Область техники
Настоящее изобретение относится к фармацевтическим композициям с быстрым высвобождением лекарственного средства, включающим чувствительное к влаге терапевтическое средство и плавкий гидрофобный компонент. Настоящее изобретение также относится к способам получения таких фармацевтических композиций с быстрым высвобождением лекарственного средства.
Предпосылки создания изобретения
Чувствительность к влаге терапевтического соединения ограничивает его коммерческую ценность. Включение гигроскопичных чувствительных к влаге терапевтических соединений в фармацевтически приемлемые композиции, предназначенные для перорального введения, приводит к значительным проблемам, так как такие терапевтические соединения характеризуются химической нестабильностью. Такая химическая нестабильность прежде всего проявляется при применении чувствительного к влаге терапевтического соединения в составе лекарственной формы, включающей экципиент с высоким равновесным содержанием влаги. В данном случае влага мигрирует из таких экципиентов и может вызывать гидролиз чувствительного к влаге терапевтического соединения.
Чтобы свести к минимуму химическую нестабильность, чувствительное к влаге терапевтическое соединение подвергают гранулированию из расплава, так называемому термоплавлению. Гранулирование из расплава включает процесс смешивания и нагревания твердых и/или полутвердых материалов в присутствии терапевтического соединения, при этом получают гранулы, содержащие частицы терапевтического соединения, покрытые твердым и/или полутвердым материалом.
Однако способ гранулирования из расплава используют также для получения фармацевтических композиций с замедленным или контролируемым высвобождением лекарственного средства. В настоящее время существует необходимость в фармацевтической композиции, которая включает гранулы, полученные гранулированием из расплава, но обладающие свойством быстрого высвобождения лекарственного средства. Настоящее изобретение относится к такой фармацевтической композиции.
Краткое изложение сущности изобретения
Настоящее изобретение относится к фармацевтической композиции, предназначенной для перорального введения чувствительного к влаге терапевтического соединения. Композиция включает гранулы чувствительного к влаге терапевтического соединения и плавкого гидрофобного компонента, полученные из расплава. Такие фармацевтические композиции позволяют повысить химическую стабильность терапевтического соединения, прежде всего, в присутствии влаги или воды. Кроме того, фармацевтические композиции по настоящему изобретению являются композициями с быстрым высвобождением лекарственного средства и не обладают свойствами композиций с замедленным или контролируемым высвобождением лекарственного средства.
Другой объект настоящего изобретения включает способ получения гранул терапевтического соединения и плавкого гидрофобного компонента, полученных гранулированием из расплава. Указанный способ, в основном, включает следующие стадии:
(а) получение смеси чувствительного к влаге терапевтического соединения по крайней мере с одним плавким гидрофобным компонентом,
(б) нагревание полученной смеси при температуре выше, вблизи или практически равной температуре плавления указанного плавкого гидрофобного компонента или в диапазоне плавления указанного компонента,
(в) гранулирование полученной смеси при перемешивании с высоким сдвигом с образованием гранул и
(г) охлаждение полученных гранул до комнатной температуры. Еще один объект настоящего изобретения относится к фармацевтической композиции, включающей частицы чувствительного к влаге терапевтического соединения, полностью или в основном полностью покрытые плавким гидрофобным компонентом. Чувствительным к влаге терапевтическим соединением является, например, ингибитор DPP-IV.
Подробное описание вариантов осуществления изобретения
Настоящее изобретение относится к способу получения лекарственных форм чувствительного к влаге терапевтического соединения с быстрым высвобождением лекарственного средства, которые включают гранулы чувствительного к влаге терапевтического соединения и плавкий гидрофобный компонент, полученные гранулированием из расплава.
Термин «фармацевтическая композиция», используемый в настоящем описании, означает смесь или раствор, содержащий чувствительное к влаге терапевтическое соединение, предназначенные для введения млекопитающему, например человеку, для профилактики или лечения конкретного заболевания или патологического состояния млекопитающего.
Термин «терапевтическое соединение», используемый в настоящем описании, означает любое соединение, субстанцию, лекарственный препарат, лекарственное средство или активный ингредиент, который обладает терапевтическими или фармакологическими свойствами и является пригодным для введения млекопитающему, например человеку, в составе композиции, которая является, прежде всего, пригодной для перорального введения.
Примеры классов терапевтических соединений включают, без ограничения перечисленным, гипотензивные средства, седативные агенты, антикоагулянты, противосудорожные средства, агенты, снижающие уровень глюкозы в крови, противоотечные средства, антигистаминные средства, противокашлевые средства, противоопухолевые средства, бета-блокаторы, противовоспалительные средства, антипсихотические агенты, усилители познавательной функции, антиатеросклеротические агенты, агенты, снижающие уровень холестерина, средства против ожирения, агенты против аутоиммунных нарушений, средства против импотенции, противобактериальные и противогрибковые средства, снотворные средства, антибиотики, антидепрессанты, противовирусные средства и их комбинации.
Фармацевтические композиции по настоящему изобретению включают терапевтическое соединение(я) в терапевтически эффективном количестве или концентрации. Термин «терапевтически эффективные количество или концентрация», известный в данной области техники, означает количество или концентрацию терапевтического соединения, которая зависит от природы терапевтического соединения и заболевания, предназначенного для лечения. Например, количество терапевтического соединения по настоящему изобретению в фармацевтической композиции составляет до приблизительно 20 мас.%, например, от приблизительно 0,05 мас.% в расчете на массу фармацевтической композиции. Количество терапевтического соединения в фармацевтической композиции может также составлять от приблизительно 0,5 мас.% до 15 мас.% в расчете на массу фармацевтической композиции, например, от приблизительно 1,5 мас.% до приблизительно 5 мас.%.
Термин «чувствительное к влаге терапевтическое соединение», используемый в настоящем описании, означает терапевтическое соединение, которое подвергается самопроизвольному разложению, например, гидролизуется в количестве по крайней мере 1 мас.% при взаимодействии с водой. Таким образом, чувствительные к влаге терапевтические соединения в большинстве случаев разлагаются при непосредственном взаимодействии с экципиентами с высоким или повышенным равновесным содержанием воды. Экципиент с высоким или повышенным равновесным содержанием воды по настоящему изобретению содержит более 5% влаги при 25°С и 75% относительной влажности (OB). Например, такие экципиенты включают, без ограничения перечисленным, микрокристаллическую целлюлозу, предварительно желатинизированный крахмал, кукурузный крахмал и повидон.
Наиболее значимыми терапевтическими соединениями по настоящему изобретению являются ингибиторы дипептидилпептидазы IV (DPP-IV), прежде всего, N-(замещенный глицил)-2-цианопирролидоны, как описано в патентах США №6011155 и 6166063 (патент '063), которые включены в данное описание в качестве ссылок. Указанные соединения и их соответствующие фармацевтически приемлемые кислотно-аддитивные соли применяют для лечения нарушений, таких как инсулинонезависимый сахарный диабет, артрит, ожирение, трансплантация аллотрансплантата и кальцитонин-остеопороз. Кроме того, предполагается, что в связи со свойствами глюкагонподобных пептидов, таких как GLP-1 и GLP-2, и их участием в процессе ингибирования DPP-IV, соединения по настоящему изобретению могут проявлять, например, седативные или анксиолитические свойства, или снижать послеоперационные катаболические изменения и гормональные ответные реакции на стресс, или снижать смертность и заболеваемость после инфаркта миокарда, или их можно использовать для лечения заболеваний, связанных с указанными выше нарушениями, опосредованными изменением уровней GLP-1 и/или GLP-2.
Более подробно, например, соединения, которые являются ингибиторами DPP-IV, по настоящему изобретению и их соответствующие фармацевтически приемлемые кислотно-аддитивные соли ускоряют продуцирование инсулина на ранней стадии в ответ на пероральное введение глюкозы и, таким образом, являются пригодными для лечения инсулинонезависимого сахарного диабета.
Пример такого соединение включает моногидрохлорид [S]-1-[2-(5-циано-2-пиридиниламино)этиламино]ацетил-2-карбонитрила формулы (I)
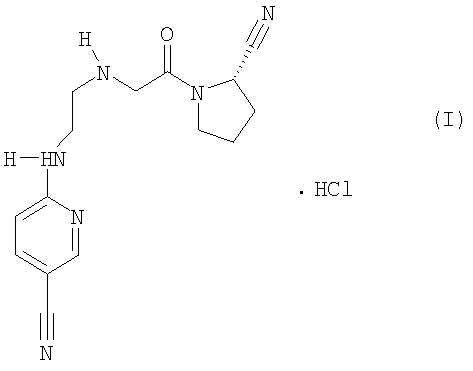
Указанный моногидрохлорид формулы (I) (в настоящем описании «соединение I») описан на схеме 2 в статье Villhauer и др., J. Med. Chem., т.45, No. 12, сс.2362-2365 (2002), которая включена в данное описание в качестве ссылки. Соединение I является чувствительным к влаге терапевтическим соединением. При влажности выше 75% соединение I абсорбирует воду и начинает растворяться в абсорбированной воде, кроме того, соединение I разлагается в кислотной и основной среде с образованием циклического продукта амидирования циклоимидата («CI»), который является основным продуктом разложения соединения I.
Другой пример N-(замещенный глицил)-2-цианопирролидинов по настоящему изобретению включает соединения формулы (II)

где
R означает замещенный адамантил и
n равно от 0 до 3,
в свободной форме или в форме кислотно-аддитивной соли.
Термин «замещенный адамантил», используемый в настоящем описании, означает 1- или 2-адамантил, замещенный одним или более заместителями, например, двумя заместителями, выбранными из группы, включающей алкил, -OR1 или -NR2R3, где R1, R2 и R3 независимо означают водород, алкил, С1-С8алканоил, карбамил или -CO-NR4R5, где R4 и R5 независимо означают алкил, незамещенный или замещенный арил, причем один из R4 и R5, кроме того, означает водород, или R4 и R5 вместе означают С2-С7алкилен.
Термин «арил», используемый в настоящем описании, означает фенил. Замещенный фенил, прежде всего, означает фенил, замещенный одним или более заместителями, например, двумя заместителями, выбранными из группы, включающей алкил, алкокси, галоген и трифторметил.
Термин «алкокси», используемый в настоящем описании, означает алкил-O-.
Термин «галоген», используемый в настоящем описании, означает фтор, хлор, бром и иод.
Термин «алкилен», используемый в настоящем описании, означает прямую углеводородную цепь, содержащую от 2 до 7 атомов углерода, например, от 3 до 6 атомов углерода или в другом варианте 5 атомов углерода.
Одна группа соединений по настоящему изобретению включает соединения формулы (II), в которых заместитель в составе адамантильной группы присоединен к основному фрагменту цепи или метилену, соседнему с основным фрагментом цепи. Соединения формулы (П), где остаток глицил-2-цианопирролидина присоединен к основному фрагменту цепи, содержат заместитель R' в составе адамантильной группы, который означает, прежде всего, 3-гидрокси. Соединения формулы (II), где остаток глицил-2-цианопирролидина присоединен к метилену, соседнему с основным фрагментом цепи, содержат заместитель R' в составе адамантильной группы, который означает, прежде всего, 5-гидрокси.
Настоящее изобретение, прежде всего, относится к соединению формулы (IIA) (в настоящем описании «соединение IIA») или формулы (ПВ) (в настоящем описании «соединение IIB»),
 или
или
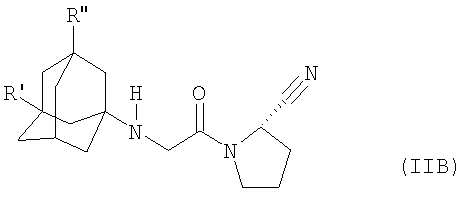
где
R' означает гидрокси, С1-С7аклокси, С1-С8алканоилокси или R5R4N-CO-O-, где R4 и R5 независимо означают С1-С7алкил или фенил, который является незамещенным или замещенным заместителем, выбранным из группы, включающей С1-С7аклил, С1-С7аклокси, галоген и трифторметил, причем R4, кроме того, означает водород, или R4 и R5 вместе означают С2-С7алкилен и
R'' означает водород или
R' и R'' независимо означают С1-С7аклил,
в свободной форме или в форме фармацевтически приемлемой кислотно-аддитивной соли.
Указанные соединения формулы (II), (IIA) или (IIB), которые являются ингибиторами DPP-IV, известны в данной области техники и описаны в патенте '063. Указанные соединения находятся в свободной форме или в форме кислотно-аддитивной соли. Фармацевтически приемлемые, т.е. нетоксичные и физиологически приемлемые соли, прежде всего, являются пригодными для применения, при этом другие соли также являются пригодными для применения, например, в процессе выделения или очистки соединений по настоящему изобретению. Кислотно-аддитивные соли включают гидрохлориды, причем соли метансульфоновой, серной, фосфорной, лимонной, молочной и уксусной кислот также являются пригодными для применения.
Прежде всего, соединения формулы (II), (IIA) или (IIB), пригодные для применения, включают (S)-1-[(3-гидрокси-1-адамантил)амино]ацетил-2-цианопирролидин формулы (в настоящем описании «соединение IIC»)
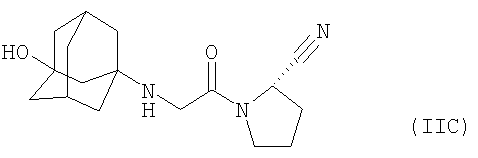
и необязательно его фармацевтически приемлемые соли.
Величина суточной дозы соединения ПС находится, например, в диапазоне от приблизительно 10 мг до приблизительно 150 мг, от приблизительно 24 мг до 100 мг и от приблизительно 50 мг до 100 мг. Величина сточной дозы при пероральном введении лекарственного средства составляет, например, 25 мг, 30 мг, 35 мг, 45 мг, 50 мг, 55 мг, 60 мг, 80 мг или 100 мг. Терапевтическое соединение вводят до трех раз в сутки или один или два раза в сутки.
Ингибиторы DPP-IV, пригодные для применения, описаны в разделе Примеры в патентах США №6124305 и №6107317 и в заявках РСТ WO 98/19998, WO 95/15309 и WO 98/18763, которые включены в данное описание в качестве ссылок. Указанные примеры включают 1-[2-[(5-цианопиридин-2-ил)аминоэтиламино]ацетил-2-циано-(S)-пирролидин и (2S)-1-[(2S)-2-амино-3,3-диметилбутаноил]-2-пирролидинкарбонитрил.
В еще одном варианте осуществления настоящего изобретения предлагается ингибитор DPP-IV по настоящему изобретению, который является N-пептидил-О-ароилгидроксиламином или его фармацевтически приемлемой солью. Ароил означает, например, нафтилкарбонил или бензоил, который является незамещенным или моно- или дизамещенным, например, группами (низш.)алкокси, (низш.)алкил, галоген или предпочтительно нитро. Остаток пептидила включает предпочтительно две α-аминокислоты, например, глицин, аланин, лейцин, фенилаланин, лизин или пролин, одна из которых присоединена непосредственно к атому азота гидроксиламина, и является прежде всего пролином.
Например, N-пептидил-О-ароилгидроксиламин является соединением формулы (IV)
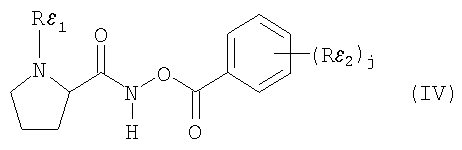
где
j равно 0, 1 или 2,
Rε1 означает боковую цепь природной аминокислоты и
Rε2 означает (низш.)алкокси, (низш.)алкил, галоген или нитро,
или его фармацевтически приемлемой солью.
В другом варианте осуществления настоящего изобретения предлагается N-пептидил-O-ароилгидроксиламин, который является соединением формулы (IVA)

или его фармацевтически приемлемой солью.
N-Пептидил-О-ароилгидроксиламины, например, формулы (IV) или (IVA) и их получение описаны в статье Demuth и др., J. Enzyme Inhibition, т.2, ее. 192-142 (1988), которая включена в данное описание в качестве ссылки.
В других вариантах осуществления настоящего изобретения предлагаются ингибиторы DPP-IV, которые включают N-замещенные адамантиламиноацетил-2-цианопирролидины, N-(замещенный глицил)-4-цианопирролидины, N-(N'-замещенный глицил)-2-цианопирролидины, N-аминоацилтиазолидины, N-аминоацилпирролидины, L-алло-изолейцилтиазолидин, L-трео-изолейцилпирролидин и L-алло-изолейцилпирролидин, 1-[2-[(5-цианопиридин-2-ил)амино]этиламино]ацетил-2-циано-(8)-пирролидин и их фармацевтически приемлемые соли.
В еще одном варианте осуществления настоящего изобретения также предлагаются ингибиторы DPP-IV по настоящему изобретению, которые включают ингибиторы DPP-IV, описанные в статье Patel и др., Exp.Opin. Invest. Drugs, т.12, No. 4, сс. 623-633 (2003), абзац 5, которая включена в данное описание в качестве ссылки, прежде всего, такие как Р32/98, К-364, FE-999011, BDPX, NVP-DDP-728 и др.
Соединение FE-999011 также описано в заявке РСТ WO 95/15309 в качестве соединения №18.
Соединение Р32/98 или Р3298 (номер CAS: 251572-86-8), также известное под названием 3-[(2S,3S)-2-амино-3-метил-1-оксопентил]тиазолидин, описанное в заявке РСТ WO 99/61431 под названием Probiodrug, можно использовать в виде смеси, 2:1, двух соединений, 3-[(28,38)-2-амино-3-метил-1-оксопентил]тиазолидина и (2Е)-2-бутендиоата, показанных ниже


В другом варианте осуществления настоящего изобретения предлагаются ингибиторы DPP-IV по настоящему изобретению, которые включают ингибиторы DPP-IV, описанные в заявках РСТ WO 04/037169 в примерах 1-48, WO 02/062764 в примерах 1-293 и WO 04/024184, которые включены в данное описание в качестве ссылок. Примеры, описанные в указанных опубликованных заявках, включают 3-(аминометил)-2-изобутил-1-оксо-4-фенил-1,2-дигидро-6-изохинолинкарбоксамид и 2-{[3-(аминометил)-2-изобутил-4-фенил-1-оксо-1,2-дигидро-6-изохинолил]окси}ацетамид, описанные на стр.7.
Другие ингибиторы DPP-IV описаны в заявке РСТ WO 03/004498 в примерах 1-33. В еще одном варианте осуществления настоящего изобретения предлагается соединение формулы (V), пример 7, известное под названием МК-0431.

Ингибиторы DPP-IV по настоящему изобретению также описаны в статье Ashton, Bioorg. Med. Chem. Lett., т.14, сс.859-863 (2004), прежде всего, пригодными являются соединение 1 или соединение 21е (см. таблицу 1) и соединения, указанные, в таблицах 1 и 2. Пригодные для применения ингибиторы DPP-IV также описаны в заявке РСТ WO 04/037181, которая включена в данное описание в качестве ссылки, прежде всего, пригодными являются соединения, представленные в примерах 1-33, и наиболее предпочтительно соединения, описанные в формуле изобретения в пп.3-5.
Термин «быстрое высвобождение лекарственного средства», используемый в настоящем описании, означает быстрое высвобождение основного количества терапевтического соединения, например, более приблизительно 50%, приблизительно 60%, приблизительно 70%, приблизительно 80% или приблизительно 90% в течение относительно короткого периода времени, например, в течение 1 ч, 40 мин, 30 мин или 20 мин, после перорального введения лекарственного средства. Прежде всего, наиболее предпочтительно скорость быстрого высвобождения лекарственного средства составляет высвобождение по крайней мере приблизительно 80% терапевтического соединения в течение 30 мин после перорального введения лекарственного средства. Скорость быстрого высвобождения лекарственного средства для определенного терапевтического соединения известна специалистам в данной области техники.
Термин «контролируемое высвобождение лекарственного средства», используемый в настоящем описании, означает постепенное, но при этом непрерывное или замедленное высвобождение терапевтического соединения после перорального введения лекарственного средства в течение относительно длительного периода времени, которое начинается после попадания фармацевтической композиции в желудок, в котором фармацевтическая композиция начинает распадаться на составные компоненты. Высвобождение лекарственного средства может продолжаться как до, так и после попадания фармацевтической композиции в кишечник. Термин «контролируемое высвобождение лекарственного средства» также означает замедленное высвобождение лекарственного средства, при котором высвобождение терапевтического соединения не начинается непосредственно после попадания фармацевтической композиция в желудок, а начинается через определенный период времени, например, после попадания фармацевтической композиции в кишечник, в котором повышение рН способствует высвобождению терапевтического соединения из фармацевтической композиции.
Термин «гидрофобный», используемый в настоящем описании в связи с термином «плавкий компонент», означает, что указанный плавкий компонент является более сопоставимым с маслами, а не с водой. Вещество, обладающее гидрофобными свойствами, является нерастворимым или практически нерастворимым в воде, но при этом легко растворяется в масле или других неполярных растворителях.
Термин «плавкий компонент», используемый в настоящем описании, означает вещества или смеси веществ, которые являются твердыми или полутвердыми веществами при комнатной температуре (при приблизительно 25°С) и имеют относительно низкие температуры плавления, например, от приблизительно 30°С до приблизительно 100°С или от приблизительно 50°С до приблизительно 80°С. Такие плавкие компоненты при нагревании приблизительно при температуре около или равной температуре плавления или в диапазоне температуры плавления переходят из твердого состояния в жидкое. Термин «диапазон температуры плавления», используемый в настоящем описании, означает температурный диапазон от более низкой температуры, при которой начинает образовываться первая капля жидкости из твердого вещества, до более высокой температуры, при которой твердое вещество полностью переходит в жидкое состояние.
Примеры гидрофобных плавких компонентов включают, без ограничения перечисленным, сложные эфиры, гидрированные масла, природные воски, синтетические воски, углеводороды, жирные спирты, жирные кислоты, моноглицериды, диглицериды, триглицериды и их смеси. Примеры сложных эфиров, таких как глицериловые эфиры включают, без ограничения перечисленным, глицерилмоностеарат, например, продукт CAPMUL GMS фирмы Abitec (Columbus, ОН), глицерилпальмитостеарат, ацетилированный глицеролмоностеарат, сорбитанмоностеарат, например, продукт ARLACEL 60 фирмы Uniqema (New Castle, DE), и цетилпальмитат, например, продукт CUTINA СР фирмы Cognis Corp.(Dьsseldorf, Германия). Примеры гидрированных масел включают, без ограничения перечисленным, гидрированное касторовое масло, например, продукт CUTINA HR фирмы Cognis Corp, гидрированное хлопковое масло, гидрированное соевое масло и гидрированное пальмовое масло. Примеры восков включают, без ограничения перечисленным, карнаубский воск, пчелиный воск и спермацетовый воск. Примеры углеводородов включают, без ограничения перечисленным, микрокристаллический воск и парафин. Примеры жирных спиртов, т.е. нелетучих спиртов с более высокой молекулярной массой, содержащих от приблизительно 14 до приблизительно 31 атомов углерода, включают, без ограничения перечисленным, цетиловый спирт, например, продукт CRODACOL С-70 фирмы Croda Corp.(Edison, NJ), стеариловый спирт, например, продукт CRODACOL S-95 фирмы Croda Corp, лауриловый спирт и миристиловый спирт. Примеры жирных кислот, которые содержат от приблизительно 10 до приблизительно 22 атомов углерода, включают, без ограничения перечисленным, стеариновую кислоту, например, продукт HYSTRENE 5016 фирмы Crompton Corp.(Middlebury, CT), декановую кислоту, пальмитиновую кислоту, лауриновую кислоту и миристиновую кислоту.
Количество плавкого гидрофобного компонента составляет, например, от приблизительно 1 мас.% до приблизительно 70 мас.% в расчете на массу фармацевтической композиции, от приблизительно 10 мас.% до приблизительно 60 мас.% и от приблизительно 20 мас.% до приблизительно 40 мас.%.
Термин «гранулирование из расплава», используемый в настоящем описании, включает следующие стадии:
(а) получение смеси чувствительного к влаге терапевтического соединения и по крайней мере одного плавкого гидрофобного компонента,
(б) нагревание полученной смеси при температуре выше, вблизи, практически вблизи температуры плавления гидрофобного плавкого компонента или в диапазоне температуры плавления, то есть в пределах 10°С, при этом при перемешивании с высоким сдвигом образуются гранулы, и
(в) охлаждение полученных гранул с контролируемой скоростью до комнатной температуры.
Нагревание и смешивание чувствительного к влаге терапевтического соединения и плавкого гидрофобного компонента с образованием гранул из расплава проводят, например, с использованием гранулятора с псевдоожиженным слоем или реактора с высокой скоростью сдвига при перемешивании. Количество гидрофобного компонента составляет, например, от приблизительно 40 мас.% до приблизительно 95 мас.% в расчете на массу композиции гранул, от приблизительно 50 мас.% до приблизительно 80 мас.% и от приблизительно 60 мас.% до приблизительно 75 мас.%. Аналогичным образом количество чувствительного к влаге терапевтического соединения составляет, например, от приблизительно 5 мас.% до приблизительно 60 мас.% в расчете на массу композиции гранул, полученных гранулированием из расплава, от приблизительно 20 мас.% до приблизительно 50 мас.% и от приблизительно 35 мас.% до приблизительно 40%.
Полученные из расплава гранулы включают, например, частицы чувствительного к влаге терапевтического соединения, покрытые или в значительной степени покрытые (например, по крайней мере 90% площади поверхности частиц) плавким гидрофобным компонентом. Полученное покрытие является, например, непрерывным или прерывистым и однородным или неоднородным барьером, покрывающим частицы терапевтического соединения.
Кроме того, в другом варианте настоящего изобретения предлагаются полученные из расплава гранулы, которые включают частицы чувствительного к влаге терапевтического соединения, инкапсулированные или в значительной степени инкапсулированные в плавкий гидрофобный компонент. Такие инкапсулированные гранулы включают частицы терапевтического соединения, совместимые с другими частицами чувствительного к влаге терапевтического соединения, покрытыми плавким гидрофобным компонентом.
Примеры смесителя с высоким сдвигом включают, без ограничения перечисленным, смеситель с высоким сдвигом, высокоскоростной смеситель, высокоскоростной гранулятор или экструдер для гранулирования из расплава. Способ гранулирования из расплава гранул более подробно описан ниже.
После образования гранул из расплава их перерабатывают в лекарственные формы, пригодные для перорального введения, например, стандартные твердые лекарственные формы для перорального введения, такие как таблетки, пилюли, пастилки, микротаблетки, капсулы или пакетики. Такие стандартные лекарственные формы для перорального введения включают стандартные экципиенты, используемые в фармацевтике. Примеры таких экципиентов включают, без ограничения перечисленным, дезинтегрирующие агенты, связующие агенты, замасливатели, глиданты, стабилизаторы, наполнители и разбавители. Специалисты в данной области техники могут выбрать один или более вышеуказанных экципиентов в зависимости от требуемых свойств стандартной твердой лекарственной формы для перорального введения после проведения стандартных экспериментов. Количество каждого используемого экципиента находится в диапазоне, известном в данной области техники. Методики и экципиенты, используемые для получения стандартных лекарственных форм для перорального введения, описаны в книгах The Handbook of Pharmaceutical Excipients, 4-е издание, ред. Rowe и др., American Pharmaceuticals Association (2003) и Remington: the Science and Practice of Pharmacy, 20-е издание, ред. Gennaro, Lippincott Williams & Wilkins (2003).
Примеры фармацевтически приемлемых дезинтегрирующих агентов включают, без ограничения перечисленным, крахмалы, глины, целлюлозы, альгинаты, камеди, сшитые полимеры, например, сшитый поливинилпирролидон или кросповидон, например, POLYPLASDONE XL фирм International Specialty Products (Wayne, NJ), натриевую соль сшитой карбоксиметилцеллюлозы или натриевую соль сшитой кроскармеллозы, например, продукт AC-DI-SOL фирмы FMC, и кальциевую соль сшитой карбоксиметилцеллюлозы, полисахариды сои и гуаровую камедь. Количество дезинтегрирующего агента составляет, например, от приблизительно 1 мас.% до приблизительно 20 мас.% в расчете на массу композиции, от приблизительно 5 мас.% до приблизительно 10 мас.%, например, приблизительно 5 мас.%.
Примеры фармацевтически приемлемых связующих агентов включают, без ограничения перечисленным, крахмалы, целлюлозы и их производные, например, микрокристаллическую целлюлозу, продукт AVICEL РН фирмы FMC (Philadelphia, PA), гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу и гидроксипропилметилцеллюлозу, продукт METHOCEL фирмы Dow Chemical Corp.(Midland, MI), сахарозу, декстрозу, кукурузную патоку, полисахариды и желатин. Количество связующего агента составляет, например, от приблизительно 5 мас.% до приблизительно 50 мас.% в расчете на массу композиции, например, от приблизительно 10 мас.% до 40 мас.%.
Примеры фармацевтически приемлемых замасливателей и фармацевтически приемлемых глидантов включают, без ограничения перечисленным, коллоидный оксид кремния, трисиликат магния, крахмалы, тальк, трехзамещенный фосфат кальция, стеарат магния, стеарат алюминия, стеарат кальция, карбонат магния, оксид магния, полиэтиленгликоль, порошкообразную и микрокристаллическую целлюлозу. Количество замасливателя составляет, например, от приблизительно 0,1 мас.% до приблизительно 5 мас.% в расчете на массу композиции, при этом количество глиданта составляет, например, от приблизительно 0,1% до приблизительно 10 мас.% в расчете на массу композиции.
Примеры фармацевтически приемлемых наполнителей и фармацевтически приемлемых разбавителей включают, без ограничения перечисленным, сахарную пудру, прессованный сахар, декстраны, декстрин, декстрозу, лактозу, маннит, микрокристаллическую целлюлозу, порошкообразную целлюлозу, сорбит, сахарозу и тальк. Количество наполнителя и/или разбавителя в композиции составляет, например, от приблизительно 15 мас.% до приблизительно 40 мас.% в расчете на массу композиции.
Для получения твердых лекарственных форм для перорального введения с быстрым высвобождением лекарственного средства из гранул, полученных гранулированием из расплава, сначала получают чувствительное к влаге терапевтическое соединение и плавкий гидрофобный компонент, которые в смеси или отдельно просеивают через сито, при этом получают частицы каждого материала максимального размера. Требуемые размеры частиц каждого материала определяются специалистами в данной области техники в зависимости от конкретной фармацевтической композиции. Например, пригодные размеры частиц составляют менее 1000 мкм, 750 мкм, 500 мкм или 250 мкм.
Чувствительное к влаге терапевтическое соединение и плавкий гидрофобный компонент смешивают в соотношении в диапазоне от 1:0,25 до 1:1 или до 1:10 (в расчете на массу в сухом состоянии) или более предпочтительно в диапазоне от 1:1 до 1:4 (в расчете на массу в сухом состоянии) в реакторе с рубашкой, снабженном, например, мешалкой с высоким сдвигом. Смесь нагревают, например, при подаче пара в рубашку до температуры вблизи, практически вблизи или равной температуре плавления плавкого гидрофобного компонента или выше диапазона температуры плавления плавкого гидрофобного компонента. Полученную смесь выдерживают при повышенной температуре и перемешивают в течение определенного периода времени, достаточного для получения в значительной степени гомогенного гранулированного продукта. После образования в значительной степени гомогенного гранулированного продукта гранулированную смесь охлаждают, например, при подаче холодной воды в рубашку, до затвердевания смеси. Затвердевшую гранулированную смесь медленно охлаждают в реакторе с рубашкой до комнатной температуры или немедленно удаляют из реактора с рубашкой и охлаждают на алюминиевой фольге (для медленного охлаждения смеси).
После охлаждения гранулы измельчают и просеивают через сито, затем полученные гранулы смешивают с экципиентами для стандартной твердой лекарственной формы, т.е. с наполнителями, связующими агентами, дезинтегрирующими агентами и замасливателями. Полученную смесь перемешивают, например, с использованием V-смесителя и затем прессуют или формуют, при этом получают таблетки, или смесью заполняют капсулы.
Пригодность всех фармацевтических композиций по настоящему изобретению оценивают методом стандартных клинических испытаний, например, при определении дозировки стандартных лекарственных форм, достаточной для достижения терапевтически эффективного уровня терапевтического соединения в крови, например, при введении лекарственного средства в дозе в диапазоне от 2,5 мг до 1000 мг/сут млекопитающему массой 75 кг, например взрослому человеку, или стандартному модельному животному.
Количество терапевтического соединения в фармацевтической композиции, например, в форме таблетки или порошка, пригодного для получения таблетки, составляет от 0,1 мг до 100 мг, например, 0,1 мг, 1 мг, 5 мг, 10 мг, 20 мг, 25 мг, 50 мг или 100 мг. Такие стандартные лекарственные формы являются пригодными для введения лекарственного средства от 1 до 5 раз в сутки в зависимости от назначенного лечения, стадии лечения и т.п.
Настоящее изобретение относится к способу лечения заболевания, состояния или нарушения у субъекта с использованием чувствительного к влаге терапевтического соединения, который включает введение терапевтически эффективного количества фармацевтической композиции по настоящему изобретению субъекту, который нуждается в таком лечении. Кроме того, настоящее изобретение относится к применению композиции по настоящему изобретению, включающей ингибитор DPP-IV, для получения лекарственного средства, предназначенного для лечения и/или профилактики заболеваний, таких как инсулинонезависимый сахарный диабет, артрит, ожирение, трансплантация аллотрансплантата и кальцитонин-остеопороз, или нарушений, опосредованных изменением уровней GLP-1 и/или GLP-2.
Следующие примеры приведены для иллюстрации настоящего изобретения и не ограничивают его объем. Примеры приведены только для описания применения способа по настоящему изобретению на практике.
Количество ингредиентов, используемое в каждом примере, представлено в массовых процентах в расчете на массу композиции и указано в соответствующих таблицах после соответствующего описания.
Пример 1
Твердая лекарственная форма для перорального введения, полученная сухим смешиванием
Сначала соединение I просеивали через сито (25 меш), при этом получали 11,2 г соединения I. В V-смеситель объемом 1 кварта добавляли соединение I и 100 г лактозы, интенсивно перемешивали в течение 5 мин. Полученную смесь удаляли, просеивали через сито (25 меш) и снова добавляли в V-смеситель, затем в V-смеситель добавляли тальк, кросповидон и остальную порцию лактозы, полученную смесь интенсивно перемешивали в течение еще 10 мин. Гидрированное касторовое масло продавливали через сито (60 меш), затем добавляли в V-смеситель и полученную смесь интенсивно перемешивали в течение 5 мин. Затем смесь прессовали на таблеточном прессе Manesty B3B с использованием круглого стандартного режущего устройства с вогнутым и скошенным краем. Устройство предварительно полировали для предотвращения образования пленки. В настоящем описании полученные таблетки массой 150 мг, содержащие приблизительно 5 мг соединения I, названы образцом 1. Образец 1 не содержит частицы соединения I, покрытые или в значительной степени покрытые гидрированным касторовом маслом.
Пример 2
Твердая лекарственная форма для перорального введения, полученная гранулированием из расплава с добавлением гидрированного касторового масла
Для сравнения с образцом 1 твердую лекарственную форму для перорального введения получали из гранулированного из расплава соединения I. Соединение I и плавкий гидрофобный компонент, т.е. гидрированное касторовое масло, каждый материал в отдельности просеивали через сито (25 меш) и сито (60 меш) соответственно, затем ингредиенты добавляли в гранулятор объемом 1 л с высоким сдвигом фирмы Key International (Englishtown, NJ), модель KG5.
Емкость гранулятора снабжена нагревающим кожухом, реостат установлен на 80°С, гранулятор снабжен лопастной мешалкой без режущих элементов, которую включали для перемешивания терапевтического соединения и плавкого гидрофобного компонента.
После перемешивания гранулы удаляли из гранулятора и равномерно распределяли на алюминиевой фольге для охлаждения, затем гранулы просеивали через сито с использованием вибратора Frewitt.
Полученные гранулы, микрокристаллическую целлюлозу и кросповидон добавляли в V-смеситель и смесь интенсивно перемешивали в течение приблизительно 10 мин, затем добавляли гидрированное касторовое масло и интенсивно перемешивали в течение еще 5 мин.
Затем полученную смесь прессовали на таблеточном прессе Manesty B3B с использованием круглого стандартного режущего устройства с вогнутым и скошенным краем. Устройство предварительно полировали для предотвращения образования пленки.
Таблетки, полученные, как описано в примере 2, названы образцом 2.
Пример 3
Твердая лекарственная форма для перорального введения, полученная гранулированием из расплава с добавлением стеариновой кислоты
Для сравнения с образцом 1 и образцом 2 другую твердую лекарственную форму для перорального введения получали из гранулированного из расплава соединения I аналогично тому, как описано в примере 2, но при замене гидрированного касторового масла на стеариновую кислоту. В данном случае стеариновую кислоту использовали в качестве плавкого гидрофобного компонента в составе полученных из расплава гранул и замасливателя в составе полученных таблеток. В настоящем описании полученные таким образом таблетки названы образцом 3.
В таблице 1 представлены композиции образцов, полученных, как описано в примерах 1, 2 и 3.
Каждый из образцов помещали во флаконы из ПЭВП, запаянные индукционным способом, и выдерживали в течение 4 недель в реальном режиме старения, т.е. при 25°С и относительной влажности (OB) 75% и в ускоренном режиме старения, т.е. при 40°С и OB 75%. Через 4 недели измеряли содержание циклического имидата в качестве продукта разложения соединения I. Результаты испытаний представлены ниже в таблице 2.
После испытаний образца 1 (таблетки, полученные сухим смешиванием) в режиме ускоренного старения в течение 4 недель наблюдалась самая высокая степень разложения терапевтического соединения. Таким образом, терапевтическое соединение в образце 1 характеризуется самой высокой химической нестабильностью и является наиболее чувствительным к влаге. Для образцов 2 и 3 получены сопоставимые результаты, которые свидетельствуют о повышении химической стабильности по сравнением с образцом 1.
Кроме испытаний на стабильность для указанных трех образцов определяли растворимость. Для всех указанных трех образцов наблюдалось практически полное высвобождение лекарственного средства через 10 мин. Образец 1, как и ожидалось, характеризовался быстрым высвобождением лекарственного средства, так как таблетки получали сухим смешиванием и прессованием. Однако неожиданно было установлено, что гранулы образцов 2 и 3, в которых терапевтическое соединение покрыто или в значительной степени покрыто плавким гидрофобным компонентом, быстро и эффективно растворялись и их можно использовать для быстрого высвобождения лекарственного средства.
Пример 4
Твердая лекарственная форма для перорального введения, полученная гранулированием из расплава, содержащая соединение I в дозе 5 мг и 20 мг
Стандартную твердую форму, содержащую соединение I в количестве 5 мг и 20 мг, получали гранулированием из расплава. Таблетки массой 125 мг и 250 мг, соответственно, получали аналогично тому, как описано в примере 2. Соотношение соединения 1 и гидрированного касторового масла в полученных гранулах составляло приблизительно 1:4.
Полученные таблетки массой 125 мг, содержащие 5 мг соединения I и лактозу, названы образцом 4А, а таблетки массой 250 мг, содержащие 20 мг соединения I и лактозу, названы образцом 4Б.
Пример 5
Твердая лекарственная форма для перорального введения, полученная гранулированием из расплава, содержащая соединение I в дозе 50 мг и 100 мг
Лекарственную форму, содержащую соединение I в дозе 50 мг и 100 мг, получали гранулированием из расплава. Таблетки массой 250 мг получали аналогично тому, как описано в примере 2. Соотношение соединения I и гидрированного касторового масла в полученных гранулах составляло приблизительно 1:1.
Полученные таблетки массой 250 мг, содержащие 50 мг соединения I и лактозу, названы образцом 5А, а таблетки массой 250 мг, содержащие 100 мг соединения I и лактозу, названы образцом 5Б.
В таблице 3 представлены композиции образцов, полученных, как описано в примерах 4 и 5. Ингредиенты, выделенные жирным шрифтом, являются компонентами гранул, полученных гранулированием из расплава.
Пример 6
Твердая лекарственная форма для перорального введения, полученная гранулированием из расплава, содержащая соединение I в дозе 20 мг, 55 мг и 100 мг и микрокристаллическую целлюлозу
Образцы 4Б, 5А и 5Б использовали для получения лекарственной формы, содержащей микрокристаллическую целлюлозу или продукт AVICEL РН фирмы FMC Corporation (Philadelphia, PA). Образцы 4Б, 5А и 5Б получали аналогично тому, как описано выше, но при замене половины количества лактозы на микрокристаллическую целлюлозу. Образцы 4Б, 5А и 5Б, содержащие микрокристаллическую целлюлозу, названы образцами 6А, 6Б и 6В соответственно.
В таблице 4 представлены композиции образцов 6А, 6Б и 6В.
В таблице 5 представлены данные о стабильности образцов 4А, 4Б, 5А, 5Б, 6А, 6Б и 6В, полученные при выдерживании в течение 4 недель при 25°С и OB 60%.
Полученные из расплава гранулы, содержащие 100 мг соединения I и
Полученные из расплава гранулы, содержащие 50 мг соединения I,
Полученные из расплава гранулы, содержащие 100 мг соединения I,
В таблице 6 представлены данные о растворимости каждого образца 4А, 4Б, 5А, 5Б, 6А, 6Б и 6В в течение 10 мин, 20 мин и 30 мин после начала проведения испытаний. Каждый образец растворяли в 0,1 н. соляной кислоте при перемешивании лопастной мешалкой (аппарат 2) при 37°С.
Полученные из расплава гранулы, содержащие 5 мг соединения I и гидрированное касторовое масло, не содержащие микрокристаллическую целлюлозу
Полученные из расплава гранулы, содержащие 20 мг соединения I и гидрированное касторовое масло, не содержащие микрокристаллическую целлюлозу
Полученные из расплава гранулы, содержащие 50 мг соединения I и гидрированное касторовое масло, содержащие микрокристаллическую целлюлозу
Полученные из расплава гранулы, содержащие 100 мг соединения I и гидрированное касторовое масло, не содержащие микрокристаллическую целлюлозу
Полученные из расплава гранулы, содержащие 50 мг соединения I, гидрированное касторовое масло и микрокристаллическую целлюлозу
При испытании каждого образца 4А, 4Б, 5А, 5Б, 6А и 6Б наблюдается растворение более 80% количества соединения I, в расчете от указанного на этикетке содержания, в течение менее 30 мин, что указывает на пригодность указанных образцов для быстрого высвобождения лекарственного средства.
Другие эксперименты (примеры 7 и 8) проводили для определения влияния экципиента с высоким равновесным содержанием воды на соединение I в присутствии плавкого гидрофобного компонента в различных концентрациях. В качестве основного экципиента использовали микрокристаллическую целлюлозу. Следующие образцы характеризуются следующим соотношением x:y:z, где x означает концентрацию соединения 1, у означает концентрацию гидрированного касторового масла и z означает концентрацию микрокристаллической целлюлозы.
Пример 7
Полученные из расплава гранулы, содержащие микрокристаллическую целлюлозу
Полученные из расплава гранулы, содержащие соединение I и гидрированное касторовое масло, получали, как описано в примере 2, при этом получали пять образцов. Все указанные образцы, кроме образца 7А, содержали полученные из расплава гранулы, которые смешивали с микрокристаллической целлюлозой при соотношении соединение 1/микрокристаллическая целлюлоза 1:1. Однако каждый образец отличался по содержанию гидрированного касторового масла. В таблице 7 представлен состав каждого образца.
7А
(1:0:1)
7Б
(1:0.5:1
(1:1:1)
7Г
(1:2:1)
7Д
(1:4:1)
В таблице 8 представлены данные о стабильности каждого образца, полученного, как описано в примере 7, при выдерживании образца при 40°С и при 40°С и OB 75%. Таблетки каждого образца выдерживали во флаконах из полиэтилена высокой плотности (ПЭВП), запаянных индукционным способом.
Данные, представленные в таблице 8, свидетельствуют об уменьшении содержания циклического имидата, образующегося в результате разложения соединения I, при этом содержание плавкого гидрофобного компонента и гидрированного касторового масла увеличивается. Неожиданно было установлено, что присутствие гидрированного касторового масла снижает чувствительность соединения I к влаге даже в присутствии экципиента с высоким равновесным содержанием влаги, т.е. микрокристаллической целлюлозы.
Пример 9
Твердая лекарственная форма для перорального введения, содержащая соединение ПС
Таблетки массой 10 мг, содержащие соединение IIС, получали по следующей методике. Соединение IIС, микрокристаллическую целлюлозу, лактозу и натриевую соль гликолята крахмала перемешивали в смесителе объемом 4 кварты до полного смешивания ингредиентов. Стеарат магния отдельно просеивали через сито (25 меш) и затем добавляли в смеситель в полученную смесь. Смесь, полученную сухим смешиванием, удаляли из смесителя и прессовали на роторном таблеточном прессе, при этом получали образец 9.
Пример 10
Твердая лекарственная форма для перорального введения, полученная гранулированием из расплава, содержащая соединение IIС
Для сравнения с примером 9 состав, содержащий 10 мг соединения IIС, получали гранулированием из расплава по следующей методике. 10 мг соединения IIС и 15 мг стеарилового спирта каждый материал в отдельности просеивали через сито (18 меш) и добавляли в смеситель Хобарта. Смеситель снабжен нагревающим кожухом, реостат установлен на 100°С. Смесь перемешивали до образования гранул в течение приблизительно 10-15 мин, затем гранулы удаляли из смесителя и размещали на алюминиевой фольге для охлаждения до комнатной температуры. После охлаждения гранулы просеивали через сито (20 меш). Половину количества полученных гранул использовали для других целей, а другую половину полученных гранул смешивали с микрокристаллической целлюлозой (128 г), лактозой (64 г), натриевой солью гликолята крахмала (12 г) и стеаратом магния (3 г) в смесителе Turbula и перемешивали в течение 10 мин, полученную смесь прессовали на роторном таблеточном прессе, при этом получали образец 10.
Каждый образец 9 и 10 помещали в индукционные герметично закрытые флаконы из ПЭВП и испытывали на стабильность в течение 3 недель при 40°С и OB 75%, в течение 6 недель при 30°С и OB 60% и в течение 6 недель при 40°С и OB 75%. В таблице 9 представлены данные о содержании продукта полного разложения в процентах, определенном после испытания на стабильность.
Данные, представленные в таблице 9, свидетельствуют о том, что твердая лекарственная форма для перорального введения, содержащая полученные гранулированием из расплава гранулы (пример 10), характеризуется повышенной химической стабильностью терапевтического соединения по сравнению с композицией, полученной сухим смешиванием (пример 9).
Кроме того, определяли растворимость таблеток, полученных, как описано в примере 10, в воде при скорости вращения 50 об/мин. После вращения в течение 30 мин наблюдалось высвобождение более 90% терапевтического соединения растворялось в воде.
Следует понимать, что настоящее изобретение подробно описано для иллюстрации и не ограничивает объем настоящего изобретения, который определен в формуле изобретения. Другие объекты, преимущества и модификации также включены в объем изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СУХОГО ГРАНУЛИРОВАНИЯ ДЛЯ ПОЛУЧЕНИЯ КОМПОЗИЦИЙ МЕТФОРМИНА В ВИДЕ ТАБЛЕТОК И ЕГО КОМПОЗИЦИИ | 2013 |
|
RU2647421C2 |
| НОВЫЙ СОСТАВ | 2006 |
|
RU2483716C2 |
| НОВЫЙ СОСТАВ | 2006 |
|
RU2821230C2 |
| ТВЕРДАЯ ДОЗИРОВАННАЯ ЛЕКАРСТВЕННАЯ ФОРМА ДЛЯ ОРАЛЬНОГО ВВЕДЕНИЯ, СОДЕРЖАЩАЯ КОМБИНАЦИЮ ВИЛДАГЛИПТИНА И ГЛИКВИДОНА | 2014 |
|
RU2585378C1 |
| ЭКСТРУЗИОННЫЙ СПОСОБ ПРИГОТОВЛЕНИЯ КОМПОЗИЦИЙ, СОДЕРЖАЩИХ ПЛОХО ПРЕССУЮЩИЕСЯ ЛЕКАРСТВЕННЫЕ СОЕДИНЕНИЯ | 2006 |
|
RU2405539C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИМАТИНИБ И ЗАМЕДЛИТЕЛЬ ВЫСВОБОЖДЕНИЯ | 2006 |
|
RU2404775C2 |
| СПОСОБ ГРАНУЛЯЦИИ ИЗ РАСПЛАВА | 2009 |
|
RU2491918C2 |
| КОМБИНИРОВАННАЯ СИСТЕМА ДОСТАВКИ С НЕМЕДЛЕННЫМ/ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ ДЛЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ С КОРОТКИМ ПЕРИОДОМ ПОЛУВЫВЕДЕНИЯ, В ТОМ ЧИСЛЕ ДЛЯ РЕМОГЛИФЛОЗИНА | 2011 |
|
RU2596787C2 |
| КОМПОЗИЦИЯ И СПОСОБ ПРЯМОГО ПРЕССОВАНИЯ | 2005 |
|
RU2391097C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ, КОМПОЗИЦИЯ И КОМБИНИРОВАННЫЙ СОСТАВ, СОДЕРЖАЩИЙ АКТИВАТОР ГЛЮКОКИНАЗЫ И ИНГИБИТОР DPP-IV, И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2770775C1 |
Фармацевтическая композиция быстрого высвобождения включает гранулы, полученные гранулированием из расплава. Гранулы содержат ингибитор DPP-IV и плавкий гидрофобный компонент при соотношении от 1:1 до 1:10 (на сухой вес). По меньшей мере 90% поверхности гранул покрыто плавким гидрофобным компонентом. Гранулы высвобождают приблизительно 50% ингибитора DPP-IV в течение 30 минут после перорального введения лекарственного средства. Ингибитором DPP-IV является N-(замещенный глицил)-2-цианопирролидин или его фармацевтически приемлемая соль. Предпочтительно ингибитором DPP-IV является (S)-1-[(3-гидрокси-1-адамантил)амино]ацетил-2-цианопирролидин. Композиции для быстрого высвобождения по изобретению обладают улучшенной стабильностью в присутствии влаги по сравнению с известными композициями для контролируемого или пролонгированного высвобождения. 4 н. и 16 з.п. ф-лы, 9 табл.
1. Фармацевтическая композиция быстрого высвобождения для перорального введения ингибитора DPP-IV, включающая гранулы, которые содержат указанный ингибитор DPP-IV, по меньшей мере 90% поверхности которых покрыто плавким гидрофобным компонентом, причем указанные гранулы включают ингибитор DPP-IV и указанный плавкий гидрофобный компонент при соотношении от 1:1 до 1:10 (сухой вес) и получены гранулированием из расплава.
2. Фармацевтическая композиция по п.1, причем из указанной фармацевтической композиции высвобождается по крайней мере приблизительно 50% ингибитора DPP-IV в течение 30 мин после перорального введения лекарственного средства.
3. Фармацевтическая композиция по п.2, причем из указанной фармацевтической композиции высвобождается по крайней мере приблизительно 80% ингибитора DPP-IV в течение 30 мин после перорального введения лекарственного средства.
4. Фармацевтическая композиция по п.1, в которой указанное соотношение составляет от 1:1 до 1:4.
5. Фармацевтическая композиция по п.1, причем указанная фармацевтическая композиция, кроме того, включает дезинтегрирующий агент, содержание которого составляет от приблизительно 1 до 20% в расчете на массу указанной композиции.
6. Фармацевтическая композиция по п.1, в которой ингибитором DPP-IV является N-(замещенный глицил)-2-цианопирролидин или его фармацевтически приемлемая соль.
7. Фармацевтическая композиция по п.6, в которой указанный N-(замещенный глицил)-2-цианопирролидин характеризуется формулой (II)

где R означает замещенный адамантил и
n равно от 0 до 3.
8. Фармацевтическая композиция по любому из пп.1-6, в которой ингибитором DPP-IV является (S)-1-[(3-гидрокси-1-адамантил)амино]ацетил-2-цианопирролидин.
9. Фармацевтическая композиция по п.8, включающая (S)-1-[(3-гидрокси-1-адамантил)амино]ацетил-2-цианопирролидин в количестве от 50 до 100 мг.
10. Способ получения гранул, по меньшей мере 90% поверхности которых покрыто плавким гидрофобным компонентом, включающий стадии:
а) получение смеси ингибитора DPP-IV и по крайней мере одного плавкого гидрофобного компонента, где соотношение ингибитора DPP-IV и указанного плавкого гидрофобного компонента составляет от 1:1 до 1:10 (сухой вес);
(б) нагревание указанной смеси при температуре, близкой к температуре плавления указанного плавкого гидрофобного компонента,
(в) гранулирование полученной смеси при перемешивании с высоким сдвигом с образованием гранул и
(г) охлаждение указанных гранул до комнатной температуры.
11. Способ по п.10, в котором указанная смесь в основном включает ингибитор DPP-IV и по крайней мере один указанный плавкий гидрофобный компонент.
12. Способ по п.10, в котором плавкий гидрофобный компонент выбирают из группы, включающей сложные эфиры, воски, углеводороды, жирные спирты, жирные кислоты, моноглицериды, диглицериды, триглицериды и их смеси.
13. Способ по п.10, в котором ингибитором DPP-IV является N-(замещенный глицил)-2-цианопирролидин или его фармацевтически приемлемая соль.
14. Способ по п.13, в котором указанным N-(замещенный глицил)-2-цианопирролидином является (S)-1-[(3-гидрокси-1-адамантил)амино]ацетил-2-цианопирролидин.
15. Фармацевтическая композиция, включающая гранулы, полученные способом по п.10.
16. Фармацевтическая композиция быстрого высвобождения для перорального введения, включающая гранулы, которые, по существу, содержат ингибитор DPP-IV и по меньшей мере один плавкий гидрофобный компонент, по меньшей мере 90% поверхности которых покрыто плавким гидрофобным компонентом, причем указанные гранулы включают ингибитор DPP-IV и указанный плавкий гидрофобный компонент при соотношении от 1:1 до 1:10 (сухой вес) и получены гранулированием из расплава.
17. Фармацевтическая композиция по п.16, причем из указанной фармацевтической композиции высвобождается по крайней мере приблизительно 50% указанного ингибитора DPP-IV в течение 30 мин после перорального введения лекарственного средства.
18. Фармацевтическая композиция по п.16, причем из указанной фармацевтической композиции высвобождается по крайней мере приблизительно 80% указанного ингибитора DPP-IV в течение 30 мин после перорального введения лекарственного средства.
19. Фармацевтическая композиция по любому из пп.15-18, в которой указанным ингибитором DPP-IV является (S)-1-[(3-гидрокси-1-адамантил)амино]ацетил-2-цианопирролидин.
20. Фармацевтическая композиция по п.19, включающая (S)-1-[(3-гидрокси-1-адамантил)амино]ацетил-2-цианопирролидин в количестве от 50 до 100 мг.
| Экономайзер | 0 |
|
SU94A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Способ управления тепломассообменным процессом | 1977 |
|
SU654263A2 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |

