Область изобретения
Данное изобретение касается производных хинолинкарбоновой кислоты и их солей, которые проявляют превосходное противомикробное действие и пероральную абсорбцию, а также противомикробных агентов, включающих их.
Предпосылки изобретения
Известно, что соединения, имеющие основной скелет хинолинкарбоновой кислоты, включают многие соединения, полезные в качестве синтетических противомикробных агентов, благодаря их превосходной противомикробной активности и широким спектрам противомикробного действия. Среди таких соединений норфлоксацин (JP 53-141286 А), эноксацин (JP 55-31042 А), офлоксацин (JP 57-46986 А), ципрофлоксацин (JP 58-74667 А), тосуфлоксацин (JP 60-228479) и аналогичные широко применяются в клинической практике в качестве терапевтических агентов от инфекционных заболеваний.
Однако данные соединения все же обладают недостаточной противомикробной активностью, абсорбцией в кишечнике и метаболической стабильностью и вызывают много проблем, подлежащих решению, таких как снижение фототоксичности и цитотоксичности, характерных для хинолинкарбоновой кислоты и ее производных. В последнее время возникала также проблема, связанная с появлением бактерий, устойчивых к данным лекарственным средствам.
Описание изобретения
Следовательно, задачей настоящего изобретения является предоставление противомикробного агента, который является клинически применимым, обладает превосходной противомикробной активностью, абсорбцией в кишечнике и метаболической стабильностью и проявляет низкие побочные эффекты.
При упомянутых выше обстоятельствах настоящие изобретатели провели широкие исследования для обеспечения клинически превосходных лекарственных агентов. В результате обнаружено, что производные пиридонкарбоновой кислоты, представленные следующей формулой (I):
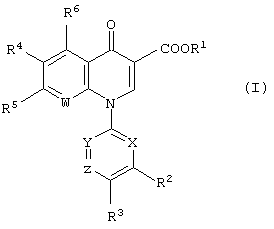
в которой R1 обозначает атом водорода или карбоксилзащитную группу, R2 обозначает гидроксильную группу, низшую алкоксигруппу или замещенную или незамещенную аминогруппу, R3 обозначает атом водорода или атом галогена, R4 обозначает атом водорода или атом галогена, R5 обозначает атом галогена или замещенную или незамещенную насыщенную циклическую аминогруппу, R6 обозначает атом водорода, атом галогена, нитрогруппу или защищенную или незащищенную аминогруппу, X, Y и Z могут быть одинаковыми или различными и каждый независимо обозначает атом азота, -СН= или -CR7=, в которой R7 обозначает низшую алкильную группу, атом галогена или цианогруппу, при условии, что, по меньшей мере, один из X, Y и Z обозначает атом азота и W обозначает атом азота или -CR8=, в которой R8 обозначает атом водорода, атом галогена или низшую алкильную группу, и их соли обладают превосходной противомикробной активностью и полезны в качестве синтетических противомикробных агентов, и на них подана международная РСТ заявка (WO 97/11068 А).
Изобретатели провели дальнейшие исследования. В результате было обнаружено, что среди описанных выше производных пиридонкарбоновой кислоты (I) 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-7-(3-этиламин-орто-азетидин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота, которая имеет 6-амино-3,5-дифторпиридинильную группу в положении 1, этиламиноазетидинильную группу в положении 7 и атом брома в положении 8 и представлена следующей формулой:
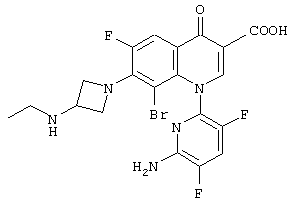
и ее соли обладают превосходными свойствами, они имеют чрезвычайно хорошую противомикробную активность и широкий спектр противомикробного действия, охватывая устойчивые бактерии, не проявляют фототоксичности, которая является характерной для хинолона, и оказывают меньшее антигипертензивное действие и побочные эффекты в отношении кожи, такие как сыпь, чем известные соединения аналогичных структур, и, кроме того, имеют большой период полураспада в крови, крайне высокую биологическую доступность и чрезвычайно полезны в качестве профилактических и терапевтических агентов при различных инфекционных заболеваниях, что привело к завершению настоящего изобретения.
Говоря конкретно, настоящее изобретение предоставляет 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-7-(3-этиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту (далее называемую здесь “соединение 1”) или ее соль.
Настоящее изобретение представляет также лекарственное средство, включающее в качестве активного ингредиента соединение 1 или его соль.
Настоящее изобретение представляет также лекарственную композицию, включающую соединение 1 или его соль и фармацевтически приемлемый носитель.
Кроме того, настоящее изобретение предлагает применение соединения 1 или его соли в качестве лекарственного средства.
Настоящее изобретение дополнительно представляет еще способ лечения инфекционного заболевания, который включает введение соединения 1 или его соли.
Лучшие варианты осуществления изобретения
Соединение 1 настоящего изобретения можно получить в виде аддитивных солей кислот и в виде аддитивных солей оснований. Следует отметить, что соединения, образующие хелаты с соединениями бора, также включены в такие соли.
Примеры аддитивных солей кислот могут включать (а) соли с минеральными кислотами, такими как соляная кислота, серная кислота и фосфорная кислота, (b) соли с органическими карбоновыми кислотами, такими как муравьиная кислота, уксусная кислота, лимонная кислота, трихлоруксусная кислота, трифторуксусная кислота, фумаровая и малеиновая кислота, и (с) соли с сульфоновыми кислотами, такими как метансульфоновая кислота, бензолсульфоновая кислота, паратолуолсульфоновая кислота, мезитиленсульфоновая кислота и нафталинсульфоновая кислота, при этом примеры аддитивных солей оснований могут включать (а’) соли со щелочными металлами, такими как натрий и калий, (b’) соли со щелочно-земельными металлами, такими как кальций и магний, (с’) соли аммония, (а’) соли с азотсодержащими органическими основаниями, такими как триметиламин, триэтиламин, трибутиламин, пиридин, N,N-диметиланилин, N-метилпиперидин, N-метилморфолин, диэтиламин, циклогексиламин, прокаин, дибензиламин, N-бензил-β-фенетиламин, 1-эфенамин и N,N’-дибензилэтилендиамин.
Иллюстрациями соединений бора являются галогениды бора, такие как фторид бора, и низший ацилоксиборон, такие как ацетоксиборон. Из них предпочитаются аддитивные соли кислот, причем особенно предпочтительны малеат, метансульфонат, паратолуолсульфонат и гидрохлорид.
Соединения 1 или их соли согласно настоящему изобретению могут существовать не только в несольватированной форме, но также в виде гидратов или сольватов. Соответственно каждое из соединений настоящего изобретения охватывает все его кристаллические формы, гидрат и его соль ваты.
Соединение 1 или соль, согласно настоящему изобретению, каждое, можно получить желательным способом.
Примерный способ можно проиллюстрировать следующим образом:
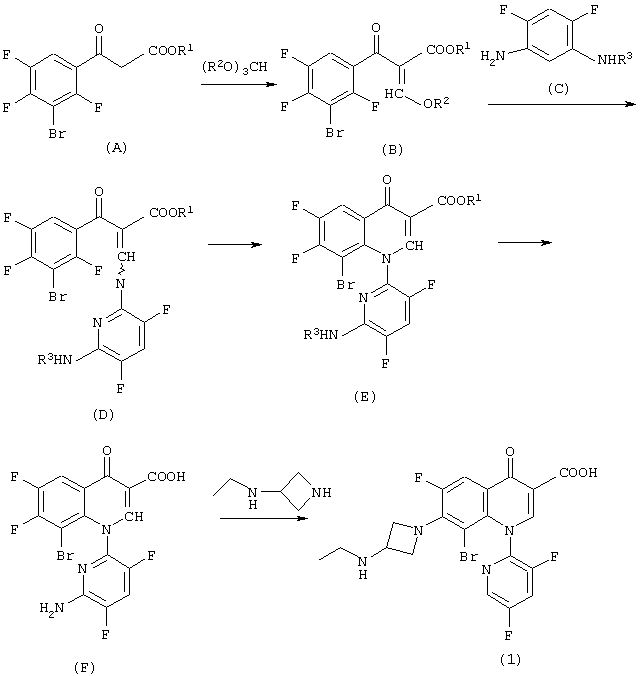
где R1 и R2 обозначают низшие алкильные группы, и R3 обозначает атом водорода или аминозащитную группу (например, трет-бутил, бензил, параметоксибензил или 1,1,3,3-тетраметилбутил).
Соединение 1 настоящего изобретения можно получить по реакции эфира ортомуравьиной кислоты, такого как этилортоформиат или метилортоформиат, с соединением (А) с образованием производного акрилатного эфира (В), реакции производного акрилатного эфира с амино-соединением (С) с получением соединения (D), реакции циклизации соединения (D) с получением соединения (Е), гидролизом соединения (Е) в соединение (F) и последующим взаимодействием соединения (F) с 3-этиламиноазетидином.
Реакцию между соединением (А) и эфиром ортомуравьиной кислоты обычно можно проводить при 0-160°С, предпочтительно при 50-150°С, и время реакции обычно может составлять от 10 минут до 48 часов, предпочтительно 1-10 часов. Эфир ортомуравьиной кислоты предпочтительно можно использовать в эквимолярном или большем количестве относительно соединения (А), особенно в молярном количестве, примерно 1-10-кратном относительно соединения (А). Предпочтительно добавлять в качестве промотора реакции ангидрид карбоновой кислоты, такой как уксусный ангидрид. Данный ангидрид карбоновой кислоты можно использовать предпочтительно в эквимолярном или большем количестве относительно соединения (А), особенно в молярном количестве, примерно 1-10-кратном относительно соединения (А).
Реакцию с соединением (С) проводят без растворителя или в подходящем растворителе. В данной реакции можно применять любой растворитель, пока он не влияет на реакцию. Примерами являются ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан, моноглим и диглим; алифатические углеводороды, такие как пентан, гексан, гептан и лигроин; галогенированные углеводороды, такие как метиленхлорид, хлороформ и четыреххлористый углерод; апротонные полярные растворители, такие как диметилформамид и диметилсульфоксид; и спирты, такие как метанол, этанол и пропанол. Данную реакцию обычно можно проводить при 0-150°С, предпочтительно при 0-100°С, и время реакции обычно составляет от 10 минут до 48 часов. Соединение (С) можно использовать в эквимолярном или большем количестве относительно соединения (А), особенно в молярном количестве, 1-2-кратном относительно соединения (А).
В альтернативном способе ацеталь, например N,N-диметилформамиддиметилацеталь или N,N-диметилформамиддиэтилацеталь, подвергают взаимодействию с соединением (А) с последующим взаимодействием с соединением (С) с получением соединения (D). В реакции можно применять любой растворитель, если только он не влияет на реакцию. Примерами являются растворители, представленные выше. Данную реакцию обычно можно проводить при 0-150°С, предпочтительно при комнатной температуре и до 100°С, и время реакции обычно составляет от 10 минут до 48 часов, предпочтительно от 1 до 10 часов.
Затем проводят реакцию, в которой соединение (D) подвергают циклизации с получением соединения (Е), в присутствии или в отсутствие основного соединения в растворителе. В данной реакции можно применять любой растворитель, пока он не влияет на реакцию. Примерами являются ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан, моноглим и диглим; галогенированные углеводороды, такие как метиленхлорид, хлороформ и четыреххлористый углерод; апротонные полярные растворители, такие как диметилформамид и диметилсульфоксид; и спирты, такие как метанол, этанол и пропанол. Соединения, используемые в качестве основного соединения, могут включать, например, щелочные металлы, такие как металлический натрий и металлический калий; гидриды металлов, такие как гидрид натрия и гидрид кальция; неорганические соли, такие как гидроксид натрия, гидроксид калия, карбонат натрия и карбонат калия; алкоголяты, такие как метилат натрия, этилат натрия и третбутилат натрия; фториды металлов, такие как фторид натрия и фторид калия; и органические основания, такие как триэтиламин и 1,8-диазабицикло[5.4.0]ундецен (DBU). Температура реакции обычно составляет от 0 до 200°С, предпочтительно от комнатной температуры до 180°С, и реакцию можно завершить за время от 5 минут до 24 часов. Основное соединение можно использовать в эквимолярном или большем количестве относительно соединения (D), особенно в молярном количестве, 1-2-кратном относительно соединения (D).
Удаление карбоксилзащитной группы R1 и аминозащитной группы R3 гидролизом соединения (Е) дает возможность получить соединение (F).
Для гидролиза применимы все условия реакций, обычно используемые при гидролизе. Гидролиз можно проводить, например, в присутствии основного соединения, такого как гидроксид натрия, гидроксид калия, карбонат натрия или карбонат калия, минеральной кислоты, такой как соляная, серная или бромистоводородная кислота, или органической кислоты, такой как пара-толуолсульфоновая кислота, в растворителе, например воде, спирте, таком как метанол, этанол или пропанол, простом эфире, таком как тетрагидрофуран или диоксан, кетоне, таком как ацетон или метилэтилкетон, в уксусной кислоте или смеси этих растворителей. Обычно реакцию можно проводить при температуре от комнатной до 180°С, предпочтительно от комнатной температуры до 140°С, и время реакции обычно может составлять от 1 до 24 часов.
Далее соединение (F) подвергают взаимодействию с 3-этиламиноазетидином, с получением соединения 1 настоящего изобретения.
Данную реакцию можно проводить в растворителе, который не влияет на реакцию, например в ароматическом углеводороде, таком как бензол, толуол или ксилол, спирте, таком как метанол или этанол, простом эфире, таком как тетрагидрофуран, диоксан или моноглим, галогенированном углеводороде, таком как метиленхлорид, хлороформ или четыреххлористый углерод, апротонном полярном растворителе, таком как диметилформамид, диметилсульфоксид или N-метилпирролидон, ацетонитриле или пиридине, в присутствии при необходимости агента, нейтрализующего кислоту, например карбоната натрия, карбоната кальция, триэтиламина или 1,8-диазабицикло[5.4,0]ундецена (DBU), при температуре от комнатной до 160°С. Время реакции может составлять от нескольких минут до 48 часов, предпочтительно от 10 минут до 24 часов. 3-Этиламиноазетин можно использовать в эквимолярном или большем количестве относительно соединения (F), предпочтительно в молярном количестве, 1-5-кратном относительно соединения (F).
Соединение 1 можно перевести в аддитивную соль кислоты или аддитивную соль основания способами, известными в данной области.
Данную реакцию можно проводить в полярном растворителе, например, спирте, таком как метанол или этанол, или воде в присутствии минеральной кислоты, такой как соляная, серная или фосфорная кислота, органической кислоты, такой как муравьиная кислота, уксусная кислота, лимонная кислота, трихлоруксусная кислота, трифторуксусная кислота, фумаровая или малеиновая кислота, органической сульфоновой кислоты, такой как метансульфоновая кислота, бензолсульфоновая кислота, паратолуолсульфоновая кислота, мезитиленсульфоновая кислота или нафталинсульфоновая кислота, основных соединений, таких как гидроксид натрия, гидроксид калия, гидроксид кальция или гидроксид магния, или азотсодержащих органических оснований, таких как аммиак, триметиламин, триэтиламин, трибутиламин, пиридин, N,N-диметиланилин, N-метилпиперидин, N-метилморфолин, диэтиламин, циклогексиламин, прокаин, дибензиламин, N-бензил-β-фенетиламин, 1-эфенамин или N,N’-дибензилэтилендиамин, при комнатной температуре или при нагревании, при необходимости.
Исходное соединение (А) может быть получено, например, по способу, раскрытому в любой из следующих публикаций, или аналогичным способом.
(1) J. Heterocyclic Chem., 22, 1033 (1985)
(2) Liebigs Ann. Chem., 29 (1987)
(3) J. Med. Chem., 31, 991 (1988)
(4) J. Org. Chem., 35, 930 (1970)
(5) JP 62-246541 А
(6) JP 63-26272 А
(7) JP 63-145268 А
(8) J. Med. Chem., 29, 2363 (1986)
(9) J. Fluorin. Chem., 28, 361 (1985)
(10) JP 63-198664 А
(11) JP 63-264461 А
(12) JP 63-104974 А
С другой стороны, реагирующее соединение (С) можно получать желаемым способом. Например, его можно получать замещением аминопроизводным атома галогена, присоединенного к атому углерода 6-членного цикла в соответствии с известной реакцией замещения галоген-амин, описанной в WO 97/11068 А или WO 97/38971 А.
Соединение настоящего изобретения, получаемое, как описано выше, можно выделять и очищать способами, известными в данной области. В зависимости от условий выделения и очистки соединение получают в виде соли или в виде свободной карбоновой кислоты или свободного амина. Данные две формы можно превращать одна в другую, при желании, и соединение настоящего изобретения можно получать в желаемом виде.
Соединение 1, которое имеет 6-амино-3,5-дифторпиридинильную группу в положении 1, этиламиноазетидинильную группу в положении 7 и атом брома в положении 8, и его соли, полученные описанным выше способом, как будет продемонстрировано в испытаниях 1-4, проявляют действие, непредсказуемое, исходя из корреляций структура-активность, принятых на сегодня в отношении производных пиридонкарбоновой кислоты, представленных формулой (I), то есть имеют большой период полураспада в крови при пероральном введении и показывают крайне высокое значение (78%) биологической доступности, рассчитанной из AUC, вплоть до 24-го часа после введения, при сохранении превосходных свойств, таких как чрезвычайно хорошая противомикробная активность и отсутствие фототоксичности, характерной для хинолона. Кроме того, соединение 1 и его соли также обладают превосходными свойствами, оказывая меньшее антигипертензивное действие и побочные эффекты в отношении кожи, такие как сыпь, по сравнению с известными соединениями аналогичных структур.
Соединение 1 и его соли, согласно настоящему изобретению, каждое, можно сформировать в качестве противомикробного агента вместе с фармацевтически приемлемыми носителями в композиции для парентерального введения, например, с помощью инъекции, ректального введения, инстилляции или перорального введения в твердом или жидком виде.
Примеры препаратов для инъекций могут включать фармацевтически приемлемые стерильные водные или неводные растворы, суспензии и эмульсии. Иллюстрациями неводных носителей, разбавителей, растворителей и наполнителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, например оливковое масло, и инъецируемые органические сложные эфиры, например этилолеат. Такие растворы, если необходимо, могут также содержать добавки, такие как консерванты, увлажняющие агенты, эмульгаторы и диспергирующие агенты. Препараты для инъекций можно стерилизовать, например, фильтрованием через бактериальные фильтры или добавлением непосредственно перед применением стерилизующих агентов, самих по себе или в виде стерильных твердых композиций, растворимых в какой-либо другой стерильной среде для инъекций.
К препаратам для введения с помощью инстилляции, если необходимо, помимо соединений настоящего изобретения можно добавлять агенты, способствующие растворению, консерванты, изотонические агенты, загустители и аналогичные.
Примеры твердых препаратов для перорального введения могут включать капсулы, таблетки, пилюли, порошки и гранулы. После составления таких твердых препаратов соединения настоящего изобретения обычно смешивают, по меньшей мере, с одним из инертных экстендеров, например сахарозой, лактозой или крахмалом. При составлении обычных препаратов можно также использовать материалы, отличные от инертных экстендеров, такие как смазывающие вещества (например, стеарат магния). В капсулах, таблетках и пилюлях можно использовать буферы. Таблетки и пилюли могут быть покрыты энтеросолюбильной оболочкой.
Примеры жидких препаратов для перорального введения могут включать фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие обычно используемые инертные разбавители, например воду. В дополнение к таким инертным разбавителям можно также добавлять добавки, такие как смачивающие агенты, эмульгаторы или суспендирующие агенты, подслащающие агенты, приправы и ароматизаторы.
Препараты для ректального применения в дополнение к соединениям настоящего изобретения могут содержать наполнители, такие как масло какао и воск для суппозиториев.
Дозировка каждого соединения настоящего изобретения варьирует в зависимости от свойств соединения, способа применения, необходимого периода лечения и других факторов. Однако обычно дневная доза предпочтительно составляет примерно от 0,1 до 1000 мг/кг, особенно предпочтительно примерно от 0,5 до 100 мг/кг. Кроме того, данную дневную дозу можно вводить 2 или 4 порциями, по желанию.
Примеры
Далее настоящее изобретение будет описано более подробно с помощью примеров и ссылочных примеров.
Ссылочный пример 1. Синтез этил 8-бром-1-[6-(трет-бутиламино)-3,5-дифторпиридин-2-ил]-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилата.
К раствору в хлороформе (5 мл), в котором растворен этил 3-этокси-2-(3-бром-2,4,5-трифторбензол) акрилат, полученный известным в данной области способом из этил 3-бром-2,4,5-трифторбензоилацетата (1,32 г), добавляют 2-амино-6-(трет-бутиламино)-3,5-дифторпиридин, при ТСХ-контроле реакции до тех пор, пока не завершится превращение в аминоакрилатное производное. Реакционную смесь концентрируют при пониженном давлении, получая желтый твердый остаток. К остатку добавляют безводный карбонат калия (1,2 г) и N,N-диметилформамид (2 мл), и смесь перемешивают при 90°С в течение 15 минут. Смеси дают охладиться. Добавляют хлороформ (30 мл) и дистиллированную воду (300 мл) и дают смеси разделиться на слои. Хлороформный слой дважды промывают дистиллированной водой (300 мл), сушат над безводным сульфатом магния, концентрируют при пониженном давлении и отставляют. Осадок собирают фильтрованием и промывают последовательно этанолом и диизопропиловым эфиром в указанном порядке, получая указанное в заголовке соединение (1,41 г) в виде бесцветного порошка.
Температура плавления: 198-203°С.
1H ЯМР (СDСl3) δ:
1,38 (с, 9Н), 1,40 (т, J=1 Гц, 3Н), 4,04 (кв, J=7 Гц, 2Н), 4,71 (ш.с., 1Н), 7,20 (дд, J=8 Гц, 10 Гц, 1Н), 8,36 (дд, J=9 Гц, 10 Гц, 1Н), 8,54 (с, 1Н).
Ссылочный пример 2. Синтез 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты.
Этил 8-бром-1-[6-(трет-бутиламино)-3,5-дифторпиридин-2-ил]-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилат (1,38 г) добавляют к жидкой смеси 12% соляной кислоты (3,5 мл) и уксусной кислоты (3,5 мл) и смесь нагревают в течение 5 часов при перемешивании и кипении с обратным холодильником. После добавления дистиллированной воды (5 мл) смеси дают охладиться. Осадок собирают фильтрованием и промывают последовательно этанолом и диизопропиловым эфиром в данном порядке, получая указанное в заголовке соединение (1,10 г) в виде бесцветного порошка.
Температура плавления: 272-278°С.
1H-ЯМР (D6-ДМCO) δ:
6,80 (с, 2Н), 7,99 (т, J=9 Гц, 1Н), 8,38 (т, J=9 Гц, 1H), 8,93 (с, 1Н).
Пример 1. Синтез 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-7-(3-этиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединение 1).
3-Этиламиноазетидин (700 мг), 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту (1,5 г), N-метилпирролидин (2,0 г) и диметилсульфоксид (4,5 г) объединяют и смесь нагревают при перемешивании при 40°С в течение 24 часов. После того, как смеси дают охладиться, добавляют изопропиловый эфир (10 мл), смесь перемешивают и удаляют прозрачный слой с поверхности смеси. Еще раз повторяют ту же процедуру и остаток концентрируют при пониженном давлении. Добавляют этанол (5 мл) и смесь нагревают при перемешивании при 70°С в течение 30 минут. Выпавшее в осадок твердое вещество собирают фильтрованием. Получают указанное в заголовке соединение (1,38 г).
Внешний вид: бесцветный порошок.
Температура плавления: 195-196°С.
1H ЯМР (D6-ДМСО) δ:
0,99 (т, J=7 Гц, 3Н), 2,48 (кв, J=7 Гц, 2Н), 4,05-4,15 (м, 2Н), 4,35-4,42 (м, 1H), 4,60-4,69 (м, 2Н), 6,74 (ш.с. 2Н), 7,88 (д, J=14 Гц, 1H), 7,93 (т, J=9 Гц, 1H), 8,69 (с, 1H).
Пример 2. Синтез малеата 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-7-(3-этиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (соединение 2).
1-(6-Амино-3,5-дифторпиридин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту (1,38 г) добавляют к этанолу (13 мл) и к смеси постепенно добавляют малеиновую кислоту (400 мг). Смесь нагревают при перемешивании при 70°С в течение 5 часов. После того, как смеси дают охладиться, твердое вещество собирают фильтрованием. Твердое вещество промывают этанолом. Получают указанное в заголовке соединение (1,33 г).
Внешний вид: бесцветный порошок.
Температура плавления: 196-199°С.
1H ЯМР (D6-ДМСО) δ:
1,16 (т, J=7 Гц, 3Н), 2,93 (кв, J=7 Гц,2Н), 3,99-4,06 (м, 1Н), 4,41-4,48 (м, 1Н), 4,50-4,56 (м, 1Н), 4,67-4,74 (м, 1Н), 4,74-4,82 (м, 1Н), 6,02 (с, 2Н), 6,76 (ш.с. 2Н), 7,95 (т, J=9 Гц, 1Н), 7,97 (д, J=14 Гц, 1Н), 8,75 (с, 1Н).
Испытания
В испытаниях 1-4 будут описаны результаты испытаний соединений настоящего изобретения на противомикробное действие, фототоксичность и in vivo распределение. В качестве сравнительных соединений используют следующие соединения, раскрытые в WO 97/11068 А и промышленно доступные, ципрофлоксацин (CPFX) и левофлоксацин (LVFX).
Сравнительное соединение 1:
1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-7-(3-метиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота.
Сравнительное соединение 2:
1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-7-(3-этиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота.
CPFX:
1-циклопропил-6-фтор-7-(1-пиперазинил)-1,4-дигидро-4-оксохинолин-3-карбоновая кислота.
LVFX:
S(-)-9-фтор-2,3-дигидро-3-метил-10-(4-метил-1-пиперазинил)-7-оксо-7Н-пиридо[1,2,3-dе][1,4]бензоксазин-6-карбоновая кислота.
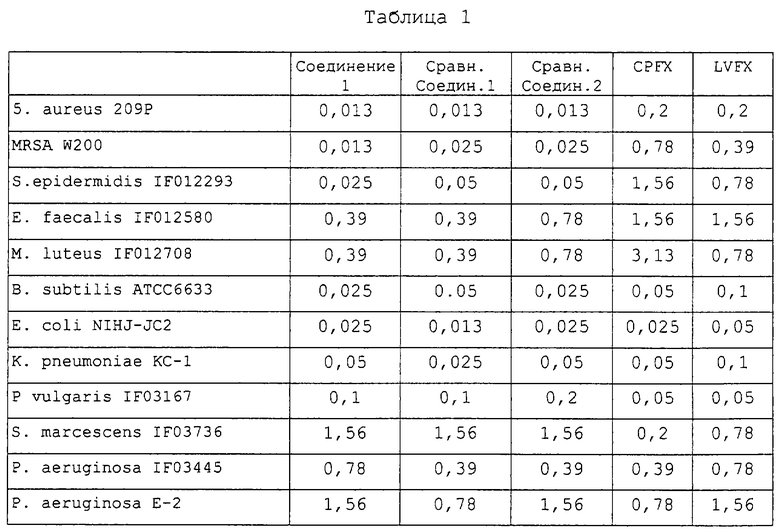
(1) Противомикробное действие
Определяют минимальные ингибирующие рост концентрации (MIC: мкг/мл) в соответствии со стандартным способом Japan Society of Chemotherapy [Chemotherapy, 29(1), 76 (1981)].
Результаты представлены в табл.1.
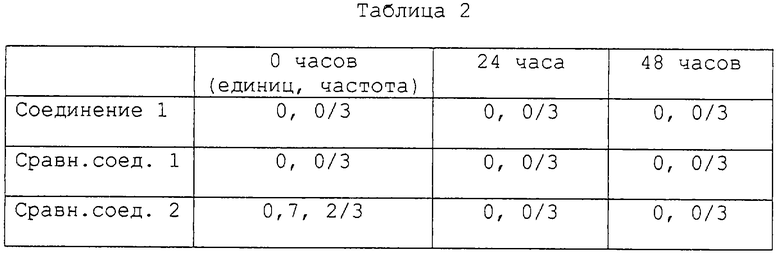
(2) Испытание на фототоксичность
Испытание на фототоксичность проводят по следующей методике. Самкам мышей ICR (5-6-недельного возраста) вводят внутривенно исследуемые соединения (40 мг/кг/10 мл) и подвергают их в течение 4 часов ультрафиолетовому облучению (от 320 до 400 нм, 1,8 мВ/см2/с). Наблюдают аномалии на их ушах в момент времени 0 ч (непосредственно после облучения) и через 24 и 48 часов.
Аномалии на ушах классифицируют по следующим стандартам:
нет аномалии (0 единиц), слабая эритема (1 единица), средняя эритема (2 единицы) и сильная эритема или отек (3 единицы).
Результаты представлены в табл.2.
(3) Противобактериальное действие на клинически выделенные устойчивые к хинолону пневмококки(pneumococci)
Используя планшеты с агаром с добавлением 5% дефибринированной овечьей крови, определяют минимальные ингибирующие рост концентрации (MIC: мкг/мл) относительно некоторых пневмококков в соответствии со стандартньм способом Japan Society of Chemotherapy [Chemotherapy, 29(1), 76 (1981)].
Результаты представлены в табл.3.

Из результатов табл.1-3 следует, что соединения настоящего изобретения демонстрируют противомикробные активности, сравнимые с активностями сравнительных соединений или выше, а также не являются фототоксичными.
(4) Фармакокинетическое исследование in vivo
Проводилось исследование абсорбции и экскреции соединений настоящего изобретения у собак.
0,5% Суспензию одного из исследуемых соединений в метилцеллюлозе (10 мг/мл/кг) принудительно вводят перорально 2-4-летним самцам гончих собак, содержавшимся в условиях голодания в течение 16-17 часов. После введения берут пробы крови через 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа и получают образцы сыворотки. Для определения скорости экскреции с мочой собирают также пробы мочи вплоть до 24-го часа после введения. Концентрации исследуемого соединения в пробах сыворотки и мочи измеряют по способу с применением бумажных дисков, используя в качестве испытываемой бактерии Bacillus subtilis ATCC6633, и классифицируют абсорбцию и экскрецию. Полученные таким образом результаты представлены в табл.4.
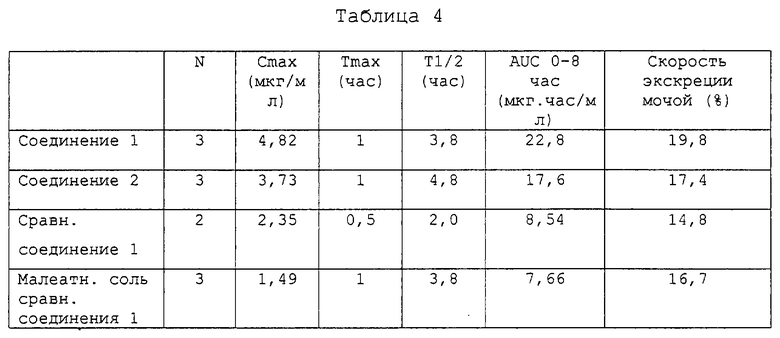
Данные табл.4 подтверждают, что соединения настоящего изобретения обладают in vivo фармакокинетическими характеристиками, существенно улучшенными относительно сравнительных соединений.
Промышленная применимость
Соединение 1 и его соли согласно настоящему изобретению обладают характерными свойствами, а именно при пероральном введении они демонстрируют длительный период полураспада в крови и исключительно высокую биологическую доступность при сохранении таких свойств, как исключительно высокие противомикробные эффекты и низкая токсичность. Соединение 1 и его соли также обладают превосходными свойствами, оказывая низкое антигипертензивное действие и побочное действие на кожу, такое как сыпь, по сравнению с известными соединениями аналогичных структур. Следовательно, соединение 1 и его соли можно широко применять в качестве профилактических и терапевтических средств против различных инфекционных заболеваний человека и животных, а также как лекарственные препараты для рыб, сельскохозяйственные химикаты, пищевые консерванты и аналогичные. Кроме того, ожидается, что соединение 1 и его соли оказывают противовирусное действие, особенно против ВИЧ (вирус иммунодефицита человека), и считается, что они эффективны для профилактики и лечения СПИД.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ | 2005 |
|
RU2379304C2 |
| ПРОИЗВОДНОЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕГО СОЛЬ | 1996 |
|
RU2177945C1 |
| ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ И АНТИБАКТЕРИАЛЬНОЕ СРЕДСТВО | 1996 |
|
RU2167873C2 |
| АМИНОСОЕДИНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ НОВЫХ ПРОИЗВОДНЫХ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛЕЙ | 1996 |
|
RU2171252C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛОНОВЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2546667C2 |
| СПОСОБ ПОЛУЧЕНИЯ (1-ЦИКЛОПРОПИЛ-6-ФТОР-1,4-ДИГИДРО-8-МЕТОКСИ-7-[(4AS,7AS)-ОКТАГИДРО-6Н-ПИРРОЛО[3,4-В]ПИРИДИН-6-ИЛ]-4-ОКСО-3-ХИНОЛИНКАРБОНОВОЙ КИСЛОТЫ | 2013 |
|
RU2641699C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 8-ХЛОРХИНОЛОНА | 1991 |
|
RU2049778C1 |
| 3-ГАЛОГЕН-6-(АРИЛ)-ИМИНОТЕТРАГИДРОПИКОЛИНАТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2010 |
|
RU2527954C2 |
| ПРОИЗВОДНОЕ 6-(ГЕТЕРОЦИКЛЗАМЕЩЕННЫЙ БЕНЗИЛ)-4-ОКСОХИНОЛИНА И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ИНТЕГРАЗЫ ВИЧ | 2007 |
|
RU2399616C1 |
| СПИРОСОЕДИНЕНИЕ ИЛИ ЕГО СОЛИ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ОБЛАДАЮЩАЯ ПРОТИВОМИКРОБНОЙ АКТИВНОСТЬЮ | 1989 |
|
RU2094432C1 |
Изобретение относится к 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-7-(3-этиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигид-рохинолин-3-карбоновой кислоте или к ее соли, а также к противомикробному лекарственному средству и композиции на основе этого соединения или его соли. Технический результат – получение новой 1-(6-амино-3,5-дифторпиридин-2-ил)-8-бром-7-(3-этиламиноазетидин-1-ил)-6-фтор-4-оксо-1,4-дигид-рохинолин-3-карбоновой кислоты или ее соли и лекарственного средства на их основе в целях профилактики и лечения различных инфекционных заболеваний человека и животных. 4 н.п. ф-лы, 4 табл.
| WO 9711068 A1, 27.03.1997 | |||
| Способ получения производных хинолинкарбоновой кислоты или их гидратов | 1981 |
|
SU1015827A3 |
| Способ получения производных 7-(пиррол-1-ил)-1-этил-1,4-дигидро-4-оксохинолеин-3-карбоновой и 7-(пиррол-1-ил)-1-этил-1,4-дигидро-4-оксо-(1,8-нафтиридин)-3-карбоновой кислот | 1984 |
|
SU1322980A3 |
| US 5910498 A, 08.06.1999. | |||

