Область техники
Настоящее изобретение относится к производному пиридонкарбоновой кислоты или его соли, которое проявляет превосходную антибактериальную активность и высокую биологическую доступность.
Описание предшествующего уровня техники
Среди соединений, имеющих пиридонкарбоновую кислоту в качестве основы структурного скелета, были известны многие соединения, которые представляют собой полезные синтетические антибактериальные средства ввиду их превосходной антибактериальной активности и широкого антибактериального спектра. Среди этих соединений такие соединения как норфлоксацин (см. патентный документ 1), эноксацин (см. патентный документ 2), офлоксацин (см. патентный документ 3), ципрофлоксацин (см. патентный документ 4) и тосуфлоксацин (патентный документ 5) широко используются в клинической практике в качестве терапевтических средств по поводу инфекционных заболеваний. Однако, эти соединения еще неудовлетворительны с точки зрения, например, антибактериальной активности, кишечного всасывания, метаболической устойчивости и побочных эффектов, в частности, фототоксичности и цитотоксичности.
Заявители ранее провели обширные исследования для разрешения указанных выше проблем и обнаружили, что производные пиридонкарбоновой кислоты, имеющие замещенную пиридильную группу в положении 1 фрагмента пиридонкарбоновой кислоты, особенно те, которые имеют аминоазетидинильную группу в положении 7 части пиридонкарбоновой кислоты, имеют особенно превосходные характеристики, т.е. они обладают сильно выраженной антибактериальной активностью и, в отличие от синтетических антибактериальных средств класса пиридонкарбоновой кислоты, не вызывают фототоксичности (патентные документы 6 и 7).
Другим требованием к препаратам для проявления ими возможности эффективного лечения инфекционных заболеваний является высокая биологическая доступность препаратов, предпочтительнее, более высокий уровень в крови свободной формы препаратов. Это последнее свойство тесно связано со связыванием препарата с сывороточными белками. То есть, препарат с более низкой скоростью связывания с сывороточными белками, приводит к более высокому содержанию свободной формы препарата в крови. Поэтому препарат, проявляющий более высокую биологическую доступность и более низкую скорость связывания с сывороточными белками, представляет собой более предпочтительное терапевтическое средство, в плане инфекционных заболеваний.
Производное пиридонкарбоновой кислоты, имеющее замещенную пиридильную группу в положении 1, и аминоазетидинильную группу в положении 7 имеет улучшенную биологическую доступность за счет введения алкильной группы в аминогруппу на азетидиновом кольце и увеличения числа атомов углерода алкильной группы, тем самым увеличивая липофильность производного. Однако такая модификация также склонна к увеличению скорости связывания производного с сывороточными белками.
Патентный документ 1: выложенная заявка на патент Японии (kokai) № 53-141286
Патентный документ 2: выложенная заявка на патент Японии (kokai) № 53-031042
Патентный документ 3: выложенная заявка на патент Японии (kokai) № 57-046986
Патентный документ 4: выложенная заявка на патент Японии (kokai) № 58-076667
Патентный документ 5: выложенная заявка на патент Японии (kokai) № 60-228479
Патентный документ 6: WO 97/11068 Публикация
Патентный документ 7: WO 01/02390 Публикация
Описание изобретения
Задачи, подлежащие решению данным изобретением
Целью настоящего изобретения является создание средства с превосходной антибактериальной активностью, низкой токсичностью, улучшенной биологической доступностью и низкой скоростью связывания с сывороточными белками.
Средства для решения проблем
С целью разрешения этих проблем, заявители всесторонне оценили указанные выше производные пиридонкарбоновой кислоты, имеющие замещенную пиридильную группу в положении 1 и аминоазетидинильную группу в положении 7, и обнаружили, что даже в случае, когда алькильная группа введена в аминогруппу азетидинового кольца с тем, чтобы увеличить липофильность, соединение, представленное следующей формулой (1), имеющее изопропильную группу или трет-бутильную группу, введенную в аминогруппу, может иметь улучшенную биологическую доступность без увеличения скорости его связывания с сывороточными белками. Заявители также обнаружили, что соединение, представленное следующей формулой (2), представляет собой полезные промежуточные соединения для синтеза соединения, представленного формулой (1).
Соответственно, настоящее изобретение направлено на производное пиридонкарбоновой кислоты, представленное формулой (1):
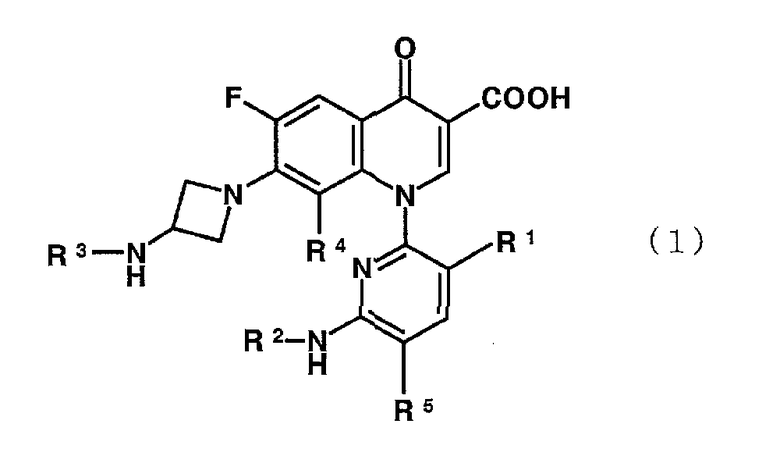
или его соль, где R1 представляет метильную группу, атом фтора или атом хлора; R2 представляет атом водорода или низшую алкильную группу; R3 представляет изопропильную группу или трет-бутильную группу; R4 представляет метильную группу или атом галогена; и R5 представляет атом фтора или атом хлора.
Настоящее изобретение также направлено на 1-(6-амино-3,5-дифторпиридин-1-ил)-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту или ее соль.
Настоящее изобретение также направлено на лекарственное средство, включающее производное пиридонкарбоновой кислоты или его соль, в качестве активного ингредиента.
Настоящее изобретение также направлено на антибактериальное средство, включающее производное пиридонкарбоновой кислоты или его соль, в качестве активного ингредиента.
Настоящее изобретение также направлено на фармацевтическую композицию, включающую производное пиридонкарбоновой кислоты или его соль, и фармацевтически приемлемый носитель.
Настоящее изобретение также направлено на применение производного пиридонкарбоновой кислоты или его соли для получения лекарственного средства.
Настоящее изобретение также направлено на способ лечения инфекционного заболевания, включающий введение производного пиридонкарбоновой кислоты или его соли.
Настоящее изобретение также направлено на производное пиридонкарбоновой кислоты, представленное формулой (2):
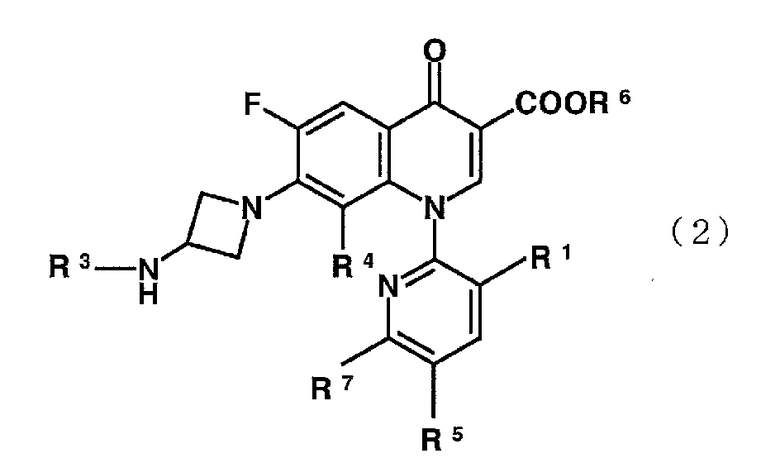
или его соль, где R1 представляет метильную группу, атом фтора или атом хлора; R3 представляет изопропильную группу или трет-бутильную группу; R4 представляет метильную группу или атом галогена, R5 представляет атом фтора или атом хлора; R6 представляет атом водорода или карбоксильную защитную группу; и R7 представляет -NR7R8, где R2 представляет атом водорода или низшую алкильную группу, и R8 представляет атом водорода или амино защитную группу, при условии, что R6 и R8 одновременно не представляют атомы водорода.
Эффекты изобретения
В соответствии с настоящим изобретением, может быть предоставлено антибактериальное средство с выраженной, превосходной антибактериальной активностью, отсутствующей фототоксичностью, высокой биологической доступностью и низкой скоростью связывания с сывороточными белками.
Наилучшие способы осуществления изобретения
Производное пиридонкарбоновой кислоты, представленное формулой (1), или его соль можно применять в качестве антибактериального средства, а производное пиридонкарбоновой кислоты, представленное формулой (2), или его соль можно применять в качестве промежуточного соединения для получения производного пиридонкарбоновой кислоты, представленного формулой (1).
Примеры низшей алкильной группы (R2) в формуле (1) или (2) включают линейные или разветвленные С1-С6 алкильные группы, такие как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, и гексил. Из них, предпочтительны линейные или разветвленные С1-С3 алкильные группы, такие как метил, этил, пропил, изопропил, причем особенно предпочтителен метил.
Примеры атома галогена (R4) включают фтор, хлор, бром, и йод, причем особенно предпочтительны хлор и бром.
Карбоксильная защитная группа (R6) относится к сложноэфирному остатку сложного эфира карбоновой кислоты и включает те остатки, которые относительно легко расщепляются с образованием соответствующей свободной карбоновой группы. Примеры защитной группы включают группы, удаляемые посредством обработки в мягких условиях (например, гидролиза и каталитического восстановления), такие как
С1-С6 низшие алкильные группы (например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил и гексил); С1-С8 алкенильные группы (например, винил, аллил, 1-пропенил, бутенил, рентинил, гексенил, и нептенил); С7-С11 аралкильные группы (например, бензил); и С6-С14 арильные группы (например, фенил и нафтинил), и группы, легко удаляемые in vivo, такие как низшие алканоилокси-низшие алкильные группы (например, ацетоксиметил и пивалоилоксиметил); низшие алкоксикарбонилокси-низшие алкильные группы (например, метоксикарбонилоксиметил и 1-этоксикарбонилоксиэтил); низшие алкокси-низшие алкильные группы (например, метоксиметил); лактонильные группы (например, фталидил); ди-низшие алкиламино-низшие алкильные группы (например, 1-диметиламиноэтил) и (5-метил-2-оксо-1,3-диоксол-4-ил)метил.
Примеры амино защитной группы (R8) включают группы, легко удаляемые вследствие гидролиза или каталитического восстановления, такие как 1-фенилэтил, трет-бутоксикарбонил, бензилоксикарбонил и бензгидрил.
Среди производных пиридонкарбоновой кислоты, представленных формулой (1), по настоящему изобретению, с точки зрения эффективности предпочтительны соединения, в которых R1 представляет собой атом фтора, R2 представляет собой атом водорода или метильную группу, R4 представляет собой атом брома или метильную группу и R5 представляет собой атом фтора, причем особенно предпочтительны те соединения, в которых R3 представляет собой изопропильную группу.
Производное пиридонкарбоновой кислоты, представленное формулой (1) или (2), может образовывать соль присоединения кислоты (кислотно-аддитивную соль) с кислотой или соль присоединения основания (основно-аддитивную соль) с основанием. Примеры кислотно-аддитивной соли могут включать: (а) соли с минеральной кислотой, такой как хлористоводородная кислота и серная кислота; (b) соли с органической карбоновой кислотой, такой как муравьиная кислота, уксусная кислота, лимонная кислота, трихлоруксусная кислота, трифторуксусная кислота, фумаровая кислота и малеиновая кислота; и (с) соли с сульфоновой кислотой, такие как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, метиленсульфоновая кислота и нафталинсульфоновая кислота. Примеры основно-аддитивной соли могут включать: (a') соли со щелочным металлом, таким как натрий и калий; (b') соли щелочноземельного металла, такого как кальций и магний; (c') соли аммония и (d') соли с содержащим азот органическим основанием, таким как триметиламин, триэтиламин, трибутиламин, пиридин, N,N-диметианилин,
N-метил-D-(-)-глюкамин, N-метилпиперидин, N-метилморфолин, диэтиламин, циклогексиламин, прокаин, дибензиламин, N-бензил-β-фенэтиламин, 1-эфенамин и N,N'-дибензилэтилендиамин. Примеры соединений бора включают галиды бора, такие как фторид бора; и низший ацилоксибор, такой как ацетоксибор.
Производное пиридонкарбоновой кислоты, представленное формулой (1) или (2) или его соль может присутствовать в не сольватированной форме или в виде гидрата или сольвата. Таким образом, настоящее изобретение охватывает все кристаллические формы, гидраты и сольваты производного пиридонкарбоновой кислоты.
Производное пиридонкарбоновой кислоты (1) и промежуточное соединение (2) можно получить любыми способами, например, по следующей схеме:
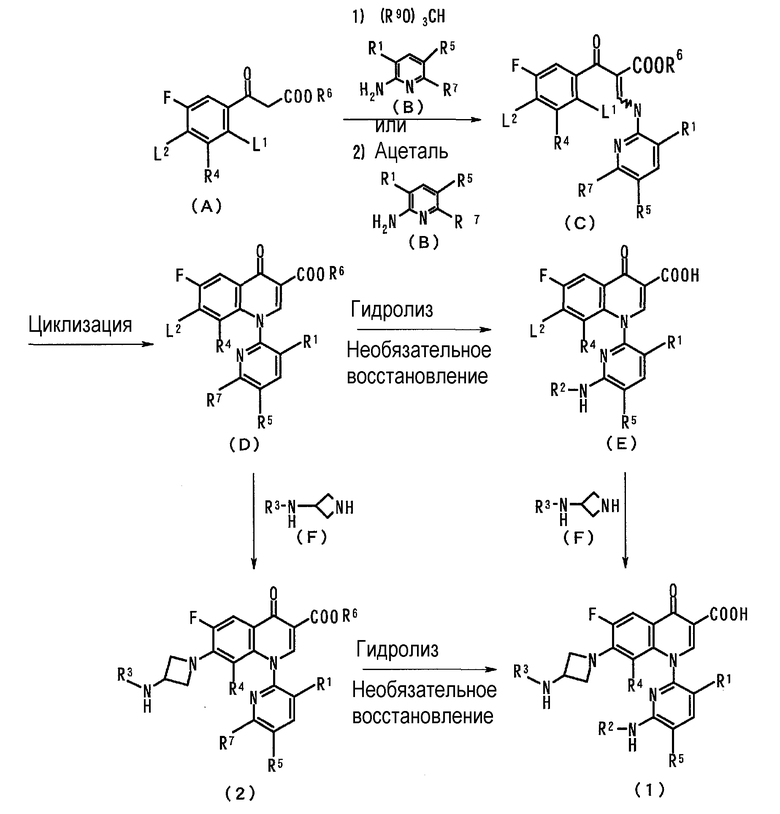
(где каждая из L1 и L2 представляет атом галогена, такой как фтор или хлор; R9 представляет C1-C5 алкильную группу; и R1, R2, R3, R4, R5, R6 и R7 представляют то же, что определено выше).
В частности, соединение (А) взаимодействует со сложным эфиром орто-муравьиной кислоты (R9O)3CH, и, далее, с амино соединением (В) для образования соединения (С). Альтернативно, соединение (А) взаимодействует с ацеталем и, далее, с амино соединением (В) для образования соединения (С). В дальнейшем, соединение (С) циклизуется для образования хинолинового кольца с получением соединения (D), которое затем гидролизуется и, необязательно, кроме того, подвергается реакции снятия защиты, такой как восстановление, с образованием соединения (Е). Соединение (Е). Соединение (Е) аминируется производным азетидина (F) с получением производного пиридонкарбоновой кислоты (1).
Альтернативно, после аминирования соединения (D) с производным азетидина (F) для получения производного пиридонкарбоновой кислоты (2), производное (2) гидролизуется и, необязательно, подвергается снятию защиты, такому как восстановление, с получением производного пиридонкарбоновой кислоты (1).
Взаимодействие между соединением (А) и сложным эфиром орто-муравьиной кислоты в целом выполняют при 0-160°С, предпочтительно, при 50-150°С, в течение времени от 10 мин до 48 ч, предпочтительно, в течение от 1 до 10 ч. Сложный эфир орто-муравьиной кислоты предпочтительно используется в эквимолярном или большем количестве в отношении соединения (А), в частности, примерно в 1-10 раз большем по молярной массе. Ангидрид карбоновой кислоты, такой как уксусный ангидрид, предпочтительно добавляется в качестве вспомогательного агента. Вспомогательный агент предпочтительно используется в эквимолярном или большем количестве относительно соединения (А), в частности, в большем примерно в 1-10 раз по молярной массе. При взаимодействии соединения (А) с амино соединением (В) в отсутствие растворителя или в присутствии соответствующего растворителя для образования соединения (С), амино соединение (В) предпочтительно используется в эквимолярном или большем количестве относительно соединения (А), в частности, примерно в количестве от эквимолярного до двух раз превышающем его по молярной массе. В отношении используемого при взаимодействии растворителя нет определенных ограничений, если растворитель не влияет на эту реакцию. Примеры растворителя включают ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как простой диэтиловый эфир, тетрагидрофуран, 1,4-диоксиан, моноглим и диглим; алифатические углеводороды, такие как пентан, гексан, гептан и лигроин; галоидуглеводороды, такие как метиленхлорид, хлороформ и тетрахлоруглерод; апротонные полярные растворители, такие как N,N-диметилформамид и диметилсульфохлорид; и спирты, такие как метанол, этанол и пропанол. Описанное выше взаимодействие в целом выполняется при 0-150°С, предпочтительно, от 0 до 100°С, в течение времени от 10 мин до 48 ч.
При другом альтернативном способе, соединение (А) может взаимодействовать с ацеталем, таким как N,N-диметилформамиддиметилацеталь и N,N-диметилформамиддиэтилацеталь, с последующим взаимодействием с амино соединением (В) с образованием соединения (С). В отношении используемого при взаимодействии с ацеталем растворителя нет определенных ограничений, если растворитель не влияет на реакцию. В частности, можно использовать такие же растворители, как указано выше. Реакцию в целом проводят при 0-150°С в течение времени от 10 мин до 48 ч, предпочтительно, при температуре от комнатной до 100°С в течение времени от 1 до 10 ч.
Циклизацию соединения (С) с образованием соединения (D) проводят в соответствующем растворителе в присутствии или отсутствие основного соединения. В отношении используемого при взаимодействии растворителя нет определенных ограничений, если растворитель не влияет на реакцию. Примеры растворителя включают ароматические углеводороды, такие как бензол, толуол и ксилол, простые эфиры, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, моноглим и диглим; галогенуглеводороды, такие как метиленхлорид, хлороформ и тетрахлоруглерод; апротонные полярные растворители, такие как диметилформамид и диметилсульфоксид; спирты, такие как метанол, этанол и пропанол. Примеры основного соединения, используемого во взаимодействии, включают щелочные металлы, такие как металлический натрий и металлический калий; гидриды металлов, такие как гидрид натрия и гидрид кальция; неорганические соли, такие как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат натрия и карбонат калия; алкоксиды, такие как метоксид натрия, этоксид натрия и трет-бутоксид калия; фториды металлов, такие как фторид натрия и фторид калия; и органические основания, такие как триэтиламин и 1,8-диазабицикло[5.4.0]ундецен (DBU). Взаимодействие в целом выполняется при 0-200°С, предпочтительно, при температуре от комнатной до 180°С, и, в целом, завершается в пределах периода от 5 мин до 24 ч. Основное соединение используется в эквимолярном или большем количестве относительно соединения (С), предпочтительно, в количестве от эквимолярного до двукратно превышающего его по молярной массе.
Посредством гидролиза соединения (D) и необязательного восстановления можно получить соединение (Е).
Гидролиз можно выполнять в обычных условиях взаимодействия. Например, гидролиз выполняется в присутствии основного соединения, такого как гидроксид натрия, гидроксид калия, карбонат натрия или карбонат калия; минеральной кислоты, такой как хлористоводородная кислота, серная кислота или бромистоводородная кислота; или органической кислоты, такой как п-толуолсульфоновая кислота, в растворителе, таком как вода; спирты (например, метанол, этанол или пропанол), простые эфиры (например, тетрагидрофуран или диоксан), кетоны (например, ацетон или метилэтилкетон) или уксусной кислоты или их смесей. Реакция в целом выполняется при температуре от комнатной до 180°С, предпочтительно, при температуре от комнатной до 140°С, в течение периода от 1 до 24 ч.
Восстановление может представлять собой каталитическое восстановление, выполняемое в присутствии, например, палладия-углерода или гидроксида палладия-углерода, использованием источника водорода, такого как водород или формиат аммония, в растворителе, таком как спирты (например, метанол или этанол) или уксусная кислота, при температуре от комнатной до 100°С, предпочтительно, от 70 до 120°С, в течение времени от 30 мин до 10 ч, предпочтительно, от 1 до 5 ч.
Когда соединение (Е), далее, взаимодействует с производным азетидина (F), то можно получить производное пиридонкарбоновой кислоты (1) по настоящему изобретению.
Взаимодействие проводят в растворителе, который не влияет на реакцию. Примеры растворителя включают ароматические углеводороды, такие как бензол, толуол и ксилол; спирты, такие как метанол или этанол; простые эфиры, такие как тетрагидрофуран, диокиан, моноглим; галогенуглеводороды, такие как метиленхлорид, хлороформ и тетрахлоруглерод; апротонные полярные растворители, такие как диметилформамид, диметилсульфоксид и N-метилпирролидон; ацетонитрил и пиридин, необязательно, в присутствии таких агентов, как карбонат натрия, карбонат кальция, гидроксид лития, триэтиламин, 1,8-дифазабицикло[5.4.0]ундецен (DBU), N-метилпирролидон или 1,1,3,3-тетраметилгуанидин и, необязательно, в присутствии добавок, таких как хлорид лития, перхлорат лития или тетрафторметансульфонат лития, при температуре от комнатной до 160°С. Время взаимодействия составляет от нескольких минут до 48 ч, предпочтительно, от 10 мин до 24 ч. Производное азетидина используется в эквимолярном или большем количестве относительно соединения (Е), предпочтительно в количестве от эквимолярного до превышающего его в 5 раз по молярной массе.
Получение производного пиридонкарбоновой кислоты (1) по настоящему изобретению из соединения (D) через производное пиридонкарбоновой кислоты (2) по настоящему изобретению можно выполнять посредством тех же взаимодействий, как указано выше.
Производное пиридонкарбоновой кислоты (1) по настоящему изобретению может быть переведено в кислотно-аддитивную соль или основно-аддитивную соль обычным способом. Реакция выполняется в полярном растворителе, таком как вода или спирт (например, метанол или этанол), при комнатной температуре или нагревании производного пиридонкарбоновой кислоты (1) по настоящему изобретению с минеральной кислотой, такой как хлористоводородная кислота или серная кислота; органической карбоновой кислотой, такой как муравьиная кислота, уксусная кислота, лимонная кислота, трихлоруксусная кислота, трифторуксусная кислота, фумаровая кислота или малеиновая кислота; сульфоновой кислотой, такой как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, мезитиленсульфоновая кислота или нафталинсульфоновая кислота или с основным соединением, таким как гидроксид натрия, гидроксид калия, гидроксид кальция или гидроксид магния; аммиаком; с содержащим азот органическим основанием, таким как триметиламин, триэтиламин, трибутиламин, пиридин, N,N-диметианилин, N-метилпиперидин, N-метилморфолин, диэтиламин, циклогексиламин, прокаин, дибензиламин, N-бензил-β-фенэтиламин, 1-эфенамин и N,N'-дибензилэтилендиамин или N-метил-D-(-)-глюкамин.
Исходное соединение (А) можно, например, получить посредством способов, раскрытых в следующих источниках или аналогичным раскрытым там способам.
(1) J. Heterocyclic Chem. 22, 1033 (1985)
(2) Liebigs Ann. Chem. 29 (1987)
(3) J. Med. Chem. 31, 991 (1988)
(4) J. Org. Chem. 35, 930 (1970)
(5) Выложенная заявка на патент Японии (kokai) № 62-246541
(6) Выложенная заявка на патент Японии (kokai) № 62-26272
(7) Выложенная заявка на патент Японии (kokai) № 63-145268
(8) J. Med. Chem. 29, 2363 (1986)
(9) J. Fluorin. Chem. 28, 361 (1985)
(10) Выложенная заявка на патент Японии (kokai) № 63-198664
(11) Выложенная заявка на патент Японии (kokai) № 63-264461
(12) Выложенная заявка на патент Японии (kokai) № 63-104974
Исходное соединение (В) можно получить посредством любых способов, например реакции, раскрытой в выложенной заявке на патент Японии (kokai) № 11-322715.
Соединения, полученные на описанных выше стадиях, можно выделить и очистить посредством обычных способов. В зависимости от условий выделения и очистки, могут быть получены продукты в форме соли, в форме свободной карбоновой кислоты, или в форме свободного амина. Эти формы могут при желании подвергаться взаимному превращению, посредством чего можно получить соединение по настоящему изобретению желательной формы.
Как показано в представленных здесь ниже примерах испытаний 1-4, производное пиридонкарбоновой кислоты (1) или его соль, имеющее замещенную пиридильную группу в положении 1, или трет-бутиламиноазетидинильную группу в положении 7, полученное в описанной выше процедуре, не только проявляет превосходную антибактериальную активность и отсутствие фототоксичности (характерной токсичности хинолоновых соединений), как соединения, раскрытые в WO 97/11068 и WO 01/02390 (т.е., сравнительные соединения 1-4), но также проявляет более высокую биологическую доступность и более низкую скорость связывания с сывороточными белками, по сравнению со сравнительными соединениями. Таким образом, производное пиридонкарбоновой кислоты (1) по настоящему изобретению или его соль полностью приобретает свою присущую ему превосходную антибактериальную активность in vivo. Следует отметить, что термин «сывороточный белок» относится к альбумину (HAS), кислотному глюкопротеиду (AFP) в крови, а термин «скорость связывания с сывороточными белками» означает отношение количества соединения, связанного с сывороточными белками, в образце крови к общему количеству соединения в образце крови.
Таким образом, производное пиридонкарбоновой кислоты (1) по настоящему изобретению или его соль в целом можно применять в качестве антибактериальных средств; например, профилактических и терапевтических средств по поводу инфекционных заболеваний, препаратов для животных, препаратов для рыб и пищевых консервантов. Для применения в качестве лекарственного средства или препаратов для животных, к производному может необязательно добавляться фармацевтически приемлемый носитель для получения фармацевтических композиций разнообразных лекарственных форм, таких как парентеральные (например, инъекционные, трансректальные, офтальмологические) и пероральные лекарственные формы.
Примеры получения препаратов для инъекций включают асептические или неводные формы, суспензии и эмульсии, которые являются фармацевтически приемлемыми. Примеры соответствующих неводных носителей, разбавителей, растворителей и носителей включают пропиленгликоль, полиэтиленгликоль, растительное масло (например, оливковое масло) и инъецируемые органические сложные эфиры (например, этилолеат). Такие фармацевтические композиции могут, кроме того, содержать адъюванты, такие как антисептическое средство, увлажнитель, эмульгирующий агент и диспергирующий агент. Эти композиции можно стерилизовать посредством фильтрации через бактериальный фильтр или посредством добавления стерилизатора непосредственно перед применением. Альтернативно, можно использовать стерилизованную твердую композицию, содержащую стерилизующий агент, который можно растворить в небольшом количестве стерилизованной инъекционной среды.
Предпочтительно, офтальмологические средства, содержащие соединение по настоящему изобретению, могут содержать солюбилизирующий агент, консервант, изотонический агент или загуститель.
Примеры твердых средств для перорального введения включают капсулы, таблетки, пилюли, порошок и гранулы. Твердые средства в целом получают посредством смешивания соединения по настоящему изобретению, по меньшей мере, с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Во время обычного получения твердых средств, можно использовать дополнительное вещество, такое как смазывающее вещество (например, стеарат магния). В случае капсул, таблеток и пилюль, можно, кроме того, использовать буфер. Таблетки и пилюли могут быть покрыты энтеросолюбильным покрытием.
Примеры жидких средств для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, которые содержат инертный разбавитель, обычно используемый в данной области, такой как вода. В дополнение к инертному разбавителю, композиции могут, кроме того, содержать адъювант, такой как увлажнитель, эмульгирующий агент, суспендирующий агент, подсластитель и ароматизирующее средство.
Предпочтительно, трансректальные средства, содержащие соединение по настоящему изобретению могут содержать носители, такие как масло какао и воск для суппозиториев.
Доза производного пиридонкарбоновой кислоты (1) по настоящему изобретению или его соли, которая варьируется, в зависимости от свойств соединения, пути введения, желательного периода лечения или других факторов, в целом составляет в целом примерно от 0,1 до 1000 мг/кг/д, особенно предпочтительно, примерно от 0,5 до 100 мг/кг/д. При желании, суточную дозу можно разделить на 2-4 порции.
Примеры
Настоящее изобретение будет подробнее описано ниже в виде примеров и ссылочных примеров, которые не следует рассматривать как ограничивающие изобретение.
Ссылочный пример 1
Синтез 1-дифенилметил-3-метилоксиазетидина
Триэтиламин (153 мл) добавляют к раствору метиленхлорида (1 л) 1-дифенилметил-3-гидроксиазетидина (239 г) при комнатной температуре, и по каплям добавляют метансульфонилхлорид (85 мл) при охлаждении льдом. Смеси дают нагреться до комнатной температуры и перемешивают в течение 2 ч. Реакционную смесь промывают водой (1 л) и органический слой сушат с безводным сульфатом натрия с последующим удалением растворителя при пониженном давлении. Осажденное твердое вещество диспергируют в гексане и собирают с использованием фильтрации для получения, таким образом, 313 г указанного в заголовке соединения.
Форма: Бесцветное твердое вещество
Температура плавления: 115-116°С
1H-ЯМР (CDCl3) δ: 2,98 (с, 3H), 3,20 (м, 2H), 3,64 (м, 2H), 4,40 (с, 1H), 5,10 (м, 1H), 7,20 (т, J=7 Гц, 2H), 7,28 (дд, J=7 Гц, 7 Гц, 4H), 7,39 (д, J=7 Гц, 4H).
Ссылочный пример 2
Синтез дигидрохлорида N-изопропилазетидин-3-иламина
1-дифенилметил-3-меpилоксиазетидин (47,6 г) и изопропиламин (59,1 г) растворяют в этаноле (800 мл), и смесь перемешивают при 50°С в течение 42 ч. Растворитель удаляют при пониженном давлении, и остаток экстрагируют этилацетатом (500 мл). Экстракт промывают 1% водным раствором карбоната натрия (500 мл) и органический слой сушат и концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии (силикагель, дихлорметан/этилацетат=4/1→дихлорметан/метанол=10/1) с получением бесцветного твердого вещества (33,5 г). Твердое вещество растворяют в дихлорметане (100 мл) и в растворе в условиях охлаждения льдом добавляют 14% хлористоводородную кислоту/диоксан (70 мл). После добавления всего количества, к смеси добавляют простой диэтиловый эфир (500 мл) с последующим тщательным перемешиванием. Образовавшееся твердое вещество собирают фильтрацией и промывают простым диизопропиловым эфиром (100 мл). Промытое твердое вещество растворяют в метаноле (500 мл) и растворитель удаляют при пониженном давлении. Остаток растворяют в метаноле (200 мл), и раствор подвергают гидрированию в присутствии Pd(OH)2/C (1,45 г) при 40°С в течение 17 ч. Катализатор отфильтровывают, и фильтрат концентрируют при пониженном давлении. К остатку добавляют простой диизопропиловый эфир (200 мл), и осадок собирают с помощью фильтрации с получением посредством этого (20,6 г) указанного в заголовке соединения.
Форма: Бесцветное твердое вещество
Температура плавления: 144-147°С
1H-ЯМР (d6-ДМСO) δ: 1,21 (с, 6H), 3,25-3,34 (м, 1H), 4,09-4,19 (м, 2H), 4,21-4,30 (м, 1H), 4,31-4,39 (м, 2H), 9,00-10,6 (шир.с, 4H).
Ссылочный пример 3
Дигидрохлорид N-трет-бутилазетидин-3-иламина
Указанное в заголовке соединение получают способом, аналогичным способу в ссылочном примере 2.
Форма: Желтое масло
1H-ЯМР (CDCl3) δ: 1,27 (с, 9H), 4,06-4,20 (м, 2H), 4,29-4,37 (м, 1H), 4,38-4,46 (м, 2H), 9,15-9,56 (шир.с, 1H), 9,59-10,00 (шир.с, 1H), 10,20-10,60 (шир.с, 2H).
Ссылочный пример 4
Синтез этил-8-бром-1-[6-(трет-бутиламино)-3,5-дифторпиридин-2-ил]-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат
К раствору этил-3-этокси-2-(3-бром-2,4,5-трифторбензоил)акрилата (1,32 г) в хлороформе (5 мл), который был получен обычным способом из этил-3-бром-2,4,5-трифторбензоилацетата, добавляют 2-амино-6-(трет-бутиламино)-3,5-дифторпиридин до полного превращения в форму аминоакрилата, в то время как ход реакции контролируют посредством TLC (тонкослойной хроматографии). Раствор концентрируют при пониженном давлении для получения, таким образом, желтого твердого остатка. Безводный карбонат калия (1,2 г) и N,N-диметилформамид (2 мл) добавляют к остатку и смесь перемешивают при 90°С в течение 15 мин. Смесь оставляют для охлаждения, добавляют хлороформ (30 мл) и дистиллированную воду (300 мл) и отделяют. Затем хлороформный слой дважды промывают дистиллированной водой (300 мл) и сушат над безводным сульфатом магния. Высушенный продукт концентрируют при пониженном давлении и оставляют для отстаивания. Осадок собирают с помощью фильтрации, последовательно промывают этанолом и простым диизопропиловым эфиром для получения, таким образом, 1,41 г указного в заголовке соединения в виде бесцветного порошка.
Температура плавления: 198-203°С
1H-ЯМР (CDCl3) δ: 1,38 (с, 9H), 1,40 (т, J=7 Гц, 3H), 4,04 (кв, J=7 Гц, 2H), 4,71 (шир.с, 1H), 7,20 (дд, J=8 Гц, 10 Гц, 1H), 8,36 (дд, J=9 Гц, 10 Гц, 1H), 8,54 (с, 1H).
Ссылочный пример 5
Способом, аналогичным способу ссылочного примера 4, получают следующие соединения с (1) по (10).
(1) Этил-[6-(трет-бутиламино)-3,5-дифторпиридин-2-ил]-8-хлор-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат
Температура плавления: 203-205°С
1H-ЯМР (d6-ДМСO) δ: 1,39 (с, 9H), 1,40 (т, J=7 Гц, 3H), 4,40 (кв, J=7 Гц, 2H), 4,70 (шир.с, 1H), 7,21 (дд, J=8 Гц, 10 Гц, 1H), 8,31 (дд, J=8 Гц, 10 Гц, 1H), 8,50 (с, 1H).
(2) Этил-1-[6-(трет-бутиламино)-3,5-дифторпиридин-2-ил]-6,7-дифтор-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоксилат
Температура плавления: 203-205°С
1H-ЯМР (d6-ДМСO) δ: 1,34-1,48 (м, 12H), 1,82 (д, J=3 Гц, 3H), 4,40 (кв, J=7 Гц, 2H), 4,75 (шир.с, 1H), 7,23 (т, J=9 Гц, 1H), 8,22 (т, J=10 Гц, 1H), 8,50 (с, 1H).
Температура плавления: 207-211°С
1H-ЯМР (CDCl3) δ: 1,20 (д, J=7 Гц, 3H), 1,24 (д, J=7 Гц, 3H), 1,40 (т, J=7 Гц, 3H), 4,11 (м, 1H), 4,40 (кв, J=7 Гц, 2H), 4,60 (шир.с, 1H), 7,22 (дд, J=8 Гц, 9 Гц, 1H), 8,32 (дд, J=8 Гц, 10 Гц, 1H), 8,49 (с, 1H).
(3) Этил-8-хлор-1-(3,5-дифтор-6-изопропиламинопиридин-2-ил)-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат
Температура плавления: 206-209°С
1H-ЯМР (CDCl3) δ: 1,40 (т, J=7 Гц, 3H), 4,40 (кв, J=7 Гц, 2H), 5,02 (шир.с, 2H), 7,57 (д, J=8 Гц, 1H), 8,30 (т, J=9 Гц, 1H), 8,48 (с, 1H).
(4) Этил-1-(6-амино-5-хлор-3-фторпиридин-2-ил)-8-хлор-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат
Температура плавления: 186-189°С
1H-ЯМР (CDCl3) δ: 1,40 (т, J=7 Гц, 3H), 2,02 (с, 3H), 4,39 (кв, J=7 Гц, 2H), 4,71 (шир.с, 2H), 7,25 (д, J=10 Гц, 1H), 8,34 (т, J=10 Гц, 1H), 8,34 (с, 1H).
(5) Этил-1-(6-амино-5-фтор-3-метилпиридин-2-ил)-8-хлор-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат
Температура плавления: 198-202°С
1H-ЯМР (CDCl3) δ: 1,38 (с, 9H), 1,41 (т, J=7 Гц, 3H), 4,41 (кв, J=7 Гц, 2H), 4,84 (шир.с, 1H), 7,32 (д, J=10 Гц, 1H), 8,32 (дд, J=8 Гц, 10 Гц, 1H), 8,45 (с, 1H).
(6) Этил-1-[6-(трет-бутиламино)-3-хлор-5-фторпиридин-2-ил]-8-хлор-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат
Температура плавления: 210-213°С
1H-ЯМР (CDCl3) δ: 1,41 (т, J=7 Гц, 3H), 2,98 (д, J=5 Гц, 3H), 4,41 (кв, J=7 Гц, 2H), 4,85 (шир.с, 1H), 7,23 (дд, J=8 Гц, 9 Гц, 1H), 8,32 (дд, J=8 Гц, 10 Гц, 1H), 8,48 (с, 1H).
(7) Этил-8-хлор-1-(3,5-дифтор-6-метиламинопиридин-2-ил)-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат
Температура плавления: 207-209°С
1H-ЯМР (d6-ДМСO) δ: 1,26 (т, J=7 Гц, 3H), 2,74 (д, J=4 Гц, 3H), 4,23 (кв, J=7 Гц, 2H), 7,17-7,23 (м, 1H), 7,94 (дд, J=8 Гц, 10 Гц, 1H), 7,96 (дд, J=10 Гц, 10 Гц, 1H), 8,62 (с, 1H).
(8) Этил-8-бром-1-(3,5-дифтор-6-метиламинопиридин-2-ил)-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат
1H-ЯМР (d6-ДМСO) δ: 1,26 (т, J=7 Гц, 3H), 1,76 (д, J=3 Гц, 3H), 2,77 (д, J=5 Гц, 3H), 4,22 (кв, J=7 Гц, 2H), 7,26-7,31 (м, 1H), 7,95 (дд, J=9 Гц, 10 Гц, 1H), 8,05 (дд, J=10 Гц, 10 Гц, 1H), 8,56 (с, 1H).
(9) Этил-1-(3,5-дифтор-6-метиламинопиридин-2-ил)-6,7-дифтор-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоксилат
1H-ЯМР (d6-ДМСO) δ: 1,09 (т, J=7 Гц, 3H), 1,25 (т, J=8 Гц, 3H), 1,77 (д, J=3 Гц, 3H), 3,21-3,29 (м, 2H), 4,22 (кв, J=7 Гц, 2H), 7,27-7,32 (м, 1H), 7,95 (дд, J=9 Гц, 10 Гц, 1H), 8,05 (дд, J=10 Гц, 10 Гц, 1H), 8,56 (с, 1H).
(10) Этил-1-(6-этиламино-3,5-дифторпиридин-2-ил)-6,7-дифтор-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоксилат
1H-ЯМР (d6-ДМСO) δ: 6,80 (с, 2H), 7,99 (т, J=9 Гц, 1H), 8,38 (т, J=9 Гц, 1H), 8,93 (с, 1H).
Ссылочный пример 6
Синтез 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты
Этил-8-бром-1-[6-(трет-бутиламино-3,5-дифторпиридин-2-ил]-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат (1,38 г) добавляют к жидкой смеси 14% хлористоводородной кислоты (3,5 мл) и уксусной кислоты (3,5 мл), с последующим перемешиванием в течение 5 ч при кипячении с обратным холодильником. К смеси добавляют дистиллированную воду (5 мл), и смесь оставляют для охлаждения. Осадок собирают с помощью фильтрации и последовательно промывают этанолом и простым диизопропиловым эфиром с получением, таким образом, 1,10 г указанного в заголовке соединения в виде бесцветного порошка.
Температура плавления: 272-278°С
1H-ЯМР (d6-ДМСO) δ: 6,80 (с, 2H), 7,99 (т, J=9 Гц, 1H), 8,38 (т, J=9 Гц, 1H), 8,93 (с, 1H).
Ссылочный пример 7
Способом, аналогичным способу ссылочного примера 6, получают следующие соединения с (1) по (10).
(1) 1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Температура плавления: ≥280°С
1H-ЯМР (d6-ДМСO) δ: 6,80 (с, 2H), 7,99 (т, J=9 Гц, 1H), 8,38 (т, J=9 Гц, 1H), 8,93 (с, 1H).
(2) 1-(6-амино-3,5-дифторпиридин-2-ил)-6,7-дифтор-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Температура плавления: 274-277°С
1H-ЯМР (d6-ДМСO) δ: 1,84 (с, 3H), 6,91 (шир.с, 2H), 8,03 (т, J=9 Гц, 1H), 8,25 (т, J=9 Гц, 1H), 8,93 (с, 1H).
(3) 8-хлор-1-(3,5-дифтор-6-изопропиламинопиридин-2-ил)-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Температура плавления: 226-230°С
1H-ЯМР (d6-ДМСO) δ: 1,10 (д, J=7 Гц, 3H), 1,16 (д, J=7 Гц, 3H), 3,94 (м, 1H), 7,02 (шир.д, J=8 Гц, 1H), 7,97 (т, J=9 Гц, 1H), 8,39 (т, J=9 Гц, 1H), 8,92 (с, 1H).
(4) 1-(6-амино-5-хлор-3-фторпиридин-2-ил)-8-хлор-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Температура плавления: ≥280°С
1H-ЯМР (d6-ДМСO) δ: 6,86 (шир.с, 2H), 8,15 (д, J=9 Гц, 1H), 8,38 (т, J=9 Гц, 1H), 8,95 (с, 1H).
(5) 1-(6-амино-5-фтор-3-метилпиридин-2-ил)-8-хлор-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Температура плавления: 279-284°С
1H-ЯМР (d6-ДМСO) δ: 1,94 (с, 3H), 6,62 (шир.с, 2H), 7,57 (д, J=11 Гц, 1H), 8,40 (т, J=9 Гц, 1H), 8,72 (с, 1H).
(6) 1-(6-амино-3-хлор-5-фторпиридин-2-ил)-8-хлор-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Температура плавления: ≥280°С
1H-ЯМР (d6-ДМСO) δ: 7,10 (шир.с, 2H), 7,99 (д, J=10 Гц, 1H), 8,40 (т, J=10 Гц, 1H), 8,89 (с, 1H).
(7) 8-хлор-1-(3,5-дифтор-6-метиламинопиридин-2-ил)-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Температура плавления: 236-242°С
1H-ЯМР (d6-ДМСO) δ: 2,76 (д, J=5 Гц, 3H), 7,21-7,28 (м, 1H), 7,98 (дд, J=9 Гц, 10 Гц, 1H), 8,39 (дд, J=9 Гц, 10 Гц, 1H), 8,92 (с, 1H).
(8) 8-бром-1-(3,5-дифтор-6-метиламинопиридин-2-ил)-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
1H-ЯМР (d6-ДМСO) δ: 2,75 (д, J=5 Гц, 3H), 7,19-7,28 (м, 1H), 7,96 (дд, J=9 Гц, 10 Гц, 1H), 8,40 (дд, J=9 Гц, 10 Гц, 1H), 8,90 (с, 1H).
(9) 1-(3,5-дифтор-6-метиламинопиридин-2-ил)-6,7-дифтор-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
1H-ЯМР (d6-ДМСO) δ: 1,80-1,85 (д, J=2 Гц, 3H), 2,77 (д, J=5 Гц, 3H), 7,29-7,38 (м, 1H), 7,97 (дд, J=9 Гц, 10 Гц, 1H), 8,22 (дд, J=9 Гц, 9 Гц, 1H), 8,89 (с, 1H).
(10) 1-(6-этиламино-3,5-дифторпиридин-2-ил)-6,7-дифтор-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
1H-ЯМР (d6-ДМСO) δ: 1,09 (т, J=6 Гц, 3H), 1,83 (д, J=2 Гц, 3H), 3,21-3,30 (м, 2H), 7,32-7,39 (м, 1H), 7,97 (дд, J=9 Гц, 10 Гц, 1H), 8,23 (дд, J=9 Гц, 9 Гц, 1H), 8,89 (с, 1H).
Пример 1
Синтез 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединения 1)
Дигидрохлорид N-изопропилазетидин-3-иламина (374 мг), 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту (518 мг), N-метилпирролидин (0,8 мл), хлорид лития (500 мг) и диметилсульфоксид (1,5 мл) смешивают и смесь перемешивают при 50°С в течение 4,5 ч. После того, как смеси дают охладиться, к смеси добавляют простой диэтиловый эфир (10 мл) и перемешивают с последующим удалением надосадочной жидкости. Такую же процедуру повторяют 3 раза. К остатку добавляют воду (3 мл) и рН смеси доводят до 6, используя водный раствор лимонной кислоты. Образовавшееся твердое вещество собирают с помощью фильтрации, промывают 3 раза водой (3 мл), затем суспендируют в этаноле (10 мл) и перемешивают при нагревании. Образовавшееся твердое вещество собирают с помощью фильтрации и суспендируют в простом диизопропиловом эфире (10 мл) с последующим перемешиванием при нагревании. Нагретую суспензию фильтруют с получением 410 мг указанного в заголовке соединения.
Форма: Бледно-желтый порошок
Температура плавления: ≥203°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 0,94 (д, J=6 Гц, 6H), 2,68-2,75 (м, 1H),3,60-3,67 (м, 1H), 4,02-4,11 (м, 2H), 4,65-4,73 (м, 2H), 6,74 (с, 2H), 7,88 (д, J=14 Гц, 1H), 7,93 (дд, J=10 Гц, 10 Гц, 1H), 8,69 (с, 1H).
Пример 2
Способом, аналогичным способу примера 1, синтезируют следующие соединения 2-11.
(1) 1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 2)
Форма: Бледно-желтый порошок
Температура плавления: ≥205°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,17 (д, J=6 Гц, 6H), 3,13-3,24 (м, 1H), 3,98-4,15 (м, 1H), 4,44-4,58 (м, 2H), 4,72-4,83 (м, 2H), 6,75 (с, 2H), 7,93 (д, J=14 Гц, 1H), 7,95 (дд, J=10 Гц, 10 Гц, 1H), 8,72 (с, 1H).
(2) 1-(6-амино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 3)
Форма: Бледно-желтый порошок
Температура плавления: ≥238°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 0,93 (д, J=7 Гц, 6H), 1,62 (с, 3H), 2,66-2,75 (м, 1H), 3,60-3,69 (м, 1H), 3,76-3,86 (м, 1H), 4,39-4,54 (м, 2H), 6,82 (с, 2H), 7,74 (д, J=14 Гц, 1H), 7,94 (дд, J=10 Гц, 10 Гц, 1H), 8,69 (с, 1H).
(3) 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-7-(3-трет-бутиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 4)
Форма: Бледно-желтый порошок
Температура плавления: ≥225°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 0,99 (с, 9H), 3,72-3,81 (м, 1H), 3,97-4,10 (м, 2H), 4,68-4,78 (м, 2H), 6,74 (с, 2H), 7,88 (д, J=14 Гц, 1H), 7,93 (дд, J=10 Гц, 10 Гц, 1H), 8,70 (с, 1H).
(4) 1-(6-амино-3,5-дифторпиридин-2-ил)-7-(3-трет-бутиламиноазетидин-1-ил)-8-хлор-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 5)
Форма: Бледно-желтый порошок
Температура плавления: ≥250°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,00 (с, 9H), 3,75-3,85 (м, 1H), 4,03-4,18 (м, 2H), 4,68-4,77 (м, 2H), 6,74 (с, 2H), 7,87 (д, J=14 Гц, 1H), 7,94 (дд, J=10 Гц, 10 Гц, 1H), 8,68 (с, 1H).
(5) 1-(6-амино-3,5-дифторпиридин-2-ил)-7-(3-трет-бутиламиноазетидин-1-ил)-6-фтор-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 6)
Форма: Бледно-желтый порошок
Температура плавления: ≥193°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,08 (с, 9H), 1,65 (с, 3H), 3,87-4,02 (м, 2H),4,06-4,18 (м, 1H), 4,49-4,62 (м, 2H), 6,84 (с, 2H), 7,80 (д, J=14 Гц, 1H), 7,96 (дд, J=10 Гц, 10 Гц, 1H), 8,72 (с, 1H).
(6) 1-(3,5-дифтор-6-метиламинопиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 7)
Форма: Бесцветный порошок
Температура плавления: ≥218°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 0,93 (д, J=6 Гц, 6H), 1,62 (с, 3H), 2,80 (д, J=4 Гц, 3H), 3,61-3,71 (м, 1H), 3,79-3,88 (м, 1H), 3,89-3,97 (м, 1H), 4,41-4,53 (м, 2H), 7,24-7,28 (м, 1H), 7,77 (д, J=14 Гц, 1H), 7,95 (дд, J=10 Гц, 10 Гц, 1H), 8,71 (с,1H).
(7) 1-(6-этиламино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 8)
Форма: Бледно-желтый порошок
Температура плавления: ≥202°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 0,94 (д, J=6 Гц, 6H), 1,14 (т, J=7 Гц, 3H), 1,63 (с, 3H), 3,62-3,73 (м, 1H), 3,83-3,97 (м, 1H), 4,41-4,53 (м, 2H), 7,24-7,29 (м, 1H), 7,77 (д, J=14 Гц, 1H), 7,95 (дд, J=10 Гц, 10 Гц, 1H), 8,70 (с, 1H).
(8) 8-хлор-1-(3,5-дифтор-6-метиламинопиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 9)
Форма: Бесцветный порошок
Температура плавления: ≥244°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,15 (шир.с, 6H), 2,77 (д, J=4 Гц, 3H), 3,99-4,14 (м, 1H), 4,36-4,57 (м, 2H), 4,71-4,82 (м, 2H), 7,16-7,25 (м, 1H), 7,91-7,98 (м, 2H), 8,73 (с, 1H).
(9) 8-хлор-1-(3,5-дифтор-6-изопропиламинопиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 10)
Форма: Бесцветный порошок
Температура плавления: ≥248°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,11-1,22 (м, 12H), 3,92-4,00 (м, 1H), 4,09-4,18 (м, 1H), 4,36-4,53 (м, 2H), 4,63-4,77 (м, 2H), 6,89 (д, J=8 Гц, 1H), 7,81 (д, J=13 Гц, 1H), 7,91 (дд, J=10 Гц, 10 Гц, 1H), 8,44 (с, 1H).
(10) 8-бром-1-(3,5-дифтор-6-метиламинопиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (соединение 11)
Форма: Бледно-желтый порошок
Температура плавления: ≥220°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,15 (шир.с, 6H), 2,77 (д, J=5 Гц, 3H), 3,96-4,17 (м, 1H), 4,32-4,55 (м, 2H), 4,70-4,84 (м, 2H), 7,15-7,24 (м, 1H), 7,90-8,00 (м, 2H), 8,75 (с, 1H).
Пример 3
Синтез соли малеиновой кислоты 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединения 12)
1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту (132 мг) и малеиновую кислоту (58 мг) добавляют к этанолу (3 мл) и смесь нагревают при 80°С. Воду постепенно добавляют к смеси до полного растворения соединений. После охлаждения раствора при отстаивании полученное твердое вещество собирают с помощью фильтрации с получением 102 мг указанного в заголовке соединения.
Форма: Бледно-желтый порошок
Температура плавления: ≥220°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,19 (д, J=7 Гц, 6H), 4,13-4,22 (м, 1H), 4,42-4,59 (м, 2H), 4,71-4,86 (м, 2H), 6,02 (с, 2H), 6,75 (с, 2H), 7,93 (дд, J=10 Гц, 10 Гц, 1H), 7,96 (д, J=14 Гц, 1H), 8,75 (с, 1H), 8,58-9,51 (шир.с, 1H).
Пример 4
Способом, аналогичным способу примера 3, синтезируют следующие соединения 13-191.
(1) Соль метансульфоновой кислоты 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединение 13)
Форма: Бледно-желтый порошок
Температура плавления: ≥224°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,24 (д, J=7 Гц, 6H), 2,30 (с, 3H),4,14-4,24 (м, 1H), 4,44-4,62 (м, 2H), 4,73-4,86 (м, 2H), 6,75 (с, 2H), 7,94 (дд, J=10 Гц, 10 Гц, 1H), 7,96 (д, J=14 Гц, 1H), 8,75 (с, 1H), 8,96-9,35 (шир.с, 0,5H).
(2) Соль п-толуолсульфоновой кислоты 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединение 14)
Форма: Бледно-желтый порошок
Температура плавления: ≥235°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,21 (д, J=6 Гц, 6H), 2,28 (с, 3H), 4,15-4,26 (м, 1H), 4,43-4,62 (м, 2H), 4,70-4,87 (м, 2H), 6,75 (с, 2H), 7,09 (д, J=8 Гц, 2H), 7,47 (д, J=9 Гц, 2H), 7,94 (дд, J=10 Гц, 10 Гц, 1H), 7,96 (д, J=14 Гц, 1H), 8,75 (с, 1H), 8,93-9,11 (шир.с, 1H).
(3) Гидрохлорид 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединение 15)
Форма: Бледно-желтый порошок
Температура плавления: ≥210°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,21 (д, J=5 Гц, 6H), 4,11-4,22 (м, 1H), 4,49-4,65 (м, 2H), 4,73-4,86 (м, 2H), 6,75 (с, 2H), 7,94 (дд, J=10 Гц, 10 Гц, 1H), 7,97 (д, J=14 Гц, 1H), 8,74 (с, 1H), 9,32-9,81 (шир.с, 2H).
(4) Соль метансульфоновой кислоты 1-(6-амино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединение 16)
Форма: Бледно-желтый порошок
Температура плавления: ≥235°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,21 (д, J=7 Гц, 6H), 1,68 (с, 3H), 2,31 (с, 3H), 3,41―3,47 (м, 1H), 4,17-4,50 (м, 2H), 4,51-4,70 (м, 2H), 6,85 (с, 2H), 7,84 (д, J=12 Гц, 1H), 7,96 (дд, J=10 Гц, 10 Гц, 1H), 8,75 (с, 1H), 9,00 (шир.с, 2H).
(5) Соль п-толуолсульфоновой кислоты 1-(6-амино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединение 17)
Форма: Желтый порошок
Температура плавления: ≥232°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,21 (д, J=6 Гц, 6H), 1,67 (с, 3H), 2,28 (с, 3H), 3,41―3,47 (м, 1H), 4,15-4,30 (м, 2H), 4,37-4,47 (м, 1H), 4,50-4,69 (м, 2H), 6,86 (с, 2H), 7,10 (д, J=8 Гц, 2H), 7,47 (д, J=8 Гц, 2H), 7,84 (д, J=14 Гц, 1H), 7,96 (дд, J=10 Гц, 10 Гц, 1H), 8,76 (с, 1H), 8,98 (шир.с, 2H).
(6) Соль малеиновой кислоты 1-(6-амино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединение 18)
Форма: Бледно-желтый порошок
Температура плавления: ≥235°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,20 (д, J=6 Гц, 6H), 1,68 (с, 3H), 4,15-4,28 (м, 2H), 4,33-4,44 (м, 1H), 4,50-4,68 (м, 2H), 6,03 (с, 2H), 6,86 (с, 2H), 7,84 (д, J=14 Гц, 1H), 7,96 (дд, J=10 Гц, 10 Гц, 1H), 8,76 (с, 1H).
(7) Гидрохлорид 1-(6-амино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-изопропиламиноазетидин-1-ил)-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединение 19)
Форма: Бесцветный порошок
Температура плавления: ≥230°С, с разложением
1H-ЯМР (d6-ДМСO) δ: 1,22 (д, J=6 Гц, 6H), 1,68 (с, 3H), 3,41-3,47 (м, 1H), 4,15-4,40 (м, 2H), 4,42-4,67 (м, 2H), 6,86 (с, 2H), 7,84 (д, J=14 Гц, 1H), 7,96 (дд, J=10 Гц, 10 Гц, 1H), 8,75 (с, 1H), 9,45 (шир.с, 1,5H).
Соединения по настоящему изобретению испытывали на их антибактериальную активность, фототоксичность, скорость связывания с сывороточными белками и фармакокинетику. Результаты этих испытаний представлены в примерах испытаний 1-4. В качестве сравнительных соединений, использовали следующие соединения, раскрытые в международных публикациях №№ 97/11068 и 01/02390.
Сравнительное соединение 1: 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6-фтор-7-(3-метиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Сравнительное соединение 2: 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6-фтор-7-(3-этиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
Сравнительное соединение 3: соль малеиновой кислоты 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6-фтор-7-(3-метиламиноазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты
Сравнительное соединение 4: соль малеиновой кислоты 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-7-(3-этиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты.
Пример испытания 1: Антибактериальная активность
Минимальную ингибирующую концентрацию (MIC: мкг/мл) соединений, показанных в таблице 1, определяли в соответствии со стандартным способом Японского Общества Химиотерапии (CHEMOTHERAPY, 29(1), 76, 1981). Результаты показаны в табл.1.
Величина MIC: (мкг/мл)
Пример испытания 2: Фототоксичность
Соединения, показанные в таблице 2, подвергали описанному ниже тесту фототоксичности.
Каждое испытуемое соединение вводили внутривенно (40 мг/10 мл/кг) самкам мышей ICR (в возрасте 5-6 нед), и мышей облучали УФ-лучами (320-400 нм, 1,8 мВт/см2/с) в течение 4 ч. Патологические отклонения, вызванные в ухе каждой мыши наблюдали сразу после (0 ч), через 24 ч и 48 ч после облучения. Патологические отклонения оценивали в соответствии со следующими критериями: нормальное (0 очков), легкая эритема (балльная оценка 1), средняя эритема (балльная оценка 2) и тяжелая эритема или отек (балльная оценка 3). Результаты показаны в таблице 2.
Пример испытания 3: Скорость связывания с белками
Скорость связывания с белками соединений, показанных в табл.3, определяли методом ультрафильтрации. В частности, 0,5 мг/мл раствора в DMSO (диметилсульфоксиде) каждого соединения разбавляли человеческой сывороткой или 0,4% фосфатным буфером (рН: 7,4, содержащим 0,5% NaCl) для получения, таким образом, раствора 5 мкг/мл. Раствор человеческой сыворотки инкубировали при 37°С в течение 30 мин и фильтровали при центрифугировании через фильтр (размер пор: 0,22 мкм). Часть сывороточного фильтрата и раствора фосфатного буфера наносили на устройство ВЭЖХ и измеряли площадь пика соединения. Скорость связывания каждого соединения с человеческими белками рассчитывали в соответствии со следующим уравнением 1:
Скорость связывания (%)=(А-В)×100/А (уравнение 1)
А: Площадь пика (в фосфатном буфере)
В: Площадь пика (в сывороточном фильтрате)
Эти результаты показывают, что соединения по настоящему изобретению проявили низкую скорость связывания с белками, без проявления увеличения скорости связывания, как предполагалось исходя из увеличения их липофильности.
Пример испытания 4: Фармакокинетика
Всасываемость соединений, показанных в таблице 4, исследовали на собаках. В частности, 0,5% суспензию метилцеллюлозы каждого испытуемого соединения (10 мг/кг) принудительно перорально вводили самцам гончих собак в возрасте 2-4 лет, которые голодали в течение 16-17 час. Кровь брали у каждой собаки через 0,25, 0,5, 1, 2, 4, 6, 8 и 24 ч после введения и уровень испытуемого соединения в сыворотке определяли для оценки всасываемости соединения. Результаты показаны в таблице 4.
(мкг/мл)
(ч)
(ч)
Как показано в таблице 4, фармакокинетика соединения 12 по настоящему изобретению значительно улучшена, по сравнению со сравнительными соединениями 3 и 4.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ И АНТИБАКТЕРИАЛЬНОЕ СРЕДСТВО | 1996 |
|
RU2167873C2 |
| АМИНОСОЕДИНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ НОВЫХ ПРОИЗВОДНЫХ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛЕЙ | 1996 |
|
RU2171252C2 |
| ПРОИЗВОДНОЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕГО СОЛЬ | 1996 |
|
RU2177945C1 |
| 1-(6-АМИНО-3,5-ДИФТОРПИРИДИН-2-ИЛ)-8-БРОМ-7(3-ЭТИЛ-АМИНОАЗЕТИДИН-1-ИЛ)-6- ФТОР-4-ОКСО-1,4-ДИГИДРОХИНОЛИН-3-КАРБОНОВАЯ КИСЛОТА, ЛЕКАРСТВЕННАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ НА ОСНОВЕ ЭТОГО СОЕДИНЕНИЯ | 2000 |
|
RU2236408C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛОНОВЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2546667C2 |
| ПРОИЗВОДНОЕ 6-(ГЕТЕРОЦИКЛЗАМЕЩЕННЫЙ БЕНЗИЛ)-4-ОКСОХИНОЛИНА И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ИНТЕГРАЗЫ ВИЧ | 2007 |
|
RU2399616C1 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ 1,4-ДИГИДРО-4-ОКСОХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИМИКРОБНОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2125046C1 |
| ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ И КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2100351C1 |
| СОЕДИНЕНИЕ 4-ОКСОХИНОЛИНА И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ВИЧ ИНТЕГРАЗЫ | 2003 |
|
RU2275361C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2556984C2 |
Настоящее изобретение относится к производному пиридонкарбоновой кислоты, представленному формулой (1): 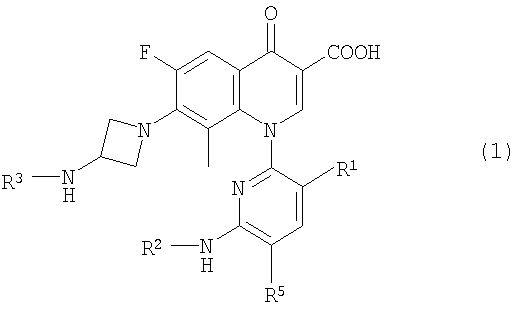
или его соли, где R представляет метильную группу, атом фтора или атом хлора; R2 представляет атом водорода или низшую алкильную группу; R3 представляет изопропильную группу или трет-бутильную группу; R4 представляет метильную группу или атом галогена; и R5 представляет атом фтора или атом хлора, а также к антибактериальному средству и фармацевтической композиции, содержащим указанное производное или его соль в качестве активного ингредиента. Кроме того, изобретение описывает применение соединения по п.1 и способ лечения инфекционного заболевания. Технический результат: получено и описано новое соединение, которое проявляет превосходную антибактериальную активность, низкую токсичность, улучшенную биологическую доступность и низкую скорость связывания с сывороточными белками. 7 н. и 3 з.п. ф-лы, 4 табл.
1. Производное пиридонкарбоновой кислоты, представленное формулой (1):
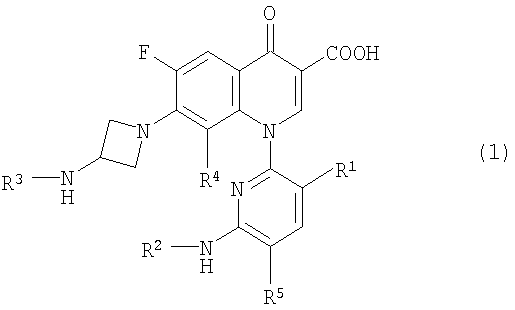
или его соль, где R1 представляет метильную группу, атом фтора или атом хлора; R2 представляет атом водорода или низшую алкильную группу; R3 представляет изопропильную группу или трет-бутильную группу; R4 представляет метильную группу или атом галогена; и R5 представляет атом фтора или атом хлора.
2. Производное пиридонкарбоновой кислоты или его соль по п.1, где каждый из R1 и R5 представляет собой атом фтора.
3. Производное пиридонкарбоновой кислоты или его соль по п.1, где R4 представляет собой атом брома или метильную группу.
4. Производное пиридонкарбоновой кислоты или его соль по любому из пп.1-3, где R2 представляет собой атом водорода или метильную группу.
5. 1-(6-Амино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-изопропил аминоазетидин-1-ил)-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота или ее соль.
6. Антибактериальное средство, включающее производное пиридонкарбоновой кислоты или его соль по любому из пп.1-5 в качестве активного ингредиента.
7. Фармацевтическая антибактериальная композиция, включающая производное пиридонкарбоновой кислоты или его соль по любому из пп.1-5, и фармацевтически приемлемый носитель.
8. Применение производного пиридонкарбоновой кислоты или его соли по п.1 для получения антибактериального лекарственного средства.
9. Способ лечения инфекционного заболевания, включающий введение производного пиридонкарбоновой кислоты или его соли по п.1.
10. Производное пиридонкарбоновой кислоты, представленное формулой (2)
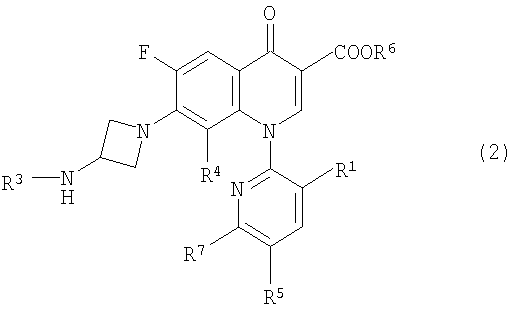
или его соль, где R1 представляет метильную группу, атом фтора или атом хлора; R3 представляет изопропильную группу или трет-бутильную группу; R4 представляет метильную группу или атом галогена; R5 представляет атом фтора или атом хлора; R6 представляет атом водорода или карбоксильную защитную группу; и R7 представляет -NR2R8, где R2 представляет атом водорода или низшую алкильную группу, и R8 представляет атом водорода или аминозащитную группу, при условии, что R6 и R8 одновременно не представляют атомы водорода.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ И АНТИБАКТЕРИАЛЬНОЕ СРЕДСТВО | 1996 |
|
RU2167873C2 |
| RU 93058417 А, 27.12.1996. | |||

