Изобретение относится к производным триазола, проявляющим противогрибковую активность.
Более конкретно, настоящее изобретение относится к 2-арил-3-(3- галопиридин-4-ил или 5-галопиримидин-4-ил)-1-(1H-1,2,4-триазол-1-ил)алкан-2-ольным производным, применимым для борьбы с грибковыми инфекциями животных, в том числе и человека.
Отдельные соединения настоящего изобретения в общем смысле описаны в нашей заявке на Европейский патент N 89307920.2 (EP-A-0357241), но ни одно из них не было специально раскрыто или подтверждено примером в указанной заявке.
Обнаружено, что соединения настоящего изобретения обладают неожиданно высоким уровнем противогрибковой активности, в частности против грибков вида Aspergillus spp., что в основном относят за счет их неожиданно хороших фармокинетических свойств, следствием чего являются более длительные периоды полураспада (значения t1/2).
Изобретением предлагаются производные триазола общей формулы ,
,
и их фармацевтически приемлемые соли,
где R представляет фенил, замещенный 1-3 галоидзаместителями;
R1 - C1-C4-алкил;
X - CH или N;
Y - F или Cl.
В вышеприведенном определении соединений формулы (I) C3 и C4 - алкилы могут иметь нормальное или изо-строение.
Рекомендуемые алкильные группы включают метил и этил.
Примеры R включают: 2-фторфенил, 4-фторфенил, 2-хлорфенил, 4- хлорфенил, 2-бромфенил, 2-йодфенил, 2,4-дихлорфенил, 2,4-дифторфенил, 2-хлор-4-фторфенил, 2-фтор-4-хлорфенил, 2,5-дифторфенил, 2,4,6- трифторфенил, 4-бром-2,5-дифторфенил.
Предпочтительно, чтобы R представлял фенил, замещенный 1 или 2 заместителями, каждый независимо выбранный из фтора и хлора.
Рекомендуемые индивидуальные воплощения R включают 2-фторфенил, 4-фторфенил, 2,4-дифторфенил, 2-хлорфенил и 2,4-дихлорфенил.
Более предпочтительно, когда R представляет 2-фторфенил, 2,4- дифторфенил, 2-хлорфенил или 2,4-дихлорфенил. Предпочтительно R1 - метил. Предпочтительно X - N. Предпочтительно Y - F.
Фармацевтически приемлемые соли соединений формулы (I) включают соли с кислотами, образованные присоединением кислот, дающих неядовитые соли, такие как хлоргидрат, бромгидрат, йодгидрат, сульфат или бисульфат, фосфат или гидрофосфат, ацетат, малеат, фумарат, лактат, тартрат, цитрат, глюконат, бензоат, метансульфонат, бензолсульфонат и п-толуолсульфонат. В качестве обзора по приемлемым фармацевтическим солям см. Berge и др., J. Pharm. Sci., 66, 1 - 19 (1977).
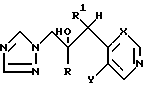
Соединения формулы (I) содержат по меньшей мере два хиральных центра (*) и, таким образом, существуют в виде по меньшей мере двух диастереомерных пар энантиомеров, например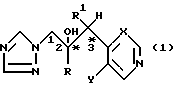 .
.
Изобретение включает как индивидуальные стереоизомеры соединений формулы (I), так и их смеси. Разделение диастереомеров может быть осуществлено обычными методами, например фракционной кристаллизацией, хроматографией или ВЭЖХ диастереомерной смеси соединения формулы (I), или его приемлемой соли, или его производного. Индивидуальный энантиомер соединения формулы (I) может быть также получен из соответствующего оптически чистого промежуточного соединения или путем разрешения рацемата либо с помощью ВЭЖХ применением приемлемого хирального носителя, либо фракционной кристаллизацией диастереомерных солей, образованных в реакции рацемата с приемлемой оптически активной кислотой, например 1R-(-)- или 1S-(+)-10-камфорсульфоновой кислотой.
Рекомендуемые соединения формулы (I), которые имеют 2R,3S-конфигурацию, представляют собой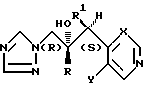 .
.
Особенно рекомендуемые индивидуальные воплощения соединений настоящего изобретения включают:
2R, 3S-2-(2,4-дифторфенил)-3-(3-фторпиридин-4-ил)-1-(1H-1,2,4- триазол-1-ил)бутан-2-ол,
2R, 3S-2-(2-хлорфенил)-3-(3-фторпиридин-4-ил)-1-(1H-1,2,4-тризл-1- ил)бутан-2-ол,
2R, 3S-2-(2-фторфенил)-3-(3-фторпиридин-4-ил)-1-(1Н-1,2,4-триазол- 1-ил)бутан-2-ол,
2R, 3S-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол,
2R, 3S-2-(2,4-дихлорфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол и их фармацевтически приемлемые соли.
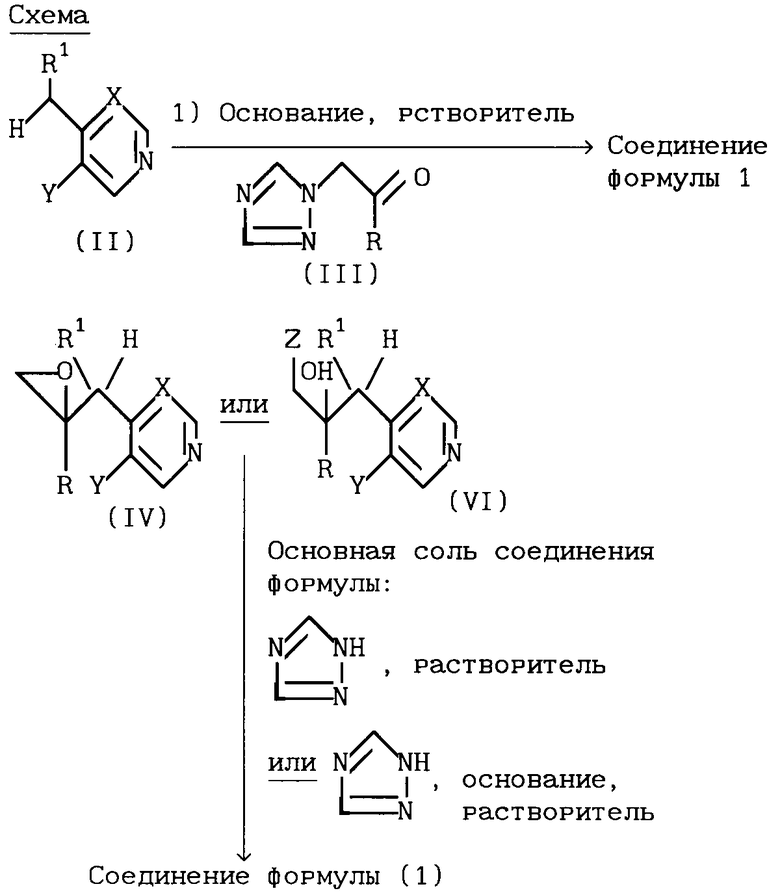
Соединения формулы (I), обеспечиваемые изобретением, могут быть получены следующими способами.
1). Все соединения формулы (I) могут быть синтезированы согласно схеме 1,
где R1, X и Y принимают значения, указанные для соединения формулы (I).
В типичной методике соединение формулы (II) депротонизируют добавлением примерно одного эквивалента приемлемого основания, например диизопропиламида лития или бис-(триметилсилил)амида натрия или калия, и образовавшуюся соль (предпочтительно соль лития, натрия или калия) вводят in situ в реакцию с кетоном формулы (III). Реакцию обычно ведут при температуре от -80 до -50oC, предпочтительно при температуре от -70 до -60oC, в приемлемом органическом растворителе, например тетрагидрофуране, толуоле или диэтиловом эфире, и в инертной атмосфере, например азоте или аргоне.
Исходные продукты формулы (II) являются либо известными соединениями (см. , например, D.J. Comins и др., Heterocycles 22, 339 (1984)), либо могут быть получены обычными методами согласно литературным методикам. Исходные продукты формулы (III) либо известны (см., например, EP-A-44605, EP-A-69442 или GB-A-1464224), либо могут быть синтезированы методами, аналогичными приведенным в указанных патентах.
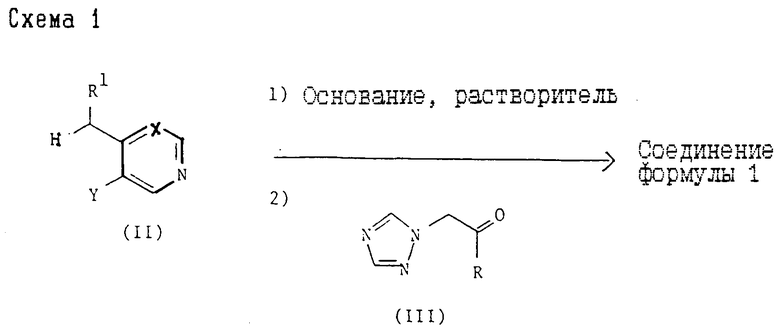
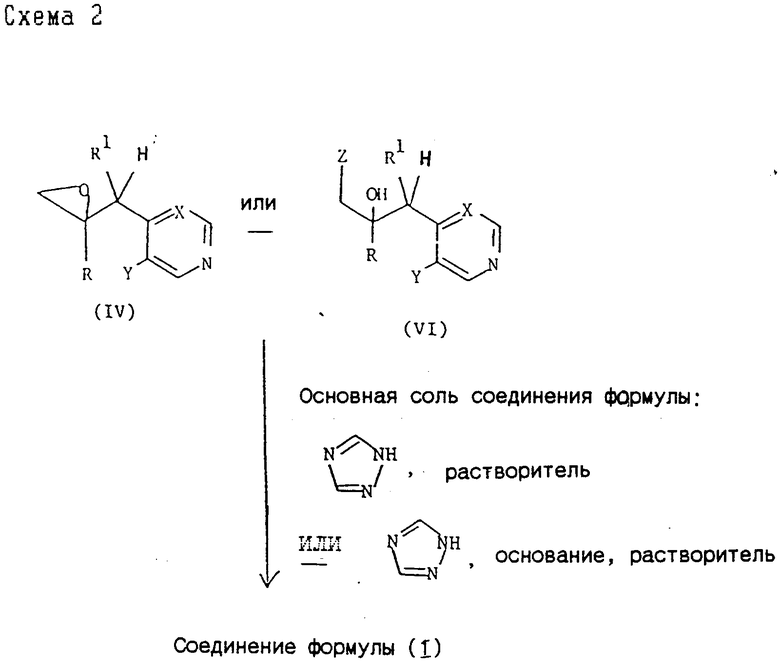
2). Все соединения формулы (I) могут быть также получены согласно схеме 2,
где R, R1, X и Y принимают значения, указанные для соединения формулы (I), и Z представляет приемлемую удаляемую группу, например, хлор, бром или C1-C4-алкансульфонилоксигруппу (такую как метансульфонилоксигруппа). Примеры приемлемых солей оснований 1H-1,2,4-триазола включают щелочнометаллические, предпочтительно натриевые и калиевые, а также тетраалкиламмониевые, предпочтительно тетрабутиламмониевые (см. патент США 4259505) соли.
Реакцию рекомендуют проводить с применением в качестве исходного продукта эпоксида формулы (IV). При использовании в процессе соединения формулы (VI) вероятно, что механизмом реакции диктуется по крайней мере частично образование в условиях реакции in situ соответствующего эпоксида формулы (IV). С этой точки зрения процесс, таким образом, аналогичен процессам, использующим в качестве исходного продукта эпоксид формулы (IV).
При использовании основной соли, 1H-1,2,4-триазола реакцию обычно проводят при температуре от комнатной до 100oC, предпочтительно при 60oC, при использовании натриевой соли 1H-1,2,4-триазола и предпочтительно при комнатной температуре при использовании соответствующей тетра-н-бутил-аммониевой соли в приемлемом органическом растворителе, например N,N-диметилформамиде или тетрагидрофуране.
Или же реакция может быть осуществлена использованием 1H-1,2,4-триазола в присутствии дополнительного приемлемого основания, например Na2CO3 или K2CO3, предпочтительно при 50 - 100oC в приемлемом растворе, например N,N-диметилформамиде, метаноле или водном ацетоне.
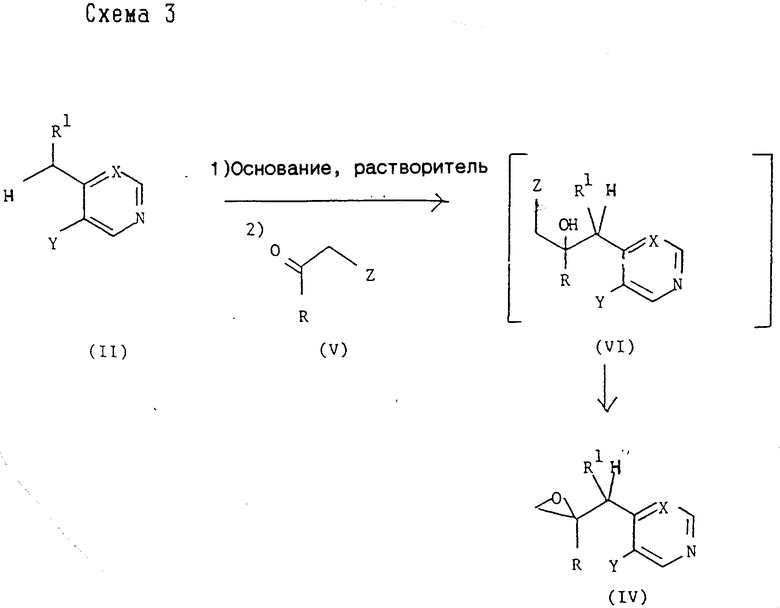
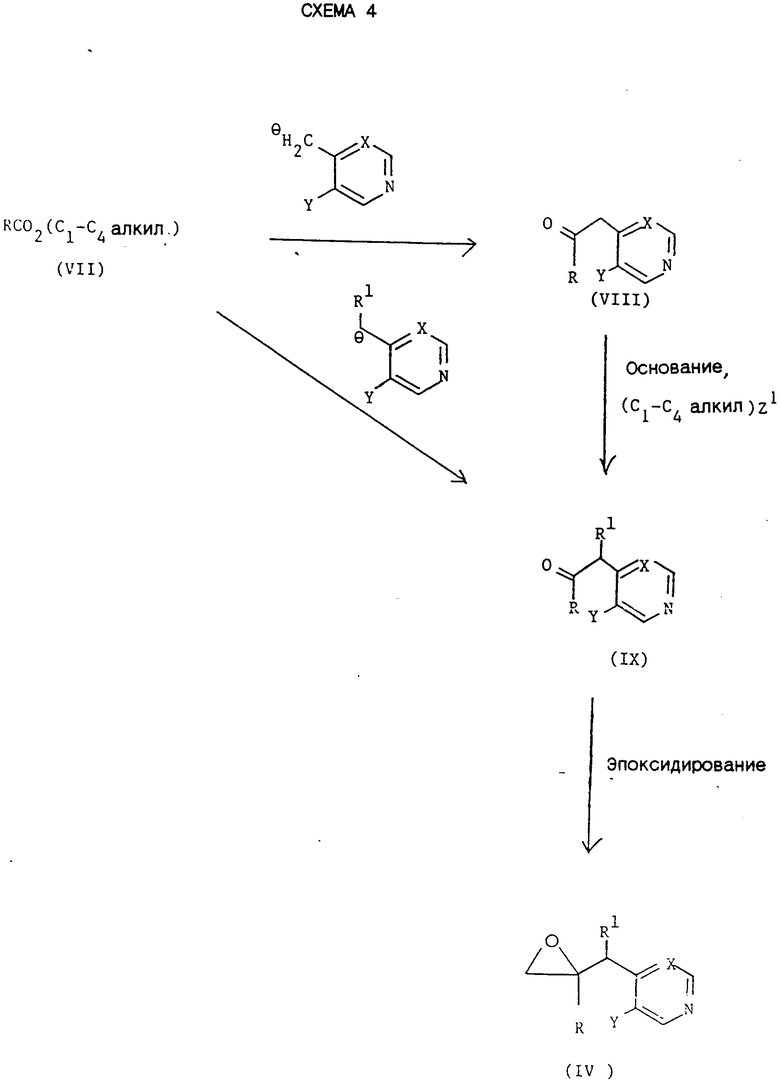
Промежуточные соединения формул (IV) и (VI) могут быть получены обычными методами, в общем виде представленными схемами 3 и 4,
где
R, R1, X и Y принимают значения, указанные для соединения формулы (I), и Z представляет удаляемую группу, предпочтительно Cl или Br.
В типичной методике соединение формулы (II) депротонизируют добавлением примерно одного эквивалента приемлемого основания, например диизопропиламида лития или бис-(триметилсилил)амида натрия или калия, и образовавшееся промежуточное металлоорганическое соединение вводят in situ в реакцию с соединением формулы (V). Обычно реакцию проводят при температуре от -80 до -50oC, предпочтительно при -70oC, в приемлемом органическом растворителе, например тетрагидрофуране, толуоле или диэтиловом эфире, и в инертной атмосфере, например азоте или аргоне. Образовавшееся соединение формулы (VI) не требует выделения и обычно его циклизуют in situ путем перемешивания какое-то время при более высокой температуре, например при комнатной температуре, с получением оксирана формулы (IV).
Соединение формулы (VI), где Z представляет хлор или бром, может быть также получено реакцией эпоксида формулы (IV) с соответствующим галоидводородом в безводных условиях.
На схеме 4 R, R', X И Y принимают значения, указанные для соединения формулы (I), и Z1 представляет приемлемую группу, например, Cl, Br, I или метансульфонилоксигруппу.
В обычной методике соединение формул (VIII), (IX) или (X) получают непосредственно из сложного эфира формулы (VII) реакцией с металлорганическим соединением, образованным депротонизацией соединения формул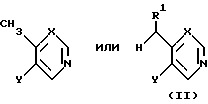 ,
,
в соответствующих условиях, где R1, X и Y принимают значения, указанные для соединения формулы (I), примерно с одним эквивалентом приемлемого основания, например диизопропиламидом лития или бис-(триметилсилил)амидом натрия или калия. Обычно реакцию проводят при температуре от -80 до -50oC, предпочтительно около -70oC,в приемлемом органическом растворителе, например тетрагидрофуране или диэтиловом эфире, и в инертной атмосфере, например азоте или аргоне.
Или же соединение формул (IX) или (X) может быть получено реакцией соответственно соединения формул (VIII) или (IX) примерно с одним эквивалентом приемлемого основания, например гидрида натрия, с последующим алкилированием in situ образовавшегося карбаниона приемлемым алкилирующим средством. Реакцию обычно проводят при температуре от 0oC до комнатной температуры в приемлемом органическом растворителе, например N,N-диметилформамиде.
Алкилирование соединения формул (VIII) или (IX) рекомендуют осуществлять в условиях переноса фаз, например, использованием системы NaOH/[CH3(CH2)]4N⊕⊖HSO4/ /H2O/CHCl3/ (C1-C4-алкил)Z1 (где Z1 предпочтительно представляет йод) при температуре от 0oC до комнатной температуры, обычно при комнатной температуре.
Эпоксидирование кетона формул (IX) или (X) осуществляют обычными методами, например использованием метилиодида диметилоксосульфония (см., например, J. A. C.S. (1965), 87, 1353) или хлорметиллития (см., например, Tet. Lett. (1986), 795).
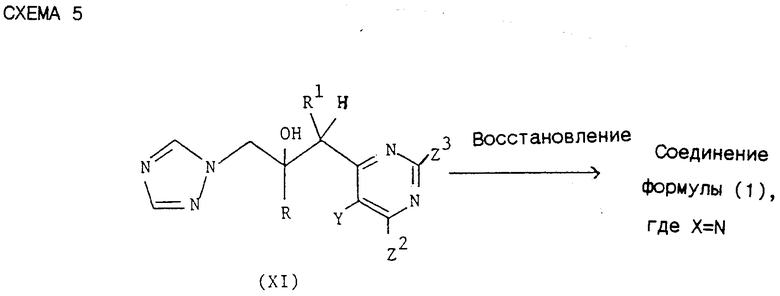
3) Соединения формулы (I), в которых R, R1 и Y принимают значения, указанные для соединения формулы (I), а Х = N могут быть синтезированы согласно схеме 5,
где R, R1 и Y принимают значения, указанные для соединения формулы (I), a Z2 и Z3 каждый независимо выбирают из водорода и группы, которая может быть селективно удалена восстановлением, при условии, что Z2 и Z3 не могут обе представлять водород. Рекомендуется, чтобы Z2 представляла группу, которая может быть селективно удалена восстановлением, a Z3 = Н. Рекомендуется, чтобы удаляемая селективным восстановлением группа была представлена галоидом (т.е. F, Cl, Br или I) и наиболее предпочтительно хлором.
Если указанная группа представлена галидом, предпочтительно хлором, тогда в качестве способа восстановления рекомендуется гидрогенолиз. Согласно обычной методике соединение формулы (XI) подвергают гидрогенолизу использованием приемлемого катализатора, например палладия на угле, и приемлемого растворителя, например этанола, возможно в присутствии дополнительного приемлемого основания, например ацетата натрия. Реакция может быть осуществлена при температуре от комнатной до температуры кипения растворителя и давлении 1 - 5 атмосфер (100 - 500 кПа), но обычно успешно протекает при комнатной температуре и атмосферном давлении.
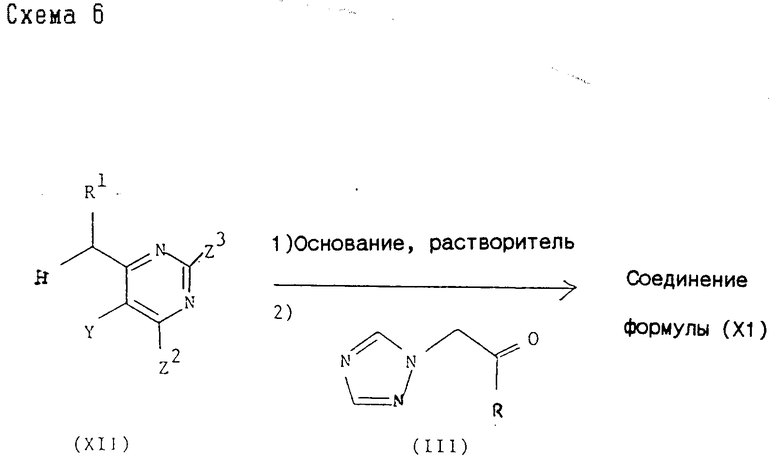
Промежуточные соединения формулы (XI), где один из Z2 и Z3 представляет водород, а другой - группу, которая может быть селективно удалена восстановлением, могут быть получены обычным образом согласно представленной схеме 6,
где R, R1 и Y принимают значения, указанные для соединения формулы (I), и один из Z2 и Z3 представляет водород, а другой - группу, которая может быть селективно удалена восстановлением. Реакция может быть осуществлена методом, аналогичным методу (1).
Промежуточные соединения формулы (XI), где один из Z2 и Z3 представляет водород, а другой - группу, которая может быть селективно удалена восстановлением, могут быть также получены методом, аналогичным методу (2).
Исходные продукты формулы (XII) могут быть синтезированы обычными методами, такими как приведенные в препаративном разделе.
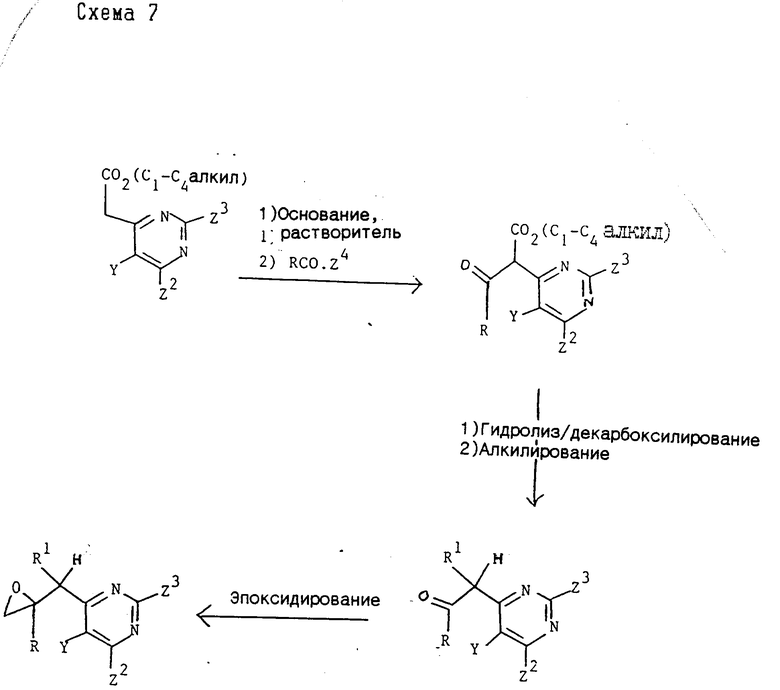
Промежуточные соединения формулы (XI), где Z2 и Z3 каждый представлен группой, которая может быть селективно удалена восстановлением, могут быть получены методом, аналогичным методу (2), использованием в качестве исходного продукта соответствующего эпоксида, который может быть синтезирован по обычной методике согласно схеме 7,
где R, R1 и Y принимают значения, указанные для соединения формулы (I), Z2 и Z3 каждый представляет группу, которая может быть селективно удалена восстановлением, и Z4 представляет хлор или C1-C4-алкоксигруппу.
Все вышеприведенные реакции являются обычными, выбор соответствующих реагентов и условий проведения этих реакций, а также методов выделения целевых продуктов специалистом может быть сделан в соответствии с литературными источниками и со ссылкой на приводимые здесь примеры.
Фармацевтически приемлемые соли, образованные добавлением кислот, могут быть легко получены совместным смешиванием растворов, содержащих свободное основание и целевую кислоту. Соль, как правило, осаждается из раствора, и ее отделяют фильтрованием или же выделяют испарением растворителя.
Соединения формулы (I) и их соли относятся к противогрибковым средствам, применимым для лечения или профилактики грибковых инфекций животных, в том числе и человека. К примеру, соединения применимы для лечения местных грибковых инфекций человека, вызванных среди прочих микроорганизмов грибками вида Candida, Trichophyton, Microsporum или Epidermophyton, или грибковых инфекций, вызванных Candida albicans (например, кандидозного стоматита или кандидоза влагалища). Соединения могут быть также использованы для лечения общих грибковых инфекций, вызванных, например, видами Candida (например, Candida albicans), Cryptococcus neoformans, Aspergillus flavus, Aspergillus fumigatus, Coccidioides, Paracoccidiodes, Histoplasma или Blastomyces.
Соединения настоящего изобретения, как было найдено, обладают неожиданно высокой активностью по отношению к клинически важным видам грибков Aspirgillus, что в первую очередь относят за счет неожиданно хороших фармокинетических свойств соединений, в результате чего они характеризуются более длительными периодами полураспада (значениями t1/2).
Выявление противогрибковой активности in vitro соединений может быть проведено путем определения минимальной ингибирующей концентрации (МИК), т. е. концентрации испытуемого соединения в приемлемой среде, при которой не происходит роста конкретного микроорганизма. На практике ряд пластинок с агаром, на которые нанесено испытуемое соединение в определенной концентрации, заражаются стандартной культурой, например Candida albicans, и каждую пластинку затем инкубируют 48 ч при 37oC. После этого отмечают наличие или отсутствие на пластинке роста грибка, на основании чего и определяют МИК. Другие микроорганизмы, применяемые в подобных испытаниях, могут включать: Aspergillus fumigatus вида Trichophyton, вида Microsporum, Epidermophyton floccosum, Coccidioides immitis и Torulopsis glabrata.
Испытания соединений in vivo может быть проведено путем внутрибрюшинных или внутривенных инъекций ряда дозировочных уровней или пероральным введением мышам, зараженных штаммом Candida albicans или Aspergillus fumigatus. Активность в этом случае основана на выживаемости группы обработанных соединением мышей после смерти группы необработанных мышей уровень дозировки, при которой соединение обеспечивает 50% защиту от летального действия инфекции (ЭД50), замечают при этом. В модели инфекции видами Aspergillus ряд мышей, вылеченных от инфекции после последовательности дозировок, позволяет осуществить дальнейшее определение активности.
Для лечения человека противогрибковые соединения формулы (I) могут быть использованы в чистом виде, но, как правило, их вводят в смеси с фармацевтическим носителем, выбранным с учетом намеченного пути введения и обычной фармацевтической практики. К примеру, соединения могут быть введены перорально в виде таблеток, содержащих такие наполнители, как крахмал и лактоза, или в капсулах, или пузырьках в чистом виде или в смеси с наполнителями, или в виде эликсиров, растворов, или суспензий, включающих вкусовые и окрашивающие добавки. Соединения могут быть иньектированы парентерально, например внутривенно, внутримышечно или подкожно. Для парентерального введения соединения лучше всего использовать в виде стерильного водного раствора, который может включать и другие вещества, например достаточное количество солей или глюкозы, для придания раствору изотоничности с кровью.
Растворимость соединения формулы (I) в водной среде может быть улучшена за счет образования комплекса с гидроксиалкильным производным циклодекстрина при получении соответствующей фармацевтической композиции. Рекомендуется, чтобы применяемым циклодекстрином был альфа-, бета- или гамма-циклодекстрин, наиболее предпочтительно бета- циклодекстрин. Рекомендуемым гидроксиалкильным производным является гидроксипропильное производное.
При пероральном и парентеральном введении больному человеку ежедневный дозировочный уровень противогрибковых соединений формулы (I) и их солей составляет 0,01 - 20 мг/кг (в единственной или поделенной дозировке). Так, таблетки или капсулы соединений содержат от 5 мг до 0,5 г активного соединения для введения одной дозировочной формы или двух или более форм за раз в зависимости от назначения. В любом случае лечащий врач сам определяет реальную дозировку, наиболее приемлемую для индивидуального больного и которая будет меняться в зависимости от возраста, веса и реакции конкретного больного. Вышеуказанные дозировки являются примерами усредненных дозировок и, разумеется, в отдельных случаях они могут быть и выше, и ниже, но тем не менее будут охватываться объемом изобретения.
Или же противогрибковые соединения формулы (I) могут быть использованы в виде свеч или вагинального суппозитория, или могут быть нанесены местно в виде лосьона, раствора, крема, мази или порошка- дуста. К примеру, соединения могут быть введены в крем, представляющий собой водную эмульсию полиэтиленгликолей или жидкого парафина, или же они могут быть введены в концентрации 1 - 10% в мазь на основе белого воска или белого мягкого парафина в смеси со стабилизаторами или консервантами, если таковые необходимы.
Таким образом, изобретением, кроме того, дается фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в смеси с фармацевтически приемлемым разбавителем или носителем.
Изобретением также дается соединение формулы (I) или его фармацевтически приемлемая соль, или его композиция, предназначенные для применения в качестве лекарства, в частности в качестве противогрибкового средства.
Изобретением далее предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли, или его композиции для создания противогрибкового средства.
Изобретением, кроме того, предлагается способ обработки животного (включая человека) с целью излечения или профилактики грибковой инфекции, заключающийся в обработке животного эффективным количеством соединения формулы (I) или, если необходимо, его фармацевтически приемлемой солью или композицией.
Изобретением также предлагаются новые промежуточные соединения формул (IV), (VI) и (XI), 4-этил-5-фторпиримидин и 4-хлор-6-этил-5- фторпиримидин.
Нижеследующие примеры иллюстрируют синтез соединений формулы (I). Полагают, что энантиомерная пара B, когда о ней упоминается в любом из последующих примеров или препаративных примерах, и в продуктах примеров 1, 3, 4 и 5 (в каждом из которых получена только одна энантиомерная пара из двух возможных), является рацемической смесью 2R,3S и 2S,3R-энантиомеров.
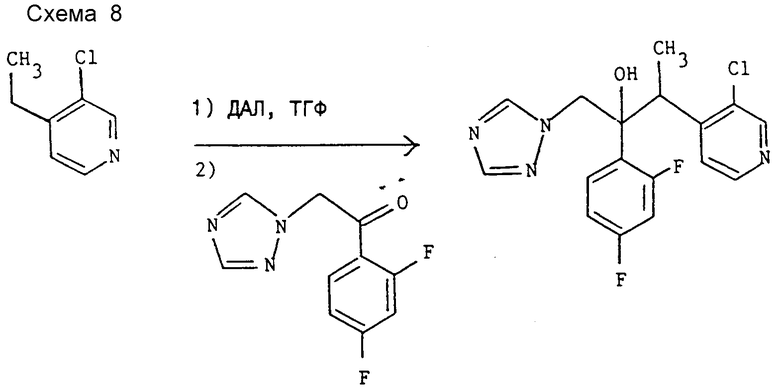
Пример 1. 3-(3-Хлорпиридин-4-ил)-2-(2,4-дифторфенил)-1-(1Н-1,2,4- триазол-1-ил)бутан-2-ол (см. схему 8).
К раствору диизопропиламина (1,01 г, 10 ммоль в сухом ТГФ (60 мл) при -60oC и в атмосфере азота по каплям прибавляют 1,6 M раствор н-бутиллития в гексане (6,25 мл, 10 ммоль). Смесь оставляют нагреваться до -20oC, после чего вновь охлаждают до -70oC и к полученному раствору диизопропиламида лития (ДАЛ) (10 ммоль) при -70oC по каплям прибавляют 1,41 г (10 ммоль) 3-хлор-4-этилпиридина (см. D.L. Comins и др. Heterocycles, 22, 339 (1984)). Полученную смесь перемешивают при той же температуре 15 мин, после чего добавляют раствор 1-(2,4-дифторфенил)-2-(1H-1,2,4- триазол-1-ил) этанола (2,23 г, 10 ммоль) в ТГФ (15 мл). Смесь оставляют нагреваться до комнатной температуры на 30 мин и реакционную смесь нейтрализуют добавлением воды (30 мл) с последующим экстрагированием этилацетатом (3•60 мл). Объединенные органические экстракты сушат над сульфатом магния, фильтруют, концентрируют при пониженном давлении и заглавное соединение выделяют "импульсной" хроматографией на окиси кремния с элюированием этилацетатом. Продукт перекристаллизован из этилацетата (выход 0,46 г), т.пл. 182 - 184oC. Найдено, %: C 55,76; H 4,15; N 15,23, вычислено для C17H15ClF2N4O, %: C 55,98, H 4,14, N 15,36.
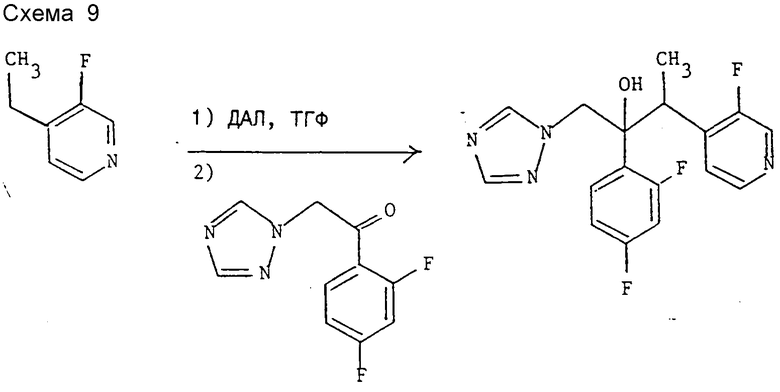
Пример 2. 2-(2,4-Дифторфенил)-3-(3-фторпиридин-4-ил)-1- (1H-1,2,4-триазол-1-ил)бутан-2-ол (см. схему 9).
Реакцию проводят по методике, аналогичной методике примера 1, но использованием 4-этил-3-фторпиридина (см. препаративный пример 1) вместо 3-хлор-4-этилпиридина в качестве исходного продукта. Колоночной хроматографией сырого продукта реакции на окиси кремния с элюированием этилацетатом вначале после объединения и испарения соответствующих фракций получают заглавное соединение (энантиомерная пара A) с т.пл. 178 -181oC, охарактеризованное с помощью 1H-ЯНР спектроскопии.
1H-ЯМР (CDCl3) δ : 1,6 (д, 3H), 3,95 (кв, 1H), 4,7 и 5,15 (AB кв, 2Н), 5,1. (c, 1H(OH)), 6,5 (м, 1H), 6,7 (м, 1H), 6,95 (м, 1H), 7,45 (т, 1H), 7,8 (c, 1H), 7,95 (c, 1H), 8,15 (c, 1H), 8,25 (д, 1H) ч/млн.
Дальнейшим элюированием смесью этилацетат-метанол (95:5) с последующим объединением и испарением соответствующих фракций получают загрязненный продукт (энантиомерную пару B). Полученный продукт подвергают дальнейшей очистке колоночной хроматографией на окиси кремния с применением в качестве элюента смеси дихлорметан-метанол- 0,88 водный аммиак (93:7:1). Целевые фракции объединяют и их испарением получают после промывания диэтиловым эфиром заглавное соединение (энантиомерная пара B), т.пл. 188 - 189oC. Найдено, %: C 57,63; H 4,32; N 15,71, вычислено для C17H15F3N4O•0,25H2O, %: C 57,87; H 4,43; N 15,88.
Энантиомерную пару B разделяют с помощью ВЭЖХ использованием хирального носителя (ХИРАЦЕЛb(R)OG) и элюированием смесью изопропанол-гексан (1:1). Соответствующие фракции объединяют и после испарения получают разделенные индивидуальные энантиомеры, каждый из которых загрязнен хиральным носителем.
Каждый загрязненный энантиомер подвергают дальнейшей очистке колоночной хроматографией на окиси кремния с элюированием смесью дихлорметан-метанол (95: 5). Соответствующие фракции объединяют и испаряют с получением после промывания смесью гексан-диэтиловый эфир очищенного индивидуального энантиомера.
Один из энантиомеров: т.пл. 57 - 59oC и [α]
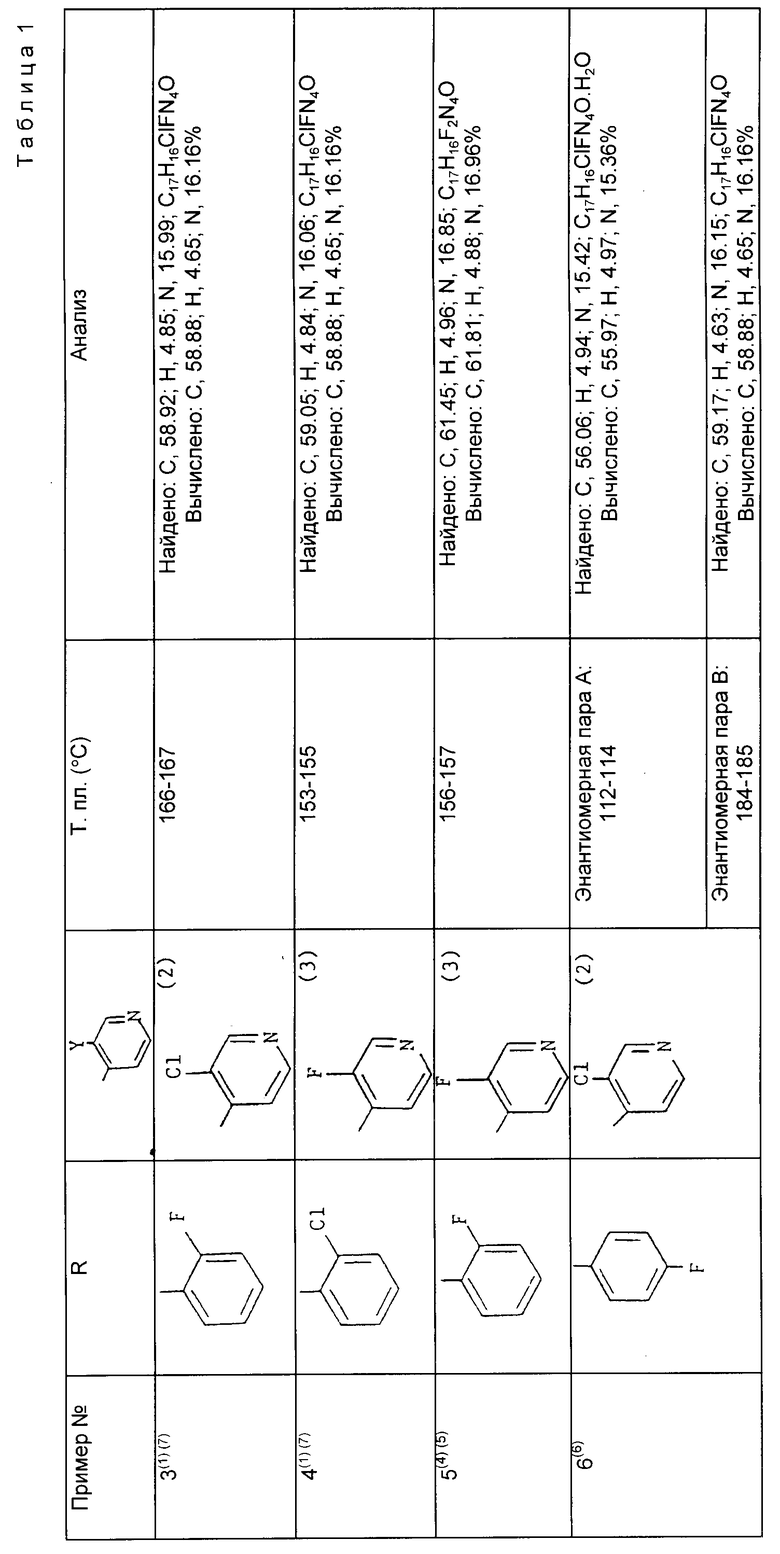
Примеры 3 - 6. По методике, аналогичной методике примера 1, с использованием в качестве исходных соединений соответствующих 4-этил-3-галоидпиридина и 1-(галоидфенил)-2-(1H-1,2,4-триазол-1-ил) этанона получают следующие соединения общей формулы (см. табл. 1).
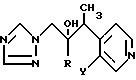 .
.
Пояснения к табл.1.
(1) Колоночную хроматографию проводят на окиси кремния с градиентным элюированием использованием вначале в качестве элюента смеси этилацетат-дихлорметан (2:1) и затем этилацетатом. Полученное твердое вещество промывают диэтиловым эфиром и получают целевой продукт.
(2) Сведения об исходном продукте см. пример 1.
(3) Сведения об исходном продукте см. препаративный пример 1.
(4) Колоночную хроматографию осуществляют на окиси кремния с градиентным элюированием использованием в качестве элюента вначале смеси этилацетат-дихлорметан (2: 1) и затем этилацетата. Соответствующие фракции объединяют, испаряют и полученный продукт подвергают дальнейшей очистке колоночной хроматографией на окиси кремния с применением в качестве элюента смеси дихлорметан-метанол- 0,88 водный аммиак (93:7:1). Соответствующие фракции объединяют, испаряют и промыванием остатка диэтиловым эфиром получают целевой продукт.
(5) Полученную энантиомерную пару разделяют с помощью ВЭЖХ методом, аналогичным приведенному в примере 2. В результате получают индивидуальные энантиомеры, один из которых имеет т.пл. 83 - 84oC и [α]
(6) Колоночную хроматографию проводят на окиси кремния с использованием в качестве элюента смеси гексан-изопропанол-0.88 водный аммиак (80:20:1,5). Соответствующие фракции объединяют, испаряют и полученный продукт подвергают дальнейшей очистке колоночной хроматографией на окиси кремния с применением в качестве элюента смеси этилацетат-этанол (97:3). Соответствующие фракции объединяют и после испарения получают разделенные пары энантиомеров. Промыванием каждой пары энантиомеров диэтиловым эфиром получают целевой продукт.
(7) Полученную пару энантиомеров разделяют с помощью ВЭЖХ методом, аналогичным приведенному в примере 2.
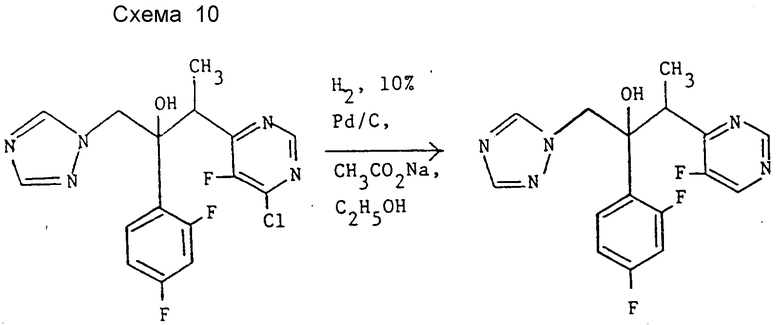
Пример 7. 2-(2,4-Дифторфенил)-3-(5-фторпиримидин-4-ил)-1- (1H-1,2,4-триазол-1-ил)бутан-2-ол (см. схему 10).
Раствор 3-(4-хлор-5-фторпиридин-6-ил)-2-(2,4- дифторфенил)-1-(1H-1,2,4-триазол)-1-ил)бутан-2-ола в виде энантиомерной пары B (см, препаративный пример 2 (III)) (0,307 г, 0,8 ммоль) в этаноле (20 мл) гидрируют при атмосферном давлении и комнатной температуре в присутствии 10% палладия на угле (30 мг) и ацетата натрия (0,082 г, 1 ммоль).
Спустя 5 ч добавляют еще 10 мг 10% палладия на угле и гидрирование продолжают еще 1 ч. Катализатор удаляют фильтрованием, фильтрат концентрируют в вакууме. "Импульсной" хроматографией остатка на окиси кремния с элюированием смесью этилацетат-метанол (97:3) получают после объединения и испарения соответствующих фракций и промывания диэтиловым эфиром заглавное соединение в виде энантиомерной пары B (0,249 г, 89%), т.пл. 127oC. Найдено, %: C 55,08, H 4,00, N 19,96; вычислено для C16H14F3N5O, %: C 55,01; H 4,01; N 20,05.
В 4 мл метанола растворяют образец заглавного соединения в виде энантиомерной пары B (0,105 г, 0,3 ммоль) и 1R-(-)-10-камфоросульфоновой кислоты (0,07 г, 0,3 ммоль) и смесь выдерживают 2 ч при 0oC. Фильтрованием образовавшегося кристаллического вещества получают 1R-(-)-10-камфоросульфонат 2R, 3S-2-(2,4-дифторфенил)-3-(5-фторпиримидин- 4-ил)-1-(1H-1,2,4-триазол-1-ил)бутан-2-ола•0,5 метанол (0,06 г), т.пл. 176oC [α]
Абсолютная конфигурация соединения подтверждена рентгеноструктурным анализом единичного кристалла.
Фильтрат от кристаллизации испаряют в вакууме и распределяют между дихлорметаном (10 мл) и насыщенным водным раствором бикарбоната натрия (5 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении. Остаток и 1S-(+)-10- камфоросульфоновую кислоту (0,46 г, 0,2 ммоль) растворяют в метаноле (3 мл) и выдерживают 2 ч при 0oC. Фильтрованием кристаллического продукта получают 1S-(+)-10-камфоросульфонат 2S, 3R- 2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)- 1-(1H-1,2,4-триазол-1- ил)бутан-2-ола•0,5 метанол (0,052 г), т.пл. 176oC [α]
Найдено, %: C 53,27; H 5,31, N 11,64; вычислено для C26H30F3H5O5S•0,5CH3OH; %: C 53,27, H 5,36, N 11,73.
Образец 1R-(-)-10-камфоросульфонатной соли (1,22 г, 2,1 ммоль), полученного вышеописанным способом, распределяют между дихлорметаном (20 мл) и насыщенным водным раствором бикарбоната натрия (3 мл). Органический слой промывают водой (5 мл), сушат над сульфатом магния, фильтруют и после испарения в вакууме получают 2R,3S-2-(2,4-дифторфенил)-3-(5- фторпиримидин-4-ил)-1-(1H-1,2,4-триазол-1-ил)бутан-2-ол (0,64 г), т.пл. 127oC [α]
Образец 1S-(+)-10-камфоросульфонатной соли (1,17 г, 2 ммоль), полученного вышеприведенным способом, обрабатывают вышеописанным способом и получают 2S, 3R-2-(2,4-дифторфенил)-3-(5- фторпиримидин-4-ил)-1-(1H-1,2,4-триазол-1-ил)бутан-2-ол (0,63 г), т.пл., 127oC, [α]
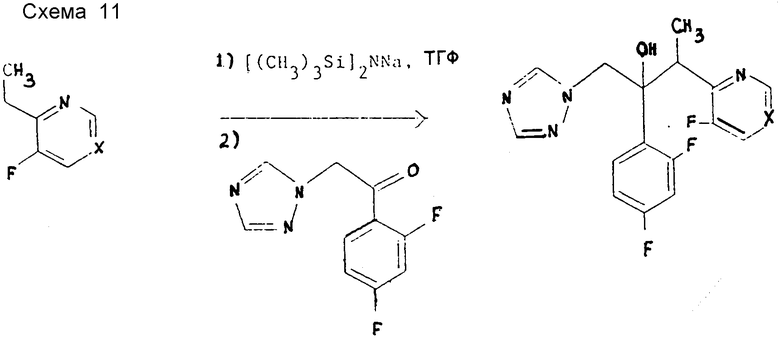
Пример 8. 2-(2,4-Дифторфенил)-3-(5-фторпиримидин-4-ил)-1- (1H-1,2,4-триазол-1-ил)бутан-2-ол, энантиомерная пара B (см. схему 11).
B ТГФ (200 мл) добавляют бис-(триметилсилил)амид натрия (79 мл 1,0 M раствора в ТГФ) и раствор охлаждают в азоте до -65oC. Затем в течение 30 мин прибавляют раствор 4-этил-5-фторпиримидина (10 г) (см. препаративный пример 8) в ТГФ (100 мл). После перемешивания 3 ч при -65oC тонкую взвесь обрабатывают раствором 1-(2,4-дифторфенил)-2-(1H-1,2,4- триазол-1-ил)этанона (17,7 г) в ТГФ (100 мл), добавляемого 30 мин по каплям. Раствор перемешивают еще 1 ч при -65oC и затем обрабатывают уксусной кислотой (20 мл). После нагревания до -20oC раствор промывают водой (200 мл), органический слой отделяют и объединяют с этилацетатом (200 мл) обратным экстрактом водной фазы. Объединенные органические слои концентрируют при пониженном давлении и получают твердое вещество, которое промывают диэтиловым эфиром (230 мл) и фильтруют. Фильтрат концентрируют при пониженном давлении и хроматографируют на окиси кремния с элюированием смесью диэтиловый эфир-этилацетат (1:1). Содержащие заглавное соединение фракции концентрируют при пониженном давлении и остаток хроматографируют на окиси кремния смесью этилацетат/гексан (1:1) в качестве элюента. Соответствующие фракции объединяют и испарением при пониженном давлении получают очищенное заглавное соединение (0,82 г), т.пл. 125 - 127oC. Найдено, %: C 54,89; H 4,06; N 19,66, вычислено для C16H14F3N5O, %: C 55,01; H 4,01; N 20,05.
Пример 9. 2-(2,4-Дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1H-1,2,4- триазол-1-ил)бутан-2-ол, энантиомерная пара A.
Заглавное соединение синтезировано способом, аналогичным способу примера 7, с использованием в качестве исходного продукта энантиомерной пары A 3-(4-хлор-5-фторпиримидин-6-ил)-2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)- бутан-2-ола (см. препаративный пример 2 (III)). Получен продукт с т.пл. 137oC. Найдено; %: C 54,89; H 4,06; N 19,82, вычислено для C16H14F3N5O, %: C 55,01; H 4,01; N 20,05.
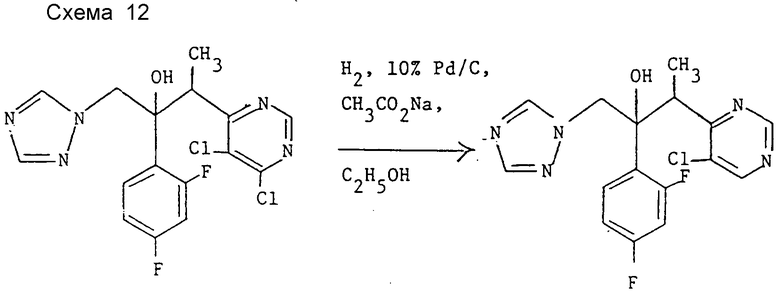
Пример 10. 3-(5-Хлорпиримидин-4-ил)-2- (2,4-дифторфенил)-1-(1H-1,2,4- триазол-1-ил)бутан-2-ол, энантиомерная пара B (см. схему 12).
Раствор энантиомерной пары B 3-(4,5-дихлорпиримидин-6-ил)-2-(2,4- дифторфенил)-1-(1H-1,2,4-триазол-1-ил)бутан-2-ола (см. препаративный пример 6 (III)) (0,58 г,1,46 ммоль) в этаноле (20 мл) гидрируют при атмосферном давлении и комнатной температуре в присутствии 10% палладия на угле (45 мг) и ацетата натрия (122 мг, 1,5 ммоль) в течение 7 ч. Затем катализатор отфильтровывают, а фильтрат концентрируют при пониженном давлении. "Импульсной" хроматографией остатка с элюированием этилацетатом получают после объединения и испарения соответствующих фракций заглавное соединение (0,35 г, 72%), т.пл. 128oC. Найдено; %: C 51,68; H 3,89; N 18,58, вычислено для C16H14ClF2N5O•0,3H2O; %: C 51,76; H 3,94; N 18,87.
Пример 11. 3-(5-Хлорпиримидин-4 ил)-2-(2,4-дифторфенил)-1-(1H- 1,2,4-триазол-1-ил)бутан-2-ол, энантиомерная пара A.
Заглавное соединение получено методом, аналогичным, методу примера 10, с использованием в качестве исходного продукта энантиомерной пары A 3-(4,5- дихлорпиримидин-6-ил)-2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1- ил)бутан-2-ола (см. препаративный пример 6 (III)). Полученный в виде смолы продукт охарактеризован с помощью 1H-ЯМР спектроскопии. 1H-ЯМР (CDCl3) δ : 1,5 (д, 3Н), 4,4 (кв, 1Н), 4,67 и 4,82 (AB кв, 2Н), 6,35 (c, 1H, (ОН)), 6,45 (м, 1H), 6,62 (м, 1Н), 7,07 (м, 1Н), 7,6 (c, 1Н), 8,05 (c, 1Н), 8,8 (c, 1Н) ч-млн.
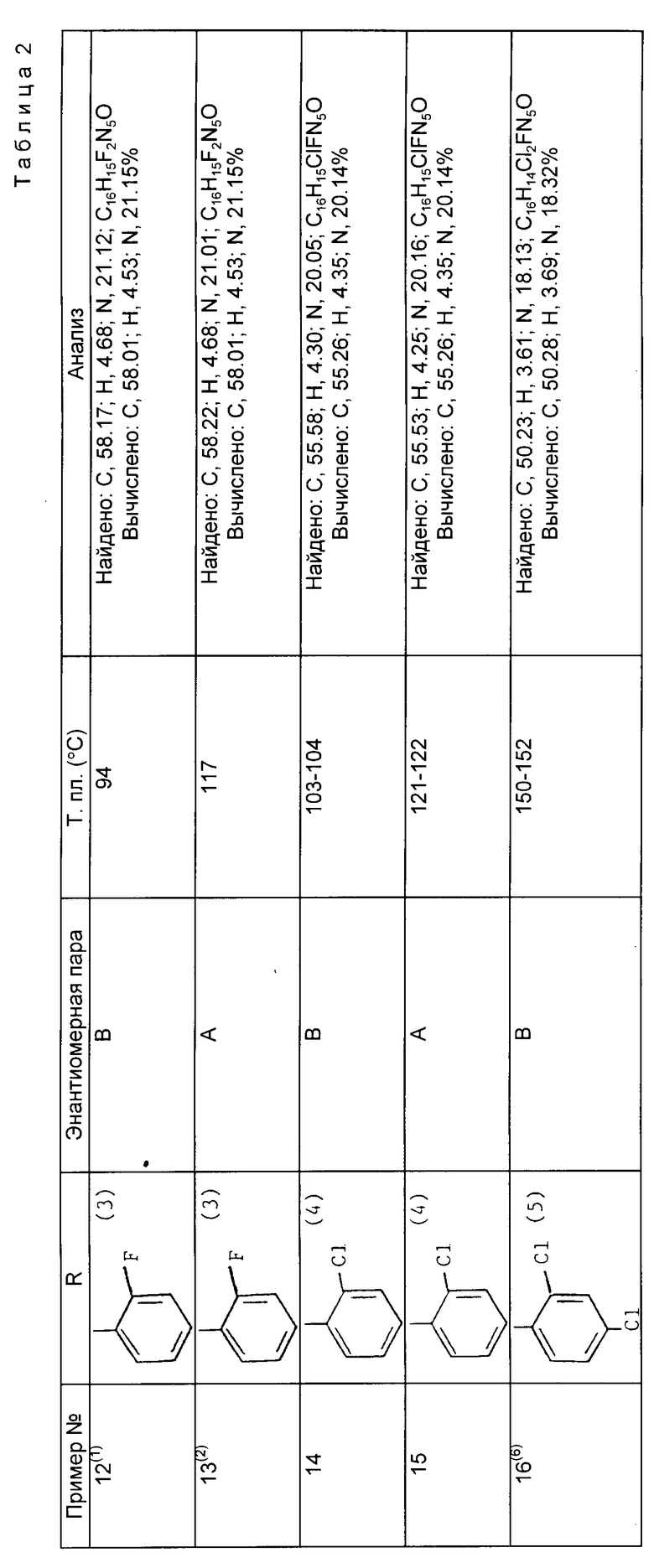
Примеры 12 - 16. Соединения общей формулы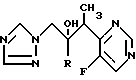 ,
,
получены способом, аналогичным способу примера 10, с использованием в качестве исходного продукта соответствующего 2-арил- 3-(4-хлор-5-фторпиримидин-6-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2- ола. (см. табл. 2).
Пояснения к таблице 2.
(1) Колоночную хроматографию проводят на окиси кремния с использованием в качестве элюента смеси этилацетат-метанол (96:4)
(2) Колоночную хроматографию проводят на окиси кремния использованием в качестве элюента изобутилметилкетона.
(3) Данные об исходном соединении см. пpeпapaтивный пример 3.
(4) Данные об исходном соединении см. препаративный пример 4.
(5) Данные об исходном соединении см. препаративный пример 5.
(6) Полученную энантиомерную пару разделяют с помощью BЭЖX методом, аналогичным методу примера 2.
Пример 17.
Водный солевой раствор 2R,3S-2-(2,4-дифтopфeнил)-3-(5- фтopпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ола и гидроксипропил- β -циклодекстрина.
В мерный колбу на 10 мл помещают гидроксипропи- β -циклодекстрин (молярное замещение = 0,41, 1 г) и растворяют в дистиллированной воде (около 7 мл). Добавляют хлорид натрия (90 мг) и растворяют, после чего дистиллированной водой объем доводят до 10 мл. К полученному раствору добавляют 2R, 3S-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)- 1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол (100 мг) (см. пример 7) в сосуде, смесь обрабатывают ультразвуком 15 мин и затем перемешивают 2 дня механическим вращением. Добавляют дополнительное количество гидроксипропил- β -циклодекстрина (200 мг) и смесь перемешивают механическим вращением сосуда 1 ч с получением целевого раствора.
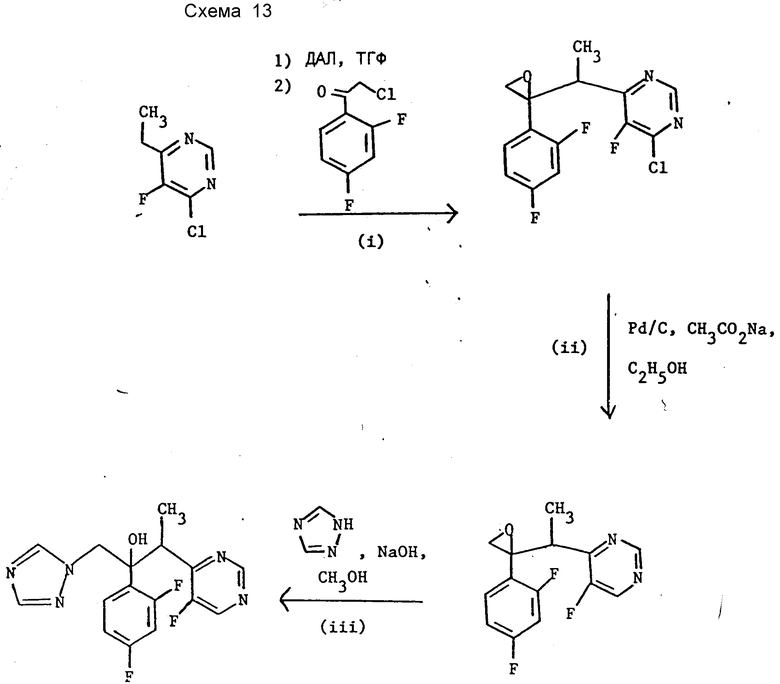
Пример 18. 2R,3S/2S,3R)-2-(2,4-дифторфенил)-3- (5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол (см. схему 13).
I. 2-(1-[4-Хлор-5-фторпиримидин-6-ил]этил)-2-(2,4-дифторфенил) оксиран.
К раствору диизопропиламина (12,89 г) в ТГФ (80 мл) при -10oC добавляют п-бутиллитий (72,6 мл 1,6 M раствора в гексане). После перемешивания в течение 30 мин при этой температуре раствор охлаждают до -67oC и добавляют раствор 4-хлор-6-этил-5-фторпиримидина (18,64 г) в ТГФ (80 мл) в течение 30 мин при температуре от -65 до -50oC. Реакционную массу перемешивают при -65oC 1 ч. Затем добавляют раствор 2-хлор-1,2,4- дифторфенил)этанона (22,1 г) в ТГФ (80 мл) при температуре от -65 до -40oC. Реакционную массу оставляют, пока она постепенно не нагревается до комнатной температуры и перемешивают при этой температуре 1 ч. В смесь добавляют насыщенный водный раствор хлорида натрия (500 мл) и экстрагируют этилацетатом (2•250 мл). Объединенные экстракты промывают насыщенным водным раствором хлорида натрия и затем упаривают при уменьшенном давлении. Остаток хроматографируют на силикагеле, элюируя смесью 4:1 гексан-дихлорметан. Обогащенные продуктом фракции объединяют, упаривают и остаток вновь хроматографируют на силикагеле, элюируя смесью 1: 1 гексан-метиленхлорид. Нужные фракции упаривают при уменьшенном давлении и получают названное соединение (6,3 г) м/е =314.
1H-ЯМР (CDCl3) (как смесь двух диастереомерных пар энантиомеров): δ = 8,7 (с, 1Н), 7,3 - 7,5 (м, 1H), 6,7 - 6,9 (м, 2Н), 3,9 (кв, 2Н), 3,0 (кв., 2H), 1,4 (д, 3H) ppm, в отношении одной энантиомерной пары; δ = 8,7 (с, 1Н), 7,1 - 7,3 (м, 1Н), 6,7 - 6,9 (м, 2Н), 3,85 (кв, 1H), 2,95 (кв, 2Н), 1,45 (д, 3H) ppm в отношении другой энантиомерной пары.
II. 2-(2,4-Дифторфенил)-2-(1-[5-фторпиримидин-4-ил]этил) оксиран.
К раствору продукта раздела I (6,37 г) в промышленном метилированном этиловом спирте (63 мл) добавляют ацетат натрия (1,66 г) и 5% палладия (угле) 50% влажности (0,64 г). Смесь гидрируют при давлении 40 psi при 40oC в течение 4 ч охлажденную реакционную смесь фильтруют для удаления катализатора и фильтрат упаривают под уменьшенным давлением. Остаток перемешивают с метиленхлоридом (30 мл) и водой (30 мл). Органическую фазу отделяют, упаривают при пониженном давлении и остаток хроматографируют на силикагеле, элюируя метиленхлоридом. Нужные фракции объединяют и упаривают при уменьшенном давлении и получают названное в заглавии соединение (2,15 г) м/е - 280.
1H-ЯМР (CDCl3) как смесь двух диастереомерных пар (энантиомеров): δ = 9,0 (с, 1Н), 8,45 (с, 1Н), 7,3 - 7,5 (м, 1Н), 6,7 - 6,9 (м, 2Н), 3,9 (кв, 2Н), 3,0 (кв, 2Н), 1,4 (д, 3Н) ppm в отношении одной энантиомерной пары;
δ = 9,0 (с, 1Н), 8,45 (с, 1Н), 7,1 - 7,3 (м, 1Н), 6,7 - 6,9 (м, 2Н), 3,85 (кв, 1Н), 2,95 (кв, 2Н), 1,45 (д, 3Н) ppm в отношении другой энантиомерной пары.
III. (2R, 3S/2S, 3R)-2-(2,4-Дифторфенил)-3-(5- фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол.
К раствору гидроокиси натрия (0,14 г) в метаноле (7 мл) добавляют 1Н-1,2,4-триазол (5 г) с последующим добавлением продукта раздела II (1 г). Перемешиваемую реакционную смесь нагревают при кипении в течение 6 ч, затем упаривают в вакууме. Остаток распределяют между метиленхлоридом (10 мл) и водой (10 мл). Метиленхлоридный слой отделяют, промывают водой (4•10 мл), высушивают (сульфат магния) и растворитель удаляют в вакууме. Остаток хроматографируют на силикагеле, элюируя смесью 1:1 этилацетат-н-гексан для разделения двух диастереомерных пар энантиомеров. Нужные фракции, содержащие названное в заглавии соединение, объединяют и упаривают с получением целевого продукта (0,1 г), температура плавления 124 - 127oC. ТХС (силикагель, этилацетат): Идентична энантиомерной паре B, полученной в пример 7.
1H-ЯМР (CDCl3): δ = 8,95 (с,1Н), 8,65 (с, 1Н), 7,97 (с, 1Н), 7,7 - 7,55 (м, 1Н). 7,55 (c, 1H), 6,9 - 6,75 (м, 2Н), 6,5 (c, 1Н), 4,75 - 4,25 (кв, 2H), 4,2 - 4,1 (кв, 1Н), 1,15 (д, 3Н) ppm.
Пример 19.
2R, 3S-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4- триазол-1-ил)бутан-2-ол бензолсульфонат.
К раствору 2R,3S-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)- 1-(1Н-1,2,4-триазол-1-ил)бутан-2-ола из примера 7 (8,0 мг) в этилацетате (120 мл) добавляют бензолсульфоновую кислоту (3,62 г) и полученную суспензию перемешивают при комнатной температуре в течение 3 ч. После охлаждения в течение 30 мин смесь фильтруют, промывают холодным этилацетатом и сушат в течение ночи при пониженном давлении. Получают соединение, указанное в названии примера, в виде белого твердого вещества (9,7 г), т.пл. 147 - 149oC.
Пример 20. Таблетки. 2R,3S-2-(2,4-дифторфенил)-3-(5- фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол (соединение примера 7) (50 мг/дозу), лактоза (62 мг/дозу), предварительно желатинированный крахмал (21 мл/дозу) и натрий кроскармеллоза (7,5 мг/дозу) смешивают вместе в течение 20 мин, просеивают и далее перемешивают в течение 20 мин.
Поливинилпирролидон (Kolliden K30, торговая марка) (7,5 мг/дозу) растворяют в очищенной воде (22,5 мг/дозу) и полученный раствор добавляют к полученной выше смеси. Полученную смесь гранулируют и, если необходимо, добавляют еще очищенную воду (22,5 мг/дозу).
Влажную массу деагломерируют, используя дробилку Fitz (торговая марка). Полученные гранулы сушат в печи при 50oC до тех пор, пока содержание влаги в гранулах не будет таким же, как первоначальное в исходной смеси (±1%). Сухие гранулы просеивают и смешивают в течение 20 мин. Гранулы затем смазывают стеаратом магния (1,5 мг/дозу) и формуют в таблетки на специальном оборудовании для таблетирования.
При необходимости таблетки покрывают специальным покрытием.
Нижеследующими препаративными примерами иллюстрируется синтез некоторых новых исходных соединений, применяемых в рабочих примерах.
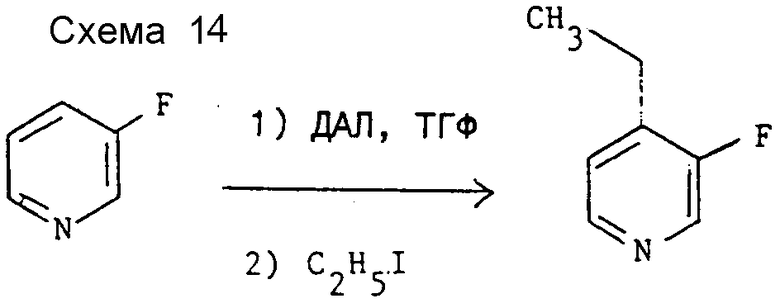
Препаративный пример 1. 4-3-Этил-3-фторпиридин (см. схему 14).
К перемешиваемому раствору ДАЛ (200 ммоль) в сухом ТГФ (400 мл) (получен по методике, аналогичной методике примера 1) при -70oC и в атмосфере азота по каплям прибавляют 3-фторпиридин (20 г, 200 ммоль). Спустя 30 мин при той же температуре к реакционной смеси по каплям добавляют этилиодид (60 г, 370 ммоль) и смесь оставляют медленно нагреваться до температуры от -10 до -5oC, при которой происходит выделение тепла с повышением температуры до 15 - 20oC. Смесь перемешивают еще 30 мин, после чего нейтрализуют добавлением воды (50 мл) и органическую фазу отделяют. Водную фазу экстрагируют эфиром (3•50 мл), объединенные органические слои сушат над сульфатом магния и концентрируют при пониженном давлении. Перегонкой полученной жидкости при атмосферном давлении получают заглавное соединение (13 г), т.кип. 154 - 158oC, охарактеризованное с помощью 1H-ЯМР спектроскопии. 1H-ЯМР (CDCl3) δ: 1,25 (т, 3Н, J = 10 Гц), 2,65 (кв, 2Н, J = 10 Гц), 7,1 (т, 1H, J = 8 Гц), 8,3 (д, 1Н, J = 8 Гц), 8,33 (c, 1H) ч/млн.
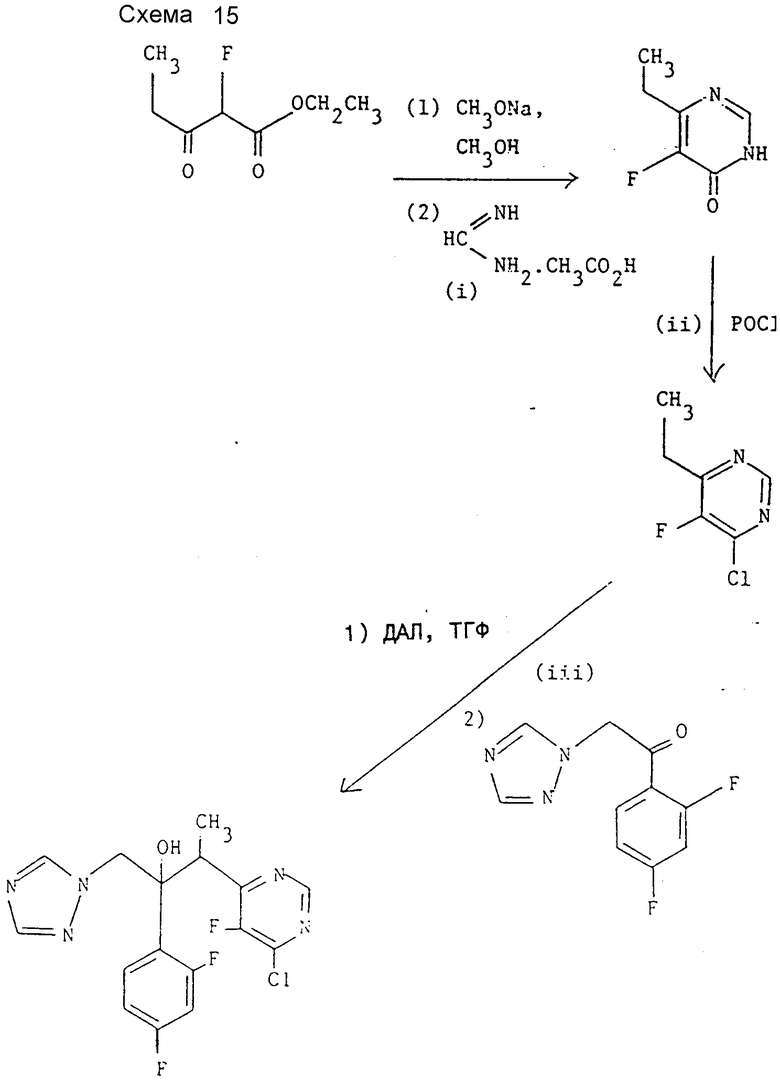
Препаративный пример 2. 3-(4-Хлор-5-фторпиримидин-6- ил)-2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)бутан-2-ол (см. схему 15).
I. 6-Этил-5- фторпиримидин-4(3Н)-он.
К раствору метоксида натрия (8,64 г, 160 ммоль) в метаноле (50 мл) при 0oC прибавляют раствор этилового эфира α -фторпропионилуксусной кислоты (см. E. D. Bergrmann и др., J. Chem. Soc., 1959, 3278 и D.J. Burton и др., Tet. Lett 30, 6113 (1989:)) (12,96 г, 80 ммоль) и ацетата формамидина (8,32 г, 80 ммоль) в метаноле (50 мл) и полученную смесь перемешивают 1 ч при 0oC, около суток при комнатной температуре и наконец 30 мин при кипении. Смесь охлаждают и избыток метоксида натрия нейтрализуют добавлением ледяной уксусной кислоты (10 г). Реакционную смесь концентрируют при пониженном давлении, остаток растворяют в горячем этилацетате, нерастворимый ацетат натрия отфильтровывают и фильтрат концентрируют при пониженном давлении. "Импульсной" хроматографией остатка с элюированием этилацетатом получают после объединения соответствующих фракций и их испарения, а также промывания диэтиловым эфиром заглавное соединение (5,5 г, 48%), т.пл. 105 - 106oC. Найдено; %: C 50,38; H 4,85; N 19,63, вычислено для C6H7FN2O; %: C 50,70; H 4,93; N 19,72.
Заглавное соединение также получено методом, приведенным в препаративном примере 7.
II. 4-Хлор-6-этил-5-фторпиримидин.
Смесь продукта стадии I (6,4 г, 45 ммоль) и фосфорилхлорида (30 мл) кипятят 3 ч. Избыток фосфорилхлорида отгоняют при пониженном давлении, а остаток переносят в ледяную воду. Полученную смесь экстрагируют хлористым метиленом (3•50 мл), объединенные органические экстракты промывают водой и сушат над сульфатом магния. Растворитель удаляют при пониженном давлении и перегонкой полученного масла при пониженном давлении получают заглавное соединение (4,81 г, 66%), т. кип. 74oC при 22 мм Hg, охарактеризованного с помощью 1H-ЯМР спектроскопии.
1H-ЯМР (CDCl3 δ : 1,3 (т, 3Н, J = 10 Гц), 2,9 (кв, 2Н, J = 10 Гц), 8,68 (c, 1Н) ч/млн.
III 3-(4-Хлор-5-фторпиримидин-6-ил)-2-(2,4-дифторфенил)- 1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол.
К раствору ДАЛ (20 ммоль) в ТГФ1 (50 мл) (получен по методике примера 1) в атмосфере азота при -70oC по каплям в течение 15 мин прибавляют раствор продукта стадии II (3,2 г, 20 ммоль) в ТГФ1 (30 мл). Полученную смесь перемешивают при той же температуре 3 ч, после чего прибавляют раствор 1-(2,4- дифторфенил)-2-(1Н-1,2,4-триазол-1-ил)этанона (4,46 г, 20 ммоль) в ТГФ (50 мл) и смесь выдерживают 1 ч при -70oC и затем еще 1 ч при -50oC. Реакционную смесь нейтрализуют добавлением раствора ледяной уксусной кислоты (1,2 г) в виде (10 мл) и смесь оставляют нагреваться до комнатной температуры. Органическую фазу отделяют, водную фазу экстрагируют этилацетатом (20 мл), объединенные органические слои сушат над сульфатом магния и концентрируют при пониженном давлении. Колоночной хроматографией остатка на окиси кремния с элюированием смесью этилацетат-диэтиловый эфир (3:2) вначале получают после объединения и испарения соответствующих фракций, а также промывания диэтиловым эфиром энантиомерную пару В заглавного соединения (0,94 г, 12%), т.пл. , 92oC. Найдено, %: C 49,93; H 3,57; N 18,17, вычислено для C16H13,ClF3N5O, %: C 50,06; H 3,39; N 18,25.
Дальнейшим элюированием после объединения и испарения соответствующих фракций получают энантиомерную пару A заглавного соединения, загрязненного исходным кетоном. Неоднократной перекристаллизацией продукта из диэтилового эфира получают продукт с т.пл. 132oC. Найдено, %: C 49,93; H 3,58; N 18,23, вычислено для C16H13ClF3N5O, %: C 50,06; H 3,39, N 18,25.
I. ТГФ может быть заменен толуолом.
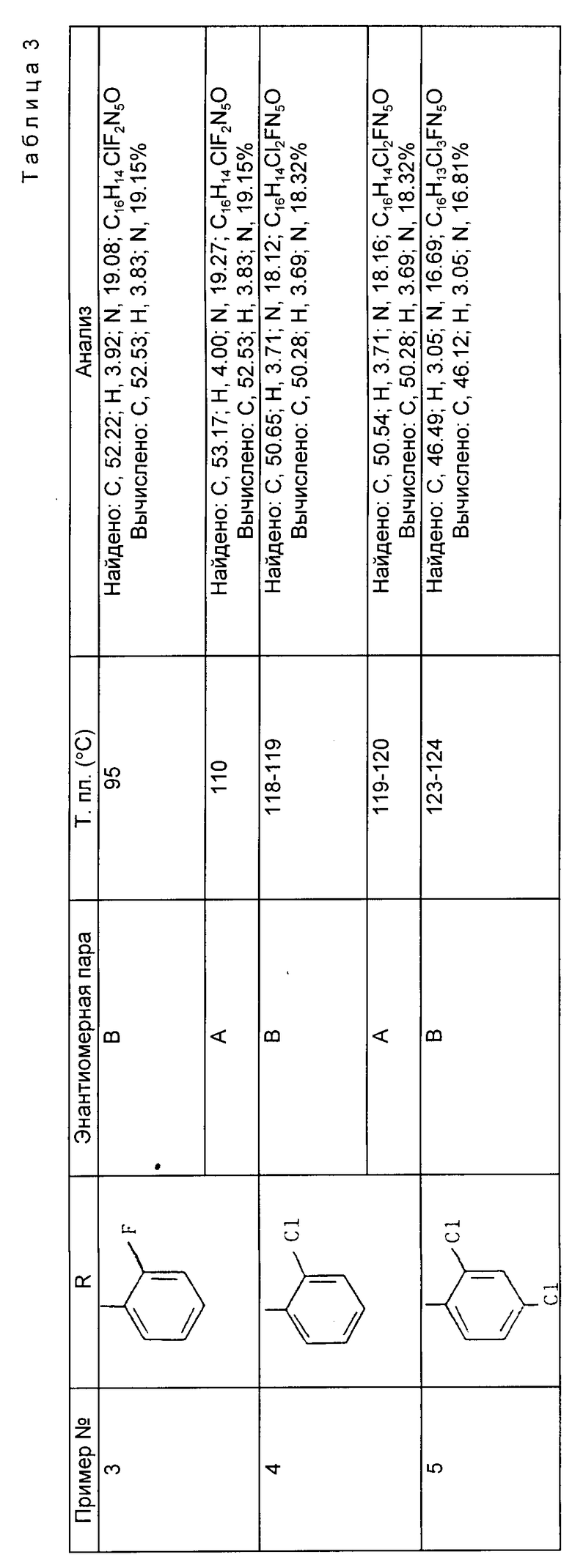
Препаративные примеры 3 - 5. Соединения общей формулы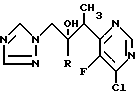
получены методом, аналогичным методу препаративного примера 2 (III), с использованием в качестве исходных продуктов 4-хлор-6-этил-5-фторпиримидина и соответствующего 1-арил-2-(1Н-1,2,4-триазол-1-ил)этанона (см. табл. 3).
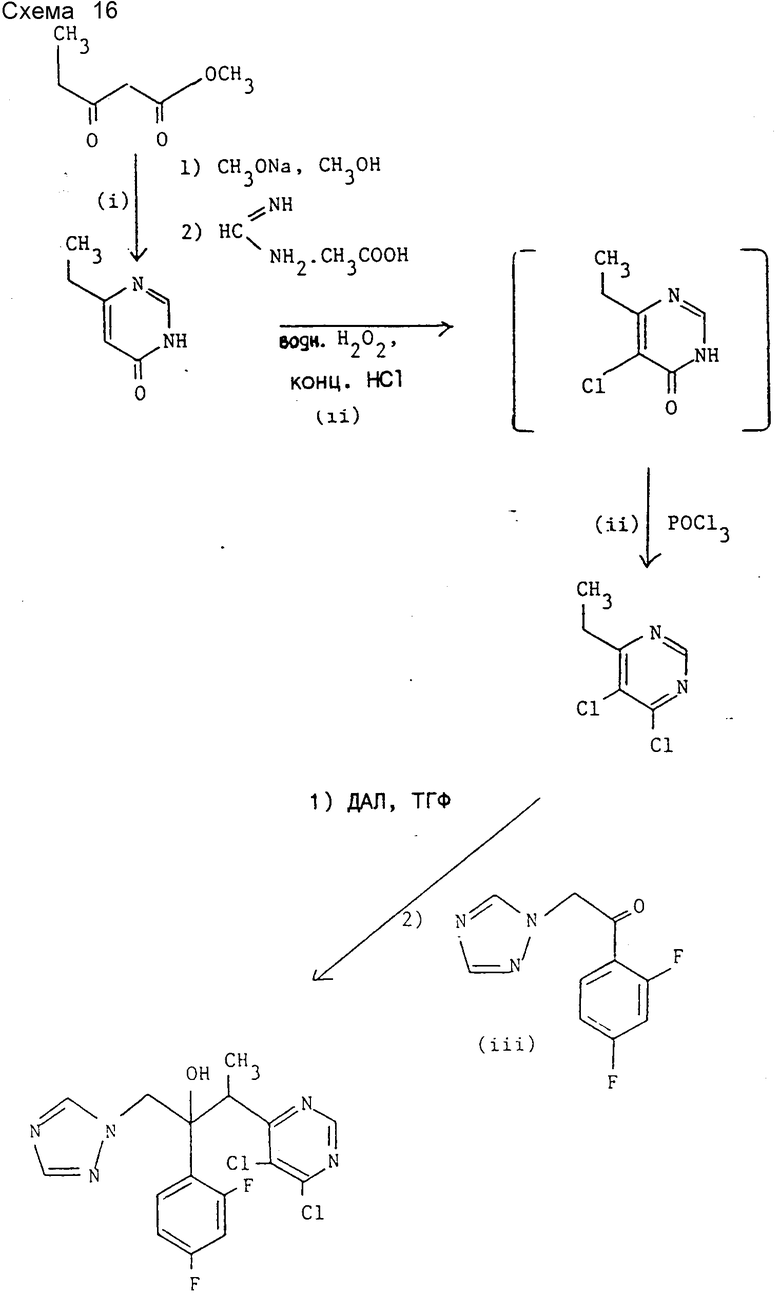
Препаративный пример 6. 3-(4,5-Дихлорпиримидин-6- ил)-2-(2,4-дифторфенил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол (см. схему 16).
I. 6-Этилпиримидин-4(3Н)-он.
К раствору метоксида натрия (4,19 кг, 77,6 моль) и ацетата формамидина (3 кг, 28,8 моль) в метаноле (45 л) при 5 - 10oC медленно добавляют раствор метилового эфира пропионилуксусной кислоты (2,5 кг, 19,2 моль) в метаноле (10 л), поддерживая температуру в ходе добавления ниже 20oC. Полученную смесь перемешивают примерно сутки при комнатной температуре, после чего добавлением концентрированной соляной кислоты устанавливают pH 7. Реакционную смесь концентрируют при пониженном давлении до объема примерно в 10 л, разбавляют водой (10 л) и экстрагируют 2-бутаноном (2•30 л). Объединенные органические экстракты концентрируют при пониженном давлении до объема примерно 2 л и разбавляют этилацетатом (4 л). Целевой продукт кристаллизуется из раствора (2,4 кг, 70%) и после перекристаллизации из изопропанола получают продукт с т.пл. 132 - 134oC. Найдено, %: C 58,45; H 6,37; N 22,41. Вычислено для C6H8N2O, %: C 58,05; H 6,50; N 22,57.
II. 4,5-Дихлор-6-этилпиримидин.
К раствору 6-этилпиримидин- 4(3H)-она (продукт стадии I (18,6 г, 150 ммоль) в концентрированной соляной кислоте (120 мл) при 30 - 40oC прибавляют в течение 30 мин 30%-ный водный раствор перекиси водорода (18 мл) (некоторое повышение температуры) и полученную смесь перемешивают около суток при 40oC. Смесь концентрируют при пониженном давлении, остаток суспендируют/растворяют в толуоле и толуол удаляют при пониженном давлении. Остаток растворяют в хлорокиси фосфора (150 мл), кипятят 3 ч, после чего избыток хлорокиси фосфора удаляют при пониженном давлении. Остаток переносят в лед с водой, экстрагируют хлористым метиленом (3•50 мл), объединенные органические экстракты промывают водой (30 мл) и сушат над сульфатом магния. Растворитель удаляют при пониженном давлении и перегонкой полученного масла при пониженном давлении получают заглавное соединение (5,4 г, 20%), т.кип. 104oC при 22 мм Hg, охарактеризованного с помощью 1H-ЯМР спектроскопии.
1H-ЯМР (CDCl3) δ : 1,3 (т, 3Н, J = 10 Гц), 3,04 (кв, 2Н, J = 10 Гц), 8,75 (c, 1H) ч/млн.
III. 3-(4,5-Дихлорпиримидин-6-ил)-2-(2,4-дифторфенил)-1-(1H- 1,2,4-триазол-1-ил)бутан-2-ол.
K раствору ДАЛ (13,6 ммоль) в ТГФ (50 мл) (получен по методике примера 1) при -70oC по каплям прибавляют 4,5-дихлор-6-этилпиримидин (продукт стадии II (2,37 г, 13,3 ммоль) и полученный раствор перемешивают при той же температуре 10 мин. К реакционной смеси добавляют раствор 1-(2,4-дифторфенил)-2- (1H-1,2,4-триазол-1-ил)этанона (2,97 г, 13,3 ммоль) в ТГФ (50 мл) с такой скоростью, что температура реакционной смеси не превышает -50oC. После перемешивания 1 ч при -70oC и еще 1 ч при -50oC реакционную смесь нейтрализуют добавлением 10%-ной водной уксусной кислоты (11 мл). Органическую фазу отделяют, водную фазу экстрагируют этилацетатом (2•20 мл) и объединенные органические слои сушат над сульфатом магния. После удаления растворителя при пониженном давлении остаток промывают диэтиловым эфиром (25 мл) и непрореагировавший исходный кетон (1,7 г) удаляют фильтрованием. Фильтрат концентрируют при пониженном давлении и "импульсной" хроматографией остатка на окиси кремния с элюированием смесью этилацетат-диэтиловый эфир (65:35) вначале получают (после объединения и испарения соответствующих фракций и промывания диэтиловым эфиром) энантиомерную пару В заглавного соединения (670 мг, 13%, т.пл. 124oC). Найдено, %: C 47,78; H 3,33: N 17,13, вычислено для C16H13Cl2F2N5O, %: 48,0; H 3,25; N 17,50.
Дальнейшим элюированием после объединения и испарения соответствующих фракций и промывания диэтиловым эфиром получают энантиомерную пару A заглавного соединения (527 мг, 10%), т.пл. 137oC. Найдено, %: C 48,02, H 3,30; N 17,39, вычислено для C16H13Cl2F2N5O, %: C 48,00; H 3,25; N 17,50.
Препаративный пример 7 (см. схему 17).
I. 2,4-Дихлор-5-фторпиримидин.
K хлорокиси фосфора (141,4 г) при 25oC добавляют 5-фторурацил (20 г) в виде порошка, полученную взвесь нагревают до 90oC и затем в течение 1 ч прибавляют N,N-диметиланилин (37,3 г). После этого реакционную смесь кипятят 5 ч и отгонкой удаляют 70 г хлорокиси фосфора. Затем смесь охлаждают до 25oC и порциями в течение 1 ч. переносят к охлажденную до 0oC 3н. HCl. Заглавное соединение экстрагируют из полученной смеси дихлорметаном (2•70 мл). Объединенные дихлорметановые слои промывают водой (50 мл) и концентрированием в вакууме получают масло (24 г), охарактеризованное с помощью 1H-ЯМР спектроскопии.
1H-ЯМР (CDCl3) δ : 8,5 (с, 1Н) ч/млн. MC: m/е = 166
II. 2,4-Дихлор-1,6-дигидро-6-этил-5-фторпиримидин.
К стружкам магния (4,27 г) в тетрагидрофуране (56 мл) в течение 5 ч прибавляют раствор бромэтана (19 г) в ТГФ (10 мл). К полученной взвеси при 0oC в течение 1 ч прибавляют раствор продукта стадии I (24 г) в 1,2-диметоксиэтане (70 мл). Реакционную смесь нейтрализуют при 10oC добавлением ледяной уксусной кислоты (10 г) и получают раствор заглавного соединения, который непосредственно используют на следующей стадии.
III. 2,4-Дихлор-6-этил-5-фторпиримидин.
К раствору, полученному в качестве продукта на стадии II, в течение 2 ч прибавляют раствор перманганата калия (23 г) в виде (260 мл), поддерживая температуру реакционной смеси ниже 20oC. Затем добавляют 5 н. соляную кислоту с последующим прибавлением раствора метанбисульфита натрия (14 г) в воде (42 мл). После обесцвечивания смеси продукт экстрагируют этилацетатом (250 мл). Последующим концентрированием органического слоя получают масло. Масло распределяют между дихлорметаном (50 мл) и 2 н. раствором гидроокиси натрия (105 мл), органический слой промывают 5%-ным рассолом (100 мл) и после концентрирования получают раствор заглавного соединения, который непосредственно используют на следующей стадии.
IV. 2-Хлор-6-этил-5-фторпиримидин-4(3Н)-он.
К полученному в качестве продукта на стадии III раствору добавляют воду (6 мл), смесь перемешивают при 80oC с медленным прибавлением в течение 2 ч 4 н. раствора гидроокиси натрия. После этого реакционную смесь охлаждают и промывают дихлорметаном (15 мл). Водный слой добавляют к дихлорметану (60 мл) и прибавлением концентрированной соляной кислоты устанавливают pH 1. Органический слой отделяют и добавлением концентрированного водного раствора аммиака устанавливают pH 3. Осадок хлорида аммония отфильтровывают, фильтрат концентрируют до объема 15 мл и разбавляют этилацетатом (150 мл). Раствор концентрируют до объема 30 мл, образовавшиеся кристаллы заглавного соединения отфильтровывают и после высушивания (8 г) характеризуют с помощью 1H-ЯМР и масс-спектрометрии.
1H-ЯМР (DMCO-d6) δ : 7,3 (обмениваемые), 2,4 (м, 2Н), 1,1 (т, 3Н) ч/млн. MC: m/е = 176.
V. 6-Этил-5-фтopпиpимидин-4(3H)-он.
К продукту стадии IV (6 г) в этаноле (60 мл) добавляют ацетат натрия (5,5 г) и 5% палладий на угле (0,6 г). Смесь гидрируют 3 ч под давлением в 3 атмосферы. Катализатор отфильтровывают, фильтрат концентрируют до объема в 10 мл и затем смешивают с водой (2 мл) и дихлорметаном (80 мл). Добавляют толуол (32 мл), раствор концентрируют до объема в 5 - 6 мл и затем смешивают с дополнительным количеством толуола (8 мл). Выделившиеся кристаллы заглавного соединения отфильтровывают и характеризуют с помощью 1H-ЯМР и масс-спектрометрии (выход 3,9 г).
1H-ЯМР (DMCO-d6) δ : 8,0 (с, 1Н), 2,5 (м, 2Н), 1,15 (т, 3H) ч/млн. MC: m/e = 142.
Препаративный пример 8. 4-Этил-5-фторпиримидин (см. схему 18).
Смесь 2,4-дихлор-6-этил-5-фторпиримидина (10 г) (см. препаративный пример 7 (III)), ацетата натрия (8,83 г), 5% палладия на угле ("влажность" 50%, 2 г) и метанола (30 мл) гидрируют 5 ч при 50oC и давлении 3 атмосферы. Полученную взвесь тщательно фильтруют через фильтрующее средство на основе целлюлозы, фильтровальный пирок промывают дополнительным количеством метанола (5 мл) и полученный оранжевый фильтрат отгоняют при 64oC и атмосферном давлении с получением бесцветного дистиллата. Дистиллат распределяют между водой (300 мл) и эфиром (400 мл) и две фазы разделяют, органический фазу промывают водой (4•50 мл), сушат над MgSO4 и удалением растворителя при комнатной температуре и пониженном давлении получают заглавное соединение в виде бледно-желтой жидкости (2,2 г).
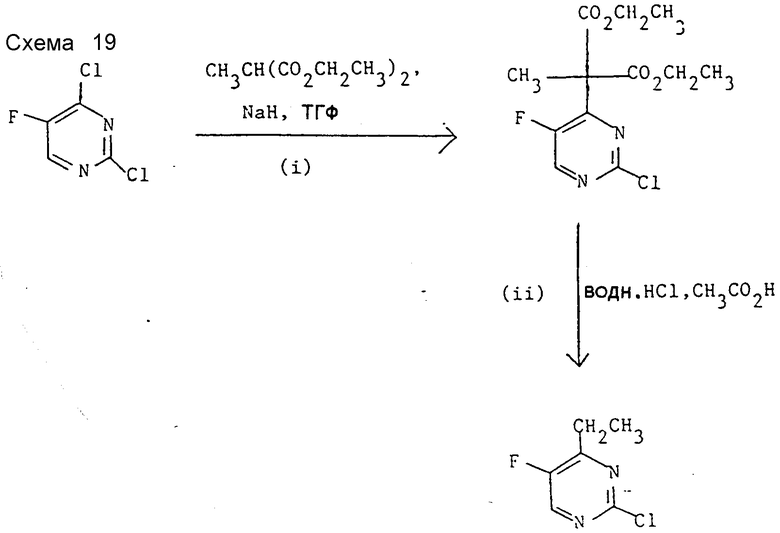
Препаративный пример 9. 2-Хлор-4-этил-5-фторпиримидин (см. схему 19).
I. Диэтиловый эфир 2-метил-2-(2-хлор-5-фторпиримидин-4-ил) пропандиевой кислоты.
Проводят реакцию при -10oC в ТГФ (200 мл) между гидридом натрия (60%-ная дисперсия в масле, 2,8 г) и диэтиловым эфиром метилмалоновой кислоты (6 г). Через 30 мин при -10oC в течение 30 мин прибавляют раствор 2,4-дихлор-5-фторпиримидина (5 г) (см, препаративный пример 7) в ТГФ (200 мл). Реакционную смесь распределяют между дихлорметаном (200 мл) и водой (200 мл), подкисляют уксусной кислотой и слои разделяют. Органический слой концентрируют при пониженном давлении и полученное масло хроматографируют на окиси кремния с элюированием дихлорметаном. В результате после объединения и испарения соответствующих фракций получают заглавное соединение (9 г), охарактеризованного с помощью 1H-ЯМР и масс-спектрометрии.
1H-ЯМР (CDCl3) δ : 8,5 (д, 1Н), 4,6 (м, 4Н), 1,9(с, 3Н), 1,3 (т, 3H) ч/млн. MC: m/e = 504.
II. 2-Хлор-4-этил-5-фторпиримидин.
Продукт стадии I (3,2 г) растворяют в уксусной кислоте (25 мл) и разбавляют 5 н. соляной кислотой (10 мл). Смесь нагревают при 100oC 16 ч, охлаждают и распределяют между водой (30 мл) и дихлорметаном (45 мл). Дихлорметановый слой отделяют, сушат и концентрированием при пониженном давлении получают масло. Заглавное соединение выделено хроматографией на окиси кремния с элюированием дихлорметаном. Продукт охарактеризован с помощью 1H-ЯМР и масс-спектрометрии (выход 350 мг).
1H-ЯМР (CDCl3) δ : 8,4 (с, 1H), 2,9 (м, 2Н), 1,5 (т, 3Н) ч/млн. MC: m/e = 160.
Выявление активности in vivo на мышах против Aspergillus fumigatus.
По общей методике испытаний группу мышей заражают штаммом Aspergillus fumigatus. Затем каждая мышь получает испытуемое соединение в стандартной дозировке в 20 мг/кг дважды в день в течение 5 дней. На десятый день определяют состояние мышей.
Активность определяется выживанием получавших лечение мышей, после смерти мышей в группе, не получавшей лечения, а также числом излеченных мышей.
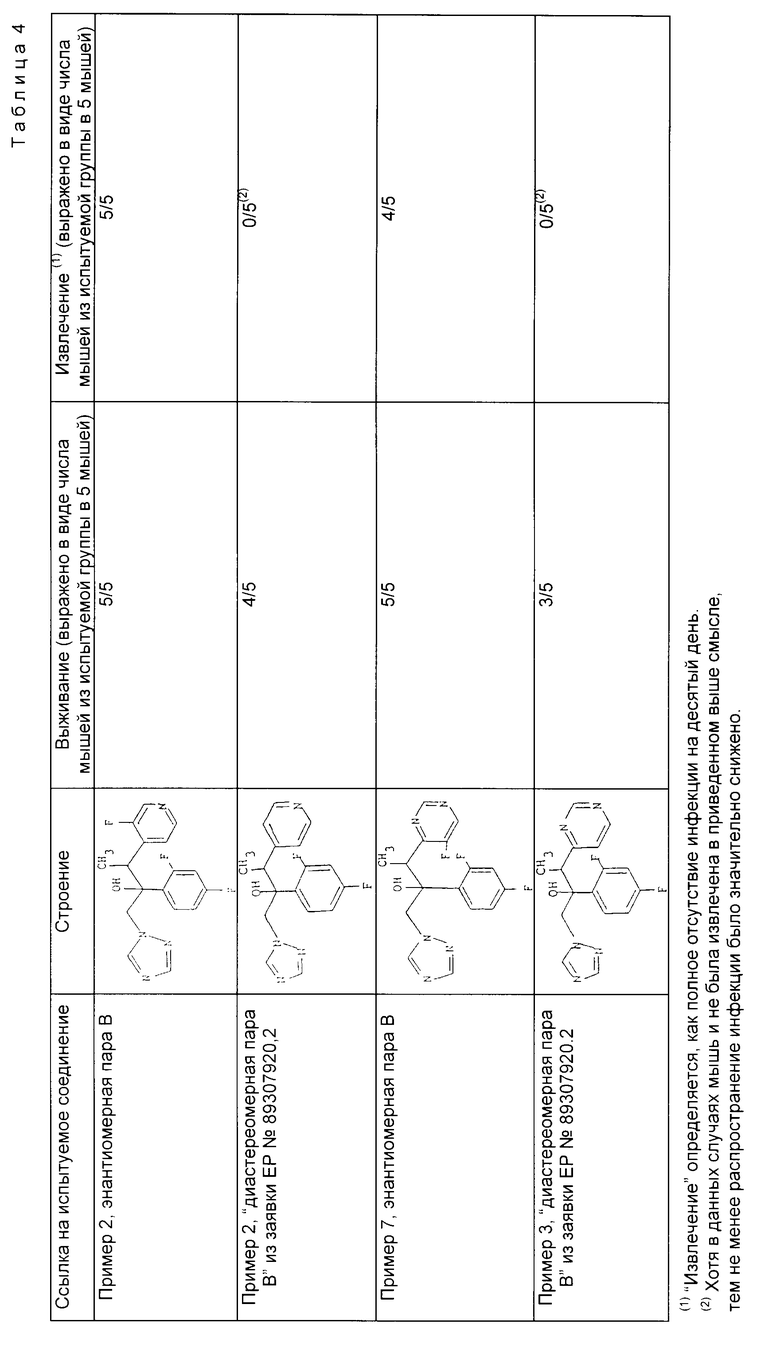
Результаты, полученные в сравнительном исследовании с применением двух соединений, охарактеризованных в рабочих примерах, и двух соединений, приведенных в заявке на Европейский патент N 89307920.2 (EP-A-0357241, приведены в табл.4.

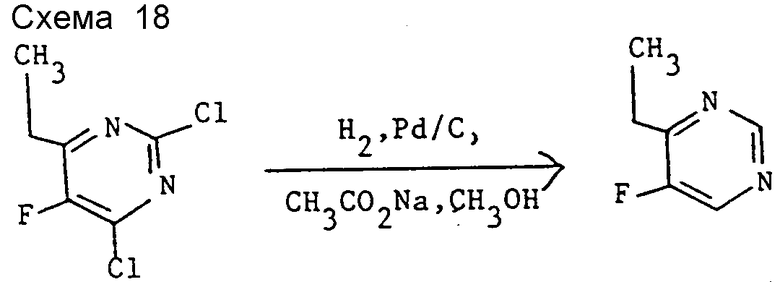
Изобретение относится к производным триазола, проявляющим противогрибковую активность. Сущность изобретения: противогрибковые соединения формулы, приведенной в описании, и их фармацевтически приемлемые соли, где R представляет фенил, замещенный 1-3 галоидзаместителями, R1 - C1-C4-алкил, X - CH или N, Y - F или Cl, фармацевтическая композиция и промежуточные продукты. 5 с. и 15 з.п.ф-лы, 5 табл.
где R - фенил, замещенный 1 - 3 галоидзаместителями;
R1 - C1-4-алкил;
X - CH или N;
Y - F или Cl,
или их фармацевтически приемлемая соль.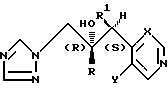
10. Соединение по п.1, представляющее собой 2R,3S-2-(2,4-дифторфенил)-3-(5-фторпиримидин-4-ил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол или его фармацевтически приемлемая соль.
где Q представляет группу формулы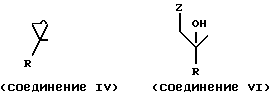
где R, R1, X и Y имеют значения, определенные в п.1;
Z - отщепляемая группа.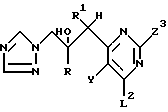
где R, R1 и Y имеют значения, указанные в п.1;
Z2 и Z3 каждый независимо выбран из водорода и группы, которая может быть селективно удалена восстановлением, при условии, что Z2 и Z3 не могут оба быть водородом.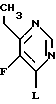
где L - означает Н или хлор.
| EP, 0069442, C 07 D 249/08, 1985 | |||
| US, патент, 1318159, C 07 D 249/08, 198 3 | |||
| GB, патент, 1464224, C 07 D 249/08, 1987 | |||
| УСТРОЙСТВО для ИЗМЕРЕНИЯ ЗАДЕРЖКИ | 0 |
|
SU332387A1 |
| УСТРОЙСТВО для ОБРАБОТКИ ТОРМОЗНБ1Х ЖЕЛЕЗНОДОРОЖНЫХ КОЛОДОК | 0 |
|
SU334035A1 |

