Изобретение относится к органическому синтезу, конкретно к новому способу получения 2-(оксиран-2-ил)-этанола формулы (1) (Схема 1)
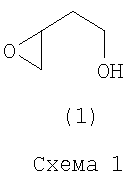
который применяется в тонком органическом синтезе для получения различных биологически активных веществ [Frick, J.A., Klassen, J.B., Bathe, A., Abramson, J. M., Rapoport, H. An efficient synthesis of enantiomerically pure (R)-(2-benzyloxyethyl)oxirane from (S)-aspartic acid. // Synthesis - 1992 - N.7. - P.621-623].
Известно несколько способов получения (S)-2-(оксиран-2-ил)-этанола (S-1), некоторые из них заключаются в энантиоселективном эпоксидировании 3-бутен-1-ола (3) [Okachi, Т., Murai, N., Onaka, M. Catalytic enantioselective epoxidation of homoallylic alcohols by chiral zirconium complexes. // Organic Letters. 2003. - Vol.5. - No.l. - P.85-87; Karjalainen, J.K., Hormi, O.E.O., Sherrington, D.C. An improved heterogeneous asymmetric epoxidation of homoallylic alcohols using polymer-supported Ti(IV) catalysts. // Tetrahedron: Asymmetry. - 1998 - Vol.9 - N.21 - P.3895-3902] (Схема 2)

Данные способы требуют проведения реакций при низкой температуре (-40°С) и соответственно использования криогенного оборудования. Из-за малых скоростей реакций для получения продукта хотя бы с небольшим выходом нужно длительное время - от суток до 2 недель. Максимальный выход продукта при этом не превышает 55%, а энантиомерный избыток - 78% [Okachi, Т., Murai, N., Onaka, M. Catalytic enantioselective epoxidation of homoallylic alcohols by chiral zirconium complexes. // Organic Letters. 2003. - Vol.5. - N.l. - P.85-87].
В другом способе используются реакции дезаминирования (R)-аспарагиновой кислоты (R-4), восстановления (R)-бромянтарной (R-5) кислоты и сольволиза бромдиола (R-6) до (S)-2-(оксиран-2-ил)-этанола (S-1) [Frick, J., Klassen, J.В., Bathe, A., Abramson, J, Rapoport, H. An efficient synthesis of enantiomerically pure (R)-(2-benzyloxyethyl)oxirane from (S)-aspartic acid. // Synthesis - 1992 - N.7. - P.621-623] (Схема 3)
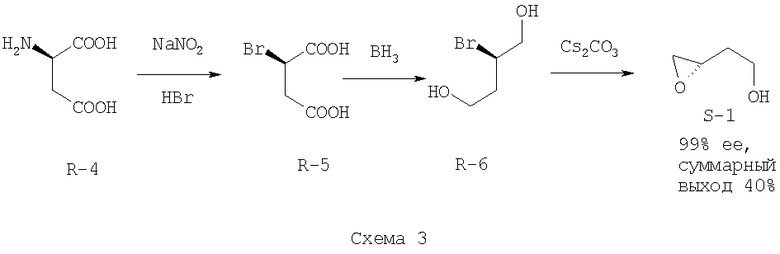
Первая стадия характеризуется небольшим выходом реакции дезаминирования (70%) и опасностью ее неконтролируемого протекания, что требует точного соблюдения условий реакции, в частности, контроля температуры реакционной среды. Кроме того, на второй стадии используется взрывоопасный восстановитель, а на последней стадии - дорогое основание карбонат цезия. Схема отличается невысоким суммарным выходом. Также вызывают сомнения сохранение стереоконфигурации в ходе реакции нуклеофильного замещения (возможна рацемизация в сильнокислой среде) и возможность получения данным способом продукта с высокой энантиомерной чистотой.
Кроме указанных выше, в литературе нами было обнаружено два схожих между собой способа с получением близкого по строению бензилоксиэпоксида (7) из (S)-яблочной кислоты (S-2). Первый способ включает восстановление яблочной кислоты (S-2) до 1,2,4-бутантриола, постановку кетальной защиты виц-диольной группировки, защиту 4-гидроксильной группы, снятие кетальной защитной группы, образование смеси сложных эфиров метансульфокислоты (мезилатов 8, 9) и их сольволиз [М.Majewski, D.Clive, P.Anderson. Synthetic studies related to compactin and mevinolin: a new synthesis of the lactone system. // Tetrahedron Letters. - 1984 - Vol.25. - N.20 - P.2101-2104] (Схема 4)

На первой стадии используется взрывоопасный восстановитель. Три последующие стадии используются для селективной защиты 4-гидроксильной группы 1,2,4-бутантриола, и их наличие драматически сказывается на суммарном выходе синтетической схемы. Реакция образования мезилатов (8, 9) идет при пониженной температуре (-30°С, это требует использования криогенного оборудования либо охлаждающих бань с CO2) с низкой селективностью образования требуемого первичного мезилата (9). Это приводит к ощутимой потере оптического выхода (соотношение энантиомеров после получения эпоксида S:R=2.3:1) и необходимости разделения первичного и вторичного мезилатов (8, 9) посредством ВЭЖХ, которое, тем не менее, не позволяет полностью разделить изомеры (8, 9) (Схема 4).
Второй способ (прототипный) схож с предыдущим и отличается от него использованием реакции с комплексом трифенилфосфина с тетрахлометаном для получения бензил-защищенного по 4-гидроксильной группе производного 2-(оксиран-2-ил)-этанола (S-7) [С.Liu, J. Coward. A facile and highly stereoselective synthesis of (R)- and (S)-(2-(phenylmethoxy)ethyl)oxirane. // J. Org. Chem. - 1991 - N.56. - P.2262-2264] (Схема 5)
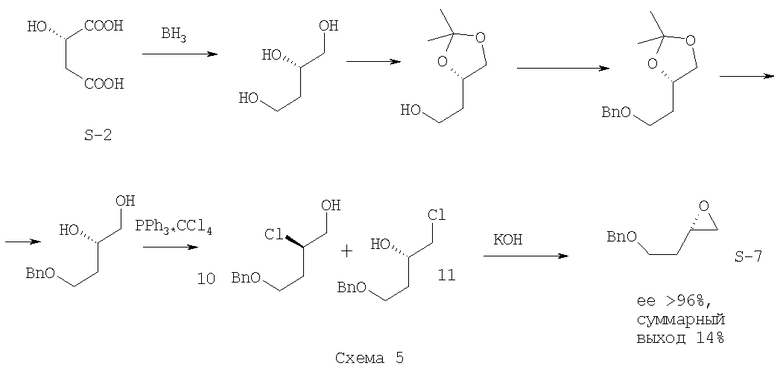
Данный способ помимо большого количества стадий и использования реакций с низкой атомной эффективностью (постановка и снятие защитных групп) имеет еще меньший суммарный выход.
Задачей изобретения является создание простого безопасного способа получения 2-(оксиран-2-ил)-этанола (1) с высоким выходом и оптической чистотой.
Поставленная задача решается способом, который включает в себя реакцию этерификации яблочной кислоты (2), нуклеофильное замещение в сложном эфире (12, 16, 17), восстановление сложного эфира (13, 15, 18-21) до галогендиола (6, 14) и сольволиз галогендиола в основной среде с образованием соединения (1) по следующей схеме (схема 6):
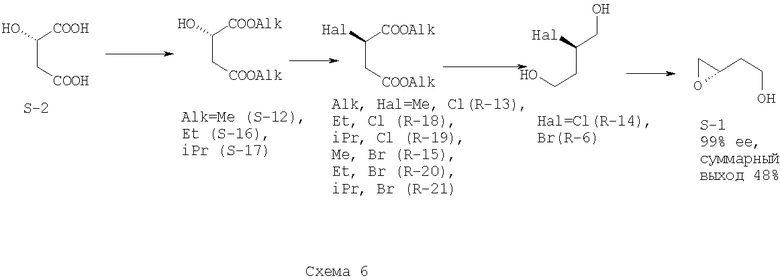
Установлено, что можно селективно и с высокой оптической чистотой получать 2-галоген-1,4-бутандиол (6, 14) при помощи последовательности реакций нуклеофильного замещения и восстановления без использования защитных групп. 2-галоген-1,4-бутандиол (6, 14), в свою очередь, при помощи доступных и дешевых оснований гладко подвергается сольволизу с образованием целевого соединения (1). Найдено, что реакция этерификации яблочной кислоты может быть проведена без кислотного катализа, что сводит риск рацемизации соединений к нулю. Установлено, что использование нитритов в кислой среде на стадии нуклеофильного замещения требует точного контроля температуры и рН, поэтому может быть заменено более простыми, технологичными и безопасными реакциями с комплексами трифенилфосфина с тетрахлорметаном или бромом. При этом пропадает необходимость в сильнокислой среде, что также устраняет риск рацемизации соединений. Установлено, что вместо боранов на стадии восстановления могут быть использованы более пожаробезопасные и менее активные восстановители (боргидриды лития и натрия), при этом существенно ускоряется процедура обработки реакционных смесей. Вместо свободных кислот можно восстанавливать их диэфиры, что выгодно с точки зрения экономии восстановителя. Это приводит к получению целевого соединения (1) с выходом, превышающим литературные в 1.2-3.5 раза, а также к значительному упрощению выполняемых процедур. Сравнением с литературными данными по вращению угла поляризации показано, что на всех стадиях данного синтетического пути сохраняется высокая степень оптической чистоты продуктов (не менее 99%).
Таким образом, патентуемый способ обладает следующими преимуществами:
- простотой выполняемых операций и доступностью реагентов;
- отсутствием пожаро-, взрывоопасных и токсичных реагентов;
- более быстрым протеканием реакций;
- высокой степенью сохранения оптической чистоты соединений по всему синтетическому пути (99%);
- высокой атомной эффективностью и отсутствием необходимости в использовании защитных групп;
- более высоким суммарным выходом конечного продукта.
Способ получения 2-(оксиран-2-ил)-этанола (1) иллюстрируется следующими примерами.
Пример 1. Получение (S)-2-(оксиран-2-ил)-этанола (S-1) из (S)-яблочной кислоты (S-2)
Стадия I. Этерификация (S)-яблочной кислоты (S-2) (Схема 7)
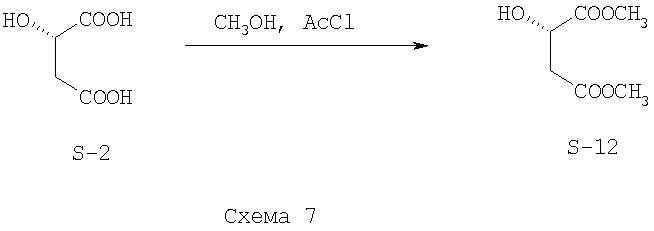
Синтез проводят на установке, состоящей из реактора и мешалки. В реактор загружают 300 мл метанола и прибавляют по каплям 15.5 мл ацетилхлорида. После перемешивания в течение 15 мин прибавляют 47 г твердой (S)-яблочной кислоты (S-2) и перемешивают в течение 18 часов. Раствор упаривают. При этом получается желтое масло, которое дальше очищают хроматографией на силикагеле при элюировании смесью дихлорметана и метанола (95:5). (S)-диметилмалат (S-12) получается с выходом 86%. Спектр ЯМР 1Н (CDCl3) (δ, м.д.): 2.70-3.00 (м, 2Н), 3.50 (с, 3H), 3.60 (с, 3H), 4.45 (м, 1Н). [α]D -8.2, (с 1.0, СН3ОН).
Стадия II. Получение (R)-диметилхлорсукцината (R-13) (Схема 8)

Синтез проводят на установке, состоящей из реактора, мешалки и обратного холодильника с водяным охлаждением. В реактор загружают 50 мл тетрахлорметана и 13.96 г (0.053 моль) трифенилфосфина. Перемешивают до полного растворения трифенилфосфина. Далее добавляют диметиловый эфир (S)-яблочной кислоты (S-12) (5.13 г, 0.031 моль) в 10 мл тетрахлорметана. Кипятят реакционную смесь с обратным холодильником в течение 2 часов. На роторном испарителе отгоняют растворитель. Оставшийся осадок тщательно промывают на пористом фильтре смесью гексана и диэтилового эфира (1:1, 5×30 мл). Маточный раствор собирают и упаривают, остаток в виде коричневого подвижного масла хроматографируют на силикагеле. Фракцию элюирования смесью гексан-диэтиловый эфир 1:1 упаривают и вакуумируют, в результате чего остается светло-желтое подвижное масло - диметиловый эфир (R)-хлорянтарной кислоты (R-13) массой 4.57 г (82%). Спектр ЯМР 1Н (CDCl3) (δ, м.д.): 2.70-3.00 (м, 2Н), 3.50 (с, 3H), 3.60 (с, 3H), 4.45 (м, 1Н). [α]D+42.8, (с 1.0, CHCl3).
Стадия III. Восстановление (R)-диметилхлорсукцината (R-13) (Схема 9)

Синтез проводят на установке, состоящей из реактора, мешалки, капельной воронки и обратного холодильника с водяным охлаждением. К суспензии 0.46 г LiBH4 (0.021 моль) в 100 мл тетрагидрофурана (ТГФ) прибавляют по каплям раствор 2.55 г (0.014 моль) (R)-диметилхлорсукцината (R-13) и 0.67 г (0.021 моль) метанола в 10 мл ТГФ. Кипятят смесь с обратным холодильником 4 часа. После охлаждения при перемешивании прибавляют 3 мл метанола или воды. После дополнительного перемешивания в течение 20 минут прибавляют по каплям разбавленную ортофосфорную кислоту до нейтральной реакции по универсальному индикатору. Осадок отфильтровывают на вакуум-фильтре, промывают ацетоном (3×10 мл). Маточный раствор упаривают, получая светло-желтое вязкое масло - (2R)-2-хлор-1,4-бутандиол (R-14) массой 1.32 г (81%). Спектр ЯМР 1Н (ДМСО d6) (δ, м.д.): 1.40-1.90 (м, 2Н), 3.50-3.95 (м, 4Н), 4.10 (м, 1Н), 6.00 (шир. с, 2Н). [α]D+36.5, (с 1.2, СН3ОН).
Стадия IV. Сольволиз (R)-2-хлор-1,4-бутандиола (R-14) (Схема 10)

К раствору 1,74 г (R)-2-хлор-1,4-бутандиола (R-14) в 20 мл ТГФ добавляют 0.94 г (0.017 моль) твердого КОН. Перемешивают 7 часов. Фильтруют суспензию на вакуум-фильтре, осадок промывают дихлорметаном (3×10 мл). Маточный раствор упаривают, маслообразный остаток пропускают через колонку с силикагелем (5 см) (элюент дихлорметан) с получением светло-желтого масла - (S)-2-(оксиран-2-ил)-этанола (S-1) массой 0.86 г (70%). Спектр ЯМР 1Н (CDCl3) (δ, м.д.): 1.50-1.60 (м, 1Н), 1.70-1.80 (м, 1Н), 2.40 (м, 1Н), 2.60-3.00 (м, 2Н), 3.60 (м, 2Н), 4.20 (шир. с, 1Н). [α]D -30.7, (с 1.0, CH2Cl2).
Пример 2.
Этерификацию (S)-яблочной кислоты осуществляют при помощи реакции с диазометаном (Схема 11)

Синтез проводят на установке, состоящей из реактора, охлаждающей бани (лед) и мешалки. (S)-Яблочную кислоту (S-2) чистоты 98+% массой 5.91 г загружают в реактор, приливают диэтиловый эфир и перемешивают до полного растворения. При охлаждении на бане добавляют по каплям эфирный раствор диазометана до появления неисчезающего желтого окрашивания раствора. Раствор перемешивается в течение 20 минут, после чего баню убирают и избыток диазометана устраняют добавлением по каплям 10% раствора уксусной кислоты в воде до обесцвечивания. Раствор упаривают и остаток обрабатывают аналогично описанному в Примере 1. Масса продукта - диметилового эфира (S)-яблочной кислоты (S-12) 6.64 г (93%). Стадии II-IV выполняются, как описано в Примере 1.
Пример 3.
Этерификацию (S)-яблочной кислоты (S-2) осуществляют при помощи реакции с диметилсульфатом (Схема 12). Синтез проводят на установке, состоящей из реактора и мешалки. (S)-Яблочную кислоту (S-2) чистоты 98+% массой 5.91 г загружают в реактор, добавляют 10 мл метанола и при перемешивании прибавляют 4.2 г гидроксида натрия, перемешивают 10 минут. Далее добавляют 13.3 г диметилсульфата. Реакционная смесь перемешивается в течение 2 часов. Реакционную смесь упаривают, разбавляют 20 мл воды и экстрагируют диметилмалат (S-12) или этилацетатом (3 раза по 30 мл). После высушивания органической фазы над сульфатом натрия раствор упаривают. При этом получается остаток - желтоватое масло, которое дальше обрабатывают аналогично описанному в Примере 1. Масса продукта - диметилового эфира (S)-яблочной кислоты (S-12) 6.64 г (85%). Стадии II-IV выполняются, как описано в Примере 1.
Пример 4.
В качестве метилирующего агента используют 15 г метилиодида (Схема 12). В качестве основания используют триэтиламин (10.7 г). Методика синтеза аналогична приведенной в Примере 3. Выход диметилового эфира (S)-яблочной кислоты (S-12) для способа с метилиодидом составляет 83%. Стадии II-IV выполняются, как описано в Примере 1.
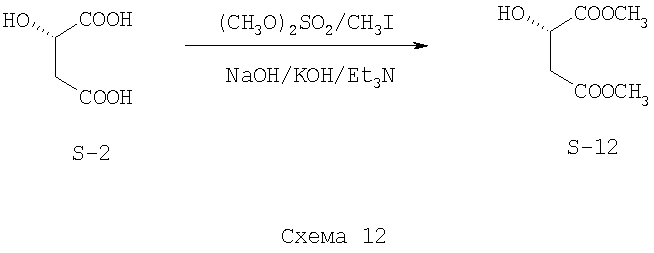
Пример 5. Стадия I выполняется по одной из методик, описанных в Примерах 1-4. На стадии II используют комплекс бром-трифенилфосфин с получением (R)-диметилбромсукцината (R-15) (Схема 13)
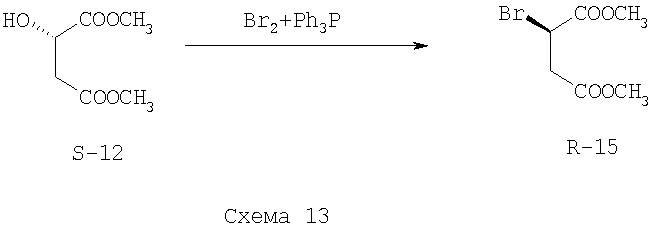
Синтез проводят на установке, состоящей из реактора, мешалки, бани со льдом и солью и обратного холодильника с водяным охлаждением. В реактор загружают 25 мл дихлорметана и 14.56 г (0.056 моль) трифенилфосфина. После полного растворения трифенилфосфина раствор охлаждают льдом с солью и прибавляют по каплям раствор 9,0 г (0,054 моль) брома в 20 мл дихлорметана. После полного добавления перемешивают еще 10 минут. Далее добавляют диметиловый эфир яблочной кислоты (S-12) (5.03 г, 0.031 моль) в 10 мл дихлорметана. Сразу после полного добавления выпавший ранее осадок полностью растворяется. Раствор кипятят с обратным холодильником в течение 2 часов. После охлаждения отгоняют растворитель на роторном испарителе. Оставшийся осадок тщательно промывают на пористом фильтре смесью гексана и диэтилового эфира (1:1, 5×30 мл). Маточный раствор собирают и упаривают, остаток в виде желтого подвижного масла хроматографируют на силикагеле. Фракцию элюирования смесью гексан-диэтиловый эфир 1:1 упаривают и вакуумируют, в результате чего остается светло-желтое подвижное масло - (R)-диметилбромсукцинат (R-15) массой 6.0 г (85%). Спектр ЯМР 1Н (CDCl3) (δ, м.д.): 2.70-3.00 (м, 2Н), 3.50 (с, 3H), 3.60 (с, 3H), 4.45 (м, 1Н). [α]D+70.3, (с 1.16, С6Н6)
На стадии III восстанавливают (R)-диметилбромсукцинат (R-15) (Схема 14)

Синтез проводят на установке, состоящей из реактора, мешалки, капельной воронки и обратного холодильника с водяным охлаждением. К суспензии 0.53 г LiBH4 (0.024 моль) в 60 мл ТГФ прибавляют по каплям раствор 3.67 г (0.016 моль) диметилбромсукцината (R-15) и 0.78 г (0.024 моль) метанола в 10 мл ТГФ. Кипятят смесь с обратным холодильником 4 часа. Охлаждают смесь и при перемешивании прибавляют 5 мл метанола или воды. После прекращения выделения газа при перемешивании прибавляют по каплям разбавленную ортофосфорную кислоту до нейтральной реакции по универсальному индикатору. Осадок отфильтровывают на вакууме, промывают ацетоном (3×10 мл). Маточный раствор упаривают, получая светло-желтое вязкое масло - (2R)-2-бром-1,4-бутандиола (R-6) массой 2.2 г (80%). Спектр ЯМР 1Н (ДМСО d6) (δ, м.д.): 1.90-2.15 (м, 2Н), 3.70-3.90 (м, 4Н), 4.20-4.40 (м, 1Н), 5.50 (шир. с, 2Н). [α]D +31.6, (с 15, CHCl3).
На стадии IV к раствору 2.37 г (2R)-2-бром-1,4-бутандиола (R-6) в 20 мл ТГФ добавляют 0.94 г (0.017 моль) твердого КОН (Схема 15)

Перемешивают 7 часов. Отфильтровывают на вакуум-фильтре, осадок промывают дихлорметаном (3×10 мл). Маточный раствор упаривают, маслообразный остаток пропускают через колонку с силикагелем (5 см) (элюент дихлорметан), получая светло-желтое масло - (S)-2-(оксиран-2-ил)-этанол (S-1) массой 0.92 г (75%). Спектр ЯМР 1Н (CDCl3) (δ, м.д.): 1.50-1.60 (м, 1Н), 1.70-1.80 (м, 1Н), 2.40 (м, 1Н), 2.60-3.00 (м, 2Н), 3.60 (м, 2Н), 4.20 (шир. с, 1Н). [α]D -30.6, (с 1.0, CH2Cl2).
Пример 6. Стадия I выполняется по одной из методик, описанных в Примерах 1-4. Стадия II выполняется по одной из методик, описанных в Примерах 1, 5. На стадии III восстановление проводят при помощи смеси боргидрида натрия и хлорида лития, взятых в 1.5-кратном избытке (по молям) по отношению к сложному эфиру (13) или (15). В результате (2R)-2-хлор-1,4-бутандиол (R-14) и (2R)-2-бром-1,4-бутандиол (R-6) получаются с выходами 80 и 78% соответственно. Стадия IV выполняется по одной из методик, описанных в Примерах 1, 5.
Пример 7. Стадия I выполняется по одной из методик, описанных в Примерах 1-4. Стадия II выполняется по одной из методик, описанных в Примерах 1, 5. На стадии III восстановление проводят с использованием 1 г этанола вместо метанола. В качестве растворителя вместо тетрагидрофурана используется серный эфир. В результате (2R)-2-хлор-1,4-бутандиол (R-14) и (2R)-2-бром-1,4-бутандиол (R-6) получаются с выходами 75 и 71% соответственно. Стадия IV выполняется по одной из методик, описанных в Примерах 1, 5.
Пример 8. Стадия I выполняется по одной из методик, описанных в Примерах 1-4. Стадия II выполняется по одной из методик, описанных в Примерах 1,5. Стадия III выполняется по одной из методик, описанных в Примерах 1, 5-7. На стадии IV вместо гидроксида калия используют равное количество гидроксида натрия. В результате из (2R)-2-хлор-1,4-бутандиола (R-14) и (2R)-2-бром-1,4-бутандиола (R-6) (S)-2-(оксиран-2-ил)-этанол (S-1) получается с выходами 68 и 70% соответственно.
Пример 9. Стадия I выполняется по одной из методик, описанных в Примерах 1-4. Стадия II выполняется по одной из методик, описанных в Примерах 1, 5. Стадия III выполняется по одной из методик, описанных в Примерах 1, 5,-7. На стадии IV вместо тетрагидрофурана в качестве растворителя может использоваться 20 мл смеси диметилсульфоксида и воды в отношении 3:1 (по объему). В этом случае реакционную смесь после окончания реакции разбавляют 20 мл воды и 2-(оксиран-2-ил)-этанол (1) экстрагируют диэтиловым эфиром (трижды по 20 мл). Эфирный раствор сушат над сульфатом натрия, отфильтровывают и упаривают. Выход (S)-2-(оксиран-2-ил)-этанола (S-1) из (2R)-2-хлор-1,4-бутандиола (R-14) и (2R)-2-бром-1,4-бутандиола (R-6) 70 и 72% соответственно.
Пример 10. Для получения (R)-2-(оксиран-2-ил)-этанола (R-1) используют (R)-яблочную кислоту (R-2) (Схема 16). На стадии I (R)-диметилмалат (R-12) ([α]D + 8.1 (с, 1.0, СН3ОН)) получен с выходом 94% по методике из Примера 2. На стадии II (S)-диметилхлорсукцинат (S-13) ([α]D -42.8 (с, 1.0, CHCl3)) получен с выходом 81% по методике из Примера 1. На стадии III (2S)-2-хлор-1,4-бутандиол (S-14) ([α]D -36.2 (с, 1.2, СН3ОН)) получен с выходом 83% по методике из Примера 1. (S)-диметилбромсукцинат (S-15) ([α]D -69.7 (с, 1.16, С6Н6)) и (2S)-2-бром-1,4-бутандиод (S-6) ([α]D -31.7 (с, 15, CHCl3)) получены по методике из Примера 5 (стадии II и III) с выходами 85 и 80% соответственно. На стадии IV (R)-2-(оксиран-2-ил)-этанол (R-1) ([α]D+31.0 (с, 1.0, CH2Cl2)) получен из (S-14) и (S-6) по методикам из Примеров 1 и 5 с выходами 75 и 70% соответственно. Спектральные характеристики всех промежуточных продуктов и (R)-2-(оксиран-2-ил)-этанола (R-1) совпадают с приведенными в Примерах 1, 5 для антиподов
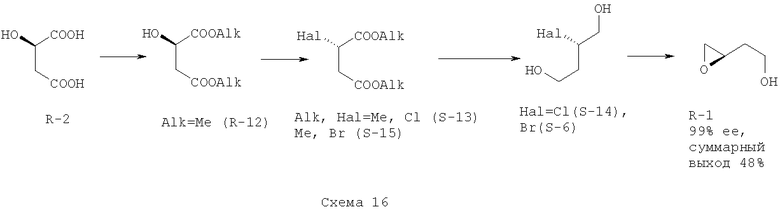
Пример 11. Для получения промежуточных продуктов - 2-хлор-1,4-бутандиола (14) и 2-бром-1,4-бутандиола (6) используют восстановление диэтилового эфира соответственно хлорянтарной и бромянтарной кислоты, получаемых из диэтилмалата (Схема 17). На I стадии диэтилмалат (16) получен с выходом 82% по способу, аналогичному описанному в Примере 1. На II стадии диэтилхлорсукцинат (18) получен с выходом 80% по способу, аналогичному описанному в Примере 1; диэтилбромсукцинат (20) получен с выходом 85% по способу, аналогичному описанному в Примере 5. На стадии III 2-хлор-1,4-бутандиол (14) был получен по методике из Примера 1 с выходом 73%, 2-бром-1,4-бутандиол (6) был получен по методике из Примера 5 с выходом 70%. Удельное вращение угла поляризации галогендиолов (6, 14) совпадает с данными, приведенными в Примерах 1, 5 и 10.
Пример 12. Для получения промежуточных продуктов - 2-хлор-1,4-бутандиола (14) и 2-бром-1,4-бутандиола (6) используют восстановление ди-н-пропилового эфира соответственно хлорянтарной и бромянтарной кислоты, получаемых из ди-н-пропилмалата (Схема 17). На I стадии ди-н-пропилмалат (17) получен с выходом 75% по способу, аналогичному описанному в Примере 1. На стадии II диэтилхлорсукцинат (19) получен с выходом 75% по способу, аналогичному описанному в Примере 1; диэтилбромсукцинат (21) получен с выходом 73% по способу, аналогичному описанному в Примере 5. На стадии III 2-хлор-1,4-бутандиол (14) был получен по методике из Примера 1 с выходом 65%, 2-бром-1,4-бутандиол (6) был получен по методике из Примера 5 с выходом 61%. Удельное вращение угла поляризации галогендиолов (6, 14) совпадает с данными, приведенными в Примерах 1, 5 и 10

| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ АРОМАТИЧЕСКИЕ АМИНОКИСЛОТЫ | 1998 |
|
RU2187506C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛА, ОПТИЧЕСКИ ИНЕРТНЫЕ ИЛИ ИМЕЮЩИЕ R- ИЛИ S-КОНФИГУРАЦИЮ C-2 И C-3 АСИММЕТРИЧНЫХ ЦЕНТРОВ, ИЛИ ИХ СОЛИ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2039050C1 |
| ЗАМЕЩЕННЫЙ 2-АМИНОПИРИДИН В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2671212C2 |
| ТРИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 1991 |
|
RU2114838C1 |
| ПРОИЗВОДНЫЕ 2-ПИПЕРИДИНО-1-АЛКАНОЛА | 1991 |
|
RU2029769C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДВУХ 4-{ [(2S)-2-{ 4-[5-ХЛОР-2-(1H-1,2,3-ТРИАЗОЛ-1-ИЛ)ФЕНИЛ]-5-МЕТОКСИ-2-ОКСОПИРИДИН-1(2H)-ИЛ} БУТАНОИЛ]АМИНО} -2-ФТОРБЕНЗАМИДНЫХ ПРОИЗВОДНЫХ | 2019 |
|
RU2804663C2 |
| ПРОИЗВОДНЫЕ МОРФОЛИН-ПИРИДИНА | 2015 |
|
RU2690154C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ АНГИОГЕНЕЗА | 2000 |
|
RU2262935C2 |
| НОВЫЕ ТИЕНОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2605403C2 |
| ПРОИЗВОДНЫЕ 2-ПИПЕРИДОНА В КАЧЕСТВЕ АГОНИСТОВ ПРОСТАГЛАНДИНА | 2004 |
|
RU2311409C2 |
Изобретение относится к новому способу получения 2-(оксиран-2-ил)-этанола формулы (1), являющемуся ценным полупродуктом для получения различных биологически активных веществ, в том числе и в энантиомерно чистом виде. Способ получения заключается в этерификации яблочной кислоты, нуклеофильном замещении гидроксильной группы диалкилмалата на атом хлора или брома, восстановлении диалкилгалосукцината и сольволизе галогендиола до 2-(оксиран-2-ил)-этанола. Данный способ позволяет получать целевой продукт (1) с не менее чем 99% сохранением энантиомерного избытка и суммарным выходом до 49% в расчете на исходную яблочную кислоту.
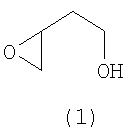
Способ получения 2-(оксиран-2-ил)-этанола формулы (1)
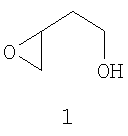
путем этерификации яблочной кислоты, нуклеофильного замещения гидроксильной группы полученного диалкилового эфира яблочной кислоты на атом хлора или брома, восстановления полученного диметилгалосукцината и последующего сольволиза галогендиола.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| TAKAHIRO OKACHI ET AL "Catalytic Enantioselective Epoxidation of Homoallylic Alcochols by Chiral Zirconium Complexes", Organic Letters, 2003, 5(1), | |||

