Настоящее изобретение относится к способу получения 4-{[(2S)-2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутаноил]амино}-2-фторбензамида (I) или 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2-фторбензамида (II) из 2,5-диметоксипиридина (III), 1-(2-бром-4-хлорфенил)-4-хлор-1Н-1,2,3-триазола (Х-Cl) или 1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазола (X-CF3), 4-амино-2-фторбензамида (XIII) и (2R)-2-аминомасляной кислоты (XVII).
Соединения 4-{[(2S)-2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутаноил]амино}-2-фторбензамид (I) и 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамид (II) известны из WO 2017/005725 и соответствуют формулам (I) и (II)

Соединения формул (I) и (II) действуют в качестве ингибиторов фактора XIa и, благодаря их специфическому механизму действия, могут после перорального введения приводить in vivo к безопасной и эффективной антикоагуляции.
В WO 2014/154794 и WO 2017/005725 описан синтез для получения соединений формул (I) и (II) в диапазоне размерности в граммах, исходя из 2,5-диметоксипиридина (III), 1-(2-бром-4-хлорфенил)-4-хлор-1Н-1,2,3-триазола (Х-Cl) или 1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазола (X-CF3), соответственно, 4-амино-2-фторбензамида (XIII) и трет-бутил- 2-бромбутаноата (VII) (Схема 1).
Схема 1

Химические названия по ИЮПАК соединений (I) - (XIV-Cl)/(XIV-CF3):
4-{[(2S)-2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутаноил]амино}-2-фторбензамид (I),
4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2-фторбензамид (II),
2,5-диметоксипиридин (III),
(2,5-диметоксипиридин-4-ил)бороновая кислота (IV),
4-бром-2,5-диметоксипиридин (V),
4-бром-5-метоксипиридин-2(1Н)-он (VI),
трет-бутил 2-бромбутаноат (VII),
трет-бутил 2-(4-бром-5-метокси-2-оксопиридин-1(2Н)-ил)бутаноат (VIII),
трет-бутил 2-[5-метокси-2-оксо-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-1(2Н)-ил]бутаноат (IX),
1-(2-бром-4-хлорфенил)-4-хлор-1Н-1,2,3-триазол (Х-Cl),
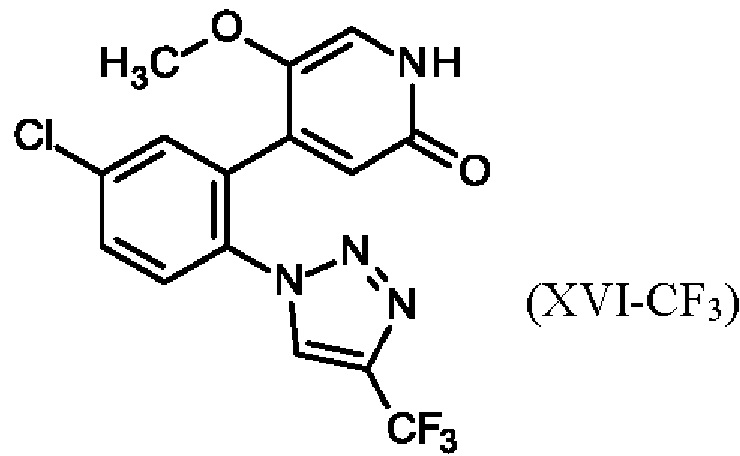
1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазол (X-CF3),
трет-бутил 2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутаноат (XI-Cl),
трет-бутил 2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноат (XI-CF3),
2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}масляная кислота (XII-Cl),
2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]масляная кислота (XII-CF3),
4-амино-2-фторбензамид (XIII),
4-(2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутанамидо)-2-фторбензамид (XIV-Cl),
4-{2-[4-{5-хлор-2-[4-(трифторметил)-4,5-дигидро-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутанамидо}-2-фторбензамид (XIV-CF3).
Синтез соединений формул (I) и (II), указанных на схеме 1, может быть разделен на три части:
a) Получение трет-бутил 2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутаноата (XI-Cl) или трет-бутил 2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноата (XI-CF3) из 2,5-диметоксипиридина (III) и 1-(2-бром-4-хлорфенил)-4-хлор-1Н-1,2,3-триазола (Х-Cl) или 1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазола (X-CF3), соответственно, через трет-бутил 2-[5-метокси-2-оксо-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-1(2Н)-ил]бутаноат (IX).
Схема 2

b) Получение 4-(2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутанамидо)-2-фторбензамида (XIV-Cl) или 4-{2-[4-{5-хлор-2-[4-(трифторметил)-4,5-дигидро-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2H)-ил]бутанамидо}-2-фторбензамида (XIV-CF3) из трет-бутил 2-{4-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1((2H)-ил}бутаноата (XI-Cl) или трет-бутил 2-[4-{5-хлор-2-[4-(трифторметил)-1H-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноата (XI-CF3), соответственно, через 2-{4-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}масляную кислоту (XII-Cl) или 2-[4-{5-хлор-2-[4-(трифторметил)-1H-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2H)-ил]масляную кислоту (XII-CF3), соответственно, и 4-амино-2-фторбензамид (XIII)
Схема 3

с) Разделение двух энантиомеров 4-(2-{4-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутанамидо)-2-фторбензамида (XIV-Cl) или 4-{2-[4-{5-хлор-2-[4-(трифторметил)-4,5-дигидро-1H-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2H)-ил]бутанамидо}-2-фторбензамида (XIV-CF3) с получением одного энантиомера 4-{[(2S)-2-{4-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2H)-ил}бутаноил]амино}-2-фторбензамид (I) или 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2-фторбензамид (II), соответственно.
Схема 4

Для промышленного внедрения и производства большего количества килограммов способы и пути получения, описанные в WO 2014/154794 и WO 2017/005725, подходят только в очень ограниченной степени. Путь длинный (9 линейных стадий) и требует утомительных процедур обработки и очистки, что приводит к низкому общему выходу. Самый большой недостаток состоит в том, что последовательность обеспечивает вещество только в рацемической форме, такой как соединение формулы (XIV-Cl)/(XIV-CF3), и ее необходимо разделить с помощью хиральной хроматографии для получения желаемых единичных энантиомеров соединения формулы (I)/(II). Последовательность стадий синтеза, описанная в WO 2014/154794 и WO 2017/005725, делает получение асимметричной версии с использованием тех же промежуточных продуктов маловероятным Приоритет в литературе указывает на то, что введение стереоцентра между пиридоновым кольцом и сложным эфиром, как в соединениях формул (VIII), (IX) и (XI-Cl)/(XI-CF3), является сложной задачей из-за высокой склонность к рацемизации этого положения в молекуле (P.S. Dragovich, et al., J. Med. Chem., 2003, 46, 4572). Кроме того, есть четкие доказательства того, что в условиях удаления защитных групп или амидного сочетания такой стереоцентр в сильно кислой позиции очень склонен к рацемизации (L. Chen, et al., Organic Process Research & Development, 2006, 10, 838). Необходимость хирального разделения представляет собой не только экономически невыгодный процесс, но также делает производство активного фармацевтического ингредиента (API) трудоемким процессом.
Таким образом, неожиданно был обнаружен новый путь синтеза, который справляется с большинством проблем для предыдущего способа, описанного выше на схеме 1. Путь, описанный на схеме 5, намного короче, с 4 стадиями в самой длинной линейной последовательности (всего 6). Достигается более высокий выход для отдельных стадий, что приводит к более высокому общему выходу для всей последовательности. Путь является конвергентным, что позволяет выполнять стадии синтеза параллельно и оптимизировать управление временем. Наиболее важно, что новый путь следует асимметричной стратегии и обеспечивает желаемое соединение формулы (I)/(II) в высоком энантиомерном избытке (ее), независимо от дорогостоящего и трудоемкого хирального разделения с помощью ВЭЖХ или SFC.
Схема 5
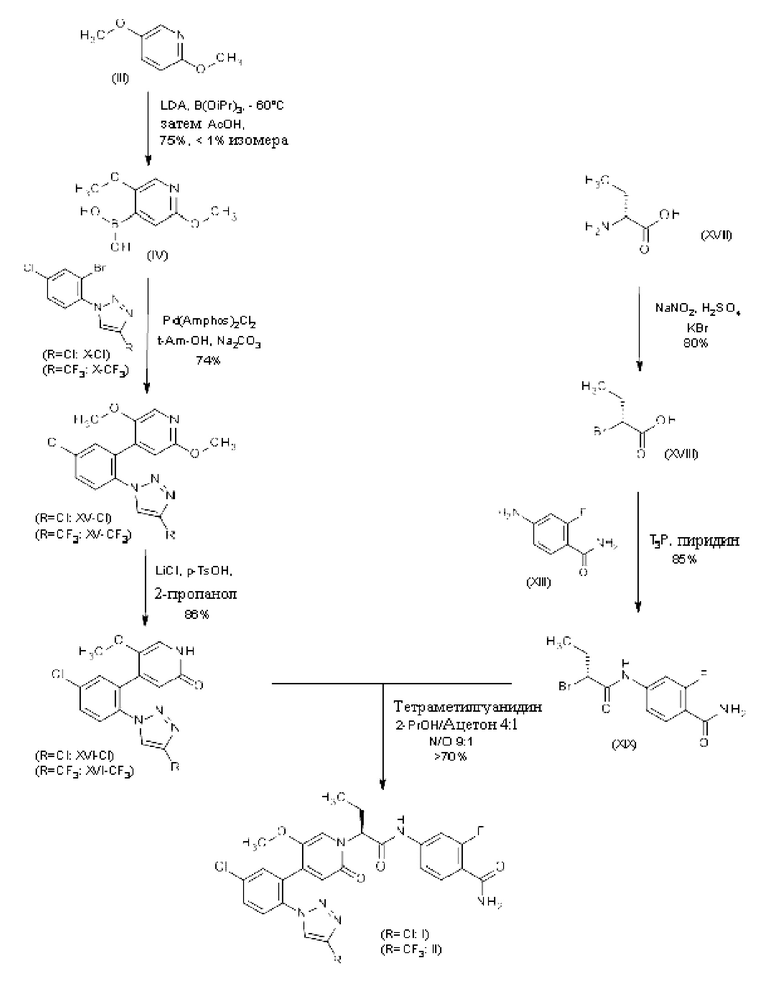
Химические названия по ИЮПАК соединений (XV-Cl)/(XV-CF3)-(XIX):
4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-2б5-диметокси-пиридин (XV-Cl)
4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметоксипиридин (XV-CF3)
4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метоксипиридин-2(1Н)-он (XVI-Cl)
4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метоксипиридин-2(1Н)-он (XVI-CF3)
(2R)-2-аминомасляная кислота (XVII)
(2R)-2-броммасляная кислота (XVIII)
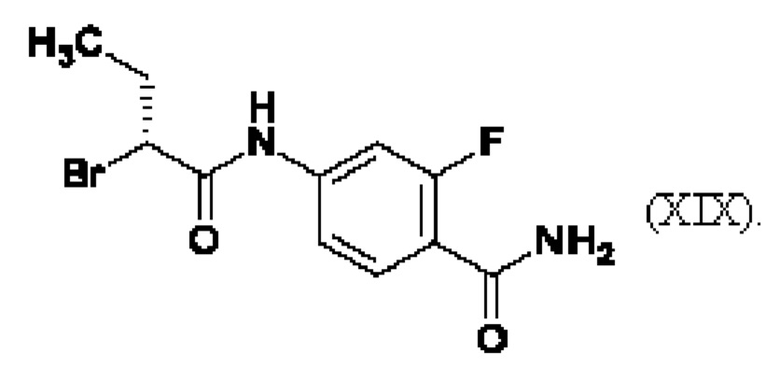
4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамид (XIX)
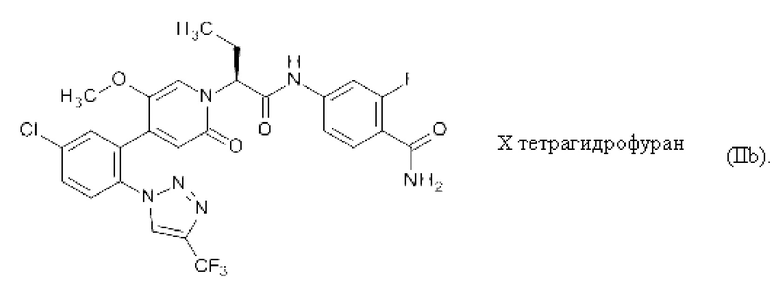
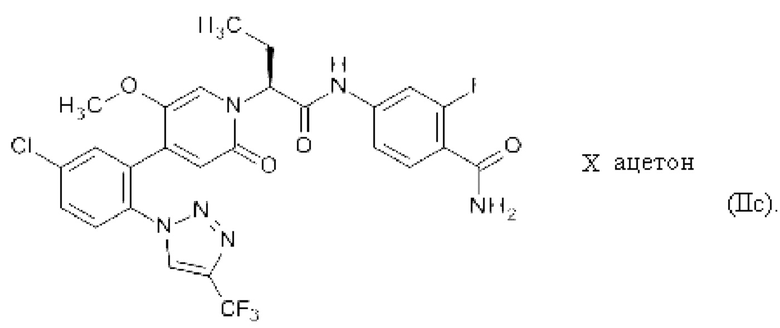
Соединение формулы (II) может быть превращено в соответствующие сольваты посредством обработки соответствующими растворителями. Растворителями являются, например, изопропилацетат, тетрагидрофуран и ацетон, приводящие к соединению 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамид изопропилацетат (IIa), 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамид тетрагидрофуран (IIb) и 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамид ацетон (IIc), соответственно.
Краткое описание чертежей:
Фиг. 1: График XRPD соединения формулы (IIa).
Фиг. 2: График XRPD соединения формулы (IIb).
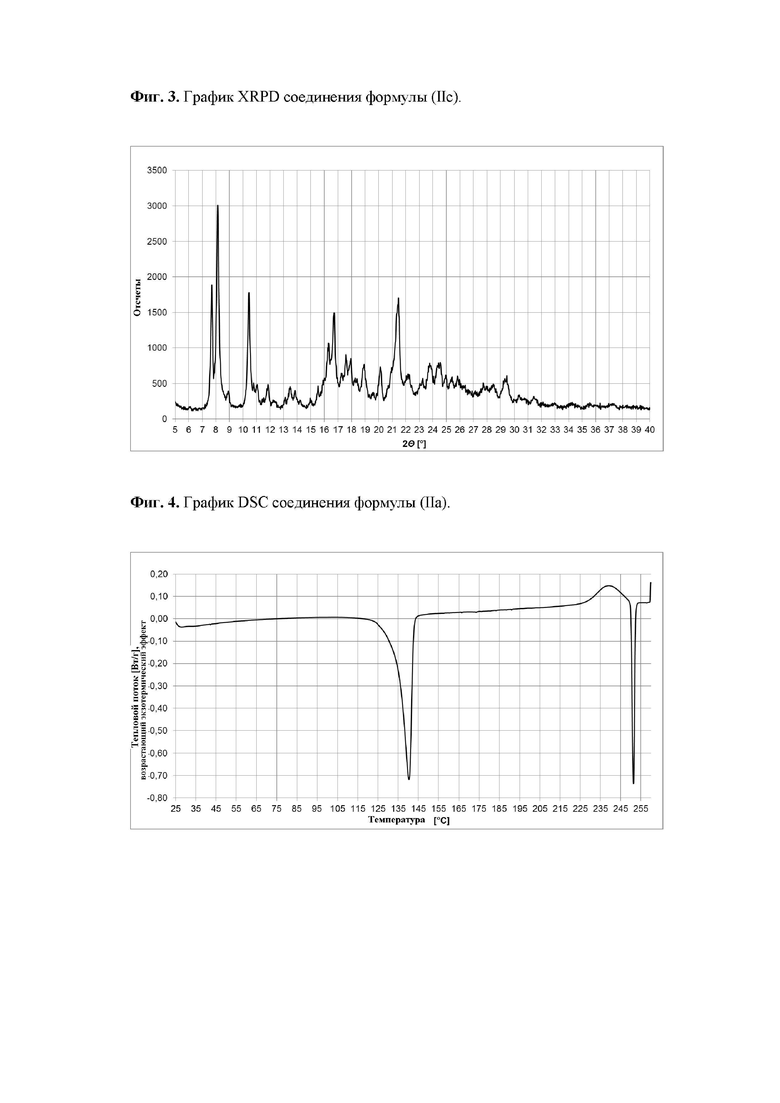
Фиг. 3: График XRPD соединения формулы (IIc).
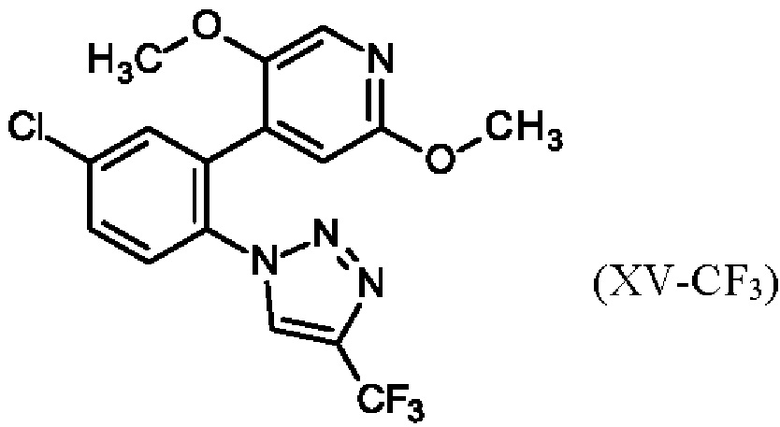
Фиг. 4: График DSC соединения формулы (IIa).
Фиг. 5: График DSC соединения формулы (IIb).
Фиг. 6: График DSC соединения формулы (IIc).
Фиг. 7: Микроснимок соединения формулы (IIa).
Фиг. 8: Микроснимок соединения формулы (IIb).
Фиг. 9: Микроснимок соединения формулы (IIc).
В случае промежуточных соединений синтеза и рабочих примеров согласно настоящему изобретению, описанных ниже, любое соединение, указанное в форме сольвата, обычно является сольватом неизвестного точного стехиометрического состава, полученным посредством соответствующего способа получения и/или очистки. Если не указано более подробно, дополнения к названиям и структурным формулам, такие как «изопропилацетат», «тетрагидрофуран» или «ацетон», не следует понимать в стехиометрическом смысле в случае таких сольватов, а имеют просто описательный характер в отношении компонентов, образующих сольват, присутствующих в них.
Предпочтительными являются сольваты со стехиометрическим составом соединение: растворитель, равным 1:1.
Сравнение последовательностей синтеза:
а) Соединение формулы (III) в соединение формулы (XI-Cl)/(XI-CF3) через соединение формулы (ГХ) (описано в WO 2017/005725) по сравнению с превращением соединения формулы (III) в соединение формулы (XVI-Cl)/(XVI-CF3) (настоящее изобретение)
Соединение формулы (III) в соединение формулы (XI-Cl)/(XI-CF3) через соединение формулы (IX) (описано в WO 2017/005725)
Последовательность, описанная в WO 2017/005725 и частично в WO 2014/154794, начинается с последовательности литирования-борилирования 2,5-диметоксипиридина (III) с обеспечением (2,5-диметоксипиридин-4-ил)бороновой кислоты (IV), и на следующей стадии бор ильную группу на пиридиновом кольце замещают на бромид с получением 4-бром-2,5-диметоксипиридина (V). 4-Бром-2,5-диметоксипиридин (V) затем деметилируют с получением 4-бром-5-метоксипиридин-2(1Н)-она (VI), который является N-алкилированным трет-бутил 2-бромбутаноатом (VII), с получением трет-бутил 2-(4-бром-5-метокси-2-оксопиридин-1(2Н)-ил)бутаноата (VIII). В соединение формулы (VIII) вводят сложный пинаколовый эфир бороновой кислоты в реакции борилирования, катализируемой Pd, с получением трет-бутил 2-[5-метокси-2-оксо-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-1(2Н)-ил]бутаноата (IX). Соединение формулы (IX) затем сочетают с 1-(2-бром-4-хлорфенил)-4-хлор-1Н-1,2,3-триазолом (Х-Cl) / 1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазолом (X-CF3) с получением промежуточного соединения трет-бутил 2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутаноата (XI-Cl) / трет-бутил 2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноата (XI-CF3). Несмотря на то, что последовательность позволяет получить целевое соединение формулы (I)/(II), она имеет некоторые недостатки для получения молекул в более крупном масштабе. Введение бромида в пиридин (соединение формулы (V)) через бороновую кислоту (соединение формулы (IV)) является относительно неэкономичной и низкопродуктивной методикой, особенно потому, что другой сложный эфир бороновой кислоты включается в том же положении позже в последовательности для получения формулы (IX). Кроме того, применение реагента (бис)пинаколатодибор на стадии из соединения формулы (VIII) в соединение формулы (IX) и катализатора Pd(dppf)Cl2 при превращении из соединения формулы (VIII) в соединение формулы (IX) и соединения формулы (IX) в соединение формулы (XI-Cl)/(XI-CF3) делает последовательность относительно затратной. Кроме того, последовательность требует нежелательных растворителей DMF и диоксан при превращениях из соединения формулы (V) в соединение формулы (XI-Cl)/(XI-CF3). Бромид меди для получения соединения формулы (V) также может создать проблемы с удалением отходов при индустриализации способа получения.
Соединение формулы (III) в соединение формулы (XVI-Cl)/(XVI-CF3) (настоящее изобретение)
Последовательность, описанная в настоящем изобретении, значительно уменьшает длину последовательности и связанные с ней проблемы за счет использования различных синтетических промежуточных соединений и более выгодных условий реакции. Первое превращение 2,5-диметоксипиридина (III) в (2,5-диметоксипиридин-4-ил)бороновую кислоту (IV) остается без изменений с улучшенным выходом благодаря улучшенным условиям реакции и методике обработки. В отличие от ранее описанных условий реакции превращение теперь можно проводить при -60°С вместо -78°С, что представляет собой преимущество для индустриализации способа. Кроме того, для качества и выхода продукта выгодно получать диизопропиламид лития непосредственно in situ и не использовать коммерчески доступный раствор диизопропиламида лития. После завершения реакции ее гасят смесью уксусной кислоты и воды, а оставшийся органический растворитель удаляют в вакууме при температуре не выше 70°С (стабильность продукта).
Полученную (2,5-диметоксипиридин-4-ил)бороновую кислоту (IV) непосредственно сочетают с 1-(2-бром-4-хлорфенил)-4-хлор-1Н-1,2,3-триазолом (Х-Cl) / 1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазолом (Х-CF3) с получением 4- [5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-2,5-диметокси-пиридина (XV-Cl) / 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметоксипиридина (XV-CF3) без прохождения через другую стадию броминирования/введения сложного эфира бороновой кислоты. Реакцию кросс-сочетания проводят в устойчивых и надежных условиях с использованием каталитической системы на основе Pd с основанием в растворителе, что обеспечивает эффективное сочетание, и процедуры обработки, которая удаляет оставшийся палладий и приводит к удобной кристаллизации целевого соединения из обработанной смеси в отличном качестве. Ключом к этой улучшенной процедуре обработки является переключение каталитической системы на основе Pd на Pd(Amphos)2Cl2 (A.S. Guram, et al., Organic Letters, 2006, 8, 1787), реализация стратегии дозирования и переключение растворителя реакции с ТГФ на трет-амиловый спирт
Реакция хорошо проходит с различными каталитическими системами на основе Pd, такими как Pd(OAc)2/PPh3, Pd(Amphos)2Cl2 или системы прекатализаторов. Особенно хорошие результаты дает применение Pd(Amphos)2Cl2 в качестве каталитической системы на основе Pd. Каталитическая система на основе Pd используется при соотношении от 0,5 мол. % до 5 мол. %, предпочтительно при соотношении от 0,7 мол. % до 1,3 мол. % и особенно предпочтительно при соотношении 1 мол. % на основе соединения формулы (Х-Cl)/(Х-CF3).
В качестве основания в этом способе могут использоваться различные неорганические основания. Особое предпочтение отдается основаниям, таким как фосфат калия, гидрофосфат калия, карбонат натрия или калия, особенно предпочтительно карбонат натрия. Соответствующее основание применяют в виде раствора в воде. Основание применяют при соотношении от 2 до 4 молярных эквивалентов на основе соединение формулы (X-Cl)/(X-CF3), предпочтительно при соотношении от 2,5 до 3,5 молярных эквивалентов и особенно предпочтительно при соотношении 3 молярных эквивалента.
(2,5-Диметоксипиридин-4-ил)бороновую кислоту (IV) применяют при соотношении от 1.0 до 1.5 молярных эквивалента. Предпочтительно применяют при соотношении от 1.2 до 1.4 молярных эквивалента на основе соединения формулы (Х-Cl) и особенно предпочтительно применяют при соотношении 1.2 молярных эквивалента на основе соединения формулы (Х-Cl). Предпочтительно ее применяют при соотношении от 1.05 до 1.15 молярных эквивалента на основе соединения формулы (X-CF3) и особенно предпочтительно применяют при соотношении 1.07 молярных эквивалента на основе соединения формулы (X-CF3).
В качестве растворителя можно использовать различные высококипящие органические растворители, такие как спирты или тетрагидрофуран. Предпочтительными спиртами являются 2-пропанол, 1-пропанол, 1-бутанол или трет-амиловый спирт (2-метил-2-бутанол), предпочтительным является трет-амиловый спирт (2-метил-2-бутанол). Особенно предпочтительно применяют трет-амиловый спирт (2-метил-2-бутанол) при соотношении 1:10 (мас/об) на основе соединения формулы (Х-Cl)/(Х-CF3). Трет-амиловый спирт (2-метил-2-бутанол) оказывается особенно полезным, так как обеспечивает отличные скорости превращения, что приводит к короткому времени реакции, а также допускает возможность высоких температур реакции и хорошего разделения фаз от водных фаз в ходе обработки.
Температура реакции предпочтительно находится в интервале от 55°С до 100°С, особенно предпочтительным является интервал температур от 63°С до 67°С для соединения формулы (XV-Cl) и от 93°С до 97°С для соединения формулы (XV-CF3).
Чтобы избежать образования побочных продуктов, а именно сочетания связывания формулы (XV-Cl)/(XV-CF3) с другой молекулой формулы (IV) через хлорид в фенильном кольце, используется стратегия дозирования.
Медленное добавление (2,5-диметоксипиридин-4-ил)бороновой кислоты (IV) к активной каталитической системе (Каталитическую систему на основе Pd с соединением формулы (Х-Cl)/(Х-CF3)) выбирают для обеспечения селективности сочетания (2,5-диметоксипиридин-4-ил)бороновой кислоты (IV) с бромидом в соединении формулы (X-Cl)/(X-CF3) по сравнению с нежелательным вторым сочетанием с первичным продуктом сочетания, которое означает реакцию с хлоридом в фенильном кольце соединения формулы (XV-Cl)/(XV-CF3).
Дозирование раствора соединения формулы (IV) и основания в воде проводят в диапазоне времени от 0.5 до 5 часов и предпочтительно от 1 до 4 часов. Особенно предпочтительным является добавление вышеуказанной смеси за 2-3 часа, предпочтительно 2.5 часа, для получения соединения формулы (XV-Cl) и за 3-4 часа, предпочтительно 4 часа, для получения соединения формулы (XV-CF3).
Полученное соединение формулы (XV-Cl)/(XV-CF3) непосредственно применяют в реакции деметилирования, которая селективно удаляет одну из двух метильных групп (метильную группу рядом с азотом) с получением пиридона, который представляет собой соединение формулы (XVI-Cl)/(XVI-CF3). Деметилирование проводят в очень предпочтительных условиях с использованием недорогого хлорида лития и п-толуолсульфоновой кислоты. Реакция деметилирования проводится в полярных и высококипящих растворителях, таких как спирты или этиленгликоль. Поскольку необходима температура реакции ≥ 75°С, требуются спирты с ≥ 3 атомами углерода, например 2-пропанол, 1-пропанол, 1-бутанол или трет-амиловый спирт (2-метил-2-бутанол). Предпочтительный температурный интервал для деметилирования составляет от 75°С до 120°С. Особенно предпочтительным является использование для реакции 2-пропанола при температуре возврата флегмы. Такой выбор растворителя обеспечивает очень удобную процедуру обработки. Простое добавление воды при температуре возврата флегмы и охлаждение до более низких температур приводит к осаждению соединения формулы (XVI-Cl)/(XVI-CF3) при отличном качестве и выходе.
Описанные стадии синтеза представляют собой важный сокращенный путь в общем синтезе сравнимых синтетических промежуточных соединений формулы (XI-Cl)/(XI-CF3) и соединений формулы (XVI-Cl)/(XVI-CF3). В обеих формулах триазол связан с ядром пиридона, и для завершения структурного образования требуется присоединение бензамида. Другими словами, оба промежуточных соединения, соединение формулы (XI-Cl)/(XI-CF3) и соединение формулы (XVI-Cl)/(XVI-CF3), представляют собой лишь еще одну стадию синтеза, образующую структуру, из промежуточного соединения, которое содержит все структурные элементы конечного целевого соединения формулы (I)/(II). В то время как путь синтеза, описанный в WO 2017/005725, требует 6 стадий с общим выходом 9,3%, тогда как для пути синтеза согласно настоящему изобретению требуется всего 3 стадии с общим выходом 47%.
b) Соединение формулы (XI-Cl)/(XI-CF3) в соединение формулы (XIV-Cl)/(XIV-CF3) (описано в WO 2017/005725) по сравнение с превращением соединения формулы (XVI-Cl)/(XVI-CF3) в соединение формулы (I)/(II) (настоящее изобретение)
Соединение формулы (XI-Cl)/(XI-CF3) в соединение формулы (XIV-Cl)/(XIV-CF3) (описано в WO 2017/005725)
В WO 2017/005725 промежуточное соединение формулы (XI-Cl)/(XI-CF3) превращают в линейной последовательности в рацемическую версию целевого соединения формулы (XVI-Cl)/(XVI-CF3). Следовательно, трет-бутиловый сложный эфир формулы (XI-Cl)/(XI-CF3) превращают в ходе гидролиза сложного эфира кислоты в соединение карбоновой кислоты формулы (XII-Cl)/(XII-CF3) с использованием 4М хлорида водорода в диоксане. Соединение формулы (XII-Cl)/(XII-CF3) затем подвергают сочетанию с 4-амино-2-фторбензамидом (XIII) с получением формулы (XIV-Cl)/(XIV-CF3). Удаление защитной группы трет-бутилового сложного эфира добавляет к последовательности еще одну непродуктивную стадию, поскольку не образуются дополнительные связи конечного соединения. Одним из самых больших недостатков всего синтеза является то, что исходный продукт последовательности, описанный в WO 2017/005725, соединение формулы (XIV-Cl)/(XIV-CF3), является полностью рацемическим, и возможность с его помощью можно получить энантиомерно чистый материал в ходе этой последовательность очень ограничена (см. литературу уровня техники, упомянутую выше).
Соединение формулы (XVI-С1)/(XVI-CF3) в соединение формулы (I)/(II) (настоящее изобретение)
Согласно настоящему изобретению всю восточную часть соединения формулы которая представляет собой соединение формулы (XIX), получают отдельно и подвергают реакции сочетания с соединением формулы (XVI-Cl)/(XVI-CF3) на последней стадии, что добавляет высокий уровень конвергентности стратегии синтеза. Для того, чтобы получить высокие уровни энантиомерной чистоты конечная стадия должна протекать в виде чистой SN2-реакции с полной инверсией стереоцентра с образованием соединения формулы (I)/(II) из энантиомерно чистого R-стереоизомера 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамида (XIX).
Получение 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамида (XIX) начинают с (2R)-2-аминомасляной кислоты (XVII), которая легко превращается в (2R)-2-броммасляную кислоту (XVIII) с бромидом калия и нитритом натрия в водной серной кислоте (Н. Rapoport, et al., J. Org. Chem., 1986, 51, 1713).
Энантиомерно чистую (2R)-2-броммасляную кислоту (XVIII) затем подвергают реакции сочетания с 4-амино-2-фторбензамидом (XIII) с получением 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамида (XIX) который вместе с пиридоном, который представляет собой соединение формулы (XVI-Cl)/(XVI-CF3), является прямым предшественником для получения соединения формулы (I)/(II). Сочетание осуществляется в случае карбоновой кислоты с карбодиимидами, такими как EDC или DIC, или с применяемой согласно настоящему изобретению системой сочетания Т3Р/пиридин (J.R. Dunetz, et al., Org. Lett., 2011, 13, 5048) в качестве реагентов для реакции сочетания, или через хлорид карбоновой кислоты, необязательно в присутствии основания, например, триэтиламина. Методика, разработанная для реакции сочетания с использованием Т3Р/пиридина в качестве системы реагентов, оказывается особенно полезной и позволяет образование амида без какой-либо рацемизации в хиральном центре соединения формулы (XIX) в ходе реакции или обработки. В способе предпочтение отдается использованию от 1,1 до 2,2 молярных эквивалентов Т3Р и от 0,5 до 3,5 молярных эквивалентов пиридина, при этом реакцию проводят в диапазоне температур от 0°С до 40°С. Предпочтительно использование от 1,2 до 1,8 молярных эквивалента Т3Р, особенно предпочтительно использование 1,5 молярных эквивалента Т3Р.
Предпочтительным является использование от 0,8 до 2,5 молярных эквивалентов пиридина, особенно предпочтительно использование 1,1 молярных эквивалента пиридина. Предпочтительная температура реакции составляет от 15°С до 30°С, особенно предпочтительная температура реакции составляет 22°С. Благодаря свойствам растворимости соединения формулы (XIX) тетрагидрофуран особенно полезен в качестве растворителя.
Для настоящего изобретения тщательно разработанная процедура обработки, основанная на добавлении воды и затравке, позволяет кристаллизацию соединения формулы (XIX) из обрабатываемой смеси с превосходным качеством и выходом без снижения энантиоселективности. Следовательно, необходимо избегать попадания противоионов галогенидов из промывочных растворов, таких как водный раствор хлорида натрия и водный раствор хлорида аммония. Процедура обработки проводится следующим образом. На первой стадии добавление воды производится предпочтительно при соотношении от 1:8 до 1:12 (мас/об) на основе соединения формулы (XVIII), наиболее предпочтительно при соотношении 1:10 (мас/об). Затем в смесь добавляют затравку соединения формулы (XIX) и еще воду, предпочтительно при соотношении от 1:4 до 1:8 (мас/об) на основе соединения формулы (XVIII), наиболее предпочтительно при соотношении 1:6 (мас/об). Удаление тетрагидрофуран перегонкой в вакууме приводит к легко фильтруемой суспензии, содержащей соединение формулы (XIX) с высоким выходом и превосходным качеством.
Наконец, соединение формулы (XIX) и соединение формулы (XVI-Cl)/(XVI-CF3) подвергают сочетанию в реакции N-алкилирования, опосредованной основанием. Задача этого превращения состоит в том, чтобы найти условия реакции, которые одновременно являются оптимальными с точки зрения превращения (времени и выхода), селективности N/O-алкилирования и энантиоселективности. Основание должно иметь pKa>13 в воде и предпочтительно является неионным органическим основанием. Более слабые основания приводят к отсутствию или недостаточному превращению, в то время как более сильные ионные основания также хуже с точки зрения превращения, энантиоселективности и селективности N/O-алкилирования. Наилучшие результаты получены с сильными неионными органическими основаниями, такими как основания амидин, гуанидин или фосфазен. Предпочтительными неионными органическими основаниями являются 1,8-диазабицикло[5.4.0]ундец-7-ен, N,N,N,N-тетраметилгуанидин и 2-трет-бутилимино-2-диэтиламино-1,3-диметилпергидро-1,3,2-диазафосфорин.
Особенно предпочтительным является основание N,N,N,N-тетраметилгуанидин, так как оно является недорогостоящим и опосредует реакцию с хорошими показателями превращения, а также хорошей энантиоселективностью и селективностью N/O-алкилирования. Кроме того, N,N,N,N-тетраметилгуанидин является смешиваемым с водой, что позволяет легко удалять его в ходе водной обработки. Основание используют при соотношении от 0,8 до 5 молярных эквивалента на основе соединения формулы (XVI-Cl)/(XVI-CF3), предпочтительно от 1,1 до 3 молярных эквивалента. Особенно предпочтительным является диапазон от 1,2 до 1,6 молярных эквивалента на основе соединения формулы (XVI-Cl)/(XVI-CF3).
В качестве растворителя можно использовать многие органические растворители в чистом виде или в смесях с тем или иным преимуществом, например, селективность N/O-алкилирования или энантиоселективность, или выход или время превращения. Спирты обеспечивают очень хорошую селективность N/O-алкилирования. Предпочтительно хорошие результаты дают трет-бутанол, 1-бутанол или 2-пропанол. Однако эти растворители неудовлетворительны с точки зрения растворимости и превращения. Ни одна из реакций не завершается через 50 часов. Время реакции составляет более 50 часов, и во многих случаях реакция не доходит до полного завершения. С другой стороны, полярные непротонные растворители, такие как тетрагидрофуран, N,N-диметилформамид, диоксан или ацетон, приводят к полному превращению менее чем за 3 часа, но страдают от плохой селективности предпочтительного N-алкилирования по сравнению с нежелательным O-алкилированием. Как следствие, смеси растворителей используют для сочетания хороших превращений, наблюдаемых с более полярными непротонными растворителями, с высокой энантиоселективностью и селективностью N/O-алкилирования, наблюдаемыми в спиртах. Хотя работают многие смеси вышеупомянутых растворителей, предпочтительной является комбинация ацетона и 1-бутанола или ацетона и 2-пропанола. Особое предпочтение отдается смеси ацетона и 2-пропанола. Смесь используется в диапазоне от 1:2 до 1:9 ацетона/2-пропанола, особенно предпочтительно соотношение ацетона/2-пропанола от 1:3 до 1:5.
Температурный диапазон реакции составляет от 0°С до 60°С для получения приемлемых превращений. Однако на более высоком конце этого температурного диапазона полученные энантиоселективности в целом неудовлетворительны. Реакции на нижнем конце описанного диапазона температур требуют более длительного времени превращения, что, в свою очередь, также отрицательно сказывается на получаемой энантиоселективности. Поэтому предпочтительный температурный диапазон определяется от 15°С до 25°С, а особенно предпочтительный температурный диапазон от 18°С до 23°С.
Таким образом, смесь протонного (т.е. спирта) и полярного непротонного растворителя при умеренных температурах (от 15°С до 25°С) является предпочтительной для получения хорошего общего превращения и хорошей энантиоселективности и селективности N/O-алкилирования. Наилучшие результаты достигаются при использовании смесей ацетона и спиртов при соотношении 1: 4 при температуре реакции 20°С. Соединение формулы (I)/(II) получают в аморфной форме с высокими значениями ее от 85% до 93% после фильтрации и выпаривания растворителей. Кроме того, предпочтительное N-алкилирование по сравнению с нежелательным O-алкилированием достигается при соотношении N-алкилирования к О-алкилированию от 9:1 до 10:1.
с) Соединение формулы (XIV-Cl)/(XIV-CF3) в соединение формулы (I)/(II) через хиральное разделение (описано в WO 2017/005725) по сравнению с обогащением желательного энантиомера соединения формулы (I)/(II) (настоящее изобретение)
Соединение формулы (XIV-Cl)/(XIV-CF3) в соединение формулы (I)/(II) через хиральное разделение (описано в WO 2017/005725)
Получение соединения формулы (I)/(II), описанная в WO 2017/005725, основано на разделении двух энантиомеров рацемического соединения формулы (XIV-Cl)/(XIV-CF3) с помощью хиральной сверхкритической жидкостной хроматографии (SFC). Это представляет собой очень дорогую и трудоемкую процедуру, которая не подходит для получения соединения формулы (I)/(II) в более крупных масштабах. Это особенно верно, поскольку время обработки стандартной лабораторной SFC уже очень низкое (3-4 г эутометра/день/устройство). Вдобавок к этому, половина произведенного материала является нежелательным энантиомером и не может быть использована сразу, но должна быть подвергнута условиям рацемизации и SFC-разделению снова.
Обогащение желаемого энантиомера соединения формулы (I)/(II) (настоящее изобретение)
Напротив, процедура, описанная в настоящем изобретении, показывает простой и масштабируемый способ достижения обогащения желаемого энантиомера до значений ее>99%. Энантиомерно чистое соединение формулы (I)/(II) присутствует в аморфной твердой форме, тогда как рацемический материал соединения формулы (I)/(II) (который в настоящем документе аналогичен соединению формулы (XIV-Cl)/(XIV-CF3)) является кристаллическим с гораздо меньшей растворимостью в органических растворителях. Органический растворитель представляет собой этилацетат, дихлорметан, метанол, 2-пропанол, ацетон и их смеси, очень предпочтительно этилацетат. На основе этого принципа различной растворимости желаемого энантиомерно чистого соединения формулы (I)/(II) и рацемического материала соединения формулы (I)/(II), продукт со значениями ее от 85% ее до 93% ее (как указано выше) растворяют в определенном количестве этилацетата, нагретого до возврата флегмы, и перемешивают. Предпочтение отдается соотношению соединения формулы (I)/(II) и этилацетата от 1:1 до 1:10 (мас/мас), особенно предпочтительно соотношению соединения формулы (I)/(II) и этилацетата от 1:2 до 1:5 (мас/мас). Менее растворимое кристаллическое рацемическое соединение формулы (I)/(II) образует суспензию, в то время как желаемый энантиомерно чистый аморфный материал соединения формулы (I)/(II) растворяется в органическом растворителе. Горячая фильтрация отделяет кристаллический рацемат от далее энантиобогащенного отдельного энантиомера. Через рацемат из продукта удаляют оставшийся нежелательный энантиомер, и получают значения ее>99%. Это означает, что энантиомерно чистое соединение формулы (I)/(II) (значения ее> 99% ее) получают посредством нагревания соединения формулы (I)/(II) со значениями ее от 85% ее до 93% ее до возврата флегмы в органическом растворителе, предпочтительным является этилацетат, и последующей фильтрации. Выпаривание растворителя из фильтрата дает энантиомерно чистое соединение формулы (I)/(II) (значения ее> 99% ее), которое дополнительно очищают с помощью колоночной хроматографии с нормальной фазой для отделения от других химических примесей. Другие химические примеси представляют собой побочные продукты, образующиеся в ходе реакции.
В альтернативной методике очистка сырого продукта соединения формулы (I)/(II) достигается путем кристаллизации энантиомерно обогащенного соединения формулы (I)/(II) в виде сольвата. Соответствующим растворителем для сольвата является этилацетат, изопропилацетат, тетрагидрофуран или ацетон, предпочтительно ацетон. В качестве сольвата могут быть получены кристаллические фазы энантиомерно обогащенного соединения формулы (I)/(II) и, в частности, энантиомерно обогащенного соединения формулы (II), что позволяет эффективно удалять органические побочные продукты, все еще присутствующие в неочищенном продукте соединения формулы (I)/(II). Таким образом, преимущество сольватов состоит в том, что стадию очистки можно проводить путем кристаллизации сольватов.
Кристаллы, образованные в виде сольватов, которые представляют собой в особенности соединения формулы (IIa), (IIb) и (IIc), лучше растворяются в органических растворителях, чем рацемические кристаллы соединения формулы (II) как таковые. Следовательно, кристаллы, образованные в виде сольватов, растворяют в органическом растворителе, таком как этанол, с последующим отфильтровыванием оставшихся рацемических кристаллов соединения формулы (II), что приводит к раствору, содержащему соединение формулы (II) с ее-обогащением. Соединение формулы (II) с ее значениями> 99% ее выделяют путем медленного дозирования раствора в холодную воду и последующей фильтрации. Таким образом, преимущество сольватов заключается в том, что очистку от побочных продуктов для соединения формулы (II) можно проводить через сольваты, и это можно комбинировать с последующим ее-обогащением путем удаления кристаллического рацемата.
Энантиомерно обогащенное соединение означает соединение с предпочтительно значениями ее от 85% до 93% ее, но энантиомерная чистота также может быть ниже значений ее 85% или выше значений ее 93% для этой стадии очистки.
В целом новый способ синтеза, описанный в настоящем изобретении, является более эффективным, экономичным и оптимизированным по времени для производства количеств соединения формулы (I)/(II) в килограммовом диапазоне. Самая длинная линейная последовательность представляет собой четыре стадии синтеза, а общий выход шести стадий в сумме составляет от 20% до 25%. Кроме того, для синтеза согласно настоящему изобретению требуется меньше стадий введения и удаления защитных групп, которые не вносят прямого вклада в построение соединения формулы (I)/(II).
Настоящее изобретение также охватывает способ получения 4-{[(2S)-2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутаноил]амино}-2-фторбензамида (I) или 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамида (II), отличающийся тем, что, соответственно, 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метоксипиридин-2(1Н)-он (XVI-Cl) или 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метоксипиридин-2(1Н)-он (XVI-CF3) вводят в реакцию с 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамидом (XIX) в присутствии основания в растворителе, и соединение формулы (I) или (II) затем выделяют.
Настоящее изобретение также охватывает способ получения 4-{[(2R)-2-бромбутаноил]амино}2-фторбензамида (XIX) посредством реакции (2R)-2-броммасляной кислоты (XVIII) с 4-амино-2-фторбензамидом (XIII).
Настоящее изобретение также охватывает способ получения 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метоксипиридин-2(1Н)-она (XVI-CF3) посредством реакции 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметоксипиридина (XV-CF3) с хлоридом лития и n-толуолсульфоновой кислотой в растворителе.
Настоящее изобретение также охватывает способ получения 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметоксипиридина (XV-CF3) посредством реакции (2,5-диметоксипиридин-4-ил)бороновой кислоты (IV) с 1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазолом (X-CF3) в присутствии каталитической системы на основе Pd с основанием в растворителе.
Настоящее изобретение также охватывает способ получения 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метоксипиридин-2(1Н)-она (XVI-Cl) посредством реакции 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-2,5-диметокси-пиридина (XV-Cl) с хлоридом лития и n-толуолсульфоновой кислотой в растворителе.
Настоящее изобретение также охватывает способ получения 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-2,5-диметокси-пиридина (XV-Cl) посредством реакции (2,5-диметоксипиридин-4-ил)бороновой кислоты (IV) с 1-(2-бром-4-хлорфенил)-4-хлор-1Н-1,2,3-триазолом (Х-Cl) в присутствии каталитической системы на основе Pd с основанием в растворителе.
Настоящее изобретение также охватывает способ получения 4-{[(2S)-2-{4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1(2Н)-ил}бутаноил]амино}-2-фторбензамида (I), отличающийся тем, что
i.) на первой стадии, (2,5-диметоксипиридин-4-ил)бороновую кислоту (IV) вводят в реакцию с 1-(2-бром-4-хлорфенил)-4-хлор-1Н-1,2,3-триазолом (X-Cl) в присутствии каталитической системы на основе Pd с основанием в растворителе с образованием 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-2,5-диметокси-пиридина (XV-Cl),
ii.) на второй стадии, 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-2,5-диметокси-пиридин (XV-Cl) вводят в реакцию с хлоридом лития и n-толуолсульфоновой кислотой в растворителе с образованием 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метоксипиридин-2(1Н)-она (XVI-Cl),
iii.) на третьей стадии, 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метоксипиридин-2(1Н)-он (XVI-Cl) вводят в реакцию с 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамидом (XIX) в присутствии основания в растворителе, и соединение формулы (I) затем выделяют.
Настоящее изобретение также охватывает способ получения 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамида (II), отличающийся тем, что
i.) на первой стадии, (2,5-диметоксипиридин-4-ил)бороновую кислоту (IV) вводят в реакцию с 1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазолом (X-CF3) в присутствии каталитической системы на основе Pd с основанием в растворителе с образованием 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметоксипиридина (XV-CF3),
ii.) на второй стадии, 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметоксипиридин (XV-CF3) вводят в реакцию с хлоридом лития и n-толуолсульфоновой кислотой в растворителе с образованием 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метоксипиридин-2(1Н)-она (XVI-CF3),
iii.) на третьей стадии, 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метоксипиридин-2(1Н)-он (XVI-CF3) вводят в реакцию с 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамидом (XIX) в присутствии основания в растворителе, и соединение формулы (II) затем выделяют.
Настоящее изобретение также охватывает способ получения 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамида (II), отличающийся тем, что последующее выделение осуществляют через 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамид ацетон (IIc).
Последовательности синтеза согласно настоящему изобретению:
Соединение формулы (XVII) превращают в соединение формулы (XVIII).
Соединение формулы (XVIII) подвергают реакции с соединением формулы (XIII) с получением соединения формулы (XIX).
Соединение формулы (III) превращают в соединение формулы (IV).
Соединение формулы (IV) подвергают реакции с соединением формулы (Х-Cl) с получением соединения формулы (XV-Cl).
Соединение формулы (IV) подвергают реакции с соединением формулы (X-CF3) с получением соединения формулы (XV-CF3).
Соединение формулы (XV-Cl) превращают в соединение формулы (XVI-Cl). Соединение формулы (XV-CF3) превращают в соединение формулы (XVI-CF3).
Соединение формулы (XVI-Cl) подвергают реакции с соединением формулы (XIX) с получением соединения формулы (I).
Соединение формулы (XVI-CF3) подвергают реакции с соединением формулы (XIX) с получением соединения формулы (II).
Настоящее изобретение также охватывает 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-2,5-диметокси-пиридин формулы

Настоящее изобретение также охватывает 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметокси-пиридин формулы
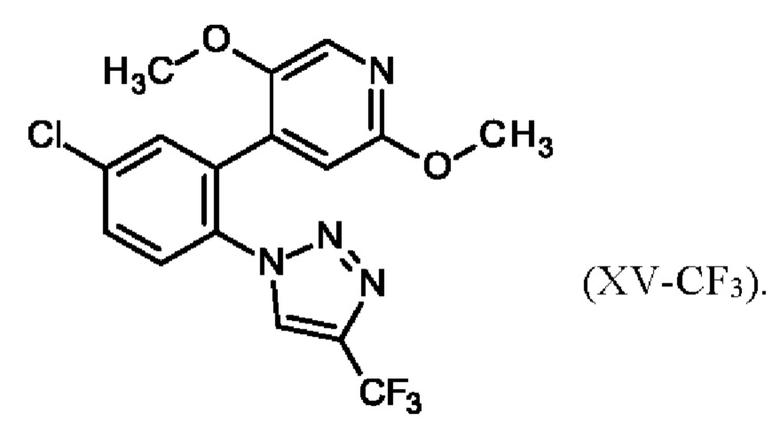
Настоящее изобретение также охватывает 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-5-метоксипиридин-2(1Н)-он формулы
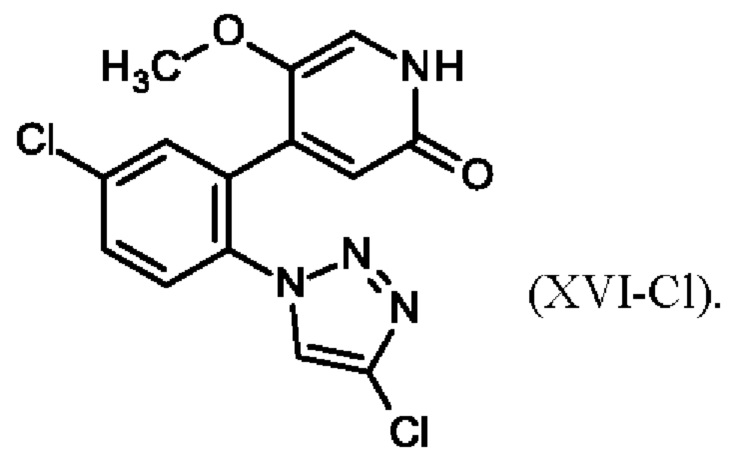
Настоящее изобретение также охватывает 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-пиридин-2(1Н)-он формулы
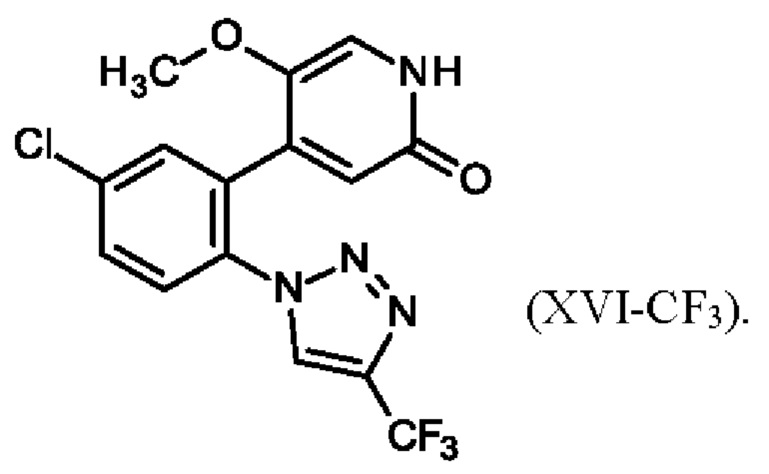
Настоящее изобретение также охватывает 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамид формулы

Настоящее изобретение также охватывает 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1H-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамид изопропилацетат формулы

Настоящее изобретение также охватывает 4-({(2S)-2-[4-{5-клор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил)-5-метокси-2-оксопиридин-1(2Н)-ил]6утаноил)-амино)-2-фтор6ензамид тетрагидрофуран формулы
Настоящее изобретение также охватывает 4-(((2S)-2-[4-(5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил)-5-метокси-2-оксопиридин-1(2Н)-ил]6утаноил)-амино)-2-фтор6ензамид ацетон формулы
Примеры
Аббревиатуры и Акронимы
Pd(amphos)2Cl2 - бис [(дициклогексил)(4-диметиламинофенил)фосфин]палладия(II)-хлорид
масс % - процент по массе
% по площади - процент по площади
% от теор. - Процент от теоретического выхода
скорр. - скорректированный
нескор. - нескорректированный
мин - минут
ч - часы
мг - миллиграмм
г - грамм
кг - килограмм
л - литр
мл - миллилитр
ESI - электронная распылительная ионизация
GC - газовая хроматография
ВЭЖХ - жидкостная хроматография высокого давления (высокоэффективная)
SFC - сверхкритическая жидкостная хроматография
Br - широкий
s - синглет
d - дублет
t - триплет
spt - сеплет
quin - квинтет
ppm - частей на миллион
m - мультиплет
Гц - Герц
М - молярный
EDC - 1-этил-3-(3-диметиламинопропил)карбодиимид
DIC - N,N'-диизопропилкарбодиимид
T3P - пропилфосфоновый ангидрид
THF - тетрагидрофуран
(м/м) - масса/масса
(мас/об) - масса/объем
XRPD - Рентгеновская порошковая дефрактометрия
DSC - Дифференциальная сканирующая калориметрия.
Термин "соединение формулы (XVI-Cl)/(XVI-CF3)" означает, что соединение формулы (XVI-Cl) или соединение формулы (XVI-CF3) применяют в соответствии с путем синтеза в соединение формулы (I), которое содержит заместитель хлор, или соединение формулы (II), которое содержит заместитель трифторметил. То же самое также относится к другим терминам, которые означают соединение формулы (X-Cl)/(X-CF3) и (XI-Cl)/(XI-CF3) и (XII-Cl)/(XII-CF3) и (XIV-Cl)/(XIV-CF3) и (XV-Cl)/(XV-CF3) и (XVI-Cl)/(XVI-CF3) и (I)/(II), а также если применяются химические названия соединений.
Если термин "соединение формулы (....)" применяют, этот термин может быть заменен названием по ИЮПАК соединения формулы (....). Названия по ИЮПАК соединений приведены выше.
Рацемическое вещество соединения формулы (I)/(II) в настоящем документе представляет собой то же самое, что соединение формулы (XIV-Cl)/(XIV-CF3).
В контексте настоящего изобретения термин «энантиомерно чистый» следует понимать как означающий, что рассматриваемое соединение в отношении абсолютной конфигурации хирального центра присутствует в энантиомерном избытке более 95%, предпочтительно более 97%. Энантиомерный избыток, ее, вычисляют в контексте настоящего изобретения посредством оценки соответствующей хроматограммы ВЭЖХ на хиральной фазе с использованием формулы ниже:
ее = [EA (% по площади) - EB (% по площади)] × 100% / [EA (% по площади) + EB (% по площади)]
(EA: основной энантиомер, EB: второстепенный энантиомер)
Рабочие примеры
Синтез (2,5 диметоксипиридин-4-ил)бороновой кислоты (IV)
66,9 г (661,1 ммоль) N,N-диизопропиламина растворяли в 380 г THF и охлаждали до температуры -60°С. 395,2 мл (632,4 ммоль) н-бутиллития (1,6 М в гексане) добавляли в течение 45 минут, поддерживая температуру ниже -50°С. Смесь перемешивали при -60°С еще 15 мин. Затем в течение 45 минут добавляли 80 г (574,9 ммоль) 2,5-диметоксипиридина, поддерживая температуру от -50 до -60 С. После завершения добавления капельную воронку промывали еще 10 мл THF. Реакционную смесь перемешивали при -60°С в течение 2 ч, перед добавлением 118.9 г (632.4 ммоль) триизопропилбората в течение 30 мин. Капельную воронку снова промывали 10 мл THF. Реакционную смесь нагревали до 20°С и перемешивали в течение 30 мин.
Затем добавляли смесь уксусной кислоты (106 г) и воды (602 г) в течение 15 минут и смесь перемешивали еще 30 минут. Затем органические растворители (650 г) выпаривали в вакууме (300 мбар) при максимальной температуре 70°С, полученную суспензию охлаждали до 20°С и фильтровали. Осадок продукта промывали холодной водой (трижды по 100 мл) и сушили при 40°С в течение примерно 16 часов при пониженном давлении в сушильном шкафу. Выход: 78,6 г (75% от теоретического выхода).
MS (ESI+): m/z=184.1 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ [ppm] = 8.15 (br s, 2H), 7.80 (s, 1H), 6.76 (s, 1H), 3.78 (d, 6H).
Синтез 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-2,5-диметокси-пиридина (XV-Cl)
5 г (17,1 ммоль) 1-(2-бром-4-хлорфенил)-4-хлор-1Н-1,2,3-триазола (Х-Cl) и 121 мг (0,17 ммоль) Pd(amphos)2Cl2 суспендировали в 40,3 г трет-амилового спирта. Реакционную смесь нагревали до 65°С, и смесь 5,4 г (51,2 ммоль) карбоната натрия и 3,8 г (20,5 ммоль) (2,5-диметоксипиридин-4-ил)бороновой кислоты (IV) в воде (35 мл) добавляли за 1 ч. Реакционную смесь перемешивали при 65°С еще 5 ч до наблюдения полного израсходования триазола (Х-Cl). Затем добавляли 0,8 г (5,1 ммоль) N-ацетилцистеина и перемешивали еще 30 мин, после чего добавляли еще 8 мл воды. Смесь охлаждали до 8°С в течение 40 минут, и полученную суспензию фильтровали. Осадок на фильтре промывали холодным этанолом (два раза по 4 мл) и водой (два раза по 5 мл) перед сушкой при 50°С в течение около 15 часов при пониженном давлении в сушильном шкафу. Выход: 4,46 г (74% от теоретического выхода).
MS (ESI+): m/z=351.0 [М+Н]+;
1Н-ЯМР (400МГц, DMSO-d6): δ [ppm] = 8.56 (s, 1Н), 7.68-7.79 (m, 4H), 6.79 (s, 1H), 3.76-3.85 (s, 3H), 3.44 (s, 3H).
Синтез 4-{5-хлор-2-[4-(трифторметил)-1H-1,2,3-триазол-1-ил]фенил}-2,5-диметокси-пиридина (XV-CF3)
5 г (15.3 ммоль) 1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазола (X-CF3) и 108 мг (0.15 ммоль) Pd(amphos)2Cl2 суспендировали в 40.3 г трет-амилового спирта. Реакционную смесь нагревали до 85°С, и смесь 4.8 г (45.9 ммоль) карбоната натрия, и 3.6 г (19.9 ммоль) (2,5-диметоксипиридин-4-ил)бороновой кислоты (IV) в воде (35 мл) добавляли за 3 ч. Реакционную смесь перемешивали при 85°С в течение еще 1 ч до полного израсходования триазола (X-CF3). Затем 0.8 г (5.1 ммоль) N-ацетил-цистеина добавляли и перемешивали еще 30 мин, прежде чем 40 мл трет-амилового спирта отгоняли, и добавляли 20 мл этанола. Смесь охлаждали до 2°С в течение 120 минут и перемешивали еще 1 час. Затем полученную суспензию фильтровали. Осадок на фильтре промывали холодным этанолом (трижды по 3 мл) и водой (два раза по 5 мл) перед сушкой при 50°С в течение примерно 15 часов при пониженном давлении в сушильном шкафу. Выход: 3,62 г (62% от теоретического выхода).
MS (ESI+): m/z=385.1 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ [ppm] = 9.14 (s, 1Н), 7.82 (s, 2H), 7.73 (s, 2H), 6.84 (s, 1H), 3.81 (s, 3H), 3.38 (s, 3H).
Синтез 4-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]-5-метоксипиридин-2(1Н)-она (XVI-CI)
9,0 г (25,6 ммоль) 4-[5-хлор-2-(4-хлор-1Н-1,2,3-триазол-1-ил)фенил]-2,5-диметоксипиридина (XV-Cl), 5,4 г (128,1 ммоль) хлорида лития и 1,8 г (46,4 ммоль) п-толуолсульфоновой кислоты растворяли в 60 мл 2-пропанола и перемешивали при температуре возврата флегмы в течение около 16 ч до полного израсходования исходного вещества. Затем добавляли 120 мл воды за 60 минут, и смесь охлаждали до 10°С в течение еще 60 минут. Суспензию фильтровали, и осадок на фильтре промывали водой (три раза по 20 мл). Затем его сушили при 50°С в течение около 15 часов при пониженном давлении в сушильном шкафу. Выход: 7,46 г (86% от теоретического выхода).
MS (ESI+): m/z=337.0 [М+Н]+;
1Н-ЯМР (500 МГц, DMSO-d6): δ [ppm] = 11.24 (br s, 1H), 8.62 (s, 1H), 7.66-7.78 (m, 3H), 6.99 (s, 1H), 6.36 (s, 1H), 3.29 (s, 3H).
Синтез 4-{5-хлор-2-[4-(трифторметил)-1H-1,2,3-триазол-1-ил]фенил}-5-метокси-пиридин-2(1Н)-она (XVI-CF3)
7.0 г (18.2 ммоль) 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметоксипиридина (XV-CF3), 3.9 г (91.0 ммоль) хлорида лития и 6.3 г (32.9 ммоль) п-толуолсульфоновой кислоты растворяли в 60 мл 2-пропанола и перемешивали при температуре возврата флегмы в течение около 16 ч до полного израсходования исходного вещества. Затем добавляли 120 мл воды за 60 минут, и смесь охлаждали до 10°С в течение еще 60 минут. Суспензию фильтровали, и осадок на фильтре промывали водой (три раза по 20 мл). Затем его сушили при 50°С в течение около 15 часов при пониженном давлении в сушильном шкафу. Выход: 6.58 г (97% от теоретического выхода).
MS (ESI+): m/z=371.0 [М+Н]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ [ppm] = 11.21 (br s, 1H), 9.18 (s, 1H), 7.81 (s, 2H), 7.72 (s, 1H), 6.95 (s, 1H), 6.41 (s, 1H), 3.23 (s, 3H).
Синтез (2R)-2-броммасляной кислоты (XVIII)
В перемешиваемом сосуде 150 г (1454,6 ммоль) (2R)-2-аминомасляной кислоты (XVII) и 605,8 г (5091,1 ммоль) бромида калия растворяли в 809 г 2,5 М водной серной кислоты. Смесь охлаждали до -10°С и добавляли водный раствор 150,4 г (2181,9 ммоль) нитрита натрия в 150 мл воды в течение 30 минут. Затем реакционную смесь перемешивали при 0°С в течение 18 ч.
После повышения температуры реакции до 20°С реакционную смесь экстрагировали этилацетатом (трижды по 500 мл), и органический слой концентрировали в вакууме с получением указанного в названии соединения. Выход: 193,9 г (80% от теоретического выхода).
MS (ESI+): m/z=166.0 [М+Н]+;
1Н-ЯМР (500МГц, DMSO-d6): δ [ppm] = 4.28 (dd, 1Н), 1.98-2.07 (m, 1H), 1.83-1.94 (m, 1H), 0.97 (t, 3H).
Синтез 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамида (XIX)
5.0 г (32.4 ммоль) 4-амино-2-фторбензамида (XIII) суспендировали в THF (50 мл) и охлаждали до 0°С. Затем 5.9 г (35.6 ммоль) (2И)-2-броммасляной кислоты (XVIII) и 2.8 г (35.6 ммоль) пиридина добавили, прежде чем добавили 31.0 г (48.6 ммоль) 50%-ого раствора Т3Р в этилацетате за 20 мин. Смесь перемешивали в течение 10 минут, а затем давали нагреться до 22°С. Смесь перемешивали еще 3 ч до полного израсходования исходных веществ. Затем за 45 минут добавляли 60 г воды и добавляли затравочные кристаллы. Дозирование прекращали на 30 мин, а затем добавляли еще 40 г воды через 15 мин. Смесь перегоняли для удаления растворителя до достижения внутренней температуры 40°С при вакууме 300 мбар. Затем ее охлаждали до комнатной температуры и фильтровали. Осадок на фильтре промывали холодной водой (10 мл) и сушили при 50°С в течение примерно 16 часов при пониженном давлении в сушильном шкафу. Выход: 8,4 г (85% от теоретического выхода).
MS (ESI+): m/z=303.0 [М+H]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ [ppm] = 10.70 (s, 1Н), 7.70 (t, 1H), 7.62-7.67 (m, 1H), 7.55 (s, 1H), 7.52 (br s, 1H), 7.35 (dd, 1H), 4.46 (t, 1H), 2.10 (spt, 1H), 1.95 (dquin, 1H), 0.96 (t, 3H).
Синтез 4-{[(2S)-2-{4-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]-5-метокси-2-оксопиридин-1-(2H)-ил}бутаноил]амино}-2-фторбензамида (I)
10.0 г (30 ммоль) 4-[5-хлор-2-(4-хлор-2,3-дигидро-1Н-1,2,3-триазол-1-ил)фенил]-5-метоксипиридин-2(1Н)-она (XVI-Cl) растворили в 2-пропанола (85 мл) и ацетоне (21 мл) при 22°С, и 10.3 г (90 ммоль) N,N,N,N-тетраметилгуанидина добавляли. После перемешивания в течение 15 мин при 22°С 9.89 г (33 ммоль) 4-{[(2R-2-бромбутаноил]амино}-2-фторбензамида (XIX) добавляли, и смесь перемешивали в течение 6 ч. Затем смесь фильтровали, и добавляли этилацетат (125 мл). Органический слой промывали насыщенным водным раствором хлорида аммония (125 мл) и насыщенным водным раствором хлорида натрия (125 мл). Затем органический слой концентрировали в вакууме. Остаток растворяли в этилацетате (140 мл), перемешивали в течение 30 мин и фильтровали. Фильтрат концентрировали в вакууме и очищали с помощью колоночной хроматографии (силикагель, градиент гексан/ацетон). Выход: 12,5 (75% от теоретического выхода).
MS (ESI+): m/z=558.1 [M]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ [ppm] = 10.78 (s, 1H), 8.62 (s, 1H), 7.62-7.81 (m, 5H), 7.53 (br d, 2H), 7.39 (dd, 1H), 7.18 (s, 1H), 6.48 (s, 1H), 5.54 (dd, 1H), 3.32 (s, 3H), 2.02-2.19 (m, 2H), 0.82 (t, 3H).
Синтез 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1H-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2- фторбензамида (II)
10.0 г (27 ммоль) 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-пиридин-2(1Н)-она (XVI-CF3) растворяли в 2-пропанола (85 мл) и ацетона (21 мл) при 22°С, и 9.2 г (81 ммоль) N,N,N,N-тетраметилгуанидина добавляли. После перемешивания в течение 15 мин при 22°С 9.0 г (30 ммоль) 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамида (XIX) добавляли, и смесь перемешивали в течение 16 ч. Реакционную смесь фильтровали и добавляли этилацетат (125 мл). Органический слой промывали насыщенным водным раствором хлорида аммония (125 мл) и насыщенным водным раствором хлорида натрия. Затем органический слой концентрировали в вакууме. Остаток растворяли в этилацетате (140 мл), перемешивали в течение 30 мин и фильтровали. Фильтрат концентрировали в вакууме и очищали с помощью колоночной хроматографии (силикагель, градиент гексан/ацетон). Выход: 11.1 г (70% от теоретического выхода).
MS (ESI+): m/z=593.1 [М]+;
1Н-ЯМР (400 МГц, DMSO-d6): δ [ppm] = 10.77 (br s, 1H), 9.13 (s, 1H), 7.58-7.95 (m, 5H), 7.53 (br d, 2H), 7.37 (dd, 1H), 7.14 (s, 1H), 6.54 (s, 1H), 5.53 (br dd, 1H), 3.26 (s, 3H), 2.02-2.22 (m, 2H), 0.79 (t, 3H).
Альтернативный способ:
25.0 г (67 ммоль) 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-пиридин-2(1Н)-она (XVI-CF3) растворяли в 2-пропаноле (125 мл), и добавляли ацетон (31.4 мл) при 22°С и 11.6 г (101 ммоль) N,N,N,N-тетраметилгуанидина. После перемешивания в течение 15 мин при 22°С 22.5 г (74 ммоль) 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамида (XIX) добавляли, и смесь перемешивали в течение 16 ч. Затем реакционную смесь медленно добавляли в ледяную (0°С) воду (661 мл). Неочищенный продукт выпадал в осадок, и его фильтровали. Неочищенный продукт затем суспендировали в ацетоне (125 мл) и перемешивали в течение 30 мин. Затем воду (98.5g) добавляли через 4 ч, в смесь добавляли затравку и перемешивали еще в течение 18 ч. Полученный сольват ацетона фильтровали, сушили и повторно растворяли в этаноле (108 мл) при 22°С. Смесь перемешивали в течение 30 минут, фильтровали, и фильтрат медленно добавляли в холодную воду (5°С, 427 г). Полученную суспензию фильтровали, осадок на фильтре промывали и сушили при 60°С в течение 16 часов при пониженном давлении в сушильном шкафу. Выход: 24,4 г (61% от теоретического выхода).
Выделение 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1-(2Н)-ил]бутаноил}амино)-2-фторбензамид изопропилацетата (IIa)
46.9 мг аморфного твердого вещества 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2-фторбензамида (II) взвешивали в стеклянном сосуде, и 100 мкл изопропилацетата добавляли. Сосуд закрывали, и содержимое перемешивали с помощью магнитной мешалки при 25°С. В ходе около 1 недели перемешивания происходило образование кристаллических частиц. Полученную суспензию сушили на воздухе всю ночь, и полученное твердое вещество применяли для последующих экспериментов.
Затем, 198.6 мг аморфного твердого вещества 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1H-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2-фторбензамида (II) взвешивали в стеклянном сосуде, и 600 мкл изопропилацетата добавляли. Сосуд закрывали, и содержимое перемешивали с помощью магнитной мешалки при 25°С в течение 30 минут. К полученному раствору добавляли небольшое количество (около 5 мг) ранее выделенного твердого вещества (как описано в абзаце выше) добавляли в качестве затравки. Содержимое далее перемешивали при 25°С, и завершение кристаллизации наблюдали в течение минуты. Полученную суспензию сушили на глинистой поверхности всю ночь. Фиг. 1 показывает XRPD полученного твердого вещества соединения формулы (IIa), Фиг. 4 показывает DSC, и Фиг. 7 показывает изображение микроскопа.
Выделение 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1-(2Н)-ил]бутаноил}амино)-2-фторбензамид-тетрагидрофурана (IIb)
53.9 мг аморфного твердого вещества 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2-фторбензамида (II) взвешивали в стеклянном сосуде, и 50 мкл THF добавляли. Сосуд закрывали, и содержимое перемешивали с помощью магнитной мешалки при 25°С. В течение около 1 недели перемешивания происходило образование кристаллических частиц. Полученную суспензию сушили на воздухе всю ночь, и полученное твердое вещество применяли для последующих экспериментов.
Затем, 198.5 мг аморфного твердого вещества 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2-фторбензамида (II) взвешивали в стеклянном сосуде, и 600 мкл THF добавляли. Сосуд закрывали, и содержимое перемешивали с помощью магнитной мешалки при 25°С в течение 30 минут. К полученному раствору добавляли небольшое количество (около 5 мг) ранее выделенного твердого вещества (как описано в абзаце выше) добавляли в качестве затравки. Содержимое далее перемешивали при 25°С, и завершение кристаллизации наблюдали в течение 30 минут. Полученную суспензию сушили на глинистой поверхности всю ночь. Фиг. 2 показывает XRPD полученного твердого вещества соединения формулы (IIb), Фиг. 5 показывает DSC, и Фиг. 8 показывает изображение микроскопа.
Выделение 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1-(2Н)-ил]бутаноил}амино)-2-фторбензамид ацетона (IIc)
50.5 мг аморфного твердого вещества 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2-фторбензамида (II) взвешивали в стеклянном сосуде, и 50 мкл ацетона добавляли. Сосуд закрывали, и содержимое перемешивали с помощью магнитной мешалки при 25°С. В течение около 1 недели перемешивания происходило образование кристаллических частиц. Полученную суспензию сушили на воздухе всю ночь, и полученное твердое вещество применяли для последующих экспериментов.
Затем, 203.9 мг аморфного твердого вещества 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1H-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}амино)-2-фторбензамида (II) взвешивали в стеклянном сосуде и 600 мкл ацетона добавляли. Сосуд закрывали, и содержимое перемешивали с помощью магнитной мешалки при 25°С в течение 30 минут. К полученному раствору добавляли небольшое количество (около 5 мг) ранее выделенного твердого вещества (как описано в абзаце выше) в качестве затравки. Содержимое далее перемешивали при 25°С, и завершение кристаллизации наблюдали в течение минуты. Полученную суспензию сушили на глинистой поверхности всю ночь. Фиг. 3 показывает XRPD полученного твердого вещества соединения формулы (IIc), Фиг. 6 показывает DSC, и Фиг. 9 показывает изображение микроскопа.
Рентгеновская порошковая дефрактометрия
Данные рентгеновской порошковой дефрактометрии (XRPD) собирали на дефрактометре Bruker D2 PHASER с детектором LynxEye с применением Cu Kα1,2 излучения (1.5418  ). Все образцы измеряли при температуре окружающей среды. Данные были собраны в горизонтальной геометрии Брэгга-Брентано (θ / 2θ) между 3.00149 и 40.0046° (2θ) при шаге 0.0264119° при 0.5 с шаг-1. Рентгеновская трубка работала при 30 кВ и 10 мА.
). Все образцы измеряли при температуре окружающей среды. Данные были собраны в горизонтальной геометрии Брэгга-Брентано (θ / 2θ) между 3.00149 и 40.0046° (2θ) при шаге 0.0264119° при 0.5 с шаг-1. Рентгеновская трубка работала при 30 кВ и 10 мА.
Дифференциальная сканирующая калориметрия (DSC)
Дифференциальную сканирующую калориметрию (DSC) проводили на Mettler Toledo DSC 2, калиброванном по индиевому стандарту. Ячейку калориметра продували азотом со скоростью 100 мл/мин. Примерно 5-10 мг каждого образца при измерении в алюминиевом тигле. Температурная программа была установлена в диапазоне 25-260°С (для сольватов изопропилацетата и тетрагидрофурана) или в диапазоне 25-250°С (для сольвата ацетона) при скорости нагрева 5°С мин-1. Данные обрабатывали с использованием системы Mettler Toledo Star System.
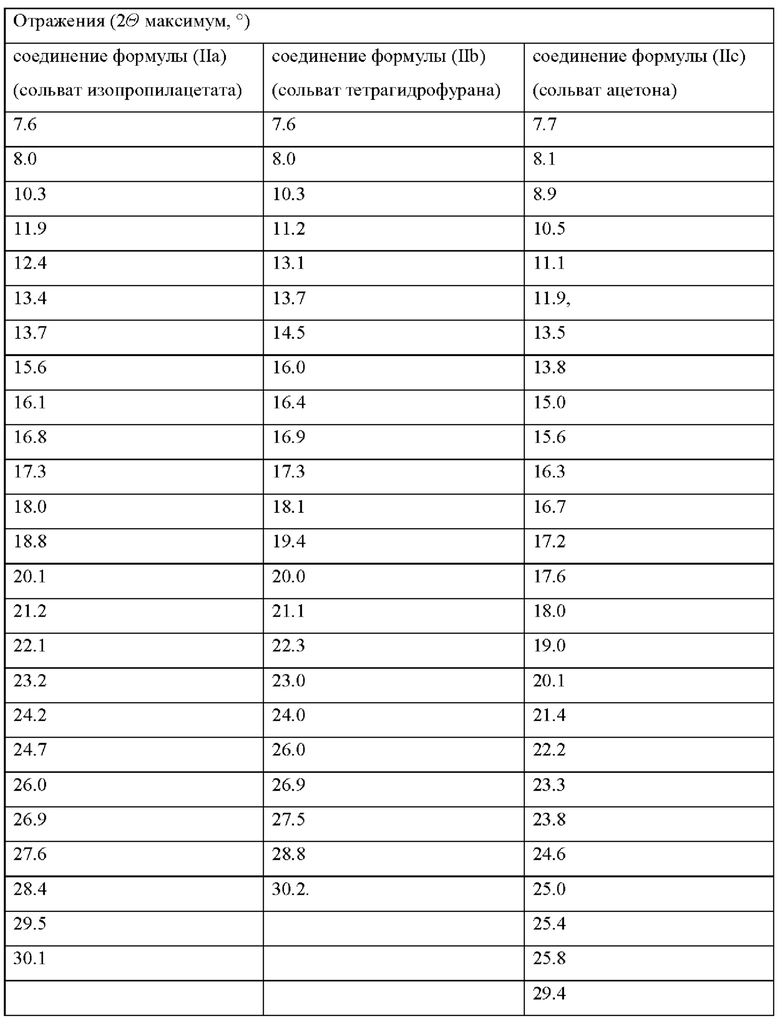
Данные рентгеновской порошковой дефрактометрии (XRPD) соединений формулы (IIa), (IIb) и (IIc).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2-[(3R)-3-МЕТИЛМОРФОЛИН-4-ИЛ]-4-(1-МЕТИЛ-1H-ПИРАЗОЛ-5-ИЛ)-8-(1H-ПИРАЗОЛ-5-ИЛ)-1,7-НАФТИРИДИНА | 2019 |
|
RU2802512C2 |
| СПОСОБ ПОЛУЧЕНИЯ (4S)-4-(4-ЦИАНО-2-МЕТОКСИФЕНИЛ)-5-ЭТОКСИ-2,8-ДИМЕТИЛ-1-4-ДИГИДРО-1,6-НАФТИРИДИН-3-КАРБОКСАМИДА И ЕГО ОЧИСТКИ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКОГО АКТИВНОГО ИНГРЕДИЕНТА | 2015 |
|
RU2729998C2 |
| ПРОИЗВОДНЫЕ [(1,5-ДИФЕНИЛ-1H-1,2,4-ТРИАЗОЛ-3-ИЛ)ОКСИ]УКСУСНОЙ КИСЛОТЫ И ИХ СОЛИ, СОДЕРЖАЩИЕ ИХ СРЕДСТВА ДЛЯ ЗАЩИТЫ ПОЛЕЗНЫХ ИЛИ КУЛЬТУРНЫХ РАСТЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЗАЩИТНЫХ СРЕДСТВ | 2020 |
|
RU2829883C1 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗОТИОФЕН-2-ИЛБОРОНАТА | 2018 |
|
RU2790014C2 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ, ТИОФЕНКАРБОНОВЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ | 2020 |
|
RU2837147C1 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И ИХ ПРОИЗВОДНЫЕ | 2020 |
|
RU2834236C2 |
| ЗАМЕЩЕННОЕ ОКСОПИРИДИНОВОЕ ПРОИЗВОДНОЕ | 2019 |
|
RU2792645C2 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И АНАЛОГИ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2019 |
|
RU2797513C2 |
| НОВЫЕ ГЕТЕРОАРИЛТРИАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПЕСТИЦИДОВ | 2020 |
|
RU2824488C2 |
| ТИЕНИЛОКСАЗОЛОНЫ И АНАЛОГИ | 2020 |
|
RU2831013C1 |




Изобретение относится к способу получения 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамида (II), который заключается в том, что 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метоксипиридин-2(1Н)-он (XVI-CF3) вводят в реакцию с 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамидом (XIX) в присутствии основания в растворителе, и с последующим выделением соединения формулы (II). Изобретение также относится к промежуточным соединениям формул (XV-CF3), (XVI-CF3), (XIX), используемым в указанном способе. Технический результат – разработан новый способ получения 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамида с высоким выходом. 4 н. и 9 з.п. ф-лы, 9 ил.
 ,
,  ,
,
1. Способ получения 4-({(2S)-2-[4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-2-оксопиридин-1(2Н)-ил]бутаноил}-амино)-2-фторбензамида (II), отличающийся тем, что 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метоксипиридин-2(1Н)-он (XVI-CF3) вводят в реакцию с 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамидом (XIX) в присутствии основания в растворителе, и соединение формулы (II) затем выделяют.
2. Способ по п. 1, отличающийся тем, что реакцию проводят с применением N,N,N,N-тетраметилгуанидина в качестве основания.
3. Способ по п. 1, отличающийся тем, что реакцию проводят с применением смеси протонного и полярного непротонного растворителя.
4. Способ по п. 1, отличающийся тем, что реакцию проводят при температуре от 15 до 25°С.
5. Способ по п. 1, отличающийся тем, что соединение формулы (II) затем выделяют в энантиомерно чистой форме посредством нагревания соединения формулы (II) со значениями ее от 85%ее до 93%ее до возврата флегмы в органическом растворителе и последующей фильтрации после выпаривания органического растворителя.
6. Способ по п. 1, отличающийся тем, что 4-{[(2R)-2-бромбутаноил]амино}-2-фторбензамид (XIX) получают посредством реакции (2R)-2-броммасляной кислоты (XVIII) с 4-амино-2-фторбензамидом (XIII).
7. Способ по п. 1, отличающийся тем, что 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метоксипиридин-2(1Н)-он (XVI-CF3) получают посредством реакции 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметоксипиридина (XV-CF3) с хлоридом лития и п-толуолсульфоновой кислотой в растворителе.
8. Способ по п. 7, отличающийся тем, что 4-{5-хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметоксипиридин (XV-CF3) получают посредством реакции (2,5-диметоксипиридин-4-ил)бороновой кислоты (IV) с 1-(2-бром-4-хлорфенил)-4-(трифторметил)-1Н-1,2,3-триазолом (X-CF3) в присутствии каталитической системы на основе Pd с основанием в растворителе.
9. Способ по п. 8, отличающийся тем, что реакцию проводят с применением Pd(Amphos)2Cl2 в качестве каталитической системы на основе Pd.
10. Способ по п. 7 или 8, отличающийся тем, что реакцию проводят с применением спирта в качестве растворителя.
11. 4-{5-Хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-2,5-диметокси-пиридин, имеющий формулу
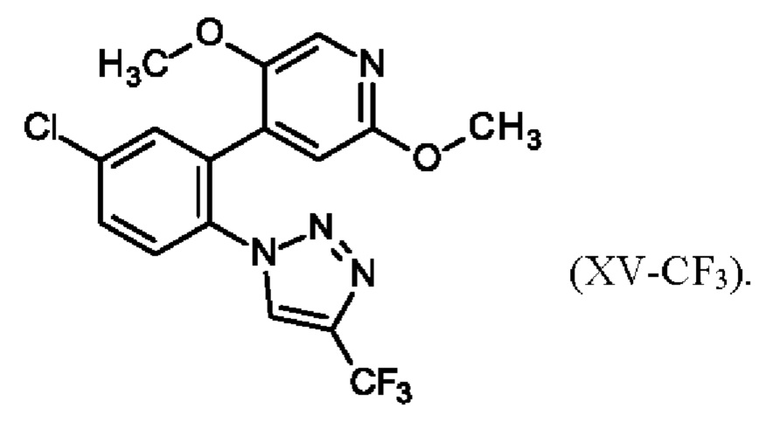
12. 4-{5-Хлор-2-[4-(трифторметил)-1Н-1,2,3-триазол-1-ил]фенил}-5-метокси-пиридин-2(1Н)-он, имеющий формулу

13. 4-([(2R)-2-Бромбутаноил]амино)-2-фторбензамид, имеющий формулу

| WO 2017005725 A1, 12.01.2017 | |||
| WO 2014154794 A1, 02.10.2014 | |||
| WO 2016046159 A1, 31.03.2016 | |||
| US 20160052884 A1, 25.02.2016 | |||
| WO 2014160592 A2, 02.10.2014 | |||
| Лента для записи со слоем нанесенной на ее рабочей поверхности копоти | 1930 |
|
SU28034A1 |
