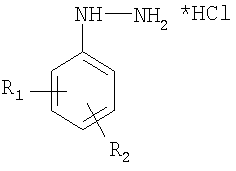
Изобретение относится к области синтетической органической химии. Предлагаемый метод позволяет получать различные замещенные арилгидразины, которые используются в органическом синтезе для получения разнообразных гетероциклических соединений, применяющихся в производстве органических красителей, биологически активных соединений, антиоксидантов и др. Например, описан синтез на основе арилгидразинов инсектицидов [1], триазолов с высокой гербицидной активностью [2-4], замещенных пиразолов [5-8], конденсированных пиразолов [9] и N-окисей пиразолов [10] с гербицидной активностью.
В качестве метода получения арилгидразинов чаще других используется восстановление солей диазония, поскольку он основан на доступных и дешевых исходных - замещенных анилинах.
В качестве восстановителей применяют хлорид олова (II), сульфитные смеси и аскорбиновую кислоту. Перечисленные методы имеют ряд недостатков и ограничений в использовании. Так, восстановление сульфитными смесями проводится, как правило, при положительных температурах [1], что предполагает использование только устойчивых к действию повышенных температур солей диазония. Восстановление сульфитными смесями, кроме того, весьма чувствительно к кислотности реакционной смеси, требует строгого выдерживания определенного значения рН в течение всего времени реакции [11]. Успешное восстановление аскорбиновой кислотой возможно также лишь при положительных температурах. Восстановление по этому способу, кроме того, требует длительного времени как для собственно восстановления, так и для последующего гидролиза образующихся в ходе восстановления производных щавелевой кислоты [5].
Наиболее близким к заявляемому решению является взятый за прототип способ восстановления солей диазония двухлористым оловом [10]. Однако этот способ предполагает использование низких температур -25°С - -50°С. Имеющиеся примеры проведения реакции при более высоких температурах крайне редки и предполагают использование только устойчивых солей диазония, содержащих в ароматическом ядре сильные электроноакцепторы, например атом фтора в о-положении к диазогруппе [3, 6, 9].
Целью изобретения является повышение выходов целевых арилгидразинов, упрощение технологии проведения реакции, сокращение времени реакции, расширение круга потенциальных исходных соединений.
Поставленная цель достигается тем, что в предлагаемом способе соль диазония, полученная из соответствующего анилина, обрабатывается сначала четыреххлористым оловом, затем выпавший осадок гексахлорстанната диазония отделяется фильтрованием и вводится во взаимодействие с 10-50% избытком двухлористого олова в концентрированной соляной кислоте при температуре -15-0°С в течение 15-40 минут. Для выделения полученного арилгидразина образовавшуюся реакционную смесь подщелачивают раствором NaOH до нейтральной реакции, полученную смесь экстрагируют бензолом, экстракт промывают насыщенным раствором хлористого аммония, водой, сушат безводным сульфатом натрия и бензол отгоняют.
Выделение синтезированного арилгидразина проводят в виде гидрохлорида, для чего бензольный раствор упаривают вдвое и при слабом охлаждении постепенно пропускают сухой хлороводород до прекращения выпадения осадка гидрохлорида арилгидразина, осадок отфильтровывают и сушат. Для выделения образовавшегося арилгидразина в виде гидрохлорида после окончания восстановления реакционную смесь нагревают при перемешивании до 80°С до растворения выпавшего осадка, реакционную смесь оставляют при комнатной температуре на 1 час, выпавший осадок отфильтровывают и сушат.
При этом не требуется специальный температурный режим и реакция проводится при температуре -15-0°С, то есть в обычных условиях для получения и использования солей диазония. Реакция сопровождается слабым экзотермическим эффектом и заканчивается, как правило, за 15-40 минут.
Целевой гидразин при описанном способе получения является практически единственным продуктом реакции, что обеспечивает его высокие выходы, определяемые лишь полнотой выделения целевого продукта и его устойчивостью. Отсутствие заметных примесей в итоговой реакционной смеси позволяет в ряде случаев выделять целевой продукт простым фильтрованием выпавшего гидрохлорида арилгидразина (см. пример 2) либо использовать полученный при стандартном выделении (пример 1) экстракт в органическом растворителе для дальнейших превращений без выделения и очистки индивидуального вещества. Способ выделения синтезированного арилгидразина определяется его растворимостью в реакционной среде и устойчивостью в виде свободного основания.
Высокие выходы, селективность, мягкие условия проведения реакции обуславливаются на наш взгляд тем, что восстановлению подвергается не хлорид диазония, а образовавшийся после его обработки тетрахлоридом олова комплексный гексахлорстаннат. При этом олово (IV) противоиона в комплексе легко восстанавливается до олова (II), которое, оставаясь в координационной сфере арилдиазоний катиона, быстро и эффективно восстанавливает последний до арилгидразина.
Суть предлагаемого решения иллюстрируется ниже приведенными примерами.
Пример 1
Синтез 3-хлор-2-(N-морфолил)гидразина (I)
В реакторе емкостью 150 мл, снабженном термометром и механической мешалкой, смешивают 10,63 г (0,05 моля) 3-хлор-2-морфолиланилина и 10,6 мл концентрированной соляной кислоты (р=1,18 г/мл) при комнатной температуре. Смесь охлаждают до -5°С (лед-соль) и постепенно по каплям добавляют 19 мл раствор нитрита натрия с концентрацией 200 г/л при температуре не выше 0°С. После окончания диазотирования к реакционной смеси добавляют при перемешивании 3,5 мл (0,03 моль) тетрахлорида олова при температуре 0-5°С. Выпавший осадок отфильтровывают. Слегка влажный осадок соответствующего гексахлорстанната небольшими порциями добавляют к раствору 25,39 г (0,1125 моль, 50%-ный мольный избыток) дигидрата двухлористого олова в 18 мл концентрированной соляной кислоты. В течение 40 минут при интенсивном перемешивании и температуре -15°С в начальный момент и не выше 0°С в конце добавления. Окончание реакции определяют взаимодействием с раствором R-соли (проба на вытек).
Выделение гидразина I (метод А): К реакционной смеси добавляют 200 мл воды и нейтрализуют концентрированным раствором едкого натра до рН=7. Полученную смесь экстрагируют бензолом (80 мл × 6). Объединенный экстракт промывают насыщенным раствором хлористого аммония и водой. Бензол упаривают до 200 мл и сушат безводным сульфатом натрия. Полученный раствор 3-хлор-2-(N-морфолил)фенилгидразина может быть использован в органическом синтезе, например, индолов по реакции Фишера без выделения и дополнительной очистки гидразина.
Выделение гидразина I виде гидрохлорида (метод Б): Через бензольный раствор при слабом охлаждении постепенно пропускают сухой хлороводород до прекращения выпадения осадка гидрохлорида 3-хлор-2-(N-морфолил)гидразина. Осадок отфильтровывают и сушат.
Пример 2
Синтез 2-метокси-5-нитрофенилгидразина (II)
Гидразин II получают из 2-метокси-5-нитроанилина, как описано в примере 1, диазотированием и последующим восстановлением двухлористым оловом, взятым с 10%-ным избытком, в течение 10 минут.
Выделение гидрохлорида гидразина II (метод В): После окончания восстановления реакционную смесь нагревают при перемешивании до 80°С до растворения выпавшего осадка. Реакционную смесь оставляют при комнатной температуре на 1 час. Выпавший осадок отфильтровывают и сушат.
Выходы синтезированных гидрохлоридов гидразинов, а также способы их выделения приведены в таблице.
Источники информации
1. Jin Haihong. Process for the preparation of insecticidal phenylhydrazine derivatives. - Pat. USA №6278022 B1. - 21.08.2001.
2. George Theodoridis. Herbicidal 1-aryl-δ2-1,2,4-triazolin-5-ones. - Pat. USA №8700730. - 12.02.1987.
3. George Theodoridis. Herbicdal aryl triazolinones. - Pat. USA №4818275. - 04.04.1989.
4. Lester L. Maravets. Herbicdal aryl triazolinones. - Pat. USA №4743291. - 10.05.1988.
5. John F. Lambert, Timothy Norris. Preparation of sodium-hydrogen exchanger type-1 inhibitors. - Pat. USA №20020082274 A1. - 27.06.2002.
6. Suresh R. Kapadia, Jinhua J. Song, Nathan K. Yee. Bis pyrazole-1H-pyrazole intermediates and their syshtesis. - Pat. USA №6492529 B1. - 10.12.2002.
7. M.V. Ramana Reddy, Stanley C. Bell. Process for the preparation of 1,5-diaryl-3-substituted-pyrazoles-Pat USA №20030109709 A1. - 12.12.2003.
8. Rafael Shapiro, licius T. Rossano, Karen L. Tenhuisen. Process for the preparation of 1,3,5-trisubstituted pyrazoles via [3+2] cycloaddition. - Pat. USA №20060069270 A1. - 30.03.2006.
9. Haga Tom, Nagano Eiki, Morita Kouchi, Sato Ryo. Indazole compounds, their production, use and intermediates. - Pat. JP. №0235567 A2. - 09.09.1987.
10. Enomoto Masayuki, Nagano Eiki, Haga Toru, Morita Kouichi, Sato Ryo, Bezoxazinyl-triazole oxides, their production and used. - Pat. JP. №0305923 A1. - 08.03.1989.
11. Jean-Manuel Mas, Christophe Rochin. Method for synthesizing aryl hydrazine through reduction of a diazo compound derivative. Pat. USA №6087534. - 11.07.2000.
| название | год | авторы | номер документа |
|---|---|---|---|
| Этил-2-(9-аминохромено[4,3-d]пиримидин-5-ил)ацетаты и способ их получения | 2020 |
|
RU2746879C1 |
| Способ получения 2-замещенных 1-гидроксипирроло[3,4-f]индол-5,7-(1Н,6Н)-дионов | 2015 |
|
RU2613582C1 |
| Способ получения леналидомида и интермедиата для его производства | 2020 |
|
RU2730858C1 |
| 4-ЗАМЕЩЕННЫЕ N-АРИЛ-1,8-НАФТАЛИМИДЫ, ПРОЯВЛЯЮЩИЕ СВОЙСТВА ФЛУОРЕСЦЕНТНЫХ СЕНСОРОВ НА КАТИОНЫ МЕТАЛЛОВ, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2012 |
|
RU2515195C1 |
| Производные 2-(хромено[4,3-d]пиримидин-5-ил)уксусной кислоты и способ их получения | 2019 |
|
RU2716597C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ГИДРОКСИ-3-R-ИНДОЛ-5,6-ДИКАРБОНИТРИЛОВ | 2013 |
|
RU2534988C1 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНОХИНОЛИНА | 1994 |
|
RU2279432C2 |
| ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ 2-ОКСАЗОЛИДИНОНА В ОДНОМ РЕАКТОРЕ | 1996 |
|
RU2167875C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРИЛГИДРАЗИНОВ | 1966 |
|
SU214542A1 |
| ПРОМЕЖУТОЧНОЕ ПРОИЗВОДНОЕ ЦЕФАЛОСПОРИНА И СПОСОБ ПОЛУЧЕНИЯ ЦЕФАЛОСПОРИНА | 1995 |
|
RU2150471C1 |
Изобретение относится к области синтетической органической химии и направлено на разработку эффективного способа получения замещенных арилгидразинов восстановлением солей арилдиазониев хлоридом олова (II), причем, соль диазония, полученная из соответствующего анилина, обрабатывается сначала четыреххлористым оловом, затем выпавший осадок гексахлорстанната диазония отделяется фильтрованием и вводится во взаимодействие с 10-50% мольным избытком двухлористого олова в концентрированной соляной кислоте при температуре -15°С - 0°С в течение 15-40 минут. Техническим результатом является повышение выходов целевых арилгидразинов. 3 з.п. ф-лы, 1 табл.
1. Способ получения замещенных арилгидразинов, отличающийся тем, что соль диазония, полученная из соответствующего анилина, обрабатывается сначала четыреххлористым оловом, затем выпавший осадок гексахлорстанната диазония отделяется фильтрованием и вводится во взаимодействие с 10-50% мольным избытком двухлористого олова в концентрированной соляной кислоте при температуре -15°С - 0°С в течение 15-40 мин.
2. Способ получения замещенных арилгидразинов по п.1, отличающийся тем, что образовавшуюся реакционную смесь подщелачивают раствором NaOH до нейтральной реакции, полученную смесь экстрагируют бензолом, экстракт промывают насыщенным раствором хлористого аммония, водой, сушат безводным сульфатом натрия и бензол отгоняют.
3. Способ получения замещенных арилгидразинов по п.2, отличающийся тем, что полученный бензольный раствор упаривают вдвое и при слабом охлаждении постепенно пропускают сухой хлороводород до прекращения выпадения осадка гидрохлорида арилгидразина, осадок отфильтровывают и сушат.
4. Способ получения замещенных арилгидразинов по п.1, отличающийся тем, что после окончания восстановления реакционную смесь нагревают при перемешивании до 80°С до растворения выпавшего осадка, реакционную смесь оставляют при комнатной температуре на 1 ч, выпавший осадок отфильтровывают и сушат.
| US 6278022 B1, 21.08.2001 | |||
| US 6087534 A, 11.07.2000 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

