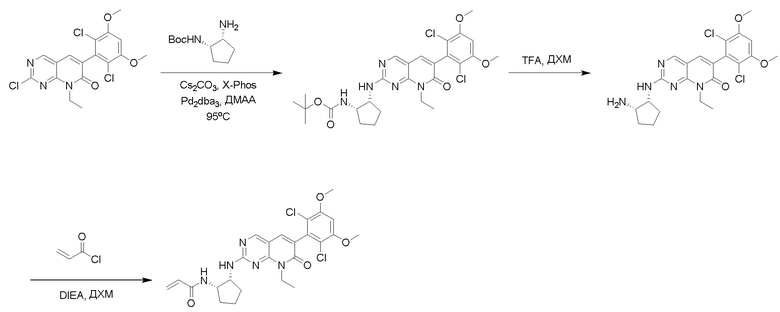 Этап 1: Синтез трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-этил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)циклопентил)карбамата
Этап 1: Синтез трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-этил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)циклопентил)карбамата

Смесь 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-8-этилпиридо[2,3-d]пиримидин-7(8H)-она (0,2 г, 0,48 ммоль), трет-бутил((1S,2R)-2-аминоциклопентил)карбамата (145 мг, 0,72 ммоль), Cs2CO3 (393 мг, 1,21 ммоль), X-Phos (23 мг, 0,048 ммоль) и Pd2dba3 (22 мг, 0,024 ммоль) в DMA (3,2 мл) дегазировали с помощью N2 в течение 5 минут и нагревали в микроволновом реакторе при 115°C в течение 60 минут. Смесь охлаждали до комнатной температуры, разбавляли с помощью EtOAc, отфильтровывали сквозь целит и промывали водой (4x), а затем насыщенным солевым раствором. Остаток очищали с помощью колоночной хроматографии на силикагеле, чтобы обеспечить трет-бутил-трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-этил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)циклопентил)карбамат (60 мг, 22%). МС (ЭР+) C27H33Cl2N5O5 требует: 577, обнаружено: 578 [M+H]+.
Этап 2: Синтез 2-(((1R,2S)-2-аминоциклопентил)амино)-6-(2,6-дихлор-3,5-диметоксифенил)-8-этилпиридо[2,3-d]пиримидин-7(8H)-она

Смесь трет-бутил-трет-бутил((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-этил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)циклопентил)карбамата (60 мг, 0,105 ммоль) и TFA (0,5 мл) в дихлорметане (2 мл) перемешивали при комнатной температуре в течение 90 минут. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь разбавляли насыщенным NaHCO3, а затем экстрагировали с помощью дихлорметана. Объединенный органические слои высушивали над Na2SO4, отфильтровывали, концентрировали до получения 2-(((1R,2S)-2-аминоциклопентил)амино)-6-(2,6-дихлор-3,5-диметоксифенил)-8-этилпиридо[2,3-d]пиримидин-7(8H)-она, который применяли на следующем этапе без дополнительной очистки.
Этап 3: Синтез N-((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-этил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)циклопентил)акриламида

К раствору 2-(((1R,2S)-2-аминоциклопентил)амино)-6-(2,6-дихлор-3,5-диметоксифенил)-8-этилпиридо[2,3-d]пиримидин-7(8H)-она (0,105 ммоль) в дихлорметане (2,1 мл) при -20°C добавляли DIEA (0,018 мл, 0,105 ммоль) и акрилоилхлорид (0,008 мл, 0,105 ммоль) и перемешивали реакционную смесь в течение 1 ч. ЖХ-МС указывала на полное расходование ИМ. Реакционную смесь очищали с помощью хроматографии на силикагеле с получением N-((1S,2R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)-8-этил-7-оксо-7,8-дигидропиридо[2,3-d]пиримидин-2-ил)амино)циклопентил)акриламида (36 мг, 65%). МС (ЭР+) C25H27Cl2N5O4 требует: 531, обнаружено: 532.
Протокол Синтеза 6
 2-Cl гетероцикл может быть замещен 1,2-транс-аминоспиртом посредством различных условиях реакции нуклеофильного замещения в ароматическом ядре с применением основания (такого как диизопропилэтиламин (DIPEA), DBU или NaHCO3) в полярном растворителе (таком как диоксан, CH3CN или NMP) или посредством медиированной палладием реакции конденсации Бухвальда для получения диамин-замещенного хиназолина. Спиртовой заместитель циклоалкана вступает в реакцию при условиях реакции нуклеофильного замещения (например, реакции Мицунобу) с получением защищенного амина. Удаление защитной группы на амине (такой как гидразин в случае фталимидной защитной группы) дает аминный заместитель циклоалкана. Амин может вступать в реакцию с пропаргиловой кислотой (с применением условий реакции конденсации амида, такого как HATU, DIPEA) или вступать в реакцию с акрилоилхлоридом для получения конечных соединений. Как приведено ниже, Соединения 81 и 82 получали с применением Протокола Синтеза 6.
2-Cl гетероцикл может быть замещен 1,2-транс-аминоспиртом посредством различных условиях реакции нуклеофильного замещения в ароматическом ядре с применением основания (такого как диизопропилэтиламин (DIPEA), DBU или NaHCO3) в полярном растворителе (таком как диоксан, CH3CN или NMP) или посредством медиированной палладием реакции конденсации Бухвальда для получения диамин-замещенного хиназолина. Спиртовой заместитель циклоалкана вступает в реакцию при условиях реакции нуклеофильного замещения (например, реакции Мицунобу) с получением защищенного амина. Удаление защитной группы на амине (такой как гидразин в случае фталимидной защитной группы) дает аминный заместитель циклоалкана. Амин может вступать в реакцию с пропаргиловой кислотой (с применением условий реакции конденсации амида, такого как HATU, DIPEA) или вступать в реакцию с акрилоилхлоридом для получения конечных соединений. Как приведено ниже, Соединения 81 и 82 получали с применением Протокола Синтеза 6.
Соединения 81 и 82
Синтез (1S,3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентанкарбоксамида и (1R,3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентан-1-карбоксамида

Этап 1: Синтез рацемического метил(3R,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)-4-гидроксициклопентан-1-карбоксилата
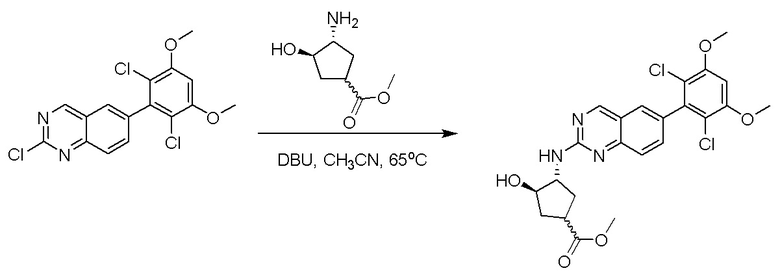
2-Хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (0,576 г, 1,558 ммоль), метил(3R,4R)-3-амино-4-гидроксициклопентан-1-карбоксилат (0,372 г, 2,337 ммоль) помещали в ацетонитрил (3 мл) и добавляли DBU (0,470 мл, 3,12 ммоль). Реакционную смесь продували N2 в течение 5 минут, а затем нагревали до 65°C в течение ночи. После охлаждения до комнатной температуры, растворитель удаляли при пониженном давлении. Остаток очищали посредством флэш-хроматографии (0-100% Hex/EtOAc; 12 г колонка) и извлекали (1S,3R,4R)-метил 3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-гидроксициклопентанкарбоксилат (0,520 г, 1,056 ммоль, 67,8% выхода). МС (ЭР+) C23H23Cl2N3O5 требует: 492, обнаружено: 493 [M+H]+.
Этап 2: Синтез рацемического метил(3R,4S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(1,3-диоксоизоиндолин-2-ил)циклопентан-1-карбоксилата

Ph3P (0,213 г, 0,812 ммоль) помещали в ТГФ (6 мл) и охлаждали до -78°C под N2. Добавляли DIAD (0,126 мл, 0,650 ммоль), после чего добавляли фталимид (0,105 г, 0,711 ммоль) и перемешивали при -78°C в течение 1 часа, после чего добавляли (1S,3R,4R)-метил-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-гидроксициклопентанкарбоксилат (0,100 г, 0,203 ммоль) в 4 мл ТГФ при -78°C. Реакционную смесь перемешивали в течение ночи, давая нагреться до комнатной температуры, после чего растворитель удаляли при пониженном давлении. Остаток очищали посредством флэш-хроматографии (0-100% Hex/EtOAc; 12 г колонка), чтобы обеспечить метил(3R,4S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(1,3-диоксоизоиндолин-2-ил)циклопентан-1-карбоксилат (0,126 г, 0,203 ммоль). МС (ЭР+) C31H26Cl2N4O6 требует: 621, обнаружено: 622 [M+H]+.
Этап 3: Синтез рацемического метил(3S,4R)-3-амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклопентан-1-карбоксилата

(1S,3R,4S)-Метил-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(1,3-диоксоизоиндолин-2-ил)циклопентанкарбоксилат (0,500 г, 0,805 ммоль) помещали в EtOH (20 мл) и добавляли моногидрат гидразина (0,079 мл, 1,61 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Белый осадок фильтровали и удаляли растворитель при пониженном давлении. Осадок растирали с эфиром, с последующим удалением растворителя при пониженном давлении до получения метил(3S,4R)-3-амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклопентан-1-карбоксилата (0,395 г, 0,805 ммоль) с количественным выходом, который применяли без дополнительной очистки. МС (ЭР+) C23H24Cl2N4O4 требует: 491, обнаружено: 492 [M+H]+.
Этап 4: Синтез рацемического метил(3S,4R)-3-((трет-бутоксикарбонил)амино)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклопентан-1-карбоксилата
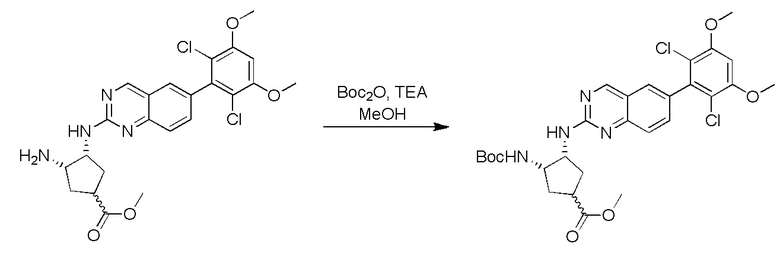
(3S,4R)-3-Амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклопентан-1-карбоксилат (0,550 г, 1,119 ммоль) помещали в метанол (10 мл), после чего добавляли Et3N (0,156 мл, 1,119 ммоль) и BOC-ангидрид (0,286 мл, 1,231 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи. После удаления растворителя под вакуумом, остаток помещали в ДХМ и промывали водой (2x), высушивали над сульфатом натрия и удаляли растворитель при пониженном давлении до получения метил(3S,4R)-3-((трет-бутоксикарбонил)амино)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклопентан-1-карбоксилата (0,662 г, 1,119 ммоль), который применяли без дополнительной очистки. МС (ЭР+) C28H32Cl2N4O6 требует: 591, обнаружено: 592 [M+H]+.
Этап 5: Синтез рацемической (3S,4R)-3-((трет-бутоксикарбонил)амино)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклопентан-1-карбоновой кислоты

Mетил(3S,4R)-3-((трет-бутоксикарбонил)амино)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)циклопентан-1-карбоксилат (0,662 г, 1,119 ммоль) помещали в метанол (10 мл), ТГФ (4 мл) и обрабатывали с помощью 10 мл 1N NaOH. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Органические растворители удаляли при пониженном давлении, затем водный слой подкисляли с помощью 1N HCl до pH ~2. Водный слой экстрагировали с помощью EtOAcx3. Органические слои объединяли, высушивали над сульфатом натрия и удаляли растворитель до получения неочищенной (3S,4R)-3-((трет-бутоксикарбонил)амино)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклопентан-1-карбоновой кислоты (0,580 г, 1,00 ммоль, 91% выхода), которую применяли с дополнительной очисткой. МС (ЭР+) C27H30Cl2N4O6 требует: 577, обнаружено: 578 [M+H]+.
Этап 6: Синтез трет-бутил((1S,2R,4S)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(диметилкарбамоил)циклопентил)карбамата и трет-бутил((1S,2R,4R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(диметилкарбамоил)циклопентил)карбамата

(3S,4R)-3-((трет-Бутоксикарбонил)амино)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)циклопентан-1-карбоновую кислоту (0,270 г, 0,468 ммоль) помещали в DMF (3 мл), HATU (0,267 г, 0,701 ммоль), добавляли 2M диметиламин в ТГФ (0,250 мл, 0,500 ммоль) и DIEA (0,245 мл, 1,403 ммоль) и перемешивали при температуре окружающей среды в течение 30 минут. Реакцию завершали после мониторинга с помощью ЖХ-МС, которая показывала два пика, содержащие правильную массу. Реакционную смесь очищали посредством обращенно-фазовой хроматографии (5-60% ацетонитрил/вода+0,01% муравьиной кислоты; 12 г колонка). Пик A: трет-бутил((1S,2R,4R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(диметилкарбамоил)циклопентил)карбамат (0,086 г, 0,142ммоль) МС (ЭР+) C29H35Cl2N5O5 требует: 604, обнаружено: 605 [M+H]+, время удерживания 3,039. Пик B: трет-бутил((1S,2R,4S)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(диметилкарбамоил)циклопентил)карбамат (0,062 г, 0,103ммоль) МС (ЭР+) C29H35Cl2N5O5 требует: 604, обнаружено: 605 [M+H]+, время удерживания 2,879. Примечание: абсолютная конфигурация была обозначена произвольно.
Этап 7a: Синтез (1S,3S,4R)-3-амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентан-1-карбоксамида

трет-Бутил((1S,2R,4R)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(диметилкарбамоил)циклопентил)карбамат (0,086 г, 0,142ммоль) помещали в ДХМ (2 мл) и обрабатывали с помощью 4 M HCl в диоксане(3 мл) и перемешивали в течение 3 часов. Растворитель удаляли до получения неочищенного (1S,3S,4R)-3-амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентан-1-карбоксамида, с количественным выходом. МС (ЭР+) C24H27Cl2N5O3 требует: 504, обнаружено: 505 [M+H]+.
Этап 8a: Синтез (1R,3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентан-1-карбоксамида

(1S,3S,4R)-3-Амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентанкарбоксамид (0,050 г, 0,099 ммоль) помещали в CH2Cl2 (25 мл) и охлаждали до 0°C, после чего добавляли DIEA (0,017 мл, 0,099 ммоль), затем медленно акрилоилхлорид (8,86 мкл, 0,109 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 минут. После того, как реакция была завершена, реакционную смесь сразу загружали на силикагель и очищали посредством флэш-хроматографии (0-10% CH2Cl2/MeOH; 12 г колонка), чтобы обеспечить(1R,3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентанкарбоксамид (0,043 г, 0,077 ммоль, 78% выхода). МС (ЭР+) C27H29Cl2N5O4 требует: 558, обнаружено: 559 [M+H]+.
Этап 7b: Синтез (1R,3S,4R)-3-амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентан-1-карбоксамида
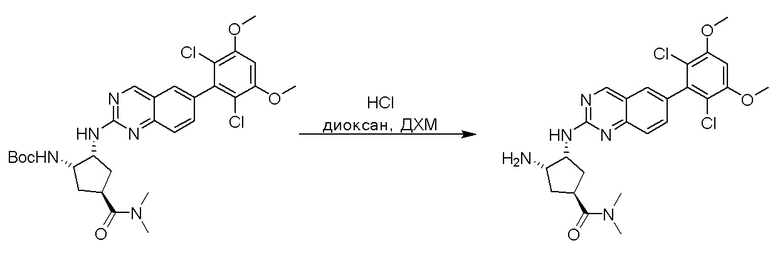
трет-Бутил((1S,2R,4S)-2-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-(диметилкарбамоил)циклопентил)карбамат (0,062 г, 0,103ммоль) помещали в ДХМ (2 мл) и обрабатывали с помощью 4 M HCl в диоксане(3 мл) и перемешивали в течение 3 часов. Растворитель удаляли до получения неочищенного (1S,3S,4R)-3-амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентан-1-карбоксамида, с количественным выходом. МС (ЭР+) C24H27Cl2N5O3 требует: 504, обнаружено: 505 [M+H]+.
Этап 8b: Синтез (1S,3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентан-1-карбоксамида
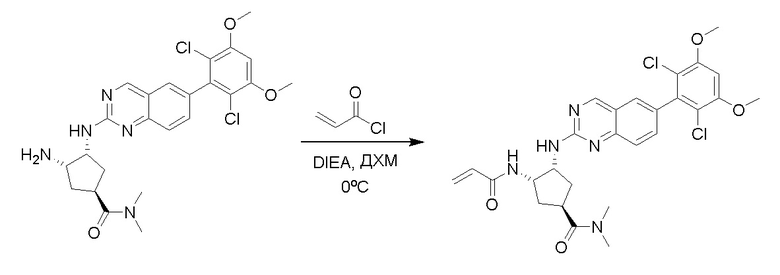
(1R,3S,4R)-3-Амино-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентанкарбоксамид (0,050 г, 0,099 ммоль) помещали в CH2Cl2 (25 мл) и охлаждали до 0°C, после чего добавляли DIEA (0,017 мл, 0,099 ммоль), затем медленно акрилоилхлорид (8,86 мкл, 0,109 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 минут. После того, как реакция была завершена, ее сразу загружали на силикагель и очищали посредством флэш-хроматографии (0-10% CH2Cl2/MeOH; 12 г колонка), чтобы обеспечить (1S,3S,4R)-3-акриламидо-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,N-диметилциклопентанкарбоксамид (0,029 г, 0,052 ммоль, 52,4% выхода). МС (ЭР+) C27H29Cl2N5O4 требует: 558, обнаружено: 559 [M+H]+.
Получение некоторых интермедиатов
Синтез трет -бутил((3R,4S)-4-аминотетрагидрофуран-3-ил)карбамата
 Этап 1: Синтез трет-бутил((3S,4R)-4-гидрокситетрагидрофуран-3-ил)карбамата
Этап 1: Синтез трет-бутил((3S,4R)-4-гидрокситетрагидрофуран-3-ил)карбамата
Интермедиат (3R,4S)-4-аминотетрагидрофуран-3-ол получали таким же образом, как и в WO 01/29013 (PCT/US00/28815; pp. 44-45; Пример 1). Раствор (3R,4S)-4-аминотетрагидрофуран-3-ола (10,6 г, 103 ммоль), триэтиламина (26 г, 257 ммоль) и BOC ангидрида (24,7 г, 113 ммоль) в метаноле (206 мл, 0,5 M) перемешивали при комнатной температуре в течение 45 часов. Растворители затем удаляли при пониженном давлении. Твердое вещество бежевого цвета обрабатывали водой (около 120 мл). Белое кристаллическое твердое вещество выделяли с помощью фильтрации и высушивали в течение ночи под вакуумом с получением трет-бутил((3S,4R)-4-гидрокситетрагидрофуран-3-ил)карбамата в виде белого твердого вещества (17,08 г, 82%).
Этап 2: Синтез трет-бутил((3R,4S)-4-(1,3-диоксоизоиндолин-2-ил)тетрагидрофуран-3-ил)карбамата
Смесь трет-бутил((3S,4R)-4-гидрокситетрагидрофуран-3-ил)карбамата (15,36 г, 76 ммоль), фталимида (13,34 г, 91 ммоль) и трифенилфосфина (23,8 г, 91 ммоль) перемешивали в ТГФ (378 мл, 0,2 M) при 0°C в течение 10 минут перед добавлением по каплям DIAD (18,34 г, 91 ммоль) за 20 минут. Реакционную смесь перемешивали около 40 минут при 0°C. Растворитель удаляли при пониженном давлении, а неочищенное масло обрабатывали с помощью менее чем 50 мл диэтилового эфира и обрабатывали ультразвуком. Образовался белый осадок. Твердое вещество выделяли с помощью фильтрации, промывали небольшими количествами эфира и высушивали с получением 10,62 г белого твердого вещества. Остывший фильтрат снова отфильтровывали с получением дополнительных 2,54 г белого твердого вещества с общим выходом 13,16 г трет-бутил((3R,4S)-4-(1,3-диоксоизоиндолин-2-ил)тетрагидрофуран-3-ил)карбамата.
Этап 3: Синтез трет-бутил((3R,4S)-4-аминотетрагидрофуран-3-ил)карбамата
трет-Бутил((3R,4S)-4-(1,3-диоксоизоиндолин-2-ил)тетрагидрофуран-3-ил)карбамат (13,08 г, 39,4 ммоль) растворяли в этаноле (98 мл, 0,4 M)). Добавляли моногидрат гидразина(1,97 г, 39,4 ммоль) и перемешивали реакционную смесь 30 минут при 50°C, а затем 2 часа при 75°C. Реакционную смесь затем охлаждали до комнатной температуры и удаляли белое твердое вещество с помощью фильтрации. Фильтрат сгущали и высушивали, затем обрабатывали этанолом (около 15 мл). Дополнительное белое твердое вещество удаляли с помощью фильтрации, затем фильтрат сгущали и высушивали с получением трет-бутил((3R,4S)-4-аминотетрагидрофуран-3-ил)карбамата в виде густого прозрачного масла (8,724 г при 90% чистоты; 99%).
Синтез (3S,4S)-4-азидотетрагидро-2H-пиран-3-амина

Этап 1: Синтез (3R,4R)-4-гидрокситетрагидро-2H-пиран-3-ил)карбамата

(3R,4R)-3-(((S)-1-Фенилэтил)амино)тетрагидро-2H-пиран-4-ол (2,0 г, 9,04 ммоль) помещали в метанол (10 мл), после чего добавляли Et3N (1,260 мл, 9,04 ммоль) и BOC-ангидрид (2,308 мл, 9,94 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворители затем удаляли in vaccuo, а остаток помещали в ДХМ (10 мл) и гексан (20 мл) и нагревали до 80°C до тех пор, пока уровень растворителя не снижали наполовину. Реакционную смесь удаляли от источников тепла и охлаждали до комнатной температуры при перемешивании. Затем добавляли 5 мл простого эфира и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Реакционную смесь отфильтровывали для удаления твердых веществ, промывали эфиром и высушивали, чтобы обеспечить трет-бутил((3R,4R)-4-гидрокситетрагидро-2H-пиран-3-ил)карбамат (1,6 г, 7,36 ммоль, 81% выхода) в виде белого твердого вещества.
Этап 2: Синтез (3R,4R)-3-((трет-бутоксикарбонил)амино)тетрагидро-2H-пиран-4-илметансульфоната

трет-Бутил((3R,4R)-4-гидрокситетрагидро-2H-пиран-3-ил)карбамат (1,6 г, 7,36 ммоль) помещали в CH2Cl2 (20 мл) и охлаждали до 0°C, после чего добавляли Et3N (1,232 мл, 8,84 ммоль). Спустя 5 минут по каплям добавляли метансульфонилхлорид (0,631 мл, 8,10 ммоль) в ДХМ (5 мл). Реакционную смесь перемешивали при 0°C в течение 30 минут и оставляли нагреваться до температуры окружающей среды при перемешивании в течение 2 часов. Реакционную смесь разбавляли водой и ДХМ и разделяли слои. Органические слои объединяли и промывали водой дважды, высушивали над Na2SO4 и удаляли растворитель in vacuo. Остаток высушивали под высоким вакуумом в течение ночи, чтобы обеспечить извлеченный (3R,4R)-3-((трет-бутоксикарбонил)амино)тетрагидро-2H-пиран-4-илметансульфонат (2,2 г, 7,45 ммоль, 100% выхода) в виде белого твердого вещества.
Этап 3: Синтез трет-бутил((3S,4S)-4-азидотетрагидро-2H-пиран-3-ил)карбамата
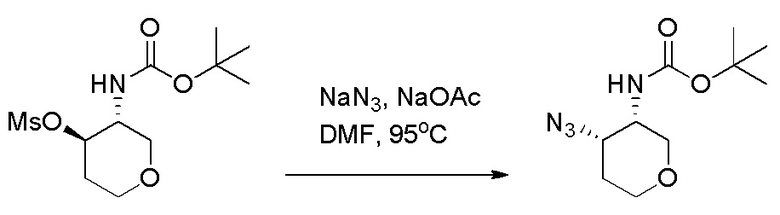
(3R,4R)-3-((трет-Бутоксикарбонил)амино)тетрагидро-2H-пиран-4-илметансульфонат (2,2 г, 7,45 ммоль), азид натрия (0,968 г, 14,90 ммоль) и ацетат натрия (1,222 г, 14,90 ммоль) помещали в DMF (15 мл). Реакционную смесь нагревали до 95°C в течение ночи. Реакционную смесь удаляли от источников тепла, добавляли 20 мл воды и перемешивали при охлаждении. Реакционную смесь экстрагировали с помощью EtOAc. Органические слои объединяли и промывали водой. Органические слои высушивали и удаляли растворитель до получения трет-бутил((3S,4S)-4-азидотетрагидро-2H-пиран-3-ил)карбамата (1,8 г, 7,43 ммоль, 100% выхода) в виде желтого масла. МС (ЭР+) C10H18N4O3 требует: 242, обнаружено: 265 [M+Na]+.
Этап 4: Синтез (3S,4S)-4-азидотетрагидро-2H-пиран-3-амина

трет-Бутил((3S,4S)-4-азидотетрагидро-2H-пиран-3-ил)карбамат (1,5 г, 6,19 ммоль) помещали в ДХМ (5 мл) и добавляли 4N HCl диоксан (4,64 мл, 18,57 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли до получения (3S,4S)-4-азидотетрагидро-2H-пиран-3-амина (1,1 г, 6,16 ммоль, 99% выхода) в виде соли HCl. МС (ЭР+) C5H10N4O требует: 142, обнаружено: 143 [M+H]+.
Синтез 2-(триметилсилил)этил(1S,2R,3S,5S)-2-аминобицикло[3.1.0]гексан-3-илкарбамата
 Этап 1: Синтез (R)-4-бензил-3-пент-4-еноилоксазолидин-2-она
Этап 1: Синтез (R)-4-бензил-3-пент-4-еноилоксазолидин-2-она
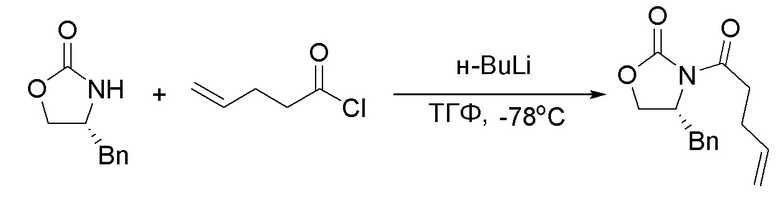
К раствору (4R)-4-(фенилметил)-1,3-оксазолидин-2-она (50 г, 282 ммоль) в ТГФ (300 мл) по каплям добавляли н-BuLi в ТГФ (2,4 M, 176 мл, 423 ммоль) под азотом при -78°C, а получаемую смесь перемешивали при -78°C в течение 1 часа. Затем по каплям добавляли 4-пентеноилхлорид (49 мл, 423 ммоль). После перемешивания при -78°C в течение еще 1 часа, реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. После разбавления водой, смесь экстрагировали этилацетатом (2 × 400 мл). Объединенные этилацетатные экстракты промывали насыщенным раствором соли, высушивали над сульфатом натрия, отфильтровывали и концентрировали при пониженном давлении. Неочищенный осадок очищали с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1: 10), чтобы обеспечить указанное в названии соединение (68 г, 93%) в виде светло-желтого масла. МС (ЭР+) C15H17NO3 требует: 259, обнаружено: 260 [M+H]+.
Этап 2: Синтез (R)-3-((2S,3S,E)-2-аллил-3-гидрокси-5-фенилпент-4-еноил)-4-бензилоксазолидин-2-она

Смесь (R)-4-бензил-3-пент-4-еноилоксазолидин-2-она (50 г, 193 ммоль), хлорида магния (18,3 г, 193 ммоль), гексафторантимоната(V) натрия (14,9 г, 58 ммоль), триэтиламина (80 мл, 579 ммоль), (транс)-коричного альдегида (30,6 г, 232 ммоль) и хлортриметилсилана (37,2 мл, 290 ммоль) в этилацетате (500 мл) перемешивали при комнатной температуре в течение 17 часов. Смесь разбавляли этилацетатом и отфильтровывали для удаления твердых веществ. Фильтрат концентрировали до малого объема, а затем разбавляли с помощью метанола (500 мл) и малого объема этилацетата. После обработки трифторуксусной кислотой (3 мл), полученный раствор перемешивали при комнатной температуре в течение 1 ч., а затем концентрировали досуха при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1:10), чтобы обеспечить указанное в названии соединение (60 г, 80%) в виде желтого мягкого вещества. МС (ЭР+) C24H25NO4 требует: 391, обнаружено: 374 [M+H - H2O]+.
Этап 3: Синтез (S)-5-бензил-1-((1S,2S)-2-гидроксициклопент-3-енкарбонил)пирролидин-2-она

Раствор (R)-3-((2S,3S,E)-2-аллил-3-гидрокси-5-фенилпент-4-еноил)-4-бензилоксазолидин-2-она (50 г, 128 ммоль) и катализатора Граббса 2го поколения (5,4 г, 6,4 ммоль) в толуоле (300 мл) дегазировали азотом три раза и перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем концентрировали досуха при пониженном давлении и очищали остаток с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1: 4), чтобы обеспечить указанное в названии соединение (32 г, 87%) в виде темно-коричневого масла, которое затвердевает при отстаивании. МС (ЭР+) C17H19NO3 требует: 285, обнаружено: 270 [M+H-H2O]+.
Этап 4: Синтез (R)-4-бензил-3-((1S,2S,3S,5S)-2-гидроксибицикло[3.1.0]гексан-3-карбонил)оксазолидин-2-она

Раствор (S)-5-бензил-1-((1S,2S)-2-гидроксициклопент-3-енкарбонил)пирролидин-2-она (25 г, 87,1 ммоль) в дихлорметане (300 мл) охлаждали на бане со льдом и обрабатывали с помощью добавления по каплям 1 M диэтилцинка в гексане (435 мл, 435 ммоль). После перемешивания при 0°C в течение 20 минут, по каплям добавляли дийодметан (69,6 мл, 871 ммоль). Полученный мутный раствор перемешивали при 0°C в течение еще 20 минут, а затем оставляли нагреваться до комнатной температуры. После перемешивания в течение 6 часов при комнатной температуре, реакционную смесь гасили насыщенным водным раствором хлорида аммония и экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, высушивали над сульфатом натрия, отфильтровывали и концентрировали досуха при пониженном давлении. Неочищенный материал очищали с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1:4), чтобы обеспечить указанное в названии соединение (308 мг, 89%) в виде светло-коричневого вязкого масла. МС (ЭР+) C17H19NO4 требует: 301, обнаружено: 284 [M+H - H2O]+.
Этап 5: Синтез (R)-4-бензил-3-((1S,2S,3S,5S)-2-(трет-бутилдиметилсилилокси)-бицикло[3.1.0]гексан-3-карбонил)оксазолидин-2-она
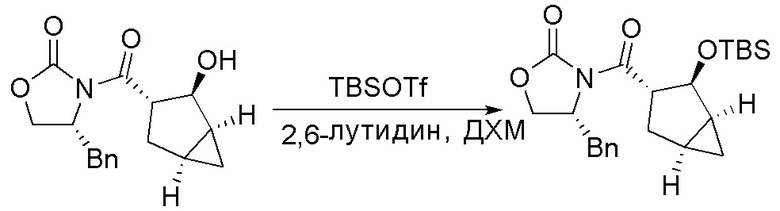
К перемешиваемому раствору (R)-4-бензил-3-((1S,2S,3S,5S)-2-гидроксибицикло[3.1.0]гексан-3-карбонил)оксазолидин-2-она (25 г, 83 ммоль) и 2,6-лутидина (38,2 мл, 332 ммоль) в дихлорметане (300 мл) добавляли трет-бутилдиметилсилилтрифторметансульфонат (47,6 мл, 207,5 ммоль) при 0°C под азотом. Получаемую смесь перемешивали при 0°C в течение 30 минут, а затем при комнатной температуре в течение 1 часа. После того, как разбавляли метанолом (25 мл), смесь выливали в воду и экстрагировали диэтиловым эфиром (2×400 мл). Объединенные эфирные экстракты промывали насыщенным раствором соли, высушивали над сульфатом натрия, отфильтровывали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1: 8), чтобы обеспечить указанное в названии соединение (29 г, 86%) в виде бесцветного масла. МС (ЭР+) C23H33NO4Si требует: 415, обнаружено: 416 [M+H - H2O]+.
Этап 6: Синтез (1S,2S,3S,5S)-2-(трет-бутилдиметилсилилокси)бицикло[3.1.0]гексан-3-карбоновой кислоты

К раствору (R)-4-бензил-3-((1S,2S,3S,5S)-2-(трет-бутилдиметилсилилокси)бицикло[3.1.0] гексан-3-карбонил)оксазолидин-2-она (40 г, 96,4 ммоль) в ТГФ (200 мл) и воде (50 мл) добавляли по каплям 30% водную перекись водорода (88 мл, 771 ммоль) при 0°C, с последующим добавлением раствора моногидрата гидроксида лития (16 г, 386 ммоль) в воде (100 мл). После перемешивания в течение 1 часа при 0°C, реакционную смесь перемешивали при комнатной температуре в течение ночи. Избыток пероксида водорода полностью израсходовали добавлением насыщенного водного бисульфата натрия. Смесь затем доводили до pH=14 с помощью 1 н. NaOH и промывали эфиром (400 мл). Водный слой затем подкисляли до pH=3 с помощью 1 M водного гидросульфата калия и экстрагировали этилацетатом (3×400 мл). Объединенные органические экстракты промывали насыщенным раствором соли, высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (22 г, 88%) в виде бесцветного масла.
Этап 7: Синтез бензил(1S,2S,3S,5S)-2-(трет-бутилдиметилсилилокси)бицикло[3.1.0]гексан-3-илкарбамата

К раствору (1S,2S,3S,5S)-2-(трет-бутилдиметилсилилокси)бицикло[3.1.0]гексан-3-карбоновой кислоты (4 г, 15,625 ммоль), триэтиламина (22 мл, 156 ммоль) и бензилового спирта (17 мл, 156 ммоль) в толуоле (50 мл) при комнатной температуре добавляли по каплям дифенилфосфорилазид (33,7 мл, 156 ммоль), а получаемую смесь перемешивали при 100°C в течение ночи. Реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом (100 мл) и промывали водой (3 × 50 мл) и соляным раствором (50 мл). Органический слой высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить неочищенный продукт, который очищали с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1: 8), чтобы обеспечить указанное в названии соединение (3,0 г, 54%) в виде белого твердого вещества. МС (ЭР+) C20H31NO3Si требует: 361, обнаружено: 362 [M+H]+.
Этап 8: Синтез бензил(1S,2S,3S,5S)-2-гидроксибицикло[3.1.0]гексан-3-илкарбамата

К раствору бензил(1S,2S,3S,5S)-2-(трет-бутилдиметилсилилокси)бицикло[3.1.0]гексан-3-илкарбамата (2,0 г, 5,540 ммоль) в ТГФ (20) при комнатной температуре добавляли 1 M тетрабутиламмонийфторид в ТГФ (55 мл, 55,4 ммоль), а смесь перемешивали при комнатной температуре в течение ночи. Реакционный раствор разбавляли этилацетатом (100 мл) и промывали водой (3 × 50 мл) и соляным раствором (50 мл). Органический слой высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (1,2 г, 92%) в виде белого твердого вещества. МС (ЭР+) C14H17NO3 требует: 247, обнаружено: 230 [M+H - H2O]+.
Этап 9: Синтез бензил(1S,2R,3S,5S)-2-(1,3-диоксоизоиндолин-2-ил)бицикло[3.1.0]гексан-3-илкарбамата

Раствор трифенилфосфина (6,4 г, 24,292 ммоль), фталимида (6,2 г, 42,511 ммоль) и бензил(1S,2S,3S,5S)-2-гидроксибицикло[3.1.0]гексан-3-илкарбамата (3,0 г, 12,146 ммоль) в толуоле (250 мл) перемешивали при -78°C в течение 30 минут под защитой азота, с последующим добавлением по каплям диизопропилазодикарбоксилата (8,6 мл, 42,511 ммоль). Получаемую смесь перемешивали при -78°C в течение еще 1 часа, а затем при комнатной температуре в течение ночи. Реакционную смесь обрабатывали с помощью 10 мл метанола и удаляли растворители при пониженном давлении. Неочищенный материал очищали с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1:2), чтобы обеспечить указанное в названии соединение (3,0 г, 65%) в виде светло-желтого масла. МС (ЭР+) C22H20N2O4 требует: 376, обнаружено: 399 [M+23]+.
Этап 10: Синтез 2-((1S,2R,3S,5S)-3-аминобицикло[3.1.0]гексан-2-ил)изоиндолин-1,3-диона
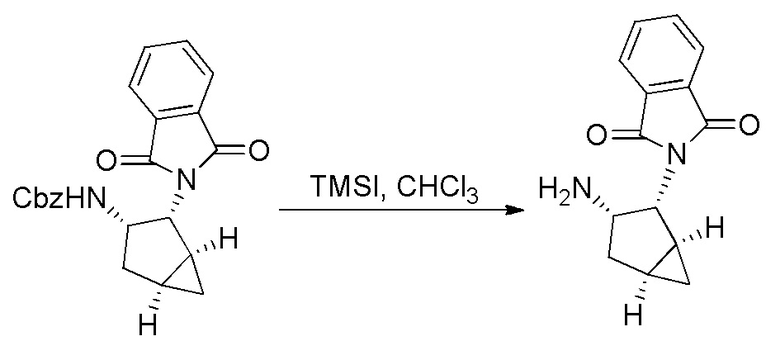
К раствору бензил(1S,2R,3S,5S)-2-(1,3-диоксоизоиндолин-2-ил)бицикло[3.1.0]гексан-3-илкарбамата (4,0 г, 10,638 ммоль) в хлороформе (30 мл) при комнатной температуре добавляли по каплям триметилсилилиодид (14,6 мл, 106,380 ммоль), а получаемую смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили метанолом (5 мл), разбавляли этилацетатом (150 мл) и промывали водой (3 × 50 мл) и соляным раствором (50 мл). Органический слой высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить неочищенное соединение, которое применяли непосредственно в следующей реакции без дополнительной очистки. МС (ЭР+) C14H14N2O2 требует: 242, обнаружено: 243 [M+H]+.
Этап 11: Синтез 2-(триметилсилил)этил(1S,2R,3S,5S)-2-(1,3-диоксоизоиндолин-2-ил)бицикло[3.1.0]гексан-3-илкарбамата

Раствор 2-((1S,2R,3S,5S)-3-аминобицикло[3.1.0]гексан-2-ил)изоиндолин-1,3-диона (2,0 г, 8,264 ммоль), 2,5-диоксопирролидин-1-ил-2-(триметилсилил)этилкарбоната (3,2 г, 12,396 ммоль) и триэтиламина (3,4 мл, 24,792 ммоль) в диоксане/воде (100 мл, об./об.=1/1) перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь затем разбавляли этилацетатом (100 мл), промывали с помощью 1 М соляной кислоты (2 × 50 мл), насыщенным раствором бикарбоната натрия (2 × 50 мл) и соляным раствором (50 мл). Органический слой концентрировали при пониженном давлении и очищали остаток с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1:4), чтобы обеспечить указанное в названии соединение (2,5 г, 78%) в виде желтого масла. МС (ЭР+) C20H26N2O4Si требует: 386, обнаружено: 410 [M+23]+.
Этап 12: Синтез 2-(триметилсилил)этил(1S,2R,3S,5S)-2-аминобицикло[3.1.0]гексан-3-илкарбамата
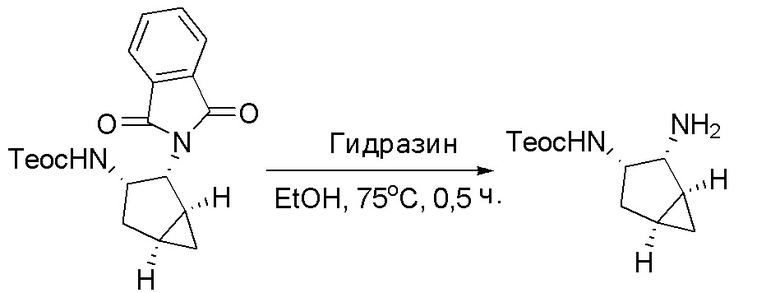
К раствору 2-(триметилсилил)этил(1S,2R,3S,5S)-2-(1,3-диоксоизоиндолин-2-ил)бицикло[3.1.0]гексан-3-илкарбамата (1,5 г, 3,886 ммоль) в этаноле (20 мл) при комнатной температуре добавляли гидразин (1,9 мл, 38,860 ммоль), а получаемую смесь перемешивали при 75°C в течение 2 часов. Реакционный раствор концентрировали и очищали остаток с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1:4), чтобы обеспечить указанное в названии соединение (800 мг, 80%) в виде светло-желтого мягкого вещества.
Синтез cis - трет -бутил-3-гидрокси-1,1-диоксогексагидро-1-тиопиран-4-илкарбамата
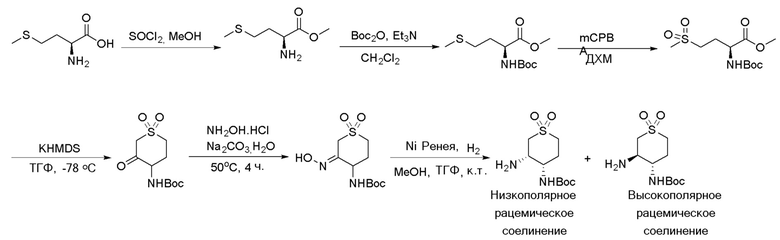 Этап 1: Синтез (S)-метил 2-амино-4-(метилтио)бутаноата
Этап 1: Синтез (S)-метил 2-амино-4-(метилтио)бутаноата

К высушенной над пламенем колбе под азотом добавляли метанол (60 мл). Перемешиваемый раствор охлаждали до 0ºC, перед тем, как по каплям добавляли тионилхлорид (7,32 мл, 100,34 ммоль). Раствор перемешивали при 0ºC в течение 10 минут, перед тем, как добавляли метионин (10 г, 33,8 ммоль) в виде одной порции. Реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего летучие вещества удаляли при пониженном давлении до получения указанного в названии соединения в виде желтоватого твердого вещества.
Этап 2: Синтез (S)-метил 2-(трет-бутоксикарбониламино)-4-(метилтио)бутаноата

К раствору (S)-метил 2-амино-4-(метилтио)бутаноата в дихлорметане (300 мл) при 0ºC добавляли триэтиламин (35 мл), с последующим добавлением ди-трет-бутилдикарбоната (26,98 г, 125 ммоль). После перемешивания при комнатной температуре в течение 3 ч., реакционную смесь разбавляли с помощью дихлорметана (200 мл) и промывали водой (2*150 мл). Объединенные органические слои высушивали (сульфат магния), отфильтровывали и концентрировали при пониженном давлении. Продукт (Rf=0,5, этилацетат:петролейный эфир, 1:4) очищали с помощью колоночной флэш-хроматографии, чтобы обеспечить указанное в названии соединение (15 г, 85% выхода) в виде прозрачного масла.
Этап 3: Синтез (S)-метил 2-(трет-бутоксикарбониламино)-4-(метилсульфонил)бутаноата

Сложный метиловый эфир N-(трет-бутоксикарбонил)-L-метионина (8,76 г, 33,3 ммоль) добавляли в 1000 мл круглодонную колбу и растворяли в дихлорметане (150 мл). Перемешиваемый раствор охлаждали до 0ºC, с последующим добавлением 3-хлорпероксибензойной кислоты (70%, 18,0 г, 7,32 ммоль) в 30 мл дихлорметана в течение 5 минут. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов, в течение которых ее разбавляли с помощью дихлорметана (200 мл) и гидрокарбоната натрия (300 мл насыщенного водного раствора). Органический слой разделяли, промывали последовательно гидрокарбонатом натрия (2 * 300 мл насыщенного водного раствора), высушивали (сульфат магния), отфильтровывали и концентрировали при пониженном давлении. Продукт очищали с помощью колоночной флэш-хроматографии (этилацетат:петролейный эфир, 6:4), чтобы обеспечить указанное в названии соединение (5 г, 51% выхода) в виде желтого твердого вещества.
Этап 4: Синтез трет-бутил(1,1-диоксидо-3-оксотетрагидро-2H-тиопиран-4-ил)карбамата
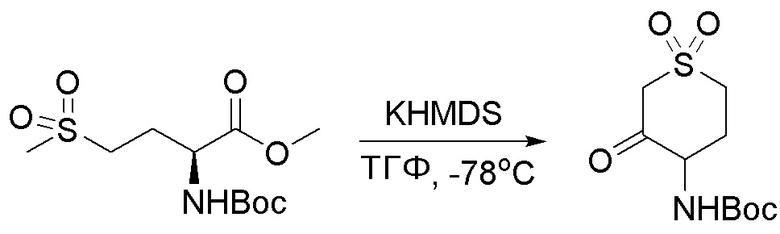
Раствор (S)-метил 2-(трет-бутоксикарбониламино)-4-(метилсульфонил)бутаноата (2 г, 6,78 ммоль) в тетрагидрофуране (50 мл) охлаждали до -78oC, к нему по каплям добавляли бис(триметилсилил)амид калия (1,0 M, раствор в толуоле, 15 мл) и перемешивали смесь при -78°C в течение 2 часов и при комнатной температуре в течение еще 2 часов. Добавляли водный раствор хлорида аммония (1 M) и перемешивали смесь. Реакционную смесь подвергали разделению в жидкости. Полученный органический слой затем промывали водой и соляным раствором и высушивали над безводным сульфатом магния. Растворитель удаляли при пониженном давлении, а образовавшееся твердое вещество собирали с помощью фильтрации для получения указанного в названии соединения. Водный слой, отделенный ранее, дважды экстрагировали этилацетатом. Полученные органические слои объединяли, промывали водой и соляным раствором и высушивали над безводным сульфатом магния. Этилацетатные экстракты объединяли, высушивали, а затем концентрировали при пониженном давлении для получения указанное в названии соединение. Объединенный продукт очищали с помощью колоночной флэш-хроматографии (этилацетат:петролейный эфир, 3:1), чтобы обеспечить указанное в названии соединение (55 мг, выход 22%) в виде желтого твердого вещества.
Этап 5: Синтез (Z)-трет-бутил(3-(гидроксиимино)-1,1-диоксидотетрагидро-2H-тиопиран-4-ил)карбамата

К смеси соединения 5 (50 мг, 0,189 ммоль) и карбоната натрия (64 мг, 0,757 ммоль) в воде (5 мл) добавляли гидрохлорид гидроксиламина (26 мг, 0,379 ммоль). После перемешивания при 50°C в течение 4 ч., реакционную смесь охлаждали до к.т. и отфильтровывали, чтобы получить указанное в названии соединение (50 мг, 95% выхода) в виде белого твердого вещества. МС (ЭР+) C10H18N2O5S требует: 278, обнаружено: 179 [M+H - 100]+, 223 [M+H - 56]+.
Этап 6: Синтез cis-трет-бутил-3-гидрокси-1,1-диоксогексагидро-1-тиопиран-4-илкарбамата

Смесь соединения (Z)-трет-бутил(3-(гидроксиимино)-1,1-диоксидотетрагидро-2H-тиопиран-4-ил)карбамата (2,5 г, 7,6ммоль) и ренеевского никелевого катализатора (избыточное количество) в метаноле (200 мл) и ТГФ (200 мл) перемешивали при комнатной температуре в емкости с атмосферой водорода в течение 24 часов. Смесь отфильтровывали и концентрировали фильтрат. Остаток очищали с помощью колоночной флэш-хроматографии (метанол:дихлорметан, 1:2), чтобы обеспечить рацемическую смесь малополярного соединения (400 мг, 16% выхода) и высокополярного соединения (600 мг, 25% выхода).
Этап 7: Синтез (3R,4S)-трет-бутил 3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата и (3S,4R)-трет-бутил 3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата
 Этап 8: Синтез транс-трет-бутил 4-(бензилоксикарбониламино)-3-гидроксипиперидин-1-карбоксилата
Этап 8: Синтез транс-трет-бутил 4-(бензилоксикарбониламино)-3-гидроксипиперидин-1-карбоксилата

К перемешиваемой смеси транс-трет-бутил 4-амино-3-гидроксипиперидин-1-карбоксилата (1,05 г, 4,86 ммоль) в 80 мл дихлорметана добавляли триэтиламин (5,89 г, 5,83 ммоль), с последующим добавлением N-(бензилоксикарбонилокси)сукцинимид (1,27 г, 5,10 ммоль) при 0oC. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, а затем разбавляли с помощью 100 мл дихлорметана. Смесь растворов промывали с помощью 5% раствора лимонной кислоты (2 X 100 мл), 5% раствора карбоната калия (2×100 мл) и соляным раствором (200 мл). Органический слой высушивали над безводным сульфатом натрия и отфильтровывали, с последующим концентрированием при пониженном давлении. Полученное маслянистое вещество очищали с помощью колоночной хроматографии на силикагеле (этилацетат: петролейный эфир=1:4~1:2), чтобы обеспечить указанное в названии соединение (1,7 г, ~100%, неочищенное) в виде бесцветного масла. МС (ЭР+) C18H26N2O5 требует: 350, обнаружено: 251 [M+H - 100]+.
Этап 9: Синтез транс-трет-бутил 4-(бензилоксикарбониламино)-3-(метилсульфонилокси)-пиперидин-1-карбоксилата
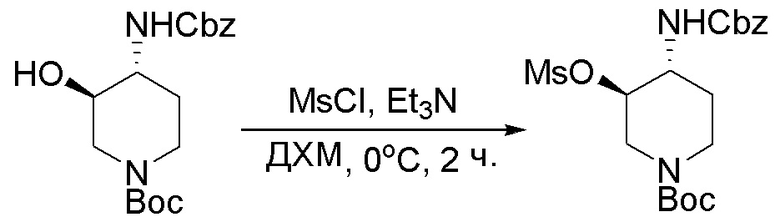
К раствору транс-трет-бутил 4-(бензилоксикарбониламино)-3-гидроксипиперидин-1-карбоксилата (5,0 г, 14,3 ммоль) и триэтиламина (4,5 г, 43,0 ммоль) в дихлорметане (100 мл) добавляли метансульфонилхлорид (4,9 г, 43,0 ммоль) при 0°C, а смесь перемешивали при 0°C в течение 2 часов. Раствор промывали водой (150 мл × 3) и соляным раствором, высушивали над безводным сульфатом натрия и отфильтровывали, с последующим концентрированием при пониженном давлении до получения указанного в названии соединения (6,0 г, неочищенное) в виде желтого масла. МС (ЭР+) C19H28N2O7S требует: 428, обнаружено: 329 [M+H - 100]+.
Этап 10: Синтез транс-трет-бутил-3-азидо-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата

К раствору транс-трет-бутил 4-(бензилоксикарбониламино)-3-(метилсульфонилокси)пиперидин-1-карбоксилата (6,0 г, 14 ммоль) в диметилсульфоксиде (40 мл) добавляли азид натрия (9,11 г, 140 ммоль), а реакционную смесь перемешивали при 90°C в течение ночи под N2. Смесь растворов охлаждали до ~30oC, разбавляли этилацетатом (~300 мл) и промывали водой (700 мл X 3) и соляным раствором. Органический слой высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали до указанного в названии соединения (3,8 г, 72%) в виде желтого масла. МС (ЭР+) C18H25N5O4 требует: 375, обнаружено: 276 [M+H - 100]+, 373 [M+Na]+.
Этап 11: Синтез (3R,4S)-трет-бутил 3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата и (3S,4R)-трет-бутил 3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата
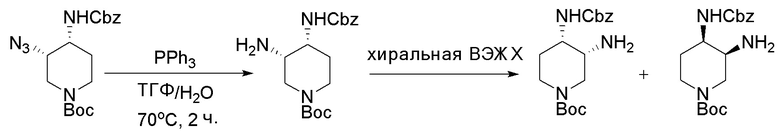
Смесь неочищенного транс-трет-бутил 3-азидо-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата (12 г, ~32 ммоль) и трифенилфосфина (41,9 г, 160 ммоль) в ТГФ (100 мл) и воде (5 мл) перемешивали при 70°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (500 мл). Органический слой промывали насыщенным раствором соли (50 мл) и сразу выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=4/1~1:1), чтобы обеспечить транс-трет-бутил 3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилат (5,0 г, 44%) в виде желтого масла. МС (ЭР+) C18H27N3O4 требует: 349, обнаружено: 350 [M+H]+.
2 г полученного выше рацемического образца разделяли с помощью хиральной ВЭЖХ, чтобы обеспечить (3R,4S)-трет-бутил 3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилат (550 мг, пик 1 в хиральной ВЭЖХ) и (3S,4R)-трет-бутил 3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилат (620 мг, пик 2 в хиральной ВЭЖХ).
Синтез cis - трет -бутил-4-амино-3-(((2-(триметилсилил)этокси)карбонил)амино)пиперидин-1-карбоксилата
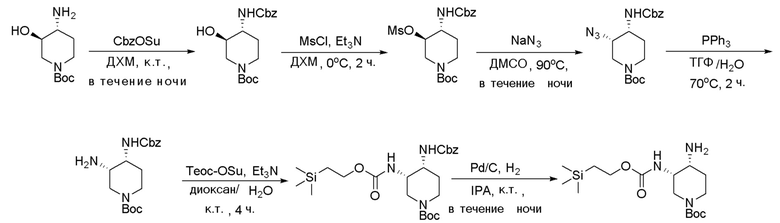
Этап 1: Синтез транс-трет-бутил 4-(бензилоксикарбониламино)-3-гидроксипиперидин-1-карбоксилата

К перемешиваемой смеси транс-трет-бутил 4-амино-3-гидроксипиперидин-1-карбоксилата (1,05 г, 4,86 ммоль) в 80 мл дихлорметана добавляли триэтиламин (5,89 г, 5,83 ммоль), с последующим добавлением N-(бензилоксикарбонилокси)сукцинимида (1,27 г, 5,10 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, а затем разбавляли с помощью 100 мл дихлорметана. Смесь растворов промывали с помощью 5% раствора лимонной кислоты (2×100 мл), 5% раствора карбоната калия (2×100 мл) и соляным раствором (200 мл). Органический слой высушивали над безводным сульфатом натрия и отфильтровывали, с последующим концентрированием при пониженном давлении. Полученное маслянистое вещество очищали с помощью колоночной хроматографии на силикагеле (этилацетат: петролейный эфир=1:4~1:2), чтобы обеспечить указанное в названии соединение (1,7 г, ~100%, неочищенное) в виде бесцветного масла. МС (ЭР+) C18H26N2O5 требует: 350, обнаружено: 251 [M+H - 100]+.
Этап 2: Синтез транс-трет-бутил 4-(бензилоксикарбониламино)-3-(метилсульфонилокси)пиперидин-1-карбоксилата

К раствору транс-трет-бутил 4-(бензилоксикарбониламино)-3-гидроксипиперидин-1-карбоксилата (5,0 г, 14,3 ммоль) и триэтиламина (4,5 г, 43,0 ммоль) в дихлорметане (100 мл) добавляли метансульфонилхлорид (4,9 г, 43,0 ммоль) при 0°C, а смесь перемешивали при 0°C в течение 2 часов. Раствор промывали водой (150 мл × 3) и соляным раствором, высушивали над безводным сульфатом натрия и отфильтровывали, с последующим концентрированием при пониженном давлении до получения указанного в названии соединения (6,0 г, неочищенное) в виде желтого масла. МС (ЭР+) C19H28N2O7S требует: 428, обнаружено: 329 [M+H - 100]+.
Этап 3: Синтез cis-трет-бутил 3-азидо-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата
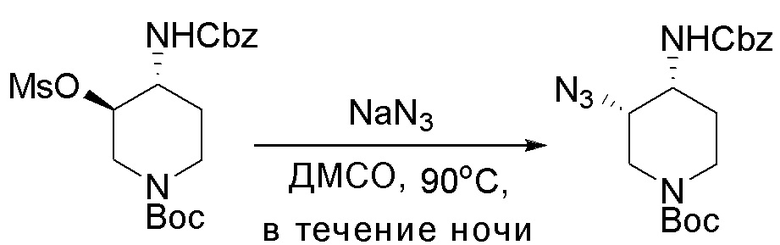
К раствору транс-трет-бутил 4-(бензилоксикарбониламино)-3-(метилсульфонилокси)пиперидин-1-карбоксилата (6,0 г, 14 ммоль) в диметилсульфоксиде (40 мл) добавляли азид натрия (9,11 г, 140 ммоль), а реакционную смесь перемешивали при 90°C в течение ночи под N2. Смесь растворов охлаждали до ~30oC, разбавляли этилацетатом (~300 мл) и промывали водой (700 мл x 3) и соляным раствором. Органический слой высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали до указанного в названии соединения (3,8 г, 72%) в виде желтого масла. МС (ЭР+) C18H25N5O4 требует: 375, обнаружено: 276 [M+H - 100]+, 373 [M+Na]+.
Этап 4: Синтез cis-трет-бутил 3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата и (3S,4R)-трет-бутил-3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата

Смесь неочищенного cis-трет-бутил 3-азидо-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата (12 г, ~32 ммоль) и трифенилфосфина (41,9 г, 160 ммоль) в ТГФ (100 мл) и воде (5 мл) перемешивали при 70°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (500 мл). Органический слой промывали насыщенным раствором соли (50 мл) и сразу выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=4/1~1:1), чтобы обеспечить указанное в названии соединение (рацемат, 5,0 г, 44%) в виде желтого масла. МС (ЭР+) C18H27N3O4 требует: 349, обнаружено: 350 [M+H]+.
Этап 5: Синтез cis-трет-бутил 4-(бензилоксикарбониламино)-3-((2-(триметилсилил)этокси)-карбониламино)пиперидин-1-карбоксилата

Раствор cis-трет-бутил 3-амино-4-(бензилоксикарбониламино)пиперидин-1-карбоксилата (3,0 г, 8,6 ммоль), 2,5-диоксопирролидин-1-ил-2-(триметилсилил)этилкарбоната (2,5 г, 9,5 ммоль) и триэтиламина в диоксане/воде(40 мл, об./об.=1/1) перемешивали при комнатной температуре в течение 4 часов. После этого раствор разбавляли этилацетатом (200 мл) и промывали с помощью 1 M соляной кислоты (50 мл), насыщенного раствора бикарбоната натрия (50 мл) и соляного раствора (50 мл). Органический слой высушивали над сульфатом натрия, отфильтровывали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=4/1), чтобы обеспечить указанное в названии соединение (3,5 г, 83%) в виде белого твердого вещества. МС (ЭР+) C24H39N3O6Si требует: 493, обнаружено: 516 [M+23]+.
Этап 6: Синтез cis-трет-бутил-4-амино-3-((2-(триметилсилил)этокси)-карбониламино)пиперидин-1-карбоксилата
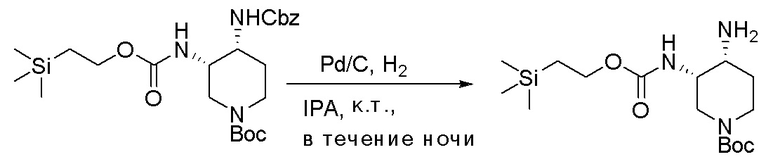
Смесь cis-трет-бутил-4-(бензилоксикарбониламино)-3-((2-(триметилсилил)этокси)карбониламино)пиперидин-1-карбоксилата (1,8 г, 3,6 ммоль) и 10% палладиевого катализатора на углеродном носителе (180 мг) в изопропаноле (60 мл) перемешивали при 1 атм в атмосфере водорода (емкость с атмосферой водорода) при комнатной температуре в течение 3 часов. После этого смесь отфильтровывали сквозь слой целита. Фильтрат концентрировали и очищали с помощью колоночной хроматографии на силикагеле (метанол/дихлорметан = от 1/30 до 1/10), чтобы обеспечить указанное в названии соединение (800 мг, 61%) в виде желтого масла. МС (ЭР+) C16H33N3O4Si требует: 359, обнаружено: 360 [M+H]+.
Синтез рацемата этил-4-амино-3-( трет -бутоксикарбониламино)циклогексанкарбоксилата
 Этап 1: Синтез рацемата 4-йод-6-окса-бицикло[3.2.1]октан-7-она
Этап 1: Синтез рацемата 4-йод-6-окса-бицикло[3.2.1]октан-7-она

К смеси циклогекс-3-енкарбоновой кислоты (рацемат, 42,0 г, 333 ммоль), йодида калия (72,0 г, 433 ммоль) и гидрокарбоната натрия (36,4 г, 433 ммоль) в метиленхлориде (750 мл) и воде (750 мл) добавляли йод (110,0 г, 433 ммоль) при внутренней температуре 5°C и перемешивали реакционную смесь при комнатной температуре в течение 3 часов. После того, как ее гасили с помощью 1 н водного тиосульфата натрия (1500 мл), получаемую смесь экстрагировали с помощью метиленхлорида (1000 мл × 2). Объединенные органические слои промывали водным гидрокарбонатом натрия (1000 мл), водой (2000 мл) и соляным раствором (1000 мл), высушивали над безводным сульфатом магния, отфильтровывали, затем концентрировали при пониженном давлении. Выпавшие в осадок кристаллы собирали с помощью фильтрации и промывали гексаном, с последующим высушиванием, чтобы тем самым получить указанное в названии соединение (80,2 г, 95%) в виде белого твердого вещества.
Этап 2: Синтез рацемата этил-7-окса-бицикло[4.1.0]гептан-3-карбоксилата

К суспензии рацемата 4-йод-6-окса-бицикло[3.2.1]октан-7-она (45,0 г, 180 ммоль) в этаноле (400 мл) добавляли 2 н водный гидроксид натрия (110 мл, 220 ммоль) при комнатной температуре при перемешивании, а получаемую смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали на бане при температуре 35°C при пониженном давлении. Воду (500 мл) добавляли к полученному маслянистому веществу, а получаемую смесь экстрагировали этилацетатом (500 мл). Органический слой промывали водой (500 мл), высушивали над безводным сульфатом натрия и отфильтровывали с последующим концентрированием при пониженном давлении. Полученное маслянистое вещество очищали с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир = 1:10 ~ 1:5), чтобы получить указанное в названии соединение (15,9 г, 52%) в виде бледно-желтого масла.
Этап 3: Синтез рацемата этил-3-азидо-4-гидроксициклогексанкарбоксилата

Смесь рацемата этил-7-окса-бицикло[4.1.0]гептан-3-карбоксилата (24,0 г, 140 ммоль), хлорида аммония (13,6 г, 210 ммоль) и азида натрия (13,7 г, 210 ммоль) в N,N-диметилформамиде (120 мл) перемешивали при 76°C в течение 13 часов. После того, как все нерастворимые вещества собирают с помощью фильтрования, фильтрат концентрировали при пониженном давлении, не позволяя растворителю испариться досуха. Остаток объединяли с твердым веществом, собранным с помощью предыдущей фильтрации, а полученную таким образом смесь растворяли в воде (500 мл). Раствор экстрагировали этилацетатом (500 мл). Экстракт промывали водой (500 мл x 5) и насыщенным соляным раствором, высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (28 г, неочищенное) в виде желтого масла. МС (ЭР+) C9H15N3O3 требует: 213, обнаружено: 214 [M+H]+, 236 [M+Na]+.
Этап 4: Синтез рацемата этил-3-(трет-бутоксикарбониламино)-4-гидроксициклогексанкарбоксилата
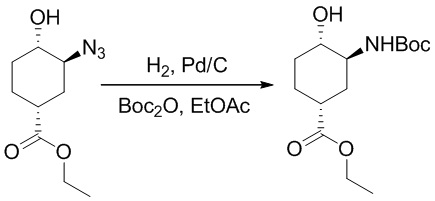
Смесь рацемата этил-3-азидо-4-гидроксициклогексанкарбоксилата (14,0 г, 66 ммоль), ди-трет-бутилдикарбоната (18,5 г, 85 ммоль) и 5% палладиевого катализатора на углеродном носителе (50% влажности, 2,5 г) в этилацетате (300 мл) перемешивали при комнатной температуре в течение ночи при давлении водорода ~1 атм. После того, как реакционную смесь отфильтровывали, фильтрат концентрировали, а полученное таким образом вещество очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=4:1-3:1). Полученное таким образом соединение кристаллизовали из гексана, чтобы тем самым получить указанное в названии соединение (12,0 г, 62%) в виде белого твердого вещества. МС (ЭР+) C14H25NO5 требует: 287, обнаружено: 188 [M+H - 100]+.
Этап 5: Синтез рацемата этил-3-(трет-бутоксикарбониламино)-4-(метилсульфонилокси)циклогексанкарбоксилата

К раствору рацемата этил-3-(трет-бутоксикарбониламино)-4-гидроксициклогексанкарбоксилата (12,0 г, 42 ммоль) и триэтиламина (12,7 г, 126 ммоль) в дихлорметане (150 мл) добавляли метансульфонилхлорид (9,5 г, 84 ммоль) по каплям при 0oC, а смесь перемешивали при 0°C в течение 3 часов. Раствор промывали водой (100 мл x 3) и соляным раствором, высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (15 г, неочищенное) в виде желтого масла. МС (ЭР+) C15H27NO7S требует: 365, обнаружено: 266 [M+H - 100]+.
Этап 6: Синтез рацемата этил-4-азидо-3-(трет-бутоксикарбониламино)-циклогексанкарбоксилата

К раствору рацемата этил-3-(трет-бутоксикарбониламино)-4-(метилсульфонилокси)циклогексанкарбоксилата (11,0 г, 30 ммоль) в диметилсульфоксиде (110 мл) добавляли азид натрия (20 г, 300 ммоль), а смесь перемешивали при 90°C в течение ночи под N2. Раствор охлаждали до ~30oC, растворяли в этилацетате (~500 мл), промывали водой (500 мл X 5) и соляным раствором, высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=4:1~2:1), чтобы тем самым получить указанное в названии соединение (4,1 г, 44%) в виде бесцветного масла. МС (ЭР+) C14H24N4O4 требует: 312, обнаружено: 213 [M+H - 100]+.
Этап 7: Синтез рацемата этил-4-амино-3-(трет-бутоксикарбониламино)-циклогексанкарбоксилата
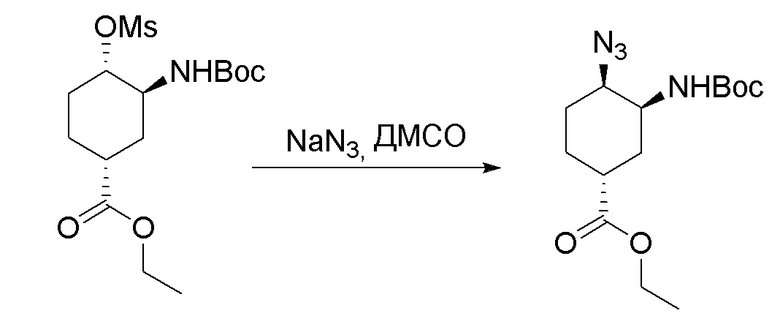
Смесь рацемата этил-4-азидо-3-(трет-бутоксикарбониламино)циклогексанкарбоксилата (14,0 г, 66 ммоль) и 5% палладиевого катализатора на углеродном носителе (50% влажности, 1,0 г) в этилацетате (100 мл) перемешивали при комнатной температуре в течение ночи при давлении водорода ~1 атм. После того, как реакционную смесь отфильтровывали, фильтрат концентрировали. Полученный маслянистый осадок очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=4:1~1:1), чтобы тем самым получить указанное в названии соединение (2,0 г, 59%) в виде желтого твердого вещества.
Синтез рацемата 4-амино-3-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-иламино)циклогексанкарбоксамида
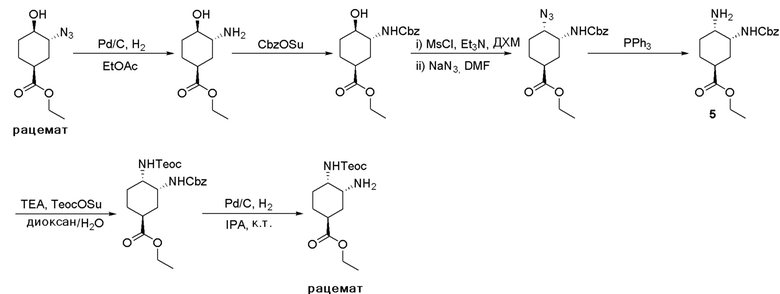 Этап 1: Синтез рацемата этил-3-амино-4-гидроксициклогексанкарбоксилата
Этап 1: Синтез рацемата этил-3-амино-4-гидроксициклогексанкарбоксилата

Суспензионная смесь рацемата этил-3-азидо-4-гидроксициклогексанкарбоксилата (8,0 г, 37,5 ммоль) и 5% палладиевого катализатора на углеродном носителе (50% влажности, 2,0 г) в этилацетате (250 мл) перемешивали под атмосферой водорода (~1 атм.) при комнатной температуре в течение ночи. После того, как реакционную смесь отфильтровывали, фильтрат концентрировали, чтобы тем самым получить указанное в названии соединение (5,8 г, 83%) в виде желтого твердого вещества. МС (ЭР+) C9H17NO3 требует: 187, обнаружено: 188 [M+H]+.
Этап 2: Синтез рацемата этил-3-(бензилоксикарбониламино)-4-гидроксициклогексанкарбоксилата

К перемешиваемой смеси рацемата этил 3-амино-4-гидроксициклогексанкарбоксилата (4,7 г, 25 ммоль) в 120 мл дихлорметана добавляли триэтиламин (3,03 г, 30 ммоль), с последующим добавлением N-(бензилоксикарбонилокси)сукцинимида (6,55 г, 26,3 ммоль) при 0oC. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, а затем разбавляли с помощью 200 мл дихлорметана. Раствор промывали 5% раствором лимонной кислоты (2×150 мл), 5% раствором карбоната калия (2×150 мл) и соляным раствором (200 мл). Органический слой разделяли, высушивали над безводным сульфатом натрия и отфильтровывали, с последующим концентрированием при пониженном давлении. Полученное маслянистое вещество очищали с помощью хроматографии на силикагеле (этилацетат:петролейный эфир=1:4 ~ 2:5), чтобы тем самым получить указанное в названии соединение (7,0 г, 87%) в виде желтого масла. МС (ЭР+) C17H23NO5 требует: 321, обнаружено: 322 [M+H]+.
Этап 3: Синтез рацемата этил-4-азидо-3-(бензилоксикарбониламино)-циклогексанкарбоксилата

К раствору рацемата этил-3-(бензилоксикарбониламино)-4-гидроксициклогексанкарбоксилата (7,0 г, 22 ммоль) и триэтиламина (6,7 г, 66 ммоль) в дихлорметане (100 мл) по каплям добавляли метансульфонилхлорид (5,1 г, 44 ммоль) при 0oC, а смесь перемешивали при этой температуре в течение 2 часов. Реакционную смесь промывали водой (200 мл x 3) и соляным раствором. Органический слой высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали, чтобы тем самым получить неочищенный продукт (8,0 г, неочищенное) в виде желтого масла. Смесь указанного выше остатка (8,0 г, 20 ммоль) и азида натрия (7,8 г, 120 ммоль) в диметилсульфоксиде (50 мл) перемешивали при 100°C в течение 18 часов. Реакционную смесь охлаждали до ~30oC, растворяли в воде (~300 мл) и экстрагировали этилацетатом (200 мл X 2). Объединенные органические слои промывали насыщенным раствором соли (~200 мл), высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали до получения указанного в названии соединения (3,5 г, 46% общего выхода двух этапов) в виде бесцветного масла. МС (ЭР+) C17H22N4O4 требует: 346, обнаружено: 347 [M+H]+, 369 [M+Na]+.
Этап 4: Синтез рацемата этил-3-(трет-бутоксикарбониламино)-4-гидроксициклогексанкарбоксилата

Смесь рацемата этил-4-азидо-3-(бензилоксикарбониламино)циклогексанкарбоксилата (3,5 г, 10 ммоль) и трифенилфосфина (10,4 г, 40 ммоль) в ТГФ (200 мл) и воде (10 мл) перемешивали при 65°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры, затем разбавляли этилацетатом (200 мл), промывали насыщенным раствором соли (200 мл) и выпаривали in vacuo. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат=2/1 ~ дихлорметан/метанол=10:1), чтобы обеспечить указанное в названии соединение (2,4 г, 75%) в виде желтого масла. МС (ЭР+) C17H24N2O4 требует: 320, обнаружено: 321 [M+H]+.
Этап 5: Синтез рацемата этил-3-(бензилоксикарбониламино)-4-((2-(триметилсилил)этокси)карбониламино)циклогексанкарбоксилата

Раствор рацемата этил-3-(трет-бутоксикарбониламино)-4-гидроксициклогексанкарбоксилата (1,6 г, 5,0 ммоль), 1,3-диоксоизоиндолин-2-ил 2-(триметилсилил)этилкарбоната (1,42 г, 5,5 ммоль) и триэтиламина (760 мг, 7,5 ммоль) в диоксане/воде (25/25 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли этилацетатом (200 мл) и промывали с помощью 1 M соляной кислоты (100 мл), насыщенного раствора бикарбоната натрия (100 мл) и соляного раствора (100 мл). Органический слой концентрировали и очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат=4/1), чтобы обеспечить указанное в названии соединение (2,3 г, 99%) в виде белого твердого вещества. МС (ЭР+) C23H36N2O6Si требует: 464, обнаружено: 487 [M+Na]+.
Этап 6: Синтез рацемата этил-3-амино-4-((2-(триметилсилил)этокси)карбониламино)циклогексанкарбоксилата
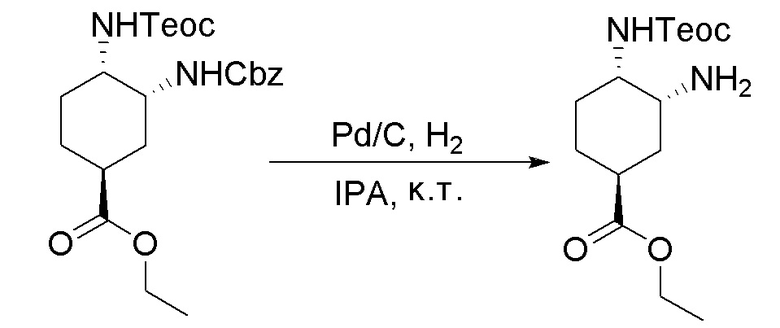
Смесь рацемата этил-3-(бензилоксикарбониламино)-4-((2-(триметилсилил)этокси)карбониламино)циклогексанкарбоксилата (1,7 г, 3,7 ммоль) и 5% палладиевого катализатора на углеродном носителе (50% влажности, 300 мг) в изопропаноле (35 мл) перемешивали под 1 атм атмосферы водорода при комнатной температуре в течение 18 часов. Смесь отфильтровывали сквозь слой целита. Фильтрат концентрировали и очищали с помощью хроматографии на силикагеле (метанол/дихлорметан=от 1/30 до 1/10), чтобы обеспечить указанное в названии соединение (1,0 г, 89%) в виде желтого масла. МС (ЭР+) C15H30N2O4Si требует: 330, обнаружено: 331 [M+H]+.
Синтез трет -бутил(4S,5S)-5-амино-2,2-диметил-тетрагидро-2H-пиран-4-илкарбамата и трет -бутил(4R,5R)-5-амино-2,2-диметил-тетрагидро-2H-пиран-4-илкарбамата
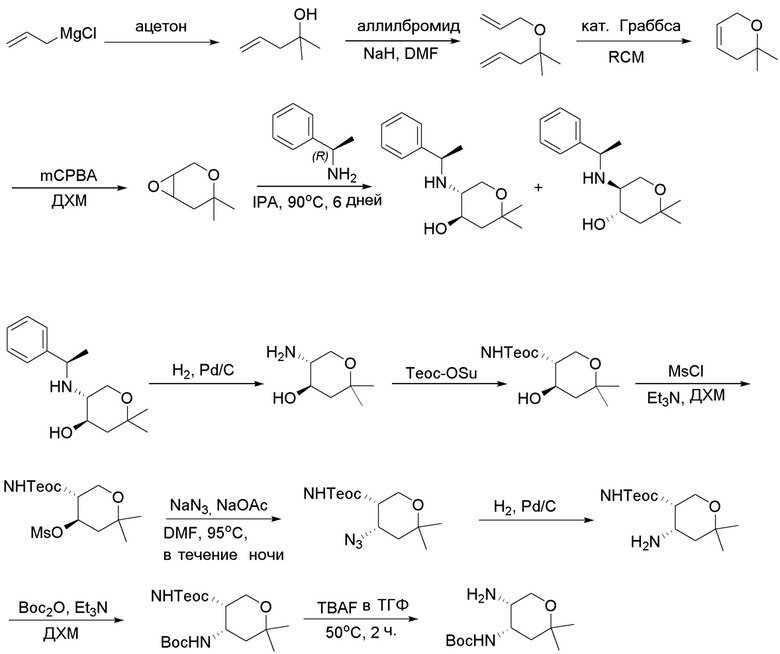
Этап 1: Синтез 2-метилпент-4-ен-2-ола
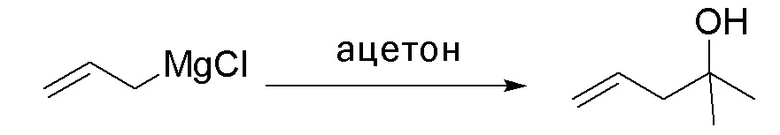
К раствору хлорида аллилмагния в безводном ТГФ (1,7 M, 200 мл, 340 ммоль) медленно добавляли ацетон (13,2 г, 227 ммоль) при 0oC. После перемешивания в течение 15 минут при 0oC, реакционную смесь перемешивали при комнатной температуре в течение еще 2 часов. Реакционную смесь гасили с помощью водн. раствора хлорида аммония и экстрагировали с помощью трет-бутилметилового эфира. Объединенные органические слои промывали водой и соляным раствором, высушивали над сульфатом натрия, отфильтровывали и концентрировали при пониженном давлении. Остаток очищали с помощью дистилляции при пониженном давлении (~10-15 бар, т.к. 50oC) до получения указанного в названии соединения (15 г, 66%) в виде бесцветного масла.
Этап 2: Синтез 4-(аллилокси)-4-метилпент-1-ена

К суспензии гидрида натрия (60%, 24 г, 60 ммоль) в N,N-диметилформамиде (150 мл) при 0°C медленно добавляли 2-метилпент-4-ен-2-ол (20,0 г, 200 ммоль). Спустя 1 час при 0°C, при 0~5°C медленно добавляли аллилбромид (48,0 г 400 ммоль), а реакционную смесь перемешивали при 0°C в течение еще 1 часа. Реакционную смесь гасили с помощью водн. раствора хлорида аммония и экстрагировали с помощью трет-бутилметилового эфира. Объединенные органические слои промывали водой и соляным раствором, высушивали над сульфатом натрия, отфильтровывали и концентрировали при пониженном давлении до получения указанного в названии соединения (44 г, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 3: Синтез 2,2-диметил-3,6-дигидро-2H-пирана

Катализатор Граббса II поколения(1,20 г, 1,43 ммоль) добавляли к раствору 4-(аллилокси)-4-метилпент-1-ена (10,0 г, 71,4 ммоль) в дихлорметане (300 мл), а реакционную смесь кипятили с обратным холодильником в течение ночи. После того, растворитель выпаривали, остаток дистиллировали при пониженном давлении до получения указанного в названии соединения (4,0 г, 50%) в виде бесцветного масла.
Этап 4: Синтез 4,4-диметил-3,7-диокса-бицикло[4.1.0]гептана
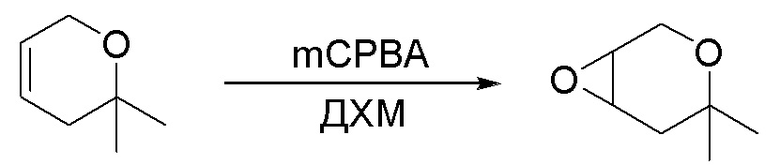
К раствору 2,2-диметил-3,6-дигидро-2H-пирана (4,0 г, 36 ммоль) в дихлорметане (20 мл) добавляли 3-хлорпероксибензойную кислоту (18,4 г, 107 ммоль), а смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем разбавляли с помощью дихлорметана и промывали насыщенным водным сульфитом натрия и соляным раствором. Органический слой высушивали над сульфатом натрия, отфильтровывали и концентрировали при пониженном давлении до получения указанного в названии соединения (5,0 г, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 5: Синтез (4R,5R)-2,2-диметил-5-((R)-1-фенилэтиламино)-тетрагидро-2H-пиран-4-ола (6a) и (4S,5S)-2,2-диметил-5-((R)-1-фенилэтиламино)-тетрагидро-2H-пиран-4-ола (6b)

Смесь 4,4-диметил-3,7-диокса-бицикло[4.1.0]гептана (7,0 г, 54 ммоль) и (R)-1-фенилэтанамина (9,9 г, 82 ммоль) в изопропаноле (50 мл) перемешивали при 80°C в течение 6 дней. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (элюат:петролейный эфир:дихлорметан, содержащий 1% аммония-метанола (7 M), 10:1 к дихлорметану, содержащему 1% аммония/метанола (7 M), чтобы обеспечить (4R,5R)-2,2-диметил-5-((R)-1-фенилэтиламино)-тетрагидро-2H-пиран-4-ол (6a) (более полярную фракцию, 1,5 г) и (4S,5S)-2,2-диметил-5-((R)-1-фенилэтиламино)-тетрагидро-2H-пиран-4-ол (6b) (менее полярную фракцию, 1,7 г) в виде желтого масла. Примечание: абсолютная конфигурация продуктов обозначали в случайном порядке. МС (ЭР+) C15H23NO2 требует: 249, обнаружено: 250 [M+H]+. Подвижная фаза для ТСХ: этилацетат/ дихлорметан=2/1.
Этап 6: Синтез (4R,5R)-5-амино-2,2-диметил-тетрагидро-2H-пиран-4-ола
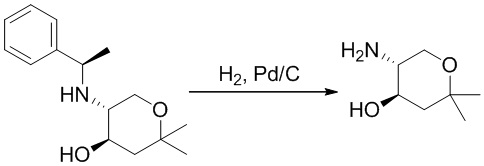
Смесь (4R,5R)-2,2-диметил-5-((R)-1-фенилэтиламино)-тетрагидро-2H-пиран-4-ола (600 мг, 2,40 ммоль) и 10% палладиевого катализатора на углеродном носителе (100 мг) в метаноле (50 мл) перемешивали при комнатной температуре при гидрогенизации в течение ночи. После этого смесь отфильтровывали сквозь слой целита, а фильтрат концентрировали, чтобы обеспечить указанное в названии соединение (550 мг, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 7: Синтез 2-(триметилсилил)этил(3R,4R)-4-гидрокси-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата

К раствору (4R,5R)-5-амино-2,2-диметил-тетрагидро-2H-пиран-4-ола (550 мг, 2,40 ммоль) и триэтиламина (484 мг, 4,80 ммоль) в диоксане (5 мл) и воде (5 мл) добавляли 2-(триметилсилил)этил 2,5-диоксопирролидин-1-карбоксилат (750 мг, 2,90 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После этого раствор разбавляли этилацетатом и промывали насыщенным раствором соли. Органический слой концентрировали, а остаток очищали на колонке с силикагелем с помощью петролейного эфира /этилацетата=от 4/1 до 1/1, чтобы обеспечить указанное в названии соединение (360 мг, 52% на 2 этапах) в виде серого твердого вещества.
Этап 8: Синтез (4R,5R)-2,2-диметил-5-((2-(триметилсилил)этокси)карбониламино)-тетрагидро-2H-пиран-4-илметансульфоната

К раствору 2-(триметилсилил)этил(3R,4R)-4-гидрокси-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата (360 мг, 1,25 ммоль) и триэтиламина (378 мг, 3,75 ммоль) в дихлорметане (5 мл) по каплям добавляли мезилхлорид (213 мг, 1,87 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч., а затем разбавляли с помощью дихлорметана (100 мл). Органический слой промывали водой (50 мл) и соляным раствором (50 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (550 мг, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 9: Синтез 2-(триметилсилил)этил(3S,4S)-4-азидо-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата
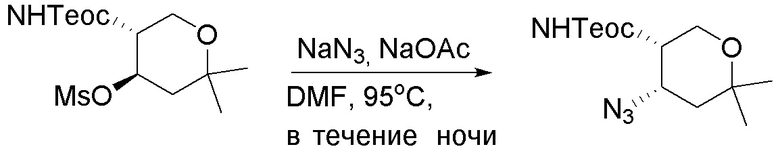
К раствору (4R,5R)-2,2-диметил-5-((2-(триметилсилил)этокси)карбониламино)-тетрагидро-2H-пиран-4-илметансульфоната (550 мг, 1,25 ммоль) в N,N-диметилформамиде (10 мл) добавляли азид натрия (812 мг, 12,5 ммоль) и ацетат натрия (1,05 мг, 12,5 ммоль) при комнатной температуре. Получаемую смесь перемешивали при 95°C в течение 2 дней. После этого смесь охлаждали до комнатной температуры и разбавляли этилацетатом (100 мл). Органическую фазу промывали водой (100 мл× 8) и соляным раствором (50 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (510 мг, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 10: Синтез 2-(триметилсилил)этил(3S,4S)-4-амино-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата

Смесь 2-(триметилсилил)этил(3S,4S)-4-азидо-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата (510 мг, 1,25 ммоль), 10% палладиевого катализатора на углеродном носителе (50 мг) в метаноле (10 мл) перемешивали при комнатной температуре под 1 атм атмосферы водорода (емкость с атмосферой водорода) в течение ночи. После этого смесь отфильтровывали сквозь слой целита. Фильтрат концентрировали для получения указанного в названии соединения (450 мг, неочищенное) в виде коричневого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 11: Синтез 2-(триметилсилил)этил(3S,4S)-(4-трет-бутоксикарбониламино)-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата
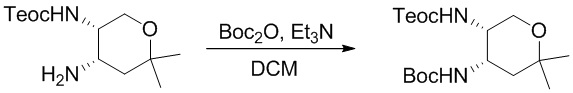
К раствору 2-(триметилсилил)этил(3S,4S)-4-амино-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата (450 мг, 1,25 ммоль) и триэтиламина (379 мг, 3,75 ммоль) в дихлорметане (10 мл) при комнатной температуре добавляли ди-трет-бутил дикарбонат (410 мг, 1,88 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После этого раствор концентрировали и очищали остаток с помощью хроматографии на силикагеле с применением петролейного эфира/этилацетата=4/1 в качестве элюента, чтобы обеспечить указанное в названии соединение (160 мг, 33% на 4 этапах) в виде желтого твердого вещества.
Этап 12: Синтез трет-бутил(4S,5S)-5-амино-2,2-диметил-тетрагидро-2H-пиран-4-илкарбамата

К раствору соединения 2-(триметилсилил)этил(3S,4S)-(4-трет-бутоксикарбониламино)-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата (160 мг, 0,41 ммоль) в тетрагидрофуране (2 мл) при комнатной температуре добавляли фторид тетрабутиламмония в тетрагидрофуране (1 M, 1,23 мл, 1,23 ммоль). Реакционную смесь перемешивали при 50°C в течение 2 часов. После этого раствор концентрировали и очищали остаток с помощью хроматографии на силикагеле этилацетатом в качестве элюента, чтобы обеспечить указанное в названии соединение (110 мг, неочищенное) в виде желтого масла.

Этап 1: Синтез (4S,5S)-5-амино-2,2-диметил-тетрагидро-2H-пиран-4-ола

Суспензионную смесь (4S,5S)-2,2-диметил-5-((R)-1-фенилэтиламино)-тетрагидро-2H-пиран-4-ола (1,0 г, 4,0 ммоль) и 10% палладиевого катализатора на углеродном носителе (200 мг) в метаноле (20 мл) перемешивали при комнатной температуре под 1атм атмосферы водорода (емкость с атмосферой водорода) в течение ночи. После этого смесь отфильтровывали сквозь слой целита и концентрировали фильтрат для получения указанного в названии соединения (1,1 г, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 2: Синтез 2-(триметилсилил)этил(3S,4S)-4-гидрокси-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата

К раствору (4S,5S)-5-амино-2,2-диметил-тетрагидро-2H-пиран-4-ола (1,1 г, 4,0 ммоль) и триэтиламина (1,1 мл, 8,0 ммоль) в смешанном растворителе, состоящем из диоксана (5 мл) и воды (5 мл), при комнатной температуре добавляли 2-(триметилсилил)этил 2,5-диоксопирролидин-1-карбоксилат (1,2 г, 4,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После этого раствор разбавляли этилацетатом и промывали насыщенным раствором соли. Органический слой разделяли и концентрировали. Получаемый остаток очищали с помощью хроматографии на силикагеле с применением петролейного эфира/этилацетата=от 4/1 до 1/1 в качестве элюента, чтобы обеспечить указанное в названии соединение (1,0 г, 86% на 2 этапах) в виде желтого масла.
Этап 3: Синтез (4S,5S)-2,2-диметил-5-((2-(триметилсилил)этокси)карбониламино)-тетрагидро-2H-пиран-4-илметансульфоната

К раствору 2-(триметилсилил)этил(3S,4S)-4-гидрокси-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата (1,0 г, 3,5 ммоль) и триэтиламина (1,4 мл, 10 ммоль) в дихлорметане (10 мл) при 0°C по каплям добавляли мезилхлорид (600 мг, 5,20 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, а затем разбавляли с помощью дихлорметана (100 мл). Органический слой промывали водой (50 мл) и соляным раствором (50 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (1,6 г, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 4: Синтез 2-(триметилсилил)этил(3R,4R)-4-азидо-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата
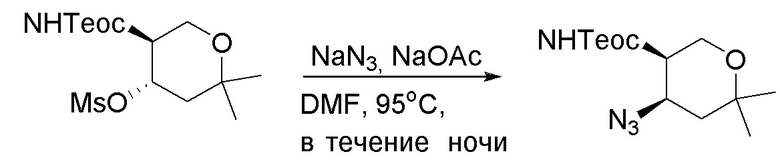
К раствору (4S,5S)-2,2-диметил-5-((2-(триметилсилил)этокси)карбониламино)-тетрагидро-2H-пиран-4-илметансульфоната (1,6 г, 3,5 ммоль) в N,N-диметилформамиде (10 мл) добавляли азид натрия (2,3 г, 35 ммоль) и ацетат натрия (2,8 г, 35 ммоль) при комнатной температуре. Получаемую смесь перемешивали при 95°C в течение 2 дней. После этого смесь охлаждали до комнатной температуры и разбавляли этилацетатом (100 мл). Органический слой промывали водой (100 мл× 8) и соляным раствором (50 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (1,3 г, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 5: Синтез 2-(триметилсилил)этил(3R,4R)-4-амино-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата

Смесь 2-(триметилсилил)этил(3R,4R)-4-азидо-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата (1,3 г, 3,5 ммоль) и 10% палладиевого катализатора на углеродном носителе (200 мг) в метаноле (10 мл) перемешивали при комнатной температуре под 1 атм атмосферы водорода (емкость с атмосферой водорода) в течение ночи. После этого смесь отфильтровывали сквозь слой целита. Фильтрат концентрировали для получения указанного в названии соединения (1,2 г, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 6: Синтез 2-(триметилсилил)этил(3R,4R)-(4-трет-бутоксикарбониламино)-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата

К раствору 2-(триметилсилил)этил(3R,4R)-4-амино-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата (1,2 г, 3,5 ммоль) и триэтиламина (1,4 мл, 10,5 ммоль) в дихлорметане (10 мл) добавляли ди-трет-бутилдикарбонат (1,1 г, 5,2 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После этого смесь сразу концентрировали и очищали с помощью хроматографии на силикагеле с применением петролейный эфир/этилацетат=4/1 в качестве элюента, чтобы обеспечить указанное в названии соединение (440 мг, 32% на 4 этапах) в виде серого твердого вещества.
Этап 7: Синтез трет-бутил(4R,5R)-5-амино-2,2-диметил-тетрагидро-2H-пиран-4-илкарбамата

К раствору 2-(триметилсилил)этил(3R,4R)-(4-трет-бутоксикарбониламино)-6,6-диметил-тетрагидро-2H-пиран-3-илкарбамата (440 мг, 1,13 ммоль) в тетрагидрофуране (2 мл) при комнатной температуре добавляли фторид тетрабутиламмония в тетрагидрофуране (1 M, 3,4 мл, 3,4 ммоль). Реакционную смесь перемешивали при 50°C в течение 2 часов. После этого раствор охлаждали до комнатной температуры, концентрировали и очищали с помощью хроматографии на силикагеле с применением этилацетата в качестве элюата, чтобы обеспечить указанное в названии соединение (80 мг, 29%) в виде желтого масла.
Синтез трет -бутил(2S,4S,5S)-5-амино-2-метилтетрагидро-2 H -пиран-4-илкарбамата
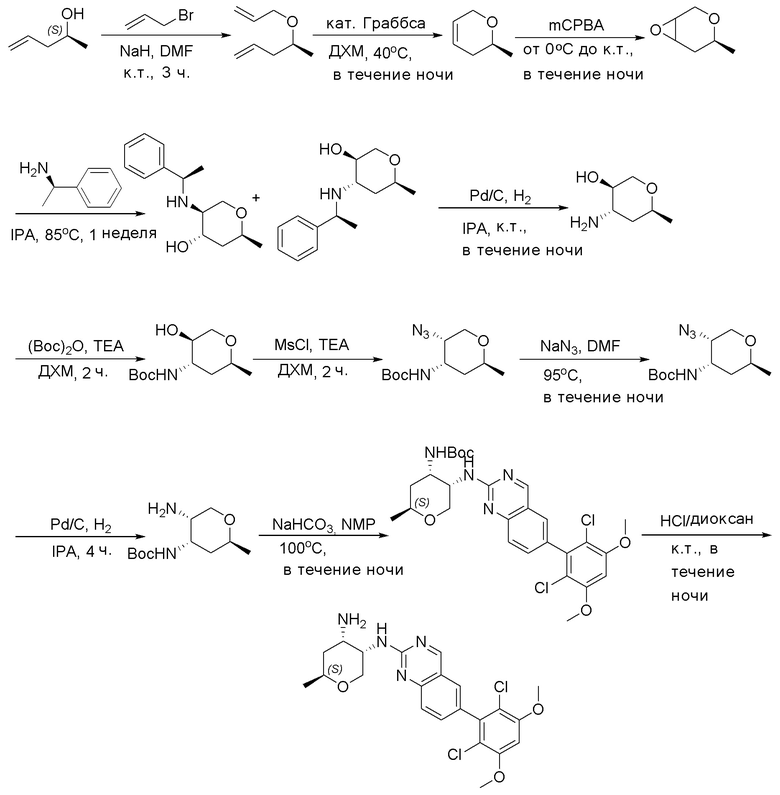
Этап 1: Синтез (S)-4-(аллилокси)пент-1-ена

К суспензии гидрида натрия (21 г, 34 ммоль) в N,N-диметилформамиде (100 мл) по каплям добавляли (S)-пент-4-ен-2-ол (10 г, 116 ммоль) при 0oC. Реакционную смесь перемешивали при 0°C в течение 1 часа. После этого, к смеси по каплям добавляли аллилбромид (14,0 г, 116,2 ммоль) при 0oC. Получаемую смесь перемешивали при 0°C в течение еще 3 ч., а смесь гасили насыщенным раствором хлорида аммония (500 мл). Водный слой экстрагировали с помощью трет-бутилметилового эфира (200 мл x 3), а объединенные органические слои промывали водой (100 мл x 3) и соляным раствором (100 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить (S)-4-(аллилокси)пент-1-ен (~20 мл раствора трет-бутилметилового эфира), который применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 2: Синтез (S)-2-метил-3,6-дигидро-2H-пирана
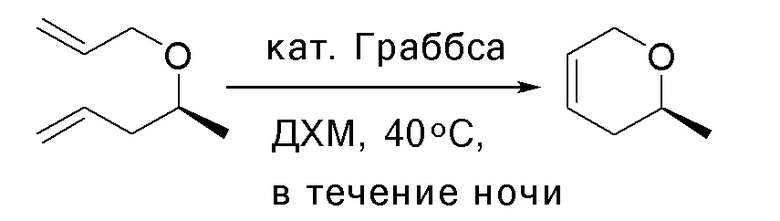
Смесь (S)-4-(аллилокси)пент-1-ена (~20 мл раствор, 116 ммоль), катализатора Граббса 2го поколения (1,8 г) в дихлорметане (500 мл) перемешивали при 40°C в течение ночи. После этого раствор охлаждали до комнатной температуры, а указанное в названии соединение (~3,5 г, 31% на 2 этапах) получали с помощью вакуумной перегонки.
Этап 3: Синтез (4S)-4-метил-3,7-диоксабицикло[4.1.0]гептана
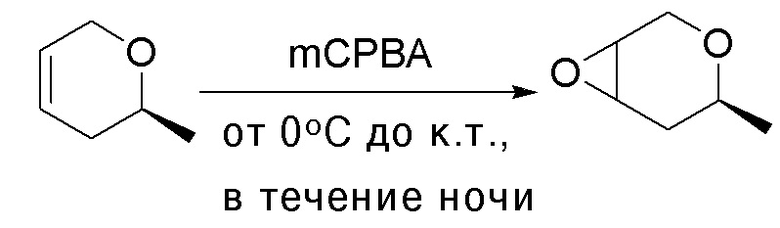
К раствору (S)-2-метил-3,6-дигидро-2H-пирана (~1 г, 10 ммоль) в дихлорметане (20 мл) добавляли 3-хлорпероксибензойную кислоту (1,8 г, 20 ммоль) при 0oC. Получаемую смесь перемешивали при комнатной температуре в течение ночи. После этого смесь промывали насыщенным раствором сульфита натрия (15 мл), карбоната натрия (15 мл) и соляным раствором (15 мл). Органический слой высушивали над сульфатом натрия и концентрировали для получения указанного в названии соединения (~3 мл раствора дихлорметана), которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 4: Синтез (3R,4S,6S)-6-метил-4-((S)-1-фенилэтиламино)тетрагидро-2H-пиран-3-ола

Смесь (4S)-4-метил-3,7-диоксабицикло[4.1.0]гептана (~3 мл раствора дихлорметана, 10 ммоль) и (R)-1-фенилэтанамина (2,4 г, 20 ммоль) в изопропиловом спирте (20 мл) перемешивали при 85°C в течение 1 недели. После этого раствор охлаждали до комнатной температуры и очищали с помощью преп-ВЭЖХ для получения указанного в названии соединения (более полярного, 120 мг, 10% на 2 этапах) в виде желтого твердого вещества и побочного продукта (менее полярного, 400 мг, 32% на 2 этапах) в виде белого твердого вещества. МС (ЭР+) C14H21NO2 требует: 235, обнаружено: 236 [M+H]+.
Этап 5: Синтез (3R,4S,6S)-4-амино-6-метилтетрагидро-2H-пиран-3-ола
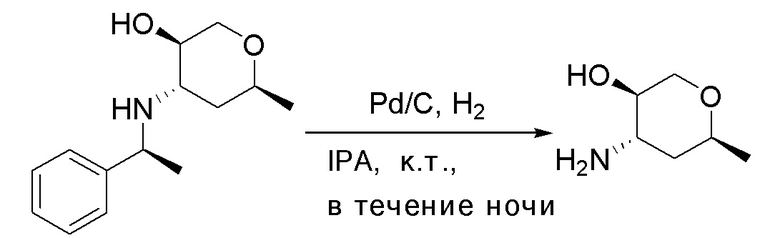
Смесь (3S,4R)-трет-бутил-трет-бутил(2S,4S,5R)-5-гидрокси-2-метилтетрагидро-2H-пиран-4-илкарбамата (200 мг, 0,85 ммоль) и 10% палладиевого катализатора на углеродном носителе (50 мг) в изопропаноле (10 мл) перемешивали при комнатной температуре при гидрогенизации в течение ночи. После этого смесь отфильтровывали сквозь слой целита и концентрировали для получения указанного в названии соединения (150 мг, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 6: Синтез трет-бутил(2S,4S,5R)-5-гидрокси-2-метилтетрагидро-2H-пиран-4-илкарбамата

К раствору (3R,4S,6S)-4-амино-6-метилтетрагидро-2H-пиран-3-ола (150 мг, 1,1 ммоль) и триэтиламина (333 мг, 3,3 ммоль) в дихлорметане (80 мл) при 0°C по каплям добавляли ди-трет-бутил дикарбонат (475 мг, 2,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После этого раствор концентрировали и очищали на колонке с силикагелем с помощью метанола/дихлорметан=от 1/30 до 1/15 в качестве элюата, чтобы обеспечить указанное в названии соединение (130 мг, 51% на 2 этапах) в виде желтого масла.
Этап 7: Синтез трет-бутил(2S,4S,5S)-5-азидо-2-метилтетрагидро-2H-пиран-4-илкарбамата

К раствору трет-бутил(2S,4S,5R)-5-гидрокси-2-метилтетрагидро-2H-пиран-4-илкарбамата 7 (130 мг, 0,6 ммоль) и триэтиламина (202 мг, 2,0 ммоль) в дихлорметане (10 мл) при 0°C по каплям добавляли мезилхлорид (194 мг, 1,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, а затем разбавляли с помощью дихлорметана (100 мл). Органические слои промывали водой (50 мл) и соляным раствором (50 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (190 мг, неочищенное) в виде желтого масла (4,0 г, 98%), которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 8: Синтез трет-бутил(2S,4S,5S)-5-азидо-2-метилтетрагидро-2H-пиран-4-илкарбамата
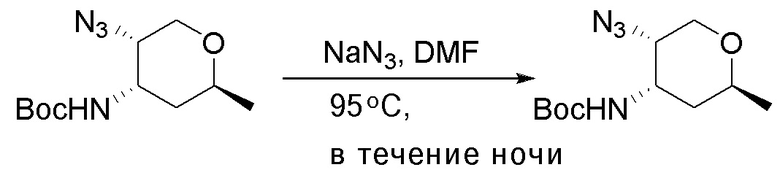
К раствору трет-бутил(2S,4S,5S)-5-азидо-2-метилтетрагидро-2H-пиран-4-илкарбамата (190 мг, 0,6 ммоль) в N,N-диметилформамиде (10 мл) добавляли азид натрия (375 мг, 5,6 ммоль) и ацетат натрия (459 мг, 5,6 ммоль) при комнатной температуре. Получаемую смесь перемешивали при 95°C в течение 2 дней. После этого смесь разбавляли этилацетатом (100 мл), промывали водой (50 мл) и соляным раствором (50 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (180 мг, неочищенное) в виде желтого масла (4,0 г, 98%), которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 9: Синтез трет-бутил(2S,4S,5S)-5-амино-2-метилтетрагидро-2H-пиран-4-илкарбамата

Смесь трет-бутил(2S,4S,5S)-5-азидо-2-метилтетрагидро-2H-пиран-4-илкарбамата (1,8 г, 3,6 ммоль) и 10% палладиевого катализатора на углеродном носителе (50 мг) в изопропаноле (10 мл) перемешивали при комнатной температуре при гидрогенизации в течение ночи. После этого смесь отфильтровывали сквозь слой целита. Концентрировали для получения указанного в названии соединения (150 мг, неочищенное) в виде желтого масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Синтез 2,8-дихлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина
 Этап 1: Синтез 2-амино-5-бром-3-хлорбензойной кислоты
Этап 1: Синтез 2-амино-5-бром-3-хлорбензойной кислоты
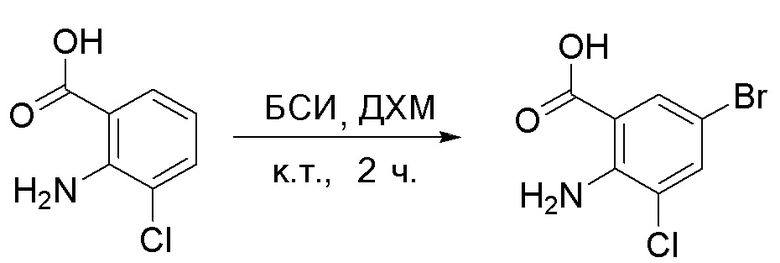
К раствору 2-амино-3-фторбензойной кислоты (10,0 г, 58,5 ммоль) в дихлорметане (150 мл) добавляли N-бромсукцинимид (10,4 г, 58,5 ммоль), а смесь перемешивали при комнатной температуре в течение 2 часов. ЖХ-МС показала, что реакция была завершена. Твердое вещество отфильтровывали и промывали с помощью дихлорметана (100 мл × 3) до получения указанного в названии соединения в виде белого твердого вещества (13,0 г, 89%), которое применяли непосредственно на следующем этапе без дополнительной очистки. МС (ЭР+) C7H5BrClNO2 требует: 249, 251, обнаружено: 250, 252 [M+H]+.
Этап 2: Синтез (2-амино-5-бром-3-хлорфенил)метанола
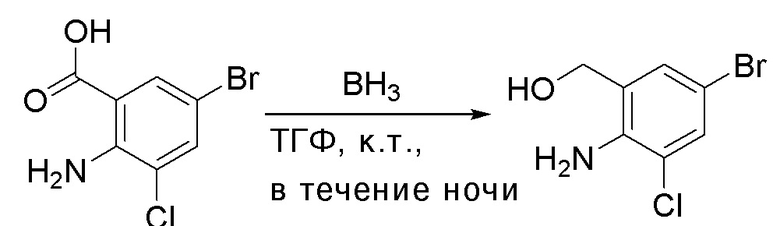
К раствору 2-амино-5-бром-3-хлорбензойной кислоты (13,0 г, 52,0 ммоль) в ТГФ (200 мл) добавляли борогидрид в ТГФ (300 мл, 1N) на водяной бане/бане со льдом, а реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь гасили метанолом (100 мл) и концентрировали до объема 50 мл. Остаток разбавляли водным бикарбонатом натрия (400 мл) и экстрагировали этилацетатом (200 мл × 3). Органические слои разделяли, объединяли, промывали соляным раствором (100 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (10,0 г, 82%). МС (ЭР+) C7H7BrClNO требует: 234, 236, обнаружено: 236, 238 [M+H]+.
Этап 3: Синтез 2-амино-5-бром-3-хлорбензальдегида

Смесь (2-амино-5-бром-3-хлорфенил)метанола (10,0 г, 42,5 ммоль) и оксида марганца (21,9 г, 255 ммоль) в дихлорметане (400 мл) перемешивали при комнатной температуре в течение ночи. Твердое вещество фильтровали, а фильтрат концентрировали до получения указанного в названии соединения в виде светло-желтого твердого вещества (9,0 г, 91%), которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 4: Синтез 6-бром-8-хлорхиназолин-2-ола

Смесь 2-амино-5-бром-3-хлорбензальдегида (9,0 г, 38,6 ммоль) и мочевины (34,7 г, 579 ммоль) нагревали до 180°C и перемешивали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, а получаемый осадок разбавляли с помощью воды (1 л) и перемешивали в течение 2 часов. Получаемый осадок отфильтровывали, а влагу в ловушке полностью удаляли с помощью трехкратного совместного выпаривания с толуолом. Указанное в названии соединение (9,0 г, 90%) получали в виде желтого твердого вещества. МС (ЭР+) C8H4BrClN2O требует: 257, 259, обнаружено: 258, 260 [M+H]+.
Этап 5: Синтез 6-бром-2,8-дихлорхиназолина

Раствор 6-бром-8-хлорхиназолин-2-ола (9,0 г, 35 ммоль) в оксихлориде фосфора (100 мл) кипятили с обратным холодильником в течение 5 часов. Большую часть оксихлорида фосфора удаляли при пониженном давлении, а остаток добавляли в перемешиваемую ледяную воду (500 мл). Получаемый осадок собирали посредством фильтрования, а затем кипятили с обратным холодильником в ТГФ. Твердое вещество фильтровали, а фильтрат концентрировали до получения указанного в названии соединения в виде желтого твердого вещества (7,0 г, 78%). МС (ЭР+) C8H4BrClN2 требует: 275, 277, обнаружено: 276, 278 [M+H]+.
Этап 6: Синтез 2,8-дихлор-6-(3,5-диметоксифенил)хиназолина

Смесь 6-бром-2,8-дихлорхиназолина (4,0 г, 14,5 ммоль), 3,5-диметоксифенилбороновой кислоты (4,23 г, 16,0 ммоль), карбоната цезия (9,42 г, 29,0 ммоль) и Pd(PPh3)2Cl2 (220 мг, 0,70 ммоль) в ТГФ (200 мл) и воде (10 мл) дегазировали азотом трижды и перемешивали при 80°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, сразу концентрировали и очищали с помощью хроматографии на силикагеле (петролейный эфир: дихлорметан=2:1~1:1) для получения указанного в названии соединения в виде желтого твердого вещества (2,0 г, 41%). МС (ЭР+) C16H12Cl2N2O2 требует: 334, 336, обнаружено: 335, 337 [M+H]+.
Этап 7: Синтез 2,8-дихлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина

К раствору 2,8-дихлор-6-(3,5-диметоксифенил)хиназолина (2,0 г, 6,0 ммоль) в сухом ТГФ (40 мл) по каплям добавляли сульфурилхлорид (1,59 г, 1,75 ммоль) при 0°C и перемешивали смесь в течение 30 минут при 0°C. Реакционную смесь гасили водой (1 мл) и собирали осадок посредством фильтрования до получения указанного в названии соединения (1,3 г, 54%) в виде желтого твердого вещества. МС (ЭР+) C16H10Cl4N2O2 требует: 402, 404, обнаружено: 403, 405 [M+H]+; 1H-ЯМР (400 МГц, CDCl3) δ ч./млн. 9,36 (с, 1H), 7,94 (с, 1H), 7,79 (с, 1H), 6,69 (с, 1H), 4,01 (с, 6H).
Синтез 2,7-дихлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина
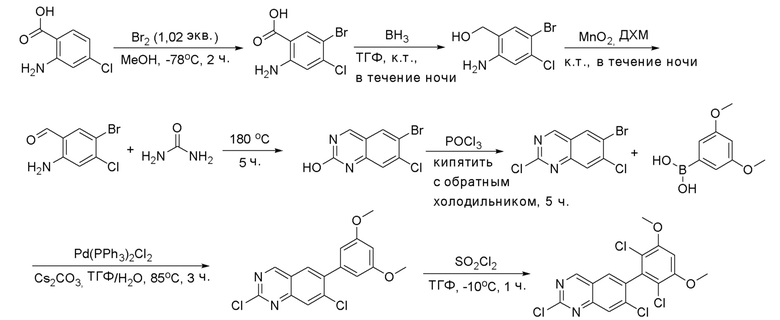 Этап 1: Синтез 2-амино-5-бром-4-хлорбензойной кислоты
Этап 1: Синтез 2-амино-5-бром-4-хлорбензойной кислоты

К раствору 2-амино-4-хлорбензойной кислоты (10,0 г, 58,5 ммоль) в метаноле (150 мл) добавляли бром (15,7 мл) при -78°C и перемешивали реакционную смесь при -78°C в течение 2 часов. Реакционную смесь гасили ледяной водой (100 мл) и водн. тиосульфатом натрия и экстрагировали этилацетатом (150 мл ×3). Органические слои разделяли, объединяли, промывали водой (100 мл) и соляным раствором (100 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (9 г, 62%).
Этап 2: Синтез (2-амино-5-бром-4-хлорфенил)метанола
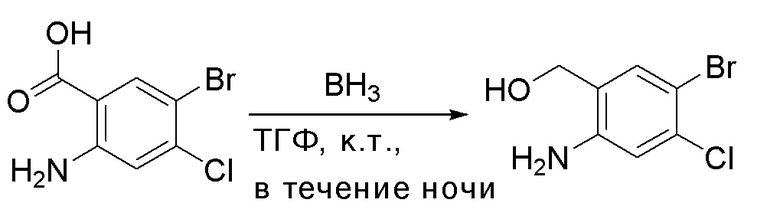
К раствору 2-амино-5-бром-4-хлорбензойной кислоты (9,0 г, 36,0 ммоль) в ТГФ (150 мл) добавляли борогидрид в ТГФ (144 мл, 1 M) при комнатной температуре и перемешивали реакционную смесь в течение ночи. Реакционную смесь гасили метанолом (50 мл) и концентрировали до объема 50 мл. Остаток разбавляли водой (100 мл) и экстрагировали этилацетатом (150 мл × 3). Органические слои разделяли, объединяли, промывали водой (100 мл) и соляным раствором (100 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (неочищенное, 6 г, 71%).
Этап 3: Синтез 2-амино-5-бром-4-хлорбензальдегида

Смесь (2-амино-5-бром-4-хлорфенил)метанола (6 г, 25,5 ммоль) и оксида марганца(IV) (15,5 г, 0,178 моль) в дихлорметане (100 мл) перемешивали при комнатной температуре в течение ночи. Твердое вещество фильтровали и концентрировали фильтрат до получения указанного в названии соединения в виде светло-желтого твердого вещества (5 г, 81%). МС (ЭР+) C7H5BrClNO требует: 233, 235, обнаружено: 234, 236 [M+H]+.
Этап 4: Синтез 6-бром-7-хлорхиназолин-2-ола
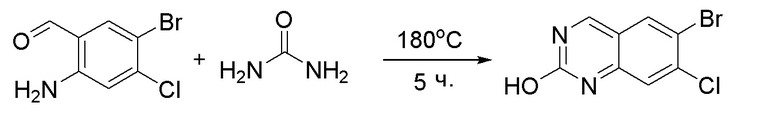
Смесь 2-амино-5-бром-4-хлорбензальдегида (5 г, 21,46 ммоль) и мочевины (18 г, 300,0 ммоль) перемешивали при 180°C в течение 5 часов. С помощью ЖХ-МС контролировали завершение реакции. Смесь охлаждали до комнатной температуры, промывали водой (100 мл × 3) и отфильтровывали. Фильтрпрессную лепешку, полученную во время фильтрования, высушивали для получения указанного в названии соединения в виде желтого твердого вещества (6 г (неочищенное, 100%). МС (ЭР+) C8H4BrClN2O требует: 258, 260, обнаружено: 259, 261 [M+H]+.
Этап 5: Синтез 6-бром-2,7-дихлорхиназолина

Раствор 6-бром-7-хлорхиназолин-2-ола (6,0 г, 23 ммоль) в оксихлориде фосфора (50 мл) кипятили с обратным холодильником в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и удаляли большую часть оксихлорида фосфора при пониженном давлении. Остаток по каплям добавляли в ледяную воду (500 мл), а получаемый осадок собирали с помощью фильтрования до получения указанного в названии соединения в виде желтого твердого вещества (3 г, 48%).
Этап 6: Синтез 2,7-дихлор-6-(3,5-диметоксифенил)хиназолина

Смесь 6-бром-2,7-дихлорхиназолина (3 г, 10,8 ммоль), 3,5-диметоксифенилбороновой кислоты (2,2 г, 11,9 ммоль), карбоната цезия (1,06 г, 32,4 ммоль) и Pd(PPh3)2Cl2 (702 мг, 1,08 ммоль) в ТГФ (50 мл) и воде (10 мл) дегазировали азотом трижды и перемешивали реакционную смесь при 85°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и сразу концентрировали. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир:этилацетат=10:1~4:1) до получения указанного в названии соединения (2,0 г, выход:55%) в виде желтоватого твердого вещества. МС (ЭР+) требует: 334, 336, C16H12Cl2N2O2, обнаружено: 335, 337 [M+H]+.
Этап 7: Синтез 2,7-дихлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолина

К раствору 2,7-дихлор-6-(3,5-диметоксифенил)хиназолина (2,0 г, 6,0 ммоль) в ТГФ (30 мл) добавляли сульфурилхлорид (1,77 г, 13,2 ммоль) при -10°C и перемешивали смесь при -10°C в течение 1 часа. Раствор гасили водой (1 мл) и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир:этилацетат=10:1~4:1) до указанного в названии соединения (1,2 г, 50%) в виде белого твердого вещества. МС (ЭР+) C16H10Cl4N2O2 требует: 402, 404, обнаружено: 403, 405 [M+H]+.
Этап 8: Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)пиридо[2,3-d]пиримидина
 Этап 9: Синтез 6-бромпиридо[2,3-d]пиримидин-2-ола
Этап 9: Синтез 6-бромпиридо[2,3-d]пиримидин-2-ола

Смесь 2-амино-5-бромникотинальдегида (2,0 г, 10,0 ммоль) и мочевины (9,0 г, 150,0 ммоль) нагревали при 180°C и энергично перемешивали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, а получаемый осадок собирали, промывали водой (3×100 мл) и выпаривали совместно с толуолом трижды, чтобы полностью удалить влагу из ловушки. Указанное в названии соединение (2,1 г, 93%) получали в виде желтого твердого вещества. МС (ЭР+) C8H5BrN2O требует: 225, 227, обнаружено: 226, 228 [M+H]+.
Этап 10: Синтез 6-бром-2-хлорпиридо[2,3-d]пиримидина

К перемешиваемой смеси 6-бромпиридо[2,3-d]пиримидин-2-ола (1,1 г, 4,9 ммоль) в 30 мл трихлорида фосфорила добавляли диизопропилэтиламин (1,6 г, 12,2 ммоль) при комнатной температуре, а реакционную смесь затем перемешивали при 120°C в течение 12 часов. Большую часть трихлорида фосфорила удаляли при пониженном давлении. Остаток разбавляли с помощью этилацетата (200 мл) и добавляли к насыщенному раствору бикарбоната натрия (300 мл) при 0oC. Смесь экстрагировали этилацетатом (200 мл x 3). Объединенные органические слои высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (800 мг, 67%) в виде желтого твердого вещества. МС (ЭР+) C7H3BrClN3 требует: 243, 245, обнаружено: 244, 246 [M+H]+.
Этап 11: Синтез 2-хлор-6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидина:

Смесь 6-бром-2-хлорпиридо[2,3-d]пиримидина (800 мг, 3,3 ммоль), 3,5-диметоксифенилбороновой кислоты (655 мг, 3,6 ммоль), бис(три-трет-бутилфосфин)палладия (83 мг, 0,16 ммоль) и карбоната цезия (1,06 г, 3,3 ммоль) в ТГФ (30 мл) и воде (6 мл) дегазировали азотом трижды, а затем нагревали при 85°C в течение 0,5 часа. Смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан/этилацетат=3/1) для получения указанного в названии соединения в виде желтого твердого вещества (460 мг, 47%) в виде желтого твердого вещества. МС (ЭР+) C15H12ClN3O2 требует: 301, 302, обнаружено: 302, 304 [M+H]+.
Этап 12: Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)пиридо[2,3-d]пиримидина
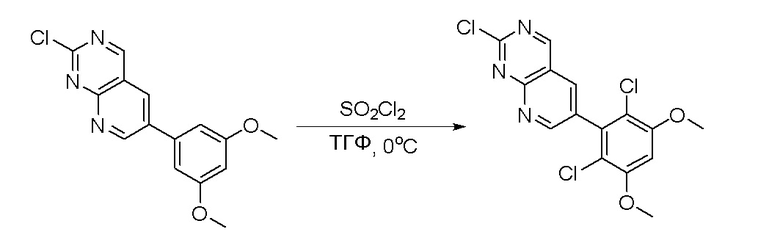
К раствору 2-хлор-6-(3,5-диметоксифенил)пиридо[2,3-d]пиримидина (300 мг, 1,0 ммоль) в ТГФ (30 мл) по каплям добавляли сульфурилхлорид (337 мг, 2,5 ммоль) при 0oC, а смесь перемешивали в течение 20 минут при 0oC. Реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (100 мл x 3). Объединенные органические слои промывали насыщенным раствором соли (100 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (дихлорметан/этилацетат=5/1) для получения указанного в названии соединения в виде желто-коричневого твердого вещества (240 мг, 65%). МС (ЭР+) C15H10Cl3N3O2 требует: 369, обнаружено: 370, 372 [M+H]+.
Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-7-фторхиназолина
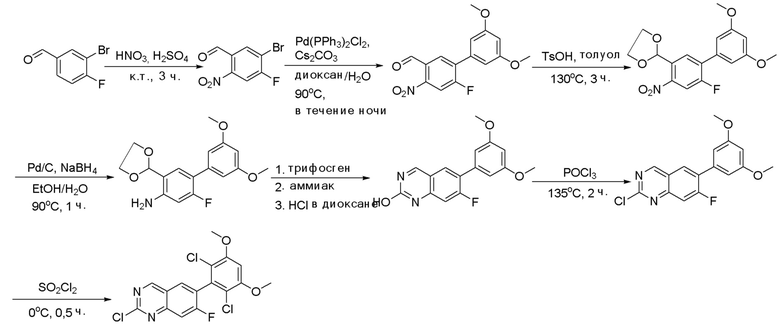 Этап 1: Синтез 5-бром-4-фтор-2-нитробензальдегида
Этап 1: Синтез 5-бром-4-фтор-2-нитробензальдегида

К перемешиваемому раствору концентрированной азотной кислоты (6,8 мл, 101,0 ммоль) в концентрированной серной кислоте (60 мл) медленно добавляли 3-бром-4-фторбензальдегид (10 г, 49,5 ммоль) при 0°C. После того, как завершали добавление, ледяную баню удаляли, а реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 3 часов. Смесь выливали в ледяную воду и экстрагировали этилацетатом (200 мл). Органический слой концентрировали до получения указанного в названии соединения в виде желтого твердого вещества (неочищенное, 12 г, 100%), которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 2: Синтез 6-фтор-3',5'-диметокси-4-нитробифенил-3-карбальдегида
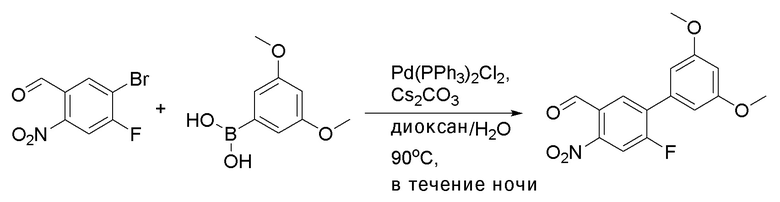
Смесь 5-бром-4-фтор-2-нитробензальдегида (10,0 г, 40,0 ммоль), 3,5-диметоксифенилбороновой кислоты (7,3 г, 40,0 ммоль), хлорида бис(трифенилфосфино)палладия(II) (1,4 г, 2,0 ммоль) и карбоната цезия (32,6 г, 100,0 ммоль) в диоксане/воде (550 мл, об./об.=10/1) дегазировали азотом трижды и нагревали при 90°C в течение 3 часов. Смесь охлаждали до комнатной температуры, концентрировали, разбавляли этилацетатом (1000 мл) и промывали водой (500 мл) и соляным раствором (500 мл). Органический слой высушивали, концентрировали, а остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=от 8/1 до 5/1), чтобы обеспечить указанное в названии соединение (9 г, 61%) в виде желтого твердого вещества. МС (ЭР+) C15H12FNO5 требует: 305, обнаружено: 306 [M+H]+.
Этап 3: Синтез 2-(6-фтор-3',5'-диметокси-4-нитробифенил-3-ил)-1,3-диоксолан

Смесь 6-фтор-3',5'-диметокси-4-нитробифенил-3-карбальдегида (1,7 г, 5,6 ммоль) и 4-толуолсульфоновой кислоты (95,8 мг, 0,6 ммоль) в 1,2-этандиоле (4,3 мл) и толуоле (60 мл) нагревали при 130°C в течение 3 часов. После этого, реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (100 мл) и промывали водой (100 мл * 3) и соляным раствором (100 мл). Объединенные органические слои высушивали над сульфатом натрия, отфильтровывали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=от 8/1 до 5/1), чтобы обеспечить указанное в названии соединение (1,8 г, 89%) в виде желтого твердого вещества. МС (ЭР+) C17H16FNO6 требует: 349, обнаружено: 350 [M+H]+.
Этап 4: Синтез 5-(1,3-диоксолан-2-ил)-2-фтор-3',5'-диметоксибифенил-4-амина
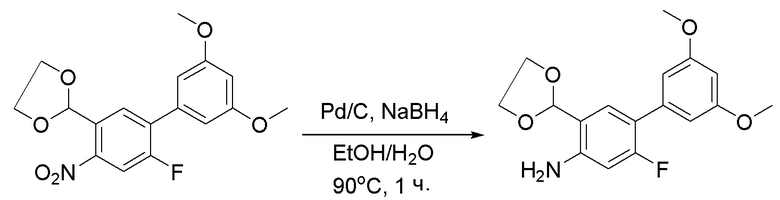
Смесь 2-(6-фтор-3',5'-диметокси-4-нитробифенил-3-ил)-1,3-диоксолана (1,8 г, 5,2 ммоль), борогидрида натрия (587,9 мг, 15,5 ммоль) и 10% палладиевого катализатора на углеродном носителе (0,2 г) в этаноле/воде (33 мл, об./об.=10/1) нагревали при 90°C в течение 1 часа. После этого смесь разбавляли этилацетатом (150 мл) и промывали водой (50 мл) и соляным раствором (50 мл). Органический слой высушивали над натрием, отфильтровывали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=от 5/1 до 4/1), чтобы обеспечить указанное в названии соединение (1,4 г, 88%) в виде желтого твердого вещества. МС (ЭР+) C17H18FNO4 требует: 319, обнаружено: 320 [M+H]+.
Этап 5: Синтез 6-(3,5-диметоксифенил)-7-фторхиназолин-2-ола
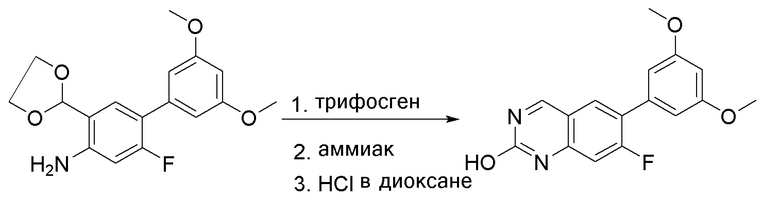
К раствору 5-(1,3-диоксолан-2-ил)-2-фтор-3',5'-диметоксибифенил-4-амина (1,9 г, 6,0 ммоль) и триэтиламина (3,0 мл, 21,4 ммоль) в ТГФ (20 мл) при 0°C добавляли трифосген (0,6 г, 2,0 ммоль) и перемешивали при 0°C в течение 0,5 часа. После этого, добавляли аммоний в метаноле (3 мл, 21 ммоль, 7 моль/л). Реакционную смесь перемешивали при 0°C в течение 30 минут и быстро нагревали до температуры окружающей среды. После перемешивания в течение дополнительных 30 минут при комнатной температуре, реакционную смесь подкисляли с помощью 4 моль/л HCl в диоксане (8,2 мл) до pH 2, а затем перемешивали при комнатной температуре в течение 1 часа. Затем получаемый раствор концентрировали и очищали с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол=от 50/1 до 10/1), чтобы обеспечить указанное в названии соединение (2,0 г, 99%) в виде желтого твердого вещества. МС (ЭР+) C16H13FN2O3 требует: 300, обнаружено: 301 [M+H]+.
Этап 6: Синтез 2-хлор-6-(3,5-диметоксифенил)-7-фторхиназолина
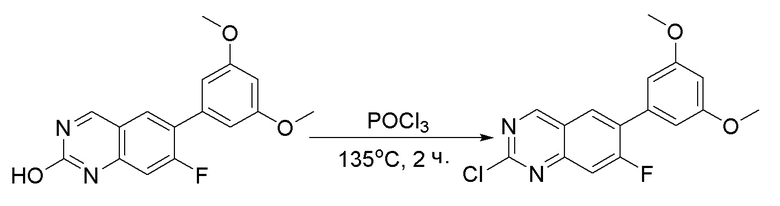
Раствор 6-(3,5-диметоксифенил)-7-фторхиназолин-2-ола (2,0 г, 6,7 ммоль) в POCl3 (30 мл) нагревали при 135°C в течение 2 часов. После этого, реакционный раствор охлаждали до комнатной температуры и по каплям добавляли к насыщенному раствору бикарбоната натрия (800 мл) при 0°C. Смесь экстрагировали этилацетатом (200 мл * 3). Объединенные органические слои высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (1,1 г, 52%) в виде бледно-желтого твердого вещества. МС (ЭР+) C16H12ClFN2O2 требует: 318, обнаружено: 319 [M+H]+.
Этап 7: Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-7-фторхиназолина

К раствору 2-хлор-6-(3,5-диметоксифенил)-7-фторхиназолина (1,2 г, 3,8 ммоль) в ацетонитриле/тетрагидрофуране (200 мл, об./об.=1/1) добавляли сульфурилхлорид (1,7 мл, 18,9ммоль) при 0oC. Получаемый раствор перемешивали при 0°C в течение 0,5 часа. После этого раствор концентрировали и разбавляли этилацетатом (500 мл). Органическую фазу промывали насыщенным раствором бикарбоната натрия (200 мл) и соляным раствором (200 мл), высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (946 мг, 65%) в виде желтого твердого вещества. МС (ЭР+) C16H10Cl3FN2O2 требует: 386, обнаружено: 387 [M+H]+.
Этап 8: Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-8-фторхиназолина

Этап 9: Синтез 2-амино-5-бром-3-фторбензойной кислоты

Раствор 2-амино-3-фторбензойной кислоты (10,85 г, 70 ммоль) в дихлорметане (175 мл) добавляли N-бромсукцинимид (12,46 г, 70 ммоль) и перемешивали смесь при комнатной температуре в течение 2 часов. Осадок отфильтровывали и промывали с помощью дихлорметана (100 мл*3) до получения указанного в названии соединения (12,7 г, 78%) в виде серого твердого вещества, которое применяли непосредственно на следующем этапе без дополнительной очистки. МС (ЭР+) C7H5BrFNO2 требует: 233, 235, обнаружено: 232, 234 [M+H]+.
Этап 10: Синтез (2-амино-5-бром-3-фторфенил)метанола

К раствору 2-амино-5-бром-3-фторбензойной кислоты (14,5 г, 62,2 ммоль) в ТГФ (150 мл) при 0°C добавляли борогидрид в ТГФ (1 M, 310 мл), а реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили метанолом (150 мл), концентрировали в вакууме, разбавляли водным бикарбонатом натрия (400 мл) и экстрагировали этилацетатом (200 мл*3). Органические слои разделяли, объединяли, промывали водой (200 мл) и соляным раствором (200 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (13,0 г, неочищенное), которое применяли непосредственно на следующем этапе без дополнительной очистки. МС (ЭР+) C7H7BrFNO требует: 219, 221, обнаружено: 220, 222 [M+H]+.
Этап 11: Синтез 2-амино-5-бром-3-фторбензальдегида
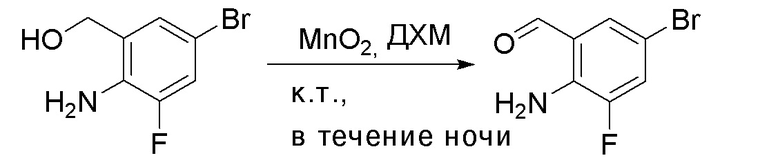
Смесь (2-амино-5-бром-3-фторфенил)метанола (13 г, 59,4 ммоль) и оксида марганца (31 г, 356,4 ммоль) в дихлорметане (400 мл) перемешивали при комнатной температуре в течение ночи. Твердое вещество фильтровали, а фильтрат концентрировали до получения указанного в названии соединения (11 г, 85%) в виде светло-желтого твердого вещества, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 12: Синтез 6-бром-8-фторхиназолин-2-ола

Перемешиваемую смесь 2-амино-5-бром-3-фторбензальдегида (2,17 г, 10 ммоль) и мочевины (9 г, 150 ммоль) нагревали при 180°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, а получаемый осадок отфильтровывали и промывали водой (500 мл *3). Влагу в ловушке полностью удаляли с помощью трехкратного совместного выпаривания с толуолом. Указанное в названии соединение (2 г, 83%) получали в виде желтого твердого вещества. МС (ЭР+) C8H4BrFN2O требует: 242, 244, обнаружено: 243, 245 [M+H]+.
Этап 13: Синтез 6-бром-2-хлорхиназолина

Раствор 6-бромхиназолин-2-ола (9,72 г, 40 ммоль) в оксихлориде фосфора (100 мл) кипятили с обратным холодильником в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и удаляли большую часть оксихлорида фосфора при пониженном давлении. Остаток по каплям добавляли в ледяную воду (500 мл), а получаемый осадок собирали с помощью фильтрования до получения указанного в названии соединения (9 г, 87%) в виде желтого твердого вещества. МС (ЭР+) C8H3BrClFN2 требует: 260, 262, обнаружено: 261, 263 [M+H]+.
Этап 14: Синтез 2-хлор-6-(3,5-диметоксифенил)-8-фторхиназолина

Смесь 6-бром-2-хлор-8-фторхиназолина (4,0 г, 15,4 ммоль), 3,5-диметоксифенилбороновой кислоты (4,47 г, 16,9 ммоль), карбоната цезия (10,0 г, 30,8 ммоль) и Pd(PPh3)2Cl2 (236 мг, 0,77 ммоль) в ТГФ (200 мл) и воде (10 мл) дегазировали азотом три раза и перемешивали при 80°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и сразу концентрировали. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир:дихлорметан=от 2:1 до 1:1), чтобы обеспечить указанное в названии соединение (2,5 г, 51%) в виде желтого твердого вещества. МС (ЭР+) C16H12ClFN2O2 требует: 318/320, обнаружено: 319/321 [M+H]+.
Этап 15: Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-8-фторхиназолина

К раствору 2-хлор-6-(3,5-диметоксифенил)-8-фторхиназолина (1,5 г, 4,7 ммоль) в сухом ТГФ (40 мл) по каплям добавляли сульфурилхлорид (1,59 г, 1,75 ммоль) при 0°C и перемешивали смесь в течение 1 часа. Реакционную смесь гасили водой (1 мл) и удаляли растворители при пониженном давлении. Остаток промывали ацетонитрилом и высушивали до получения указанного в названии соединения (700 мг, 38%) в виде белого твердого вещества. (МС (ЭР+) C16H10Cl3FN2O2 требует: 386, 388, обнаружено: 387, 389 [M+H]+.
Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-5-фторхиназолина

Этап 1: Синтез 6-амино-3-бром-2-фторбензойной кислоты

К раствору 2-амино-6-фторбензойной кислоты (12,0 г, 77,35 ммоль) в метаноле (150 мл) добавляли бром (15,7 мл) при -780C, и перемешивали смесь 2 часа при -78 oC. Реакционную смесь гасили ледяной водой (100 мл) и водным раствором сульфотиоата натрия и экстрагировали этилацетатом (150 мл × 3). Органические слои разделяли, объединяли, промывали водой (100 мл) и соляным раствором (100 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанный в названии неочищенный продукт (9,0 г, 50%). МС (ЭР+) C7H5BrFNO2 требует: 232, обнаружено: 233, 235 [M+H]+.
Этап 2: Синтез (6-амино-3-бром-2-фторфенил)метанола

К раствору 6-амино-3-бром-2-фторбензойной кислоты (9,0 г, 38,46 ммоль) в ТГФ (150 мл) добавляли BH3-ТГФ (1 M, 193 мл) при 0°C и перемешивали смесь при комнатной температуре в течение ночи. Реакционную смесь медленно гасили метанолом (50 мл) и удаляли растворители при пониженном давлении. Остаток разбавляли с помощью 200 мл этилацетата, промывали водой (200 мл) и соляным раствором (200 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение (8,3 г, 98%), которое применяли непосредственно на следующем этапе без дополнительной очистки. МС (ЭР+) C7H7BrFNO требует: 219, обнаружено: 220, 222 [M+H]+.
Этап 3: Синтез 6-амино-3-бром-2-фторбензальдегида

Суспензионную смесь (6-амино-3-бром-2-фторфенил)метанола (8,3 г, 37,72 ммоль) и оксида марганца(IV) (19,68 г, 226,32 ммоль) в дихлорметане (400 мл) перемешивали при комнатной температуре в течение ночи. Твердое вещество фильтровали, а фильтрат концентрировали до получения указанное в названии соединение в виде светло-желтого твердого вещества (6,0 г, 73%), которое применяли непосредственно на следующем этапе без дополнительной очистки. МС (ЭР+) C7H5BrFNO требует: 217, обнаружено: 218, 220 [M+H]+.
Этап 4: Синтез 6-бром-5-фторхиназолин-2-ола

Смесь 6-амино-3-бром-2-фторбензальдегида (3,0 г, 13,76 ммоль) и мочевины (12,40 г, 206,40 ммоль) нагревали до 180°C и перемешивали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры. Получаемый осадок собирали, промывали водой (3×100 мл) и выпаривали совместно с толуолом трижды, чтобы полностью удалить влагу в ловушке. Указанное в названии соединение (3,3 г, 99%) получали в виде желтого твердого вещества. МС (ЭР+) C8H4BrFN2O требует: 242, обнаружено: 243, 245 [M+H]+.
Этап 5: Синтез 6-бром-2-хлор-5-фторхиназолина

Раствор 6-бром-5-фторхиназолин-2-ола (3,0 г, 12,34 ммоль) в трихлориде фосфорила (10 мл) кипятили с обратным холодильником при 135°C в течение 5 часов. Большую часть трихлорида фосфорила удаляли при пониженном давлении, а остаток по каплям добавляли в ледяную воду (200 мл). Получаемый осадок собирали посредством фильтрования в виде желтого твердого вещества (3,1 г, 96%). МС (ЭР+) C8H3BrClFN2 требует: 260, обнаружено: 261, 263 [M+H]+.
Этап 6: Синтез 2-хлор-6-(3,5-диметоксифенил)-5-фторхиназолина
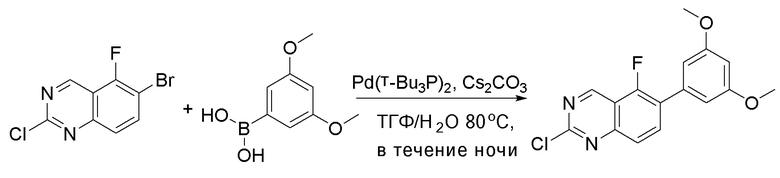
Смесь 6-бром-2-хлор-5-фторхиназолина (1,5 г, 5,74 ммоль), 3,5-диметоксифенилбороновой кислоты (1,15 г, 6,31 ммоль), карбоната церия (1,87 г, 5,74 ммоль) и бис(три-трет-бутилфосфин)палладия (148 мг, 0,29 ммоль) в ТГФ (30 мл) и воде (3 мл) дегазировали азотом три раза и перемешивали при 80°C в течение ночи. Смесь охлаждали до комнатной температуры и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали водой и соляным раствором, высушивали над сульфатом натрия, отфильтровывали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир:этилацетат=8:1) для получения указанного в названии продукта в виде белого твердого вещества (1,3 г, 70%). МС (ЭР+) C16H12ClFN2O2 требует: 318, обнаружено: 319, 321 [M+H]+.
Этап 7: Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-5-фторхиназолина

К раствору 2-хлор-6-(3,5-диметоксифенил)-5-фторхиназолина (1,25 г, 3,92 ммоль) в сухом ацетонитриле/ТГФ (20 мл/10 мл) по каплям добавляли сульфурилхлорид (1,32 г, 9,80 ммоль) при -20°C и перемешивали смесь в течение 1 часа. Реакционную смесь гасили водой (1 мл) и удаляли растворители при пониженном давлении. Осадок промывали ацетонитрилом и высушивали до получения указанного в названии соединения (886,5 мг, 56%) в виде белого твердого вещества. МС (ЭР+) C16H10Cl3FN2O2 требует: 386, обнаружено: 387, 389 [M+H]+.
Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-7-метоксихиназолина
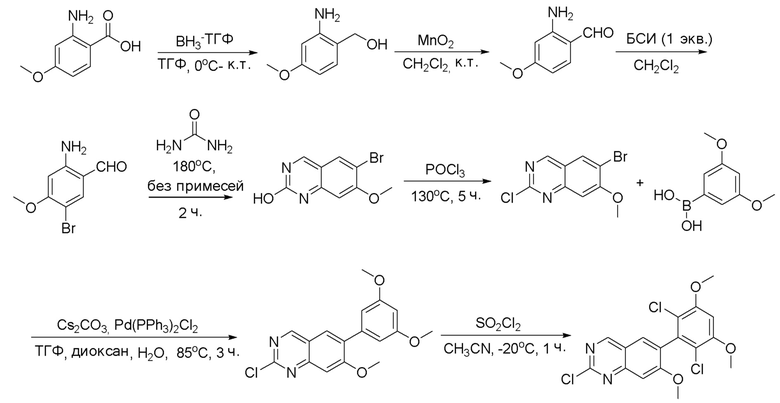 Этап 1: Синтез (2-амино-4-метоксифенил)метанола
Этап 1: Синтез (2-амино-4-метоксифенил)метанола
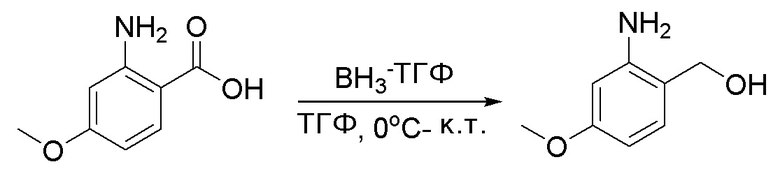
К раствору 2-амино-4-метоксибензойной кислоты (15,0 г, 89,8 ммоль) в ТГФ (300 мл) добавляли борогидрид в ТГФ (450 мл, 450 ммоль) при 0°C и перемешивали реакционную смесь при комнатной температуре в течение ночи. Реакционную смесь гасили водой (150 мл) и экстрагировали этилацетатом (500 мл ×3). Органические слои разделяли, объединяли, промывали водой (200 мл) и соляным раствором (200 мл), высушивали над сульфатом натрия, отфильтровывали и концентрировали, чтобы обеспечить указанное в названии соединение. МС (ЭР+) C8H11NO2 требует: 153, обнаружено: 154 [M+H]+.
Этап 2: Синтез 2-амино-4-метоксибензальдегида

Смесь (2-амино-4-метоксифенил)метанола (20 г, 131,0 ммоль) и оксида марганца (68 г, 786,0 ммоль) в дихлорметане (300 мл) перемешивали при комнатной температуре в течение ночи. Твердое вещество фильтровали, а фильтрат концентрировали. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир:этилацетат=6:1) до получения указанного в названии соединения (7 г, 35%) в виде желтого твердого вещества. МС (ЭР+) C8H9NO2 требует: 151, обнаружено: 152 [M+H]+.
Этап 3: Синтез 2-амино-5-бром-4-метоксибензальдегида

К перемешиваемому раствору 2-амино-4-метоксибензальдегида (6 г, 39,7 ммоль) в дихлорметане (100 мл) добавляли N-бромсукцинимид (7 г, 39,7 ммоль). Реакционную смесь разбавляли с помощью дихлорметана и воды. Отделенный органический слой высушивали с помощью сульфата натрия, отфильтровывали и концентрировали до получения указанного в названии соединения (5 г, 56%) в виде желтого твердого вещества. МС (ЭР+) C8H8BrNO2 требует: 229, 231, обнаружено: 230, 232 [M+H]+.
Этап 4: Синтез 6-бром-7-метоксихиназолин-2-ола
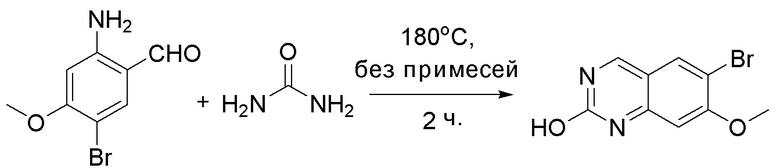
Смесь 2-амино-5-бром-4-метоксибензальдегида (3 г, 13,1 ммоль) и мочевины (12 г, 196,5 ммоль) перемешивали при 180°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и промывали водой (3×100 мл). Осадок собирали и высушивали до получения указанного в названии соединения (3 г, неочищенное) в виде желтого твердого вещества. МС (ЭР+) C8H7BrN2O2 требует: 254, 256, обнаружено: 255, 257 [M+H]+.
Этап 5: Синтез 6-бром-2-хлор-7-метоксихиназолина

Раствор 6-бром-7-метоксихиназолин-2-ола (3,0 г, 11,8 ммоль) в трихлориде фосфорила (30 мл) кипятили с обратным холодильником при 130°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и выпаривали большую часть трихлорида фосфорила. Остаток по каплям добавляли в ледяную воду (100 мл), а получаемый осадок собирали посредством фильтрования до получения указанного в названии соединения в виде желтого твердого вещества (2,4 г, 75%). МС (ЭР+) C9H6BrClN2O требует: 272, 274, обнаружено: 273, 275 [M+H]+.
Этап 6: Синтез 2-хлор-6-(3,5-диметоксифенил)-7-метоксихиназолина
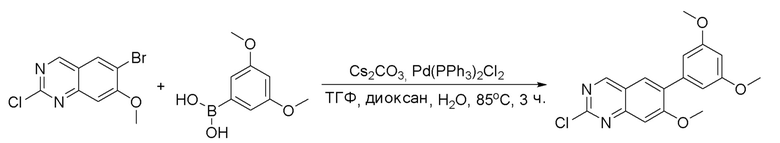 Смесь 6-бром-2-хлор-7-метоксихиназолина (2,4 г, 8,82 ммоль), 3,5-диметоксифенилбороновой кислоты (1,6 г, 8,82 ммоль), карбоната церия (8,6 г, 26,46 ммоль) и Pd(PPh3)2Cl2 (1,4 г, 2,1 ммоль) в ТГФ (10 мл), диоксане (10 мл) и воде (2 мл) дегазировали азотом трижды и перемешивали при 85°C в течение 3 часов. Смесь охлаждали до комнатной температуры и экстрагировали с помощью дихлорметана (3×50 мл). Органические слои разделяли, объединяли, промывали водой и соляным раствором, высушивали над сульфатом натрия, отфильтровывали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир: этилацетат=1:4) до получения указанного в названии соединения (1,1 г, 38%) в виде белого твердого вещества. МС (ЭР+) C17H15ClN2O3 требует: 330, 332, обнаружено: 331, 333 [M+H]+.
Смесь 6-бром-2-хлор-7-метоксихиназолина (2,4 г, 8,82 ммоль), 3,5-диметоксифенилбороновой кислоты (1,6 г, 8,82 ммоль), карбоната церия (8,6 г, 26,46 ммоль) и Pd(PPh3)2Cl2 (1,4 г, 2,1 ммоль) в ТГФ (10 мл), диоксане (10 мл) и воде (2 мл) дегазировали азотом трижды и перемешивали при 85°C в течение 3 часов. Смесь охлаждали до комнатной температуры и экстрагировали с помощью дихлорметана (3×50 мл). Органические слои разделяли, объединяли, промывали водой и соляным раствором, высушивали над сульфатом натрия, отфильтровывали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (петролейный эфир: этилацетат=1:4) до получения указанного в названии соединения (1,1 г, 38%) в виде белого твердого вещества. МС (ЭР+) C17H15ClN2O3 требует: 330, 332, обнаружено: 331, 333 [M+H]+.
Этап 7: Синтез 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)-7-метоксихиназолина

К раствору 2-хлор-6-(3,5-диметоксифенил)-7-метоксихиназолина (200 мг, 0,61 ммоль) в ацетонитриле (5 мл) добавляли сульфурилхлорид (205 мг, 1,52 ммоль) и перемешивали смесь при -20°C в течение 1 часа. Реакционную смесь гасили водой (1 мл) и концентрировали при пониженном давлении. Осадок промывали ацетонитрилом и высушивали до получения указанного в названии соединения в виде белого твердого вещества (120 мг, 50%). МС (ЭР+) C17H13Cl3N2O3 требует: 398, обнаружено: 399, 401 [M+H]+.
Данные ЯМР и ЖХ-МС для некоторых соединений приведены в таблице ниже. Приведен также протокол синтеза, применяемый для получения соединений.
8,3 Гц, 1H), 7,55 (д, J=8,8 Гц, 1H), 7,12 (д, J=8,1 Гц, 1H), 4,45 (с, 1H), 4,34 (с, 1H), 3,61 (дд, J=18,4, 11,5 Гц, 1H), 3,15 (дд, J=7,3, 4,4 Гц, 2H), 2,86 (тд, J=7,3, 3,7 Гц, 2H), 1,95 (д, J=8,2 Гц, 3H), 1,87-1,61 (м, 3H), 1,56 (с, 1H).
Оценка биохимической активности
Для того, чтобы оценить активность химических соединений по отношению к соответствующей киназе, представляющей интерес, применяли технологическую платформу сдвига электрофоретической подвижности Caliper LifeSciences. Флуоресцентно-меченный пептид субстрата инкубируют в присутствии дозированных уровней соединений, набора концентрации киназы и АТФ таким образом, что фосфорилируют отражающую часть пептида. В конце реакции смесь фосфорилированных (продукт) и нефосфорилированных (субстрат) пептидов пропускают сквозь микрофлюидную систему Caliper LabChip® EZ Reader II, под приложенной разностью потенциалов. Присутствие фосфатной группы на пептиде продукта обеспечивает разницу в массе и заряде между пептидом продукта и пептидом субстрата, что приводит к разделению пулов субстрата и продукта в образце. По мере того как пулы проходили LEDS внутри прибора, эти пулы обнаруживали и разрешали в виде отдельных пиков. Следовательно, соотношение между этими пиками отражает активность химического вещества в этой концентрации в этой лунке при этих условиях.
Анализ FGFR-4 дикого типа при Км: В каждой лунке 384-луночного планшета инкубировали 0,5 нг/мкл FGFR-4 дикого типа (Carna Biosciences, Inc.) в общем объеме 12,5 мкл буфера (100 мM HEPES pH 7,5, 0,015% Brij 35, 10 мM MgCl2, 1 мM DTT) с 1 мкM CSKtide (5-FAM- KKKKEEIYFFFG-NH2) и 400 мкM АТФ при 25 C в течение 90 минут в присутствии или отсутствии серий дозированных концентраций соединения (конечная концентрация ДМСО 1%). Реакционную смесь останавливали с помощью добавления 70 мкл останавливающего буфера (100 мM HEPES pH 7,5, 0,015% Brij 35, 35 мM ЭДТА и 0,2% Coating Reagent 3 (Caliper Lifesciences)). Затем планшет считывали на Caliper LabChip® EZ Reader II (настройки протокола: -1,9 фунтов на квадратный дюйм, напряжение на входе -700, напряжение на выходе -3000, стерилизации после образца 35 с).
Обнаружение pMAPK (Thr202/Tyr204) с применением Alpha Elisa
Клетки MDA-MB453 или DMS 114 высевали в 96‐луночные планшеты для культивирования клеток при плотности 1x105 клеток или 3x104 клеток, соответственно. Клеткам позволяли закрепиться, а ростовую среду заменяли бессывороточной средой. Добавляли соединения в указанных концентрациях. Спустя 1 ч. инкубации в присутствии соединения клетки собирали. В случае клеток DMS 114, добавляли 100 нг/мл FGF2 в течение 10 минут до сбора клеток. Получали клеточные лизаты и обрабатываются согласно инструкции производителя (AlphaScreen® SureFire™ Phospho-ERK 1/2 Kit (Perkin Elmer).
В таблице ниже представлены биохимические данные для Соединений 1-92. В приведенной ниже таблице, в случае FGFR4 и pERK alphaLISA: «A» означает, что IC50 составляет менее10 нмоль; «B» означает, что IC50 составляет более или равно 10 и менее 100 нмоль; «C» означает, что IC50 составляет более или равно 100 и менее 1000 нмоль; «D» означает, что IC50 составляет более 1000 нмоль.
Эффективность в модели in vivo
Изучали эффекты Соединения 27 на ингибирование роста модели подкожного ксенотрансплантата клеток рака печени Нер3В с различными дозировками.
Применяли самок бестимусных мышей (Mus Musculus) в возрасте от 6 до 8 недель. Культура опухолевых клеток и прививка: клетки Нер3В культивировали со средой EMEM (Invitrogen, США), дополненной 10% FBS (Gibco, Австралия). Клетки собирали при 90% конфлуентности, а жизнеспособность составляла не менее 90%. Мышам в правый бок в начале исследования подкожно имплантировали (п/к) 200 мкл 10 × 106 клеток Hep3B в 50% Matrigel.
Группирование животных и график дозирования: Спустя десять дней после имплантации клеток, когда опухоли достигали среднего объема 284 мм3, на основе объема опухоли выбирали 36 мышей и случайным образом распределяли на 5 групп лечения (n=9). День рандомизации обозначали как день 0 и лечение начинали с него.
Измерения объема опухоли и массы тела: Размер опухоли измеряли два раза в неделю в двух измерениях с применением штангенциркуля, а объем выражали в мм3 с применением формулы: V=0,5 a × b2, где a и b представляли собой длинные и короткие диаметры опухоли, соответственно. Массу тела измеряли по меньшей мере два раза в неделю.
Объемы опухолей несущих Hep3B бестимусных мышей: Фиг. 1 представляет собой линейный график, отражающий ингибирование роста в обработанных Соединением 27 группах относительно опухолей ксенотрансплантатов Hep3B у бестимусных мышей. Статистически значимое снижение объемов опухолей наблюдалось в группах с эффективностью 30 и 100 мг/кг п/о дважды в сутки, по сравнении с контрольной группой, получающей носитель. Увеличение дозировки Соединения 27 повышало эффективность ингибирования опухоли. Опухоли в обработанной Соединением 27 группе (100 мг/кг п/о дважды в сутки) регрессировали.
Изменение массы тела (%) несущих Hep3B бестимусных мышей: Фиг. 2 представляет собой линейный график, отображающий изменение массы тела (%) в течение всего периода исследования. Все мыши, за исключением мышей обработанных Соединением 27 групп (100 мг/кг п/о дважды в сутки) показывали значительную потерю массы тела. Масса тела мышей в группе, получавшей носитель, сократилась примерно на 15% к 10-ому Дню опухолевой нагрузки. Этот результат указывает на то, что Соединение 27 хорошо переносилось при текущих дозировках и режиме дозирования у бестимусных мышей и что Соединение 27 может облегчить потерю массы тела путем ингибирования роста опухоли.
У мышей, обработанных с помощью Соединения 27, наблюдалось значительное уменьшение объема опухоли по сравнению с группой, получавшей носитель, в течение всего периода исследования. Увеличение дозировки Соединения 27 от 10 мг/кг до 100 мг/кг повышало эффективность ингибирования опухоли. Опухоли у мышей в обработанной Соединением 27 группе (100 мг/кг п/о дважды в сутки) регрессировали и почти исчезли. Все мыши, за исключением мышей обработанных Соединением 27 групп (100 мг/кг п/о дважды в сутки) теряли массу тела. Масса тела мышей в группе, получавшей носитель, сократилась примерно на 15% к 10-ому Дню опухолевой нагрузки. Эти результаты указывают на то, что Соединение 27 хорошо переносилось при текущих дозировках и режиме дозирования у бестимусных мышей и что Соединение 27 может облегчить потерю массы тела путем ингибирования роста опухоли.
Включение посредством ссылки
Все публикации и патенты, упомянутые в данном документе, включены посредством ссылки во всей их полноте, как если бы каждая отдельная публикация или патент были конкретно и отдельно указаны как включенные посредством ссылки.
Эквиваленты
Специалист в данной области техники поймет или будет в состоянии определить множество эквивалентов конкретных вариантов реализации изобретения, описанных в данном документе, с применением не более чем рутинных экспериментов. Такие эквиваленты, как предполагается, охвачены следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2013 |
|
RU2679130C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2732127C1 |
| ИНГИБИТОРЫ FGFR4 | 2014 |
|
RU2715708C2 |
| КРИСТАЛЛИЧЕСКИЙ ИНГИБИТОР FGFR4 И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2763328C2 |
| ИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ FGFR, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2719428C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| Производные хинолона как ингибиторы рецептора фактора роста фибробластов | 2015 |
|
RU2721723C2 |
| НИТРИЛСОДЕРЖАЩИЕ ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2021 |
|
RU2786722C1 |
| НИТРИЛСОДЕРЖАЩИЕ ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2021 |
|
RU2820150C2 |
| УЛУЧШЕННЫЕ АГОНИСТЫ АПЕЛИНОВОГО РЕЦЕПТОРА (APJ) И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2766148C1 |


Изобретение относится к новому соединению формулы I и к соединению формулы II или их фармацевтически приемлемой соли, которые обладают свойствами ингибитора FGFR-4. Соединения могут найти применение при лечении заболевания или состояния опосредованного FGFR-4, например гепатоклеточной карциномы или гиперлипедимии. В соединении формулы I
 (I)
(I)  (II)
(II)
головная часть представляет собой  , где каждый из Ra, Rb и Rc независимо выбран из H, C1-4 алкила, C1-4 циклоалкила или циано; кольцо A представляет собой тетрагидрофуранил или тетрагидропиранил; каждый из R1 и R2 независимо выбран из галогена, циано, C1-6 алкокси, гидрокси, оксо, амино, амидо и C1-6 алкила, где каждый из C1-6 алкокси и C1-6 алкила независимо замещен 0-5 случаями R4; R3 представляет собой галоген; каждый R4 независимо выбран из C1-6 алкила, C1-6 алкокси, галогена, гидрокси, оксо, амино и циано; m равен 0-3; n равен 0-4; и p равен 0-2. 7 н. и 14 з.п. ф-лы, 2 ил., 3 табл., 92 пр.
, где каждый из Ra, Rb и Rc независимо выбран из H, C1-4 алкила, C1-4 циклоалкила или циано; кольцо A представляет собой тетрагидрофуранил или тетрагидропиранил; каждый из R1 и R2 независимо выбран из галогена, циано, C1-6 алкокси, гидрокси, оксо, амино, амидо и C1-6 алкила, где каждый из C1-6 алкокси и C1-6 алкила независимо замещен 0-5 случаями R4; R3 представляет собой галоген; каждый R4 независимо выбран из C1-6 алкила, C1-6 алкокси, галогена, гидрокси, оксо, амино и циано; m равен 0-3; n равен 0-4; и p равен 0-2. 7 н. и 14 з.п. ф-лы, 2 ил., 3 табл., 92 пр.
1. Соединение Формулы I или его фармацевтически приемлемая соль:
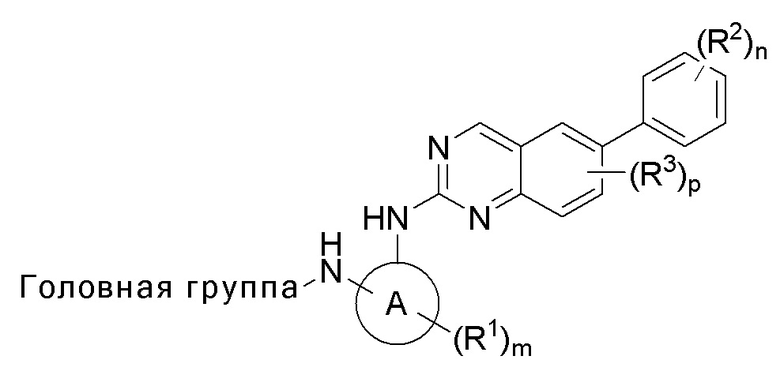
Формула I,
где
головная часть представляет собой  , где каждый из Ra, Rb и Rc независимо выбран из H, C1-4 алкила, C1-4 циклоалкила или циано;
, где каждый из Ra, Rb и Rc независимо выбран из H, C1-4 алкила, C1-4 циклоалкила или циано;
кольцо A представляет собой тетрагидрофуранил или тетрагидропиранил;
каждый из R1 и R2 независимо выбран из галогена, циано, C1-6 алкокси, гидрокси, оксо, амино, амидо и C1-6 алкила, где каждый из C1-6 алкокси и C1-6 алкила независимо замещен 0-5 случаями R4;
R3 представляет собой галоген;
каждый R4 независимо выбран из C1-6 алкила, C1-6 алкокси, галогена, гидрокси, оксо, амино и циано;
m равен 0-3;
n равен 0-4; и
p равен 0-2.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где головная часть представляет собой  .
.
3. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, где каждый из R1 и R2 независимо выбран из галогена, циано, C1-6 алкокси, гидрокси, оксо, амино, амидо и C1-6 алкила.
4. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, где:
R2 независимо выбран из галогена, циано, C1-6 алкокси, гидрокси и C1-6 алкила;
R3 представляет собой галоген;
m равен 0;
n равен 4; и
p равен 0 или 1.
5. Соединение Формулы II или его фармацевтически приемлемая соль:

Формула II,
где:
кольцо A представляет собой тетрагидрофуранил или тетрагидропиранил;
R1 выбран из галогена, циано, C1-6 алкокси, гидрокси, оксо, амино, амидо и C1-6 алкила;
каждый R2 независимо выбран из галогена и C1-6 алкокси;
R3 представляет собой галоген;
m равен 0-1;
n равен 0-4; и
p равен 0-1.
6. Соединение по п. 5 или его фармацевтически приемлемая соль, где:
m равен 0; и
n равен 1-4.
7. Соединение по п. 5 или 6 или его фармацевтически приемлемая соль, где:
m равен 0; и
n равен 4.
8. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль, где кольцо А представляет собой тетрагидрофуранил.
9. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль, где кольцо А представляет собой тетрагидропиранил.
10. Соединение по любому из пп. 1-9 или его фармацевтически приемлемая соль, где часть соединения, представленная  , представляет собой
, представляет собой  .
.
11. Соединение, выбранное из группы, состоящей из:
,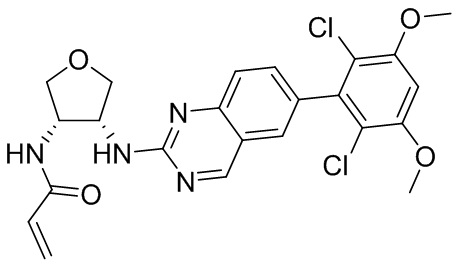 ,
, ,
, 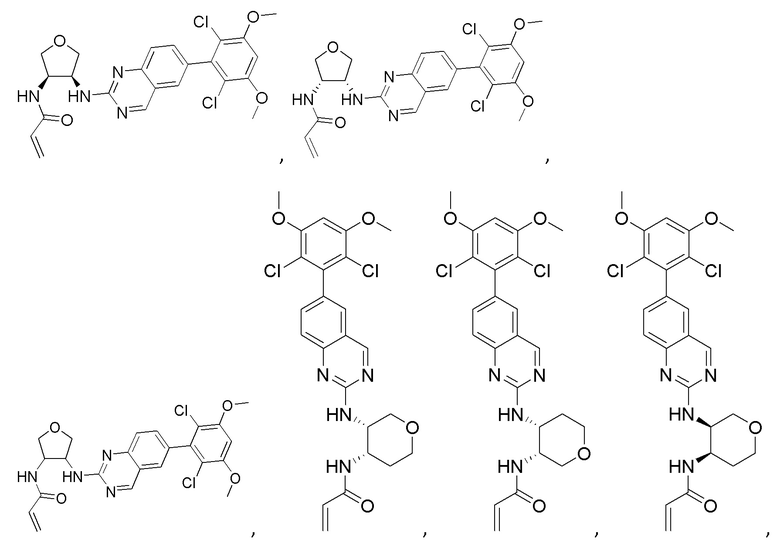
 и
и 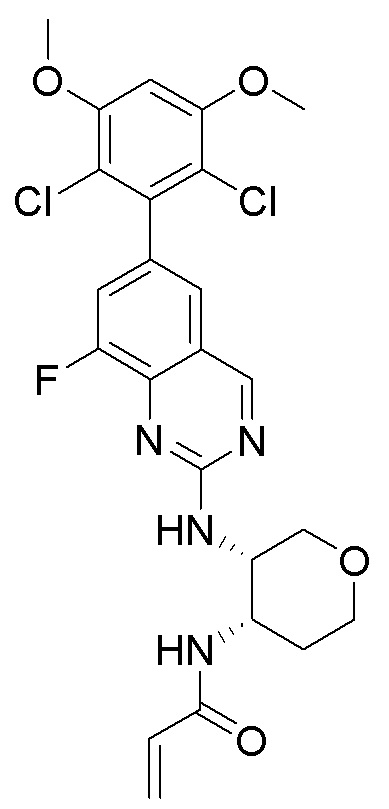
или его фармацевтически приемлемой соли.
12. Соединение, представляющее собой

или его фармацевтически приемлемая соль.
13. Соединение, представляющее собой

или его фармацевтически приемлемая соль.
14. Фармацевтическая композиция, обладающая FGF-19 модулирующей активностью, содержащая:
фармацевтически приемлемый носитель; и
терапевтически эффективное количество соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли.
15. Способ лечения состояния, опосредованного FGFR-4, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 14.
16. Способ лечения состояния, характеризующегося чрезмерной экспрессией FGFR-4, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 14.
17. Способ лечения состояния, характеризующегося амплификацией FGF-19, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 14.
18. Способ лечения состояния, характеризующегося чрезмерной экспрессией FGF-19, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 14.
19. Способ лечения рака, опосредованного FGFR-4, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 14, причем рак, опосредованный FGFR-4, выбран из рака печени, рака молочной железы, рака легких, рака яичника и саркомы.
20. Способ лечения гепатоклеточной карциномы, опосредованной FGFR-4, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 14.
21. Способ лечения гиперлипидемии, опосредованной FGFR-4, включающий введение терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 14.
| Жидкоподвижная смесь для изготовления литейных форм и стержней | 1991 |
|
SU1836174A3 |
| RU 2015104342 A, 27.08.2016 & WO 2014011900 A2, 16.01.2014 | |||
| WO 2011034907 A2, 24.03.2011 & RU 2012114902 A1, 27.10.2013 | |||
| WO 2010028236 A1, 11.03.2010 & RU 2542963 C2, 27.02.2015. | |||

