Область техники, к которой относится изобретение
Настоящее изобретение относится к фармацевтическим составам, пригодным для перорального введения в твердой лекарственной форме, содержащей эффективное количество продукта соли лекарственного вещества и композицию с контролируемой скоростью высвобождения. Более конкретно, соль продукта лекарственного вещества является основной солью карбоксамида гидроксипиримидинона, а композиция с контролируемой скоростью высвобождения содержит солюбилизирующий агент, гелеобразующий агент и растворимый в воде наполнитель.
Уровень техники
Карбоксамиды гидроксипиримидинона, описанные в WO 03/035077, и карбоксамиды гидрокситетрагидропиридопиримидинона и родственные карбоксамиды, описанные в WO 2004/058756, являются ингибиторами ВИЧ (HIV) интегразы, пригодными для лечения ВИЧ инфекции и СПИДа (AIDS). Некоторые из этих карбоксамидных соединений обнаруживают относительно низкую растворимость в воде, что может привести к слабому всасыванию соединения в желудочно-кишечном (GI) тракте после перорального введения. Растворимость этих соединений может быть улучшена путем введения лекарственных веществ в форме основных солей (то есть, солей, образованных реакцией соединений с основными солями, такими как гидроксиды металлов), но растворимость некоторых из получаемых солей может меняться в зависимости от pH. Более конкретно, основные соли могут быть относительно растворимыми в нейтральной или основной водной среде, но могут превращаться в менее растворимые формы в кислых условиях. Представителем таких солей является калиевая соль соединения A:
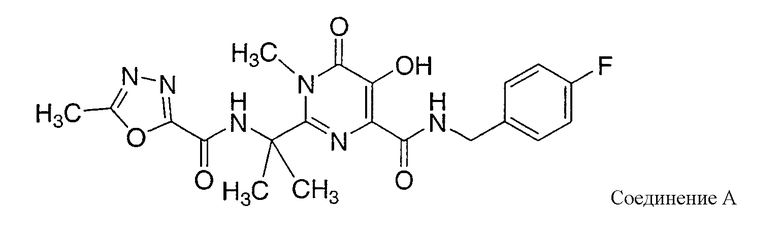
Калиевая соль соединения A является относительно растворимой в нейтральных или основных водных растворах, но в кислых растворах она имеет тенденцию к диспропорционированию до относительно нерастворимой формы свободного основания. Когда калиевую соль соединения А вводят перорально в твердой лекарственной форме, соединение может демонстрировать слабое всасывание в системной циркуляции вследствие потери или значительного снижения растворимости соли в кислых условиях, обычно имеющихся в желудке.
Удовлетворительная пероральная биодоступность может быть достигнута путем формирования этих солей вместе с агентом антинуклеации. Например, спрессованные таблетированные формы калиевой соли соединения А, содержащие гидроксипропилметилцеллюлозу (например, HPMC 2910) в качестве агента антинуклеации, демонстрировали улучшенную растворимость, в сравнении с аналогичными составами, не содержащими агента антинуклеации, в тестах на растворение in vitro, и улучшенную фармакокинетику (PK) при исследовании на животных. При пероральном введении вместе с HPMC таблетированные формы калиевой соли соединения А также позволили получить удовлетворительную фармакокинетику (PK) у человека. Полагают, что агент антинуклеации, используемый в этих составах, может удовлетворительно ингибировать и/или замедлять осаждение (или может иначе обеспечивать длительное перенасыщение) соединения лекарственного вещества в условиях кислой среды желудка или кишечника, так чтобы дать возможность лекарственному веществу более эффективно всасываться в системной циркуляции.
С другой стороны, в твердых лекарственных формах солей соединения, содержащих агент антинуклеации, соединение может обладать относительно быстрым всасыванием в системную циркуляцию (то есть, относительно коротким Tmax, временем, проходящим от момента введения дозы до момента достижения Cmax, максимальной концентрации соединения в плазме), с возможным, быстрым его снижением. Например, спрессованная таблетка, содержащая HPMC калиевую соль соединения А, упомянутую в предыдущем абзаце, демонстрировала относительно высокие величины Cmax, малые значения Tmax (например, от, приблизительно, 30 до 90 минут) и относительно низкие плазменные концентрации впоследствии. Высокие значения отношений пиковых и минимальных плазменных концентраций могут быть связаны с неблагоприятными эффектами; и низкие плазменные концентрации, следующие за Tmax, могут привести к малому всасыванию или отсутствию всасывания лекарственного вещества вне пределов желудка и перед устранением лекарственного вещества из желудочно-кишечного тракта (то есть, незначительному всасыванию или отсутствию всасывания в тонкой кишке или толстой кишке). Соответственно, существует потребность в твердых формах пероральных лекарственных составов данных соединений с контролируемым высвобождением, таким образом, чтобы обеспечить изменение профиля PK относительно профиля твердых лекарственных форм на основе агентов антинуклеации, (то есть, получение более длительного Tmax, более низкого отношения пиковых плазменных концентраций и минимальных плазменных концентраций, и/или получение более высоких минимальных плазменных концентраций после Tmax).
Раскрытие изобретения
Настоящее изобретение относится к фармацевтическим составам для перорального введения, содержащим карбоксамид гидроксипиримидинона или родственные конденсированные циклические карбоксамиды и композицию, которая после введения контролирует высвобождение карбоксамида в системной циркуляции. Более конкретно, настоящее изобретение включает фармацевтический состав для перорального введения в виде твердой дозы, которая содержит эффективное количество основной соли соединения формулы I (альтернативно и более просто обозначаемое здесь как "Соединение I") и композицию, контролирующую скорость высвобождения, содержащую солюбилизирующий агент, гелеобразующий агент и, необязательно, растворимый в воде наполнитель; где формула I представляет собой:
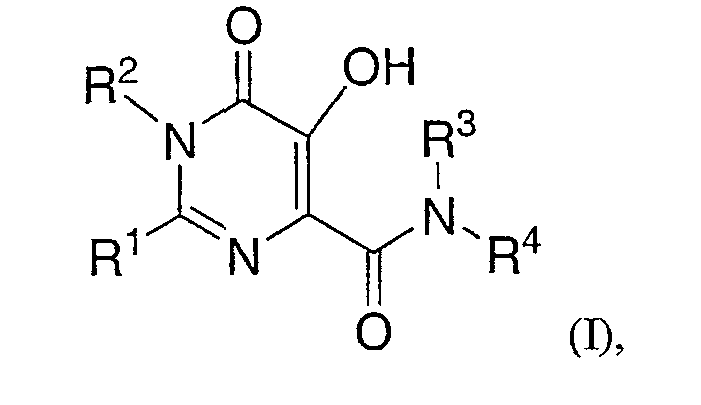
где Rl представляет собой С1-6 алкил, замещенный:
(1) N(RA)-C(=O)-N(RC)RD,
(2) N(RA)-C(=O)-С1-6 алкилен-N(RC)RD,
(3) N(RA)SO2RB,
(4) N(RA)SO2N(RC)RD,
(5) N(RA)-C(=O)-С1-6 алкилен-SO2RB,
(6) N(RA)-C(=O)-С1-6 алкилен-SO2N(RC)RD,
(7) N(RA)C(=O)C(=O)N(RC)RD;
(8) N(RA)-C(=O)-HetA,
(9) N(RA)C(=O)C(=O)-HetA, или
(10) HetB;
R2 представляет собой - С1-6 алкил;
или, альтернативно, Rl and R2 соединены вместе так, что соединение Формулы I становится соединением Формулы ІІ:
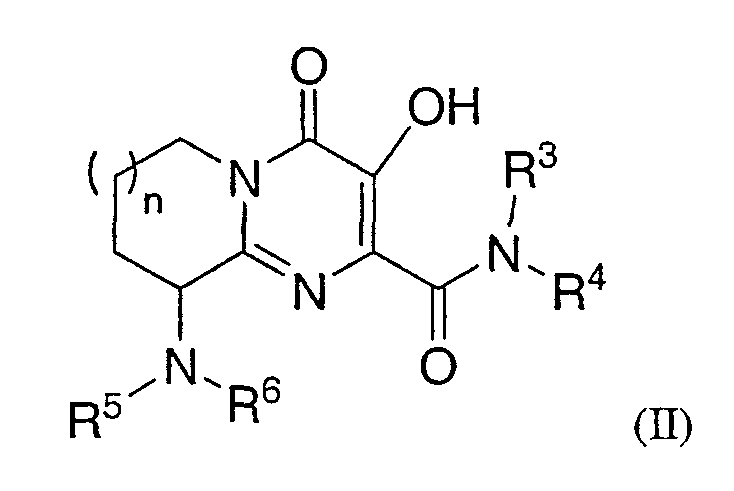
R3 представляет собой -H или - С1-6 алкил;
R4 представляет собой -С1-6алкил, замещенный арилом (например, фенил), который, необязательно, замещен заместителями, в количестве от 1 до 4, каждый из которых независимо представляет собой галоген, -OH, -С1-4 алкил, -С1-4 алкил-ORA,
-С1-4 галогеналкил, -O-С1-4 алкил, -O-С1-4 галогеналкил, -CN, -NO2, -N(RA)RB; -С1-4алкил-N(RA)RB, -C(=O)N(RA)RB, -C(=O)RA, -CO2RA, -С1-4алкил-CO2RA, -OCO2RA, -SRA, -S(=O)RA, -SO2RA, -N(RA)SO2RB, -SO2N(RA)RB, -N(RA)C(=O)RB, -N(RA)CO2RB, -С1-4алкил-N(RA)CO2RB, метилендиокси, присоединенный к двум соседним кольцевым атомам углерода, фенил или -С1-4 алкилфенил;
R5 представляет собой:
(1) N(RA)-C(=O)-N(RC)RD;
(2) N(RA)-C(=O)-С1-6 алкилен-N(RC)RD,
(3) N(RA)SO2RB,
(4) N(RA)SO2N(RC)RD,
(5) N(RA)-C(=O)-С1-6 алкилен-SO2RB,
(6) N(RA)-C(=O)-С1-6 алкилен-SO2N(RC)RD,
(7) N(RA)C(=O)C(=O)N(RC)RD
(8) N(RA)-C(=O)-HetA или
(9) N(RA)C(=O)C(=O)-HetA;
R6 представляет собой -H или -С1-6 алкил;
n является целым числом, равным 1 или 2;
каждый RA независимо представляет собой -H или -С1-6 алкил;
каждый RB независимо представляет собой -H или -С1-6 алкил;
RC и RD, каждый независимо, представляют собой -H или -С1-6 алкил, или вместе с атомом азота, к которому они присоединены, образуют насыщенное 5- или 6-членное гетероциклическое кольцо, необязательно содержащее гетероатом, в дополнение к атому азота, присоединенному к RC и RD, выбранным из N, O и S, где S необязательно окислена до S(O) до S(O2), и где насыщенное гетероциклическое кольцо необязательно замещено 1 или 2 С1-6 алкильными группами;
HetA представляет собой 5- или 6-членное гетероароматическое кольцо, содержащее от 1 до 4 гетероатомов, независимо выбранных из N, О и S, где гетероароматическое кольцо необязательно замещено 1 или 2 заместителями, каждый из которых независимо представляет собой - С1-4 алкил, - С1-4 галогеналкил, -O-С1-4 алкил,
-O-С1-4 галогеналкил, или -CO2RA; и
HetB представляет собой 5-7-членное насыщенное гетероциклическое кольцо, содержащее от 1 до 4 гетероатомов, независимо выбранных из N, О и S, где каждый атом S необязательно окислен до S(O) или S(O)2, и гетероциклическое кольцо необязательно замещено 1-3 заместителями, каждый из которых независимо представляет собой галоген, -С1-4алкил, -С1-4 фторалкил, -C(O)-С1-4 алкил, или -С1-4 алкил, замещенный OH.
Один вариант настоящего изобретения представляет собой обозначенный выше фармацевтический состав, в котором в соединении I: R2 является метилом; R3 является -H; и R4 представляет собой CH2-фенил, где фенил необязательно замещен 1 или 2 заместителями, каждый из которых независимо представляет собой бром, хлор, фтор, СН3, CF3, C(O)NH2, C(O)NH(СН3), C(O)N(СН3)2, SCH3, SO2CH3, или SO2N(СН3)2; а все другие варианты определены выше. Особенностью этого аспекта изобретения является то, что R4 представляет собой 4-фторбензил, 3,4-дихлорбензил, 3-хлор-4-фторбензил или 4-фтор-3-метилбензил. Еще одной особенностью является то, что R4 представляет собой 4-фторбензил.
Фармацевтические составы по настоящему изобретению могут обеспечить изменение PK профиля для соединения I, по сравнению с другими твердыми лекарственными формами, вводимыми перорально. Например, фармацевтический состав по настоящему изобретению, содержащий калиевую соль соединения A, демонстрирует при пероральном введении более длительное Tmax, более низкое отношение пиковой плазменной концентрации к минимальной плазменной концентрации и более высокие минимальные плазменные концентрации после прохождения Tmax. Сравнение проводилось с аналогичными составами, использующими агенты антинуклеации, вместо композиции с контролируемой скоростью высвобождения. Не привязываясь к какой-либо конкретной теории, все же полагают, что композиция с контролируемой скоростью высвобождения следующим образом ответственна за изменение профиля PK: солюбилизирующий агент действует так, чтобы препятствовать осаждению или минимизировать осаждение соединения I (которое, как указывали выше, может иметь низкую растворимость, в частности, в кислых условиях, существующих в желудке) в GI (желудочно-кишечном) тракте в течение нескольких часов после введения, посредством сохранения его в растворенной форме. Гелеобразующий агент воздействует, образуя гель вокруг частиц соединения I, где гель действует как диффузионный барьер, который замедляет высвобождение соединения I для всасывания в системном кровообращении. Растворимый в воде наполнитель после введения растворяется относительно быстро и действует, вытягивая воду в гелевый слой, образованный гелеобразующим агентом, и тем самым ускоряет диффузию лекарственного вещества и высвобождение. Два компонента (или три компонента, когда композиция с контролируемой скоростью высвобождения, содержит наполнитель, растворимый в воде) используют таким образом и в таких количествах, чтобы сохранять активное соединение в растворе и продлить высвобождение соединения так, чтобы безопасное и эффективное количество лекарственного вещества всасывалось в системную циркуляцию на протяжении длительного периода времени и из желудка, и из кишечного тракта.
Настоящее изобретение также включает в себя способы получения инкапсулированных и таблетированных форм фармацевтических составов по изобретению. Настоящее изобретение, кроме того, включает использование фармацевтического состава по изобретению для ингибирования ВИЧ-интегразы, для лечения или профилактики ВИЧ-инфекции или для лечения, замедления возникновения или профилактики СПИДа.
Разнообразные варианты осуществления, аспекты и особенности настоящего изобретения или будут описаны в дальнейшем или будут очевидны в результате описания, примеров и прилагаемой формулы изобретения.
Краткое описание чертежей
Фиг.1 представляет собой порошковую рентгенограмму калиевой соли соединения A, получаемого в примере 2.
Фиг.2 представляет собой кривую дифференциальной сканирующей калориметрии (DSC) для калиевой соли соединения A, получаемой в примере 2.
Фиг.3 представляет собой график по данным растворимости, полученный при исследовании растворимости, описанном в примере 8; то есть, график зависимости процента растворенного соединения A от времени растворения при исследовании растворимости полоксамерсодержащих таблеток, содержащих 400 мг соединения A.
Фиг.4 представляет собой график данных по растворимости, полученный при исследовании растворимости, описанный в примере 8; то есть, график зависимости процента растворенного соединения A от времени растворения, при исследовании растворимости лактозосодержащих таблеток, содержащих 100 мг соединения A.
Подробное описание изобретения
Доза перорально вводимых фармацевтических составов по настоящему изобретению включает эффективное количество основной соли соединения Формулы I в твердом виде. Соединения формулы I являются ингибиторами интегразы ВИЧ. Более конкретно, репрезентативные соединения, отображаемые Формулой I, тестировали в опыте по ингибированию интегразы, в котором перенос нити катализируется рекомбинантной интегразой, и обнаружили, что они являются активными ингибиторами интегразы ВИЧ. Активность ингибирования интегразы, например, можно определять, используя тест, описанный в Hazuda et al., J. Virol. 1997, 71: 7005-7011. Было обнаружено также, что репрезентативные соединения оказываются активными в тесте на ингибирование острой ВИЧ-инфекции T-лимфоидных клеток, который проводили в соответствии с Vacca et al., Proc. Natl. Acad. Sci. USA 1994, 91.: 4096-4100. Дальнейшее описание репрезентативных соединений, охватываемых Формулой I, способы их получения и тесты по измерению степени ингибирования ими активности интегразы и ингибирования ими репликации ВИЧ можно найти в WO 03/035077, описание которого полностью включено сюда в виде ссылки.
Используемый здесь термин "состав" предназначен для обозначения вводимого перорально твердого дозированного продукта, содержащего обозначенные ингредиенты, а также любого продукта, который получают в результате прямого или косвенного объединения указанных ингредиентов.
Используемый здесь термин "эффективное количество" означает количество соединения I (или другого фармацевтического агента), которое выявляет биологическую или медицинскую ответную реакцию в ткани, системе, животном или человеке, которая требуется для исследователя, ветеринара, лечащего врача или другого клинициста. Эффективное количество может быть "терапевтически эффективным количеством", применяемым для смягчения симптомов заболевания или состояния, подвергаемого лечению. Эффективное количество также может быть "профилактически эффективным количеством", применяемым для профилактики симптомов заболевания или состояния, которое предупреждают. Термин также относится к количеству соединения формулы I, достаточному для ингибирования интегразы ВИЧ и, тем самым, выявления требуемой ответной реакции (то есть, "эффективное количество для ингибирования").
Понятно, что основная соль соединения I, используемая в фармацевтических составах, входящих в объем настоящего изобретения, представляет собой фармацевтически приемлемую соль.
Термин "фармацевтически приемлемая соль" относится здесь к основной соли, которая обладает эффективностью первичного соединения и которая не является биологически или иным образом нежелательной (например, не является ни токсичной, ни действующей отрицательно на реципиента иным образом). Пригодные соли включают соли, образованные в результате реакции соединения I с основанием, включая, например, соли щелочных металлов (например, соли натрия или калия), соли щелочноземельных металлов (например, соли кальция или магния) и соли аммония. Соли щелочных металлов соединений могут быть получены обработкой соединения, растворенного в пригодном растворителе, водным раствором гидроксида щелочного металла (например, NaOH или KOH).
Один вариант осуществления настоящего изобретения представляет собой фармацевтический состав, первоначально описанный выше (то есть, первоначально описанный в части «Раскрытие изобретения»), где основная соль соединения I представляет собой соль щелочного металла соединения I (например, натриевая или калиевая соль соединения I).
Фармацевтические составы по настоящему изобретению включают композицию с контролируемой скоростью высвобождения, содержащую солюбилизирующий агент, гелеобразующий агент и, необязательно, растворимый в воде наполнитель. Пригодные солюбилизирующие агенты включают полоксамеры и макроголглицериды жирных кислот. Полоксамеры представляют собой блоксополимеры этиленоксида и пропиленоксида. Пригодные полоксамеры включают, например, полоксамеры, имеющие среднюю молекулярную массу в интервале от, приблизительно, 1000 до, приблизительно, 20000 при содержании оксиэтилена, составляющем от, приблизительно, 40 до, приблизительно, 90 мас.%. Репрезентативные полоксамеры, пригодные для использования в настоящем изобретении, включают полоксамер 188, полоксамер 237, полоксамер 338 и полоксамер 407. Пригодным макроголглицеридом жирной кислоты является стеароилмакроголглицерид, такой как GELUCIRE® 50/13 (распространяемый фирмой Gattefosse, Paramus, NJ), который представляют собой смесь моно-, ди- и триглицеридов и эфиров полиэтиленгликоля и моно- и ди-жирных кислот с температурами плавления в интервале от 46,0 до 51,0°C и величиной HLB, составляющей 13.
Пригодные гелеобразующие агенты включают эфиры глицерина и жирных кислот, такие как глицерилбегенат (например, Compritol® 888ATO, который является глицерилбегенатом, распространяемым фирмой Gattefosse) и высоковязкие HPMC. Термин "высоковязкие" HPMC относится к HPMC (гидроксипропилметилцеллюлоза), которая дает 2 мас.% (то есть, масса полимера/масса воды) водный раствор, имеющий вязкость, составляющую при 20°C, по меньшей мере, приблизительно, 2900 сП (cps) (1 сП=1 мПа·сек). Высоковязкая HPMC обычно дает 2 мас.% раствор, имеющий вязкость, по крайней мере, составляющую, приблизительно, 3100 сП (например, от, приблизительно, 3100 до, приблизительно, 100,000 сП) при 20°C. Пригодные высоковязкие HPMC включают такие HPMC, которые продают под торговой маркой METHOCEL® (Dow Chemical) (например, METHOCEL класса K4M, K15M, K100M) и METOLOSE® (Shin-Etsu). Высоковязкие HPMC могут быть использованы по-отдельности или в смесях из двух и более компонентов, где полимерная смесь представляет 2 мас.% раствор со средней вязкостью, составляющей, по меньшей мере, приблизительно, 2900 сП, и обычно составляющей по меньшей мере, приблизительно, 3100 сП. Средняя вязкость полимерной смеси обычно отличается от вязкости каждого составляющего полимера.
Пригодные растворимые в воде наполнители включают сахара, такие как лактоза, глюкоза, фруктоза, маннит и декстроза. В частности, пригодными являются лактоза и маннит. Лактоза является предпочтительным растворимым в воде наполнителем.
Еще один вариант осуществления настоящего изобретения представляет собой фармацевтический состав, определенный выше, в котором солюбилизирующий агент содержит полоксамер; гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу; а необязательный растворимый в воде наполнитель содержит лактозу.
Еще один вариант осуществления настоящего изобретения представляет собой фармацевтический состав, определенный выше, в котором основную соль соединения I используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 75 мас.% по содержанию свободного фенола; солюбилизирующий агент содержит полоксамер, который используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 25 мас.%; гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу, которую используют в количестве, находящемся в интервале от, приблизительно, 2 до, приблизительно, 15 мас.%; и необязательный водорастворимый наполнитель содержит лактозу, которую используют в количестве, находящемся в интервале от нуля до, приблизительно, 15 мас.%.
Фармацевтические составы по настоящему изобретению могут содержать дополнительные компоненты, включающие разбавители, смазывающие вещества, разрыхлители, антиоксиданты и тому подобное. Соответственно, еще один вариант осуществления настоящего изобретения представляет собой фармацевтический состав, определенный первоначально или определенный в любом из вышеприведенных вариантов осуществления, в которых состав дополнительно содержит разбавитель и смазывающее вещество.
Другой вариант осуществления настоящего изобретения представляет собой фармацевтический состав, первоначально определенный выше или определенный в любом из предшествующих вариантов осуществления, где состав является инкапсулированным или спрессованным в таблетку.
Еще один вариант осуществления настоящего изобретения представляет собой фармацевтический состав, определенный первоначально, в котором соединение I является соединением A. Этот состав здесь альтернативно обозначают как " состав Fl" или "Fl состав ".
Еще один вариант осуществления настоящего изобретения представляет собой Fl состав, определенный выше, в котором основная соль соединения I представляет собой соль щелочного металла соединения A.
Еще один вариант осуществления настоящего изобретения представляет собой Fl состав, определенный выше, в котором основная соль соединения I представляет собой калиевую соль соединения A. В одном аспекте этого варианта осуществления, калиевая соль соединения A является формой 1 кристаллической калиевой соли соединения A; где форма 1 калиевой соли является безводной кристаллической солью, характеризуемой по порошковой рентгенограмме, которую получают, используя излучение меди Кα (то есть, источником излучения является излучение, представляющее комбинацию CuKα1 и CuKα2), которое охватывает значения 2θ (то есть, отражения при значениях 2θ), составляющие 5,9, 12,5, 20,0, 20,6 и 25,6 градусов.
Еще один вариант осуществления настоящего изобретения представляет собой Fl состав, первоначально определенный выше, где солюбилизирующий агент содержит полоксамер; гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу; а необязательный водорастворимый наполнитель содержит лактозу. В одном аспекте этого варианта осуществления, основная соль соединения A является калиевой солью соединения A, которую используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 75 мас.%, исходя из содержания свободного фенола. Полоксамер используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 25 мас.%. Высоковязкую гидроксипропилметилцеллюлозу используют в количестве, находящемся в интервале от, приблизительно, 2 до, приблизительно, 15 мас.%. Лактозу используют в количестве, находящемся в интервале от, приблизительно, 15 мас.%. В предпочтительном аспекте этого варианта осуществления, основная соль соединения A является калиевой солью соединения A, которую используют в количестве, заключенном в интервале от, приблизительно, 25 до, приблизительно, 75 мас.%, исходя из содержания свободного фенола. Полоксамер используют в количестве, заключенном в интервале от, приблизительно, 10 до, приблизительно, 20 мас.%. Высоковязкую гидроксипропилметилцеллюлозу используют в количестве, заключенном в интервале от, приблизительно, 3 до, приблизительно, 9 мас.%. Лактозу используют в количестве, находящемся в интервале от, приблизительно, 3 до, приблизительно, 9 мас.%. В этом варианте осуществления и предыдущих его аспектах, предпочтительным полоксамером является полоксамер 407 (особенно, полоксамер 407, измельченный до среднего размера частицы, находящемся в интервале от, приблизительно, 50 до, приблизительно, 150 микрон и, предпочтительно, измельченный до среднего размера частицы, заключенного в интервале от, приблизительно, 50 до, приблизительно, 105 микрон). Высоковязкая гидроксипропилметилцеллюлоза представляет собой HPMC K4M; а лактоза представляет собой высушенный спрей водного раствора лактозы. В частности, было обнаружено, что измельченный полоксамер дает более однородные и гомогенные смеси с частицами калиевой соли соединения А (в особенности, формы 1 кристаллической калиевой соли). В предшествующих аспектах этого варианта осуществления, калиевая соль соединения A, предпочтительно, является формой 1 кристаллической калиевой соли соединения A.
Еще один вариант осуществления настоящего изобретения представляет собой Fl состав, первоначально определенный выше или определенный в любом из предшествующих вариантов осуществления, где Fl состав дополнительно содержит разбавитель и смазывающее вещество.
Еще один вариант осуществления настоящего изобретения представляет собой Fl состав, первоначально определенный выше или определенный в любом из предшествующих вариантов осуществления, где состав является инкапсулированным или спрессованным в таблетку. В одном аспекте этого варианта осуществления, состав Fl является инкапсулированным для того, чтобы получить капсулу, содержащую основную соль соединения A (например, калиевую соль соединения А) в количестве, находящемся в интервале от, приблизительно, 5 мг до, приблизительно, 1000 мг (например, от, приблизительно, 5 мг до, приблизительно, 900 мг, или от, приблизительно, 5 мг до, приблизительно, 600 мг, или от, приблизительно, 10 мг до, приблизительно, 400 мг). В еще одном аспекте этого варианта осуществления Fl является спрессованным в таблетку, содержащую основную соль соединения A (например, калиевую соль соединения А) в количестве, находящемся в интервале от, приблизительно, 5 мг до, приблизительно, 1000 мг (например, от, приблизительно, 5 мг до, приблизительно, 900 мг или от, приблизительно, 5 мг до, приблизительно, 600 мг, или от, приблизительно, 10 мг до, приблизительно, 400 мг).
Заметим, что любая приводимая здесь ссылка на количество основной соли соединения I обозначает количество соединения I в свободном состоянии, не в форме соли. Таким образом, например, фраза «таблетка композиции, содержащая основную соль соединения I в количестве, находящемся в интервале от, приблизительно, 5 мг до, приблизительно, 1000 мг», обозначает таблетку композиции, содержащую количество лекарственного вещества соли, эквивалентное, приблизительно, количеству от 5 мг до, приблизительно, 1000 мг исходного соединения I (свободного фенола).
Еще один вариант осуществления настоящего изобретения представляет собой фармацевтический состав для перорального введения в форме твердой дозы (альтернативно обозначаемый здесь как "Состав F2" или "F2 состав "), которая содержит: (i) эффективное количество калиевой соли соединения A; (ii) композицию с контролируемой скоростью высвобождения, содержащую солюбилизирующий агент, гелеобразующий агент и водорастворимый наполнитель; (iii) разбавитель и (iv) смазывающее вещество; где солюбилизирующий агент содержит полоксамер; гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу; водорастворимый наполнитель содержит лактозу; разбавитель содержит микрокристаллическую целлюлозу и, необязательно, фосфат кальция; а смазывающее вещество содержит стеарат металла и стеарилрилфумарат металла. В одном аспекте этого варианта осуществления, калиевую соль соединения A используют в количестве, находящемся в интервале от, приблизительно, 40 до, приблизительно, 60 мас.%, исходя из содержания свободного фенола; полоксамер используют в количестве, находящемся в интервале от, приблизительно, 10 до, приблизительно, 20 мас.%; высоковязкую гидроксипропилметилцеллюлозу используют в количестве, находящемся в интервале от, приблизительно, 3 до, приблизительно, 9 мас.%; лактозу используют в количестве, находящемся в интервале от, приблизительно, 3 до, приблизительно, 9 мас.%; микрокристаллическую целлюлозу используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 30 мас.%; фосфат кальций используют в количестве, находящемся в интервале от, приблизительно, нуля до, приблизительно, 15 мас.%; и стеарат металла и стеарилрилфумарат металла, каждый независимо, используют в количестве, находящемся в интервале от, приблизительно, 1 до, приблизительно, 3 мас.%. Предшествующий аспект отличает то, что полоксамер является полоксамером 407, измельченным до частиц, средний размер которых находится в интервале от приблизительно, 50 до 150 микрон; высоковязкая гидроксипропилметилцеллюлоза представляет собой HPMC K4M; лактоза представляет собой высушенный спрей водного раствора лактозы; микрокристаллическая целлюлоза представляет собой AVICEL PH-102; фосфат кальций является двухосновным фосфатом кальция; стеарат металла является стеаратом магния; и стеарилфумарат металл представляет собой стеарилфумарат натрия. В этом варианте осуществления и его предшествующих аспектах, калиевая соль соединения A предпочтительно, является формой 1 кристаллической калиевой соли соединения A.
Другой вариант осуществления по настоящему изобретению является F2 составом, только что определенным или определенным, исходя из его особенностей, где состав является инкапсулированным или спрессованным в таблетку, например, в капсулу или таблетку, содержащую калиевую соль соединения A, в количестве, которое находится в интервале от, приблизительно, 100 мг до, приблизительно, 600 мг, исходя из содержания свободного фенола.
Если не оговорено особо, массовые проценты, приведенные здесь, считают от общей массы всех компонентов композиции (учитывая, что, как было обозначено ранее, массовый процент основной соли соединения I выражают как массовый процент формы свободного основания соединения).
Как описано выше, фармацевтические составы по настоящему изобретению могут включать в себя разбавитель и смазывающее вещество. Разбавитель (в данной области техники обозначаемый также как "наполнитель") представляет собой вещество, используемое для придания объема композиции. Разбавитель может быть использован, например, для получения достаточного объема и/или сжимаемости, чтобы дать возможность композиции быть спрессованной в таблетку, имеющую удобный размер. Пригодные разбавители включают в себя безводный двухосновный фосфат кальция, дигидрат двухосновного фосфата кальция, трехосновный фосфат кальция, сульфат кальция, кальцийкарбоксиметилцеллюлозу, микрокристаллическую целлюлозу и порошкообразную целлюлозу. Предпочтительным разбавителем для использования в составах Fl и F2 является микрокристаллическая целлюлоза, необязательно, в комбинации с фосфатом кальция.
Пригодные формы микрокристаллической целлюлозы для использования в фармацевтических составах по изобретению включают в себя, кроме прочих, материалы, продаваемые как AVICEL PH-101, AVICEL PH-102, AVICEL PH-103 и AVICEL PH-105 (все распространяются фирмой FMC Corporation) и их смеси. Таким образом, например, микрокристаллическая целлюлоза, используемая в составах Fl и F2, может представлять собой AVICEL PH-102 или AVICEL PH-105 или их смесь.
Смазывающее вещество может иметь одну или несколько функций, зависящих от лекарственной формы композиции. Смазывающее вещество может, например, препятствовать прилипанию спрессованных таблеток к компрессорному оборудованию. Оно может улучшить движение гранул, получаемых гранулированием композиции перед компрессией или капсулированием, и/или оно может улучшить движение негранулированного порошка при наполнении капсулы. Пригодные смазывающие вещества включают стеарат кальция, глицерилмоностеарат, глицерилпальмитостеарат, гидрированное касторовое масло касторовое масло, гидрированное растительное масло, легкое минеральное масло, стеарат магния, минеральное масло, полиэтиленгликоль, стеариновая кислота, тальк, стеарат цинка, стеарилфумарат натрия. В одном аспекте изобретения, смазывающее вещество, используемое в составе по изобретению, является стеаратом магния, стеарилфумаратом натрия или комбинацией этих двух соединений. Когда фармацевтический состав является составом Fl или F2, смазывающее вещество обычно представляет собой комбинацию стеарата магния и стеарилфумарата натрия.
Фармацевтический состав по изобретению может также содержать разрыхлитель, который представляет собой вещество или смесь веществ, используемых для облегчения разделения или диспергирования состава после введения. Пригодные разрыхлители включают в себя альгиновую кислоту, кальций- карбоксиметилцеллюлозу, натрий-карбоксиметилцеллюлозу, коллоидную двуокись кремния, натрий-кроскармеллозу, кросповидон, гуаровую камедь, алюмосиликат магния, метилцеллюлозу, микрокристаллическую целлюлозу, полиакрилин калия, повидон, альгинат натрия, натрийкрахмалгликолат и крахмал. Разрыхлитель, используемый в фармацевтическом составе по изобретению, может быть “супер-разрыхлителем”, таким как натрий-кроскармеллоза, кросповидон или натрийкрахмалгликолат.
Антиоксидант может быть использован в фармацевтическом составе по изобретению для того, чтобы препятствовать окислительной деструкции активного ингредиента и/или других компонентов фармацевтического состава или минимизировать ее. Пригодный антиоксидант включает в себя токоферол или его сложный эфир, алкилгаллат (например, пропилгаллат), бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), аскорбиновую кислоту, аскорбат натрия, лимонную кислоту и метабисульфит натрия. Фармацевтические составы по настоящему изобретению могут, например, включать в себя BHA.
Фармацевтические составы по настоящему изобретению могут быть сформированы в спрессованные таблетки или капсулы. Спрессованные таблетки могут быть получены посредством гранулирования, где общий размер частиц состава увеличивается путем постоянного агрегирования более мелких частиц. Может быть использовано влажное или сухое гранулирование. Влажное гранулирование может быть выполнено, например, смачивая хорошо перемешанную смесь сухих ингредиентов (например, соли соединения I, композиции, контролирующей скорость высвобождения, разбавителя, необязательно разрыхлителя, и, необязательно антиоксиданта) с удовлетворительным растворителем (например, водой или водой вместе со спиртовым сорастворителем) для увлажнения сухой смеси, так чтобы частицы в смеси соединялись друг с другом, образуя более крупные частицы, и затем просеивая, измельчая, или иным образом, воздействуя на размер частиц. Как только получаемый жидкий гранулят образован, он может затем быть высушен и измельчен до частиц нужного размера (то есть, гранул). Гранулы смешивают со смазывающим веществом, и смазанные гранулы спрессовывают в таблетки.
Для чувствительных к влаге композиций, гранулирование может быть выполнено или посредством влажного гранулирования с неводным растворителем или путем сухого гранулирования. Сухое гранулирование может также быть перспективной альтернативой влажному гранулированию, когда композиция является термально чувствительной и подвержена деструкции при температурах, используемых во время высушивания влажных гранул. Сухое гранулирование может быть выполнено, например, путем сухого смешивания соли соединения I, композиции, контролирующей скорость высвобождения, первой части смазывающего вещества и, необязательно, других ингредиентов (например, разбавителя, или разбавителя и разрыхлителя), затем прессуя смешанную композицию в бруски или вальцуя смешанную композицию в брикет. Бруски или брикет затем могут быть откалиброваны по размеру (например, пропуская через сито или измельчающую мельницу) для получения сухих гранул, которые могут быть смешаны с остающейся частью смазывающего вещества, и смазанные гранулы спрессовывают в таблетки.
Спрессованные таблетки могут быть покрыты сахаром для маскировки любого неприятного вкуса или покрыты пленкой для защиты таблетки от разрушения под действием атмосферных условий. Оболочка не должна оказывать отрицательного воздействия на высвобождение лекарственного вещества после его перорального введения. Пригодной суспензией для пленочного покрытия является состав Opadry ІІ HP (распространяемый фирмой Colorcon, West Point, PA), который представляет собой частично гидролизованный поливиниловый спирт и полимер на основе смеси макрогол/полиэтиленгликоль (PEG), 3350. Пленки могут наносить, напыляя суспензию на таблетки и затем высушивая. Техника пленочного покрытия, пригодная для использования в настоящем изобретении, описана в Remington's Pharmaceutical Sciences, 18th edition, edited by A.R.Gennaro, 1990, Mack Publishing Co., pp.1665-1675.
Инкапсулированные фармацевтические составы по настоящему изобретению могут быть сформированы, например, гранулированием ингредиентов состава (то есть, основной соли соединения I, композиции, контролирующей скорость высвобождения, и, необязательно, одного или нескольких других ингредиентов, таких как разбавитель и/или смазывающее вещество) путем влажного или сухого гранулирования, описанного выше, наполняя капсулы (например, твердые желатиновые капсулы) соответствующим количеством гранул и герметизируя капсулы.
Технология и оборудование, пригодные для получения твердых лекарственных форм фармацевтических составов по настоящему изобретению (например, капсул и спрессованных таблеток), описаны в Remington's Pharmaceutical Sciences, 18th edition, edited by A.R.Gennaro, 1990, Chapter 89.
Настоящее изобретение включает в себя способ (альтернативно, обозначаемый здесь как "способ P1" или "P1 способ") получения спрессованного в таблетки фармацевтического состава, содержащего эффективное количество основной соли соединения I, солюбилизирующий агент, гелеобразующий агент, необязательно, водорастворимый наполнитель, разбавитель и смазывающее вещество; где способ предусматривает:
(A) перемешивание смеси основной соли соединения I, солюбилизирующего агента, гелеобразующего агента, необязательно, водорастворимого наполнителя, разбавителя и первой части смазывающего вещества;
(B) просеивание перемешанной смеси и затем дальнейшее перемешивание просеянной смеси;
(C) вальцевание просеянной и перемешанной смеси с образованием брикета, и затем калибровка полученного брикета с образованием гранул;
(D) смешивание гранул с остающейся частью смазывающего вещества; и
(E) прессование смазанных гранул стадии D для получения таблетки.
Варианты осуществления способа PI включают в себя только что описанный способ, включая одну или несколько нижеследующих (от (i) до (xiv)) особенностей:
(i-a) основная соль соединения I является солью щелочного металла соединения I;
(i-b) основная соль соединения I является натриевой солью или калиевой солью соединения I;
(i-c) основная соль соединения I является основной солью соединения A;
(i-d) основная соль соединения I является солью щелочного металла соединения A;
(i-e) основная соль соединения I является калиевой солью соединения A; или
(i-f) основная соль соединения I представляет собой форму 1 кристаллической калиевой соли соединения A;
(ii-a) основную соль соединения I (например, калиевую соль соединения A) используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 75 мас.%, исходя из содержания свободного фенола; или
(ii-b) основную соль соединения I (например, калиевую соль соединения A) используют в количестве, находящемся в интервале от, приблизительно, 25 до, приблизительно, 75 мас.% (или от, приблизительно, 40 до, приблизительно, 60 мас.%), исходя из содержания свободного фенола; или
(iii-a) солюбилизирующий агент содержит полоксамер;
(iii-b) солюбилизирующий агент содержит полоксамер 407; или
(iii-c) солюбилизирующий агент содержит полоксамер 407, измельченный до среднего размера частицы, находящегося в интервале от, приблизительно, 50 до, приблизительно, 150 микрон (или в интервале от, приблизительно, 50 до, приблизительно, 105 микрон);
(iv-a) солюбилизирующий агент содержит полоксамер (например, полоксамер 407, необязательно измельченный до среднего размера частицы, находящегося в интервале от, приблизительно, 50 до, приблизительно, 150 микрон), который используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 25 мас.%; или
(iv-b) солюбилизирующий агент содержит полоксамер (например, полоксамер 407, необязательно, измельченный до среднего размера частицы, находящегося в интервале от, приблизительно, 50 до, приблизительно, 150 микрон), который используют в количестве, находящемся в интервале от, приблизительно, 10 до, приблизительно, 20 мас.%;
(v-a) гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу; или
(v-b) гелеобразующий агент содержит HPMC K4M
(vi-a) гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу (например, HPMC K4M), которую используют в количестве, находящемся в интервале от, приблизительно, 2 до, приблизительно, 15 мас.% (или в интервале от, приблизительно, 3 до, приблизительно, 9 мас.%); или
(vi-b) гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу (например, HPMC K4M), которую используют в количестве, находящемся в интервале от, приблизительно, 3 до, приблизительно, 9 мас.%);
(vii-a) необязательный, растворимый в воде наполнитель содержит лактозу; или
(vii-b) необязательный, растворимый в воде наполнитель содержит высушенный спрей водного раствора лактозы;
(viii-a) необязательный, растворимый в воде наполнитель содержит лактозу (например, высушенный спрей водного раствора лактозы), которую используют в количестве, находящемся в интервале от, приблизительно, нуля до, приблизительно, 15 мас.%; или
(viii-b) необязательный, растворимый в воде наполнитель содержит лактозу (например, высушенный спрей водного раствора лактозы), которую используют в количестве, находящемся в интервале от, приблизительно, 3 до, приблизительно, 9 мас.%;
(ix-a) разбавитель содержит микрокристаллическую целлюлозу; или
(ix-b) разбавитель содержит AVICEL PH-102;
(x-a) разбавитель содержит микрокристаллическую целлюлозу (например, AVICEL PH-102) которую используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 50 мас.%; или
(x-b) разбавитель содержит микрокристаллическую целлюлозу (например, AVICEL PH-102) которую используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 40 мас.%;
(xi-a) смазывающее вещество содержит стеарат металла; или
(xi-b) смазывающее вещество содержит стеарат магния;
(xii-a) смазывающее вещество содержит стеарат металла (например, стеарат магния), которое используют в количестве, находящемся в интервале от, приблизительно, 0,5 до, приблизительно, 5 мас.%; или
(xii-b) смазывающее вещество содержит стеарат металла (например, стеарат магния) который используют в количестве, находящемся в интервале от, приблизительно, 0,5 до, приблизительно, 3 мас.%;
(xiii-a) способ, кроме того, предусматривает: (F) покрытие спрессованной таблетки; или
(xiii-b) способ, кроме того, включает в себя: (F) покрытие спрессованной таблетки эмульсией, образующей пленочное покрытие, (например, состав Opadry ІІ HP) для получения таблетки в оболочке, где покрытие составляет от, приблизительно, 2 до, приблизительно, 4% от массы спрессованной таблетки; и
(xiv-a) основную соль соединения I (например, калиевую соль соединения A) используют в количестве, приходящемся на одну таблетку, находящемся в интервале от, приблизительно, 100 мг до, приблизительно, 600 мг, исходя из содержания свободного фенола; или
(xiv-b) основную соль соединения I (например, калиевую соль соединения A) используют в количестве, приходящемся на одну таблетку, составляющем, приблизительно, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, или 600 мг, исходя из содержания свободного фенола.
Настоящее изобретение также включает спрессованный таблетированный фармацевтический состав, получаемый способом PI, первоначально изложенным выше или изложенным в любом из упомянутых выше вариантов осуществления PI способа.
Настоящее изобретение включает способ (альтернативно обозначаемый здесь как "способ P2" или "P2 способ") получения спрессованного таблетированного фармацевтического состава, содержащего эффективное количество калиевой соли соединения A, солюбилизирующий агент, гелеобразующий агент, водорастворимый наполнитель, первый разбавитель, второй разбавитель, первое смазывающее вещество и второе смазывающее вещество; где способ предусматривает:
(A) перемешивание смеси калиевой соли соединения A, солюбилизирующего агента, гелеобразующего агента, водорастворимого наполнителя, первого разбавителя, второго разбавителя, первой части первого смазывающего вещества и второго смазывающего вещества;
(B) просеивание перемешиваемой смеси и затем дальнейшее перемешивание просеянной смеси;
(C) вальцевание просеянной и перемешанной смеси для образования брикета и затем калибрование полученного брикета с образованием гранул;
(D) перемешивание гранул с остающейся частью первого смазывающего вещества; и
(E) прессование промазанных гранул стадии D для получения таблетки.
Один вариант осуществления P2 способа представляет собой только что описанный P2 способ, где солюбилизирующий агент содержит полоксамер; гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу; водорастворимый наполнитель содержит лактозу; первый разбавитель представляет собой микрокристаллическую целлюлозу; второй разбавитель является фосфатом кальция; первое смазывающее вещество представляет собой стеарат металла; а второе смазывающее вещество является стеарилфумаратом металла.
Другой вариант осуществления P2 способа представляет собой P2 способ, первоначально описанный, где калиевую соль соединения A используют в количестве, находящемся в интервале от, приблизительно, 40% до, приблизительно, 60 мас.% исходя из содержания свободного фенола; солюбилизирующий агент представляет собой полоксамер, который используют в количестве, находящемся в интервале от, приблизительно, 10 до, приблизительно, 20 мас.%; гелеобразующий агент представляет собой высоковязкую гидроксипропилметилцеллюлозу, которую используют в количестве, находящемся в интервале от, приблизительно, 3 до, приблизительно, 9 мас.%; водорастворимый наполнитель является лактозой, которую используют в количестве, находящемся в интервале от, приблизительно, 3 до, приблизительно, 9 мас.%; первый разбавитель является микрокристаллической целлюлозой, которую используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 25 мас.%; второй разбавитель является фосфатом кальция, который используют в количестве, находящемся в интервале от, приблизительно, 5 до, приблизительно, 25 мас.%; первое смазывающее вещество является стеаратом металла, который используют в количестве, находящемся в интервале от, приблизительно, 1 до, приблизительно, 3 мас.%; и второе смазывающее вещество является стеарилфумаратом металла, который используют в количестве, находящемся в интервале от, приблизительно, 1 до, приблизительно, 3 мас.%.
Еще один вариант осуществления P2 способа представляет собой P2 способ, описанный в любом из двух предшествующих вариантов его осуществления, в которых полоксамер представляет собой полоксамер 407, измельченный до среднего размера частицы, находящегося в интервале от, приблизительно, 50 до 150 микрон; высоковязкая гидроксипропилметилцеллюлоза является HPMC K4M; лактоза представляет собой высушенный спрей водного раствора лактозы; микрокристаллическая целлюлоза представляет собой AVICEL PH 102; фосфат кальция является двухосновным фосфатом кальция; стеарат металла представляет собой стеарат магния; а стеарилфумарат металла представляет собой стеарилфумарат натрия.
Еще один вариант осуществления P2 способа представляет собой P2 способ, первоначально описанный или описанный в любом из предшествующих вариантах его осуществления, где калиевая соль соединения A представляет собой форму 1 кристаллической калиевой соли соединения A.
Еще один вариант осуществления P2 способа представляет собой P2 способ, первоначально описанный или описанный в любом из предшествующих вариантах его осуществления, где способ, кроме того, включает в себя: (F) покрытие спрессованной таблетки. Согласно одному аспекту этого варианта осуществления спрессованную таблетку покрывают образующей пленочное покрытие эмульсией, (например, составом Opadry ІІ HP) для получения таблетки в оболочке, где покрытие составляет от, приблизительно, 2 до, приблизительно, 4% массы спрессованной таблетки.
Еще один вариант осуществления P2 способа представляет собой P2 способ, первоначально описанный или описанный в любом из предшествующих вариантов его осуществления, где калиевую соль соединения A, используют в количестве, приходящемся на одну таблетку, которое находится в интервале от, приблизительно, 100 мг до, приблизительно, 600 мг, исходя из содержания свободного фенола.
Согласно одному аспекту этого варианта осуществления калиевую соль соединения A используют в количестве, приходящемся на одну таблетку, составляющем, приблизительно, 100 мг, приблизительно, 200 мг, приблизительно, 300 мг, приблизительно, 400 мг, приблизительно, 500 мг или приблизительно, 600 мг.
Настоящее изобретение, кроме того, включает спрессованный таблетированный фармацевтический состав, получаемый способом P2, первоначально изложенным выше или изложенным в любом из предшествующих вариантов осуществления P2 способа.
Фармацевтические составы по настоящему изобретению применимы при ингибировании интегразы ВИЧ, лечении или профилактике ВИЧ инфекции и лечении, профилактике или замедлении возникновения последующих патологических состояний, таких как СПИД. Лечение СПИДа, профилактика СПИДа, замедление возникновения СПИДа, лечение ВИЧ-инфекции или профилактика ВИЧ-инфекции определяют как включающие в себя, кроме прочих, лечение или профилактику широкого разнообразия состояний при ВИЧ-инфицировании: СПИД, СПИД-ассоциированный комплекс (ARC), как симптоматичный, так и бессимптомный, и действительное или возможное воздействие ВИЧ. Например, композиции по данному изобретению применимы при лечении или профилактике инфицирования ВИЧ после предполагаемого бывшего воздействия ВИЧ таким путем, как переливание крови, замена жидкой среды организма, укусы, случайный укол иглой, воздействие крови больного во время оперативного вмешательства.
Настоящее изобретение включает способ ингибирования ВИЧ-интегразы у субъекта, нуждающегося в этом, который предусматривает введение субъекту фармацевтического состава по настоящему изобретению, первоначально определенного выше. Изобретение также включает способ лечения или профилактики ВИЧ-инфекции или лечения, профилактики или замедления возникновения СПИДа у субъекта, нуждающегося в этом, который предусматривает введение субъекту фармацевтического состава по изобретению, первоначально определенного выше. В этих способах фармацевтический состав по настоящему изобретению необязательно может быть использован в комбинации с одним или несколькими анти-ВИЧ-агентами, выбранными из антивирусных агентов, направленных против ВИЧ, антиинфекционных агентов и иммуномодуляторов. Варианты осуществления этих способов включают в себя только что описанные способы, в которых фармацевтический состав по изобретению является составом, предложенным в любом из предшествующих вариантах их осуществления, описанных выше (включая, среди прочих, Fl состав и спрессованные таблетированные составы, получаемые способами P1 и P2).
Термин "субъект" (используемый здесь как взаимозаменяемый с термином "пациент") обозначает животное, предпочтительно, млекопитающее, наиболее предпочтительно, человека, который является объектом лечения, наблюдения или эксперимента.
Когда фармацевтический состав по настоящему изобретению используют или вводят в комбинации с еще одним агентом (например, когда Fl вводят в комбинации с анти-ВИЧ-агентом), состав и агент могут быть введены по-отдельности или совместно. И когда их вводят по-отдельности, состав и агент могут быть введены одновременно или в разное время (например, попеременно).
Настоящее изобретение также включает в себя фармацевтический состав для перорального введения в виде твердой лекарственной формы, которая содержит основную соль соединения Формулы 1 и композицию, контролирующую скорость высвобождения, первоначально определенную и описанную в части, озаглавленной «Раскрытие изобретения»; (i) при использовании для, (ii) для использования в качестве лекарственного средства для, или (iii) для использования при получении лекарственного средства для: (a) ингибирования интегразы ВИЧ, (b) лечения или профилактики инфицирования ВИЧ, или (c) лечения, профилактики или замедления возникновения СПИДа. Варианты осуществления таких применений включают в себя только что описанные варианты использования, в которых фармацевтический состав по изобретению, определенный первоначально, заменяют вышеописанными вариантами его осуществления (которые включают в себя, среди прочего, Fl состав и спрессованные таблетированные составы, получаемые по P1 и P2 способам). При таких вариантах использования, фармацевтические составы по настоящему изобретению могут, необязательно, использовать в комбинации с одним или несколькими анти-ВИЧ-агентами, выбранными из антивирусных агентов, направленных против ВИЧ, антиинфекционными агентами, иммуномодуляторами.
Термин "анти-ВИЧ-агент" обозначает агента (не являющегося соединением Формулы I), который является эффективным при одном или нескольких нижеследующих вариантах использования: ингибирования интегразы или другого фермента, необходимого для репликации или инфицирования ВИЧ, профилактики ВИЧ-инфекции, лечения ВИЧ-инфекции, замедления возникновения СПИДа, профилактики СПИДа или лечения СПИДа.
Антивирусные агенты, направленные против ВИЧ, применимые для использования в комбинации с фармацевтическим составом по изобретению, включают в себя, например, ингибиторы протеазы ВИЧ (например, индинавир, лопинавир, необязательно, с ритонавиром, саквинавиром, или нелфинавиром), ингибиторы нуклеозидной обратной транскриптазы ВИЧ (например, абакавир, ламивудин (3TC), зидовудин (AZT) или тенофовир), и ингибиторы ненуклиозидной обратной транскриптазы ВИЧ (например, ифавиренц или невирапин). Эти агенты могут быть использованы в свободной форме или в форме фармацевтическии приемлемой соли. Эти агенты также могут быть использованы по-отдельности, но обычно их включают в пригодные фармацевтические композиции.
Фармацевтические составы по изобретению могут быть введены в твердой форме, пригодной для перорального введения. Композиции могут, например, быть введены в виде капсул или таблеток. Композиции могут быть введены таким образом, чтобы получить активный ингредиент в дозировке, находящейся в интервале от, приблизительно, 0,001 до, приблизительно, 1000 мг/кг массы тела млекопитающего (например, человека) в день в однократной дозе или в разделенных дозах. Один предпочтительный интервал дозирования составляет от, приблизительно, 0,01 до, приблизительно, 500 мг/кг массы тела в день в однократной дозе или в разделенных дозах. Другой предпочтительный интервал дозирования составляет от, приблизительно, 0,1 до, приблизительно, 100 мг/кг массы тела в день в однократной дозе или в разделенных дозах.
Фармацевтические составы по изобретению могут, соответственно, быть получены в виде таблеток или капсул для перорального введения, где каждая таблетка или капсула содержит от, приблизительно, 1 до, приблизительно, 1000 миллиграмм активного ингредиента, в частности, 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 700, 800, 900 и 1000 миллиграмм активного ингредиента для симптоматичного регулирования дозирования лекарственного средства пациенту, подвергаемому лечению. В частности, фармацевтические составы по настоящему изобретению, содержащие калиевую соль соединения A (например, форму 1) для взрослых пациентов предпочтительно, дозируют в капсулах или таблетках, доза в которых составляет от 100 мг до 600 мг соединения A, дважды в день. Определенный уровень и частота дозирования для любого отдельного больного будет зависеть от разных факторов, включая активность конкретного используемого соединения лекарственного вещества, метаболическую стабильность и продолжительность действия данного соединения, возраст, массу тела, общее состояние здоровья, пол, диету больного, способ и время введения, скорость экскреции, комбинацию лекарственных средств, тяжесть конкретного состояния, и организм-хозяин, подвергаемый терапии. Подходящий уровень дозы отдельного лекарственного средства, пригодный для конкретного больного, может быть установлен без излишнего экспериментирования специалистом среднего звена в данной области.
Используемый здесь термин "алкил" обозначает любую алкильную группу с линейной или разветвленной цепью, с числом углеродных атомов, заключенном в определенном интервале. Так, например, "C1-6 алкил" (или "C1-С6 алкил") обозначает любой из гексильных и пентильных алкилизомеров, так же, как н-, изо-, втор- и т-бутил, н- и изопропил, этил и метил. В то время как в другом примере, "C1-4 алкил " обозначает н-, изо-, втор- и т-бутил, н- и изопропил, этил и метил.
Термин "алкилен" обозначает любую алкиленовую группу с линейной или разветвленной цепью (или, альтернативно, "алкандиил"), с числом углеродных атомов, заключенном в определенном интервале. Таким образом, например, «-C1-6 алкилен-» обозначает любой из линейных или разветвленных алкиленов от C1 до C6. Класс алкиленов, представляющий собой -(CH2)l-6-, имеет особый интерес для изобретения; и особый интерес представляет подкласс, который содержит -(CH2)l-4-, -(CH2)l-3-,
-(CH2)l-2- и -CH2-. Кроме того, интерес представляет алкилен - CH(СН3)-.
Термин "галоген" (или "гало") обозначает фтор, хлор, бром и иод (альтернативно, обозначают как фторо, хлоро, бромо и иодо).
Термин "галогеналкил" обозначает алкильную группу, определенную выше, в которой один или несколько атомов водорода замещены галогеном (то есть, F, Cl, Br и/или I). Таким образом, например, "C1-6 галогеналкил" (или "C1-С6 галогеналкил") обозначает линейную или разветвленную C1-С6 алкильную группу, определенную выше, с одним или несколькими галогеновыми заместителями. Термин "фторалкил" имеет аналогичный смысл, кроме того, что галогеновые заместители ограничены атомом фтора. Пригодные фторалкилы включает в себя ряд (CH2)0-4CF3 (то есть, трифторметил, 2,2,2-трифторэтил, 3,3,3-трифтор-н-пропил, и так далее).
Термин "арил" обозначает (i) фенил или (ii) 9- или 10-членную бициклическую, конденсированную карбоциклическую циклическую систему, в которой, по крайней мере, одно кольцо является ароматическим. Арил обычно является фенилом или нафтилом, и чаще - фенилом.
Термин "HetA" обозначает, необязательно, замещенный 5- или 6-членный гетероароматический цикл, содержащий от 1 до 4 гетероатомов, независимо выбираемых из N, О и S. В одном варианте осуществления HetA является необязательно замещенным гетероароматическим циклом, выбираемым из группы, состоящей из пиридинила, пирролила, пиразинила, пиримидинила, пиридазинила, триазинила, фуранила, тиенила, имидазолила, пиразолила, триазолила, тетразолила, оксазолила, изооксазолила, тиазолила, изотиазолила и оксадиазолила; в которых имеет место необязательное замещение 1 или 2 заместителями, каждый из которых независимо представляет собой -C1-4 алкил, -C1-4 галогеналкил, -O-C1-4 алкил, -O-C1-4 галогеналкил или -CO2-C1-4 алкил. Понятно, что HetA может быть присоединен к оставшейся части соединения формулы I у любого атома цикла (то есть, любого углеродного атома или любого гетероатома), при условии, что в результате получают устойчивое соединение.
Термин "HetB" обозначает необязательно замещенное 5-7-членное насыщенное гетероциклическое кольцо, содержащее от 1 до 4 гетероатомов, независимо выбираемых из N, О и S. В одном варианте осуществления, HetB представляет собой необязательно замещенное насыщенное гетероциклическое кольцо, выбираемое из группы, состоящей из пирролидинила, имидазолидинила, пиперидинила, пиперазинила, морфолинила, тиоморфолинила, тиазинанила и тетрагидропиранила, в которых имеется необязательное замещение 1 или 2 заместителями, каждый из которых независимо представляет собой -C1-4 алкил, -C1-4 галогеналкил,, -C(O)CF3, -С(О)СН3 или -CH2CH2OH. Понятно, что HetA может быть присоединен к оставшейся части соединения Формулы I у любого атома кольца (то есть, любого углеродного атома или любого гетероатома) при условии, что в результате получают устойчивое соединение. В другом варианте осуществления, HetB выбирают из группы состоящей из

где (*) указывает точку присоединения к остальной части молекулы.
В соединении формулы I, RC и RD вместе с атомом азота, к которому они присоединены, могут образовывать насыщенное 5- или 6-членное гетероциклическое кольцо, необязательно, содержащее гетероароматическое кольцо, в дополнение к атому азота, присоединенному к RC и RD, выбираемый из N, O, и S, где S необязательно, окислена до S(O) или S(O)2, и где насыщенное гетероциклическое кольцо необязательно, замещено 1 или 2 С1-6 алкильными группами. В одном варианте осуществления насыщенное гетероциклическое кольцо, образованное
RC и RD вместе с атомом азота, к которому они присоединены, выбирают из группы состоящей из 4-морфолинила, 4-тиоморфолинила, 1-пиперидинила, 1-пиперазинила, необязательно замещенного C1-4 алкилом (например, метилом), и 1-пирролидинилом.
До тех пор пока намеренно не утверждают противоположное, все интервалы, упоминаемые здесь, являются включительными. Например, при описании гетероциклического кольца, как содержащего от "1 до 4 гетероатомов", подразумевают, что кольцо может содержать 1, 2, 3 или 4 гетероатома. Еще один пример, формулировка «фармацевтический состав, содержащий основную соль соединения I в интервале от приблизительно, 25 до, приблизительно, 75 мас.%» означает, что композиция может содержать, приблизительно, 25 мас.% соединения I, приблизительно, 75 мас.% соединения I, или любое его количество, заключенное между этими величинами.
Когда любая переменная (например, RA и RB) встречается более чем один раз в Формуле I или в любой другой формуле, отображающей и описывающей соединение, чья соль может быть использована в фармацевтических составах по изобретению, ее определение при каждом появлении является независимым от ее определения при каждом другом появлении. Кроме того, комбинации заместителей и/или переменных являются допустимыми в той степени, пока такие комбинации приводят к стабильным соединениям.
Термин "стабильное" соединение определяет соединение, которое может быть получено и выделено, и чья структура и характеристики сохраняются или могут поддерживаться, по существу, неизменными в течение достаточного периода времени, позволяющего использовать соединение, в описанных здесь целях, (например, использовать в виде соли в фармацевтическом составе по изобретению).
В результате выбора заместителей и структур заместителей, некоторые из соединений Формулы I, чьи соли могут быть использованы в настоящем изобретении, могут иметь асимметричные центры и могут существовать в виде смеси стереоизомеров, или как отдельные диастереомеры или энантиомеры. Соли всех изомерных форм этих соединений, или отдельно или в смесях могут быть использованы в фармацевтических композициях по настоящему изобретению.
Соединения Формулы I могут также существовать в виде таутомеров, вследствие кето-енольной таутомерии. Соли всех таутомеров соединений гидроксипиримидинона Формулы I, и по-отдельности, и в смесях, могут быть использованы в фармацевтических составах по настоящему изобретению. Аббревиатура, используемая здесь, включает в себя нижеследующее:
ACN = ацетонитрил
AIDS = синдром приобретенного иммунодефицита
APCI = хемоионизация при атмосферном давлении
ARC = СПИД ассоциированный комплекс
Cbz = бензилоксикарбонил
DIEA = диизопропилэтиламин
DMADC = диметилацетилендикарбоксилат
DMF = N,N-диметилформамид
DMSO = диметилсульфоксид
DSC = дифференциальная сканирующая калориметрия
EDTA = этилендиаминтетрауксусная кислота
EtOH = этанол
Eq. = эквивалент(ы)
GI = желудочно-кишечный
HIV = вирус иммунодефицита человека
HPLC = высокоэффективная жидкостная хроматография
HPMC = гидроксипропилметилцеллюлоза
IPA = изопропиловый спирт
KF = титрование водных растворов по Карлу Фишеру
LC = жидкостная хроматография (ЖХ)
LCAP = процент по площади пика на хроматограмме в методе ЖХ
LCWP = ЖХ, массовый процент
Me = метил
MeOH = метанол
MRM = множественный мониторинг реакции
MS = масс-спектрометрия
MSA = метансульфоновая кислота
MTBE = трет-бутилметиловый эфир
MW = молекулярную масса
NMM = N-метилморфолин
ЯМР = ядерно-магнитный резонанс
PK = фармакокинетика
SDS = додецилсульфат натрия
TG = термогравиметрический
THF = тетрагидрофуран
XRPD = порошковая диффракция рентгеновских лучей
Нижеследующие примеры служат только для иллюстрации изобретения и его применения. Примеры не следует рассматривать как ограничивающие объем или сущность изобретения.
ПРИМЕР 1
Получение соединения A и его кристаллической калиевой соли
Стадия 1:
Образование амина по методу Strecker
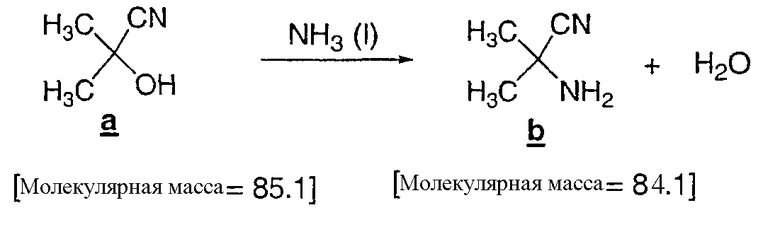
Ацетонцианогидрин (11,5 кг, 12,3 л) загружали в автоклав, емкостью 5 галлон, и резервуар помещали в атмосферу азота с давлением в 5 футов/дюйм2. Автоклав охлаждали до 10°C и в резервуар подавали газообразный аммиак (~3,44 кг), сжатый до 30 футов/дюйм2, пока реакция не проходила до полного превращения, что определяли по ГХ (GC) (менее чем 0,5% a ). Полученную суспензию перемещали в полиэтиленовую емкость, и автоклав промывали MTBE (приблизительно, 17 л). Реакционную смесь и промывочную жидкость затем загружали в 100 л экстрактор, с последующим добавлением MTBE (15 л); смесь перемешивали и слои осторожно разделяли. Водный слой подвергали обратному экстрагированию MTBE (5 л) и слои осторожно разделяли. Органические слои объединяли и загружали в 100 л колбу, оборудованную концентратором загружаемой порции, совмещенным с фильтром, и порцию концентрировали (15-20°C, низкий вакуум) до, приблизительно, 20 л, чтобы удалить любой избыток аммиака. Аминонитрил получали с 97% выходом (11,1 кг), согласно ЯМР анализу, в виде раствора в MTBE.
Стадия 2:
Добавление бензилоксикарбонильной (CBz) защитной группы
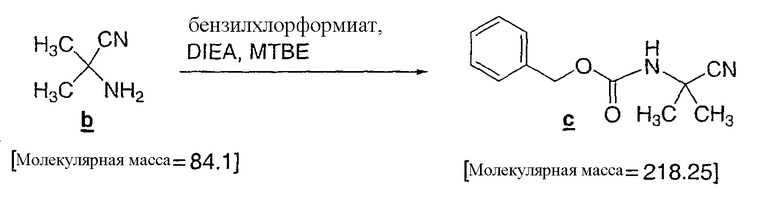
В визуально чистую 100 л колбу, снабженную 5 л капельной воронкой, термопарой и входным отверстием для азота загружали 59 мас.% раствора цианоамина b в MTBE (4,44 кг на опыт). Раствор затем разбавляли MTBE (62,5 л), чтобы довести концентрацию до приблизительно, 15 мл/г. Затем на протяжении 15 минут загружали бензилхлорформиат (1,20 экв, 10,42 кг, 61,10 моль) через капельную воронку с такой скоростью, чтобы поддерживать температуру порции ниже 35°C. Затем к желтой суспензии на протяжении 1,5 часов добавляли DIEA (1,3 экв, 8,88 кг, 68,70 моль), поддерживая температуру порции ниже 35°C. При добавлении DIEA растворимость взвеси несколько возросла, но когда перемешивание остановили, наблюдали присутствие двух фаз. Реакционную смесь выдерживали 16 часов при 20-25°C, после чего в порцию загружали DI (деионизированную) воду (20 л, 4,5 мл/г). Затем порцию перемещали в 100 л экстрактор и фазы разделяли. Органический слой затем промывали водой, 3 раза по 10 л и затем 15 л соляного раствора. Органический слой затем перемещали через встроенный в систему 10 мкм фильтр в 100 л круглодонную колбу и затем «растворитель» переключили на «смесь 90:10 гептан/MTBE». Кристаллизация происходила во время смены растворителя; и полученный белый кристаллический продукт фильтровали и 3 раза промывали 5 л смеси 90:10 гептан/MTBE. Всего получали 10,1 кг продукта (с выходом 88%) и более 99 A% (процент по площади пика на хроматограмме) в методе ВЭЖХ. Всего для трех порций получили 26,7 кг продукта со средним выходом отделенного продукта, составляющим 86%.
Стадия 3:
Образование амидоксима

аминонитрил (c)
Раствор аминонитрила (15 г) в IPA (40 мл) нагревали с перемешиванием до 60°C, и при этой температуре на протяжении 20 минут добавляли NH2OH в воде (5,05 мл). Прозрачную смесь затем выдерживали при 60°C в течение 3 часов, где после 2 часов нахождения при данной температуре продукт начинал выкристаллизовываться из раствора. Взвесь затем охлаждали до 0°-5°C и на протяжении 20 минут по каплям добавляли н-гептан (40 мл). После перемешивания в течение 2 часов при 0°-5°C взвесь фильтровали и осадок на фильтре промывали 20% IPA в растворе гептана (60 мл) и затем сушили в вакууме в токе азота при комнатной температуре, получая чистый амидоксим с выходом 88%.
Стадия 4:
Образование гидроксипиримидинона

К взвеси амидоксима (2,90 кг) в метаноле (12 л) на протяжение 20 минут добавляли диметилацетилендикарбоксилат (1,77 кг). В результате происходило незначительное выделение тепла, такое, что температура взвеси в течение 15-20 минут повышалась от 20°C до 30°C. После 1,5 часа ВЭЖХ показала, что превращение до интермедиатных цис/транс продуктов присоединения превышает 95%. Затем при пониженном давлении «растворитель» переключали на «ксилолы» (максимальная температура составляла 50°C) и добавляли 2 объема растворителя [2 раза по 7,5 л]; затем конечный объем уменьшали до 7,5 л. Реакционную смесь нагревали до 90°C и выдерживали при этой температуре в течение 2 часов, при этом вымывая остающийся MeOH струей азота. Затем температуру повышали до 125°C с шагом 10°C на протяжении 3,5 часов и поддерживали эту температуру в течение 2 часов. Затем температуру окончательно повысили до 135°C в течение 5 часов. Затем реакционную смесь охлаждали до 60°C и добавляли MeOH (2,5 л). После 30 минут медленно добавляли MTBE (9 л), чтобы создать слой затравочных кристаллов. Порцию затем в течение 14 часов охлаждали до 0°C и затем дополнительно охлаждали до -5°C и выдерживали 1 час перед фильтрованием. Твердые кристаллы подвергали вытеснительному промыванию 10% смесью MeOH/MTBE (предварительно охлажденной до 0°C; сначала 6 л, затем 4 л;) и сушили на фильтре в токе азота для получения 2,17 кг (откорректированный выход 51,7%; 99,5 мас.%).
Способ ВЭЖХ: колонка Zorbax C-8, 4,6 мм × 250 мм; смесь от 40% ACN/60% 0,1% H3PO4 до 90% ACN /10% 0,1% H3PO4 пропускают на протяжении 12 минут, удерживают 3 минуты, затем в течение 1 минуты возвращаются обратно к 40% ACN. Времена удерживания: амидоксим d - 2,4 минуты, DMAD- 6,7 минут, интермедиатные продукты присоединения - 8,4 и 8,6 минут (пик у 8,4 минуты соответствует более быстрой циклизации), продукт e - 5,26 минут, ксилолы - несколько пиков в окрестности 10,4 -10,7 минут.
Стадия 5:
N-Метилирование

(DMSO)
5 мас.% в воде
К раствору пиримидиндиола e (2 кг) в (DMSO) (16 л) добавляли раствор Mg(OMe)2 в MeOH (11,95 кг), после чего избыточный MeOH выпаривали в вакууме (30 мм Hg) при 40°C в течение 30 минут. Смесь затем охлаждали до 20°C, после чего добавляли метилиодид (MeI) (1,38 л), и смесь перемешивали при 20-25°C в течение 2 часов и затем в течение 5 часов - при 60°C в закрытой колбе под давлением. Данные ВЭЖХ показали, что реакция прошла полностью. Смесь затем охлаждали до 20°C, после чего добавляли MeOH (14 л), с последующим медленным добавлением 2 M HCl (20 л) (выделение тепла) на протяжении 60 минут. Затем добавляли бисульфит натрия (5 мас.%, 2 л), чтобы погасить избыточный I2, при этом раствор приобрел белую окраску. Затем на протяжении 40 минут добавляли воду (40 л) и взвесь перемешивали в течение 40 минут на ледяной бане и затем фильтровали. Фильтровальный осадок промывали сначала водой (20 л) и затем смесью 9/1 MTBE/MeOH (30 л) для удаления O-метилированного побочного продукта. После промывания ВЭЖХ показала менее, чем 0,5 A% O-метилированного продукта. Твердое вещество сушили на протяжении ночи при комнатной температуре в вакууме в токе N2 для получения 1,49 кг N-метилпиримидона (с выходом 70%, откорректированным на чистоту исходных веществ и продукта).
Стадия 6:
Реакция сочетания амина
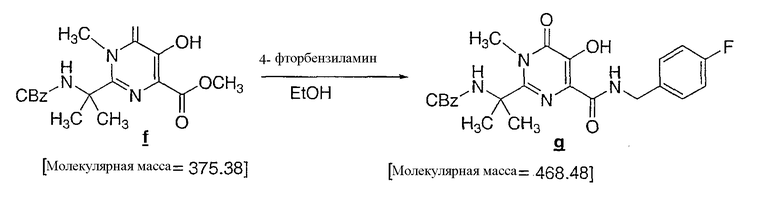
К взвеси N-метилированного пиримидинона f (1,4 кг) в EtOH (14 л) при 4°C медленно добавляли 4-фторбензиламин (1,05 кг) на протяжении 15 минут, где во время добавления первого 1 мольного эквивалента амина наблюдали выделение тепла с повышением температуры до 9°C. Взвесь становилась очень густой, и было необходимо энергичное перемешивание. Реакционную смесь нагревали до 72°C на протяжении 2 часов и поддерживали при этой температуре в течение 1 час 45 минут. При 45°C раствор сделался чрезвычайно вязким, наблюдалось незначительное экзотермичное нагревание до 50°C, после чего взвесь медленно освобождалась от чрезмерной вязкости и после 1 часа нахождения при 72°C делалась гомогенной. Исследование образца методом ВЭЖХ (ВЭЖХ способ был аналогичен способу, используемому на стадии 4, описанному выше) в конце реакции показало менее 0,5 A% N-метилированного пиримидинона. Затем реакционную смесь охлаждали до 60°C и на протяжении 30 минут добавляли уксусную кислоту (0,55 л), с последующим добавлением воды (6,7 л) на протяжении 30 мин, и добавлением затравочного кристалла (3,0 г) для инициирования кристаллизации. После 30 мин нахождения при 60°C на протяжении 30 минут добавляли еще воды (7,3 л) и реакционную смесь оставляли охлаждаться при температуре окружающей среды на протяжении ночи. После 13 часов температура была 20°C; при этой температуре реакционную смесь фильтровали, и взвесь промывали 50% смесью вода/EtOH (2 раза по 4 л). Твердые кристаллы сушили на фильтре в вакууме и в токе N2 до постоянной массы и получения белого твердого продукта (1,59 кг; с откорректированным выходом 90%; 99% по LCWP и 99,7% по LCAP; определение осуществляли ВЭЖХ методом, аналогичным методу, используемому на стадии 4, описанному выше).
Стадия 7: Гидрирование бензилоксикарбониламида

(смесь 4:1 МеОН:Н2О)
Сосуд для гидрирования из нержавеющей стали предварительно обрабатывали MeOH, Pd/C катализатором и MSA в условиях протекания реакции, описанных ниже. Затем сbzамид g (10 г) суспендировали в MeOH (80 мл) в предварительно обработанном сосуде. При комнатной температуре к взвеси добавляли MSA (1,45 мл), всю порцию за один раз. В сосуд для гидрирования также добавляли 5% Pd/C (0,15 г, 50% увлажнение). Водород загружали в сосуд тремя последовательными циклами вакуум/водородной продувки, после чего смесь в течение 3-4 часов гидрировали при избыточном давлении 40 футов / дюйм2 и при 50°C. Следуя схеме гидрирования, воду (8 мл) добавляли к реакционной смеси, смесь перемешивали и катализатор отфильтровывали и промывали смесью 4:1 MeOH/вода (20 мл). pH объединенных фильтратов устанавливали у pH 7-8,0 путем медленного добавления 1н. NaOH (22,4 мл), который осаждал твердое вещество. Взвесь перемешивали при 0-5°C в течение 4 часов. Твердое вещество фильтровали, промывали водой (30 мл), собирали и сушили в вакууме при 50°C. Продукт амина (в форме гидрата) получали в виде белого кристаллического твердого вещества (7,7 г), с выходом 96% (откорректированным по KF), 89% по LCWP, 99,8% по LCAP, KF=11 мас.%.
ВЭЖХ Способ A (исследование продукта): колонка: 25 см × 4,6 мм Zorbax RX-C8; подвижная фаза: A=0,1% H3PO4, В=CH3CN, 0 минут (80% A/20% B), 20 минут (20% A/80% B), 25 минут (20% A/80%B); расход: 1,0 мл/минута; длина волны: 210 нм; температура колонки: 40°C; времена удерживания: побочный продукт дезфторамина - 5,5 мин, продукт амина - 5,85 минут, толуол -16,5 минут, Cbzамид - 16,82 минуты.
ВЭЖХ Способ В (чистота продукта): колонка: 25 см × 4,6 мм YMC-основной; подвижная фаза: A=25 ммоль KH2PO4, отрегулированного до pH=6,1, В=CH3CN, 0 минут (90% A/ 10% B), 30 минут (30% A/70% B), 35 минут (30% A/70% B); расход: 1 мл/минута; длина волны: 210 нм; температура колонки: 3°C; времена удерживания: дезфторамин - 9,1 минут, амин - 10,1 минут, толуол - 24,2 минуты, Cbzамид - 25,7 минут.
Стадия 8: Оксадиазольное сочетание
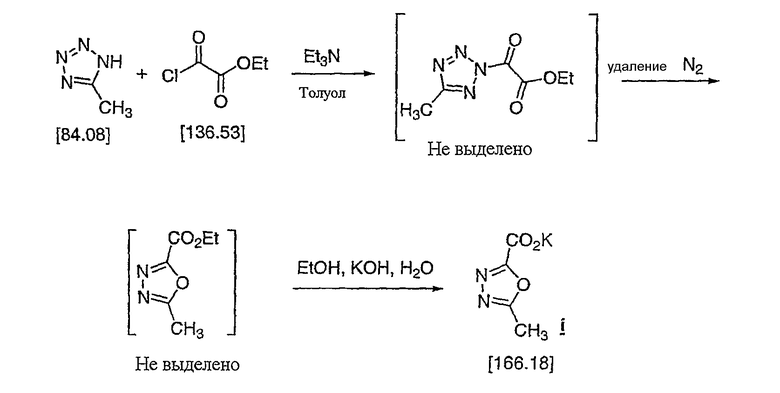
Часть A: Получение калиевой соли оксадиазола
валент
(96 мас.%)
(2,4 кг)
Этилоксалилхлорид (4,01 кг) медленно добавляли к смеси 5-метилтетразола (2,50 кг), триэтиламина (3,03 кг) в толуоле (32 л) при 0°C с такой скоростью, что температура оставалась ниже 5°C.
Полученную взвесь перемешивали в течение 1 часа при 0-5°C, затем соль триэтиламин/HCl отфильтровывали. Твердое вещество промывали 27 л холодного толуола (5°C). Объединенные фильтраты сохраняли при 0°C и медленно на протяжении 40-50 минут добавляли в горячий раствор толуола (50°C, 15 л) (выделение газообразного N2), затем раствор выдерживали 1 час при 60-65°C. После охлаждения до 20°C раствор толуола промывали 5 л 10% соляного раствора, затем «растворитель» переключали на «этанол» (уменьшали до 8 л, затем добавляли 17 л EtOH, затем концентрировали до 8 л, затем добавляли 33 литра EtOH для доведения конечного объема до 41 л). Раствор этанола охлаждали до 10°C и на протяжении 30 минут добавляли водный KOH (8,0 л), и затем полученную густую взвесь перемешивали 40 минут при комнатной температуре, при этом выкристаллизовывалась калиевая соль оксадиазола. Твердое вещество отфильтровывали, промывали 11 л EtOH и окончательно - 15 л MTBE. Твердое вещество всю ночь сушили в вакууме при 20°C в токе азота, чтобы получить выход 4,48 кг калиевой соли i (90,8%).
Часть В: Оксадиазольное сочетание

(96,1 мас.%)
(99 мас.%)
В 500 мл круглодонную колбу загружали калиевую соль оксадиазола i (33,8 г), затем загружали ACN (280 мл), ДМФА (0,33 мл), сопровождая энергичным перемешиванием. Затем полученную взвесь охлаждали до 0-5°C и на протяжении 20 минут добавляли оксалилхлорид (23,7 г), чтобы внутренняя температура оставалась менее 5°C. Затем полученную взвесь, содержащую ацилхлорид, выдерживали 1 час.
В 2 л круглодонную колбу добавляли свободный амин h (30 г) с последующим добавлением ТГФ (821 мл). Полученную взвесь охлаждали до 0-5°C, после чего добавляли NMM (21,56 г), и взвесь, полученную таким образом, перемешивали 10 минут при низкой температуре. Полученную ранее взвесь, содержащую ацилхлорид, на протяжении 20 минут медленно добавляли к взвеси свободного амина, так чтобы температура не превышала 5°C. Взвесь затем выдерживали в течение 1,5 часа при 0-5°C. В это время ВЭЖХ показала отсутствие амина h (менее 0,5% LCAP, 100% превращение). Затем реакционную смесь гасили NH4OH (30% в воде) (69 мл), который добавляли на протяжении 3 минут. Затем полученную желтую взвесь дополнительно перемешивали еще час при температурах ниже 10°C. Затем желтую взвесь подкисляли с помощью HC1 (2н.) (500 мл) до pH 2-3. К полученному винно-красному раствору, добавляли IPA (920 мл). Затем органические растворители с низкими температурами кипения выпаривали при пониженном давлении (40 торр) и комнатной температуре до конечного объема раствора, составляющего 1100 мл; при этом объеме начинает осаждаться кристаллическое Соединение A. Затем к этой новой взвеси на протяжении 10 минут добавляли воду (400 мл), и взвесь выдерживали при комнатной температуре на протяжении ночи. Выдержанную взвесь фильтровали и полученное твердое вещество промывали водой (170 мл), с последующим промыванием струей холодного MeOH (300 мл, предварительно охлажденные на ледяной бане), и окончательным промыванием струей воды (700 мл). Твердое вещество, полученное таким образом, сушили в вакууме и в токе азота на протяжении ночи, чтобы получить 35,5 г соединение A (с выходом 91%).
Стадия 9: Образование кристаллической калиевой соли соединения A
Ацетонитрил (50 мл) и безводное соединение A (5,8 г, 97,4 мас.%) загружали при комнатной температуре в 125 мл круглодонную колбу с двойными стенками, снабженную механической мешалкой и оборудованную впускным отверстием для азота (то есть, кристаллизацию проводили в атмосфере азота). Полученную взвесь перемешивали при 45°C, пока твердое вещество полностью не перешло в раствор. Затем в качестве затравочного кристалла в раствор загружали форму 1 кристаллической калиевой соли соединения A (0,184 г, 3 мас.% - к теоретическому количеству калиевой соли). Затем добавляли водный 30 мас.%/об.(w/v) раствор KOH (0,98 экв., 2,33 мл, 0,0125 моль), с нижеследующим профилем загрузки, при сохранении температуры загрузочной порции у 45°C,:
0,466 мл на протяжении 5 часов, 0,0932 мл/час (20% моль)
1,864 мл на протяжении 7 часов, 0,2663 мл/час (80% моль)
Полученную взвесь охлаждали до 20°C и выдерживали при 20°C, до тех пор пока измеренная концентрация соединения A в маточном растворе не оказалась меньше 4 г/л. Порцию фильтровали, осадок на фильтре промывали ACN (3 раза по 12 мл) и затем сушили в вакууме при 45°C и в слабом токе азота, пока количество присутствующих ACN и воды, определяемое термогравиметрическим анализом, не оказалось меньше, чем 1 мас.%. Получали более 99 A% по методу ВЭЖХ калиевой соли соединения A.
ПРИМЕР 2
ФОРМА 1 КРИСТАЛЛИЧЕСКОЙ КАЛИЕВОЙ СОЛИ СОЕДИНЕНИЯ A
ЧАСТЬ A: ПОЛУЧЕНИЕ
Этанол (147 мл), вода (147 мл) и соединение A (97,9 г образца, как определено по ВЭЖХ) загружали в 1 л круглодонную колбу, снабженную механической мешалкой, капельной воронкой, впускным отверстием для азота (то есть, опыт проводили в атмосфере азота), и термопарой. Водный KOH (45% масс/масс, 0,98 экв., 18,5 мл, 216 ммоль) добавляли к суспензии при 21°C на протяжении 10 минут. Полученную суспензию перемешивали 0,5 часа, в результате получая растворение большей части кристаллов, после чего порцию фильтровали через 1 мкм фильтр непосредственно в 5 л круглодонную колбу, снабженную механической мешалкой, капельной воронкой, впускным отверстием для азота, и термопарой. 1 л Колбу промывали смесью 1:1 (об./об.) вода/EtOH (48 мл), и промывную жидкость фильтровали в 5 л кристаллизатор. В отфильтрованный раствор при комнатной температуре вносили затравочный кристалл кристаллической формы 1 калиевой соли соединения А (200 мг) и затем выдерживали 1 час для образования надежного затравочного кристаллического слоя, после чего суспензию на протяжении 1,5 часа разбавляли EtOH (1,57 л) при 20°C. Затем порцию охлаждали до, приблизительно, 4°C и выдерживали, пока измеренная концентрация соединения A в маточном растворе не составила 4,7 г/л. Порцию фильтровали. Кристаллизатор промывали над фильтром, используя 50 мл EtOH. Осадок на фильтре промывали EtOH (4 раза по 100 мл) и затем сушили в вакууме в атмосфере азота, пока количество присутствующего EtOH, определяемое с помощью ЯМР, не составило, приблизительно, 0,4% моль калиевой соли. Калиевую соль соединения A получали с выходом 88% (91,5 г образец исследовали методом ВЭЖХ, получая 99% по площади пиков ВЭЖХ).
ЧАСТЬ B: ХАРАКТЕРИСТИКА
Порошковую рентгенограмму калиевой соли, полученной способом, описанным в части A, получали на рентгеновском порошковом дифрактометре модели Philips Analytical X'Pert Pro, используя непрерывную развертку 2θ от 2,5 до 40 градусов на протяжении, приблизительно, 12 минут (то есть, с шагом в 0,02, при соотношении 40 секунд/шаг), при вращении предметного столика со скоростью 2 об/сек (RPS) и оси развертки угла. В качестве источника излучения использовали медное излучение K-альфа 1 (Kα1) и K-альфа 2 (Kα2). Эксперимент проводили в условиях окружающей среды. Характеристические значения для 2θ на порошковой рентгенограмме (показаны на Фиг.1) и соответствующие d-расстояния включает нижеследующие:
Калиевую соль соединения А (АК соль), полученную способом, описанным в части A, анализировали также на приборах для термического анализа (ТА), на дифференциальном сканирующем калориметре DSC 2910, при скорости нагрева 10°C/мин от комнатной температуры до 350°C в гофрированном алюминиевом тигле с точечным отверстием в атмосфере азота. Кривая ДСК (DSC) (дифференциальная сканирующая калориметрия), показанная на фиг.2, обнаруживает отдельный, резкий эндотермический температурный пик у, приблизительно, 279°C и связанную с ним теплоту плавления, составляющую приблизительно, 230,0 Дж/грамм. Полагают, что поглощение тепла обусловлено плавлением.
Термогравиметрический анализ осуществляли на приборе фирмы Perkin-Elmer, модели TGA 7 в атмосфере азота, при скорости нагревания от комнатной температуры до, приблизительно, 350°C, составляющей 10°C/мин. (TG) ТГ кривая показала 0,3% потерю массы во время нагревания до 250°C.
Данные по гигроскопичности получали на симметричном анализаторе сорбции пара VTI, модели SGA-1. Данные собирали при комнатной температуре, при величине относительной влажности, составляющей от 5 до 95% и наоборот, шагом, соответствующим 5% изменению относительной влажности. Равновесные условия представляли собой изменение массы на 0,01 процент в течение 5 минут; максимальное время установления равновесия составляло 180 минут. Данные указывали на то, что в состоянии равновесия при 25°C и 95% относительной влажности (RH), вещество имело 1,8 мас.% прироста. Когда равновесие устанавливали, возвращаясь к 5% RH, вещество возвращалось, приблизительно, к своей сухой массе. Анализ XRPD вещества после проведения эксперимента на гигроскопичность показал, что фазы вещества не изменились.
Калиевую соль, полученную, как описано в части A, кроме того, анализировали, подвергая титрованию HCL, используя Brinkmann Metrohm 716 DMS Titrino. Результаты исследования показали, что соль представляла собой монокалиевую соль.
ПРИМЕР 3
ПОЛУЧЕНИЕ СПРЕССОВАННЫХ ТАБЛЕТОК, СОДЕРЖАЩИХ КАЛИЕВУЮ СОЛЬ СОЕДИНЕНИЯ A
(мас.%)
Спрессованные таблетки, содержащие 400 мг соединения A, исходя из содержания свободного фенола, получали с помощью уплотнения вальцеванием и ряда способов прессования таблеток. Полоксамер 407, стеарат магния и натрий стеарилфумарат предварительно последовательно просеивали через сита с размерами ячеек No. 30 и No. 60 и затем в течение 5 минут смешивали со всеми другими ингредиентами, за исключением гранулированного (экстра) стеарата магния, в смесителе-V фирмы Patterson-Kelly (PK). Перемешанные вещества затем пропускали через сито с ячейками No. 35 для разрушения агломератов, и просеянный материал затем смешивали дополнительно в том же PK смесителе в течение, приблизительно, 15-20 минут. Затем смесь подвергали уплотнению вальцеванием, используя минивальцевый пресс типа TF фирмы Freund, при давлении вальцев 40 кгс/см2, скорости вращения вальцев 3 об/мин и скорости вращения шнека 10 об/мин. Полученную полосу измельчали в небольшом устройстве фирмы Quadro Comil, оборудованном круглым ротором и ситом с размером 39R (то есть, размер круглого отверстия составлял 0,039 дюйма, что, приблизительно, соответствует размеру ячейки No. 20) и функционирующем со скоростью 1700 об/мин. Полученные гранулы затем смешивали с 0,5% гранулированного (экстра) стеарата магния в PK смесителе в течение 5 минут для получения конечной смеси. Затем смазанные гранулы прессовали в таблетки, используя роторный пресс для таблетирования с плоским резцом овальной формы, при необходимой силе сжатия для достижения прочности таблетки от 16 до 20 килопонд (то есть, от 156,9 до 196,1 ньютон), измеренной при использовании основной модели HT-300 установки определения прочности.
ПРИМЕР 4
ПОЛУЧЕНИЕ ПОКРЫТЫХ ПЛЕНКОЙ СПРЕССОВАННЫХ ТАБЛЕТОК, СОДЕРЖАЩИХ КАЛИЕВУЮ СОЛЬ СОЕДИНЕНИЯ A
Спрессованные таблетки, содержащие нижеследующую композицию, получали по процедуре, описанной в примере 5:
ПРИМЕР 5
ПОЛУЧЕНИЕ ПОКРЫТЫХ ПЛЕНКОЙ СПРЕССОВАННЫХ ТАБЛЕТОК, СОДЕРЖАЩИХ КАЛИЕВУЮ СОЛЬ СОЕДИНЕНИЯ A
Спрессованные таблетки, содержащие 400 мг соединения A, исходя из содержания свободного фенола, получали, смешивая все перечисленные выше ингредиенты, за исключением гранулированной (экстра) микрокристаллической целлюлозы, стеарата магния и состава Opadry White, в смесителе (смеситель фирмы Patterson-Kelly V, в дальнейшем обозначаемый как "V-смеситель") в течение 10 минут, с последующим смазыванием в том же смесителе внутрикристаллитным стеаратом магния в течение 5 минут. Затем смесь подвергали уплотнению вальцеванием в полосы в вальцевом прессе Alexanderwerk WP 120, используя 25 мм накаточный ролик, при давлении ролика в 60 бар. Затем полосы измельчали в гранулы, используя ротационный высококачественный гранулятор (составная часть вальцевого пресса WP 120), оборудованный ситами с размерами ячеек 2,0 мм и 0,8 мм. Затем гранулы смешивали с экстрагранулированной микрокристаллической целлюлозой в V-смесителе в течение 10 минут, с последующим 5 минутным смазыванием в том же смесителе экстрагранулированным стеаратом магния. Затем смазанные гранулы прессовали на ротационном прессе для таблетирования (Korsch) до 800 мг образца таблетки, используя 2×16/32" стандартные округленные вогнутые резцы. Измеренная прочность внутренней части таблеток находилась между 10 и 15 килопонд (kp = 1 кгс). Затем внутренние части таблеток покрывали составом Opadry White в машине для нанесения покрытий Vector (1,3 л тигель) для получения таблеток в оболочке, с, приблизительно, 4% приращением массы по отношению к внутренней части таблетки.
ПРИМЕР 6
ПОЛУЧЕНИЕ СПРЕССОВАННЫХ ТАБЛЕТОК, СОДЕРЖАЩИХ КАЛИЕВУЮ СОЛЬ СОЕДИНЕНИЯ A
Часть A
Спрессованные таблетки, содержащие 100 мг соединения A, исходя из содержания свободного фенола, получали, смешивая все ингредиенты, перечисленные выше, за исключением экстрагранулированного стеарата магния, в смесителе (вибрационный смеситель T2F, типа Turbula®, Базель, Швейцария) в течение 10 минут. Часть смешанного материала с массой, приблизительно, 1 грамм прессовали в брикеты или бруски в настольном прессе (Auto Carver Model Auto "C", Catalog No. 3888, Carver, Inc., Wabash, Indiana), используя 1×0,5 дюймовые прямоугольные резцы, с давлением до l2 МПа (4 КН). Затем порции калибровали в гранулы, пропуская через сито с 1 мм отверстиями. Гранулы в течение 5 минут смешивали с экстрагранулированным стеаратом магния в смесителе Turbula. И смазанные гранулы прессовали в таблетки, используя пресс Auto Carver с 13/32-дюймовыми стандартными вогнутыми округленными резцами.
ЧАСТЬ В
Спрессованные таблетки, имеющие состав, описанный в приведенной выше таблице, получали, используя процедуру аналогичную процедуре, предлагаемой в части A.
ПРИМЕР 7
ФАРМАКОКИНЕТИЧЕСКОЕ ИССЛЕДОВАНИЕ, ПРОВОДИМОЕ НА ЗДОРОВЫХ ПАЦИЕНТАХ МУЖСКОГО ПОЛА.
4-этапное, частично рандомизированное, перекрестное, исследование с открытой меткой, изучающее фармакокинетику однократных оральных доз составов, содержащих калиевую соль соединения A, проводили на здоровых людях мужского пола в состоянии голодания. Каждый субъект последовательно получал однократную дозу, состоящую из:
(A) спрессованной таблетки, содержащей 400 мг соединения A (исходя из содержания свободного фенола), состав и способ получения которой, аналогичны описанным в примере 3;
(B) спрессованной таблетки, содержащей 400 мг соединения A (исходя из содержания свободного фенола), состав и способ получения которой, аналогичны описанным в примере 4;
(C) спрессованной таблетки, содержащей 400 мг соединения A (исходя из содержания свободного фенола), состав и способ получения которой аналогичны описанным в примере 5; и
(D) четырех спрессованных таблеток, где каждая содержит 100 мг соединение A (исходя из содержания свободного фенола) и имеет состав и способ получения, аналогичные описанным в части В примера 6.
Пробы крови отбирали перед введением дозы и на 0,5, 1, 1,5, 2, 3, 4, 5, 6, 8, 10, 12, 16, 24, 32, 48 и 72 часу после ведения дозы. Имелся, по крайней мере, 4-дневный период вымывания между каждыми А, В, С и D дозами, начиная от введения дозы предшествующего этапа. Безопасность субъектов контролировали до и после каждого дозирования посредством клинической оценки неблагоприятных опытов и путем исследования других параметров безопасности, включая лабораторные анализы мочи и крови на безопасность, исследование показателей жизненно важных функций, физикальные обследования и электрокардиограммы.
Получение и анализ образцов:
Образцы плазмы экстрагировали, используя 96-лунковую экстракцию жидкость-жидкость. Экстракт плазмы инжектировали на колонку ВЭЖХ Ace C18 (50×3.0 мм, 3 мкм, титановый наполнитель) и анализировали в изократических условиях с подвижной фазой, состоящей из 42,5/57,5 (об./об.%) смеси 0, 1 мМ EDTA в 0,1% муравьиная кислота/метанол, при скорости течения 0,5 мл/минута. Экстракты образцов ионизировали, используя APCI поверхности, и контролировали посредством MRM в режиме положительной ионизации. Динамический диапазон анализа ЖХ/МС/МС составлял 2-1000 нг/мл, основываясь на аликвоте 200 мкл плазмы человека.
PK Расчеты:
Площадь под кривой на графике зависимости концентрации плазмы от времени, где концентрацию отсчитывали до последней определяемой концентрации (AUC0-посл.), рассчитывали, используя некамерную модель и способ расчета «Linear Up / Log Down» Версии 4.1 программы WinNonLin. Данные точек, идущих после Cmax подгоняли к биэкспоненциальному уравнению (A*exp(αt)+B*exp(-βt)), используя версию 4.1 программы WinNonlin, а значения AUC(площади под кривой) экстраполировали к бесконечности по нижеследующему уравнению:
AUC0-(=AUC0 посл. + Спосл/β, где Спосл. представляет собой последнюю определяемую концентрацию, а β берут из описанного выше биэкспоненциального уравнения. Наблюдаемые максимальная плазменная концентрация (Cmax), время достижения Cmax, (Tmax) и плазменные концентрации через 12 часов после дозирования (C12 час) определяли путем обследования.
Результаты исследование были следующими:
1. Таблетки дозы A позволяли получить на усредненной кривой величину Cmax, которая, приблизительно, была на 58% ниже, и Tmax, которое было, приблизительно, на 2 часа дольше, чем соответствующие значения, полученные для таблеток дозы D. Не было изменений в средних значениях для C12 час для таблеток дозы A, по сравнению с таблетками дозы D, и средняя величина AUC0-oo для таблеток дозы A было, приблизительно, на 40% ниже, чем для таблеток дозы D.
2. Таблетки дозы A позволяли получить на усредненной кривой величину Cmax, которая приблизительно, была такой же, и Tmax, которое было, приблизительно, на 1 час дольше, чем соответствующие значения, полученные для таблеток дозы В. Наблюдали, приблизительно, 20% возрастание средних величин C12 час для таблеток дозы A, по сравнению с таблетками дозы В; и средняя величина AUC0-oo для таблеток дозы A было, приблизительно, на 14% выше, чем для таблеток дозы В.
3. Таблетки дозы A позволяли получить на усредненной кривой величину Cmax, которая была, приблизительно, на 45% ниже, и Tmax, которое было, приблизительно, на 1,5 часа дольше, чем соответствующие значения, полученные для таблеток дозы С. Наблюдали, приблизительно, 20% возрастание средних величин C12 час для таблеток дозы A, по сравнению с таблетками дозы С; и средняя величина AUC0-oo для таблеток дозы A было, приблизительно, на 20% ниже, чем для таблеток дозы С.
По сравнению с таблетками доз С и D [составы компаратора на основе лактозы с 25 мас.% (100 мг соединения A/таблетка) и 50 мас.% (400 мг соединения A/таблетка) лекарственного вещества, соответственно, дозируемого, исходя из содержания свободного фенола], таблетки дозы A [состав на основе полоксамера по настоящему изобретению с 46 мас.% (400 мг соединение A/таблетка) лекарственного вещества, дозируемого, исходя из содержания свободного фенола] приводили к более низкой
Cmax и увеличенному Tmax. Такие характеристики могут приводить к улучшенной переносимости при дозировании хроническим больным, когда должны проявиться любые свойства токсичности, связанные с Cmax. По сравнению с составом на основе лактозы с такой же дозировкой лекарственного вещества (доза C), доза A также приводила к некоторому повышению уровней C12 час. Наиболее низкие концентрации (например, в режиме ежедневного дозирования по два раза в день), поэтому могут быть умеренно улучшены с помощью дозы A, давая возможное преимущество в эффективности. Возможным недостатком таблеток дозы A является их сниженная биодоступность, по сравнению с таблетками дозы С и дозы D, но полагают, что минимальные концентрации являются более релевантными для эффективности, и форма профиля плазменной концентрации для дозы A (более низкая Cmax, одинаковая или повышенная C12 час), как полагают, является более благоприятной для балансировки переносимости и эффективности, по сравнению с дозой С и дозой D. По сравнению с дозой В (состав компаратора на основе фосфата кальция с 50 мас.% (400 мг соединения A/таблетка лекарственного вещества, при дозировании, исходя из содержания свободного фенола], доза A имеет преимущество в увеличенном Tmax, несколько увеличенной величине C12 час и несколько более высокой биодоступности.
ПРИМЕР 8
ИССЛЕДОВАНИЕ РАСТВОРЕНИЯ IN VITRO
Тестирование характеристик растворения таблеток, получаемых способом, описанным в примере 3 (то есть, полоксамерсодержащие таблетки с 400 мг соединения A, исходя из содержания свободного фенола), осуществляли нижеследующим способом. Отдельную таблетку добавляли в сосуд для растворения USP тип II, содержащий в качестве растворяющей среды 900 мл 0,025 M натрийфосфатного буфера (pH=6,8). Температуру среды регулировали у 37°C. После того как поплавок в виде корзинки, содержащий одну таблетку, оставляли погружаться на дно сосуда, среду перемешивали при 100 об/мин в течение 10 часов. Образцы (1,0 мл) удаляли из среды на 0,5, 1, 2, 3, 4, 5, 6, 8 и 10 часу. Каждый образец затем анализировали с помощью ВЭЖХ для определения концентрации соединения A в растворе. Калиевую соль соединения A использовали в качестве эталона сравнения; таким образом, переводной коэффициент, составляющий 0,9211, использовали для получения результатов, исходя из содержания формы свободного фенола. Используя эту процедуру определяли затем характеристики растворения пяти дополнительных таблеток. На Фиг.3 представлены результаты в виде графика зависимости среднего процента растворенного соединения A от времени растворения.
ВЭЖХ: колонка = Merck KGaA Chromolith Performance RP-18e (100×4,6 мм); подвижная фаза=38:62 (об:об) смесь ACN:0,01M фосфат калия (pH=3,0); скорость течения = 5,0 мл/минута; температура колонки 40°C; инжектируемый объем = 10 мкл; длина волны детектирования 303 нм; время опыта = 1 минута.
Характеристики растворения таблеток, получаемых способом, описанным в части В примера 6 (то есть, лактозасодержащие таблетки со 100 мг соединения A, исходя из содержания свободного фенола), определяли нижеследующим способом: отдельную таблетку добавляли в сосуд для растворения USP тип II, содержащий в качестве растворяющей среды 900 мл 0,025 M натрийфосфатного буфера (pH=6,8)/0,5% SDS. Температуру среды регулировали у 37°C. После того как одна таблетка погрузилась на дно сосуда, среду перемешивали при 75 об/мин в течение 60 минут. Образцы (5,0 мл) удаляли из среды на 10, 15, 20, 30, 60 минуте. Каждый образец затем анализировали с помощью ВЭЖХ для определения концентрации соединения A в растворе. Калиевую соль соединения A затем использовали как эталон сравнения, так что переводной коэффициент, составляющий 0,9211, использовали для получения результатов при пересчете на содержание свободного фенола. Затем, используя эту процедуру, определяли характеристики растворения пяти дополнительных таблеток. На Фиг.4 в виде графика представлены результаты зависимости процента растворенного соединения A от времени растворения.
ВЭЖХ: колонка = Waters Atlantis dC18 (150×4,6 мм, 5 мкм); подвижная фаза = 45:55 (об:об)смесь ACN:0,01M фосфат калия (pH=3,0); скорость течения = 1,5 мл/минута; температура колонки 40°C; инжектируемый объем = 40 мкл; длина волны детектирования 303 нм; время опыта = 5 минут.
Характеристики растворения таблеток, получаемых способом, описанным в примере 5 (то есть, лактозасодержащие таблетки с 400 мг соединения A, исходя из содержания свободного фенола, определяли аналогично способу получения таблеток в примере 6, кроме того, что буферная среда с pH 6,8 не содержала SDS (который может способствовать растворению), скорость вращения лопастей мешалки составляла 100 об/мин, и на временных точках удаляли только 1,0 мл образцов растворяющей среды. Две таблетки тестировали и анализировали с помощью ВЭЖХ способом, использующим колонку Chromolith.
Профиль растворения таблеток примера 3 показал более медленную скорость высвобождения лекарственного средства, по сравнению с таблетками примера 6. Более конкретно, для таблеток примера 3, среднее время, необходимое для достижения 80% растворения Соединения A, находилось между 2 и 3 часами, тогда как среднее время, необходимое для достижения 80% растворения соединения A таблеток примера 6, составляло, приблизительно, 20 минут. Таблетки примера 5 демонстрируют профиль, соответствующий быстрому растворению (то есть, 80% растворение занимает менее чем 20 минут), аналогично профилю растворения таблеток примера 6, даже при отсутствии SDS в среде.
Хотя предшествующее описание объясняет основные положения настоящего изобретения, с примерами, приводимыми с целью иллюстрации, осуществление изобретения охватывает все обычные изменения, усовершенствования и/или модификации, которые входят в объем нижеследующей формулы изобретения.


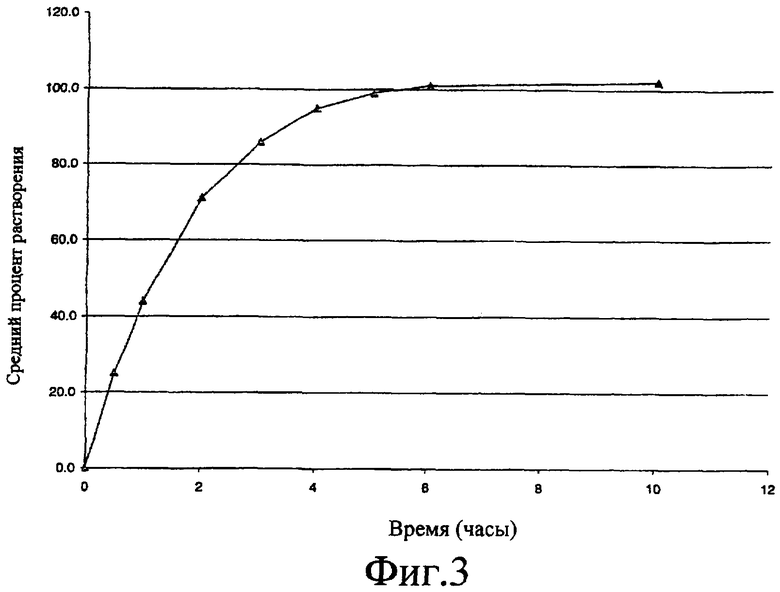

Изобретение относится к области фармакологии и касается фармацевтического состава, пригодного для перорального введения в виде твердой лекарственной формы, содержащего эффективное количество основной соли соединения формулы 1 и композицию с контролируемой скоростью высвобождения, содержащую солюбилизирующий агент, гелеобразующий агент и водорастворимый наполнитель. Составы пригодны для использования при ингибировании интегразы ВИЧ, лечении и профилактике ВИЧ-инфекции, и при лечении, профилактике и замедлении возникновения СПИДа. 4 н. и 12 з.п. ф-лы, 4 ил.
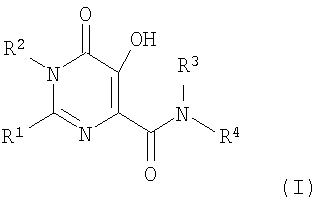
1. Фармацевтический состав для перорального введения в виде твердой дозы, который содержит эффективное количество основной соли соединения А и композицию, контролирующую скорость высвобождения, содержащую солюбилизирующий агент, гелеобразующий агент и, необязательный, водорастворимый наполнитель, где соединение А представляет собой
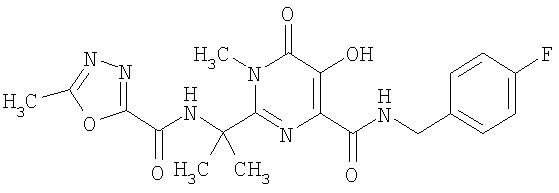
2. Состав по п.1, в котором основная соль соединения А является калиевой солью соединения А.
3. Состав по п.2, в котором солюбилизирующий агент содержит полоксамер; гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу; а необязательный водорастворимый наполнитель содержит лактозу.
4. Состав по п.3, в котором:
калиевая соль соединения А используется в количестве от приблизительно 25 до приблизительно 75 мас.% исходя из содержания свободного фенола;
полоксамер используется в количестве от приблизительно 10 до приблизительно 20 мас.%;
высоковязкая гидроксипропилметилцеллюлоза используется в количестве от приблизительно 3 до приблизительно 9 мас.% и
лактоза используется в количестве от приблизительно 3 до приблизительно 9 мас.%.
5. Состав по п.4, в котором полоксамер представляет собой полоксамер 407, измельченный до среднего размера частиц от приблизительно 50 до приблизительно 150 мкм; высоковязкая гидроксипропилметилцеллюлоза представляет собой НРМС К4М; лактоза представляет собой высушенный спрей водного раствора лактозы.
6. Состав по п.5, в котором калиевая соль соединения А является формой 1 кристаллической калиевой соли соединения А.
7. Состав по п.1, который дополнительно содержит разбавитель и смазывающее вещество и в котором основная соль соединения А является калиевой солью соединения А; солюбилизирующий агент содержит полоксамер; гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу; водорастворимый наполнитель содержит лактозу; разбавитель содержит микрокристаллическую целлюлозу и, необязательно, фосфат кальция; а смазывающее вещество содержит стеарат металла и стеарилфумарат металла.
8. Состав по п.7, в котором: калиевая соль соединения А используется в количестве от приблизительно 40 до приблизительно 60 мас.%, исходя из содержания свободного фенола;
полоксамер используется в количестве от приблизительно 10 до приблизительно 20 мас.%;
высоковязкая гидроксипропилметилцеллюлоза используется в количестве от приблизительно 3 до приблизительно 9 мас.%;
лактоза используется в количестве от приблизительно 3 до приблизительно 9 мас.%;
микрокристаллическая целлюлоза используется в количестве от приблизительно 5 до приблизительно 30 мас.%;
фосфат кальция используется в количестве от приблизительно нуля до приблизительно 15 мас.%;
стеарат металла и стеарилфумарат металла, каждый независимо используется в количестве от приблизительно 1 до приблизительно 3 мас.%;
и где калиевая соль соединение А является формой 1 кристаллической калиевой соли соединения А; полоксамер представляет собой полоксамер 407, измельченный до среднего размера частиц от приблизительно 50 до приблизительно 150 мкм; высоковязкая гидроксипропилметилцеллюлоза представляет собой НРМС К.4М; лактоза представляет собой высушенный спрей водного раствора лактозы; микрокристаллическая целлюлоза представляет собой AVICEL РН-102; фосфат кальция представляет собой двухосновный фосфат кальция; стеарат металла является стеаратом магния; а стеарилфумарат металла является стеарилфумаратом натрия.
9. Состав по любому из пп.2-8, где калиевая соль соединения А используется в количестве от приблизительно 100 до приблизительно 600 мг исходя из содержания свободного фенола.
10. Состав по п.9, где состав является инкапсулированным или спрессованным в таблетку.
11. Способ получения спрессованного в таблетки фармацевтического состава, содержащего эффективное количество калиевой соли соединения А, солюбилизирующий агент, гелеобразующий агент, водорастворимый наполнитель, первый разбавитель, второй разбавитель, первое смазывающее вещество и второе смазывающее вещество, где способ предусматривает:
(A) перемешивание смеси калиевой соли соединения А, солюбилизирующего агента, гелеобразующего агента, водорастворимого наполнителя, первого разбавителя, второго разбавителя, первой части первого смазывающего вещества и второго смазывающего вещества;
(B) просеивание перемешанной смеси и затем дополнительное перемешивание просеянной смеси;
(C) вальцевание просеянной и перемешанной смеси для образования брикета и затем калибрование полученного брикета для образования гранул;
(D) смешивание гранул с остающейся частью первого смазывающего вещества и
(Е) прессование смазанных гранул стадии D для получения таблеток.
12. Способ по п.11, в котором:
солюбилизирующий агент содержит полоксамер;
гелеобразующий агент содержит высоковязкую гидроксипропилметилцеллюлозу;
водорастворимый наполнитель содержит лактозу;
первый разбавитель представляет собой микрокристаллическую целлюлозу;
второй разбавитель представляет собой фосфат кальция;
первое смазывающее вещество является стеаратом металла и
второе смазывающее вещество является стеарилфумаратом металла.
13. Способ по п.12, в котором:
калиевая соль соединения А используется в количестве от приблизительно 40 до приблизительно 60 мас.% исходя из содержания свободного фенола;
полоксамер используется в количестве от приблизительно 10 до приблизительно 20 мас.%;
высоковязкая гидроксипропилметилцеллюлоза используется в количестве от приблизительно 3 до приблизительно 9 мас.%;
лактоза используется в количестве от приблизительно 3 до приблизительно 9 мас.%;
микрокристаллическая целлюлоза и фосфат кальция каждый независимо используется в количестве от приблизительно 5 до приблизительно 25 мас.% и
стеарат металла и стеарилфумарат металла каждый независимо используется в количестве от приблизительно 1 до приблизительно 3 мас.%;
и где полоксамер представляет собой полоксамер 407, измельченный до среднего размера частицы от приблизительно 50 до приблизительно 150 мкм; высоковязкая гидроксипропилметилцеллюлоза представляет собой НРМС К4М; лактоза представляет собой высушенный спрей водного раствора лактозы; микрокристаллическая целлюлоза представляет собой AVICEL РН-102; фосфат кальция представляет собой двухосновный фосфат кальция; стеарат металла является стеаратом магния; а стеарилфумарат металла является стеарилфумаратом натрия.
14. Способ по п.13, где способ дополнительно включает: (F) покрытие спрессованной таблетки составом Opadry II HP для получения таблетки в оболочке, в которой покрытие составляет от приблизительно 2 до приблизительно 4% от массы спрессованной таблетки.
15. Способ лечения или профилактики ВИЧ-инфекции у субъекта, нуждающегося в этом, который предусматривает введение субъекту фармацевтического состава по любому из пп.1-10.
16. Способ лечения, профилактики или замедления возникновения СПИДа у субъекта, нуждающегося в этом, который предусматривает введение субъекту фармацевтического состава по любому из пп.1-10.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| ЗАМЕЩЕННЫЕ ПИРИМИДИНТИОАЛКИЛЬНЫЕ ИЛИ АЛКИЛЭФИРНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИНГИБИРОВАНИЯ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИРУСОВ | 1996 |
|
RU2167155C2 |

