Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения фармацевтических композиций в виде быстрораспадающихся оральных таблеток, предпочтительно, для трансбуккального или сублингвального введения в числе прочих путей введения, содержащих биологически активное действующее лекарственное вещество, по существу, свободное от кристаллической формы, предпочтительно направленных на лечение панического расстройства или острых приступов выраженной тревоги. Представлены также фармацевтические композиции, полученные этим способом.
Предпосылки создания изобретения
Бензодиазепины представляют собой гетерогенную группу химических соединений, имеющую общее основное 5-арил-1,4-бензодиазепиновое ядро. Среди них можно выделить алпразолам, клоназепам и бромазепам.
Фармакодинамические и фармакокинетические различия определяют различные терапевтические применения в соответствии с интенсивностью проявления седативной, снотворной миорелаксирующей, анксиолитической или противосудорожной активности.
Алпразолам, 8-хлор-1-метил-6-фенил-4H-1,2,4-триазоло(4,3-a)(l,4)бензодиазепин, часто назначают в качестве анксиолитического средства. В последние годы исследовали его применение при дистресс состояниях, таких как паническое расстройство. Существует множество научных работ по этой тематике и среди них:
Ballenger JC, Burrows D, DuPont, RL Jr. - ARCH. GEN. PSYCHIATRY 45: 413-422, 1988.
Spiegel DA-PSYCOPHARAMOL. BULL. 1988, 34 (2): 191.
Noyes R., Burrows GD, Reich JH, Judd FK, Gravy MJ et al - J. CLIN. PSYCHIATRY 1996, 57 (8): 349.
Gleman James H, - Treatment of Panic Disorder with Alprazolan - патент США 4508726 (1985).
Вследствие того что расстройства, вызываемые у больных этим типом патологии, являются такими, как чувство беспокойства, ощущение надвигающейся смерти, дезориентировка, существенно, чтобы терапевтическое воздействие наступало по возможности максимально быстро в том времени и месте, где начался кризис.
Однако до настоящего времени отсутствует соответствующий препарат алпразолама, который производит терапевтическое воздействие настолько быстро, насколько этого требуют симптомы болезни.
Исследования, выполненные несколькими авторами, показали, что сублингвальное введение могло бы оказаться альтернативным и эффективным путем введения для некоторых бензодиазепинов. Поэтому именно такую возможность оценивали при использовании лоразепама (Gale GD, Gallon S, Porter WR. Sublingual Lorasepam: a better premedication. - BR. J. ANAETH. 1983: 55: 761-5), Greenblatt DJ, Divoll M, Harmatz JS et al. Pharmacokinetic comparisons of sublingual lorazepam with intravenous, intramuscular and oral lorazepam, J. PHARM. SCL. 1982; 71: 248-252); триазолама (Scavone JM, Greenblatt DJ, Friedman H, et al. Enhanced bioavailability of triazolam following sublingual versus oral administration. J. CLIN.PHARMACOL. 1986; 26: 208-10) и флунитразепама (Huttel MS, Bang U. Sublingual flunitrazepam for premedication, ACTA ANAETH. SCAND. 1985; 29: 209-11).
В 1987 г. Joseph Scavone, David Greenbaltt and Richard Shader (J. CLIN. PSUCHOPAHARMACOLOGY- Vol. 7 №5, Oct. 1987) продемонстрировали на здоровых пациентах, добровольно подвергшихся лечению доступными выпускаемыми промышленностью таблетками для орального введения, что сублингвальное всасывание алпразолама возможно. Такие доступные выпускаемые промышленностью таблетки, находясь в течение 15 минут под языком добровольцев, которые голодали предшествующие 8 часов и затем голодали 3 часа после введения лекарственного средства, как было показано, всасываются сублингвально также быстро и без статистически значимых различий, как и при оральном всасывании после голодания.
Пики плазменных концентраций после сублингвального введения были выше, чем наблюдаемые после опыта при оральном введении (17,3 по сравнению с 14,9 нг/мл). Время, необходимое для достижения максимальной концентрации, составляло 1,17 часов при сублингвальном введении и 1,73 часа при нормальном или дигестивном пути введения.
В 1992 г. те же авторы (Scavone JM, Greenblatt DJ, Goddard JE, Friedman H, Harmatz JS and Shader RI - EUR. J. CLIN. PHARMACOL. (1992) 42: 439-443) исследовали влияние питания на оральное или сублингвальное всасывание алпразолама. Они подтвердили, что сублингвальное всасывание, происходящее после приема пищи, при удержании таблетки алпразолама, массой 1 мг, под языком в течение 15 минут было ниже, чем всасывание, наблюдаемое теми же авторами у голодающих пациентов. Они наблюдали, что при введении, происходящем после приема пищи, плазменные концентрации достигают пиковых значений при сублингвальном пути введения позднее, чем при оральном пути введения (2,8 часов, по сравнению с 1,8 часами, P<0,01). Более того, они подтвердили, что через 15 минут под языком не было обнаружено оставшихся частей таблеток.
Хорошо известно, что распадаемость является существенным свойством таблеток, предназначенных для орального введения, и это даже в большей степени верно для сублингвальных таблеток, потому что активные действующие лекарственные вещества, предпочтительно, всасываются в полости рта через трансбуккальную или сублингвальную слизистые оболочки.
Кроме того, известно, что чем выше скорость распадаемости, тем быстрее произойдет усвоение лекарственного средства, которое всасывается окружающей средой. Для лекарственных средств широкого спектра применения в терапии человека различные фармакопеи устанавливают максимальное время распадаемости таблетки, для того чтобы убедиться в ее эффективности. Например, для сублингвальных таблеток изосорбида или нитроглицерина динитрата фармакопея США XXIII устанавливает максимальное время распадаемости, не превышающее 2 минуты, а для эрготамина тартрата время распадаемости составляет не более чем 5 минут. Также приведены другие продукты с более длительным временем распадаемости. Описанный выше уровень техники подчеркивает практическую уместность времени распадаемости, непосредственно связанную с терапевтической активностью и эффективностью каждого продукта.
Результаты, полученные исследователями в приведенных выше работах, указывают на то, что сублингвальный путь введения фактически является альтернативным путем, который заслуживает изучения. Однако эти исследования, кроме того, подтверждают тот факт, что использование оральной таблетки, которую необходимо удерживать под языком в течение 15 минут, для того чтобы достигнуть всасывания активного действующего вещества, не находит практического применения в области терапии. Так же как и в случае, когда такая таблетка требует условий длительного голодания как до, так и после ее введения для всасывания бензодиазепина.
В частности, когда бензодиазепин, например алпразолам, используют при лечении панического расстройства или острых приступов выраженной тревоги, жесткие условия голодания, которые предшествуют введению продукта или следуют за его введением, для того чтобы убедиться в эффективности продукта, и, более того, практическое неудобство удерживания таблетки в щечном кармане в течение длительного периода времени (от 10 до 15 минут) до ее полного распада, как это описывают в приведенной выше научной литературе, затрудняют выполнение больным схемы и режима лечения.
Suck Won Kim et al. (ANNALS OF CLINICAL PSYCHIATRY- Vol. 4 №2, June 1992) полагают, что благотворное действие на больного окажет быстрое воздействие продукта, позволяющее нейтрализовать симптомы, которые предшествуют панической атаке (продромальные симптомы), включая само паническое расстройство.
Поэтому, в случае пациентов, страдающих паническими атаками или острыми приступами выраженной тревоги, желательно быстрое воздействие лекарственного препарата. Такое воздействие может быть достигнуто посредством использования орального введения лекарственного средства, предпочтительно сублингвального пути введения.
Аналогичный преимущественный критерий применяют к другим терапевтическим рецептурам бензодиазепинов, когда фармацевтическая композиция обеспечивает быструю доставку и воздействие содержащегося в ней активного действующего лекарственного вещества.
Хорошо известно, что для большинства действующих лекарственных веществ аморфное состояние является наиболее удобной физической формой, способствующей их растворению и всасыванию.
Твердые кристаллические вещества имеют определенную форму, соответствующую установленным кристаллографическим системам, которую они сохраняют даже после мелкого дробления, как было показано Aiache JM et al {BIOFARMACIA - Ed. El Manuel Moderno, Mexico, 1983), которые считают, что расположение молекул в структурной сетке остается неизменным.
С другой стороны, вещества в аморфном состоянии не имеют определенной структуры, они обладают неупорядоченной структурой по трем измерениям и термодинамически менее стабильны, однако такое состояние представляет собой обладающую наибольшей растворимостью форму действующего лекарственного вещества. Существует некоторое количество ссылок, которые подтверждают приведенную выше информацию. Mullins and Macek (Mullins, JD, Macek, TJ - J. PHARM. SCI., 49, 425 (I960)) раскрывают, что аморфный новобиоцин в десять раз более растворим, чем его кристаллическая форма, и обладает оральным всасыванием. Кристаллическая форма не является биодоступной при таком пути введения.
Higuchi and col. (Higuchi, WI, Lau, PK, Higuchi, T, Schell, JW - J. PHARM. SCI., 52, 150 (1963)) указывают на то, что растворимость аморфного метилпреднизолона в 20 раз выше, чем растворимость того же самого вещества в кристаллической форме.
В 1972 г. Monkhouse et al. (Monkhouse, DC and Lach, JL - PHARM. SCI. 1431- vol. 61(9) 1972) описывают способ увеличения растворимости слаборастворимых лекарственных средств посредством увеличения поверхности контактирования активного действующего лекарственного вещества со средой. Они смешивают и встряхивают лекарственное средство с инертном веществом (диоксид кремния) и растворителем в течение некоторого периода времени, затем удаляют растворитель и смесь измельчают. Продемонстрированный способ оказывается пригодным для значительного увеличения растворения веществ.
Однако способ имеет недостаток, который заключается в отсутствии линейного соотношения между количеством использованного инертного вещества и количеством свободного активного действующего лекарственного вещества, предварительно осажденного на его поверхности. Более того, инертный носитель не всегда оказывается пригодным для фармацевтической композиции.
Поэтому все еще существует необходимость в получении таблеток для орального введения, предпочтительно сублингвального введения, для лечения панического расстройства или острых приступов выраженной тревоги, которые обладают лучшей растворимостью и биодоступностью активного действующего лекарственного вещества композиции, предпочтительно алпразолама, что делает возможным его быстрое воздействие.
Сущность изобретения
Поэтому целью данного изобретения является обеспечение способа получения фармацевтических композиций в виде таблеток для орального, предпочтительно трансбуккального или сублингвального введения, для лечения дистресс состояний, панического расстройства или острых приступов выраженной тревоги.
Другой целью настоящего изобретения является обеспечение простого и экономичного способа получения фармацевтических композиций в виде таблеток для орального введения, предпочтительно сублингвального введения, обладающих быстрой распадаемостью, содержащих биологически активное действующее лекарственное вещество, которое, по существу, свободно от кристаллической формы.
Еще одной целью настоящего изобретения является обеспечение способа получения фармацевтических композиций для орального, предпочтительно трансбуккального или сублингвального путей введения, обладающих быстрой распадаемостью и содержащих биологически активное действующее лекарственное вещество, которое, по существу, свободно от кристаллической формы и в которое не добавляют агенты, ингибирующие кристаллизацию.
Дополнительной целью данного изобретения является обеспечение способа получения фармацевтических композиций в виде таблеток для орального, предпочтительно трансбуккального или сублингвального путей введения, содержащих биологически активное действующее лекарственное вещество, которое, по существу, свободно от кристаллической формы, обладающих быстрой распадаемостью и общей усвояемостью биологически активного действующего лекарственного вещества.
Другой целью данного изобретения является обеспечение способа получения фармацевтических композиций в виде таблеток для орального, предпочтительно трансбуккального или сублингвального путей введения, обладающих быстрой распадаемостью, содержащих биологически активное действующее лекарственное вещество, которое, по существу, свободно от кристаллической формы, гомогенно распределенное по всей массе таблетки.
Другой целью данного изобретения является обеспечение способа получения фармацевтических композиций в виде таблеток для орального, предпочтительно трансбуккального или сублингвального путей введения, уменьшенного размера, который способствует малой прочности таблетки в ротовой полости.
Еще одной целью данного изобретения является обеспечение способа получения фармацевтических композиций в виде таблеток для орального, предпочтительно трансбуккального или сублингвального путей введения, содержащих алпразолам, который, по существу, свободен от кристаллической формы, для лечения дистресс состояний, панических расстройств или острых приступов выраженной тревоги.
Дополнительной целью данного изобретения является обеспечение способа получения фармацевтических композиций в виде таблеток, обладающих быстрой распадаемостью для орального, предпочтительно трансбуккального или сублингвального путей введения, содержащих биологически активное действующее лекарственное вещество, которое, по существу, свободно от кристаллической формы.
Другой целью данного изобретения является обеспечение способа получения фармацевтических композиций в виде таблеток, обладающих быстрой распадаемостью, для орального, предпочтительно трансбуккального или сублингвального путей введения, содержащих алпразолам, который, по существу, свободен от кристаллической формы.
Удивительно, что, как было обнаружено, в случае, когда последовательность добавления эксципиентов при получении фармацевтического препарата меняют, можно получить таблетки, содержащие биологически активное действующее лекарственное вещество, предпочтительно алпразолам, который, по существу, свободен от кристаллической формы, и обладающие требуемыми характеристиками растворимости и биодоступности без необходимости добавления дополнительных агентов, таких как ингибиторы кристаллизации.
В настоящее время было обнаружено, что такие таблетки получают, когда активное действующее лекарственное вещество фармацевтической композиции для получения раствора растворяют в растворителе или соответствующей смеси растворителей вместе со связывающим веществом и частью смазывающего вещества, от 20% до 60%. Оставшиеся компоненты композиции пропитывают этим раствором, затем растворитель удаляют и, в конечном счете, ароматизированную массу прессуют.
Таким образом, способствуют гомогенному диспергированию активного действующего лекарственного вещества по поверхности всех компонентов фармацевтической композиции и, что удивительно, было обнаружено, что присутствие в растворителе агглютинирующего и смазывающего агентов действует аналогично действию ингибиторов кристаллизации, предотвращая полное восстановление исходной кристаллической формы активного действующего лекарственного вещества в получаемой таблетке.
Обе характеристики, однородность распределения и некристаллическое состояние, замечательно улучшают растворимость и биодоступность активного действующего лекарственного вещества композиции.
Такая последовательность не только является новой, но, кроме того, и очень удобной, так как она препятствует тому, чтобы активное действующее лекарственное вещество восстанавливало исходную кристаллическую форму после его растворения в выбранном растворителе.
Более того, эти таблетки обнаруживают существенное преимущество по сравнению с традиционными таблетками, составленными с такими же активными действующими лекарственными веществами, из-за их малого размера, их замечательной скорости распадаемости и пониженной устойчивости всей таблетки или ее фрагментов в ротовой полости или при сублингвальной локализации. Такие характеристики также способствуют эффективному соблюдению пациентом предписанного терапевтического лечения.
Более того, фармацевтические композиции, полученные способом по настоящему изобретению, вследствие их уменьшенного размера и быстрого растворения не требуют использования жидкостей, например воды, при их введении, что дает возможность пациенту использовать продукт в любое время и в любом месте.
Краткое описание фигур
Фиг.1-5, включенные в качестве иллюстративных примеров, показывают и подтверждают изменения в кристаллической структуре активного агента, алпразолама, используемого при получении фармацевтических композиций по настоящему изобретению.
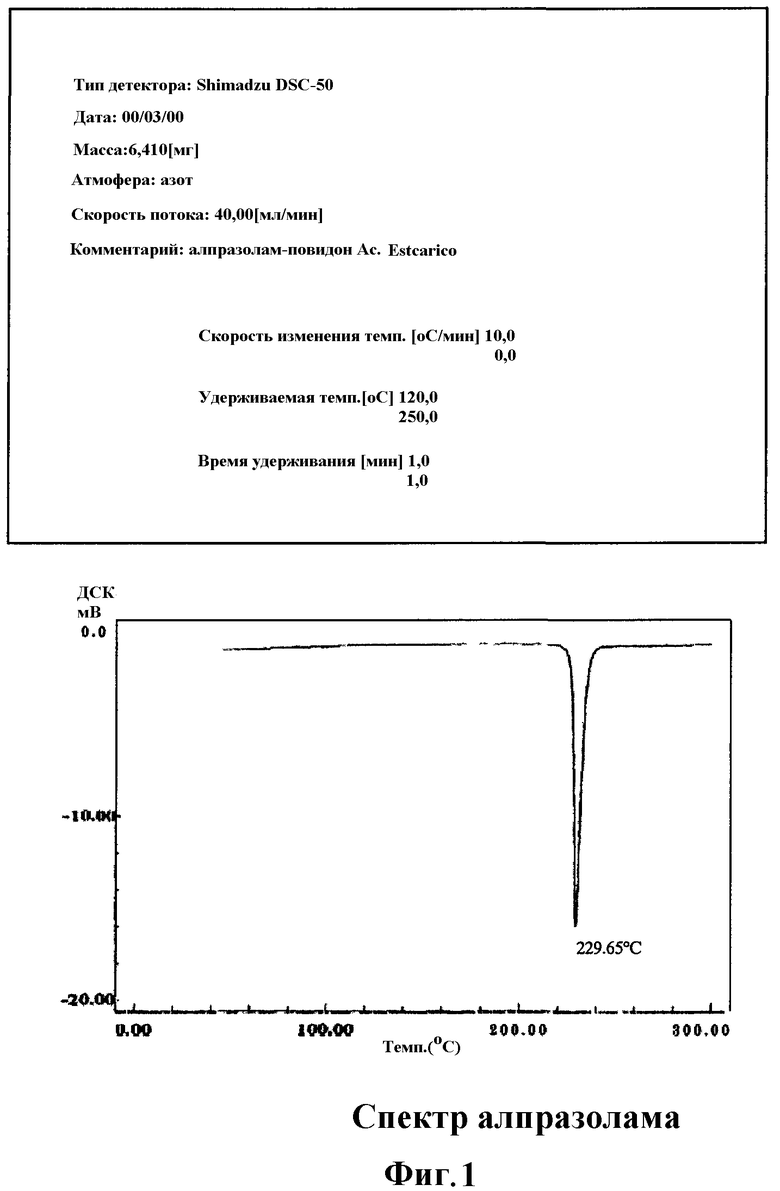
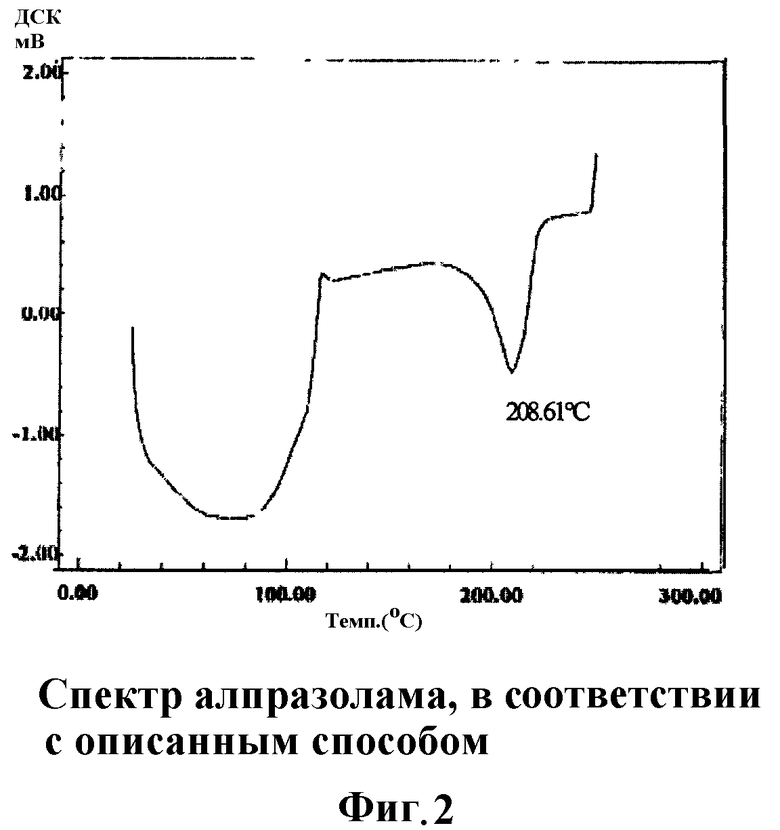
Фиг.1 и 2 соответствуют кривым дифференциального термического анализа, полученным с помощью ДСК (дифференциальной сканирующей калориметрии) алпразолама (Фиг.1) и алпразолама, обработанного смазывающим и связывающим веществами, в соответствии с получением, используемым в нижеследующих примерах I и II по данному изобретению соответственно.
Фиг.3 соответствуют кривым дифференциального термического анализа для физических смесей тех же компонентов (алпразолам, повидон и стеариновая кислота), с таким же соотношением масс, что и использованные для получения кривых фиг.2.
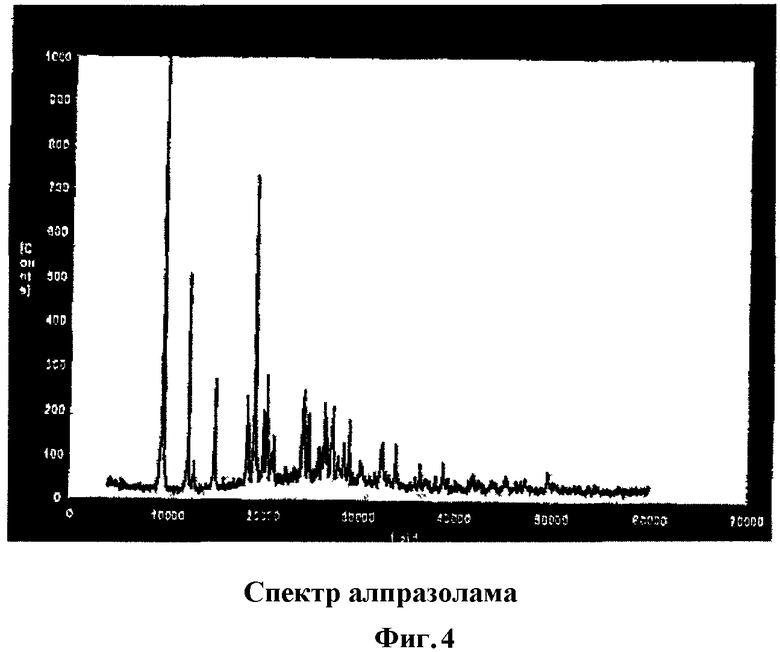
Фиг.4 демонстрирует спектр дифракции рентгеновских лучей алпразолама.
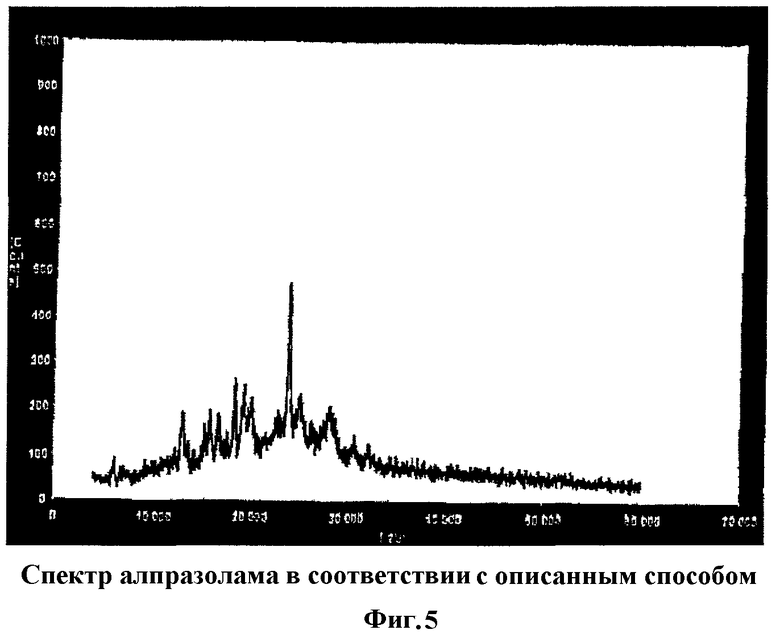
Фиг.5 показывает спектр дифракции рентгеновских лучей смеси алпразолама, обработанного смазывающим и связывающим веществами, в соответствии с методикой, представленной в настоящем изобретении и описанной в примерах I и II, в которых, по сравнению с Фиг.4, показан спектр с широкими линиями дифракции и диффузными максимумами.
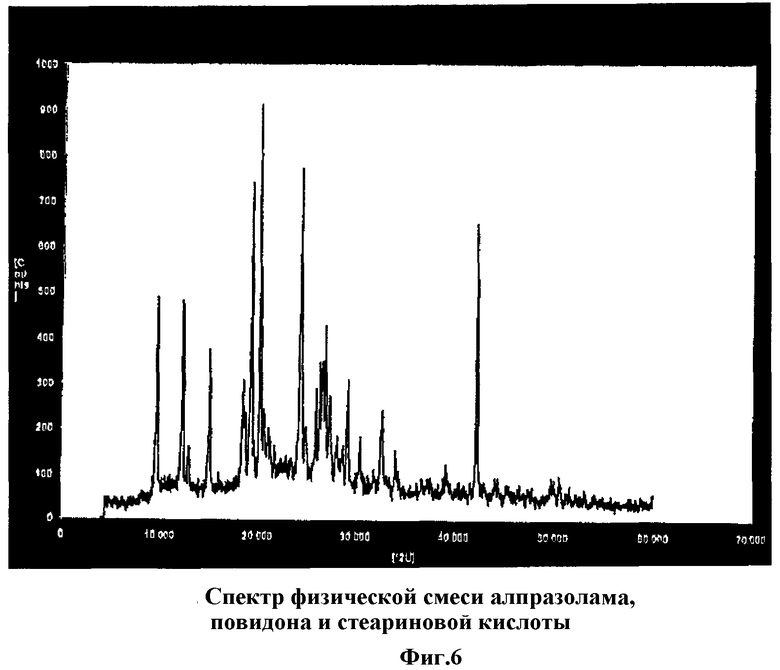
Фиг.6 показывает спектр дифракции рентгеновских лучей физической смеси алпразолама, повидона и стеариновой кислоты, полученной простым перемешиванием тех же компонентов, которые были использованы в образце, для которого была получена Фиг.5.
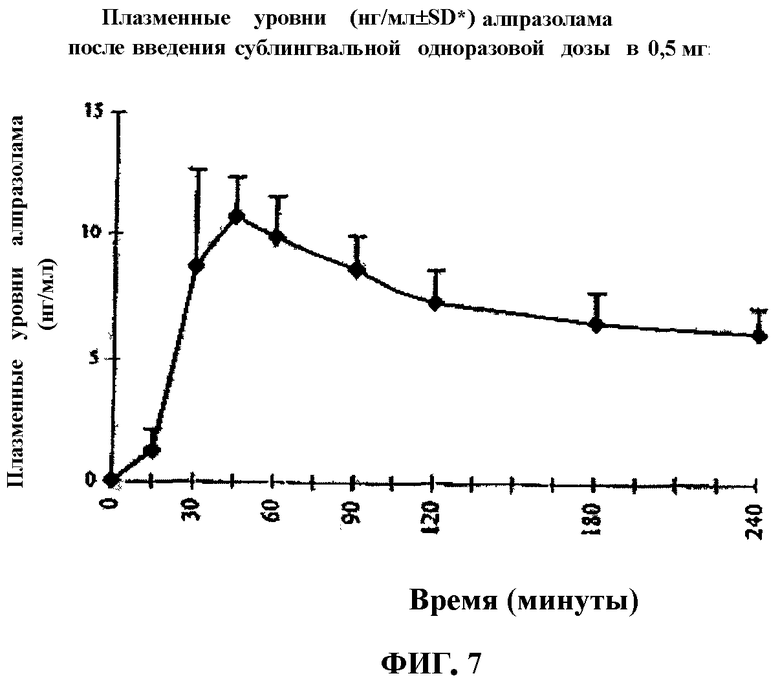
Фиг.7 представляет характерную кривую плазменного уровня (мг/мл±SD*) таблеток алпразолама, содержащих одноразовую дозу (0,5 мг (сублингвальный путь введения)), полученных в соответствии со способом по настоящему изобретению.
*) SD обозначает стандартное отклонение
Подробное описание изобретения
Настоящее изобретение основано на неожиданном открытии того, что возможно предотвратить или замедлить кристаллизацию биоактивного компонента при получении лекарственных таблеток и получить таблетки, обладающие более быстрой распадаемостью, более быстрой доставкой и более быстрой биодоступностью активного компонента, чем таблетки, выпускаемые промышленностью и имеющиеся в продаже, если предварительную операцию гранулирования такого растворенного компонента осуществляют в комбинации со связывающим веществом и частью смазывающего вещества; а при второй операции гранулирования полученное гранулированное вещество, предварительно измельченное, обрабатывают в смеси с другими компонентами и оставшейся частью смазывающего вещества.
В предпочтительном варианте осуществления по настоящему изобретению способ получения фармацевтических композиций таблеток, предназначенных для орального введения, обладающих быстрой распадаемостью, которые содержат биологически активное действующее лекарственное вещество, которое, по существу, свободно от кристаллической формы, включает:
- получение раствора биологически активного действующего лекарственного вещества в фармацевтически приемлемом растворителе вместе с ингибирующим кристаллизацию агентом, который получают смешиванием связывающего вещества и от 20 до 60 мас.% от общего количества смазывающего вещества;
- пропитывание указанным выше раствором смеси загружаемых компонентов и эксципиентов композиции до получения однородной гранулированной массы;
- измельчение предварительно высушенной гранулированной массы до получения измельченной массы однородного гранулометрического состава;
- добавление к высушенной измельченной массе 100% остающегося смазывающего вещества и остающихся компонентов и необязательных ароматизирующих добавок; перемешивание и прессование таких ароматизированных масс.
В еще одном предпочтительном варианте осуществления по настоящему изобретению способ получения фармацевтических композиций таблеток, обладающих быстрой распадаемостью, предназначенных для орального пути введения, которые содержат алпразолам, который, по существу, свободен от кристаллической формы, включает:
- получение раствора биологически активного действующего лекарственного вещества в фармацевтически приемлемом растворителе вместе с ингибирующим кристаллизацию агентом, полученным смешиванием связывающего вещества и от 20 до 60 мас.% от общего количества смазывающего вещества;
- пропитывание указанным выше раствором смеси загружаемых компонентов и эксципиентов композиции до получения однородной гранулированной массы;
- измельчение предварительно высушенной гранулированной массы до получения измельченной массы однородного гранулометрического состава;
- добавление к высушенной измельченной массе 100% остающегося смазывающего вещества и остающихся компонентов, необязательных ароматизирующих добавок; перемешивание и прессование таких ароматизированных масс.
Способ по настоящему изобретению позволяет получить и использовать фармацевтические композиции, нижеследующие характеристики которых совершенно отличают их от композиций предыдущего уровня техники.
Первое различие заключается в том, что те же самые наполнители фармацевтической композиции функционируют в качестве носителей активных действующих лекарственных веществ. Благодаря предварительному растворению активного агента в растворителе и последующему пропитыванию всех эксципиентов фармацевтического препарата гарантировано гомогенное распределение действующего лекарственного вещества по всей исходной массе.
Другое существенное различие заключается в том, что введение связывающего вещества и части смазывающего вещества на предварительной стадии гранулирования, которые действуют как ингибиторы кристаллизации в среде растворителя, предотвращая восстановление исходной кристаллической формы и структуры активного агента на поверхности гранулированных компонентов, которые образуют массу каждой таблетки.
Изменения, наблюдаемые среди кривых дифференциального термического анализа, подтверждают неупорядоченность и частичную потерю кристаллической структуры алпразоламом в таблетках, полученных в соответствии со способом по настоящему изобретению (Фиг.2), по сравнению с алпразоламом отдельно (Фиг.1).
Более того, сравнивая кривые, полученные на Фиг.2 и 3, можно заметить более сильное возмущающее влияние на кристаллическую структуру алпразолама, обработанного согласно способу по настоящему изобретению, поскольку эндотермический пик алпразолама смещен от 228,64°C (Фиг.1) к 208,61°C (Фиг.2); в то время как для простой физической смеси алпразолама, повидона и стеариновой кислоты наблюдают сдвиг от 228,64°C к 213,03°С (Фиг.3).
Потерю кристаллической структуры алпразоламом в таблетках, полученных согласно способу по настоящему изобретению, также можно наблюдать по спектрам дифракции рентгеновских лучей (Фиг.4, 5 и 6). Алпразолам не утрачивает кристаллической структуры в физической смеси компонентов, имеющей спектр (Фиг.6), аналогичный по своим характеристикам (четко определенные резкие пики) спектрам алпразолама как такового (Фиг.4). Тем не менее алпразолам, обработанный согласно способу по настоящему изобретению, обнаруживает спектр с большими искажениями, с широкими линиями дифракции и диффузными максимумами.
Такие характеристики процесса, гомогенное распределение активного действующего лекарственного вещества, способствуют его общей “усвояемости” в ротовой полости в течение замечательно короткого периода времени и поэтому гарантируют всасывание лекарственного средства, предпочтительно, через ротовую и сублингвальную слизистые оболочки. Такое всасывание стимулируют предложением больному положить таблетку под язык. Более того, как можно будет наблюдать в нижеследующих примерах вариантов осуществления, менее чем через 15 секунд целая таблетка практически отсутствует в ротовой полости. Оба факта гарантируют максимальную возможность всасывания описанной оригинальной композиции через сублингвальную и ротовую слизистые оболочки. Такая характеристика всасывания дает еще одно дополнительное преимущество перед другими оральными таблетками, содержащими такое же активное действующее лекарственное вещество, поскольку пациента освобождают от необходимости соблюдения режима голодания, рекомендуемого до и после введения продукта, для того чтобы гарантировать эффективность продукта, и, кроме того, отсутствует недостаток, заключающийся в необходимости держать таблетку в ротовой полости в течение длительного периода времени (от 10 до 15 минут) до ее полного распада, как это было описано в приведенных выше научных литературных трудах.
Усвояемость активного действующего лекарственного вещества, находящегося в некристаллическом состоянии, также увеличена. Как было указано выше, такое состояние способствовало бы его растворимости и биодоступности.
Таблетки, полученные согласно способу по настоящему изобретению, обладают “коротким периодом распадаемости”, меньшим, чем 1 минута и, предпочтительно, находящимся в интервале от 8 до 15 секунд. Замечательно, как это можно будет наблюдать в нижеследующих примерах вариантов осуществления, что выпускаемые промышленностью и имеющиеся в продаже таблетки обладают более длительным временем распадаемости, чем полученные таблетки, содержащие фармацевтические композиции, являющиеся объектом настоящего изобретения. Оральные, трансбуккальные и сублингвальные таблетки, являющиеся объектом настоящего изобретения, выделяет их значительно короткое время распадаемости. Так, например, полная усвояемость лекарственного средства, которое подлежит всасыванию, предпочтительно, через слизистую оболочку, происходит за время, менее чем 1 минута и, предпочтительно, за период от 8 до 15 секунд, после того как лекарственное средство помещено в рот, в каждом случае время распадаемости меняется в соответствии с природой содержащегося действующего вещества, но находится в обозначенных выше пределах.
Тот факт, что те же самые компоненты действуют как препятствующие кристаллизации биологически активного компонента, устраняет необходимость введения дополнительных ингибирующих агентов, тем самым упрощая способ получения.
Другое преимущество, которое продемонстрировали таблетки, полученные согласно способу по настоящему изобретению, заключается в том, что таблетки имеют “очень маленький размер”. Они имеют массу, составляющую приблизительно 75 мг и, предпочтительно, 30 и 40 мг. Они содержат минимально возможное количество эксципиентов, не являющихся активными веществами. Это минимально допустимое количество, тем не менее, оказывается необходимым и достаточным, для того чтобы гарантировать отвечающую требованиям стабильную фармацевтическую композицию. Это подтверждают выполненные физико-химические определения, которые приведены в примерах вариантов осуществления по настоящему изобретению. Существенное уменьшение содержания эксципиентов обеспечивает наилучшее взаимодействие между активным действующим лекарственным веществом и сублингвальной или оральной слизистыми оболочками после распада таблетки, которое способствует всасыванию активного действующего лекарственного вещества. Окончательная масса таблеток, которые являются объектом настоящего изобретения, фактически в 2-6 раз меньше, чем масса других традиционных таблеток, содержащих то же самое активное действующее лекарственное вещество.
Кроме того, таблетки по настоящему изобретению обнаруживают лучшую “биодоступность” биологически активного действующего вещества. Как это подробно продемонстрировано в нижеследующих биологических анализах, биологически активное действующее вещество, алпразолам, попадает в плазму быстрее, чем через 15 минут после его введения, а плазменный пик получают раньше (при 45 минутах), чем с фармацевтическими композициями. Плазменные уровни, полученные с таблетками по настоящему изобретению, содержащими 0,5 мг алпразолама, эквивалентны плазменным уровням, полученным с таблетками, описанными в литературе, но содержащими 1 мг алпразолама.
Следующие компоненты необходимы при получении таблеток, обладающих быстрой распадаемостью: разбавитель, связывающее вещество, дезинтегрирующие и смазывающие агенты и растворитель для гранулирования.
Растворители могут быть неорганическими, такими как вода, или органическими, такими как дихлорметан или этанол. Последние можно легко удалить. Этанол является предпочтительным растворителем, потому что после его удаления маловероятно, чтобы остались какие-либо загрязняющие примеси.
Кроме того, возможно использовать комбинации этанола и воды, или этанола и дихлорметана в соответствии с природой активного действующего лекарственного вещества, которое растворяют.
Микрокристаллическая целлюлоза, моногидрат лактозы, сорбит, маннит и ксилит могут быть использованы в качестве разбавителей.
Наиболее обычными связывающими агентами являются акация, желатин, сахароза, повидон, метилцеллюлоза, карбоксиметилцеллюлоза, микрокристаллическая целлюлоза, крахмальная паста. Предпочтительно, когда в способе по настоящему изобретению в качестве связывающего вещества выбран повидон.
Дезинтегрирующими агентами являются крахмал, модифицированный крахмал, альгиновая кислота, микрокристаллическая целлюлоза, коллоидные силикаты.
Смазывающие вещества включают стеараты металлов, такие как стеарат магния, стеариновую кислоту, эфиры стеариновой кислоты, такие как натрийстеарилфумарат, гидрированные растительные масла и тальк.
Фармацевтическая композиция, описанная в настоящем изобретении, применима к лекарственным средствам, предназначенным для трансбуккального и сублингвального всасывания, совместимым с эксципиентами разработанной композиции и описанной технологией получения. Она специально предназначена для эффективных лекарственных средств, даже в очень малых дозах.
Примеры
Следующие примеры представлены для дальнейшей иллюстрации получения по заявленному изобретению. В примерах I и II описаны предпочтительные способы осуществления изобретения и некоторые свойства фармацевтической композиции, такие как время распадаемости, масса, ломкость, прочность и содержание действующего лекарственного вещества в фармацевтической композиции, полученной способом по настоящему изобретению. Пример III описывает получение трансбуккальных или сублингвальных таблеток в отсутствии биологически активного действующего лекарственного вещества при использовании лактозы в качестве инертного вещества.
Пример 1
Состав для получения 10000 таблеток сублингвального или трансбуккального алпразолама
К раствору 5 г алпразолама и 3,5 г повидона в смеси этанол:дихлорметан (1:1) добавляли 60% от количества стеариновой кислоты, указанной в составе (Раствор A).
Смесь 0,880 г ретикулярной карбоксиметилцеллюлозы (25% от количества, приведенного в составе) и указанные количества микрокристаллической целлюлозы и лактозы пропитывали раствором A в смесителе-грануляторе (типа Collete) до получения однородной гранулированной массы. Гранулированную массу сушили в сушилке с псевдоожиженным слоем (типа Glatt) до полного удаления растворителя.
Затем гранулированную массу перемещали в тот же самый сборник смесителя-гранулятора и пропитывали 30 мл этанола. Растворитель снова удаляли высушиванием в сушилке с псевдоожиженным слоем. Высушенную гранулированную массу измельчали в устройстве типа Quadro Comil до получения частиц однородного размера. Добавляли ароматизирующий агент и 75% от рассчитанного количества ретикулярной карбоксиметилцеллюлозы и в течение 20 минут компоненты перемешивали. После добавления оставшейся стеариновой кислоты и перемешивания в течение 5 минут массу подвергали прессованию.
Тесты
Пример II
Состав для получения 10000 таблеток сублингвального или трансбуккального алпразолама
Получали раствор 5 г алпразолама, 3,5 г повидона и 60% количества стеариновой кислоты из предыдущего состава (1,02 г) в 270 мл этанола. Растворитель удаляли и массу растворяли в 60 мл этанола (раствор A).
Смесь 25% от рассчитанного количества ретикулярной карбоксиметилцеллюлозы и указанных количеств микрокристаллической целлюлозы и лактозы пропитывали раствором A в смесителе-грануляторе (типа Collete) до получения однородной гранулированной массы. Гранулированную массу сушили в сушилке с псевдоожиженным слоем (типа Glatt) до полного удаления растворителя.
Высушенную гранулированную массу измельчали в устройстве типа Quadro Comil до получения частиц однородного размера. Добавляли ароматизирующий агент и 75% от рассчитанного количества ретикулярной карбоксиметилцеллюлозы и компоненты перемешивали в течение 20 минут. После добавления оставшейся стеариновой кислоты и перемешивания в течение 5 минут массу подвергали прессованию.
ТЕСТ
Пример III
Состав для получения 10000 таблеток сублингвальный или трансбуккальной лактозы
Получали раствор 3,0 г повидона и 60% от рассчитанного количества стеариновой кислоты в 50 мл этанола и 10 мл воды (раствор A).
Смесь 25% от рассчитанного количества ретикулярной карбоксиметилцеллюлозы и указанных количеств микрокристаллической целлюлозы и лактозы пропитывали раствором A в смесителе-грануляторе (типа Collete) до получения однородной гранулированной массы. Гранулированную массу сушили в сушилке с псевдоожиженным слоем (типа Glatt) до полного удаления растворителя.
Высушенную гранулированную массу измельчали в устройстве типа Quadro Comil до получения частиц однородного размера. Добавляли ароматизирующий агент и 75% от рассчитанного количества ретикулярной карбоксиметилцеллюлозы и компоненты перемешивали в течение 20 минут. После добавления оставшейся стеариновой кислоты и перемешивания в течение 5 минут массу подвергали прессованию.
ТЕСТЫ
Сравнительные анализы
Нижеследующая таблица I показывает сопоставление различий по массе и времени распадаемости оральных таблеток и таблеток для трансбуккального или сублингвального применения, полученных с использованием способа по настоящему изобретению. Средняя масса и время распадаемости таблетки, не содержащей активного действующего лекарственного вещества, полученной согласно способу, показанному в примере III, приведены в таблице II.
Таблетки, содержащие 0,5 мг алпразолама
сублингвальная
сублингвальная
Нижеследующая таблица II показывает средние массу и время распадаемости таблеток, полученных в соответствии с примером III.
Таблетки, содержащие лактозу
сублингвальная
Кроме того, было продемонстрировано, что фармацевтическая композиция и способ получения могут быть использованы для получения таблеток большего размера, предназначенных исключительно для орального применения. В таких таблетках максимальное время распадаемости было менее чем 10 минут и, предпочтительно, менее чем 5 минут.
Биологические анализы
I. Изучение плазменных уровней
Целью данного исследования было определение плазменных уровней алпразолама у 12 здоровых добровольцев, которых наблюдали после введения однократной дозы алпразолама (0,5 мг) в фармацевтической композиции по настоящему изобретению, получение которой было описано в приведенных выше примерах вариантов осуществления.
Исследование осуществляли на 12 здоровых добровольцах в возрасте от 21 до 50 лет, имеющих опыт нерегулярного приема дозы бензодиазепина, без возникновения вредных побочных эффектов, с нормальными показателями психофизического обследования, и биохимическим контролем без отклонений от нормы (формула крови, наличие глюкозы в крови, креатинемия, уремия, протеинограмма, билирубинемия, TGO, TGP, щелочная фосфотаза, холестерин, общий анализ мочи, реакция Вассермана, реакция на вирус гепатита Ag, реакция на ВИЧ).
Таблетку, содержащую 0,5 мг алпразолама, специально изготовленную для быстрой распадаемости, соответствующим образом ароматизированную, вводили сублингвально здоровым добровольцам после подписания соответствующего документа об информированном согласии.
20 мл крови забирали у каждого добровольца в следующие периоды времени: 0; 5; 15; 45; 60; 120; 180 и 240 минут.
Время, отмеченное нулем, считают моментом, непосредственно предшествующим введению сублингвальной таблетки.
После отделения плазмы центрифугированием ее хранили при температуре -20°C до проведения анализа, который осуществляли посредством ВЭЖХ (хроматограф HP 1100 фирмы Hewlett Packard).
Полученные результаты показаны на Фиг.7 и представлены в нижеследующей таблице III:
Время в минутах (n)
0,90
Площадь под кривой, в интервале от 0 до 4 часов, составляла 2608.85 мг/мл.ч-1
Как можно видеть на Фиг.7, плазменные уровни определяли через 15 минут после сублингвального введения, получая максимальный пик на 45 минутах (10,73 мг/мл).
Здоровые добровольцы заявляли, что менее чем через 30 секунд во рту у них уже не было целой таблетки.
Эти величины сравнивали с величинами максимальных плазменных пиков алпразолама, о которых сообщено в литературе. Так, например, после введения оральных таблеток, содержащих 1 мг алпразолама, Garzone, PD et al. получали уровни от 14,4 до 19 нг/мл в интервале от 1,17 до 1,73 часов после введения (Garzone, PD and Kroboth PD ”Pharmacokinetics of the newer benzodiazepines” CLIN. PHARMACOKIN.; 1989; 16: 337-364).
Было проведено сравнение с данными, полученными после введения 1 мг алпразолама сублингвальным путем, происходящего после приема пищи, но при использовании обычного фармацевтического препарата для орального применения. В этом случае, величина плазменного пика составляла 11,3 нг/мл при 2,8 часах после введения препарата (Scavone, JM et al. - The Pharmacokinetics and Pharmacodynamics of Sublingual and Oral alprazolam in the Postprandial State - EUR. J. CLIN. PHARMACOL.; 1992; 42: 439-443).
Кроме того, изучали стандартную фармацевтическую композицию для орального введения, содержащую 1 мг алпразолама, после ее введения в условиях режима голодания сублингвальным путем введения.
В этом случае плазменный пик составлял 17,3 нг/мл при 1,17 часах (Scavone,JM at al - Alprazolam Kinetic Following Sublingual and Oral Administration - J. CLIK PSYCHOPHARMACOL; 1987; 7:332-334).
Из такого сравнения следует, что новая фармацевтическая композиция алпразолама по настоящему изобретению дает возможность:
i. лекарственному средству попадать в плазму ранее, чем через 15 минут после его введения;
ii. плазменному пику (получаемый на 45 минутах) появляться быстрее по сравнению с плазменными пиками, получаемыми при трансбуккальном или сублингвальном введении оральной фармацевтической композиции, на которую ранее ссылались в литературе;
iii. в соответствии с клиническими результатами плазменным уровням, получаемым с новой фармацевтической композицией, оказаться терапевтически эффективными;
iv. указанным плазменным уровням являться фактически эквивалентными плазменным уровням, описанным в приведенных в данном описании исследованиях, поскольку новая фармацевтическая композиция содержит только 0,5 мг алпразолама, а уровни, приведенные в литературе, относятся, в частности, к оральным таблеткам, содержащим 1 мг алпразолама, но введенным сублингвальным путем. Результаты изучения описанных уровней подтверждают преимущества новой композиции по настоящему изобретению и существенное различие между новой композицией (в которой концентрация алпразолама составляет только 0,5 мг на одну таблетку) и опубликованной композицией, предназначенной для таблеток, содержащих 1 мг алпразолама.
II. Исследование терапевтического эффекта
Клиническое применение таблеток алпразолама, введенного сублингвальным путем группе пациентов, позволило подтвердить, что:
i. негативные мысли, которые инициируют классические симптомы и признаки, ассоциированные с паническим расстройством, исчезают в течение короткого периода времени;
ii. общие симптомы панической атаки, такие как тахикардия, повышенное артериальное давление, сердцебиение, одышка, чрезмерное потоотделение, сухость во рту и так далее, также ослабевают или исчезают в течение короткого периода времени;
iii. через приблизительно 15 минут больные преодолевают состояние кризиса или паническую атаку, будучи способными вернуться к обязанностям, которые они выполняли.
Подавление приступа кризиса или панической атаки и ассоциированных симптомов в течение такого короткого периода времени (около 15 минут) подтверждает чрезвычайно высокую скорость, с которой достигают полезных терапевтических уровней активного действующего лекарственного вещества при использовании фармацевтической композиции по настоящему изобретению. Этот терапевтический результат сочетается с присутствием плазменных уровней алпразолама, определяемых на 5 минутах, и максимального уровня алпразолама, достигаемого в интервале от 30 до 45 минут после введения таблетки.
В заключение, исследования выполняли с привлечением здоровых добровольцев и испытуемых больных, использующих фармацевтическую композицию по настоящему изобретению, содержащую 0,5 мг алпразолама в одной таблетке, применение которой описано ниже, позволяет утверждать что:
Менее чем через 30 секунд во рту отсутствуют остатки таблетки. Это означает, что все активно действующее лекарственное вещество фармацевтической композиции усваивается, всасываясь через слизистую оболочку рта.
Плазменный уровень бензодиазепина определяют на 15 минутах после введения фармацевтической формы.
Пик максимального всасывания получают на 45 минутах после введения фармацевтической композиции.
Клиническое результаты были удовлетворительными.
Действующие лекарственные вещества, предпочтительные способы и направления действий по настоящему изобретению были описаны в настоящем документе.
Однако подлежащее защите изобретение не ограничено описанными отдельными вариантами осуществления, поэтому их следует рассматривать как иллюстративные, а не как ограничительные. Специалистами в данной области могут быть внесены изменения и модификации, не отходя от существа и объема данного изобретения.


Способ получения оральных таблеток, содержащих алпразолам, свободный от кристаллической формы, включает на предварительной стадии операции гранулирования получение раствора алпразолама в фармакологически приемлемом растворителе вместе с ингибирующим кристаллизацию агентом, образованным смесью связывающего вещества и от 20 до 60 мас.% от общего количества смазывающего вещества, пропитывание полученным раствором смеси разбавителя и 25% сетчатой карбоксиметилцеллюлозы в качестве дезинтегрирующего агента до получения однородной гранулированной массы и измельчение полученной гранулированной массы, предварительно высушенной, до получения измельченной гранулометрически однородной массы. На второй стадии операции гранулирования добавляют к высушенной и измельченной массе, полученной на предварительной стадии операции гранулирования, 100% оставшегося смазывающего агента и оставшейся сетчатой карбоксиметилцеллюлозы и ароматизирующих добавок, окончательно смешивают и прессуют ароматизированную массу. Некристаллическое состояние алпразолама улучшает его растворимость и биодоступность. Таблетки алпразолама имеют меньший размер, чем традиционные таблетки, и время распадаемости менее чем 1 минута, предпочтительно менее чем 30 секунд. 2 н. и 9 з.п. ф-лы, 3 табл., 7 ил.
1. Способ получения фармацевтической композиции оральных таблеток, обладающих быстрой распадаемостью, содержащих алпразолам, свободный от кристаллической формы, отличающийся тем, что операция гранулирования состоит из
А) предварительной стадии операции гранулирования, состоящей из а) получения раствора алпразолама в фармакологически приемлемом растворителе, вместе с ингибирующим кристаллизацию агентом, образованным смесью связывающего вещества и от 20% до 60 мас.% от общего количества смазывающего вещества; b) пропитывания указанным выше раствором смеси разбавителя и 25% сетчатой карбоксиметилцеллюлозы в качестве дезинтегрирующего агента до получения однородной гранулированной массы; с) измельчения полученной гранулированной массы, предварительно высушенной, до получения измельченной гранулометрически однородной массы;
В) второй стадии операции гранулирования, состоящей из добавления к высушенной и измельченной массе, полученной на предварительной стадии операции гранулирования А, 100% оставшегося смазывающего агента и оставшейся сетчатой карбоксиметилцеллюлозы, и ароматизирующих добавок; и окончательного смешивания и прессования этой ароматизированной массы.
2. Способ по п.1, отличающийся тем, что растворитель выбирают из воды, этанола, дихлорметана, смеси этанол-дихлорметан и смеси этанол-вода.
3. Способ по п.1, отличающийся тем, что связывающий компонент выбран из карбоксиметилцеллюлозы, полиэтиленгликоля, желатина, крахмала, повидона, метилцеллюлозы, этилцеллюлозы, гидроксиэтилцеллюлозы, гидроксипропилцеллюлозы и производных целлюлозы.
4. Способ по п.1, отличающийся тем, что ароматизирующий агент выбран из порошкообразных эссенций, аспартама или устранителя горечи, и смазывающий компонент представляет собой стеариновую кислоту, стеарат магния и натрийстеарилфумарат, и разбавители выбраны из лактозы, микрокристаллической целлюлозы, ксилита, маннита и сорбита.
5. Способ по п.1, отличающийся тем, что в дополнение к последовательности стадий следующие ингредиенты вносят вклад в некристаллизацию алпразолама: 20-60% смазывающего вещества и 100% связывающего вещества.
6. Способ по п.1, отличающийся тем, что получают таблетки для сублингвального введения, имеющие массу менее чем 50 мг.
7. Способ по п.6, отличающийся тем, что полученные таблетки при оральном введении имеют время распадаемости менее чем 1 мин, предпочтительно менее чем 30 с.
8. Способ по п.7, отличающийся тем, что биологически активное вещество алпразолам присутствует в количестве от 0,125 до 2 мг на одну таблетку.
9. Фармацевтическая композиция таблеток, обладающих быстрой распадаемостью, полученная способом по п.1, отличающаяся тем, что она, по существу, свободна от кристаллов биологически активного действующего вещества алпразолама, полученного путем непосредственного превращения в процессе получения.
10. Фармацевтическая композиция по п.9, отличающаяся тем, что полученная таблетка для сублингвального введения имеет массу менее 50 мг и время распадаемости менее 30 с.
11. Фармацевтическая композиция по п.9, отличающаяся тем, что алпразолам присутствует в количестве от 0,125 до 2 мг на одну таблетку.
| РАСТВОРЯЮЩИЕСЯ ВНУТРИ РОТОВОЙ ПОЛОСТИ ИЗДЕЛИЯ, СФОРМОВАННЫЕ ПРЕССОВАНИЕМ, И СПОСОБ ИХ ПРОИЗВОДСТВА | 1995 |
|
RU2147227C1 |
| ТАБЛЕТКА, БЫСТРО ДЕЗИНТЕГРИРУЮЩАЯСЯ В ЩЕЧНОМ КАРМАНЕ, И СПОСОБ ЕЕ ПРОИЗВОДСТВА | 2002 |
|
RU2244543C2 |
| RU 2004122919 A, 10.04.2005 | |||
| ТАБЛЕТКА ДЛЯ ОЧЕНЬ БЫСТРОГО И ПРОЛОНГИРОВАННОГО ВЫСВОБОЖДЕНИЯ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ | 1998 |
|
RU2212885C2 |
| БЫСТРО ДИСПЕРГИРУЕМЫЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ, СОДЕРЖАЩИЕ РЫБИЙ ЖЕЛАТИН | 2000 |
|
RU2242969C2 |
| БЫСТРОДИСПЕРГИРУЮЩАЯСЯ ЛЕКАРСТВЕННАЯ ФОРМА, НЕ СОДЕРЖАЩАЯ ЖЕЛАТИН | 2000 |
|
RU2242968C2 |

