ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям общей формулы I, действующим и как антагонисты мускариновых рецепторов, и как агонисты бета-2-адренергических рецепторов, к способам их получения, к композициям, содержащим их, к терапевтическому применению и комбинациям с другими фармацевтически активными ингредиентами.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Легочные расстройства, такие как астма и хроническая обструктивная болезнь легких (ХОБЛ), обычно лечат бронходилататорами. Известный класс бронходилататоров состоит из агонистов бета-2-адренергических рецепторов, таких как сальбутамол, фенотерол, формотерол и сальметерол. Эти соединения обычно вводят посредством ингаляции.
Другой известный класс бронходилататоров состоит из антагонистов мускариновых рецепторов (антихолинергические соединения), таких как ипратропий и тиотропий. Эти соединения обычно также вводят посредством ингаляции.
Ингалируемые препараты как бета-2-агонистов, так и антагонистов мускариновых рецепторов являются чрезвычайно полезными агентами при лечении астмы и ХОБЛ, при этом оба класса агентов обеспечивают симптоматическое облегчение вследствие их способности расслаблять суженные дыхательные пути. Данные наблюдений о том, что бронходилататорные эффекты двух классов агентов были аддитивными, побудили к исследованиям комбинаций двух агентов. В 1975 году было показано, что эти положительные эффекты могут быть достигнуты путем объединения двух ингредиентов, таких как фенотерол и ипратропий бромид, в одном аэрозоле. Это повлекло за собой разработку комбинированных препаратов ипратропия бромида с фиксированными дозами сначала с фенотеролом (беродуал, выпущенный в 1980), а затем с сальбутамолом (комбивент, выпущенный в 1994).
В последнее время доступность как антагонистов мускариновых рецепторов длительного действия, так и бета-2-агонистов длительного действия повлекла за собой разработку комбинаций этих агентов. Например, в WO 00/69468 раскрыты композиции лекарственных средств, содержащие антагонист мускариновых рецепторов, такой как тиотропий бромид, и агонисты бета-2-адренергических рецепторов, такие как фумарат формотерола или сальметерол, а в WO 2005/115467 раскрыта комбинация, которая содержит бета-2-агонист и антагонист мускариновых рецепторов подтипа М3, который представляет собой соль 3(R)-(2-гидрокси-2,2-дитиен-2-илацетокси)-1-(3-феноксипропил)-1-азониабицикло[2.2.2]октана.
Альтернативным подходом к разработке комбинированных препаратов с фиксированными дозами является выявление молекул, которые сочетают в себе активность как мускариновых антагонистов, так и бета-2-агонистов. Действительно, соединения обладающие как активностью агонистов бета-2-адренергических рецепторов, так и активностью антагонистов мускариновых рецепторов, чрезвычайно востребованы, поскольку такие бифункциональные соединения могут обеспечивать бронходилатацию посредством двух независимых механизмов действия, имея одну молекулярную фармакокинетику.
Такой вид соединений был описан в некоторых публикациях международных заявок, таких как WO 2004/074246, WO 2004/074812, WO 2005/051946, WO 2006/023457, WO 2006/023460, WO 2010/123766, WO 2011/048409 и находящаяся на одновременном рассмотрении заявка WO 2012/168359.
В настоящее время обнаружено, что некоторые конкретные карбаматные производные, помимо того, что они обладают как активностью агонистов бета-2-адренергических рецепторов, так и активностью антагонистов мускариновых рецепторов, обладают повышенным сродством к мускариновым рецепторам подтипа М3 и бронходилатирующей активностью длительного действия.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям общей формулы I, действующим и как антагонисты мускариновых рецепторов, и как агонисты бета-2-адренергических рецепторов, к способам их получения, к композициям, содержащим их, к терапевтическому применению и комбинациям с другими фармацевтически активными ингредиентами, среди которых имеются, например, те, которые используются в настоящее время при лечении респираторных нарушений, среди них бета-2-агонисты, антимускариновые агенты, ингибиторы митоген-активируемых протеинкиназ (MAP киназы р38), ингибиторы бета-субъединицы киназы ядерного фактора каппа-В (IKK2), ингибиторы эластазы нейтрофилов человека (HNE), ингибиторы фосфодиэстеразы-4 (PDE4), модуляторы лейкотриенов, нестероидные противовоспалительные агенты (NSAID), противокашлевые агенты, регуляторы секреции слизи, муколитические агенты, отхаркивающие средства/мукокинетические модуляторы, пептидные муколитические агенты, антибиотики, ингибиторы JAK (Янус-киназ), ингибиторы SYK (тирозинкиназы селезенки), ингибиторы РI3Кдельта или РI3Кгамма, кортикостероиды и М3-антагонисты/РDЕ4-ингибиторы (MAPI).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
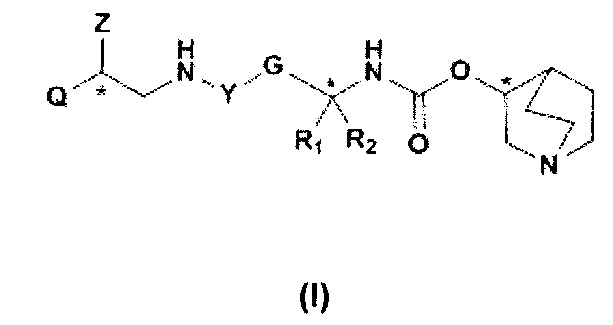
В частности, изобретение относится к соединениям общей формулы I
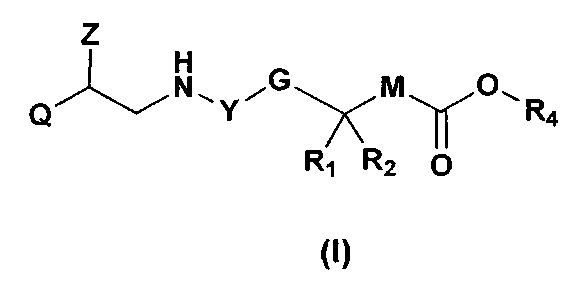 ,
,
где
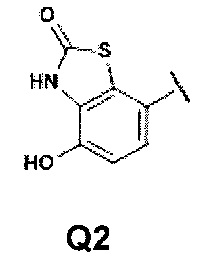
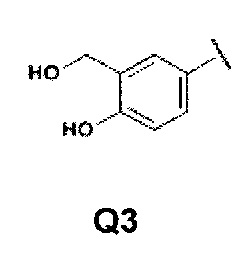
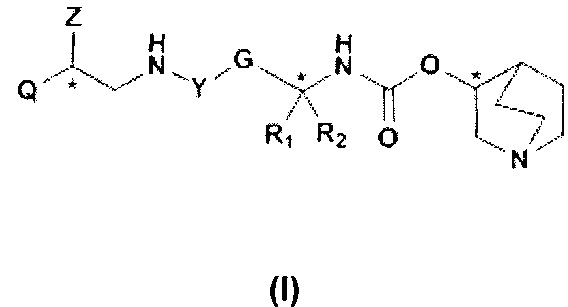
Q представляет собой группу формулы Q1, Q2 и Q3

Z представляет собой Н или ОН;
Y выбран из Y' и Y1, которые представляют собой двухвалентные группы формулы
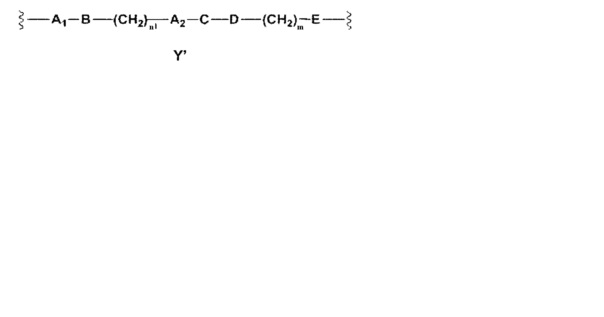
или
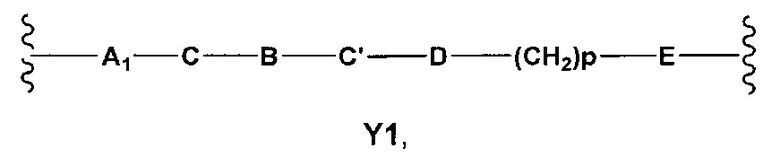
где
A1 и А2 независимо отсутствуют или выбраны из группы, состоящей из (C1-C6)алкилена, (C3-C8)циклоалкилена и (C3-C8)гетероциклоалкилена, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из (C1-C6)алкила, арил(C1-C6)алкила и гетероарил(C1-C6)алкила;
В отсутствует или выбран из группы, состоящей из (C3-C8)циклоалкилена, (C3-C8)гетероциклоалкилена, арилена и гетероарилена, возможно замещенных одной или более чем одной группой, выбранной из галогенов, нитрила, прямого или разветвленного (C1-C6)алкила, прямого или разветвленного (C1-C6)галогеноалкила, (C1-C6)алкокси, арила, арил(C1-C6)алкила, -NR7(R8) и гетероарила;
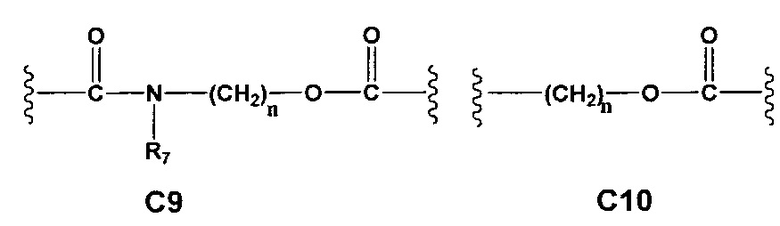
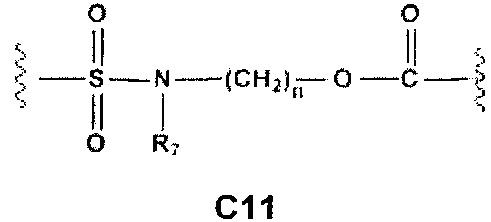
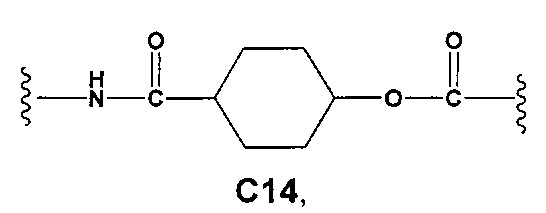
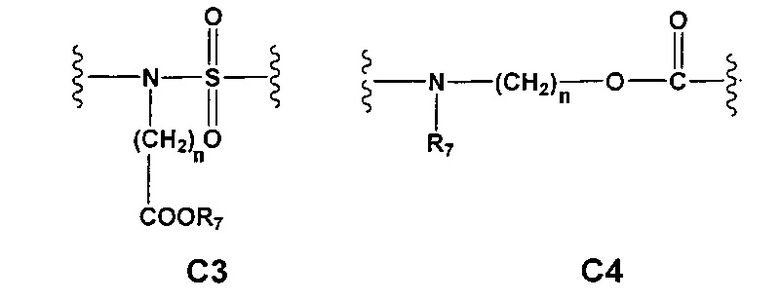
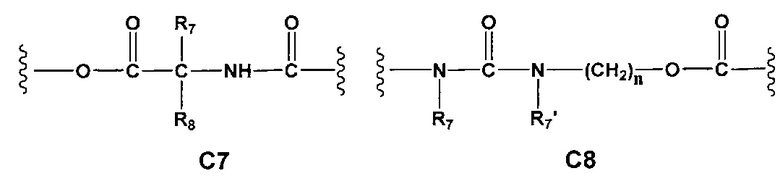
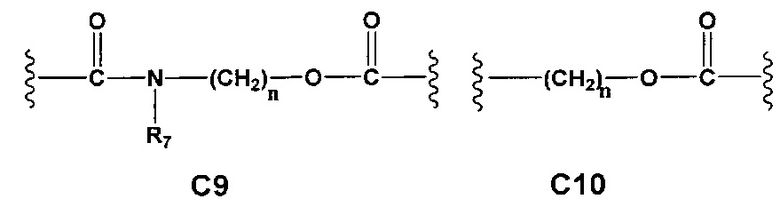
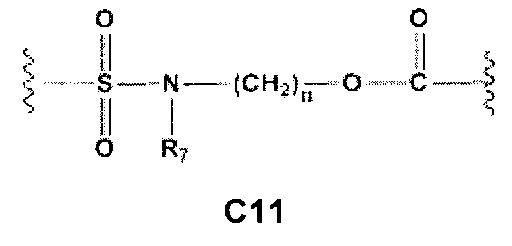
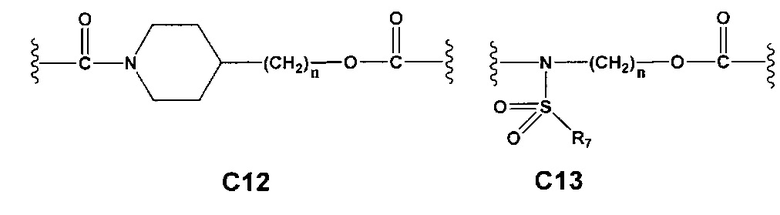
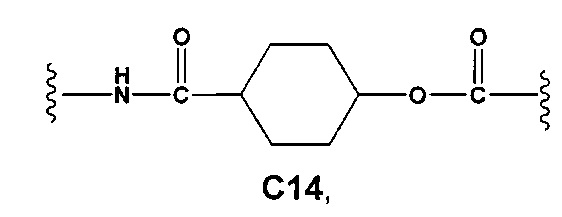
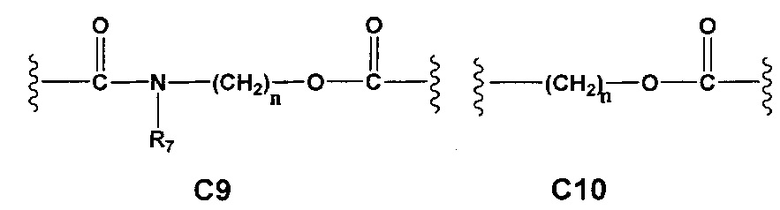
С и C' отсутствуют или независимо выбраны из группы, состоящей из -O-, -CO-, -OC(O)- и -C(OO)-, или представляют собой одну из следующих групп С1-С14
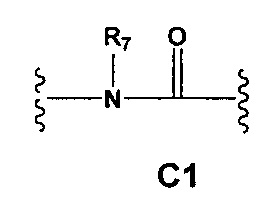
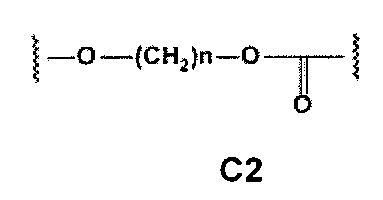
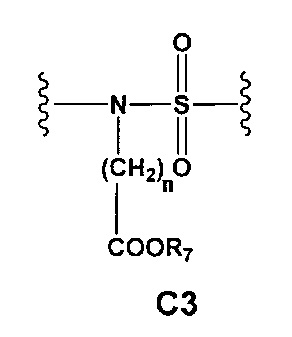
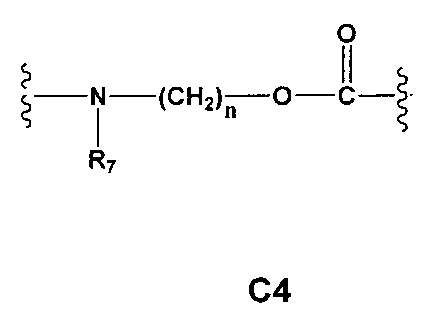
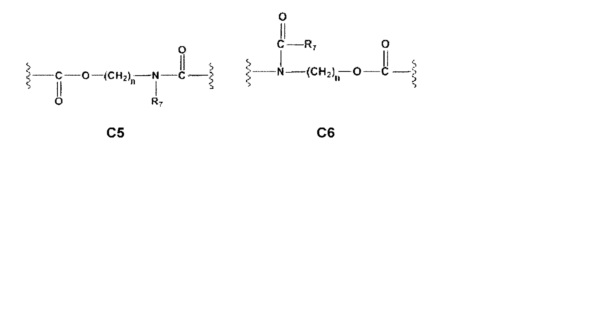
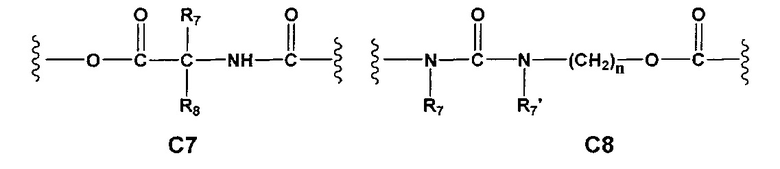
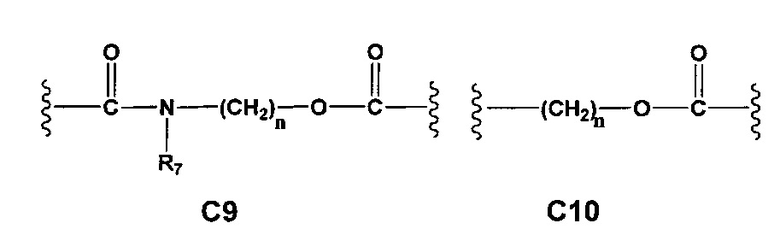
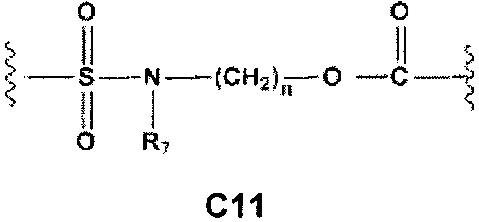
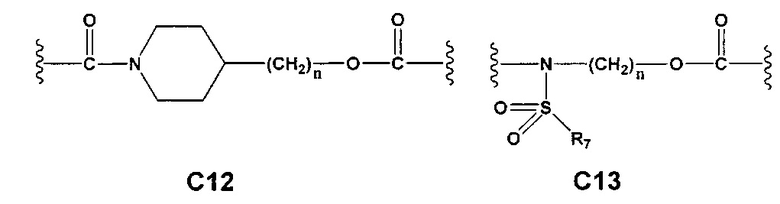
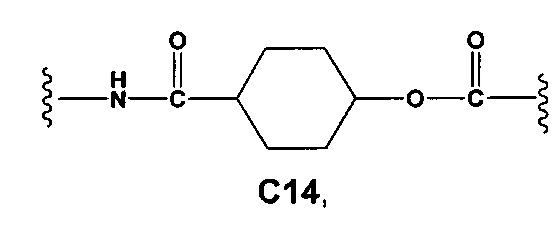
где R7, R7' и R8 независимо представляют собой Н или выбраны из группы, состоящей из прямого или разветвленного (С1-С6)алкила, (C3-C8)циклоалкила, (C3-C8)циклоалкил(C1-C6)алкила, (C3-C8)гетероциклоалкил(C1-C6)алкила, арила и арил(C1-C6)алкила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из (C1-C6)алкила, галогено(C1-C6)алкила, атомов галогена, (C1-C6)алкокси и галогено(C1-C6)алкокси;
D отсутствует или выбран из группы, состоящей из (C1-C6)алкилена, арилена, гетероарилена и (C3-C8)гетероциклоалкилена, возможно замещенных одной или более чем одной (C1-C6)алкильной группой;
n, n', m и p независимо равны 0 или целому числу от 1 до 3;
Е отсутствует или выбран из -O- и -OC(O)-;
G представляет собой арилен, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена, -OH, оксо(=O), -SH, -NO2, -CN и -NH2;
R1 и R2 независимо представляют собой Н или выбраны из группы, состоящей из (C1-C6)алкила и арила, возможно замещенных одним или более чем одним атомом галогена;
М представляет собой -N(R3)-;
R3 представляет собой Н или (C1-C6)алкил;
R4 представляет собой группу формулы J1
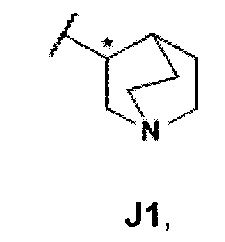
и их фармацевтически приемлемым солям или сольватам.
Выражение "(C1-Cx)алкил" относится к алкильным группам с прямой или разветвленной цепью, где число атомов углерода составляет от 1 до x. Примерами групп являются метил, этил, н-пропил, изопропил, трет-бутил, пентил, гексил, октил, нонил, децил, ундецил, додецил и им подобные.
Аналогичным образом, выражение "(C1-Cx)алкилен" относится к двухвалентным группам, таким как метилен, этилен, н-пропилен, изопропилен, трет-бутилен, пентилен, гексилен, октилен, нонилен, децилен, ундецилен, додецилен и им подобные.
Выражение "(C1-C6)галогеноалкил" относится к вышеупомянутой группе "(C1-C6)алкил", где один или более чем один атом водорода заменен одним или более чем одним из атомов галогена, которые могут быть одинаковыми или отличаться друг от друга.
Примеры указанных (C1-C6)галогеноалкильных групп включают галогенированные, полигалогенированные и полностью галогенированные алкильные группы, где один или более чем один атом водорода заменен атомами галогена, например, трифторметильную группу.
Выражение "(C1-C6)алкокси" относится к группам алкил-окси (например алкокси), причем алкильная часть является такой, как определено выше. Примеры указанных групп включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, пентокси, гексокси и им подобные.
Выражение "галогено(C1-C6)алкокси" включает галогенированные, полигалогенированные и полностью галогенированные группы алкил-окси (например алкокси), причем алкоксильная часть является такой, как определено выше, где один или более чем один атом водорода заменен атомами галогена, например, трифторметоксигруппу.
Выражение "(C3-C8)циклоалкил" относится к моно- или бициклоалифатическим углеводородным группам с 3-8 атомами углерода. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, бицикло[2.2.1]гепт-2-ил и им подобные.
Выражение "(C3-C8)гетероциклоалкил" относится к (C3-C8)циклоалкильным группам, в которых по меньшей мере один кольцевой атом углерода заменен гетероатомом или гетероароматической группой (например N, NH, S или О). Примеры включают хинуклидинил, n и pролидинил, пиперидинил, азабицикло[3.2.1]октан-3-ил и азониабицикло[2.2.2]октанил и им подобные.
Аналогичным образом, выражения "(C3-C8)циклоалкилен" и "(C3-C8)гетероциклоалкилен" относятся к двухвалентным группам, таким как, соответственно, циклопропилен, циклобутилен, циклопентилен, циклогексилен, циклогептилен, бицикло[2.2.1]гепт-2-илен и хинуклидинилен, пиpролидинилен, пиперидинилен, азабицикло[3.2.1]октан-3-илен, азониабицикло[2.2.2]октанилен и им подобным.
Выражение "арил" относится к моно-, би- или трициклическим кольцевым системам, имеющим от 5 до 20, предпочтительно от 5 до 15, кольцевых атомов, и где по меньшей мере одно кольцо является ароматическим.
Выражение "гетероарил" относится к моно-, би- или трициклическим системам с 5-20 кольцевыми атомами, предпочтительно 5-15, в которых по меньшей мере одно кольцо является ароматическим и в которых по меньшей мере один кольцевой атом углерода является гетероатомом или гетероароматической группой (например, N, NH, S или О).
Примеры подходящих арильных или гетероарильных моноциклических систем включают, например, радикалы тиофена, бензола, n и pрола, пиразола, имидазола, изоксазола, оксазола, изотиазола, тиазола, пиpидина, имидазолидина, фурана и им подобные.
Примеры подходящих арильных или гетероарильных бициклических систем включают радикалы нафталина, бифенилена, пурина, птеридина, бензотриазола, хинолина, изохинолина, индола, изоиндола, бензотиофена, дигидробензодиоксина, дигидроиндена, дигидробензодиоксепина, бензооксазина и им подобные.
Примеры подходящих арильных или гетероарильных трициклических систем включают флуореновые радикалы, а также бензоконденсированные производные вышеуказанных гетероарильных бициклических систем.
Аналогичным образом, выражения "арилен" и "гетероарилен" относятся к двухвалентным группам, таким какфенилен, бифенилен и тиенилен.
Выражения "арил(C1-C6)алкил", "гетероарил(C1-C6)алкил", "(C3-C8)гетероциклоалкил(C1-C6)алкил" и "(C3-C8)циклоалкил(C1-C6)алкил" относятся к (C1-C6)алкилу, замещенному соответственно одной или более чем одной арильной, гетероарильной, (C3-C8)гетероциклоалкильной или (C3-C8)циклоалкильной группой, как определено выше.
Примеры арил(C1-C6)алкила включают трифенилметил.
В каждом случае, когда основные амино- или четвертичные аммониевые группы присутствуют в соединениях формулы I, могут присутствовать и физиологически приемлемые анионы, выбранные из хлорида, бромида, иодида, трифторацетата, формиата, сульфата, фосфата, метансульфоната, нитрата, малеата, ацетата, цитрата, фумарата, тартрата, оксалата, сукцината, бензоата, пара-толуолсульфоната, памоата и нафталиндисульфоната. Подобным образом, в присутствии кислотных групп, таких как СООН группы, также могут присутствовать соответствующие физиологические катионы солей, например, включая ионы щелочных или щелочноземельных металлов.
Будет очевидно, что соединения общей формулы I могут содержать ассиметрические центры. Поэтому, изобретение также включает любые из оптических стереоизомеров, диастереоизомеров и их смесей в любом соотношении.
В частности, атом углерода, связанный с R1, R2, G и М группами, в зависимости от обозначений, данных для R1 и R2 из числа ранее представленных, может представлять собой хиральный центр.
В одном воплощении конфигурацией является (S).
В другом воплощении абсолютной конфигурацией этого хирального центра является предпочтительно (R).
В другом предпочтительном воплощении соединения общей формулы I, описанные в настоящем изобретении, присутствуют в виде смесей диастереоизомеров.
В другом воплощении, когда в соединениях общей формулы I
R4 представляет собой группу формулы J1

атом углерода, помеченный звездочкой, представляет собой хиральный центр.
В предпочтительном воплощении этот хиральной центр имеет (R) конфигурацию.
Специалисту в данной области техники совершенно ясно, что соединения общей формулы I, где R4 представляет собой J1, содержат три стереогенных центра, которые указаны ниже с помощью звездочек (*). Это означает, что структура формулы I характеризуется восемью различными стереоизомерами.
Следует понимать, что все предпочтительные группы или воплощения, описанные ниже для соединений формулы I, могут быть объединены друг с другом и применяться также с учетом необходимых изменений.
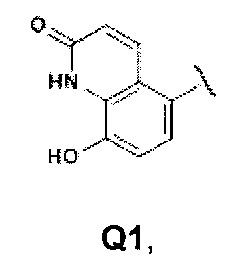
Первой предпочтительной группой соединений являются соединения общей формулы I, где Q представляет собой группу формулы Q1, Q2 и Q3
Z представляет собой Н или ОН;
Y выбран из Y' и Y1, которые являются двухвалентными группами формулы
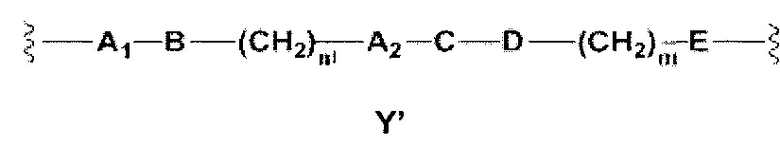
или
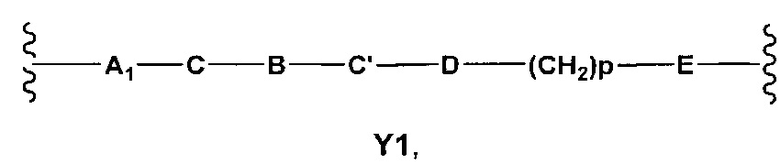
где
A1 и А2 независимо отсутствуют или выбраны из группы, состоящей из (C1-C6)алкилена, (C3-C8)циклоалкилена и (C3-C8)гетероциклоалкилена, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из (C1-C6)алкила, арил(C1-C6)алкила и гетероарил(C1-C6)алкила; В отсутствует или выбран из группы, состоящей из (C3-C8)циклоалкилена, (C3-C8)гетероциклоалкилена, арилена и гетероарилена, возможно замещенных одной или более чем одной группой, выбранной из галогенов, нитрила, прямого или разветвленного (C1-C6)алкила, прямого или разветвленного (C1-C6)галогеноалкила, (C1-C6)алкокси, арила, арил(C1-C6)алкила, -NR7(R8) и гетероарила; С и C' отсутствуют или независимо выбраны из группы, состоящей из -O-, -CO-, -OC(O)- и -C(OO)-, или представляют собой одну из следующих групп С1-С14
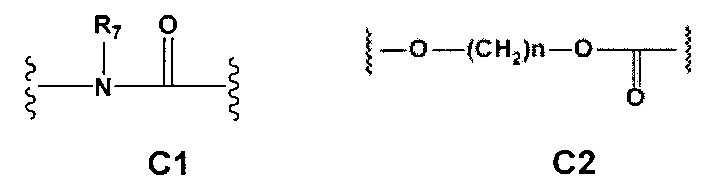
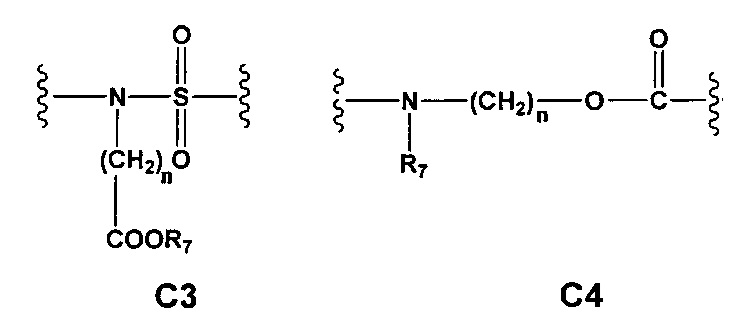
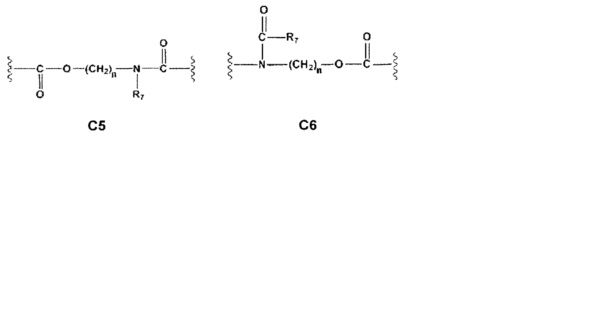
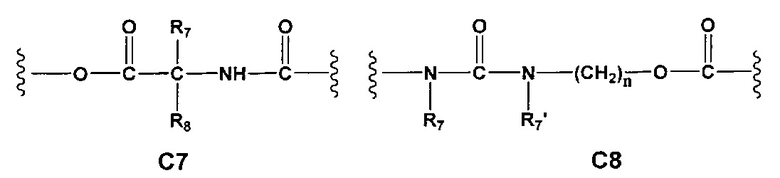

где R7, R7' и R8 независимо представляют собой Н или выбраны из группы, состоящей из прямого или разветвленного (C1-C6)алкила, (C3-C8)циклоалкила, (С3-С8)циклоалкил(С1-С6)алкила, (C3-C8)гетероциклоалкил(C1-C6)алкила, арила и арил(С1-С6)алкила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из (C1-C6)алкила, галогено(C1-C6)алкила, атомов галогена, (C1-C6)алкокси и галогено(C1-C6)алкокси;
D отсутствует или выбран из группы, состоящей из (C1-C6)алкилена, арилена, гетероарилена и (C3-C8)гетероциклоалкилена, возможно замещенных одной или более чем одной (C1-C6)алкильной группой; n, n', m и p независимо равны 0 или целому числу от 1 до 3; Е отсутствует или выбран из -O- и -OC(O)-; G представляет собой арилен, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -CN и -NH2; R1, R2, М, R4 и R6 являются такими, как определено выше.
Еще более предпочтительными в этой первой группе являются соединения общей формулы I, где Q представляет собой Q1
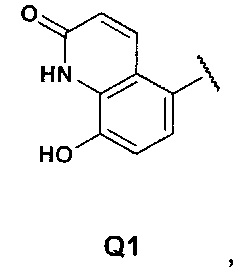
Z представляет собой Н или ОН;
Y представляет собой Y1, который является двухвалентной группой формулы
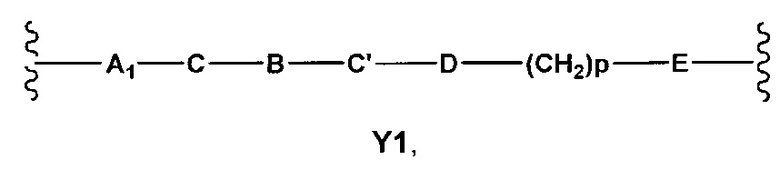
где
A1 отсутствует или выбран из группы, состоящей из (C1-C6)алкилена, (C3-C8)циклоалкилена и (C3-C8)гетероциклоалкилена, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из (C1-C6)алкила, арил(C1-C6)алкила и гетероарил(C1-C6)алкила; В отсутствует или выбран из группы, состоящей из (C3-C8)циклоалкилена, (С3-С8)гетероциклоалкилена, арилена и гетероарилена, возможно замещенных одной или более чем одной группой, выбранной из галогенов, нитрила, прямого или разветвленного (C1-C6)алкила, прямого или разветвленного (C1-C6)галогеноалкила, (C1-C6)алкокси, арила и гетероарила; С и C' отсутствуют или независимо выбраны из группы, состоящей из -O-, -CO-, -OC(O)- и -C(OO)-, или представляют собой одну из следующих групп С1-С14
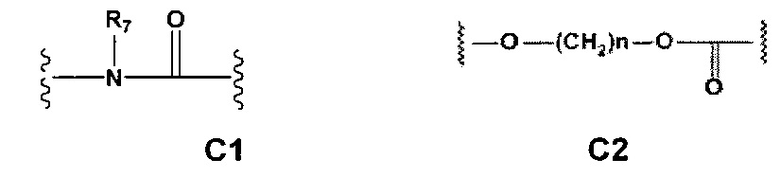
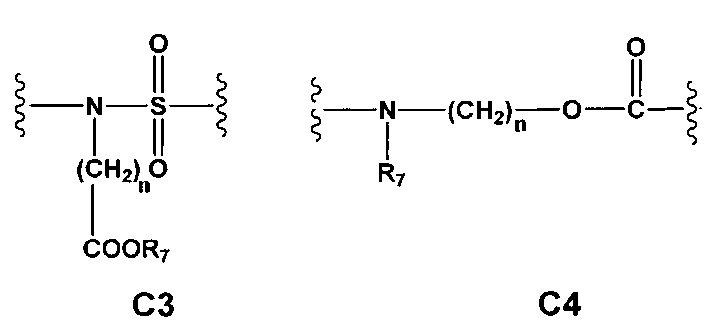

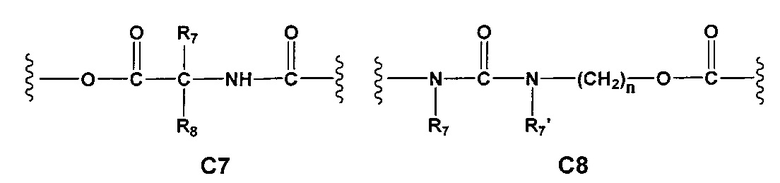
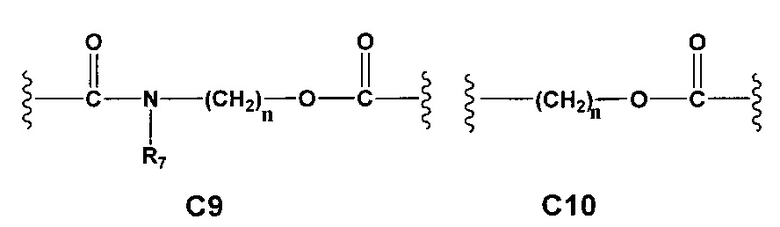
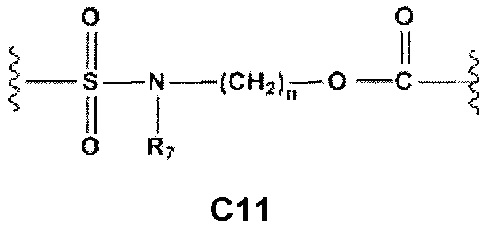
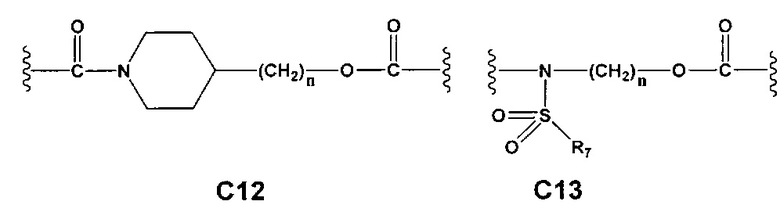
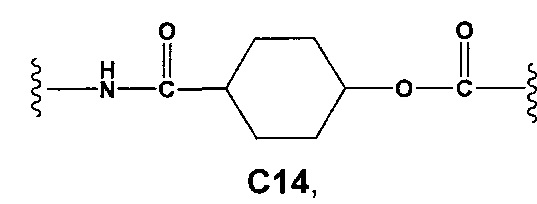
где R7, R7' и R8 независимо представляют собой Н или выбраны из группы, состоящей из прямого или разветвленного (C1-C6)алкила, (C3-C8)циклоалкила, (C3-C8)циклоалкил(C1-C6)алкила, (C3-C8)гетероциклоалкил(С1-C6)алкила, арила и арил(C1-C6)алкила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из (C1-C6)алкила, галогено(C1-C6)алкила, атомов галогена, (C1-C6)алкокси и галогено(C1-C6)алкокси; D отсутствует или выбран из группы, состоящей из (C1-C6)алкилена и арилена, возможно замещенных одной или более чем одной (C1-C6)алкильной группой; n и p независимо равны 0 или целому числу от 1 до 3; Е отсутствует или выбран из -O- и -OC(O)-; G представляет собой арилен, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -CN и -NH2.
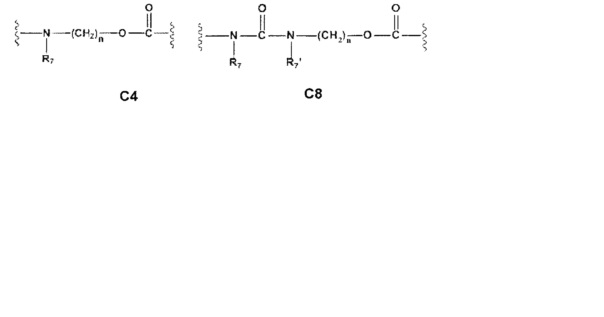
Еще более предпочтительными в этой первой группе являются соединения общей формулы I, где A1 представляет собой (C1-C6)алкилен; В отсутствует или выбран из группы, состоящей из (C3-C8)циклоалкилена, (C3-C8)гетероциклоалкилена, арилена и гетероарилена, возможно замещенных одной или более чем одной группой, выбранной из галогенов, нитрила, прямого или разветвленного (C1-C6)алкила, прямого или разветвленного (C1-C6)галогеноалкила и (C1-C6)алкокси; С и C' отсутствуют или независимо выбраны из группы, состоящей из -O-, -CO-, -OC(O)- и -C(OO)-, или представляют собой одну из следующих групп C4, С7-С13
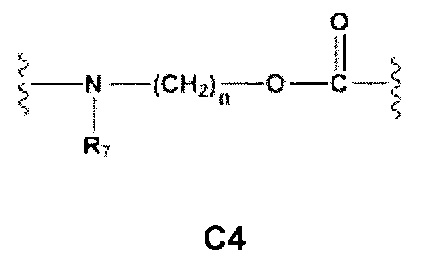
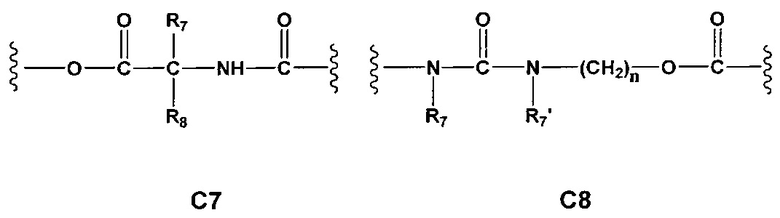

где R7, R7' и R8 независимо представляют собой Н или выбраны из группы, состоящей из прямого или разветвленного (C1-C6)алкила, (C3-C8)циклоалкила, арила и арил(C1-C6)алкила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена и (C1-C6)алкокси; D отсутствует или выбран из группы, состоящей из (C1-C6)алкилена и арилена, возможно замещенных одной или более чем одной (C1-C6)алкильной группой; n и p независимо равны 0 или целому числу от 1 до 3; Е отсутствует или представляет собой -O-; G представляет собой арилен, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -CN и -NH2.
Еще более предпочтительными в этой первой группе являются соединения общей формулы I, где A1 выбран из группы, состоящей из метилена, пропилена и бутилена; В отсутствует или выбран из группы, состоящей из пиперидинилена, фенилена, пиpидиндиила, фурандиила, тиофендиила и циклогексилена, возможно замещенных одной или более чем одной группой, выбранной из метокси, трифторметила, фтора и хлора; С отсутствует или выбран из группы, состоящей из -OС(O)-, C4, С7-С13
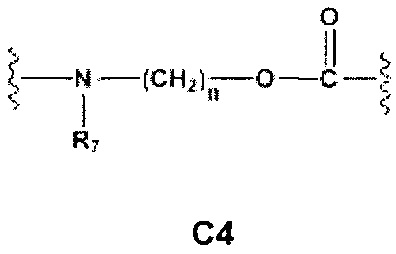
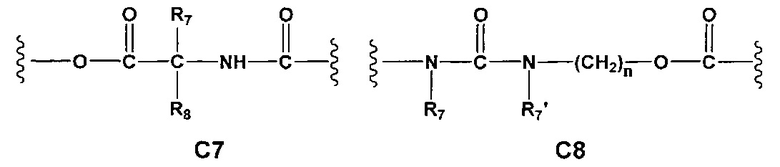

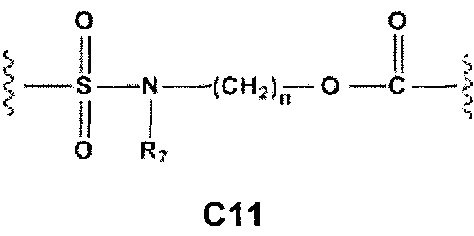
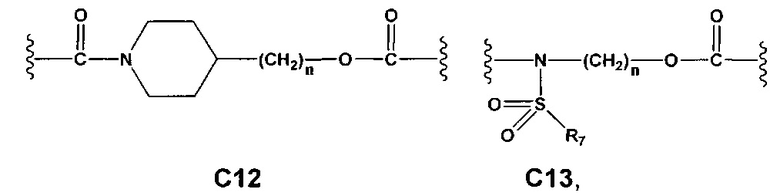
и C' отсутствует или представляет собой -CO-; R7, R7' и R8 независимо представляют собой Н или выбраны из группы, состоящей из прямого или разветвленного (C1-C6)алкила, (C3-C8)циклоалкила, арила и арил(C1-C6)алкила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена и (C1-C6)алкокси; D отсутствует или выбран из группы, состоящей из (C1-C6)алкилена и арилена; n равен 2 или 3, и p равен 1; Е представляет собой -O-; G представляет собой фенилен.
Второй группой предпочтительных соединений общей формулы I являются соединения, где Q представляет собой группу формулы Q1, Q2 и Q3
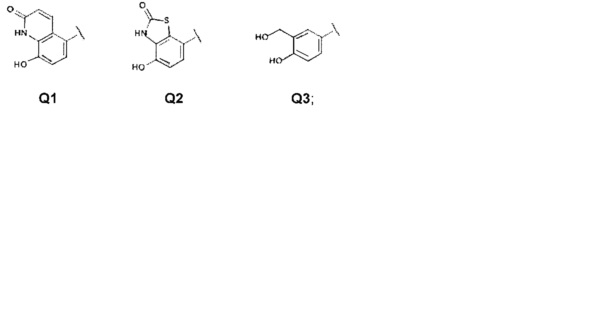
Z представляет собой Н или ОН;
Y представляет собой Y', который является двухвалентной группой формулы
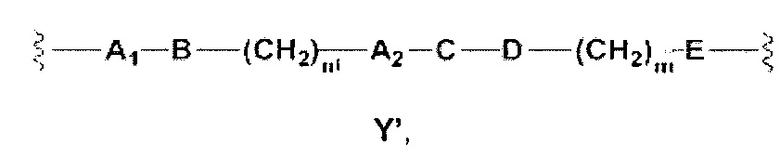
где
А1 и A2 независимо отсутствуют или выбраны из группы, состоящей из (C1-C6)алкилена, (C3-C8)циклоалкилена и (C3-C8)гетероциклоалкилена, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из (C1-C6)алкила, арил(C1-C6)алкила и гетероарил(C1-C6)алкила; В отсутствует или выбран из группы, состоящей из (C3-C8)циклоалкилена, (C3-C8)гетероциклоалкилена, арилена и гетероарилена, возможно замещенных одной или более чем одной группой, выбранной из галогенов, нитрила, прямого или разветвленного (C1-C6)алкила, прямого или разветвленного (C1-C6)галогеноалкила, (C1-C6)алкокси, арила, арил(C1-C6)алкила, -NR7(R8) и гетероарила; С и C' отсутствуют или независимо выбраны из группы, состоящей из -O-, -CO-, -OC(O)- и -C(OO)-, или представляют собой одну из следующих групп С1-С14
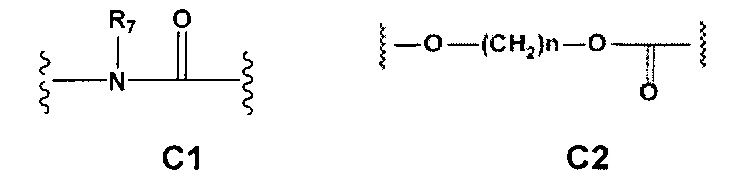
где R7, R7' и R8 независимо представляют собой Н или выбраны из группы, состоящей из прямого или разветвленного (C1-C6)алкила, (C3-C8)циклоалкила, арила и арил(C1-C6)алкила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена и (C1-C6)алкокси; D отсутствует или выбран из группы, состоящей из (C1-C6)алкилена, арилена, гетероарилена и (C3-C8)гетероциклоалкилена, возможно замещенных одной или более чем одной (C1-C6)алкильной группой; n, n' и m независимо равны 0 или целому числу от 1 до 3; Е отсутствует или выбран из -O- и -OC(O)-; G представляет собой арилен, возможно замещенный одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена, -OH, оксо (=O), -SH, -NO2, -CN и -NH2; R1, R2, М и R4 являются такими, как определено выше.
Еще более предпочтительными в этой второй группе являются соединения общей формулы I, где Q представляет собой группу формулы Q1
Z представляет собой Н или ОН;
Y представляет собой Y', который является двухвалентной группой формулы
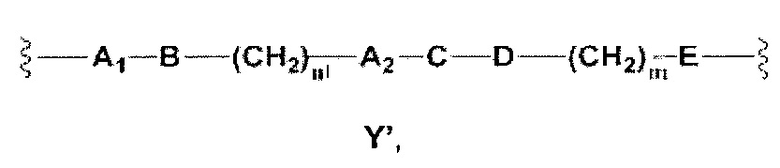
где
A1 и А2 независимо отсутствуют или выбраны из группы, состоящей из (C1-C6)алкилена и (C3-C8)гетероциклоалкилена, возможно замещенных одним или более чем одним (C1-C6)алкилом; В отсутствует или выбран из группы, состоящей из (C3-C8)гетероциклоалкилена, арилена и гетероарилена, возможно замещенных одной или более чем одной группой, выбранной из галогенов, прямого или разветвленного (C1-C6)алкила, прямого или разветвленного (C1-C6)галогеноалкила, (C1-C6)алкокси и арила; С выбран из группы, состоящей из -O-, -CO-, -OC(O)- и -C(OO)-, или представляет собой одну из следующих групп С4, С8-С12
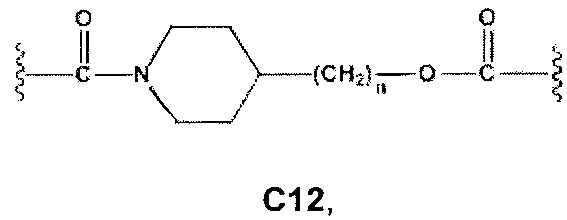
где R7 и R7' независимо представляют собой Н или выбраны из группы, состоящей из прямого или разветвленного (C1-C6)алкила, (C3-C8)циклоалкила, арила и арил(C1-C6)алкила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена и (C1-C6)алкокси; D отсутствует или выбран из группы, состоящей из (C1-C6)алкилена и арилена; n, n' и m независимо равны 0 или целому числу от 1 до 3; Е отсутствует или выбран из -O- и -OC(O)-; G представляет собой арилен; R1, R2, М, R4 и R6 являются такими, как определено выше.
Еще более предпочтительными в этой второй группе являются соединения общей формулы I, где A1 представляет собой (C1-C6)алкилен, и А2 отсутствует или представляет собой (C3-C8)гетероциклоалкилен; В отсутствует или выбран из группы, состоящей из (C3-C8)гетероциклоалкилена, арилена и гетероарилена, возможно замещенных одной или более чем одной группой, выбранной из галогенов, прямого или разветвленного (C1-C6)алкила, прямого или разветвленного (C1-C6)галогеноалкила и (C1-C6)алкокси; С выбран из группы, состоящей из -O-, -CO-, -OC(O)- и -C(OO)-, или представляет собой одну из следующих групп С4, С8-С12
где R7 и R7' независимо представляют собой Н или выбраны из группы, состоящей из прямого или разветвленного (C1-C6)алкила, (C3-C8)циклоалкила, арила и арил(C1-C6)алкила, возможно замещенных одним или более чем одним заместителем, выбранным из группы, состоящей из атомов галогена и (C1-C6)алкокси; D отсутствует или выбран из группы, состоящей из (C1-C6)алкилена и арилена; n, n' и m независимо равны 0 или целому числу от 1 до 3; Е отсутствует или выбран из -O- и -OC(O)-; и G представляет собой арилен.
Еще более предпочтительными в этой второй группе являются соединения общей формулы I, где A1 выбран из группы, состоящей из метилена, пропилена и бутилена, (C1-C6)алкилена, и А2 отсутствует или выбран из группы, состоящей из метилена и пиперидинилена; В отсутствует или выбран из группы, состоящей из фенилена, пиридиндиила, фурандиила, тиофендиила, циклогексилена, возможно замещенных одной или более чем одной группой, выбранной из метокси, трифторметила, фтора и хлора; С выбран из группы, состоящей из -O- и -OC(O)-, или представляет собой одну из следующих групп C4, С8-С13
где R7 и R7' независимо представляют собой Н или выбраны из группы, состоящей из метила, этила, бензила, фенила, изопропила, циклогексила, хлорбензила, фторбензила; D отсутствует или представляет собой фенил; n равен 2 или 3; n' равен 1, m равен; Е отсутствует или представляет собой -O-; и G представляет собой фенилен.
Согласно настоящему изобретению также предложены фармацевтические композиции только соединений формулы I или в комбинации или в смеси с одним или более чем одним фармацевтически приемлемым носителем и/или эксципиентом.
Согласно настоящему изобретению также предложено применение соединений формулы I для изготовления лекарственного средства.
В дополнительном аспекте согласно изобретению предложено применение соединений формулы I для предупреждения и/или лечения любого бронхообструктивного или воспалительного заболевания, предпочтительно астмы или хронического бронхита или хронической обструктивной болезни легких (ХОБЛ).
В дополнительном аспекте согласно изобретению предложено применение соединений формулы I для изготовления лекарственного средства для предупреждения и/или лечения любого бронхообструктивного или воспалительного заболевания, предпочтительно астмы или хронического бронхита или хронической обструктивной болезни легких (ХОБЛ).
Согласно настоящему изобретению дополнительно предложен способ предупреждения и/или лечения любого бронхообструктивного или воспалительного заболевания, предпочтительно астмы или хронического бронхита или хронической обструктивной болезни легких (ХОБЛ), включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения общей формулы I.
Согласно настоящему изобретению также предложены фармацевтические композиции, подходящие для введения посредством ингаляции.
Ингалируемые препараты включают ингалируемые порошки, пропеллент-содержащие дозированные аэрозоли или не содержащие пропеллентов ингаляционные препараты.
Изобретение также относится к устройству, которое может представлять собой одно- или многодозовый ингалятор сухого порошка, дозирующий ингалятор и небулайзер мягкого тумана, содержащему соединения формулы I.
Изобретение также относится к набору, содержащему фармацевтические композиции только соединений формулы I одних или в комбинации или в смеси с одним или более чем одним фармацевтически приемлемым носителем и/или эксципиентом и устройство, которое может представлять собой одно- или многодозовый ингалятор сухого порошка, дозирующий ингалятор и небулайзер мягкого тумана, содержащее соединения общей формулы I.
В соответствии с конкретными воплощениями согласно настоящему изобретению предложены соединения, представленные ниже: 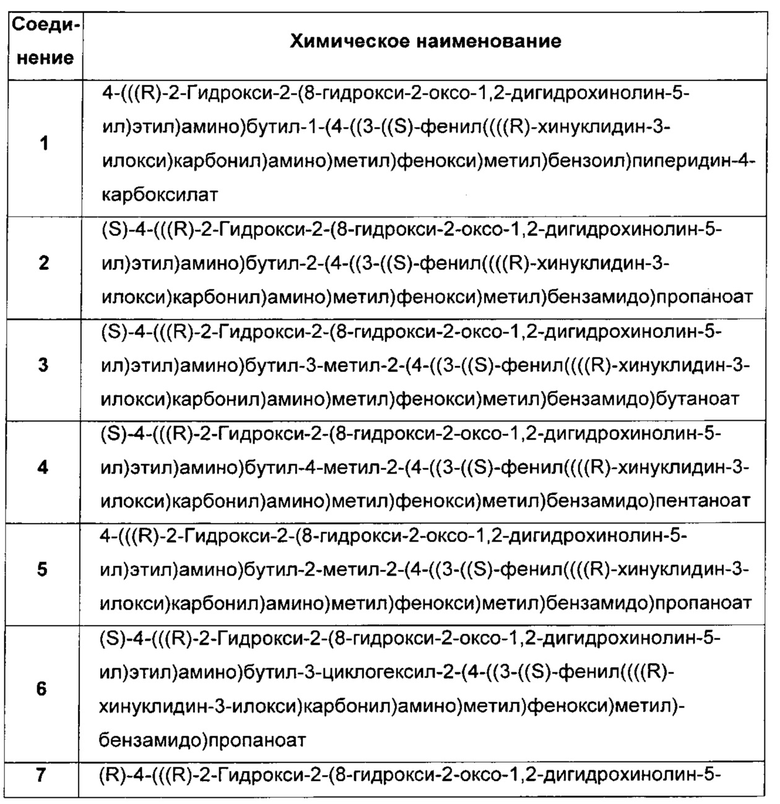










Согласно настоящему изобретению также предложены фармацевтические композиции, содержащие соединение по изобретению, либо как таковое, либо в виде фармацевтически приемлемой соли, и один или более чем один фармацевтически приемлемый носитель и/или эксципиент.
Соединения по изобретению могут быть введены в качестве единственного активного агента или в комбинации с другими фармацевтически активными ингредиентами, включая те, которые используются в настоящее время при лечении респираторных нарушений, например, бета-2-агонисты, антимускариновые агенты, ингибиторы митоген-активируемых протеинкиназ (MAP киназы р38), ингибиторы бета-субъединицы киназы ядерного фактора каппа-В (IKK2), ингибиторы эластазы нейтрофилов человека (HNE), ингибиторы фосфодиэстеразы-4 (PDE4), модуляторы лейкотриенов, нестероидные противовоспалительные агенты (NSAID), противокашлевые агенты, регуляторы секреции слизи, муколитические агенты, отхаркивающие средства/мукокинетические модуляторы, муколитические агенты, воздействующие на пептиды, антибиотики, ингибиторы JAK, ингибиторы SYK, ингибиторы Р13Кдельта или Р13Кгамма, кортикостероиды и М3-антагонисты/РОЕ4-ингибиторы (MAPI).
Согласно настоящему изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с кортикостероидом, выбранным из группы, состоящей из дексаметазона, флутиказона, фуроата флутиказона, пропионата флутиказона, преднизолона, бетаметазона, будесонида, мометазона, фуроата мометазона, ацетонида триамцинолона, циклесонида, TPI-1020, дипропионата беклометазона, преднизона, дефлазакорта, гидрокортизона, QAE-397 и флунизолида.
Согласно настоящему изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с β2-агонистом, выбранным из группы, состоящей из кармотерола, GSK-642444, индакатерола, милветерола, арформотерола, тартрата арформотерола, формотерола, фумарата формотерола, салметерола, ксинафоата салметерола, сальбутамола, альбутерола, левальбутерола, тербуталина, индакатерола (QAB-149), AZD-3199, BI-1744-CL, LAS-100977, GSK159797, GSK59790, GSK159802, GSK642444, GSK678007, GSK96108, бамбутерола, изопротеренола, прокатерола, кленбутерола, репротерола, фенотерола, битолтерола, бродксателора и ASF-1020 и их солей.
Согласно настоящему изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с антимускариновым агентом, выбранным из группы, состоящей из аклидиния, тиотропия, тиотропия бромида (Spiriva®), ипратропия, ипратропия бромида, троспия, гликоn и pролата, NVA237, LAS34273, GSK656398, GSK233705, GSK57319, LAS35201, QAT370 и солей окситропия.
Согласно настоящему изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с ингибитором PDE4, выбранным из группы, состоящей из AN-2728, AN-2898, CBS-3595, апремиласта, ELB-353, KF-66490, К-34, LAS-37779, IBFB-211913, AWD-12-281, ципамфиллина, циломиласта, рофлумиласта, BAY19-8004 и SCH-351591, AN-6415, indus-82010, TPI-PD3, ELB-353, СС-11050, GSK-256066, оглемиласта, ОХ-914, тетомиласта, МЕМ-1414 и RPL-554.
Согласно настоящему изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с ингибитором MAP киназы р38, выбранным из группы, состоящей из семапимода, телмапимода, пирфенидона, РН-797804, GSK-725, GSK856553, GSK681323, минокина и лосмапимода и их солей
В предпочтительном воплощении согласно настоящему изобретению предложены комбинации соединения по изобретению с ингибитором IKK2.
Согласно изобретению также предложены комбинации соединения по изобретению с ингибитором HNE, выбранным из группы, состоящей из ААТ, ADC-7828, аэривы (aeriva), TAPI, АЕ-3763, KRP-109, АХ-9657, POL-6014, AER-002, AGTC-0106, респривы (respriva), AZD-9668, земаиры, ААТ IV, PGX-100, элафина, SPHD-400, проластина C и ингаляционного проластина.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с модулятором лейкотриенов, выбранным из группы, состоящей из монтелукаста, зафирлукаста и пранлукаста.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с NSAID, выбранным из группы, состоящей из ибупрофена и кетопрофена.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с противокашлевым агентом, выбранным из группы, состоящей из кодеина и декстраморфана.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с муколитическим агентом, выбранным из группы, состоящей из N-ацетилцистеина и фудостеина.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с отхаркивающим средством/мукокинетическим модулятором, выбранным из группы, состоящей из амброксола, гипертонических растворов (например физиологического раствора или маннита) и сурфактанта.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с муколитическим агентом, воздействующим на пептиды, выбранным из группы, состоящей из рекомбинантной человеческой дезоксирибонуклеазы I (дорназа альфа и рчДНКаза) и гелицидина.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с антибиотиком, выбранным из группы, состоящей из азитромицина, тобрамицина и азтреонама.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с регулятором образования слизи, выбранным из группы, состоящей из INS-37217, диквафозола, сибенадета, CS-003, талнетанта, DNK-333, MSI-1956 и гефитиниба.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с ингибитором JAK, выбранным из группы, состоящей из СР-690550 и GLPG0634.
Согласно изобретению также предложены комбинации соединения по изобретению, либо как такового, либо в виде фармацевтически приемлемой соли, с ингибитором SYK, выбранным из группы, состоящей из R406, R343 и PRT062607.
Соединения по изобретению могут быть получены из легкодоступных исходных материалов при использовании следующих общих способов и методик или при использовании других сведений, легкодоступных специалистам в данной области техники. Хотя здесь может быть показано и описано конкретное воплощение настоящего изобретения, специалистам в данной области техники будет понятно, что все воплощения или аспекты настоящего изобретения могут быть получены при использовании способов, описанных здесь, или при использовании других способов, реагентов и исходных материалов, известных специалистам в данной области техники. Следует понимать, что там, где даны типовые или предпочтительные условия процесса (то есть температуры реакций, время, молярные соотношения реагентов, растворители, давление и так далее), также могут быть использованы другие условия процесса, если не оговорено особо. Хотя оптимальные условия реакции могут изменяться в зависимости от конкретных реагентов или используемых растворителей, такие условия могут быть легко определены специалистом в данной области техники посредством общепринятой методики оптимизации.
Соединения общей формулы I могут быть получены согласно следующей схеме синтеза.
Схема
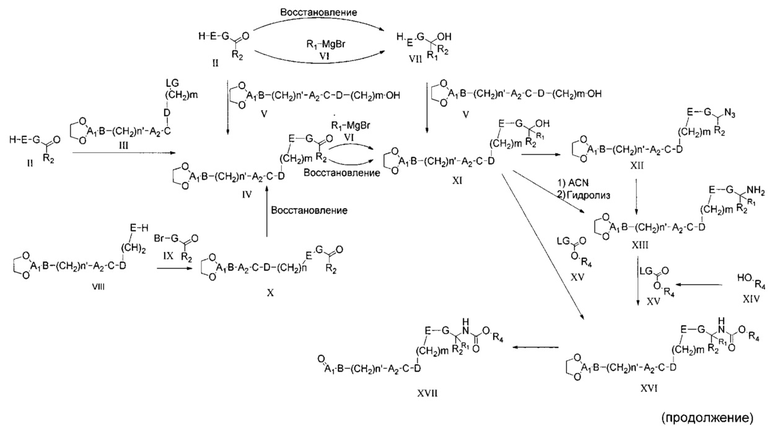
Общая методика получения соединений формулы I
Соединения общей формулы VIII представляют собой соединение, где A1 представляет собой алкилен, замещенный оксо, что приводит к образованию защищенного альдегида или кетона в виде циклического ацеталя.
Для синтеза соединений общей формулы I может требоваться защита возможных реакционных функциональных групп в дополнение к тем способам, которые уже описаны. В этом случае примеры совместимых защитных групп (PG) и конкретные способы защиты и снятия защиты с их помощью описаны в "Protecting groups in organic Synthesis" by T.W. Green и P. Wutz (Wiley-Interscience publication, 1999). Соединения общей формулы I могут быть получены, например путем взаимодействия соединения общей формулы XVII с соединением общей формулы XVIII. Эта реакция восстановительного аминирования может быть проведена согласно нескольким различным протоколам, описанным в литературе и известным специалистам в данной области техники. Например, она может быть проведена в растворителе, таком как метанол, этанол, тетрагидрофуран (ТГФ) или дихлорметан (ДХМ), при использовании восстановителя, такого как NaBH4, NaCNBH3 или NaBAcO3H. Перед добавлением восстановителя может быть полезным получение имина. Реакция протекает спокойно при комнатной температуре (RT) в течение 1-12 часов.
Промежуточное соединение общей формулы XVII может быть легко получено путем взаимодействия соединения общей формулы XIII с соединением общей формулы XV. Реакция протекает спокойно при RT или при более низкой температуре в растворителе, таком как ДХМ или пиpидин, в течение 1-16 часов, приводя к получению соединений формулы XVI, с которых легко может быть снята защита в водном кислотном растворе, что приводит к получению соединения общей формулы XVII.
Соединения общей формулы XV либо имеются в продаже, либо могут быть получены путем взаимодействия спиpта общей формулы XIV, например, с дифосгеном в растворителе, таком как ДХМ, ТГФ или ацетонитрил (ACN), при RT или при более низкой температуре в течение периода времени в пределах от 0,5 до 12 часов, что приводит к получению соединения общей формулы XV, где уходящая группа LG представляет собой хлор. Альтернативно, спирт общей формулы XIV может быть подвергнут взаимодействию, например, с карбонилдиимидазолом (CDI), что приводит к такому же промежуточному соединению, где LG представляет собой имидазол. Другие возможные промежуточные соединения с другими известными LG могут быть получены, как описано в литературе.
Соединение общей формулы XIII может быть получено из соединения общей формулы XI реакцией Риттера (ацетонитрил и серная кислота при RT) с последующим гидролизом промежуточного ацетамида, проводимым в щелочных условиях.
Альтернативно, соединения общей формулы XIII могут быть получены путем восстановления азида формулы XII гидрированием в атмосфере водорода или в условиях переноса водорода. Взаимодействие проводят в спиpтах при RT или при более высокой температуре и прекращают через 1-12 часов. Альтернативным способом восстановления может являться реакция Штаудингера, которая включает обработку азида сначала, например, трифенилфосфином, с последующим гидролизом иминофосфоранового промежуточного соединения водой. Это взаимодействие проводят при RT в смешивающемся с водой растворителе, таком как, например, ТГФ. Использованием сильного восстановителя, такого как, например, LiAlH4 в ТГФ или эфире, при -40°C или при более низкой температуре легко может обеспечить проведение требуемого превращения соединения XIV в соединение XIII.
Азид XII получают из соединения формулы XI путем взаимодействия с дифенилфосфорилазидом. Взаимодействие проводят в высококипящем растворителе, таком как толуол или ксилол, в присутствии сильного основания, такого как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), но не ограничиваясь им, при температуре в пределах от 80 до 120°C, и его завершают через 12-24 часа. Альтернативно, гидроксильная групn и pовка промежуточного соединения формулы XI может быть превращена в подходящую уходящую группу (LG), такую как, например, мезил, тозил или галоген, и затем подвергнута взаимодействию с азидом щелочных металлов в полярном растворителе, таком как ацетонитрил, ДМФА (диметилформамид) или N-метил-2-пиpролидон (NMP) при RT или более высокой температуре.
Промежуточные соединения общей формулы XI могут быть получены несколькими различными путями. Например, они могут быть получены в результате взаимодействия соединения общей формулы VII, где E представляет собой -O-, и альдегида общей формулы V, несущего подходящую гидроксильную группу, которые могут быть легко подвергнуты взаимодействию в стандартных условиях реакции Мицунобу. Взаимодействие проводят в растворителях, таких как ТГФ или N-метилморфолин (NMM), при температуре от -10°C до RT и завершают через 1-24 часа. Его осуществляют в присутствии диэтилазодикарбоксилата (DEAD) или диизопропилазодикарбоксилата (DIAD) и органического фосфина, такого как трифенилфосфин, но не ограничивается им.
Спирт общей формулы VII либо имеется в продаже, либо может быть получен из соединения формулы II путем добавления реактива Гриньяра формулы VI. Взаимодействие обычно проводят в апротонном растворителе, таком как простой эфир или ТГФ, при RT или при более низкой температуре и завершают через 0,5-12 часов. Альтернативно, он может быть получен путем восстановления соединения общей формулы II, где R2 не является водородом, восстановителем, таким как NaBH4, но не ограничиваясь им, что приводит в этом случае к получению соединения формулы VII, где R1 представляет собой водород. Взаимодействие проводят в растворителе, таком как метанол, этанол или ТГФ, и завершают в течение периода времени в пределах от 1 до 12 часов. Сходный протокол синтеза может быть использован для получения промежуточного соединения XI из соединений общей формулы IV.
Специалисту в данной области техники ясно, что получение соединения общей формулы VII или XI может быть достигнуто с помощью обратной реакции Гриньяра, в которой реактив Гриньяра формулы G-MgBr взаимодействует с соединением формулы R1C(O)R2 в таких же условиях реакции, как описано выше.
Соединения общей формулы IV, где E представляет собой -O-, могут быть получены из соединения общей формулы II, следуя способу, аналогичному описанному для получения соединений формулы XI из соединений VII. Альтернативно, соединения общей формулы IV могут быть получены путем алкилирования соединения общей формулы II соединением общей формулы III, где LG представляет собой подходящую уходящую группу, такую как тозилат, мезилат или галоген. Взаимодействие обычно проводят в полярных растворителях, таких как ацетонитрил или ДМФА, его осуществляют в присутствии основания, такого как, например, карбонат щелочных металлов, бикарбонаты щелочных металлов или органические основания, и завершают в течение периода времени в пределах от 1 до 24 часов.
Получение соединений формулы X может быть достигнуто путем взаимодействия соединения общей формулы IX или его аналога, где бром замещен иодом или трифлатом, с соединением общей формулы VIII, где n равен 2, в условиях реакции кросс-сочетания, катализируемой переходными металлами. Полученный в итоге алкен VIII может быть подвергнут взаимодействию, например, в условиях реакции Хека, с соединением IX, что приводит к образованию алкениленового промежуточного соединения X, которое может быть легко восстановлено путем классического каталитического гидрирования двойной связи с получением соединений формулы IV. Большое число протоколов, реагентов и катализаторов может быть легко использовано для достижения требуемого превращения, как известно специалисту в данной области техники.
Альтернативно, соединения общей формулы I, несущие сложноэфирную группировку в линкере Y, могут быть получены путем обработки соединения общей формулы XXXI соединением общей формулы XXXII, где А2 функционализирован ОН, в условиях реакции конденсации для получения сложных эфиров. Возможно получение соединения общей формулы I, где C является точно таким же, как C1, путем взаимодействия соединения общей формулы XXXI с соединением XXXII, где A2 замещен группой -NR3, в известных реакционных условиях для получения амида, начиная с карбоновой кислоты и аминов.
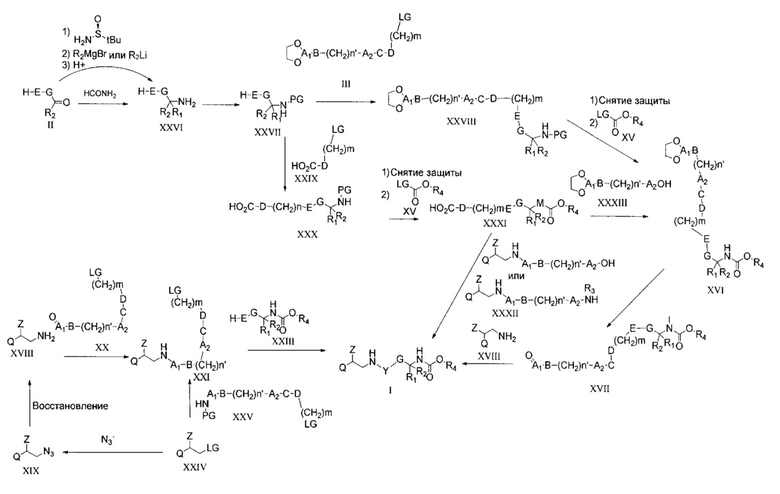
В другом воплощении настоящего изобретения соединения общей формулы I могут быть получены путем взаимодействия соединения формулы XXI с соединением формулы XXIII в условиях, описанных выше для взаимодействия соединения формулы II с соединением формулы III.
Промежуточные соединения формулы XXI могут быть получены путем взаимодействия соединения формулы XVIII в условиях реакции восстановительного аминирования, описанных выше для взаимодействия соединения формулы XVII с соединением XVIII, начиная с соединения формулы XX.
Соединения общей формулы XVIII могут быть получены путем простого восстановления азида формулы XIX. Взаимодействие может быть достигнуто посредством каталитического гидрирования в присутствии палладиевого катализатора. Взаимодействие проводят в полярном растворителе, таком как метанол или этанол, в атмосфере водорода или в условиях переноса водорода при использовании, например, 1,4-циклогексадиена или 1-метил-1,4-циклогексадиена в качестве источника водорода. Взаимодействие проводят при RT. В случае его проведения в условиях переноса водорода может потребоваться более высокая температура.
Азид XIX может быть легко получен из соединения XXIV путем известного нуклеофильного замещения алкилбромида азидом щелочных металлов. Взаимодействие проводится при температуре в пределах от 50 до 80°C и в полярном растворителе, таком как, например, ДМФА или NMP, и может быть ускорено в присутствии иодида щелочных металлов.
В другом воплощении настоящего изобретения соединения общей формулы XXI могут быть получены путем взаимодействия промежуточного соединения общей формулы XXIV с амином общей формулы XXV. Это взаимодействие представляет собой простое алкилирование амина, в котором уходящая группа LG (обычно хлор, бром или сульфат) заменена нуклеофилом, подобного амину XXV как таковому или защищенному по аминной группиpовке. В литературе описаны несколько способов проведения этого взаимодействия, которое обычно осуществляют в полярном растворителе при температуре выше RT. Сходное взаимодействие может быть использовано для получения соединения общей формулы XXXII.
Для специалистов в данной области техники очевидно, что соединения общей формулы I, где R4 представляет собой J1, содержат три стереогенных центра, которые обозначены ниже символом *. Это означает, что структура формулы I характеризуется восемью различными стереоизомерами.
Каждый диастереоизомер теоретически может быть получен путем хроматографического разделения смеси, полученной путем взаимодействия рацемических смесей требуемых промежуточных соединений. Понятно, что такой способ не подходит и, что он может быть использован только для разделения смесей, содержащих небольшое число диастереоизомеров.
В более подходящем способе синтез каждого отдельного стереоизомера может быть достигнут при использовании во взаимодействиях, описанных выше, только энантиомерно чистых промежуточных соединений.
Энантиомерно чистые спирты, требуемые для получения соединений общей формулы I, где R4 представляет собой J1, имеются в продаже.
Получение отдельных энантиомерно чистых соединений общей формулы XXIV, где LG представляет собой бром, описано в WO 2005/080324, US 2005-2222128, WO 2004/032921, US 2005/215590 и WO 2005/092861 (цитируемых в WO 2007/107228). Энантиомерно чистые соединения общей формулы XXVII могут быть получены путем хирального хроматографического разделения рацемической смеси или начиная с энантиомерно чистых аминов общей формулы XXVI. Промежуточные соединения формулы XXVI содержат основную группу, возможно получение двух энантиомеров посредством кристаллизации диастереоизомерной соли, полученной путем образования соли рацемической смеси с помощью энантиомерно чистой карбоновой кислоты. Широко используемыми карбоновыми кислотами, используемыми для этой цели, являются, например, миндальная кислота, винная кислота и их производные. Основание XXVI растворяют в подходящем полярном растворителе и затем обрабатывают энантиомерно чистой карбоновой кислотой, вызывая осаждение одной из двух диастереоизомерных солей. Для получения требуемого уровня энантиомерного избытка может потребоваться повторить процесс несколько раз.
Альтернативно, амины формулы XXVI могут быть получены путем энантиоселективного синтеза согласно, например, способу, описанному в литературе (Tetrahedron: Asymmetry 13 (2002) 303-310), в котором альдегид формулы II, где R2 представляет собой Н, обрабатывают сначала энантиомерно чистым трет-бутилсульфонамидом и затем R2MgBr или R2Li (где R2 не является Н) с последующим гидролизом промежуточного соединения, что приводит к образованию энантиомерно обогащенных соединений формулы XXVI, которые могут быть использованы как есть или дополнительно очищены для повышения энантиомерного избытка.
Рацемический амин общей формулы XXVI может быть получен несколькими различными путями, например, путем добавления гидроксиламина к соединению общей формулы II с последующим восстановлением полученного промежуточного оксима, которое может быть проведено в различных реакционных условиях, известных специалистам в данной области техники. Например, каталитическое гидрирование или использованием восстановителя, такого как LiAlH4 или Zinc, в присутствии формиата аммония являются очень эффективными способами достижения восстановления оксима в амин.
Имеющийся амин формулы XXVI далее может быть легко дериватизирован в условиях реакции, описанных выше. Например, он может быть обработан защищенным альдегидом формулы III в условиях, описанных для алкилирования соединений формулы II соединениями формулы III, что приводит к образованию соединения общей формулы XXVIII. Снятие защиты с аминогруппы и взаимодействие с соединениями формулы XV приводят к получению соединения общей формулы XVI.
Альтернативно, соединение общей формулы I может быть получено путем сочетания соединения общей формулы XXXI с соединением общей формулы XXXII, что приводит к образованию соединения общей формулы I, где C представляет собой -OCO- или C1. Этот эфир или амид может быть получен в различных реакционных условиях, известных специалистам в данной области техники. Для взаимодействия требуется активация кислоты XXXI реагентом, таким как N,N'-дициклогексилкарбодиимид (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU), гексафторфосфат (O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), или она может быть превращена в соответствующий ацилхлорид. Активированный эфир может быть легко подвергнут взаимодействию в ДХМ, пиридине или других апротонных растворителях с соединением формулы XXXII.
Соединение формулы XXXI может быть получено, начиная с соединения XXVII, путем алкилирования соединением формулы XXIX, снятия защиты и взаимодействия с соединением формулы XV. Условия реакции для этого превращения описаны выше и описаны в литературе. Кислота XXXI может быть легко подвергнута взаимодействию с соединением формулы XXXIII, как описано выше.
Соединение общей формулы XXXII может быть получено путем взаимодействия соединения общей формулы XXIV с амином формулы -NH2-A1-(СН2)n'-В-А2-ОН или -NH2-A1-(CH2)n'-B-A2-NHR4 в условиях реакции, описанных для взаимодействия соединений общей формулы XXIV с соединениями общей формулы XXV.
Из всего вышесказанного следует, что синтез соединений общей формулы I может быть проведен, следуя нескольким различным способам. В частности, следует отметить, что последовательность требуемых реакций строго зависит от природы линкеров Y и Y1 и от функциональных групп, присутствующих в линкере. Примеры, данные выше для получения соединений формулы I, где C представляет собой -OСO- или C1, дают возможность специалисту в данной области техники принять во внимание этот аспект изобретения.
Способы ЖХМС (жидкостной хроматомасс-спектрометрии) А, B и C, используемые для характеристики соединений по настоящему изобретению, описаны ниже.
Способ A (10 см _ESCI_Муравьиная кислота)


Типовые инъекции 2-7 мкл (концентрация примерно 0,2-1 мг/мл).
УФ-детектирование (ультрафиолетового излучения) с помощью DAD (диодно-матричного детектора) HP или Waters
Другие записи длин волн получают из данных DAD.
Возможное детектировние ELS (испарительного светорассеяния) при использовании Polymer Labs ELS-1000.
МС (масс-спектрометрическое) детектирование: Micromass ZQ, одноквадрупольный ЖХ-МС (жидкостная хроматография-масс-спектрометрия) или Quattro Micro ЖХ-МС-МС (жидкостная хроматография-тандемная масс-спектрометрия).
Разделитель потока дает приблизительно 300 мкл/мин для масс-спектра.
Диапазон сканирования для данных МС (m/z)
с переключением +ve/-ve.
Ионизация включает установленную опцию ESCI (электрораспыление/химическая ионизация), которая дает как ESI (электрораспылительная ионизация), так и APCI (химическая ионизация при атмосферном давлении) данные с одного потока.
Типовые напряжения и температуры ESI составляют:
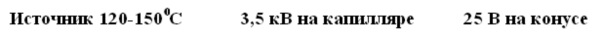
Типовые напряжения и температуры APCI составляют:
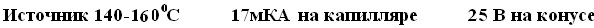
Способ B (условия ВЭЖХ - 15 см_Муравьиная кислота_Ascentis_ВЭЖХ_СН3CN)
Параметры ВЭЖХ

Типовые инъекции 0,2-10 мкл.
Максимальное устанавливаемое давление 400 бар (40 МПа).
Прибор: жидкостной хроматограф Agilent 1100, бинарный насос, пробоотборник Agilent и DAD детектор Agilent.
Диодно-матричное детектирование: (300 нм, ширина полос 200 нм; контрольн. 450 нм, ширина полос 100 нм).
Способ C (условия ВЭЖХ - 10 см_Муравьиная кислота_АСЕ-АР_ВЭЖХ_CH3CN) 

Типовые инъекции 0,2-10 мкл.
Максимальное устанавливаемое давление 400 бар (40 МПа).
Прибор: жидкостной хроматограф Agilent 1100, бинарный насос, пробоотборник Agilent и DAD детектор Agilent.
Диодно-матричное детектирование: (300 нм, ширина полос 200 нм; контрольн. 450 нм, ширина полос 100 нм).
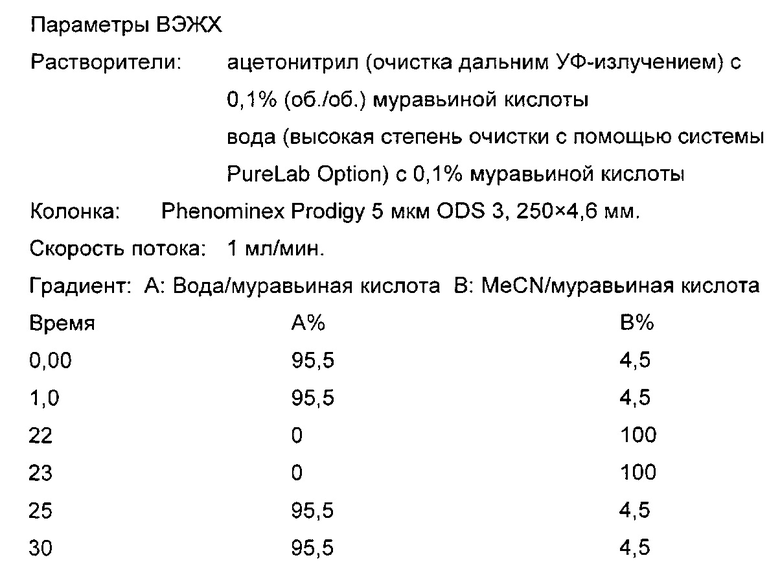
Способ D (условия ВЭЖХ - 25 см_кислотная_Prodigy_ВЭЖХ)
Типовые инъекции 2-7 мкл.
Прибор: жидкостной хроматограф Agilent 1100, бинарный насос, пробоотборник Agilent и DAD детектор Agilent.
Согласно изобретению также предложены фармацевтические композиции соединений формулы I в смеси с одним или более чем одним фармацевтически приемлемым носителем, например таким, который описан в Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Введение соединений по настоящему изобретению можно осуществлять в соответствии с потребностями пациентов, например перорально, интраназально, парентерально (подкожно, внутрикожно, внутримышечно, интрастернально и посредством инфузии), посредством ингаляции, ректально, вагинально, местно, локально, трансдермально и посредством введения в глаз.
Для введения соединений по изобретению могут быть использованы различные твердые лекарственные формы для перорального применения, включая такие твердые формы, как таблетки, желатиновые капсулы, капсулы, каплеты, гранулы, лепешки или нерасфасованные порошки. Соединения по настоящему изобретению могут быть введены отдельно или в комбинации с различными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмалы) и известными эксципиентами, включая суспендирующие агенты, солюбилизаторы, буферные агенты, связующие вещества, разрыхлители, консерванты, красители, ароматизаторы, смазывающие вещества и им подобные. При введении соединений по настоящему изобретению также преимущество имеют капсулы, таблетки и гели с высвобождением по времени.
Для введения соединений по изобретению также могут быть использованы различные жидкие лекарственные формы для перорального применения, включая водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие лекарственные формы также могут содержать подходящие известные инертные разбавители, такие как вода, и подходящие известные эксципиенты, такие как консерванты, смачивающие агенты, подсластители, корригенты, а также агенты для эмульгирования и/или суспендирования соединений по изобретению. Соединения по настоящему изобретению могут быть введены, например, внутривенно в форме изотонического стерильного раствора. Другие препараты также возможны.
Суппозитории для ректального введения соединений по изобретению могут быть приготовлены посредством смешивания соединения с подходящим эксципиентом, таким как масло какао, салицилаты или полиэтиленгликоли.
Препараты для вагинального введения могут иметь форму крема, геля, пасты, пены или аэрозоля, содержащего, в дополнение к активному ингредиенту, такие подходящие носители, которые также известны.
Для местного введения фармацевтические композиции могут иметь форму кремов, мазей, линиментов, лосьонов, эмульсий, суспензий, гелей, растворов, паст, порошков, аэрозолей и капель, подходящих для применения на кожу, глаз, а также в ухо и в нос. Местное введение может также включать трансдермальное введение с помощью таких средств, как трансдермальные пластыри.
Для лечения заболеваний дыхательных путей соединения по изобретению предпочтительно вводят посредством ингаляции.
Ингаляционные препараты включают ингалируемые порошки, пропеллент-содержащие дозированные аэрозоли или не содержащие пропеллентов ингаляционные препараты.
Для введения в виде сухого порошка могут быть использованы известные в данной области техники однодозовые или многодозовые ингаляторы. В этом случае порошком могут быть наполнены желатиновые, пластиковые или другие капсулы, картриджи или блистерные упаковки, или резервуар.
К порошкообразным соединениям по изобретению может быть добавлен разбавитель или носитель, как правило нетоксичный и химически инертный в отношении соединений по изобретению, например, лактоза или любое другое вспомогательное вещество, подходящее для улучшения вдыхаемой фракции.
Ингаляционные аэрозоли, содержащие пропеллент, такой как гидрофторалканы, могут содержать соединения по изобретению либо в виде раствора, либо в дисперсной форме. Пропеллент-содержащие препараты могут также содержать другие ингредиенты, такие как сорастворители, стабилизаторы и возможно другие эксципиенты.
Не содержащие пропеллент ингаляционные препараты, содержащие соединения по изобретению, могут иметь форму растворов или суспензий в водной, спиpтовой или водно-спиpтовой среде, и они могут быть доставлены с помощью струйных или ультразвуковых небулайзеров, известных в данной области техники, или небулайзеров мягкого тумана, таких как Respimat®.
Соединения по изобретению могут быть введены в качестве единственного активного агента или в комбинации с другими фармацевтически активными ингредиентами, включая ингредиенты, используемые в настоящее время для лечения респираторных заболеваний, например, кортикостероиды, ингибиторы MAP киназы р38, ингибиторы IKK2, ингибиторы HNE, ингибиторы PDE4, модуляторы лейкотриенов, NSAID и регуляторы секреции слизи.
Дозировки соединений по изобретению зависят от ряда факторов, включая конкретное заболевание, подлежащее лечению, тяжесть симптомов, способ введения, частоту применения, конкретное используемое соединение, эффективность, токсикологический профиль и фармакокинетический профиль соединения.
Преимущественно, соединения формулы I могут быть введены, например, в дозировке, составляющей от 0,001 до 1000 мг/сутки, предпочтительно от 0,1 до 500 мг/сутки.
При введении соединений формулы I ингаляционным путем их предпочтительно принимают в дозировке, составляющей от 0,001 до 500 мг/сутки, предпочтительно от 0,1 до 200 мг/сутки.
Соединения формулы I могут быть введены для предупреждения и/или лечения бронхообструктивных или воспалительных заболеваний, таких как астма, хронический бронхит, хроническая обструктивная болезнь легких (ХОБЛ), бронхиальная гиперреактивность, кашель, эмфизема или ринит; урологические нарушения, такие как недержание мочи, поллакиурия, цистоспазм, хронический цистит и гиперактивный мочевой пузырь (ГМП); расстройства желудочно-кишечного тракта, такие как синдром раздраженного кишечника, спастический колит, дивертикулит, пептическая язва, нарушение моторики желудочно-кишечного тракта или секреции желудочного сока; сухость во рту; мидриаз, тахикардия; сердечно-сосудистые расстройства после офтальмологических вмешательств, такие как вагусная синусовая брадикардия.
Настоящее изобретение проиллюстрировано ниже следующими примерами.
Промежуточные соединения для синтеза конечных соединений общей формулы (I) получали способами, описанными далее.
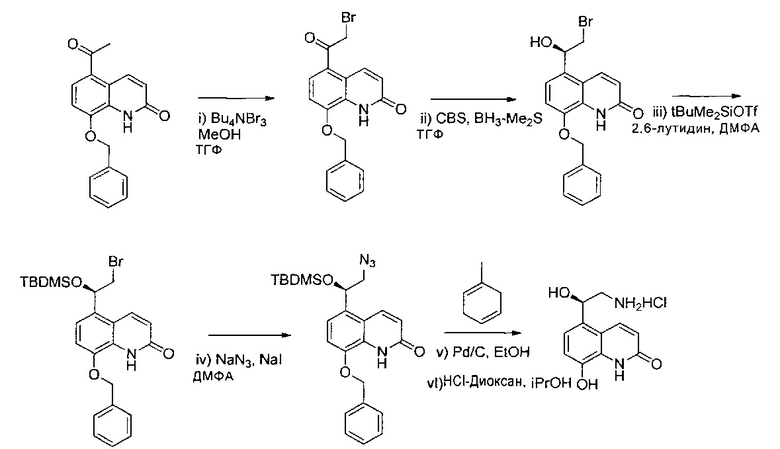
Получение гидрохлорида (R)-5-(2-амино-1-гидроксиэтил)-8-гидроксихинолин-2(1Н)-она
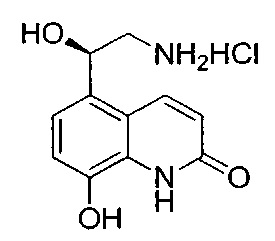
Стадия 1: 8-(бензилокси)-5-(2-бромацетил)хинолин-2(1Н)-он
К суспензии 5-ацетил-8-(бензилокси)хинолин-2(1Н)-она (19,4 г; 66,4 ммоль) в безводном ТГФ (240 мл) и безводном метаноле (165 мл) по каплям добавляли раствор трибромида тетра-н-бутиламмония (54,5 г; 113,0 ммоль) в безводном ТГФ (130 мл) в течение 1,5 часа. Полученный раствор перемешивали при RT в течение ночи, а затем концентрировали при пониженном давлении без нагревания. Остаток повторно растворяли в метаноле (200 мл). Насыщенный водный раствор хлорида аммония (390 мл) добавляли при охлаждении льдом. Полученную суспензию фильтровали и твердое вещество промывали водой и сушили под вакуумом. Твердое вещество суспендировали в ДХМ и метаноле (1:1, об./об.; 100 мл) в течение 90 минут. Твердое вещество собирали посредством фильтрации, промывали ДХМ и сушили на воздухе с получением указанного в заголовке соединения (18,0 г; 73%).
1Н ЯМР (400 МГц, ДМСО-d6): δ 11.07 (s, 1Н); 8.51 (d, J=10,0 Гц, 1Н); 7.94-7.83 (m, 1Н); 7.60 (d, J=7,5 Гц, 2Н); 7.44-7.27 (m, 4Н); 6.79-6.65 (m, 1Н); 5.53-5.39 (s, 2Н); 4.93 (s, 2Н).
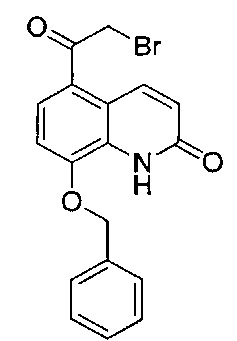
Стадия 2: (R)-8-(бензилокси)-5-(2-бром-1-гидроксиэтил)хинолин-2(1H)-он
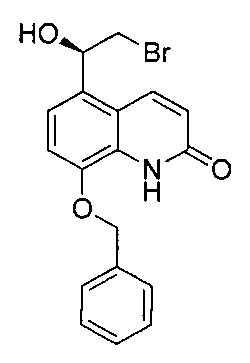
8-(Бензилокси)-5-(2-бромацетил)хинолин-2(1Н)-он (26,0 г; 69,9 ммоль) и (R)-3,3-дифенил-1-метилтетрагидро-3H-пиpроло[1,2-с][1,3,2] оксазаборол (21,3 г; 76,8 ммоль) подвергали азеотропной перегонке с толуолом (×3), затем суспендировали в безводном ТГФ (400 мл) в атмосфере азота. Суспензию охлаждали до -20°C (температура окружающей среды) и добавляли раствор комплекса боран-диметилсульфид (45,4 мл; 90,8 ммоль; 2,0 М раствор в ТГФ) посредством шприцевого насоса в течение 3 часов. После завершения добавления реакционную смесь перемешивали в течение одного часа, а затем гасили метанолом (25 мл). Реакционную смесь нагревали до RT в течение 20 минут. Смесь концентрировали при пониженном давлении и остаток суспендировали в водной соляной кислоте (500 мл; 1 М раствор) и перемешивали при RT в течение 18 часов. По прошествии этого времени твердое вещество собирали посредством фильтрации и промывали водой (3×100 мл). Твердое вещество частично растворяли в этилацетате и нагревали с обратным холодильником в течение 2 часов. Оставшееся твердое вещество удаляли посредством горячей фильтрации и фильтрат упаривали с получением указанного в заголовке соединения. Твердое вещество, собранное из горячего этилацетата, снова частично растворяли в этилацетате и нагревали с обратным холодильником в течение 2 часов, затем фильтровали с получением фильтрата, содержащего чистый продукт. Этот процесс повторяли еще четыре раза. Объединенное твердое вещество перекристаллизовывали из этилацетата и петролейного эфира с получением указанного в заголовке соединения (20,0 г; 76%).
1Н ЯМР (400 МГц, ДMCO-d6): δ 10.68 (s, 1Н); 8.19 (d, J=9,9 Гц, 1Н); 7.58 (d, J=7,5 Гц, 2Н); 7.41-7.36 (m, 2Н); 7.34-7.29 (m, 1Н); 7.23-7.19 (m, 2Н); 6.57 (d, J=9,8 Гц, 1Н); 5.94 (d, J=4,7 Гц, 1Н); 5.31 (s, 2Н); 5.25-5.19 (m, 1Н); 3.71-3.58 (m, 2Н).
Стадия 3: (R)-8-(бензилокси)-5-(2-бром-1-((трет-бутилдиметилсилил)окси)этил)хинолин-2(1Н)-он
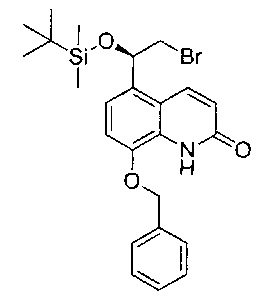
2,6-Лутидин (6,9 мл; 59,5 ммоль) добавляли к раствору (R)-8-(бензилокси)-5-(2-бром-1-гидроксиэтил)хинолин-2(1H)-она (10,1 г; 27,0 ммоль) в ДХМ (100 мл) при 0°C. Реакционную смесь перемешивали в течение 5 минут, затем по каплям добавляли трет-бутилдиметилсилил-трифторметансульфонат (13,0 мл; 56,8 ммоль) в течение 15 минут. Смесь перемешивали при 0°C в течение 30 минут с последующим перемешиванием при RT в течение ночи. По прошествии этого времени реакционную смесь гасили насыщенным водным раствором бикарбоната натрия и экстрагировали ДХМ (×3). Объединенные органические экстракты сушили (сульфат магния), фильтровали и концентрировали при пониженном давлении. К неочищенному веществу добавляли изогексан (500 мл), и полученное твердое вещество собирали посредством фильтрации. Твердое вещество перекристаллизовывали из этилацетата и петролейного эфира (40:60) с получением указанного в заголовке соединения (11,3 г; 85%).
1Н ЯМР (400 МГц, CDCl3): δ 9.19 (s, 1Н); 8.23 (dd, J=9,9, 4,4 Гц, 1Н); 7.43 (d, J=4,6 Гц, 5Н); 7.17 (dd, J=8,3, 4,5 Гц, 1Н); 7.03 (dd, J=8,2, 4,4 Гц, 1Н); 6.71 (dd, J=9,9, 3,7 Гц, 1Н); 5.18 (d, J=4,5 Гц, 3Н); 3.63-3.56 (m, 1Н); 3.49 (dd, J=10,4, 4,8 Гц, 1Н); 0.88 (t, J=4,4 Гц, 9Н); 0.14 (d, J=4,4 Гц, 3Н); -0.11 (d, J=4,4 Гц, 3Н).
Стадия 4: (R)-5-(2-азидо-1-((трет-бутилдиметилсилил)окси)этил)-8-(бензилокси)хинолин-2(1Н)-он
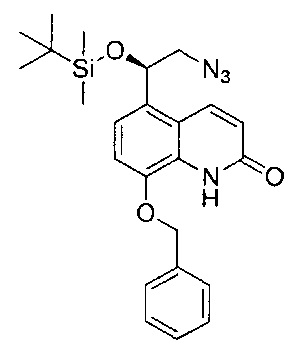
(R)-8-(Бензилокси)-5-(2-бром-1-((трет-бутилдиметилсилил)окси)-этил)хинолин-2(1H)-он (10,0 г; 20,5 ммоль) растворяли в диметилформамиде (180 мл) и воде (20 мл). Последовательно добавляли йодид натрия (3,39 г; 22,6 ммоль) и азид натрия (1,47 г; 22,6 ммоль). Реакционную смесь перемешивали при RT до тех пор, пока все твердое вещество не перешло в раствор. Раствор нагревали при 80°C в течение 40 часов, затем охлаждали до RT и разбавляли этилацетатом (300 мл). Смесь промывали водой, рассолом (×2) и органический экстракт сушили (сульфат магния), фильтровали и концентрировали при пониженном давлении. Неочищенный остаток растирали с изогексаном с получением требуемого соединения (8,16 г; 88%). Вещество использовали без дополнительной очистки на следующей стадии.
1Н ЯМР (400 МГц, CDCl3): δ 9.19 (s, 1Н), 8.18 (d, J=9,9 Гц, 1Н), 7.45-7.36 (m, 4Н), 7.20 (d, J=8,3 Гц, 1Н), 7.04 (d, J=8,3 Гц, 1Н), 6.70 (dd, J=9,9, 2,2 Гц, 1Н), 5.19-5.13 (m, 3Н), 3.48 (dd, J=12,7, 8,1 Гц, 1Н), 3.26 (dd, J=12,7, 3,8 Гц, 1Н), 0.89 (s, 9Н), 0.14 (s, 3Н), -0.11 (s, 3Н).
Стадия 5: гидрохлорид (R)-5-(2-амино-1-гидроксиэтил)-8-гидроксихинолин-2(1Н)-она
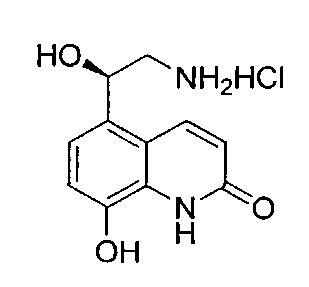
К раствору (R)-5-(2-азидо-1-((трет-бутилдиметилсилил)окси)-этил)-8-(бензилокси)хинолин-2(1H)-она (4,50 г; 10,0 ммоль) в этаноле (50 мл) добавляли 10%-ный палладий на угле (4,50 г) с последующим добавлением1-метил-1,4-циклогексадиена (11,0 мл; 97,9 ммоль). Реакционную смесь нагревали до 60°C (ОСТОРОЖНО - ВОЗМОЖЕН ЭКЗОТЕРМИЧЕСКИЙ ЭФФЕКТ) и затем перемешивали при 60°C в течение 2 часов. Реакционную смесь оставляли охлаждаться и фильтровали через набивку из целита. Осадок на фильтре промывали дополнительным количеством этанола и фильтрат упаривали при пониженном давлении. Остаток упаривали из изопропанола (×2) и растворяли в изопропаноле (30 мл). Добавляли HCl-диоксан (4 М; 50 мл; 200 ммоль) и реакционную смесь перемешивали при RT в течение 18 часов. Полученную суспензию фильтровали, осадок на фильтре промывали простым эфиром и твердое вещество сушили под вакуумом в присутствии P2O5 с получением указанного в заголовке соединения (1,65 г; 62%).
1Н ЯМР (400 МГц, MeOD): δ 7.71 (d, J=9,8 Гц, 1Н), 6.57 (d, J=8,2 Гц, 1Н), 6.31 (d, J=8,2 Гц, 1Н), 6.02 (dd, J=9,8, 6,5 Гц, 1Н), 4.58 (dd, J=9,6, 3,5 Гц, 1Н), 2.47-2.31 (m, 2Н).
Синтез соединений с 1 по 9
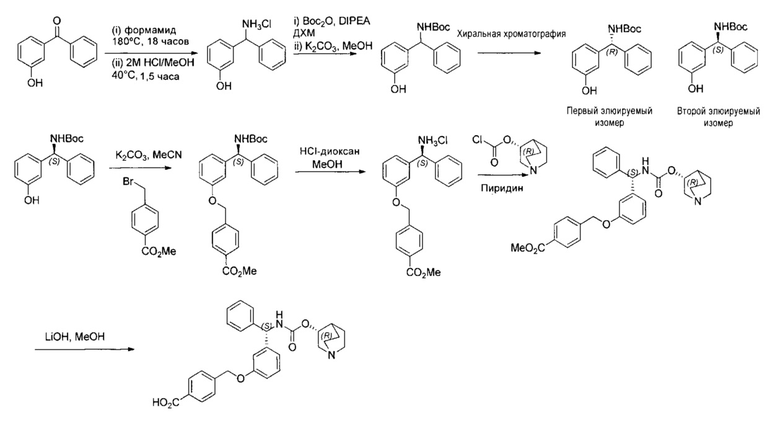
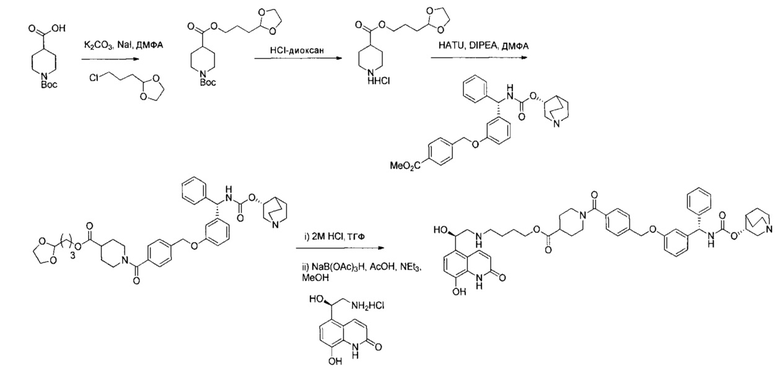
Пример 1
4-(((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)-этил)амино)бутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)-амино)метил)фенокси)метил)бензоил)пиперидин-4-карбоксилат (соединение 1)
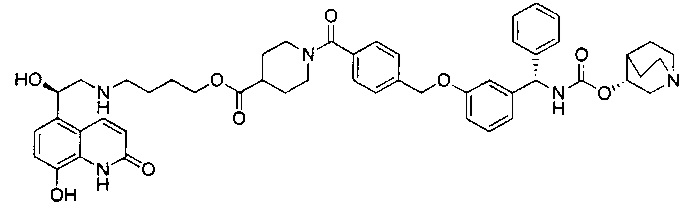
Стадия 1: N-((3-гидроксифенил)(фенил)метил)формамид
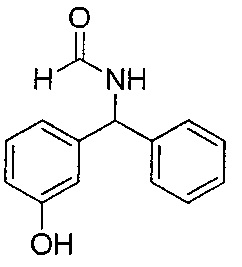
3-Гидроксибензофенон (25 г; 126,1 ммоль) в формамиде (130 мл; 3,3 ммоль) нагревали до 180°C в течение 18 часов. Реакционную смесь оставляли немного охладиться, затем выливали в ледяную воду, перемешивали в течение 30 минут, фильтровали и промывали водой. Твердое вещество перемешивали в воде (60 мл) и этаноле (60 мл) и нагревали до 50°C в течение 1 часа, затем оставляли охлаждаться. Твердое вещество фильтровали и промывали водой с получением указанного в заголовке соединения в виде коричневого твердого вещества (33,94 г; 118%).
1Н ЯМР (400 МГц, CD3OD): δ 7.39-7.28 (m, 5Н); 7.21-7.13 (m,1Н); 6.79 (d, J=7,78 Гц, 1Н); 6.73-6.68 (m, 2Н); 5.45 (s, 1Н).
Стадия 2: гидрохлорид 3-(амино(фенил)метил)фенола
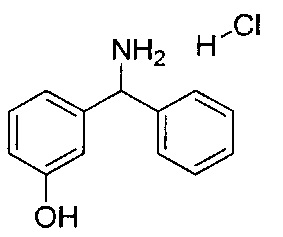
Метанол (125 мл) охлаждали до 0°C и по каплям добавляли ацетилхлорид (17,8 мл) с получением 2 М метанольного раствора хлористого водорода. N-((3-гидроксифенил)(фенил)метил)формамид перемешивали при 40°C в течение 1,5 часа с 2 М метанольным раствором хлористого водорода. Растворитель удаляли при пониженном давлении, остаток повторно растворяли в метаноле и растворитель удаляли при пониженном давлении. Этот процесс повторяли три раза с получением указанного в заголовке соединения в виде коричневого твердого вещества (29,09 г; 97,9%).
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.76 (s, 1Н); 9.07 (s, 3Н); 7.59-7.53 (m, 2Н); 7.51-7.37 (m, 3Н); 7.26 (t, J=7,89 Гц, 1Н); 6.99 (d, J=7,75 Гц, 1Н); 6.90 (t, J=1,97 Гц, 1Н); 6.81 (dd, J=8,10, 2,32 Гц, 1Н); 5.58 (d, J=5,82 Гц, 1Н).
Стадия 3: трет-бутил-((3-гидроксифенил)(фенил)метил)карбамат
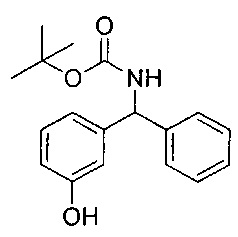
Гидрохлорид 3-(амино(фенил)метил)фенола (29,09 г; 123,4 ммоль) в дихлорметане (450 мл) охлаждали до 0°C и медленно добавляли диизопропилэтиламин (65,9 мл; 370,2 ммоль) и ди-трет-бутилдикарбонат (59,2 г; 271,5 ммоль). Реакционную смесь перемешивали при 0°C в течение 2 часов, затем нагревали до RT в течение 16 часов. Растворитель удаляли и остаток очищали с помощью набивки из диоксида кремния, элюируя 0-20% этилацетата в изогексане с получением черного масла. К этой смеси в метаноле (300 мл) добавляли карбонат калия (51 г; 370,2 ммоль) и перемешивали при RT в течение 16 часов. Суспензию фильтровали, фильтрат упаривали при пониженном давлении и остаток повторно растворяли в этилацетате (370 мл). Добавляли диоксид кремния (73 г) и суспензию перемешивали в течение 30 минут, фильтровали и осадок на фильтре промывали дополнительным количеством этилацетата. Фильтрат упаривали досуха. Темный твердый остаток растворяли в этилацетате (200 мл), добавляли древесный уголь и суспензию нагревали с обратным холодильником в течение 1 часа. Суспензию фильтровали через целит и растворитель удаляли при пониженном давлении. Темное твердое вещество растворяли в дихлорметане, добавляли изогексан и растворитель выпаривали (повторяя 3 раза) с получением указанного в заголовке соединения в виде желтого твердого вещества (34,81 г; 92%).
1Н ЯМР (400 МГц, CDCl3): δ 7.36-7.16 (m, 6Н); 6.80 (d, J=7,79 Гц, 1Н); 6.74-6.69 (m, 2H); 5.83 (s, 1H); 5.15 (s, 1H); 1.53-1.30 (s, 9H).
Стадия 4: (S)-трет-бутил-((3-гидроксифенил)(фенил)метил)-карбамат
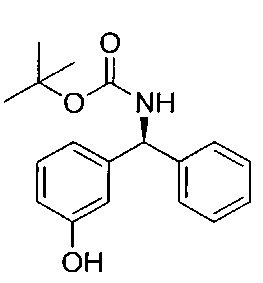
Рацемическую смесь со стадии 3 очищали посредством СФХ (сверхкритической флюидной хроматографией) при использовании колонки CHIRALPAK(R) AD 20 мкМ размером 250×110 мм с использованием смеси н-гептан/2-пропанол/диэтиламин (60/40/0,1) в качестве элюента со скоростью потока 570 мл/мин при 25°C. Из 54,1 г неочищенного вещества получили (S)-трет-бутил-((3-гидроксифенил)(фенил)метил)карбамат (Rt (время удерживания) составляет 8,5-8,6 мин; 23,9 г; 99,2 е.е. (энантиомерный избыток)).
Стадия 5: (S)-метил-4-((3-(((трет-бутоксикарбонил)амино)(фенил)-метил)фенокси)метил)бензоат
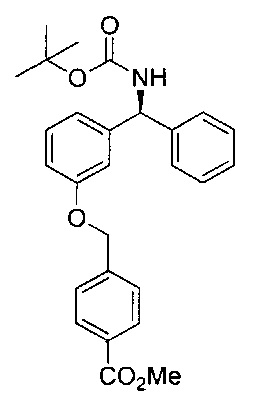
Смесь (S)-трет-бутил-((3-гидроксифенил)(фенил)метил)карбамата
(3,20 г; 10,7 ммоль), метил-4-(бромметил)бензоата (2,70 г; 11,8 ммоль) и карбоната калия (2,20 г; 16,1 ммоль) в ацетонитриле (54 мл) перемешивали при RT в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении и остаток распределяли между этилацетатом и водой. Водную фазу экстрагировали дополнительным количеством этилацетата и объединенные органические экстракты объединяли, сушили безводным сульфатом магния, фильтровали и растворитель выпаривали при пониженном давлении. Остаток перекристаллизовывали из этилацетата и изогексана с получением указанного в заголовке соединения в виде белого твердого вещества (3,25 г; 68%).
1Н ЯМР (400 МГц, CDCl3): δ 8.04 (d, J=8,2 Гц, 2Н); 7.46 (d, J=8,2 Гц, 2Н); 7.34-7.20 (m, 6Н); 6.90-6.81 (m, 3Н); 5.87 (s, 1Н); 5.13 (s, 1Н); 5.07 (s, 2Н); 3.92 (s, 3Н); 1.44 (s, 9Н).
Стадия 6: гидрохлорид (S)-метил-4-((3-(амино(фенил)метил)-фенокси)метил)бензоата
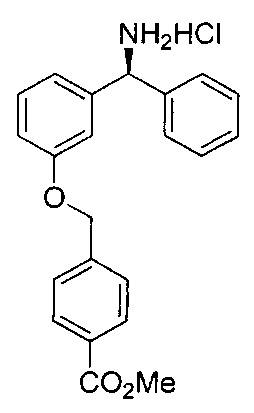
К раствору (S)-метил-4-((3-(((трет-бутоксикарбонил)амино)(фенил)-метил)фенокси)метил)бензоата (3,21 г; 7,20 ммоль) в метаноле (36 мл) добавляли хлорид водорода в диоксане (4 М, 9,0 мл; 36 ммоль). Реакционную смесь перемешивали при RT в течение 16 часов. Растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения (2,65 г; более 95%).
1Н ЯМР (400 МГц, CDCl3): δ 9.21 (s, 2Н); 8.03 (d, J=8,1 Гц, 2Н); 7.64 (d, J=8,1 Гц, 2Н); 7.59 (d, J=7,6 Гц, 2Н); 7.49-7.34 (m, 5Н); 7.17 (d, J=7,7 Гц, 1Н); 7.06 (dd, J=8,3, 2,4 Гц, 1Н); 5.64 (s, 1Н); 5.28 (s, 2Н); 3.91 (s, 3Н).
Стадия 7: метил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)бензоат
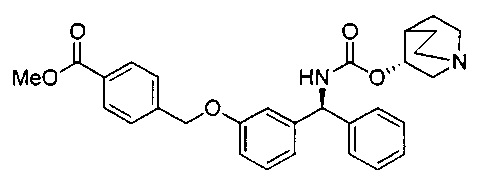
К перемешиваемому раствору гидрохлорида (S)-метил-4-((3-(амино-(фенил)метил)фенокси)метил)бензоата (12,0 г; 31,3 ммоль) в пиpидине (100 мл) при 0°C порциями добавляли (R)-хинуклидин-3-иловый эфир хлоругольной кислоты (8,50 г; 37,5 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа и затем оставляли нагреваться до RT в течение 16 часов. В реакционную смесь добавляли воду и экстрагировали этилацетатом (×3). Объединенные экстракты промывали рассолом, сушили (сульфат натрия), фильтровали и растворитель выпаривали при пониженном давлении. Неочищенное вещество очищали посредством хроматографии на картридже KP-NH Biotage, элюируя 0-20% метанола в этилацетате, с получением указанного в заголовке соединения (10,3 г; 66%).
Стадия 8: 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)-амино)метил)фенокси)метил)бензойная кислота
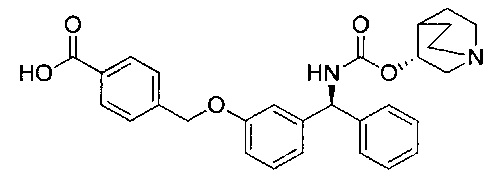
К перемешиваемому раствору метил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата (2,27 г; 4,50 ммоль) в ТГФ (23 мл) добавляли водный раствор гидроксида лития (2,0 М; 9,0 мл; 18,0 ммоль). Смесь перемешивали при RT в течение 16 часов. Значение pH реакционной смеси доводили до 6 посредством добавления 4 М водной соляной кислоты. Смесь затем экстрагировали 10%-ным раствором этилацетата в метаноле (×2) и объединенные органические экстракты упаривали при пониженном давлении. Остаток затем растворяли в этаноле и повторно упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (1,85 г; 84%).
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.41 (d, J=9,4 Гц, 1Н); 7.99 (d, J=7,9 Гц, 2Н); 7.58 (d, J=8,0 Гц, 2Н); 7.42-7.26 (m, 6Н); 7.09 (s, 1Н); 7.02-6.91 (m, 2Н); 5.87 (d, J=9 Гц, 1Н); 5.21 (s, 2Н); 4.76 (s, 1Н); 3.98-2.72 (m, 6Н); 2.12-1.54 (m, 5Н).
Стадия 9: 4-(3-(1,3-диоксолан-2-ил)пропил)-1-1-трет-бутил-пиперидин-1,4-дикарбоксилат
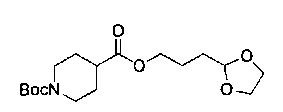
К перемешиваемому раствору N-трет-бутоксикарбонил-пиперидин-4-карбоновой кислоты (1,86 г; 8,11 ммоль) в ДМФА (20 мл) добавляли карбонат калия (1,68 г; 12,2 ммоль). Реакционную смесь перемешивали при RT в течение 20 минут и затем добавляли раствор 2-(3-хлорпропил)-1,3-диоксолана (0,815 г; 5,41 ммоль) в ДМФА (5 мл) и йодид натрия (0,973 г; 6,49 ммоль). Полученную смесь нагревали при 80°C в течение 18 часов. Реакционную смесь оставляли охлаждаться и разбавляли этилацетатом и водой. Органическую фазу удаляли, промывали рассолом (×2), сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии, элюируя смесью от 0 до 25% этилацетата/изогексан, с получением указанного в заголовке соединения (0,719 г; 39%).
1Н ЯМР (400 МГц, CDCl3): δ 4.90-4.88 (m, 1Н); 4.14-4.11 (m, 2Н); 4.02-3.83 (m, 6Н); 2.86-2.80 (m, 2Н); 2.47-2.40 (m, 1Н); 1.89-1.60 (m, 8Н); 1.45 (s, 9Н).
Стадия 10: гидрохлорид 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата
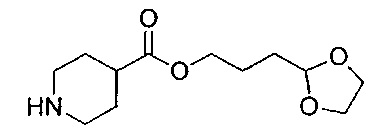
HCl-диоксан (4 М; 5 мл; 20 ммоль) добавляли к 4-(3-(1,3-диоксолан-2-ил)пропил)-1-трет-бутил-пиперидин-1,4-дикарбоксилату (0,71 г; 2,07 ммоль) и реакционную смесь перемешивали при RT в течение 1,5 часа. Растворитель выпаривали при пониженном давлении. Неочищенное вещество использовали непосредственно без очистки.
Стадия 11: 3-(1,3-диоксолан-2-ил)пропил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)-пиперидин-4-карбоксилат
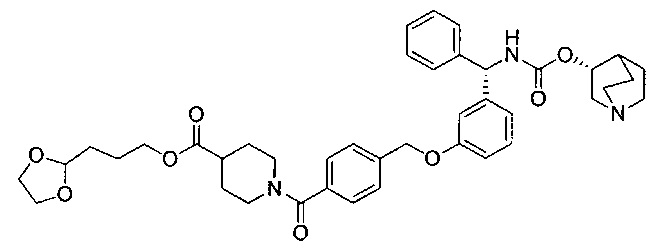
К перемешиваемому раствору 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензойной кислоты (0,300 г; 0,57 ммоль) в ДМФА (2,5 мл) добавляли диизопропилэтиламин (0,40 мл; 2,29 ммоль) и HATU (0,262 г; 0,69 ммоль) и смесь перемешивали при RT в течение 30 минут. К полученному раствору добавляли раствор гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата (0,241 г; 0,86 ммоль) в ДМФА (1,8 мл). Смесь перемешивали при RT в течение 18 часов. Смесь разбавляли этилацетатом, промывали 10%-ным водным карбонатом калия, рассолом (×2), сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 12: 4-оксобутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)пиперидин-4-карбоксилат
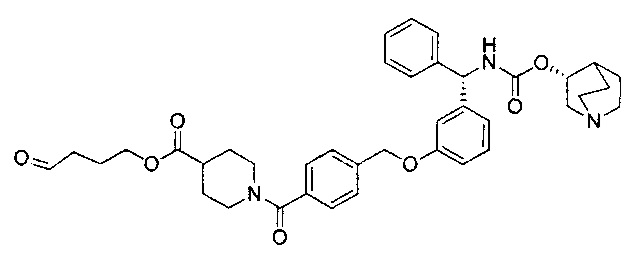
К раствору 3-(1,3-диоксолан-2-ил)пропил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)-пиперидин-4-карбоксилата в ТГФ (4,7 мл) добавляли 2 М водной соляной кислоты (4,7 мл). Реакционную смесь перемешивали при RT в течение 4 часов. Реакционную смесь распределяли между насыщенным гидрокарбонатом натрия и этилацетатом. Органическую фазу удаляли и водную фазу экстрагировали дополнительным количеством этилацетата (×2). Объединенные органические фазы промывали рассолом, сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 13: 4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)-пиперидин-4-карбоксилат (соединение 1)
К суспензии гидрохлорида (R)-5-(2-амино-1-гидроксиэтил)-8-гидроксихинолин-2(1Н)-она (0,136 г; 0,53 ммоль) в метаноле (3,5 мл) добавляли триэтиламин (0,148 мл; 1,06 ммоль). Смесь перемешивали в течение 10 минут и затем добавляли раствор 4-оксобутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоил)пиперидин-4-карбоксилата (0,337 г; 0,50 ммоль) в метаноле (3,4 мл). Смесь перемешивали при RT в течение 1 часа. Добавляли триацетоксиборгидрид натрия (0,267 г; 1,26 ммоль) с последующим добавлением уксусной кислоты (0,121 мл; 2,12 ммоль) и взаимодействие продолжали в течение еще 18 часов. Реакционную смесь гасили водой и упаривали при пониженном давлении. Остаток растворяли в изобутаноле и промывали водой. Органическую фазу упаривали при пониженном давлении и неочищенное вещество очищали посредством обращенно-фазовой препаративной ВЭЖХ с получением указанного в заголовке соединения (0,072 г; 16%).
1Н ЯМР (400 МГц, ДМСО-d6, 85°C): δ 8.23-8.17 (m, 3Н); 7.78-7.70 (m, 1Н); 7.48 (d, J=7,9 Гц, 2Н); 7.41-7.20 (m, 8Н); 7.08 (d, J=8,1 Гц, 1Н); 7.05-7.01 (m, 1Н); 6.97-6.88 (m, 3Н); 6.49 (d, J=9,9 Гц, 1Н); 5.83 (d, J=8,8 Гц, 1Н); 5.12 (s, 2Н); 5.04 (dd, J=7,7, 4,9 Гц, 1Н); 4.63-4.57 (m, 1Н); 4.06 (t, J=6,5 Гц, 2Н); 3.98-3.86 (m, 1Н); 3.13-3.03 (m, 3Н); 2.83-2.52 (m, 9Н); 1.94-1.83 (m, 3Н); 1.80-1.70 (m, 1Н); 1.67-1.37 (m, 9Н); 1.36-1.25 (m, 1Н). 1Н скрыт за сигналом воды.
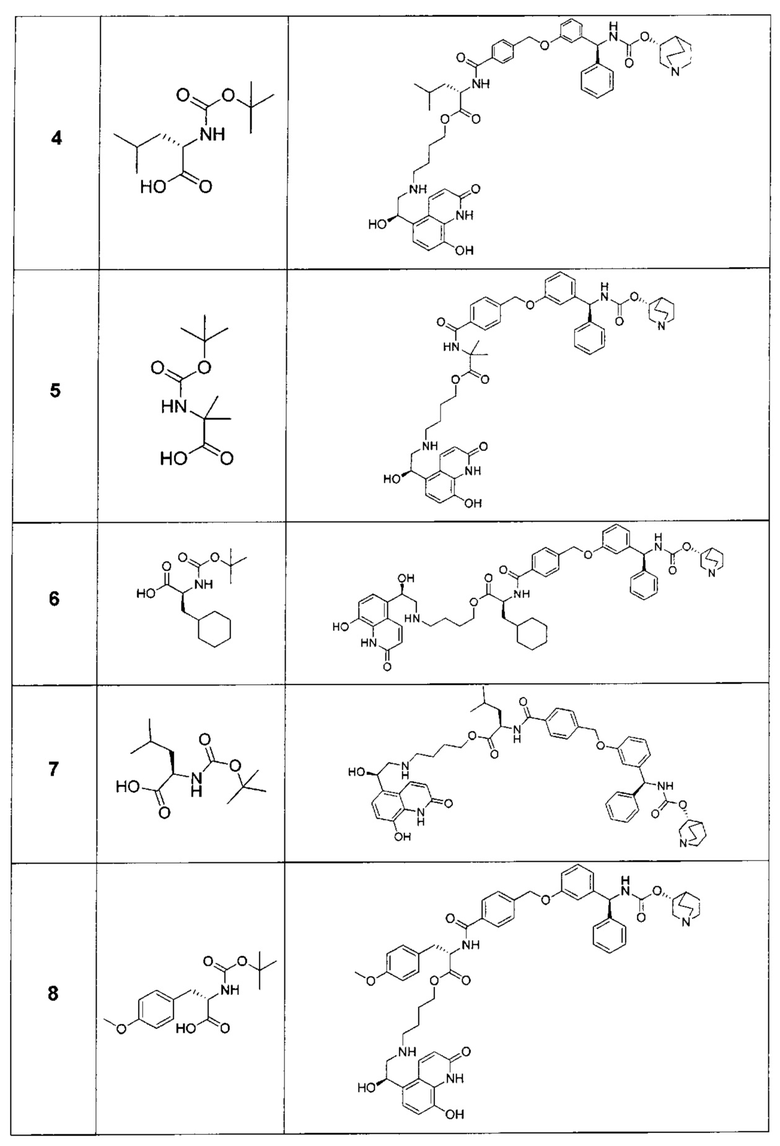
Следующие соединения получали, как описано в примере 1, с использованием соответствующей трет-бутоксикарбониламинокислоты на стадии 9 и продукта на последующих стадиях. 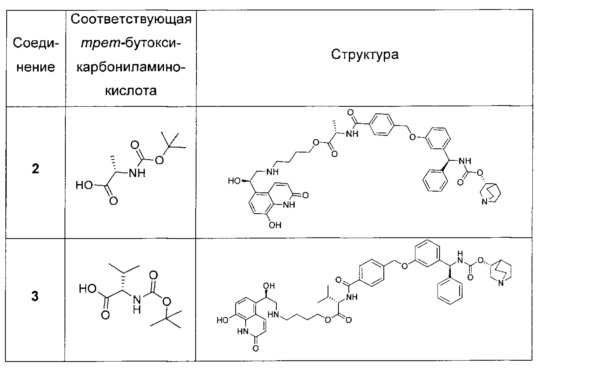
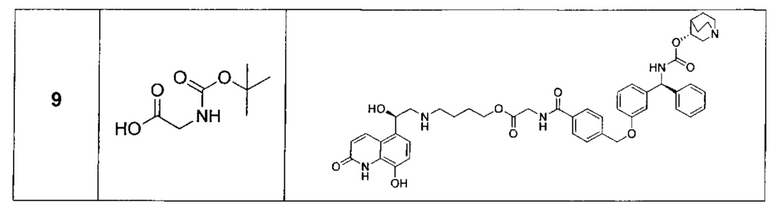
Пример 1А
(R)-Хинуклидин-3-ил-((S)-(3-((4-(4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пиперидин-1-карбонил)-бензил)окси)фенил)(фенил)метил)карбамат (соединение 9А)
Указанное в заголовке соединение получали, как описано в примере 1, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата на стадии 11 на 1,4-диокса-8-азаспироpо[4.5]декан, и полученные продукты использовали на стадиях 12-13.
1Н ЯМР (400 МГц, ДМСО-d6, 105°C): δ 8.25-8.17 (m, 3Н); 7.57 (d, J=8,8 Гц, 1Н); 7.47 (d, J=7,9 Гц, 2Н); 7.37-7.27 (m, 6Н); 7.28-7.20 (m, 2Н); 7.09 (d, J=8,1 Гц, 1Н); 7.03 (d, J=2,2 Гц, 1Н); 6.98-6.88 (m, 3Н); 6.49 (d, J=9,9 Гц, 1Н); 5.83 (d, J=8,7 Гц, 1Н); 5.12 (s, 2Н); 5.04-4.97 (m, 1Н); 4.64-4.58 (m, 1Н); 3.87 (s, 2Н); 3.12-3.00 (m, 3Н); 2.83 (t, J=6,2 Гц, 2Н); 2.79-2.56 (m, 6Н); 1.93-1.89 (m, 1Н); 1.79 (d, J=14,6 Гц, 3Н); 1.64-1.56 (m, 1Н); 1.52-1.45 (m, 1Н); 1.34-1.22 (m, 3Н).
Пример 1В
(R)-Хинуклидин-3-ил-((S)-(3-((4-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)пиперидин-1-карбонил)бензил)окси)фенил)(фенил)метил)карбамат (соединение 9В)
Указанное в заголовке соединение получали, как описано в примере 1, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата на стадии 11 на 4-(1,3-диоксолан-2-ил)пиперидин, и полученные продукты использовали на стадиях 12-13.
1Н ЯМР (400 МГц, ДMCO-d6, 110°C): δ (d, J=9,9 Гц, 1Н); 7.84 (d, J=8,6 Гц, 1Н); 7.49 (d, J=7,9 Гц, 2Н); 7.39-7.20 (m, 8Н); 7.16 (d, J=8,2 Гц, 1Н); 7.04-7.00 (m, 2Н); 6.99-6.90 (m, 2Н); 6.57 (d, J=9,9 Гц, 1Н); 5.85 (d, J=8,7 Гц, 1Н); 5.40 (dd, J=8,0, 5,0 Гц, 1Н); 5.13 (s, 2Н); 4.97-4.91 (m, 1Н); 4.04 (d, J=13,5 Гц, 2Н); 3.66 (ddd, J=14,0, 8,4, 2,6 Гц, 1Н); 3.33-3.11 (m, 6Н); 3.12-2.95 (m, 5Н); 2.25 (d, J=4,4 Гц, 1Н); 2.16-1.71 (m, 7Н); 1.28 (dd, J=24,1, 12,0 Гц, 2Н).
Пример 1С
4-(((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)-этил)амино)бутил-1-(3-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)бензоил)пиперидин-4-карбоксилат (соединение 9С)
Стадия 1: 3-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)-амино)метил)фенокси)метил)бензойная кислота
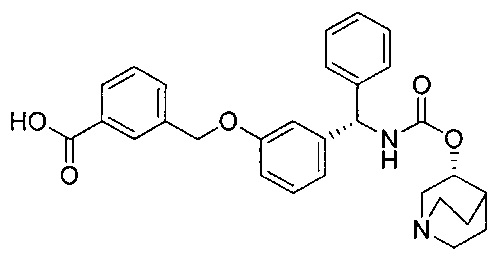
Указанное в заголовке соединение получали, как описано в примере 1 со стадии 5 до стадии 8, с заменой метил-4-(бромметил)бензоата на метил-3-(бромметил)бензоат на стадии 5, и продукты использовали на последующих стадиях.
1Н ЯМР (400 МГц, ДMCO-d6/D2O): δ 8.27 (s, 1Н); 7.96 (s, 1Н); 7.84 (d, J=7,7 Гц, 1Н); 7.55 (d, J=7,7 Гц, 1Н); 7.46-7.40 (m, 1Н); 7.31-7.18 (m, 7Н); 7.02 (s, 1Н); 6.88 (t, J=9,5 Гц, 2 Н); 5.78 (s, 1Н); 5.14 (s, 2Н); 4.72 (s, 1Н); 3.36 (m, 1Н); 3.06-2.72 (m, 5Н); 2.09-2.02 (m, 1Н), 1.94 (s, 1Н); 1.77 (s, 1Н); 1.70-1.51 (m, 2Н).
Стадия 2: 4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-1-(3-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)-пиперидин-4-карбоксилат (соединение 9С)
Указанное в заголовке соединение получали, как описано в примере 1 со стадии 11 до стадии 13, с заменой 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензойной кислоты на 3-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)-фенокси)метил)бензойную кислоту на стадии 11, и продукты использовали на последующих стадиях.
1Н ЯМР (400 МГц, ДМСО-d6): δ 10.50 (s, 2Н); 9.69 (s, 1Н); 8.65 (s, 3Н); 8.45 (d, J=9,2 Гц, 1Н); 8.16 (d, J=10,0 Гц, 1Н); 7.55-7.41 (m, 3Н); 7.35-7.20 (m, 7 Н); 7.15 (d, J=8,2 Гц, 1Н); 7.05-6.92 (m, 3Н); 6.91 (dd, J=8,2, 2,4 Гц, 1Н); 6.58 (d, J=9,9 Гц, 1Н); 6.19 (s, 1Н); 5.83 (d, J=9,1 Гц, 1Н); 5.32 (d, J=9,7 Гц, 1Н); 5.12 (s, 2Н); 4.88-4.83 (m, 1Н); 4.33 (s, 1Н); 4.10-4.04 (m, 2Н); 3.69-3.51 (m, 2Н); 3.36-2.90 (m, 8Н); 2.70-2.60 (m, 1Н); 2.23 (s, 1Н); 2.12-2.01 (m, 1Н); 1.97-1.52 (m, 12Н).
Пример 1D
4-(((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-1-(5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)фуран-2-карбонил)пиперидин-4-карбоксилат (соединение 9D)
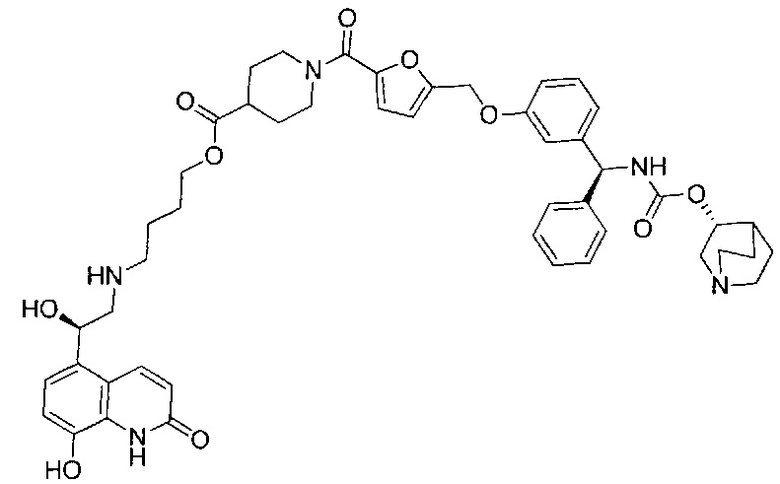
Стадия 1: 5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)-амино)метил)фенокси)метил)фуран-2-карбоновая кислота
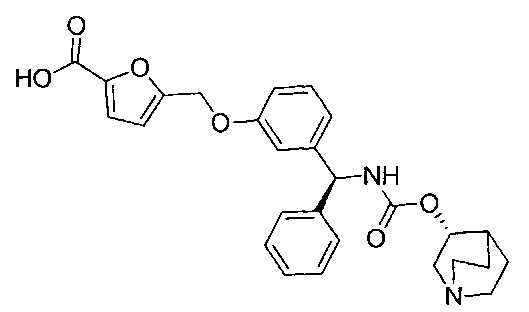
Указанное в заголовке соединение получали, как описано в примере 1 со стадии 5 до стадии 8, с заменой метил-4-(бромметил)бензоата на метил-5-(хлорметил)фуран-2-карбоксилат на стадии 5, и продукты использовали на последующих стадиях.
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.32 (d, J=9,3 Гц, 1Н); 7.36-7.28 (m, 4Н); 7.28-7.20 (m, 2Н); 7.08 (s, 1Н); 6.91 (t, J=9,4 Гц, 2Н); 6.83 (d, J=3,3 Гц, 1Н); 6.58 (d, J=3,3 Гц, 1Н); 5.82 (d, J=9,2 Гц, 1Н); 5.06 (s, 2Н); 4.73 (s, 1Н); 2.98 (s, 2Н); 2.88 (s, 2Н); 2.76 (d, J=14,8 Гц, 1Н); 2.07 (s, 1Н); 1.94 (s, 1Н); 1.73 (s, 1Н); 1.65 (s, 2Н); 1.55 (s, 1Н).
Стадия 2: 4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-1-(5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)фуран-2-карбонил)пиперидин-4-карбоксилат (соединение 9D)
Указанное в заголовке соединение получали, как описано в примере 1 со стадии 11 до стадии 13, с заменой 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензойной кислоты на 5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)-фенокси)метил)фуран-2-карбоновую кислоту на стадии 11, и продукты использовали на последующих стадиях.
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.36-8.08 (m, 4Н); 7.33-7.19 (m, 6Н); 7.11 (d, J=8,2 Гц, 1Н); 7.03 (s, 1Н); 6.97-6.90 (m, 4Н); 6.68 (d, J=3,4 Гц, 1Н); 6.53 (d, J=9,9 Гц, 1Н); 5.82 (d, J=9,2 Гц, 1Н); 5.20 (dd, J=8,0, 4,6 Гц, 1Н); 5.10 (s, 2Н); 4.60 (s, 1Н); 4.16 (d, J=13,1 Гц, 2Н); 4.05 (t, J=5,9 Гц, 2Н); 3.18-3.07 (m, 1Н); 2.90-2.55 (m, 9Н); 1.88 (d, J=17,3 Гц, 4Н); 1.67-1.31 (m, 12Н).
Пример 1Е
4-(((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)-этил)амино)бутил-1-(1-метил-5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-1Н-пиразол-3-карбонил)пиперидин-4-карбоксилат (соединение 9Е)
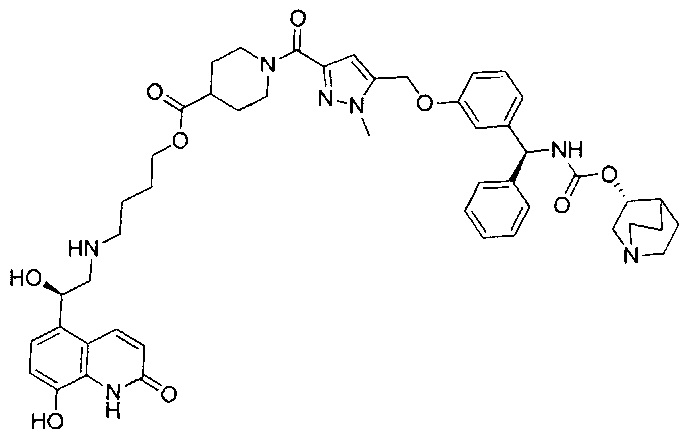
Стадия 1: 1-метил-5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)-1Н-пиразол-3-карбоновая кислота
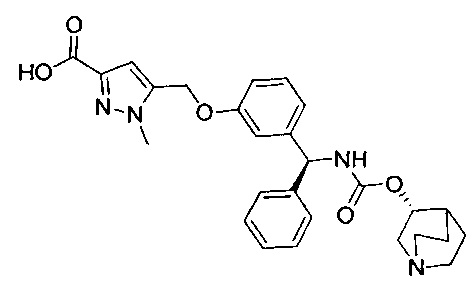
Указанное в заголовке соединение получали, как описано в примере 1 со стадии 5 до стадии 8, с заменой метил-4-(бромметил)бензоата на метиловый эфир 5-бромметил-1-метил-1Н-пиразол-3-карбоновой кислоты на стадии 5, и продукты использовали на последующих стадиях.
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.26 (d, J=9,3 Гц, 1Н); 7.33-7.18 (m, 6Н); 7.07 (s, 1Н); 6.99-6.91 (m, 2Н); 6.74 (s, 1Н); 5.82 (d, J=9,2 Гц, 1Н); 5.18 (s, 2Н); 4.60 (s, 2Н); 3.88 (s, 3Н); 3.18-3.07 (m, 1Н); 2.74 (m, 4Н); 1.94 (s, 1Н); 1.83 (s, 1Н); 1.62 (s, 1H); 1.52 (s, 1H); 1.39 (s, 1H).
Стадия 2: 4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-1-(1-метил-5-((3-((S)-фенил-((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-1Н-пиразол-3-карбонил)пиперидин-4-карбоксилат (соединение 9Е)
Указанное в заголовке соединение получали, как описано в примере 1 со стадии 11 до стадии 13, с заменой 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензойной кислоты на 1-метил-5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)-метил)фенокси)метил)-1Н-пиразол-3-карбоновую кислоту на стадии 11, и продукты использовали на последующих стадиях.
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.34-8.18 (m, 3Н); 8.18 (d, J=9,9 Гц, 1Н); 7.34-7.18 (m, 6Н); 7.13-7.04 (m, 2Н); 6.99-6.90 (m, 3Н); 6.67 (s, 1Н); 6.53 (d, J=9,9 Гц, 1Н); 5.83 (d, J=9,1 Гц, 1Н); 5.20-5.11 (m, 3Н); 4.59 (s, 1Н); 4.50 (d, J=12,8 Гц, 1Н); 4.34 (s, 1Н); 4.08-4.01 (m, 2Н); 3.87 (s, 3Н); 3.34-3.02 (m, 2Н); 2.89-2.55 (m, 10Н); 1.95-1.79 (m, 4Н); 1.69-1.36 (m, 10Н).
Пример 1F
Хинуклидин-3-ил-((S)-(3-((3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)пиперидин-1-карбонил)-1-метил-1Н-пиразол-5-ил)метокси)фенил)(фенил)метил)-карбамат (соединение 9F)
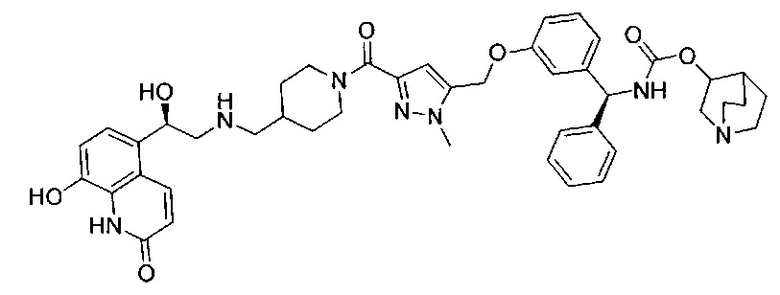
Указанное в заголовке соединение получали, как описано в примере 1, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата на 4-(1,3-диоксолан-2-ил)пиперидин и 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-бензойной кислоты на 1-метил-5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-1Н-пиразол-3-карбоновую кислоту на стадии 11, и полученные продукты использовали на стадиях 12-13.
1Н ЯМР (400 МГц, ДМСО, 110°C): δ 8.20 (d, J=9,9 Гц, 1Н); 7.84 (d, J=8,7 Гц, 1Н); 7.34-7.21 (m, 6Н); 7.16 (d, J=8,2 Гц, 1Н); 7.04-6.92 (m, 4Н); 6.61-6.55 (m, 2Н); 5.86 (d, J=8,6 Гц, 1Н); 5.39 (dd, J=8,0, 5,0 Гц, 1Н); 5.18 (s, 2Н); 4.96-4.91 (m, 1Н); 4.52 (d, J=13,0 Гц, 2Н); 3.88 (s, 3Н); 3.65-2.94 (m, 10Н); 2.26 (d, J=4,5 Гц, 1Н); 2.18-1.76 (m, 9Н); 1.27 (dd, J=12,2, 4,1 Гц, 2Н).
Пример 1G
(R)-Хинуклидин-3-ил-((3-((3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)пиперидин-1-карбонил)бензил)окси)фенил)(фенил)метил)карбамат (соединение 9G)
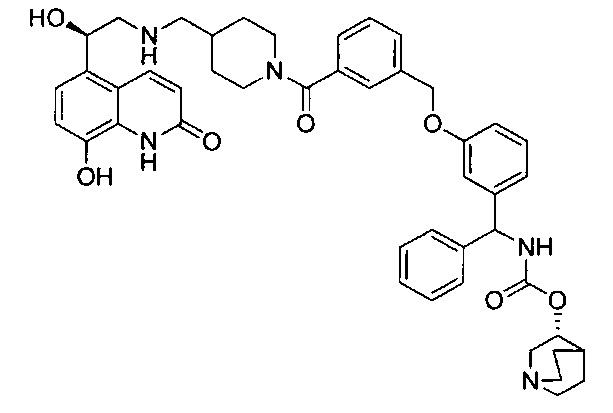
Указанное в заголовке соединение получали, как описано в примере 1, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата на 4-(1,3-диоксолан-2-ил)пиперидин и заменой 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензойной кислоты на 3-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензойную кислоту на стадии 11, и полученные продукты использовали на стадиях 12-13.
1Н ЯМР (400 МГц, ДМСО-d6, 110°C); 8.22 (d, J=9,9 Гц, 1Н); 8.15 (s, 2Н); 7.54 (d, J=8,3 Гц, 1Н); 7.49-7.39 (m, 3Н); 7.32-7.30 (m, 5Н); 7.23 (t, J=8,2 Гц, 2Н); 7.09 (d, J=8,2 Гц, 1Н); 7.02 (dd, J=2,1, 2,1 Гц, 1Н); 6.98-6.89 (m, 3Н); 6.49 (d, J=9,8 Гц, 1Н); 5.83 (d, J=8,7 Гц, 1Н); 5.13 (s, 2Н); 5.05 (dd, J=4,8, 7,6 Гц, 1Н); 4.65-4.61 (m, 1Н); 4.02-3.91 (m, 2Н); 3.12 (dd, J=8,3, 14,4 Гц, 1Н); 2.90-2.61 (m, 8Н); 2.60-2.51 (m, 3Н); 1.93 (dd, J=3,2, 6,3 Гц, 1Н); 1.83-1.45 (m, 6Н); 1.37-1.30 (m, 1Н); 1.18-1.07 (m, 2Н).
Синтез соединений с 10 по 15
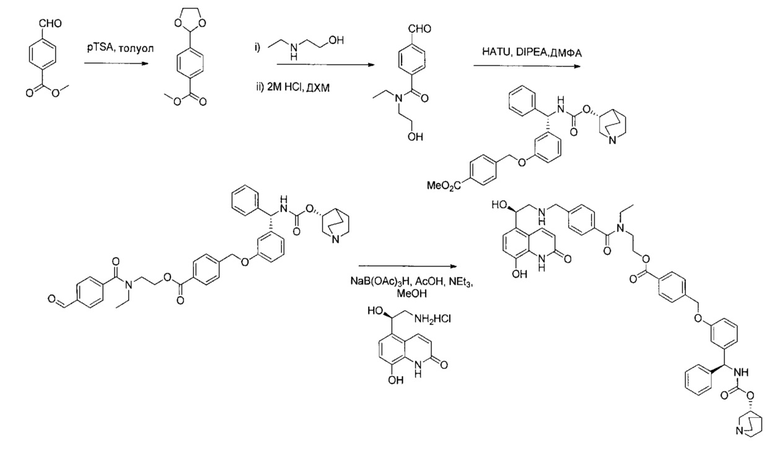
Пример 2
2-(N-Этил-4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат (соединение 10)
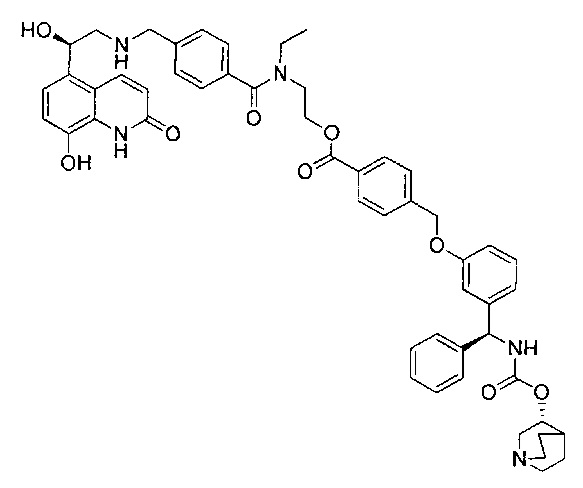
Стадия 1: метил-4-(1,3-диоксолан-2-ил)бензоат
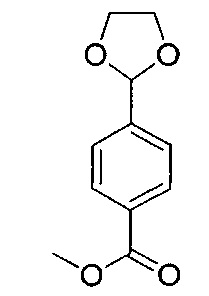
К перемешиваемому раствору метил-4-формилбензоата (5,0 г; 30,46 ммоль) в толуоле (196 мл) добавляли этиленгликоль (8,49 мл; 152,3 ммоль) и моногидрат пара-толуолсульфоновой кислоты (0,58 г; 3,05 ммоль). Реакционную смесь нагревали с обратным холодильником в условиях Дина-Старка в течение 1,5 часа. Растворитель выпаривали при пониженном давлении и остаток распределяли между этилацетатом и насыщенным водным гидрокарбонатом натрия. Органическую фазу удаляли и промывали дополнительным количеством насыщенного водного гидрокарбоната натрия, рассолом и сушили (сульфат магния). Смесь фильтровали и растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (6,29 г; 99%).
1Н ЯМР (400 МГц, CDCl3): δ 8.09-8.02 (m, 2Н); 7.58-7.51 (m, 2Н); 5.86 (s, 1Н); 4.16-4.01 (m, 4Н); 3.92 (s, 3Н).
Стадия 2: N-этил-4-формил-N-(2-гидроксиэтил)бензамид
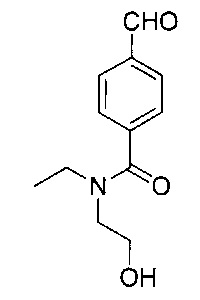
Раствор метил-4-(1,3-диоксолан-2-ил)бензоата (0,63 г; 3,0 ммоль) в 2-(этиламино)этаноле (2,93 мл; 30,0 ммоль) нагревали в микроволновой печи при 130°C в течение 2 часов. Реакционную смесь распределяли между ДХМ и 1 М водной соляной кислотой и интенсивно перемешивали в течение 1 часа. Органическую фазу удаляли, промывали рассолом и пропускали через гидрофобную фритту. Растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (0,334 г; 50%).
1Н ЯМР (400 МГц, CDCl3): δ 10.06 (s, 1Н); 7.95 (d, J=7,8 Гц, 2Н); 7.58 (d, J=7,9 Гц, 2Н); 3.76 (t, J=57,5 Гц, 4Н); 3.44-3.16 (m, 3Н); 1.18-1.11 (m, 3Н).
Стадия 3: 2-(N-этил-4-формилбензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат
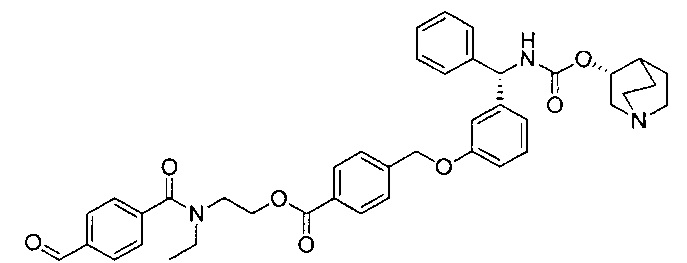
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 11, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропилпиперидин-4-карбоксилата на N-этил-4-формил-N-(2-гидроксиэтил)бензамид. Продукт использовали непосредственно без дополнительной очистки.
Стадия 4: 2-(N-этил-4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат (соединение 10)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 13, с заменой 4-оксобутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)-пиперидин-4-карбоксилата на 2-(N-этил-4-формилбензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат.
1Н ЯМР (400 МГц, ДМСО-d6): δ 10.30 (bs, 1Н); 8.28-8.21 (m, 2Н); 8.11 (d, J=9,9 Гц, 1Н); 8.04-7.84 (m, 2Н); 7.58 (d, J=8,0 Гц, 2Н); 7.40-7.18 (m, 10Н); 7.09-7.02 (m, 2Н); 6.97-6.86 (m, 3Н); 6.48 (d, J=9,9 Гц, 1Н); 5.82 (d, J=9,1 Гц, 1Н); 5.18 (s, 2Н); 5.07 (dd, J=8,0, 4,3 Гц, 1Н); 4.62-4.30 (m, 3Н); 3.85-3.07 (m, 7Н); 2.81-2.54 (m, 7Н); 1.95-1.74 (m, 2Н); 1.66-1.29 (m, 3Н); 1.23-0.98 (m, 3Н).
Следующие соединения получали, как описано в примере 2, с использованием соответствующего амина на стадии 2 и продукта на последующих стадиях.
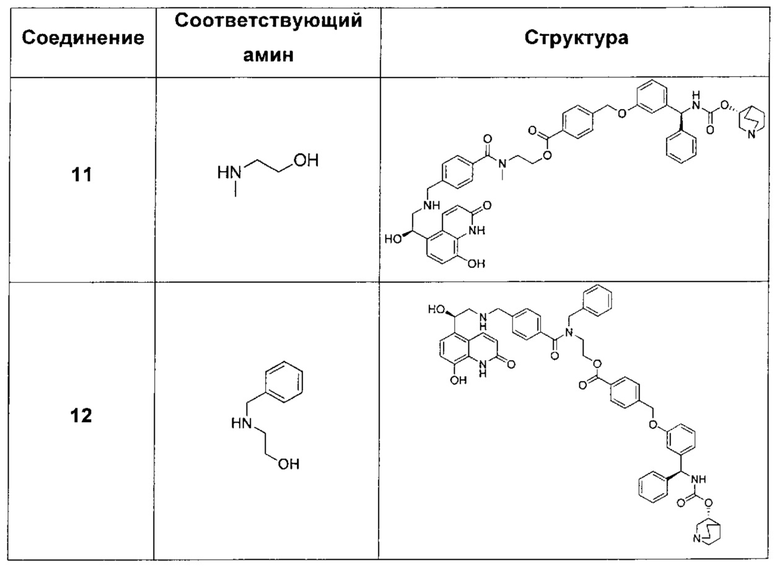

Пример 3
2-(3-((((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-N-метилбензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоат (соединение 16)
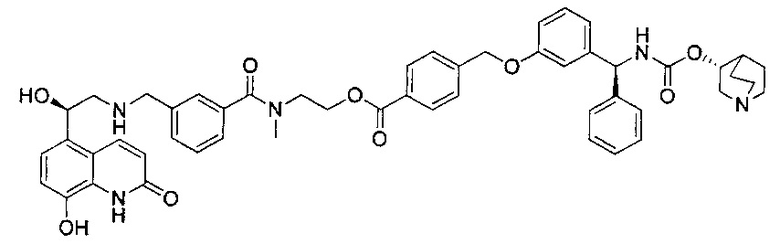
Стадия 1: метил-3-(1,3-диоксолан-2-ил)бензоат
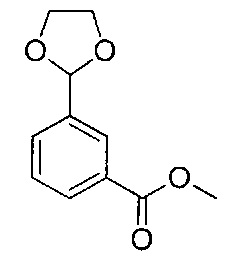
Указанное в заголовке соединение получали, как описано в примере 2 на стадии 1, с заменой метил-4-формилбензоата на метил-3-формилбензоат.
Стадия 2: 3-формил-N-(2-гидроксиэтил)-N1-метилбензамид
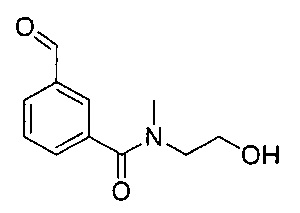
Указанное в заголовке соединение получали, как описано в примере 2 на стадии 2, с заменой метил-4-(1,3-диоксолан-2-ил)бензоата и N-этилэтаноламина на метил-3-(1,3-диоксолан-2-ил)бензоат и N-метилэтаноламин, соответственно.
Стадия 3: 2-(3-формил-N-метилбензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат
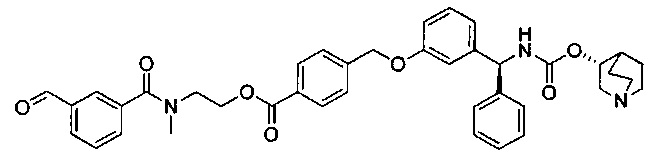
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 11, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропилпиперидин-4-карбоксилата на 3-формил-N-(2-гидроксиэтил)-N-метилбензамид. Продукт использовали непосредственно без дополнительной очистки.
Стадия 4: 2-(3-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-N-метилбензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат (соединение 16)
Указанное в заголовке соединение получали, как описано в Примере 1 на стадии 13.
Следующие соединения получали, как описано в примере 2, с заменой метил-4-формилбензоата на соответствующий альдегид на стадии 1 и замене 2-(этиламино)этанола на этаноламин на стадии 2. Последующие стадии являются такими, как описано в примере 2. 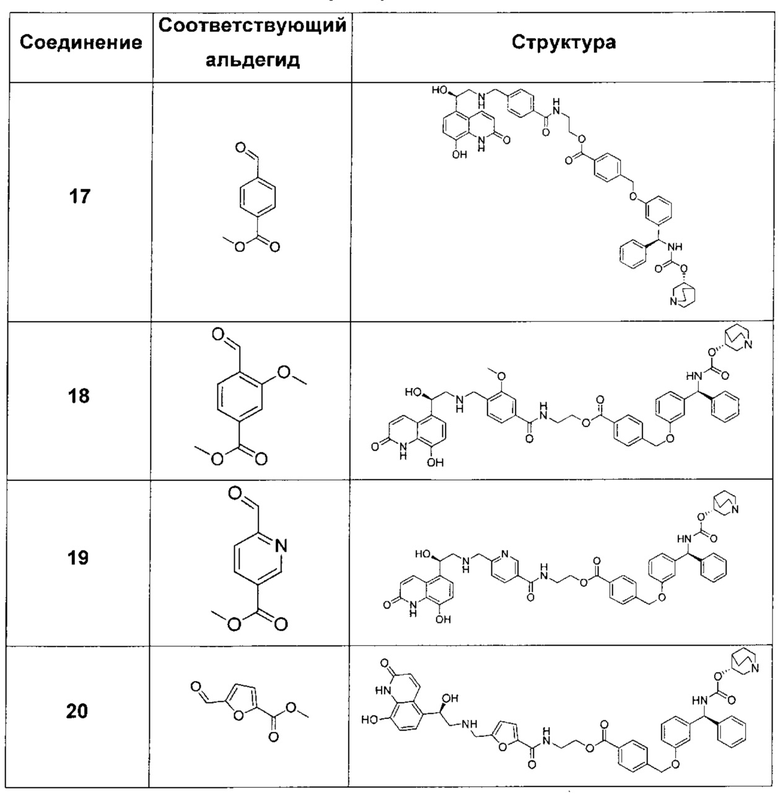
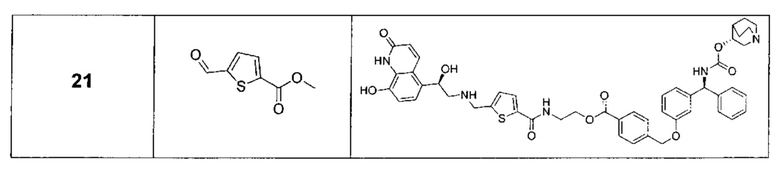
Следующие соединения получали, как описано в примере 2, с заменой метил-4-формилбензоата на соответствующий альдегид на стадии 1 и замене 2-(этиламино)этанола на пропаноламин на стадии 2. Последующие стадии являются такими, как описано в примере 2.
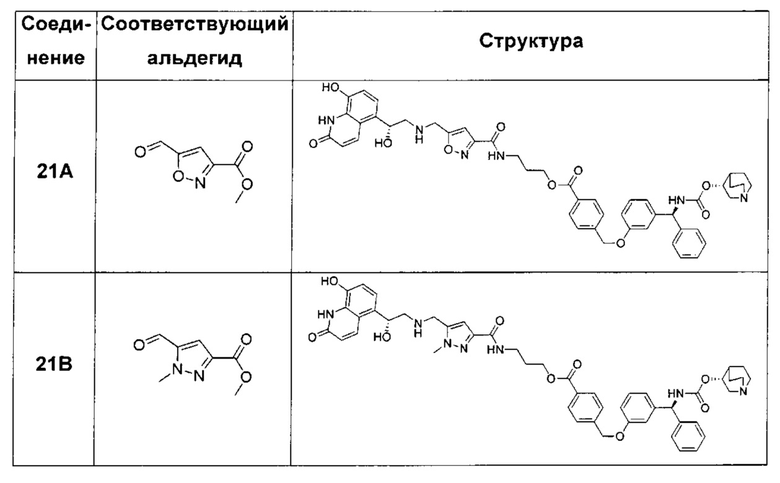
Пример 4
4-(((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-2-метокси-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоат (соединение 22)
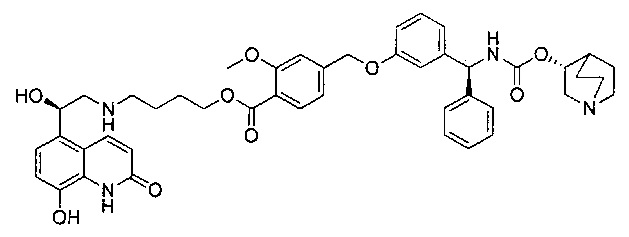
Стадия 1: метил-4-(бромметил)-2-метоксибензоат
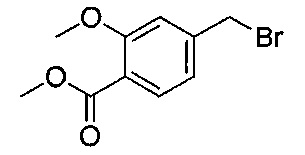
К раствору метил-2-метокси-4-метилбензоата (2,07 г; 11,5 ммоль) в хлороформе (30 мл) добавляли бензоилпероксид (0,14 г; 0,57 ммоль) и N-бромсукцинимид (2,04 г; 11,5 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 3 часов. Реакционную смесь промывали насыщенным гидрокарбонатом натрия, органическую фазу пропускали через гидрофобную фритту и растворитель выпаривали при пониженном давлении. Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Стадия 2: (S)-метил-4-((3-(((трет-бутоксикарбонил)амино)-(фенил)метил)фенокси)метил)-2-метоксибензоат
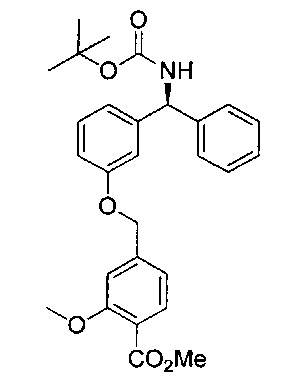
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 5, с заменой метил-4-(бромметил)бензоата на метил-4-(бромметил)-2-метоксибензоат.
1Н ЯМР (400 МГц, CDCl3): δ 7.84-7.76 (m, 1Н); 7.36-7.18 (m, 6Н); 7.09-6.77 (m, 5Н); 5.87 (s, 1Н); 5.17 (s, 1Н); 5.03 (s, 2Н); 3.92-3.87 (m, 6Н); 1.49 (s, 9Н).
Стадия 3: (S)-4-((3-(((трет-бутоксикарбонил)амино)(фенил)-метил)фенокси)метил)-2-метоксибензойная кислота
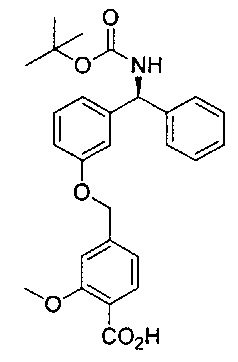
К раствору (S)-метил-4-((3-(((трет-бутоксикарбонил)амино)-(фенил)метил)фенокси)метил)-2-метоксибензоата (0,90 г; 1,89 ммоль) в смеси ТГФ (9 мл) и метанола (9 мл) добавляли 2 М водный гидроксид натрия (9 мл). Реакционную смесь перемешивали при RT в течение 18 часов. 10%-ную водную лимонную кислоту добавляли в реакционную смесь до pH 3. Смесь экстрагировали этилацетатом (×2). Объединенные органические фазы промывали рассолом, сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (0,82 г; 87%).
1Н ЯМР (400 МГц, CDCl3): δ 8.20-8.16 (m, 1Н); 7.37-7.19 (m, 6Н); 7.13 (d, J=6,7 Гц, 2Н); 6.93-6.83 (m, 3Н); 5.87 (s, 1Н); 5.16-5.03 (m, 3Н); 4.12-4.03 (m, 3Н); 1.43 (s, 9Н).
Стадия 4: (S)-3-(1,3-диоксолан-2-ил)пропил-4-((3-(((трет-бутоксикарбонил)амино)(фенил)метил)фенокси)метил)-2-метоксибензоат
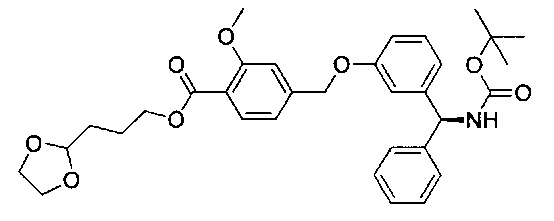
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 9, с заменой N-трет-бутоксикарбонил-пиперидин-4-карбоновой кислоты на (S)-4-((3-(((трет-бутоксикарбонил)амино)-(фенил)метил)-фенокси)метил)-2-метоксибензойную кислоту.
Стадия 5: 3-(1,3-диоксолан-2-ил)пропил-2-метокси-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат
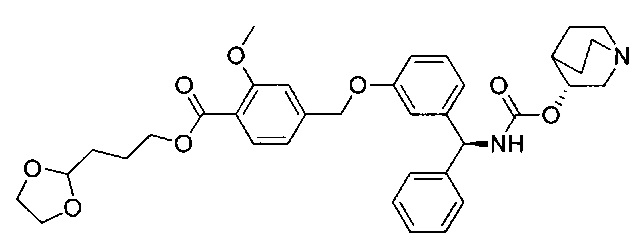
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 6 и стадии 7, с заменой (S)-метил-4-((3-(((трет-бутоксикарбонил)-амино)(фенил)метил)фенокси)метил)бензоата на (S)-3-(1,3-диоксолан-2-ил)пропил-4-((3-(((трет-бутоксикарбонил)амино)- (фенил)метил)фенокси)метил)-2-метоксибензоат на стадии 6, и полученный продукт использовали на стадии 7.
Стадия 6: 4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-2-метокси-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоат (соединение 22)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 12 и стадии 13, с заменой 3-(1,3-диоксолан-2-ил)пропил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-бензоил)пиперидин-4-карбоксилата на 3-(1,3-диоксолан-2-ил)пропил-2-метокси-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)-метил)фенокси)метил)бензоат на стадии 12, и полученный продукт использовали на стадии 13.
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.29 (s, 1Н); 8.24 (d, J=9,6 Гц, 1Н); 8.17 (d, J=9,9 Гц, 1Н); 7.64 (d, J=7,9 Гц, 1Н); 7.34-7.18 (m, 8Н); 7.12-7.02 (m, 2Н); 6.97-6.87 (m, 3Н); 6.51 (d, J=9,9 Гц, 1Н); 5.82 (d, J=9,0 Гц, 1Н); 5.13 (s, 2Н); 4.57 (s, 1Н); 4.21 (t, J=6,2 Гц, 2Н); 3.80 (s, 3Н); 3.10 (s, 1Н); 2.81 (d, J=6,3 Гц, 2Н); 2.74 (d, J=7,7 Гц, 4Н); 2.67 (s, 1Н); 2.62 (s, 2Н); 1.97-1.72 (m, 3Н); 1.74-1.66 (m, 2Н); 1.64-1.56 (m, 2Н); 1.48 (s, 2Н); 1.34 (s, 1Н).
Следующие соединения получали, как описано в примере 4, с заменой метил-2-метокси-4-метилбензоата на соответствующий толуол на стадии 1.
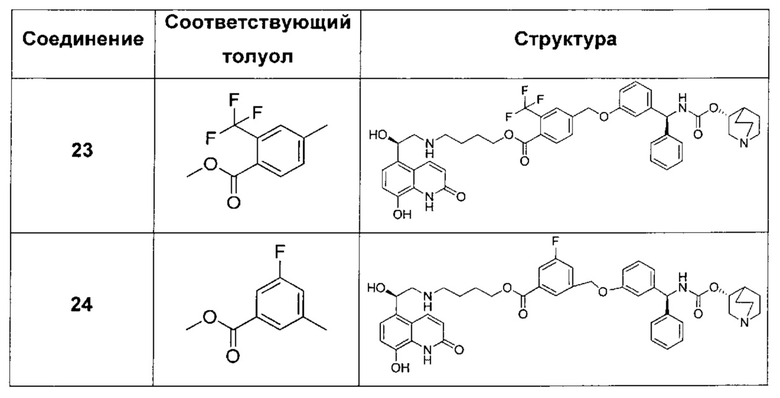
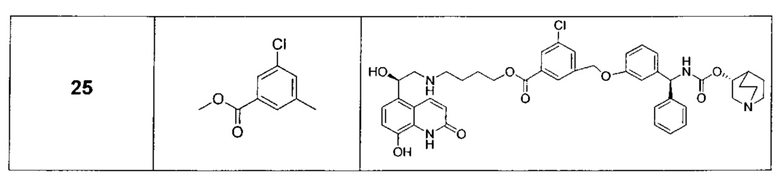
Пример 5
2-(N-(3-(((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)ацетамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоат (соединение 26)
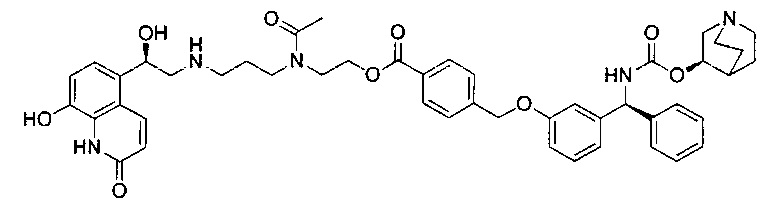
Стадия 1: 2-((2-(1,3-диоксолан-2-ил)этил)(бензил)амино)этанол
Раствор 2-бензилэтаноламина (4,00 г; 26,5 ммоль), 2-бромэтил-1,3-диоксалана (5,27 г; 29,1 ммоль) и диизопропилэтиламина (6,12 мл; 34,45 ммоль) в ацетонитриле (100 мл) нагревали с обратным холодильником в течение 18 часов. Реакционную смесь разбавляли ДХМ, промывали водой и органическую фазу пропускали через гидрофобную фритту. Растворитель выпаривали при пониженном давлении и остаток загружали на картридж SCX-2. Колонку элюировали этанолом (5 объемов колонки) с последующим элюированием 10% триэтиламина в этаноле (5 объемов колонки). Продукт, содержащий фракции, объединяли и растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (3,15 г; 47%).
1Н ЯМР (400 МГц, CDCl3): δ 7.34-7.21 (m, 5Н); 4.93-4.82 (m, 1Н); 3.99-3.77 (m, 4Н); 3.66-3.50 (m, 4Н); 2.69-2.55 (m, 4Н); 1.93-1.79 (m, 2Н); 1.27-1.17 (m, 1Н).
Стадия 2: N-(2-(1,3-диоксолан-2-ил)этил)-N-бензил-2-((трет-бутилдиметилсилил)окси)этанамин
К перемешиваемому раствору 2-((2-(1,3-диоксолан-2-ил)этил)-(бензил)амино)этанола (2,81 г; 11,2 ммоль) в ДМФА (10 мл) добавляли имидазол (1,67 г; 24,61 ммоль) и трет-бутилдиметилсилилхлорид (3,35 г; 22,39 ммоль). Реакционную смесь перемешивали при RT в течение 18 часов. Реакционную смесь разбавляли этилацетатом и промывали водой и рассолом. Органическую фазу сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении (3,87 г; 95%).
1Н ЯМР (400 МГц, CDCl3): δ 7.32-7.17 (m, 5Н); 4.90-4.85 (m, 1Н); 3.98-3.75 (m, 4Н); 3.69-3.61 (m, 4Н); 2.72-2.52 (m, 4Н); 1.88-1.76 (m, 2Н); 0.96-0.77 (m, 9Н); 0.05-0.01 (m, 6Н).
Стадия 3: N-(2-(1,3-диоксолан-2-ил)этил)-2-(трет-бутилдиметилсилил)окси)этанамин
К перемешиваемому раствору N-(2-(1,3-диоксолан-2-ил)этил)-N-бензил-2-((трет-бутилдиметилсилил)окси)этанамина (3,87 г; 10,6 ммоль) в этаноле (100 мл) добавляли 10%-ный палладий на угле (1,93 г) и 1-метил-1,4-циклогексадиен (5,93 мл; 53,0 ммоль). Суспензию нагревали с обратным холодильником в течение 30 минут и оставляли охлаждаться. Суспензию фильтровали через целит и осадок на фильтре промывали дополнительным количеством этанола. Растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (2,64 г; 91%).
1Н ЯМР (400 МГц, CDCl3): δ 4.91-4.81 (m, 1Н); 3.96-3.71 (m, 4Н); 3.67-3.56 (m, 2Н); 2.74-2.61 (m, 4Н); 1.86-1.72 (m, 2Н); 1.68 (s, 2Н); 0.83-0.78 (m, 9Н); 0.09-0.13 (m, 6Н).
Стадия 4: N-(2-(1,3-диоксолан-2-ил)этил)-N-(2-((трет-бутилдиметилсилил)окси)этил)ацетамид
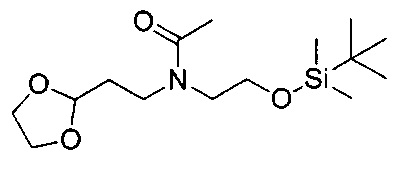
К охлаждаемому льдом раствору N-(2-(1,3-диоксолан-2-ил)этил)-2-((трет-бутилдиметилсилил)окси)этанамина (0,85 г; 3,09 ммоль) и диизопропилэтиламина (0,82 мл; 4,63 ммоль) добавляли ацетилхлорид (0,26 мл; 3,71 ммоль). Реакционную смесь перемешивали при охлаждении льдом в течение 1 часа, затем охладитель удаляли и перемешивание продолжали при RT в течение 18 часов. Реакционную смесь промывали насыщенным водным гидрокарбонатом натрия и органическую фазу пропускали через гидрофобную фритту. Растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (0,64 г; 65%).
1Н ЯМР (400 МГц, CDCl3): δ 4.84 (dt, J=6,3, 4,5 Гц, 1Н); 3.97-3.76 (m, 4Н); 3.75-3.63 (m, 2Н); 3.55-3.29 (m, 4Н); 2.10-2.04 (m, 3Н); 1.97-1.84 (m, 2Н); 0.84 (s, 9Н); 0.00 (s, 6Н).
Стадия 5: N-(2-(1,3-диоксолан-2-ил)этил)-N-(2-гидроксиэтил)-ацетамид
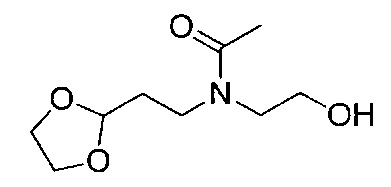
К перемешиваемому раствору N-(2-(1,3-диоксолан-2-ил)этил)-N-(2-((трет-бутилдиметилсилил)окси)этил)ацетамида (0,65 г; 2,04 ммоль) в ТГФ (5 мл) добавляли раствор фторида тетрабутиламмония (1,0 М в ТГФ; 2,24 мл). Реакционную смесь перемешивали при RT в течение 18 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом (×3). Объединенные органические экстракты сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии, элюируя смесью от 0 до 100% этилацетата/изогексан, с получением указанного в заголовке соединения (0,287 г; 69%), которое использовали непосредственно на следующей стадии.
Стадия 6: 2-(N-(2-(1,3-диоксолан-2-ил)этил)ацетамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-бензоат

Указанное в заголовке соединение получали, как описано в примере 1 на стадии 11, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата на N-(2-(1,3-диоксолан-2-ил)этил)-N-(2-гидроксиэтил)ацетамид.
Стадия 7: 2-(N-(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)ацетамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат (соединение 26)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 12 и стадии 13, с заменой 3-(1,3-диоксолан-2-ил)пропил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-бензоил)пиперидин-4-карбоксилата на 2-(N-(2-(1,3-диоксолан-2-ил)этил)-ацетамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)-амино)метил)фенокси)метил)бензоат на стадии 12, и полученный продукт использовали на стадии 13.
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.27 (s, 2Н); 8.20 (d, J=9,9 Гц, 1Н); 7.94 (d, J=7,9 Гц, 2Н); 7.61-7.50 (ш, 3Н); 7.32-7.29 (m, 4Н); 7.28-7.19 (m, 2Н); 7.08 (d, J=8,1 Гц, 1Н); 7.04-6.99 (m, 1Н); 6.98-6.88 (m, 3Н); 6.47 (d, J=9,9 Гц, 1Н); 5.85-5.79 (m, 1Н); 5.17 (s, 2Н); 5.04-4.99 (m, 1Н); 4.63-4.56 (m, 1Н); 4.46-4.34 (m, 2Н); 3.70-3.61 (m, 2Н); 3.43-3.35 (m, 2Н); 3.08-3.03 (m, 1Н); 2.82-2.55 (m, 9Н); 2.01 (s, 3Н); 1.93-1.84 (m, 1Н); 1.75-1.58 (m, 4Н); 1.52-1.42 (m, 1Н); 1.36-1.23 (m, 1Н).
Следующие соединения получали, как описано в примере 5, с соответствующим спиpтом на стадии 1 и ацилирующим агентом на стадии 4.
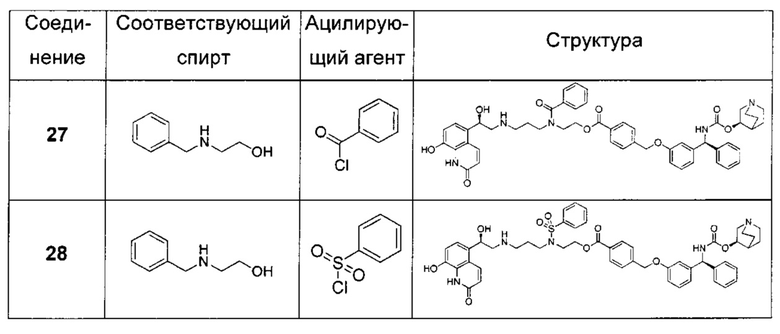
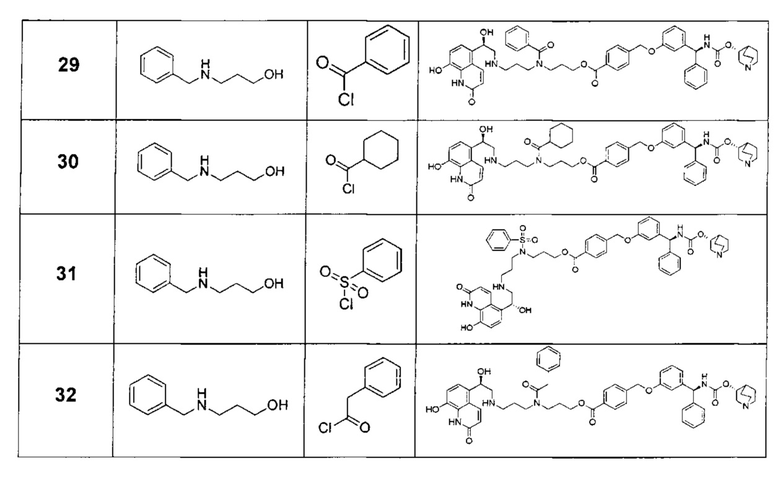
Пример 6
транс-4-(((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)циклогексанкарбоксилат (соединение 33)
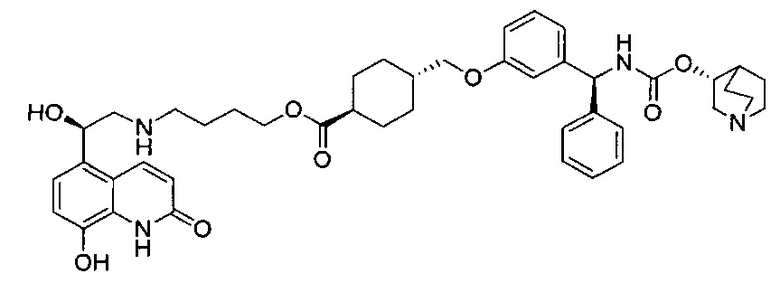
Стадия 1: транс-4-(((трет-бутилдиметилсилил)окси)метил)-циклогексанкарбоновая кислота
К перемешиваемому раствору транс-4-(1-гидроксиметил)-циклогексанкарбоновой кислоты (1,00 г; 6,32 ммоль) и 2,6-лутидина (2,95 мл; 25,29 ммоль) в ДХМ (61 мл) по каплям добавляли трет-бутилдиметилсилилтрифлат (4,36 мл; 18,96 ммоль). Реакционную смесь перемешивали при RT в течение 1,5 часа. Реакционную смесь гасили водой и органическую фазу удаляли. Водную фазу экстрагировали дополнительным количеством ДХМ (×2). Объединенные органические экстракты объединяли и промывали рассолом, сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток растворяли в ТГФ (30 мл) и метаноле (90 мл) и добавляли 10%-ный водный карбонат калия (30 мл). Реакционную смесь перемешивали при RT в течение 2 часов. Растворитель выпаривали при пониженном давлении и остаток распределяли между водой и эфиром. Органическую фазу отбрасывали. Водную фазу промывали дополнительным количеством эфира (×2). pH водного экстракта доводили до 5 и экстрагировали этилацетатом (×3). Объединенные этилацетатные экстракты промывали рассолом, сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (1,59 г; 93%).
1Н ЯМР (400 МГц, CDCl3): δ 3.37 (d, J=6,2 Гц, 2Н); 2.27-2.17 (m, 1Н); 2.00 (d, J=11,3 Гц, 2Н); 1.81 (dd, J=13,2, 3,7 Гц, 2Н); 1.50-1.33 (m, 3Н); 1.04-0.72 (m, 11Н); -0.00 (t, J=3,1 Гц, 6Н).
Стадия 2: транс-3-(1,3-диоксолан-2-ил)пропил-4-(((трет-бутилдиметилсилил)окси)метил)циклогексанкарбоксилат

Указанное в заголовке соединение получали, как описано в примере 1 на стадии 9, с заменой N-трет-бутоксикарбонил-пиперидин-4-карбоновой кислоты на транс-4-(((трет-бутилдиметилсилил)окси)-метил)-циклогексанкарбоновую кислоту.
1Н ЯМР (400 МГц, CDCl3): δ 4.91-4.87 (m, 1Н); 4.15-4.07 (m, 2Н); 3.99-3.82 (m, 4Н); 3.40 (d, J=6,2 Гц, 2Н); 2.26-2.16 (m, 1Н); 1.99 (d, J=13,1 Гц, 2Н); 1.84-1.67 (m, 6Н); 1.49-1.34 (m, 3Н); 1.06-0.80 (m, 11Н); 0.00 (s, 6Н).
Стадия 3: транс-3-(1,3-диоксолан-2-ил)пропил-4-(гидроксиметил)-циклогексанкарбоксилат

Указанное в заголовке соединение получали, как описано в примере 5 на стадии 5 с заменой N-(2-(1,3-диоксолан-2-ил)этил)-N-(2-((трет-бутилдиметилсилил)окси)этил)ацетамида на транс-3-(1,3-диоксолан-2-ил)пропил-4-(((трет-бутилдиметилсилил)окси)метил)-циклогексанкарбоксилат.
1Н ЯМР (400 МГц, CDCl3): δ 4.91-4.87 (m, 1Н); 4.14-4.07 (m, 2Н); 4.01-3.81 (m, 4Н); 3.50-3.43 (m, 2Н); 2.29-2.19 (m, 1Н); 2.06-1.98 (m, 2Н); 1.87 (dd, J=13,1, 3,7 Гц, 2Н); 1.80-1.69 (m, 3Н); 1.53-1.39 (m, 3Н); 1.29-1.21 (m, 2Н); 1.06-0.90 (m, 2Н).
Стадия 4: транс-3-(1,3-диоксолан-2-ил)пропил-4-((тозилокси)метил)-циклогексанкарбоксилат
К охлаждаемому льдом раствору транс-3-(1,3-диоксолан-2-ил)пропил-4-(гидроксиметил)циклогексанкарбоксилата (0,306 г; 1,13 ммоль) в пиpидине (0,9 мл) добавляли пара-толуолсульфонилхлорид (0,236 г; 1,24 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа и охладитель удаляли. Реакционную смесь перемешивали при RT в течение 18 часов. Реакционную смесь разбавляли этилацетатом и промывали водой, рассолом, сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток использовали непосредственно без очистки.
Стадия 5: транс-3-(1,3-диоксолан-2-ил)пропил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-циклогексанкарбоксилат
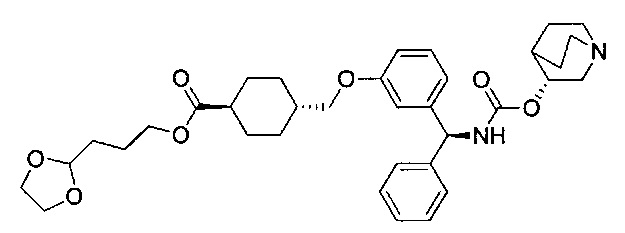
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 5, стадии 6 и стадии 7, с заменой метил-4-(бромметил)бензоата на транс-3-(1,3-диоксолан-2-ил)пропил-4-((тозилокси)метил)-циклогексанкарбоксилат на стадии 6, и полученные продукты использовали на стадии 6 и стадии 7.
Стадия 6: транс-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)згил)амино)бутил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)циклогексанкарбоксилат (соединение 33)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 12 и стадии 13, с заменой 3-(1,3-диоксолан-2-ил)пропил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)-фенокси)метил)-бензоил)пиперидин-4-карбоксилата на транс-3-(1,3-диоксолан-2-ил)-пропил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)-метил)фенокси)метил)циклогексанкарбоксилат на стадии 12, и полученный продукт использовали на стадии 13.
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.28-8.16 (m, 4Н); 7.36-7.28 (m, 4Н); 7.25-7.18 (m, 2Н); 7.09 (d, J=8,2 Гц, 1Н); 6.96-6.87 (m, 3Н); 6.81-6.77 (m, 1Н); 6.53 (d, J=9,9 Гц, 1Н); 5.81 (d, J=9,0 Гц, 1Н); 5.13 (t, J=6,3 Гц, 1Н); 4.61-4.55 (m, 1Н); 4.02 (t, J=6,1 Гц, 2Н); 3.74 (d, J=6,3 Гц, 2Н); 3.16-3.06 (m, 1Н); 2.84-2.54 (m, 9Н); 2.30-2.21 (m, 1Н); 1.97-1.27 (m, 16Н); 1.14-1.02 (m, 2Н).
Следующие соединения получали, как описано в примере 6, за исключением того, что транс-4-(((трет-бутилдиметилсилил)окси)-метил)циклогексанкарбоновую кислоту связывали с требуемым амином с использованием способа по примеру 1, стадия 11.
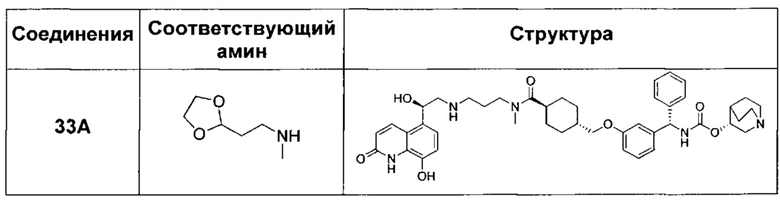
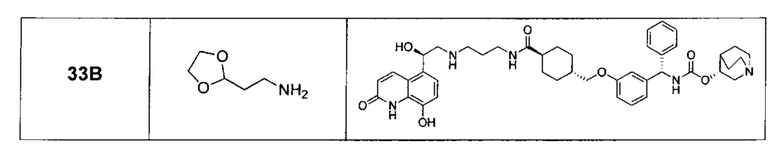
Пример 7
2-(3-(4-((((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)-1-метилуреидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат (соединение 34)
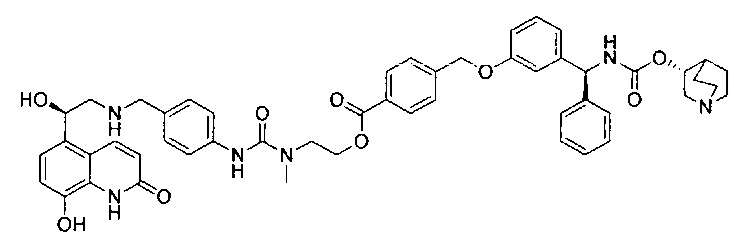
Стадия 1: метил-4-(3-(2-гидроксиэтил)-3-метилуреидо)бензоат
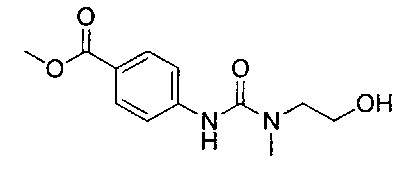
К охлаждаемому льдом раствору N-метилэтаноламина (0,600 г; 7,99 ммоль) в ДХМ (80 мл) добавляли метил-4-изоцианатобензоат (1,56 г; 8,79 ммоль). Через 20 минут охладитель удаляли и реакционную смесь перемешивали в течение 2 часов. Растворитель выпаривали при пониженном давлении и остаток очищали посредством колоночной флэш-хроматографии, элюируя смесью от 0 до 100% этилацетата/изогексан, с получением указанного в заголовке соединения (2,26 г; 112%).
1Н ЯМР (400 МГц, CDCl3): δ 8.74 (s, 1Н); 7.89 (m, 2 Н): 7.50 (m, 2Н); 5.02 (m, 1Н); 3.80 (s, 3Н); 3.59 (m, 2Н); 3.44 (m, 2Н); 3.00 (s, 3Н).
Стадия 2: 1-(2-гидроксиэтил)-3-(4-(гидроксиметил)фенил)-1-метилмочевина
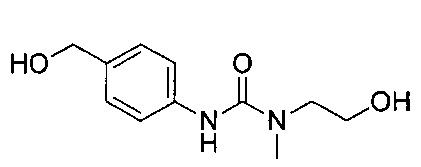
К охлаждаемому (-78°C) раствору метил-4-(3-(2-гидроксиэтил)-3-метилуреидо)бензоата (2,02 г; 8,00 ммоль) в ТГФ (80 мл) добавляли раствор алюмогидрида лития (2,0 М раствор в ТГФ; 6,0 мл; 12,0 ммоль). Через 20 минут охладитель удаляли и реакционную смесь перемешивали при RT в течение 18 часов. Реакционную смесь гасили водой (0,46 мл), 2 М водным гидроксидом натрия (0,46 мл) и водой (3×0,46 мл). Смесь разбавляли этилацетатом и добавляли сульфат магния. Смесь перемешивали в течение 1 часа и затем фильтровали. Фильтрат упаривали при пониженном давлении и остаток очищали посредством колоночной флэш-хроматографии, элюируя смесью от 0 до 100% этилацетата/изогексан, с получением указанного в заголовке соединения (1,09 г; 60%).
1Н ЯМР (400 МГц, ДМСО-d6): δ 8.30 (s, 1Н); 7.36 (d, J=8,3 Гц, 2Н); 7.16 (d, J=8,2 Гц, 2Н); 5.05-4.94 (m, 2Н); 4.40 (d, J=5,6 Гц, 2Н); 3.56 (dd, J=10,5, 5,3 Гц, 2Н); 2.95 (s, 3Н).
Стадия 3: 3-(4-формилфенил)-1-(2-гидроксиэтил)-1-метилмочевина
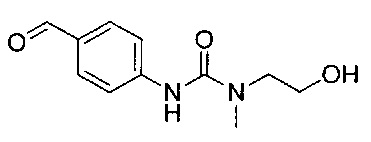
К раствору 1-(2-гидроксиэтил)-3-(4-(гидроксиметил)фенил)-1-метилмочевины (0,50 г; 2,23 ммоль) в ДХМ (30 мл) добавляли оксид марганца(IV) (0,78 г; 8,92 ммоль). Реакционную смесь перемешивали при RT в течение 3 часов. Суспензию фильтровали через целит и осадок на фильтре промывали дополнительным количеством ДХМ. Фильтрат упаривали при пониженном давлении с получением указанного в заголовке соединения (0,23 г; 49%).
1Н ЯМР (400 МГц, ДMCO-d6): δ 9.82 (s, 1Н); 8.86 (s, 1Н); 7.82-7.75 (m, 2Н); 7.66 (d, J=8,5 Гц, 2Н); 5.03 (d, J=5,8 Гц, 1Н); 3.61-3.55 (m, 2Н); 3.45-3.37 (m, 2Н); 2.99 (s, 3Н).
Стадия 4: 2-(3-(4-формилфенил)-1-метилуреидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-бензоат
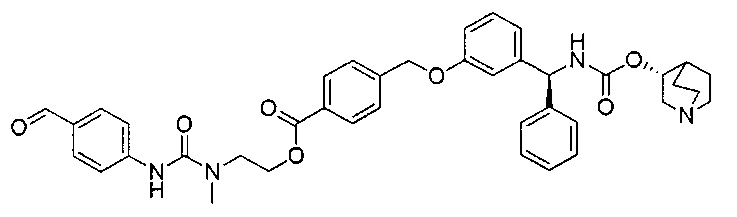
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 11, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата на 3-(4-формилфенил)-1-(2-гидроксиэтил)-1-метилмочевину.
Стадия 5: 2-(3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)-1-метилуреидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат (соединение 34)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 13, с заменой 4-оксобутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)пиперидин-4-карбоксилата на 2-(3-(4-формилфенил)-1-метилуреидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)бензоат.
1Н ЯМР (400 МГц, ДМСО-d6): δ 10.28 (br s, 1Н); 8.28 (s, 1Н); 8.23 (s, 2Н); 8.09 (d, J=9,95 Гц, 1Н); 7.96 (d, J=8,04 Гц, 2Н); 7.52 (d, J=7,99 Гц, 2Н); 7.39-7.13 (m, 10Н); 7.09-7.00 (m, 2Н); 6.97-6.83 (m, 3Н); 6.47 (d, J=9,88 Гц, 1Н); 5.82 (d, J=8,82 Гц, 1Н); 5.15 (s, 2Н); 5.07 (dd, J=7,96, 4,45 Гц, 1Н); 4.57 (s, 1Н); 4.45-4.39 (m, 2Н); 3.76-3.69 (m, 4Н); 3.16-3.05 (m, 1Н); 3.04 (s, 3Н); 2.77-2.61 (m, 6Н); 2.36-2.32 (m, 1Н); 1.94-1.75 (m, 2Н); 1.62-1.31 (m, 4Н).
Следующие соединения получали, как описано в Примере 7, с соответствующим амином на стадии 1.
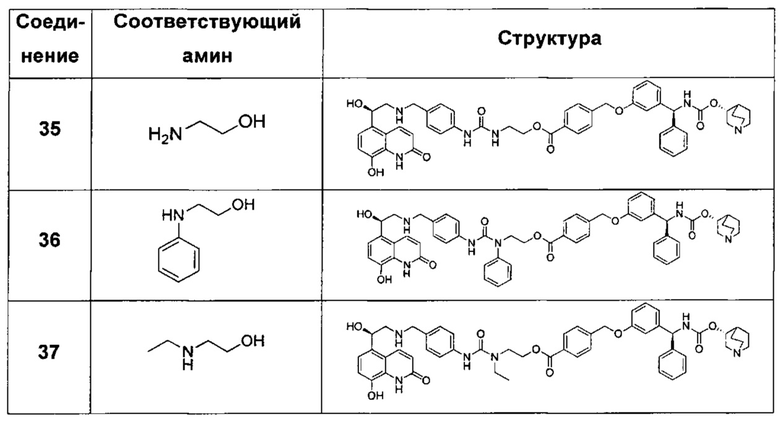
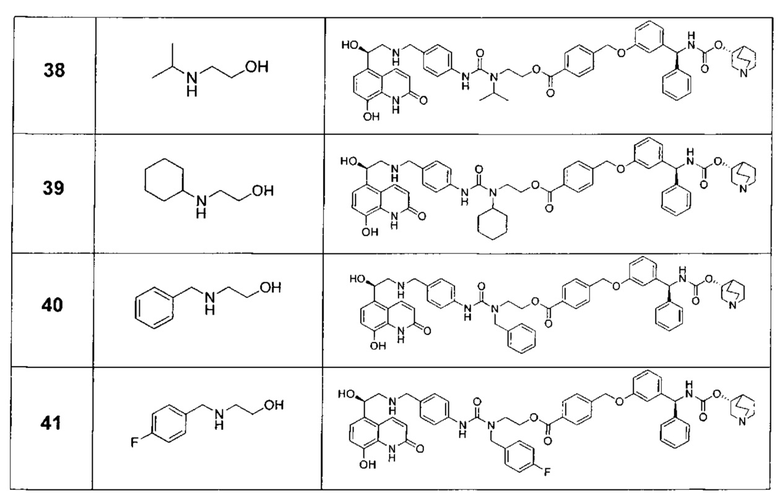
Следующие соединения получали, как описано в примере 2 на стадии 3 и стадии 4, с заменой N-этил-4-формил-N-(2-гидроксиэтил)бензамида на имеющийся в продаже спиpт на стадии 2.
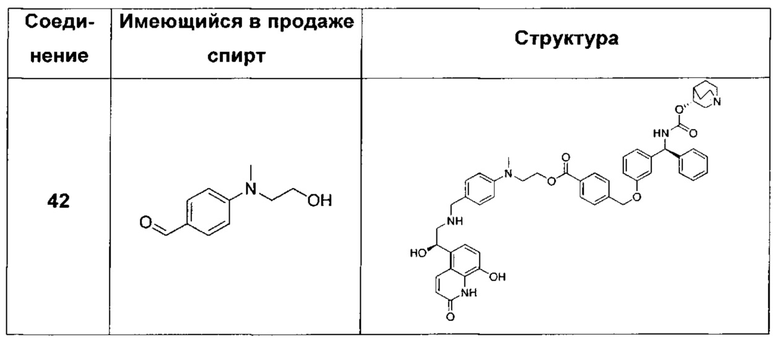
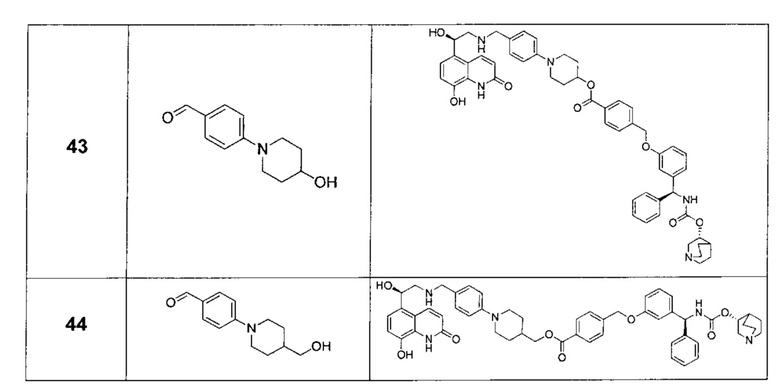
Пример 8
2-(4-((((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-N-метилфенилсульфонамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат (соединение 45)
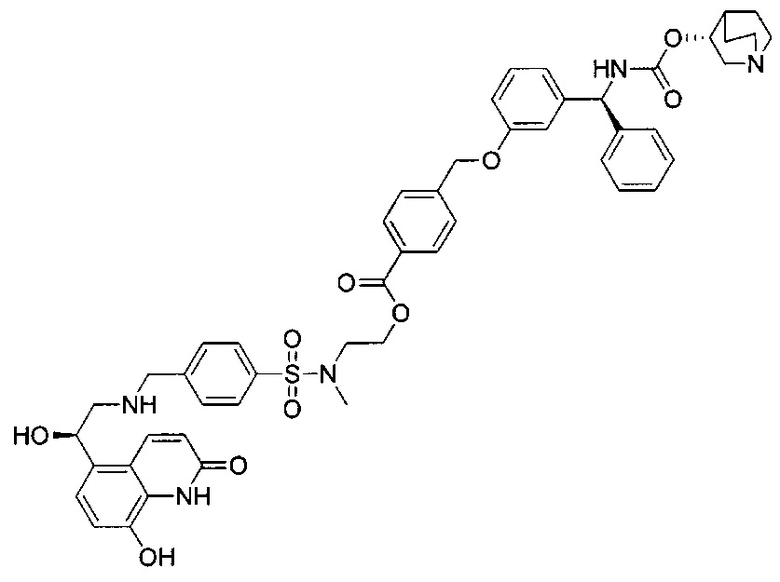
Стадия 1: 4-формил-N-(2-гидроксиэтил)-N-метилбензол-сульфонамид
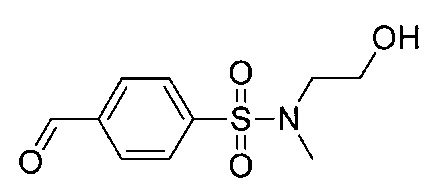
К охлаждаемому льдом раствору 2-(метиламино)этанола (0,196 мл; 2,44 ммоль) и триэтиламина (0,407 мл; 2,93 ммоль) в ДХМ (10 мл) добавляли 4-формилбензолсульфонилхлорид (0,500 г; 2,44 ммоль). Реакционную смесь медленно нагревали до RT и перемешивали при RT в течение 80 часов. Реакционную смесь разбавляли ДХМ и промывали 10%-ным водным гидросульфатом калия и сушили (сульфат магния). Смесь фильтровали и растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (0,548 г; 92%).
1Н ЯМР (400 МГц, CDCl3): δ 10.12 (s, 1Н); 8.09-8.04 (m, 2Н); 8.00-7.97 (m, 2Н); 3.82-3.78 (m, 2Н); 3.24 (t, J=5,2 Гц, 2Н); 2.90 (s, 3Н); 1.93 (t, J=5,2 Гц, 1Н).
Стадия 2: 2-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-N-метилфенилсульфонамидо)-этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)-фенокси)метил)бензоат (соединение 45)
Указанное в заголовке соединение получали, как описано в примере 2 на стадии 3 и стадии 4, с заменой N-этил-4-формил-N-(2-гидроксиэтил)-бензамида на 4-формил-N-(2-гидроксиэтил)-N-метилбензолсульфонамид на стадии 3, и полученный продукт использовали на стадии 4.
Пример 9
2-(4-(4-((((R)-2-Гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)-амино)метил)бензил)пиперазин-1-ил)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоат (соединение 46)
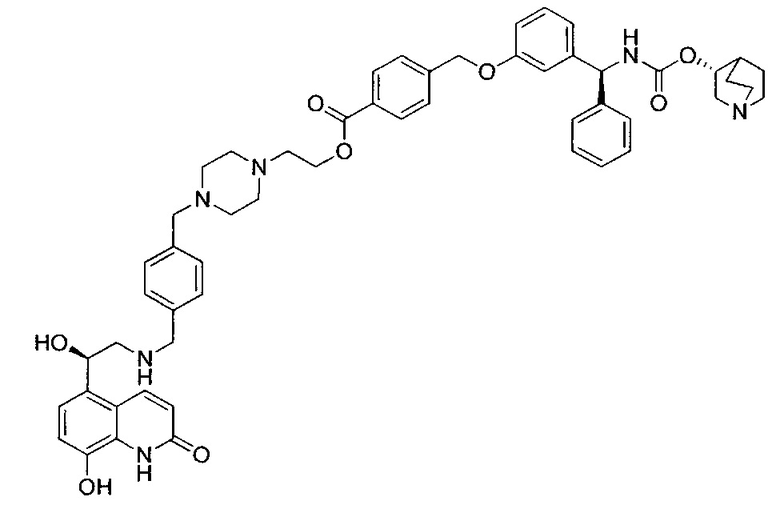
Стадия 1: 4-((4-(2-гидроксиэтил)пиперазин-1-ил)метил)бензальдегид
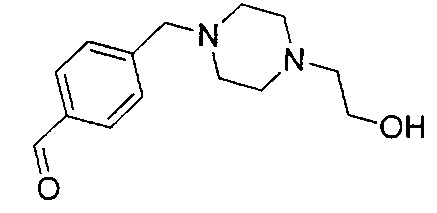
К раствору 4-(диметоксиметил)бензальдегида (2,08 г; 10,0 ммоль) в метаноле (30 мл) добавляли 1-(2-гидроксиэтил)пиперидин (1,02 мл; 8,31 ммоль). Реакционную смесь перемешивали при RT в течение 30 минут и затем добавляли триацетоксиборгидрид натрия (2,97 г; 14,0 ммоль). Реакционную смесь перемешивали при RT в течение 16 часов. Реакционную смесь разбавляли этилацетатом и промывали 10%-ным водным карбонатом калия, водой и рассолом. Органическую фазу сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток загружали на картридж SCX-2 и колонку элюировали этанолом (5 объемов колонки) с последующим элюированием 10%-ным триэтиламином в этаноле (5 объемов колонки). Содержащие продукт фракции объединяли и растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в ТГФ (20 мл) и добавляли 2 М водную соляную кислоту (20 мл). Реакционную смесь перемешивали при RT в течение 18 часов. К реакционной смеси добавляли 10%-ный водный карбонат калия и смесь экстрагировали ДХМ. Органическую фазу пропускали через гидрофобную фритту и растворитель выпаривали с получением указанного в заголовке соединения (1,28 г; 48%).
1Н ЯМР (400 МГц, CDCl3): δ 10.0 (s, 1Н); 7.85-7.83 (m, 2Н); 7.52 (d, J=8,8 Гц, 2Н); 3.62-3.56 (m, 4Н); 2.75-2.50 (m, 10Н); 1.43 (br s, 1Н).
Стадия 2: 2-(4-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)-этил)амино)метил)бензил)пиперазин-1-ил)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)-фенокси)метил)бензоат (соединение 46)
Указанное в заголовке соединение получали, как описано в примере 2 на стадии 3 и стадии 4, с заменой N-этил-4-формил-N-(2-гидроксиэтил)бензамида на 4-((4-(2-гидроксиэтил)пиперазин-1-ил)-метил)бензальдегид на стадии 3, и полученный продукт использовали на стадии 4.
Следующие соединения получали, как описано в Примере 9 на стадии 1 и стадии 2, с заменой 1-(2-гидроксиэтил)пиперидина на соответствующий аминоспиpт на стадии 1.
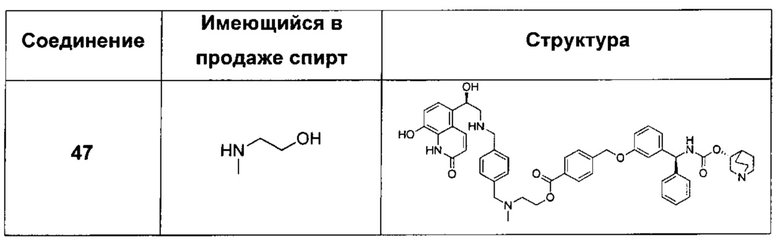
Синтез соединений с 48 по 51
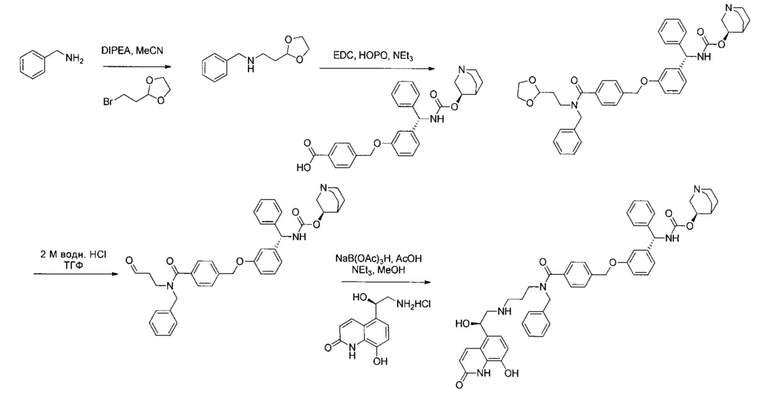
Пример 10
(R)-Хинуклидин-3-ил-((S)-(3-((4-(бензил(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)-карбамоил)бензил)окси)фенил)(фенил)метил)карбамат (соединение 48)
Стадия 1: N-бензил-2-(1,3-диоксолан-2-ил)этанамин
К смеси бензиламина (2,18 мл; 20,0 ммоль) и диизопропилэтиламина (2,61 мл; 15,0 ммоль) в ацетонитриле (30 мл) добавляли 2-(2-бромэтил)-1,3-диоксолан (1,17 мл; 10,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 72 часов. Реакционную смесь разбавляли этилацетатом и промывали водой и рассолом. Органическую фазу сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии, элюируя смесью от 0 до 4% метанола/дихлорметан, с получением указанного в заголовке соединения (0,836 г; 40%).
1Н ЯМР (400 МГц, CDCl3): δ 7.52-7.22 (m, 5Н); 4.95-4.88 (m, 1Н); 3.96-3.79 (m, 7Н); 2.80-2.76 (m, 2Н); 1.93-1.88 (m, 2Н).
Стадия 2: (R)-хинуклидин-3-ил-((S)-(3-((4-((2-(1,3-диоксолан-2-ил)-этил)(бензил)карбамоил)бензил)окси)фенил)(фенил)метил)карбамат
Триэтиламин (0,278 мл; 2,00 ммоль) добавляли к раствору 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензойной кислоты (0,418 г; 0,80 ммоль) в ДМФА (2 мл). Эту смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли 2-гидроксипиpидин-N-оксид (0,107 г; 0,96 ммоль) и EDC (0,184 г; 0,96 ммоль) и смесь перемешивали в течение еще 10 минут. Добавляли раствор N-бензил-2-(1,3-диоксолан-2-ил)этанамина (0,331 г; 1,6 ммоль) в ДМФА (2 мл). Реакционную смесь нагревали при 40°C в течение 4 часов с последующим перемешиванием при комнатной температуре в течение 48 часов. Реакционную смесь разбавляли этилацетатом и промывали 10%-ным водным карбонатом калия и дважды рассолом. Органическую фазу сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии, элюируя растворителями от 100% этилацетата до смеси 8%-ный метанольный раствор аммиака/этилацетат, с получением указанного в заголовке соединения (0,471 г; 87%). Это вещество использовали на следующей стадии без дополнительной характеризации.
Стадия 3: (R)-хинуклидин-3-ил-((S)-(3-((4-(бензил(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)-карбамоил)бензил)окси)фенил)(фенил)метил)карбамат
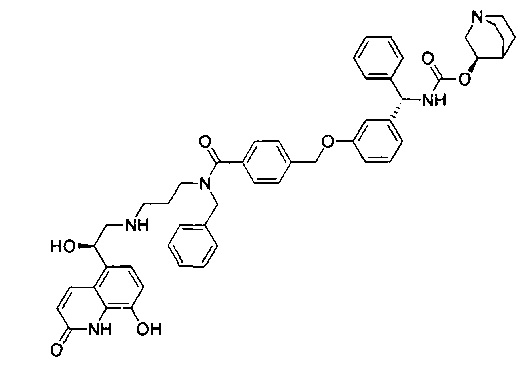
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 12 и стадии 13, с заменой 3-(1,3-диоксолан-2-ил)пропил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоил)пиперидин-4-карбоксилата на (R)-хинуклидин-3-ил-((S)-(3-((4-((2-(1,3-диоксолан-2-ил)этил)(бензил)карбамоил)бензил)окси)фенил)-(фенил)метил)карбамат на стадии 12, и полученный продукт использовали на стадии 13.
1Н ЯМР (400 МГц, ДМСО-d6, 105°C): δ 8.21-8.15 (m, 3Н); 7.58 (s, 1Н); 7.46 (d, J=7,9 Гц, 2Н); 7.41-7.29 (m, 7Н); 7.29-7.20 (m, 6Н); 7.07-7.00 (m, 2Н); 6.97-6.85 (m, 3Н); 6.47 (d, J=9,9 Гц, 1Н); 5.83 (d, J=7,2 Гц, 1Н); 5.11 (s, 2Н); 4.98 (dd, J=7,7, 4,9 Гц, 1Н); 4.66-4.58 (m, 3Н); 3.40-3.25 (m, 2Н); 3.08 (dd, J=14,4, 8,3 Гц, 1Н); 2.78-2.56 (m, 9Н); 1.93-1.89 (m, 1Н); 1.81-1.70 (m, 1Н); 1.70-1.56 (m, 3Н); 1.53-1.44 (m, 1Н); 1.36-1.27 (m, 1Н).
Следующие соединения получали, как описано в Примере 10, с заменой бензиламина на соответствующий амин на стадии 1, и полученные продукты использовали на стадии 2 и стадии 3.
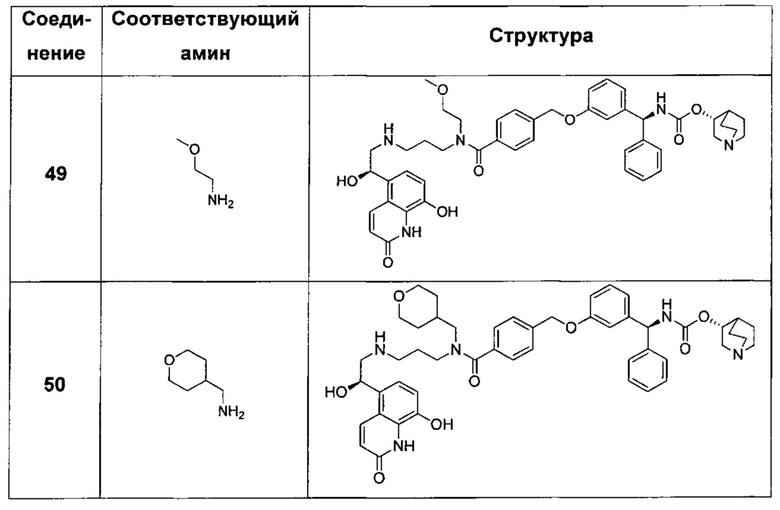
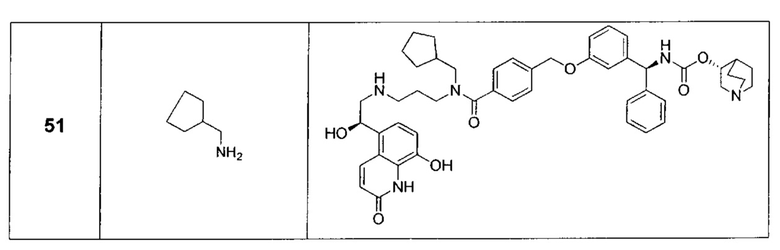
Следующие соединения получали, как описано в Примере 10, с заменой 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)-фенокси)метил)бензойной кислоты на соответствующую кислоту на стадии 2, и полученные продукты использовали на стадии 3.
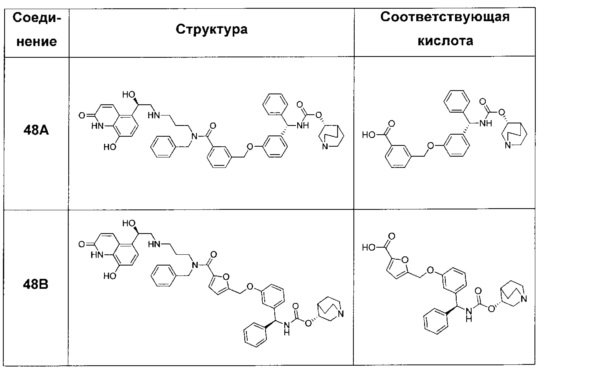
Синтез соединений с 52 по 68
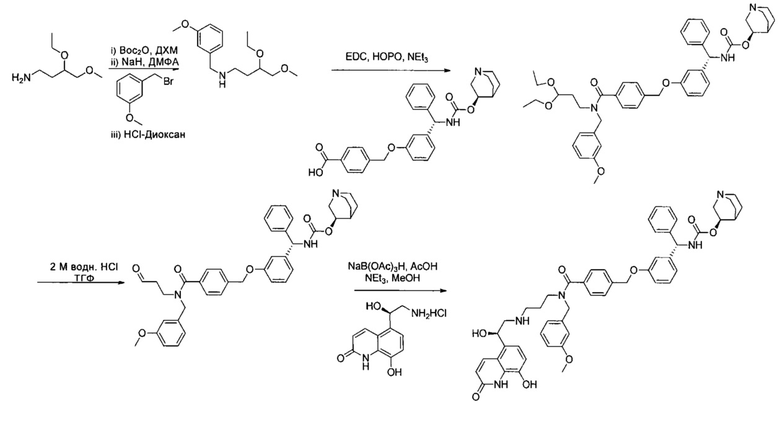
Пример 11
(R)-Хинуклидин-3-ил-((S)-(3-((4-((3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)(3-метоксибензил)-карбамоил)бензил)окси)фенил)(фенил)метил)карбамат (соединение 52)
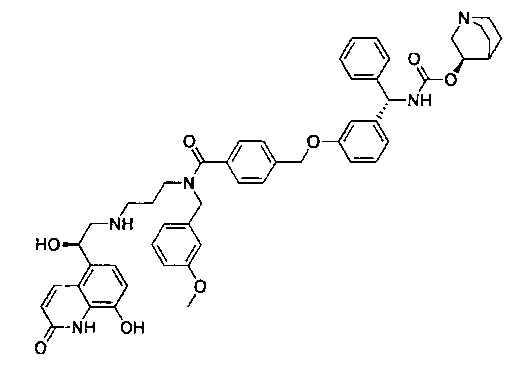
Стадия 1: трет-Бутил-(3,3-диэтоксипропил)карбамат
Раствор ди-трет-бутилдикарбоната (6,74 г; 31,0 ммоль) в ДХМ (50 мл) добавляли к раствору 1-амино-3,3-диэтоксипропана (5,0 г; 34,0 ммоль) в ДХМ (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь промывали 10%-ным водным гидросульфатом калия и органическую фазу сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении с получением указанного в заголовке продукта (8,13 г; 100%).
1Н ЯМР (400 МГц, CDCl3): δ 5.00 (s, 1Н), 4.56-4.53 (m, 1Н); 3.69-3.62 (m, 2Н); 3.54-3.46 (m, 2Н); 3.24-3.20 (m, 2Н); 1.84-1.80 (m, 2Н); 1.49 (s, 9Н); 1.23-1.19 (m, 6Н).
Стадия 2: 2-(1,3-диоксолан-2-ил)-N-(3-метоксибензил)этанамин
К перемешиваемому раствору трет-бутил-(3,3-диэтоксипропил)-карбамата (1,0 г; 4,05 ммоль) в ДМФА (15 мл) добавляли гидрид натрия (0,25 г; 6,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и добавляли 3-метоксибензилбромид (0,875 мл; 6,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 72 часов. Реакционную смесь разбавляли этилацетатом и промывали водой и дважды рассолом. Органическую фазу сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток растворяли в этаноле (10 мл) и добавляли 4 М хлорид водорода в диоксане (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и растворитель выпаривали при пониженном давлении. Остаток загружали на картридж SCX-2 и элюировали этанолом (4 объема колонки) и затем смесью 10%-ный триэтиламин/этанол (4 объема колонки). Фракции, содержащие продукт, объединяли и растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения (0,543 г; 50%).
1Н ЯМР (400 МГц, CDCl3): δ 7.25-7.21 (m, 1Н); 6.90-6.80 (m, 2Н); 6.78-6.77 (m, 1Н); 4.60 (t, J=5,6 Гц, 1Н); 3.81 (s, 3Н); 3.76 (s, 2Н); 3.55-3.43 (m 2Н); 2.72 (t, J=6,8 Гц, 2Н); 1.87-1.79 (m, 2Н); 1.50 (s, 1Н); 1.29-1.09 (m, 6Н).
Стадия 3: (R)-хинуклидин-3-ил-((S)-(3-((4-((2-(1,3-диоксолан-2-ил)этил)(3-метоксибензил)карбамоил)бензил)окси)фенил)(фенил)метил)-карбамат
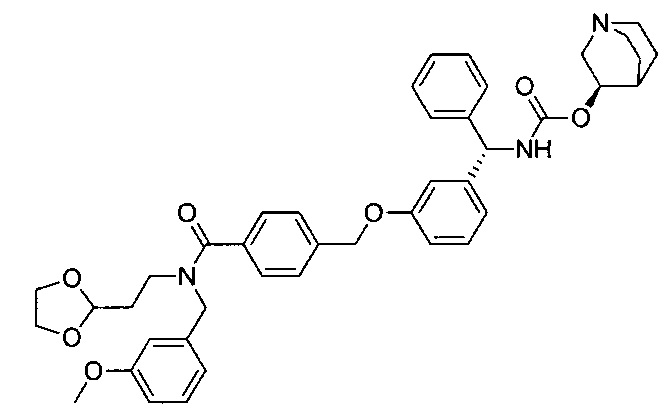
Указанное в заголовке соединение получали, как описано в примере 10 на стадии 2, с заменой N-бензил-2-(1,3-диоксолан-2-ил)этанамина на 2-(1,3-диоксолан-2-ил)-N-(3-метоксибензил)этанамин.
Стадия 4: (R)-хинуклидин-3-ил-((S)-(3-((4-((3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)(3-метоксибензил)карбамоил)бензил)окси)фенил)(фенил)метил)карбамат
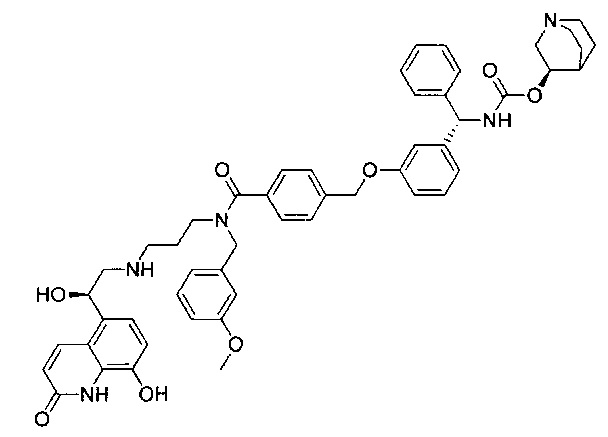
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 12 и стадии 13, с заменой 3-(1,3-диоксолан-2-ил)пропил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоил)пиперидин-4-карбоксилата на (R)-хинуклидин-3-ил-((S)-(3-((4-((2-(1,3-диоксолан-2-ил)этил)(3-метоксибензил)карбамоил)бензил)-окси)фенил)(фенил)метил)карбамат на стадии 12, и полученный продукт использовали на стадии 13.
1Н ЯМР (400 МГц, ДМСО-d6, 85°C): δ 8.23-8.13 (m, 3Н); 7.72 (s, 1Н); 7.47 (d, J=7,9 Гц, 2Н); 7.39 (d, J=7,9 Гц, 2Н); 7.33-7.18 (m, 7Н); 7.07-7.01 (m, 2Н); 6.96-6.80 (m, 6Н); 6.47 (d, J=9,9 Гц, 1Н); 5.82 (d, J=8,1 Гц, 1Н); 5.10 (s, 2Н); 5.00-4.94 (m, 1Н); 4.63-4.55 (m, 3Н); 3.76 (s, 3Н); 3.38-3.28 (m, 2Н); 3.08 (m, 1Н); 2.74-2.47 (m, 9Н); 1.90 (s, 1Н); 1.73-1.54 (m, 4Н); 1.52-1.43 (m, 1Н); 1.31 (t, J=10,8 Гц, 1Н).
Следующие соединения получали, как описано в примере 11, с заменой 3-метоксибензилбромида на соответствующий бензилгалогенид на стадии 2, и полученные продукты использовали на стадиях 3-4.
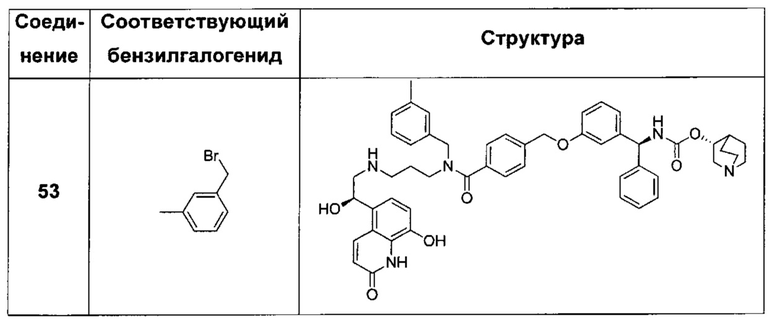
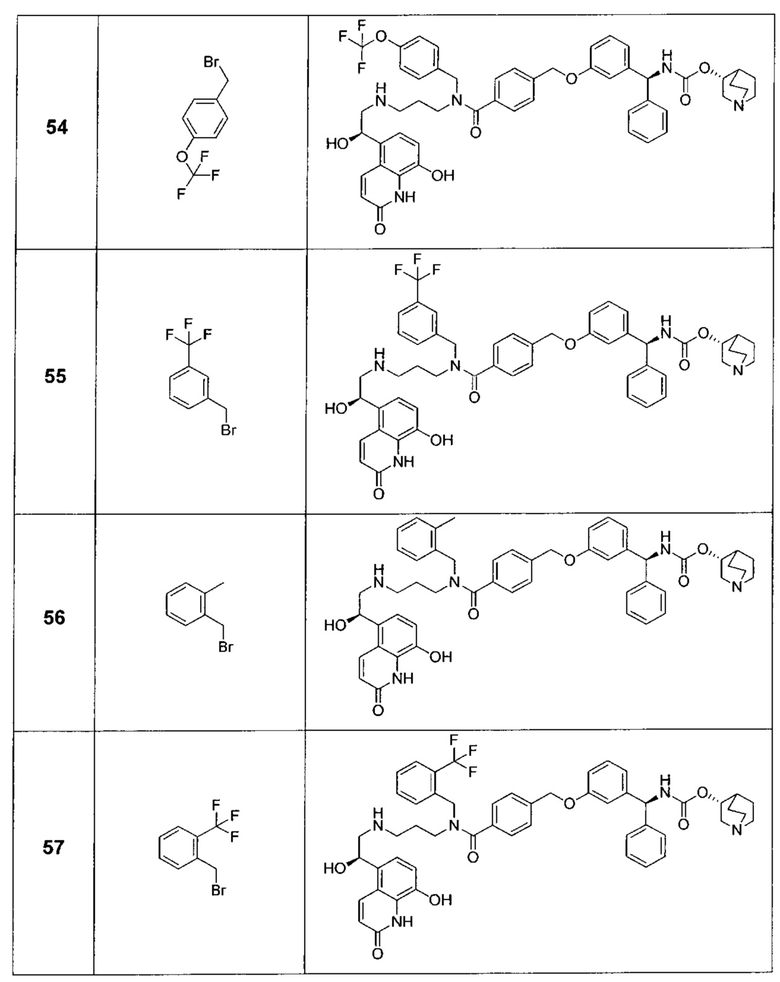
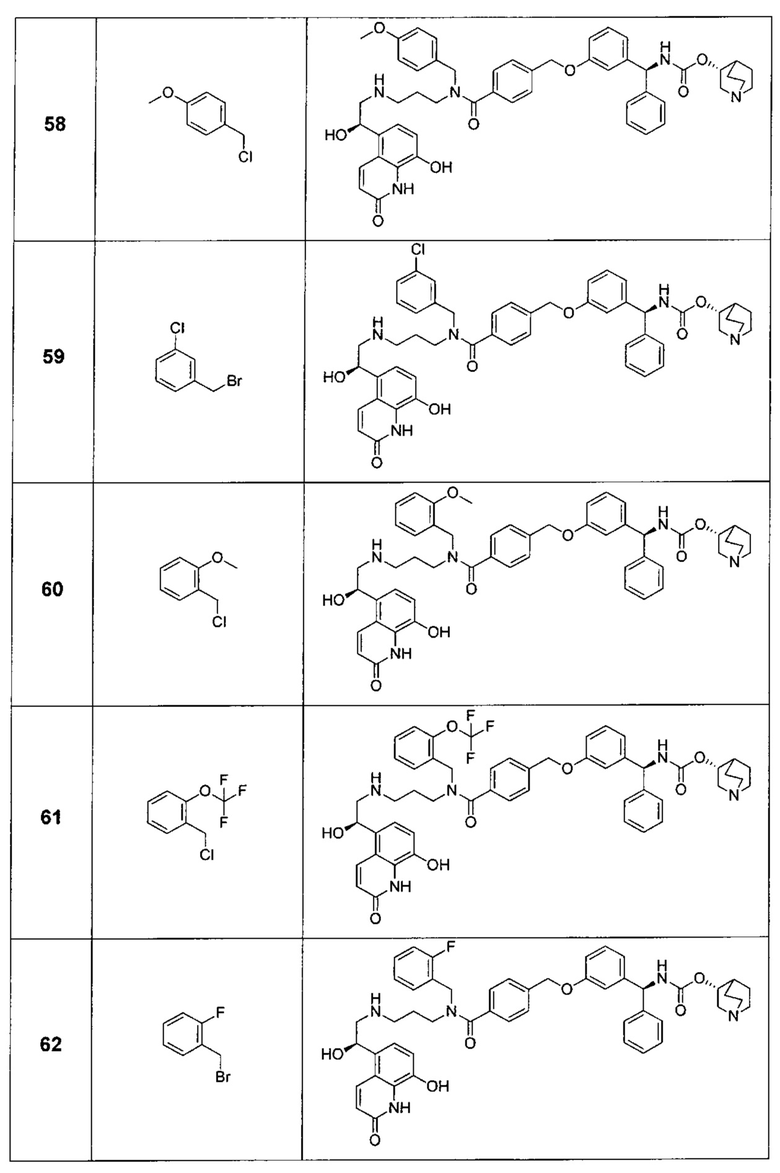

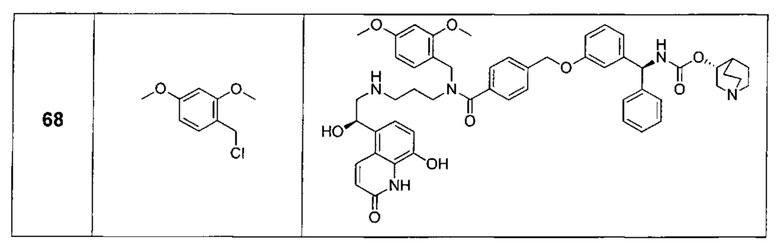
Пример 12
(R)-Хинуклидин-3-ил-((S)-(3-((4-(циклопентил(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)-карбамоил)бензил)окси)фенил)(фенил)метил)карбамат (соединение 69)

Стадия 1: N-(2-(1,3-диоксолан-2-ил)этил)циклопентанамин
К суспензии гидрохлорида N-бензилциклопентанамина (0,986 г; 4,66 ммоль) в ацетонитриле (10 мл) добавляли диизопропилэтиламин (2,0 мл; 11,5 ммоль) и смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли раствор 2-(2-бромэтил)-1,3-диоксолана (1,01 г; 5,58 ммоль) в ацетонитриле (5 мл) и смесь нагревали при 80°C в течение 48 часов. Реакционную смесь разбавляли ДХМ и промывали водой, сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток загружали на картридж SCX-2 и элюировали ацетонитрилом (4 объема колонки) и затем смесью 10% триэтиламин/ацетонитрил (4 объема колонки). Фракции, содержащие продукт, объединяли и растворитель выпаривали при пониженном давлении. Остаток растворяли в этаноле (10 мл) и добавляли 10%-ный палладий на угле (0,90 г). Смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли 1-метил-1,4-циклогексадиен (1,9 мл; 16,9 ммоль). Реакционную смесь нагревали до температуры дефлегмации и затем реакционную смесь перемешивали при температуре дефлегмации в течение 1 часа. Реакционную смесь оставляли охлаждаться и суспензию фильтровали. Фильтрат упаривали при пониженном давлении с получением указанного в заголовке соединения (0,249 г; 40%).
1Н ЯМР (400 МГц, CDCl3): δ 4.96-4.90 (m, 1Н); 4.01-3.92 (m, 2Н); 3.89-3.80 (m, 2Н); 3.12-3.01 (m, 1Н); 2.81-2.72 (m, 2Н); 1.99-1.81 (m, 5Н); 1.75-1.47 (m, 4Н); 1.39-1.26 (m, 2Н).
Стадия 2: (R)-хинуклидин-3-ил-((S)-(3-((4-((2-(1,3-диоксолан-2-ил)-этил)(циклопентил)карбамоил)бензил)окси)фенил)(фенил)метил)-карбамат
Указанное в заголовке соединение получали, как описано в примере 10 на стадии 2, с заменой N-(2-(1,3-диоксолан-2-ил)этил)-циклопентанамина на 2-(1,3-диоксолан-2-ил)-N-(3-метоксибензил)этанамин.
Стадия 3: (R)-хинуклидин-3-ил-((S)-(3-((4-(циклопентил(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)-пропил)карбамоил)бензил)окси)фенил)(фенил)метил)карбамат (соединение 69)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 12 и стадии 13, с заменой 3-(1,3-диоксолан-2-ил)пропил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоил)пиперидин-4-карбоксилата на (R)-хинуклидин-3-ил-((S)-(3-((4-((2-(1,3-диоксолан-2-ил)этил)(циклопентил)карбамоил)бензил)окси)-фенил)(фенил)метил)карбамат на стадии 12, и полученный продукт использовали на стадии 13.
1Н ЯМР (400 МГц, ДМСО-d6, 110°C): δ 8.25-8.12 (m, 3Н); 7.53 (s, 1Н); 7.46 (d, J=7,9 Гц, 2Н); 7.32 (d, J=5,8 Гц, 6Н); 7.28-7.20 (m, 2Н); 7.09 (d, J=8,1 Гц, 1Н); 7.04 (s, 1Н); 6.98-6.86 (m, 3Н); 6.48 (d, J=9,9 Гц, 1Н); 5.84 (d, J=7,8 Гц, 1Н); 5.11 (s, 2Н); 5.06-4.99 (m, 1Н); 4.65-4.60 (m, 1Н); 4.05 (d, J=8,6 Гц, 1Н); 3.32-3.21 (, 2Н); 3.10 (dd, J=14,4, 8,4 Гц, 1Н); 2.82-2.53 (m, 9Н); 1.91 (s, 1Н); 1.78-1.56 (m, 11Н); 1.49-1.43 (m, 2Н); 1.32 (m, 1Н).
Следующие соединения получали, как описано в примере 12, с заменой гидрохлорида N-бензилциклопентанамина на соответствующий амин на стадии 1, и полученные продукты использовали на стадиях 2-3.
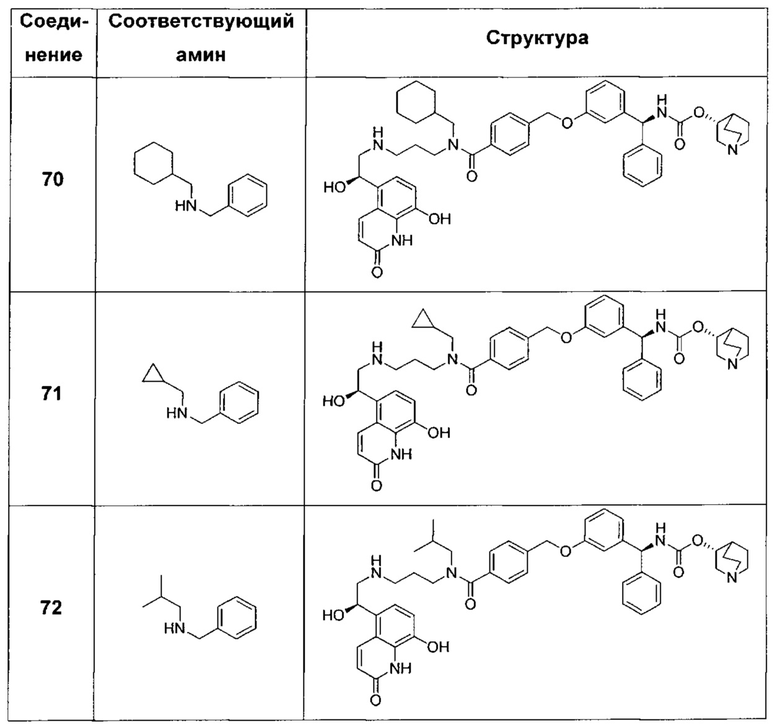
Общий синтез для соединений 73-77
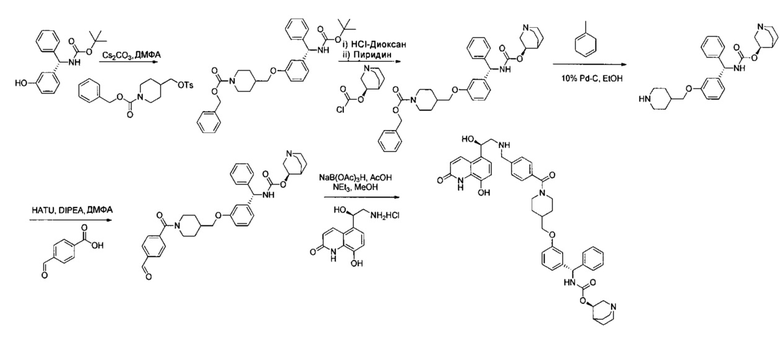
Пример 13
(R)-Хинуклидин-3-ил-((S)-(3-((1-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензоил)пиперидин-4-ил)метокси)фенил)(фенил)метил)карбамат (соединение 73)
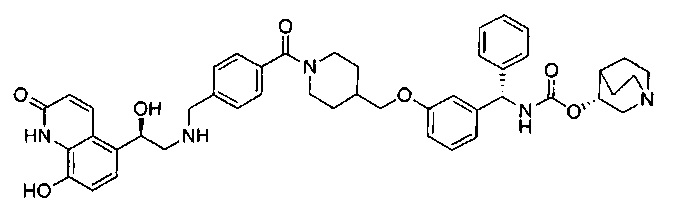
Стадия 1: (S)-бензил-4-((3-(((трет-бутоксикарбонил)амино)(фенил)-метил)фенокси)метил)пиперидин-1-карбоксилат
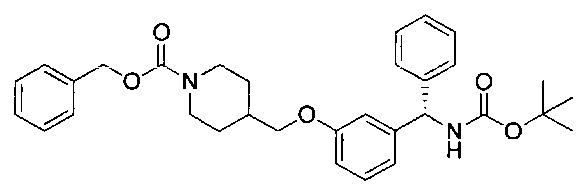
К смеси (S)-трет-бутил-((3-гидроксифенил)(фенил)метил)карбамата (8,0 г; 26,7 ммоль) и карбоната цезия (13,1 г; 37,4 ммоль) в ДМФА (130 мл) добавляли бензиловый эфир 4-(толуол-4-сульфонилоксиметил)-пиперидин-1-карбоновой кислоты (12,94 г; 32,1 ммоль). Полученную реакционную смесь перемешивали при 50°C в течение 17 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом (×3). Объединенные органические экстракты промывали рассолом (×3), сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии, элюируя смесью от 0 до 50% этилацетата/изогексан, с получением указанного в заголовке соединения (13,7 г; 93%).
1Н ЯМР (400 МГц, CDCl3): δ 7.38-7.18 (m, 11Н); 6.84-6.72 (m, 3Н); 5.86 (s, 1Н); 5.13 (s, 3Н); 4.23 (s, 2Н); 3.76 (d, J=6,3 Гц, 2Н); 2.82 (s, 2Н); 1.99-1.88 (m, 1Н); 1.82 (d, J=13,2 Гц, 2Н); 1.44 (s, 9Н); 1.26 (t, J=7,1 Гц, 2Н).
Стадия 2: бензил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)пиперидин-1-карбоксилат
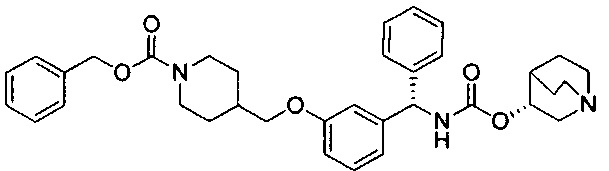
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 6 и стадии 7, с заменой (S)-метил-4-((3-(((трет-бутоксикарбонил)амино)(фенил)метил)фенокси)метил)бензоата на (S)-бензил-4-((3-(((трет-бутоксикарбонил)амино)(фенил)метил)фенокси)-метил)пиперидин-1-карбоксилат на стадии 6, и полученный продукт использовали на стадии 7.
Стадия 3: (R)-хинуклидин-3-ил-((S)-фенил(3-(пиперидин-4-илметокси)фенил)метил)карбамат
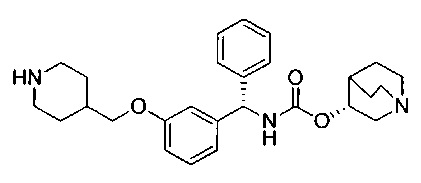
К раствору бензил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)пиперидин-1-карбоксилата (1,32 г; 2,26 ммоль) в этаноле (100 мл) добавляли палладий на угле (0,66 г) и перемешивали при комнатной температуре в течение 10 минут. Добавляли 1-метил-1,4-циклогексадиен (1,27 мл; 11,3 ммоль) и реакционную смесь нагревали до температуры дефлегмации. Реакционную смесь перемешивали при температуре дефлегмации в течение 1 часа. Реакционную смесь оставляли охлаждаться и суспензию фильтровали. Фильтрат упаривали при пониженном давлении с получением указанного в заголовке соединения (1,05 г; 100%).
1Н ЯМР (400 МГц, ДМСО-d6): δ 9.78 (s, 1Н); 8.44 (d, J=9,4 Гц, 1Н); 7.33 (d, J=4,5 Гц, 4Н); 7.28-7.21 (m, 2Н); 6.97 (d, J=7,8 Гц, 1Н); 6.90 (s, 1Н); 6.83 (dd, J=8,2, 2,4 Гц, 1Н); 5.83 (d, J=9,1 Гц, 1Н); 4.88-4.83 (m, 1Н); 3.82 (d, J=6,5 Гц, 3Н); 3.35-3.06 (m, 6Н); 2.90 (m, 2Н); 2.23 (s, 1Н); 2.12-1.64 (m, 5Н); 1.52-1.37 (m, 2Н).
Стадия 4: (R)-хинуклидин-3-ил-((S)-(3-((1-(4-формилбензоил)-пиперидин-4-ил)метокси)фенил)(фенил)метил)карбамат
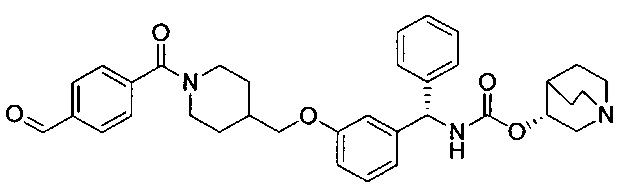
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 11, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата и 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензойной кислоты на (R)-хинуклидин-3-ил-((S)-фенил(3-(пиперидин-4-илметокси)фенил)-метил)карбамат и 4-формилбензойную кислоту, соответственно.
1Н ЯМР (400 МГц, CDCl3): δ 10.08-10.03 (m, 1Н); 7.97-7.90 (m, 2Н); 7.56 (d, J=8,0 Гц, 2Н); 7.35-7.20 (m, 7Н); 6.84 (d, J=7,7 Гц, 1Н); 6.79-6.72 (m, 2Н); 5.86 (d, J=7,7 Гц, 1Н); 5.14 (s, 1Н); 4.79 (d, J=12,9 Гц, 1Н); 3.80 (s,2 Н); 3.69 (d, J=13,3 Гц, 1Н); 3.07 (s, 1Н); 2.84 (s, 1Н); 2.15-1.02 (m, 16Н).
Стадия 5: 13 (R)-хинуклидин-3-ил-((S)-(3-((1-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-бензоил)пиперидин-4-ил)метокси)фенил)(фенил)метил)карбамат (соединение 73)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 13, с заменой 1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)бензоил)пиперидин-4-карбоксилата на (R)-хинуклидин-3-ил-((S)-(3-((1-(4-формилбензоил)-пиперидин-4-ил)метокси)фенил)(фенил)метил)карбамат.
1Н ЯМР (400 МГц, ДМСО-d6, 110°C): δ 8.21-8.13 (m, 2Н); 7.55 (d, J=9,0 Гц, 1Н); 7.38-7.28 (m, 8Н); 7.28-7.20 (m, 2Н); 7.09 (d, J=8,1 Гц, 1Н); 6.99-6.89 (m, 3Н); 6.85-6.80 (m, 1Н); 6.46 (d, J=9,9 Гц, 1Н); 5.83 (d, J=8,5 Гц, 1Н); 5.08 (dd, J=7,6, 4,7 Гц, 1Н); 4.67-4.61 (m, 1Н); 4.05 (d, J=13,2 Гц, 3Н); 3.91-3.85 (m, 2Н); 3.82 (s, 2Н); 3.12 (dd, J=14,4, 8,3 Гц, 1Н); 3.01-2.92 (m, 2Н); 2.91-2.52 (m, 5Н); 2.09-2.01 (m, 1Н); 1.95-1.91 (m, 1Н); 1.83-1.75 (m, 3Н); 1.69-1.59 (m, 1Н); 1.56-1.46 (m, 1Н); 1.38-1.24 (m, 4Н).
Следующие соединения получали, как описано в примере 13, с заменой 4-формилбензойной кислоты на соответствующую кислоту на стадии 4, и полученные продукты использовали на стадии 5.
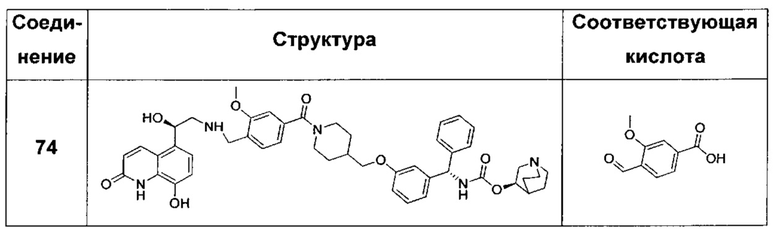
Пример 14
(R)-Хинуклидин-3-ил-((S)-(3-((1-(4-(2-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)этил)бензоил)пиперидин-4-ил)-метокси)фенил)(фенил)метил)карбамат (соединение 75)
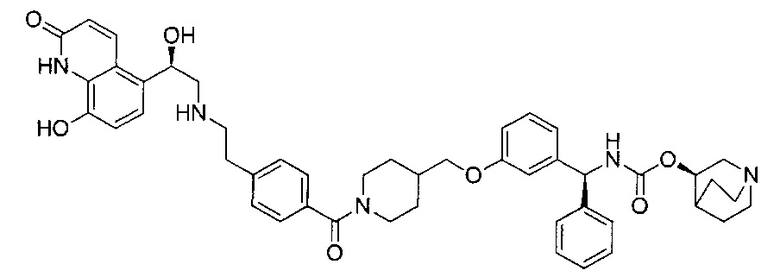
Стадия 1: (R)-хинуклидин-3-ил-((S)-(3-((1-(4-((1,3-диоксолан-2-ил)-метил)бензоил)пиперидин-4-ил)метокси)фенил)(фенил)метил)карбамат
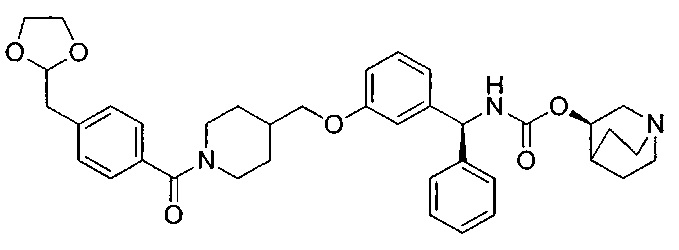
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 11, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата и 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)бензойной кислоты на (R)-хинуклидин-3-ил-((S)-фенил(3-(пиперидин-4-илметокси)фенил)-метил)-карбамат и 4-(1,3-диоксолан-2-илметил)бензойную кислоту, соответственно.
Стадия 2: (R)-хинуклидин-3-ил-((S)-(3-((1-(4-(2-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)этил)бензоил)-пиперидин-4-ил)метокси)фенил)(фенил)метил)карбамат (соединение 75)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 12 и стадии 13, с заменой 3-(1,3-диоксолан-2-ил)пропил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)-метил)бензоил)пиперидин-4-карбоксилата на (R)-хинуклидин-3-ил-((S)-(3-((1-(4-((1,3-диоксолан-2-ил)метил)бензоил)пиперидин-4-ил)метокси)фенил)-(фенил)метил)карбамат на стадии 12, и продукт использовали на последующих стадиях.
1Н ЯМР (400 МГц, ДМСО-d6, 110°C): δ 8.19 (d, J=9,9 Гц, 1Н); 7.83 (d, J=8,7 Гц, 1Н); 7.38-7.29 (m, 7Н); 7.30-7.22 (m, 3Н); 7.16 (d, J=8,2 Гц, 1Н); 7.02 (d, J=8,1 Гц, 1Н); 6.96-6.89 (m, 2Н); 6.84 (dd, J=8,2, 2,5 Гц, 1Н); 6.57 (d, J=9,9 Гц, 1Н); 5.84 (d, J=8,7 Гц, 1Н); 5.37 (dd, J=7,9, 5,0 Гц, 1Н); 4.96-4.91 (m, 1Н); 4.05 (d, J=12,3 Гц, 2Н); 3.88 (d, J=6,2 Гц, 2Н); 3.66 (ddd, J=13,9, 8,4, 2,6 Гц, 1Н); 3.36-2.90 (m, 11Н); 2.28-2.24 (m, 1Н); 2.08-1.77 (m, 8Н); 1.37-1.27 (m, 3Н).
Следующие соединения получали, как описано в примере 14, с заменой 4-(1,3-диоксолан-2-илметил)бензойной кислоты на соответствующую кислоту на стадии 2, и полученные продукты использовали на стадии 2.
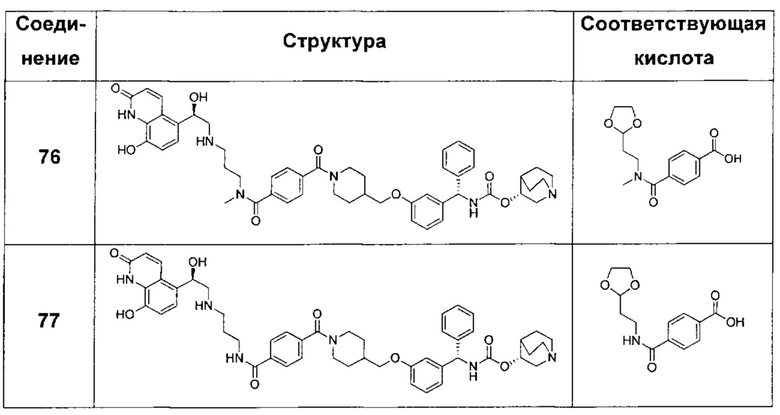
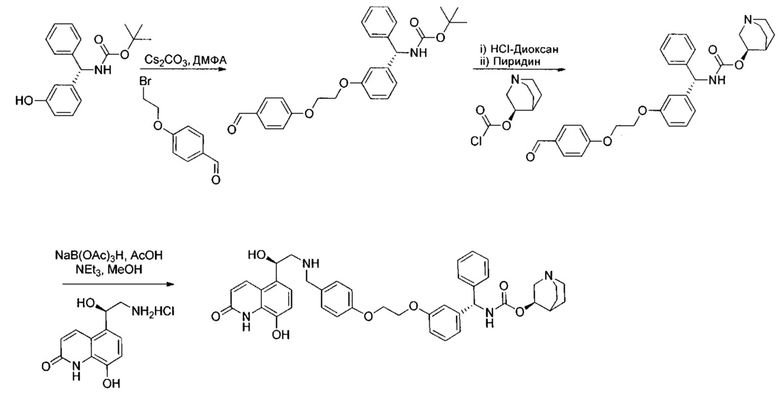
Пример 15
(R)-Хинуклидин-3-ил-((S)-(3-(2-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенокси)этокси)-фенил)(фенил)метил)карбамат (соединение 78)
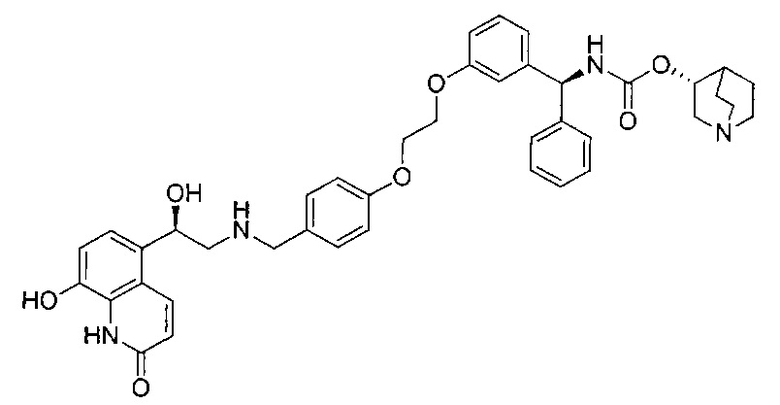
Стадия 1: (S)-трет-бутил-((3-(2-(4-формилфенокси)этокси)фенил)-(фенил)метил)карбамат
К перемешиваемому раствору (S)-трет-бутил-((3-гидроксифенил)-(фенил)метил)карбамата (1,76 г; 5,9 ммоль) в ДМФА (10 мл) добавляли карбонат цезия (4,0 г; 12,0 ммоль). Реакционную смесь затем перемешивали при комнатной температуре в течение 20 минут. Добавляли 4-(2-бромэтокси)бензолкарбоксальдегид (1,35 г; 5,9 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом (×3). Объединенные органические экстракты промывали рассолом (×3), сушили (сульфат магния), фильтровали и растворитель выпаривали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии, элюируя смесью от 0 до 50% этилацетата/изогексан, с получением указанного в заголовке соединения (1,6 г; 61%).
1Н ЯМР (400 МГц, ДМСО): δ 9.89 (s, 1Н); 7.97-7.83 (m, 3Н); 7.36-7.13 (m, 8Н); 7.01-6.89 (m, 2Н); 6.85 (dd, J=8,2, 2,5 Гц, 1Н); 5.80 (d, J=9,5 Гц, 1Н); 4.44 (t, J=4,1 Гц, 2Н); 4.35-4.28 (m, 2Н); 1.40 (s, 9Н).
Стадия 2: (R)-хинуклидин-3-ил-((S)-(3-(2-(4-формилфенокси)этокси)-фенил)(фенил)метил)карбамат
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 6 и стадии 7, с заменой (S)-метил-4-((3-(((трет-бутоксикарбонил)-амино)(фенил)метил)фенокси)метил)бензоата на ((S)-трет-бутил-((3-(2-(4-формилфенокси)этокси)фенил)(фенил)метил)карбамат на стадии 6, и полученный продукт использовали на стадии 7.
Стадия 3: (R)-хинуклидин-3-ил-((S)-(3-(2-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенокси)-этокси)фенил)(фенил)метил)карбамат (соединение 78)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 13, с заменой 4-оксобутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)-пиперидин-4-карбоксилата на (R)-хинуклидин-3-ил-((S)-(3-(2-(4-формилфенокси)этокси)-фенил)(фенил)метил)карбамат.
1Н ЯМР (400 МГц, ДМСО-d6, 110°C): δ 8.10 (d, J=9,9 Гц, 1Н); 7.83 (d, J=8,6 Гц, 1Н); 7.45 (d, J=8,2 Гц, 2Н); 7.35-7.22 (m, 6Н); 7.12 (d, J=8,2 Гц, 1Н); 7.03-6.96 (m, 5Н); 6.88 (d, J=8,3 Гц, 1Н); 6.53 (d, J=9,9 Гц, 1Н); 5.85 (d, J=8,6 Гц, 1Н); 5.39-5.32 (m, 1Н); 4.94 (d, J=7,6 Гц, 1Н); 4.38-4.27 (m, 4Н); 4.20 (s, 2Н); 3.70-3.61 (m, 1Н); 3.30-3.07 (m, 7Н); 2.25 (s, 1Н); 2.03 (s, 1Н); 1.97-1.90 (m, 1Н); 1.86 (s, 1Н); 1.76 (s, 1Н).
Следующие соединения получали, как описано в примере 15, с заменой 4-(2-бромэтокси)бензолкарбоксальдегида на 3-(2-бромэтокси)-бензолкарбоксальдегид на стадии 2, и полученные продукты использовали на стадии 2.
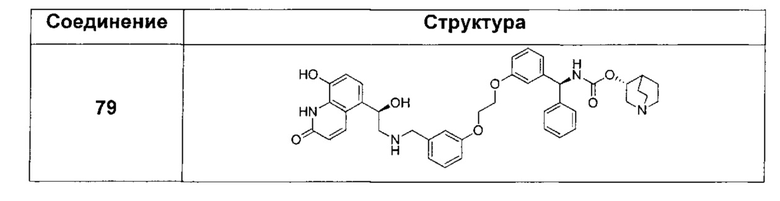
Пример 16
(R)-Хинуклидин-3-ил-((S)-(3-(2-((3-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)амино)-2-оксоэтокси)фенил)(фенил)метил)карбамат (соединение 80)
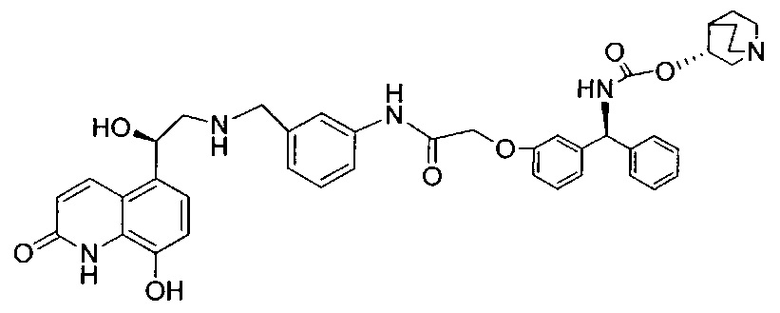
Стадия 1: 2-(3-((S)-фенил-((((R)-хинуклидин-3-илокси)карбонил)-амино)метил)фенокси)уксусная кислота
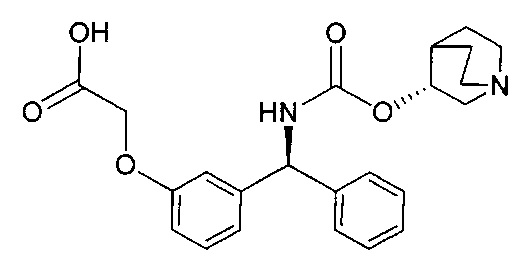
Указанное в заголовке соединение получали, как описано в примере 1 со стадии 5 до стадии 8, с заменой метил-4-(бромметил)бензоата на метилбромацетат на стадии 5, и продукты использовали на последующих стадиях.
1Н ЯМР (400 МГц, ДMCO-d6): δ 8.32-8.24 (m, 1Н); 7.34-7.12 (m, 6Н); 6.89 (s, 1Н); 6.84 (d, J=7,6 Гц, 1Н); 6.69 (dd, J=8,2, 2,5 Гц, 1Н); 5.79 (d, J=9,2 Гц, 1Н); 4.72-4.70 (m, 1Н); 4.37 (s, 2Н); 3.30-3.29 (m, 1Н); 2.93 (s, 2Н); 2.89-2.69 (m, 4Н); 2.05-2.03 (m, 1Н); 1.92-1.88 (m, 1Н); 1.70-1.50 (m, 3Н).
Стадия 2: (R)-хинуклидин-3-ил-((S)-(3-(2-((3-(гидроксиметил)-фенил)амино)-2-оксоэтокси)фенил)(фенил)метил)карбамат
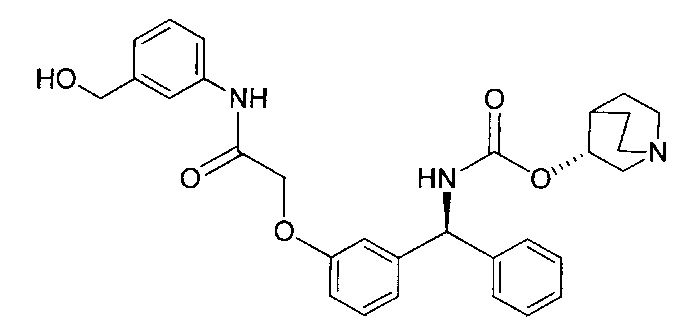
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 11, с заменой гидрохлорида 3-(1,3-диоксолан-2-ил)пропил-пиперидин-4-карбоксилата и 4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)бензойной кислоты на 3-аминобензиловый спиpт и 2-(3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)уксусную кислоту, соответственно. Неочищенное вещество использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 3: (R)-хинуклидин-3-ил-((S)-(3-(2-((3-формилфенил)амино)-2-оксоэтокси)фенил)(фенил)метил)карбамат
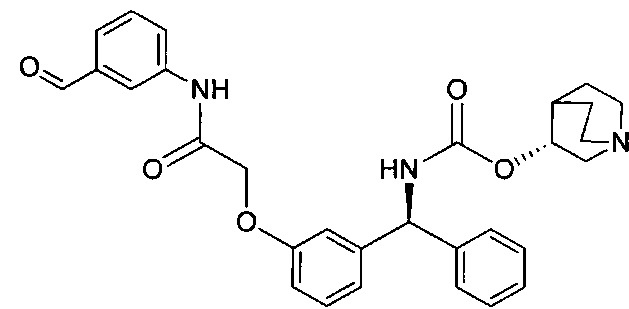
К раствору (R)-хинуклидин-3-ил-((S)-(3-(2-((3-(гидроксиметил)-фенил)амино)-2-оксоэтокси)фенил)(фенил)метил)карбамата (0,35 г; 0,68 ммоль) в 1,4-диоксане (25 мл) добавляли оксид марганца(IV) (0,58 г; 6,8 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем при 50°C в течение 15 часов. Суспензию фильтровали через набивку из целита. Растворитель выпаривали при пониженном давлении с получением неочищенного вещества. Это вещество использовали на следующей стадии без дополнительной очистки.
Стадия 4: (R)-хинуклидин-3-ил-((S)-(3-(2-((3-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)-амино)-2-оксоэтокси)фенил)(фенил)метил)карбамат (соединение 80)
Указанное в заголовке соединение получали, как описано в примере 1 на стадии 13, с заменой 4-оксобутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)-пиперидин-4-карбоксилата на (R)-хинуклидин-3-ил-((S)-(3-(2-((3-(гидроксиметил)фенил)-амино)-2-оксоэтокси)фенил)(фенил)метил)карбамат.
1Н ЯМР (400 МГц, ДМСО-d6): δ 10.02 (s, 1Н); 8.25 (d, J=9,3 Гц, 1Н); 8.11 (d, J=9,9 Гц, 1Н); 7.61 (s, 1Н); 7.49 (d, J=8,2 Гц, 1Н); 7.35-7.17 (m, 7Н); 7.08-7.01 (m, 3Н); 6.99-6.81 (m, 3Н); 6.46 (d, J=9,9 Гц, 1Н); 5.83 (d, J=8,3 Гц, 1Н); 5.06 (dd, J=8,0, 4,2 Гц, 1Н); 4.66 (s, 2Н); 4.56 (s, 1Н); 3.73 (s, 2Н); 3.07 (t, J=10,6 Гц, 1Н); 2.73-2.59 (m, 6Н); 1.89 (s, 1Н); 1.78 (s, 1Н); 1.57 (s, 1Н); 1.45 (s, 2Н); 1.31 (s, 1Н).
Крупномасштабный синтез гидрохлорида (S)-3-(амино(фенил)-метил)фенола
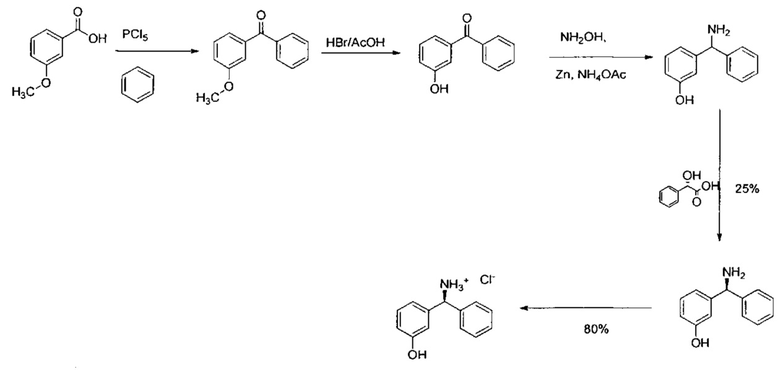
Стадия 1: (3-метоксифенил)(фенил)метанон
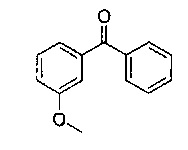
К смеси пентахлорида фосфора (3763 г; 18,1 моль) в 7500 мл бензола порциями добавляли 3-метоксибензойную кислоту (2500 г; 16,4 моль). Смесь перемешивали в течение 50 минут до тех пор, пока она не становилась гомогенной. Образование хлорангидрида контролировали посредством ТСХ (тонкослойной хроматографии). После завершения смесь охлаждали до 10°C, реактор покрывали алюминиевой фольгой и порциями добавляли трихлорид алюминия (4820 г; 36,1 моль) (внутреннюю температуру поддерживали на уровне 30°C максимум). Перемешивание продолжали в течение 18 часов при RT. Взаимодействие контролировали посредством ТСХ (AcOEt:гекс. в соотношении 1:9). После завершения реакционную смесь выливали в лед и разбавляли AcOEt (7 л). Органический слой затем отделяли и водный слой экстрагировали AcOEt (2×10 л; 1×6 л). Объединенные органические слои промывали водой (5×3 л) до pH приблизительно 6-7, насыщенным водным раствором гидрокарбоната натрия (15 л), сушили (сульфат натрия), фильтровали и растворитель выпаривали при пониженном давлении с получением неочищенного масла. Продукт очищали посредством вакуумной перегонки (130-139°C, 2 мбар (0,2 кПа)) с получением указанного в заголовке соединения (2637 г; 76%) в виде бледно-желтого масла.
1Н ЯМР (600 МГц, CDCl3); δ 7.80 (m, 2Н); 7.57 (m, 1Н); 7.46 (m, 2Н); 7.32-7.37 (m, 3Н); 7.12 (m, 1Н); 3.83 (s, 3Н).
Стадия 2: (3-гидроксифенил)(фенил)метанон
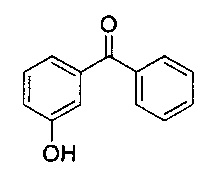
1458 г (6,9 моль) (3-метоксифенил)(фенил)метанона растворяли в 2090 мл АсОН. К этому раствору добавляли 2320 мл (20,6 моль) 48% HBr и смесь перемешивали при 90°C в течение 18 часов. Взаимодействие контролировали посредством ТСХ (AcOEt:гекс. в соотношении 1:9). После завершения взаимодействия смесь охлаждали до RT и выливали в лед при перемешивании. Осажденное твердое вещество отфильтровывали, промывали водой и сушили с получением указанного в заголовке соединения в виде белого твердого вещества (1234 г; 91%).
1Н ЯМР (600 МГц, CDCl3); δ 7.80 (m, 2Н); 7.58 (m, 1Н); 7.47 (m, 2Н); 7.39 (m, 1Н); 7.28-7.34 (m, 2Н); 7.11 (m, 1Н); 5.59 (br s, 1Н).
Стадия 3: 3-(амино(фенил)метил)фенол
(3-Гидроксифенил)(фенил)метанон (400 г; 2 моль) растворяли в метаноле (4 л). К полученному раствору добавляли гидрохлорид гидроксиламина (168 г; 2,4 моль) и ацетат натрия (331 г; 4 моль). Смесь нагревали с обратным холодильником в течение 18 часов. После охлаждения до RT растворитель выпаривали при пониженном давлении, затем к остатку добавляли воду (3 л). Продукт экстрагировали этилацетатом (3×3 л). Объединенные органические экстракты промывали насыщенным водным гидрокарбонатом натрия, рассолом, сушили (сульфат натрия), фильтровали и растворитель выпаривали при пониженном давлении. Неочищенный остаток (1085 г) использовали на следующей стадии без очистки.
362 г неочищенного оксима (287 г; 1,3 моль чистого оксима (на основании анализа)) растворяли в этаноле (860 мл) и 25%-ном водном аммиаке (3000 мл). К этой смеси добавляли ацетат аммония (52 г; 0,7 моль) с последующим добавлением порциями цинковой пыли (440 г; 6,7 моль) для поддержания внутренней температуры не выше 40°C. Смесь перемешивали без нагревания в течение 18 часов, затем фильтровали через набивку из целита. Осадок на фильтре промывали этилацетатом. Фильтрат собирали, и образованные слои разделяли. Водный слой экстрагировали этилацетатом (5×5 л). Объединенные органические слои промывали рассолом (×2) и растворитель выпаривали при пониженном давлении. Продукт сушили под вакуумом (35°C, 18 часов).
1Н ЯМР (600 МГц, ДМСО-d6); δ 9.25 (br s, 1Н); 7.36 (m, 2Н); 7.25 (m, 2Н); 7.15 (m, 1Н); 7.03 (m, 1Н); 6.79 (m, 2Н); 6.54 (m, 1Н);4.98 (s, 1Н); 2.17 (br s, 2Н).
Стадия 4: кристаллизация (S)-3-(амино(фенил)метил)фенол-(S)-манделата
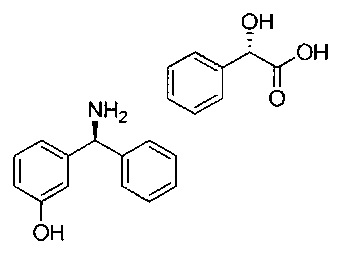
Образование соли: 3-(амино(фенил)метил)фенол (1081 г; 5,4 моль) растворяли в изопропаноле (21,62 л) и нагревали до температуры дефлегмации. К смеси по каплям добавляли раствор S-миндальной кислоты (908 г; 6 моль) в изопропаноле (2160 мл). Смесь нагревали с обратным холодильником в течение 1 часа и затем оставляли охлаждаться до 10°C (в течение 18 часов). Образовавшийся осадок отфильтровывали, промывали холодным изопропанолом и сушили под вакуумом при 35°C.
Полученную соль нагревали с обратным холодильником в 95%-ном изопропаноле в течение 1 часа. Смесь оставляли охлаждаться до 10°C в течение 18 часов. Твердое вещество отфильтровывали, промывали холодным изопропанолом и сушили в вакуумной печи при 35°C. Процесс кристаллизации повторяли два или более раз до тех пор, пока ее (энантиомерный избыток) не составил более 98% согласно анализу хиральной ВЭЖХ.
Стадия 5: гидрохлорид (S5)-3-(амино(фенил)метил)фенола
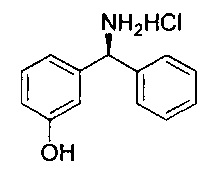
(S)-3-(Амино(фенил)метил)фенол-(S)-манделат (1027 г; 2,9 моль) суспендировали в этилацетате. По каплям добавляли раствор гидрокарбоната натрия (737 г; 8,8 моль) в воде (11,05 л) и смесь перемешивали при RT в течение 18 часов. Смесь разделяли и водный слой экстрагировали этилацетатом (5×10 л). Объединенные органические экстракты объединяли и растворитель выпаривали при пониженном давлении с получением 464 г (85%) амина в виде бледно-желтых кристаллов.
Амин (464 г; 2,3 моль) суспендировали в метаноле и по каплям добавляли 4 М HCl в AcOEt (3500 мл; 14 моль). Смесь перемешивали в течение 18 часов и растворитель выпаривали при пониженном давлении. Остаток растирали с эфиром (2740 мл) в течение 18 часов. Суспензию фильтровали, осадок на фильтре промывали эфиром и сушили.
1Н ЯМР (600 МГц, ДMCO-d6); δ 9.74 (s, 1Н); 9.19 (s, 3Н); 7.54 (m, 2Н); 7.40 (m, 2Н); 7.33 (m, 1Н); 7.19 (m, 1Н); 7.00 (m, 1Н); 6.89 (m, 1Н); 6.78 (m, 1Н); 5.49 (s, 1Н).

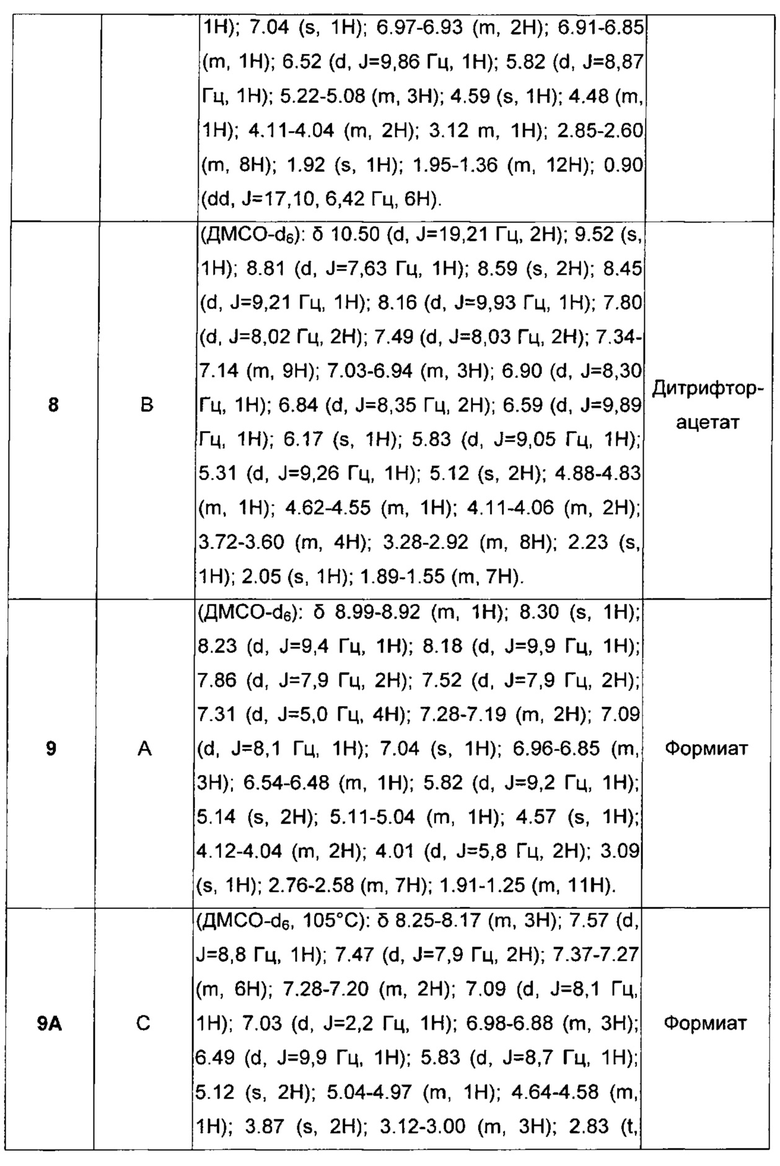
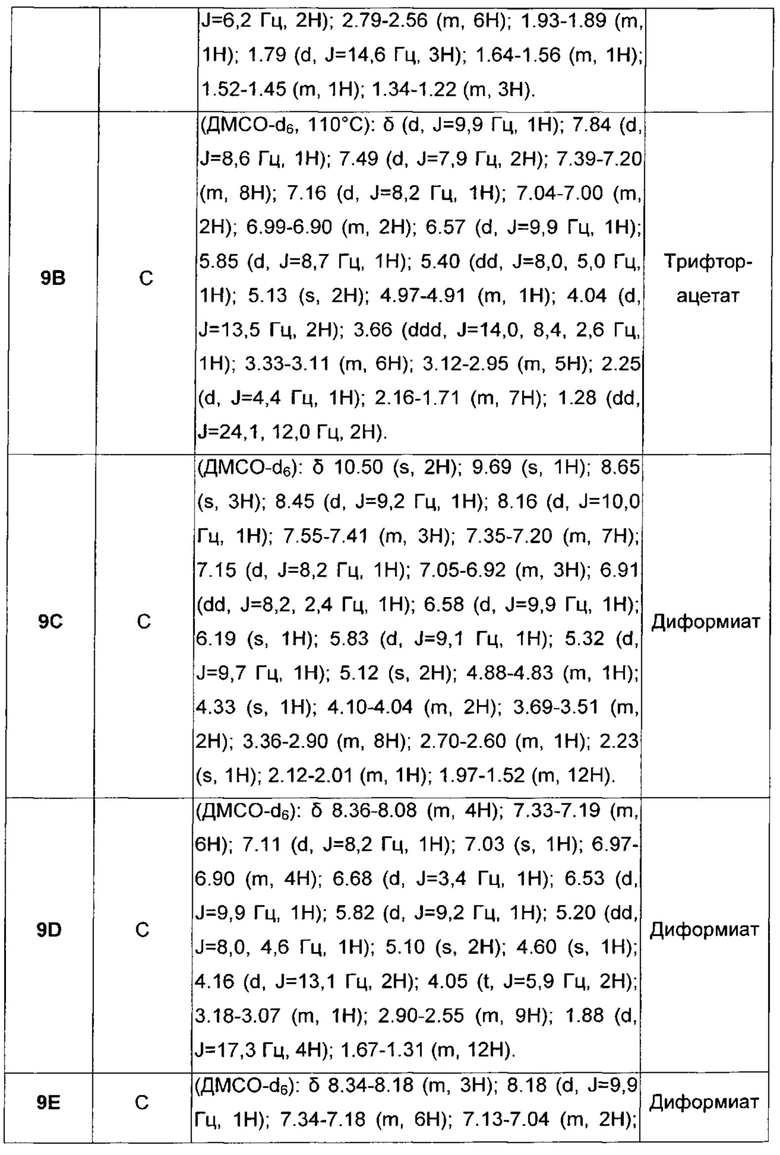
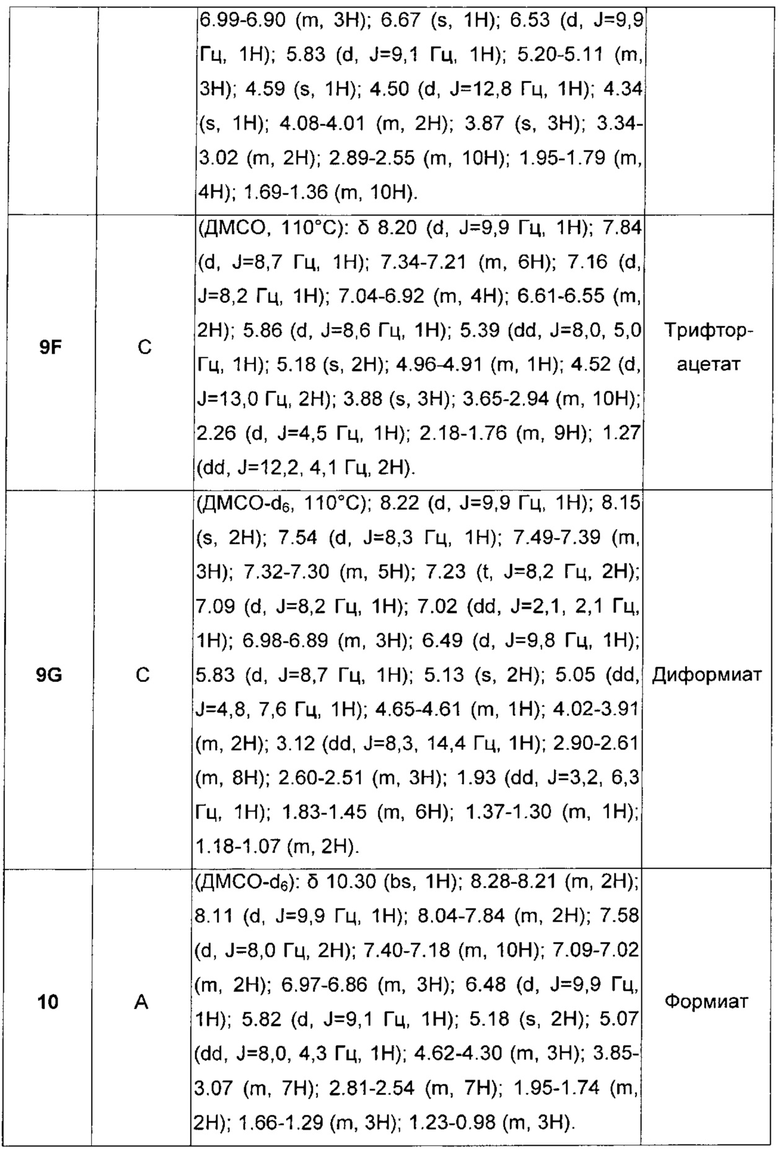
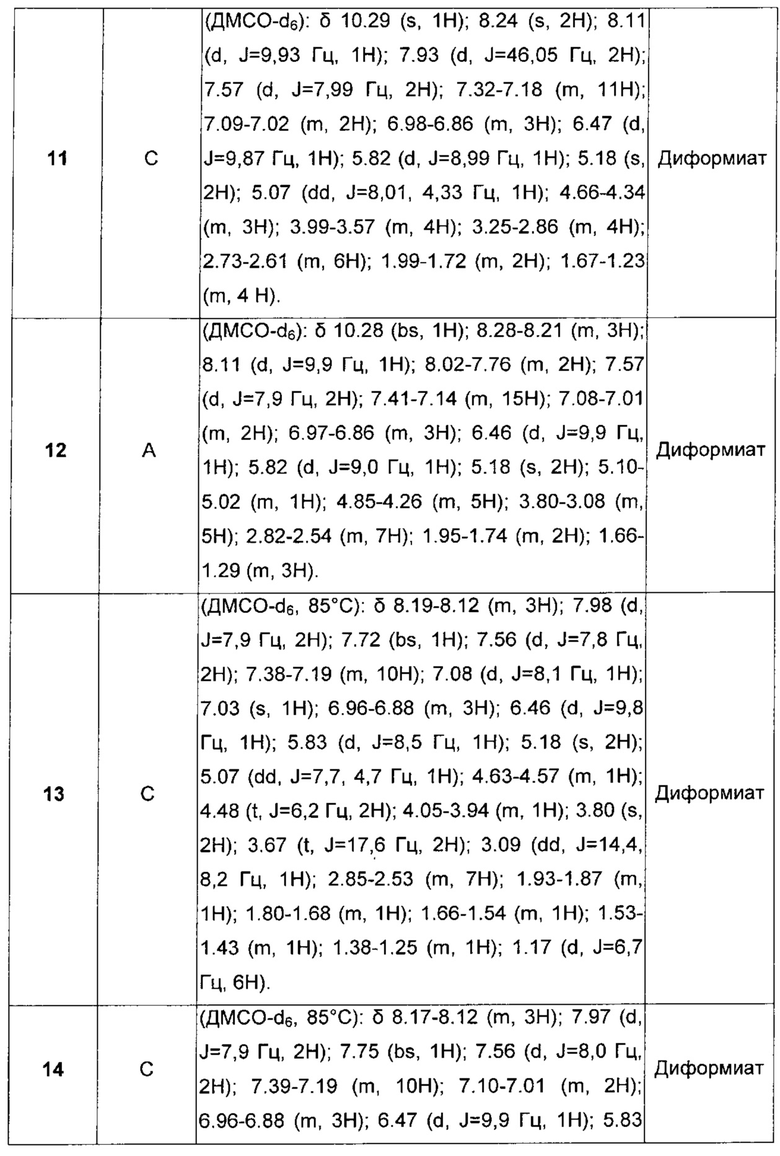
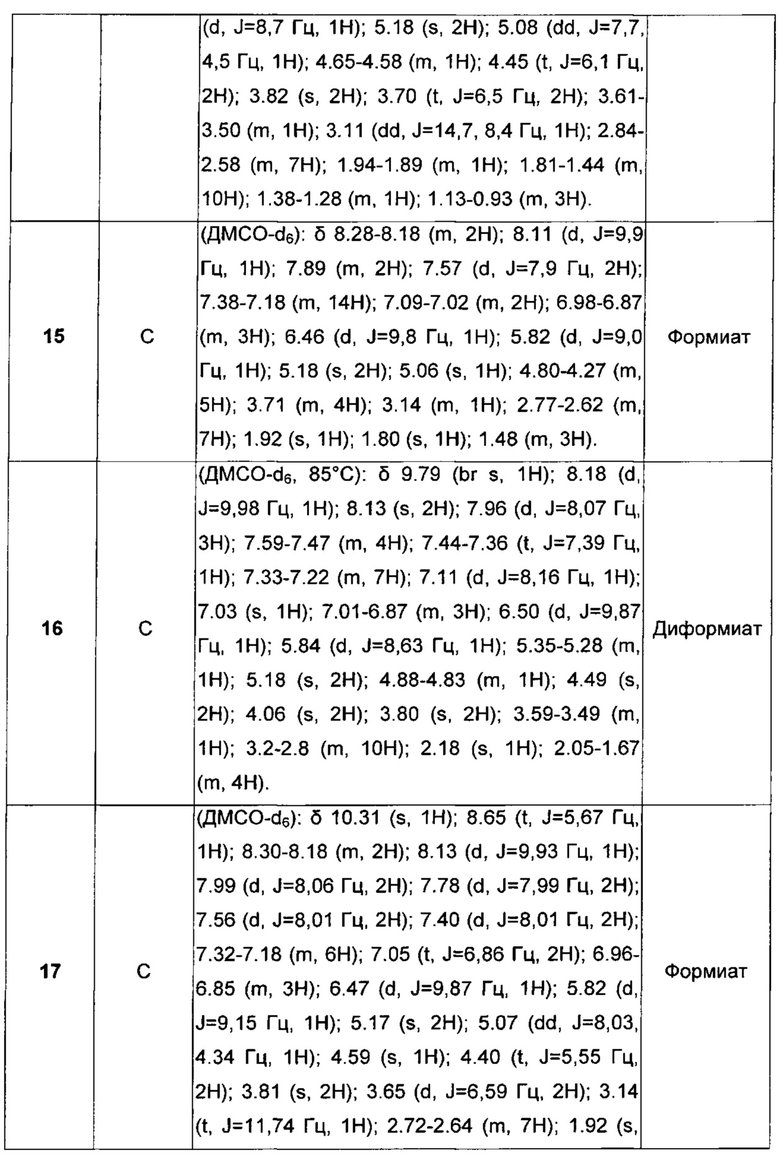
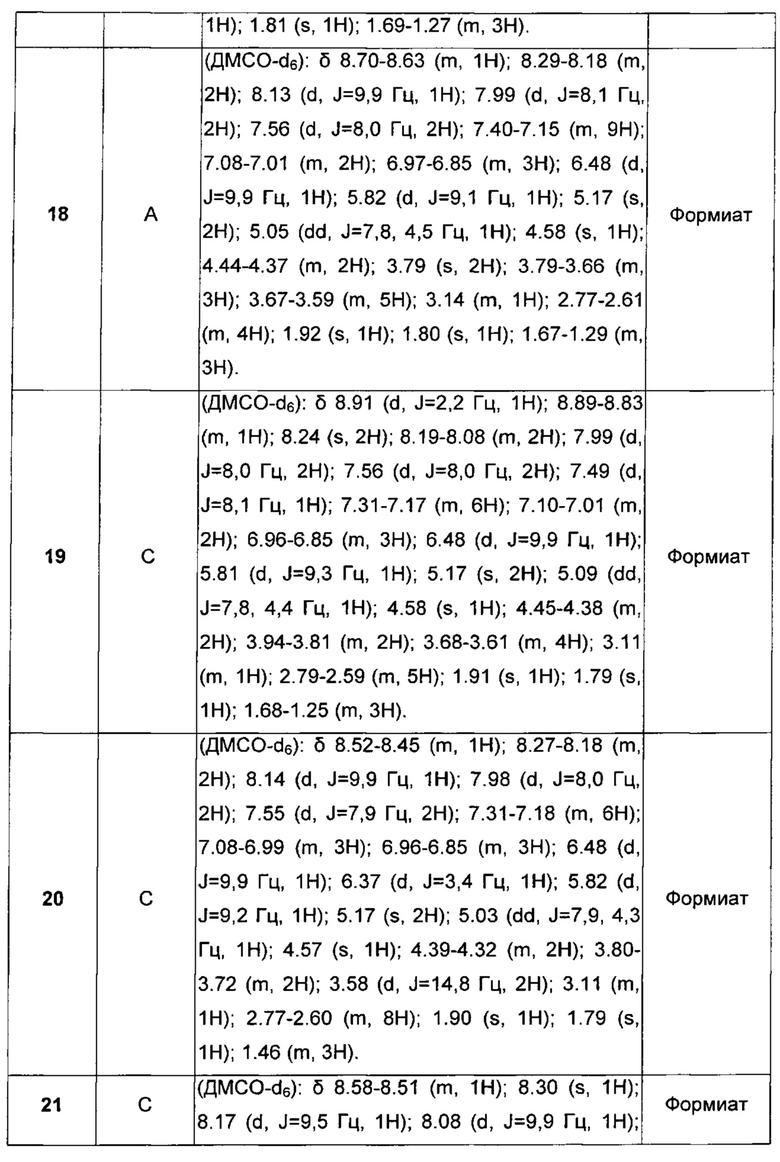
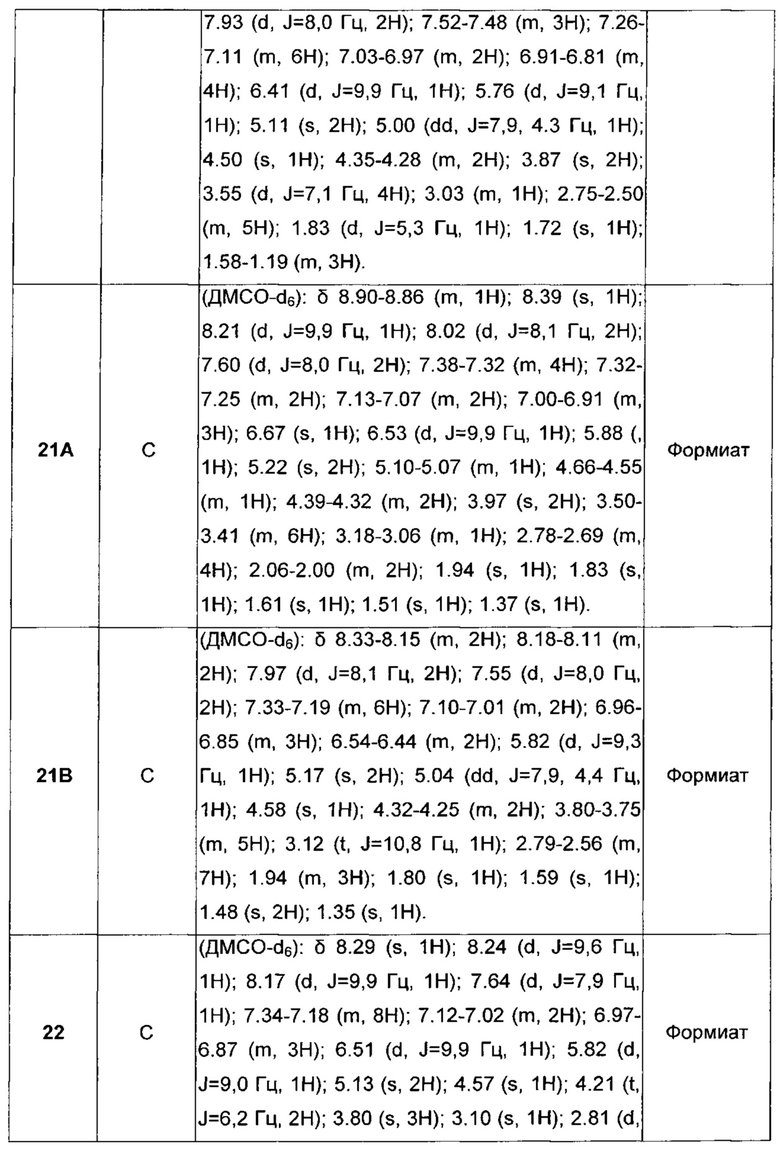
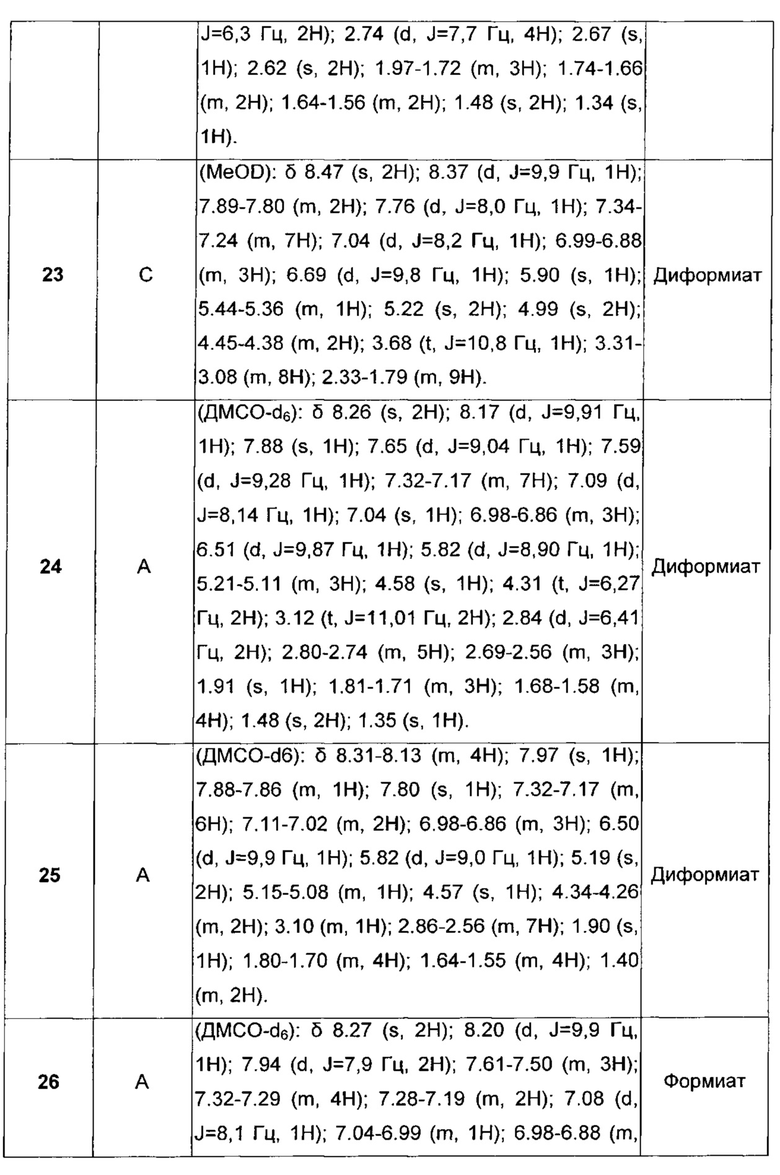
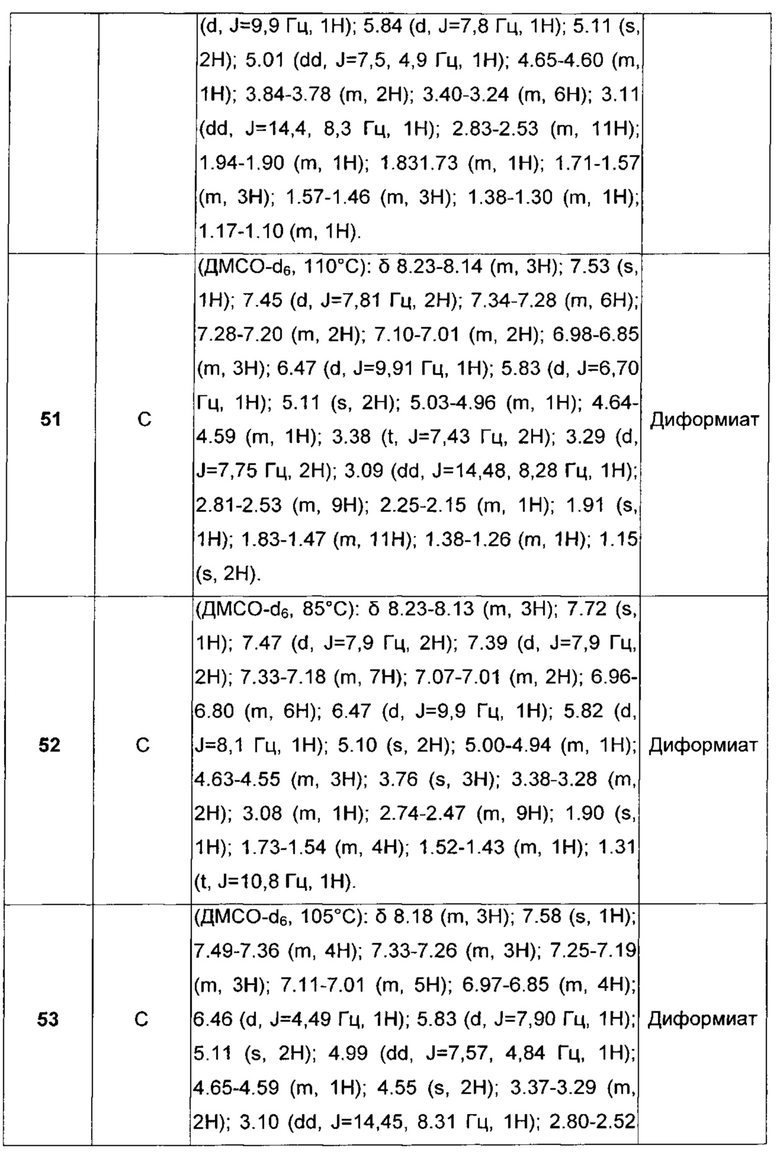
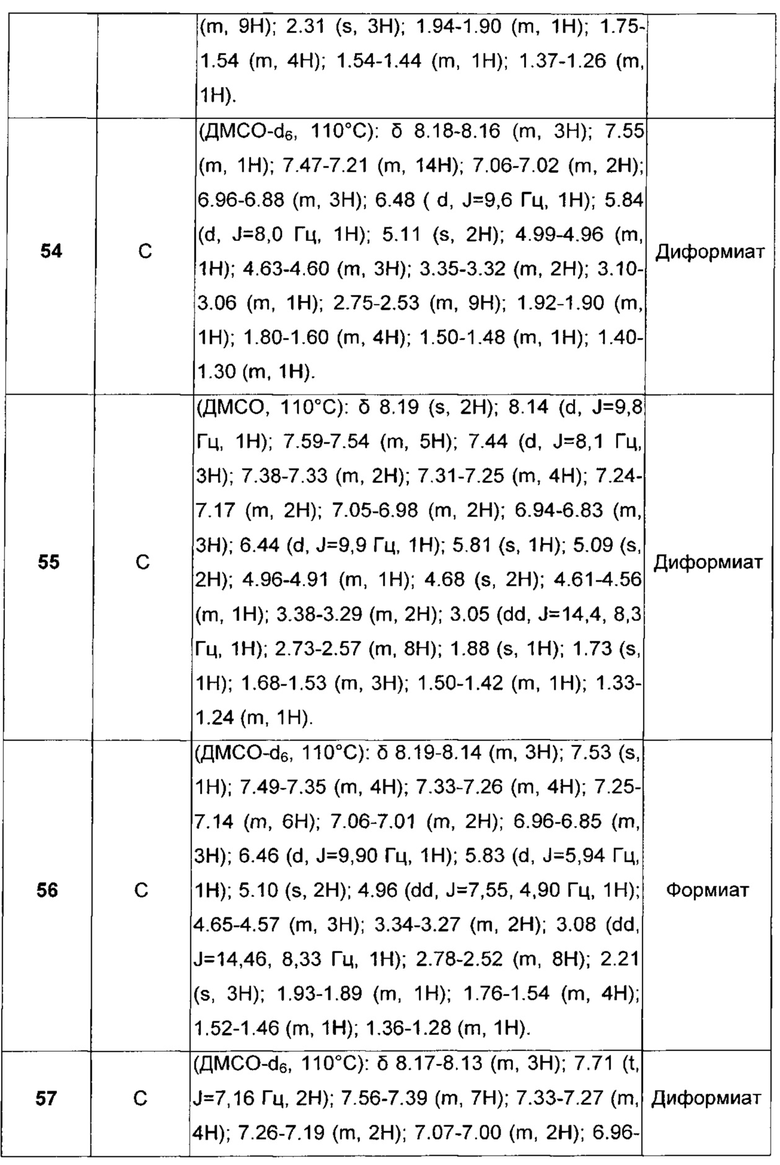
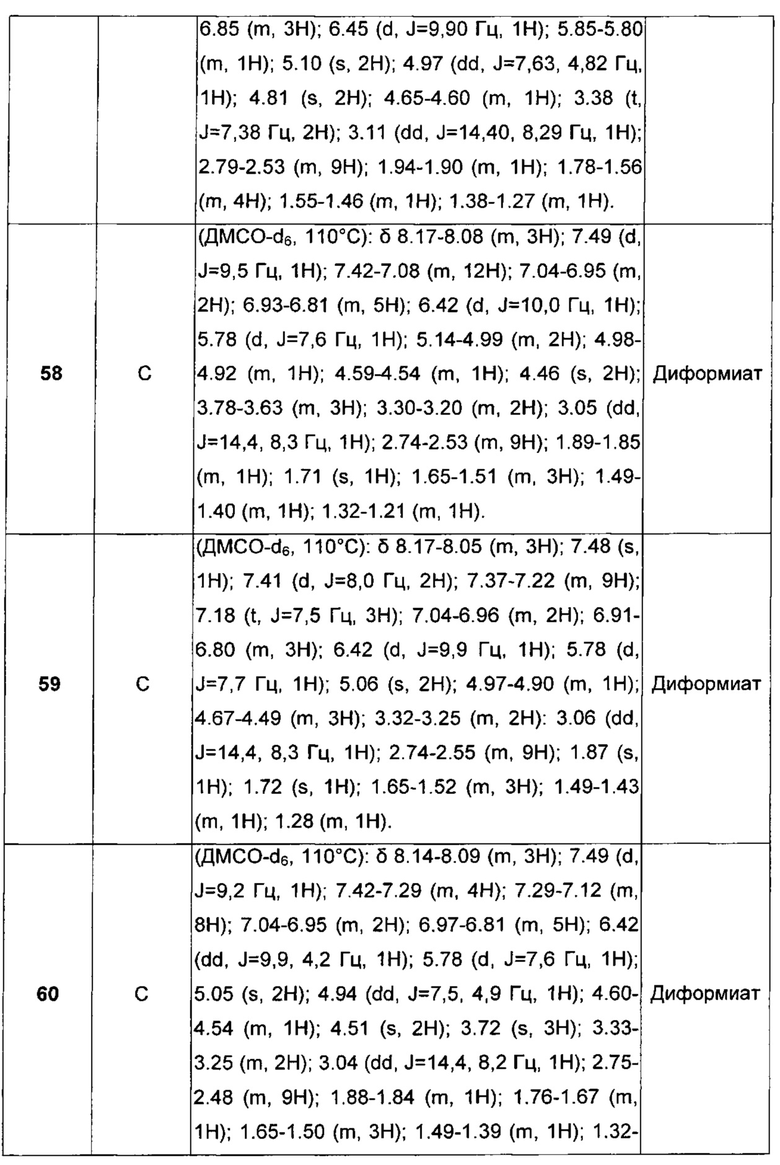
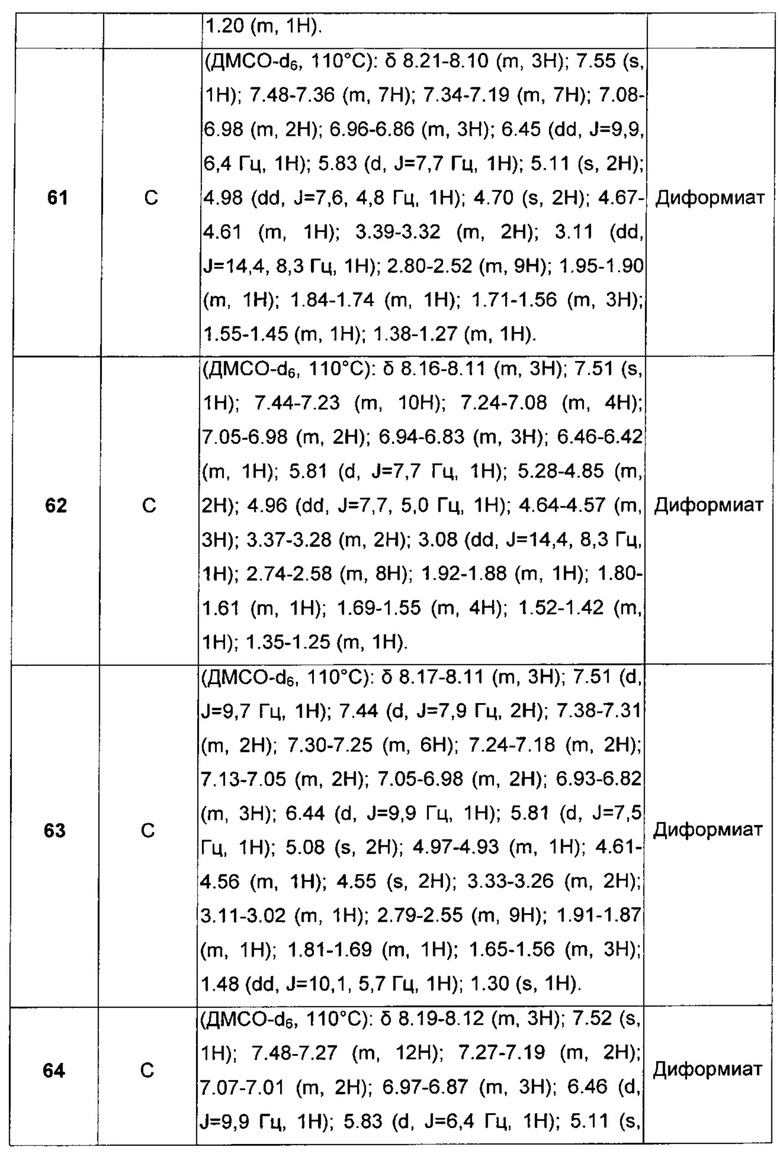
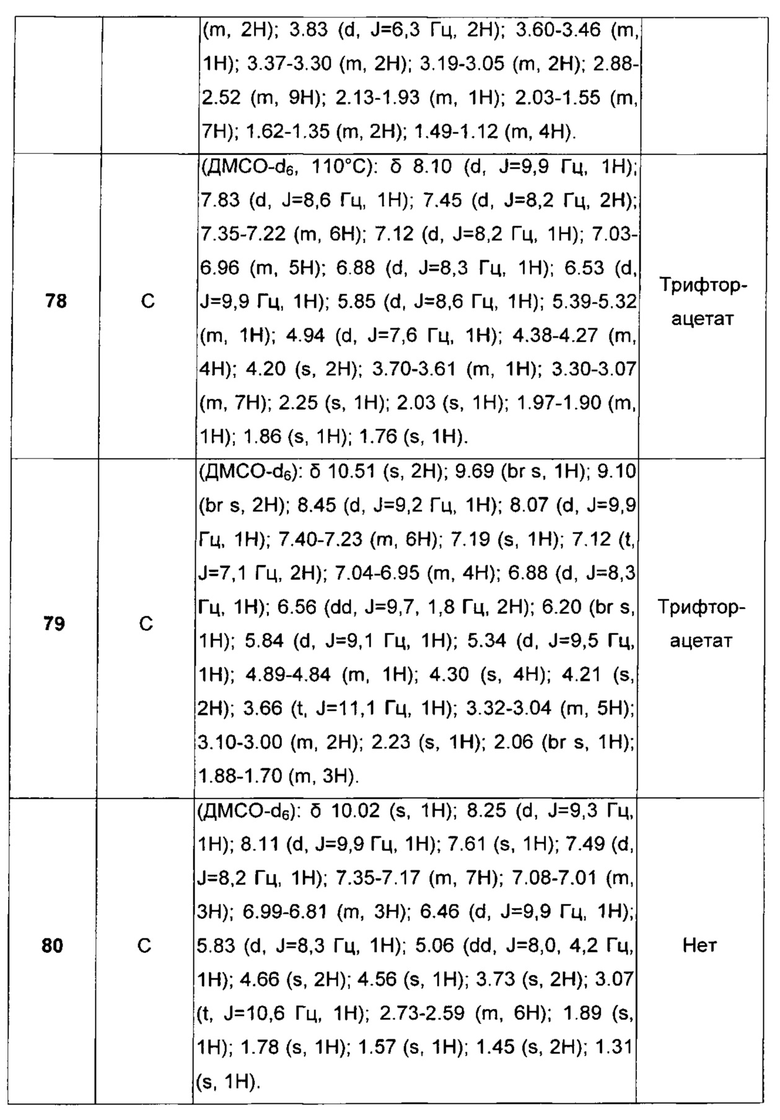
Определение биологических свойств
Пример 17
Анализ связывания радиолигандов с рецепторами подтипа М3
Мембраны, содержащие человеческий М3-рецептор (15 мкг/лунка), от Perkin Elmer инкубировали с 0,52 нМ N-метил-[3Н]-скополамина метилхлорида с исследуемыми соединениями или без них, или атропином в концентрации насыщения (5 мкМ) для определения неспецифического связывания. Анализ проводили в 96-луночных полипропиленовых планшетах в объеме 250 мкл. Используемый буфер для анализа представлял собой 50 мМ Трис-HCl, 154 мМ NaCl (pH 7,4). Конечная концентрация ДМСО в анализе составляла 0,5% (об./об.). Планшеты герметизировали и инкубировали в течение 2 часов при RT на орбитальном шейкере (с малым числом оборотов). Мембраны собирали на 96-луночных фильтровальных планшетах Unifilter GF/C, предварительно обработанных 0,5%-ным полиэтиленимином (об./об.), при использовании коллектора фильтра, промывали четыре раза 200 мкл буфера для анализа. Планшеты сушили перед добавлением 50 мкл сцинтилляционной жидкости microscint-0, герметизировали, затем считывали на сцинтилляционном счетчике Microbeta Trilux. Значения IC50 определяли по кривым сравнения при использовании программы нелинейной аппроксимации. Значения Ki подсчитывали из значений IC50 с помощью уравнения Ченга-Прусоффа.
Значения Ki большинства соединений по примерам составляют менее 10 нМ.
Пример 18
Анализ связывания радиолигандов с β2-адренорецептором
Мембраны, содержащие человеческий β2-адренорецептор (7,5 мкг/лунка), от Perkin Elmer инкубировали с 0,3 нМ 125-I-цианопиндололом с исследуемыми соединениями или без них, или s-пропранололом в концентрации насыщения (2 мкМ) для определения неспецифического связывания. Анализ проводили в 96-луночных полипропиленовых планшетах в объеме 200 мкл. Используемый буфер для анализа представлял собой 25 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазин-этансульфоновая кислота), 0,5% BSA (бычий сывороточный альбумин) (мас./об.), 1 мМ ЭДТА (этилендиаминтетрауксусная кислота), 0,02% аскорбиновой кислоты (об./об.), (pH 7,4). Конечная концентрация ДМСО в анализе составляла 0,5% (об./об.). Планшеты герметизировали и инкубировали в течение 1 часа при RT на орбитальном шейкере (с малым числом оборотов). Мембраны собирали на 96-луночных фильтровальных планшетах Unifilter GF/C, предварительно обработанных 0,5%-ным полиэтиленимином (об./об.), при использовании коллектора фильтра, промывали шесть раз 200 мкл промывочного буфера, содержащего 10 мМ HEPES и 500 мМ NaCl. Планшеты сушили перед добавлением 50 мкл сцинтилляционной жидкости microscint-0, герметизировали, затем считывали на сцинтилляционном счетчике Microbeta Trilux. Значения IC50 определяли по кривым сравнения при использовании программы нелинейной аппроксимации. Значения Ki подсчитывали из значений IC50 с помощью уравнения Ченга-Прусоффа.
Значения Ki большинства соединений по примерам составляют менее 10 нМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединения, обладающие активностью антагонистов мускариновых рецепторов и агонистов бета-2-адренергических рецепторов | 2016 |
|
RU2709777C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ МУСКАРИНОВЫХ РЕЦЕПТОРОВ И АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ БЕТА2-АДРЕНОРЕЦЕПТОРОВ | 2012 |
|
RU2606121C2 |
| КОМПОЗИЦИИ ТЕТРАГИДРОХИНОЛИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНА ВЕТ | 2014 |
|
RU2727169C2 |
| ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ИНГИБИТОР НАТРИЙЗАВИСИМОГО ПЕРЕНОСЧИКА ФОСФАТА | 2015 |
|
RU2811864C1 |
| Соединение на основе оксазолидина и содержащий его селективный агонист андрогенового рецептора | 2015 |
|
RU2702001C2 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 2000 |
|
RU2269525C2 |
| ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ИНГИБИТОР НАТРИЙЗАВИСИМОГО ПЕРЕНОСЧИКА ФОСФАТА | 2015 |
|
RU2740008C2 |
| ПРОИЗВОДНЫЕ ФЕНИЛКАРБАМАТА В КАЧЕСТВЕ МОДУЛЯТОРОВ ФОРМИЛПЕПТИДНОГО РЕЦЕПТОРА | 2014 |
|
RU2703725C1 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2653500C2 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
Изобретение относится к ряду соединений, выбранных из группы, состоящей из 4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)пиперидин-4-карбоксилата; (S)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пропаноата; (S)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-3-метил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)бутаноата; (S)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-4-метил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пентаноата; 4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-2-метил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пропаноата; (S)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-3-циклогексил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пропаноата и т.д, которые обладают действием как антагониста мускариновых рецепторов, так и агониста бета-2-адренергических рецепторов. Изобретение относится также к фармацевтической композиции, применению указанных соединений для изготовления лекарственного средства для предупреждения и/или лечения бронхообструктивных или воспалительных заболеваний, их комбинации с другими активными ингредиентами и устройству, содержащему указанную фармацевтическую композицию. 5 н. и 1 з.п. ф-лы, 18 пр.
1. Соединение, выбранное из группы, состоящей из:
4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-1-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоил)пиперидин-4-карбоксилата;
(S)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пропаноата;
(S)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-3-метил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)бутаноата;
(S)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-4-метил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пентаноата;
4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-2-метил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пропаноата;
(S)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-3-циклогексил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пропаноата;
(R)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-4-метил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пентаноата;
(S)-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-3-(4-метоксифенил)-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)пропаноата;
4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-2-(4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензамидо)ацетата;
2-(N-этил-4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-N-метилбензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(N-бензил-4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-N-изопропилбензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(N-циклогексил-4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(N-(4-хлорбензил)-4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(3-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-N-метилбензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-3-метоксибензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(6-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)никотинамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(5-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фуран-2-карбоксамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(5-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)тиофен-2-карбоксамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-2-метокси-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)-2-(трифторметил)бензоата;
4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-3-фтор-5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-3-хлор-5-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(N-(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)ацетамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(N-(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)бензамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(N-(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)фенилсульфонамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
3-(N-(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)бензамидо)пропил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
3-(N-(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)циклогексанкарбоксамидо)пропил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
3-(N-(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)фенилсульфонамидо)пропил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
3-(N-(3-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)пропил)-2-фенилацетамидо)пропил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
транс-4-(((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)бутил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)-карбонил)амино)метил)фенокси)метил)циклогексанкарбоксилата;
2-(3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)-1-метилуреидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)уреидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)-1-фенилуреидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(1-этил-3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)уреидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)-1-изопропилуреидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(1-циклогексил-3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)уреидо)этил-4-((3-((S)-фенил-((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(1-бензил-3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)уреидо)этил-4-((3-((S)-фенил-((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(1-(4-фторбензил)-3-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)уреидо)этил-4-((3-((S)-фенил-((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-((4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)(метил)амино)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
1-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)пиперидин-4-ил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
(1-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)фенил)пиперидин-4-ил)метил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)-N-метилфенилсульфонамидо)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
2-(4-(4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензил)пиперазин-1-ил)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата; и
2-((4-((((R)-2-гидрокси-2-(8-гидрокси-2-оксо-1,2-дигидрохинолин-5-ил)этил)амино)метил)бензил)(метил)амино)этил-4-((3-((S)-фенил((((R)-хинуклидин-3-илокси)карбонил)амино)метил)фенокси)метил)бензоата;
и их фармацевтически приемлемых солей.
2. Фармацевтическая композиция, обладающая действием как антагониста мускариновых рецепторов, так и агониста бета-2-адренергических рецепторов, содержащая в эффективном количестве соединение по п.1 либо как таковое, либо в виде фармацевтически приемлемой соли с одним или более чем одним фармацевтически приемлемым носителем и/или эксципиентом.
3. Применение соединения по п.1 для изготовления лекарственного средства для предупреждения и/или лечения бронхообструктивных или воспалительных заболеваний, предпочтительно астмы, или хронического бронхита, или хронической обструктивной болезни легких (ХОБЛ).
4. Комбинация соединения по п.1 с одним или более чем одним активным ингредиентом, выбранным из классов, состоящих из бета-2-агонистов, антимускариновых агентов, ингибиторов митоген-активируемых протеинкиназ (MAP киназы р38), ингибиторов бета-субъединицы киназы ядерного фактора каппа-В (IKK2), ингибиторов эластазы нейтрофилов человека (HNE), ингибиторов фосфодиэстеразы-4 (PDE4), модуляторов лейкотриенов, нестероидных противовоспалительных агентов (NSAID), противокашлевых агентов, регуляторов секреции слизи, муколитических агентов, отхаркивающих средств/мукокинетических модуляторов, пептидных муколитических агентов, антибиотиков, ингибиторов Янус-киназ (JAK), ингибиторов тирозинкиназы селезенки (SYK), ингибиторов фосфатидилинозитол-3-киназы (PI3K) дельта или PI3K гамма, кортикостероидов и М3-антагонистов/PDE4-ингибиторов (MAPI), для предупреждения и/или лечения бронхообструктивных или воспалительных заболеваний, таких как астма, хронический бронхит или хроническая обструктивная болезнь легких.
5. Фармацевтическая композиция по п.2 для введения посредством ингаляции, такая как ингалируемые порошки, пропеллентсодержащие дозированные аэрозоли или не содержащие пропеллентов ингаляционные препараты.
6. Устройство, содержащее фармацевтическую композицию по п.5, которое представляет собой одно- или многодозовый ингалятор сухого порошка, дозирующий ингалятор и небулайзер мягкого тумана.
| Прибор для определения неравномерности хода машин | 1929 |
|
SU22342A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| A | |||
| D | |||
| HUGHES ET AL., Discovery of muscarinic acetylcholine receptor antagonist and beta 2 adrenoceptor agonist (MABA) dual pharmacology molecules, BIOORG | |||
| MED | |||
| CHEM | |||
| LETT., 2011, vol | |||
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ХИНУКЛИДИНА И МЕДИЦИНСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 1995 |
|
RU2143432C1 |

