ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к области получения противоинфекционных соединений. Более конкретно, настоящее изобретение относится к получению семейства хинолоновых соединений, пригодных в качестве противоинфекционных средств. Настоящее изобретение относится к способу получения хинолонового соединения, согласно которому получают менее чем приблизительно 0,40% димерной примеси хинолона.
Предшествующий уровень техники
С момента открытия пенициллина в 1920-ых и стрептомицина в 1940-ых получено или специально сконструировано много новых соединений для применения в качестве антибактериальных средств. Когда-то считали, что применяя данные терапевтические средства можно бороться или полностью искоренить инфекционные заболевания. Появились устойчивые штаммы грамположительных бактерий, такие как устойчивые к метициллину стафилококки, устойчивые к пенициллину стрептококки и устойчивые к ванкомицину энтерококки, которые могут вызывать серьезные и даже смертельные случаи у пациентов, инфицированных данными устойчивыми бактериями. Появились бактерии, которые являются устойчивыми к макролидным антибиотикам, т.е. антибиотикам на основе 14-16-членного лактонового кольца. Кроме того, обнаружены устойчивые штаммы грамотрицательных бактерий, такие как H. influenzae и M. catarrhalis. См., например, F. D. Lowry, "Antimicrobial Resistance: The Examples of Staphylococcus aureus," J. Clin. Invest., 2003, 7/7(9), 1265-1273 и Gold, H.S. and Moellering, R.C., Jr., "Antimicrobial-Drug Resistance," N. Engl. J. Med, 1996, 335, 1445-53.
Несмотря на данную проблему повышения устойчивости к антибиотикам новые основные классы антибиотиков для клинического применения не разрабатывались с момента санкционирования в Соединенных Штатах в 2000 году антибиотика, содержащего оксазолидиноновое кольцо, N-[[(5S)-3-[3-фтор-4-(4-морфолинил)фенил]-2-оксо-5-оксазолидинил]метилацетамида, который известен как линезолид и продается под торговым названием Zyvox® (см. соединение A). См. R.C. Moellering, Jr., "Linezolid: The First Oxazolidinone Antimicrobial," Annals of Internal Medicine, 2003, 735(2), 135-142.
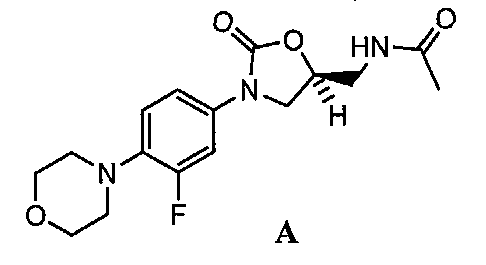
Линезолид является одобренным для применения в качестве антибактериального средства, активного против грамположительных организмов. К несчастью, уже сообщалось об устойчивых к линезолиду организмах. См. Tsiodras et al., Lancet, 2001, 358, 207; Gonzales et al., Lancet, 2001, 357, 1 179; Zurenko et al., Proceedings Of The 39th Annual lnterscience Conference On Antibacterial Agents and Chemotherapy (ICAAC); San Francisco, CA, USA, (September 26-29, 1999).
Несмотря на вышеизложенное существует постоянная необходимость в новых противоинфекционных средствах и в способах их получения.
Краткое описание фигур
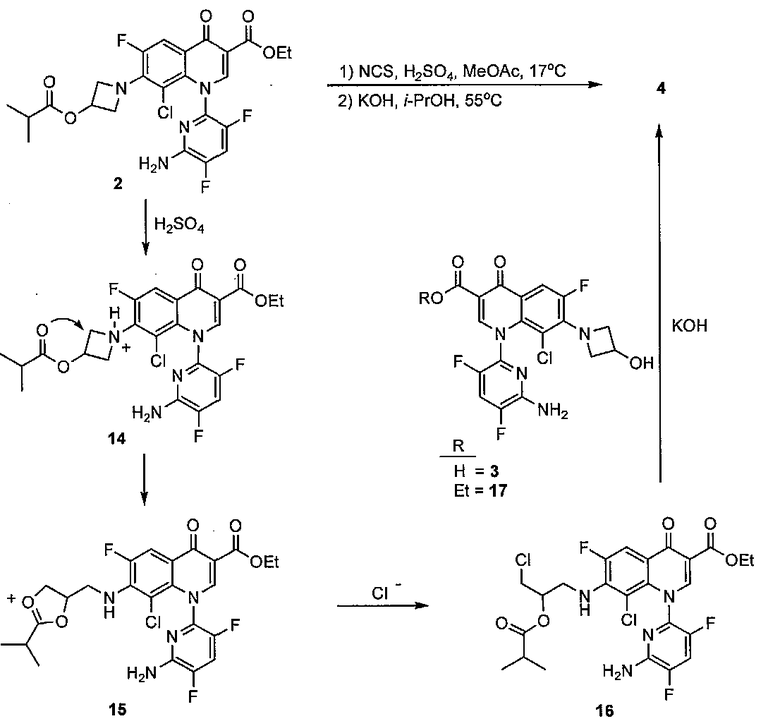
На Фиг.1 показаны предсказанные профили для количества димерной примеси 4, когда этилацетат (EtOAc) является растворителем. Это основано на первоначальном дизайне экспериментов. Центральная линия каждого графика показывает прогнозируемые значения и две линии, фланкирующие центральную линию, представляют приблизительно +95 процентов степень достоверности. Горизонтальная пунктирная линия показывает концентрацию димера 4 0,235094 процентов. Вертикальные пунктирные линии показывают переменные для 1,05 эквивалентов N-хлорсукцинимида (NCS), 3,5 мольных процентов серной кислоты, 17°C, 0,05 процентов содержания воды в растворителе и скорости добавления NCS 0,1 объема в минуту. 95-процентный предел достоверности представляет собой ±0,040991 для величин, показанных в предыдущем предложении.
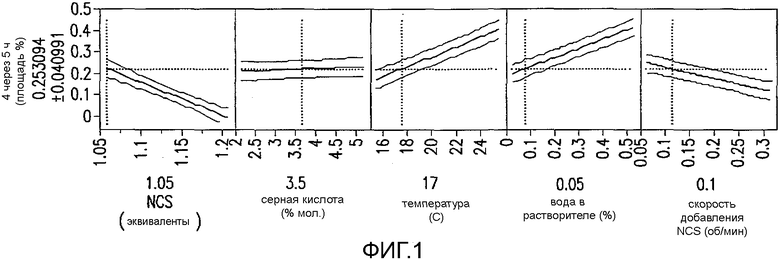
На Фиг.2 показано влияние H2SO4 и времени на концентрацию димерной примеси 4.
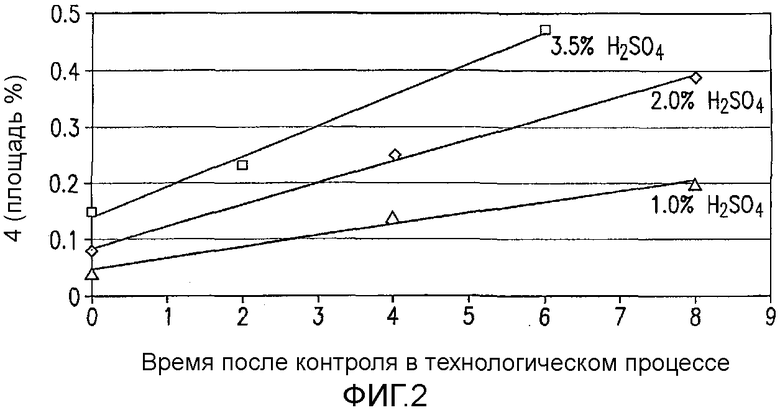
На Фиг.3 показан худший вариант для предсказанного профиля для количества димерной примеси 4 для устойчивости DoE, т.е. для второго дизайна экспериментов. Центральная линия каждого графика показывает прогнозируемые значения и две линии, фланкирующие центральную линию, представляют приблизительно ±95 процентов степень достоверности. Горизонтальная пунктирная линия показывает концентрацию димера 4 0,1045 процентов. Вертикальные пунктирные линии показывают переменные для 1,04 эквивалентов N-хлорсукцинимида (NCS), 21°C, скорости добавления NCS 30 минут и 0,8 мольных процентов серной кислоты. 95-процентный предел достоверности представляет собой +0,009339 для величин, показанных в предыдущем предложении.
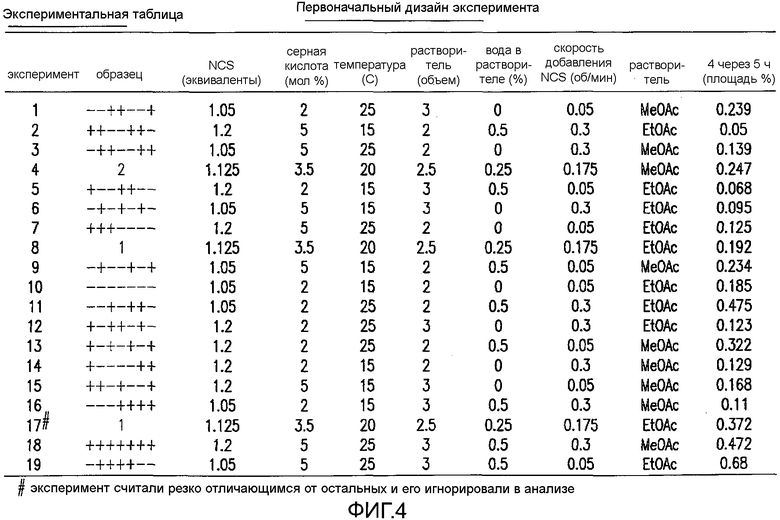
На Фиг.4 показан первоначальный дизайн экспериментальной таблицы экспериментов.
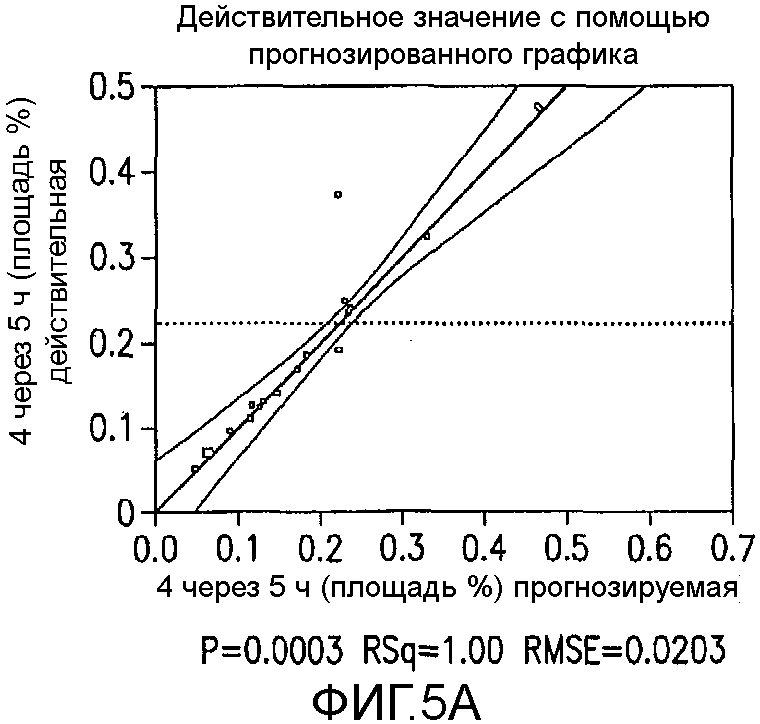
На Фиг.5a показаны истинные значения с помощью предсказанного графика для первоначального дизайна экспериментов фиг.4.
На Фиг.5b показан итог аппроксимации для первоначального дизайна экспериментов фиг.4.
На Фиг.5c показан анализ дисперсии для первоначального дизайна фиг.4.
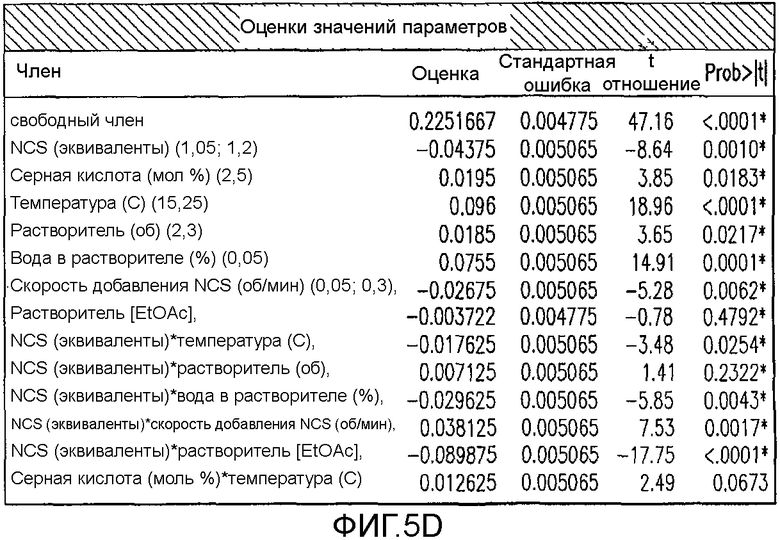
На Фиг.5d показаны оценки значения параметров для первоначального дизайна экспериментов фиг.4.
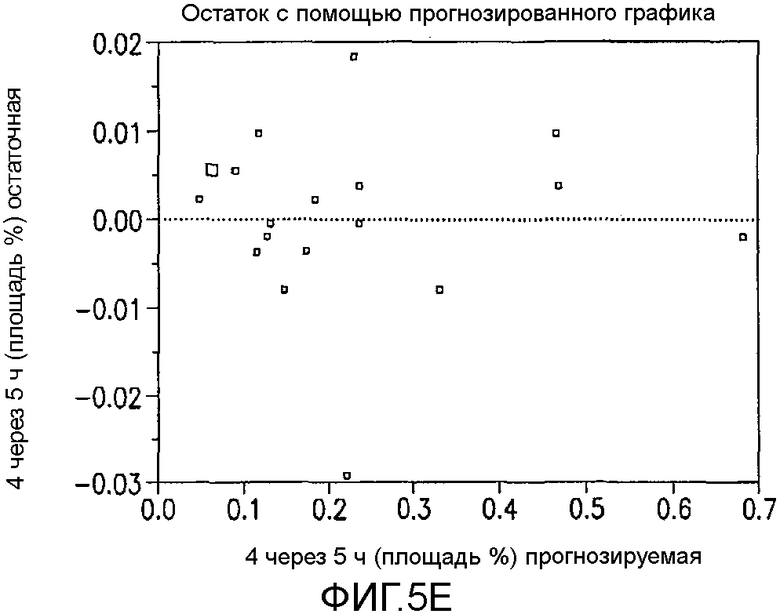
На Фиг.5e показан остаток с помощью предсказанного графика для первоначального дизайна экспериментов фиг.4.
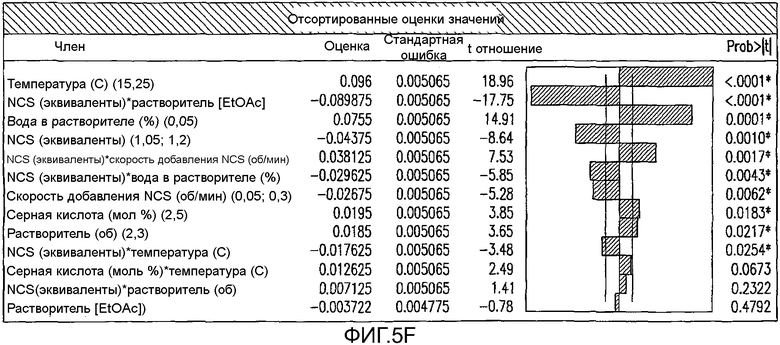
На Фиг.5f показаны отсортированные оценки значения параметров для первоначального дизайна экспериментов фиг.4.
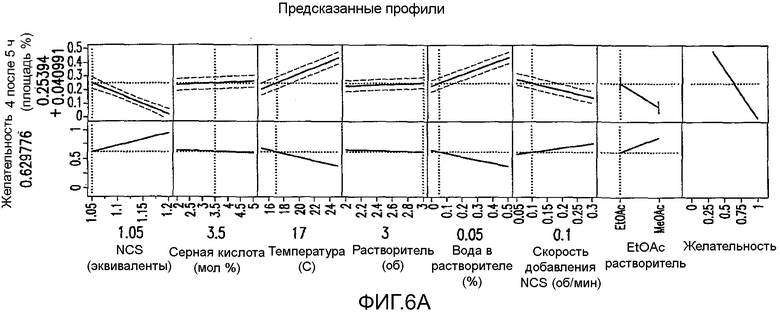
На Фиг.6a показаны предсказанные профили для первоначального дизайна экспериментов фиг.4.
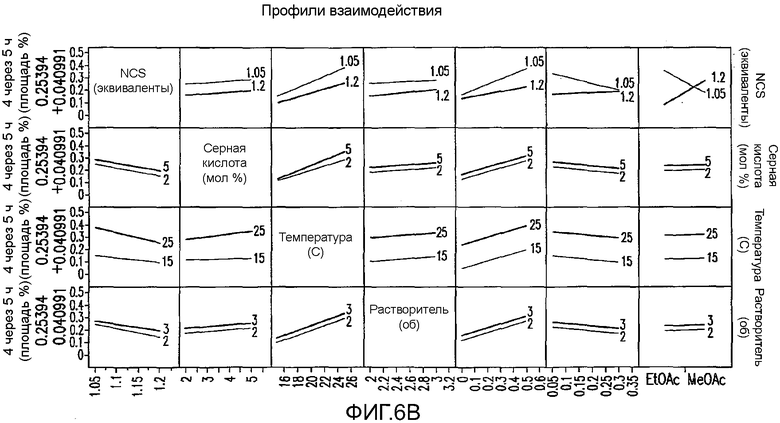
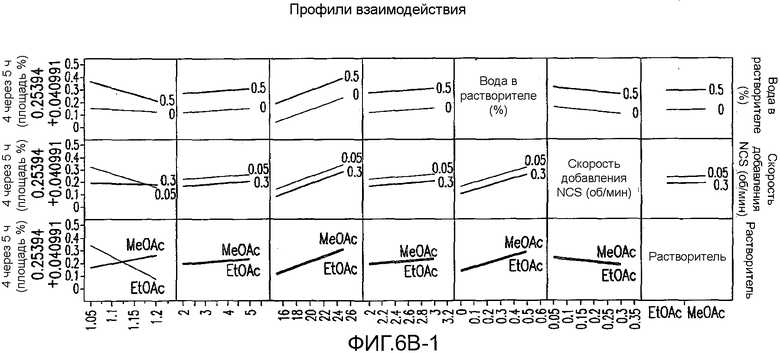
На Фиг.6b показаны профили взаимодействия для первоначального дизайна экспериментов фиг.4.
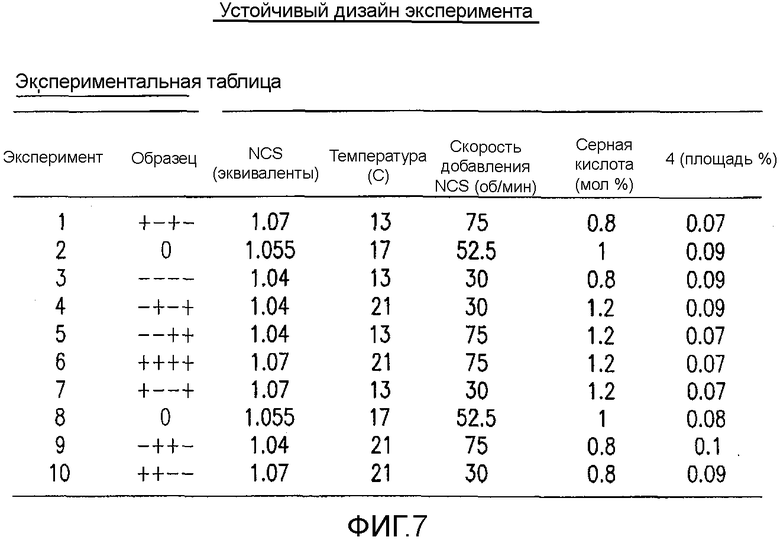
На Фиг.7 показан устойчивый дизайн экспериментальной таблицы экспериментов для второго дизайна экспериментов.
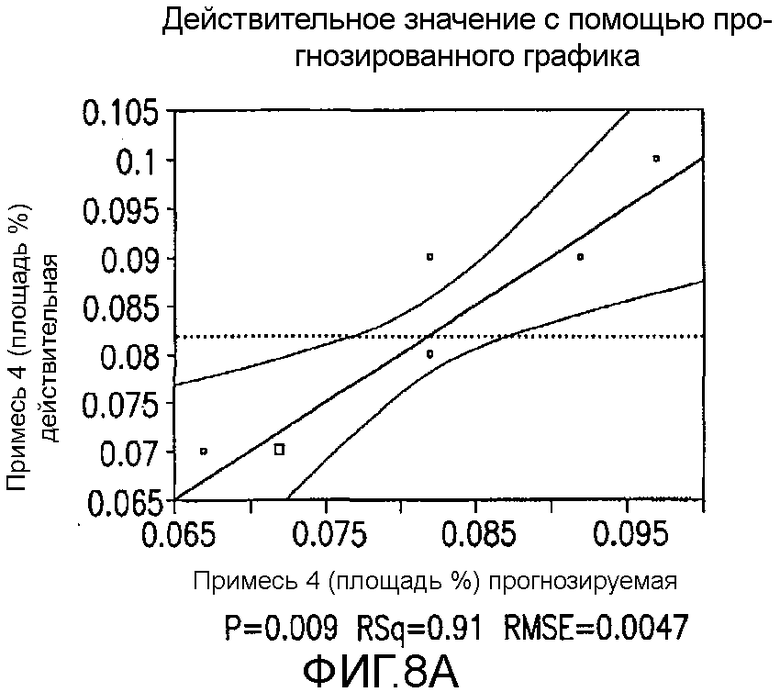
На Фиг.8a показаны истинные значения с помощью предсказанного графика для второго дизайна экспериментов фиг.7.
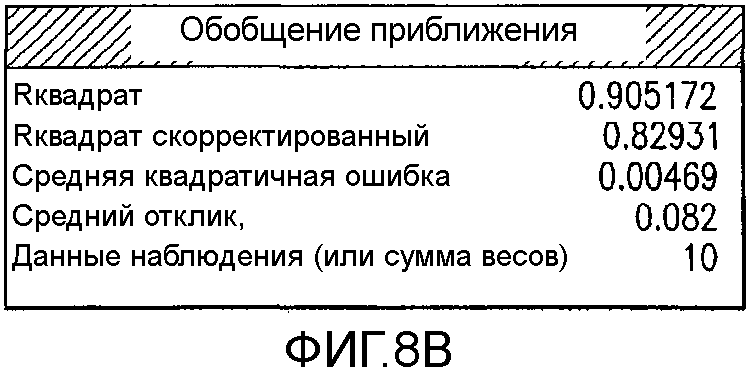
На Фиг.8b показан итог аппроксимации для второго дизайна экспериментов фиг.7.
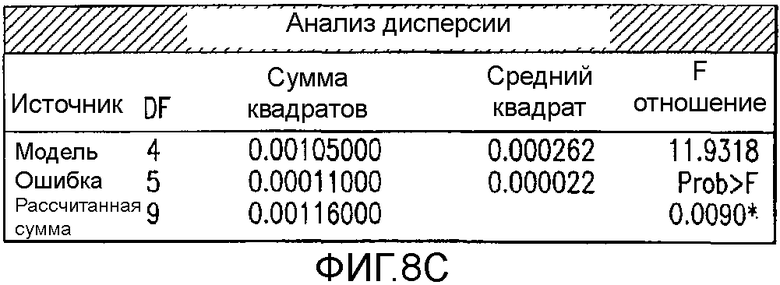
На Фиг.8c показан анализ дисперсии для второго дизайна экспериментов фиг.7.
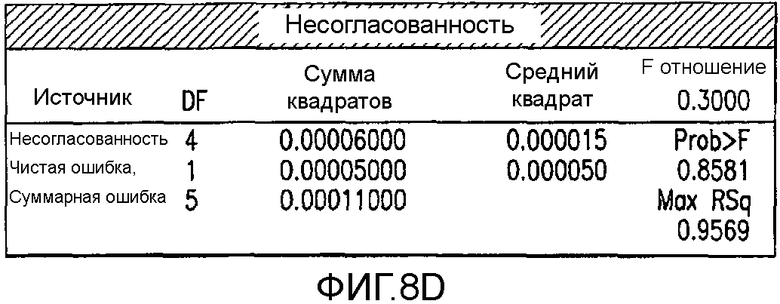
На Фиг.8d показана рассогласованность для второго дизайна экспериментов фиг.7.
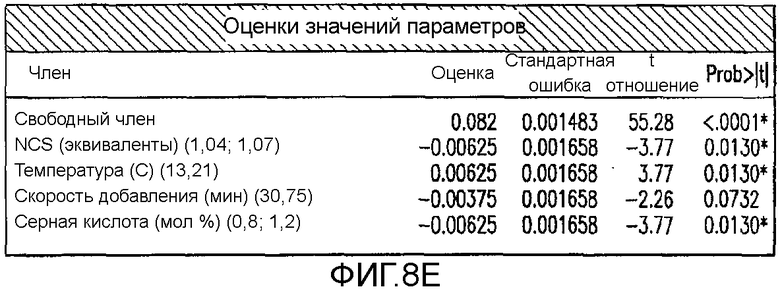
На Фиг.8e показаны оценки значения параметров для второго дизайна экспериментов фиг.7.
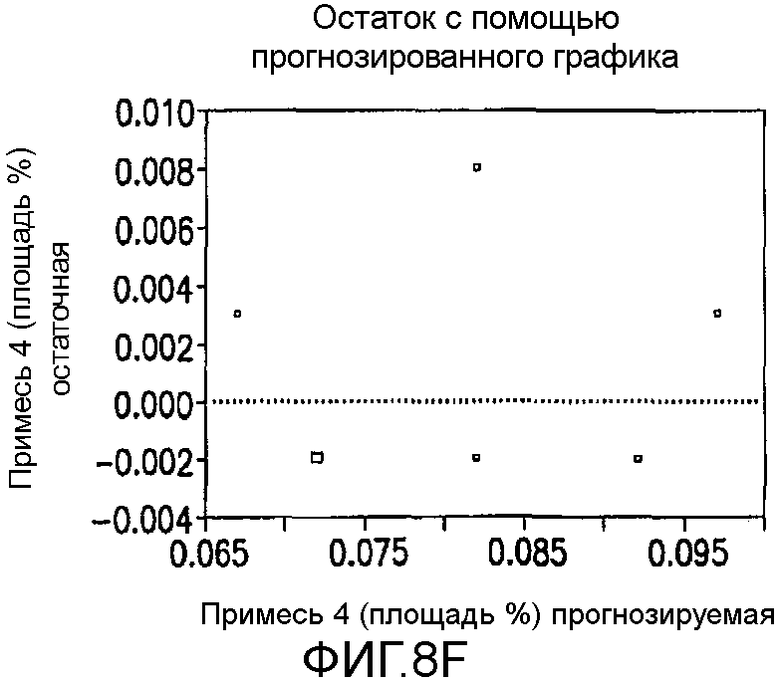
На Фиг.8f показан остаток с помощью предсказанного графика для второго дизайна экспериментов фиг.7.
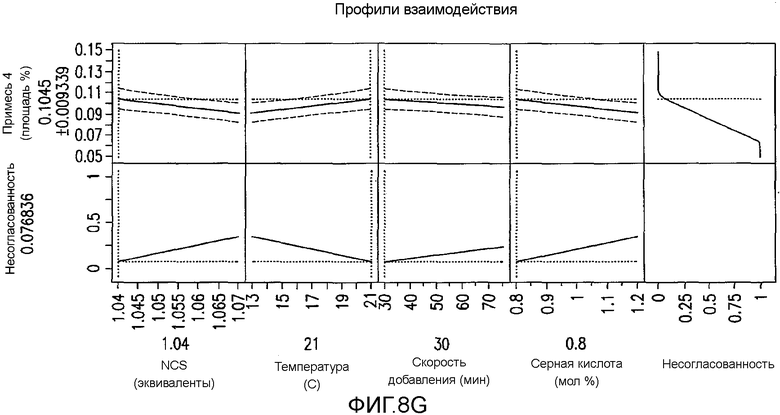
На Фиг.8g показаны предсказанные профили для второго дизайна экспериментов фиг.7.
Сущность настоящего изобретения
Настоящее изобретение относится к области получения противоинфекционных соединений. Более конкретно, настоящее изобретение относится к получению семейства хинолоновых соединений, пригодных в качестве противоинфекционных средств.
Настоящее изобретение относится к способу получения хинолонового соединения, включающему стадию взаимодействия дехлорхинолонового соединения, или его фармацевтически приемлемой соли, или эфира с хлорирующим агентом и кислотой, в котором получают, менее чем приблизительно 0,40% в расчете на проценты площади, как определено количественно аналитической ВЭЖХ, димерной примеси хинолона.
В других вариантах осуществления настоящее изобретение относится к способу, в котором дэхлорхинолоновое соединение представляет собой 1-(6-амино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-гидрокси-азетидин-1-ил)-4-оксо-l,4-дигидрохинолин-3-карбоновую кислоту, или ее фармацевтически приемлемую соль, или эфир, хинолоновое соединение представляет собой 1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-6-фтор-7-(3-гидроксиазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту, или ее фармацевтически приемлемую соль, или эфир.
В других вариантах осуществления настоящее изобретение относится к способу, в котором димерная примесь представляет собой соединение 1-амино-3-(азетидин-3-илокси)пропан-2-олбис(N,N'-хинолонкарбоновую кислоту), или ее фармацевтически приемлемую соль, или эфир. В других вариантах осуществления настоящее изобретение относится к способу, в котором димерная примесь представляет собой моноэфир. В других вариантах осуществления настоящее изобретение относится к способу, в котором димерная примесь представляет собой диэфир.
В других вариантах осуществления настоящее изобретение относится к способу, в котором хлорирующий агент представляет собой N-хлорсукцинимид.
В других вариантах осуществления настоящее изобретение относится к способу, в котором кислоту выбирают из группы, состоящей из серной кислоты, хлористоводородной кислоты, бромистоводородной кислоты, фосфорной кислоты, трифторуксусной кислоты, трифторметансульфокислоты, метансульфокислоты, п-толуолсульфокислоты или перхлорной кислоты и их смесей.
В других вариантах осуществления настоящее изобретение относится к способу, в котором кислота представляет собой серную кислоту.
В других вариантах осуществления настоящее изобретение относится к способу, в котором взаимодействие проводят при температуре от приблизительно 0°C до приблизительно 30°C.
В других вариантах осуществления настоящее изобретение относится к способу, в котором взаимодействие проводят при температуре от приблизительно 15°C до приблизительно 25°C.
В других вариантах осуществления настоящее изобретение относится к способу, в котором взаимодействие проводят при температуре от приблизительно 13°C до приблизительно 21°C.
В других вариантах осуществления настоящее изобретение относится к способу, в котором молярное отношение N-хлорсукцинимида к дэхлорхинолону составляет более чем приблизительно 1.
В других вариантах осуществления настоящее изобретение относится к способу, в котором молярное отношение N-хлорсукцинимида к дэхлорхинолону составляет от приблизительно 1,05 до 1,2.
В других вариантах осуществления настоящее изобретение относится к способу, в котором молярное отношение N-хлорсукцинимида к дэхлорхинолону составляет от приблизительно 1,04 до 1,07.
В других вариантах осуществления настоящее изобретение относится к способу, в котором молярное отношение серной кислоты к дэхлорхинолону составляет от приблизительно 0,005 до приблизительно 0,05.
В других вариантах осуществления настоящее изобретение относится к способу, в котором молярное отношение серной кислоты к дэхлорхинолону составляет от приблизительно 0,007 до приблизительно 0,02.
В других вариантах осуществления настоящее изобретение относится к способу, в котором молярное отношение серной кислоты к дэхлорхинолону составляет от приблизительно 0,008 до приблизительно 0,012.
В других вариантах осуществления настоящее изобретение относится к способу, в котором применяют эфир ацетата в качестве растворителя.
В других вариантах осуществления настоящее изобретение относится к способу, в котором эфир ацетата выбирают из группы, состоящей из метилацетата, этилацетата и их смесей.
В других вариантах осуществления настоящее изобретение относится к способу, в котором упомянутый эфир ацетата представляет собой метилацетат.
В других вариантах осуществления настоящее изобретение относится к способу, включающему дополнительную стадию взаимодействия хинолонового соединения с основанием.
В других вариантах осуществления настоящее изобретение относится к способу, в котором основание представляет собой гидроксид.
В других вариантах осуществления настоящее изобретение относится к способу, в котором гидроксид выбирают из группы, состоящей из гидроксида натрия, гидроксида калия, гидроксида лития, гидроксида бария и их смесей.
В других вариантах осуществления настоящее изобретение относится к способу, в котором гидроксид представляет собой гидроксид калия.
В других вариантах осуществления настоящее изобретение относится к способу, в котором применяют смесь C1-C6 спирта и воды в качестве растворителя.
В других вариантах осуществления настоящее изобретение относится к способу, в котором C1-C6 спирт представляет собой изопропанол.
В других вариантах осуществления настоящее изобретение относится к способу, в котором способ представляет собой способ в коммерческом масштабе.
В других вариантах осуществления настоящее изобретение относится к композиции, содержащей хинолоновое соединение или его соль или эфир, содержащий менее чем приблизительно 0,40% димерной примеси хинолонового соединения.
В других вариантах осуществления настоящее изобретение относится к композиции, в которой хинолоновое соединение представляет собой 1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-6-фтор-7-(3-гидрокси-азетидин-1-ил)-4-оксо-l,4-дигидрохинолин-3-карбоновую кислоту, или ее фармацевтически приемлемую соль, или эфир.
В других вариантах осуществления настоящее изобретение относится к композиции, в которой димерная примесь представляет собой 1-амино-3-(азетидин-3-илокси)пропан-2-олбис(N,N'-хинолонкарбоновую кислоту), или ее фармацевтически приемлемую соль, или эфир.
В других вариантах осуществления настоящее изобретение относится к композиции, в которой композиция представляет собой композицию в коммерческом масштабе.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,35%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,30%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,25%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,20%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,15%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,10%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,05%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,04%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,03%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой димерная примесь составляет менее чем приблизительно 0,02%.
В других вариантах осуществления настоящее изобретение относится к способу или композиции, в которой упомянутая димерная примесь составляет менее чем приблизительно 0,01%.
Подробное описание настоящего изобретения
Хинолоны
Способы и композиции настоящего изобретения включают хинолоновое соединение.
Хинолоновые соединения, такие как производные пиридонкарбоновой кислоты, пригодные в настоящем изобретении, описывают, включая их получение, формулирование и применение, в патенте США № 6156903, Yazaki et al., опубликованном 5 декабря 2000 и его список замеченных опечаток 13 ноября 2001 и 11 декабря 2001; в патенте США № 6133284, Yazaki et al., опубликованном 17 октября 2000; в патенте США № 5998436, Yazaki et al., опубликованном 7 декабря 1999 и его список замеченных опечаток 23 января 2001, 30 октября 2001 и 17 декабря 2002; в PCT заявке № WO 2006/1 10815, Abbott Laboratories, опубликованной 1 октября 2006; в PCT заявке № WO 2006/042034, Abbott Laboratories, опубликованной 20 апреля 2006, в PCT заявке № WO 2006/015194, Abbott Laboratories, опубликованной 9 февраля 2006; в PCT заявке № WO 01/34595, Wakunaga Pharmaceutical Co., Ltd., опубликованной 17 мая 2001, и в PCT заявке № WO 97/1 1068, Wakunaga Pharmaceutical Co., Ltd., опубликованной 27 марта 1997.
Производные пиридонкарбоновой кислоты настоящего изобретения включают соединения, соответствующие следующей структуре. (Производное 1 пиридонкарбоновой кислоты)
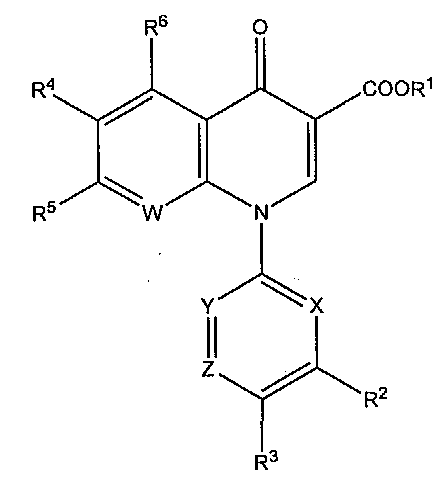
Производное 1 пиридонкарбоновой кислоты, в которой R1 представляет собой атом водорода или защитную группу карбоксильной функции; R2 представляет собой гидроксильную группу, низшую алкоксигруппу, или замещенную, или незамещенную аминогруппу; R3 представляет собой атом водорода или атом галогена; R4 представляет собой атом водорода или атом галогена; R5 представляет собой атом галогена или необязательно замещенную насыщенную циклическую аминогруппу; R6 представляет собой атом водорода, атом галогена, нитрогруппу или необязательно защищенную аминогруппу; X, Y и Z могут быть одинаковыми или различными и соответственно представлять собой атом азота, CH или CR7 (в которой R7 представляет собой низшую алкильную группу, атом галогена или цианогруппу), при условии, что по меньшей мере один из X, Y и Z представляет собой атом азота, и W представляет собой атом азота или CR8 (в которой R8 представляет собой атом водорода, атом галогена или низшую алкильную группу), и при условии, что когда R1 представляет собой атом водорода, R2 представляет собой аминогруппу, R3 и R4 представляют собой атом фтора, R6 представляет собой атом водорода, X представляет собой атом азота, Y представляет собой CR7 (в которой R7 представляет собой атом фтора), Z представляет собой CH, и W представляет собой CR8 (в которой R8 представляет собой атом хлора), тогда R5 не представляет собой 3-гидроксиазетидин-1-ильную группу или их фармацевтически приемлемую соль, эфир или пролекарство.
Как описано выше, когда R1 представляет собой защитную группу карбоксильной функции, она может представлять собой любой остаток эфира карбоновой кислоты, который относительно легко расщепляется, давая соответствующую свободную карбоксильную группу. Примеры защитных групп карбоксильной функции включают защитные группы, которые отщепляются гидролизом, каталитическим восстановлением и другими обработками в мягких условиях, такие как низшие алкильные группы, такие как метильная группа, этильная группа, н-пропильная группа, изопропильная группа, н-бутильная группа, изобутильная группа, трет-бутильная группа, пентильная группа, гексильная группа и гептильная группа; низшие алкенильные группы, такие как винильная группа, аллильная группа, 1-пропенильная группа, бутенильная группа, пентенильная группа, гексенильная группа и гептенильная группа; аралкильные группы, такие как бензильная группа; и арильные группы, такие как фенильная группа и нафтильная группа; и группы, которые можно легко удалить в несколько этапов, такие как низшие алканоилокси низшие алкильные группы, такие как ацетоксиметильная группа и пивалоилоксиметильная группа; низшие алкоксикарбонилокси низшие алкильные группы, такие как метоксикарбонилоксиметильная группа и 1-этоксикарбонилоксиэтильная группа; низшая алкоксиметильная группа, такая как метоксиметильная группа; лактонильная группа, такая как фталидил; ди- низшие алкиламино низший алкильная группа, такая как 1-диметиламиноэтильная группа; и (5-метил-2- оксо-1,3-диоксол-4-ил)метильная группа.
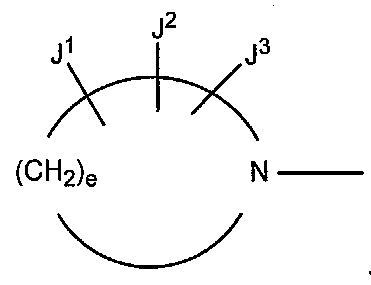
Следует отметить, что заместители R1, R2, R3, R4, R5, R6, R7, R8, R9, A, J1, J2, J3, W, X, Y, Z, e, f и g определяют в настоящем изобретении для удобства относительно химической структуры производных пиридонкарбоновой кислоты.
В других вариантах осуществления настоящее изобретение относится к способу получения производного пиридонкарбоновой кислоты, имеющего структуру производного 1 пиридонкарбоновой кислоты, в котором W представляет собой CR8, в которой R8 представляет собой атом водорода, атом галогена или низшую алкильную группу.
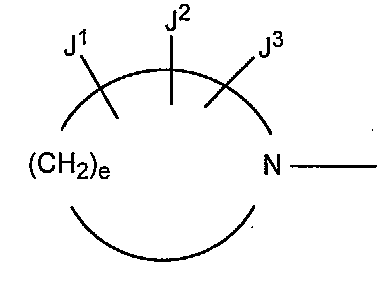
В других вариантах осуществления настоящее изобретение относится к способу получения производного пиридинкарбоновой кислоты, имеющего структуру производного 1 пиридинкарбоновой кислоты, в котором R5 представляет собой группу, представленную следующими формулами (a) или (b):
(a)
(b)
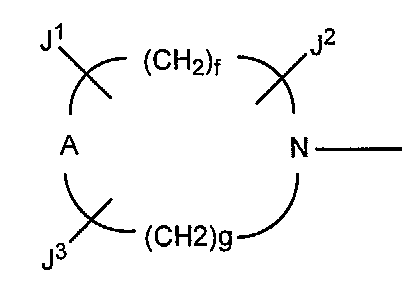
в которых A представляет собой атом кислорода, атом серы или NR9 (в которой R9 представляет собой атом водорода или низшую алкильную группу), e представляет собой число от 3 до 5, f представляет собой число от 1 до 3, g представляет собой число от 0 до 2, J1, J2 и J3, которые могут быть одинаковыми или отличными друг от друга, представляют собой атом водорода, гидроксильную группу, низшую алкильную группу, амино низшую алкильную группу, аминогруппу, низшую алкиламиногруппу, низшую алкоксигруппу или атом галогена.
В других вариантах осуществления настоящее изобретение относится к способу получения производного пиридинкарбоновой кислоты, имеющего структуру производного 1 пиридинкарбоновой кислоты, в котором R5 представляет собой группу, представленную формулой (a).
(a)
В других вариантах осуществления настоящее изобретение относится к способу получения производного пиридинкарбоновой кислоты, имеющего структуру производного 1 пиридинкарбоновой кислоты, в котором e в формуле (a) равно 3 или 4.
(a)
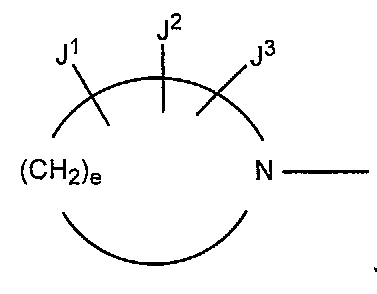
В других вариантах осуществления настоящее изобретение относится к способу получения производного пиридинкарбоновой кислоты, имеющего структуру производного 1 пиридинкарбоновой кислоты, в котором R1 представляет собой атом водорода; R2 представляет собой аминогруппу, низшую алкиламиногруппу или ди- низшую алкиламиногруппу; R3 представляет собой атом галогена; R4 представляет собой атом галогена; R представляет собой атом водорода; X представляет собой атом азота; Y и Z представляют собой CH или CR7 (в которой R7 представляет собой низшую алкильную группу или атом галогена); и W представляет собой CR8 (в которой R8 представляет собой атом галогена или низшую алкильную группу).
В других вариантах осуществления настоящее изобретение относится к способу получения производного пиридинкарбоновой кислоты, имеющего структуру производного 1 пиридинкарбоновой кислоты, в котором R2 представляет собой аминогруппу; R3 представляет собой атом фтора; R4 представляет собой атом фтора; Y представляет собой CF; Z представляет собой CH; W представляет собой CR8 (в которой R8 представляет собой атом хлора, атом брома или метильную группу), и e в формуле (a) равно 3.
(a)
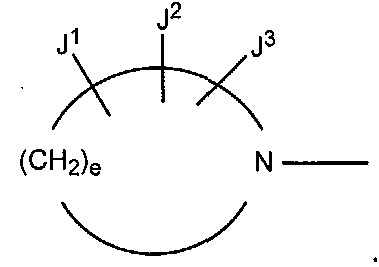
В других вариантах осуществления настоящее изобретение относится к способу получения пиридонкарбоновой кислоты, где упомянутая пиридонкарбоновая кислота соответствует следующей структуре:
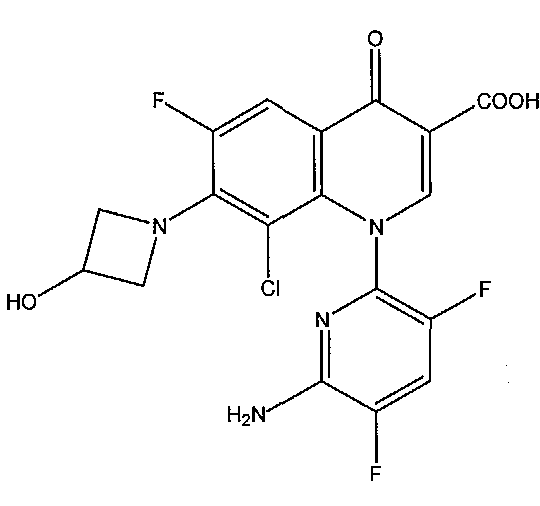
или ее фармацевтически приемлемой соли, эфира или пролекарства. Данная вышеуказанная пиридонкарбоновая кислота также известна под общедоступным кодовыми наименованиями Abbott Laboratories ABT-492, Wakunaga Pharmaceutical Co., Ltd. WQ 3034, Rib-X Pharmaceuticals, Inc., RX-3341, USAN делафлоксацин и также под химическими названиями 1-(6-амино-3,5-дифтор-2-пиридинил)-8-хлор-6-фтор-1,4-дигидро-7-(3-гидрокси-1-азетидинил)-4-оксо-3-хинолинкарбоновая кислота, 1-(6-амино-3,5-дифтор-2-пиридинил)-8-хлор-6-фтор-1,4-дигидро-7-(3-гидроксиазетидин-1-ил)-4-оксо-3-хинолинкарбоновая кислота, 3-хинолинкарбоновая кислота, 1-(6-амино-3,5-дифтор-2-пиридинил)-8-хлор-6-фтор-1,4-дигидро-7-(3-гидрокси-1-азетидинил)-4-оксо и 1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-6-фтор-7-(3-гидроксиазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота. Данная форма соединения карбоновой кислоты соответствует CAS регистрационному номеру 189279-58-1. Кроме того, WO 2006/042034, цитируемая выше, описывает D-глюцитольную соль данного соединения [D-глюцитол 1-(6-амино-3,5-дифтор-2-пиридинил)-8-хлор-6-фтор-1,4-дигидро-7-(3-гидрокси-1-азетидинил)-4-оксо-3-хинолинкарбоксилат (соль)] и тригидрат D-глюцитольной соли данного соединения [D-глюцитол 1-(6-амино-3,5-дифтор-2-пиридинил)-8-хлор-6-фтор-1,4-дигидро-7-(3-гидрокси-1-азетидинил)-4-оксо-3-хинолинкарбоксилат тригидрат (соль)]. D-глюцитольная соль и тригидрат D-глюцитольной соли соответствуют CAS регистрационным номерам 352458-37-8 и 883105-02-0 соответственно. D-глюцитол соответствует CAS регистрационному номеру 6284-40-8. WO 2006/042034 также описывает кристаллическую форму D-глюцитольной соли, охарактеризованную при измерении при приблизительно 25°C с Cu-Ka облучением, порошковой рентгенограммой, показанной на фиг.1 (см. WO 2006/042034), и кристаллическую форму тригидрата D-глюцитольной соли при измерении при приблизительно 25°C с Cu-Ka облучением, порошковой рентгенограммой, показанной на фиг.2 (см. WO 2006/042034). Данные D-глюцитольные соли являются пригодными в настоящем изобретении. Кроме того, см. A. R. Haight et al., "Synthesis of Quinolona ABT-492: Crystallizations for Optimal Processing", Organic Process Research & Development (2006), 10(4), 751-756.
Термины "способ в коммерческом масштабе" и "композиция в коммерческом масштабе" относится к способу и композиции, соответственно, который осуществляют или которую получают за один цикл в количестве по меньшей мере приблизительно 100 грамм.
Обнаружение и подавление образования димерной примеси при получении делафлоксацина
См. Hanselmann, R., et al., "Identification and Suppression of a Dimer Impurity in the Development of Delafloxacin", Organic Process Research & Development, vol. 13, pages 54-59 (2009).
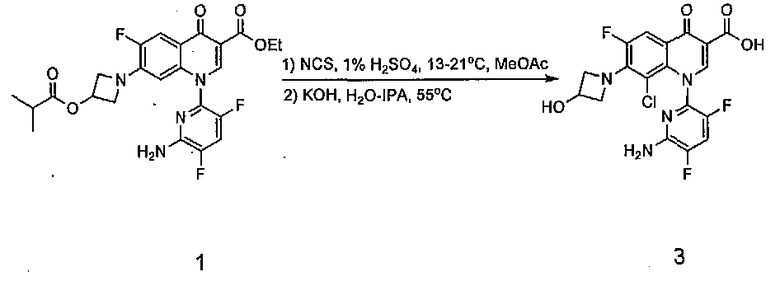
Делафлоксацин представляет собой 6-фторхинолоновый антибиотик, который разрабатывается в Rib-X Pharmaceuticals, Inc. В процессе первоначальных попыток масштабирования по получению делафлоксацина образовывалось вплоть до 0,43% новой примеси на предпоследней стадии хлорирования. Она была идентифицирована как димерный аддукт делафлоксацина. Последующее применение дизайна экспериментов (DoE) привело к обнаружению факторов, ответственных за образование данной примеси. Использование знаний, полученных у DoE, позволило воспроизводимо подавлять данную примесь до приемлемого уровня.
Противомикробная устойчивость в привычной социальной среде и в больничных условиях вызывает возрастающую озабоченность в здравоохранении в результате непрерывного появления бактериальных штаммов, устойчивых ко многим лекарственным средствам. См. (a) Cosgrove, S. E.; Carmeli, Y. Clin. Infect. Dis. 2003, 36, 1433. (b) Seybold, U.; Kourbatova, E. V.; Johnson, J. G.; Halvosa, S. J.; Wang, Y. F.; King, M. D.; Ray, S. M.; Blumberg, H. M. Clin. Infect. Dis. 2006, 42, 647 и (c) Tenover, F. C; McDougal, L. K.; Goering, R. V.; Killgore, G.; Projan, S. J.; Patel, J. B.; Dunman, P. M. J. Clin. Microbiol. 2006, 44, 108.
Метициллин-устойчивый золотистый стафилококк (MRSA) занимает первое место как наиболее часто выделяемый патогенный организм в больничных блоках интенсивной терапии в Соединенных Штатах и частота MRSA случаев увеличилась с 35,9% в 1992 до 64,4% в 2003. См. Klevens, R.M.; Edwards, J. R.; Tenover, F. C; McDonald, L. C; Horan, T.; Gaynes, R. Clin. Infect. Dis. 2006, 42, 389.
С момента введения налидиксовой кислоты приблизительно 40 лет назад, хинолоновые антибиотики занимают главное место в списке антибиотиков. 6-Фторхинолоны, такие как ципрофлоксацин, приобрел особенно возросшую роль при лечении инфекций благодаря его широкому спектру применения. См. (a) Bush, K. Clin. Microbiol. Infect. 2004, 10 (Suppl. 4), 10. и (b) Emmerson, A. M.; Jones, A. M. J. Antimicrob. Chemother. 2003, 51 (Suppl. Sl), 13.
Делафлоксацин представляет собой 6-фторхинолоновый антибиотик с превосходной антибактериальной активностью против грамположительных организмов, включая и метициллин-чувствительные S aureus и MRSA. В настоящее время он проходит вторую фазу клинических испытаний. Делафлоксацин первоначально получен Wakunaga Pharmaceuticals и Abbott Laboratories и был впоследствии запатентован Rib-X Pharmaceuticals, Inc.
Получение делафлоксацина первоначально было разработано Abbott Laboratories (Схема 1) и ключевой стадией в данной схеме является селективное хлорирование 8-положения функционализированного хинолона 1 (дэхлорхинолон). См. (a) Haight, A. R.; Ariman, S. Z.; Barnes, D. M.; Benz, N. J.; Gueffier, F. X.; Henry, R. F.; Hsu, M. C; Lee, E. C; Morin, L.; Pearl, K. B.; Peterson, M. J. ; Plata, D. J.; Willcox, D. R. Org. Process Res. Dev. 2006, 4, 751. и (b) Barnes, D. M.; Christesen, A. C; Engstrom, K. M.; Haight, A. R.; Hsu, M. C; Lee, E. C; Peterson, M. J.; Plata, D. J.; Raje, P. S.; Stoner, E. J.; Tedrow, J. S.; Wagaw, S. Org. Process Res. Dev. 2006, 4, 803.
В данном способе раствор 1 в смеси метилацетата (MeOAc) и этилацетата хлорировали, применяя NCS в присутствии 3,5 моль % H2SO4, получая 2. За этим следовала замена растворителя и омыление KOH для того, чтобы получить 3. Делафлоксацин получали после образования соли с N-метил-D-глюкамином.
Схема 1: Получение делафлоксацина
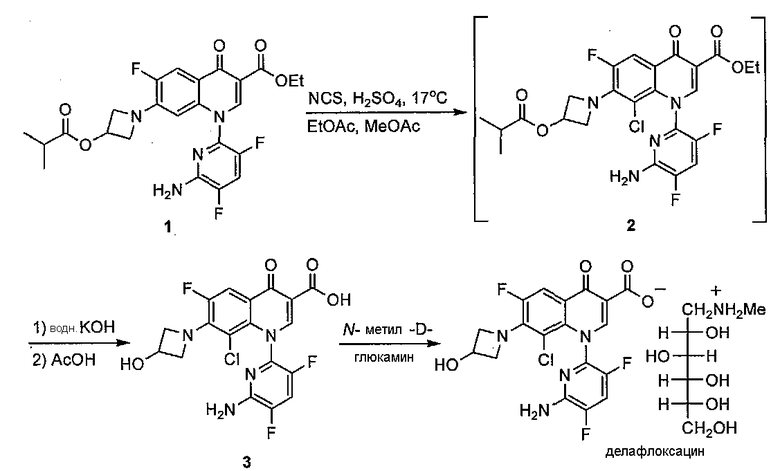
Несмотря на первоначальный успех при внедрении данного способа авторы изобретения столкнулись с трудностями при масштабировании данной стадии, заключающимися в том, что вплоть до 0,43 % по площади новой примеси обнаружили с помощью ВЭЖХ и RRT 1.60 после выделения 3. Кроме того, оказалось, что данную новую примесь трудно удалить в процессе конечного образования соли. Таким образом, авторы изобретения решили начать исследования по установлению данной примеси, понять, как она образуется, и подавить ее образование.
Результаты и обсуждение
Несмотря на многочисленные попытки выделить данную новую примесь препаративной ВЭЖХ авторам изобретения не удалось сделать это и был установлен только молекулярный вес с помощью HPLC-MS 880 Да. Измеренный молекулярный вес данной примеси точно в два раза больше молекулярного веса кислоты 3, что предполагает димерное производное данного соединения. Тщательное исследование профиля чистоты субстрата реакции хлорирования не привели к обнаружению какой-либо примеси, которой можно было бы аналогично приписать димерную структуру и ее образование, следовательно, было отнесено к последовательности хлорирование-гидролиз. Был рассмотрен ряд потенциальных димерных аддуктов, которые могли бы образовываться на данной стадии, включая 4, который мог быть результатом расщепления азетидиновой группировки в одной молекуле 3 и реакции гидроксильной группы второй молекулы. Для того чтобы дополнительно исследовать данную возможность, авторы изобретения приступили к получению 4.
Схема 2: Ретросинтез предполагаемой примеси 4
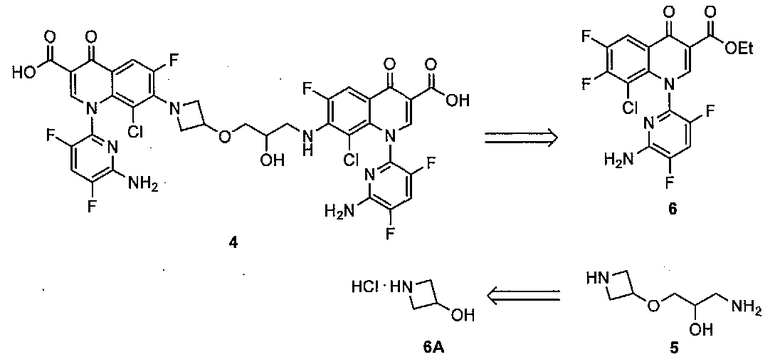
Ретросинтетически (Схема 2) молекула 4 легко размыкается на подходящим образом защищенный аминоспирт 5 и хинолон 6; последний является известным соединением. Фрагмент 5 можно получить из имеющегося в продаже гидрохлорида азетидин-3-ола 7. См. (a) Yazaki, A.; Niino, Y.; Ohshita, Y.; Hirao, Y.; Amano, H.; Hayashi, N.; Kuramoto, Y. PCT международная заявка WO 9711068, 1997. CAN: 126, 305587 и (b) Yazaki, A.; Aoki, S. PCT международная заявка WO 2001034595, 2001. CAN: 134, 366811.
Таким образом, синтез начинали с 7, в котором атом азота защищали в виде бензилкарбамата для того, чтобы получить 8 с количественным выходом. Данный аддукт алкилировали рацемическим эпихлоргидрином для того, чтобы получить 9 с выходом 84%. Раскрытие эпоксида 9 аммиаком давало 10, которое конденсировали без очистки с 6 для того, чтобы получить 11 c суммарным выходом 83%. Удаление Cbz группы в условиях гидрирования давало 12 с выходом 93%. Вторая конденсация с 6 приводила к образованию димерного соединения 13 с выходом 71%. После омыления предполагаемую примесь 4 получали с выходом 98%.
Схема 3: Синтез примеси 4
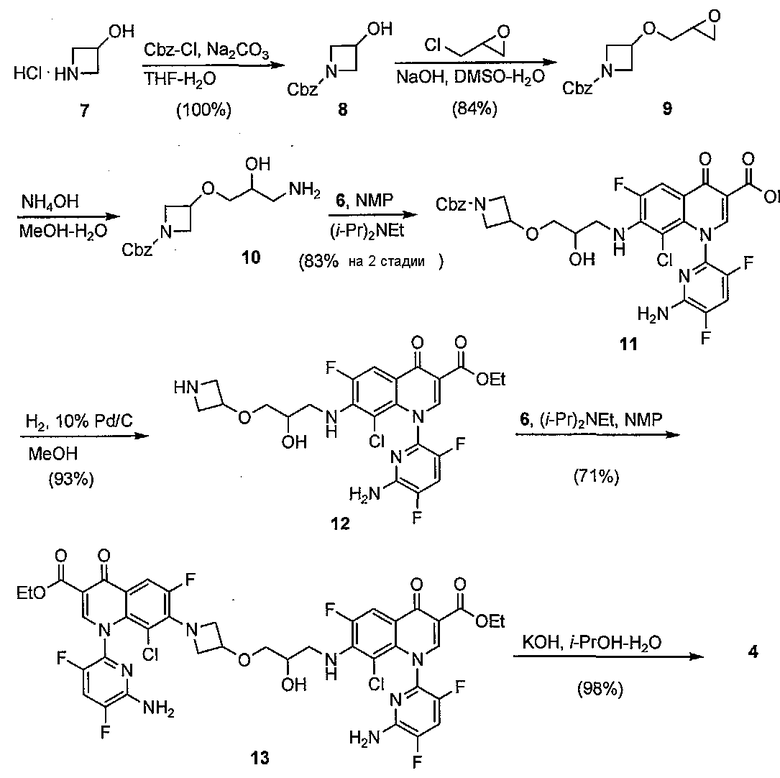
С синтезированным 4 в наличии неизвестную примесь в загрязненной порции делафлоксацина сравнивали с полученным 4 с помощью приведенных экспериментов и сравнивали с помощью HPLC-MS и HPLC-UV. Оказалось, что синтетическое 4 однозначно соответствовало неизвестной примеси, наблюдаемой в ранее полученных порциях делафлоксацина.
Для того чтобы понять динамику образования примеси 4, авторы изобретения решили дополнительно исследовать реакцию в исследованиях дизайна экспериментов (DoE). Следующие факторы выбирали для изучения в DoE исследовании разрушения IV, в диапазонах, как показано: температура (15-25°C), количество NCS (1,05-1,2 экв.), количество H2SO4 (2-5 моль %), содержание воды в растворителе (0-0,5%), объем растворителя (2-3 об.), растворитель (метилацетат/этилацетат) и скорость добавления NCS (0,05-0,3 об/мин). См. фиг.4, фиг.5a, 5b, 5c, 5d, 5e и 5f, фиг.6a и 6b, фиг.7 и фиг.8a, 8b, 8c, 8d, 8e, 8f и 8g. Проводили всего 19 реакций хлорирования в MultiMaxTM реакторе, полученном у Mettler-Toledo, Inc., 1900 Polaris Parkway, Columbus, OH, 43240.
В каждом случае образцы из реакций гасили через 5 ч, омыляли KOH и неочищенные реакционные смеси анализировали ВЭЖХ. Для того чтобы определить количество 4, хлорированные образцы 2 омыляли до 3. Величину площади в процентах для примеси 4, которая была получена в каждом случае, обрабатывали и анализировали, применяя DoE программное обеспечение. Дизайн эксперимента и анализ проводили, применяя JMP, Design of Experiments, Version 7, SAS Institute Inc., Cary, NC, 1989-2007, применяя пошаговое приближение, с последующим стандартным способом наименьших квадратов.
Превосходную корреляцию R2 0,997 получали, следуя обработке данных. Основные эффекты: большие количества NCS, снижение температуры и более быстрое добавление раствора NCS, а также применение сухих растворителей, оказывали наиболее благоприятный эффект на подавление количества примеси 4 (фиг.1). Применяли метилацетат, содержащий менее чем 500 частей на миллион воды, перед корректировкой, как требуется в подходящем эксперименте в DoE. Кроме того, сильное взаимодействие наблюдали между количеством NCS и растворителя в том, что метилацетат был предпочтительным, когда применяли только небольшой избыток NCS. Для того чтобы подавить какое-либо избыточное хлорирование 2, 1,05 эквивалента NCS было предпочтительным и, следовательно, метилацетат выбирали в качестве предпочтительного растворителя для данной стадии. Более подробный анализ можно найти на фиг.4, фиг.5a, 5b, 5c, 5d, 5e и 5f, фиг.6a и 6b, фиг.7 и фиг.8a, 8b, 8c, 8d, 8e, 8f и 8g.
С точки зрения механизма реакции, авторы изобретения пришли к выводу, что примесь 4 могла бы возникать в результате первоначальной катализируемой кислотой активации азетидинового кольца, которая вызывает изобутировый эфир/хлорид индуцированную последовательность раскрытия до 16. В процессе последующего омыления 16 реагирует с промежуточным соединением гидролиза 17 или 3 до 4 (Схема 4). Омыление и последующее образование эпоксида 16 перед конденсацией с 3 или 17 нельзя исключать. Обоснованность данной последовательности дополнительно усиливалась последующим HPLC-MS анализом неочищенной реакционной смеси хлорирования перед омылением. В нем примесь с молекулярным весом 574 Да, которая соответствует 16, обнаруживали в приблизительно равных количествах по сравнению с 4 после омыления.
Схема 4: Предлагаемый механизм образования примеси 4
На основании данного гипотетического механизма, временная зависимость для образования 4 в процессе хлорирования не может исключаться, и так как время реакции сохраняется постоянным в DoE исследовании, было решено оценить данный параметр независимо. Реакцию хлорирования проводили, применяя 3,5% H2SO4 и метилацетат в качестве растворителя при 15°C, и образец гасили после того, как считали, что реакция завершилась. Дополнительные образцы гасили через 2 ч и 6 ч, омыляли и анализировали ВЭЖХ. Вполне закономерно, наблюдали непрерывное увеличение количества примеси 4 с течением времени. Данный результат влияет на контроль процесса хлорирования в том, что адекватное изменение относительно времени для ВЭЖХ контроля данной реакции могло бы быть необходимо для того, чтобы снизить до минимума образование 4. Однако последующие эксперименты показали, что снижение количества H2SO4 до 1% уменьшает количество примеси 4, полученной с течением времени без значительного влияния на время реакции хлорирования или качество 3 (фиг.2). Таким образом, приемлемое изменение времени для технологического контроля можно достигнуть, когда концентрацию 1% H2SO4 применяют в качестве катализатора.
После установления критических параметров, касающихся образования данной примеси, проводили второе DoE изучение для исследования устойчивости реакции в предполагаемом эксплуатационном диапазона способа. В нем DoE изучение разложения IV конструировали следующими факторами, подвергающимися изменению: температура (13-21°C), количество NCS (1,04-1,07 экв.), скорость добавления NCS (30-75 мин) и H2SO4 (0,8-1,2 моль %). Проводили всего 10 реакций хлорирования в MultiMaxTM реакторе. В каждом случае образцы гасили и омыляли после прохождения контроля в процессе обработки. Полученную в результате площадь в процентах 4 обрабатывали и анализировали, применяя DoE программное обеспечение. Как и ожидалось, температура, количество NCS и H2SO4 имели статистически значимый эффект на количество примеси 4 в изучаемых диапазонах параметров. Однако принимая наихудший вариант в предполагаемом профиле, примесь 4 имела величину 0,11 площади в процентах ±0,01%, что находится в пределах приемлемого диапазона, который установлен из токсикологической порции делафлоксацина (фиг.3).
Две последовательные лабораторные серии данной реакции в килограммовых количествах подтвердили эффективность изменений параметров, и вещество с высокой чистотой с концентрацией примеси 4 0,07% получали после омыления.
В заключении, авторы изобретения с успехом идентифицировали димерную примесь, которая была обнаружена в процессе масштабирования получения делафлоксацина. Последующие DoE эксперименты позволили нам обнаружить способы снижения образования данной примеси до приемлемой концентрации в небольшом масштабе, также как в лабораторных сериях в килограммовых масштабах.
Примеры
ПРИМЕР 1
1-(6-Амино-3,5-дифторпиридин-2-ил)-8-хлор-6-фтор-7-(3-гидроксиазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота, 3, улучшенная методика.
К суспензии 1 (3,1 кг, 6,15 моль) в метилацетате (8,6 кг) добавляли раствор H2SO4 (5,9 г, 62 ммоль) и NCS (0,88 кг, 6,46 моль) в метилацетате (14,4 кг) при 10-17°C в течение 45 мин. Раствор перемешивали при 13-19°C в течение 2 часов, гасили 1,6% водным NaHCO3 (12,6 кг) и органический слой промывали 11% водным Na2SO3 (7 кг). Метилацетатный раствор был заменен на 2-пропанольный раствор при 50°C/вакуум, затем добавляли раствор KOH (1,1 кг, 19,7 моль) в воде (24,8 кг) и смесь перемешивали при 55°C в течение 3 часов. Добавляли при 40°C 13% водную уксусную кислоту (2,6 кг) и раствор затравливали 3 (27 г, 61 ммоль). Суспензию перемешивали в течение 1 часа при 40°C и затем медленно добавляли 13% водную уксусную кислоту (11,7 кг). После перемешивания в течение дополнительного часа при 40°C суспензию охлаждали до комнатной температуры, фильтровали, промывали водой (41 кг) и сушили при 60°C/вакуум для того, чтобы получить 3 в виде желтых кристаллов (2,5 кг, 91%). Выделенный 3 имел те же спектроскопические свойства, как сообщалось.
ПРИМЕР 2
1-Амино-3-(азетидин-3-илокси)пропан-2-олбис(N,N'-хинолонкарбоновая кислота), 4.
Бензиловый эфир 3-гидроксиазетидин-1-карбоновой кислоты, 8.
К раствору гидрохлорида азетидин-3-ола 7 (25 г, 0,23 моль) в воде (150 мл) и THF (300 мл) добавляли K2CO3 (63,1 г, 0,46 моль). Смесь перемешивали в течение 30 мин при 20-25°C. Затем добавляли в течение 30 мин бензилхлорформиат (40,9 г, 0,24 моль) при 0-5°C, с последующим перемешиванием смеси в течение ночи при 20-25°C. THF удаляли на роторном испарителе при 30°C / вакуум и смесь экстрагировали этилацетатом (2×150 мл). Объединенный органический слой промывали водой (1×50 мл), сушили над Na2SO4 и концентрировали. Остаток очищали колоночной флеш-хроматографией на силикагеле, элюируя этилацетат-гептан 1:1 и 4:1 для того, чтобы получить 8 в виде прозрачного масла (47,3 г, 100%).
1H NMR (300 МГц, CDCl3): δ 3,72 (1H, д, J=6,2 Гц), 3,85 (2H, дд, J=9,5, 4,4 Гц), 4,17 (2H, дд, J=9,5, 6,7 Гц), 4,49-4,57 (1H, м), 5,06 (2H, с), 7,31-7,38 (5H, м);
13C NMR (75 МГц, CDCl3): δ 59,2, 61,6, 66,9, 127,9, 128,1, 128,5, 136,5, 156,6; IR: (пленка) 3406, 1686, 1438 см-1; ES-HRMS m/z: (M+ +1H) рассчитан для C11H14NO3 208,0968, найдено 208,0967.
Бензиловый эфир 3-оксиранилметокси-азетидин-1-карбоновой кислоты, 9.
К раствору 8 (30 г, 0,15 моль) в DMSO (250 мл) медленно добавляли раствор NaOH (9,9 г, 0,25 моль) в воде (195 мл) при 15-25°C. Добавляли эпихлоргидрин (93,8 г, 1,01 моль) и смесь перемешивали при 20-25°C в течение 24 часов. Смесь разбавляли водой (300 мл) и экстрагировали этилацетатом (2×150 мл). Объединенный органический слой промывали водой (2×50 мл), сушили над Na2SO4 и концентрировали. Остаток очищали колоночной флеш-хроматографией на силикагеле, элюируя этилацетат-гептан 3:2, получая 9 в виде прозрачного масла (32,1 г, 84%).
1H NMR (300 МГц, CDCl3): δ 2,60 (1H, дд, J=4,8, 2,6 Гц), 2,81 (1H, дд, J=4,9, 4,2 Гц), 3,09-3,16 (1H, м), 3,25 (1H, дд, J=11,4, 6,2 Гц), 3,68 (1H, дд, J=11,5, 2,5 Гц), 3,89-3,97 (2H, м), 4,15-4,24 (2H, м), 4,29-4,37 (1H, м), 5,09 (2H, с), 7,28-7,36 (5H, м);
13C NMR (75 МГц, CDCl3): δ 44,2, 50,4, 56,7, 56,9, 66,7, 68,6, 70,0, 128,0, 128,1, 128,5, 136,6, 156,5; IR: (пленка) 2951, 1709, 1420 см-1; ES-HRMS m/z: (M+ +1H) рассчитан для C14H18NO4 264,1230, найдено 264,1230.
Этиловый эфир 1-(6-амино-3,5-дифторпиридин-2-ил)-7-[3-(1-бензилоксикарбонилазетидин-3-илокси)-2-гидроксипропиламино]-8-хлор-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты, 11.
Смесь 9 (19 г, 72,2 ммоль) в конц. NH4OH (380 мл) и 7M NH3 в MeOH (86 мл) перемешивали в течение 5 ч при комнатной температуре. Прозрачный раствор концентрировали и азеотропно сушили с толуолом. Остаток в виде прозрачного масла и 6 (20 г, 48,1 ммоль) растворяли в NMP (150 мл). Добавляли N,N-диизопропилэтиламин (12,4 г, 96,2 ммоль) и раствор перемешивали при 70°C в течение 3 часов. Раствор выливали в смесь 1N лимонная кислота/лед (300 мл) и экстрагировали этилацетатом (2×150 мл). Объединенный органический слой промывали водой (2×100 мл), сушили над Na2SO4 и концентрировали. Остаток очищали колоночной флеш-хроматографией на силикагеле, элюируя этилацетат-гептан 1:1, с последующим элюированием этилацетат-MeOH 95:5, получая 11 в виде желтой пены (27,1 г, 83%).
1H NMR (300 МГц, CDCl3): δ 1,35 (3H, т, J=7,1 Гц), 3,35-3,52 (4H, м), 3,62-3,77 (1H, м), 3,84-3,91 (2H, м), 3,95-4,08 (1H, м), 4,15 (2H, дд, J=9,3, 6,5 Гц), 4,23-4,30 (1H, м), 4,35 (2H, кв, J=7,1 Гц), 4,85-5,13 (3H, уш. с), 5,08 (2H, с), 7,18-7,25 (1H, м), 7,31-7,35 (5H, м), 7,99 (1H, дд, J=13,7, 3,1 Гц), 8,31 (1H, с);
13C NMR (75 МГц, CDCl3): δ 14,4, 48,5 (д, JF=10 Гц), 56,6, 61,1, 66,9, 68,6, 69,3, 70,8, 107,2, 111,5, 112,6 (д, JF=24 Гц), 113,2 (м), 120,6, 128,0, 128,1 , 128,5, 134,1 (д, JF=5 Гц), 134,7 (м), 136,5, 139,2 (д, JF=13 Гц), 144,9 (д, JF=253 Гц), 144,4 (д, JF=13 Гц), 145,6 (дд, JF=262,4 Гц), 149,9 (д, JF=246 Гц), 150,0, 156,5, 164,7, 172,9; IR: (KBr) 2949, 1700, 1615 см-1 ; ES-HRMS m/z: (M+ +1H) рассчитан. для C31H30ClF3N5O7 676,1780, найдено 676,1762.
Этиловый эфир 1-(6-амино-3,5-дифторпиридин-2-ил)-7-[3-(азетидин-3-илокси)-2-гидроксипропиламино]-8-хлор-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты, 12.
К суспензии 10% Pd на угле (2,1 г) в MeOH (20 мл) добавляли раствор 11 (13,7 г, 20,3 ммоль) в MeOH (230 мл). Смесь гидрировали при 1 атм. в течение 1 часа, фильтровали через Hyflo и упаривали, получая 12 в виде бежевых кристаллов (10,3 г, 93%). Тпл 148-152°C;
1H NMR (300 МГц, DMSO-d6): δ 1,27 (3H, т, J=7,1 Гц), 3,27 (1H, д, J=5,0 Гц), 3,28-3,80 (10H, м), 4,19 (1H, уш. с), 4,21 (2H, кв, J=7,1 Гц), 5,86 (1H, с), 6,74 (2H, с), 7,84 (1H, д, J=13,8 Гц), 7,94 (1H, дд, J=9,7, 9,0 Гц), 8,43 (1H, с);
13C NMR (75 МГц, CDC13): δ 14,1, 48,4 (д, JF=10 Гц), 53,6, 60,2, 68,4, 70,4 (д, JF=4 Гц), 72,1 , 106,4 (д, JF=6 Гц), 111,0, 111,3 (д, JF=23 Гц), 113,6 (дд, JF=23,21 Гц), 118,9 (д, JF=6 Гц), 133,8 (д, JF=13 Гц), 134,2, 139,5 (д, JF=12 Гц), 143,3 (дд, JF=248,4 Гц), 145,0 (дд, JF=259,5 Гц), 145,6 (д, JF=14 Гц), 149,3 (д, JF=245 Гц), 149,5, 163,5, 171,0; IR: (KBr) 1697, 1614, 1496, 1457 см-1; ES-HRMS m/z: (M+ +1H) рассчитан для C23H24ClF3N5O5 542,1413, найдено 542,1391.
1-Амино-3-(азетидин-3-илокси)пропан-2-олбис(N,N'-хинолоновый диэфир), 13.
Раствор 12 (9,6 г, 17,7 ммоль), 6 (7,8 г, 18,6 ммоль) и N,N- диизопропилэтиламин (4,6 г, 35,4 ммоль) в NMP (150 мл) перемешивали при 55°C в течение 3 часов. Раствор выливали в смесь 1N лимонная кислота/лед (300 мл) и экстрагировали этилацетатом (3×100 мл). Объединенный органический слой промывали водой (2×100 мл), сушили над Na2SO4 и концентрировали. Остаток очищали колоночной флеш-хроматографией на силикагеле, элюируя этилацетат-MeOH 95:5. Полученная желтая пена кристаллизовалась в CH2Cl2-MeOH 9:1 (160 мл), давая 13 в виде бежевых кристаллов (11,8 г, 71%). Тпл 184-187°C;
1H NMR (300 МГц, DMSO-d6): δ 1,26 (6H, т, J=7,1 Гц), 3,29-3,48 (3H, м), 3,49-3,62 (1H, м), 3,73-3,82 (1H, м), 4,12-4,30 (3H, м), 4,21 (4H, кв, J=7,1 Гц), 4,52-4,65 (2H, м), 5,13-5,22 (1H, м), 5,83-5,92 (1H, м), 6,72 (4H, с), 7,73 (1H, д, J=13,9 Гц), 7,82 (1H, д, J=13,9 Гц), 7,92 (1H, т, J=9,6 Гц), 7,93 (1H, т, J=8,7 Гц), 8,41 (2H, с);
13C NMR (75 МГц, CDCl3): δ 12,3 (2x), 46,4 (д, JF=11 Гц), 58,4 (2x), 61,9 (2x), 66,7, 67,3 (д, JF=4 Гц), 69,1, 103,4 (д, JF=6 Гц), 104,6 (д, JF=6 Гц), 108,7 (д, JF=23 Гц), 109,2, 109,4 (д, JF=23 Гц), 109,5, 111,7 (дд, JF=25, 24 Гц), 111,8 (дд, JF=25,24 Гц), 117,1 (д, JF=7 Гц), 1 17,8 (д, JF=6 Гц), 132,1 (дд, JF=17,4 Гц), 132,2, 132,5, 133,5, 137,7 (д, JF=12 Гц), 139,4 (д, JF=12 Гц), 141,0 (дд, JF=247,5 Гц), 141,5 (дд, JF=248,5 Гц), 143,0 (дд, JF=259,5 Гц), 143,3 (дд, JF=259,5 Гц), 143,8 (2x, д, JF=15 Гц), 147,5 (д, JF=245 Гц), 147,7, 147,8, 148,1 (д, JF=247 Гц), 161,7 (2x), 169,1, 169,2; IR: (KBr) 1728, 1615, 1491, 1448 см-1 ; ES-HRMS m/z: (M+ +1H) рассчитан для C40H33Cl2F6N8O8 937,1697, найдено 937,1696.
1-Амино-3-(азетидин-3-илокси)пропан-2-олбис(N,N'-хинолонкарбоновая кислота), 4.
К суспензии 13 (17,0 г, 18,1 ммоль) в 2-пропаноле (75 мл) добавляли 1N раствор KOH (127 мл, 126,7 ммоль). После перемешивания смеси при 55°C в течение 3,5 часов раствор охлаждали до 30°C и добавляли в течение 1 часа раствор AcOH (12,4 г, 206,5 ммоль), растворенный в воде (94 мл). Суспензию перемешивали при комнатной температуре в течение 2 часов, фильтровали, промывали водой (3×40 мл) и сушили при 50°C/вакуум, получая 4 в виде желтых кристаллов (15,7 г, 98%). Тпл 198-205°C (разлаг.);
1H NMR (300 МГц, DMSO-d6): δ 3,28-3,45 (2H, м), 3,45-3,78 (2H, м), 3,79-3,88 (1H, м), 4,16-4,33 (3H, м), 4,61-4,75 (2H, м), 5,25 (1H, уш. с), 6,23-6,35 (1H, м), 6,76 (4H, с), 7,79 (1H, д, J=13,7 Гц), 7,90 (1H, д, J=13,8 Гц), 7,93 (2H, дд, J=9,7, 2,4 Гц), 8,70 (1H, с), 8,71 (1H, с), 14,59 (2H, уш. с);
13C NMR (75 МГц, CDCl3): δ 48,1 (д, JF=11 Гц), 63,8, 68,4, 69,0 (д, JF=5 Гц), 70,6 (д, JF=6 Гц), 104,5 (д, JF=6 Гц), 105,9 (д, JF=7 Гц), 107,8, 108,2, 109,8 (д, JF=23 Гц), 110,8 (д, JF=23 Гц), 113,4 (д, JF=23 Гц), 113,7 (д, JF=23 Гц), 115,8 (д, JF=8 Гц), 116,6 (д, JF=8 Гц), 133,3 (дд, JF=14,3 Гц), 133,5 (дд, JF=14,4 Гц), 134,8, 135,9, 141,0 (д, JF=12 Гц), 142,1 (д, JF=12 Гц), 142,8 (дд, JF=249,5 Гц), 143,3 (дд, JF=249,5 Гц), 145,1 (дд, JF=259,5 Гц), 145,4 (дд, JF=260,5 Гц), 145,6 (2x, д, JF=15 Гц), 149,5 (д, JF=248 Гц), 150,1 (2x), 150,2 (д, JF=249 Гц), 164,7, 164,8, 175,8 (д, JF=3 Гц), 175,9 (д, JF=3 Гц); IR: (KBr) 1727, 1622, 1489, 1439 см-1; ES-HRMS m/z: (M+ +1H) рассчитан для C36H25Cl2F6N8O8 881,1071, найдено 881,1090.
Дополнительные экспериментальные материалы
Экспериментальные таблицы и анализы DoE исследований далее представлены на фиг.4, фиг.5a, 5b, 5c, 5d, 5e и 5f, фиг.6a и 6b, фиг.7 и фиг.8a, 8b, 8c, 8d, 8e, 8f и 8g.
Формулирование и введение
Соединения настоящего изобретения можно использовать на практике, доставляя их с применением подходящего носителя. Доза активного соединения, способ введения и применение подходящего носителя будут зависеть от предполагаемого пациента или субъекта и таргетного микроорганизма, например бактериального таргетного организма. Композиции соединений согласно настоящему изобретению как для медицинского применения на людях, так и для ветеринарного применения, обычно содержат данные соединения в сочетании с фармацевтически приемлемым носителем.
Носитель должен быть "приемлемым" в смысле быть совместимым с соединениями настоящего изобретения и не должен быть вредным для реципиента. Предполагается, что фармацевтически приемлемые носители, в этом смысле, включают любые и все растворители, дисперсные среды, покрытия, агенты, задерживающие поглощение, и подобные, совместимые с фармацевтическим введением. Применение данных сред и средств для фармацевтически активных веществ является известным в данной области техники. За исключением случаев, когда какая-нибудь общепринятая среда или агент является несовместимым с активным соединением, его применение в композициях предполагается. Дополнительные активные соединения (установленные или разработанные согласно настоящему изобретению и/или известные в данной области техники) также можно вводить в композиции. Композиции можно удобно предоставлять в стандартной лекарственной форме и их можно получить любым из способов, хорошо известных в области фармации/микробиологии. В общем, некоторые композиции получают смешением соединения с жидким носителем или мелкоизмельченным твердым носителем или обоими и затем, в случае необходимости, формованием продукта в требуемую композицию.
Фармацевтическую композицию настоящего изобретения нужно формулировать так, чтобы она была совместима с предполагаемым путем введения. Растворы или суспензии могут содержать следующие компоненты: стерильный разбавитель, такой как вода, соляной раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. pH можно регулировать кислотами или основаниями, такими как хлорводородная кислота или гидроксид натрия.
Широкий спектр композиций и способов введения, включая, например, внутривенные композиции и способы введения можно найти в S. K. Niazi, ed., Handbook of Pharmaceutical Composition, Vols. 1-6 [Vol. 1 Compressed Solid Products, Vol. 2 Uncompressed Drug Products, Vol. 3 Liquid Products, Vol. 4 Semi-Solid Products, Vol. 5 Over the Counter Products, and Vol. 6 Sterile Products], CRC Press, April 27, 2004.
Пригодные растворы для перорального или парентерального введения можно получить любым из способов, известным в области фармацевтики, описанным, например, в Remington's Pharmaceutical Sciences, 10th ed. (Mack Publishing Company, 1990). Композиции для парентерального введения могут также содержать гликохолат для буккального введения, метоксисалицилат для ректального введения или лимонную кислоту для вагинального введения. Парентеральный препарат можно заключать в капсулы, шприцы одноразового применения или флаконы для многократного применения, полученные из стекла или пластика. Суппозитории для ректального введения можно также получить смешением лекарственного средства с нераздражающим вспомогательным веществом, таким как масло какао, другие глицериды, или другими композициями, которые являются твердыми при комнатной температуре и жидкими при температурах тела. Композиции могут также содержать, например, полиалкиленгликоли, такие как полиэтиленгликоль, масла растительного происхождения и гидрогенезированные нафталины. Композиции для прямого введения могут содержать глицерин и другие композиции с высокой вязкостью. Другие потенциально пригодные парентеральные носители для данных лекарственных средств включают сополимерные частицы этиленвинил-ацетат, осмотические насосы, имплантируемые системы для вливания и липосомы. Композиции для введения ингаляцией могут содержать в качестве вспомогательных веществ, например, лактозу, или могут представлять собой водные растворы, содержащие, например, полиоксиэтилен-9-лауриловый эфир, гликохолат и дезоксихолат, или масляные растворы для введения в виде назальных капель, или в виде геля для применения внутриназально. Удерживающие клизмы можно также применять для ректальной доставки.
Композиции настоящего изобретения, подходящие для перорального введения, могут быть в форме: дискретных единиц, таких как капсулы, желатиновые капсулы, саше, таблетки, пастилки или таблетки для рассасывания, причем каждая содержит заранее определенное количество лекарственного средства; порошка или гранулярных композиций; растворов или суспензий в водной жидкости или неводной жидкости; или эмульсии масло в воде или эмульсии вода в масле. Лекарственное средство можно также вводить в форме болюса, электуария или пасты. Таблетку можно получить сдавливанием или прессованием лекарственного средства необязательно с одним или более вспомогательными ингредиентами. Сдавленные таблетки можно получить сдавливанием в подходящей машине лекарственного средства в свободно текучей форме, такой как порошок или гранулы, необязательно смешанной со связующим, смазывающим агентом, инертным разбавителем, поверхностно-активным или диспергирующим агентом. Прессованные таблетки можно получить прессованием в подходящей машине смеси порошкообразного лекарственного средства и подходящего носителя, смоченного инертным жидким разбавителем.
Пероральные композиции обычно содержат инертный разбавитель или пищевой носитель. Для целей перорального терапевтического введения, активное соединение можно вводить с вспомогательными веществами. Пероральные композиции, полученные, применяя жидкий носитель, для применения в качестве жидкости для полоскания рта, содержат соединение в жидком носителе, и их применяют перорально и ими полоскают рот и выплевывают или проглатывают. Фармацевтически совместимые связующие агенты и/или вспомогательные материалы можно включать в качестве части композиции. Таблетки, пилюли, капсулы, пастилки и подобные могут содержать любой из следующих ингредиентов или соединения с аналогичными свойствами: связующее, такое как микрокристаллическая целлюлоза, трагакантовую камедь или желатин; вспомогательное вещество, такое как крахмал или лактозу; разрыхлитель, такой как альгиновая кислота, примогель или кукурузный крахмал; смазывающее вещество, такое как стеарат магния или стеараты; регулятор сыпучести, такой как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или ароматизатор, такой как мятное масло, метилсалицилат или апельсиновый ароматизатор.
Фармацевтическии композиции, подходящие для применения инъекций, содержат стерильные водные растворы (когда они водорастворимые) или дисперсии и стерильные порошки, приготовленные для немедленного приема препарата стерильных растворов или дисперсий для инъекции. Для внутривенного введения, подходящие носители включают физиологический соляной раствор, бактериостатическую воду, Cremophor ELTM (BASF, Parsippany, NJ) или фосфатно-буферный солевой раствор (PBS). Они должны быть стабильными в условиях получения и хранения и должны быть сохранены от загрязняющего действия микроорганизмов, таких как бактерии и грибки. Носитель может представлять собой растворитель или дисперсную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль) и их подходящие смеси. Подходящую текучесть можно поддерживать, например, применением покрытия, такого как лецитин, поддержанием требуемого размера частиц в случае дисперсии и применением поверхностно-активных веществ. Во многих случаях будет предпочтительно включать изотонические агенты, например, сахара, полиспирты, такие как манитол, сорбитол, хлорид натрия в композиции. Продолжительное поглощение инъецируемой композиции можно вызвать включением в композицию агента, который замедляет поглощение, например, моностеарата алюминия и желатина.
Стерильные растворы для инъекции можно получить введением активного соединения в требуемом количестве в подходящий растворитель с одним или комбинацией ингредиентов, перечисленных выше, при необходимости, с последующей стерилизацией фильтрацией. Обычно, дисперсии получают введением активного соединения в стерильную среду, которая содержит основную дисперсную среду и требуемые другие ингредиенты из ингредиентов, перечисленных выше. В случае стерильных порошков для получения стерильных растворов для инъекции способы получения включают вакуумную сушку и лиофилизацию, которая дает порошок активного ингредиента плюс дополнительный требуемый ингредиент из их ранее стерилизованного фильтрованием раствора.
Композиции, подходящие для внутрисуставного введения, могут быть в форме стерильного водного препарата лекарственного средства, который может быть в микрокристаллической форме, например в форме водной микрокристаллической суспензии. Липосомные композиции или биоразлагаемые порошковые системы можно также применять для предоставления лекарственного средства и для внутрисуставного и офтальмического введения.
Композиции, подходящие для местного введения, включая обработку глаз, включают жидкие или полужидкие препараты, такие как линименты, лосьоны, гели, примочки, эмульсии масла в воде или воды в масле, такие как крема, мази или пасты; или растворы, или суспензии, такие как капли. Композиции для местного введения на поверхность кожи можно получить диспергированием лекарственного средства с дерматологически приемлемым носителем, таким как лосьон, крем, мазь или мыло. Пригодными являются носители, способные образовывать пленку или слой на коже для локализации применения и замедления удаления. Для местного введения на поверхности внутренних тканей, агент можно диспергировать в жидком тканевом клее или другом веществе, о котором известно, что он усиливает адсорбирование на поверхности ткани. Например, растворы гидроксипропилцеллюлозы или фиброгена/тромбина можно применять для получения преимуществ. Альтернативно, можно применять растворы для покрытия тканей, такие как пектин-содержащие композиции.
Для обработок ингаляцией можно применять ингаляцию порошка (самораспыляющиеся или аэрозольные композиции), распределяемого с помощью аэрозольного баллончика, распылителя или аэрозольного ингалятора. Данные композиции могут быть в форме мелкого порошка для легочного введения из устройства для порошковой ингаляции или самораспыляющейся распыляющей порошок композиции. В случае самораспыляющегося раствора и аэрозольной композиции эффекта можно достигнуть или подбором клапана, имеющего требуемые характеристики аэрозоля (т.е., совместимый с получением спрея, имеющего требуемый размер частиц) или введением активного ингредиента в виде суспендированного порошка с контролируемым размером частиц. Для введения ингаляцией соединения можно также доставлять в форме аэрозольного спрея из емкости под давлением или распылителя, который содержит подходящий пропеллент, например, газ, такой как диоксид углерода, или аэрозольного баллончика.
Системное введение также может осуществляться трансмукозальным или трансдермальным способами. Для трансмукозального или трансдермального введения в композиции применяют пенетранты, подходящие для проникновения через барьер. В общем, данные пенетранты известны в данной области техники и включают, например, для трансмукозального введения детергенты и соли желчной кислоты. Трансмукозальное введение может осуществляться посредством применения назальных спреев или суппозиторий. Для трансдермального введения, активные соединения обычно формулируют в мази, бальзамы, гели или крема, как обычно известно в данной области техники.
Активные соединения можно получить с носителями, которые будут защищать соединение от быстрого выведения из тела, такие как композиция с контролируемым высвобождением, включая имплантанты и микроинкапсулированные системы для доставки. Можно применять биоразлагаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Способы получения данных композиций будут очевидны специалистам в данной области техники. Липосомальные суспензии можно также применять в качестве фармацевтически приемлемых носителей. Их можно получить согласно способам, известным специалистам в данной области техники, например, как описано в патенте США № 4522811.
Пероральные или парентеральные композиции можно формулировать в стандартную лекарственную форму для простоты введения и однородности дозирования. Стандартная лекарственная форма относится к физически дискретным единицам, подходящим в качестве единиц дозирования для субъекта, который подвергается лечению; причем каждая единица содержит предварительно определенное количество активного соединения, рассчитанное для получения требуемого терапевтического эффекта в сочетании с требуемым фармацевтическим носителем. Спецификация стандартных лекарственных форм настоящего изобретения определяется и непосредственно зависит от уникальных характеристик активного соединения и терапевтического эффекта, который нужно достигнуть, и ограничений, присущих данной области техники получения данного активного соединения для лечения индивидуумов. Кроме того, введение может осуществляться периодической инъекцией болюса, или его можно осуществлять более непрерывно внутривенным, внутримышечным или внутрибрюшинным введением из внешней емкости (например, контейнера для внутривенного введения).
Когда требуется адгезия на поверхности ткани, композиция может содержать лекарственное средство, диспергированное на фиброген-тромбиновой композиции или другом биоадгезиве. Затем соединение можно наносить, распылять или, другими словами, применять на требуемой поверхности ткани. Альтернативно, лекарственное средство можно формулировать для парентерального или перорального введения людям или другим млекопитающим, например, в эффективных количествах, например количествах, которые дают подходящие концентрации лекарственного средства в целевой ткани в течение времени, достаточном для того, чтобы вызвать требуемый эффект.
Когда активное соединение предполагается применять в качестве части процесса трансплантации, его можно доставлять к живой ткани или органу, которые будут трансплантировать, перед удалением ткани или органа у донора. Соединение можно предоставлять хозяину, являющемуся донором. Альтернативно или, кроме того, после удаления у донора, орган или живую ткань можно помещать в раствор для сохранения, содержащий активное соединение. Во всех случаях, активное соединение можно вводить непосредственно на требуемую ткань, как инъекцией в ткань, или оно может быть предоставлено системно, или пероральным, или парентеральным введением, применяя любой из способов и композиций, описанных в настоящем изобретении и/или известных в данной области техники. Когда лекарственное средство содержит часть раствора для хранения ткани или органа, для получения преимуществ можно применять любой имеющийся в продаже раствор для хранения. Например, подходящие растворы, известные в данной области техники, включают раствор Коллинза, раствор Винсконсина, раствор Бельцера, «Eurocollins» раствор и лактированный раствор Рингера.
В сочетании со способами настоящего изобретения можно принять во внимание фармакогеномику (т.е., исследование связи генотипа индивидуума и ответной реакции индивидуума на чужеродное соединение или лекарственное средство). Различия в метаболизме лекарственных средств могут приводить к сильной токсичности или терапевтической неудаче за счет изменения соотношения между дозой и концентрацией в крови фармакологически активного лекарственного средства. Таким образом, врач или терапевт может учитывать применение знаний, полученных в соответствующих фармакогеномных исследованиях, для определения того, вводить ли лекарственное средство, а также варьировать ли дозирование и/или терапевтический режим лечения лекарственным средством.
Обычно, эффективное количество дозы активного соединения будет в диапазоне от приблизительно 0,1 до приблизительно 100 мг/кг веса тела/день, более предпочтительно от приблизительно 1,0 до приблизительно 50 мг/кг веса тела/день. Вводимое количество будет также вероятно зависеть от таких переменных, как тип операции или инвазивная медицинская процедура, общее состояние здоровья пациента, относительная биологическая эффективность доставляемого соединения, композиция лекарственного средства, присутствие и типы вспомогательных веществ в композиции, и способа введения. Кроме того, должно быть ясно, что первоначальную вводимую дозу можно увеличивать за пределы верхнего уровня для того, чтобы быстро достигать требуемую концентрацию в крови или ткани, или первоначальное дозирование может быть меньшим, чем оптимальное.
Неограничивающие дозы активного соединения включают от приблизительно 0,1 до приблизительно 1500 мг на дозу. Неограничивающие примеры доз, которые можно формулировать в виде единичной дозы для общепринятого введения пациенту, включают: приблизительно 25 мг, приблизительно 50 мг, приблизительно 75 мг, приблизительно 100 мг, приблизительно 125 мг, приблизительно 150 мг, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг, приблизительно 325, приблизительно 350 мг, приблизительно 375 мг, приблизительно 400 мг, приблизительно 425 мг, приблизительно 450 мг, приблизительно 475 мг, приблизительно 500 мг, приблизительно 525 мг, приблизительно 550 мг, приблизительно 575 мг, приблизительно 600 мг, приблизительно 625 мг, приблизительно 650 мг, приблизительно 675 мг, приблизительно 700 мг, приблизительно 725 мг, приблизительно 750 мг, приблизительно 775 мг, приблизительно 800 мг, приблизительно 825 мг, приблизительно 850 мг, приблизительно 875 мг, приблизительно 900 мг, приблизительно 925 мг, приблизительно 950 мг, приблизительно 975 мг, приблизительно 1000 мг, приблизительно 1025 мг, приблизительно 1050, мг, приблизительно 1075 мг, приблизительно 1100 мг, приблизительно 1125 мг, приблизительно 1150 мг, приблизительно 1175 мг, приблизительно 1200 мг, приблизительно 1225 мг, приблизительно 1250 мг, приблизительно 1275 мг, приблизительно 1300 мг, приблизительно 1325 мг, приблизительно 1350 мг, приблизительно 1375 мг, приблизительно 1400 мг, приблизительно 1425 мг, приблизительно 1450 мг, приблизительно 1475 мг и приблизительно 1500 мг. Вышеуказанные дозы являются пригодными для введения соединений настоящего изобретения согласно способам настоящего изобретения.
Как ясно специалистам в данной области техники, обычно, когда описывают дозирование для фармацевтически активного соединения, дозирование дают относительно исходной или активной молекулы. Следовательно, если применяют соль, гидрат или другую форму исходной молекулы, осуществляют соответствующий пересчет веса соединения, хотя доза все еще приводится относительно доставляемой исходной или активной молекулы. В качестве неограничивающего примера, если интересующая исходная или активная молекула представляет собой монокарбоновую кислоту, имеющую молекулярный вес 250, и если требуется доставить мононатриевую соль кислоты при той же дозировке, затем осуществляют пересчет, учитывая, что мононатриевая соль будет иметь молекулярный вес приблизительно 272 (т.е. минус 1H или 1,008 атомных массовых единиц и плюс 1 Na или 22,99 атомных массовых единиц). Следовательно, 250 мг доза исходного или активного соединения будет соответствовать приблизительно 272 мг мононатриевой соли, которая также будет доставлять 250 мг исходного или активного соединения. Говоря другими словами, приблизительно 272 мг мононатриевой соли будут эквивалентны 250 мг дозе исходного или активного соединения.
Все проценты и отношения, применяемые в настоящем изобретении, если не указано особо, даны по весу. Процент димерной примеси дается в расчете на проценты площади, обычно как количественно получено аналитической ВЭЖХ.
Во всем описании, при описании композиций как имеющих, содержащих или включающих конкретные компоненты, предполагается, что композиции также состоят по существу, или состоят из перечисленных компонентов. Аналогично, при описании способов или процессов как имеющих, включающих или содержащих конкретные стадии процесса, процессы также состоят по существу или состоят из перечисленных стадий процесса. Кроме того, должно быть ясно, что порядок стадий или порядок проведения определенных действий не является существенным, поскольку настоящее изобретение остается работоспособным. Более того, две или более стадий или действий можно проводить одновременно.
Примеры композиций
Композиция для внутривенного введения
Данную композицию для внутривенного введения формулировали нагреванием воды для инъекции до приблизительно 60°C. Затем добавляли цитрат натрия, лимонную кислоту и декстрозу и перемешивали до растворения. Добавляли к полученной смеси раствор или водную суспензию противомикробного соединения и перемешивали до растворения. Смесь охлаждали до 25°C при перемешивании. Измеряли pH и регулировали ее в случае необходимости. Смесь доводили до требуемого объема, при необходимости, водой для инъекции. Смесь фильтровали, наполняли в требуемый контейнер (пузырек, шприц, контейнер для вливания и т.д.), обворачивали и термически стерилизовали влажным паром.
Данная композиция является пригодной для внутривенного введения, или болюса, или вливания, пациенту.
Таблетки для перорального введения
Противомикробное соединение (любое из соединений, эквивалентных требуемому вводимому количеству, например, 50-1500 мг на таблетку) смешивали с 1/3 микрокристаллической целлюлозы NF и 1/2 безводной лактозы NF в ленточном смесителе в течение 5 минут при 20 об/мин. К смеси добавляли оставшиеся 2/3 микрокристаллической целлюлозы NF и оставшуюся 1/2 безводной лактозы NF. Смесь перемешивали в течение 10 минут при 20 об/мин. Добавляли к перемешанному порошку кросскармелозу натрия и перемешивали в течение 5 минут при 20 об/мин. Добавляли к смеси стеарат магния пропусканием через 90 меш сито и перемешивали в течение дополнительных 5 минут при 20 об/мин. Смазанную смесь сдавливали для получения таблеток с 500 мг активного ингредиента.
Данные таблетки являются пригодными для перорального введения пациенту.
Введение с помощью ссылки
Полное описание каждого из патентных документов, включая удостоверения об исправлении ошибок в описании, патентные заявки, научные статьи, отчеты, предусмотренные государственными органами, вебсайты и другие материалы, на которые ссылаются в настоящем изобретении, вводится с помощью ссылки полностью во всех отношениях. В случае конфликта в терминологии следует руководствоваться настоящим описанием.
Эквиваленты
Настоящее изобретение можно воплотить в других конкретных формах, не выходя за его пределы или существенных характеристик. Следовательно, вышеуказанные варианты осуществления следует рассматривать во всех аспектах как иллюстративные, а не ограничивающие настоящее изобретение, описанное в настоящем описании. Таким образом, объем настоящего изобретения определяется прилагаемой формулой изобретения, а не вышеуказанным описанием, и предполагается, что все изменения, которые подпадают под значения и диапазоны равноценности формулы изобретения, включены в нее.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2013 |
|
RU2632203C2 |
| ДЕГАЛОГЕНИРОВАННЫЕ СОЕДИНЕНИЯ, АНТИБАКТЕРИАЛЬНОЕ СРЕДСТВО И ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ, СПОСОБ ПОЛУЧЕНИЯ АНТИБАКТЕРИАЛЬНОГО ИЛИ ЛЕКАРСТВЕННОГО СРЕДСТВА, ПРИМЕНЕНИЕ ДЕГАЛОГЕНИРОВАННЫХ СОЕДИНЕНИЙ ДЛЯ ПОЛУЧЕНИЯ АНТИБАКТЕРИАЛЬНОГО ИЛИ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2001 |
|
RU2298006C2 |
| СЛОЖНЫЕ АРИЛАЛКИЛОВЫЕ ЭФИРЫ 4-АМИНО-6-(ЗАМЕЩЕННЫЙ ФЕНИЛ)ПИКОЛИНАТОВ И 6-АМИНО-2-(ЗАМЕЩЕННЫЙ ФЕНИЛ)-4-ПИРИМИДИНКАРБОКСИЛАТОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2012 |
|
RU2566760C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2437886C2 |
| БИОЛОГИЧЕСКИ ДОСТУПНАЯ ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ КОФЕЙНАЯ КИСЛОТА, ОТНОСЯЩАЯСЯ К ПРОТИВООПУХОЛЕВЫМ ЛЕКАРСТВЕННЫМ СРЕДСТВАМ | 2007 |
|
RU2456265C2 |
| 6-АМИНО-2-ЗАМЕЩЕННЫЕ-5- ВИНИЛСИЛИЛПИРИМИДИН-4-КАРБОНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ И 4-АМИНО-6-ЗАМЕЩЕННЫЕ-3-ВИНИЛСИЛИЛПИРИДИН-ПИКОЛИНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ КАК ГЕРБИЦИДЫ | 2012 |
|
RU2556000C2 |
| СЕЛЕКТИВНЫЕ ЛИГАНДЫ-РАЗРУШИТЕЛИ АНДРОГЕННЫХ РЕЦЕПТОРОВ (SARD) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2689988C2 |
| ТРИАЗОЛОПИРИДИНОВОЕ СОЕДИНЕНИЕ И ЕГО ДЕЙСТВИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОЛИЛГИДРОКСИЛАЗЫ И ИНДУКТОРА ВЫРАБОТКИ ЭРИТРОПОЭТИНА | 2010 |
|
RU2538963C2 |
| L-НУКЛЕОЗИДЫ, ОБЛАДАЮЩИЕ АНТИ-HBV ИЛИ АНТИ-EBV АКТИВНОСТЬЮ, СПОСОБ ИНГИБИРОВАНИЯ HBV ИЛИ EBV ИНФЕКЦИИ | 1995 |
|
RU2171809C2 |
| ПРОЛЕКАРСТВА КАРБИДОПА И L-DOPA И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2015 |
|
RU2743347C2 |


Изобретение относится к области органической химии, а именно к способу получения хинолонового соединения, включающему стадию взаимодействия дехлорхинолонового соединения, или его фармацевтически приемлемой соли, или эфира с хлорирующим агентом и кислотой, в котором молярное отношение кислоты к дехлорхинолоновому соединению составляет от 0,008 до 0,012 и в котором получают менее чем 0,40% димерной примеси в процентах площади в расчете на полученное хинолоновое соединение, и где хинолоновое соединение представляет собой 1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-6-фтор-7-(3-гидроксиазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту, или ее фармацевтически приемлемую соль, или эфир, дехлорхинолоновое соединение представляет собой 1-(6-амино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-гидроксиазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту, или ее фармацевтически приемлемую соль, или эфир и димерная примесь представляет собой 1-амино-3-(азетидин-3-илиокси)пропан-2-олбис(Ν,Ν′-хинолонкарбоновую кислоту), или ее фармацевтически приемлемую соль, или эфир. Технический результат: разработан улучшенный способ получения производного хинолона, полезного в качестве противоинфекционного средства. 14 з.п. ф-лы, 21 ил., 2 пр., 2 табл.
1. Способ получения хинолонового соединения, включающий стадию взаимодействия дехлорхинолонового соединения, или его фармацевтически приемлемой соли, или эфира с хлорирующим агентом и кислотой, в котором молярное отношение кислоты к дехлорхинолоновому соединению составляет от 0,008 до 0,012 и в котором получают менее чем 0,40% димерной примеси в процентах площади в расчете на полученное хинолоновое соединение, и где хинолоновое соединение представляет собой 1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-6-фтор-7-(3-гидроксиазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту, или ее фармацевтически приемлемую соль, или эфир, дехлорхинолоновое соединение представляет собой 1-(6-амино-3,5-дифторпиридин-2-ил)-6-фтор-7-(3-гидроксиазетидин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту, или ее фармацевтически приемлемую соль, или эфир и димерная примесь представляет собой 1-амино-3-(азетидин-3-илиокси)пропан-2-ол-бис(Ν,Ν′-хинолонкарбоновую кислоту), или ее фармацевтически приемлемую соль, или эфир.
2. Способ по п. 1, в котором хлорирующий агент представляет собой N-хлорсукцинимид.
3. Способ по п. 1, в котором кислоту выбирают из группы, состоящей из серной кислоты, хлористоводородной кислоты, бромистоводородной кислоты, фосфорной кислоты, трифторуксусной кислоты, трифторметансульфокислоты, метансульфокислоты, п-толуолсульфокислоты или перхлорной кислоты и их смесей.
4. Способ по п. 1, в котором кислота представляет собой серную кислоту.
5. Способ по п. 1, в котором реакцию проводят при температуре от 0°С до 30°С.
6. Способ по п. 1, в котором молярное отношение хлорирующего агента к дезхлорхинолону составляет более чем 1.
7. Способ по п. 1, где ацетатный эфир применяют в качестве растворителя.
8. Способ по п. 7, в котором ацетатный эфир выбирают из группы, состоящей из метилацетата, этилацетата и их смесей.
9. Способ по п. 8, в котором ацетатный эфир представляет собой метилацетат.
10. Способ по п. 1, включающий дополнительную стадию взаимодействия хинолонового соединения с основанием.
11. Способ по п. 10, в котором основание представляет собой гидроксид.
12. Способ по п. 11, в котором гидроксид выбирают из группы, состоящей из гидроксида натрия, гидроксида калия, гидроксида лития, гидроксида бария и их смесей.
13. Способ по п. 12, в котором гидроксид представляет собой гидроксид калия.
14. Способ по п. 10, согласно которому применяют смесь изопропанола и воды в качестве растворителя.
15. Способ по п. 1, предназначенный для промышленного применения.
| Haight A.R | |||
| Катодный усилитель | 1923 |
|
SU492A1 |
| Barnes D.M | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |

