По настоящей заявке испрашивается приоритет на основании предварительной заявки США № 60/632945 (поданной 3 декабря 2004 года), раскрытие которой полностью включено в настоящее описание в качестве ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение направлено на способ улучшения фармакокинетических характеристик пероральных лекарственных средств, метаболизируемых изоформой UDP-глюкуронозилтрансферазы 1A1 (UGT1A1), где лекарственные средства вводят в комбинации с атазанавиром. Кроме того, настоящее изобретение направлено на способы ингибирования интегразы ВИЧ, лечения и профилактики инфекции ВИЧ, а также лечения, профилактики и замедления развития СПИДа, где указанные способы предусматривают пероральное введение ингибитора интегразы ВИЧ, метаболизируемого UGT1A1, в комбинации с атазанавиром.
УРОВЕНЬ ТЕХНИКИ
UDP-глюкуронозилтрансферазы (UGT) относятся к семейству ферментов, катализирующих глюкуронидацию эндогенных и ксенобиотических химических веществ; то есть UGT катализируют перенос остатка глюкуроновой кислоты от кофактора уридиндифосфатглюкуроновой кислоты к субстрату. Перенос осуществляется к нуклеофильному гетероатому O, N или S. Субстраты включают ксенобиотики, функции которых осуществляются через реакции фазы I (например, P450-зависимый окислительный метаболизм), а также эндогенные соединения, такие как билирубин, стероидные гормоны и гормоны щитовидной железы. Хотя глюкуронидацию обычно классифицируют как метаболизм фазы II - фазы, наступающей после P450-зависимого окислительного метаболизма, - многие соединения не требуют предварительного окисления, поскольку они уже содержат функциональные группы, по которым можно проводить глюкуронидацию. Продукты глюкуронидации выводятся с мочой, если субстрат обладает низкой молекулярной массой (менее приблизительно 250 граммов), а более крупные глюкуронидированные субстраты выводятся с желчью.
UGT играют ключевую роль в осуществлении некоторых важных метаболических функций, таких как: экскреция лекарственных средств (например, нестероидных противовоспалительных средств, опиоидов, антигистаминных средств, антипсихотических средств и антидепрессантов); обезвреживание веществ, загрязняющих окружающую среду, таких как бензо(a)пирены; регуляция уровня гормонов, таких как андрогены, эстрогены, прогестины и ретиноиды; и выведение продукта деградации гема в билирубин.
UGT расположены в микросомах печени, почек, кишечника, кожи, мозга, селезенки и слизистой оболочки носовой полости, где они находятся на той же стороне мембраны эндоплазматического ретикулума, что и ферменты цитохрома P450 и флавинсодержащие монооксигеназы, и, следовательно, имеют идеальный доступ к продуктам метаболизма лекарственных средств фазы I. UGT, участвующие в метаблизме лекарственных средств, кодируются двумя семействами генов, UGT1 и UGT2. Члены семейства UGT1, которые экспрессируются в печени человека, где происходит основной метаболизм ксенобиотических веществ, включают UGT 1A1, 1A3, 1A4, 1A6 и 1A9. Изоформа UDP-глюкуронозилтрансферазы 1A1 (UGT1A1) катализирует глюкуронидацию билирубина.
Некоторые пероральные лекарственные средства, в том числе некоторые ингибиторы интегразы ВИЧ, непосредственно метаболизируются UGT1A1, что может ухудшать фармакокинетические характеристики и приводить к потребности более частого введения и/или введения более высоких доз, чем это необходимо или желательно. Необходимость частого введения доз (например, 3 раза в сутки или чаще) может приводить к преднамеренному или непреднамеренному несоблюдению пациентом режима приема лекарственного средства. Введение более высоких доз может приводить к увеличению побочных реакций и/или токсических эффектов. Применение таких лекарственных средств вместе со средством, ингибирующим метаболизм UGT1A1, может улучшить фармакокинетические характеристики лекарственного средства и, следовательно, снизить частоту его введения. Улучшение фармакокинетических характеристик в результате совместного введения с ингибитором UGT1A1 также позволяет использовать более низкие дозы, что, в свою очередь, уменьшает или устраняет побочные реакции и токсические эффекты. Соответственно, существует потребность в разработке соединений, которые могут улучшать фармакокинетические характеристики лекарственных средств, метаболизируемых UGT1A1.
Уровень техники также раскрывается в нижеследующих ссылках:
US 2003/0215462 A1, где описаны способы увеличения биодоступности некоторых перорально вводимых фармацевтических соединений путем совместного введения соединений с ингибиторами UDP-глюкуронозилтрансферазы.
WO 03/35076 и соответствующем патенте США 2005/0075356, каждый из которых раскрывает некоторые 5,6-дигидроксипиримидин-4-карбоксамиды как ингибиторы интегразы ВИЧ, а также WO 03/35077 и соответствующем патенте США 2005/0025774, каждый из которых раскрывает некоторые N-замещенные 5-гидрокси-6-оксо-1,6-дигидропиримидин-4-карбоксамиды как ингибиторы интегразы ВИЧ. В каждой из указанных ссылок также раскрывается применение описанных в них карбоксамидных соединений в комбинации с одним или несколькими средствами, пригодными для лечения инфекции ВИЧ или СПИДа, где в списке подходящих средств присутствует атазанавир.
WO 2004/058756, где в качестве ингибиторов интегразы ВИЧ описаны некоторые гидрокситетрагидропиридопиримидинон-карбоксамиды и родственные карбоксамиды. В указанной ссылке также раскрывается применение описанных в ней карбоксамидных соединений в комбинации с одним или несколькими средствами, пригодными для лечения инфекции ВИЧ или СПИДа, и отмечается, что подходящие средства включают вещества, перечисленные в таблице WO 02/30930, в том числе атазанавир.
WO 2005/087768, где в качестве ингибиторов интегразы ВИЧ описаны некоторые гидроксиполигидро-2,6-нафтиридиндионовые соединения. В указанной ссылке также раскрывается применение указанных соединений в комбинации с одним или несколькими средствами, пригодными для лечения инфекции ВИЧ или СПИДа, и отмечается, что подходящие средства включают атазанавир.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Обнаружено, что совместное введение атазанавира с лекарственным средством, которое непосредственно метаболизируется UGT1A1, улучшает фармакокинетические характеристики лекарственного средства. Более конкретно, настоящее изобретение включает способ улучшения фармакокинетических характеристик перорального лекарственного средства, непосредственно метаболизируемого UGT1A1, который предусматривает пероральное введение млекопитающиему (особенно человеку), нуждающемуся в лечении указанным лекарственным средством, эффективного количества комбинации лекарственного средства, или его фармацевтически приемлемой соли, с атазанавиром или его фармацевтически приемлемой солью.
Разные воплощения, аспекты и признаки настоящего изобретения дополнительно описаны или разъяснены в приведенных ниже описании, примерах и прилагающейся формуле изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 показана диаграмма рентгеновской порошковой дифрактометрии калиевой соли соединения A, полученного по способу примера 2.
На фиг.2 показана кривая DSC калиевой соли соединения A, полученного по способу примера 2.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение включает пероральное введение эффективного количества комбинации лекарственного средства, непосредственно метаболизируемого UGT1A1, и атазанавира. Следует понимать, что лекарственное средство и атазанавир можно вводить отдельно или совместно. В случае раздельного введения их можно вводить одновременно или в разное время (например, поочередно). В случае совместного введения их можно вводить в виде отдельных композиций, которые могут быть упакованы совместно или отдельно, или их можно вводить в составе одной композиции.
Лекарственные средства, подходящие для применения в настоящем изобретении, представляют соединения, в значительной степени метаболизируемые UGT1A1. В настоящем контексе термин "в значительной степени" означает, что по меньшей мере приблизительно 20% перорального лекарственного средства непосредственно метаболизируется UGT1A1. Для применения в способе настоящего изобретения в особенности подходят такие лекарственные средства, первичным путем метаболизма которых после перорального введения является непосредственный метаболизм под действием UGT1A1. Термин "непосредственный метаболизм" и его варианты (например, "непосредственно метаболизируемый") в настоящем описании означает, что метаболизм включает непосредственную глюкуронидацию лекарственного средства; то есть предварительное окисление лекарственного средства в фазе I практически отсутствует.
Атазанавир (также означаемый BMS-232632) представляет азапептидный ингибитор протеазы ВИЧ-1, эффективный для лечения инфекции ВИЧ. Атазанавир имеет следующую структурную формулу:
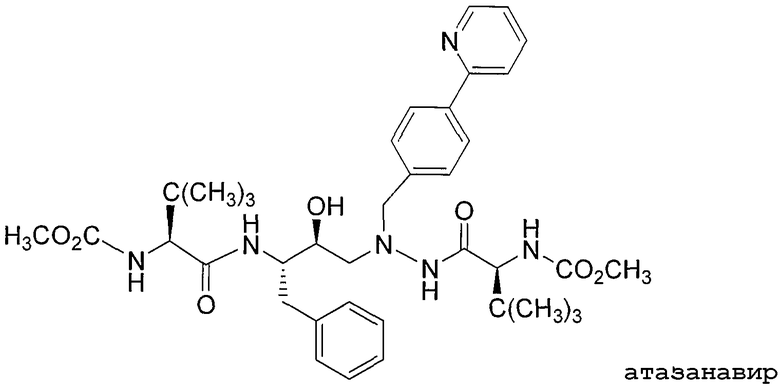
и химическое название сложный диметиловый эфир [3S-(3R*,8'R*,9'R*,12R*)]-3,12-бис(1,1-диметилэтил)-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенилметил]-2,5,6,10,13-пентаазатетрадекандикарбоновой] кислоты. Атазанавира сульфат разрешен к применению для лечения инфекции ВИЧ и имеется в продаже в виде капсул под торговым названием REYATAZTM (Bristol-Myers Squibb). Атазанавир раскрыт в US 5849911, а сульфат атазанавира раскрыт в US 6087383. В Physician's Desk Reference, редакция 2004 года (смотрите стр.1082), описано, что атазанавир является ингибитором изоформы UDP-глюкуронозилтрансферазы 1A1 (UGT1A1).
Улучшение фармакокинетических характеристик (PK) лекарственного средства в настоящем описании означает увеличение одного или нескольких из нижеследующих параметров PK в результате совместного введения лекарственного средства и атазанавира по сравнению с соответствующим значением, полученным при введении лекарственного средства в отсутствие атазанавира: максимальная концентрация в плазме (Cmax), минимальная концентрация в плазме (Cmin), количество лекарственного средства в кровотоке, измеряемое по площади под кривой зависимости концентрации в плазме от времени (AUC0-last, где "last" относится ко времени взятия последнего образца - например, 24 часа), и период полужизни (T1/2).
Лекарственное средство и атазанавир можно вводить независимо и поочередно в виде фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" относится к соли, которая обладает эффективностью исходного средства и не оказывает нежелательных биологических или иных эффектов (например, не является токсичной для реципиента или не оказывает иного вредного воздействия). Подходящие соли включают кислотно-аддитивные соли, которые могут быть получены, например, путем смешивания раствора исходного средства с раствором фармацевтически приемлемой кислоты, такой как хлористо-водородная кислота, серная кислота, уксусная кислота, трифторуксусная кислота или бензойная кислота. Если лекарственное средство содержит кислотный фрагмент (например, -COOH или фенольную группу), его фармацевтически приемлемые соли могут включать соли щелочных металлов (например, соли натрия или калия), соли щелочноземельных металлов (например, соли кальция или магния) и соли, образованные подходящими органическими лигандами, такие как соли четвертичного аммония. Предпочтительной солевой формой атазанавира является сульфат атазанавира, раскрытый в US 6087383.
Если не указано иначе, ссылки на количества лекарственных средств и/или атазанавира осуществляются в отношении их свободных несолевых форм.
Термин "эффективное количество" в применении к комбинации, используемой в настоящем изобретении, относится к совместному введению UGT1A1-метаболизируемого лекарственного средства и атазанавира в количествах, обеспечивающих биологический или медицинский ответ на лекарственное средство, желательный с точки зрения исследователя, врача или другого клинициста. Термин "эффективное количество" относится к "терапевтически эффективному количеству", то есть к количеству UGT1A1-метаболизируемого лекарственного средства и атазанавира, которое при их совместном введении вызывает уменьшение симптомов заболевания или состояния, подлежащего лечению указанным лекарственным средством. Термин "эффективное количество" также относится к "профилактически эффективному количеству", то есть к количеству лекарственного средства и атазанавира, которое при их совместном введении обспечивает профилактику симптомов заболевания или состояния, развитие которого можно предотвращать с помощью указанного лекарственного средства. Указанный термин также включает количество активного соединения, достаточное для ингибирования фермента (например, интегразы ВИЧ) и, следовательно, вызывающее желательный эффект (то есть "количество, эффективное для ингибирования").
В настоящем изобретении лекарственное средство и атазанавир можно совместно вводить в любых соотношениях при условии, что достигается желательный биологический или медицинский ответ на лекарственное средство. Например, в случае совместного введения лекарственное средство можно вводить в таком количестве, которое при раздельном введении не позволяет достигнуть желательного ответа (например, обеспечивает неудовлетворительные значения PK для лекарственного средства и/или неудовлетворительный уровень лекарственного средства в кровотоке, приводя к низкой эффективности, или к отсутствию эффективности), но которое при совместном введении с атазанавиром обеспечивает желательный ответ. В другом примере в случае совместного введения лекарственное средство можно вводить в таком количестве, которое при отдельном введении позволяет достигать подходящего ответа (например, значений PK и/или уровня в кровотоке, которые обеспечивают эффективность препарата), но которое при совместном введении с атазанавиром обеспечивает более высокую эффективность (то есть более высокие значения PK, например, более высокие значения AUC0-last и/или Cmin, или более высокий уровень в кровотоке).
Первое воплощение настоящего изобретения относится к способу улучшения PK перорального лекарственного средства, непосредственно метаболизируемого UGT1A1, как описано выше (то есть как описано в разделе "Краткое описание изобретения"), где атазанавир вводят в комбинации с лекарственным средством в количестве, достаточном для улучшения фармакокинетических характеристик лекарственного средства, по меньшей мере, на приблизительно 10% по отношению к фармакокинетическим характеристикам лекарственного средства, вводимого в отсутствие атазанавира (например, для 10% улучшения AUC0-last или Cmin, или Cmax, или T1/2, или их комбинации).
Второе воплощение настоящего изобретения относится к способу улучшения PK, как описано выше, или как описано в предыдущем воплощении, где млекопитающим, нуждающимся в лечении лекарственным средством, является человек.
Третье воплощение настоящего изобретения относится к способу улучшения PK, как описано выше, или как описано в первом воплощении, где млекопитающим, нуждающимся в лечении лекарственным средством, является человек, а лекарственное средство, непосредственно метаболизируемое UGT1A1, выбрано из группы, состоящей из эзетимиба, ралоксифена, эстрадиола и их фармацевтически приемлемых солей. Эзетимиб селективно ингибирует абсорбцию холестерина в кишечнике и является активным ингредиентом таблеток ZETIATM (поставляемых Merck-Schering Plough Pharmaceuticals). Эзетимиб и симвастатин являются активными ингредиентами таблеток VYTORINTM (поставляемых Merck-Schering Plough Pharmaceuticals). Эзетимиб описан в US 5846966 и повторном издании US 37721. Ралоксифен является селективным модулятором рецептора эстрогенов. Гидрохлорид ралоксифена является активным ингредиентом таблеток EVISTA® (поставляемых Eli Lilly), которые назначают для лечения и профилактики остеопороза у женщин после менопаузы. Ралоксифен описан в US 6458811. Эстрадиол является активным ингредиентом нескольких продуктов, утвержденных для лечения различных заболеваний и состояний, таких как атрофия женских наружных половых органов и влагалища, остеопороз и развитый рак простаты.
Четвертое воплощение настоящего изобретения относится к способу улучшения PK, как описано выше, или как описано в первом или втором воплощении, где лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет ингибитор интегразы ВИЧ.
Пятое воплощение настоящего изобретения относится к способу улучшения PK, как описано выше, или как описано в первом или втором воплощении, где лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет соединение формулы I, или его фармацевтически приемлемую соль:
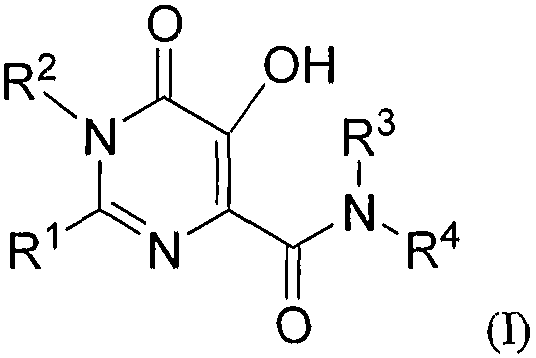
где R1 означает C1-6алкил, замещенный:
(1) N(RA)-C(=O)-N(RC)RD,
(2) N(RA)-C(=O)-C1-6алкилен-N(RC)RD,
(3) N(RA)SO2RB,
(4) N(RA)SO2N(RC)RD,
(5) N(RA)-C(=O)-C1-6алкилен-SO2RB,
(6) N(RA)-C(=O)-C1-6алкилен-SO2N(RC)RD,
(7) N(RA)C(=O)C(=O)N(RC)RD,
(8) N(RA)-C(=O)-HetA,
(9) N(RA)C(=O)C(=O)-HetA или
(10) HetB;
R2 означает -C1-6алкил;
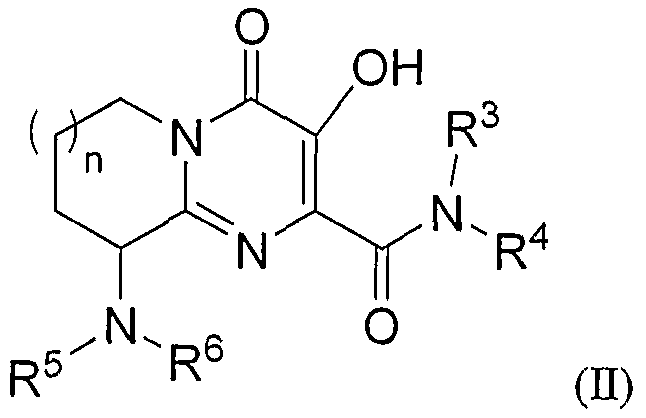
или, альтернативно, R1 и R2 соединены вместе так, что соединение формулы I представляет соединение формулы II:
R3 означает -H или -C1-6алкил;
R4 означает C1-6алкил, замещенный арилом (например, фенилом), который необязательно замещен 1-4 заместителями, каждый из которых независимо означает галоген, -OH, -C1-4алкил, -C1-4алкил-ORA, -C1-4галогеналкил, -O-C1-4алкил,
-O-C1-4галогеналкил, -CN, -NО2, -N(RA)RB, -C1-4алкил-N(RA)RB, -C(=O)N(RA)RB;
-C(=O)RA, -CO2RA, -C1-4алкил-CО2RA, -OCO2RA, -SRA, -S(=O)RA, -SO2RA,
-N(RA)SО2RB, -SO2N(RA)RB, -N(RA)C(=O)RB, -N(RA)CО2RB, -C1-4алкил-N(RA)CO2RB, метилендиокси, присоединенный к двум соседним циклическим атомам углерода, фенил или
-C1-4алкилфенил;
R5 означает:
(1) N(RA)-C(=O)-N(RC)RD,
(2) N(RA)-C(=O)-C1-6алкилен-N(RC)RD,
(3) N(RA)SO2RB,
(4) N(RA)SO2N(RC)RD,
(5) N(RA)-C(=O)-C1-6алкилен-SO2RB,
(6) N(RA)-C(=O)-C1-6алкилен-SO2N(RC)RD,
(7) N(RA)C(=O)C(=O)N(RC)RD,
(8) N(RA)-C(=O)-HetA или
(9) N(RA)C(=O)C(=O)-HetA;
R6 означает -Н или -C1-6алкил;
n означает целое число, равное 1 или 2;
каждый RA независимо означает -H или -C1-6алкил;
каждый RB независимо означает -H или -C1-6алкил;
каждый из RC и RD независимо означает -H или -C1-6алкил, или вместе с атомом азота, к которому они присоединены, образуют насыщенный 5- или 6-членный гетероциклический фрагмент, необязательно содержащий гетероатом, помимо атома азота, соединенного с RC и RD, выбранный из N, O и S, где S необязательно окислен до S(O) или S(O)2, и где насыщенный гетероциклический фрагмент необязательно замещен 1 или 2 C1-6алкильными группами;
HetA означает 5- или 6-членный гетероароматический цикл, содержащий от 1 до 4 гетероатомов, независимо выбранных из N, O и S, где гетероароматический цикл необязательно замещен 1 или 2 заместителями, каждый из которых независимо означает -C1-4алкил, -C1-4галогеналкил, -O-C1-4алкил, -O-C1-4галогеналкил или
-CО2RA; и
HetB означает 5-7-членный насыщенный гетероциклический фрагмент, содержащий от 1 до 4 гетероатомов, независимо выбранных из N, O и S, где каждый S необязательно окислен до S(O) или S(O)2, и где гетероциклический фрагмент необязательно замещен 1-3 заместителями, каждый из которых независимо означает галоген, -C1-4алкил, -C1-4фторалкил, -C(O)-C1-4алкил или -C1-4алкил, замещенный OH.
В аспекте предыдущего воплощения в соединении формулы I R2 означает метил; R3 означает -H; а R4 означает CH2-фенил, где фенил необязательно замещен 1 или 2 заместителями, каждый из которых независимо представляет бром, хлор, фтор, CH3, CF3, C(О)NH2, C(О)NH(CH3), C(O)N(CH3)2, SCH3, SO2CH3 или SO2N(CH3)2; а все другие переменные имеют указанные выше значения. В одном признаке настоящего аспекта R4 означает 4-фторбензил, 3,4-дихлорбензил, 3-хлор-4-фторбензил или 4-фтор-3-метилбензил. В одном признаке настоящего аспекта R4 означает 4-фторбензил.
В настоящем описании термин "алкил" относится к любой линейной или разветвленной алкильной группе, которая содержит число атомов углерода, находящееся в указанном интервале. Так, например, термин "C1-6алкил" (или "C1-C6 алкил") относится ко всем изомерам гексилалкила и пентилалкила, а также к н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу. В другом примере термин "C1-4алкил" относится к н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу.
Термин "алкилен" относится к любой линейной или разветвленной алкиленовой группе (или, альтернативно, "алкандиил"), которая содержит число атомов углерода, находящееся в указанном интервале. Так, например, термин "-C1-6алкилен-" относится ко всем C1-C6 линейным или разветвленным алкиленам. С точки зрения настоящего изобретения особый интерес представляет класс алкиленов -(CH2)1-6-, в частности подклассы -(CH2)1-4-, -(CH2)1-3-, -(CH2)1-2- и -CH2-. Также представляет интерес алкилен -CH(CH3)-.
Термин "галоген" (или "гало-") относится ко фтору, хлору, брому и йоду (альтернативно называемым фтор, хлор, бром и йод).
Термин "галогеналкил" относится к алкильной группе, определенной выше, в которой один или несколько атомов водорода замещены галогеном (то есть F, Cl, Br и/или I). Так, например, "C1-6галогеналкил" (или "C1-C6 галогеналкил") относится к C1-C6 линейной или разветвленной алкильной группе, определенной выше, содержащей один или несколько галогеновых заместителей. Термин "фторалкил" имеет аналогичное значение, за исключением того, что галогеновые заместители ограничены фтором. Подходящие фторалкилы включают серии (CH2)0-4CF3 (то есть трифторметил, 2,2,2-трифторэтил, 3,3,3-трифтор-н-пропил и другие).
Термин "арил" относится к (i) фенилу или (ii) 9- или 10-членной бициклической, сопряженной карбоциклической системе, в которой по меньшей мере один цикл является ароматическим. Как правило, арил представляет фенил или нафтил, и чаще всего - фенил.
Термин "HetA" относится к необязательно замещенному 5- или 6-членному гетероароматическому циклу, содержащему от 1 до 4 гетероатомов, независимо выбранных из N, O и S. В одном воплощении HetA означает необязательно замещенный гетероароматический цикл, выбранный из группы, состоящей из пиридинила, пирролила, пиразинила, пиримидинила, пиридазинила, триазинила, фуранила, тиенила, имидазолила, пиразолила, триазолила, тетразолила, оксазолила, изооксазолила, тиазолила, изотиазолила и оксадиазолила; где указанный цикл необязательно замещен 1 или 2 заместителями, каждый из которых независимо означает -C1-4алкил, -C1-4галогеналкил, -O-C1-4алкил, -O-C1-4галогеналкил или -CO2-C1-4алкил. Следует понимать, что HetA может быть присоединен к остальной части соединения формулы I по любому циклическому атому (то есть по любому атому углерода или гетероатому) при условии, что получается стабильное соединение.
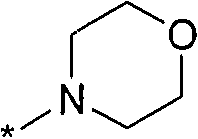
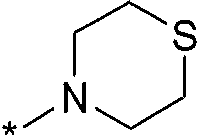
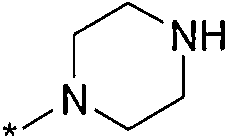
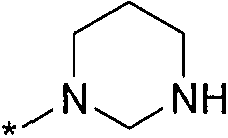
Термин "HetB" относится к необязательно замещенному 5-7-членному насыщенному гетероциклическому фрагменту, содержащему от 1 до 4 гетероатомов, независимо выбранных из N, O и S. В одном воплощении HetB означает необязательно замещенный насыщенный гетероциклический фрагмент, выбранный из группы, состоящей из пирролидинила, имидазолидинила, пиперидинила, пиперазинила, морфолинила, тиоморфолинила, тиазинанила и тетрагидропиранила, где указанный фрагмент необязательно замещен 1 или 2 заместителями, каждый из которых независимо означает -C1-4алкил, -C1-4галогеналкил, -C(O)CF3, -C(O)CH3 или -CH2CH2OH. Следует понимать, что HetA может быть присоединен к остальной части соединения формулы I по любому циклическому атому (то есть по любому атому углерода или по любому гетероатому) при условии, что получается стабильное соединение. В другом воплощении HetB выбран из группы, состоящей из  ,
, 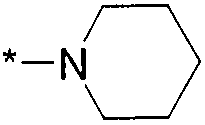 ,
, 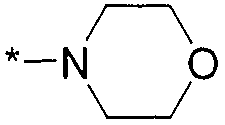 ,
, 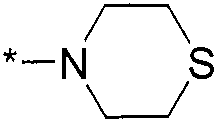 ,
, 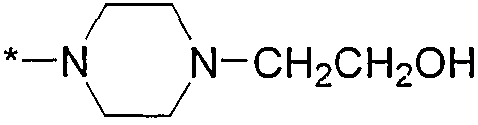 ,
, 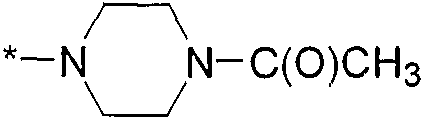 ,
, 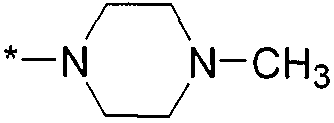 и
и 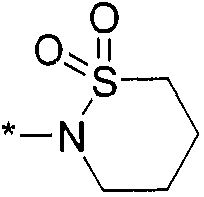 , где * означает точку присоединения к остатку молекулы.
, где * означает точку присоединения к остатку молекулы.
В соединении формулы I RC и RD вместе с атомом азота, к которому они присоединены, могут образовывать насыщенный 5- или 6-членный гетероциклический фрагмент, необязательно содержащий гетероатом, помимо атома азота, соединенного с RC и RD, выбранный из N, O и S, где S необязательно окислен до S(O) или S(O)2, причем насыщенный гетероциклический фрагмент необязательно замещен 1 или 2 C1-6алкильными группами. В одном воплощении насыщенный гетероциклический фрагмент, образованный RC, RD и атомом азота, к которому они присоединены, выбран из группы, состоящей из 4-морфолинила, 4-тиоморфолинила, 1-пиперидинила, 1-пиперазинила, необязательно замещенного C1-4алкилом, и 1-пирролидинила.
Если какая-либо переменная (например, RA и RB) встречается более одного раза в формуле I или в любой другой формуле, изображающей и описывающей соединение, подходящее для применения в настоящем изобретении, его определение в каждом случае не зависит от его определения в каждом другом случае. Кроме того, комбинации заместителей и/или переменных допускаются при условии, что такие комбинации приводят к получению стабильных соединений.
"Стабильное" соединение - это соединение, которое можно получить и выделить и у которого структура и свойства сохраняются или могут сохраняться практически неизменными в течение периода времени, достаточного для применения соединения с целью, описанной в настоящем описании.
В зависимости от выбора заместителей и характера замещения некоторые соединения формулы I, соли которых можно применять в настоящем изобретении, могут иметь ассиметрические центры и могут существовать в виде смесей стереоизомеров или в виде индивидуальных диастереомеров или энантиомеров. Соли всех изомерных форм указанных соединений, либо индивидуальных, либо их смесей, можно использовать в настоящем изобретении.
Соединения формулы I также могут существовать в виде таутомеров вследствие кето-енольной таутомерии. Соли всех таутомеров гидроксипиримидиноновых соединений формулы I, как индивидуальных, так и их смесей, можно использовать в настоящем изобретении.
Соединения, охватываемые формулой I, являются ингибиторами интегразы ВИЧ. Типичные соединения формулы I, отличные от соединений формулы II, раскрыты в WO 03/035077. Типичные соединения формулы I, которые являются соединениями формулы II, раскрыты в WO 2004/058757 и WO 2004/058756.
Шестое воплощение настоящего изобретения относится к способу улучшения PK, как описано выше или как описано в первом или втором воплощении, где лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет соединение A, или его фармацевтически приемлемую соль, где соединение A представляет N-(4-фторбензил)-5-гидрокси-1-метил-2-(1-метил-1-{[(5-метил-1,3,4-оксадиазол-2-ил)карбонил]амино}этил)-6-оксо-1,6-дигидропиримидин-4-карбоксамид. Соединение A имеет следующую структуру:
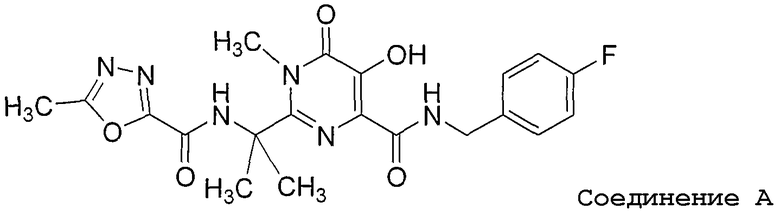
Соединение A, раскрытое в международной публикации № WO 03/035077, является эффективным ингибитором интегразы ВИЧ.
Шестое воплощение имеет нижеследующие аспекты, каждый из которых представляет способ улучшения PK, как описано ранее в шестом воплощении, где:
(1) количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, находится в интервале от приблизительно 2 мг/кг до приблизительно 10 мг/кг массы тела;
(2) количество соединения A, вводимое в сутки, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг;
(3) атазанавир вводят в комбинации в количестве, которое в случае введения только атазанавира меньше колчества, эффективного для лечения инфекции ВИЧ или СПИДа;
(4) количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, находится в интервале от приблизительно 2 мг/кг до приблизительно 5 мг/кг массы тела;
(5) количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг, а количество атазанавира, вводимое в сутки в комбинации, составляет менее 400 мг (например, от приблизительно 100 мг до приблизительно 350 мг в сутки или от приблизительно 100 мг до приблизительно 250 мг в сутки, или от приблизительно 100 мг до приблизительно 200 мг в сутки);
(6) количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 200 мг до приблизительно 1200 мг (например, от приблизительно 100 мг до приблизительно 600 мг два раза в сутки), а количество атазанавира, вводимое в сутки в комбинации, составляет менее 400 мг (например, от приблизительно 100 мг до приблизительно 350 мг в сутки или от приблизительно 100 мг до приблизительно 250 мг в сутки, или от приблизительно 100 мг до приблизительно 200 мг в сутки).
Следует понимать, что каждый из соединения A и атазанавира, или и тот, и другой, можно альтернативно использовать в вышеописанных аспектах шестого воплощения в виде фармацевтически приемлемых солей. В настоящих аспектах количество соединения A указано для его несолевой свободной фенольной формы, а количество атазанавира указано для его несолевой свободной основной формы.
Седьмое воплощение настоящего изобретения относится к способу улучшения PK, как описано выше или как описано в первом или втором воплощении, где лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет соединение A в виде калиевой соли. Аспекты настоящего воплощения включают аспекты, аналогичные аспектам (1)-(6), описанным выше для шестого воплощения. В настоящем воплощении и его аспектах калиевая соль соединения A предпочтительно представляет кристаллическую калиевую соль соединения A, более предпочтительно кристаллическую калиевую соль соединения A типа 1, где K соль типа 1 представляет безводную кристаллическую соль, охарактеризованную с помощью порошковой рентгенограммы, полученную с использованием медного излучения Kα (то есть источником излучения является комбинация излучения Cu Kα1 и Kα2) со значениями 2Θ (то есть отражения при значениях 2Θ) 5,9, 12,5, 20,0, 20,6 и 25,6 градусов.
Восьмое воплощение настоящего изобретения относится к способу улучшения PK, как описано выше или как описано в первом или втором воплощении, где лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет гидроксиполигидро-2,6-нафтиридиндионовое соединение формулы III, или его фармацевтически приемлемую соль:
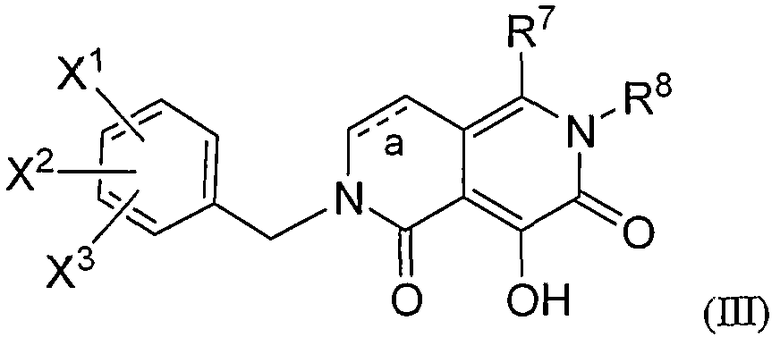
где:
связь " " в цикле представляет одинарную или двойную связь (например, одинарную связь);
" в цикле представляет одинарную или двойную связь (например, одинарную связь);
каждый из Х1 и Х2 независимо означает:
(1) -H,
(2) -C1-6алкил,
(3) -OH,
(4) -O-C1-6алкил,
(5) -C1-6галогеналкил,
(6) -О-C1-6галогеналкил,
(7) галоген,
(8) -CN,
(9) -N(Ra)Rb,
(10) -C(=O)N(Ra)Rb,
(11) -SRa,
(12) -S(O)Ra,
(13) SO2Ra,
(14) -N(Ra)SO2Rb,
(15) -N(Ra)SO2N(Ra)Rb,
(16) -N(Ra)C(=O)Rb,
(17) -N(Ra)C(=O)-C(=O)N(Ra)Rb,
(18) -HetK,
(19) -C(=O)- HetK или
(20) HetL;
где каждый HetK независимо означает C4-5азациклоалкил или C3-4диазациклоалкил, каждый из которых необязательно замещен 1 или 2 заместителями, каждый из которых независимо означает оксо или C1-6алкил; при условии, что, если HetK присоединен к остатку соединения через фрагмент -C(=О)-, HetK присоединяется к -C(=О)- через циклический атом N; и
каждый HetL независимо означает 5- или 6-членный гетероароматический цикл, содержащий от 1 до 4 гетероатомов, независимо выбранных из N, O и S, где гетероароматический цикл необязательно замещен 1-4 заместителями, каждый из которых независимо означает галоген, -C1-6алкил, -C1-6галогеналкил,
-O-C1-6алкил, -O-C1-6галогеналкил или гидрокси;
или, альтернативно, X1 и Х2 соответственно расположены на соседних атомах углерода фенильного цикла и вместе образуют метилендиокси или этилендиокси;
X3 означает:
(1) -H,
(2) -C1-6алкил,
(3) -O-C1-6алкил,
(4) -C1-6галогеналкил,
(5) -O-C1-6галогеналкил или
(6) галоген;
R7 означает:
(1) -C1-6алкил,
(2) -CО2Ra,
(3) -C(=O)N(Ra)Rb,
(4) -C(=O)-N(Ra)-(CH2)2-3-ORb,
(5) -N(Ra)C(O)Rb,
(6) -N(Ra)SO2Rb,
(7) -C3-6циклоалкил, который необязательно замещен 1-4 заместителями, каждый из которых независимо означает галоген, -C1-6алкил, -CF3, -O-C1-6алкил или -OCF3,
(8) -HetK,
(9) -C(=O)-HetK,
(10) -C(O)N(Ra)-HetK,
(11) -C(=О)N(Ra)-(CH2)0-2-(C3-6циклоалкил), где циклоалкил необязательно замещен 1-4 заместителями, каждый из которых независимо означает галоген,
-C1-6алкил, -CF3, -O-C1-6алкил или -OCF3, или
(12) -C(=О)N(Ra)-CH2-фенил, где фенил необязательно замещен 1-4 заместителями, каждый из которых независимо означает -C1-6алкил, -O-C1-6алкил, -CF3, -OCF3 или галоген;
(13) -HetL,
(14) -C(O)N(Ra)Rc или
(15) галоген;
где HetK означает 5- или 6-членный насыщенный гетероциклический фрагмент, содержащий всего от 1 до 4 гетероатомов, независимо выбранных из 1-4 атомов N, 0-2 атомов O и 0-2 атомов S, где гетероциклический фрагмент необязательно замещен (i) 1-4 заместителями, каждый из которых независимо означает -C1-6алкил, оксо, галоген, -C(=O)N(Ra)Rb, -C(=O)(=O)N(Ra)Rb, -C(=O)Ra, -CО2Ra, -SО2Ra или -SО2N(Ra)Rb и (ii) 0-1 C3-6циклоалкилом; при условии, что, если HetK присоединен к остатку соединения через фрагмент -C(=O)-, HetK присоединен к -C(=O)- через циклический атом N;
где HetL означает 5- или 6-членный гетероароматический цикл, содержащий от 1 до 4 гетероатомов, независимо выбранных из N, O и S, где гетероароматический цикл необязательно замещен 1-4 заместителями, каждый из которых независимо означает
-C1-6алкил или -OH;
R8 означает:
(1) -H,
(2) -C1-6алкил,
(3) -C3-6циклоалкил,
(4) -(CH2)1-2-C3-6циклоалкил,
(5) -CH2-фенил, где фенил необязательно замещен 1-4 заместителями, каждый из которых независимо означает галоген, C1-6алкил, C1-6галогеналкил, -О-C1-6алкил или -O-C1-6галогеналкил,
(6) -(CH2)1-2-HetM, где HetM означает 4-7-членный насыщенный гетероциклический фрагмент, содержащий от 1 до 2 гетероатомов, независимо выбранных из 1-2 атомов N, 0-1 атома O и 0-1 атома S, где гетероциклический фрагмент присоединен к остатку молекулы через циклический атом N, и гетероциклический фрагмент необязательно замещен 1-4 заместителями, каждый из которых независимо означает
-C1-6алкил, -C1-6галогеналкил, -O-C1-6алкил, -O-C1-6галогеналкил, оксо,
-C(=O)N(Ra)Rb, -C(=O)Ra, -CО2Ra, -SО2Ra или -SО2N(Ra)Rb,
(7) фенил, который необязательно замещен 1-4 заместителями, каждый из которых независимо означает -C1-6алкил, -O-C1-6алкил, -C1-6галогеналкил,
-O-C1-6галогеналкил, -OH, галоген, -CN, -NO2, -C(=O)Ra, -CО2Ra, -SО2Ra,
-N(Ra)C(=O)-C1-6галогеналкил, -N(Ra)C(=O)Rb, -N(Ra)C(=O)N(Ra)Rb, -N(Ra)CО2Rb,
-N(Ra)SO2Rb, -C(=O)N(Rd)Re или -SO2N(Rd)Re;
(8) 5- или 6-членный гетероароматический цикл, содержащий от 1 до 4 гетероатомов, независимо выбранных из N, O и S, где гетероароматический цикл необязательно замещен 1-4 заместителями, каждый из которых независимо означает
-C1-6алкил, -C1-6галогеналкил, -O-C1-6алкил, -O-C1-6галогеналкил или -OH,
(9) C1-6алкил, замещенный -O-C1-6алкилом, -CN, -N(Ra)Rb, -C(=O)N(Ra)Rb, -C(=O)Ra, -CО2Ra, -SО2Ra или -SО2N(Ra)Rb или
(10) -C1-6галогеналкил;
каждый Ra независимо означает H или C1-6алкил;
каждый Rb независимо означает H или C1-6алкил;
Rс означает C1-6галогеналкил или C1-6алкил, замещенный -CО2Ra, -SО2Ra,
-SО2N(Ra)Rb или N(Ra)Rb; и
каждый из Rd и Re независимо означает H или C1-6алкил, или вместе с атомом N, к которому они присоединены, образуют 4-7-членный насыщенный гетероциклический фрагмент, необязательно содержащий гетероатом, помимо атома азота, присоединенного к Rd и Re, выбранный из N, O и S, где S необязательно окислен до S(O) или S(O)2, и где насыщенный гетероциклический фрагмент необязательно замещен 1-4 заместителями, каждый из которых независимо означает галоген, -CN,
-C1-6алкил, -OH, оксо, -O-C1-6алкил, -C1-6галогеналкил, -C(=O)Ra, -CО2Ra, -SО2Ra или
-SО2N(Ra)Rb.
Девятое воплощение настоящего изобретения относится к способу улучшения PK, как описано выше или как описано в первом или втором воплощении, где лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет гидрокси полигидро-2,6-нафтиридиндионовое соединение формулы IV, или его фармацевтически приемлемую соль:
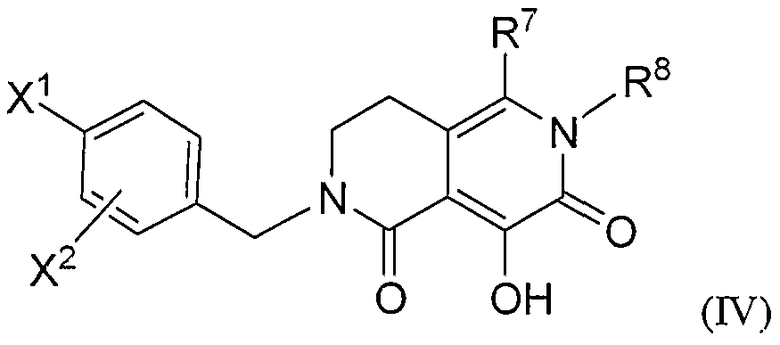
где:
X1 означает: (1) -H, (2) бром, (3) хлор, (4) фтор или (5) метокси;
X2 означает: (1) -H, (2) бром, (3) хлор, (4) фтор, (5) метокси, (6) -C1-4алкил, (7) -CF3, (8) -OCF3, (9) -CN или (10) -SO2(C1-4алкил);
R7 означает: (1) -CO2H, (2) -C(=O)-O-C1-4алкил, (3) -C(=О)NH2,
(4) -C(=O)NH-C1-4алкил, (5) -C(=О)N(C1-4алкил)2, (6) -C(=O)-NH-(CH2)2-3-O-C1-4алкил,
(7) -C(=О)-N(C1-4алкил)-(CH2)2-3-O-C1-4алкил, (8) -NHC(=O)-C1-4алкил,
(9) -N(C1-4алкил)C(=O)-C1-4алкил, (10) -NHSO2-C1-4алкил, (11) -N(C1-4алкил)SO2-C1-4алкил,
(12) -C(=O)-HetK, где HetK означает:
 ,
,  ,
,  ,
,  ,
,  ,
,  или
или 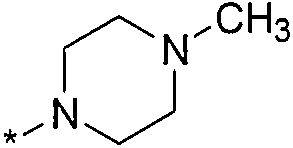 ,
,
где символ * означает точку присоединения к остатку соединения,
(13) -C(=O)NH-(CH2)0-1-(C3-6циклоалкил),
(14) -C(=O)N(C1-4алкил)-(CH2)0-1-(C3-6циклоалкил), (15) -C(=O)NH-CH2-фенил или
(16) -C(=O)N(C1-4алкил)-CH2-фенил; и
R8 означает: (1) -H, (2) -C1-4алкил, (3) циклопропил, (4) циклобутил,
(5) -CH2-циклопропил, (6) -CH2-циклобутил или (7) -CH2-фенил.
В одном аспекте девятого воплощения Х1 означает фтор; Х2 означает -H или хлор; R7 означает:
(1) -C(=О)N(C1-3алкил)2,
(2) -C(=O)-HetK, где HetK означает:
, , , , или ,
где символ * означает точку присоединения к остатку соединения,
(3) -C(O)N(C1-3алкил)-(CH2)0-1-циклопропил или
(4) -C(O)N(C1-3алкил)-(CH2)0-1-циклобутил; и
R8 означает -C1-4алкил.
Десятое воплощение настоящего изобретения относится к способу улучшения PK, как описано выше или как описано в первом или втором воплощении, где лекарственное средство, непосредственно метаболизируемое UGT1A1, выбрано из группы, состоящей из:
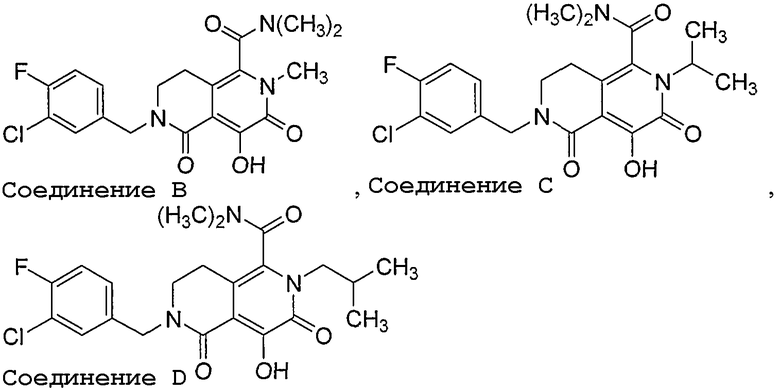 , , ,
, , ,
и их фармацевтически приемлемых солей.
В одном аспекте десятого воплощения соединение представляет соединение B. В другом аспекте десятого воплощения соединение представляет соединение C. В следующем аспекте десятого воплощения соединение представляет соединение D.
Соединения, охватываемые формулой III и формулой IV, и соединения B, C и D являются ингибиторами интегразы ВИЧ. Указанные соединения, их получение и применение дополнительно описаны в WO 2005/087768.
Настоящее изобретение также относится к способу повышения уровня перорального лекарственного средства, непосредственно метаболизируемого UGT1A1, в кровотоке, который предусматривает пероральное введение млекопитающему, нуждающемуся в лечении лекарственным средством, эффективного количества комбинации лекарственного средства, или его фармацевтически приемлемой соли, и атазанавира, или его фармацевтически приемлемой соли. Повышение уровня лекарственного средства в кровотоке в настоящем описании означает повышение уровня лекарственного средства в большом круге кровообращения (например, в кровотоке человека) по сравнению с соответствующим значением, полученным при введении лекарственного средства в отсутствие атазанавира. Указанный способ включает нижеследующие воплощения, каждое из которых относится к способу повышения уровня в кровотоке, как описано ранее, где:
(1) атазанавир вводят в комбинации в количестве, достаточном для повышения уровня лекарственного средства в кровотоке, по меньшей мере, на приблизительно 10% по отношению к уровню в кровотоке лекарственного средства, вводимого в отсутствие атазанавира;
(2) млекопитающим, нуждающимся в лечении лекарственным средством, является человек;
(3) млекопитающим, нуждающимся в лечении лекарственным средством, является человек, а лекарственное средство, непосредственно метаболизируемое UGT1A1, выбрано из группы, состоящей из эзетимиба, ралоксифена, эстрадиола и их фармацевтически приемлемых солей;
(4) лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет ингибитор интегразы ВИЧ;
(4a) способ, описанный в абзаце (4), где млекопитающим, нуждающимся в лечении лекарственным средством, является человек;
(4b) способ, описанный в абзаце (4), где атазанавир вводят в комбинации в количестве, достаточном для повышения уровня в кровотоке ингибитора интегразы, по меньшей мере, на приблизительно 10% по отношению к уровню в кровотоке соединения I, вводимого в отсутствие атазанавира;
(4c) способ, описанный в абзаце (4), где атазанавир вводят в комбинации в количестве, которое, в случае введения только атазанавира, меньше количества, эффективного для лечения инфекции ВИЧ или СПИДа;
(4d) способ, описанный в абзаце (4), где способ включает признак (4a) и один из признаков (4b) и (4c);
(4e) способ, описанный в абзаце (4), где способ включает признаки (4a), (4b) и (4c);
(5) лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет соединение формулы I, как указано выше, или его фармацевтически приемлемую соль;
(5a) способ, описанный в абзаце (5), где млекопитающим, нуждающимся в лечении лекарственным средством, является человек;
(5b) способ, описанный в абзаце (5), где атазанавир вводят в комбинации в количестве, достаточном для повышения уровня в кровотоке соединения I, по меньшей мере, на приблизительно 10% по отношению к уровню в кровотоке соединения I, вводимого в отсутствие атазанавира;
(5c) способ, описанный в абзаце (5), где атазанавир вводят в комбинации в количестве, которое, в случае введения только атазанавира, меньше количества, эффективного для лечения инфекции ВИЧ или СПИДа;
(5d) способ, описанный в абзаце (5), где способ включает признак (5a) и один из признаков (5b) и (5c);
(5e) способ, описанный в абзаце (5), где способ включает признаки (5a), (5b) и (5c);
(6) лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет соединение A, как указано выше, или его фармацевтически приемлемую соль;
(6a) способ, описанный в абзаце (6), где млекопитающим, нуждающимся в лечении лекарственным средством, является человек;
(6b) способ, описанный в абзаце (6), где атазанавир вводят в комбинации в количестве, достаточном для повышения уровня в кровотоке соединения A, по меньшей мере, на приблизительно 10% по отношению к уровню в кровотоке соединения A, вводимого в отсутствие атазанавира;
(6c) способ, описанный в абзаце (6), где количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, находится в интервале от приблизительно 2 мг/кг до приблизительно 10 мг/кг (или от приблизительно 5 мг/кг до приблизительно 10 мг/кг) массы тела;
(6d) способ, описанный в абзаце (6), где атазанавир вводят в комбинации в количестве, которое, в случае введения только атазанавира, меньше количества, эффективного для лечения инфекции ВИЧ или СПИДа;
(6e) способ, описанный в абзаце (6), где количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, находится в интервале от приблизительно 2 мг/кг до приблизительно 5 мг/кг массы тела;
(6f) способ, описанный в абзаце (6), где количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, составляет менее 400 мг (например, от приблизительно 100 мг до приблизительно 350 мг в сутки или от приблизительно 100 мг до приблизительно 250 мг в сутки, или от приблизительно 100 мг до приблизительно 200 мг в сутки);
(6g) способ, описанный в абзаце (6), где количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 200 мг до приблизительно 1200 мг (например, от приблизительно 100 мг до приблизительно 600 мг два раза в сутки), а количество атазанавира, вводимое в сутки в комбинации, составляет менее 400 мг (например, от приблизительно 100 мг до приблизительно 350 мг в сутки или от приблизительно 100 мг до приблизительно 250 мг в сутки, или от приблизительно 100 мг до приблизительно 200 мг в сутки);
(6h) способ, описанный в абзаце (6), где способ включает признак (6a) и один из признаков (6b)-(6g);
(7) лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет калиевую соль соединения A (предпочтительно кристаллическую калиевую соль соединения A, более предпочтительно первую форму кристаллической калиевой соли соединения A);
(7a)-(7h) каждый из указанных способов подобен описанному в абзаце (7) и соответственно включает признаки, аналогичные указанным в приведенных выше абзацах (6a)-(6h);
(8) лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет соединение формулы III, как указано выше, или его фармацевтически приемлемую соль;
(8a)-(8e) каждый из указанных способов подобен описанному в абзаце (8) и соответственно включает признаки, аналогичные указанным в приведенных выше абзацах (5a)-(5e);
(9) лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет соединение формулы IV, как указано выше, или его фармацевтически приемлемую соль;
(9a)-(9e) каждый из указанных способов подобен описанному в абзаце (9) и соответственно включает признаки, аналогичные указанным в приведенных выше абзацах (5a)-(5e);
(10) лекарственное средство, непосредственно метаболизируемое UGT1A1, представляет соединение, выбранное из группы, состоящей из соединения B, соединения C и соединения D, или его фармацевтически приемлемую соль;
(10a)-(10e) каждый из указанных способов подобен описанному в абзаце (10) и соответственно включает признаки, аналогичные указанным в приведенных выше абзацах (5a)-(5e).
Настоящее изобретение также включает способ ингибирования интегразы ВИЧ, который предусматривает введение млекопитающему, нуждающемуся в таком ингибировании, эффективного количества комбинации ингибитора интегразы ВИЧ, непосредственно метаболизируемого UGT1A1, или его фармацевтически приемлемой соли, и атазанавира, или его фармацевтически приемлемой соли. Указанный способ включает нижеследующие воплощения, каждое из которых относится к способу ингибирования интегразы ВИЧ, как описано ранее, где:
(1) атазанавир вводят в комбинации в количестве, достаточном для улучшения PK ингибитора интегразы ВИЧ, по меньшей мере, на приблизительно 10% по отношению к PK ингибитора интегразы ВИЧ, вводимого в отсутствие атазанавира;
(2) млекопитающим, нуждающимся в лечении ингибитором интегразы ВИЧ, является человек;
(3) ингибитор интегразы ВИЧ, непосредственно метаболизируемый UGT1A1, представляет соединение формулы I, описанное выше, или его фармацевтически приемлемую соль;
(3a) способ, описанный в абзаце (3), где млекопитающим, нуждающимся в лечении лекарственным средством, является человек;
(3b) способ, описанный в абзаце (3), где атазанавир вводят в комбинации в количестве, достаточном для улучшения PK ингибитора интегразы ВИЧ, по меньшей мере, на приблизительно 10% по отношению к PK ингибитора интегразы ВИЧ, вводимого в отсутствие атазанавира;
(3c) способ, описанный в абзаце (3), где атазанавир вводят в комбинации в количестве, которое, в случае введения только атазанавира, меньше количества, эффективного для лечения инфекции ВИЧ или СПИДа;
(3d) способ, описанный в абзаце (3), где способ включает признак (3a) и один или оба из признаков (3b) и (3c);
(4) ингибитор интегразы ВИЧ, непосредственно метаболизируемый UGT1A1, представляет соединение A, описанное выше, или его фармацевтически приемлемую соль;
(4a) способ, описанный в пункте (4), где млекопитающим, нуждающимся в лечении лекарственным средством, является человек;
(4b) способ, описанный в пункте (4), где атазанавир вводят в комбинации в количестве, достаточном для улучшения PK соединения A, по меньшей мере, на приблизительно 10% по отношению к PK соединения A, вводимого в отсутствие атазанавира;
(4c) способ, описанный в абзаце (4), где количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, находится в интервале от приблизительно 2 мг/кг до приблизительно 10 мг/кг (или от приблизительно 5 мг/кг до приблизительно 10 мг/кг) массы тела;
(4d) способ, описанный в абзаце (4), где атазанавир вводят в комбинации в количестве, которое, в случае введения только атазанавира, меньше количества, эффективного для лечения инфекции ВИЧ или СПИДа;
(4e) способ, описанный в абзаце (4), где количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, находится в интервале от приблизительно 2 мг/кг до приблизительно 5 мг/кг массы тела;
(4f) способ, описанный в абзаце (4), где количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, составляет менее 400 мг (например, от приблизительно 100 мг до приблизительно 350 мг в сутки или от приблизительно 100 мг до приблизительно 250 мг в сутки, или от приблизительно 100 мг до приблизительно 200 мг в сутки);
(4g) способ, описанный в абзаце (4), где количество соединения A, вводимое в сутки в комбинации, находится в интервале от приблизительно 200 мг до приблизительно 1200 мг (например, от приблизительно 100 мг до приблизительно 600 мг два раза в сутки), а количество атазанавира, вводимое в сутки в комбинации, составляет менее 400 мг (например, от приблизительно 100 мг до приблизительно 350 мг в сутки или от приблизительно 100 мг до приблизительно 250 мг в сутки, или от приблизительно 100 мг до приблизительно 200 мг в сутки);
(4h) способ, описанный в абзаце (4), где способ включает признак (4a) и один из признаков (4b)-(4g);
(5) ингибитор интегразы ВИЧ, непосредственно метаболизируемый UGT1A1, представляет калиевую соль соединения A (предпочтительно кристаллическую калиевую соль соединения A, более предпочтительно первую форму кристаллической калиевой соли соединения A;
(5a)-(5h) каждый из указанных способов подобен описанному в пункте (5) и соответственно включает признаки, аналогичные указанным в приведенных выше абзацах (4a)-(4h);
(6) ингибитор интегразы ВИЧ, непосредственно метаболизируемый UGT1A1, представляет соединение формулы III, или его фармацевтически приемлемую соль;
(6a)-(6d) каждый из указанных способов подобен описанному в абзаце (6) и соответственно включает признаки, аналогичные указанным в приведенных выше абзацах (3a)-(3d);
(7) ингибитор интегразы ВИЧ, непосредственно метаболизируемый UGT1A1, представляет соединение формулы IV, или его фармацевтически приемлемую соль;
(7a)-(7d) каждый из указанных способов подобен описанному в пункте (7) и соответственно включает признаки, аналогичные указанным в приведенных выше абзацах (3a)-(3d);
(8) ингибитор интегразы ВИЧ, непосредственно метаболизируемый UGT1A1, представляет соединение, выбранное из группы, состоящей из соединения B, соединения C и соединения D, или его фармацевтически приемлемую соль;
(8a)-(8d) каждый из указанных способов подобен описанному в абзаце (8) и соответственно включает признаки, аналогичные указанным в приведенных выше абзацах (3a)-(3d).
Настоящее изобретение также включает способ лечения инфекции ВИЧ или СПИДа, способ профилактики инфекции ВИЧ или СПИДа или способ замедления развития СПИДа, который предусматривает пероральное введение млекопитающему, нуждающемуся в таком лечении, профилактике или замедлении развития, эффективного количества комбинации ингибитора интегразы ВИЧ, непосредственно метаболизируемого UGT1A1, или его фармацевтически приемлемой соли, и атазанавира, или его фармацевтически приемлемой соли. Указанный способ включает воплощения, аналогичные воплощениям (1), (2), (3)-(3d), (4)-(4h), (5)-(5h), (6)-(6d), (7)-(7d) и (8)-(8d), описанным выше для способа ингибирования интегразы ВИЧ.
Настоящее изобретение также включает фармацевтическую комбинацию для перорального введения млекопитающему, включающую лекарственное средство, используемое для лечения или профилактики заболевания или состояния и непосредственно метаболизируемое UGT1A1, или его фармацевтически приемлемую соль, и атазанавир, или его фармацевтически приемлемую соль, где каждый из лекарственного средства и атазанавира используется в количестве, которое обеспечивает терапевтическую или профилактическую эффективность лекарственного средства. Указанная комбинация включает нижеследующие воплощения, каждое из которых представляет описанную выше комбинацию, где:
(1) млекопитающим, которому вводят комбинацию, является человек;
(2) атазанавир вводят в комбинации в количестве, достаточном для улучшения фармакокинетических характеристик лекарственного средства, по меньшей мере, на приблизительно 10% по отношению к фармакокинетическим характеристикам лекарственного средства, вводимого в отсутствие атазанавира;
(3) млекопитающим, которому вводят комбинацию, является человек, а лекарственное средство выбрано из группы, состоящей из эзетимиба, ралоксифена, эстрадиола и их фармацевтически приемлемых солей;
(4) комбинация входит в состав одной фармацевтической композиции, которая также содержит фармацевтически приемлемый носитель;
(5) комбинация включает признак (1) и один или оба из признаков (2) и (4);
(6) комбинация включает признак (2) и один или оба из признаков (3) и (4);
(7) комбинация включает признаки (3) и (4).
Настоящее изобретение также включает фармацевтическую комбинацию для перорального введения млекопитающему, содержащую ингибитор интегразы ВИЧ, непосредственно метаболизируемый UGT1A1, или его фармацевтически приемлемую соль, и атазанавир, или его фармацевтически приемлемую соль, где каждый из ингибитора интегразы ВИЧ и атазанавира используются в количестве, которое обеспечивает эффективность ингибитора интегразы для (i) лечения инфекции ВИЧ или СПИДа, (ii) профилактики инфекции ВИЧ или СПИДа, или (iii) ингибирования интегразы ВИЧ. Настоящая комбинация включает воплощения (1), (2), (3)-(3d), (4)-(4h), (5)-(5h), (6)-(6d), (7)-(7d) и (8)-(8d), описанные выше для способа ингибирования интегразы ВИЧ. Другие воплощения настоящей комбинации включают комбинацию, описанную выше в каждом из вышеуказанных воплощений, где комбинация входит в состав одной фармацевтической композиции, которая также содержит фармацевтически приемлемый носитель.
Настоящее изобретение также включает применение атазанавира или его фармацевтически приемлемой соли в комбинации с пероральным лекарственным средством, непосредственно метаболизируемым UGT1A1, или его фармацевтически приемлемой солью, для улучшения фармакокинетических характеристик (или уровня в кровотоке) лекарственного средства у млекопитающего, нуждающегося в лечении лекарственным средством. Настоящее изобретение также относится к применению атазанавира, или его фармацевтически приемлемой соли, в комбинации с пероральным лекарственным средством, непосредственно метаболизируемым UGT1A1, или его фармацевтически приемлемой солью, в производстве лекарственного средства, улучшающего фармакокинетические характеристики (или уровень в кровотоке) лекарственного средства у млекопитающего, нуждающегося в лечении таким лекарственным средством. Воплощения указанных применений аналогичны воплощениям, описанным выше в соответствующих пунктах способов.
Кроме того, настоящее изобретение включает применение атазанавира, или его фармацевтически приемлемой соли, в комбинации с пероральным ингибитором интегразы ВИЧ, непосредственно метаболизируемым UGT1A1, или его фармацевтически приемлемой солью, для ингибирования интегразы ВИЧ у млекопитающего, нуждающегося в таком ингибировании. Настоящее изобретение также включает применение атазанавира, или его фармацевтически приемлемой соли, в комбинации с пероральным ингибитором интегразы ВИЧ, непосредственно метаболизируемым UGT1A1, или его фармацевтически приемлемой солью, в производстве лекарственного средства, ингибирующего интегразу ВИЧ у млекопитающего, нуждающегося в таком ингибировании. Воплощения указанных применений аналогичны воплощениям, описанным выше в соответствующих пунктах способов.
Настоящее изобретение также включает применение атазанавира, или его фармацевтически приемлемой соли, в комбинации с пероральным ингибитором интегразы ВИЧ, непосредственно метаболизируемым UGT1A1, или его фармацевтически приемлемой солью, для лечения инфекции ВИЧ или СПИДа, для профилактики инфекции ВИЧ или СПИДа или для замедления развития СПИДа у млекопитающего, нуждающегося в таком лечении, профилактике или замедлении развития. Настоящее изобретение также включает применение атазанавира, или его фармацевтически приемлемой соли, в комбинации с пероральным ингибитором интегразы ВИЧ, непосредственно метаболизируемым UGT1A1, или его фармацевтически приемлемой солью, в производстве лекарственного средства для лечения инфекции ВИЧ или СПИДа, для профилактики инфекции ВИЧ или СПИДа или для замедления развития СПИДа у млекопитающего, нуждающегося в таком лечении, профилактике или замедлении развития. Воплощения указанных применений аналогичны воплощениям, описанным выше в соответствующих пунктах способов.
Комбинацию атазанавира и лекарственного средства, непосредственно метаболизируемого UGT1A1, в составе одной или разных композиций, вводят перорально. Можно использовать жидкие композиции, включающие, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие композиции можно получить с помощью известных в данной области методов и с использованием любой традиционной среды, такой как вода, гликоли, масла, спирты и тому подобное. Можно также использовать твердые композиции, включающие, например, порошки, гранулы, пиллюли, капсулы и таблетки. Твердые композиции можно получить с помощью известных в данной области методов и с использованием таких твердых наполнителей, как крахмалы, сахара, каолин, смазывающие средства, связующие средства, разрыхляющие средства и тому подобное.
Суточная доза атазанавира, вводимая человеку или другому млекопитающему в комбинации с UGT1A1-метаболизируемым лекарственным средством, обычно представляет количество, достаточное для улучшения фармакокинетических характеристик лекарственного средства, по меньшей мере, на приблизительно 10% по отношению к фармакокинетическим характеристикам лекарственного средства, вводимого в отсутствие атазанавира. Руководство по установлению подходящей пероральной дозы атазанавира можно найти в US 5849911, а также на этикетке разрешенного к применению лекарственного продукта REYATAZTM (капсулы сульфата атазанавира; смотрите, например, Physicians' Desk Reference, 2004 edition, pp. 1080-1088). Суточная доза перорального лекарственного средства, метаболизируемого UGT1A1, вводимого в комбинации с атазанавиром, представляет количество, эффективное против конкретного заболевания или состояния, подлежащего лечению или профилактике. Способы установления подходящих суточных доз для таких лекарственных средств известны в данной области. Для многих лекарственных средств такие способы можно найти, например, в издании 2004 г. Physicians' Desk Reference. Уровни доз также можно найти в патентной литературе; например, информацию по уровням доз для эзетимиба и ралоксифена можно найти в US RE37721 и US 6458811 соответственно. Конкретные уровни доз атазанавира и лекарственного средства зависят от ряда факторов, включающих (i) активность конкретного лекарственного средства, используемого в комбинации; (ii) возраст, массу тела, общее состояние здоровья, пол и диету субъекта (человека или другого млекопитающего); (iii) способа перорального введения, (iv) скорости выведения и (v) тяжести конкретного заболевания или состояния, подлежащего лечению. Подходящие пероральные дозы атазанавира и лекарственного средства для лечения или профилактики конкретного заболевания или состояния у конкретного субъекта (то есть человека или другого млекопитающего) может определить специалист в данной области, не прибегая к излишнему экспериментированию.
Соединения, охватываемые формулой I, формулой III и формулой IV, можно вводить в дозировочном интервале от приблизительно 0,001 до приблизительно 1000 мг/кг массы тела млекопитающего (например, человека) в сутки однократно или в несколько приемов. Предпочтительный дозировочный интервал составляет от приблизительно 0,01 до приблизительно 500 мг/кг массы тела в сутки однократно или в несколько приемов. Другой предпочтительный дозировочный интервал составляет от приблизительно 0,1 до приблизительно 100 мг/кг массы тела в сутки однократно или в несколько приемов. Композиции, содержащие соединение формулы I, можно получать в виде таблеток или капсул для перорального введения, где каждая таблетка или капсула содержит от приблизительно 1 до приблизительно 1000 миллиграмм активного ингредиента, в частности 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 700, 800, 900 и 1000 миллиграмм активного ингредиента, для симптоматической коррекции дозы у пациента, подлежащего лечению. Конечно, конкретный уровень дозы и частоту введения для конкретного пациента можно варьировать в зависимости от ряда факторов, включающих факторы (i)-(v), описанные в предыдущем абзаце.
Ниже приведены подходящие суммарные суточные дозы соединения A и атазанавира:
от приблизительно 5 мг/кг до приблизительно 10 мг/кг
(то есть от приблизительно 100 мг до приблизительно 350 мг, от приблизительно 100 мг до приблизительно 250 мг, или от приблизительно 100 мг до приблизительно 200 мг)
от приблизительно 200 мг до приблизительно 1200 мг
(то есть от приблизительно 100 мг до приблизительно 600 мг два раза в сутки)
(то есть от приблизительно 100 мг до приблизительно 350 мг, от приблизительно 100 мг до приблизительно 250 мг, или от приблизительно 100 мг до приблизительно 200 мг)
(то есть приблизительно 400 мг два раза в сутки)
(то есть от приблизительно 100 мг до приблизительно 350 мг, от приблизительно 100 мг до приблизительно 250 мг, или от приблизительно 100 мг до приблизительно 200 мг)
Соединение A предпочтительно вводят в виде калиевой соли (особенно в виде первой формы кристаллической калиевой соли). В предпочтительном воплощении калиевую соль соединения A вводят перорально в составе фармацевтической композиции, содержащей K соль соединения A и гидроксипропилметилцеллюлозу (например, HPMC 2910), где композицию получают в виде таблеток. В другом предпочтительном воплощении калиевую соль соединения A вводят перорально в составе фармацевтической композиции, содержащей K соль соединения A полоксамер (например, полоксамер 407), гидроксипропилметилцеллюлозу (например, HPMC K4M) и лактозу (например, высушенную распылением водосодержащую лактозу), где композицию получают в виде таблеток.
Если специально не указано иначе, все приводимые в настоящем описании интервалы являются включающими. Например, если указано, что гетероциклический фрагмент содержит "от 1 до 4 гетероатомов", это означает, что цикл может содержать 1, 2, 3 или 4 гетероатома. В другом примере, если указано, что суточная доза соединения A находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, это означает, что доза может составлять приблизительно 5 мг/кг или приблизительно 10 мг/кг, или она может находиться между этими значениями.
В настоящем описании используются следующие сокращения: ACN = ацетонитрил; СПИД = синдром приобретенного иммунодефицита; ARC = СПИД-ассоциированный комплекс; Bz = бензоил; CBz = бутилоксикарбонил; DIEA = диизопропилэтиламин; DMADC = диметилацетилен дикарбоксилат; ДМФА = N,N-диметилформамид; ДМСО = диметилсульфоксид; DSC = дифференциальная сканирующая калориметрия; ЭДТА = этилендиаминтетрауксусная кислота; экв. = эквивалент(ы); EtOH = этанол; ВИЧ = вирус иммунодефицита человека; ВЭЖХ = высокоэффективная жидкостная хроматография; HPMC = гидроксипропилметилцеллюлоза; IPA = изопропиловый спирт; KF = титрование Карла-Фишера для воды; ЖХ = жидкостная хроматография; LCAP = процент площади ЖХ; LCWP = массовый процент ЖХ; Me = метил; MeOH = метанол; МС = масс-спектроскопия; MSA = метансульфоновая кислота; MTBE = простой метил-трет-бутиловый эфир; мол.масса = молекулярная масса; NMM = N-метилморфолин; ЯМР = ядерный магнитный резонанс; PK = фармакокинетические характеристики; TG = термогравиметрия; ТГФ = тетрагидрофуран; UDPGA = уридин 5'-дифосфоглюкуроновая кислота; XRPD = рентгеновская порошковая дифрактометрия.
Нижеследующие примеры служат только для иллюстрации изобретения и его осуществления. Приведенные примеры не следует понимать как ограничивающие объем или сущность настоящего изобретения. Соединение B в примерах 4 и 5 представляет 5-(1,1-диоксидо-1,2-тиазинан-2-ил)-N-(4-фторбензил)-8-гидрокси-1,6-нафтиридин-7-карбоксамид, описанный в US 2003/055071.
ПРИМЕР 1
Получение соединения A и его кристаллической калиевой соли
Стадия 1: Получение стрекер-амина (Strecker amine)
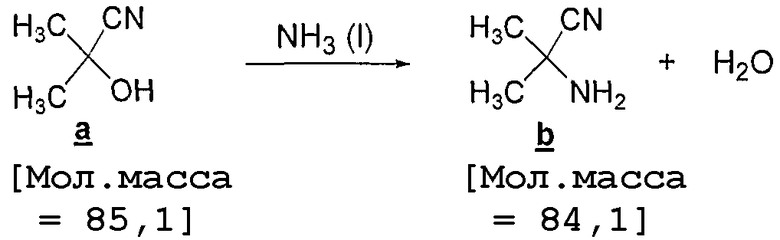
Ацетонциангидрин (11,5 кг, 12,3 л) загружают в 5-галлоновый автоклав и сосуд помещают в атмосферу азота под давлением 5 фунт/кв.дюйм. Автоклав охлаждают до 10°C и в сосуд подают газообразный аммиак (~3,44 кг) под давлением 30 фунт/кв.дюйм до достижения полного превращения, которое устанавливают методом ГХ (менее 0,5% a ). Полученную суспензию переносят в полимерную емкость и автоклав промывают MTBE (приблизительно 17 л). Затем реакционную смесь и смыв загружают в экстрактор объемом 100 л, затем туда вносят MTBE (15 л), смесь перемешивают и слои осторожно разделяют. Водный слой снова экстрагируют MTBE (5 л) и слои осторожно разделяют. Органические слои объединяют, загружают в 100-литровую колбу, оборудованную периодическим концентратором со встроенным фильтром, и содержимое концентрируют (15-20°C, низкий вакуум) до 20 л, чтобы удалить любой избыток аммиака. Аминонитрил получают с 97% выходом (11,1 кг) по результатам анализа ЯМР в виде раствора в MTBE.
Стадия 2: Присоединение защитной группы бензилоксикарбонила (CBz)
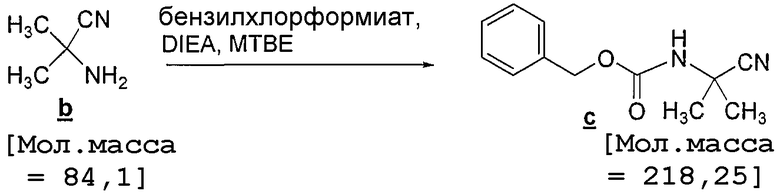
В визуально прозрачную 100-литровую колбу, содержащую дополнительную воронку объемом 5 л, термопару и впускное отверстие для азота, загружают 59% (по массе) раствор цианоамина b в MTBE (4,44 кг по результатам анализа). Затем раствор разбавляют MTBE (62,5 л), чтобы довести концентрацию приблизительно до приблизительно 15 мл/г. Затем в течение 15 минут через дополнительную воронку загружают бензилхлорформиат (1,20 экв., 10,42 кг, 61,10 моль) с такой скоростью, чтобы поддерживать температуру содержимого ниже 35°C. Затем к желтой взвеси в течение 1,5 часа добавляют DIEA (1,3 экв., 8,88 кг, 68,70 моль), поддерживая температуру загрузки ниже 35°C. При добавлении DIEA растворимость взвеси немного увеличивается, однако после прекращения перемешивания наблюдаются две фазы. Реакционную смесь выдерживают в течение 16 часов при 20-25°C, после чего к загрузке добавляют деионизованную воду (20 л, 4,5 мл/г). Затем загрузку переносят в 100-литровый экстрактор и фазы разделяют. Органический слой промывают водой, 3×10 л, и затем насыщенным соляным раствором, 15 л. Органический слой переносят через встроенный фильтр с размером пор 10 мкм в 100-литровую круглодонную колбу и затем растворитель меняют на смесь гептан:MTBE 90:10. В процессе смены растворителя происходит кристаллизация и полученный белый кристаллический подукт фильтруют и промывают 3×5 л смеси гептан:MTBE 90:10. Получают всего 10,1 кг продукта (выход 88%) с чистотой более 99% по данным ВЭЖХ A. От 3 загрузок получают в сумме 26,7 кг продукта со средним выходом после выделения 86%.
Стадия 3: Получение амидоксима
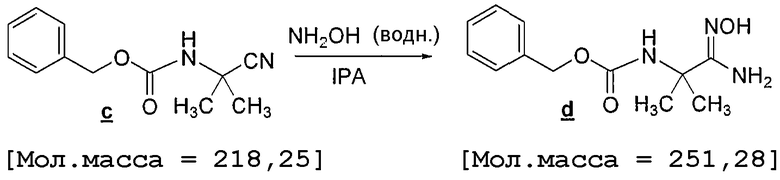
Раствор аминонитрила (15 г) в IPA (40 мл) нагревают до 60°C при перемешивании и при этой же температуре в течение 20 минут добавляют NH2OH в воде (5,05 мл). Затем прозрачную смесь выдерживают при 60°C в течение 3 часов, при указанной температуре через 2 часа продукт начинает выкристаллизовываться из раствора. Затем взвесь охлаждают до 0-5°C и в течение 20 минут по каплям добавляют н-гептан (40 мл). После перемешивания в течение 2 часов при 0-5°C взвесь фильтруют, кек промывают 20% раствором IPA в гептане (60 мл) и сушат в вакууме в потоке азота при комнатной температуре, получая чистый амидоксим с выходом 88%.
Стадия 4: Получение гидроксипиримидинона
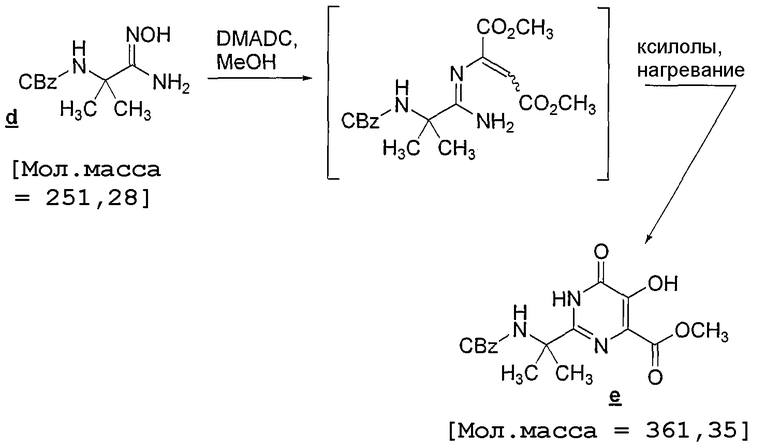
Ко взвеси амидоксима (2,90 кг) в метаноле (12 л) добавляют диметилацетилендикарбоксилат (1,77 кг) в течение 20 минут. Наблюдается медленный экзотермический эффект, так что температура взвеси повышается от 20 до 30°C в течение 15-20 минут. Через 1,5 часа анализ ВЭЖХ показывает, что превращение в промежуточные цис-/транс-аддукты произошло на более 95%. Затем при пониженном давлении растворитель заменяют на ксилолы (максимальная температура 50°C), при этом добавляют 2 объема [2×7,5 л] и упаривают до конечного объема 7,5 л. После этого реакционную смесь нагревают до 90°C и выдерживают при указанной температуре в течение 2 часов, удаляя остаток MeOH в потоке азота. Затем температуру увеличивают с шагом 10°C в течение 3,5 часа до 125°C и смесь выдерживают при указанной температуре в течение 2 часов. Наконец, температуру повышают до 135°C в течение 5 часов. После этого реакционную смесь охлаждают до 60°C и добавляют MeOH (2,5 л). Через 30 минут медленно добавляют MTBE (9 л), чтобы получить зародыши кристаллизации. Затем загрузку охлаждают до 0°C в течение 14 часов, после чего еще охлаждают до -5°C и отстаивают в течение 1 часа перед фильтрацией. Твердые вещества подвергают заместительному промыванию 10% MeOH/MTBE (6 л, затем 4 л; предварительно охлажденным до 0°C) и сушат на фильтровальном тигле в потоке азота, получая 2,17 кг (скорректированный выход 51,7%; 99,5 мас.%).
Метод ВЭЖХ: Колонка: Zorbax C-8 4,6 мм × 250 мм; от 40% ACN/60% 0,1% H3PO4 до 90% ACN/10% 0,1% H3PO4 в течение 12 минут, выдерживают 3 минуты, после чего снова используют 40% ACN в течение 1 минуты. Время удерживания: амидоксим d - 2,4 минуты, DMAD - 6,7 минуты, промежуточные аддукты - 8,4 и 8,6 минуты (пик, соответствующий 8,4 минуты, циклизуется быстрее), продукт e - 5,26 минуты, ксилолы - несколько пиков в интервале приблизительно 10,4-10,7 минуты.
Стадия 5: N-метилирование
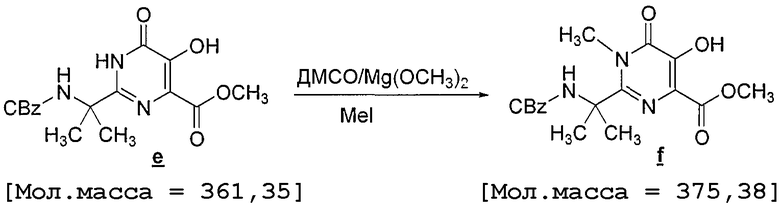
К раствору пиримидиндиола e (2 кг) в ДМСО (16 л) добавляют раствор Mg(ОMe)2 в MeOH (11,95 кг), после чего избыток MeOH упаривают в вакууме (30 мм Hg) при 40°C в течение 30 минут. Затем смесь охлаждают до 20°C, после чего добавляют MeI (1,38 л), смесь перемешивают при 20-25°C в течение 2 часов и затем при 60°C в течение 5 часов под давлением в закрытой колбе. Анализ ВЭЖХ показывает, что реакция завершилась. Смесь охлаждают до 20°C, добавляют MeOH (14 л) и затем медленно в течение 60 минут добавляют 2 M HCl (20 л) [наблюдается экзотермический эффект]. Чтобы нейтрализовать избыток I2, добавляют бисульфит натрия (5 мас.%, 2 л), при этом раствор становится белым. Затем в течение 40 минут добавляют воду (40 л), взвесь перемешивают в течение 40 минут на бане со льдом и фильтруют. Фильтровальный кек промывают вначале водой (20 л), затем смесью MTBE:MeOH 9/1 (30 л), чтобы удалить O-метилированный побочный продукт. Анализ ВЭЖХ показывает, что после промывания присутствует менее 0,5 A% O-метилированного продукта. Твердое вещество сушат в течение ночи при комнатной температуре в вакууме в потоке N2, получая 1,49 кг N-метилпиримидона (70% выход, скорректированный по чистоте исходного вещества и продукта).
Стадия 6: Присоединение амина
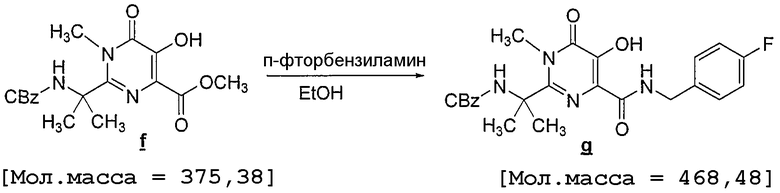
Ко взвеси N-метилированного пиримидинона f (1,4 кг) в EtOH (14 л) при 4°C медленно добавляют 4-фторбензиламин (1,05 кг) в течение 15 минут, при этом в процессе добавления первого мольного эквивалента амина наблюдается экзотермический эффект, и смесь нагревается до 9°C. Взвесь становится очень густой и требуется энергичное перемешивание. Реакционную смесь нагревают до 72°C в течение 2 часов и выдерживают при указанной температуре в течение 1 часа и 45 минут. При 45°C раствор становится крайне вязким, наблюдается небольшой экзотермический эффект с нагреванием до 50°C, а через 1 час при 72°C взвесь медленно растворяется с образованием гомогенной смеси. Анализ образца методом ВЭЖХ (используют такой же метод ВЭЖХ, как и на стадии 4) в конце реакции показывает, что присутствует менее 0,5 A% N-метилированного пиримидинона. Реакционную смесь охлаждают до 60°C, в течение 30 минут добавляют уксусную кислоту (0,55 л), после чего в течение 30 минут добавляют воду (6,7 л) и затем кристаллы-затравку (3,0 г), чтобы инициировать кристаллизацию. Через 30 минут при 60°C добавляют еще воду (7,3 л) в течение 30 минут и реакционную смесь оставляют охлаждаться до температуры окружающей среды в течение ночи. Через 13 часов температура достигает 20°C, в этот момент реакционную смесь фильтруют и взвесь промывают смесью 50% вода/EtOH (2×4 л). Твердые вещества сушат на фильтровальном тигле в вакууме/потоке N2 до достижения постоянной массы и получают белый твердый продукт (1,59 кг; 90% скорректированный выход; 99% LCWP и 99,7% LCAP по данным анализа ВЭЖХ, проведенного по способу, описанному выше на стадии 4).
Стадия 7: Гидрирование Cbz-амида
Сосуд для гидрирования из нержавеющей стали предварительно обрабатывают MeOH, катализатором Pd/C и MSA в условиях реакции, описанных ниже. Затем Cbz-амид g (10 г) суспендируют в MeOH (80 мл) в предварительно обработанном сосуде. Ко взвеси при комнатной температуре одной порцией добавляют MSA (1,45 мл). В сосуд для гидрирования также добавляют 5% Pd/C (0,15 г, 50% влажн.). Водород загружают в сосуд тремя последовательными циклами продувки вакуум/водород, после чего смесь гидрируют под давлением 40 фунт/кв.дюйм в течение 3-4 часов при 50°C. После гидрирования к реакционной смеси добавляют воду (8 мл), смесь перемешивают, катализатор фильтруют и промывают смесью 4:1 MeOH:вода (20 мл). pH объединенных фильтратов доводят до 7-8,0 путем медленного добавления 1 н. NaOH (22,4 мл), в результате чего осаждается твердое вещество. Взвесь перемешивают при 0-5°C в течение 4 часов, затем твердое вещество фильтруют, промывают водой (30 мл), собирают и сушат в вакууме при 50°C. Продукт амин (в виде гидрата) получают в виде белого кристаллического твердого вещества (7,7 г) с выходом 96% (скорректированным по KF), 89% LCWP, 99,8% LCAP, KF=11 мас.%. ВЭЖХ, метод A (анализ продукта): колонка: 25 см × 4,6 мм Zorbax RX-C8; подвижная фаза: A=0,1% H3PO4, B=CH3CN, 0 минут (80% A/20% B), 20 минут (20% A/80% B), 25 минут (20% A/80% B); скорость потока: 1,0 мл/мин; длина волны: 210 нм; температура колонки: 40°C; время удерживания: дефтораминовый побочный продукт - 5,5 минуты, аминовый продукт - 5,85 минуты, толуол - 16,5 минуты, Cbz-амид - 16,82 минуты.
ВЭЖХ, метод B (чистота продукта): колонка: 25 см × 4,6 мм YMC-basic; подвижная фаза: A=25 ммоль KH2PO4, доведенный до pH=6,1, B=CH3CN, 0 минут (90% A/10% B), 30 минут (30% A/70% B), 35 минут (30% A/70% B); скорость потока: 1 мл/мин; длина волны: 210 нм; температура колонки: 30°C; время удерживания: дефторамин - 9,1 минуты, амин - 10,1 минуты, толуол - 24,2 минуты, Cbz-амид - 25,7 минуты.
Стадия 8: Присоединение оксадиазола
Часть A: Получение K соли оксадиазола
(Примечание: Альтернативная процедура приведена в скобках. Если не указано иначе, альтернативная процедура практически идентична основной процедуре).
Этилоксалилхлорид (4,01 кг) медленно добавляют к смеси 5-метилтетразола (2,50 кг), триэтиламина (3,03 кг) в толуоле (32 л) при 0°C с такой скоростью, чтобы температура оставалась ниже 5°C. Полученную взвесь перемешивают в течение 1 часа при 0-5°C, после чего соль триэтиламин/HCl отфильтровывают. (Примечание: в альтернативной процедуре после перемешивания в течение 1 часа взвесь нагревают до 60-65°C в течение 1 часа с выделением газообразного N2, после чего отстаивают при 60-65°C в течение 1 часа и затем охлаждают до 20-25°C перед извлечением соли триэтиламин/HCl.) Твердое вещество промывают 27 л холодного толуола (5°C) (примечание: в процедуре, альтернативной указанной выше, используют неохлажденный толуол). Объединенные фильтраты выдерживают при 0°C, затем медленно добавляют к горячему раствору толуола (50°C, 15 л) в течение 40-50 минут (с образованием газообразного N2) и раствор выдерживают при 60-65°C в течение 1 часа. После охлаждения до 20°C толуольный раствор промывают 5 л 10% насыщенного соляного раствора (примечание: в альтернативной процедуре объединенные фильтраты промывают только насыщенным соляным раствором), затем растворитель заменяют на этанол (упаривают до 8 л, затем добавляют 17 л EtOH и концентрируют до 8 л, после чего добавляют 33 литра EtOH, чтобы довести конечный объем до 41 л). Этанольный раствор охлаждают до 10°C и в течение 30 минут добавляют водный раствор KOH (8,0 л), после чего полученную густую взвесь перемешивают в течение 40 минут при комнатной температуре, при этом выкристаллизовывается K соль оксадиазола. Твердое вещество отфильтровывают, промывают 11 л EtOH и в конце 15 л MTBE. Твердое вещество сушат в течение ночи в вакууме при 20°C в потоке азота и получают 4,48 кг (90,8%) K соли i .
Часть B: Присоединение оксадиазола
(Примечание: Альтернативная процедура приведена в скобках. Если не указано иначе, альтернативная процедура практически идентична основной процедуре.)
В круглодонную колбу объемом 500 мл загружают K соль оксадиазола i (33,8 г), затем при интенсивном перемешивании добавляют ACN (280 мл) и ДМФА (0,33 мл). Полученную взвесь охлаждают до 0-5°C и в течение 20 минут добавляют оксалилхлорид (23,7 г), чтобы поддерживать внутреннюю температуру ниже 5°C. Полученную взвесь, содержащую хлорангидрид, отстаивают в течение 1 часа.
В круглодонную колбу объемом 2 л добавляют свободный амин h (30 г), затем ТГФ (821 мл). Полученную взвесь охлаждают до 0-5°C, после чего добавляют NMM (21,56 г) и перемешивают в течение 10 минут при низкой температуре. Ко взвеси, содержащей свободный амин, медленно, в течение 20 минут, добавляют полученную ранее взвесь, содержащую хлорангидрид, так, чтобы температура при этом не поднималась выше 5°C. Затем взвесь отстаивают в течение 1,5 часа при 0-5°C. После этого проводят анализ ВЭЖХ, который показывает, что амин h отсутствует (<0,5% LCAP, 100% превращение). Затем реакционную смесь гасят NH4OH (30% раствор в воде) (69 мл), который добавляют в течение 3 минут (в альтернативном способе вместо NH4OH используют водный раствор KOH). Полученную желтую взвесь перемешивают в течение еще одного часа при температуре менее 10°C. Затем желтую взвесь подкисляют до pH 2-3 с помощью HCl (2 н.) (500 мл). К полученному раствору цвета красного вина добавляют IPA (920 мл). Низкокипящие органические растворители упаривают при пониженном давлении (40 Торр) при комнатной температуре до конечного объема раствора 1100 мл, по достижении указанного объема начинает осаждаться кристаллическое соединение A. К полученной взвеси в течение 10 минут добавляют воду (400 мл), после чего взвесь отстаивают в течение ночи при комнатной температуре (в альтернативном способе взвесь охлаждают до 0-10°C и отстаивают в течение 2 часов). Отстоявшуюся взвесь фильтруют, полученное твердое вещество промывают водой (170 мл) с последующим быстрым промыванием холодным MeOH (300 мл, предварительно охлажденный на бане со льдом) и в конце быстро промывают водой (700 мл). (В альтернативном способе твердое вещество, полученное фильтрованием отстоявшейся взвеси, дважды промывают водой; то есть MeOH не используют.) Полученное твердое вещество сушат в течение ночи в вакууме в потоке азота, получая 35,5 г соединения A (выход 91%). (В альтернативном способе получают соединение A с выходом 95%.)
Стадия 9: Получение кристаллической калиевой соли соединения A
Ацетонитрил (50 мл) и безводное соединение A (5,8 г, 97,4 мас.%) загружают при комнатной температуре в закрытую кожухом круглодонную колбу объемом 125 мл, оборудованную механической мешалкой и впускным отверстием для азота (то есть кристаллизацию проводят в атмосфере азота). Полученную взвесь перемешивают при 45°C, пока твердое вещество полностью не растворится. Кристаллическую K соль соединения A формы 1 добавляют в раствор в качестве затравки (0,184 г, 3 мас.% по отношению к теоретическому количеству K соли). Затем добавляют 30% (мас./об.) водный раствор KOH (0,98 экв., 2,33 мл, 0,0125 моль), используя приведенный ниже режим добавления и поддерживая температуру загрузки при 45°C:
0,466 мл в течение 5 часов, 0,0932 мл/час (20 мол.%);
1,864 мл в течение 7 часов, 0,2663 мл/час (80 мол.%).
Полученную взвесь охлаждают до 20°C и отстаивают при 20°C, пока измеряемая концентрация соединения A в маточном растворе не станет меньше 4 г/л. Загрузку фильтруют, кек промывают ACN (3×12 мл) и сушат в вакууме при 45°C в небольшом потоке азота, пока количество присутствующих ACN и воды по данным термогравиметрического анализа не станет меньше 1 мас.%. K соль соединения A получают с выходом 99 A% по данным анализа ВЭЖХ.
ПРИМЕР 2
Кристаллическая калиевая соль соединения A формы 1
Часть A: Получение
Этанол (147 мл), воду (147 мл) и соединение A (97,9 г по данным анализа ВЭЖХ) загружают в круглодонную колбу объемом 1 л, оборудованную механической мешалкой, дополнительной воронкой, впускным отверстием для азота (то есть реакцию проводят в атмосфере азота) и термопарой. К суспензии в течение 10 минут при 21°C добавляют водный раствор KOH (45% мас./мас., 0,98 экв., 18,5 мл, 216 ммоль). Полученную суспензию перемешивают в течение 0,5 часа, при этом основная часть твердых веществ растворяется, и затем загрузку фильтруют через фильтр 1 мкм сразу в круглодонную колбу объемом 5 л, оборудованную механической мешалкой, дополнительной воронкой, впускным отверстием для азота и термопарой. Колбу объемом 1 л ополаскивают смесью 1:1 (об./об.) вода/EtOH (48 мл) и смыв фильтруют в сосуд для кристаллизации объемом 5 л. В отфильтрованный раствор при комнатной температуре вносят кристаллическую K соль соединения A формы 1 (200 мг) в качестве затравки и затем раствор оставляют на 1 час, чтобы образовались хорошие зародыши кристаллизации, после чего суспензию разбавляют EtOH (1,57 л) при 20°C в течение 1,5 часа. Затем загрузку охлаждают до 4°C и отстаивают до тех пор, пока измеряемая концентрация соединения A в маточном растворе не станет равна 4,7 г/л. Загрузку фильтруют, сосуд для кристаллизации ополаскивают 50 мл EtOH в фильтр, кек промывают EtOH (4×100 мл), сушат в вакууме в потоке азота до тех пор, пока количество EtOH, обнаруживаемое методом ЯМР, не составит приблизительно 0,4 мол.% по отношению к калиевой соли. Калиевую соль соединения A получают с выходом 88% (91,5 г по результатам анализа ВЭЖХ, 99% площади по результатам анализа ВЭЖХ).
Часть B: Характеристика
Диаграмму XRPD для K соли, полученной по способу, описанному в части A, получают с помощью порошкового рентгеновского дифрактометра Philips Analytical X'Pert Pro с использованием непрерывного сканирования от 2,5 до 40 градусов 2Θ в течение приблизительно 12 минут (то есть размер шага 0,02° при продолжительности шага 40 секунд), ротацией шага 2 RPS, и осью гониометрического сканирования. В качестве источника используют медное излучение K-альфа 1 (Kα1) и K-альфа 2 (Kα2). Эксперимент проводят в условиях окружающей среды. Ниже приведены характеристические значения 2Θ в диаграмме XRPD (показанной на фиг.1) и соответствующие d-интервалы:
K соль, полученную по способу, описанному в части A, также анализируют с помощью дифференциального сканирующего калориметра TA Instruments DSC 2910 со скоростью нагревания 10°C/мин от комнатной температуры до 350°C в гофрированном алюминиевом лотке с микроотверстиями в атмосфере азота. Кривая DSC (показанная на фиг.2) демонстрирует один острый эндотерм с пиком температуры приблизительно 279°C и ассоциированной теплотой слияния приблизительно 230,0 Дж/г. Считается, что эндотерм обусловлен плавлением.
Термогравиметрический анализ проводят с помощью Perkin-Elmer, модель TGA 7, в атмосфере азота со скоростью нагревания 10°C/мин от комнатной температуры до 350°C. Кривая TG показывает в течение нагревания до 250°С уменьшение массы на 0,3 мас.%.
Гигроскопичность определяют на VTI Symmetrical Vapor Sorption Analyzer, модель SGA-1. Данные получают при комнатной температуре и изменении относительной влажности от 5 до 95%, и наоборот, с шагом 5% относительной влажности. Равновесными условиями являются изменение массового процента на 0,01 за 5 минут с максимальным временем уравновешивания 180 минут. Результаты показывают, что при уравновешивании при 95% относительной влажности и 25°C масса вещества увеличивается на 1,8%. При снижении относительной влажности до 5% масса вещества уменьшается приблизительно до сухой массы. Анализ XRPD после эксперимента по определению гигроскопичности показывает, что фаза вещества не изменилась.
Калиевую соль, полученную по способу, описанному в части A, также анализируют путем HCl-титрования с помощью Brinkmann Metrohm 716 DMS Titrino. Результаты анализа показывают, что полученная соль является монокалиевой солью.
ПРИМЕР 2-A
Кристаллическая калиевая соль соединения A формы 1
Соединение A (400 г) растворяют в 4 литрах смеси 60:40 этанол:ацетонитрил при 45°C, получая раствор соединения A с концентрацией 95 г/л. Этанол (1201 г) добавляют к 300 г 24% (по массе) раствора этоксида калия в этаноле и получают 4,8% (по массе) раствор KOEt в этаноле. Зародыши кристаллизации получают путем добавления кристаллической калиевой соли соединения A формы 1 (78 г) к 1,08 литра смеси 70:30 этанол:ацетонитрил. Зародыши кристаллизации подвергают влажному размалыванию с помощью миксера Ultra Turrax IKA T-50 в течение 45 минут при 10000 об/мин, получая ~50000 частиц (1-500 мкм) со средним размером 10 мкм, определенным с помощью анализатора размеров частиц Lasentec FBRM, модель S400.
Взвесь затравочных кристаллов (1,16 литра) загружают в кристаллизатор с температурой кожуха 35°C. Затем ко взвеси затравочных кристаллов в кристаллизаторе при 45°C добавляют раствор соединения A. При перемешивании смеси раствор соединения А-взвесь затравочных кристаллов при 250 об/мин в кристаллизатор загружают полученный ранее раствор KOEt поверх поверхности раздела раствор-взвесь затравочных кристаллов с постоянной скоростью 4,7 мл/мин в течение 6 часов и 40 минут. Температуру кожуха кристаллизатора устанавливают на 35°C в течение первых 6 часов и затем снижают ее до 20°C, при этом в течение последних 40 минут загружают оставшиеся ~9% этоксида. Загрузку отстаивают при 20°C в течение 30 минут, фильтруют и полученный фильтровальный кек промывают 2,8 л этанола. Затем промытый кек продувают азотом в течение 1 часа, переносят в вакуумную печь и сушат в течение ночи при 45°C, получая указанную в заголовке соль.
ПРИМЕР 3
Получение прессованных таблеток, содержащих калиевую соль соединения A
Часть A
(в пересчете на свободный фенол)
(100)
(25,0)
Прессованные таблетки, содержащие 100 мг соединения A в пересчете на свободный фенол, получают путем смешивания всех перечисленных выше ингредиентов, за исключением экстрагранулярного стеарата магния, в блендере (шейкер-миксер Turbula® типа T2F, Basel, Швейцария) в течение 10 минут. Порции полученной смеси массой приблизительно 1 грамм прессуют в брикеты (или слэги) в настольном прессе (Auto Carver, модель Auto "C", № по каталогу 3888, Carver, Inc., Wabash, Indiana) с прямоугольной обработкой 1×0,5 дюйма до 12 МПа (4 кН). Слэги затем сортируют, получая гранулы, путем пропускания через сито с отверстиями размером 1 мм. Гранулы смешивают с экстрагранулярным стеаратом магния в блендере Turbula в течение 5 минут, после чего смазанные гранулы прессуют в таблетки с использованием пресса Auto Carver, позволяющего получать таблетки со стандартной вогнуто-круглой формой размером 13/32 дюйма.
Часть B
(в пересчете на свободный фенол)
(100)
(25,0)
Прессованные таблетки, содержащие композицию, указанную в приведенной выше таблице, получают по способу, описанному в части А.
ПРИМЕР 4
Получение прессованных таблеток, содержащих калиевую соль соединения A
(400)
(46,0)
1 Кристаллическая монокалиевая соль соединения A формы 1; коэффициент превращения (с учетом чистоты) = 1,086.
2 Полученный от BASF. Средний размер частиц = 50 мкм.
Прессованные таблетки, содержащие 400 мг соединения A в пересчете на свободный фенол, получают с помощью роллерного прессования и серии процессов прессования таблеток. Вначале полоксамер 407, стеарат магния и стеарилфумарат натрия последовательно просеивают через сита 30 и 60 меш, затем смешивают их с другими ингредиентами, за исключением экстрагранулярного стеарата магния, в V-блендере Patterson-Kelly (PK) в течение 5 минут. Полученную смесь просеивают через сито 35 меш, чтобы раздробить агломераты, после чего просеянное вещество снова перемешивают в таком же блендере PK в течение приблизительно 15-20 минут. Затем смесь подвергают роллерному прессованию с помощью роллерного минипресса Freund типа TF при давлении валка 40 кгс/см2, скорости валка 3 об/мин и скорости вращения шнека 10 об/мин. Полученную ленту размалывают в маленьком Quadro Comil, оборудованном турбинной мешалкой и ситом размером 39R (то есть с круглыми отверстиями размером 0,039 дюйма; приблизительно 20 меш), со скоростью 1700 об/мин. Затем полученные гранулы смешивают с 0,5% экстрагранулярного стеарата магния в блендере PK в течение 5 минут с получением конечной смеси. Смазанные гранулы прессуют в таблетки с помощью ротационного таблеточного пресса, позволяющего получать таблетки с гладкой овальной формой, при усилии прессования, обеспечивающем твердость таблетки от 16 до 20 килограмм-сила (то есть от 156,9 до 196,1 Ньютона), которую определяют с помощью прибора для определения твердости Key, модель HT-300.
ПРИМЕР 5
Исследования in vitro
Значение IC50 атазанавира для ингибирования глюкуронидации соединения A в микросомах печени человека определяют, используя пул микросом печени человека (полученный от Xenotech LLC, Lenexa, KS). Соединение A (в виде калиевой соли) добавляют к микросомам печени человека (1,0 мг/мл) в буфере, содержащем 0,1 M калий-фосфатный буфер (pH=7,4), 5 мM хлорид магния, 10 мкг/мл аламетицина и 10 мM 1,4-лактон D-сахарной кислоты. Смесь соединения A и микросом в буфере (0,5 мл) инкубируют при Km соединения A (200 мкM). К инкубируемому образцу добавляют UDPGA до концентрации 4 мM, чтобы инициировать реакцию глюкуронидации, которую останавливают через 25 минут (37°C) добавлением 2 объемов (то есть 1 мл) ацетонитрила, содержащего 1,5 мкM соединения B в качестве внутреннего стандарта для последующего анализа ЖХ/МС. Затем все образцы центрифугируют, полученный супернатант разбавляют 1:1 0,1% раствором муравьиной кислоты в воде и аликвоту объемом 10 мкл вводят в ЖХ/МС, чтобы определить количество образовавшегося глюкуронида. Получают аналогичные образцы соединения A, содержащие атазанавир в концентрации от 0,1 до 50 мкM, инкубируют их и анализируют таким же способом.
Значение IC50 атазанавира для ингибирования глюкуронидации соединения A в присутствии микросом печени крысы определяют по способу, описанному выше для микросом печени человека.
Обнаружено, что значение IC50 атазанавира для ингибирования соединения A в микросомах печени человека составляет 0,5 мкM. Также обнаружено, что атазанавир ингибирует глюкуронидацию соединения A в микросомах печени крысы (41% в концентрации 50 мкM).
ПРИМЕР 6
Исследования на крысах in vivo
Самцы крыс Sprague-Dawley (по 4 крысы в группе) массой приблизительно 300 граммов каждый получают перорально либо 0,5% раствор метилцеллюлозы в воде (контрольная группа), либо атазанавир в 0,5% растворе метилцеллюлозы один раз в сутки в течение трех суток. Суточная доза атазанавира составляет 50 мг/кг, ее вводят в 0,5% растворе метилцеллюлозы в количестве 5 мл/кг. На четвертые сутки контрольные крысы получают 10 мг/кг соединения A в виде калиевой соли в 0,5% растворе метилцеллюлозы, а опытная группа получает атазанавир с последующим введением 10 мг/кг соединения A (в виде калиевой соли) в 0,5% растворе метилцеллюлозы. Образцы крови берут у всех крыс из опытных и контрольных групп через 0, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа после введения дозы на 4 сутки. Уровни соединения A в плазме определяют методом ЖХ-МС/МС следующим способом:
Активность UDP-глюкуронозилтрансферазы определяют путем измерения образования глюкуронида соединения A. Анализ ВЭЖХ проводят на градиентной системе Agilent HP1100, используя следующие параметры: колонка = Phenomenex Luna C18-2 (2 мм × 150 мм, 5 мкм); подвижная фаза = 0,1% раствор муравьиной кислоты в воде (растворитель A) и 0,1% раствор муравьиной кислоты в ацетонитриле (растворитель B); скорость потока = 0,2 мл/мин; метод = вначале используют смесь растворителей, содержащую 10% B, затем содержание В увеличивают до 80% в течение 10 минут, указанный состав смеси используют неизменным в течение 3 минут, после чего его возвращают к исходному в течение 6 минут. Система ВЭЖХ соединена с тандемным масс-спектрометром Finnigan TSQ Quantum. Масс-спектрометрические анализы проводят с использованием ионизации электрораспылением (ESI) в режиме положительных ионов. Во всех анализах температура в трубке переноса ионов составляет 320°C, а напряжение ионизации ESI поддерживают при 4,4 кВ. Тандемная масс-спектрометрия (МС/МС) основана на индуцированной столкновениями диссоциации (CID) ионов, входящих в высокочастотную октаполярную область, где в качестве коллизионного газа используют аргон под давлением 0,8 мТорр. Для анализа МС/МС используют напряжение столкновения -22 эВ. Используемые CID-переходы составляют m/z=621,1→445,1 (глюкуронид соединения A) и 431,2→306,1 (соединение B).
Значения Cmax и AUC для крыс, получающих перорально атазанавир (50 мг/кг) и соединение A (10 мг/кг), на 4 сутки составляют 7,2±6,1 и 9,9±3,7 мкM·час соответственно. Соответствующие значения для соединения A в контрольной группе составляют 2,3±0,9 и 2,9±0,6 мкM·час соответственно. Таким образом, атазанавир увеличивает уровень соединения A в плазме приблизительно в 3 раза.
ПРИМЕР 7
Исследования на людях in vivo
Настоящий способ представляет исследование на здоровых мужчинах-добровольцах, состоящее из 2 периодов с фиксированной последовательностью, целью которого является определение влияния многократного введения атазанавира на однократную дозу соединения A. В 1 периоде 12 субъектов получают перорально однократную дозу (100 мг) соединения A (то есть таблетку, описанную в части B примера 3) (N=10) или плацебо (N=2). Во 2 периоде те же 12 субъектов получают 400 мг атазанавира один раз в сутки по способу открытого исследования (капсулы) в течение 9 суток. На 7 сутки субъектам вводят атазанавир в комбинации с однократной пероральной дозой 100 мг соединения A (таблетка) или с плацебо (в обоих периодах исследования плацебо получают одни и те же субъекты). Все дозы вводят после получения пищи средней жирности. Образцы плазмы PK собирают в течение 72 часов после введения соединения A в обоих периодах.
Получение и анализ образца: Образцы плазмы экстрагируют с использованием 96-луночной экстракции жидкости жидкостью. Экстракты плазмы вводят на колонку ВЭЖХ Ace C18 (50×3,0 мм, 3 мкм, титановые фильтры) и анализируют в изократических условиях с использованием подвижной фазы, содержащей 42,5/57,5 (об./об.%) 0,1 мM ЭДТА в 0,1% муравьиная кислота/метанол, со скоростью потока 0,5 мл/мин. Экстракты образцов ионизируют, используя интерфейс APCI, и регистрируют с помощью MRM в режиме положительной ионизации. Динамический интервал анализа ЖХ/МС/МС составляет 2-1000 нг/мл при объеме аликвоты плазмы человека 200 мкл.
Вычисление PK: Площадь под кривой зависимости концентрации в плазме от времени до последней детектируемой концентрации (AUC0-last) рассчитывают, используя некомпартментную модель и метод линейных вычислений Up/Log Down из WinNonLin, версия 4.1. Точки значений после Cmaх подгоняют к биэкспоненциальному уравнению (A*exp(-αt)+B*exp(-βt)), используя WinNonlin v4.1, а значения AUC экстраполируют к бесконечности в соответствии со следующим уравнением: AUC0-∞=AUC0-last+Clast/β, где Clast означает последнюю детектируемую концентрацию, а β определяют из приведенного выше биэкспоненциального уравнения. Наблюдаемую максимальную концентрацию в плазме (Cmax), время, соответствующее Cmax (Tmax), и концентрацию в плазме через 12 часов после введения соединения (C12ч) определяют эмпирически.
Результаты: В присутствии атазанавира наблюдаются более высокие уровни соединения A в плазме, чем в его отсутствие. Отношение средних геометрических значений (GMR) концентраций через 12 часов после введения в присутствии и в отсутствие атазанавира составляет 1,96. В присутствии атазанавира также наблюдаются более высокие значения AUC и Cmax для соединения A, чем в отсутствие атазанавира [для AUC0-last GMR=1,73; для Cmax GMR=1,53]. Также существует тенденция к небольшому удлинению альфа-фазы периода полужизни соединения A в присутствии атазанавира по сравнению с отсутствием атазанавира (1,4 часа в присутствии атазанавира по сравнению с 1,1 часа в присутствии только соединения A).
В то время как приведенное выше описание разъясняет принципы настоящего изобретения вместе с примерами, приведенными для иллюстрации, осуществление изобретения включает традиционные вариации, адаптации и/или модификации, входящие в объем нижеследующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ КАРБОКСАМИДНЫХ ИНГИБИТОРОВ ИНТЕГРАЗЫ ВИЧ, СОДЕРЖАЩИЙ КОМПОЗИЦИЮ С КОНТРОЛИРУЕМОЙ СКОРОСТЬЮ ВЫСВОБОЖДЕНИЯ | 2005 |
|
RU2382648C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИММУНОМОДУЛЯТОРЫ В КАЧЕСТВЕ ИНГИБИТОРА КОНТРОЛЬНЫХ ТОЧЕК ИММУННОГО ОТВЕТА PDL1 | 2020 |
|
RU2824127C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРИМИДИНА, ИНГИБИРУЮЩИЕ ВИЧ | 2005 |
|
RU2403254C2 |
| ПРОИЗВОДНЫЕ АЗАПЕПТИДОВ | 2008 |
|
RU2448958C2 |
| 1,5,6-ЗАМЕЩЕННЫЕ 2-ОКСО-3-ЦИАНО-1,6А-ДИАЗАТЕТРАГИДРОФЛУОРАНТЕНЫ | 2006 |
|
RU2389730C2 |
| 6,7,8,9-ЗАМЕЩЕННЫЕ 1-ФЕНИЛ-1,5-ДИГИДРОПИРИДО (3,2-b) ИНДОЛ-2-ОНЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТИИНФЕКЦИОННЫХ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2005 |
|
RU2377243C2 |
| 5-ЗАМЕЩЕННЫЕ 1-ФЕНИЛ-1,5-ДИГИДРОПИРИДО[3,2-b]ИНДОЛ-2-ОНЫ И АНАЛОГИ КАК ПРОТИВОВИРУСНЫЕ ПРЕПАРАТЫ | 2005 |
|
RU2362776C2 |
| 5-ГЕТЕРОЦИКЛИЛПИРИМИДИНЫ, ИНГИБИРУЮЩИЕ ВИЧ | 2005 |
|
RU2405778C2 |
| КОМБИНИРОВАННЫЕ ПРОДУКТЫ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ ТИРОЗИНКИНАЗ, И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2660354C2 |
| АНТИВИРУСНЫЕ АЛИФАТИЧЕСКИЕ СЛОЖНОЭФИРНЫЕ ПРОЛЕКАРСТВА ТЕНОФОВИРА | 2017 |
|
RU2759902C2 |
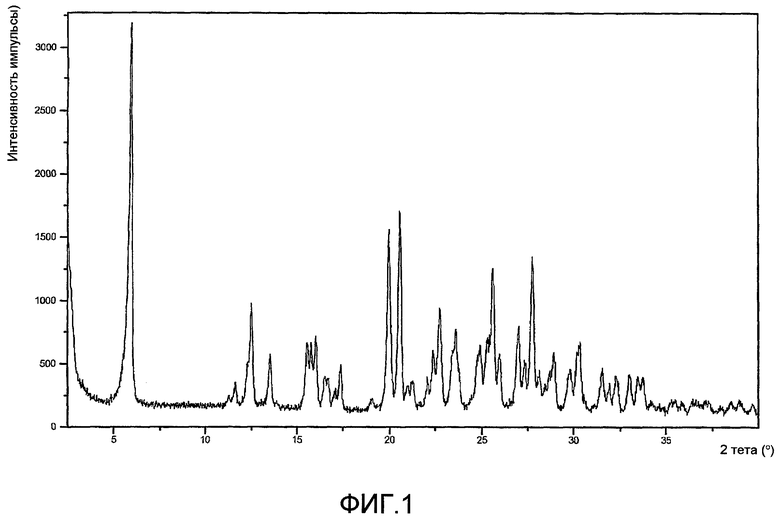
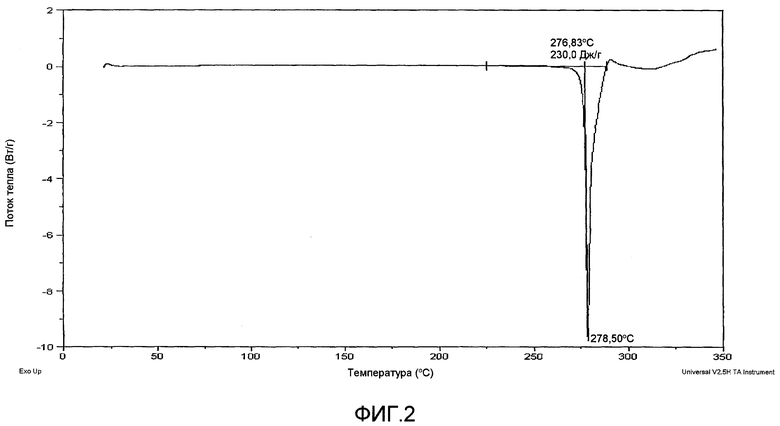
Изобретение относится к области фармацевтики и медицины и касается способа улучшения фармакокинетических характеристик перорального лекарственного средства, непосредственно метаболизируемого UGT1A1, формулы (1), который предусматривает пероральное введение млекопитающему, нуждающемуся в лечении лекарственным средством, комбинации лекарственного средства, или его фармацевтически приемлемой соли, и атазанавира, или его фармацевтически приемлемой соли. 2 н. и 11 з.п. ф-лы, 2 ил., 14 табл.
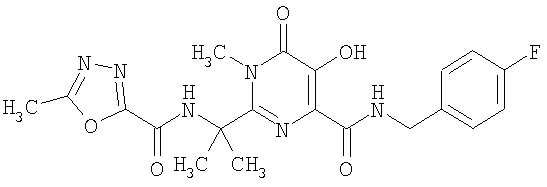
1. Способ улучшения фармакокинетических характеристик перорального лекарственного средства, непосредственно метаболизируемого UGT1A1, который предусматривает пероральное введение млекопитающему, нуждающемуся в лечении лекарственным средством, эффективного количества комбинации лекарственного средства, или его фармацевтически приемлемой соли, и атазанавира, или его фармацевтически приемлемой соли, где лекарственное средство представляет собой соединение А или его фармацевтически приемлемую соль, где соединение А имеет формулу
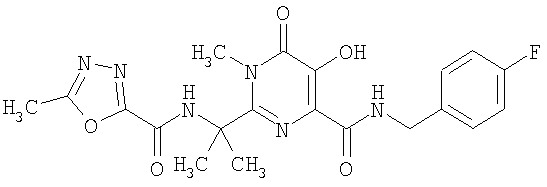 .
.
2. Способ по п.1, где атазанавир вводят в комбинации в количестве, достаточном для улучшения фармакокинетических характеристик соединения А, по меньшей мере, на приблизительно 10% по отношению к фармакокинетическим характеристикам соединения А, вводимого в отсутствии атазанавира.
3. Способ по п.1, где количество соединения А, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, находится в интервале от приблизительно 2 мг/кг до приблизительно 10 мг/кг массы тела.
4. Способ по п.1, где атазанавир присутствует в комбинации в количестве, которое, в случае введения только атазанавира, меньше количества, эффективного для лечения инфекции ВИЧ или СПИДа.
5. Способ по п.1, где количество соединения А, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, находится в интервале от приблизительно 2 мг/кг до приблизительно 5 мг/кг массы тела.
6. Способ по п.1, где количество соединения А, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг, а количество атазанавира, вводимое в сутки в комбинации, составляет менее 400 мг.
7. Способ по любому из пп.1-6, где соединение А присутствует в форме калиевой соли.
8. Фармацевтическая комбинация для перорального введения млекопитающему, содержащая ингибитор интегразы ВИЧ, непосредственно метаболизируемый UGT1A1, и атазанавир или его фармацевтически приемлемую соль, где ингибитор интегразы ВИЧ представляет собой соединение А или его фармацевтически приемлемую соль, где соединение А имеет формулу
.
9. Комбинация по п.8, где атазанавир вводится в комбинации в количестве, достаточном для улучшения фармакокинетических характеристик соединения А, по меньшей мере, на приблизительно 10% по отношению к фармакокинетическим характеристикам соединения А, вводимого в отсутствии атазанавира.
10. Комбинация по п.8, где количество соединения А, вводимое в сутки в комбинации, находится в интервале от приблизительно 5 мг/кг до приблизительно 10 мг/кг массы тела, а количество атазанавира, вводимое в сутки в комбинации, находится в интервале от приблизительно 2 мг/кг до приблизительно 10 мг/кг массы тела.
11. Комбинация по п.8, где атазанавир вводится в комбинации в количестве, которое, в случае введения только атазанавира, меньше количества, эффективного для лечения инфекции ВИЧ или СПИДа.
12. Комбинация по любому из пп.8-11, где соединение А присутствует в форме калиевой соли.
13. Комбинация по любому из пп.8-12, где комбинация представляет единую фармацевтическую композицию, которая дополнительно содержит фармацевтически приемлемый носитель.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US 6087383 A, 11.07.2000 | |||
| US 5849911 A, 15.12.1998. | |||

